Embed Size (px)
Citation preview

An heteroduplex mobility analysis assay based on capillaryelectrophoresis for the study of HCV quasispecies
Laura Rossi a,*, Michela Leveri a, Daniela Marinelli a, Luca Belli c, Emilio Civardi a,Enrico M. Silini a,b
a ASAEV*/Associazione Studio Avanzato Epatiti Virali, via G. Garibaldi 13, 24040 Bonate Sotto, BG, Italyb Department of Pathology, Universita and IRCCS-Policlinico S. Matteo, Pavia, Italy
c Unita di Epato-gastroenterologia ‘‘Crespi’’, Ospedale Niguarda, Milan, Italy
Received 10 September 2002; received in revised form 26 February 2003; accepted 27 February 2003
Abstract
The quasispecies nature of the hepatitis C virus (HCV) genome is central to the transmission, persistence and pathogenesis of the
infection. Heteroduplex mobility analysis (HMA) is a simple and an inexpensive technique for the qualitative and quantitative
analysis of genetic variation of viral quasispecies. An original HMA for the HVR1 region of HCV was developed, based on a semi-
automated, non-radioactive capillary electrophoresis system, which allows the processing of large numbers of samples in short times,
the accurate measure of mobility shifts and the quantitation of heteroduplexes. A set of 120 HVR1 clones of known sequence was
used to develop the assay, which was tested on HVR1 sequences amplified directly from sera of 17 HCV-infected patients. HVR1
sequence divergence directly correlated with the heteroduplex mobility ratio (HMR) of hybrid molecules between six and 40
mismatches. Heteroduplexes between one and six mismatches were resolved, although HMRs were not proportional to base
changes, likely due to an effect of type and position of the substitutions. The assay sensitivity was 1% of the total sample size. This
assay may allow the application of quasispecies analysis to a wider range of clinical and basic investigations.
# 2003 Elsevier Science B.V. All rights reserved.
Keywords: Heteroduplex mobility assay; Hepatitis C virus; Quasispecies; Semi-automated analysis
1. Introduction
Hepatitis C virus (HCV) displays a high genetic
variation both in populations and within infected
individuals where it exists as a complex mixture of
related molecular species known as viral quasispecies
(Martel et al., 1992).
Quasispecies typically contain at least one major
variant, known as master sequence, and a variable
number of minor variants (Choo et al., 1989; Weiner
et al., 1991; Hijikata et al., 1991; Martel et al., 1992;
Coffin, 1992). Quasispecies composition can be de-
scribed by two variables: complexity, defined as the
number of different variants present at any time, and
divergence, defined as the overall sequence variation
between variants.
In the chronically infected host, HCV quasispecies
change over time as a consequence of the continuous
generation of new variants fuelled by the high produc-
tion rates of viral particles, the low fidelity of the
replication machinery and the selective pressure of the
immune responses (Weiner et al., 1992; Farci et al.,
1994). This provides the viral genome with an extra-
ordinary plasticity and adaptability to environmental
changes such as passage to new hosts, immune selection
or anti-viral therapy (Holland et al., 1992; Domingo,
1996; Domingo and Holland, 1997). The highly hetero-
geneous nature of viral populations plays a central role
in the transmission, persistence and pathogenesis of
HCV infection (Brambilla et al., 1998; Farci et al.,
2000).
To maximize the detection of HCV quasispecies,
sequences of the hypervariable region 1 of second
* Corresponding author. Tel.: �/39-035-499-7039; fax: �/39-035-99-
2828.
E-mail address: [email protected] (L. Rossi).
Journal of Virological Methods 110 (2003) 37�/49
www.elsevier.com/locate/jviromet
0166-0934/03/$ - see front matter # 2003 Elsevier Science B.V. All rights reserved.
doi:10.1016/S0166-0934(03)00096-X

envelope gene (E2-HVR1) are usually analyzed, as this
region displays the highest genetic variation (Choo et
al., 1991). HVR1 nucleotide diversities were observed
previously as high as 30% over a 1-year period within asingle host, although with marked differences between
individuals (Brambilla et al., 1998).
Several methods have been used to characterize HCV
quasispecies, such as direct sequencing (Holland et al.,
1998), restriction fragment length polymorphism analy-
sis (McOmish et al., 1993) or genotype-specific probes
(Stuyver et al., 1996). Overall, these procedures are
difficult technically, expensive and time-consuming, andthey have a low sensitivity in the detection of minor
variants.
Electrophoretic techniques based on differential mo-
bility shift between different DNA sequences, such as
‘‘single strand conformation polymorphism’’ (SSCP)
(Enomoto et al., 1994; Moribe et al., 1995) and
‘‘heteroduplex mobility analysis’’ (HMA) (Delwart et
al., 1993, 1994; Wilson et al., 1995; Polyak et al., 1997;Sullivan et al., 2001) are relatively simple, sensitive and
less expensive alternatives for the analysis of genetic
variation. SSCP analysis is easy to perform, but mobility
shifts are extremely sensible to changes in electrophore-
tic conditions and they are not proportional to sequence
divergence between molecules. HMA has been particu-
larly useful for the study of quasispecies in different viral
models because it allows the determination of bothcomplexity and diversity of sequence mixtures (Delwart
et al., 1993; Gretch et al., 1996; Polyak et al., 1998;
Sullivan et al., 2001). This technique relies on the
formation of hybrid molecules between divergent
DNA strands when these are mixed, denatured and
allowed to anneal. Homoduplex (perfectly matched) and
heteroduplex (mismatched) molecules run differently on
electrophoretic gels as mobility is reduced proportion-ally to the nucleotide divergence between strands.
A variety of HMA assays have been developed to
analyze HCV quasispecies, usually based on polyacrila-
mide slab sequence gels. These methods are not easy to
standardize, require the use of radioactive probes and
remain costly and time-consuming for a routine use in a
clinical setting.
In an effort to improve the ability to analyze HCVviral quasispecies on a large scale, a new HMA assay for
the HVR1 region was developed, based on a capillary
electrophoresis instrument which allows the semi-auto-
mated, non-radioactive analysis of a large number of
samples in short times, the accurate measure of mobility
shifts and the quantitation of heteroduplexes.
The specific aims of the study were: (i) to validate the
use of this assay in the analysis of HCV quasispeciescomplexity and diversity, (ii) to test its performance in
the detection of qualitative and quantitative changes in
HVR1 sequence in selected patients undergoing inter-
feron therapy or liver transplantation.
2. Materials and methods
2.1. Patients
Seventeen anti-HCV positive Caucasian patients were
considered for the study (Table 1).
Ten patients (nine males; mean age 45 years) hadchronic hepatitis C and received interferon plus ribavirin
antiviral treatment for 6 months. Serum samples were
obtained before, at 1 and 3 months of therapy and 6
months after discontinuation. Normalization of bio-
chemical indices or loss of circulating HCV RNA after
therapy were used to define patients as responders, non
responders or relapsers.
Seven patients underwent orthotopic liver transplan-tation (OLT) for HCV-related liver cirrhosis (five males;
mean age 53 years) and they were monitored immedi-
ately prior to transplantation and at regularly scheduled
post-transplant intervals. Four patients experienced
hepatitis recurrence. All patients had sera analyzed at
baseline and 1 and 6 months after transplantation.
Three patients (AL, CO and AR) were also examined
at 12 months after transplantation.Cloned HVR1 sequences (mean of five clones for each
time point) were available from sera at corresponding
times for patients of both categories.
2.2. HCV RNA extraction, detection and typing
HCV RNA was extracted from 140 ml of serum using
the ‘‘QIAamp Viral RNA Mini Kit’’ (Qiagen GmbH,
Hilden, Germany) and it was detected by nested RT-
PCR using conserved primers localized in the 5? non-coding region of the viral genome (Silini et al., 1993).
HCV genotyping was performed by type-specific pri-
mers of the core gene according to Okamoto et al. (1992)
and subsequent modifications (Silini et al., 1993).
2.3. HCV-HVR1 sequence analysis
A 179 nucleotide sequence of the E2-HVR1 region of
HCV (aminoacids 364�/422) (Choo et al., 1991) was
amplified by nested RT-PCR. Oligonucleotide primers
used in the first round of PCR amplification were: senseouter primer 5?-CGCATGGCATGGGACATGAT-3?(nts. 1278�/1297) and anti-sense outer primer 5?-GG(AG)GTGAA(GA)CAATACAC(TC)GG-3? (nts.
1842�/1861). The second round PCR product was
generated using the sense inner primer 5?-ATGCTGGG-
TACGTGGGCTAAGGT-3? (nts. 1418�/1441) and the
anti-sense inner primer 5?-TTGATGTGCCAACTGC-
CATTGGT-3? (nts. 1575�/1597).First round PCR was carried out using the ‘‘Titan
One Tube RT-PCR System’’ (Roche Diagnostic, Man-
nheim, Germany) on a GeneAmp PCR system 9700
L. Rossi et al. / Journal of Virological Methods 110 (2003) 37�/4938

thermal cycler (Applied Biosystem, Foster City, CA) for
45 cycles at 30 s at 95 8C, 2 min at 45 8C and 2 min at
68 8C. In the second round PCR, 10 ml of the first
reaction products were re-amplified in 50 mM MgCl2,
0.2 mmol dNTPs, 10 mM Tris�/HCl pH 8.3, 15 mM KCl
and 100 pmol of each internal primer. Amplification was
at 30 s at 95 8C, 2 min at 45 8C and 2 min at 68 8C for 25
cycles. PCR products were separated on a 2% agarose
gel, stained with ethidium bromide, and visualized under
UV light.
HVR1 PCR products were cloned into the plasmid
vector pCRIITM using the ‘‘TA Cloning Kit’’ (Invitro-
gen, San Diego, CA). Sequencing was performed using
the ‘‘BigDyeTM Terminator Cycle Sequencing Ready
Reaction Kit’’ (Applied Biosystem) with M13 universal
primers. Sequencing reactions were run and analyzed on
an ABI PRISM Genetic Analyzer, model 310 (Applied
Biosystem).
2.4. Heteroduplex mobility analysis (HMA)
Second round PCR products were used for HMA.
Hybrid molecules were generated mixing labeled
‘‘probe’’ sequences with an excess of unlabeled ‘‘target’’
sequences.
Probe sequences were produced using the sense inner
primer labeled at 5?-end with the TET fluorochrome and
the unlabelled anti-sense inner primer, whereas the two
unlabelled primers were used to generate target se-quences. For the set-up of the assay, probes were
generated by amplification of uncut plasmid DNA
(‘clonal’ HMA); when applied to the study of quasis-
Table 1
Relevant clinical features of the two patient groups
(A ) Ten chronic hepatitis C patients who received antiviral treatment
Patient Sex Age Response HMA heteroduplex profile
Intra sample analysis Inter sample analysis
Pre-treatment During and post treatment Pre�/post treatment
3 M 36 Relapser Barely detectable, many peaks Detectable, few peaks Detectable, many peaks
4 M 56 Sustained responder Barely detectable, many peaks Detectable, few peaks Detectable, few peaks
11 F 55 Non responder Barely detectable, few peaks Barely detectable Non detectable
12 M 56 Sustained reponder Barely detectable, few peaks Detectable, few peaks Non detectable
13 M 36 Non responder Non detectable Non detectable Non detectable
21 M 45 Sustained responder Barely detectable, many peaks Barely detectable, many
peaks
Detectable, few peaks
22 M 32 Non responder Detectable, few peaks Detectable, few peaks Detectable, few peaks
31 M 43 Non responder Barely detectable, many peaks Barely detectable, many
peaks
Detectable, few peaks
32 M 36 Non responder Barely detectable, few peaks Detectable, few peaks Barely detectable, few peaks
35 M 53 Non responder Barely detectable, many peaks Barely detectable, many
peaks
Barely detectable, many peaks
(B ) Seven patients who underwent OLT for HCV-related liver cirrhosis
Patient Sex Age OLT date Hepatitis
recurrence
HMA heteroduplex profile
Intra samples analysis Inter sample analysis
Pre-OLT Post-OLT Pre�/post-OLT
AL M 49 10.09.92 Yes Detectable, many peaks Barely detectable,
many peaks
Detectable, few
peaks
CO F 52 23.10.92 No Barely detectable many
peaks
Detectable, many
peaks
Barely detectable,
few peaks
AR M 54 18.03.95 Yes Barely detectable many
peaks
Detectable, many
peaks
Detectable, few
peaks
AN M 54 01.05.00 No Non detectable Non detectable Non detectable
NA M 54 05.07.99 Yes Non detectable Non detectable Non detectable
RO M 52 19.05.99 Yes Barely detectable many
peaks
Barely detectable
many peaks
Non detectable
TO F 53 19.04.00 No Barely detectable many
peaks
Detectable, few
peaks
Detectable, many
peaks
Results of HMA intra- and inter-sample analysis on HVR1 sequences directly amplified from serum RNA are summarized.
L. Rossi et al. / Journal of Virological Methods 110 (2003) 37�/49 39

pecies ‘in vivo’, probes were obtained by direct ampli-
fication of total HCV RNA extracted from the patients
sera (‘serum’ HMA).
Hydrid molecules were produced by mixing 9 ml (15pmol of DNA) of undiluted ‘target’ PCR products, to 1
ml (1.5 pmol of DNA) of probe PCR products diluted 1�/
10 in water, to obtain a final probe to target ratio of 1/
100. PCR products were semi-quantified on gel before
annealing; exact DNA quantitation by spectrophoto-
metry was not necessary. Sample mixtures were dena-
tured at 95 8C for 5 min and allowed to re-anneal at
55 8C for 2 h. The resulting hybrid molecules were runon a capillary electrophoresis system, ABI PRISM 310
Genetic Analyzer (Applied Biosystem) using a capillary
length of 47 cm, in 2.5% GeneScan PolymerTM and 1�/
Gene Analyzer BufferTM with EDTA (Applied Biosys-
tem). The run was carried out at 60 8C for 10 min for
each sample. Mismatched hybrids (heteroduplexes) dis-
played retarded electrophoretic mobility as compared
with perfectly matched hybrids (homoduplexes), whichwas measured by the time of peak detection from the
start of the run.
Mobility shifts were expressed as heteroduplex mobi-
lity ratio (HMR) given by the migration time of the
heteroduplex peak divided by the migration time of the
homoduplex peak. Correlation between HMR and
nucleotide divergence was determined by linear regres-
sion analysis. Peak height was taken as a measure of thequantity of the different molecules.
The qualitative and quantitative analysis of electro-
phoretic data was performed by the GENESCAN Analysis
Software 3.1.2 (Applied Biosystem).
3. Results
3.1. Set up of the HMA (assay) and correlation between
sequence divergence and heteroduplex mobility shifts
The set up of the procedure took advantage of the
availability of a large set of HVR1 clones of knownsequence. ‘Clonal’ HMA allowed to control for the
number and type of heteroduplexes and their degree of
divergence; therefore, the best conditions for annealing
and electrophoresis could be identified. As the assay
should detect heteroduplex mixtures of unknown com-
position over a wide range of sequence divergence,
several hybridization and electrophoresis conditions
were explored varying probe/target ratios, temperatures,
polymer percentages, buffer concentrations and collec-
tion times.To evaluate whether the assay allowed the separation
of heteroduplexes according to their nucleotide diver-
gence, 120 HVR1 clones differing 2�/60 nucleotides
within the 179 bp sequence were amplified and hybri-
dized to generate heteroduplexes of known diversity and
the resulting products were analyzed by HMA.
All homoduplex peaks showed migration times corre-
sponding to a 179 bp sequence as expressed by aninternal size standard, whereas heteroduplex peaks
displayed retarded mobility. Heteroduplex mobility
decreased proportionally to the divergence of the paired
strands, as exemplified in Fig. 1 for selected clones.
The profile analysis showed detectable signals for
heteroduplexes with base changes up to 40 mismatches,
whereas hybrid molecules with less than six mismatches
displayed mobility shifts not proportional to the numberof base changes. Sixty pairs of sequences with 6�/40
mismatches were, therefore, considered to construct a
calibration curve. Their mobility shifts, expressed as
HMR, were calculated, plotted versus nucleotide
changes and analyzed by linear regression. A significant
positive correlation (R2: 0.92; P B/0.001) between HMR
values and number of nucleotide mismatches was
observed, as shown in panel A of Fig. 2.The assay allowed to detect heteroduplex up to two
base pair of divergence between any two HVR1
sequences corresponding to a 1.1% resolution. All
experiments were carried out in triplicate with good
reproducibility of the observed HMRs as shown by the
low value of mean standard deviation (0.004).
To assess the accuracy of the assay, the predicted
value calculated according to the equation (y (nucleo-tide changes)�/116.87x (HMR)�/107.39), was com-
pared with the observed value obtained by direct
comparison of the cloned sequences. The average
difference between the predicted value and observed
value was 1.9%.
3.2. Sensitivity and specificity of the HMA
To assess the limit of detection of minor quasispecies
variants, a titration analysis was carried out in which
Fig. 1. Heteroduplex mobility assay using capillary electrophoresis on ABI Prism 310 Genetic Analyzer. (A) Different target HVR1 clones were
hybridized to probes (*) derived from the same patients. Homoduplex peaks (Ho), with migration time of 179 bp sequence, correspond to the
annealing of templates with homologous probes. Heteroduplex peaks (He) correspond to hybrid molecules between templates and heterologous
probes. Clear peaks represent GS350 DNA size standard (Applied Biosystem) with fragment lengths between 35 and 350 bp; filled peaks represent
sample data. Horizontal scale represents DNA size in base pair. Fluorescence intensity is represented as data points along vertical scale. n.d.,
Nucleotide divergence between pair of sequences. (B) Nucleotide sequences of the HVR1 insert of the nine clones shown in panel A. Target sequences
have been aligned to the probe sequences. Identical nucleotides are shown as line and nucleotide substitutions are indicated. Primer sequences are
shown in italics.
L. Rossi et al. / Journal of Virological Methods 110 (2003) 37�/4940

Fig. 1
L. Rossi et al. / Journal of Virological Methods 110 (2003) 37�/49 41
two different probe sequences with a divergence of ten
and 20 nucleotides were co-hybridized to the same
target. The minor clone was detectable at a concentra-
tion of 3% of total probe sequences, corresponding to
0.03% of total mixture. The heteroduplex peak heights
were proportional (R2: 0.9; P B/0.001) to the quantity of
probe present in the mixture (data not shown).
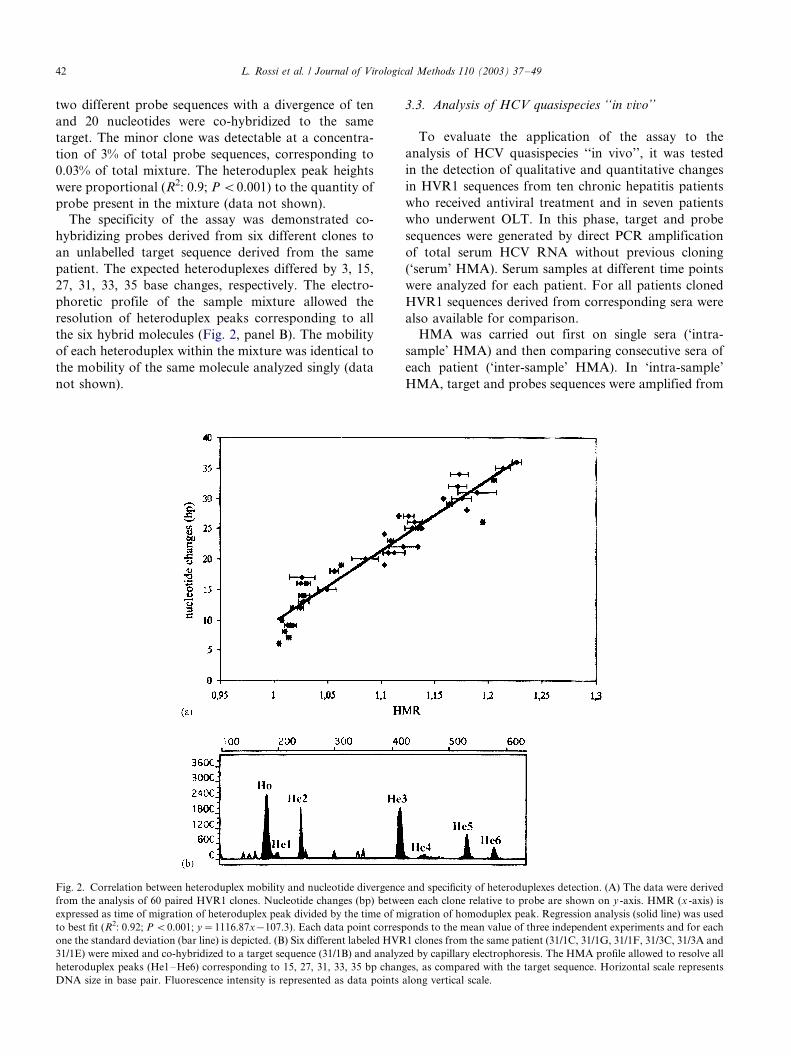
The specificity of the assay was demonstrated co-
hybridizing probes derived from six different clones to
an unlabelled target sequence derived from the same
patient. The expected heteroduplexes differed by 3, 15,
27, 31, 33, 35 base changes, respectively. The electro-
phoretic profile of the sample mixture allowed the
resolution of heteroduplex peaks corresponding to all
the six hybrid molecules (Fig. 2, panel B). The mobility
of each heteroduplex within the mixture was identical to
the mobility of the same molecule analyzed singly (data
not shown).
3.3. Analysis of HCV quasispecies ‘‘in vivo’’
To evaluate the application of the assay to the
analysis of HCV quasispecies ‘‘in vivo’’, it was tested
in the detection of qualitative and quantitative changes
in HVR1 sequences from ten chronic hepatitis patients
who received antiviral treatment and in seven patients
who underwent OLT. In this phase, target and probe
sequences were generated by direct PCR amplification
of total serum HCV RNA without previous cloning
(‘serum’ HMA). Serum samples at different time points
were analyzed for each patient. For all patients cloned
HVR1 sequences derived from corresponding sera were
also available for comparison.
HMA was carried out first on single sera (‘intra-
sample’ HMA) and then comparing consecutive sera of
each patient (‘inter-sample’ HMA). In ‘intra-sample’
HMA, target and probes sequences were amplified from
Fig. 2. Correlation between heteroduplex mobility and nucleotide divergence and specificity of heteroduplexes detection. (A) The data were derived
from the analysis of 60 paired HVR1 clones. Nucleotide changes (bp) between each clone relative to probe are shown on y -axis. HMR (x -axis) is
expressed as time of migration of heteroduplex peak divided by the time of migration of homoduplex peak. Regression analysis (solid line) was used
to best fit (R2: 0.92; P B/0.001; y�/1116.87x�/107.3). Each data point corresponds to the mean value of three independent experiments and for each
one the standard deviation (bar line) is depicted. (B) Six different labeled HVR1 clones from the same patient (31/1C, 31/1G, 31/1F, 31/3C, 31/3A and
31/1E) were mixed and co-hybridized to a target sequence (31/1B) and analyzed by capillary electrophoresis. The HMA profile allowed to resolve all
heteroduplex peaks (He1�/He6) corresponding to 15, 27, 31, 33, 35 bp changes, as compared with the target sequence. Horizontal scale represents
DNA size in base pair. Fluorescence intensity is represented as data points along vertical scale.
L. Rossi et al. / Journal of Virological Methods 110 (2003) 37�/4942

total HCV RNA of the same serum and the analysis
allowed to assess the genetic variability present within
each quasispecie. In ‘inter-sample’ HMA, HVR1 se-
quences from the baseline serum were used as target and
sequences from the follow-up sera as probes and this
allowed to assess the divergence of one patient’s
quasispecies over time.Selected clinical feature of the patients are shown in
Table 1, in which relevant results of the HMA are also
summarized. Results of two representative patients are
shown in detail.
Patient 4 was a 36-year-old male with chronic
hepatitis, who received interferon and ribavirin therapy
three times a week for 3 months. He was classified as a
sustained biochemical responder showing normalization
of transaminases but persistent serum HCV RNA at the
end of the treatment and 3 months after therapy.
Panel A of Fig. 3 illustrates the ‘intra-sample’ HMA
electropherograms of the HVR1 sequences of patient 4
obtained from sera at baseline, 1 and 3 months of
treatment and 3 months after therapy discontinuation.
The amino acid sequence of clones isolated from the
corresponding serum samples are shown in panel B of
Fig. 3.
The heteroduplex profile of the pre-treatment sample
was characterized by a major homoduplex peak and
several minor heteroduplexes, corresponding to variant
molecules from 129/2 to 179/2 nucleotide changes, as
calculated according to the calibration curve. During
treatment, the ‘intra-sample’ HMA profiles changed
considerably, showing the appearance of two new
heteroduplex peaks corresponding to molecules with
129/2 and 149/2 nucleotide divergence, and several
minor variants, ranging from 179/2 to 249/2 bp
changes. These differences were consistent with the
variation observed in the cloned sequences from the
same sera (panel B).
Fig. 4 shows the ‘inter-sample’ HMA profiles of
follow-up sera of patient 4. A high heteroduplex peak
was evident starting at 1 month of treatment corre-
sponding to a new HVR1 sequence (panel A). The
HMR value calculated for this heteroduplex corre-
sponded to a variant exhibiting a 169/2 nucleotide
difference from the original master sequence. The
mobility of this new variant corresponded exactly to
that of the more represented sequence cloned from post-
treatment sera, hybridized to total baseline HVR1
sequences and run under the same conditions (panel
B). The new variant increased progressively over the
observation time as shown by the ratio with the original
master sequence (homoduplex peak). Several minor
quasispecies, represented by small heteroduplex peaks,
were also detected.
Patient AL was a 59-year-old male who underwent
liver transplantation for HCV-related cirrhosis and
developed early recurrent hepatitis with progression to
cirrhosis within 2 years.
The ‘intra-sample’ HMA profiles of HVR1 sequences
from sera of patient AL drawn before and after 1, 6 and12 months post-transplantation are shown in panel A of
Fig. 5. Pre-transplantation sequences showed a complex
electropherogram characterized by several heteroduplex
peaks with migration times corresponding to 109/2�/
389/2 nucleotide divergence. The three major hetero-
duplexes had estimated nucleotide changes of 149/2,
289/2 and 389/2 bp, respectively. Post-transplantation
HVR1 sequences revealed simpler electropherogramswith a predominant homoduplex peak along with
several barely detectable heteroduplexes with mobility
shifts ranging from 119/2 to 289/6 nucleotide diver-
gences. The cloned sequences (panel B) at corresponding
times also showed a simplification of the genetic
composition of the quasispecies at 1 and 6 months
post-transplantation, whereas divergence increased at 12
months.The inter-sample HMA (panel C) showed the appear-
ance at 1 month post-transplantation of a heteroduplex
characterized by a HMR value corresponding to a
molecule with 109/2 nucleotide divergences. At 6
months a second, more represented heteroduplex with
a 319/2 nucleotide divergence appeared which was
maintained and progressively increased at 12 months.
The deduced divergences of the two main heterodu-plexes corresponded to nucleotide changes observed in
the HVR1 clones predominantly isolated at 1 and 6
months post-transplantation (panel B).
The remaining patients showed different patterns of
quasispecies variation and their number was too limited
to allow comparison with clinical variables. Overall, a
good correspondence was observed between the HMA
profiles and the HVR1 variation shown by the clonedsequences. It is notable that in the group of patients who
underwent antiviral treatment most subjects who ex-
perienced sustained or transient responses had detect-
able inter-sample HVR1 variation as compared with
non-responders (three out of four vs. two out of six). In
both groups, HVR1 changes occurred early either after
therapy or transplantation and tended to remain stable
afterwards.
4. Discussion
The HCV genome behaves as a rapidly evolving
quasispecies in the infected human host, however, the
clinical significance of this complex phenomenon in
terms of viral persistence, transmission and pathogenesis
is largely unknown (Weiner et al., 1992; Holland et al.,1992; Kato et al., 1993; Farci et al., 1994).
The analysis of HCV quasispecies in infected patients
has to date mainly relied on cloning of PCR products,
L. Rossi et al. / Journal of Virological Methods 110 (2003) 37�/49 43

Fig. 3
L. Rossi et al. / Journal of Virological Methods 110 (2003) 37�/4944

sequencing of an arbitrary number of clones and
comparison of their sequence. This process is labor-
intensive and requires large amounts of reagents and
specific equipments.
HMA provides an easy and relatively inexpensive
method of assessing sequence diversity of quasispecies
without the need for DNA cloning and sequencing,
simply by PCR amplification of total serum HCV RNA
followed by denaturation, annealing and electrophor-
esis. In the present study, an improvement of the HMA
for the detection of HCV quasispecies in HVR1 region
was described based on the use of a semi-automated
capillary electrophoresis instrument.
In comparison with methods reported previously, the
present technique was: (i) simpler, as loading, running
and analysis of samples did not require the operator’s
intervention, (ii) faster, as the whole procedure could be
performed in less than 2 h and 30 min post-PCR, thus
allowing the screening of a large number of samples, and
(iii) it did not require the use of radioactive probes. The
assay was at least as sensitive as previous methods and
allowed to measure both complexity and diversity of the
quasispecies.
Viral quasispecies are complex mixtures of molecules
and the number and type of heteroduplexes cannot be
determined beforehand. Therefore, the use of HMA in
this setting requires to optimize the conditions of
hybrids formation and maximize their differential mi-
gration. To develop the assay, ‘clonal’ HMA using 120
different HVR1 clones of known nucleotide sequence
was performed. A direct linear association with a high
degree of correlation between HMR and number of
nucleotide substitutions was demonstrated. Heterodu-
plex migration times increased with sequence divergence
for hybrid molecules within the range of 6�/40 mis-
matches. Hybrid molecules with less than 3% divergence
were also detectable as minimal shifts, but their mobility
was not proportional to the number of base changes.
Slab gel HMA assays have also been reported to detect
nucleotide divergences between 3�/4% (White et al.,
1999) and 1.7�/1.4%, with differences related to the total
length of the sequence and the electrophoretic condi-
tions (Wilson et al., 1995; Calvo et al., 1998). The
present assay could not reproducibly identify hetero-
duplexes over 40 mismatches, corresponding to approxi-
mately 25% divergence, likely due to the instability of
the hybrids in the electrophoretic conditions used.
Similar limits were found in previous studies, in which
it was not possible to resolve heteroduplexes over 30%
of nucleotide divergence (Polyak et al., 1997).
Several, non-predictable factors influence the mobility
and the thermodynamic stability of the hybrids in
HMA, which may vary according to the sequence
considered and the type and position of the nucleotide
substitution (Wilson et al., 1995). Nucleotide insertions
and deletions appear to have greater effects on the
reduction of the heteroduplex mobility as compared
Fig. 4. Inter-sample HMA analysis of HVR1 sequences amplified
from the serum of a HCV positive patient treated with interferon plus
ribavirin. (A) Heteroduplexes were obtained hybridizing labeled pre-
treatment sequences (probe) to target sequence from post-treatment
serum samples of patient 4. (B) HMA profile obtained by the annealing
of major HVR1 clone obtained after 3 months of treatment (4/3F) and
total HCV RNA sample from untreated serum sample (4/1). Hor-
izontal scale represents DNA size in base pair. Fluorescence intensity is
represented as data points along vertical scale.
Fig. 3. Intra-sample HMA analysis of HVR1 sequences amplified from the serum of a HCV positive patient treated with interferon plus ribavirin.
(A) Electropherograms of HVR1 sequences from patient 4 obtained before (4/1) and after 1 (4/2) and 3 (4/3) months of treatment with interferon plus
ribavirin and at 3 months after therapy discontinuation (4/4). Horizontal scale represents DNA size in base pair. Fluorescence intensity is represented
as data points along vertical scale. Hes: heteroduplexes. (B) Alignments of deduced amino acidic sequences of representative HVR1 clones isolated
from serum samples at corresponding times. Identical amino acids are shown as line; amino acid substitutions and insertions are indicated.
L. Rossi et al. / Journal of Virological Methods 110 (2003) 37�/49 45

Fig. 5. Intra- and inter-sample HMA analysis of HVR1 sequences amplified from the serum of a HCV positive liver transplant recipient. (A) Intra-
sample HMA analysis of HVR1 sequences from sera of patient AL drawn before (pre-OLT) and after 1, 6, 12 months post-transplantation. (B)
Alignments of deduced amino acidic sequences of representative HVR1 clones isolated from serum samples before transplantation (OLT2/pre) and
after 1 (OLT2/1M), 6 (OLT2/6M), 12 (OLT2/12M) months. Sequences are compared with clone A. Identical amino acids are shown as lines. (C)
Inter-sample HMA analysis of sera from patient AL. Horizontal scale represents DNA size in base pair. Fluorescence intensity is represented as data
points along vertical scale. Arrows indicate the highest heteroduplex peaks.
L. Rossi et al. / Journal of Virological Methods 110 (2003) 37�/4946

with substitutions (Delwart et al., 1993). The position of
the mismatches can also influence electrophoretic mo-
bility whether in the middle or close to the end of the
molecule. Heteroduplexes that have the same number of
mismatches with different bases in the same positions
(transitions or transversions) or the same number of
mismatches with bases in different positions demon-
strate unique shift patterns relative to one another
(Wilson et al., 1995).
It is not known whether the present assay or other
similar assays can reliably quantify the different variants
present in a quasispecies and provide a truly quantita-
tive view of HVR1 variation that should consider not
only the degree of divergence between single molecules
but also their relative abundance. A good correlation
between peak height and amount of an heteroduplex
was obtained in titration experiments of different HVR1
clones, but it was not established whether this could also
be applied to HMA assays performed on highly complex
sequence mixtures such as those derived from direct
amplification of total serum HCV RNA.
Indeed, the assay has several intrinsic limitations that
can in principle affect its quantitative application. First,
the representation of quasispecies by PCR can be
influenced by the choice of primers, which may bias
the amplification of different sequences due to affinity
of binding or thermodynamic stability of the amplicons.
Secondly, the efficiency of heteroduplex formation
within complex mixtures may vary according to their
sequence and their relative concentration. Thirdly,
major variants may saturate the signal and the presence
of several variants can lead to the formation of multiple
overlapping heteroduplexes peaks and interfere with the
interpretation of the electrophoretic profiles. These
limitations, however, did not seem to affect the potential
applications of the assay, which allowed the character-
ization of quasispecies composition and its evolution
over time in two different in vivo models, antiviral
therapy of chronic hepatitis and hepatitis recurrence
after liver transplantation. In both models, HMA
profiles showed a good qualitative and quantitative
correspondence with the sequence modifications ob-
served in selected clones from the same patients and
provided a picture of quasispecies variation consistent
with the current interpretation of this phenomenon.
The accepted view is that viral genetic variation under
major selective events is adaptive in nature and reflects
the intensity of the antiviral immune response (Tanigu-
chi et al., 1993; Kato et al., 1994; Mondelli et al., 1999).
Quasispecies changes, therefore, may contribute to viral
persistence through evasion of the immune surveillance
and be a potential mechanism of treatment failure
through selection and/or emergence of resistant variants
(Weiner et al., 1992; Taniguchi et al., 1993).
The results of the present study showed that HCV
quasispecies during therapy and transplantation under-
went relevant changes, although these varied consider-
ably between patients excluding any meaningful
conclusion. These individual differences might underlie
variable courses of the disease, either spontaneous or
therapy-induced, and be of clinical significance as
indicated by the observed differences between respon-
ders and non-responders patients which are consistent
Fig. 5 (Continued)
L. Rossi et al. / Journal of Virological Methods 110 (2003) 37�/49 47

with an enhancement of the selection of viral sequences
under antiviral treatment.
Previous researches applied HMA to the study of
HCV quasispecies in liver transplantation (Gretch et al.,1996; Sullivan et al., 1998) and under interferon therapy
(Polyak et al., 1997, 1998; Gerotto et al., 1999; Hassoba
et al., 1999) with different results and interpretations.
Technical limitations in the analysis may have contrib-
uted significantly to the discrepancies among different
studies, such as: (i) the low sensitivity of the techniques
used to measure HCV variation; (ii) the different
methods of assessing and defining divergence andcomplexity; (iii) the analysis of quasispecies over time
rather than at single time points; (iv) the limited number
of patients or clones per patients analyzed.
The application of a semi-automated, large through-
output HMA assay should contribute to overcome some
of the limitations of current methods of analysis, to
address the issue of quasispecies variation in a quanti-
tative way at variance with previous descriptive ap-proaches and to allow the extension of these studies to a
wider range of clinical and basic applications.
Acknowledgements
Supported in part by the project grant 030RFM93/01
of the Italian Ministry of Health to the IRCCS
Policlinico San Matteo, Pavia, Italy.
References
Brambilla, S., Bellati, G., Asti, M., Lisa, A., Candusso, M.E.,
D’Amico, M., Grassi, G., Giacca, M., Franchini, A., Bruno, S.,
Ideo, G., Mondelli, M.U., Silini, E.M., 1998. Dynamics of
hypervariable region 1 variation in hepatitis C virus infection and
correlation with clinical and virological features of liver disease.
Hepatology 27, 1678�/1686.
Calvo, P.L., Kansopon, J., Sra, K., Quan, S., DiNello, R., Guaschino,
R., Calabrese, G., Danielle, F., Brunetto, M.R., Bonino, F.,
Massaro, A.L., Polito, A., Houghton, M., Weiner, A.J., 1998.
Hepatitis C virus heteroduplex tracking assay for genotype
determination reveals diverging genotype 2 isolates in Italian
hemodialysis patients. J. Clin. Microbiol. 36, 227�/233.
Choo, Q.L., Kuo, G., Weiner, A.J., Overby, L.R., Bradley, D.W.,
Houghton, M., 1989. Isolation of cDNA clone derived from a
blood-borne non-A, non-B viral hepatitis genome. Science 244,
359�/362.
Choo, Q.L., Richman, K.H., Han, J.H., Berger, K., Lee, C., Dong, C.,
Gallegos, C., Coit, D., Medina-Selby, R., Bar, P.J., et al., 1991.
Genetic organization and diversity of the hepatitis C virus. Proc.
Natl. Acad. Sci. USA 88, 2451�/2455.
Coffin, J.M., 1992. Genetic diversity and evolution of retroviruses.
Curr. Top. Microbiol. Immunol. 176, 143�/164.
Delwart, E.L., Shpaer, E.G., Louwagie, J., McCutchan, F.E., Grez,
M., Rubsamen-Waigmann, H., Mullins, J.I., 1993. Genetic rela-
tionships determined by a DNA heteroduplex mobility assay:
analysis of HIV-1 env genes. Science 262, 1257�/1261.
Delwart, E.L., Sheppard, H.W., Walker, B.D., Goudsmit, J., Mullins,
J.I., 1994. Human immunodeficiency virus type 1 evolution in vivo
tracked by DNA heteroduplex mobility assays. J. Virol. 68, 6672�/
6683.
Domingo, E., 1996. Biological significance of viral quasispecies. Viral
Hep. Rev. 2, 247�/261.
Domingo, E., Holland, J.J., 1997. RNA virus mutations and fitness for
survival. Annu. Rev. Microbiol. 51, 151�/178.
Enomoto, N., Kurosaki, M., Tanaka, Y., Marumo, F., Sato, C., 1994.
Fluctuation of hepatitis C virus quasispecies in persistent infection
and interferon treatment revealed by single-strand conformation
polymorphism analysis. J. Gen. Virol. 75, 1361�/1369.
Farci, P., Alter, H.J., Wong, D.C., Miller, R.H., Govindarajan, S.,
Engle, R., Shapiro, M., Purcell, R.H., 1994. Prevention of hepatitis
C virus infection in chimpanzees after antibody-mediated in vitro
neutralization. Proc. Natl. Acad. Sci. USA 91, 7792�/7796.
Farci, P., Shimoda, A., Coiana, A., Diaz, G., Peddis, G., Melpolder,
J.C., Strazzera, A., Chien, D.Y., Munoz, S.J., Balestrieri, A.,
Purcell, R.H., Alter, H.J., 2000. The outcome of acute hepatitis C
predicted by the evolution of the viral quasispecies. Science 288,
339�/344.
Gerotto, M., Sullivan, D.J., Polyak, S.J., Chemello, L., Cavalletto, L.,
Pontisso, P., Alberti, A., Gretch, D.R., 1999. Effect of retreatment
with interferon alone or interferon plus ribavirin on hepatitis C
virus quasispecies diversification in nonresponder patients with
chronic hepatitis C. J. Virol. 73, 7241�/7247.
Gretch, D.R., Polyak, S.J., Wilson, J.J., Carithers, R.L., Perkins, J.D.,
Corey, L., 1996. Tracking hepatitis C virus quasispecies major and
minor variants in symptomatic and asymptomatic liver transplant
recipients. J. Virol. 70, 7622�/7631.
Hassoba, H.M., Bzowej, N., Berenguer, M., Kim, M., Zhou, S.,
Phung, Y., Grant, R., Pessoa, M.G., Wright, T.L., 1999. Evolution
of viral quasispecies in interferon-treated patients with chronic
hepatitis C virus infection. J. Hepatol. 31, 618�/627.
Hijikata, M., Kato, N., Ootsuyama, Y., Nakagawa, M., Ohkoshi, S.,
Shimotohno, K., 1991. Hypervariable regions in the putative
glycoprotein of hepatitis C virus. Biochem. Biophys. Res. Com-
mun. 175, 220�/228.
Holland, J.J., De La Torre, J.C., Steinhauer, D.A., 1992. RNA virus
populations as quasispecies. Curr. Top. Microbiol. Immunol. 176,
1�/20.
Holland, J., Bastian, I., Ratcliff, R.M., Beers, M.Y., Hahesy, P.,
Harley, H., Shaw, D.R., Higgins, G.D., 1998. Hepatitis C
genotyping by direct sequencing of the product from the Roche
AMPLICOR test: methodology and application to a South
Australian population. Pathology 30, 192�/195.
Kato, N., Sekiya, H., Ootsuyama, Y., Nakazawa, T., Hijikata, M.,
Ohkoshi, S., Shimotohno, K., 1993. Humoral immune response to
hypervariable region 1 of the putative envelope glycoprotein (gp70)
of hepatitis C virus. J. Virol. 67, 3923�/3930.
Kato, N., Nakazawa, T., Ootsuyama, Y., Sugiyama, K., Ohkoshi, S.,
Shimotohno, K., 1994. Virus isolate-specific antibodies against
hypervariable region 1 of the hepatitis C virus second envelope
protein, gp70. Jpn. J. Cancer Res. 85, 987�/991.
Martel, M., Esteban, J.I., Quer, J., Genesca, J., Weiner, A., Esteban,
R., Guardia, J., Gomez, J., 1992. Hepatitic C virus (HCV)
circulates as a population of different but closely related genomes:
quasispecies nature of HCV genome distribution. J. Virol. 66,
3225�/3229.
McOmish, F., Chan, S.W., Dow, B.C., Gillon, J., Frame, W.D.,
Crawford, R.J., Yap, P.L., Follett, E.A., Simmonds, P., 1993.
Detection of three types of hepatitis C virus in blood donors:
investigation of type-specific differences in serologic reactivity and
rate of alanine aminotransferase abnormalities. Transfusion 33, 7�/
13.
Mondelli, M.U., Cerino, A., Lisa, A., Brambilla, S., Segagni, L.,
Cividini, A., Bissolati, M., Missale, G., Bellati, G., Meola, A.,
L. Rossi et al. / Journal of Virological Methods 110 (2003) 37�/4948

Bruniercole, B., Nicosia, A., Galfre, G., Silini, E., 1995. Antibody
responses to hepatitis C virus hypervariable region 1: evidence for
cross-reactivity and immune-mediated sequence variation. Hepa-
tology 30, 537�/545.
Moribe, T., Hayashi, N., Kanazawa, Y., Mita, E., Fusamoto, H.,
Negi, M., Kaneshige, T., Igimi, H., Kamada, T., Uchida, K., 1995.
Hepatitis C viral complexity detected by single-strand conforma-
tion polymorphism and response to interferon therapy. Gastro-
enterology 108, 789�/795.
Okamoto, H., Sugiyama, Y., Okada, S., Kurai, K., Akahane, Y.,
Sugai, Y., Tanaka, T., Sato, K., Tsuda, F., Miyakawa, Y., et al.,
1992. Typing hepatitis C virus by polymerase chain reaction with
type-specific primers: application to clinical surveys and tracing
infectious sources. J. Gen. Virol. 73, 673�/679.
Polyak, S.J., Faulkner, G., Carithers, R.L., Corey, L., Gretch, D.R.,
1997. Assessment of hepatitis C virus quasispecies heterogeneity by
gel shift analysis: correlation with response to interferon therapy. J.
Infect. Dis. 175, 1101�/1107.
Polyak, S.J., McArdle, S., Liu, S., Sullivan, D.G., Chung, M.,
Hofgartner, W.T., Carithers, R.L., McMahon, B.J., Mullins, J.I.,
Corey, L., Gretch, D.R., 1998. Evolution of hepatitis C virus
quasispecies in hypervariable region 1 and the putative interferon
sensitivity-determining region during interferon therapy and nat-
ural infection. J. Virol. 72, 4288�/4296.
Silini, E., Bono, F., Cerino, A., Piazza, V., Solcia, E., Mondelli, M.U.,
1993. Virological features of hepatitis C virus infection in
haemodialysis patients. J. Clin. Microbiol. 31, 2913�/2917.
Stuyver, L., Wyseur, A., van Arnhem, W., Hernandez, F., Maertens,
G., 1996. Second-generation line probe assay for hepatitis C virus
genotyping. J. Clin. Microbiol. 34, 2259�/2266.
Sullivan, D.G., Wilson, J.J., Carithers, R.L., Perkins, J.D., Gretch,
D.R., 1998. Multigene tracking of hepatitis C virus quasispecies
after liver transplantation: correlation of genetic diversification in
the envelope region with asymptomatic or mild disease patterns. J.
Virol. 72, 10036�/10043.
Sullivan, D.G., Kim, S.S., Wilson, J.J., Stehman-Breen, C., Gretch,
D.R., 2001. Investigating hepatitis C virus heterogeneity in a high
prevalence setting using heteroduplex tracking analysis. J. Virol.
Methods 96, 5�/16.
Taniguchi, S., Okamoto, H., Sakamoto, M., Kojima, M., Tsuda, F.,
Tanaka, T., Munekata, E., Muchmore, E.E., Peterson, D.A.,
Mishiro, S., 1993. A structurally flexible and antigenically variable
N-terminal domain of the hepatitis C virus E2/NS1 protein:
implication for an escape from antibody. Virology 195, 297�/301.
Weiner, A.J., Brauer, M.J., Rosenblatt, J., Richman, K.H., Tung, J.,
Crawford, K., Bonino, F., Saracco, G., Choo, Q.L., Houghton,
M., et al., 1991. Variable and hypervariable domains are found in
the regions of HCV corresponding to the flavivirus envelope and
NS1 proteins and the pestivirus envelope glycoproteins. Virology
180, 842�/848.
Weiner, A.J., Geysen, H.M., Christopherson, C., Hall, J.E., Mason,
T.J., Saracco, G., Bonino, F., Crawford, K., Marion, C.D.,
Crawford, K.A., Brunetto, M., Barr, P.J., Miyamura, T.,
McHutchinson, J., Houghton, M., 1992. Evidence of immune
selection of hepatitis C virus (HCV) putative envelope glycoprotein
variants: potential role in chronic HCV infections. Proc. Natl.
Acad. Sci. USA 89, 3468�/3472.
White, P.A., Li, Z., Rawlinson, W.D., 1999. Sequence diversity in the
5?-UTR region of GB virus C/hepatitis G virus assessed using
sequencing, heteroduplex mobility analysis and single-strand con-
formation polymorphism. J. Virol. Methods 83, 91�/101.
Wilson, J.J., Polyak, S.J., Day, T.D., Gretch, D.R., 1995. Character-
ization of simple and complex hepatitis C virus quasispecies by
heteroduplex gel shift analysis: correlation with nucleotide sequen-
cing. J. Gen. Virol. 76, 1763�/1771.
L. Rossi et al. / Journal of Virological Methods 110 (2003) 37�/49 49





























