Embed Size (px)
Citation preview

ORIGINAL ARTICLE 658J o u r n a l o fJ o u r n a l o f
CellularPhysiologyCellularPhysiology
BMP-3 Promotes MesenchymalStem Cell Proliferation Throughthe TGF-b/Activin SignalingPathway
AARON STEWART, HAIYAN GUAN AND KAIPING YANG*Departments of Obstetrics and Gynaecology and Physiology and Pharmacology, The University of Western Ontario,
Children’s Health Research Institute-Lawson Health Research Institute, London, Ontario, Canada
Adipogenesis plays a key role in the pathogenesis of obesity. It begins with the commitment of mesenchymal stem cells (MSCs) to theadipocyte lineage, followed by terminal differentiation of preadipocytes to mature adipocytes. A critical, but poorly understood,component of adipogenesis involves proliferation of MSCs and preadipocytes. The present study was undertaken to examine thehypothesis that bone morphogenetic protein-3 (BMP-3) promotes adipogenesis using C3H10T1/2 MSCs and 3T3-L1 preadipocytes as invitro model systems. We demonstrated that although it did not promote the commitment of MSCs to the adipocyte lineage or thedifferentiation of preadipocytes to adipocytes, BMP-3-stimulated proliferation by threefold in both cell types. Owing to a lack ofinformation on MSC proliferation, we then delineated the molecular mechanisms underlying BMP-3-stimulated MSC proliferation. Weshowed that BMP-3 activated the transforming growth factor-b (TGF-b)/activin but not ERK1/2, p38 MAPK, or JNK signaling pathways inC3H10T1/2 cells. Furthermore, the TGF-b/activin receptor kinase inhibitor SB-431542 blocked BMP-3-stimulated proliferation.Importantly, siRNA-mediated knockdown of the key TGF-b/activin signaling pathway components, ActRIIB, ALK4, or Smad2, abrogatedthe mitogenic effects of BMP-3 on MSCs. Together, these results demonstrate that BMP-3 stimulates MSC proliferation via the TGF-b/activin signaling pathway, thus revealing a novel role for this divergent and poorly understood member of the TGF-b superfamily inregulating MSC proliferation.
J. Cell. Physiol. 223: 658–666, 2010. � 2010 Wiley-Liss, Inc.
Contract grant sponsor: Heart & Stroke Foundation of Ontario;Contract grant number: NA-6049.Contract grant sponsor: Canadian Institutes of Health Research;Contract grant number: MOP-79484.
*Correspondence to: Kaiping Yang, Children’s Health ResearchInstitute, Room A5-132, 800 Commissioners Road East, London,Ontario, Canada N6C 2V5. E-mail: [email protected]
Received 23 August 2009; Accepted 14 December 2009
Published online in Wiley InterScience(www.interscience.wiley.com.), 8 February 2010.DOI: 10.1002/jcp.22064
Obesity is becoming a global health crisis, not only because of itsincreasing prevalence but also because it significantly impairsquality of life and increases the risk of cardiovascular andmetabolic diseases as well as certain cancers (Dixon, 2009;Eckel et al., 2005). Obesity is characterized by excess adiposetissue expansion in the fat depots in the body. Visceral or intra-abdominal obesity is the best predictor of obesity-associatedmorbidity and mortality (Montague and O’Rahilly, 2000).
Although the etiology of visceral obesity remains largelyunknown, accumulating epidemiological evidence indicates thatlow birth-weight is associated with increased visceral adiposityin adult life, suggesting an early-life origin for this disorder(Osmond and Barker, 2000). Recently, we have demonstratedthat poor early nutrition, evoked by maternal proteinrestriction (MPR) during pregnancy and lactation, leads to lowbirth weight and subsequent development of increased visceraladiposity in adult male rat offspring (Guan et al., 2005).Moreover, we have obtained evidence suggesting that increasedvisceral adiposity in our rat model is characterized by adipocytehyperplasia. Indeed, adipocyte precursor cells derived fromMPR offspring exhibit accelerated rate of proliferation, even afew days after removal from their in vivo environment (Zhanget al., 2007). This suggests that MPR permanently altersadipocyte development, but the factors and molecularmechanisms that are responsible for programming this aberrantphenotype remain largely unknown.
As a first step in identifying the causal factors involved, weutilized a candidate gene approach by capitalizing on ourpreviously published visceral adipose tissue gene expressionprofiling database generated with our early-life programmed ratmodel of increased visceral adiposity (Guan et al., 2005). Ourprimary objective was to identify novel factors that promoteadipogenesis. Consequently, candidate genes were selectedbased on the following criteria. (a) They are known to be
� 2 0 1 0 W I L E Y - L I S S , I N C .
involved in the commitment of mesenchymal stem cells (MSCs)to specific cell lineages, because adipogenesis begins with thecommitment of MSCs to the adipocyte lineage (Rosen andMacDougald, 2006), the regulation of which is largely unknown.(b) They stimulate non-adipose cell proliferation and, ordifferentiation, since the second step in adipogenesis is terminaldifferentiation of preadipocytes to mature adipocytes (Rosenand MacDougald, 2006). Importantly, a critical but poorlyunderstood component of adipogenesis involves self-renewal/proliferation of MSCs and preadipocytes (Zhang and Li, 2005).(c) Their expression is up-regulated in our rat model. One ofsuch candidates is the gene encoding bone morphogeneticprotein-3 (BMP-3), the expression of which is increased�2-fold (Guan et al., 2005). BMP-3 is a member of thetransforming growth factor-b (TGF-b) superfamily(Bahamonde and Lyons, 2001). Although it was originallythought to be a trophic factor for bone growth because it waspresent in large quantities in bone (Sampath et al., 1987),subsequent gene targeting studies revealed that BMP-3 wasanti-osteogenic (Bahamonde and Lyons, 2001).

B M P - 3 P R O M O T E S S T E M C E L L P R O L I F E R A T I O N 659
Given that there is a reciprocal relationship betweenosteogenesis and adipogenesis whereby factors that promoteosteogenesis inhibit adipocyte formation and vice versa (Jeonet al., 2003; Lecka-Czernik et al., 2002), coupled with the recentfindings that BMP-3 expression was increased 1.9-fold in theadipose tissue of a diet-induced mouse model of obesity and theenhanced BMP-3 expression proceeded increases in fat mass(Koza et al., 2006), the present study was undertaken toexamine the hypothesis that BMP-3 stimulates adipogenesis. Inaddition, we explored the possibility that BMP-3 may stimulateproliferation of MSCs and/or preadipocytes, because othermembers of the TGF-b superfamily are known to promoteproliferation of stem cells and other progenitor cells (Feng andDerynck, 2005). The C3H10T1/2 stem cells and 3T3-L1preadipocytes were used as in vitro model systems.
Materials and MethodsCell culture
The 3T3-L1 preadipocyte line and the C3H10T1/2 MSC line wereobtained from the American Type Culture Collection (Manassas,VA). C2C12 cells were a generous gift from Dr. Timothy Regnault(University of Western Ontario, London, ON, Canada). Primarypreadipocytes were isolated from epididymal and perirenal fatpads, which were collected from male Wistar rats (Charles RiverLaboratories, Wilmington, MA) at 8 weeks of age, and their purityverified by >95% conversion to adipocytes as determined by OilRed O staining following an established differentiation protocol, asdescribed previously (Zhang et al., 2007). 3T3-L1 preadipocyteswere cultured in standard growth medium consisting of Dulbecco’sModified Eagle’s Medium (DMEM; Sigma–Aldrich Canada Ltd,Oakville, ON, Canada) and 10% newborn calf serum (NCS; Sigma).Primary preadipocytes were cultured DMEM/F-12 medium(Invitrogen Canada Inc., Burlington, ON) supplemented with 10%fetal bovine serum (FBS) (Sigma). C3H10T1/2 C2C12 cells werecultured in standard growth medium consisting of minimal essentialmedium (MEM; Sigma) and 10% FBS. All cultures were maintained ina humidified incubator at 378C and 5% CO2. Growth medium wasreplaced every other day.
Commitment to the adipocyte lineage
To determine if BMP-3 promotes the commitment of MSCs to theadipocyte lineage, C3H10T1/2 stem cells were subjected to apublished treatment protocol, as described previously (Tang et al.,2004). Briefly, C3H10T1/2 stem cells were plated at low density(20–30% confluence) on 12-well plates, and cultured in standardgrowth medium for 2 days. Cells were then treated with increasingconcentrations of recombinant human BMP-3 and/or BMP-4 (R&DSystems, Minneapolis, MN) until 2 days post-confluence (day 0), atwhich time they were induced to differentiate by the addition of astandard differentiation cocktail containing 0.5 mM of 3-isobutyl-1-methylxanthine (IBMX; Sigma), 0.25mM of dexamethasone(SABEX, Boucherville, QC, Canada), and 1mg/ml of insulin (Eli LillyCanada, Inc., Toronto, ON, Canada). After 48 h (day 2), themedium was replaced with growth medium supplemented with1mg/ml insulin. Subsequently, the medium was changed at days 4and 6 with fresh growth medium. Both positive (100 ng/ml BMP-4)and negative (cocktail only) controls were also included. At day 8,differentiation was assessed by Oil Red O staining of lipid droplets,as described below.
Adipocyte differentiation
3T3-L1 preadipocytes were cultured and differentiated intoadipocytes, as described previously (Yang et al., 2008). Briefly, cellswere grown in growth medium and allowed to reach confluence.At 2 days post-confluence (day 0), cells were induced todifferentiate by the addition of a differentiation cocktail containing
JOURNAL OF CELLULAR PHYSIOLOGY
0.5 mM IBMX, 0.25mM dexamethasone, and 1mg/ml insulin, asdescribed above. After 48 h (day 2), the medium was replaced withgrowth medium supplemented with 1mg/ml insulin. Subsequently,the medium was changed at days 4 and 6 with fresh growth medium.On day 8, >90% of cells had acquired adipocyte phenotype. Tostudy the effects of BMP-3 on adipocyte differentiation, 3T3-L1preadipocytes were subjected to the same differentiation protocolexcept that insulin was replaced with BMP-3 (50, 100, and200 ng/ml). Positive (standard cocktail) and negative (IBMX anddexamethasone only) controls were also included. At day 8,differentiation was assessed by Oil Red O staining of lipid droplets,as described below.
Oil Red O staining
Oil Red O staining was performed as described previously (Yanget al., 2008). Briefly, differentiated adipocyte monolayers werewashed with DPBS, fixed for 1 h with 10% formalin at roomtemperature, and incubated in 60% isopropanol for 5 min. Oil RedO (3 g/L; Sigma) in 99% isopropanol was diluted with water, filtered,and added to the fixed cell monolayers for 5 min. Nuclei were thenstained with hematoxylin for 10 sec. Cell monolayers were thenwashed with water, and the stained triglyceride droplets werevisualized and photographed.
Proliferation assay-[3H]-thymidine incorporation
Proliferation capacity of C3H10T1/2 and C2C12 cells as well as3T3-L1 and rat primary preadipocytes was assessed by measuring[3H]-thymidine incorporation, as described previously (Yang et al.,2008). Briefly, cells were plated at low density (20–30% confluence)on 24-well plates and cultured in growth medium for 2–3 days until50–60% confluence. Cells were growth arrested in serum-freemedium for 20–24 h and were then treated in the serum-freemedium with increasing concentrations of BMP-3 (1–100 ng/ml) for24 h. For inhibitor studies, cells were pretreated for 1 h with U0126(Calbiochem, EMD Chemicals, Inc., San Diego, CA), SB-202190(Alexis Biochemicals, Plymouth Meeting, PA), or SB-431542(Sigma). Subsequently, cells were treated with or without 100 ng/ml BMP-3 for 24 h. During the last 4 h of treatment, cells werepulsed labeled with [3H]-thymidine (0.5mCi/well; 75.2 Ci/mmol,PerkinElmer Life and Analytical Sciences, Woodbridge, ON,Canada). Cells were washed twice with ice-cold phosphate-buffered saline (PBS), once with 5% trichloroacetic acid (TCA) andtwice with 95% ethanol. Cells were then solubilized by the additionof 400ml of 0.5 M NaOH. The solubilized cell lysate (300ml) wasadded to 4 ml of scintillation fluid, and the incorporation of [3H]-thymidine into DNA was determined by scintillation counting.Results were expressed as a percentage of control.
Analysis of TGF-b/activin pathway componentexpression—RT-PCR
Expression of ActRIIB, ALK-4, Smad2, Smad3, and Smad4 wasanalyzed by standard RT-PCR. Briefly, total RNA was isolated fromC3H10T1/2 cells using RNeasy Mini Kit (Qiagen, Mississauga, ON,Canada) coupled with on-column DNase digestion with theRNase-Free DNase Set (Qiagen) according to the manufacturer’sinstructions. One microgram of total RNA was reverse transcribedin a volume of 20ml with the High Capacity cDNA Archive Kit(Applied Biosystems, Foster City, CA), following themanufacturer’s instructions. For every RT reaction, one RNAsample was set-up without reverse transcriptase enzyme toprovide a negative control against possible genomic DNAcontamination. The primers specific for mouse ActRIIB, ALK-4,Smad2, Smad3, and Smad4 as well as their expected product sizesare shown in Table 1. PCR reactions were performed for 32 cycleswith denaturing at 958C, annealing at 588C, and extension at 728C.PCR products were confirmed with standard restriction enzymedigestions and sequencing analysis.

TABLE 1. Primers for the TGF-b/activin pathway components RT-PCR
Gene Primer sequence Product size (bp)
ActRIIB Forward: 50-ATCGTCATCGGAAACCTCCC; reverse: 50-CAGCCAGTGATCCTTAATC 929ALK-4 Forward: 50-ACCGCTACACAGTGACCATT; reverse: 50-TCTTCACATCTTCCTGCACG 630Smad2 Forward: 50-CGGAGATTCTAACAGAACTG; reverse: 50-TGCTTGAGCATCGCACTGAA 846Smad3 Forward: 50-AGCACACAATAACTTGGACC; reverse: 50-TAAGACACACTGGAACAGCGGATG 636Smad4 Forward: 50-GATCTATGCCCGTCTGTGGAGGTG; reverse: 50-AATACTGGCCGGCTGACTTGTGGA 403
Fig. 1. Effects of BMP-3 on proliferation of C3H10T1/2 stem cellsand 3T3-L1 preadipocytes. C3H10T1/2 stem cells (A) and 3T3-L1preadipocytes (B) were cultured in standard growth medium (10%FBS) until 50–60% confluence, at which time cells were starved for24 h in serum-free medium. Cells were then treated with increasingconcentration of BMP-3 (1–100 ng/ml) in serum-free medium for 24 h.During the last 4 h of treatment, cells were pulse labeled with[3H]-thymidine (0.5mCi/well) and the rate of [3H]-thymidineincorporation was determined. Data are means W SEM, n U 4–5independent experiments, each performed in triplicate (MP < 0.05,MMP < 0.01, MMMP < 0.001 vs. control).
660 S T E W A R T E T A L .
siRNA-mediated knockdown of the key TGF-b/activinpathway components
To provide additional evidence for the involvement of the TGF-b/activin signaling pathway in mediating BMP-3 stimulation of MSCproliferation, a siRNA-mediated knockdown approach was utilized(Sharma et al., 2009). Briefly, C3H10T1/2 stem cells were plated on24-well plates and cultured under standard conditions for 48 h.Cells were then transfected with 100 nM of the SilencerPredesigned siRNA targeting mouse ActRIIB (Ambion, Austin,Texas, Cat# S61935), ALK-4 (Ambion, Cat# S61928), and Smad2(Ambion, Cat# S69491), respectively, in Opti-MEM I medium(Invitrogen) containing 1ml/well of siPORTTM Amine TransfectionAgent (Ambion), following the manufacturer’s instructions. Cellswere also transfected in an identical manner with 100 nM of thenegative control #1 siRNA (Ambion, Cat# 4624) or with thetransfection agent alone to serve as controls. At 72 h post-transfection, cells were either collected for Western blot analysis(as described below), or treated for 24 h with 100 ng/ml BMP-3 andpulsed with [3H]-thymidine (0.5mCi/well) for measuring [3H]-thymidine incorporation, as described above.
Western blot analysis
Western blot analysis was used to determine various proteinlevels, as described previously (Yang et al., 2008). Briefly,C3H10T1/2 stem cells were lysed in SDS sample buffer (62.5 mMTris–HCl, pH 6.8, 2% (w/v) SDS, 10% (v/v) glycerol, 50 mM DTT,and 0.01% (w/v) bromophenol blue). Equal concentrations of thewhole cell lysates were subjected to a standard 10% SDS–PAGE.After electrophoresis, proteins were transferred to PVDF transfermembrane (Amersham HybondTM-P, Cat# RPN303F) using aBio-Rad Mini Transfer Apparatus. Non-specific antibody bindingwas blocked with 5% (w/v) milk in TTBS (0.1%, v/v Tween-20 inTBS) for 1 h at room temperature. Membranes were thenhybridized with primary antibody (human phospho-Smad2(Ser465/467), Cell Signaling, Danvers, MA, Cat# 3101; mouseSmad2 (L16D3), Cell Signaling, Cat# 3103; human phospho-p38MAP kinase (Thr180/Tyr182), Cell Signaling, Cat# 9211; humanp38 MAP kinase, Cell Signaling, Cat# 9212; human phospho-p44/42MAP kinase (Thr202/Tyr204), Cell Signaling, Cat# 9101; humanp44/42 MAP kinase, Cell Signaling, Cat# 9102; phospho-SAPK/JNK,Cell Signaling, Cat# 9251S; human SAPK/JNK, Cell Signaling, Cat#9252; human ActRIIB, Santa Cruz Biotechnology Inc., Santa Cruz,CA, Cat #SC-25453; human ALK-4, Santa Cruz, Cat #SC-25450;human GAPDH, Imgenex Corporation, San Diego, CA, Cat# IMG-5567) overnight at 48C. All primary antibodies were used at 1:1000dilutions. Membranes were always probed with anti-phospho-protein antibody first. They were then stripped by incubation instripping buffer (100 mM 2-Mercaptoethanol, 2% SDS, and62.5 mM Tris–HCl, pH 6.7) for 30 min at 558C, blocked andre-probed with the corresponding anti-total-protein antibody.After 3� 10 min washes with TTBS, the membrane was incubatedwith the appropriate HRP-labeled secondary antibody, eitheranti-rabbit (R&D Systems, Cat# HAF008, 1:500) or anti-mouse(R&D Systems, Cat# G-202-C, 1:10,000) for 1 h at roomtemperature. Following another 3� 10 min washes in TTBS,proteins were detected by chemiluminescence (WesternLightningTM Plus-ECL, PerkinElmer Life and Analytical Sciences,
JOURNAL OF CELLULAR PHYSIOLOGY
Cat# NEL103001). The membrane was then exposed to X-ray film(Eastman Kodak, Rochester, NY) for 1–60 min.
ResultsBMP-3 does not promote the commitment of C3H10T1/2 stem cells to the adipocyte lineage
Given that BMP-3 inhibits osteogenesis (Daluiski et al., 2001)and there is evidence for a reciprocal relationship betweenosteogenesis and adipogenesis wherein factors that induceosteogenesis inhibit the formation of adipocytes and vice versa(Lecka-Czernik et al., 2002; Jeon et al., 2003), we hypothesizedthat BMP-3 promotes adipogenesis. As a first step in examining

B M P - 3 P R O M O T E S S T E M C E L L P R O L I F E R A T I O N 661
this hypothesis, we studied the effects of BMP-3 on thecommitment of MSCs to the adipocyte lineage using C3H10T1/2 stem cells as an in vitro model system. Treatment ofC3H10T1/2 stem cells with BMP-4, which is known to promotethe commitment of MSCs to the adipocyte lineage, led toadipocyte formation as evidenced by lipid accumulationfollowing the induction of adipogenesis with a standarddifferentiation cocktail. In contrast, treatment of these cellswith various concentrations of BMP-3 was ineffective (data notshown). We also examined the possibility that BMP-3 mightfunction in concert with BMP-4 to promote the commitment tothe adipocyte lineage. To do so, we treated C3H10T1/2 stemcells with 200 ng/ml BMP-3 and 10 ng/ml BMP-4; aconcentration that was shown to be insufficient to drive thesecells towards the adipocyte lineage when administered alone.There was no evidence of lipid accumulation when these cellswere subjected to a standard adipogenic protocol following thecombined treatment (data not shown).
BMP-3 does not promote adipocyte differentiation
The second step in adipogenesis is the terminal differentiationof preadipocytes to mature adipocytes. To determine if BMP-3promotes adipocyte differentiation, we treated 3T3-L1preadipocytes with an established protocol (Yang et al., 2008) inwhich insulin, one of the three components of a standarddifferentiation cocktail, was substituted with the testcompound BMP-3 at various concentrations. Using thisprotocol,>95% of preadipocytes were converted to lipid-filledmature adipocytes in the presence of insulin (served as a
Fig. 2. A: Expression of TGF-b/activin pathway components byC3H10T1/2 stem cells. Total cellular RNA was prepared fromcultured C3H10T1/2 stem cells and the mRNAs encoding mouseactivin receptor type I (ALK-4), type II (ActRIIB), Smad2, Smad3, andSmad4 were assessed with standard RT-PCR. B: BMP-3 activatesSmad2 in C3H10T1/2 stem cells. C3H10T1/2 stem cells were culturedin standard growth medium (10% FBS) until 50–60% confluence, atwhich time cells were starved for 24 h in serum-free medium. Cellswere then treated with 100 ng/ml BMP-3 in serum-free medium, andat indicated times thereafter, cell lysates were prepared andsubjected to standard Western blot analysis with antibodies specificfor phosphorylated Smad2 and total Smad2 proteins. Results fromone representative Western blot are shown. Similar results wereobtained from two independent experiments.
JOURNAL OF CELLULAR PHYSIOLOGY
positive control). In the absence of insulin, <40% of adipocyteswere formed and this rate of adipocyte differentiation was notaltered by the addition of BMP-3 (data not shown).
BMP-3 stimulates proliferation of C3H10T1/2 stem cellsand 3T3-L1 preadipocytes
A critical but poorly understood step in adipogenesis involvesproliferation of MSCs and preadipocytes, because terminallydifferentiated adipocytes are unable to undergo mitosis.Consequently, we examined the effects of BMP-3 on MSC andpreadipocyte proliferation using a standard [3H]-thymidineincorporation assay. As shown in Figure 1A,B, treatment ofC3H10T1/2 stem cells and 3T3-L1 preadipocytes with BMP-3led to a concentration-dependent increase in [3H]-thymidineincorporation reaching 300% of the control at 100 ng/ml BMP-3(P< 0.001). Similar results were obtained using C2C12 cellsand rat primary preadipocytes (data not shown).
Fig. 3. Effects of TGF-b/activin pathway inhibitor SB-431542 onbasal and BMP-3-stimulated C3H10T1/2 stem cell proliferation.C3H10T1/2 stem cells were cultured in standard growth medium(10% FBS) until 50–60% confluence, at which time cells were starvedfor 24 h in serum-free medium. A: Cells were treated with increasingconcentrations of SB-431542 (0.5–2mM) for 1 h in serum-freemedium, followed by treatment with 100 ng/ml BMP-3 for 1 h inserum-free medium. Cell lysates were prepared and subjected tostandard Western blot analysis with antibodies specific forphosphorylated Smad2 and total Smad2 proteins. Results from onerepresentative Western blot are shown. Similar results wereobtained from two independent experiments. B: Alternatively, cellswere pretreated for 1 h with or without increasing concentrations ofSB-431542 (0.5–2mM), followed by treatment with 100 ng/ml BMP-3in serum-free medium for 24 h. During the last 4 h of treatment, cellswere pulse labeled with [3H]-thymidine (0.5mCi/well) and the rate of[3H]-thymidine incorporation was determined. Data aremeans W SEM, n U 3–5 independent experiments, each performedin triplicate (MP < 0.05, MMP < 0.01, MMMP < 0.001 vs. control). NS,non-significant.

Fig. 4. EffectsofsiRNA-mediatedknockdownofexpressionofActRIIB,ALK-4,andSmad2onBMP-3stimulationofMSCproliferation.C3H10T1/2 stem cells were cultured in standard growth medium (10% FBS) until 30–40% confluence. They were then transfected with 100 nM of ActRIIBsiRNA,ALK-4siRNA,Smad2siRNA,oranegativecontrol(i.e., scrambled)siRNA.Seventy-twohoursaftertransfection,cellswerelysed,andlevelsof ActRIIB, ALK-4, and Smad2 proteins were determined by Western blotting. GAPDH was used as a control to show the specificity of siRNA-mediatedknockdownofActRIIB,ALK-4,andSmad2(A–F).Alternatively,cellsweretreatedfor24 hwithorwithout100 ng/mlBMP-3inserum-freemedium.Duringthe last4 hof treatment,cellswerepulse labeledwith[3H]-thymidine (0.5mCi/well) andtherateof[3H]-thymidine incorporationwasdetermined(G–I).Dataarepresentedasmeans W SEMoffour independentexperimentseachperformedintriplicate(MMMP < 0.001vs.control).
662 S T E W A R T E T A L .
Expression of the key TGF-b/activin signalingcomponents in C3H10T1/2 stem cells
Owing to a lack of information on the regulation of MSCproliferation and considering that BMP-3 has similar effects onC3H10T1/2 stem cells and 3T3-L1 preadipocytes, we chose tostudy the molecular mechanisms that underlie BMP-3stimulation of C3H10T1/2 stem cell proliferation. Based on ourcritical analysis of the pertaining literature, we investigated theinvolvement of the TGF-b/activin signaling pathway. As a firststep, we examined the expression of the key TGF-b/activinsignaling components with standard RT-PCR. Our resultsrevealed that C3H10T1/2 stem cells express mRNAs encoding
JOURNAL OF CELLULAR PHYSIOLOGY
the type I receptor (ALK-4), type II receptor (ActRIIB), andSmad proteins (Smad2, Smad3, Smad4) (Fig. 2A).
BMP-3 activates Smad2 in C3H10T1/2 stem cells
Having established the intrinsic ability of C3H10T1/2 stem cellsto signal through the TGF-b/activin signal transductionpathway, we then determined if BMP-3 activates this pathway.Given that the receptor-activated protein Smad2 isphosphorylated upon the sequential ligand-dependentactivation of ActRIIB and ALK-4, we determined thephosphorylation status of Smad2 following treatment ofC3H10T1/2 stem cells with BMP-3. As shown in
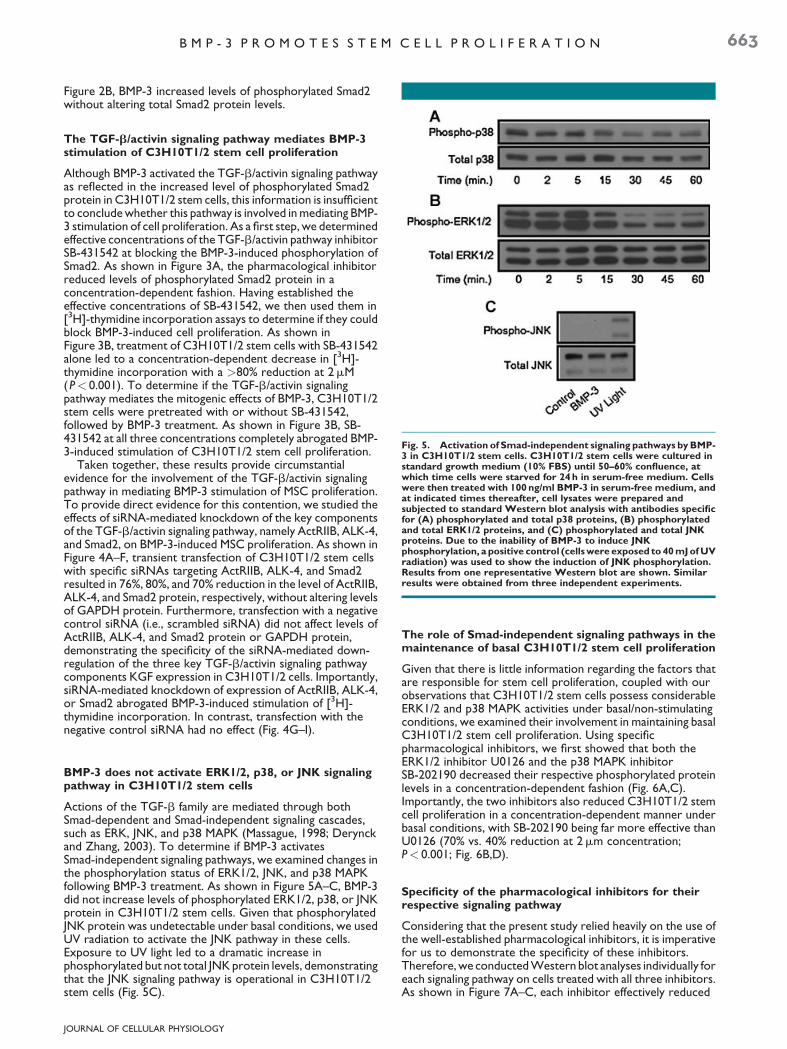
Fig. 5. Activation of Smad-independent signaling pathways by BMP-3 in C3H10T1/2 stem cells. C3H10T1/2 stem cells were cultured instandard growth medium (10% FBS) until 50–60% confluence, atwhich time cells were starved for 24 h in serum-free medium. Cellswere then treated with 100 ng/ml BMP-3 in serum-free medium, andat indicated times thereafter, cell lysates were prepared andsubjected to standard Western blot analysis with antibodies specificfor (A) phosphorylated and total p38 proteins, (B) phosphorylatedand total ERK1/2 proteins, and (C) phosphorylated and total JNKproteins. Due to the inability of BMP-3 to induce JNKphosphorylation, a positive control (cells were exposed to 40 mJ of UVradiation) was used to show the induction of JNK phosphorylation.Results from one representative Western blot are shown. Similarresults were obtained from three independent experiments.
B M P - 3 P R O M O T E S S T E M C E L L P R O L I F E R A T I O N 663
Figure 2B, BMP-3 increased levels of phosphorylated Smad2without altering total Smad2 protein levels.
The TGF-b/activin signaling pathway mediates BMP-3stimulation of C3H10T1/2 stem cell proliferation
Although BMP-3 activated the TGF-b/activin signaling pathwayas reflected in the increased level of phosphorylated Smad2protein in C3H10T1/2 stem cells, this information is insufficientto conclude whether this pathway is involved in mediating BMP-3 stimulation of cell proliferation. As a first step, we determinedeffective concentrations of the TGF-b/activin pathway inhibitorSB-431542 at blocking the BMP-3-induced phosphorylation ofSmad2. As shown in Figure 3A, the pharmacological inhibitorreduced levels of phosphorylated Smad2 protein in aconcentration-dependent fashion. Having established theeffective concentrations of SB-431542, we then used them in[3H]-thymidine incorporation assays to determine if they couldblock BMP-3-induced cell proliferation. As shown inFigure 3B, treatment of C3H10T1/2 stem cells with SB-431542alone led to a concentration-dependent decrease in [3H]-thymidine incorporation with a >80% reduction at 2mM(P< 0.001). To determine if the TGF-b/activin signalingpathway mediates the mitogenic effects of BMP-3, C3H10T1/2stem cells were pretreated with or without SB-431542,followed by BMP-3 treatment. As shown in Figure 3B, SB-431542 at all three concentrations completely abrogated BMP-3-induced stimulation of C3H10T1/2 stem cell proliferation.
Taken together, these results provide circumstantialevidence for the involvement of the TGF-b/activin signalingpathway in mediating BMP-3 stimulation of MSC proliferation.To provide direct evidence for this contention, we studied theeffects of siRNA-mediated knockdown of the key componentsof the TGF-b/activin signaling pathway, namely ActRIIB, ALK-4,and Smad2, on BMP-3-induced MSC proliferation. As shown inFigure 4A–F, transient transfection of C3H10T1/2 stem cellswith specific siRNAs targeting ActRIIB, ALK-4, and Smad2resulted in 76%, 80%, and 70% reduction in the level of ActRIIB,ALK-4, and Smad2 protein, respectively, without altering levelsof GAPDH protein. Furthermore, transfection with a negativecontrol siRNA (i.e., scrambled siRNA) did not affect levels ofActRIIB, ALK-4, and Smad2 protein or GAPDH protein,demonstrating the specificity of the siRNA-mediated down-regulation of the three key TGF-b/activin signaling pathwaycomponents KGF expression in C3H10T1/2 cells. Importantly,siRNA-mediated knockdown of expression of ActRIIB, ALK-4,or Smad2 abrogated BMP-3-induced stimulation of [3H]-thymidine incorporation. In contrast, transfection with thenegative control siRNA had no effect (Fig. 4G–I).
BMP-3 does not activate ERK1/2, p38, or JNK signalingpathway in C3H10T1/2 stem cells
Actions of the TGF-b family are mediated through bothSmad-dependent and Smad-independent signaling cascades,such as ERK, JNK, and p38 MAPK (Massague, 1998; Derynckand Zhang, 2003). To determine if BMP-3 activatesSmad-independent signaling pathways, we examined changes inthe phosphorylation status of ERK1/2, JNK, and p38 MAPKfollowing BMP-3 treatment. As shown in Figure 5A–C, BMP-3did not increase levels of phosphorylated ERK1/2, p38, or JNKprotein in C3H10T1/2 stem cells. Given that phosphorylatedJNK protein was undetectable under basal conditions, we usedUV radiation to activate the JNK pathway in these cells.Exposure to UV light led to a dramatic increase inphosphorylated but not total JNK protein levels, demonstratingthat the JNK signaling pathway is operational in C3H10T1/2stem cells (Fig. 5C).
JOURNAL OF CELLULAR PHYSIOLOGY
The role of Smad-independent signaling pathways in themaintenance of basal C3H10T1/2 stem cell proliferation
Given that there is little information regarding the factors thatare responsible for stem cell proliferation, coupled with ourobservations that C3H10T1/2 stem cells possess considerableERK1/2 and p38 MAPK activities under basal/non-stimulatingconditions, we examined their involvement in maintaining basalC3H10T1/2 stem cell proliferation. Using specificpharmacological inhibitors, we first showed that both theERK1/2 inhibitor U0126 and the p38 MAPK inhibitorSB-202190 decreased their respective phosphorylated proteinlevels in a concentration-dependent fashion (Fig. 6A,C).Importantly, the two inhibitors also reduced C3H10T1/2 stemcell proliferation in a concentration-dependent manner underbasal conditions, with SB-202190 being far more effective thanU0126 (70% vs. 40% reduction at 2mm concentration;P< 0.001; Fig. 6B,D).
Specificity of the pharmacological inhibitors for theirrespective signaling pathway
Considering that the present study relied heavily on the use ofthe well-established pharmacological inhibitors, it is imperativefor us to demonstrate the specificity of these inhibitors.Therefore, we conducted Western blot analyses individually foreach signaling pathway on cells treated with all three inhibitors.As shown in Figure 7A–C, each inhibitor effectively reduced

Fig. 6. EffectsofSmad-independentpathway inhibitorsonC3H10T1/2stemcellproliferationunderbasal/non-stimulatingconditions.C3H10T1/2 stem cells were cultured in standard growth medium (10% FBS) until 50–60% confluence, at which time cells were starved for 24 h in serum-freemedium. Cells were then treated for 1 h in serum-free medium with increasing concentration (0.5–2mM) of the p38 MAPK inhibitor SB-202190 orthe ERK1/2 inhibitor U0126. At the end of treatment, cell lysates were prepared and subjected to standard Western blot analysis with antibodiesspecificfor(A)phosphorylatedandtotalp38proteins,and(C)phosphorylatedandtotalERK1/2proteins.Results fromonerepresentativeWesternblot are shown.Similar results were obtained from two independentexperiments. Alternatively, cells were treated for24 h in serum-free mediumwith increasing concentration (0.5–2mM) of the p38 MAPK inhibitor SB-202190 (B), or the ERK1/2 inhibitor U0126 (D). During the last 4 h oftreatment, cells were pulse labeled with [3H]-thymidine (0.5mCi/well) and the rate of [3H]-thymidine incorporation was determined. Data aremeans W SEM, n U 4–6 independent experiments, each performed in triplicate (MMP < 0.01, MMMP < 0.001 vs. control).
664 S T E W A R T E T A L .
phosphorylation of their intended target protein without non-specifically decreasing phosphorylation of other proteins.
DISCUSSION
The present study was undertaken to test this hypothesis thatBMP-3 promotes adipogenesis using the well-characterized invitro model systems, C3H10T1/2 MSCs and 3T3-L1preadipocytes, which have been shown to recapitulate many ofthe physiological events that occur in vivo (Student et al., 1980;Katagiri et al., 1990; Tang et al., 2004; Zhang and Stott, 2004;Jasuja et al., 2005). We demonstrated for the first time thatalthough BMP-3 did not promote the commitment of MSCs tothe adipocyte lineage or the differentiation of preadipocytes tomature adipocytes, it stimulated proliferation of both MSCsand preadipocytes. We also provided evidence that theTGF-b/activin signaling pathway mediated the mitogenic effectsof BMP-3 on MSCs.
Accelerated adipogenesis is a hallmark of obesity, but thefactors and the molecular mechanisms that orchestrate thisimportant event are poorly understood. Adipogenesis beginswith the commitment of MSCs to the adipocyte lineage,followed by terminal differentiation of preadipocytes tolipid-filled mature adipocytes (Rosen and MacDougald, 2006).Several transcription factors have been identified to play a key
JOURNAL OF CELLULAR PHYSIOLOGY
role in controlling adipocyte differentiation, which includePPARg and members of the C/EBP family (Farmer, 2006).However, the molecular determinants of MSCs to adipocyteprecursor cells remain largely unknown. MSCs can give rise tofour distinct cell types, including adipocytes, chondrocytes,myocytes, and osteocytes (Rosen and MacDougald, 2006).Among the four cell lineages, there is evidence for an intimaterelationship between the formation of adipocytes andosteocytes. For instance, a number of in vitro studies using bonemarrow-derived MSCs have demonstrated that factors thatinduce adipogenesis inhibit osteoblast formation and,conversely, factors that promote osteogenesis inhibitadipocyte formation (Lecka-Czernik et al., 2002; Jeon et al.,2003).
Considering that BMP-3 is a negative regulator ofosteogenesis (Daluiski et al., 2001), we first examined thepossibility that BMP-3 may promote the commitment of MSCsto the adipocyte lineage. To do so, we utilized an establishedprotocol in which C3H10T1/2 MSCs were treated withrecombinant BMP-3 during their proliferative phase until 2 dayspost-confluence when cells were induced for adipogenesis. Weused BMP-4 as a positive control because it is known to driveMSCs toward the adipocyte lineage (Tang et al., 2004). Ourresults revealed that BMP-3 treatment did not result inadipocyte formation. We also explored, but found no evidence

Fig. 7. Specificity of the pharmacological inhibitors for theirtargeted signaling pathways. C3H10T1/2 stem cells were cultured instandard growth medium (10% FBS) until 50–60% confluence, atwhich time cells were starved for 24 h in serum-free medium. Cellswere then pretreated for 1 h with 2mM of SB-431542, SB-202190, orU0126 prior to treatment for 1 h with 100 ng/ml BMP-3 in serum-freemedium (A). Alternatively, cells were treated for 1 h with 2mM of SB-431542, SB-202190, or U0126 in serum-free medium (B,C). Celllysates were prepared, and subjected to standard Western blotanalysis with antibodies specific for phosphorylated and total Smad2(A), p38 (B), and ERK1/2 (C) proteins. Results from onerepresentative Western blot are shown. Similar results wereobtained from three independent experiments.
B M P - 3 P R O M O T E S S T E M C E L L P R O L I F E R A T I O N 665
to support, the contention that BMP-3 may act in concert withBMP-4 to promote the commitment of MSCs to the adipocytelineage. Furthermore, we determined the effects of BMP-3 onadipocyte differentiation, and showed that BMP-3 did not alterthe differentiation of 3T3-L1 preadipocytes. Together, our dataindicate that BMP-3 has no effect on the commitment or thedifferentiation steps of adipogenesis.
A key but poorly understood component of adipogenesisinvolves proliferation of MSCs and preadipocytes. Using astandard [3H]-thymidine incorporation assay, wedemonstrated that treatment of both C3H10T1/2 stem cellsand 3T3-L1 preadipocytes with BMP-3 led to a threefoldincrease in DNA synthesis, a surrogate marker of cellproliferation. We also showed that BMP-3 exerted similarmitogenic effects on C2C12 cells and rat primarypreadipocytes. Together, these results demonstrate thatBMP-3 is a potent mitogen for both MSCs and preadipocytes.
Members of the BMP family typically signal through the BMPsignaling pathway. BMP ligands bind to the BMP type II receptor(BMPRII), which then binds and activates the type I receptor(BMPRI). The activated type I receptor phosphorylates Smad1,Smad5, and/or Smad8. The receptor-activated Smads form atrimeric complex with the common Smad, Smad4, which then
JOURNAL OF CELLULAR PHYSIOLOGY
translocates into the nucleus where it regulates the expressionof its target genes (Kitisin et al., 2007). In contrast, several linesof evidence suggest that actions of BMP-3 are mediated throughthe TGF-b/activin signaling pathway (Bahamonde and Lyons,2001; Allendorph et al., 2007). First, BMP-3 activatesexpression of a TGF-b/activin-responsive reporter but not theBMP-responsive reporter (Daluiski et al., 2001). Second, BMP-3displays a much higher affinity for ActRIIB, the type II receptorfor ligands that activate the TGF-b/activin pathway (Allendorphet al., 2007). This pathway consists of the type II receptorActRIIB and the type I receptor ALK-4. Together, this receptorcomplex activates Smad2/Smad3 (Kitisin et al., 2007).
Several lines of evidence suggested that the TGF-b/activinpathway mediated the mitogenic effects of BMP-3 on MSCs.First, all the major components of the TGF-b/activin signalingpathway were highly expressed in C3H10T1/2 stem cells,including ActRIIB, ALK-4, Smad2, Smad3, and Smad4. Thissuggested that these cells are capable of responding to factorsthat activate the TGF-b/activin signaling pathway. Second,BMP-3 dramatically increased levels of phospho-Smad2 protein,and the TGF-b/activin inhibitor SB-431542 blocked BMP-3stimulation of MSC proliferation. It is noteworthy that weempirically determined the effective concentrations of theinhibitor to be in the lower micromolar range (i.e., 0.5, 1, and2mM), as shown by their concentration-dependent inhibition ofBMP-3-induced Smad2 phosphorylation. These concentrationsof SB-431542 were much lower than those (�10mM) reportedby the majority of others in the literature. This is an importantissue because at higher concentrations, SB-431542 could non-specifically inhibit Smad-independent pathways, such as p38MAPK, ERK1/2 and, or JNK. Importantly, siRNA-mediatedknockdown of ActRIIB, ALK-4, and Smad2 abrogated themitogenic effects of BMP-3 on C3H10T1/2 stem cellproliferation.
In addition to activating the BMP pathway, several BMPs havebeen shown to signal through MAPK signaling pathways, namelyp38, ERK, and JNK. For example, BMP-4 has been shown toactivate both p38 MAPK (Fiori et al., 2006) and ERK1/2 (Yanget al., 2007), and BMP-2 can activate ERK1/2 (Lou et al., 2000;Xing et al., 2002). In comparison, the JNK pathway is lessdefined in relation to BMP signaling (Massague, 1998).Therefore, we also investigated the possibility that BMP-3 mayactivate p38 MAPK, ERK1/2, and/or JNK signaling pathways.We showed that BMP-3 had no effect on ERK1/2, p38, or JNKprotein phosphorylation. It is noteworthy that phosphorylatedJNK protein was undetectable in C3H10T1/2 stem cells underbasal conditions. Consequently, we used UV radiation, a knownactivator of JNK, to show that our cells were capable ofresponding to JNK activation, and perhaps more importantly todemonstrate that our inability to detect JNK phosphorylationunder both basal and BMP-3-stimulated conditions was notattributed to any technical difficulties. Collectively, our resultssuggested that MAPK signaling pathways were not involved inmediating BMP-3 stimulation of MSC proliferation.
It is noteworthy that there were considerable levels ofphosphorylated p38 MAPK and ERK1/2 proteins in C3H10T1/2stem cells under basal conditions. Although the precise factorsresponsible for maintaining basal p38 and ERK1/2 activitiesremain to be identified, it is tempting to speculate that thesestem cells must produce a variety of growth factors andsignaling molecules under non-stimulating conditions, some ofwhich may activate p38 and ERK1/2 signaling pathways. Usingwell-established pharmacological inhibitors, we demonstratedthat both p38 MAPK and ERK1/2 signaling pathways playimportant roles in maintaining basal C3H10T1/2 stem cellproliferation, although the former has a more prominent rolebecause the p38 MAPK inhibitor SB-202190 caused a far greaterreduction (70%) in DNA synthesis than the ERK1/2 inhibitorU0126 (40%). Owing to the lack of detectable levels of

666 S T E W A R T E T A L .
phosphorylated JNK protein under both basal and BMP-3-stimulated conditions, we did not pursue this pathway further.
Knowing that there is potential for pharmacologicalinhibitors to non-specifically target other signaling pathways, itis imperative that the specificity as well as the effectiveconcentrations of the inhibitors used in the present study beempirically determined and demonstrated in our cell modelsystem. Indeed, we conducted such experiments, and ourresults showed that each inhibitor was able to attenuatephosphorylation of its target protein in a concentration-dependent fashion. Importantly, they also demonstrated thatthere was no cross-inhibition among the inhibitors. Thus, eachinhibitor used in the present study specifically targeted itscorresponding signaling pathway.
In summary, the present study demonstrates that BMP-3stimulates MSC proliferation via the TGF-b/activin signalingpathway, thus revealing a novel role for this divergent andpoorly understood member of the TGF-b superfamily inregulating MSC proliferation.
Acknowledgments
Heart & Stroke Foundation of Ontario (Grant-In-Aid #NA-6049) and Canadian Institutes of Health Research (OperatingGrant #MOP-79484).
Literature Cited
Allendorph GP, Isaacs MJ, Kawakami Y, Belmonte JC, Choe S. 2007. BMP-3 and BMP-6structures illuminate the nature of binding specificity with receptors. Biochemistry46:12238–12247.
Bahamonde ME, Lyons KM. 2001. BMP3: To be or not to be a BMP. J Bone Joint Surg Am 83-A:S56–S62.
Daluiski A, Engstrand T, Bahamonde ME, Gamer LW, Agius E, Stevenson SL, Cox K, Rosen V,Lyons KM. 2001. Bone morphogenetic protein-3 is a negative regulator of bone density.Nat Genet 27:84–88.
Derynck R, Zhang YE. 2003. Smad-dependent and Smad-independent pathways in TGF-betafamily signalling. Nature 425:577–584.
Dixon JB. 2009. The effect of obesity on health outcomes. Mol Cell Endocrinol. [Epub aheadof print]
Eckel RH, Grundy SM, Zimmet PZ. 2005. The metabolic syndrome. Lancet 365:1415–1428.Farmer SR. 2006. Transcriptional control of adipocyte formation. Cell Metab 4:263–273.Feng XH, Derynck R. 2005. Specificity and versatility in tgf-beta signaling through Smads.
Annu Rev Cell Dev Biol 21:659–693.Fiori JL, Billings PC, de la Pena LS, Kaplan FS, Shore EM. 2006. Dysregulation of the BMP-p38
MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva(FOP). J Bone Miner Res 21:902–909.
Guan H, Arany E, van Beek JP, Chamson-Reig A, Thyssen S, Hill DJ, Yang K. 2005. Adiposetissue gene expression profiling reveals distinct molecular pathways that define visceral
JOURNAL OF CELLULAR PHYSIOLOGY
adiposity in offspring of maternal protein-restricted rats. Am J Physiol Endocrinol Metab288:E663–E673.
Jasuja R, Catlin DH, Miller A, Chang YC, Herbst KL, Starcevic B, Artaza JN, Singh R, Datta G,Sarkissian A, Chandsawangbhuwana C, Baker M, Bhasin S. 2005. Tetrahydrogestrinone isan androgenic steroid that stimulates androgen receptor-mediated, myogenicdifferentiation in C3H10T1/2 multipotent mesenchymal cells and promotes muscleaccretion in orchidectomized male rats. Endocrinology 146:4472–4478.
Jeon MJ, Kim JA, Kwon SH, Kim SW, Park KS, Park SW, Kim SY, Shin CS. 2003. Activation ofperoxisome proliferator-activated receptor-gamma inhibits the Runx2-mediatedtranscription of osteocalcin in osteoblasts. J Biol Chem 278:23270–23277.
Katagiri T, Yamaguchi A, Ikeda T, Yoshiki S, Wozney JM, Rosen V, Wang EA, Tanaka H,Omura S, Suda T. 1990. The non-osteogenic mouse pluripotent cell line, C3H10T1/2, isinduced to differentiate into osteoblastic cells by recombinant human bone morphogeneticprotein-2. Biochem Biophys Res Commun 172:295–299.
Kitisin K, Saha T, Blake T, Golestaneh N, Deng M, Kim C, Tang Y, Shetty K, Mishra B, Mishra L.2007. TGF-beta signaling in development. Sci STKE 2007:cm1.
Koza RA, Nikonova L, Hogan J, Rim JS, Mendoza T, Faulk C, Skaf J, Kozak LP. 2006. Changes ingene expression foreshadow diet-induced obesity in genetically identical mice. PLoS Genet2:e81.
Lecka-Czernik B, Moerman EJ, Grant DF, Lehmann JM, Manolagas SC, Jilka RL. 2002.Divergent effects of selective peroxisome proliferator-activated receptor-gamma2 ligands on adipocyte versus osteoblast differentiation. Endocrinology 143:2376–2384.
Lou J, Tu Y, Li S, Manske PR. 2000. Involvement of ERK in BMP-2 induced osteoblasticdifferentiation of mesenchymal progenitor cell line C3H10T1/2. Biochem Biophys ResCommun 268:757–762.
Massague J. 1998. TGF-beta signal transduction. Annu Rev Biochem 67:753–791.Montague CT, O’Rahilly S. 2000. The perils of portliness: Causes and consequences of
visceral adiposity. Diabetes 49:883–888.Osmond C, Barker DJ. 2000. Fetal, infant, and childhood growth are predictors of coronary
heart disease, diabetes, and hypertension in adult men and women. Environ HealthPerspect 108:545–553.
Rosen ED, MacDougald OA. 2006. Adipocytedifferentiation from the inside out. Nat Rev MolCell Biol 7:885–896.
Sampath TK, Muthukumaran N, Reddi AH. 1987. Isolation of osteogenin, an extracellularmatrix-associated, bone-inductive protein, by heparin affinity chromatography. Proc NatlAcad Sci USA 84:7109–7113.
Sharma A, Guan H, Yang K. 2009. The p38 mitogen-activated protein kinase regulates 11beta-hydroxysteroid dehydrogenase type 2 (11beta-HSD2) expression in human trophoblastcells through modulation of 11beta-HSD2 messenger ribonucleic acid stability.Endocrinology 150:4278–4286.
Student AK, Hsu RY, Lane MD. 1980. Induction of fatty acid synthetase synthesis indifferentiating 3T3-L1 preadipocytes. J Biol Chem 255:4745–4750.
Tang QQ, Otto TC, Lane MD. 2004. Commitment of C3H10T1/2 pluripotent stem cells tothe adipocyte lineage. Proc Natl Acad Sci USA 101:9607–9611.
Xing X, Manske PR, Li YY, Lou J. 2002. The role of Sp1 in BMP2-up-regulated Erk2 geneexpression. Biochem Biophys Res Commun 297:116–124.
Yang X, Lee PJ, Long L, Trembath RC, Morrell NW. 2007. BMP4 induces HO-1 via a Smad-independent, p38MAPK-dependent pathway in pulmonary artery myocytes. Am J RespirCell Mol Biol 37:598–605.
Yang K, Guan H, Arany E, Hill DJ, Cao X. 2008. Neuropeptide Y is produced in visceraladipose tissue and promotes proliferation of adipocyte precursor cells via the Y1 receptor.FASEB J 22:2452–2464.
Zhang J, Li L. 2005. BMP signaling and stem cell regulation. Dev Biol 284:1–11.Zhang WV, Stott NS. 2004. BMP-2-modulated chondrogenic differentiation in vitro
involves down-regulation of membrane-bound beta-catenin. Cell Commun Adhes 11:89–102.
Zhang T, Guan H, Arany E, Hill DJ, Yang K. 2007. Maternal protein restriction permanentlyprograms adipocyte growth and development in adult male rat offspring. J Cell Biochem101:381–388.















![The therapeutic potential of targeting the endothelial-to-mesenchymal transition · 2019. 2. 2. · SNAI1/2 [22]. In addition, certain TGF-β family members (TGF-β2, BMP2, and BMP4)](https://img.dokumen.tips/doc/110x75/6127d04e9164a1191a27f7a4/the-therapeutic-potential-of-targeting-the-endothelial-to-mesenchymal-transition.jpg)













