Embed Size (px)
Citation preview

Base quality and read quality: How should data quality be measured?
Gabor T. MarthBoston College Biology Department
1000 Genomes MeetingCold Spring Harbor LaboratoryMay 5-6. 2008

Read accuracy / quality values
• quality values help distinguish between sequencing error and allelic difference• some aligners (e.g. MAQ) use quality values to find correct read mapping position
• the more accurate the read, the easier and faster it is to map• the more errors the aligner must tolerate, the less reads can be uniquely aligned0 1 2
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
Fra
ctio
n of
gen
ome
Number of mismatches allowed
Read accuracy
Well-calibrated quality values

How to tabulate sequencing error rates?
…atggatgagtataacgtcaggctaaactgtagtatatggataaaatgacca*acga…
tatgcataaaatgaccatacg
S Itggatgagtataa*gtcagg
D
measured fragment length (L)
• Align a set of reads to the corresponding organismal reference genome sequences
PE read
reference sequence
• Register positions of mismatches / gaps

Caveat #1 – paralogous mapping
aaccttactttgccttgtactgaaattactacgtacaccttactttaccttgtactgc
accttactttaccttgtactg
correct map location
spurious error
• can be avoided by using exhaustive alignment that reveals the fact of multiple possible map locations. Reads that don’t map uniquely should not be used for error analysis…
accttactttaccttgtactg
incorrect map location

Caveat #2 – local misalignment
aaccttactttgccttgtactgaaattactacgtacaccttactttaccttgtactgc
accttactttgccttgtact*a
correct alignment
D
spurious S
• typically happens at the ends of reads• consequence of the scoring scheme… difficult to fix…
accttactttgccttgtacta
incorrect alignment

Caveat #3 – polymorphic dataset / ref errors
aaccttactttgccttgtactgaaattactacgtacaccttactttaccttgtactgc
aaccttactttgccttgcactgaaattactacgtacaccttactttaccttgtactgc
reference sequence
resequenced individualactttgccttgcactgaaatt
spurious S
• important source of error: Θ ≈ 1/1,000 for humans• use resequencing data from haploid DNA source (e.g. BAC)• in polymorphic datasets, maybe do SNP calling, and exclude reads that overlap a SNP (this should also work for errors in the reference sequence)
SNP

Caveat #4 – very low quality reads
aaccttactttgccttgtactgaaattactacgtacaccttactttaccttgtactgc
acaatgcgttgca***agatt
• these reads are hard to even align elude error statistics• only tabulate error stats for reads that will be aligned?

Study design
• Took 3 random lanes each from 3 random runs of PE Illumina reads from Sanger (100,000 random pairs per lane)
• Mapped the reads to NCBI build36 using MosaikPE
• Alignment conditions: paired-end alignments, unique-unique end-readsmaximum 4 mismatches per end-read
• Fraction aligned:on average, X fraction of the reads used
Analysis by Derek Barnett

Fragment length distribution

Base error rate and error type

Base error rate by substitution type

Per-read error rates
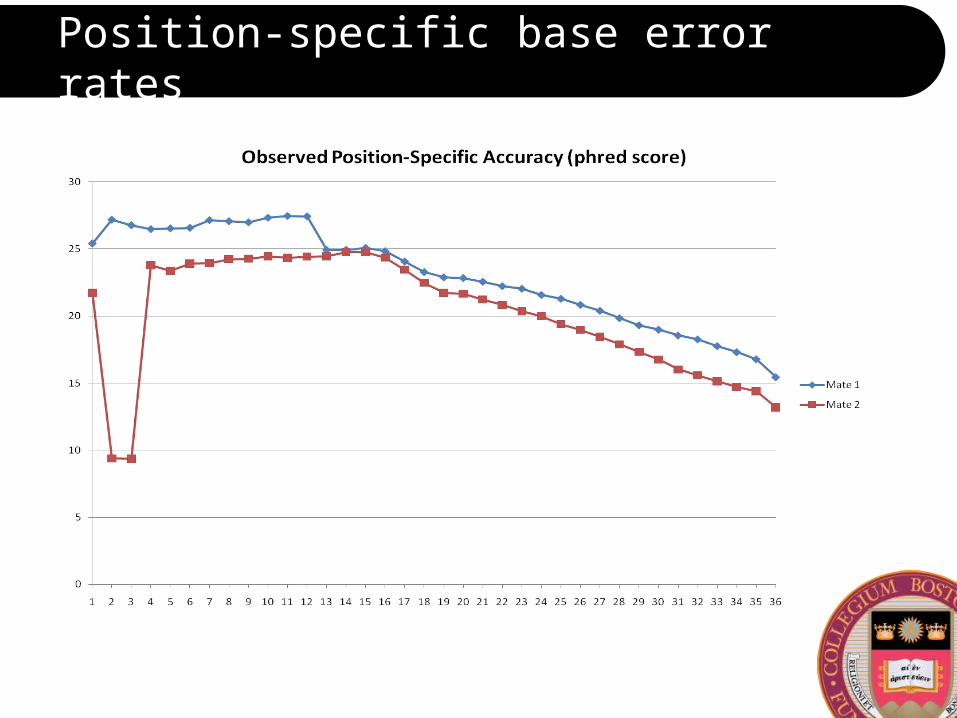
Position-specific base error rates

Accuracy across lanes and runs
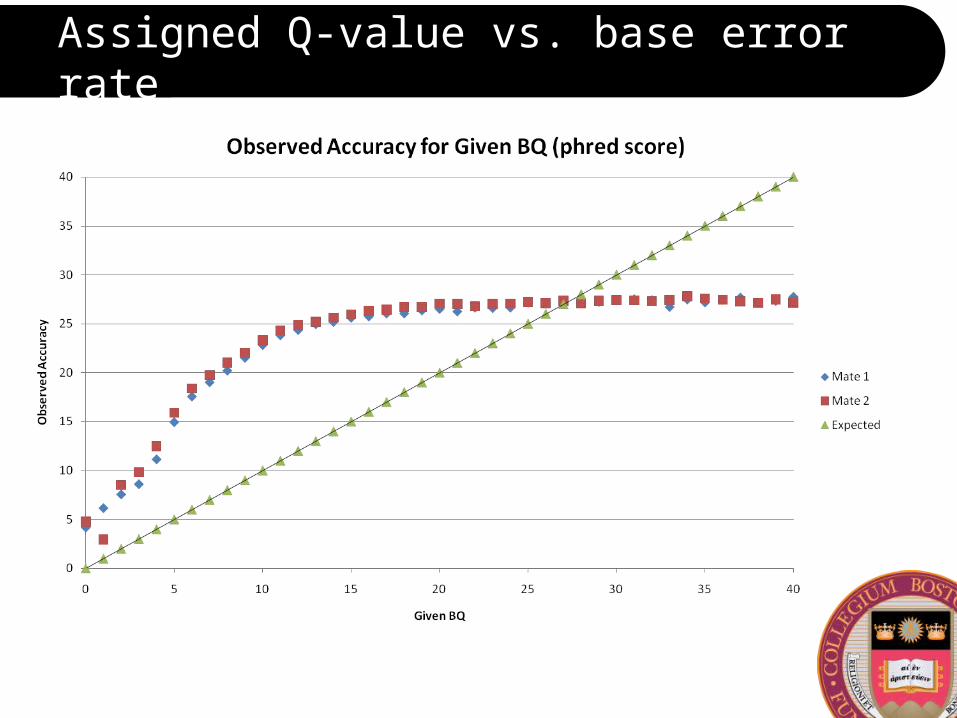
Assigned Q-value vs. base error rate

Does the Q-value depend on base cycle?
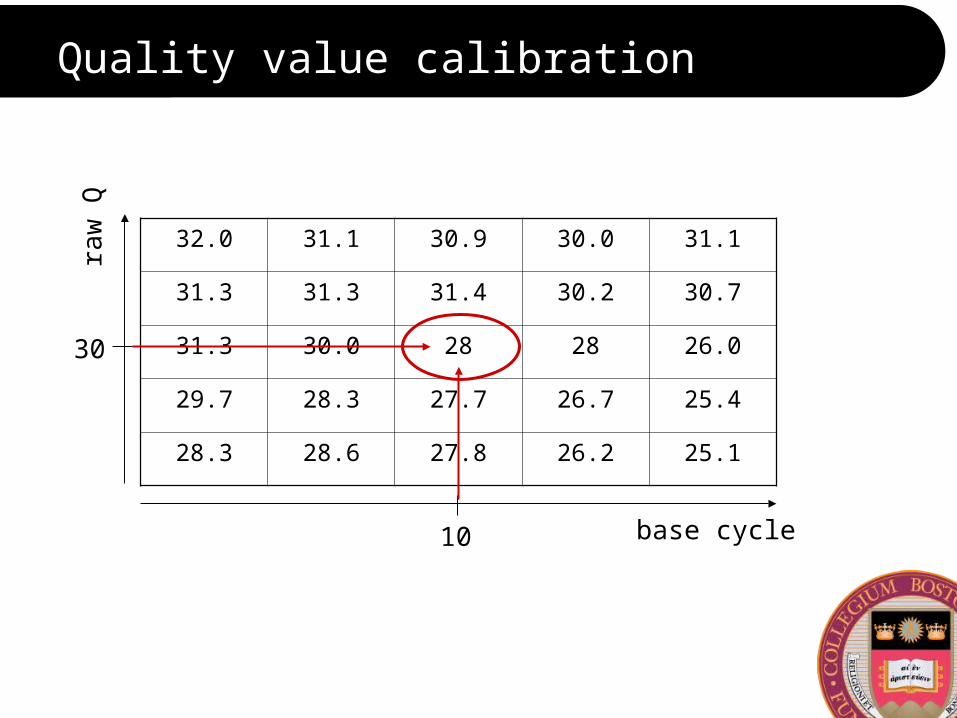
Quality value calibration
base cycle
raw
Q
30
10
32.0 31.1 30.9 30.0 31.1
31.3 31.3 31.4 30.2 30.7
31.3 30.0 28 28 26.0
29.7 28.3 27.7 26.7 25.4
28.3 28.6 27.8 26.2 25.1

Q-values for read simulations
0.7 0.9 0.8 0.8 0.1
3.6 2.3 2.1 2.0 1.0
20.1 19.4 18.3 18.2 16.3
9.0 10.1 9.2 7.9 7.5
7.9 6.7 7.1 5.4 4.3
Weichun Huang, see poster at Genome Meeting
base cycle
Q
30
10
introduce error in read at base cycle 10 with P=0.001

Thanks
Derek Aaron Weichun
Michael Chip





























