Embed Size (px)
Citation preview

1. Introduction
2. NSAID-induced hepatotoxicity
assessment
3. Conclusion
4. Expert opinion
Review
Assessment of nonsteroidalanti-inflammatory drug-inducedhepatotoxicityJose AG Ag�undez†, Marıa Isabel Lucena, Carmen Martınez, Ra�ul J Andrade,Miguel Blanca, Pedro Ayuso & Elena Garcıa-Martın†Medical School University of Extremadura, Department of Pharmacology, Badajoz, Spain
Introduction: Liver toxicity related to NSAIDs is of outstanding importance
because of the wide use of these drugs. NSAIDs are responsible for roughly
10% of the total of cases of drug-induced hepatotoxicity. The assessment of
NSAID-induced hepatotoxicity, presently based on clinical and analytical bio-
markers, is critical for early diagnosis and immediate withdrawal of the
causing drug.
Areas covered: The review presents an overview of current knowledge of the
assessments of NSAID-induced hepatotoxicity with emphasis on the causative
drugs, the NSAID-specific mechanisms involved, and a summary of genetic
and non-genetic risk factors. Additionally, the authors discuss genetic factors
which show NSAID-specific risk, namely CYP2C, UGT2B7, GSTM1 and GSTT1, as
well as HLA alleles. The paper includes a list of the NSAID ‘usual suspects’ that
cause hepatotoxicity based on the integrated information of drug-induced
hepatotoxicity databases.
Expert opinion: The ultimate goal of this research is pre-prescription testing.
Unfortunately, genetic testing, alone, is not sufficient to predict NSAID-
induced hepatotoxicity. The development of genetic biomarkers capable of
identifying at-risk individuals will not be complete until we develop the
ability to fully characterize patients’ phenomes and the phenome--genome
interaction in patients with NSAID-induced hepatotoxicity. Additionally, a
characterization of the metabolic profile of the causative drug in patients
with NSAID-induced hepatotoxicity would add crucial information which is
presently disregarded in most studies. The full development of robust bio-
markers will require the combination of several disciplines including causal
statistics, phenomics, genomics, transcriptomics and metabonomics.
Keywords: bioactivation, genetic markers, hepatotoxicity, metabolism, NSAIDs
Expert Opin. Drug Metab. Toxicol. (2011) 7(7):817-828
1. Introduction
NSAIDs are among the most widely used drugs both as prescription and over-the-counter drugs. Six percent of the adult population in the US report the use of atleast one prescription of NSAID a month [1]. Over 30 million people receiveNSAIDs daily [2] and nearly 25% of the population have experienced NSAID-related side effects that require medical care [3]. Several studies and databases havereported patients who developed fatal hepatic toxicity related to NSAID use [4-7].Although it is generally accepted that liver toxicity is a relatively rare adverse reac-tion to NSAIDs [4,8], liver toxicity related to NSAIDs is of outstanding importancebecause of the wide use of these drugs. In fact, NSAIDs are responsible for roughly10% of the total of cases of drug-induced hepatotoxicity [8-15].
10.1517/17425255.2011.574613 © 2011 Informa UK, Ltd. ISSN 1742-5255 817All rights reserved: reproduction in whole or in part not permitted
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f L
aval
on
07/1
6/14
For
pers
onal
use
onl
y.

Initially spontaneous reports of NSAID-related hepato-toxicity signaled a potential problem. Then, systematicreviews indicated that NSAID-related hepatotoxicity is nota rare event and that the incidence of NSAID-related liverinjuries resulting in hospitalization ranged from 3.1 to23.4/100,000 patient-years [4]. Moreover, about one outof four patients who develop jaundice die as a result ofNSAID-induced liver failure [14]. A comprehensive assessmentof NSAID-induced hepatotoxicity, based on clinical and ana-lytical biomarkers, is critical for early diagnosis and immedi-ate withdrawal of the causing drug. In this invited review,we analyze relevant factors in the assessment of NSAID-induced hepatotoxicity and potential biomarkers whichmay be useful in the prediction of serious clinical outcomesand may be used in pre-prescription testing and primaryprevention of this potentially fatal adverse hepatic reaction.
2. NSAID-induced hepatotoxicity assessment
The diagnosis of NSAID-related liver hepatotoxicity is not aneasy task. It includes the identification of a temporal relation-ship between the use of the drug and the clinical event,although the time to onset is typically delayed by weeks ormonths in idiosyncratic drug-induced liver injury [16]. Otherfactors relevant for diagnosis are the type of the drug used(that is, whether the drug has been related to liver hepato-toxicity in spontaneous reports or in published databases [9]),the biochemical pattern, the presence of known risk factorsand the exclusion of alternative causes. Unfortunately, mildcases are underreported and it is likely that many of
these mild hepatotoxicity events related to NSAID use areunnoticed by the scientific community.
It should be stated that the frequency of NSAID-relatedhepatotoxicity is too low to assess accurately in clinicaltrials [17]. Spontaneous reports do not allow the determinationof incidence or relative risk, and available databases are scarceand based on heterogeneous criteria. An attempt to integratediverse databases has resulted in a unified list based on inter-national collaborative work. This list of drugs includes severalNSAIDs, which constitute a prominent pharmacologicalgroup in drug-induced hepatotoxicity, although differencesin the association of NSAIDs across databases are observed [9].Factors that have been proposed to influence the risk of deve-loping drug-induced liver injury include age, gender, chronicalcohol consumption, concomitant drugs, underlying diseasestates, obesity, diabetes mellitus type 2 and insulin resis-tance [18]. However, when age and gender were analyzed in aseries of 66 patients with NSAID-induced hepatotoxicity,none of these factors significantly influenced the risk althougholder age was a predictor of cholestatic expression of drug-induced hepatotoxicity while younger age was related to hepa-tocellular pattern [19]. Known risk factors for drug-inducedhepatotoxicity may be, at least in part, drug-specific. Riskfactors for drug-induced hepatotoxicity have been recentlyreviewed and are not discussed here in detail because inmost cases the associations were identified in patients withdrug-induced hepatotoxicity related to drugs other thanNSAIDs [16,19]. It is worthy of mention, however, thatNSAID-related hepatotoxicity is notably more frequent inpatients with concomitant medication with other potentiallyhepatotoxic drugs [20].
Regarding the type of the drug, Table 1 summarizes theNSAIDs and other non-NSAID analgesics of common usewhich caused hepatotoxicity in the unified list including threemajor registries in Spain, Sweden and the US, as well as otherdata sources [4,9,13,21]. Besides the non-NSAID analgesicacetaminophen, which is the commonest cause of intrinsichepatotoxicity, seven NSAIDs account for the vast majorityof NSAID-induced hepatotoxicity. These seven NSAIDstogether make up about 99% of all the cases related to NSAIDuse. According to their chemical structures, these drugs belongto five groups, namely, from higher to lower frequency: aceticacid derivatives (46.5%), propionic acid derivatives (25.7%),salicylates (12%), enolic acid derivatives (9.3%) and sulfonani-lides (5.8%). The average of patients with NSAID-inducedhepatotoxicity who develops acute liver failure (ALF) is 6%.The highest risk of developing NSAID-induced ALF is relatedto the use of ibuprofen (9.4%), aspirin (8.2%), naproxen(8.1%) and nimesulide (6.6%) whereas lower risk is observedwith diclofenac (5.1%), piroxicam (4.1%) and sulindac(2.5%). Whereas most NSAID-induced hepatotoxicity casesare related to individual susceptibility, aspirin is a dose-related hepatotoxin [22], thus, suggesting that the mechanismsunderlying NSAID-induced hepatotoxicity are not identicalfor all NSAIDs. Figure 1 summarizes the structure of the
Article highlights.
. Clinical and biochemical assessment of NSAID-inducedhepatotoxicity is crucial for early diagnosis andimmediate withdrawal of the causing drug.
. At present, diagnosis relies on clinical features and on alist of NSAIDs which are the ‘usual suspects’ ofcausing hepatotoxicity.
. Over 99% of cases reported of NSAID-inducedhepatotoxicity are caused by only seven NSAIDs.
. Acetic acid derivatives account for about 50% of theNSAID-induced hepatotoxicity cases reported.
. NSAIDs which cause hepatocellular damage includeaceclofenac, diclofenac, droxicam, ibuprofen,indomethacin, ketoprofen, ketorolac, meloxicam,nimesulide, piroxicam, rofecoxib and sulindac.
. NSAIDs which cause hepatotoxicity with a cholestaticpattern include celecoxib, droxicam, ibuprofen,naproxen, piroxicam, rofecoxib and sulindac.
. NSAIDs which cause mixed hepatotoxicity includedroxicam, ibuprofen, piroxicam, rofecoxib and sulindac.
. NSAID-specific genetic risk factors for developinghepatotoxicity include CYP2C, UGT2B7, GSTM1 andGSTT1 polymorphisms.
This box summarizes key points contained in the article.
Assessment of NSAID-induced hepatotoxicity
818 Expert Opin. Drug Metab. Toxicol. (2011) 7(7)
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f L
aval
on
07/1
6/14
For
pers
onal
use
onl
y.

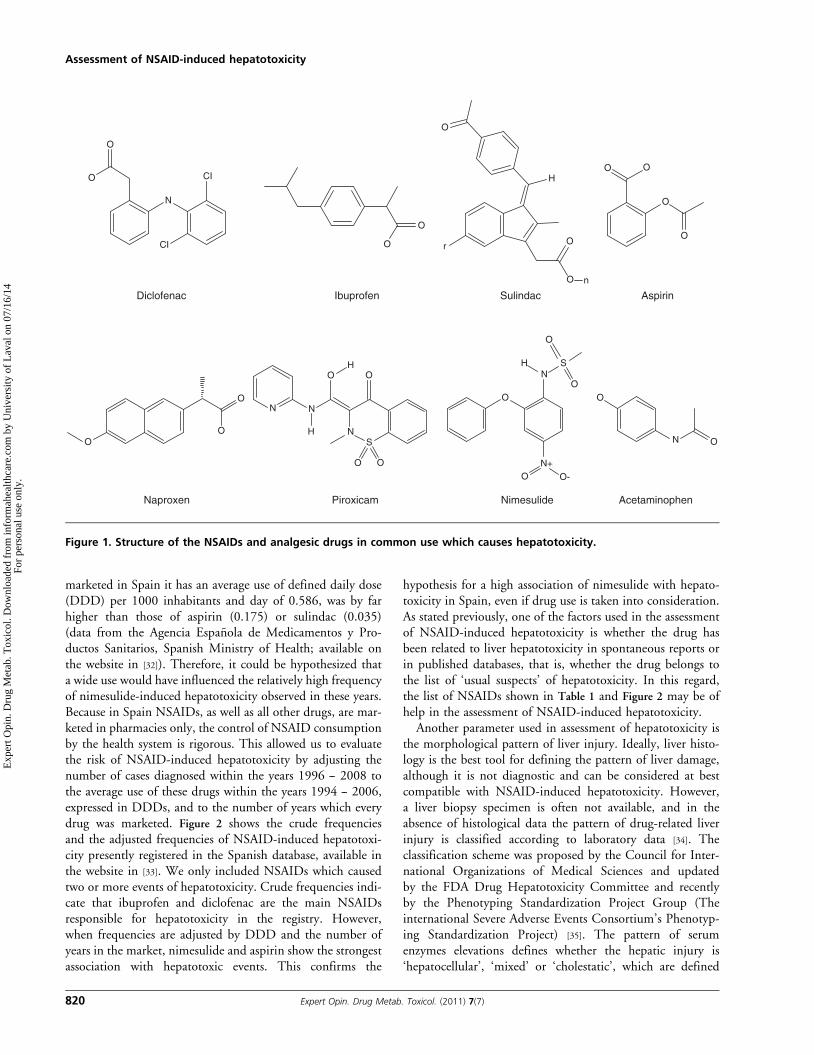
NSAIDs and analgesics identified as a major source ofdrug-induced hepatotoxicity. The chemical structures of theNSAIDs involved in hepatotoxicity are heterogeneous and,therefore, the chemical group is a poor index for assessingthe risk. For instance, within the group of acetic acid deriva-tives virtually all the risk is related to only two drugs (diclofe-nac and sulindac) whereas the rest of the NSAIDs in thischemical group are related with an extremely low frequency(aceclofenac, acemetacin, indomethacin or ketorolac) or nocases at all have been reported. This example is valid for allchemical groups of NSAIDs, and in most chemical groupsthe vast majority of cases are related to one or two drugs(Table 1). It is of note that, with regard to drug-inducedhepatotoxicity, selective COX-2 inhibitors and fenamic acidderivatives are relatively safe, as compared to other chemicalgroups of NSAIDs. Besides the data shown in Table 1, isolatedcases of hepatotoxicity have been attributed to other NSAIDssuch as diflunisal [23], etodolac [5], fenoprofen [24], flurbipro-fen [25], fosfosal [26], lumiracoxib [27], mefenamic acid [28] andniflumic acid [29]. From a search in MEDLINE and Embasesources with the terms ‘hepatotoxicity’, ‘liver injury’ and‘hepatic complications’ we identified no reports of hepatoto-xicity cases with the following NSAIDs: aloxiprin, butibufen,dexibuprofen, etoricoxib, etarsalate, fenbufen, glucametacin,ibuproxam, lornoxicam, meclofenamic acid, morniflumate,
nabumetone, oxametacin, piproxen, proglumetacin, tiaprofenor tolmetin.
The identification of the NSAID causing hepatotoxicityand the use of unified lists would be of great help in the diag-nosis, but it should be remembered that the occurrence ofNSAID hepatotoxicity with a particular drug will be influ-enced by the hepatotoxic potential and also to the prescriptionpattern of such a drug. Therefore, the presence of widely usedNSAIDs such as diclofenac, ibuprofen or aspirin among thoseNSAIDs commonly related to hepatotoxicity is not surpris-ing. Ideally, the evaluation of the hepatotoxic potential of aparticular NSAID should be adjusted to figures of drugprescription that allow an approximation to population’sexposure to a particular drug. This is a major cause of hetero-geneity in studies on the frequency of association of a partic-ular NSAID with hepatotoxicity which were carried out indiverse countries [13,30]. A good example of this heterogeneityis nimesulide. In 2002, Finland and Spain suspended themarketing of nimesulide because it was associated with ahigh frequency of hepatotoxicity [21,31]. Today, nimesulidehas been withdrawn in Europe, but after the discontinuationof use in Spain and Finland, nimesulide continued to be mar-keted in several countries and for many years it was the mostprescribed NSAID in Italy and in Portugal. In this regard, itshould be stated that in the years in which nimesulide was
Table 1. NSAIDS and analgesic drugs in common use which cause hepatotoxicity.
Group Generic name Percentage of total
cases of liver injury
(NSAID + analgesics)
Percentage of total
cases of liver injury
(NSAIDs)
Salicylates Aspirin 6.9% 12%Salsalate < 0.1% < 0.1%
Acetic acid derivatives Aceclofenac < 0.1% < 0.1%Acemetacin < 0.1% < 0.1%Diclofenac 19.5% 34.1%Indomethacin < 0.1% < 0.1%Ketorolac < 0.1% < 0.1%Sulindac 7.1% 12.4%
Propionic acid derivatives Dexketoprofen < 0.1% < 0.1%Ibuprofen 8.4% 14.6%Ketoprofen < 0.1% < 0.1%Naproxen 6.4% 11.1%Oxaprozin < 0.1% < 0.1%
Enolic acid derivatives Droxicam < 0.1% < 0.1%Meloxicam < 0.1% < 0.1%Piroxicam 5.4% 9.3%Tenoxicam < 0.1% < 0.1%
Selective COX-2 inhibitors Celecoxib < 0.1% < 0.1%Rofecoxib < 0.1% < 0.1%Valdecoxib < 0.1% < 0.1%
Sulfonanilides Nimesulide 3.3% 5.8%Non-NSAID analgesics Acetaminophen 42.7% -
Methamizole < 0.1% -
Frequencies are based on the WHO Program for International Drug Monitoring, Vigibase� and on drug-induced liver injury registries from Europe and the US, as
well as other published data sources [4,9,13,21]. Liver injury cases were patients with clinically significant liver injury according to standard diagnostic criteria (see
the text for details). Total cases (100%) for NSAID + analgesics = 10,506. Total cases for NSAIDs = 6023.
Ag�undez, Lucena, Martınez, Andrade, Blanca, Ayuso & Garcıa-Martın
Expert Opin. Drug Metab. Toxicol. (2011) 7(7) 819
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f L
aval
on
07/1
6/14
For
pers
onal
use
onl
y.
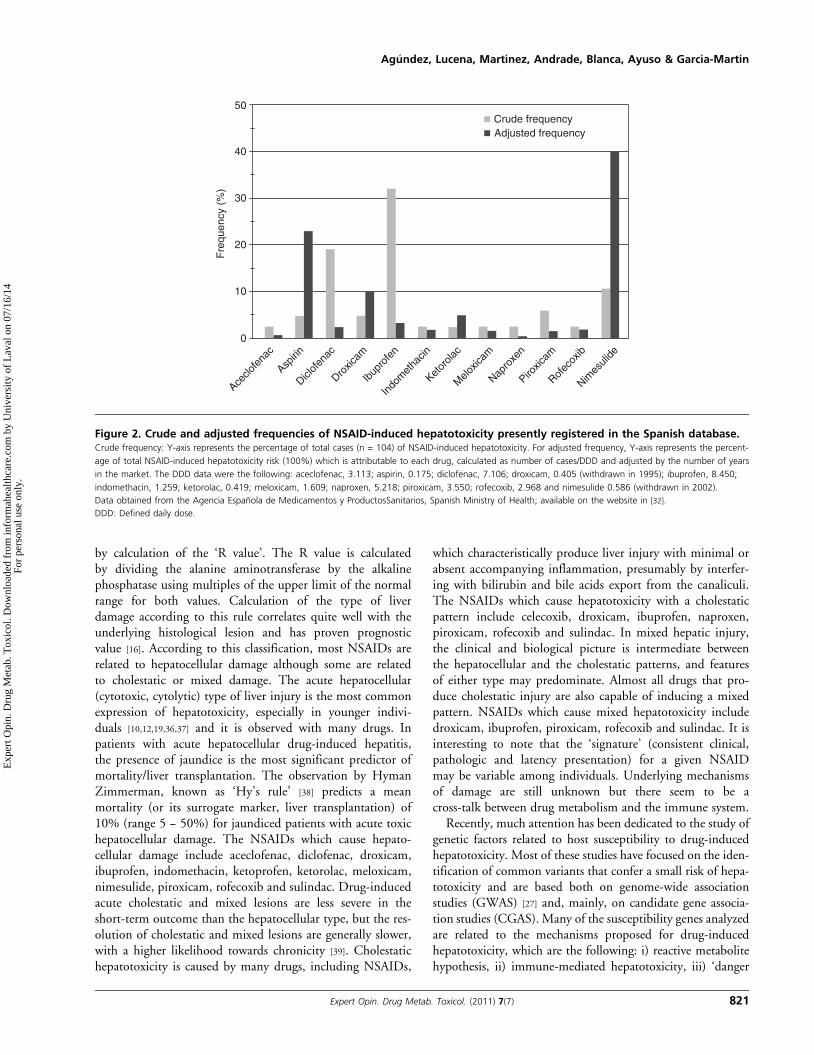
marketed in Spain it has an average use of defined daily dose(DDD) per 1000 inhabitants and day of 0.586, was by farhigher than those of aspirin (0.175) or sulindac (0.035)(data from the Agencia Espanola de Medicamentos y Pro-ductos Sanitarios, Spanish Ministry of Health; available onthe website in [32]). Therefore, it could be hypothesized thata wide use would have influenced the relatively high frequencyof nimesulide-induced hepatotoxicity observed in these years.Because in Spain NSAIDs, as well as all other drugs, are mar-keted in pharmacies only, the control of NSAID consumptionby the health system is rigorous. This allowed us to evaluatethe risk of NSAID-induced hepatotoxicity by adjusting thenumber of cases diagnosed within the years 1996 -- 2008 tothe average use of these drugs within the years 1994 -- 2006,expressed in DDDs, and to the number of years which everydrug was marketed. Figure 2 shows the crude frequenciesand the adjusted frequencies of NSAID-induced hepatotoxi-city presently registered in the Spanish database, available inthe website in [33]. We only included NSAIDs which causedtwo or more events of hepatotoxicity. Crude frequencies indi-cate that ibuprofen and diclofenac are the main NSAIDsresponsible for hepatotoxicity in the registry. However,when frequencies are adjusted by DDD and the number ofyears in the market, nimesulide and aspirin show the strongestassociation with hepatotoxic events. This confirms the
hypothesis for a high association of nimesulide with hepato-toxicity in Spain, even if drug use is taken into consideration.As stated previously, one of the factors used in the assessmentof NSAID-induced hepatotoxicity is whether the drug hasbeen related to liver hepatotoxicity in spontaneous reports orin published databases, that is, whether the drug belongs tothe list of ‘usual suspects’ of hepatotoxicity. In this regard,the list of NSAIDs shown in Table 1 and Figure 2 may be ofhelp in the assessment of NSAID-induced hepatotoxicity.
Another parameter used in assessment of hepatotoxicity isthe morphological pattern of liver injury. Ideally, liver histo-logy is the best tool for defining the pattern of liver damage,although it is not diagnostic and can be considered at bestcompatible with NSAID-induced hepatotoxicity. However,a liver biopsy specimen is often not available, and in theabsence of histological data the pattern of drug-related liverinjury is classified according to laboratory data [34]. Theclassification scheme was proposed by the Council for Inter-national Organizations of Medical Sciences and updatedby the FDA Drug Hepatotoxicity Committee and recentlyby the Phenotyping Standardization Project Group (Theinternational Severe Adverse Events Consortium’s Phenotyp-ing Standardization Project) [35]. The pattern of serumenzymes elevations defines whether the hepatic injury is‘hepatocellular’, ‘mixed’ or ‘cholestatic’, which are defined
OO
ON N
O OH
H NS
OO
O
SN
N+O O-
O
O
H
N O
O
CI
Diclofenac Ibuprofen Sulindac Aspirin
Naproxen Piroxicam Nimesulide Acetaminophen
N
O CI
O
O
O
O
O
O
H
r
O
O
O
O
n
Figure 1. Structure of the NSAIDs and analgesic drugs in common use which causes hepatotoxicity.
Assessment of NSAID-induced hepatotoxicity
820 Expert Opin. Drug Metab. Toxicol. (2011) 7(7)
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f L
aval
on
07/1
6/14
For
pers
onal
use
onl
y.
by calculation of the ‘R value’. The R value is calculatedby dividing the alanine aminotransferase by the alkalinephosphatase using multiples of the upper limit of the normalrange for both values. Calculation of the type of liverdamage according to this rule correlates quite well with theunderlying histological lesion and has proven prognosticvalue [16]. According to this classification, most NSAIDs arerelated to hepatocellular damage although some are relatedto cholestatic or mixed damage. The acute hepatocellular(cytotoxic, cytolytic) type of liver injury is the most commonexpression of hepatotoxicity, especially in younger indivi-duals [10,12,19,36,37] and it is observed with many drugs. Inpatients with acute hepatocellular drug-induced hepatitis,the presence of jaundice is the most significant predictor ofmortality/liver transplantation. The observation by HymanZimmerman, known as ‘Hy’s rule’ [38] predicts a meanmortality (or its surrogate marker, liver transplantation) of10% (range 5 -- 50%) for jaundiced patients with acute toxichepatocellular damage. The NSAIDs which cause hepato-cellular damage include aceclofenac, diclofenac, droxicam,ibuprofen, indomethacin, ketoprofen, ketorolac, meloxicam,nimesulide, piroxicam, rofecoxib and sulindac. Drug-inducedacute cholestatic and mixed lesions are less severe in theshort-term outcome than the hepatocellular type, but the res-olution of cholestatic and mixed lesions are generally slower,with a higher likelihood towards chronicity [39]. Cholestatichepatotoxicity is caused by many drugs, including NSAIDs,
which characteristically produce liver injury with minimal orabsent accompanying inflammation, presumably by interfer-ing with bilirubin and bile acids export from the canaliculi.The NSAIDs which cause hepatotoxicity with a cholestaticpattern include celecoxib, droxicam, ibuprofen, naproxen,piroxicam, rofecoxib and sulindac. In mixed hepatic injury,the clinical and biological picture is intermediate betweenthe hepatocellular and the cholestatic patterns, and featuresof either type may predominate. Almost all drugs that pro-duce cholestatic injury are also capable of inducing a mixedpattern. NSAIDs which cause mixed hepatotoxicity includedroxicam, ibuprofen, piroxicam, rofecoxib and sulindac. It isinteresting to note that the ‘signature’ (consistent clinical,pathologic and latency presentation) for a given NSAIDmay be variable among individuals. Underlying mechanismsof damage are still unknown but there seem to be across-talk between drug metabolism and the immune system.
Recently, much attention has been dedicated to the study ofgenetic factors related to host susceptibility to drug-inducedhepatotoxicity. Most of these studies have focused on the iden-tification of common variants that confer a small risk of hepa-totoxicity and are based both on genome-wide associationstudies (GWAS) [27] and, mainly, on candidate gene associa-tion studies (CGAS). Many of the susceptibility genes analyzedare related to the mechanisms proposed for drug-inducedhepatotoxicity, which are the following: i) reactive metabolitehypothesis, ii) immune-mediated hepatotoxicity, iii) ‘danger
Aceclo
fena
c
Aspirin
Diclof
enac
Droxic
am
Ibup
rofe
n
Indo
met
hacin
Ketor
olac
Melo
xicam
Napro
xen
Piroxic
am
Rofec
oxib
Nimes
ulide
Crude frequencyAdjusted frequency
50
40
30
20
10
0
Fre
quen
cy (
%)
Figure 2. Crude and adjusted frequencies of NSAID-induced hepatotoxicity presently registered in the Spanish database.Crude frequency: Y-axis represents the percentage of total cases (n = 104) of NSAID-induced hepatotoxicity. For adjusted frequency, Y-axis represents the percent-
age of total NSAID-induced hepatotoxicity risk (100%) which is attributable to each drug, calculated as number of cases/DDD and adjusted by the number of years
in the market. The DDD data were the following: aceclofenac, 3.113; aspirin, 0.175; diclofenac, 7.106; droxicam, 0.405 (withdrawn in 1995); ibuprofen, 8.450;
indomethacin, 1.259; ketorolac, 0.419; meloxicam, 1.609; naproxen, 5.218; piroxicam, 3.550; rofecoxib, 2.968 and nimesulide 0.586 (withdrawn in 2002).
Data obtained from the Agencia Espanola de Medicamentos y ProductosSanitarios, Spanish Ministry of Health; available on the website in [32].
DDD: Defined daily dose.
Ag�undez, Lucena, Martınez, Andrade, Blanca, Ayuso & Garcıa-Martın
Expert Opin. Drug Metab. Toxicol. (2011) 7(7) 821
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f L
aval
on
07/1
6/14
For
pers
onal
use
onl
y.

signal’ hypothesis and iv) alterations in mitochondrial func-tion. These mechanisms have recently been revised [16] andare not described in detail. An interesting mechanistic classifi-cation for genetic factors related to drug-induced hepatotoxi-city was that formulated by Kaplowitz, which classified theevents leading to drug-induced hepatotoxicity as ‘upstream’,that is, drug-specific mechanisms involved in the initial injury,and ‘downstream’, which are general mechanisms that balanceinjurious versus protective cellular pathways [40]. This modelwas implemented in an elegant model with three consecutivesteps [41]. The first step is initial injury, caused by the parentdrug or more frequently by its reactive metabolites, the secondstep is mitochondrial permeability transition and the third stepis hepatocyte death [41,42]. The first step is drug-specificbecause in this step the drug or their metabolites cause cellstress, mitochondrial impairment or specific immune reac-tions. Further steps are not drug-specific as these involvegeneral mechanisms of mitochondrial function and apoptosis.Because the present review focuses on the assessment of
NSAID-induced hepatotoxicity, only drug-specific mecha-nisms related to NSAIDs and general ‘downstream’ mecha-nisms which are not drug-specific are discussed here.Further information on genes related to non-NSAID drugscan be accessed in recent reviews on pharmacogenetics andpharmacogenomics in drug-induced hepatotoxicity [16,42,43].With regard to drug-specific mechanisms, drug biotransfor-
mation is believed to be a relevant process in NSAID-inducedhepatotoxicity. Most NSAIDs are extensively metabolized inhumans and it has been shown that the activity of NSAID-metabolizing enzymes shows significant interindividual andinterethnic variation [44]. Genetic and environmental factorsare responsible for this variability. It has been recently shownthat drugs with a high degree of hepatic metabolism, and par-ticularly that mediated through cytochrome P450 (CYP)CYP2C enzymes, are more likely to cause hepatotoxicitythan those with a low metabolism [45]. Most enzymes thatmetabolize NSAIDs are polymorphic. Variant genes causeabolished, reduced, altered or increased enzyme activitybecause of complete gene deletions, single nucleotidepolymorphisms that occur isolated or combined, and geneduplications. Individuals carrying enzyme-inactivating poly-morphisms display higher drug plasma concentrations andlower clearance rates when treated at standard doses. There-fore, an increased prevalence and severity of adverse drugreactions, including hepatotoxicity, could be expected amongsubjects carrying enzyme-inactivating mutations, whenreceiving NSAIDs that are substrates of the defect enzyme.Although NSAIDs causing hepatotoxicity constitute a
chemically heterogeneous group of drugs that differ in theirtherapeutic efficacy and toxicity (Figure 1), two enzymes,namely CYP2C8 and CYP2C9, are the major enzymesinvolved in the first steps of the metabolism or participate insecondary metabolic steps of most NSAIDs shown in Table 1
[44] with few exceptions. Regarding aspirin, the hepatotoxicsalicylic acid is partly metabolized by CYP enzymes, mainly
CYP2E1 and CYP3A4, although controversial informationsuggest a participation of CYP2C9, together with the xenobi-otic/medium chain fatty acid:CoA ligase (ACSM2), andUDP-glucuronosyltransferase enzymes [46,47] and, therefore,CYP enzymes are likely to play a role, though secondary, inthe bioactivation of aspirin metabolites. Besides the putativeimplications of CYP2C enzymes in other diseases [44,48-52], ithas been shown that genetic variations in CYP2C enzymescause variations in NSAID metabolism. CYP2C8 has a partialoverlapping in substrate specificity with CYP2C9, and grow-ing evidence indicates that NSAIDs initially thought to beCYP2C9 substrates are also metabolized by CYP2C8 [53,54].The CYP2C8*3 variant allele causes impaired biodispositionof ibuprofen [54]. Other variant CYP2C8 alleles areCYP2C8*2, which is related to increased Km of theenzyme [55], CYP2C8*4, which causes a marginal decrease inthe metabolic capacity, and CYP2C8*5, which causes a frameshift and an early stop codon [56]. The most commonCYP2C8 variant allele in caucasian subjects, designated asCYP2C8*3, is in linkage disequilibrium with CYP2C9*2.This linkage is disrupted in some pathologies [57], but is con-sistently observed in healthy subjects. The presence ofCYP2C8* 3 plus CYP2C9*2 variant alleles has beenassociated with decreased metabolism of ibuprofen [54,58].The presence of variations in the CYP2C9 gene is related toimpairment in the metabolism of several NSAIDs [44]. More-over, the enzyme activity can be modified by concomitanttherapy; for instance, CYP2C8 can be inhibited by trimetho-prim [59] and induced by paclitaxel, a CYP2C8 substrate [60,61].Therefore, it is unlikely that the analysis of single nucleotidepolymorphisms alone would give an absolutely true pic-ture of the CYP2C metabolic capacity in vivo. Based onthe hypothesis of genetically-determined susceptibility toNSAID-induced hepatotoxicity linked to drug biotransforma-tion, various studies CGAS were conducted to test whetherCYP2C8 and/or CYP2C9 polymorphisms can be consideredas risk factors for NSAID-induced hepatotoxicity [62,63].Aithal et al. investigated the influence of CYP2C9*2 andCYP2C9*3 in hepatotoxicity caused by diclofenac in 24patients, with negative findings [62]. Another study byPachkoria et al. had a similar sample size and did not identifya positive association [63]. Daly et al. analyzed the poly-morphisms of several enzymes involved in the metabolismof diclofenac with relation to the risk of developing hepato-toxicity and found a positive association for CYP2C8variant alleles, although the association was marginal [8].One case report indicated the presence of homozygousCYP2C9*3 genotype associated with severe hepatotoxicityduring the use of the disease-modifying antirheumatic drugleflunomide [64].
CYP2E1 is the major enzyme involved in the production ofhepatotoxic metabolites from general anesthetics [65] and hasbeen related to the production of the hepatotoxic aceta-minophen metabolite N-acetyl-p-benzquinone imine [66].In agreement with the hypothesis of CYP2E1-mediated
Assessment of NSAID-induced hepatotoxicity
822 Expert Opin. Drug Metab. Toxicol. (2011) 7(7)
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f L
aval
on
07/1
6/14
For
pers
onal
use
onl
y.

bioactivation of acetaminophen is the fact that deletion ofCYP2E1 and CYP1A2 prevented paracetamol toxicity in amouse line [67]. Among the 13 variant CYP2E1 allelesdescribed so far, the most common is the variant CYP2E1*5allele, initially designated as C2 allele, which is located inthe 5¢ flanking region [68]. Another variant allele, designatedas CYP2E1*2, causes a substitution of the amino acid 76,and is related with reduced enzyme activity in vitro [69],although it has recently been shown that CYP2E1*2 is anextremely rare allelic variant [70]. Because CYP2E1 allelic var-iants related to altered enzyme activity are rare, no CGAS stud-ies on the effect of paracetamol-induced hepatotoxicity havebeen carried out, and evidence of a link between CYP2E1genetic variants and drug-induced hepatotoxicity are limitedto anti-tuberculosis drugs (for a recent meta-analysis see [71]).Other CYP enzymes related to drug-induced hepatotoxicityare CYP2D6, CYP3A4 and CYP3A5. However, the associa-tion of variations in these genes with drug-induced hepato-toxicity is related to non-NSAID drugs. Because no cases ofNSAID-induced hepatotoxicity have been reported to be asso-ciated with variations in these CYP genes, these will not bedetailed here. For recent reviews, see [16,42].
With regard to other NSAID-metabolizing enzymes relatedto hepatotoxicity, uridine diphosphate glucuronosyltransfer-ase 2B7 (UGT2B7) was investigated with regard to diclofenachepatotoxicity. Diclofenac metabolism is carried out bydiverse hydroxylation pathways and by acyl glucuronidation.About 50% of diclofenac is eliminated as 4-hydroxydiclofe-nac, a CYP2C9 product [16]. Daly et al. [8] found that thevariant UGT2B7*2 allele is more common in patientswith hepatotoxicity caused by diclofenac, compared withhealthy controls and with patients receiving diclofenac with-out developing hepatotoxicity. With regard to glutathioneS-transferases (GSTs), besides their relevance in cancerrisk and in the risk of ethanol-induced liver damage [72,73]
these enzymes are likely to play a double role on defenseagainst NSAID-induced hepatotoxicity, because GSTs metab-olize reactive drug metabolites and GSTs play a general‘downstream’ protection mechanism against drug-inducedhepatotoxicity. In a CGAS study involving 154 cases ofdrug-induced hepatotoxicity, the presence of the double-null genotype for GSTT1 and GSTM1 is associated with a2.7-fold increased risk of developing drug-induced hepato-toxicity regardless of the type of drug involved, thus suggest-ing a general drug-unspecific mechanism [74]. Other studieson the role of GST and drug-induced hepatotoxicity supportthis general role [75-78]. Interestingly, in the Spanish studythe association of the genetic risk with NSAID-inducedhepatotoxicity was strikingly high with an 8.8-fold increasedrisk for carriers of the double-null genotype [74]. This indicatesthat besides the general ‘downstream’ effect, GSTs polymor-phisms are likely to play a drug-specific ‘upstream’ rolein NSAID-induced hepatotoxicity and points out that com-mon genetic variations may contribute to susceptibility ofdeveloping hepatotoxicity with multiple drugs.
The role of the immune system in the susceptibility to drug-induced hepatotoxicity is emerging from GWAS studies. TheHLA-DRB1*1501-DQB1*0602-DRB5*0101-DQA1* 0102haplotype has been recently found a risk factor for the selectiveNSAID COX-2 inhibitor lumiracoxib [27]. If the mechanisminvolved in the association of the haplotype and the risk is,as proposed by the authors, an immune reaction triggered bylumiracoxib or by adducts formed with reactive lumiracoxibmetabolites, it is likely that this mechanism may be relevantto other NSAIDs. However, the same haplotype was associatedwith amoxicillin and clavulanate-induced hepatotoxicity [79],two drugs which have no structural similarity with lumira-coxib, thus suggesting that the mechanism underlying theassociation of the haplotype risk and hepatotoxicity is notdrug-specific. Further studies aimed to analyze whether therisk haplotype is related to hepatotoxicity caused by NSAIDsother than lumiracoxib, particularly other coxibs, are urgentlyrequired to have a better understanding of such interestingassociation. Diclofenac-induced hepatotoxicity in a northernEuropean population has been related to polymorphisms ingenes coding for cytokines (IL-4 and -10) and to ATP-binding cassette transporters (ABCC2) although these findingscould not be replicated in a Spanish drug-induced hepatotox-icity cohort [42,80,81]. Interestingly, IL-10 polymorphisms havebeen also related to the risk of advanced chronic alcoholic liverdisease thus suggesting similarities between alcoholic anddrug-induced hepatotoxicity [82]. For recent reviews ongeneral mechanisms involved in the pharmacogenomics ofdrug-induced hepatotoxicity, see [16,42,43].
Particular attention should be paid to the mechanismsinvolving mitochondrial oxidative stress management inNSAID-induced hepatotoxicity. Because most NSAIDs aremetabolized by Phase I enzymes, these drugs or their majormetabolites produce reactive species [44]. Molecular damagefrom reactive species is important in the pathogenesis of toxicliver injury, as has been recently demonstrated with theNSAID sulindac [83]. The mitochondrial enzyme manganesesuperoxide dismutase (MnSOD, SOD2) is the major scaven-ger of mitochondrial superoxide. It has been shown thatSOD2 knockout mice have increased susceptibility to hepato-toxicity caused by the NSAID nimesulide [84]. In addition toSOD2, glutathione peroxidases can modulate the intracellularlevel of hydrogen peroxide. Glutathione peroxidase 1 (GPX1)is part of the cellular antioxidant defense system by catalyzingthe reduction of hydrogen peroxide and various organichydroperoxides using reduced glutathione as a co-substrate.GPX1 is the main glutathione peroxidase in the mammalianliver and plays a significant role in preventing mitochondrialoxidative stress.
Two independent CGAS studies have revealed that theMnSOD mutant C allele is associated with drug-inducedhepatotoxicity. This mutant allele encodes a protein withthe amino-acid substitution Val16Ala (rs4880). In thefirst study NSAIDs were not independently analyzed and,therefore, only a general association with hepatotoxic drugs
Ag�undez, Lucena, Martınez, Andrade, Blanca, Ayuso & Garcıa-Martın
Expert Opin. Drug Metab. Toxicol. (2011) 7(7) 823
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f L
aval
on
07/1
6/14
For
pers
onal
use
onl
y.

could be detected, with a 2.4-fold increased risk for carriers ofthe variant allele [76]. Another study analyzed the same poly-morphism and found a 2.3-fold increased risk for homozy-gous carriers of the variant allele [85]. The risk of developinghepatotoxicity associated with the SOD2 polymorphism washigher in patients with cholestatic/mixed hepatotoxicity, andwith patients taking drugs that are mitochondria hazardousor that produce reactive metabolites, but no particularlyhigh risk was observed among patients with NSAID-inducedhepatotoxicity [85]. The same study analyzed two GXP1 andone GPX4 polymorphisms. The GPX1 polymorphism causingthe amino-acid substitution Pro200Leu (rs1050450) wasassociated with a 5.1-fold increased risk for cholestatic hepaticdamage. Moreover, when risk was analyzed with regard to thecombination of GPX1 polymorphism and GST null alleles, acumulative effect on hepatotoxicity was observed. When casesof NSAID-induced hepatotoxicity are separately analyzed, theincreased risk related to the presence of the GPX1 Leu allele is2.3-fold, although the statistical significance was not conclu-sive (p = 0.065) due to the sample size of patients withNSAID-induced hepatotoxicity (n = 21). Taken together,these findings support a genetic basis for susceptibility todrug-induced hepatotoxicity related to mitochondrial dam-age, but do not suggest that this is due to a NSAID-specificmechanism [85]. Recently, it has been described that an associ-ation of the keratin variants KRT8 and KRT18 predisposed toALF among patients with acetaminophen-induced hepatotox-icity [86]. Whether these genetic associations are specificallyrelated to NSAID-induced hepatotoxicity remains to be eluci-dated. Diminished expression of the hepatocellular trans-porter ABCB11 (BSEP), which mediates the elimination ofbile salts from hepatocytes into the bile canaliculi, mightinfluence the level of cellular exposure to reactive metabolitesand cytotoxic bile salts. Patients with NSAID-induced hepa-totoxicity who are homozygous for the C allele (ABCB111331T > C polymorphism) are at increased risk of developingliver toxicity with a hepatocellular pattern (odds ratio = 2.995% CI 1.6 -- 5.4; P corrected for multiple comparison(Pc) > 0.002), as a manifestation of accumulation of toxicbile salts in hepatocytes. This study suggests that the BSEPtransporter might represent a novel pharmacological site ofinteraction for NSAIDs [87].All these gene variations, although not directly associated
with NSAIDs, should be taken into consideration in theassessment of NSAID-induced hepatotoxicity because thesegenes influence general mechanisms that may modify thedevelopment or the clinical presentation and outcomeof hepatotoxicity.
3. Conclusion
Clinical and analytical assessment of NSAID-induced hepato-toxicity is crucial for early diagnosis and immediate with-drawal of the causing drug. In the absence of robustbiomarkers, the diagnosis is based on a temporal relationship,
clinical and biochemical parameters and estimation of proba-bilities with a list of NSAIDs which are ‘usual suspects’ fordrug-induced hepatotoxicity. Genetic association studies arepromising for producing biomarkers, but much additionalwork is required before we can develop pre-prescriptiontesting and primary prevention based in biomarkers.
4. Expert opinion
Besides a rapid and correct diagnosis, genetic and non-geneticrisk factors should be considered in the assessment of NSAID-induced hepatotoxicity. Milestone findings in this field are thedevelopment of a rule for the evaluation of the morphologicalpattern of the liver damage without biopsies [16,34], thedetailed evaluation of the non-genetic risk factors whichmay help in the diagnosis [18], elaboration of a list of causativedrugs, which are of great help in the estimation of a cause---effect probability [9], elaboration of mechanistic models ofthe disease [40,41] and identification and characterization ofgenetic risk factors in association studies. Major weaknessesinclude the fact that mild cases are often underreported, andan even more important is the fact that variability in diagno-sis, therapy and clinical evaluation greatly differs across differ-ent countries. This makes it difficult to obtain consistentassociations between risk factors and NSAID-induced hepato-toxicity. In our opinion, another major weakness in this fieldis related to phenomics; our ability to characterize the full setof phenotypes of an individual (phenome) lags behind ourability to characterize genomes [88], and this hampers thedevelopment of robust biomarkers.
The ultimate goal in NSAID-induced hepatotoxicityresearch should include the development of biomarkers forpre-prescription testing and primary prevention. Howevergenetic testing alone, although promising, is not sufficientto predict NSAID hepatotoxicity. The analysis of non-genetic risk factors [18] (age, gender, daily dose, the metabolicprofile of the NSAID, drug interactions, alcohol intake andco-morbidities) should be combined in the assessment withthe analysis of genetic factors (genetic polymorphisms inPhase I and II enzymes, drug transporters, HLA class antigens,cytokines or genes related to mitochondrial function).A crucial point that should be taken into consideration isthat genetic associations discovered in one population arenot often replicated in another population. A study compar-ing the French and the Spanish databases on NSAID-induced hepatotoxicity revealed that some NSAIDs showeddifferent risk levels in the two countries. These differenceswere attributed to differences in drugs use and genetic or envi-ronmental factors [30]. A potential cause for these discrepan-cies is the occurrence of interethnic and intraethnicvariability in polymorphisms of drug-metabolizing enzymessuch as CYP2C, GSTs enzymes and, to a lesser degree ofassociation, NAT2 [53,89,90], as well as the difficulties inherentto haplotype assignation when several polymorphisms areanalyzed in the same gene, as is the case of NAT2 [91]. These
Assessment of NSAID-induced hepatotoxicity
824 Expert Opin. Drug Metab. Toxicol. (2011) 7(7)
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f L
aval
on
07/1
6/14
For
pers
onal
use
onl
y.

factors should be taken into consideration in the geneticassessment of NSAID-induced hepatotoxicity.
The combination of GWAS and CGAS studies indicateclear associations of HLA genes with drug-induced hepato-toxicity, thus suggesting an immune response [43]. However,many mechanisms involved in the immune response to drugsremain to be explored in mechanistically-based CGAS.Phenomic and genomic factors involved in hypersensitivityreactions to NSAIDs have recently been reviewed [92,93] andthese should be assessed in NSAID-induced hepatotoxicitybecause, as stated in the mechanistic models, immune reactionsof NSAID metabolites may trigger the hepatotoxicityevents [41,42]. Drug-induced hepatitis shares many of the fea-tures of cell-mediated reactions, such as eosinophilia, atypicalperipheral blood lymphocytes and a liver T-cell infiltrate.Drugs involved include methamizole, diclofenac, ibuprofenand occasionally any NSAID [92]. Alterations in genes codingfor enzymes involved in histamine homeostasis, which arerelated to the development or the clinical presentation ofimmune diseases, such as those of the enzymes diamine oxidaseor histidine decarboxylase [94-98], remain to be analyzed.
Two particular areas of the research of interest at presentare transcriptomics and metabonomics. It has recently beenshown that acetaminophen causes a transcriptome signaturecharacterized by downregulation of oxidative phosphorylationgenes [99]. The availability of rapid and affordable analyses of
blood transcriptome in patients with drug-induced hepato-toxicity opens up new and exciting possibilities in their assess-ment. In addition to genomic, transcriptomics and phenomicanalyses, because NSAIDs are extensively metabolized inhumans, the metabonomic [100] approach to biological analy-sis in NSAID-induced hepatotoxicity should contribute sig-nificantly to improving our knowledge of the mechanismsinvolved in the first stages of liver injury.
Acknowledgements
The authors thank J McCue for assistance in language editing.
Declaration of interest
The work of the authors’ laboratory is funded by a numberof grants. JAG Ag�undez is supported by Grants PS09/00943 and RETICS RD07/0064/0016; E Garcia-Martin issupported by PS09/00469 while RT Andrade is supportedby PS 09/01384 and CIBERehD from the Instituto de SaludCarlos III, Madrid, Spain. JAG Ag�undez is also supportedby GR10068 from Junta de Extremadura, Spain while RTAndrade is supported by grant CTS-6470 from Consejeriade Salud, Junta de Andalucıa, Spain. The authors arealso financed in part with FEDER funds from theEuropean Union.
BibliographyPapers of special note have been highlighted as
either of interest (�) or of considerable interest(��) to readers.
1. Paulose-Ram R, Hirsch R, Dillon C,
et al. Prescription and non-prescription
analgesic use among the US adult
population: results from the third
National Health and Nutrition
Examination Survey (NHANES III).
Pharmacoepidemiol Drug Saf
2003;12:315-26
2. Singh G, Triadafilopoulos G.
Epidemiology of NSAID induced
gastrointestinal complications.
J Rheumatol Suppl 1999;56:18-24
3. Bloom BS. Direct medical costs of
disease and gastrointestinal side effects
during treatment for arthritis. Am J Med
1988;84:20-4
4. Rubenstein JH, Laine L. Systematic
review: the hepatotoxicity of
non-steroidal anti-inflammatory
drugs. Aliment Pharmacol Ther
2004;20:373-80
5. Mabee CL, Mabee SW, Baker PB, et al.
Fulminant hepatic failure associated with
etodolac use. Am J Gastroenterol
1995;90:659-61
6. McCormick PA, Kennedy F, Curry M,
et al. COX 2 inhibitor and
fulminant hepatic failure. Lancet
1999;353:40-1
7. Teoh NC, Farrell GC. Hepatotoxicity
associated with non-steroidal
anti-inflammatory drugs. Clin Liver Dis
2003;7:401-13
8. Daly AK, Aithal GP, Leathart JB, et al.
Genetic susceptibility to
diclofenac-induced hepatotoxicity:
contribution of UGT2B7, CYP2C8, and
ABCC2 genotypes. Gastroenterology
2007;132:272-81.. Description of genetic factors related
to diclofenac hepatotoxicity.
9. Suzuki A, Andrade RJ, Bjornsson E,
et al. Drugs associated with
hepatotoxicity and their reporting
frequency of liver adverse events in
VigiBase: unified list based on
international collaborative work.
Drug Saf 2010;33:503-22.. Unified list of drugs in common use
which cause hepatotoxicity.
10. Andrade RJ, Lucena MI, Fernandez MC,
et al. Drug-induced liver injury:
an analysis of 461 incidences submitted
to the Spanish registry over a 10-year
period. Gastroenterology
2005;129:512-21
11. Bessone F. Non-steroidal
anti-inflammatory drugs: what
is the actual risk of liver
damage? World J Gastroenterol
2010;16:5651-61
12. Bjornsson E. Review article:
drug-induced liver injury in clinical
practice. Aliment Pharmacol Ther
2010;32:3-13
13. Sgro C, Clinard F, Ouazir K, et al.
Incidence of drug-induced hepatic
injuries: a French population-based
study. Hepatology 2002;36:451-5
14. Bjornsson E, Jerlstad P, Bergqvist A,
et al. Fulminant drug-induced
hepatic failure leading to death or liver
transplantation in Sweden.
Scand J Gastroenterol
2005;40:1095-101
15. Bjornsson E, Olsson R. Outcome
and prognostic markers in severe
drug-induced liver disease. Hepatology
2005;42:481-9
16. Andrade RJ, Agundez JA, Lucena MI,
et al. Pharmacogenomics in drug
Ag�undez, Lucena, Martınez, Andrade, Blanca, Ayuso & Garcıa-Martın
Expert Opin. Drug Metab. Toxicol. (2011) 7(7) 825
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f L
aval
on
07/1
6/14
For
pers
onal
use
onl
y.

induced liver injury. Curr Drug Metab
2009;10:956-70.. A recent review on pharmacogenetic
and genomic basis of
drug-induced hepatotoxicity.
17. Laine L, Goldkind L, Curtis SP, et al.
How common is diclofenac-associated
liver injury? Analysis of 17,289 arthritis
patients in a long-term prospective
clinical trial. Am J Gastroenterol
2009;104:356-62
18. Chalasani N, Bjornsson E. Risk factors for
idiosyncratic drug-induced liver injury.
Gastroenterology 2010;138:2246-59.. A review of nongenetic risk factors for
drug-induced hepatotoxicity.
19. Lucena MI, Andrade RJ, Kaplowitz N,
et al. Phenotypic characterization of
idiosyncratic drug-induced liver injury:
the influence of age and sex. Hepatology
2009;49:2001-9
20. Henry D, Lim LL, Garcia Rodriguez LA,
et al. Variability in risk of gastrointestinal
complications with individual
non-steroidal anti-inflammatory drugs:
results of a collaborative meta-analysis.
BMJ 1996;312:1563-6
21. Traversa G, Bianchi C, Da Cas R, et al.
Cohort study of hepatotoxicity associated
with nimesulide and other non-steroidal
anti-inflammatory drugs. BMJ
2003;327:18-22
22. Tolman KG. Hepatotoxicity of
non-narcotic analgesics. Am J Med
1998;105:13S-19S
23. Geoffroy P, Carteret E, Salagnac V, et al.
Hepatitis caused by diflunisal (Dolobis).
Apropos of a case. Therapie 1987;42:253
24. Stennet DJ, Simonson W, Hall CA.
Fenoprofen-induced hepatotoxicity. Am J
Hosp Pharm 1978;35:901
25. Lacroix I, Lapeyre-Mestre M, Bagheri H,
et al. Nonsteroidal anti-inflammatory
drug-induced liver injury: a case-control
study in primary care.
Fundam Clin Pharmacol 2004;18:201-6
26. Delgadillo Duarte J, Videla Ces S,
Navas Ramirez J, et al. Fosfosal-induced
hepatitis. Rev Clin Esp 1994;194:203-4
27. Singer JB, Lewitzky S, Leroy E, et al.
A genome-wide study identifies
HLA alleles associated with
lumiracoxib-related liver injury.
Nat Genet 2010;42:711-14. Genome-wide association study on
hepatotoxicity related to the
NSAID lumiracoxib.
28. Somchit N, Sanat F, Gan EH, et al.
Liver injury induced by the non-steroidal
anti-inflammatory drug mefenamic acid.
Singapore Med J 2004;45:530-2
29. Boboc B, Larrey D, Bernuau J, et al.
Acute hepatitis probably caused by
niflumic acid. Gastroenterol Clin Biol
1987;11:175-6
30. Lapeyre-Mestre M, de Castro AM,
Bareille MP, et al. Non-steroidal
anti-inflammatory drug-related hepatic
damage in France and Spain: analysis
from national spontaneous reporting
systems. Fundam Clin Pharmacol
2006;20:391-5
31. Macia MA, Carvajal A, del Pozo JG,
et al. Hepatotoxicity associated with
nimesulide: data from the Spanish
Pharmacovigilance System.
Clin Pharmacol Ther 2002;72:596-7
32. Available from: http://www.aemps.es/
profHumana/observatorio/docs/AINE.pdf
33. Available from: http://www.spanishdili.
uma.es/index.php?option=com_content&
view=article&id=92&Itemid=83&lang=en
34. Benichou C. Criteria of drug-induced
liver disorders. Report of an international
consensus meeting. J Hepatol
1990;11:272-6
35. Available from: http://www.
saeconsortium.org/?q=node/23
36. Chalasani N, Fontana RJ,
Bonkovsky HL, et al. Causes, clinical
features, and outcomes from a
prospective study of drug-induced liver
injury in the United States.
Gastroenterology
2008;135:1924-34, 34 e1-4
37. Ghabril M, Chalasani N, Bjornsson E.
Drug-induced liver injury: a clinical
update. Curr Opin Gastroenterol
2010;26:222-6
38. Zimmerman HJ. Drug-induced liver
disease. Clin Liver Dis 2000;4:73-96, vi
39. Andrade RJ, Lucena MI, Kaplowitz N,
et al. Outcome of acute idiosyncratic
drug-induced liver injury: long-term
follow-up in a hepatotoxicity registry.
Hepatology 2006;44:1581-8
40. Kaplowitz N. Idiosyncratic drug
hepatotoxicity. Nat Rev Drug Discov
2005;4:489-99.. A mechanistic model for
drug-induced hepatotoxicity.
41. Russmann S, Kullak-Ublick GA,
Grattagliano I. Current concepts of
mechanisms in drug-induced
hepatotoxicity. Curr Med Chem
2009;16:3041-53
42. Russmann S, Jetter A,
Kullak-Ublick GA. Pharmacogenetics of
drug-induced liver injury. Hepatology
2010;52:748-61.. A recent review on pharmacogenetic
and genomic basis of
drug-induced hepatotoxicity.
43. Daly AK. Drug-induced liver injury:
past, present and future.
Pharmacogenomics 2010;11:607-11.. A recent review on pharmacogenetic
and genomic basis of
drug-induced hepatotoxicity.
44. Agundez JA, Garcia-Martin E,
Martinez C. Genetically based
impairment in CYP2C8- and
CYP2C9-dependent NSAID metabolism
as a risk factor for gastrointestinal
bleeding: is a combination of
pharmacogenomics and metabolomics
required to improve personalized
medicine? Expert Opin Drug
Metab Toxicol 2009;5:607-20. A review of the role of metabonomics
in NSAID-induced adverse reactions.
45. Lammert C, Einarsson S, Saha C, et al.
Relationship between daily dose of oral
medications and idiosyncratic
drug-induced liver injury: search for
signals. Hepatology 2008;47:2003-9
46. Dupont I, Berthou F, Bodenez P, et al.
Involvement of cytochromes P-450
2E1 and 3A4 in the 5-hydroxylation of
salicylate in humans. Drug Metab Dispos
1999;27:322-6
47. Agundez JA, Martinez C, Perez-Sala D,
et al. Pharmacogenomics in aspirin
intolerance. Curr Drug Metab
2009;10:998-1008
48. Agundez JA. Cytochrome P450 gene
polymorphism and cancer.
Curr Drug Metab 2004;5:211-24
49. Agundez JA, Martinez C,
Garcia-Martin E, et al. Cytochrome
P450 CYP2C9 polymorphism and
NSAID-related acute gastrointestinal
bleeding. Gastroenterology
2007;133:2071-2
50. Blanco G, Martinez C, Ladero JM, et al.
Interaction of CYP2C8 and
CYP2C9 genotypes modifies the risk for
nonsteroidal anti-inflammatory
drugs-related acute gastrointestinal
bleeding. Pharmacogenet Genomics
2008;18:37-43
Assessment of NSAID-induced hepatotoxicity
826 Expert Opin. Drug Metab. Toxicol. (2011) 7(7)
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f L
aval
on
07/1
6/14
For
pers
onal
use
onl
y.

51. Garcia-Martin E, Martinez C,
Ladero JM, et al. Influence of
cytochrome P450 CYP2C9 genotypes in
lung cancer risk. Cancer Lett
2002;180:41-6
52. Martinez C, Garcia-Martin E,
Ladero JM, et al. Association of
CYP2C9 genotypes leading to high
enzyme activity and colorectal cancer
risk. Carcinogenesis 2001;22:1323-6
53. Garcia-Martin E, Martinez C,
Ladero JM, et al. Interethnic and
intraethnic variability of CYP2C8 and
CYP2C9 polymorphisms in healthy
individuals. Mol Diagn Ther
2006;10:29-40
54. Garcia-Martin E, Martinez C, Tabares B,
et al. Interindividual variability in
ibuprofen pharmacokinetics is
related to interaction of cytochrome
P450 2C8 and 2C9 amino acid
polymorphisms. Clin Pharmacol Ther
2004;76:119-27
55. Dai D, Zeldin DC, Blaisdell JA, et al.
Polymorphisms in human CYP2C8
decrease metabolism of the anticancer
drug paclitaxel and arachidonic acid.
Pharmacogenetics 2001;11:597-607
56. Soyama A, Saito Y, Komamura K, et al.
Five novel single nucleotide
polymorphisms in the CYP2C8 gene,
one of which induces a frame-shift.
Drug Metab Pharmacokinet
2002;17:374-7
57. Martinez C, Garcia-Martin E,
Alonso-Navarro H, et al. Changes at the
CYP2C locus and disruption of
CYP2C8/9 linkage disequilibrium in
patients with essential tremor.
Neuromolecular Med 2007;9:195-204
58. Martinez C, Garcia-Martin E, Blanco G,
et al. The effect of the cytochrome
P450 CYP2C8 polymorphism on the
disposition of (R)-ibuprofen enantiomer
in healthy subjects. Br J Clin Pharmacol
2005;59:62-9
59. Wen X, Wang JS, Backman JT, et al.
Trimethoprim and sulfamethoxazole are
selective inhibitors of CYP2C8 and
CYP2C9, respectively.
Drug Metab Dispos 2002;30:631-5
60. Garcia-Martin E, Pizarro RM,
Martinez C, et al. Acquired resistance to
the anticancer drug paclitaxel is
associated with induction of cytochrome
P450 2C8. Pharmacogenomics
2006;7:575-85
61. Martinez C, Garcia-Martin E,
Pizarro RM, et al. Expression of
paclitaxel-inactivating CYP3A activity in
human colorectal cancer: implications for
drug therapy. Br J Cancer 2002;87:681-6
62. Aithal GP, Day CP, Leathart JB, et al.
Relationship of polymorphism in
CYP2C9 to genetic susceptibility to
diclofenac-induced hepatitis.
Pharmacogenetics 2000;10:511-18
63. Pachkoria K, Lucena MI,
Ruiz-Cabello F, et al. Genetic
polymorphisms of CYP2C9 and
CYP2C19 are not related to
drug-induced idiosyncratic
liver injury (DILI). Br J Pharmacol
2007;150:808-15
64. Sevilla-Mantilla C, Ortega L,
Agundez JA, et al. Leflunomide-induced
acute hepatitis. Dig Liver Dis
2004;36:82-4
65. Restrepo JG, Garcia-Martin E,
Martinez C, et al. Polymorphic drug
metabolism in anaesthesia.
Curr Drug Metab 2009;10:236-46
66. Raucy JL, Lasker JM, Lieber CS, et al.
Acetaminophen activation by human
liver cytochromes P450IIE1 and
P450IA2. Arch Biochem Biophys
1989;271:270-83
67. Zaher H, Buters JT, Ward JM, et al.
Protection against acetaminophen toxicity
in CYP1A2 and CYP2E1 double-null
mice. Toxicol Appl Pharmacol
1998;152:193-9
68. Hayashi S, Watanabe J, Kawajiri K.
Genetic polymorphisms in the
5¢-flanking region change transcriptional
regulation of the human cytochrome
P450IIE1 gene. J Biochem
1991;110:559-65
69. Hu Y, Oscarson M, Johansson I, et al.
Genetic polymorphism of human
CYP2E1: characterization of two
variant alleles. Mol Pharmacol
1997;51:370-6
70. Martinez C, Galvan S, Garcia-Martin E,
et al. Variability in ethanol biodisposition
in whites is modulated by
polymorphisms in the ADH1B and
ADH1C genes. Hepatology
2010;51:491-500
71. Sun F, Chen Y, Xiang Y, et al.
Drug-metabolising enzyme
polymorphisms and predisposition to
anti-tuberculosis drug-induced liver
injury: a meta-analysis. Int J Tuberc
Lung Dis 2008;12:994-1002
72. Ladero JM, Martinez C,
Garcia-Martin E, et al. Polymorphisms
of the glutathione S-transferases mu-1
(GSTM1) and theta-1 (GSTT1) and the
risk of advanced alcoholic liver disease.
Scand J Gastroenterol 2005;40:348-53
73. Martinez C, Martin F, Fernandez JM,
et al. Glutathione S-transferases mu 1,
theta 1, pi 1, alpha 1 and mu 3 genetic
polymorphisms and the risk of colorectal
and gastric cancers in humans.
Pharmacogenomics 2006;7:711-18
74. Lucena MI, Andrade RJ, Martinez C,
et al. Glutathione S-transferase m1 and
t1 null genotypes increase susceptibility
to idiosyncratic drug-induced liver injury.
Hepatology 2008;48:588-96.. Candidate gene association study
describing association between
glutathione S-transferase null alleles
and NSAID-induced hepatotoxicity.
75. Roy B, Chowdhury A, Kundu S, et al.
Increased risk of antituberculosis
drug-induced hepatotoxicity in
individuals with glutathione
S-transferase M1 ‘null’ mutation.
J Gastroenterol Hepatol 2001;16:1033-7
76. Huang YS, Su WJ, Huang YH, et al.
Genetic polymorphisms of manganese
superoxide dismutase, NAD(P)H:quinone
oxidoreductase, glutathione S-transferase
M1 and T1, and the susceptibility to
drug-induced liver injury. J Hepatol
2007;47:128-34.. Candidate gene association study
describing association between
glutathione S-transferase null alleles
and drug-induced hepatotoxicity.
77. Leiro V, Fernandez-Villar A, Valverde D,
et al. Influence of glutathione
S-transferase M1 and T1 homozygous
null mutations on the risk of
antituberculosis drug-induced
hepatotoxicity in a Caucasian population.
Liver Int 2008;28:835-9
78. Ueda K, Ishitsu T, Seo T, et al.
Glutathione S-transferase M1 null
genotype as a risk factor for
carbamazepine-induced mild
hepatotoxicity. Pharmacogenomics
2007;8:435-42
79. Andrade RJ, Lucena MI, Alonso A, et al.
HLA class II genotype influences the
type of liver injury in drug-induced
idiosyncratic liver disease. Hepatology
2004;39:1603-12
Ag�undez, Lucena, Martınez, Andrade, Blanca, Ayuso & Garcıa-Martın
Expert Opin. Drug Metab. Toxicol. (2011) 7(7) 827
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f L
aval
on
07/1
6/14
For
pers
onal
use
onl
y.

80. Aithal GP, Ramsay L, Daly AK, et al.
Hepatic adducts, circulating antibodies,
and cytokine polymorphisms in patients
with diclofenac hepatotoxicity.
Hepatology 2004;39:1430-40
81. Pachkoria K, Lucena MI, Crespo E,
et al. Analysis of IL-10, IL-4 and
TNF-alpha polymorphisms in
drug-induced liver injury (DILI) and its
outcome. J Hepatol 2008;49:107-14
82. Ladero JM, Fernandez-Arquero M,
Tudela JI, et al. Single nucleotide
polymorphisms and microsatellite alleles
of tumor necrosis factor alpha and
interleukin-10 genes and the risk of
advanced chronic alcoholic liver disease.
Liver 2002;22:245-51
83. Zou W, Roth RA, Younis HS, et al.
Oxidative stress is important in the
pathogenesis of liver injury induced by
sulindac and lipopolysaccharide
cotreatment. Toxicology 2010;272:32-8
84. Ong MM, Wang AS, Leow KY, et al.
Nimesulide-induced hepatic
mitochondrial injury in heterozygous
Sod2(+/-) mice. Free Radic Biol Med
2006;40:420-9
85. Lucena MI, Garcia-Martin E,
Andrade RJ, et al. Mitochondrial
superoxide dismutase and glutathione
peroxidase in idiosyncratic drug-induced
liver injury. Hepatology 2010;52:303-12.. Candidate gene association study
describing association of genes related
to mitochondrial function and with
drug-induced hepatotoxicity.
86. Strnad P, Zhou Q, Hanada S, et al.
Keratin variants predispose to acute liver
failure and adverse outcome: race and
ethnic associations. Gastroenterology
2010;139:828-35, 35 e1-3
87. Andrade RJ, Crespo E, Ulzurrun E, et al.
Polymorphic bile salt export pump
transporter is a major determinant of
hepatocellular Drug-Induced Liver
Injury. Hepatology 2008;48:468A
88. Houle D, Govindaraju DR, Omholt S.
Phenomics: the next challenge.
Nat Rev Genet 2010;11:855-66
89. Garcia-Martin E. Interethnic and
intraethnic variability of NAT2 single
nucleotide polymorphisms.
Curr Drug Metab 2008;9:487-97
90. Agundez JA, Ladero JM. Glutathione
S-transferase GSTT1 and GSTM1
allozymes: beyond null alleles.
Pharmacogenomics 2008;9:359-63
91. Agundez JA, Golka K, Martinez C, et al.
Unraveling ambiguous NAT2
genotyping data. Clin Chem
2008;54:1390-4
92. Cornejo-Garcia JA, Blanca-Lopez N,
Dona I, et al. Hypersensitivity reactions
to non-steroidal anti-inflammatory drugs.
Curr Drug Metab 2009;10:971-80
93. Dona I, Blanca-Lopez N,
Cornejo-Garcia JA, et al. Characteristics
of subjects experiencing hypersensitivity
to non-steroidal anti-inflammatory drugs:
patterns of response. Clin Exp Allergy
2010;41:86-95. An article investigating the phenomic
aspects of NSAID hypersensitivity.
94. Garcia-Martin E, Ayuso P, Martinez C,
et al. Histamine pharmacogenomics.
Pharmacogenomics 2009;10:867-83
95. Garcia-Martin E, Garcia-Menaya J,
Sanchez B, et al. Polymorphisms of
histamine-metabolizing enzymes and
clinical manifestations of asthma and
allergic rhinitis. Clin Exp Allergy
2007;37:1175-82
96. Garcia-Martin E, Mendoza JL,
Martinez C, et al. Severity of ulcerative
colitis is associated with a polymorphism
at diamine oxidase gene but not at
histamine N-methyltransferase gene.
World J Gastroenterol 2006;12:615-20
97. Gervasini G, Agundez JA,
Garcia-Menaya J, et al. Variability of the
L-Histidine decarboxylase gene in allergic
rhinitis. Allergy 2010;65:1576-84
98. Garcia-Martin E, Ayuso P, Martinez C,
et al. Improved analytical sensitivity
reveals the occurrence of gender-related
variability in diamine oxidase enzyme
activity in healthy individuals.
Clin Biochem 2007;40:1339-41
99. Fannin RD, Russo M, O’Connell TM,
et al. Acetaminophen dosing of humans
results in blood transcriptome and
metabolome changes consistent with
impaired oxidative phosphorylation.
Hepatology 2010;51:227-36
100. Barton RH. A decade of advances in
metabonomics. Expert Opin Drug
Metab Toxicol 2011;7:129-36. A comprehensive review
of metabonomics.
AffiliationJose AG Ag�undez†1,2 MD PhD,
Marıa Isabel Lucena3,4, Carmen Martınez1,2,
Ra�ul J Andrade4,5, Miguel Blanca2,6,
Pedro Ayuso2,7 & Elena Garcıa-Martın2,7
†Author for correspondence1Medical School University of Extremadura,
Department of Pharmacology,
Avda. de Elvas s/n, E-06071,
Badajoz, Spain
Tel: +34924289458; Fax: +34924289676;
E-mail: [email protected] de Investigacion de Reacciones Adversas a
Alergenos y Farmacos (RIRAAF)3Clinical Pharmacology Service,
Hospital Universitario Virgen de la Victoria,
Facultad de Medicina,
Malaga, Spain4Centro de Investigacion Biomedica en Red de
Enfermedades Hepaticas y Digestivas
(CIBERehd)5Liver Unit, Hospital Universitario Virgen
de la Victoria,
Facultad de Medicina,
Malaga, Spain6Allergy Unit, Hospital Carlos Haya,
Malaga, Spain7University of Extremadura,
Department of Biochemistry& Molecular
Biology,
Avda. de Elvas s/n, E-06071,
Badajoz, Spain
Assessment of NSAID-induced hepatotoxicity
828 Expert Opin. Drug Metab. Toxicol. (2011) 7(7)
Exp
ert O
pin.
Dru
g M
etab
. Tox
icol
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f L
aval
on
07/1
6/14
For
pers
onal
use
onl
y.