Embed Size (px)
Citation preview

AcZDJ
FEa
Bbcrmoc
O1
Mch
RSai(
ISpUsatStvsssf
cHrtFm
1
ssociation of TGFBR2 polymorphism with risk of suddenardiac arrest in patients with coronary artery diseaseian H. Tseng, MD, MAS,* Eric Vittinghoff, PhD, MPH,† Stacy L. Musone, BA,‡ Feng Lin, MS,†
ean Whiteman, BS,* Ludmila Pawlikowska, PhD,‡� Pui-Yan Kwok, MD, PhD,‡§
effrey E. Olgin, MD, FHRS,*§ Bradley E. Aouizerat, PhD, MAS‡¶
rom the *Section of Cardiac Electrophysiology, Division of Cardiology, Department of Medicine, †Department ofpidemiology and Biostatistics, ‡Institute for Human Genetics, §Cardiovascular Research Institute, �Department of Anesthesia
¶
nd Perioperative Care, and Department of Physiologic Nursing, University of California, San Francisco, California.CpIT
Ktf
AiOcpad
(
ACKGROUND Transforming growth factor ß (TGFß) signaling haseen shown to promote myocardial fibrosis and remodeling withoronary artery disease (CAD), and previous studies show a majorole for fibrosis in the initiation of malignant ventricular arrhyth-ias (VA) and sudden cardiac arrest (SCA). Common single nucle-tide polymorphisms (SNPs) in TGFß pathway genes may be asso-iated with SCA.
BJECTIVE We examined the association of common SNPs among2 candidate genes in the TGFß pathway with the risk of SCA.
ETHODS SNPs (n � 617) were genotyped in a case-control studyomparing 89 patients with CAD and SCA caused by VA to 520ealthy control subjects.
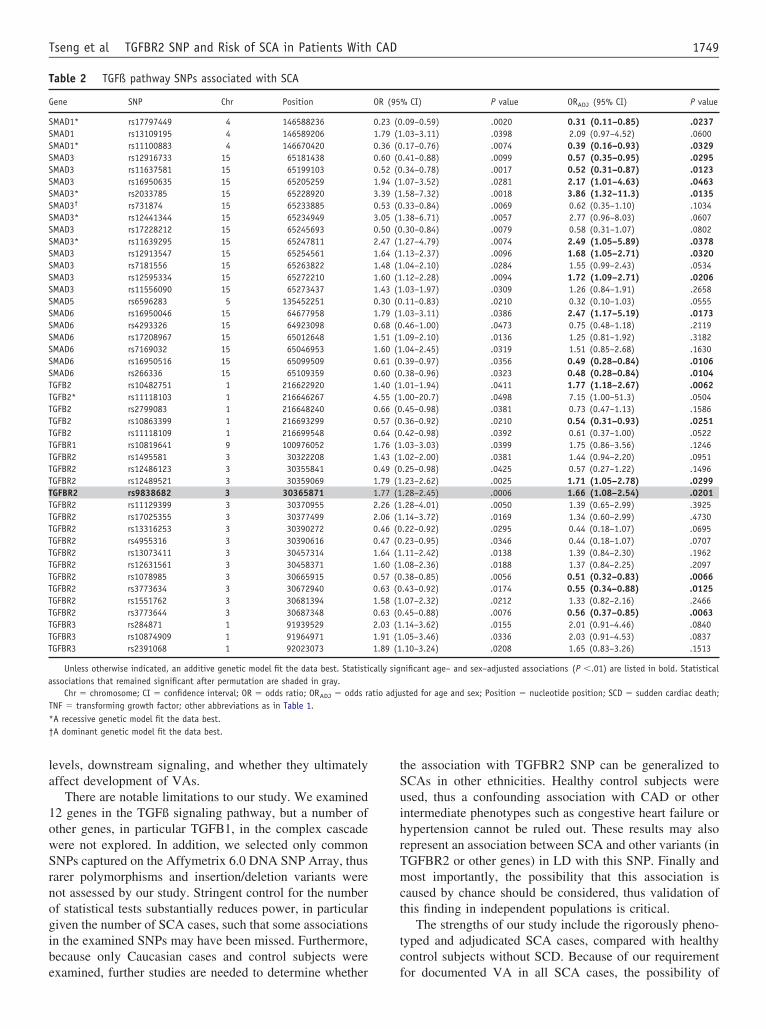
ESULTS Nineteen SNPs among 5 genes (TGFB2, TGFBR2, SMAD1,MAD3, SMAD6) were associated with SCA after adjustment for agend sex. After permutation analysis to account for multiple test-ng, a single SNP in TGFBR2 (rs9838682) was associated with SCA
catrfllmaRgnatdaotsiedicine.ucsf.edu. (Received July 30, 2009; accepted August 23, 2009.)
547-5271/$ -see front matter © 2009 Heart Rhythm Society. All rights reserved
ONCLUSION We show an association between a common TGFBR2olymorphism and risk of SCA caused by VA in the setting of CAD.f validated, these findings support the role of genetic variation inGFß signaling in SCA susceptibility.
EYWORDS Sudden cardiac arrest; Ventricular tachycardia; Ven-ricular fibrillation; Coronary artery disease; Transforming growthactor ß; Genetics
BBREVIATIONS CAD � coronary artery disease; CI � confidencenterval; LD � linkage disequilibrium; MI � myocardial infarction;R � odds ratio; PCA � principal component analysis; SCA � suddenardiac arrest; SCD � sudden cardiac death; SNP � single nucleotideolymorphism; TGF � transforming growth factor; VA � ventricularrrhythmia; VF � ventricular fibrillation; VT � ventricular tachycar-ia
Heart Rhythm 2009;6:1745–1750) © 2009 Heart Rhythm Society. All
odds ratio: 1.66, 95% confidence interval: 1.08 to 2.54, P � .02). rights reserved.ntroductionudden cardiac arrest (SCA) remains a major public healthroblem, accounting for up to 450,000 deaths per year in the.S.1 Previous studies suggest that approximately 80% of
udden cardiac deaths (SCDs) occur in the setting of coronaryrtery disease (CAD).2 Ventricular tachycardia (VT) or ven-ricular fibrillation (VF) is the initiating event in the majority ofCA cases.3 Ejection fraction remains the only major criterion
o stratify patients for risk of SCD and implantable cardio-erter-defibrillator implantation, but this strategy alone is in-ensitive and nonspecific.4 Genetic susceptibility to SCA in theetting of CAD is supported by several epidemiologic studieshowing that a family history of SCD is an independent riskactor for SCD and primary VF.5–7
Funded by grants from the National Center for Research Resources, aomponent of the National Institutes of Health and the National Institutes ofealth Roadmap for Medical Research (KL2 RR024130). Address reprint
equests and correspondence: Dr. Zian H. Tseng, Section of Cardiac Elec-rophysiology, 500 Parnassus Avenue, Box 1354, University of California, Sanrancisco, San Francisco, California 94143-1354. E-mail: zhtseng@
Transforming growth factor ß (TGFß) is a potent profibroticytokine. Three structurally similar isoforms (TGFß1, TGFß2,nd TGFß3) encoded by 3 distinct genes transduce their signalhrough combinations of transmembrane type I and type IIeceptors (TGFBR1 and TGFBR2) and their downstream ef-ectors, the SMAD proteins.8,9 TGFß is expressed at highevels in the heart during embryonic development and adultife, localizing in both cardiomyocytes and the extracellularatrix.10 TGFß2 knockout mice show perinatal mortality andwide range of developmental, including cardiac, defects.11
egulatory mutations in the TGFß3 gene cause arrhythmo-enic right ventricular cardiomyopathy, an autosomal-domi-ant clinical syndrome of ventricular arrhythmias (VAs), SCD,nd fibrofatty replacement of the right ventricle.12 Members ofhe TGFß family are markedly induced in infarcted myocar-ium and play a central role in infarct healing, cardiac repair,nd left ventricular remodeling through mechanisms of cardi-myocyte growth, fibroblast activation, and extracellular ma-rix deposition.13 Although these basic and genetic studiesuggest a central role for the TGFß signaling pathway in
schemic remodeling, genetic variation in members of the path-. doi:10.1016/j.hrthm.2009.08.031

wV
gwSc
MTHwp
SCCpCaaVsomCtSwiH
TsccStl((Cn2dpfwp
GFSpfst
teirtSengtsaTSaino
BBD(
FrSSbttTASoSoSaTSav(c
1746 Heart Rhythm, Vol 6, No 12, December 2009
ay have not been investigated for association with risk ofAs and SCA in the setting of CAD.We examined the potential role of polymorphisms in 12
enes in the TGFß signaling pathway in a group of patientsith a history of myocardial infarction (MI) and abortedCD with documented VT/VF compared with 3 healthyontrol groups without SCA or VAs.
ethodshe University of California at San Francisco Committee onuman Research approved all protocols. Informed consentas obtained from all participants for DNA isolation andlasma collection.
tudy designonsecutive cases of SCA presenting to the University ofalifornia at San Francisco Medical Center Emergency De-artment or in-patient cases of SCA at the University ofalifornia at San Francisco Medical Center between Janu-ry 2000 and June 2008 were screened. SCA was defined ascardiac arrest with documented sustained monomorphicT or VF requiring cardioversion or defibrillation, exclu-
ive of torsades de pointes (drug-induced QT prolongationr otherwise). Because we were interested in the most com-on phenotype of SCA, that occurring in the setting ofAD, only patients with a history of MI were included in
his study. Although all SCA cases had a history of MI, 36CA cases occurred in the setting of acute ischemia,hereas 53 SCA cases occurred in the absence of active
schemia. Thus, SCA cases consisted of 89 Caucasian non-ispanic patients with aborted SCD and a history of MI.The control population originated from 3 resources.14
hese were randomly selected, healthy Caucasian controlubjects without specific information regarding CAD. Re-ent large genome-wide association studies using sharedontrol subjects of this type have met with success.14–16
ixty unrelated control subjects employed by the Interna-ional HapMap Project (http://www.HapMap.org) were se-ected from the Coriell Institute for Medical Researchhttp://ccr.coriell.org) Human Genetic Data Collectionsample identifiers are available on request); 216 healthyaucasian control subjects were derived from a large ge-ome-wide association study of narcolepsy.14 The remaining44 control subjects were healthy Caucasian renal transplantonors from an ongoing study of the genomics of renal trans-lantation (National Institutes of Health U19 AI063603). In-ormed consent was obtained from all participants. Protocolsere approved by the local institution review boards at allarticipating institutions.
ene selectionigure 1 describes the TGFß-related signaling pathway. TheMAD mediators can be divided into 2 main intracellularathways: SMAD2/3 or SMAD1/5.17 Members of the TGFßamily (TGFß1/2/3) bind TGFBR2, type 2 receptor, apecific serine/threonine receptor kinase that catalyzes
he phosphorylation of type 1 receptor, TGFBR1, activa- ting the protein. TGFß type 3 receptor, TGFBR3, maynhance the binding of TGFß ligands to TGFBR2 by bind-ng TGFß and presenting it to TGFBR2. Activated type Ieceptors phosphorylate specific receptor-regulated R-SMADs,he intracellular effectors of TGFß signaling, eitherMAD2/3 or SMAD1/5. Activated R-SMADs form het-romeric complexes with SMAD4 that translocate to theucleus, where they regulate the expression of targetenes in conjunction with transcription factors. Inhibi-ory SMADs, SMAD6 and SMAD7, antagonize TGFßignaling by inhibiting activation of R-SMADs. We ex-mined 12 genes (TGFB2, TGFB3, TGFBR1, TGFBR2,GFBR3, SMAD1, SMAD2, SMAD3, SMAD4, SMAD5,MAD6, SAMD7) in the TGFß signaling pathway to ex-mine for association with risk of SCA. TGFB1 was notncluded because the only variant on the array (see later) didot meet our criteria for a common variant (allele frequencyf �0.05) and thus was not analyzed.
lood collection and genotypinglood samples were obtained by venipuncture, and genomicNA was extracted from peripheral blood lymphocytes
Invitrogen, Carlsbad, California) for the SCA cases. Geno-
igure 1 Gene selection in the TGFß signaling pathway. Schematicepresentation of genes examined in the TGFß signaling pathway. TheMAD mediators can be divided into 2 main intracellular pathways:MAD2/3 or SMAD1/5.17 Members of the TGFß family (TGFB1/2/3)ind TGFBR2, type 2 receptor, a specific serine/threonine receptor kinasehat catalyzes the phosphorylation of type 1 receptor, TGFBR1, activatinghe protein. TGFß type 3 receptor, TGFBR3, may enhance the binding ofGFß ligands to TGFBR2 by binding TGFß and presenting it to TGFBR2.ctivated type I receptors phosphorylate specific receptor regulated R-MADs, the intracellular effectors of TGFß signaling, either SMAD2/3r SMAD 1/5. Activated R-SMADs form heteromeric complexes withMAD4 that translocate to the nucleus, where they regulate the expressionf target genes in conjunction with transcription factors. InhibitoryMADs, SMAD6 and SMAD7, antagonize TGFß signaling by inhibitingctivation of R-SMADs. We examined 12 genes (TGFB2, TGFB3,GFBR1, TGFBR2, TGFBR3, SMAD1, SMAD2, SMAD3, SMAD4,MAD5, SMAD6, SAMD7) in the TGFß signaling pathway to examine forssociation with risk of SCA. TGFB1 was not included because the onlyariant on the platform did not meet our criteria for a common variantallele frequency of �0.05) and thus was not analyzed. SCA � suddenardiac arrest; TGF � transforming growth factor.
yping was performed blinded to clinical status; positive and

nq(imgmfagiAadp
SCqse“grCwS�wgSS71ifeS2fss
STfvw
gblcmuHs
aiittmpowac
enniutmop
rwctPtf
RSCopcagmwn
GwGddd4STtawSj
1747Tseng et al TGFBR2 SNP and Risk of SCA in Patients With CAD
egative control subjects were included. DNA samples wereuantitated with a Nanodrop spectrophotometer (ND-1000),Thermo Fisher Scientific, Wilmington, DE) and normal-zed to a concentration of 50 ng/�l (diluted in 10 mM Tris/1
M ethylene-diamine-tetra-acetic acid). Samples wereenotyped using the genome-wide single nucleotide poly-orphism (SNP) array 6.0 (Affymetrix, Santa Clara, Cali-
ornia) and processed according to the standard Affymetrixutomated protocol. This involved fragmentation, whole-enome amplification, precipitation, resuspension in hybrid-zation buffer, and hybridization to the Genome-Wide SNPrray 6.0. After hybridization, the arrays were processed
nd imaged on an Affymetrix GeneChip Scanner. Genotypeata for the control subjects were obtained using the samerotocols in the same laboratory as described for SCA cases.
NP selectionommon variants (defined as having a minor allele fre-uency �0.05 in the combined sample of cases and controlubjects) mapping to the candidate genes of interest werextracted from the array .cel files. Data files (Affymetrix.cel” files) were obtained for all samples (n � 608), andenotype calls were assigned using the Birdseed-dev algo-ithm for Affy 6.0 (Affymetrix Power Tools apt-1.8.5, Santalara, CA). To ensure robust genetic association analyses,e performed quality control filtering of both samples andNPs. For each Birdseed calling run, SNPs with call rates95%, or Hardy-Weinberg P �.001 in control subjectsere excluded. A total of 617 SNPs among the 12 candidateenes (SMAD1: 25 SNPs; SMAD2: 22 SNPs; SMAD3: 65NPs; SMAD4: 7 SNPs; SMAD5: 18 SNPs; SMAD6: 147NPs; SMAD7: 21 SNPs; TGFB2: 47 SNPs; TGFB3:SNPs; TGFBR1: 24 SNPs; TGFBR2: 125 SNPs; TGFBR3:
09 SNPs) passed all quality control filters and were includedn the genetic association analyses. SNP selection was per-ormed using the HelixTree SNP and Variation Suite 7 (Gold-nHelix, Bozeman, Montana). Potential functional roles ofNPs associated with SCA were examined using PUPASuite.0,18 a comprehensive search engine that tests a series ofunctional effects (i.e., nonsynonymous changes, altered tran-cription factor binding sites, exonic splicing enhancing orilencing, splice site alterations, microRNA target alterations).
tatistical analyseso examine for differences in the distribution of demographic
actors between cases and control subjects, we used the unequal-ariance t test for age and a chi-square test for sex. These analysesere carried out using STATA 9.0 (College Station, TX).
Allele and genotype frequencies were determined byene counting. Hardy-Weinberg equilibrium was assessedy the chi-square exact test. Measures of linkage disequi-ibrium (LD), D= and r2, were computed from the case andontrol genotypes with Haploview 4.1 (http://www.broad.it.edu/mpg/haploview/) and heat maps of pairwise D= val-
es are presented in Supplementary Figures 1 through 11.omogeneity in ancestry between cases and shared control
ubjects was verified by cluster and principal component o
nalysis (PCA).19 To investigate other biases that could bentroduced by using control subjects shared with other stud-es such as batch effects due to differences in instrumenta-ion,20 we assessed the potential effect of substructure withhe genomic-control method21 and with PCA, as imple-
ented in HelixTree (GoldenHelix). Briefly, the number ofrincipal components were sought that minimized the valuef the genome control test statistic. Population substructureas found to be negligible, with no discernible batch effects
nd PCA-adjusted tests of association that varied little asompared with unadjusted tests (data not shown).
For association tests, 4 genetic models were assessed forach SNP: dominant, recessive, log additive, and codomi-ant. Barring trivial improvements (delta �10%), the ge-etic model that best fit the data by maximizing the signif-cance of the P value was selected for each SNP. Bothnadjusted and adjusted associations were calculated; logis-ic regression was used to control for age and sex. Geneticodel fit and both unadjusted and age- and sex-adjusted
dds ratios (ORs) were estimated using the SAS softwareackage, version 9.1.3 (SAS Inc., Cary, North Carolina).
Permutation tests were used to protect the type-I errorate against inflation caused by testing of multiple SNPsithin each gene. To account for multiple comparisons,
ase-control status was permuted 10,000 times to determinehe likelihood that our findings were attributable to chance.ermutated significance tests are presented in Supplemen-
ary Tables 1 through 12. Permutation analyses were per-ormed using the PLINK software package (version 1.06).22
esultstudy populationompared with control subjects (Table 1), SCA cases werelder (73.0 � 12.7 vs 52.7 � 15.6 years; P �.0001) andredominantly male (90.9% vs 51.9%, P �.0001). SCAases had a mean body mass index of 25.7 � 4.4 kg/m2 andmean ejection fraction of 35.7 � 14.5%. History of con-
estive heart failure was present in 76.5% of cases, diabetesellitus was present in 28.4% of cases, and hypertensionas present in 81.6% of cases. Clinical characteristics wereot available for the anonymous healthy control subjects.
enetic association of TGFBR2 rs9838682ith SCAenotype frequencies for each SNP for each of the 12 candi-ate genes are shown in Online Tables 1 through 12. No SNPeviated from Hardy-Weinberg expectations. Initially, evi-ence of genetic association with SCA was identified for5 SNPs among 8 genes (SMAD1: 3 SNPs; SMAD3: 12NPs; SMAD5: 1 SNP; SMAD6: 6 SNPs; TGFB2: 5 SNPs;GFBR1: 1 SNP; TGFBR2: 14 SNPs; TGFBR3: 3 SNPs) in
he unadjusted analyses (Table 2). After adjustment for agend sex, 19 SNPs among 5 genes retained their associationith SCA (SMAD1: 2 SNPs; SMAD3: 7 SNPs; SMAD6: 3NPs; TGFB2: 2 SNPs; TGFBR2: 5 SNPs). Of note, ad-
ustment for age and sex generally resulted in modest effects
n the OR point estimates. Permutation was used to account
fnTw(at
pisaaIa1cnd2i.a
ipaihtq2nw
DImcposm
atmT3ciasv
MzrtiarMls
crwbinefsmt
wptmHtm
T
C
AAMCEAHDCB
t.
1748 Heart Rhythm, Vol 6, No 12, December 2009
or the number of false positives that are likely given theumber of comparisons made, resulting in a single SNP inGFBR2 (rs9838682) remaining statistically significantith an adjusted P �.05. An additional SNP in SMAD3
rs11637581) showed a permuted P value of .056 that,lthough suggestive, did not meet the a priori significancehreshold for additional analyses (Online Table 3).
TGFBR2 rs9838682 is a G-to-A transition polymor-hism with the frequency of the minor allele (A) of 33.1%n control subjects. The frequency of the A allele wasignificantly elevated in SCA cases (46.6%, P �.001). Thedditive genetic model fit the data best. The A allele wasssociated with SCA with an OR of 1.77 (95% Confidencenterval [CI]: 1.28 to 2.45; P � .0006). Adjustment for agend sex yielded a similar OR point estimate: 1.66 (95% CI:.08 to 2.54, P � .02). The association remained statisti-ally significant after permutation testing (P � .045). Ofote, limiting the analysis to the 36 SCA cases occurringuring active ischemia (adjusted OR: 1.58, 95% CI: 0.89 to.80, P � .12) or the 53 SCA cases in the absence ofschemia (adjusted OR: 1.71, 95% CI: 0.99 to 2.95, P �054) reduced power but resulted in similar point estimatesnd overlapping CIs for the OR.
TGFBR2 rs9838682 lies in a region of high LD (Figure 2)n a haplotype block spanning more than 43.6 thousand baseairs (kB). Although other SNPs in this region of LD weressociated with SCA (Table 2), none reached statistical signif-cance after permutation testing and were thus not examined inaplotype analyses, because such analyses have limited powero detect meaningful effects.23 The functional impact of se-uence variation in rs9838682 was assessed using PUPASuite.0, with no impact predicted for the minor allele.24 Similarly,one of the tested SNPs in LD (D= � 0.80) with rs9838682ere predicted to impact function.
iscussionn the present study, we show an association between a com-on TGFBR2 polymorphism (rs9838682) and risk of SCA
aused by malignant VA in the setting of CAD. Rigorouslyhenotyped cases were collected, with documented presencer absence of CAD and VA. Compared with healthy controlubjects, adjusted for sex and age differences, the rs9838682
able 1 Demographic characteristics of SCA cases and healthy
haracteristics SCA cases All control subjects Na
N 89 520 21ge (yrs) 73.0 � 12.7 52.7 � 15.6 59ale (%) 90.9 51.9 5ongestive heart failure (%) 76.5% N/Ajection fraction (%) 35.7 � 14.5 N/Age at index MI (yrs) 56.7 � 13.6 N/Aypertension (%) 84.2 N/Aiabetes (%) 28.4 N/Aurrent smoker (%) 10.9 N/Aody mass index (kg/m2) 25.7 � 4.4 N/A
MI � myocardial infarction; N/A � not applicable; SCA � sudden cardiac arres
inor allele was associated with increased odds of SCA. v
The TGFBR2 gene lies on chromosome 3p22 with 7 exonsnd encodes for the type II TGFß receptor, a 565-amino acidransmembrane tyrosine kinase with a calculated molecularass of approximately 60 kD.25 In gastrointestinal tissues,GFBR2 acts as a tumor suppressor gene and approximately0% of colorectal cancers carry TGFBR2 mutations.26 In theardiovascular system, mutations resulting in abnormal splic-ng in TGFBR2 lead to loss of function of TGFß signalingctivity on extracellular matrix formation and cause Marfanyndrome.27 Interestingly, tissues derived from affected indi-iduals show increased expression of collagen.
An important substrate for VAs that leads to SCD afterI is altered conduction, particularly in the infarct border
one, caused by structural remodeling. TGFß plays a keyole in the development of cardiac fibrosis.28 In experimen-al models, ventricular fibrosis increases the incidence of VFnitiated by triggered activity.29 After MI, TGFß signaling isltered and plays a central role in infarct healing and cardiacemodeling.13 TGFß1 and TGFß2 are induced early after
I, whereas TGFß3 shows delayed and prolonged upregu-ation.30,31 SMAD2, SMAD3, and SMAD4 protein expres-ion is upregulated in the scar and border zone area.32
Recent work from our laboratory shows mitigation ofardiac fibrosis and reduction in vulnerability to atrial ar-hythmias and VAs with an antifibrotic agent (pirfenidone),hich is known to inhibit TGFß1 signaling.33 TGFBR2inds all 3 TGFß ligands, and represents the first criticalntracellular step in this complex signaling cascade.17 Ge-etic variation in TGFBR2 that affects function may bexpected to play a central role in altered downstream ef-ects, such as enhancing collagen and extracellular matrixynthesis,34 and in inducing conversion of fibroblasts toyofibroblasts,35 and thus risk for VT/VF, particularly in
he setting of CAD.The TGFBR2 rs9838682 SNP found to be associated
ith SCA in the setting of CAD is a common polymor-hism. This SNP is intronic, lying between exons 3 and 4,hus it may be in LD with the actual functional variant, or itay impact gene splicing, transcription, or regulation.owever, limited data are available on its specific func-
ional effects, therefore further study is necessary to fine-ap the association signal, identify causative functional
l subjects
control subjects Renal donor control subjects HapMap control subjects
244 605 41.9 � 13.8 74.0 � 7.9
52.1 50.0
contro
rcolepsy
6.0 � 7.2.3
ariants, and elucidate their impact on TGFBR2 function or
la
1owSrnogibe
tSuihrTmct
tc
T
G
SSSSSSSSSSSSSSSSSSSSSSTTTTTTTTTTTTTTTTTTTTTTT
a
T*†
1749Tseng et al TGFBR2 SNP and Risk of SCA in Patients With CAD
evels, downstream signaling, and whether they ultimatelyffect development of VAs.
There are notable limitations to our study. We examined2 genes in the TGFß signaling pathway, but a number ofther genes, in particular TGFB1, in the complex cascadeere not explored. In addition, we selected only commonNPs captured on the Affymetrix 6.0 DNA SNP Array, thusarer polymorphisms and insertion/deletion variants wereot assessed by our study. Stringent control for the numberf statistical tests substantially reduces power, in particulariven the number of SCA cases, such that some associationsn the examined SNPs may have been missed. Furthermore,ecause only Caucasian cases and control subjects were
able 2 TGFß pathway SNPs associated with SCA
ene SNP Chr Position
MAD1* rs17797449 4 146588236MAD1 rs13109195 4 146589206MAD1* rs11100883 4 146670420MAD3 rs12916733 15 65181438MAD3 rs11637581 15 65199103MAD3 rs16950635 15 65205259MAD3* rs2033785 15 65228920MAD3† rs731874 15 65233885MAD3* rs12441344 15 65234949MAD3 rs17228212 15 65245693MAD3* rs11639295 15 65247811MAD3 rs12913547 15 65254561MAD3 rs7181556 15 65263822MAD3 rs12595334 15 65272210MAD3 rs11556090 15 65273437MAD5 rs6596283 5 135452251MAD6 rs16950046 15 64677958MAD6 rs4293326 15 64923098MAD6 rs17208967 15 65012648MAD6 rs7169032 15 65046953MAD6 rs16950516 15 65099509MAD6 rs266336 15 65109359GFB2 rs10482751 1 216622920GFB2* rs11118103 1 216646267GFB2 rs2799083 1 216648240GFB2 rs10863399 1 216693299GFB2 rs11118109 1 216699548GFBR1 rs10819641 9 100976052GFBR2 rs1495581 3 30322208GFBR2 rs12486123 3 30355841GFBR2 rs12489521 3 30359069GFBR2 rs9838682 3 30365871GFBR2 rs11129399 3 30370955GFBR2 rs17025355 3 30377499GFBR2 rs13316253 3 30390272GFBR2 rs4955316 3 30390616GFBR2 rs13073411 3 30457314GFBR2 rs12631561 3 30458371GFBR2 rs1078985 3 30665915GFBR2 rs3773634 3 30672940GFBR2 rs1551762 3 30681394GFBR2 rs3773644 3 30687348GFBR3 rs284871 1 91939529GFBR3 rs10874909 1 91964971GFBR3 rs2391068 1 92023073
Unless otherwise indicated, an additive genetic model fit the data best. Statistissociations that remained significant after permutation are shaded in gray.
Chr � chromosome; CI � confidence interval; OR � odds ratio; ORADJ � odds raNF � transforming growth factor; other abbreviations as in Table 1.A recessive genetic model fit the data best.A dominant genetic model fit the data best.
xamined, further studies are needed to determine whether f
he association with TGFBR2 SNP can be generalized toCAs in other ethnicities. Healthy control subjects weresed, thus a confounding association with CAD or otherntermediate phenotypes such as congestive heart failure orypertension cannot be ruled out. These results may alsoepresent an association between SCA and other variants (inGFBR2 or other genes) in LD with this SNP. Finally andost importantly, the possibility that this association is
aused by chance should be considered, thus validation ofhis finding in independent populations is critical.
The strengths of our study include the rigorously pheno-yped and adjudicated SCA cases, compared with healthyontrol subjects without SCD. Because of our requirement
% CI) P value ORADJ (95% CI) P value
.09–0.59) .0020 0.31 (0.11–0.85) .0237
.03–3.11) .0398 2.09 (0.97–4.52) .0600
.17–0.76) .0074 0.39 (0.16–0.93) .0329
.41–0.88) .0099 0.57 (0.35–0.95) .0295
.34–0.78) .0017 0.52 (0.31–0.87) .0123
.07–3.52) .0281 2.17 (1.01–4.63) .0463
.58–7.32) .0018 3.86 (1.32–11.3) .0135
.33–0.84) .0069 0.62 (0.35–1.10) .1034
.38–6.71) .0057 2.77 (0.96–8.03) .0607
.30–0.84) .0079 0.58 (0.31–1.07) .0802
.27–4.79) .0074 2.49 (1.05–5.89) .0378
.13–2.37) .0096 1.68 (1.05–2.71) .0320
.04–2.10) .0284 1.55 (0.99–2.43) .0534
.12–2.28) .0094 1.72 (1.09–2.71) .0206
.03–1.97) .0309 1.26 (0.84–1.91) .2658
.11–0.83) .0210 0.32 (0.10–1.03) .0555
.03–3.11) .0386 2.47 (1.17–5.19) .0173
.46–1.00) .0473 0.75 (0.48–1.18) .2119
.09–2.10) .0136 1.25 (0.81–1.92) .3182
.04–2.45) .0319 1.51 (0.85–2.68) .1630
.39–0.97) .0356 0.49 (0.28–0.84) .0106
.38–0.96) .0323 0.48 (0.28–0.84) .0104
.01–1.94) .0411 1.77 (1.18–2.67) .0062
.00–20.7) .0498 7.15 (1.00–51.3) .0504
.45–0.98) .0381 0.73 (0.47–1.13) .1586
.36–0.92) .0210 0.54 (0.31–0.93) .0251
.42–0.98) .0392 0.61 (0.37–1.00) .0522
.03–3.03) .0399 1.75 (0.86–3.56) .1246
.02–2.00) .0381 1.44 (0.94–2.20) .0951
.25–0.98) .0425 0.57 (0.27–1.22) .1496
.23–2.62) .0025 1.71 (1.05–2.78) .0299
.28–2.45) .0006 1.66 (1.08–2.54) .0201
.28–4.01) .0050 1.39 (0.65–2.99) .3925
.14–3.72) .0169 1.34 (0.60–2.99) .4730
.22–0.92) .0295 0.44 (0.18–1.07) .0695
.23–0.95) .0346 0.44 (0.18–1.07) .0707
.11–2.42) .0138 1.39 (0.84–2.30) .1962
.08–2.36) .0188 1.37 (0.84–2.25) .2097
.38–0.85) .0056 0.51 (0.32–0.83) .0066
.43–0.92) .0174 0.55 (0.34–0.88) .0125
.07–2.32) .0212 1.33 (0.82–2.16) .2466
.45–0.88) .0076 0.56 (0.37–0.85) .0063
.14–3.62) .0155 2.01 (0.91–4.46) .0840
.05–3.46) .0336 2.03 (0.91–4.53) .0837
.10–3.24) .0208 1.65 (0.83–3.26) .1513
nificant age– and sex–adjusted associations (P �.01) are listed in bold. Statistical
sted for age and sex; Position � nucleotide position; SCD � sudden cardiac death;
OR (95
0.23 (01.79 (10.36 (00.60 (00.52 (01.94 (13.39 (10.53 (03.05 (10.50 (02.47 (11.64 (11.48 (11.60 (11.43 (10.30 (01.79 (10.68 (01.51 (11.60 (10.61 (00.60 (01.40 (14.55 (10.66 (00.57 (00.64 (01.76 (11.43 (10.49 (01.79 (11.77 (12.26 (12.06 (10.46 (00.47 (01.64 (11.60 (10.57 (00.63 (01.58 (10.63 (02.03 (11.91 (11.89 (1
cally sig
tio adju
or documented VA in all SCA cases, the possibility of

aBnl
CITCtnt
ATDgt
SSi
R
1
1
1
1
1
1
1
1
1
1
2
2
2
2
2
2
2
2
2
2
3
3
3
3
3
3
Fdw(lrgs
1750 Heart Rhythm, Vol 6, No 12, December 2009
ssociation with other causes of sudden death is avoided.ecause we used shared control subjects, principal compo-ents analysis was used to exclude the possibility of popu-ation stratification as a cause of spurious association.
onclusionn summary, our results suggest that genetic variation inGFBR2 may be associated with risk for SCA associated withAD. These findings contribute to accumulating evidence for
he influence of TGFß signaling in fibrosis and risk for malig-ant VAs. If these results are validated, further investigation ofhe TGFß pathway in the development of VAs is warranted.
cknowledgmentshe authors thank Emmanuel Mignot, MD, PhD, andaniel Salomon, MD, for providing anonymous controlenotypes, and Annie Poon and Matthew Akana for assis-ance with genotyping experiments.
upplementary dataupplementary data associated with this article can be found,
n the online version, at doi:10.1016/j.hrthm.2009.08.031.
eferences1. Zheng ZJ, Croft JB, Giles WH, Mensah GA. Sudden cardiac death in the United
States, 1989 to 1998. Circulation 2001;104:2158–2163.2. Zipes DP, Wellens HJ. Sudden cardiac death. Circulation 1998;98:2334–2351.3. Bayes de Luna A, Coumel P, Leclercq JF. Ambulatory sudden cardiac death:
mechanisms of production of fatal arrhythmia on the basis of data from 157cases. Am Heart J 1989;117:151–159.
4. Huikuri HV, Castellanos A, Myerburg RJ. Sudden death due to cardiac arrhyth-mias. N Engl J Med 2001;345:1473–1482.
5. Friedlander Y, Siscovick DS, Weinmann S, et al. Family history as a risk factorfor primary cardiac arrest. Circulation 1998;97:155–160.
6. Jouven X, Desnos M, Guerot C, Ducimetiere P. Predicting sudden death in thepopulation: the Paris Prospective Study I. Circulation 1999;99:1978–1983.
7. Dekker LR, Bezzina CR, Henriques JP, et al. Familial sudden death is animportant risk factor for primary ventricular fibrillation: a case-control study inacute myocardial infarction patients. Circulation 2006;114:1140–1145.
8. Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and
igure 2 Linkage disequilibrium map of TGFBR2 gene. D= values areisplayed within each diamond. Color scheme gradient indicates r2 valuesith red indicating complete LD (D= � 1.0) to shades of red to pink
D= � 1.0). Blue and white diamonds indicate LD values with suboptimalog of odds (LOD � 2) scores. Haplotype blocks are defined in black.s9838682 is an intronic SNP located within the first haplotype block of theene and is indicated by the arrow. LD � linkage disequilibrium; SNP �ingle nucleotide polymorphism; other abbreviations as in Figure 1.
gene regulation: consequences for extracellular matrix remodeling and woundhealing. J Dermatol Sci 2004;35:83–92.
9. Piek E, Heldin CH, Ten Dijke P. Specificity, diversity, and regulation inTGF-beta superfamily signaling. FASEB J 1999;13:2105–2124.
0. Thompson NL, Flanders KC, Smith JM, Ellingsworth LR, Roberts AB, SpornMB. Expression of transforming growth factor-beta 1 in specific cells and tissuesof adult and neonatal mice. J Cell Biol 1989;108:661–669.
1. Sanford LP, Ormsby I, Gittenberger-de Groot AC, et al. TGFbeta2 knockoutmice have multiple developmental defects that are non-overlapping with otherTGFbeta knockout phenotypes. Development 1997;124:2659–2670.
2. Beffagna G, Occhi G, Nava A, et al. Regulatory mutations in transforminggrowth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopa-thy type 1. Cardiovasc Res 2005;65:366–373.
3. Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardialinfarction and cardiac remodeling. Cardiovasc Res 2007;74:184–195.
4. Hallmayer J, Faraco J, Lin L, et al. Narcolepsy is strongly associated with theT-cell receptor alpha locus. Nat Genet 2009;41:708–711.
5. Liu Y, Helms C, Liao W, et al. A genome-wide association study of psoriasisand psoriatic arthritis identifies new disease loci. PLoS Genet 2008;4:e1000041.
6. Wellcome Trust Case Control C. Genome-wide association study of 14,000cases of seven common diseases and 3,000 shared control subjects. Nature2007;447:661–678.
7. ten Dijke P, Arthur HM. Extracellular control of TGFbeta signalling in vasculardevelopment and disease. Nat Rev Mol Cell Biol 2007;8:857–869.
8. Conde L, Vaquerizas JM, Dopazo H, et al. PupaSuite: finding functional singlenucleotide polymorphisms for large-scale genotyping purposes. Nucleic AcidsRes 2006;34:W621–625.
9. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D.Principal components analysis corrects for stratification in genome-wide asso-ciation studies. Nat Genet 2006;38:904–909.
0. Plenge RM, Padyukov L, Remmers EF, et al. Replication of putative candidate-gene associations with rheumatoid arthritis in �4,000 samples from NorthAmerica and Sweden: association of susceptibility with PTPN22, CTLA4, andPADI4. Am J Hum Genet 2005;77:1044–1060.
1. Devlin B, Roeder K, Wasserman L. Genomic control, a new approach togenetic-based association studies. Theor Popul Biol 2001;60:155–166.
2. Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome associ-ation and population-based linkage analyses. Am J Hum Genet 2007;81:559–575.
3. Junyent M, Tucker KL, Smith CE, et al. The effects of ABCG5/G8 polymor-phisms on plasma HDL cholesterol concentrations depend on smoking habit inthe Boston Puerto Rican Health Study. J Lipid Res 2009;50:565–573.
4. PUPA Suite 2.0. Available at: http://pupasuite.bioinfo.cipf.es/index.jsf. Ac-cessed June 4, 2009.
5. Lin HY, Wang XF, Ng-Eaton E, Weinberg RA, Lodish HF. Expression cloningof the TGF-beta type II receptor, a functional transmembrane serine/threoninekinase. Cell 1992;68:775–785.
6. Munoz NM, Upton M, Rojas A, et al. Transforming growth factor beta receptortype II inactivation induces the malignant transformation of intestinal neoplasmsinitiated by Apc mutation. Cancer Res 2006;66:9837–9844.
7. Mizuguchi T, Collod-Beroud G, Akiyama T, et al. Heterozygous TGFBR2mutations in Marfan syndrome. Nat Genet 2004;36:855– 860.
8. Lijnen PJ, Petrov VV, Fagard RH. Induction of cardiac fibrosis by transforminggrowth factor-beta(1). Mol Genet Metab 2000;71:418–435.
9. Morita N, Hayashi H, Ogawa M. Mechanism of ventricular fibrillation in theaged hearts exposed to glycolytic inhibition. A new model of sudden cardiacdeath. J Am Coll Cardiol 2007;49 Suppl A:420A.
0. Deten A, Holzl A, Leicht M, Barth W, Zimmer HG. Changes in extracellularmatrix and in transforming growth factor beta isoforms after coronary arteryligation in rats. J Mol Cell Cardiol 2001;33:1191–1207.
1. Dewald O, Ren G, Duerr GD, et al. Of mice and dogs: species-specific differ-ences in the inflammatory response following myocardial infarction. Am JPathol 2004;164:665–677.
2. Hao J, Ju H, Zhao S, Junaid A, Scammell-La Fleur T, et al. Elevation ofexpression of Smads 2, 3, and 4, decorin and TGF-beta in the chronic phaseof myocardial infarct scar healing. J Mol Cell Cardiol 1999;31:667– 678.
3. Lee KW, Everett THt, Rahmutula D, et al. Pirfenidone prevents the developmentof a vulnerable substrate for atrial fibrillation in a canine model of heart failure.Circulation 2006;114:1703–1712.
4. Eghbali M, Tomek R, Sukhatme VP, Woods C, Bhambi B. Differential effectsof transforming growth factor-beta 1 and phorbol myristate acetate on cardiacfibroblasts. Regulation of fibrillar collagen mRNAs and expression of earlytranscription factors. Circ Res 1991;69:483–490.
5. Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growthfactor-beta 1 induces alpha-smooth muscle actin expression in granulation tissuemyofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol
1993;122:103–111.








![Associations of TGFBR1 and TGFBR2 gene polymorphism s with ... · testis [2]. Hypospadias can be classified as distal, medial and proximalsubtypes according to the Hypospadias can](https://img.dokumen.tips/doc/110x75/5d497cae88c99347278b63df/associations-of-tgfbr1-and-tgfbr2-gene-polymorphism-s-with-testis-2-hypospadias.jpg)
![java1-lecture6.ppt [호환 모드]dis.dankook.ac.kr/lectures/java20/wp-content/... · Polymorphism 다형성(Polymorphism) 다형성(polymorphism)이란객체들의타입이다르면똑같은](https://img.dokumen.tips/doc/110x75/5fcfbaad9d9260016a636609/java1-eeoedisdankookackrlecturesjava20wp-content-polymorphism.jpg)


















