Embed Size (px)
Citation preview

Biochemistry- Paper II Feb 2009
I. Essay questions: (2 x 15=30)
1. Name the compounds derived from glycine. Explain any two in detail.
Ans.
Glycine is the simplest aminoacid. It is a non essential aminoacid derived from serine,
threonine and from CO2 and NH3 by glycine synthase.
Products derived from glycine:
1. methylene tetrahydrofolate by glycine cleavage system.
2. glucose-it is glucogenic
3. Creatine, Creatine phosphate and Creatine.
4. heme
5. Purine nucleotides
6. Glutathione
7. conjugation for xenobiotics
8. constituent of proteins like collagen.
Creatine production from glycine:
Glycine combines with arginine and a methyl group is transferred forming guanidoacetic
acid and ornithine by glycine arginine methyl transferase
Guanidoacetic acid receives a methyl group from Sam to form Creatine.
Creatine kinase in muscle converts Creatine to Creatine phosphate which acts as a high
energy molecule involved in resynthesis of ATP from ADP during muscle contraction

Creatine loses water to form creatinine which is excreted by the kidneys.
Fig. creatine production
Creatinine is a marker of renal failure. Creatinine clearance test is used to measure GFR
Creatine kinase has many isoenzymes and is elevated in muscular dystrophies and
myocardial infarction.
Production of heme:
Succinyl CoA combines with glycine in presence of pyridoxal phosphate by the enzyme
ALA synthase in mitochondria to form alpha amino beta keto adipic acid which is converted by
the same enzyme to delta amino levulanic acid.
ALA synthase deficiency leads to X linked sideroblastic anemia, which is a mild condition.

2. Describe in detail the mechanism of regulation of blood pH
Ans.
Normal blood pH is btween7.38-7.42. it is maintained by:
I. Role of buffers in body fluids:
Buffers resist changes in pH when small quantities of an acid or an alkali are added. Various
buffers in body are:
1. Bicarbonate buffer system
it is the most important buffer in plasma and is formed by (NaHCO3/H2CO3)
the base HCO3- is the metabolic component as it is regulated by kidney and the acid
H2CO3 is called respiratory component since it is regulated by the lungs.
The normal bicarbonate level in plasma is 24mmol/L. It has a pKa of 6.1-so it is a poor
buffer. But the high blood concentration and the ratio of base to salt is high (20:1),
which makes it an effective buffer.
When acid(H+) is added
o H+ + HCO3- H2CO3 H20 + CO2 excreted by lungs and kidney.
When alkali is (HCO3-) added
o H+ + HCO3- H2CO3 H20 + CO2 excreted by lungs and kidney.
2. phosphate buffer system
It is made of NaHPO4/NaH2PO4. It has a pKa of 6.8.
In acidosis
NaHPO4 + H+ NaH2PO4 - excreted by the kidneys
In alkalosis
NaH2PO4 - NaHPO4 + H+
3. Protein buffer system
The Histidine molecules in albumin acts as a buffer.

In acidosis
H+ + Pr- - HPr
In alkalosis
HPr H+ + Pr-
II. Kidneys regulate acid base balance by:
1. Excretion of H+ -In PCT cells CO2 combines with water to form carbonic acid using carbonic
anhydrase. Then it becomes H+ and HCO3- . this H+ is then excreted into lumen in exchange for
Na+ .
2. reabsorption of HCO3- - sodium bicarbonate in the lumen becomes sodium and bicarbonate.
Sodium is taken up by PCT cell in exchange of hydrogen ions. H+ combines with HCO3- to form
carbonic acid, which forms CO2 and water and both are reclaimed into the cell and converted
back to carbonic acid and again to H+ and HCO3- . HCO3
- is taken into blood with sodium.
. fig. Excretion of H+ fig. reabsorption of HCO3-
3. Excretion of titrable acid- The Na2 HPO4 becomes Na+ and NaHPO4 - . sodium is exchanged
with H+ ions and H+ combines with NaHPO4 – to become Na H2PO4 and gets excreted.

Fig. Excretion of titrable acid and ammonium ions
4. Excretion of NH4 + - Glutamine in DCT becomes glutamate and ammonia. This ammonia is
secreted into the lumen which combines with hydrogen ions to become ammonium ions and
gets excreted.
III. Role of lungs in Acid base balance
When there is fall in pH the respiratory rate is stimulated resulting in hyperventilation.
This would eliminate more CO2 thereby lowering H2CO3 .
In tissues pCO2 is high and pH is low to the formation of acids by the cells like lactate
and production of CO2 by cells. CO2 diffuses into RBC. It combines with water to form carbonic
acid by carbonic anhydrase. And dissociates into H+ and HCO3-. So RBC traps H+ from the tissues.
Some of the HCO3- diffuses out of the cell in exchange for chloride.
In the lungs H+ combines with HCO3- to form H2CO3 which becomes H2O and CO2. This
CO2 is released into the lungs. So lungs reduce the acid load of H2CO3 by excretion of CO2.
In metabolic acidosis lungs hyperventilate to excrete more acid. In Metabolic alkalosis
the reverse happens.
Fig. Reactions in tissues fig. Reaction in lungs

II. Write short notes on: (10x5 = 50)
1. Phenylketonuria
Ans.
Phenyl ketonuria
-it is an autosomal recessive disease with an incidence of 1:1500 births. It is due to
deficiency of phenylalanine hydroxylase. So phenylalanine is not converted into tyrosine and it
accumulates.
- the excess of phenylalanine is converted to phenyl pyruvate, phenyl lactate, and
phenyl acetate and phenyl acetyl glutamine. Phenyl pyruvate, phenyl lactate, phenyl acetate
are excreted in urine.
- the child is mentally retarded and convulsions, tremors agitation, hyperactivity may
present. The child often has hypo pigmentation due to reduced availability of tyr for melanin
production.
- phenyl lactate causes mousy odor of urine.
- blood levels of phenyl alanine are elevated, Guthrie’s test is confirmative. Urine FeCl3
test is positive.
- tapioca based diet which have less phe is the treatment of choice. Gene therapy is
under trial.

2.Formation of Uric acid
Ans.
Purine catabolism leads to the production of Uric acid.
Step1. AMP deaminase converts AMP to IMP.
Step 2. IMP becomes Inosine by phosphomonoesterase losing phosphate.
Step 3. Adenosine via adenosine deaminase becomes Inosine
Step 4. Inosine loses R1P to form hypoxanthine
Step 5. Guanosine loses R1P and becomes Guanine which loses ammonium to form xanthine
Step 6. Hypoxanthine by xanthine oxidase becomes xanthine.
Step 7. Xanthine becomes uric acid by the same enzyme. The last two steps produces hydrogen
peroxide

3.Porphyria
Ans.
Porphyrias are a group of inborn errors of metabolism associated with biosynthesis of
heme. These are characterized by increased production and excretion of porphyrins and/or
their precursors.
Porphyrias are:
1. X linked sideroblastic anemia- ALAS deficiency
2. ALAD deficiency
3. Acute intermittent porphyria-Uroporphyrinogen I synthase deficiency
This leads to secondary increase in activity of ALAs. The levels of ALA and PBG are
elevated in blood and urine. Urine is dark on voiding due to photo oxidation of PBG to
phorphobilin. Symptoms appear intermittently. Patients will have acute abdominal pain.
An attack is precipitated by starvation and symptoms are relieved by glucose infusion.
Patients may have neurological abnormalities like sensory and motor disturbances,
agitation and confusion. Patients may have neuropsychiatric problems.
4. Congenital erythropoietic porphyria- Uroporphyrinogen I synthase deficiency .
autosomal recessive disorder. Photosensitivity and dark urine are the symptoms.
5. Porphyria cutanea tarda -uroporphyrinogrn decarboxylase deficiency. Here patients are
more prone for photosensitivity. The urobilinogens accumulate and when patients come
under light they spontaneously form urobilin which is a potent oxidant and destroys skin
cells and causes scarring. Patients have gross skin malformations leading to monster like
appearance, and they prefer night. Sunscreens are mildly effective.
6. Hereditary coproporphyria-coproporphyrinogen oxidase deficiency
7. Variegate porphyria - protoporphyrinogen oxidase deficiency
8. Erythropoietic proto porphyria- ferrochelatase deficiency

Fig: Porphyrias

4. Urea cycle
Ans.
The urea cycle is the first metabolic pathway to be elucidated in 1932.
This cycle is also called as KREBS- HESELIT cycle.
The two nitrogen atoms of urea derived from two different sources, one from ammonia and
the directly from aspartic acid (amino group).
STEP 1: Formation of carbomyl phosphate:
One molecule of ammonia condenses with CO2in the presence of two molecules of ATP to
form carbomyl phosphate by the enzyme carbomyl phosphate synthase I.
STEP II: Formation of citrulline:
The carbomyl group is transferred to the NH2 group of ornithine by ornithine
transcarbomylase.
STEP III: Formation of argino succinate:
One molecule of aspartic acid adds to citrulline forming a carbon to nitrogen which provides
second nitrogen of urea by arginosuccinate synthetase.
Two high energy phosphate bonds utilized.
STEP IV: Formation of arginine:
Arginosuccinate cleaved by argino succinate lyase to arginine and fumarate.
STEP V: Formation of urea:
Hydrolysis of arginine to urea and ornithine by arginase.
Ornithine return to the mitochondria to react with other molecule of carbomyl phosphate
for proceeds of cycle.

Fig. Urea cycle

5. ELISA
Ans.
ELISA is Enzyme linked immune sorbent assay. ELISA is used widely to measure
hormone, growth factors, tumor markers, bacterial and viral antigens, etc.,
Antigen detection: sandwich ELISA
1. specific antibody to the antigen coated well is taken
2. sample is added. The antigen in patient’s sample binds to the antibody.
3. an antibody attached to an enzyme like peroxidase, which will bind to other part of the
antigen is added(secondary Ag). Remaining is washed.
4. Now the Ab-Ag-Ab-Enz complex obtained. Now a substrate for the enzyme which will form a
colored product is added. The color developed is directly proportional to the concentration of
the analyte antigen.
Antibody detection: Indirect ELISA:
1. specific antigen to the antibody coated well is taken
2. sample is added. The antibody in patient’s sample binds to the antigen.
3. an antibody attached to an enzyme like peroxidase, which will bind to antibody is
added(secondary Ab). Remaining is washed.
4. Now the Ag-Ab-Ab-Enz complex obtained. Now a substrate for the enzyme which will form a
colored product is added. The color developed is directly proportional to the concentration of
the analyte antibody.


6. Active methionine
Ans.
S-adenosyl methionine is the active form of methionine
Transmethylation reaction is acceptance of a methyl group from a donor like S-adenosyl
methionine (SAM) by a compound resulting in another compound.
Transmethylation reaction requires SAM which is obtained by accepting adenosyl group
from ATP by methionine by methionine adenosyl transferase.
The transmethylation reactions are
Methyl acceptor Methylated product
Guanidoacetic acid Creatine
Serine Choline
Epinephrine Metanephrine
Nor epinephrine Epinephrine
tRNA Methylated tRNA

7. Flame Photometer
Ans.
This is an analytical instrument used for the quantitative analysis of Na, K, Ca, and Li in
biological fluids.
Principle: when introduced to flame the analyte element like Na emits a light at characteristic
wavelength. The intensity of the light is measured at the particular wavelength of the material
and it is directly proportional to the concentration of the element.
Equipment: it contains
1. Atomizer: where sample is introduced.
2. a compressor which pumps air at high pressure which mixes with fuel and is fed into the
flame
3. A diaphragm which functions as a slit
4. a monochromator which will allow only the selected wavelength of the particular element.
5. A lens for condensation of light.
6. A photo sensor which converts light energy to electrical energy. The intensity of light
compared with that of standard and the result calculated.
Fig. Flame photometer

8. Bilirubin formation and excretion
Ans.
Formation of Bilirubin: (microsomal heme oxygenase system)
From hemoglobin the globin chains are separated and they are reduced to aminoacids
and join the aminoacid pool. The ferric heme in the reticulo endothelial system is reduced to
ferrous heme by NADPH and oxygen is introduced to form a hydroxy group between 1st and 2nd
pyrolle rings. One more molecule of oxygen added and one C is removed as CO and iron is
released, breaking the cyclic tetrapyrolle to linear tetra pyrolle called Biliverdin. 2H are added
to form bilirubin.

Excretion:
Bilirubin is bound to serum albumin and reaches the liver. In liver it is taken up and
binds to ligandin, since it is not water soluble. UGT(UDP glucuronyl transferase) acts on the
bilirubin and attaches two glucuronic acids to form Bilirubin Diglucuronide which is the
conjugated bilirubin. MOAT (Multispecific organic anion transporter) and MRP-2(Multidrug
resistance like protein) secretes bilirubin into the biliary canaliculi reaching the bile duct. In the
intestine. Intestinal bacteria contain beta glucuronidases which converts bilirubin into
urobilinogens. 99% of urobilinogens is taken to liver by enterohepatic circulation, some of it
reaching urine. Urobilinogen in intestine gets converted into stercobilinogen and both get
excreted in the feces.
Fig. formation, transport, uptake, excretion of bilirubin

9. Plasma proteins
Ans.
The total protein content of plasma is 6-8g/dl.
They are:
1. albumin- it is produced by the liver and of 69,000 D Mol.wt.
Functions of albumin:
Transport protein- it transports various substances like bilirubin, free fatty acid, drugs
like aspirin, hormones like thyroxine, steroid hormones, minerals like calcium, copper,
etc.,
Colloid osmotic pressure- albumin cannot pass between intracellular and extracellular
compartment. So exerts a net osmotic pressure. The osmotic pressure of plasma is
about 278-305mosm/kg, which is necessary for movement of water from ECF to ICF in
arteriolar end and reverse in venular end of capillaries
Buffer-the 16 Histidine residues in albumin can bind to H+ and can function as a buffer
Nutrition- liver takes up aminoacids from diet and converts it into albumin. It is then
released in blood and taken up by other tissues by pinocytosis. So albumin is a source of
aminoacids for the cells. PEM is characterized by hypoalbuminemia which results in
growth retardation and edema
2. Globulin-
Globulins are transport proteins like TBG (thyroxine binding globulin), haptoglobin which binds
Hb and immunoglobulins like Ig which acts as antibody.
3. Clotting factors- Clotting factors I to XIII are produced by the liver.
4. Transport proteins- Following are the plasma transport proteins:
- Prealbumin –steroid hormones, Thyroxine, Retinol.
- Retinol Binding protein- Retinol
- Thyroxine biding Globulin- Thyroxine
- TransCortin- Cortisol

- Transferrin – Iron
-Haptoglobin- Hb
- Hemopexin- Heme
HDL, LDL- Cholesterol
5. Acute phase reactants- CRP, Ceruloplasmin, Alpha1 Antitrypsin, Alpha 2 macroglobulin are
acute phase reactants. Their levels increase in the blood during acute reactions like fever,
inflammation, etc.

10. Replication
Ans.
DNA replication:
During cell division two daughter cells receives DNA from mother cell. The DNA divides
by the process called replication. In daughter cell, only one strand is from mother, other is
newly synthesized. This is called semiconsevative method of replication.
Steps of replication:
Each strand serves as template for new complementary strand synthesis. . The base
pairing rule is always followed.
1. Initiation: At the origin of replication (ori), there is an association of sequence-specific
dsDNA-binding proteins with a series of direct repeat DNA sequences. This leads to the local
denaturation and unwinding of an adjacent A+T-rich region of DNA.
2. Unwinding of DNA: The interaction of proteins with ori defines the start site of replication
and provides a short region of ssDNA essential for initiation of synthesis of the nascent DNA
strand. A critical step is provided by a DNA helicase that allows for processive unwinding of
DNA. Single-stranded DNA-binding proteins (SSBs) stabilize this complex.
3. Formation of replication fork: A replication fork consists of four components that form in the
following sequence:
(1) The DNA helicase unwinds a short segment of the parental duplex DNA;
(2) A primase initiates synthesis of an RNA molecule that is essential for priming DNA
synthesis;
(3) The DNA polymerase initiates nascent, daughter strand synthesis; and
(4) SSBs bind to ssDNA and prevent premature reannealing of ssDNA to dsDNA.
The polymerase III holoenzyme binds to template DNA and synthesizes DNA in the 5’ to 3’
direction. Because the DNA strands are antiparallel, the polymerase functions asymmetrically.
On the leading (forward) strand, the DNA is synthesized continuously. On the lagging
(retrograde) strand, the DNA is synthesized in short (1–5 kb) fragments, the so-called Okazaki
fragments.
4. Initiation and elongation: The initiation of DNA synthesis requires priming by a short length
of RNA, about 10–200 nucleotides long. This priming process involves the nucleophilic attack by
the 3’-hydroxyl group of the RNA primer on the α-phosphate of the first entering
deoxynucleoside triphosphate with the splitting off of pyrophosphate. The 3’-hydroxyl group of
the recently attached deoxyribonucleoside monophosphate is then free to carry out a
nucleophilic attack on the next entering deoxyribonucleoside triphosphate again at it’s α
phosphate moiety, with the splitting off of pyrophosphate.

5. Gap filling :In mammals, after many Okazaki fragments are generated, the replication
complex begins to remove the RNA primers, to fill in the gaps left by their removal with the
proper base paired deoxynucleotide, and then to seal the fragments of newly synthesized DNA
by enzymes referred to as DNA ligases.

III. Short answer questions (10 x 2 = 20)
1. Detoxification by conjugation
Phase 2 reactions: conjugation
A xenobiotic that undergone a phase 1 reaction is now a new metabolite that contains a
chemical group like OH, NH2, COOH groups. Phase 2 reactions lead to conjugation (addition) of
conjugating agents like:
Glucuronic acid: Bilirubin conjugated with glucuronic acid to form Bilirubin
Diglucuronide and excreted in bile.
Sulfate conjugation: phenol converted to phenol sulphate using PAPS(phosphor
adenosyl phosphor sulphate-active sulphate)
Cysteine and Glutathione: alkyl or aryl halide, epoxide are detoxified in this manner.
Acetylation: acetic acid s is conjugated to sulfanilamide, INH
Glycine: Benzoic acid is conjugated with glycine to form hippuric acid
Glutamine: phenyl acetic acid is conjugated to form Phenyl acetyl glutamine

2. Glutathione
Formation:
Glutamate combines with cysteine to form gamma glutamyl cysteine, which combines
with glycine to form Glutathione.
Fig. Formation of glutathione
-Glutathione is involved in meister cycle which is needed for neutral aminoacid
transport in intestines kidney tubules and brain. 5-oxoprolinase enzyme deficiency will cause
oxoprolinuria. Hartnup’s disease, Iminoglycinuria, Cystinuria, Oasthouse syndrome are the
other inborn errors in this cycle.
Fig. Meister cycle

- Glutathione is needed as a cofactor in
-maleyl acetoacetate isomerase,
-cysteic acid taurine,
-iodine to hydrogen iodide.
- Glutathione is needed for glutathione peroxidase, glutathione reductase to protect
RBCs from free radical induced damage.
Fig. Antioxidant function of Glutathione
- Conversion of met Hb to normal Hb.
-glutathione is involved in conjugation reactions in detoxification reactions
Fig. Detoxification by glutathione

3. Metabolic acidosis
Ans.
Acidosis is reduction of pH less than 7.38. It is classified into metabolic and respiratory
acidosis.
Metabolic acidosis is primarily due to base deficit. The bicarbonate deficit may occur
due to excess acid production or depletion of bicarbonate.
Anion gap: it is difference between measured cations and measured anions. Usually it shows
the unmeasured anions. The normal value is 12 mmol/L.
Metabolic acidosis is classified into:
1. High anion gap metabolic acidosis- accumulation of acid
Renal failure- H+ excretion is less.
DKA- ketoacid production is more
Lactic acidosis- hypoxia, circulatory failure, many drugs, and bacterial metabolism
increases lactic acid.
Methanol, ethanol also causes lactic acidosis
2. Normal anion gap metabolic acidosis- both anions and cations lost but acidosis present
Diarrhea- loss of bicarbonate from intestinal secretions
Hyperchloremic metabolic acidosis- in renal tubular acidosis- which may be due to
either failure to secrete acid or conserve bicarbonate; acetazolamide treatment
Compensation:
Metabolic acidosis is compensated by hyperventilation so that pCO2 comes down.
Features:
pH will be low, Bicarbonate will be low. PCO2 starts decreasing due to respiratory compensation

4. Codons
The letters A, G, T, and C correspond to the nucleotides found in DNA. They are organized into three letter code words called codons, and the collection of these codons makes up the genetic code.
Twenty different amino acids are required for the synthesis of the cellular complement of proteins; thus, there must be at least 20 distinct codons that make up the genetic code. Since there are only four different nucleotides in mRNA, each codon must consist of more than a single purine or pyrimidine nucleotide. Codons consisting of two nucleotides each could provide for only 16 (42) specific codons, whereas codons of three nucleotides could provide 64 (43) specific codons. It is now known that each codon consists of a sequence of three nucleotides; ie, it is a triplet code.
Three of the 64 possible codons do not code for specific amino acids; these have been termed nonsense codons. These nonsense codons are utilized in the cell as termination signals; they specify where the polymerization of amino acids into a protein molecule is to stop.
The remaining 61 codons code for 20 amino acids. Thus, there must be “degeneracy” in the genetic code—ie, multiple codons must decode the same amino acid. Some amino acids are encoded by several codons; for example, six different codons specify serine. Other amino acids, such as methionine and tryptophan, have a single codon.
In general, the third nucleotide in a codon is less important than the first two in determining the specific amino acid to be incorporated, and this accounts for most of the degeneracy of the code.
However, for any specific codon, only a single amino acid is indicated; with rare exceptions, the genetic code is unambiguous—ie, given a specific codon, only a single amino acid is indicated. With few exceptions, given a specific codon, only a specific amino acid will be incorporated— although, given a specific amino acid, more than one codon may be used.
It has now been shown that the set of tRNA molecules in mitochondria reads four codons differently from the tRNA molecules in the cytoplasm. The codon AUA is read as Met, and UGA codes for Trp in mammalian mitochondria. In addition, in mitochondria, the codons AGA and AGG are read as stop or chain terminator codons rather than as Arg.

5. Renal function tests
1. Glomerular function: Glomerulus acts as a sieve filtering blood but prevents cells and
proteins making a glomrular filtrate.
Glomerular filtration rate: Normal GFR is 120-125ml/min. This is reduced in renal failure. This
test is done by using either Urea or creatinine or other clearance tests like Inulin.
Clearance = mg of substance excreted per min / mg of substance per ml of plasma
2. Tubules reabsorb, secrete substances, concentrate the urine, and acidifies the urine so it is
important for maintaining specific gravity and Osmolality of urine.
The following are the tubular function tests:
a. specific gravity of urine- specific gravity depends on the concentration of the solutes. In early
stages of renal failure SG may be low due to kidney’s inability to excrete solutes.
b. Concentration test- in Diabetes insipidus and other conditions patients cannot concentrate
the urine
c. Osmolality-simultaneous measurement of serum and urine Osmolality done and if the ratio
between urine/plasma is around 3-4.5 then the patient is normal.
d. Dilution tests- this is done to check whether kidneys can excrete an excess water load.
e. Urinary acidification-. In renal tubular acidosis decreased pH is not achieved.
3. Blood Urea: Blood urea is elevated in renal conditions like Acute Glomerulo Nephritis, early
stages of nephrosis, malignant hypertension, and pyelonephritis. Elevation of blood urea is call
uremia. It is a crude indicator of renal function
4. Serum creatinine: this is a better marker of renal function than blood urea. It is elevated in
chronic renal failure.
5. Makers of Glomerular permeability: proteinuria: Glomerular sieve won’t permit molecules
more than 67000D. But in Glomerular damage that occurs in diseases like Diabetic nephropathy
the higher molecular weight proteins are filtered and appears in urine. Albumin is one of the
first proteins to appear in urine due to Glomerular damage.
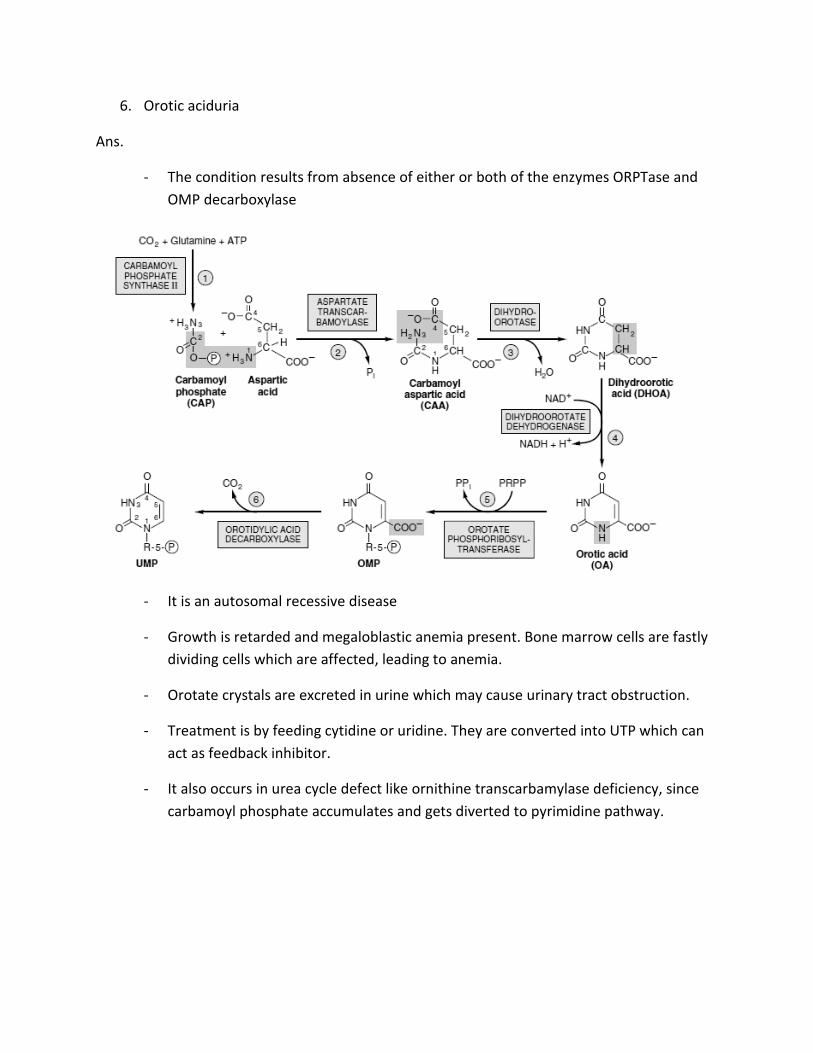
6. Orotic aciduria
Ans.
- The condition results from absence of either or both of the enzymes ORPTase and
OMP decarboxylase
- It is an autosomal recessive disease
- Growth is retarded and megaloblastic anemia present. Bone marrow cells are fastly
dividing cells which are affected, leading to anemia.
- Orotate crystals are excreted in urine which may cause urinary tract obstruction.
- Treatment is by feeding cytidine or uridine. They are converted into UTP which can
act as feedback inhibitor.
- It also occurs in urea cycle defect like ornithine transcarbamylase deficiency, since
carbamoyl phosphate accumulates and gets diverted to pyrimidine pathway.

7. Wobble hypothesis
61 codons code for 20 amino acids. Thus, there must be “degeneracy” in the genetic code—ie, multiple codons must decode the same amino acid. The degeneracy of the genetic code resides mostly in the last nucleotide of the codon triplet, suggesting that the base pairing between this last nucleotide and the corresponding nucleotide of the anticodon is not strictly by the Watson-Crick rule. This is called wobble.
For example, the two codons for arginine, AGA and AGG, can bind to the same anticodon having a uracil at its 5’ end (UCU). Similarly, three codons for glycine—GGU, GGC, and GGA—can form a base pair from one anticodon, CCI.

8. Van den Bergh test
Ans.
The serum bilirubin estimated by Van den Bergh reaction, where diazotised sulfanilic
acid reacts with bilirubin to form a purple colored complex, azobilirubin. Normal serum does
not give a positive van den Bergh test.
When bilirubin is conjugated, the purple color is produced immediately on mixing with
the reagent, the response is said to be van den Bergh direct positive.
When the bilirubin is unconjugated, the color appears only after addition of alcohol, so
it is said to be van den Bergh indirect positive.
When both conjugated and unconjugated bilirubins are present, it produces an
immediate color, which intensifies on adding alcohol. It is then said to be biphasic.
In hemolytic jaundice- unconjugated bilirubin elevated- so indirect positive
In obstructive jaundice-conjugated bilirubin elevated- so direct positive
In hepatic jaundice- both conjugated and unconjugated bilirubin elevated- so biphasic

9. Rickets
Ans. The deficiency of Vit D in children causes Rickets.
Poor exposure to sunlight like people living in high altitudes, winter seasons, women wearing
purdah, steattorhoea, liver and renal diseases are the causes of Vit D deficient.
Clinical features are
Bones become soft and pliable
Rickety rosary
Harrison’s sulcus
Knock knee
Bossing of frontal bones
Bow legs

10. γ-globulins
Ans. Immunoglobulins are γ- globulins which act as antibodies. The antibody has a high
specificity for a particular antigen only. The antibody’s structure is complementary to that
specific antigen.
Classification and structure:
Different types of Igs are A, D, E, G, M
1. IgG: IgG consist of 2 heavy chains and 2 light chains. VL and CL are variable and constant
regions in light chains and VH and CH in heavy chains. The variable regions are important for
antigen binding and recognition. It is the major antibody. They are produced by B cells and are
involved in secondary immune response and indicate chronicity of the disease. It crosses
placenta and is a reason for Rh isoimmunisation.
Other Igs in comparison with IgG
2. IgM: it has five subunits. It is involved in primary response.
3. IgA: these are secretory antibodies and gives protection to skin, intestine, eyes, urogenital
tract. It is also secreted in breast milk protecting the baby against intestinal infections.
4. IgE: they are produced by mast cells and are the cause for allergy and anaphylaxis.





























