Embed Size (px)
Citation preview

Receptor Tyrosine Kinase c-Kit Signalling in
Hematopoietic Progenitor Cells
Charlotte Edling
Department of Medical Biosciences, Pathology, Umeå University, Sweden
Umeå 2006

Umeå University Medical Dissertations
New Series no. 1058 ISSN: 0346-6612 ISBN: 91-7264-180-0
Edited by the Dean of the Faculty of Medicine
Copyright © 2006 by Charlotte Edling
Printed in Sweden by Solfjädern Offset AB, Umeå 2006

RTK C-KIT SIGNALLING IN HEMATOPOIETIC PROGENITOR CELLS _______________________________________________________________________
ABSTRACT
The receptor tyrosine kinase (RTK) c-Kit is expressed in hematopoietic stem and progenitor cells,
mast cells and in several non-hematopoietic tissues. In the hematopoietic system c-Kit and its
ligand Steel Factor (SF, aka Stem Cell Factor) are critical for proliferation, survival and
differentiation. Mutations in either receptor or ligand lead to lethal anaemia, hematopoietic stem
cell defects, mast cell deficiency and a series of non-hematological defects.
The aims of the studies included in this thesis are to describe the signalling pathways
downstream c-Kit in hematopoietic stem/progenitor cells and to further analyse the role of c-Kit
signalling in fundamental biological functions.
To study c-Kit signalling in the hematopoietic system we have employed hematopoietic
stem cell-like cell lines which share many properties with primary hematopoietic stem cells in
vitro and in vivo, including surface markers, multipotentiality, capacity for self-renewal and long
term repopulation. In paper I we demonstrate that upon SF activation the RTK c-Kit is
autophosphorylated and downstream signalling mediators are transiently activated. Surprisingly
we find that the c-Kit mediated activation of the MAPK pathway is dependent on the activation
of phosphoinositide 3-kinase (PI3K) in hematopoietic progenitor cells and that differentiation of
these progenitors to mast cells results in a signalling switch where Raf activation changes from
PI3K dependent to PI3K independent. We here establish that PI3K activity is required for
viability and proliferation of hematopoietic progenitor cells. In paper II we studied the
conventional protein kinase C (cPKC) involvement in c-Kit signalling. We observe that the
cPKCs can phosphorylate c-Kit on serine 746 and that this phosphorylation negatively regulates
the activation of the receptor. We demonstrate that inhibition of this negative phosphorylation
results in dramatically increased protein kinase B (PKB) activation and as a consequence
inhibition of cPKCs rescues cells from starvation induced apoptosis. Moreover we exhibit that the
cPKCs are necessary for full activation of extracellular signal-regulated kinase (Erk) and that
impaired PKC activity leads to hampered proliferation. In paper III we demonstrate that in
addition to the cPKCs also the novel PKCδ is required for Erk activation and proliferation.
Furthermore we present results indicating that PKCδ negatively regulates differentiation of bone
marrow.
In conclusion, with the studies in this thesis we display details in the signalling pathways
induced upon RTK c-Kit activation and we demonstrate that c-Kit has significant effects on
hematopoietic cell-physiology.
1

CHARLOTTE EDLING 2006 _______________________________________________________________________
TABLE OF CONTENTS
ABSTRACT ..................................................................................................................... 1 PAPERS IN THIS THESIS............................................................................................... 4 ABBREVIATIONS ........................................................................................................... 5 INTRODUCTION.............................................................................................................. 7
Phosphorylation mediated signalling ....................................................................................7 Protein phosphorylation .................................................................................................................... 7 Receptor Tyrosine Kinases................................................................................................................ 8 The Src homology 2 domain.............................................................................................................. 8 Phosphatases...................................................................................................................................... 9 Endocytosis......................................................................................................................................... 9 Regulation of signal transduction..................................................................................................... 9 Protein kinase inhibitors .................................................................................................................. 10
Receptor tyrosine kinase c-Kit .............................................................................................10 Identification of receptor and ligand............................................................................................... 10 Expression and Mutations ............................................................................................................... 11 Imatinib mesylate.............................................................................................................................. 11 SF, the c-Kit ligand ........................................................................................................................... 12 C-Kit structure ................................................................................................................................... 12 Isoforms and splice variants ........................................................................................................... 13 Activation of c-Kit ............................................................................................................................. 13 Protein binding sites on c-Kit .......................................................................................................... 14
APS and Cbl .................................................................................................................................. 14 Src .................................................................................................................................................. 15 Shp1/2 ............................................................................................................................................ 15 Grb2/7 and Shc ............................................................................................................................. 15 P85, PI3K regulatory subunit....................................................................................................... 15 PLCγ ............................................................................................................................................... 16
Signalling mediators downstream of c-Kit ..........................................................................16 Ras and the Raf/Mek/Erk cascade................................................................................................... 16
Ras activation ............................................................................................................................... 16 Raf activation and phosphorylation sites .................................................................................. 17 Mek and Erk activation................................................................................................................. 18 Regulation of the Raf/Mek/Erk cascade ..................................................................................... 19
The PI3K pathway ............................................................................................................................. 19 The lipid kinase PI3K.................................................................................................................... 19 PKB activation .............................................................................................................................. 20 PKB substrates ............................................................................................................................. 20 Regulation of the PI3K pathway.................................................................................................. 21
The Protein Kinase C family ............................................................................................................ 22 The hematopoietic system ....................................................................................................23
The hematopoietic stem cell............................................................................................................ 23 Defining the Hematopoietic stem cell ........................................................................................ 23 Regulation of HSC self-renewal .................................................................................................. 24
The hematopoietic hierarchy ........................................................................................................... 25 The mast cell lineage ................................................................................................................... 25 The roles of c-Kit and SF in the hematopoietic system ........................................................... 27
Model systems for hematopoietic cells.......................................................................................... 27 The Hematopoietic progenitor cell lines .................................................................................... 28
AIMS .............................................................................................................................. 29
2

RTK C-KIT SIGNALLING IN HEMATOPOIETIC PROGENITOR CELLS _______________________________________________________________________
RESULTS AND DISCUSSION ...................................................................................... 30
Paper I .....................................................................................................................................30 Erk activation is PI3K dependent in progenitor cells but not in differentiated cells ................. 30
Crosstalks reported in the literature .......................................................................................... 32 Erk is required for proliferation of HPCs........................................................................................ 33 Activation of the PI3K pathway is crucial for survival .................................................................. 34
Paper II ....................................................................................................................................35 PKC activates Raf, but not Ras or PKB in the HPC lines ............................................................. 35 PKC is required for Erk activation and proliferation ..................................................................... 36 The cPKC mediates serine phosphorylation of c-Kit .................................................................... 36 Conventional PKCs inhibit PKB activation and promote apoptosis ........................................... 37
Paper III ...................................................................................................................................38 Inhibition of PKCδ does not affect c-Kit ......................................................................................... 38 Rottlerin prevents starvation induced apoptosis .......................................................................... 38 Inhibition of PKCδ causes G1 cell cycle arrest.............................................................................. 39 PKCδ influences differentiation of bone marrow........................................................................... 40
CONCLUSIONS............................................................................................................. 41 ACKNOWLEDGEMENTS.............................................................................................. 42 REFERENCES............................................................................................................... 43 ORIGINAL PAPERS………………………………………………………………………….I-III
3

PAPERS IN THIS THESIS _______________________________________________________________________
PAPERS IN THIS THESIS This thesis is based on the following papers referred to in the text by their
roman numerals (I-III).
Paper I
“Activation of the MAP-kinase pathway by c-Kit is PI-3-kinase dependent in hematopoietic progenitor/stem cell lines”
Wandzioch E*, Edling CE*, Palmer RH, Carlsson L and Hallberg B.
Blood. 2004 Jul 1;104(1):51-7
*contributed equally Paper II
“Hematopoietic progenitor cells and mast cells utilize PKC to activate Erk and suppress PKB/Akt activity in response to c-Kit stimulation.”
Edling CE, Pedersen M, Carlsson L, Rönnstrand L, Palmer RH and Hallberg B.
Submitted to British Journal of Haematology Paper III
"PKCδ is required for proliferation and can negatively regulate differentiation in c-Kit expressing hematopoietic cells"
Edling CE, Rönnstrand L and Hallberg B. manuscript
4

ABBREVIATIONS _______________________________________________________________________
ABBREVIATIONS
aPKC atypical PKCs APS adaptor containing PH and SH2 domains ATP adenosine triphosphate CMP common myeloid progenitors cPKC conventional PKCs CSF-1 colony-stimulating factor-1 DAG diacylglycerol Erk extracellular signal regulated kinase ES embryonic stem FACS fluorescence-activated cell sorting FoxO forkhead box, class O GAP GTPase activating protein GDP guanine diphosphate GEF guanine nucleotide exchange factor GIST gastrointestinal stromal tumour GM-CSF granulocyte-macrophage colony stimulating factor GNNK glycine-asparagine-asparagine-lysine Grb2 growth factor receptor-bound protein 2 GSK3 glycogen synthase kinase-3 GTP guanine triphosphate GTPase guanosine triphosphatase HEK human embryonic kidney HPC hematopoietic progenitor cell HSC hematopoietic stem cell HZ4-FeSV Hardy-Zuckerman 4 feline sarcoma virus IGF insulin-like growth factor Ig-like immunoglobulin-like IL-3 interleukin-3 IP3 inositol-1,4,5-triphosphate JM juxtamembrane KSR kinase suppressor of Ras Lhx2 LIM (Lin11, Isl1 and Mec3) homeobox 2 lin- lineage negative LSK lin- Sca+ c-Kit+ LT long-term MAPK mitogen-activated protein kinase MCP mast cell progenitor Mek MAPK/Erk kinase MGF mast cell growth factor MP1 Mek partner-1 MPP multipotential progenitor mTor mammalian target of Rapamycin nPKC novel PKC PDGF platelet-derived growth factor
5

ABBREVIATIONS _______________________________________________________________________
PDK phosphoinositide-dependent protein kinase PH pleckstrin homology PHLPP PH domain leucine-rich repeat protein phosphatase PI3K phosphoinositide-3 kinase PIP2 phosphatidylinositol 4,5 diphosphate PIP3 phosphatidylinositol 3,4,5 triphosphate PKA protein kinase A PKB protein kinase B PKC protein kinase C PLCγ phospholipase C γ PMA phorbol 12-myristate 13-acetate PP2A protein phosphatase-2A PTB phosphotyrosine binding PTEN phosphatase and tensin homologue deleted on chromosome ten Raf rapidly growing fibrosarcomas Ras rat sarcoma RBD Ras-binding domain RKIP Raf kinase inhibitor protein RTK receptor tyrosine kinase S serine Sca stem cell antigen SF steel factor SFK Src family kinases SH2 Src homology 2 Shc SH2-containing transforming protein C1 SHIP SH2-containing phosphatase Shp1/2 SH2 domain-containing phosphatase 1 and 2 Sl steel Sos son-of-sevenless ST short-term T threonine W white spotting Y tyrosine
6

INTRODUCTION _______________________________________________________________________
INTRODUCTION
Phosphorylation mediated signalling Protein phosphorylation Eukaryotic cell functions are to a large extent regulated by protein kinases. Protein
phosphorylation is involved in intracellular and intercellular communication which controls
pivotal processes such as metabolism, cell cycle progression, differentiation, apoptosis,
cytoskeletal architecture and gene expression in the cell.
Phosphate in proteins was described for the first time one hundred years ago by Levene and
Alsberg [1], however the enzymatic phosphorylation reaction was not described until fifty years
later by Kennedy and Burnett [2]. In the 1970s studies by Krebs, Cohen and others had
established that serine/threonine phosphorylation was a rapid post-translational key mechanism in
signal transduction [3] and in 1979 Hunter and colleagues identified tyrosine phosphorylation.
They furthermore demonstrated that this new form of phosphorylation could be linked to
malignant transformation [4].
Today, the protein kinases constitute a large protein family. In fact, almost 2% of the eukaryotic
genes contain protein kinase domains [5]. The protein kinase family members are well conserved
through the molecular evolution based on sequence and structure. The importance of the protein
kinases are further underlined by the number of diseases that are caused by mutations in protein
kinase genes [6].
Protein kinases act by catalysing the transfer of an ATP (adenosine triphosphate) gamma-
phosphate to hydroxyl groups of the amino acids serines, threonines or tyrosines on target
proteins. The phosphorylation of a target protein in most cases results in a modification of the
conformation that changes the enzyme activity or facilitates physical interaction with other
molecules. Phosphorylation of proteins is a reversible mechanism enabling proteins to switch
from an inactive state to an active state in the course of a millisecond [7,8].
7

INTRODUCTION _______________________________________________________________________
Receptor Tyrosine Kinases Following the discovery that proteins could be tyrosine phosphorylated Hunter and others
displayed that the viral protein Src transformed cells through tyrosine phosphorylation.
Continuation on this line of research has established that the receptors for several growth factors
are in fact tyrosine kinases and that abnormal activation or expression can induce cellular
transformation [9]. It is now predicted that, based on the sequences of the catalytic domains, of
the total 518 protein kinases 90 are tyrosine kinases. These are further divided in 58 receptor
tyrosine kinases (RTK) and 32 non-receptor tyrosine kinases [6].
The RTKs are transmembrane protein tyrosine kinases with an extracellular part comprising the
ligand binding domain and an intracellular part containing the kinase domain. The extracellular
and intracellular domains are connected via a single helix through the cell membrane. The
intracellular domains are much conserved among the RTKs, contrary to the extracellular regions
which display high divergence and are able to interact with different ligands. All RTKs, except
the Insulin receptor, are monomers in the cell membrane. However ligand binding induces
dimerisation of the receptors which is followed by autophosphorylation of their cytoplasmic
domains. The dimerisation induced activation differs among the RTKs, some are activated by a
single ligand binding to two receptors simultaneously, and then additional receptors can bind to
the complex for further stabilisation. Other receptor-ligand complexes are constructed by two
ligands and two receptors to enable full activation [10].
The Src homology 2 domain The autophosphorylation of the receptor results in intracellular proteins containing Src homology
2 (SH2) domains recognizing and transiently interacting with specific phosphotyrosine motifs on
the receptor. The SH2 domain was first identified by Pawson and colleagues in 1986 as a non-
catalytic region that could modify both kinase activity and substrate recognition [11]. A few years
later Kuriyan and Eck solved the structure for the Src SH2 domain and described in detail how it
could recognise and bind phosphorylated tyrosines [12,13]. Individual SH2 domains can
distinguish between the specific autophosphorylation sites because of their ability to recognise
residues C-terminally of the phosphotyrosine and depending on preference they will bind [14]. In
addition to the SH2 domain it has been revealed that proteins can contain a phosphotyrosine
binding (PTB) domain in connection to the SH2 domain that also enables binding to
phosphorylated tyrosines, although, with less specificity. Upon activation and intrinsic
8

INTRODUCTION _______________________________________________________________________
phosphorylation of the RTK, cytoplasmic mediators containing SH2 domains are recruited to the
phosphotyrosines on the receptor and these interactions activate the effector proteins which
subsequently mediate the signal via downstream pathways. These signals mediate the effects of,
for example; growth factors, hormones, or antigens which are vital for the cell [9].
Phosphatases Activation by phosphorylation is closely regulated by different mechanisms. The protein
phosphatases play a key role in regulating the action of kinases since they catalyze the removal of
phosphate groups from phosphorylated substrates. The first protein tyrosine phosphatase, PTP1B,
was isolated and described by Tonks et al. in 1988 [15,16]. Since then the family of phosphatases
has been shown to constitute a wide range of enzymes with different specificity. The
phosphatases are divided into two major classes: protein serine/threonine phosphatases and
tyrosine phosphatases. In contrast to the kinases there is no sequence similarity in the catalytic
domain between the two classes. The protein tyrosine phosphatases are defined by the signature
motif, (H/V)C(X)5R(S/T), in the catalytic domain. Mechanistically they act by transferring the
phosphate group to the cysteine residue before hydrolysation with water. In addition to enzyme
regulation, phosphates can also be removed in a nonenzymatic hydrolytic reaction [17,18].
Endocytosis Phosphorylation and dephosphorylation mechanisms are crucial to regulate activity and inactivity
of proteins. Beside this instant mechanism of regulation, proteins, and especially receptors,
become down regulated after activation via internalisation by endocytosis. Upon activation of a
RTK a clathrin coated vesicle is formed at the signalling site and the receptor is internalised in
the vesicle and transferred via the early and late endosomes for degradation in the lysosome. The
endocytosis process is ongoing constantly in the cell. Activated receptors get rapidly degraded
and inactive receptors get recycled at a low rate [19].
Regulation of signal transduction A major question in the field of cell regulation is how signals can be transduced with the required
specificity. Despite the fact that many receptors are expressed in several tissue types and that
different receptors can utilise the same downstream pathways, the effect of the receptor activation
can still induce a specific biological response. The answer probably lays in that the system of cell
types, receptors, ligands and pathways comprise an extremely complex network that enables
enough variation to fulfil specificity for all purposes. Factors involved in determining the signal
9

INTRODUCTION _______________________________________________________________________
transduction specificity include the balance between kinase and phosphatase activity, cellular
localisation of proteins, binding of scaffold proteins, multisite versus single site phosphorylation,
duration and strength of signals, number of available receptors and combinations of receptors and
signalling molecules [9,10,20].
Protein kinase inhibitors Since all kinases require ATP for there enzymatic activity they can all be inhibited by blocking
the domain where the ATP binds. The catalytic domain, including the ATP binding pocket, is
highly conserved among the protein kinases. Despite this fact, it has proven possible to develop
specific inhibitors because, especially in the inactive state, the kinases present distinct features
that can be targeted by small binding molecules. The available protein kinase inhibitors are well
used tools for biochemical analyses of protein functions and some are now successfully used in
cancer therapy [21].
Receptor tyrosine kinase c-Kit Identification of receptor and ligand In 1986 Besmer et al. identified a new viral oncogene in the Hardy-Zuckerman 4 feline sarcoma
virus (HZ4-FeSV) that had been isolated from a feline fibrosarcoma. The amino acid sequence
revealed homology with the tyrosine kinase gene family and it was shown to be closely related to
v-fms. This oncogene was designated v-Kit [22]. A year later, the human cellular homologue, c-
Kit, was identified and characterized by Yarden et al. [23]. They reported that the c-Kit gene
product is structurally related to the receptor for macrophage colony-stimulating factor-1 (CSF-1
or c-fms) and the receptor for platelet-derived growth factor (PDGF) that both had been recently
identified [24,25]. They further demonstrated that the c-Kit gene encoded a transmembrane
glycoprotein of 145kDa which was capable of autophosphorylation on tyrosine residues and
suggested, based on the structural likeness to the PDGF and CSF-1 receptors, that c-Kit (also
referred to as stem cell factor receptor or CD117) was a growth factor receptor for an unidentified
ligand [23]. In 1988 c-Kit was exhibited to be encoded by the white spotting (W) locus on
chromosome 5 in mice (chromosome 4 in human [26]) [27,28]. A couple of years later a novel
mast cell growth factor (MGF) was identified and demonstrated to be a ligand for the c-Kit
receptor [29] and at the same time the MGF coding sequence was cloned and mapped to the Steel
(Sl) locus in the distal region of mouse chromosome 10 [30]. The c-Kit ligand has been assigned
10

INTRODUCTION _______________________________________________________________________
several different names: kit ligand, MGF, stem cell factor and Steel Factor (SF). In this text it will
be referred to as Steel Factor (SF).
Expression and Mutations The mapping of c-Kit and SF explained the similar phenotypes caused by mutations in the W and
Sl locuses that had been described several years earlier [31]. Normal c-Kit expression is observed
in several tissues including: mast cells, hematopoietic stem and progenitor cells, interstitial cells
of Cajal in the gastrointestinal tract, melanocytes, germ cells and neurons [32]. The expression in
the hematopoietic system will be described more in a later section. Various types of loss-of-
function and gain-of function mutations have been reported at the W locus. The loss-of-function
mutations in mice are clearly connected to the c-Kit expressing tissues. Abnormalities due to
impaired c-kit function in mice are depletion of mast cells [33] and interstitial cells of Cajal [34],
severe anaemia due to damaged erythropoiesis [31], piebaldism [31] and reduced hearing ability
[35] due to lack of melanocytes and sterility in both males and females [31].
There is a range of different types of gain-of-function mutations of c-Kit including point
mutations, deletions and duplications. Frequently occurring point mutated residues are valine 560
in the juxtamembrane (JM) domain and asparagine 816 in the kinase domain [36]. The activating
mutants of c-Kit confer with constitutional tyrosine phosphorylation and downstream activation
independent of ligand binding. These mutations expose a strong oncogenic potential of c-Kit.
Tumours caused by constitutively activated c-Kit are different types of mast cell neoplasms,
gastrointestinal stromal tumours (GISTs), germ cell tumours and some leukemias. In addition
aberrant expression of c-Kit has been observed in some tumours such as small cell lung
carcinomas [32].
Imatinib mesylate As discussed above there are several tyrosine kinase inhibitors available. Interruption of
signalling pathways are said to be the new generation of tumour therapy. One such inhibitor that
is used with remarkable success is imatinib mesylate, formerly known as STI571 (commercially
Gleevec/Glivec). Initially this drug was developed at Ciba-Geigy (now Novartis) as a specific
inhibitor of the tyrosine kinases Bcr-Abelson (Abl) and the PDGF receptor [37]. Extended
research has revealed that imatinib mesylate is also a potent inhibitor of c-Kit, but not of the
closely related c-fms, flt and tek tyrosine kinases [38]. Imatinib mesylate is approved and
11

INTRODUCTION _______________________________________________________________________
indicated for treatment of chronic myeloid leukaemia (CML) that is caused by expression of Bcr-
Abl and of c-Kit expressing GISTs [US and European drug administrations].
SF, the c-Kit ligand SF exists in a soluble and a membrane-anchored isoform, due to alternative RNA splicing and
proteolytic processing [39]. The soluble form consists of 165 amino acids and it functions as a
noncovalent homodimer. SF dimers bind soluble or membrane bound c-Kit with high specificity
and affinity [40].
Figure 1. Schematic structure of c-Kit, amino acids apply to human c-Kit
C-Kit structure signal sequence
C-terminal tail
transmembrane segment
juxtamembrane domain
activation loop
distal kinase domain
kinase insert domain
Ig-like motifs
proximal kinase domain
1
5
4
3
2
1-22
685-761
521-543
Y568
Y936
Y721
Y570544-581
Y703
Y823
Y730762-937
582-684
938-976
810-839
signal sequence
C-terminal tail
transmembrane segment
juxtamembrane domain
activation loop
distal kinase domain
kinase insert domain
Ig-like motifs
proximal kinase domain
1
5
4
3
2
1-22
685-761
521-543
Y568Y568
Y936Y936
Y721Y721
Y570Y570544-581
Y703Y703
Y823Y823
Y730Y730762-937
582-684
938-976
810-839
The c-Kit receptor is categorised as a type III
receptor tyrosine kinase. The members in
this subfamily features unique
characteristics; an extracellular part
containing five immunoglobulin-like (Ig-
like) motifs and a tyrosine kinase domain
that is divided in a proximal and a distal part
by an insert sequence of variable length. In
addition to these characteristics the c-Kit
structure is composed of domains that are
common among most RTKs. See figure 1.
A signal sequence is located in the N-
terminal end. This sequence is followed by
the five Ig-like motifs. The second and third
Ig-like motifs together constitute the ligand
binding pocket [41]. The fourth Ig-like motif
contains the dimerisation site. Deletion of
the fourth Ig-like motif completely abolishes
dimerisation and the following signal
transduction. A defective dimerisation site
results in accelerated ligand dissociation,
indicating that the ligand affinity is
dependent on dimerisation of the receptors
[42].
12

INTRODUCTION _______________________________________________________________________
The intracellular part of c-Kit is composed of the autoinhibitory juxta-membrane domain and the
two kinase domains with the insert between them. Deletion of the juxtamembrane domain causes
increased induction time for activation of c-Kit and addition of an exogenous juxtamembrane
domain peptide inhibits the trans phosphorylation [43]. The kinase domains, with the activation
loop located in the distal kinase domain, are responsible for catalysing the transfer of a phosphate
group from ATP to the substrate [44].
Isoforms and splice variants In addition to the above described transmembrane form of c-Kit there is a soluble truncated form
of c-Kit. The truncated c-kit is constituted of a single distal kinase domain and it is expressed in
male germ cells [45]. Since this isoform is severely truncated it lacks functional kinase activity.
Even so, it can still mediate signals and has been shown to be of importance for spermatogenesis
[46].
In both human and mouse alternative mRNA splicing has resulted in c-Kit isoforms that differ in
a four amino acid sequence, Glycine-Asparagine-Asparagine-Lysine (GNNK), located
extracellulary near the transmembrane segment [47]. The ratio between the two isoforms varies in
different tissues, but the GNNK- isoform is usually more abundant [48]. A difference in
transformation ability and downstream signalling depending on isoform has been suggested. The
GNNK- form appears to induce anchorage-independent growth and tumourigenicity to a higher
extent than the GNNK+ from [49]. Moreover, the SF stimulated c-Kit GNNK- form is more
highly tyrosine phosphorylated, and Erk and Shc are more strongly activated due to increased and
more rapid Src activation [50]. In addition to these splice forms, human c-Kit splice variants that
differ in the presence or absence of a single serine residue in the kinase insert domain have been
described [48], although whether this has any impact on signalling is not known.
Activation of c-Kit Activation of c-Kit is initiated when a SF dimer simultaneously binds two monomeric c-Kit and
thereby promote dimer formation [41]. Dimerisation enables autophosphorylation in trans on
tyrosine residues in the intracellular domain. Commonly the first tyrosine residues to be
phosphorylated are those located in the activation loop. This results in stabilisation of the receptor
in its most active enzyme form [51]. However, in the RTK type III subfamily the tyrosine
residues in the juxtamembrane are phosphorylated first. Studies of the crystal structure of c-kit
have revealed why tyrosines 568 and 570 in the juxtamembrane domain are phosphorylated
13

INTRODUCTION _______________________________________________________________________
before the tyrosine 823 in the activation loop. These initial phosphorylations of the
juxtamembrane domain alter the static configuration that sterically blocks the activation loop
from converting to its extended active conformation. Subsequently the tyrosine 823 is
phosphorylated to stabilise the activation loop in the active enzyme form [52,53]. When c-Kit is
fully activated the phosphorylated tyrosine residues in the juxtamembrane domain, kinase
domains and the kinase insert domain facilitate docking sites for substrates encompassing SH2.
The conformation change of the activation loop allows access to the bound ATP and the gamma
phosphate group from ATP is transferred to hydroxyl groups on the substrate [10].
Protein binding sites on c-Kit Upon activation the RTK c-Kit is phosphorylated at specific intracellular tyrosine (Y) residues to
which downstream signalling mediators bind and subsequently get activated. See table 1. In
addition to the effectors included in Table 1, there are many different proteins that are recruited to
the activated receptor and take part in constituting signalling complexes, but do not directly
associate with the receptor (for reference see Roskoski, 2005 [54]).
Phospho-site Recruited proteins Mechanism Y568/570 APS, Src family kinases, Shp1/2, Cbl, Shc autophosphorylation Y703 Grb2 autophosphorylation Y721 p85 subunit of PI3K autophosphorylation Y730 PLCγ autophosphorylation Y823 activation loop autophosphorylation Y900 p85/Crk phosphorylated by Src Y936 Grb2/7, APS, Cbl autophosphorylation
Table 1. Phospho-sites in human c-Kit, protein name and phosphorylation mechanism
APS and Cbl
APS (adaptor containing PH and SH2 domains) is an adaptor protein which associates with c-Kit
on Y568 and Y936. APS activation is involved in induction of internalisation and degradation of
c-Kit by interacting with Cbl [55]. Cbl acts as an E3 ubiquitin-protein ligase, transferring
ubiquitin from ubiquitin-conjugating enzymes to substrates promoting their degradation by the
proteasome [56].
14

INTRODUCTION _______________________________________________________________________ Src
The Src family kinases (SFK) are suggested to be involved in many different biological
functions, including survival, proliferation, differentiation and motility [57]. They associate with
Y568 and Y570, either directly or via the adaptor proteins APS. Activation of c-Kit induces
rapidly increased SFK kinase activity [58,59]. They have further been shown to be responsible
for phosphorylating Shc (SH2-containing transforming protein C1) and thereby enabling
recruitment of the Grb2/Sos complex (growth factor receptor-bound protein 2/Son-of-Sevenless)
[60]. Recruitment and activation of this complex subsequently results in activation of Ras [61].
Moreover, recent data demonstrates that Cbl can bind directly to Y568 and Y936, but it requires
phosphorylation by SFK members for activation leading to down-regulation of c-Kit [62].
Shp1/2
Shp1 and 2 (SH2 domain-containing phosphatase 1 and 2) are cytosolic phosphotyrosyl
phosphatases which negatively regulate growth factor signalling. Shp1 is mainly expressed in
hematopoietic and epithelial cells, while Shp2 is ubiquitously expressed [63]. In vivo studies
indicate that Shp1 binds to c-Kit at Y570 while Shp2 binds at Y568. They act by
dephosphorylating the receptor directly, or inhibit c-kit signalling indirectly by
dephosphorylating other receptor-associated protein-tyrosine kinases [64].
Grb2/7 and Shc
Grb2 and 7 are adaptor proteins. Grb2 binds c-Kit both at Y703 and Y936, and Grb7 only at
Y936 [65]. Grb2, in complex with Sos and Shc, is the link that mediates the growth factor signal
from the receptor to Ras (rat sarcoma) [61]. Grb2 is also shown to be involved in the recruitment
of Cbl to c-Kit [66].
P85, PI3K regulatory subunit
Phosphoinositide-3 Kinase (PI3K) is a heterodimeric lipid kinase constituting two subunits: p85
and p110 [67]. PI3K signalling mechanisms and effects will be discussed in a later section. SF
stimulation of cells leads to rapid activation of PI3K, and PI3K has been demonstrated to interact
with the kinase insert domain of c-Kit [68]. More exactly, the regulatory subunit (p85) of PI3K
binds to Y721 of c-Kit [69] causing increased kinase activity of the catalytic subunit. The p85-
receptor interaction is suggested to be negatively regulated by the SFK member Lyn since PI3K
is constitutively associated with c-Kit in Lyn-deficient bone marrow mast cells [70].
15

INTRODUCTION _______________________________________________________________________
Furthermore, the p85 subunit is also known to associate with the adapter proteins Cbl and Crk
[71]. This complex, possibly without Cbl, is thought to bind, via p85, the Y900 residue after it
has been phosphorylated by Src or a downstream mediator. Mutation of this site results in weaker
induction of DNA synthesis. [72].
PLCγ
Phospholipase C γ (PLCγ) belongs to an enzyme family that catalyses the hydrolysis of
phosphatidylinositol-4,5-biphosphate (PIP2) to generate the second messengers diacylglycerol
(DAG) and inositol-1,4,5-triphosphate (IP3). DAG activates protein kinase C (PKC) family
members and IP3 triggers the release of Ca2+ by binding specific receptors on the endoplasmatic
reticulum [73]. Rottapel et al. showed that both PLCγ and PI3K were activated upon SF
stimulation of murine mast cells [74] and overexpression studies have suggested Y730 to be the
site for PLCγ association with c-Kit [75,76].
Signalling mediators downstream of c-Kit Ras and the Raf/Mek/Erk cascade Ras activation
Ras belongs to the Ras superfamily that contains over 150 human members. Ras is a small
guanosine triphosphatase (GTPase), like all members in the Ras superfamily it binds guanine
nucleotides with high affinity and it possesses low intrinsic GTPase activity. Ras is active when
bound to GTP (guanine triphosphate) and it is inactive when bound to GDP (guanine
diphosphate). Exchange from GDP to GTP causes conformational changes that result in the GTP-
bound form exhibiting a higher affinity for effector targets. The switch from inactive to active
and back is tightly regulated by GEFs (guanine nucleotide exchange factors) and GAPs (GTPase
activating proteins). GEFs promote the switch from GDP to GTP, hence result in activation.
GAPs increase the intrinsic GTPase activity resulting in inactivation. [77].
Upon activation of c-Kit the adapter proteins discussed above are recruited to the phosphorylated
receptor. Grb2 is constitutively associated with Sos, which is a GEF for Ras. The colocalisation
and complex-building of Grb2, Sos, Shc and possibly other adaptor proteins increase Ras activity
[78].
16

INTRODUCTION _______________________________________________________________________
The Ras superfamily members can induce mitogen-activated protein kinase (MAPK) cascades.
The MAPKs constitute a network of cascades transmitting stimuli-induced signals. The MAPK
pathways contain a kinase cascade comprising a MAP kinase kinase kinase, a MAP kinase
kinase, and a MAP kinase. In this thesis we have mainly focused on the classical MAPK cascade
that is commonly activated downstream of RTKs. This pathway constitutes Raf (rapidly growing
fibrosarcomas), Mek (MAPK/Erk kinase), and Erk (extracellular signal regulated kinase) [79].
Raf activation and phosphorylation sites
Activation of Raf is complex and not fully understood. However it involves membrane
recruitment, dimerisation or oligomerisation, protein-binding, conformational changes and
phosphorylation [80]. The activation process is initiated by the binding of GTP-bound Ras to the
Ras-binding domain (RBD) of Raf [81]. This interaction recruits Raf to the plasma membrane
where it gets activated [80,82]. There are three isoforms of Raf; a-Raf, b-Raf and c-Raf. C-Raf
was cloned first [83] and has been studied most intensively.
C-Raf is tightly regulated by phosphorylation. Protein kinase A (PKA) is believed to negatively
regulate c-Raf by phosphorylation on three different serine (S) residues; S43, S233 and S259.
PKA is a cyclic-AMP-dependent kinase and these sites have been demonstrated to be
phosphorylated upon increase of cyclic-AMP levels in the cell [84]. S259 is also suggested to be
phosphorylated by protein kinase B (PKB, also known as Akt) resulting in suppression of c-Raf
activity [85]. Phosphorylation of S43 sterically hinders binding of Ras to Raf which inhibits
activation [86]. Phosphorylated S233 and S259 both create binding sites for the scaffold protein
14-3-3. Binding at these sites blocks the activation process of c-Raf [84,87].
In addition to the three negative sites there are five sites, located within or near the kinase
domain, that stimulate c-Raf activity. Phosphorylation of S338 is required for activation of c-Raf,
however, there is a discrepancy in the literature regarding which kinase is targeting this site.
Some studies suggest that it is the serine/threonine(S/T)-specific protein kinase PAK [80].
However, the PAKs are demonstrated to phosphorylate c-Raf located in the cytosol, in a Ras
independent manner, and are therefore probably not responsible for the growth-factor stimulated
activation at the membrane. Furthermore growth-factor stimulated S338 phosphorylation is not
inhibited by dominant negative PAK [88]. The SFKs have been shown to phosphorylate Y341, a
site which is required for activation [82,89,90]. Phosphorylation of T491 and S494 are essential
since they are located in the activation loop of c-Raf and mutations of these sites have been
17

INTRODUCTION _______________________________________________________________________
observed to inhibit activation [91]. Finally, phosphorylation of S621 mediates binding of 14-3-3
to the C-terminal end which is also necessary for activation [87,92].
Mek and Erk activation
The Raf proteins are serine/threonine kinases with just one established substrate; Mek1/2 (two
isoforms). Active Raf transmits the signal from Ras to Mek1/2 by phosphorylating Mek1/2 at two
sites, S217 and S221, in the activation loop [93]. Mek1/2 is a dual specificity kinase, targeting
both serine/threonine and tyrosine residues [94-96]. In addition, Mek1/2 is very specific, it targets
only one substrate, the serine/threonine kinase Erk1/2, which is activated by phosphorylation of
both T202 and Y204 in the threonine-glutamine-tyrosine motif in its activation loop [97]. Erk1/2
has an extensive range of substrates including nuclear transcription factors, cytoskeletal proteins,
kinases and phosphatases. Activation of the Raf/Mek/Erk cascade therefore regulates many cell
functions. Its main effects are the promotion of proliferation and differentiation [98].
Shc
KSR
RafP P P
14-3-3
MekPP
ErkP P
RasGTP
MP1
Nuclear and cytosolic substrates
PPPP
PPP
Grb2 Sos
SF SF
P
14-3-3
Shc
KSR
RafP P P
14-3-3
MekPP
ErkP P
RasGTP
MP1
Nuclear and cytosolic substrates
PPPP
PPP
Grb2 Sos
SF SFSF SF
P
14-3-3
Figure 2. Activation of Ras and the Raf/Mek/Erk cascade.
18

INTRODUCTION _______________________________________________________________________
Regulation of the Raf/Mek/Erk cascade
The Raf/Mek/Erk cascade is regulated and stabilised by various proteins. The kinase suppressor
of Ras (KSR) is a scaffold protein that is constitutively associated with Mek, but can also bind
Raf and Erk upon extracellular stimulation [99]. KSR acts by organising the kinases together in a
module. Following activation, KSR gets dephosphorylated by the protein phosphatase-2A (PP2A)
and the KSR-Raf/Mek/Erk-module is localised in proximity to Ras at the plasma membrane
enabling mediation of the signal [100]. The interaction between Mek and Erk is further
coordinated by the scaffold protein MEK partner-1 (MP1), resulting in enhanced activation. MP1
forms heterodimers with a small protein called p14, which targets the MP1-Mek-Erk complex to
endosomes. Erk is initially activated at the membrane, however the translocation to the
endosomes is believed to sustain the activation [101].
There are some proteins that have been described to negatively regulate the Raf/Mek/Erk
cascade. The 14-3-3 proteins are expressed in abundance and can bind several different proteins.
They mainly act by sequestering proteins, for example Raf and KSR, in the cytosol when the cell
is not under the influence of extracellular stimuli [100]. The interaction between Raf and Mek is
negatively regulated by the Raf kinase inhibitor protein (RKIP). RKIP inhibits Mek
phosphorylation by binding to both Raf and Mek and thereby preventing their physical
interaction [102].
Research over the last few years suggests that MAPK cascades are more complex than has
previously been thought. For example, the Raf isoforms are now suggested to have quite different
functions. It has been demonstrated that b-Raf actually has much higher kinase activity than c-
Raf and a-Raf, and that b-Raf might activate c-Raf. Furthermore, c-Raf is suggested to have other
targets than Mek1/2 and that it might itself act as a scaffold protein for the MAPK cascade
[80,100]. In addition, recent research connect b-Raf, but not c-Raf, with several types of
malignancies which is suggested to be explained by the higher kinase activity of b-Raf [103,104].
The PI3K pathway The lipid kinase PI3K
There are several classes of phosphoinositide-3 kinases. Here the class IA PI3Ks will be
discussed, since these are the PI3Ks that are linked to RTK activation. The PI3Ks are
heterodimers consisting of a catalytic and a regulatory subunit. In mammals there are three genes
19

INTRODUCTION _______________________________________________________________________
encoding the class IA catalytic subunit isoforms, p110α, p110β and p110δ, and three genes
encoding the regulatory subunit isoforms p85α, p85β and p55γ [105].
PI3K is a lipid kinase that catalyses the phosphorylation of phosphatidylinositol 4,5 diphosphate
(PIP2) to phosphatidylinositol 3,4,5 triphosphate (PIP3). These inositol phospholipids are located
in the cell membrane and acts as second messengers by facilitating binding sites for proteins
containing Pleckstrin homology (PH) domains [105]. Some of the PH domain containing proteins
bind phospholipids with high affinity. The residues for binding phospholipids effectively are
located in the N-terminal part and the motif includes basic amino acids which directly interact
with the inositol phosphate groups of the phosphatidylinositol [106].
PKB activation
Protein kinase B and phosphoinositide-dependent protein kinase (PDK) are serine/threonine
kinases containing PH domains. Upon receptor autophosphorylation and subsequent PI3K
catalysed generation of PIP3, PKB and PDK are attracted to the membrane where the interaction
with PIP3 occurs via their PH domains [107,108]. PKB gets activated by phosphorylation at two
sites; T308 in the activation loop and S473 in the carboxy-terminal regulatory region [109]. PDK,
which is localised in close proximity to PKB due to the interaction with PIP3, phosphorylates
PKB on T308 [110]. The kinase that is responsible for phosphorylating PKB on S473 is still not
fully clarified. However, a recent study suggests that the complex mTor (mammalian target of
Rapamycin)-Rictor directly phosphorylates S473 of PKB, further it is demonstrated that
inhibition or downregulation of mTor inhibits PKB activation [111,112].
PKB substrates
In resting cells PKB is localised in the cytosol, and upon activation it becomes localised to the
cell membrane. When activated, PKB detaches from the phospholipid and is translocated to the
cytosol and nucleus [113,114]. Active PKB phosphorylates several different downstream targets,
mainly involving metabolism and cell survival [105]. The first protein to be defined as a PKB
substrate was Glycogen synthase kinase-3 (GSK3) [115]. Phosphorylation of GSK3 inactivates
the protein and thereby glycogen synthesis is promoted [116]. PKB substrates affecting cell
survival include BAD (Bcl2/BclX-antagonist, causing cell death), and the FoxO (Forkhead box,
class O) transcription factors. Both these targets are inactivated by PKB phosphorylation and
hence are inhibited from inducing apoptosis [117].
20

INTRODUCTION _______________________________________________________________________
P
PKBP
P
Survival and metabolism
SF SF
PPPP
PPPP
PP
PP
P
PDK
mTorRictor
?
FoxOBAD
P
GSK3
PP
P
PTENSHIP
PI3K
P
14-3-3
P
P
PKBP
P
Survival and metabolism
SF SFSF SF
PPPP
PPPP
PP
PP
P
PDK
mTorRictor
mTorRictor
?
FoxOBAD
P
GSK3
PP
P
PTENSHIP
PI3K
P
14-3-3
P
Figure 3. Activation of the PI3K pathway via the c-Kit receptor.
Regulation of the PI3K pathway
The activation of the PI3K pathway is suppressed, or terminated, by two different phosphatases,
PTEN (phosphatase and tensin homologue deleted on chromosome ten) and SHIP (SH2-
containing phosphatase). PTEN counteracts the activity of PI3K by hydrolysing PI(3,4,5)P3 back
to PI(4,5)P2. PTEN is ubiquitously expressed and a major regulator of the PI3K pathway,
furthermore PTEN is known as a tumour suppressor implicated in a wide variety of human
cancers [118]. SHIP acts by hydrolysing PI(3,4,5)P3 to PI(3,4)P2 and thereby suppressing the
PI3K activity. SHIP expression is primarily restricted to hematopoietic cells and stem cells,
which makes it particularly important for c-Kit signalling [119]. Furthermore, the PI3K pathway
is suppressed by PHLPP (PH domain leucine-rich repeat protein phosphatase), a phosphatase that
targets the S473 phosphorylation on PKB. This dephosphorylation results in triggered apoptosis
and inhibited tumour growth [120].
21

INTRODUCTION _______________________________________________________________________
The Protein Kinase C family The PKCs are serine/threonine kinases that are involved in a multitude of signalling pathways.
The PKC family comprises at least ten related isoforms which are classified into three subgroups
depending on structural and biochemical properties; the conventional PKCs (cPKC), the novel
PKCs (nPKC), and the atypical PKCs (aPKC) [121]. The table below describes how the isoforms
are subgrouped, and summarises how they are regulated (Table 2).
Subgroup: Classical PKCs Novel PKCs Atypical PKCs Isoforms: α, βΙ, βΙΙ, γ δ, ε, η, θ ζ, ι Regulated
by: Phosphatidylserines DAG/Phorbol esters
Ca2+ Phosphorylation
Phosphatidylserines DAG/Phorbol esters
Phosphorylation
PhosphatidylserinesPhosphorylation
Table 2. The PKC isoforms and their regulation All PKC isoforms require the negatively-charged phospholipid phosphatidylserine for activation.
Phosphatidylserine resides within the plasma membrane and, in the presence of DAG, PKC binds
phosphatidylserine with higher affinity than other anionic lipids [122]. DAG and IP3 are second
messengers which are generated by RTK activated PLCγ catalysed cleavage of PI(4,5)P2, as
mentioned in a previous section [73]. The regulatory domain of cPKCs and nPKCs constitute
binding-sites for DAG and other phorbol esters. Following interaction, formation of a
hydrophobic surface occurs that enables part of the protein to penetrate the lipid bilayer of the
membrane where the phosphatidylserines are located [122]. The aPKCs lack essential residues in
the binding-site for DAG and are therefore ‘atypical’ [123]. IP3 causes release of Ca2+ which
leads to increased Ca2+ levels in the cytosol [73]. The cPKCs contain five conserved aspartic acid
residues which constitute a Ca2+ binding site [124]. Binding to Ca2+ increases the cPKC affinity
for phosphatidylserines, and thereby further promotes activation [125]. In order to become fully
activated PKC is phosphorylated at three sites [126]. PDK catalyses the phosphorylation of the
threonine in the activation loop in most PKC isoforms. This initial phosphorylation is not
activating, however, it promotes autophosphorylation at two other sites which stabilises the
protein in a protease and phosphatase resistant conformation. The fully phosphorylated protein is
catalytically competent but inactive due to an inhibitory conformation. Upon binding to
phosphatidylserines the conformation is altered, which enables PKC to interact and phosphorylate
substrates [127]. The wide range of substrates include receptors, enzymes, cytoskeletal proteins,
nuclear proteins and transcription factors [128].
22

INTRODUCTION _______________________________________________________________________
The hematopoietic system The hematopoietic system functions to continuously provide mature blood cells from the
embryonic stages and on throughout the life-time. Prior to birth the development of the
hematopoietic system is initiated in the yolk sac, and then progresses in the fetal liver. Shortly
after birth the bone marrow (BM) becomes the principal site for blood development. The
hematopoietic activity in the BM is extensive since mature blood cells are relatively short-lived
and need replacement continuously. In the BM billions of new mature blood cells are generated
every day [129].
The hematopoietic stem cell The blood comprises a variety of cells with diverse functions, however, they all originate from
the small pool of hematopoietic stem cells (HSC) that reside in the BM. Stem cells are rare in
most tissue types and the HSCs are no exception, in the BM less than 0.05% are estimated to be
HSCs [130].
Defining the Hematopoietic stem cell
HSCs are, by definition, cells that possess the capability to self-renew and to generate mature
progeny of all blood cell lineages by differentiation [131]. The HSCs are divided in long-term
(LT) and short-term (ST) HSCs depending on self-renewal capacity. The LT-HSCs are capable of
unlimited self-renewal, while the ST-HSCs regeneration time seems to be more constrained,
based on experiments analysing the ability of reconstituting the hematopoietic lineages in lethally
irradiated mice [132,133]. The HSC phenotype is characterised by physical and functional
properties. The method of fluorescence-activated cell sorting (FACS) has revealed a set of
surface markers that discriminate the HSCs from their differentiating progeny. The isolation is
based on a combination of negative and positive selection. HSCs express low or undetectable
levels of surface markers associated with committed mature cells which are described as lineage
negative (lin-). The positive selection is based on different markers, most consistently stem cell
antigen (Sca) and c-Kit, which by themselves are not HSC specific, but in combination coincide
with the HSC phenotype. Selection for lin- Sca+ c-Kit+ (LSK) cells results in a population highly
enriched for HSCs with long-term activity [134]. Functionally, the established method to define
HSCs is to demonstrate sustained capacity for regeneration of the hematopoietic system
following transplantation into immuno-compromised recipients. Transplantation experiments
23

INTRODUCTION _______________________________________________________________________
indicating the presence of stem cell properties were successfully performed already some 50
years ago by Ford et al [135] and, Till and McCulloch [136].
Regulation of HSC self-renewal
Hematopoiesis is tightly regulated by intrinsic and extrinsic events that control the lineage
commitment and maturation of the hematopoietic cells. Fundamental for this control is the
regulation of the HSC that is the initiating factor for the blood development. The question in
focus is how the balance between self-renewal and differentiation of the HSCs is maintained and
regulated.
The process of differentiation of HSCs is suggested to be based on asymmetric cell division,
where a single division results in the formation of one daughter cell that maintains the stem cell
capacity and one daughter cell that is committed to differentiation. This hypothesis is supported
by several in vitro and in vivo studies showing that HSCs and multipotential progenitors can
divide asymmetrically [137]. Recent data demonstrate that the most primitive hematopoietic cells
to a high extent divide asymmetrically, while the more committed progenitors have a tendency to
foremost expand their own cell fate by forming two equal daughter cells with the same
developmental capacity [138].
During development, the amount of stem cells increases, suggesting that the system, at the time,
favours symmetric division to expand the HSC pool. In adults, however, the asymmetric division
results in sufficient amount of self-renewing cells to sustain the hematopoiesis throughout life
and to produce enough mature blood cells. During physiological stress, like bleeding, this balance
is hypothesised to be shifted towards more symmetric division favouring differentiation and
production of mature blood [139].
The stem cell niche has shown to be an important extrinsic factor that regulates how the HSC
divides. The concept of the niche is based on primitive cells, restricted to a specific location,
being regulated by physical interaction with the surrounding cells and matrix components. The
sustained contact between the stem cells and the supporting cells can determine the fate of the
daughter cells [139].
Identification of specific extrinsic factors that promote self-renewal of HSCs is a hot topic. Such
factors would enable long-term ex vivo expansion of HSCs, which indeed has extensive clinical
24

INTRODUCTION _______________________________________________________________________
potential. One of the more promising suggestions to play such a role is Wnt. Members of the Wnt
family can be produced by both hematopoietic cells and their stromal cell environment, i.e. the
niche [140]. Furthermore, Wnt proteins can promote self-renewal of HSCs in vitro, and increase
the ability to reconstitute the hematopoietic system in lethally irradiated mice [141].
The hematopoietic system in general is largely regulated by different cytokines and transcription.
These include, for example, interleukins, erythropoietin and SF, and are normally expressed by
stromal cells within the hematopoietic tissues and they exert their function through receptors on
the hematopoietic cells [142].
The hematopoietic hierarchy The hematopoietic system is organised as a hierarchy where the long-term HSCs give rise to the
short-term HSCs, which in turn give rise to the multipotent progenitors, which subsequently
generate the mature cells of the different lineages via more committed precursor cells (see figure
4). As the HSCs progress from long-term to short-term to multipotent progenitor they lose their
self-renewal capability but become more responsive to growth factors [133].
The mast cell lineage
Mast cells retain a high expression of c-Kit during differentiation from HSCs. As mentioned in a
previous section, c-Kit expression is required for mast cell development. Mast cells play a central
role in immediate hypersensitivity and chronic allergic reactions. Following recognition of an
antigen, the reaction occurs within minutes and multiple signalling pathways are activated via the
IgE receptor (FcεR1). This response leads to degranulation and production of various cytokines
[143]. Unlike other mature blood cells, mast cells normally circulate through the vascular system
as progenitors and complete their development within the connective tissues. For a long time it
was actually believed that the mast cells were a component of the connective tissue. However, it
is now clear that the mast cells develop from the HSCs [143]. How this differentiation process is
lineated has until recently been unclear. It has been proposed that the mast cells are myeloid cells
and hence are derived from the common myeloid progenitors (CMP). Recent evidence though
indicates that the mast cell progenitor (MCP) arises from the multipotential progenitor (MPP) as a
separate lineage [144]. See figure 4.
25

INTRODUCTION _______________________________________________________________________
LT-HSC ST-HSC
B cell
Megakaryocyte
NK cell
Macrophage
Granulocyte
Trombocytes
T cell
Erytrocytes
Mast cell
MPPCMP
MCP
CLP
Pro-B
Pro-T
GMP
MEP
Monocyte
Figure 4. Organisation of the mammalian hematopoietic hierarchy. LT-HSC, long-term
hematopoietic stem cell, ST-HSC, short-term hematopoietic stem cell, MMP,
multipotential progenitor, CLP, common lymphoid progenitor, CMP, common myeloid
progenitor, MCP, mast cell-committed progenitor, GMP, granulocyte/macrophage
progenitor, MEP, megakaryocyte/erythrocyte progenitor. Adopted from Reya 2001 [130]
and Chen 2005 [144].
26

INTRODUCTION _______________________________________________________________________ The roles of c-Kit and SF in the hematopoietic system
In the hematopoietic system c-Kit is expressed on HSCs and progenitors at all developmental
stages [145], on mast cells [146,147], and to some extent on granulocytes and lymphoid cells
[148]. SF, though, is expressed by mast cells [149], fibroblasts [147], stromal cells [150], and on
several cell types in the airways [148]. Signalling via c-Kit influences hematopoiesis in numerous
different ways, extensively reviewed by Broudy 1997 [151]. Inhibition of SF/c-Kit interaction
with a monoclonal antibody abrogates the hematopoietic development, and diminishes the
progenitor pool in the BM, indicating that SF signalling is required for self-renewal of
progenitors at different stages [145,152] and SF is also shown to promote progenitor cell survival
[153]. In HSCs SF promotes survival [154] and sustained self-renewal [155]. Furthermore, SF in
combination with interleukin-11 is shown to favour the long-term repopulating activity of BM
cells [156] and SF also synergises with other cytokines to support colony growth of both
granulocyte-macrophage and erythroid units [151]. In mast cells SF is demonstrated to regulate
survival, proliferation, migration and mediator release [148].
Model systems for hematopoietic cells To investigate the effects of c-kit signalling in hematopoietic cells in a physiologically relevant
environment it would be preferable to use non-transformed primitive cells of human origin. Since
that is problematic for practical and technical reasons different model systems are used.
Studying c-Kit in mast cells is fairly uncomplicated since cultures can be derived from both
embryonic stem (ES) cells and BM and then cultured and expanded for a few weeks without
transforming the cells [157,158]. However, to find suitable systems to study HSC or progenitors
is more complex considering that factors like surface marker expression, cytokine response and
ability to self-renew and differentiate should be fulfilled. In a recent paper Olsen et al. have
reviewed several specific hematopoietic progenitor lines [159]. Additionally there are a few
HSC-like cell lines that have been used to study c-Kit signalling. Bernstein et al. have generated
HCS-like cell lines able to self-renew and commit to both lymphoid and myeloid lineage by
immortalizing ES cells with constitutive Notch signalling [160]. In our studies we have used cell
lines known as hematopoietic progenitor cells (HPC) developed in the laboratory of Leif Carlsson
at Umeå University, and described below.
27

INTRODUCTION _______________________________________________________________________ The Hematopoietic progenitor cell lines
The hematopoietic progenitor cell (HPC) lines are immortalised SF-dependent hematopoietic
stem cell-like cell lines. They are generated by transduction of the Lhx2 (LIM (Lin11, Isl1 and
Mec3) homeobox 2) gene into murine embryonic stem (ES) cells [161] or HSCs derived from
adult BM [162]. The Lhx2 gene encodes a transcription factor that is expressed in the fetal liver
during active hematopoiesis [163] and Lhx2 homozygous mutant mice die before birth due to
severe anaemia [164]. Lhx2 is also believed to play a role in the control of asymmetric cell
division and differentiation of specific cell types, reviewed and discussed in the thesis by
Wandzioch 2004 [165]. The experimental procedure for generating the cell lines is reviewed in
detail by Carlsson et al. [166] and discussed further in the thesis by Pinto do Ó 2002 [167]. The
HPC lines feature several of the characteristics common for early hematopoietic progenitor/stem
cells, including response to cytokines, transcription factor expression and cell surface markers
[161,162,168]. Like wild type HSCs they also display the capacity to differentiate into several
different mature blood cells (e.g. erythrocytes, megakaryocytes, macrophages, granulocytes and
mast cells) in response to cytokine stimulation in vitro [161] and the BM derived HPCs can
reconstitute the hematopoietic system in lethally irradiated mice [162]. Furthermore, they self-
renew by an unknown cell nonautonomous mechanism [168]. These features, in combination
with the possibility for long-term in vitro culture, make the HPC lines a useful model system for
studying c-Kit signalling.
28

AIMS _______________________________________________________________________
AIMS
The aim of this thesis was to analyse the signalling downstream the receptor tyrosine kinase c-Kit
in immature and differentiated hematopoietic cells and to investigate the effects of the activated
signalling pathways on proliferation, survival and differentiation.
The specific aims were:
• To identify what proteins are initially activated upon SF stimulation of c-Kit.
• To study if, and how, the known signalling pathways interact.
• To compare c-Kit signalling in hematopoietic progenitor cells with differentiated
hematopoietic cells.
• To further investigate the effects of PKC in hematopoietic cells.
• To gain insights in the mechanisms behind c-Kit-dependent proliferation and survival of
hematopoietic cells.
29

RESULTS AND DISCUSSION _______________________________________________________________________
RESULTS AND DISCUSSION Paper I “Activation of the MAP kinase pathway by c-Kit is PI3 kinase dependent in hematopoietic progenitor/stem cell lines”
• SF induces transient activation of c-Kit, receptor binding proteins (Grb2, Shc), Ras and
MAPK pathway (Raf, Mek, Erk), and PI3K pathway (PKB).
• SF stimulated Erk activation is PI3K dependent in HPCs, but not in differentiated
hematopoietic cells.
• The PI3K pathway interconnects with the MAPK cascade at the level of Raf.
• Erk and PI3K activity are required for proliferation of HPCs.
• Inhibition of PI3K induces apoptosis.
Erk activation is PI3K dependent in progenitor cells but not in differentiated cells To investigate c-Kit signalling in hematopoietic cells we have used the hematopoietic stem cell-
like cell lines, denoted HPCs. The characteristics of these cells are described in the introduction
section. In Paper I we firstly depict the proteins that are activated upon SF stimulation in this
HSC-like model system with endogenously expressed proteins. To study the signalling
downstream c-Kit in further detail we have used different inhibitors to block the activity of
specific proteins in the known signalling pathways. Surprisingly, we found that activation of Erk
was dependent on PI3K activity since usage of the PI3K specific inhibitors LY294002 [169] and
Wortmannin [170] effectively blocked Erk phosphorylation as well as PKB phosphorylation.
Further analysis showed that the MAPK cascade was targeted at the level of Raf, since Ras
activation was not influenced by PI3K inhibition. Thus, SF stimulated activation of Raf/Mek/Erk
in the HPCs requires PI3K signalling.
In order to find clues to explain the purpose of this crosstalk between the pathways we compared
the c-Kit signalling in these hematopoietic progenitor cell lines with more mature hematopoietic
cells. The mature cells we used were generated by differentiation of the HPCs (ES or BM
generated HPCs) to myeloid committed cells or mast cells. We also used mast cells differentiated
directly from wild type BM. Comparison of the different cell types revealed that the crosstalk was
30
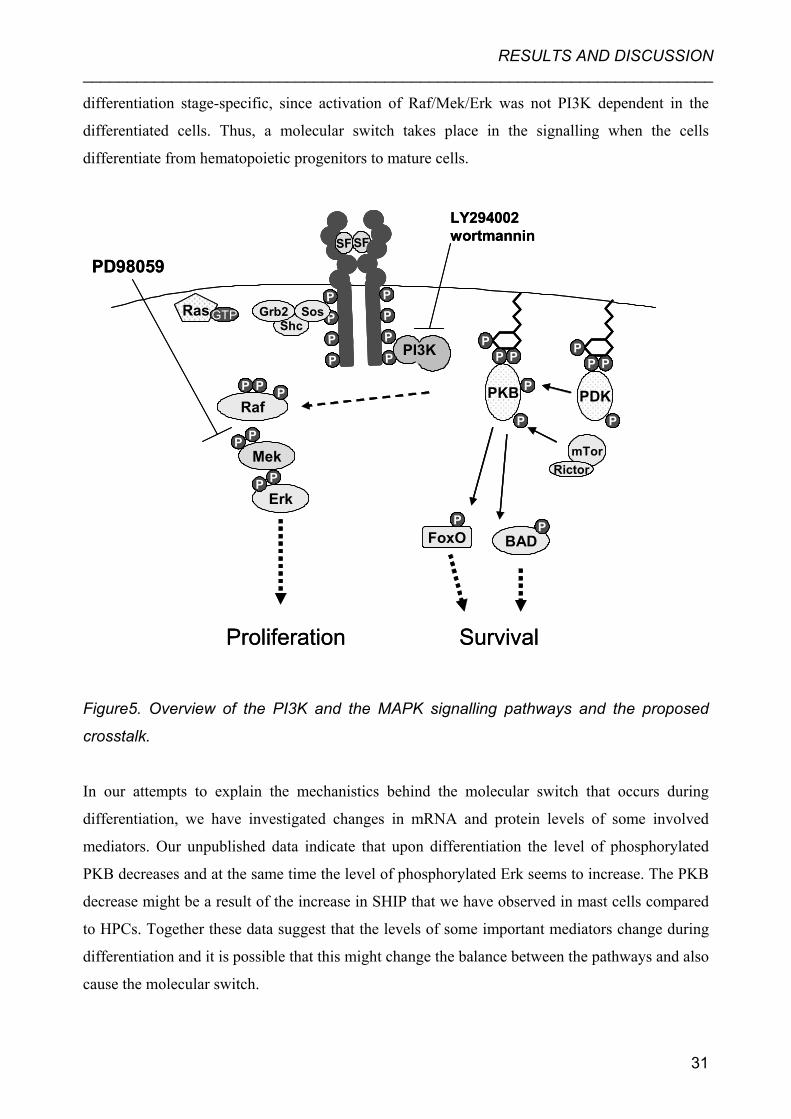
RESULTS AND DISCUSSION _______________________________________________________________________
differentiation stage-specific, since activation of Raf/Mek/Erk was not PI3K dependent in the
differentiated cells. Thus, a molecular switch takes place in the signalling when the cells
differentiate from hematopoietic progenitors to mature cells.
SF SF
PPPP
PPPP P
PP P
PP
PKB P
P
PDKP
PI3K
LY294002wortmannin
Ras GTP
RafP P P
MekPP
ErkP P
ShcGrb2 Sos
mTorRictor
PFoxO BAD
P
SurvivalProliferation
PD98059SF SF
PPPP
PPPP
SF SFSF SF
PPPP
PPPP P
PP P
PP P
PP P
PP
PKB P
P
PDKP
PI3KPI3K
LY294002wortmannin
Ras GTPRas GTP
RafP P P
MekPP
ErkP P
ShcGrb2 Sos
mTorRictor
mTorRictor
PFoxO BAD
PBAD
P
SurvivalProliferation
PD98059
Figure5. Overview of the PI3K and the MAPK signalling pathways and the proposed
crosstalk.
In our attempts to explain the mechanistics behind the molecular switch that occurs during
differentiation, we have investigated changes in mRNA and protein levels of some involved
mediators. Our unpublished data indicate that upon differentiation the level of phosphorylated
PKB decreases and at the same time the level of phosphorylated Erk seems to increase. The PKB
decrease might be a result of the increase in SHIP that we have observed in mast cells compared
to HPCs. Together these data suggest that the levels of some important mediators change during
differentiation and it is possible that this might change the balance between the pathways and also
cause the molecular switch.
31

RESULTS AND DISCUSSION _______________________________________________________________________
As discussed in the introduction section, SF, together with the stem cell niche, is required for
HSC self-renewal and maintenance of the HSC pool [145,152,154,155]. The HPCs though, are
transduced with Lhx2, which maintains the cells in an immature state. It is plausible that Lhx2
induces expression of an unknown stem cell specific factor that in combination with SF, induces
self-renewal. Furthermore, such factor might also be responsible for maintaining Erk activation
PI3K dependent in the immature cells.
A possible function of the differentiation-stage specific crosstalk might be to enable enhanced
control of the balance between proliferation and differentiation. In undifferentiated cells the
crosstalk might function to ensure adequate expansion and prevent pre-mature differentiation of
HSC/HPCs. However, the exact mechanism and function of the crosstalk presented in this study
is yet to be explained.
Crosstalks reported in the literature
Reviewing the literature on the subject of crosstalks between the PI3K and MAPK pathways
reveals that interconnections are actually observed in several different cell types, however with
contrasting mechanisms and outcomes.
Already in 1994 it was reported that PI3K could be a direct target for Ras activation in in vitro
experiments and in COS cells [171,172]. In c-Kit signalling this does not seem to be the case and
PI3K has clearly been demonstrated to become activated upon direct binding to the receptor [69].
However, Zhang et al. have recently made observations in erythroid progenitor cells indicating a
role for K-Ras in PKB activation [173]. Downstream of Ras several mechanistically different
interconnections have been reported. Essentially the field can be divided into two parts; negative
and positive regulation of the MAPK pathway by PI3K.
The groups of Moelling and Glass presented evidence in 1999 that PKB can directly
phosphorylate Raf on serine 259 in human embryonic kidney (HEK) 293 cells stimulated with
insulin-like growth factor (IGF) and that this has an inhibitory effect on Raf activation [85].
Furthermore, they demonstrated that this inhibitory phosphorylation was differentiation stage-
specific, since PKB inhibition of Raf was observed in myotubes, but not in myoblast precursors
[174]. In continuation this crosstalk has also been observed in PDGF stimulated vascular smooth
muscle cells where abrogation of PI3K/PKB lead to a phenotypic modulation [175]. Furthermore,
32

RESULTS AND DISCUSSION _______________________________________________________________________
Moelling et al. have presented results implicating that this type of crosstalk is regulated by ligand
type, concentration and time course and that the function might be to regulate the balance
between proliferation and differentiation [176]. In enterocytic cells it has been demonstrated that
inhibition of PI3K enhances Erk activation when the cells are confluent but not subconfluent.
This correlates with differentiation since upon confluence the cells switch from proliferating to
differentiating [177].
Contrary to the above findings that PKB has an inhibitory effect on Raf there are several reports,
in addition to our study, demonstrating that PI3K activity is required for activation of the MAPK
pathway. Partly confirming our results Chiu et al. have tested a range of hematopoietic cell lines
and found that in some cells MAPK activation was blocked by the PI3K inhibitors LY294002
and wortmannin, but the result differed depending on inhibitor. In this series of experiments the
cells were stimulated with granulocyte-macrophage colony stimulating factor (GM-CSF),
interleukin-3 (IL-3) or serum which might explain the varying results [178]. In erythroid
progenitor cells Ras and the downstream MAPKs were blocked by PI3K inhibitors upon
erythropoietin stimulation, however in contrast to our results, this was not the case upon SF
stimulation in the same cells [179]. In a myeloid progenitor cell line it has been demonstrated that
Mek and A-Raf, but not C-Raf require PI3K for activation upon IL-3 stimulation. This distinction
between the Raf isoforms suggests an additional regulation of the signalling [180]. In T cells a
variant of the crosstalk has been reported, here PI3K overexpression enhances Erk activation and
wortmannin inhibits Erk activation. However, these effects were not observed in signalling
components upstream of Erk [181].
In non-hematopoietic cells there are a several reports that concur with the above findings even
though cell types, stimulation and exact mechanisms diverge [182-191]. Contrary to our findings
in HPCs there are observations indicating that the crosstalk may be receptor concentration
dependent, so that low receptor concentration results in PKB dependent Erk activation, but not if
the receptor is expressed in abundance [185,189].
Erk is required for proliferation of HPCs As authentic HSCs, the HPC lines are cytokine dependent, withdrawal of SF leads to inhibited
proliferation and ultimately cell death ([161] and unpublished data). To investigate the biological
consequences of interfering with the MAPK pathway we used a specific Mek inhibitor and
monitored proliferation and survival. In Paper I we demonstrate that blocking the activity of Mek
33

RESULTS AND DISCUSSION _______________________________________________________________________
results in a dramatic decrease of the rate of growth indicating that activation of the MAPK
pathway is required for proliferation. Further we demonstrate that Mek inhibition only slightly
increases the death rate. Thus, even though the MAPK pathway is required for proliferation it is
not necessary for survival of the HPCs. These data confirm that a main function of the MAPK
pathway is to regulate cell growth [98]. However, in HSCs SF is not enough to induce self-
renewal [154]. Since the HPC lines are transduced with Lhx2 which keep the cells immature, it is
possible that MAPK signalling together with the effects of the transformation promotes self-
renewal in the HPCs.
Activation of the PI3K pathway is crucial for survival The PI3K pathway in hematopoietic cells is, as discussed in the introduction section, an anti-
apoptotic pathway, crucial for the viability of hematopoietic cells [192-194]. In addition to being
involved in survival, PI3K activation has been demonstrated to be a regulator of different mast
cell functions [195,196], macrophage growth [197] and stem cell renewal [198,199]. In Paper I
we utilised the Trypan blue exclusion assay to analyse the survival upon PI3K inhibition with
LY294002. Our results demonstrate a dramatic and rapid increase of cell death under such
conditions. Actually, the rate of survival after PI3K inhibition was as low as upon full withdrawal
of SF (data not shown). The data presented here supports a critical role for PI3K in HPC
physiology. The severe effects following inhibition of PI3K in the HPCs might be a consequence
of the crosstalk since in practice both MAPK and PI3K pathways are inhibited with the single
inhibitor. However, inhibition of PI3K in mast cells induces apoptosis to the same extent as
starvation which suggests that the PI3K pathway alone is necessary for survival of these cells. It
is noticeable though that the effect of starvation and PI3K inhibition is much delayed in mast
cells compared to HPCs (unpublished data).
34

RESULTS AND DISCUSSION _______________________________________________________________________
Paper II “Hematopoietic progenitor cells and mast cells utilize conventional PKC to suppress PKB/Akt activity in response to c-Kit stimulation”
• PKCs are activated upon SF and PMA stimulation of HPCs.
• Raf and downstream mediators, but not Ras and PKB, are activated by PMA.
• PMA induced Raf activation is PI3K independent in the HPCs.
• The cPKCs are required for full Erk activation.
• The cPKCs mediate phosphorylation of S746 on c-Kit.
• Inhibition of the cPKCs leads to increased tyrosine phosphorylation of c-Kit.
• Inhibition of the cPKCs leads to increased PKB phosphorylation and activation in both
HPCs and mast cells.
• The cPKCs are required for HPC proliferation.
• Inhibition of the cPKCs increases the survival rate upon starvation induced apoptosis.
PKC activates Raf, but not Ras or PKB in the HPC lines To investigate the role of PKC in HPCs we started by analysing what proteins are activated by
PMA (phorbol 12-myristate 13-acetate). PMA is a DAG analogue and therefore able to activate
the conventional and novel PKCs [200]. In Paper II we show that PMA treatment of starved
HPCs activates the Raf/Mek/Erk cascade independently of c-Kit and Ras. Furthermore, the PI3K
pathway is not activated by PMA. This is in agreement with studies from other cell systems
showing that PKC phosphorylates Raf directly and thereby activates Raf independently of Ras
[201-204]. As an extension to Paper I we further studied the PI3K dependence of Raf activation.
Here we show that PMA induced Raf activation is PI3K independent in HPCs. This is in contrast
to the SF induced activation, at the level of phosphorylation status of S338 on Raf, which is
known to be important for activation [80]. In addition, comparison of PMA and SF induced
activation of Raf can be used to evaluate the specificity of the crosstalk presented in Paper I.
Since only RTK induced Raf activation is PI3K dependent it is clear that the crosstalk is c-Kit
signalling dependent and not due to unspecific inhibitor effects.
35

RESULTS AND DISCUSSION _______________________________________________________________________
PKC is required for Erk activation and proliferation As discussed above, it is evident that PKCs are activated both by PMA and SF in the HPCs.
However, to study the effects of PKC on c-Kit signalling we have utilised some of the inhibitors
available to block PKC activity. In Paper II we have focused on the conventional PKCs (cPKC)
and in Paper III the effects of the novel PKCδ are investigated. In Paper II we demonstrate that
not only can PKC activate Raf/Mek/Erk, but also that PKC is required for Erk activation.
Ro318220 is a pharmacological inhibitor, which specifically targets the cPKCs [205,206]], and
we demonstrate that pre-treatment with Ro318220 prior to SF stimulation blocks Erk
phosphorylation. We have also used long-term treatment of PMA to inhibit the PKCs and study
the effects on c-Kit signalling. While PMA is known as an activator of PKCs, long-term
treatment depletes the PKC pool and can therefore be used to inhibit the pathway [207]. In
concurrence with Ro318220 treatment we have observed that long-term treatment with PMA
blocks Erk activation in the HPCs. Contrary to this result, Blume-Jensen et al. did not observe an
decrease in Erk phosphorylation upon PKC inhibition in SF stimulated cells [208]. This
discrepancy might be explained by the fact that they use porcine aortic endothelial cells
transfected with c-Kit instead of a model system that examines the endogenous c-Kit receptor.
In Paper I we establish an important role of the Raf/Mek/Erk cascade in the regulation of
proliferation of HPCs. Since we in this study observed that PKC is required for Erk
phosphorylation we decided to monitor proliferation of the HPCs under the influence of
Ro318220. PKC inhibition of HPCs in culture results in total block of growth, but does not seem
to induce significant cell death. This result is similar to what we observed with the Mek inhibitor
in Paper I, which is not surprising since a common effect of these inhibitors is inhibition of Erk
activation.
The cPKC mediates serine phosphorylation of c-Kit Blume-Jensen et al suggested that PKC can phosphorylate c-Kit on serine residues [209]. Further,
they identified S741 and S746 as the major sites targeted by PKC [210]. However, these data are
based on in vitro studies and experiments on the porcine aortic endothelial cells transfected with
and therefore overexpressing c-Kit. In order to investigate whether PKC could phosphorylate c-
Kit in the HPCs as well, we used a novel phospho-specific antibody targeting S746 on c-Kit. In
Paper II we demonstrate that this site is phosphorylated upon SF stimulation and that PKC
inhibition abrogates this phosphorylation. Furthermore, we demonstrate that inhibition of PKC
36

RESULTS AND DISCUSSION _______________________________________________________________________
mediated serine phosphorylation leads to increased overall tyrosine phosphorylation of the c-Kit
receptor, which is in agreement with the studies of Blume-Jensen et al. [209]. Together these data
indicate that PKC and c-Kit constitute a negative feedback loop where c-Kit activates PKC which
subsequently phosphorylates c-Kit on serine residues which negatively regulate c-Kit activation.
A similar type of feedback loop has recently been reported in studies on a chimeric form of the
RTK RET, where they show that PKCα associates with Y1062 on RET resulting in decreased
tyrosine phosphorylation and decreased mitogenicity [211]. Thus, this feedback loop appears to
be a novel mechanism for regulating RTK activation.
Conventional PKCs inhibit PKB activation and promote apoptosis In addition to the results discussed above we have observed that PKC inhibition dramatically
increases PKB activation. Initially we analysed the phosphorylation status of PKB following
inhibition with Ro318220 or depletion of PKCs with PMA long-term treatment and observed that
phosphorylation of PKB is increased, and prolonged, upon SF stimulation of HPCs. To confirm
the effect of PKC on PKB we performed in vitro PKB activity assays, which demonstrate that
PKC severely hampers PKB activation since PKC inhibition with Ro318220 increases PKB
activity 5-6 times in both HPCs and mast cells. This finding might be a consequence of increased
c-Kit activation that is caused by abrogation of the proposed PKC/c-Kit negative feedback loop.
We have observed increased phosphorylation of Y719 in c-Kit and the hypothesis is further
supported by experiments in the porcine aortic endothelial cells indicating increased association
efficiency between c-Kit and PI3K upon PKC inhibition [208]. To further analyse the possible
biological effects of PKC inhibition, and specifically the dramatically increased PKB activity, we
decided to monitor the viability of HPCs. FACS analyses of propidium labelled HPCs show that
Ro318220 treatment rescues cells from starvation induced apoptosis. We believe that this is an
effect of the increased PKB activity in combination with decreased Erk activity, since PKB
signalling is known to block activation of proapoptotic mediators and Erk promotes proliferation
(see introduction and discussion above).
It is noteworthy that the limited numbers of studies on the subject of PKC effects on PKB are
quite contradictory. In a recent report Grandage et al. demonstrated that one PKC inhibitor
(Gö6976) actually inhibits IL-3 and GM-CSF induced signalling, proliferation and survival.
Further they put forward the hypothesis that this inhibitor has no effect on c-Kit signalling [212].
However, in the non-hematopoietic cells A549 and HEK293, it is reported that three different
PKC inhibitors (Ro318220, bisindolylmaleimide VIII, and LY379196), increase PKB activity
37

RESULTS AND DISCUSSION _______________________________________________________________________
and that PMA attenuates the same [213]. Another recent report supports PKC as a negative
regulator of PI3K signalling, but, contrary to our results, suggests that PKC targets PI3K directly
and not via a receptor [214]. It is evident that PKC can influence PKB activity; however it is also
clear that the mode of operation is dependent upon cell system, and possibly, the inhibitor used.
Paper III “PKCδ is required for proliferation and can negatively regulate differentiation in c-
Kit expressing hematopoietic cells”
• Inhibition of PKCδ rescues HPCs from starvation induced apoptosis.
• Inhibition of PKCδ causes G1 arrest in HPCs within 4 hours.
• Long-term treatment with Rottlerin results in cell death of the HPCs.
• Low dose Rottlerin treatment of BM cells accelerates the differentiation process.
• PKCδ is required for SF stimulated Erk activation.
• Inhibition of PKCδ does not affect c-Kit phosphorylation.
Inhibition of PKCδ does not affect c-Kit
In paper II we studied the effects of the conventional PKCs on c-Kit signalling in HPCs. In paper
III we used Rottlerin, an inhibitor that specifically targets PKCδ [215] to investigate how a
member of the novel PKCs affect hematopoietic cells.
Contrary to the conventional PKCs, the novel PKCδ is not required for phosphorylation of S746
on c-Kit, and inhibition with Rottlerin does not increase the overall tyrosine phosphorylation
(data not shown). Furthermore, we show that PKCδ inhibition does not seem to increase PKB
activity, which is concurrent with the lack of effect on the receptor. These differences underline
that effects of the PKC family of serine/threonine kinases are highly diverse.
Rottlerin prevents starvation induced apoptosis
Even though PKCδ is not indicated in c-Kit regulation, we demonstrate that inhibition of PKCδ
with Rottlerin in hematopoietic cells has surprising effects. At first, Rottlerin treatment is
observed to greatly prevent starvation induced apoptosis as measured by FACS analysis of
propidium iodide incorporated cells. However, even though the FACS analysis indicates very low
38

RESULTS AND DISCUSSION _______________________________________________________________________
numbers of apoptotic cells, morphological analysis of the cells tells otherwise. Under the
microscope it is clear that cells are affected by the treatment. HPCs that are starved normally
remain quite small and uniform, with a visibly disintegrated cell membrane, however Rottlerin
treated cells vary more in size and contain large vacuoles. This might indicate that the treatment
induces a mitotic catastrophe (described in [216]) with delayed cell death that is not apparent
with FACS analysis. Hence, we demonstrate that treatment with Rottlerin initially prevents
apoptosis, but that long-term treatment kills the cells. Our results suggest that even though
Rottlerin is a well used inhibitor, it might provide results that are deceiving if not properly used.
In other hematopoietic cells (HL60, Jurkat and RAW264.7 cell lines) Liao et al. have reported
that Rottlerin indeed induces apoptosis in a time and dose dependent manner [217]. In their report
they show that the induced apoptosis is mediated by dissipation of membrane potential,
redistribution of cytochrome C and induction of the caspase cascade [217].
Inhibition of PKCδ causes G1 cell cycle arrest
The rescue from starvation induced apoptosis seen on the histograms is accompanied by a clear
G1 arrest, which might explain why the cells seem viable since it is possible that cell division is a
prerequisite for induction of apoptosis under these circumstances. Since no increase in PKB
phosphorylation was observed upon Rottlerin treatment the apoptotic inhibition observed cannot
be explained by increased PKB activity, as with inhibition of the cPKCs (Paper II). Thus, in
Paper III we show that inhibition of PKCδ induces a PKB independent G1 arrest that blocks
proliferation and prevents induction of apoptosis for up to about ten hours. The observation that
Rottlerin induces G1 arrest is supported by a recent report that shows that PKCδ stimulates G1
phase cell cycle progression in thyroid cells, but subsequently induces S phase arrest and caspase-
dependent apoptosis [218]. Their presentation of a PKCδ induced cell cycle-dependent cell death
supports our hypothesis that proliferation is a prerequisite for apoptosis. The study from Liao et
al. also supports a proliferation repressing effect of Rottlerin [217].
When monitoring the proliferation rate of HPCs under the influence of Rottlerin the cell numbers
were at first stable, but started to decrease after about ten hours, indicating that PKCδ is required
for proliferation and that long-term treatment at the used dose is toxic for the HPCs. In trying to
explain the observed G1 arrest and blocked proliferation we analysed the effects of Rottlerin on
the MAPK pathway downstream of c-Kit. As expected PKCδ, like the cPKCs, was required for
Erk activation.
39

RESULTS AND DISCUSSION _______________________________________________________________________
PKCδ influences differentiation of bone marrow
Our data and others suggest that proliferation and survival are closely linked and it is presumable
that differentiation is a third event that is somehow linked to the first two. To test if PKCδ might
be involved in the differentiation of hematopoietic cells we cultured freshly purified BM
according to protocols for mast cell differentiation (see Paper I), but with the addition of a low
dose of Rottlerin. Surprisingly, we observed that the cells seemed to differentiate faster than in
the absence of Rottlerin. Observations like this have not been reported previously, and the
mechanism behind the accelerated differentiation of BM cells is unknown. It is possible that it is
associated with the inhibited proliferation. Seemingly, the low dose that was used in this
experiment was non-toxic to the cells, but promoted differentiation over proliferation perhaps due
to the inactivation of Erk. This finding further adds to the complexity of PKCδ function.
40

CONCLUSIONS _______________________________________________________________________
CONCLUSIONS
• The canonical signalling pathways, MAPK pathway and PI3K pathway, interconnect in a
differentiation stage-dependent manner in hematopoietic progenitor cells.
• Activation of the PI3K pathway is crucial for the viability of hematopoietic progenitor
cells and mast cells.
• Conventional and novel PKC family members are important regulators of c-Kit signalling
and of hematopoietic cell physiology.
In conclusion, the data presented in this thesis demonstrate that the signalling downstream c-Kit,
and possibly receptor tyrosine kinases in general, is more complex than earlier studies have
suggested. Our data contribute to the idea that receptor tyrosine kinase signalling can be regulated
in a cell stage specific manner in addition to ligand stimulation, concentration and expression
patterns. Furthermore, we show that the receptor itself can be targeted by kinases which add to
the number of regulatory mechanisms. Consequently, these data furthermore underline the
significance of employing appropriate model systems to study signalling downstream receptor
tyrosine kinases. In the hematopoietic cells, where c-Kit plays an important role in progenitors
but not in mature cells, this has great relevance for the understanding of the balance between self-
renewal and differentiation.
41

ACKNOWLEDGEMENTS _______________________________________________________________________
ACKNOWLEDGEMENTS
Finally, the thesis is written and all that remains is to acknowledge the people that made the whole PhD journey possible or just more enjoyable. Great thanks to my supervisor, Bengt, who convinced me to start in the group way long ago. Thanks for teaching me a whole lot about hardcore signalling and for always being enthusiastic about the research and for always trying to see good results no matter the blackness of the film. Moreover, you are the only one I know that find western blots hot, even if they’re not radio-labelled. And thanks for giving me super skies to vasaloppet, that really helped! Ruth, my co-supervisor, many thanks for coming up with smart ideas for the project during the years and special thanks for reading and correcting the thesis and the papers I finally managed to write. Leif Carlsson group, and specially Ewa, who taught me how to manage the cells, provided me with all sorts of factors and for collaborating on our fab blood paper! Lars Rönnstrand and Malin Pedersen for collaboration and for providing antibodies. The Hallberg group; Maria, Anna, Emma, Lubna, Hailing; Thanks a lot, you are a great bunch to have in the lab and as friends! Please disturb me any time now… And thanks to all other students, that passed during the years and made the lab an interesting place to work to say the least. Especially Pawel who actually helped with the project. The RHP group who made Friday morning meetings more populated and more fun. I have to admit that I’m now almost convinced; fruitflies are cool! Caroline, you’re soon finished too! And yes, I think I now know a little about telomerase and bugs as well, thanks Sofie and Margareta! The Med Bio Dep – thanks for creating a very nice place to do research. Folket i korridoren som gör att det är trevligt att gå och fika även på icke-torsdagar☺. Även molbiol folket som man får hänga med fast man blev förflyttad! Sekreterarna/intendenten/diskpersonalen som verkligen tagit hand om den Hallbergska gruppen. Mången administrativt problem och högar med disk har ni fixat bort åt oss. Tack. Stort tack till en samling fantastiska vänner som gjort mitt liv de senaste åren så mycket roligare och som man kan lita på vid alla tänkbara tillfällen. Helga och Marie, jag är tacksam att ni ser till att vi håller kontakten trots att jag blev fast lite längre i norr än vad som var tanken. Linda, tack för att du roat och supportat mig genom flertal mail och besök. Sofia T, nu är vi äntligen färdiga! Nu ska det bli svänga av! Maja, tack för att du varit en sån bra jobb-orelaterad vän. Så till gänget, jag är otroligt tacksam att jag har er till vänner. Ni gör verkligen helgen till fest och vardagen uthärdlig (när det behövs). Sofia, alltid på min sida (förutom i spel), Sara-Rara, du är super! Anna, tur att du inte blev läkare på en gång, för då hade jag missat en fin kompis. Christina, tack detsamma för delade intressen mm☺ Petra, jobba inte ihjäl dig, jag skulle bli så ledsen då . Maria E, det är roligt att ha dig som vän, Gussing också och Gunnarn fast du alltid envisas. Jeanette, du är alltid snäll med mig, David, ack så retsam (men det får man ta om man vill leka med de stora fiskarna). Kicki och Erik, ni är hyvens ni! Magnus, Martina, Johan, Linda, (London lockar lite nu trots allt) Niklas, Maria, Håkan, Henke, Katrin, CharlotteP, KristinaL, jag är glad att många ännu är kvar, saknar dock er som redan flyttat. Min familj, som alltid stöttar mig i alla avseenden och åtminstone låtsas förstå vad jag gör. Alla ni nämda, och en del onämnda, som hjälpt mig fixa de här åren. Tack.
42

REFERENCES _______________________________________________________________________
REFERENCES 1. Levene PA and Alsberg CL (1906) The cleavage products of Vitellin. J Biol Chem 2:127- 133
2. Burnett G and Kennedy EP (1954) The enzymatic phosphorylation of proteins. J Biol Chem 211:969-980
3. Pawson T and Scott JD (2005) Protein phosphorylation in signaling--50 years and counting. Trends Biochem Sci 30:286-290
4. Eckhart W, Hutchinson MA and Hunter T (1979) An activity phosphorylating tyrosine in polyoma T antigen immunoprecipitates. Cell 18:925-933
5. Rubin GM, Yandell MD, Wortman JR, Gabor Miklos GL, Nelson CR, Hariharan IK, Fortini ME, Li PW, Apweiler R, Fleischmann W, Cherry JM, Henikoff S, Skupski MP, Misra S, Ashburner M, Birney E, Boguski MS, Brody T, Brokstein P, Celniker SE, Chervitz SA, Coates D, Cravchik A, Gabrielian A, Galle RF, Gelbart WM, George RA, Goldstein LS, Gong F, Guan P, Harris NL, Hay BA, Hoskins RA, Li J, Li Z, Hynes RO, Jones SJ, Kuehl PM, Lemaitre B, Littleton JT, Morrison DK, Mungall C, O'Farrell PH, Pickeral OK, Shue C, Vosshall LB, Zhang J, Zhao Q, Zheng XH and Lewis S (2000) Comparative genomics of the eukaryotes. Science 287:2204-2215
6. Manning G, Whyte DB, Martinez R, Hunter T and Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298:1912-1934
7. Pawson T (1994) Introduction: protein kinases. Faseb J 8:1112-1113
8. Papin JA, Hunter T, Palsson BO and Subramaniam S (2005) Reconstruction of cellular signalling networks and analysis of their properties. Nat Rev Mol Cell Biol 6:99-111
9. Pawson T (2004) Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 116:191-203
10. Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103:211-225
11. Sadowski I, Stone JC and Pawson T (1986) A noncatalytic domain conserved among cytoplasmic protein-tyrosine kinases modifies the kinase function and transforming activity of Fujinami sarcoma virus P130gag-fps. Mol Cell Biol 6:4396-4408
12. Sicheri F, Moarefi I and Kuriyan J (1997) Crystal structure of the Src family tyrosine kinase Hck. Nature 385:602-609
43

REFERENCES _______________________________________________________________________
13. Xu W, Harrison SC and Eck MJ (1997) Three-dimensional structure of the tyrosine kinase c-Src. Nature 385:595-602
14. Cantley LC and Songyang Z (1994) Specificity in recognition of phosphopeptides by src- homology 2 domains. J Cell Sci Suppl 18:121-126
15. Tonks NK, Diltz CD and Fischer EH (1988) Characterization of the major protein- tyrosine-phosphatases of human placenta. J Biol Chem 263:6731-6737
16. Tonks NK, Diltz CD and Fischer EH (1988) Purification of the major protein-tyrosine- phosphatases of human placenta. J Biol Chem 263:6722-6730
17. Zhang ZY (1998) Protein-tyrosine phosphatases: biological function, structural characteristics, and mechanism of catalysis. Crit Rev Biochem Mol Biol 33:1-52
18. Zhang ZY (2005) Functional studies of protein tyrosine phosphatases with chemical approaches. Biochim Biophys Acta 1754:100-107
19. Wiley HS and Burke PM (2001) Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic 2:12-18
20. Cohen P (2000) The regulation of protein function by multisite phosphorylation-a 25 year update. Trends Biochem Sci 25:596-601
21. Noble ME, Endicott JA and Johnson LN (2004) Protein kinase inhibitors: insights into drug design from structure. Science 303:1800-1805
22. Besmer P, Murphy JE, George PC, Qiu FH, Bergold PJ, Lederman L, Snyder HW, Jr., Brodeur D, Zuckerman EE and Hardy WD (1986) A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature 320:415-421
23. Yarden Y, Kuang WJ, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U and Ullrich A (1987) Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. Embo J 6:3341-3351
24. Sherr CJ, Rettenmier CW, Sacca R, Roussel MF, Look AT and Stanley ER (1985) The c- fms proto-oncogene product is related to the receptor for the mononuclear phagocyte growth factor, CSF-1. Cell 41:665-676
25. Yarden Y, Escobedo JA, Kuang WJ, Yang-Feng TL, Daniel TO, Tremble PM, Chen EY, Ando ME, Harkins RN, Francke U et al. (1986) Structure of the receptor for platelet- derived growth factor helps define a family of closely related growth factor receptors. Nature 323:226-232
44

REFERENCES _______________________________________________________________________
26. Spritz RA, Strunk KM, Lee ST, Lu-Kuo JM, Ward DC, Le Paslier D, Altherr MR, Dorman TE and Moir DT (1994) A YAC contig spanning a cluster of human type III receptor protein tyrosine kinase genes (PDGFRA-KIT-KDR) in chromosome segment 4q12. Genomics 22:431-436
27. Chabot B, Stephenson DA, Chapman VM, Besmer P and Bernstein A (1988) The proto- oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature 335:88-89
28. Geissler EN, Ryan MA and Housman DE (1988) The dominant-white spotting (W) locus of the mouse encodes the c-kit proto-oncogene. Cell 55:185-192
29. Williams DE, Eisenman J, Baird A, Rauch C, Van Ness K, March CJ, Park LS, Martin U, Mochizuki DY, Boswell HS et al. (1990) Identification of a ligand for the c-kit proto- oncogene. Cell 63:167-174
30. Copeland NG, Gilbert DJ, Cho BC, Donovan PJ, Jenkins NA, Cosman D, Anderson D, Lyman SD and Williams DE (1990) Mast cell growth factor maps near the steel locus on mouse chromosome 10 and is deleted in a number of steel alleles. Cell 63:175-183
31. Russell ES (1979) Hereditary anemias of the mouse: a review for geneticists. Adv Genet 20:357-459
32. Miettinen M and Lasota J (2005) KIT (CD117): a review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl Immunohistochem Mol Morphol 13:205-220
33. Kitamura Y, Go S and Hatanaka K (1978) Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood 52:447-452
34. Maeda H, Yamagata A, Nishikawa S, Yoshinaga K, Kobayashi S, Nishi K and Nishikawa S (1992) Requirement of c-kit for development of intestinal pacemaker system. Development 116:369-375
35. Cable J, Barkway C and Steel KP (1992) Characteristics of stria vascularis melanocytes of viable dominant spotting (Wv/Wv) mouse mutants. Hear Res 64:6-20
36. Furitsu T, Tsujimura T, Tono T, Ikeda H, Kitayama H, Koshimizu U, Sugahara H, Butterfield JH, Ashman LK, Kanayama Y et al. (1993) Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest 92:1736-1744
37. Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Druker BJ and Lydon NB (1996) Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2- phenylaminopyrimidine derivative. Cancer Res 56:100-104
45

REFERENCES _______________________________________________________________________
38. Buchdunger E, Cioffi CL, Law N, Stover D, Ohno-Jones S, Druker BJ and Lydon NB (2000) Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther 295:139-145
39. Lu HS, Clogston CL, Wypych J, Fausset PR, Lauren S, Mendiaz EA, Zsebo KM and Langley KE (1991) Amino acid sequence and post-translational modification of stem cell factor isolated from buffalo rat liver cell-conditioned medium. J Biol Chem 266:8102- 8107
40. Lemmon MA, Pinchasi D, Zhou M, Lax I and Schlessinger J (1997) Kit receptor dimerization is driven by bivalent binding of stem cell factor. J Biol Chem 272:6311-6317
41. Zhang Z, Zhang R, Joachimiak A, Schlessinger J and Kong XP (2000) Crystal structure of human stem cell factor: implication for stem cell factor receptor dimerization and activation. Proc Natl Acad Sci U S A 97:7732-7737
42. Blechman JM, Lev S, Barg J, Eisenstein M, Vaks B, Vogel Z, Givol D and Yarden Y (1995) The fourth immunoglobulin domain of the stem cell factor receptor couples ligand binding to signal transduction. Cell 80:103-113
43. Chan PM, Ilangumaran S, La Rose J, Chakrabartty A and Rottapel R (2003) Autoinhibition of the kit receptor tyrosine kinase by the cytosolic juxtamembrane region. Mol Cell Biol 23:3067-3078
44. Roskoski R, Jr. (2005) Structure and regulation of Kit protein-tyrosine kinase--the stem cell factor receptor. Biochem Biophys Res Commun 338:1307-1315
45. Rossi P, Marziali G, Albanesi C, Charlesworth A, Geremia R and Sorrentino V (1992) A novel c-kit transcript, potentially encoding a truncated receptor, originates within a kit gene intron in mouse spermatids. Dev Biol 152:203-207
46. Sette C, Dolci S, Geremia R and Rossi P (2000) The role of stem cell factor and of alternative c-kit gene products in the establishment, maintenance and function of germ cells. Int J Dev Biol 44:599-608
47. Reith AD, Ellis C, Lyman SD, Anderson DM, Williams DE, Bernstein A and Pawson T (1991) Signal transduction by normal isoforms and W mutant variants of the Kit receptor tyrosine kinase. Embo J 10:2451-2459
48. Crosier PS, Ricciardi ST, Hall LR, Vitas MR, Clark SC and Crosier KE (1993) Expression of isoforms of the human receptor tyrosine kinase c-kit in leukemic cell lines and acute myeloid leukemia. Blood 82:1151-1158
49. Caruana G, Cambareri AC and Ashman LK (1999) Isoforms of c-KIT differ in activation of signalling pathways and transformation of NIH3T3 fibroblasts. Oncogene 18:5573- 5581
46

REFERENCES _______________________________________________________________________
50. Voytyuk O, Lennartsson J, Mogi A, Caruana G, Courtneidge S, Ashman LK and Ronnstrand L (2003) Src family kinases are involved in the differential signaling from two splice forms of c-Kit. J Biol Chem 278:9159-9166
51. Huse M and Kuriyan J (2002) The conformational plasticity of protein kinases. Cell 109:275-282
52. Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, Snell GP, Zou H, Sang BC and Wilson KP (2004) Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem 279:31655-31663
53. Mol CD, Lim KB, Sridhar V, Zou H, Chien EY, Sang BC, Nowakowski J, Kassel DB, Cronin CN and McRee DE (2003) Structure of a c-kit product complex reveals the basis for kinase transactivation. J Biol Chem 278:31461-31464
54. Roskoski R, Jr. (2005) Signaling by Kit protein-tyrosine kinase--the stem cell factor receptor. Biochem Biophys Res Commun 337:1-13
55. Wollberg P, Lennartsson J, Gottfridsson E, Yoshimura A and Ronnstrand L (2003) The adapter protein APS associates with the multifunctional docking sites Tyr-568 and Tyr- 936 in c-Kit. Biochem J 370:1033-1038
56. Zeng S, Xu Z, Lipkowitz S and Longley JB (2005) Regulation of stem cell factor receptor signaling by Cbl family proteins (Cbl-b/c-Cbl). Blood 105:226-232
57. Roskoski R, Jr. (2004) Src protein-tyrosine kinase structure and regulation. Biochem Biophys Res Commun 324:1155-1164
58. Price DJ, Rivnay B, Fu Y, Jiang S, Avraham S and Avraham H (1997) Direct association of Csk homologous kinase (CHK) with the diphosphorylated site Tyr568/570 of the activated c-KIT in megakaryocytes. J Biol Chem 272:5915-5920
59. Linnekin D, DeBerry CS and Mou S (1997) Lyn associates with the juxtamembrane region of c-Kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J Biol Chem 272:27450-27455
60. Lennartsson J, Blume-Jensen P, Hermanson M, Ponten E, Carlberg M and Ronnstrand L (1999) Phosphorylation of Shc by Src family kinases is necessary for stem cell factor receptor/c-kit mediated activation of the Ras/MAP kinase pathway and c-fos induction. Oncogene 18:5546-5553
61. Lowenstein EJ, Daly RJ, Batzer AG, Li W, Margolis B, Lammers R, Ullrich A, Skolnik EY, Bar-Sagi D and Schlessinger J (1992) The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 70:431-442
47

REFERENCES _______________________________________________________________________
62. Masson K, Heiss E, Band H and Ronnstrand L (2006) Direct binding of Cbl to Y568 and Y936 of the stem cell factor receptor/c-Kit is required for ligand-induced ubiquitination, internalization and degradation. Biochem J
63. Neel BG, Gu H and Pao L (2003) The 'Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28:284-293
64. Kozlowski M, Larose L, Lee F, Le DM, Rottapel R and Siminovitch KA (1998) SHP-1 binds and negatively modulates the c-Kit receptor by interaction with tyrosine 569 in the c-Kit juxtamembrane domain. Mol Cell Biol 18:2089-2099
65. Thommes K, Lennartsson J, Carlberg M and Ronnstrand L (1999) Identification of Tyr- 703 and Tyr-936 as the primary association sites for Grb2 and Grb7 in the c-Kit/stem cell factor receptor. Biochem J 341 (Pt 1):211-216
66. Brizzi MF, Dentelli P, Lanfrancone L, Rosso A, Pelicci PG and Pegoraro L (1996) Discrete protein interactions with the Grb2/c-Cbl complex in SCF- and TPO-mediated myeloid cell proliferation. Oncogene 13:2067-2076
67. Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296:1655-1657
68. Lev S, Givol D and Yarden Y (1992) Interkinase domain of kit contains the binding site for phosphatidylinositol 3' kinase. Proc Natl Acad Sci U S A 89:678-682
69. Serve H, Hsu YC and Besmer P (1994) Tyrosine residue 719 of the c-kit receptor is essential for binding of the P85 subunit of phosphatidylinositol (PI) 3-kinase and for c-kit- associated PI 3-kinase activity in COS-1 cells. J Biol Chem 269:6026-6030
70. Shivakrupa R and Linnekin D (2005) Lyn contributes to regulation of multiple Kit- dependent signaling pathways in murine bone marrow mast cells. Cell Signal 17:103-109
71. Sattler M, Salgia R, Shrikhande G, Verma S, Pisick E, Prasad KV and Griffin JD (1997) Steel factor induces tyrosine phosphorylation of CRKL and binding of CRKL to a complex containing c-kit, phosphatidylinositol 3-kinase, and p120(CBL). J Biol Chem 272:10248-10253
72. Lennartsson J, Wernstedt C, Engstrom U, Hellman U and Ronnstrand L (2003) Identification of Tyr900 in the kinase domain of c-Kit as a Src-dependent phosphorylation site mediating interaction with c-Crk. Exp Cell Res 288:110-118
73. Carpenter G and Ji Q (1999) Phospholipase C-gamma as a signal-transducing element. Exp Cell Res 253:15-24
74. Rottapel R, Reedijk M, Williams DE, Lyman SD, Anderson DM, Pawson T and Bernstein A (1991) The Steel/W transduction pathway: kit autophosphorylation and its association with a unique subset of cytoplasmic signaling proteins is induced by the Steel factor. Mol Cell Biol 11:3043-3051
48

REFERENCES _______________________________________________________________________
75. Gommerman JL, Sittaro D, Klebasz NZ, Williams DA and Berger SA (2000) Differential stimulation of c-Kit mutants by membrane-bound and soluble Steel Factor correlates with leukemic potential. Blood 96:3734-3742
76. Herbst R, Lammers R, Schlessinger J and Ullrich A (1991) Substrate phosphorylation specificity of the human c-kit receptor tyrosine kinase. J Biol Chem 266:19908-19916
77. Wennerberg K, Rossman KL and Der CJ (2005) The Ras superfamily at a glance. J Cell Sci 118:843-846
78. Linnekin D (1999) Early signaling pathways activated by c-Kit in hematopoietic cells. Int J Biochem Cell Biol 31:1053-1074
79. Qi M and Elion EA (2005) MAP kinase pathways. J Cell Sci 118:3569-3572
80. Wellbrock C, Karasarides M and Marais R (2004) The RAF proteins take centre stage. Nat Rev Mol Cell Biol 5:875-885
81. Moodie SA, Willumsen BM, Weber MJ and Wolfman A (1993) Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 260:1658-1661
82. Marais R, Light Y, Paterson HF and Marshall CJ (1995) Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. Embo J 14:3136-3145
83. Rapp UR, Goldsborough MD, Mark GE, Bonner TI, Groffen J, Reynolds FH, Jr. and Stephenson JR (1983) Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc Natl Acad Sci U S A 80:4218-4222
84. Dumaz N and Marais R (2003) Protein kinase A blocks Raf-1 activity by stimulating 14- 3-3 binding and blocking Raf-1 interaction with Ras. J Biol Chem 278:29819-29823
85. Zimmermann S and Moelling K (1999) Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 286:1741-1744
86. Wu J, Dent P, Jelinek T, Wolfman A, Weber MJ and Sturgill TW (1993) Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3',5'-monophosphate. Science 262:1065-1069
87. Light Y, Paterson H and Marais R (2002) 14-3-3 antagonizes Ras-mediated Raf-1 recruitment to the plasma membrane to maintain signaling fidelity. Mol Cell Biol 22:4984-4996
88. Chiloeches A, Mason CS and Marais R (2001) S338 phosphorylation of Raf-1 is independent of phosphatidylinositol 3-kinase and Pak3. Mol Cell Biol 21:2423-2434
49

REFERENCES _______________________________________________________________________
89. Fabian JR, Daar IO and Morrison DK (1993) Critical tyrosine residues regulate the enzymatic and biological activity of Raf-1 kinase. Mol Cell Biol 13:7170-7179
90. Chow YH, Pumiglia K, Jun TH, Dent P, Sturgill TW and Jove R (1995) Functional mapping of the N-terminal regulatory domain in the human Raf-1 protein kinase. J Biol Chem 270:14100-14106
91. Chong H, Lee J and Guan KL (2001) Positive and negative regulation of Raf kinase activity and function by phosphorylation. Embo J 20:3716-3727
92. Tzivion G, Luo Z and Avruch J (1998) A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature 394:88-92
93. Alessi DR, Saito Y, Campbell DG, Cohen P, Sithanandam G, Rapp U, Ashworth A, Marshall CJ and Cowley S (1994) Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. Embo J 13:1610-1619
94. Crews CM, Alessandrini A and Erikson RL (1992) The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science 258:478-480
95. Crews CM and Erikson RL (1992) Purification of a murine protein-tyrosine/threonine kinase that phosphorylates and activates the Erk-1 gene product: relationship to the fission yeast byr1 gene product. Proc Natl Acad Sci U S A 89:8205-8209
96. Rossomando A, Wu J, Weber MJ and Sturgill TW (1992) The phorbol ester-dependent activator of the mitogen-activated protein kinase p42mapk is a kinase with specificity for the threonine and tyrosine regulatory sites. Proc Natl Acad Sci U S A 89:5221-5225
97. Payne DM, Rossomando AJ, Martino P, Erickson AK, Her JH, Shabanowitz J, Hunt DF, Weber MJ and Sturgill TW (1991) Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). Embo J 10:885-892
98. Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, Franklin RA and McCubrey JA (2003) Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia 17:1263-1293
99. Therrien M, Michaud NR, Rubin GM and Morrison DK (1996) KSR modulates signal propagation within the MAPK cascade. Genes Dev 10:2684-2695
100. Kolch W (2005) Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol 6:827-837
101. Schaeffer HJ, Catling AD, Eblen ST, Collier LS, Krauss A and Weber MJ (1998) MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science 281:1668-1671
50

REFERENCES _______________________________________________________________________
102. Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, Sedivy JM and Kolch W (1999) Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature 401:173-177
103. Emuss V, Garnett M, Mason C and Marais R (2005) Mutations of C-RAF are rare in human cancer because C-RAF has a low basal kinase activity compared with B-RAF. Cancer Res 65:9719-9726
104. Garnett MJ and Marais R (2004) Guilty as charged: B-RAF is a human oncogene. Cancer Cell 6:313-319
105. Vanhaesebroeck B and Alessi DR (2000) The PI3K-PDK1 connection: more than just a road to PKB. Biochem J 346 Pt 3:561-576
106. Isakoff SJ, Cardozo T, Andreev J, Li Z, Ferguson KM, Abagyan R, Lemmon MA, Aronheim A and Skolnik EY (1998) Identification and analysis of PH domain-containing targets of phosphatidylinositol 3-kinase using a novel in vivo assay in yeast. Embo J 17:5374-5387
107. James SR, Downes CP, Gigg R, Grove SJ, Holmes AB and Alessi DR (1996) Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem J 315 (Pt 3):709-713
108. Franke TF, Kaplan DR, Cantley LC and Toker A (1997) Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 275:665-668
109. Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P and Hemmings BA (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. Embo J 15:6541-6551
110. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB and Cohen P (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7:261-269
111. Sarbassov DD, Guertin DA, Ali SM and Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098-1101
112. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL and Sabatini DM (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22:159-168
113. Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM and Hemmings BA (1997) Role of translocation in the activation and function of protein kinase B. J Biol Chem 272:31515-31524
51

REFERENCES _______________________________________________________________________
114. Meier R, Alessi DR, Cron P, Andjelkovic M and Hemmings BA (1997) Mitogenic activation, phosphorylation, and nuclear translocation of protein kinase Bbeta. J Biol Chem 272:30491-30497
115. Cross DA, Alessi DR, Cohen P, Andjelkovich M and Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785-789
116. Shaw M, Cohen P and Alessi DR (1997) Further evidence that the inhibition of glycogen synthase kinase-3beta by IGF-1 is mediated by PDK1/PKB-induced phosphorylation of Ser-9 and not by dephosphorylation of Tyr-216. FEBS Lett 416:307-311
117. Downward J (2004) PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol 15:177-182
118. Cantley LC and Neel BG (1999) New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A 96:4240-4245
119. Kalesnikoff J, Sly LM, Hughes MR, Buchse T, Rauh MJ, Cao LP, Lam V, Mui A, Huber M and Krystal G (2003) The role of SHIP in cytokine-induced signaling. Rev Physiol Biochem Pharmacol 149:87-103
120. Gao T, Furnari F and Newton AC (2005) PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell 18:13- 24
121. Oliva JL, Griner EM and Kazanietz MG (2005) PKC isozymes and diacylglycerol- regulated proteins as effectors of growth factor receptors. Growth Factors 23:245-252
122. Newton AC and Johnson JE (1998) Protein kinase C: a paradigm for regulation of protein function by two membrane-targeting modules. Biochim Biophys Acta 1376:155-172
123. Hurley JH, Newton AC, Parker PJ, Blumberg PM and Nishizuka Y (1997) Taxonomy and function of C1 protein kinase C homology domains. Protein Sci 6:477-480
124. Shao X, Davletov BA, Sutton RB, Sudhof TC and Rizo J (1996) Bipartite Ca2+-binding motif in C2 domains of synaptotagmin and protein kinase C. Science 273:248-251
125. Verdaguer N, Corbalan-Garcia S, Ochoa WF, Fita I and Gomez-Fernandez JC (1999) Ca(2+) bridges the C2 membrane-binding domain of protein kinase Calpha directly to phosphatidylserine. Embo J 18:6329-6338
126. Keranen LM, Dutil EM and Newton AC (1995) Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr Biol 5:1394-1403
127. Newton AC (2001) Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev 101:2353-2364
52

REFERENCES _______________________________________________________________________
128. Liu JP (1996) Protein kinase C and its substrates. Mol Cell Endocrinol 116:1-29
129. Moore MA (1997) Stem cell proliferation: ex vivo and in vivo observations. Stem Cells 15 Suppl 1:239-248; discussion 248-251
130. Reya T, Morrison SJ, Clarke MF and Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414:105-111
131. Siminovitch L, McCulloch EA and Till JE (1963) The Distribution Of Colony-Forming Cells Among Spleen Colonies. J Cell Physiol 62:327-336
132. Morrison SJ and Weissman IL (1994) The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity 1:661-673
133. Morrison SJ, Wandycz AM, Hemmati HD, Wright DE and Weissman IL (1997) Identification of a lineage of multipotent hematopoietic progenitors. Development 124:1929-1939
134. Spangrude GJ, Heimfeld S and Weissman IL (1988) Purification and characterization of mouse hematopoietic stem cells. Science 241:58-62
135. Ford CE, Hamerton JL, Barnes DW and Loutit JF (1956) Cytological identification of radiation-chimaeras. Nature 177:452-454
136. Till JE and McCulloch EA (1961) A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat Res 14:213-222
137. Ho AD (2005) Kinetics and symmetry of divisions of hematopoietic stem cells. Exp Hematol 33:1-8
138. Giebel B, Zhang T, Beckmann J, Spanholtz J, Wernet P, Ho AD and Punzel M (2006) Primitive human hematopoietic cells give rise to differentially specified daughter cells upon their initial cell division. Blood 107:2146-2152
139. Attar EC and Scadden DT (2004) Regulation of hematopoietic stem cell growth. Leukemia 18:1760-1768
140. Rattis FM, Voermans C and Reya T (2004) Wnt signaling in the stem cell niche. Curr Opin Hematol 11:88-94
141. Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R and Weissman IL (2003) A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 423:409-414
142. Kaushansky K (2006) Lineage-specific hematopoietic growth factors. N Engl J Med 354:2034-2045
53

REFERENCES _______________________________________________________________________
143. Okayama Y and Kawakami T (2006) Development, migration, and survival of mast cells. Immunol Res 34:97-115
144. Chen CC, Grimbaldeston MA, Tsai M, Weissman IL and Galli SJ (2005) Identification of mast cell progenitors in adult mice. Proc Natl Acad Sci U S A 102:11408-11413
145. Ogawa M, Matsuzaki Y, Nishikawa S, Hayashi S, Kunisada T, Sudo T, Kina T, Nakauchi H and Nishikawa S (1991) Expression and function of c-kit in hemopoietic progenitor cells. J Exp Med 174:63-71
146. Mayrhofer G, Gadd SJ, Spargo LD and Ashman LK (1987) Specificity of a mouse monoclonal antibody raised against acute myeloid leukaemia cells for mast cells in human mucosal and connective tissues. Immunol Cell Biol 65 (Pt 3):241-250
147. Nocka K, Buck J, Levi E and Besmer P (1990) Candidate ligand for the c-kit transmembrane kinase receptor: KL, a fibroblast derived growth factor stimulates mast cells and erythroid progenitors. Embo J 9:3287-3294
148. Da Silva CA, Reber L and Frossard N (2006) Stem cell factor expression, mast cells and inflammation in asthma. Fundam Clin Pharmacol 20:21-39
149. Zhang S, Anderson DF, Bradding P, Coward WR, Baddeley SM, MacLeod JD, McGill JI, Church MK, Holgate ST and Roche WR (1998) Human mast cells express stem cell factor. J Pathol 186:59-66
150. Heinrich MC, Dooley DC, Freed AC, Band L, Hoatlin ME, Keeble WW, Peters ST, Silvey KV, Ey FS, Kabat D et al. (1993) Constitutive expression of steel factor gene by human stromal cells. Blood 82:771-783
151. Broudy VC (1997) Stem cell factor and hematopoiesis. Blood 90:1345-1364
152. Okada S, Nakauchi H, Nagayoshi K, Nishikawa S, Nishikawa S, Miura Y and Suda T (1991) Enrichment and characterization of murine hematopoietic stem cells that express c-kit molecule. Blood 78:1706-1712
153. Keller JR, Ortiz M and Ruscetti FW (1995) Steel factor (c-kit ligand) promotes the survival of hematopoietic stem/progenitor cells in the absence of cell division. Blood 86:1757-1764
154. Li CL and Johnson GR (1994) Stem cell factor enhances the survival but not the self- renewal of murine hematopoietic long-term repopulating cells. Blood 84:408-414
155. Miller CL, Rebel VI, Helgason CD, Lansdorp PM and Eaves CJ (1997) Impaired steel factor responsiveness differentially affects the detection and long-term maintenance of fetal liver hematopoietic stem cells in vivo. Blood 89:1214-1223
54

REFERENCES _______________________________________________________________________
156. Holyoake TL, Freshney MG, McNair L, Parker AN, McKay PJ, Steward WP, Fitzsimons E, Graham GJ and Pragnell IB (1996) Ex vivo expansion with stem cell factor and interleukin-11 augments both short-term recovery posttransplant and the ability to serially transplant marrow. Blood 87:4589-4595
157. Tsai M, Tam SY, Wedemeyer J and Galli SJ (2002) Mast cells derived from embryonic stem cells: a model system for studying the effects of genetic manipulations on mast cell development, phenotype, and function in vitro and in vivo. Int J Hematol 75:345-349
158. Valent P, Sillaber C and Bettelheim P (1991) The growth and differentiation of mast cells. Prog Growth Factor Res 3:27-41
159. Olsen AL, Stachura DL and Weiss MJ (2006) Designer blood: creating hematopoietic lineages from embryonic stem cells. Blood 107:1265-1275
160. Varnum-Finney B, Xu L, Brashem-Stein C, Nourigat C, Flowers D, Bakkour S, Pear WS and Bernstein ID (2000) Pluripotent, cytokine-dependent, hematopoietic stem cells are immortalized by constitutive Notch1 signaling. Nat Med 6:1278-1281
161. Pinto do OP, Kolterud A and Carlsson L (1998) Expression of the LIM-homeobox gene LH2 generates immortalized steel factor-dependent multipotent hematopoietic precursors. Embo J 17:5744-5756
162. Pinto do OP, Richter K and Carlsson L (2002) Hematopoietic progenitor/stem cells immortalized by Lhx2 generate functional hematopoietic cells in vivo. Blood 99:3939- 3946
163. Xu Y, Baldassare M, Fisher P, Rathbun G, Oltz EM, Yancopoulos GD, Jessell TM and Alt FW (1993) LH-2: a LIM/homeodomain gene expressed in developing lymphocytes and neural cells. Proc Natl Acad Sci U S A 90:227-231
164. Porter FD, Drago J, Xu Y, Cheema SS, Wassif C, Huang SP, Lee E, Grinberg A, Massalas JS, Bodine D, Alt F and Westphal H (1997) Lhx2, a LIM homeobox gene, is required for eye, forebrain, and definitive erythrocyte development. Development 124:2935-2944
165. Wandzioch E (2004) The role of Lhx2 in the hematopoietic stem cell function, liver development and disease. Thesis
166. Carlsson L, Wandzioch E, Pinto do OP and Kolterud A (2003) Establishment of multipotent hematopoietic progenitor cell lines from ES cells differentiated in vitro. Methods Enzymol 365:202-214
167. Pinto do Ó P (2002) Generation of hematopoietic stem cell-like cell lines, characterisation in vitro and in vivo. Thesis
55

REFERENCES _______________________________________________________________________
168. Pinto do OP, Wandzioch E, Kolterud A and Carlsson L (2001) Multipotent hematopoietic progenitor cells immortalized by Lhx2 self-renew by a cell nonautonomous mechanism. Exp Hematol 29:1019-1028
169. Vlahos CJ, Matter WF, Hui KY and Brown RF (1994) A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem 269:5241-5248
170. Powis G, Bonjouklian R, Berggren MM, Gallegos A, Abraham R, Ashendel C, Zalkow L, Matter WF, Dodge J, Grindey G et al. (1994) Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res 54:2419-2423
171. Kodaki T, Woscholski R, Hallberg B, Rodriguez-Viciana P, Downward J and Parker PJ (1994) The activation of phosphatidylinositol 3-kinase by Ras. Curr Biol 4:798-806
172. Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD and Downward J (1994) Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 370:527-532
173. Zhang J and Lodish HF (2005) Identification of K-ras as the major regulator for cytokine- dependent Akt activation in erythroid progenitors in vivo. Proc Natl Acad Sci U S A 102:14605-14610
174. Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K, Moelling K, Yancopoulos GD and Glass DJ (1999) Differentiation stage-specific inhibition of the Raf- MEK-ERK pathway by Akt. Science 286:1738-1741
175. Reusch HP, Zimmermann S, Schaefer M, Paul M and Moelling K (2001) Regulation of Raf by Akt controls growth and differentiation in vascular smooth muscle cells. J Biol Chem 276:33630-33637
176. Moelling K, Schad K, Bosse M, Zimmermann S and Schweneker M (2002) Regulation of Raf-Akt Cross-talk. J Biol Chem 277:31099-31106
177. Laprise P, Langlois MJ, Boucher MJ, Jobin C and Rivard N (2004) Down-regulation of MEK/ERK signaling by E-cadherin-dependent PI3K/Akt pathway in differentiating intestinal epithelial cells. J Cell Physiol 199:32-39
178. Chiu D, Ma K, Scott A and Duronio V (2005) Acute activation of Erk1/Erk2 and protein kinase B/akt proceed by independent pathways in multiple cell types. Febs J 272:4372- 4384
179. Schmidt EK, Fichelson S and Feller SM (2004) PI3 kinase is important for Ras, MEK and Erk activation of Epo-stimulated human erythroid progenitors. BMC Biol 2:7
56

REFERENCES _______________________________________________________________________
180. Sutor SL, Vroman BT, Armstrong EA, Abraham RT and Karnitz LM (1999) A phosphatidylinositol 3-kinase-dependent pathway that differentially regulates c-Raf and A-Raf. J Biol Chem 274:7002-7010
181. Von Willebrand M, Jascur T, Bonnefoy-Berard N, Yano H, Altman A, Matsuda Y and Mustelin T (1996) Inhibition of phosphatidylinositol 3-kinase blocks T cell antigen receptor/CD3-induced activation of the mitogen-activated kinase Erk2. Eur J Biochem 235:828-835
182. Cross DA, Alessi DR, Vandenheede JR, McDowell HE, Hundal HS and Cohen P (1994) The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin, but not by rapamycin: evidence that wortmannin blocks activation of the mitogen-activated protein kinase pathway in L6 cells between Ras and Raf. Biochem J 303 (Pt 1):21-26
183. Hu Q, Klippel A, Muslin AJ, Fantl WJ and Williams LT (1995) Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science 268:100-102
184. Standaert ML, Bandyopadhyay G and Farese RV (1995) Studies with wortmannin suggest a role for phosphatidylinositol 3-kinase in the activation of glycogen synthase and mitogen-activated protein kinase by insulin in rat adipocytes: comparison of insulin and protein kinase C modulators. Biochem Biophys Res Commun 209:1082-1088
185. Duckworth BC and Cantley LC (1997) Conditional inhibition of the mitogen-activated protein kinase cascade by wortmannin. Dependence on signal strength. J Biol Chem 272:27665-27670
186. Grammer TC and Blenis J (1997) Evidence for MEK-independent pathways regulating the prolonged activation of the ERK-MAP kinases. Oncogene 14:1635-1642
187. Lopez-Ilasaca M, Crespo P, Pellici PG, Gutkind JS and Wetzker R (1997) Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase gamma. Science 275:394-397
188. Bondeva T, Pirola L, Bulgarelli-Leva G, Rubio I, Wetzker R and Wymann MP (1998) Bifurcation of lipid and protein kinase signals of PI3Kgamma to the protein kinases PKB and MAPK. Science 282:293-296
189. Wennstrom S and Downward J (1999) Role of phosphoinositide 3-kinase in activation of ras and mitogen-activated protein kinase by epidermal growth factor. Mol Cell Biol 19:4279-4288
190. Yart A, Laffargue M, Mayeux P, Chretien S, Peres C, Tonks N, Roche S, Payrastre B, Chap H and Raynal P (2001) A critical role for phosphoinositide 3-kinase upstream of Gab1 and SHP2 in the activation of ras and mitogen-activated protein kinases by epidermal growth factor. J Biol Chem 276:8856-8864
57

REFERENCES _______________________________________________________________________
191. Campbell M, Allen WE, Sawyer C, Vanhaesebroeck B and Trimble ER (2004) Glucose- potentiated chemotaxis in human vascular smooth muscle is dependent on cross-talk between the PI3K and MAPK signaling pathways. Circ Res 95:380-388
192. Young SM, Cambareri AC and Ashman LK (2006) Role of c-KIT expression level and phosphatidylinositol 3-kinase activation in survival and proliferative responses of early myeloid cells. Cell Signal 18:608-620
193. Engstrom M, Karlsson R and Jonsson JI (2003) Inactivation of the forkhead transcription factor FoxO3 is essential for PKB-mediated survival of hematopoietic progenitor cells by kit ligand. Exp Hematol 31:316-323
194. Blume-Jensen P, Janknecht R and Hunter T (1998) The kit receptor promotes cell survival via activation of PI 3-kinase and subsequent Akt-mediated phosphorylation of Bad on Ser136. Curr Biol 8:779-782
195. Serve H, Yee NS, Stella G, Sepp-Lorenzino L, Tan JC and Besmer P (1995) Differential roles of PI3-kinase and Kit tyrosine 821 in Kit receptor-mediated proliferation, survival and cell adhesion in mast cells. Embo J 14:473-483
196. Vosseller K, Stella G, Yee NS and Besmer P (1997) c-kit receptor signaling through its phosphatidylinositide-3'-kinase-binding site and protein kinase C: role in mast cell enhancement of degranulation, adhesion, and membrane ruffling. Mol Biol Cell 8:909- 922
197. Munugalavadla V, Borneo J, Ingram DA and Kapur R (2005) p85alpha subunit of class IA PI-3 kinase is crucial for macrophage growth and migration. Blood 106:103-109
198. Haneline LS, White H, Yang FC, Chen S, Orschell C, Kapur R and Ingram DA (2006) Genetic reduction of class IA PI-3 kinase activity alters fetal hematopoiesis and competitive repopulating ability of hematopoietic stem cells in vivo. Blood 107:1375- 1382
199. Paling NR, Wheadon H, Bone HK and Welham MJ (2004) Regulation of embryonic stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J Biol Chem 279:48063-48070
200. Liu WS and Heckman CA (1998) The sevenfold way of PKC regulation. Cell Signal 10:529-542
201. Cai H, Smola U, Wixler V, Eisenmann-Tappe I, Diaz-Meco MT, Moscat J, Rapp U and Cooper GM (1997) Role of diacylglycerol-regulated protein kinase C isotypes in growth factor activation of the Raf-1 protein kinase. Mol Cell Biol 17:732-741
202. Schonwasser DC, Marais RM, Marshall CJ and Parker PJ (1998) Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol Cell Biol 18:790-798
58

REFERENCES _______________________________________________________________________
203. Sozeri O, Vollmer K, Liyanage M, Frith D, Kour G, Mark GE, 3rd and Stabel S (1992) Activation of the c-Raf protein kinase by protein kinase C phosphorylation. Oncogene 7:2259-2262
204. Ueda Y, Hirai S, Osada S, Suzuki A, Mizuno K and Ohno S (1996) Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J Biol Chem 271:23512-23519
205. Way KJ, Chou E and King GL (2000) Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol Sci 21:181-187
206. Davies SP, Reddy H, Caivano M and Cohen P (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351:95-105
207. Krug E, Biemann HP and Tashjian AH, Jr. (1987) Evidence for increased synthesis as well as increased degradation of protein kinase C after treatment of human osteosarcoma cells with phorbol ester. J Biol Chem 262:11852-11856
208. Blume-Jensen P, Ronnstrand L, Gout I, Waterfield MD and Heldin CH (1994) Modulation of Kit/stem cell factor receptor-induced signaling by protein kinase C. J Biol Chem 269:21793-21802
209. Blume-Jensen P, Siegbahn A, Stabel S, Heldin CH and Ronnstrand L (1993) Increased Kit/SCF receptor induced mitogenicity but abolished cell motility after inhibition of protein kinase C. Embo J 12:4199-4209
210. Blume-Jensen P, Wernstedt C, Heldin CH and Ronnstrand L (1995) Identification of the major phosphorylation sites for protein kinase C in kit/stem cell factor receptor in vitro and in intact cells. J Biol Chem 270:14192-14200
211. Andreozzi F, Melillo RM, Carlomagno F, Oriente F, Miele C, Fiory F, Santopietro S, Castellone MD, Beguinot F, Santoro M and Formisano P (2003) Protein kinase Calpha activation by RET: evidence for a negative feedback mechanism controlling RET tyrosine kinase. Oncogene 22:2942-2949
212. Grandage VL, Everington T, Linch DC and Khwaja A (2006) Go6976 is a potent inhibitor of the JAK 2 and FLT3 tyrosine kinases with significant activity in primary acute myeloid leukaemia cells. Br J Haematol
213. Wen HC, Huang WC, Ali A, Woodgett JR and Lin WW (2003) Negative regulation of phosphatidylinositol 3-kinase and Akt signalling pathway by PKC. Cell Signal 15:37-45
214. Sipeki S, Bander E, Parker PJ and Farago A (2006) PKCalpha reduces the lipid kinase activity of the p110alpha/p85alpha PI3K through the phosphorylation of the catalytic subunit. Biochem Biophys Res Commun 339:122-125
59

REFERENCES _______________________________________________________________________
215. Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G and Marks F (1994) Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun 199:93- 98
216. Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, Zhivotovsky B, Blagosklonny MV, Malorni W, Knight RA, Piacentini M, Nagata S and Melino G (2005) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ 12 Suppl 2:1463-1467
217. Liao YF, Hung YC, Chang WH, Tsay GJ, Hour TC, Hung HC and Liu GY (2005) The PKC delta inhibitor, rottlerin, induces apoptosis of haematopoietic cell lines through mitochondrial membrane depolarization and caspases' cascade. Life Sci 77:707-719
218. Santiago-Walker AE, Fikaris AJ, Kao GD, Brown EJ, Kazanietz MG and Meinkoth JL (2005) Protein kinase C delta stimulates apoptosis by initiating G1 phase cell cycle progression and S phase arrest. J Biol Chem 280:32107-32114
60





























