Embed Size (px)
Citation preview

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
CANCER
1Translational Clinical ResearchDivision, Chugai Pharmaceutical Co., Ltd., 1-1Nihonbashi-Muromachi 2-Chome Chuo-ku, Tokyo 103-8324, Japan. 2Research Division, ChugaiPharmaceutical Co., Ltd., 200 Kajiwara, Kamakura, Kanagawa 247-8530, Japan. 3Re-search Division, Chugai Pharmaceutical Co., Ltd., 1-135 Komakado, Gotemba, Shizuoka412-8513, Japan. 4Companion Diagnostics Pharma Services, Ventana Medical SystemsInc., 1910 E Innovation Parkway, Tucson, AZ 85755, USA. 5Chugai Pharmabody Re-search Pte. Ltd., 3 Biopolis Drive, Synapse 138623, Singapore.*Corresponding author. Email: [email protected]
Ishiguro et al., Sci. Transl. Med. 9, eaal4291 (2017) 4 October 2017
Copyright © 2017
The Authors, some
rights reserved;
exclusive licensee
American Association
for the Advancement
of Science. No claim
to original U.S.
Government Works
by guehttp://stm
.sciencemag.org/
Dow
nloaded from
An anti–glypican 3/CD3 bispecific T cell–redirectingantibody for treatment of solid tumorsTakahiro Ishiguro,1 Yuji Sano,2 Shun-ichiro Komatsu,3 Mika Kamata-Sakurai,3 Akihisa Kaneko,3
Yasuko Kinoshita,2 Hirotake Shiraiwa,3 Yumiko Azuma,2 Toshiaki Tsunenari,2 Yoko Kayukawa,2
Yukiko Sonobe,2 Natsuki Ono,3 Kiyoaki Sakata,2 Toshihiko Fujii,2 Yoko Miyazaki,2
Mizuho Noguchi,2 Mika Endo,2 Asako Harada,3 Werner Frings,3 Etsuko Fujii,2 Eitaro Nanba,3
Atsushi Narita,3 Akihisa Sakamoto,3 Tetsuya Wakabayashi,3 Hiroko Konishi,3 Hiroaki Segawa,3
Tomoyuki Igawa,3 Takashi Tsushima,2 Hironori Mutoh,2 Yukari Nishito,2 Mina Takahashi,2
Lorraine Stewart,4 Ehab ElGabry,4 Yoshiki Kawabe,2,3 Masaki Ishigai,3 Shuichi Chiba,3
Masahiro Aoki,2 Kunihiro Hattori,2 Junichi Nezu5*
Cancer care is being revolutionized by immunotherapies such as immune checkpoint inhibitors, engineered T celltransfer, and cell vaccines. The bispecific T cell–redirecting antibody (TRAB) is one such promising immunotherapy,which can redirect T cells to tumor cells by engagingCD3on a T cell and an antigenon a tumor cell. Because T cells canbe redirected to tumor cells regardless of the specificity of T cell receptors, TRAB is considered efficacious for lessimmunogenic tumors lacking enough neoantigens. Its clinical efficacy has been exemplified by blinatumomab,a bispecific T cell engager targeting CD19 and CD3, which has shown marked clinical responses against hemato-logical malignancies. However, the success of TRAB in solid tumors has been hampered by the lack of a targetmolecule with sufficient tumor selectivity to avoid “on-target off-tumor” toxicity. Glypican 3 (GPC3) is a highlytumor-specific antigen that is expressed during fetal development but is strictly suppressed in normal adult tissues.We developed ERY974, a whole humanized immunoglobulin G–structured TRAB harboring a common light chain,which bispecifically binds to GPC3 and CD3. Using a mouse model with reconstituted human immune cells, werevealed that ERY974 is highly effective in killing various types of tumors that have GPC3 expression comparableto that in clinical tumors. ERY974 also induced a robust antitumor efficacy even against tumorswith nonimmunogenicfeatures,which are difficult to treat by inhibiting immune checkpoints such as PD-1 (programmed cell deathprotein–1)and CTLA-4 (cytotoxic T lymphocyte–associated protein–4). Immune monitoring revealed that ERY974 convertedthe poorly inflamed tumor microenvironment to a highly inflamed microenvironment. Toxicology studies in cyno-molgusmonkeys showed transient cytokine elevation, but this wasmanageable and reversible. No organ toxicity wasevident. These data provide a rationale for clinical testing of ERY974 for the treatment of patientswithGPC3-positivesolid tumors.
st
on June 17, 2020INTRODUCTIONOver the past decade, extraordinary clinical benefits of cancer immu-notherapies have been demonstrated, such as durable responses evenfor late-stage cancers. In particular, the clinical outcomes of immunecheckpoint inhibitors such as anti–CTLA-4 (cytotoxic T lymphocyte–associated protein–4) and anti–PD-1 (programmed cell death protein–1) antibodies have been impressive, and they are changing the paradigmof cancer treatment (1, 2). Although these therapies have shownpromising clinical responses when used for a broad range of solid tu-mors, the number of patients who benefit from them is limited (3, 4),and it is necessary to predict which patients will be responsive to suchtreatments. Many ongoing studies to identify predictive biomarkershave suggested the usefulness of tumor mutation-derived antigens(neoantigens) to select patients who can benefit from treatment withcheckpoint inhibitors (5). For example, Rizvi et al. (6) reported thatneoantigen burden was positively correlated with clinical benefit in pa-
tients with non–small cell lung cancer receiving anti–PD-1 antibody.These studies have provided evidence that neoantigen-reactive T cellsare the most important players in the responses to checkpoint inhibi-tors. However, these advances do not address a fundamental problemwith checkpoint inhibitors: immune checkpoint inhibitorswill notworkif endogenous T cells cannot recognize the cancer cells via T cell recep-tors (TCRs) because of a lack of neoantigens. Tumors with this char-acteristic are called “nonimmunogenic” tumors, and targeting them isthe next challenge for cancer immunotherapy (7).
One strategy to mitigate this problem is T cell–redirecting antibody(TRAB) technology, which bispecifically engages CD3 on T cells andantigens on cancer cells; this binding activates T cells to kill cancer cells(8–10). TRAB technology is highly potent because it can theoreticallyredirect all T cells in the body to cancer cells regardless of the intrinsicantigen specificity of the TCR. Moreover, it has been reported thatTRABs can harness not only CD8+ T cells but also CD4+ T cells, includ-ing regulatory T cells, as effector cells to kill cancer cells (11). The ther-apeutic potential of TRABs is exemplified by blinatumomab, a bispecificT cell engager (BiTE) targeting CD19 and CD3 for the treatment ofhematological malignancies (12). The U.S. Food and Drug Administra-tion granted approval of blinatumomab based on a phase 2 study fortreatment of patients with Philadelphia chromosome–negative (Ph−)relapsed or refractory B cell precursor acute lymphoblastic leukemia(13). Several other promising TRABs such as anti-CD20/CD3 and
1 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on June 17, 2020http://stm
.sciencemag.org/
Dow
nloaded from
anti-CD123/CD3 are being developed to target hematological malig-nancies (14, 15). TRABs targeting solid tumors are also under clinicaldevelopment, including solitomab, which targets the epithelial cell ad-hesionmolecule (EpCAM) (16), or those against carcinoembryonic an-tigen (17) or prostate-specific membrane antigen (18). However, theseTRABs are still not at the stage of showing clinical benefits. Given thepotent cytotoxic potential of TRABs, the key to achieving clinical benefitis to avoid “on-target off-tumor” toxicity, which is caused by low expres-sion of the target (on-target) in normal tissues (off-tumor). It is wellknown that classical tumor antigens such as EpCAM, epidermal growthfactor receptor (EGFR), and human EGFR 2 (HER2) are not tumor-specific; therefore, on-target off-tumor toxicity could be a problem.The administration of solitomab was limited by toxicity induced bythe triggering of a response in EpCAM+ normal tissues such as bowelepithelia and bile ducts (19). In addition, an anti-EGFR BiTE was nottolerated in cynomolgus monkeys (20). Therefore, the development ofTRABs targeting solid tumors has been hampered by the lack of an ap-propriate antigen that has high tumor specificity.
Glypican 3 (GPC3) is a highly tumor-specific antigen. GPC3belongsto a family of heparin sulfate proteoglycans that are tethered to the cellsurface via a glycosylphosphatidylinositol anchor (21). An analysisusing GPC3-deficient mice suggested that GPC3 is involved in the reg-ulation of Wnt, hedgehog, and fibroblast growth factor pathways tocontrol cell growth and apoptosis in particular cell types during devel-opment (22–24). GPC3 is a fetal protein that is expressed in a wide va-riety of tissues during embryonic development (25, 26); however, itsexpression is strictly suppressed in most adult tissues (27). In contrast,elevated GPC3 expression has been reported in a wide variety of tumortypes such as liver (28, 29), lung (30), gastric (31), ovarian (32, 33),esophageal (34), and others (35). These features suggest that GPC3 isan ideal target for TRABs in these solid tumors.
Here, we generated an anti-GPC3 TRAB, ERY974, which is a fullyhumanized immunoglobulinG (IgG)–based bispecific antibody. Usingthis agent, we observed striking antitumor efficacy in vitro and in invivo xenograft models. We also examined its antitumor efficacy inan immunocompetent mouse model that mimics cancer patientswho are insensitive to checkpoint inhibitors. Using this approach, werevealed that ERY974 is effective against nonimmunogenic tumors. Inaddition, we report the results of pharmacokinetic (PK) and toxicolog-ical studies performed in cynomolgus monkeys, which suggested clin-ical safety for ERY974.
RESULTSGeneration of the anti-GPC3/CD3 bispecificantibody ERY974ERY974was generated by combining two antibodies, specifically an anti-GPC3 antibody (clone GC33) that we previously obtained (36) and ananti-CD3 antibody (clone CE115) that was newly obtained by immu-nizing a rat with human CD3e (CD3E) recombinant protein (Fig. 1A).A common light chain was generated by complementarity-determiningregion and framework region (CDR/FR) shuffling as previously de-scribed (37). First, light-chain CDRs from both parental antibodies(anti-GPC3 or anti-CD3) were shuffled and combined with humangermline FRs to form an assembly of light chains. Screening of theassembly by binding assay identified a lead common light chain thatconfers an antibody of the strongest affinity against GPC3 or CD3Ewhen combinedwith the heavy chains from the respective parental anti-bodies. The variable domains of the two heavy chains were humanized
Ishiguro et al., Sci. Transl. Med. 9, eaal4291 (2017) 4 October 2017
as previously described (38, 39). Various sets of anti-GPC3 and anti-CD3 arms having different affinities against each antigen were gener-ated and produced in a bispecific antibody format. These were assessedby in vitro T cell–dependent cellular cytotoxicity (TDCC) and in vivoantitumor efficacy in xenograft models described below. The combina-tion of arms that showed the strongest antitumor efficacy was finallyselected for ERY974 (Fig. 1A). For the heavy-chain constant region,L235R/S239K/N297A (in EU numbering) mutations were introducedinto the CH2 region of human IgG4 to abolish binding to Fcg receptors(Fig. 1A). This was to avoid GPC3-independent cytokine release byengaging Fcg receptors and CD3. To facilitate heterodimerization ofthe two heavy chains by changing the CH3 interface electrical charge(40), we introduced E356K and K439E mutations on each heavy chain.AK196Qmutationwas also introduced on the heavy chain of theGPC3arm to reduce the isoelectric point (pI) to facilitate separation of thehomodimers of each heavy chain by ion chromatography during themanufacturing process (39). In addition, other mutations to achievepI difference between the two heavy chains, decrease nonspecificbinding, reduce deamidation, and minimize immunogenicity were alsointroduced to facilitate bispecific antibody manufacturing and improvephysicochemical and safety properties (37). The resulting molecule,termed ERY974, was confirmed to specifically bind membranous hu-man GPC3 and CD3E by flow cytometry analysis using cell lines over-expressing either antigen (Fig. 1B).
GPC3 expression in tumors and normal tissuesTo investigate theGPC3protein expressionprofile in tumors andnormaltissues,weperformed immunohistochemistry (IHC)using an anti-GPC3mouse antibody (mGC33, mouse IgG2a) that specifically detects GPC3.Tissuemicroarray (TMA) cores from31hepatocellular carcinomas, 40 lungsquamous cell carcinomas, 69 lung small cell carcinomas, 87 esophagussquamous cell carcinomas, 30 cardiac adenocarcinomas, 40 gastriccancers, and 68 head and neck cancers (Fig. 2 and tables S1 to S7), aswell as the cores from 30 different normal tissues (Fig. 2 and table S8),were analyzed. In tumor tissues, 90% (28 of 31) of hepatocellularcarcinomas were positive for GPC3 in the cytoplasm or plasma mem-brane. Similarly, 65% (26 of 40) of lung squamous cell carcinomas, 64%(44 of 69) of lung small cell carcinomas, 28% (24 of 87) of esophagussquamous cell carcinomas, 30% (9 of 30) of cardia adenocarcinomas,20% (8 of 40) of gastric cancers, and 28% (19 of 68) of head and neckcancers appeared positive forGPC3. In contrast, GPC3was not detectedin normal tissues, except for the endometrium and placenta.
In vitro pharmacological profiles of ERY974In vitro pharmacological profiles of ERY974 were evaluated using SK-HEP-1 cells, a GPC3-negative cancer cell line, and SK-HEP-1/hGPC3cells, which were SK-HEP-1 cells transduced to express human GPC3on their cell surface, with human peripheral blood mononuclear cells(PBMCs) as effector cells. Concentration-dependent TDCC was seenagainst SK-HEP-1/hGPC3 cells but not against SK-HEP-1 cells(Fig. 3A). The same results were also confirmed bymonitoring cancercell growth for 72 hours by using an xCELLigence Real-Time CellAnalysis system (fig. S1). We also observed that the expression ofCD69, an early marker of T cell activation, and that of CD25, a latemarker of T cell activation, were both elevated only in T cells incu-bated with SK-HEP-1/hGPC3 cells (Fig. 3B). The aforementionedGPC3-dependent activation of T cells was also seen when tested withPC-10 cells in which GPC3 is endogenously expressed, but not withSNU-16 cells in whichGPC3 expression was not detected (fig. S2).We
2 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on June 17, 2020http://stm
.sciencemag.org/
Dow
nloaded from
next examined the characteristics of T cell activation by ERY974 usingpurifiedCD4+ orCD8+T cells from twodifferent donors (Fig. 3C).Wefound that not only CD8+ T cells but also CD4+ T cells induced clearTDCC, although the degree of CD4+ T cell–mediated TDCCwas low-er than that with CD8+ T cells. After 3 days of activation by ERY974,the absolute number of CD4+ or CD8+ T cells increased (Fig. 3D), anda carboxyfluorescein diacetate succinimidyl ester (CFSE) cell divisionassay revealed that ERY974 induces proliferation in almost all popula-tions of T cells, which suggests that ERY974 can induce polyclonal ac-tivation of T cells independently of TCR specificity (Fig. 3E).
Ishiguro et al., Sci. Transl. Med. 9, eaal4291 (2017) 4 October 2017
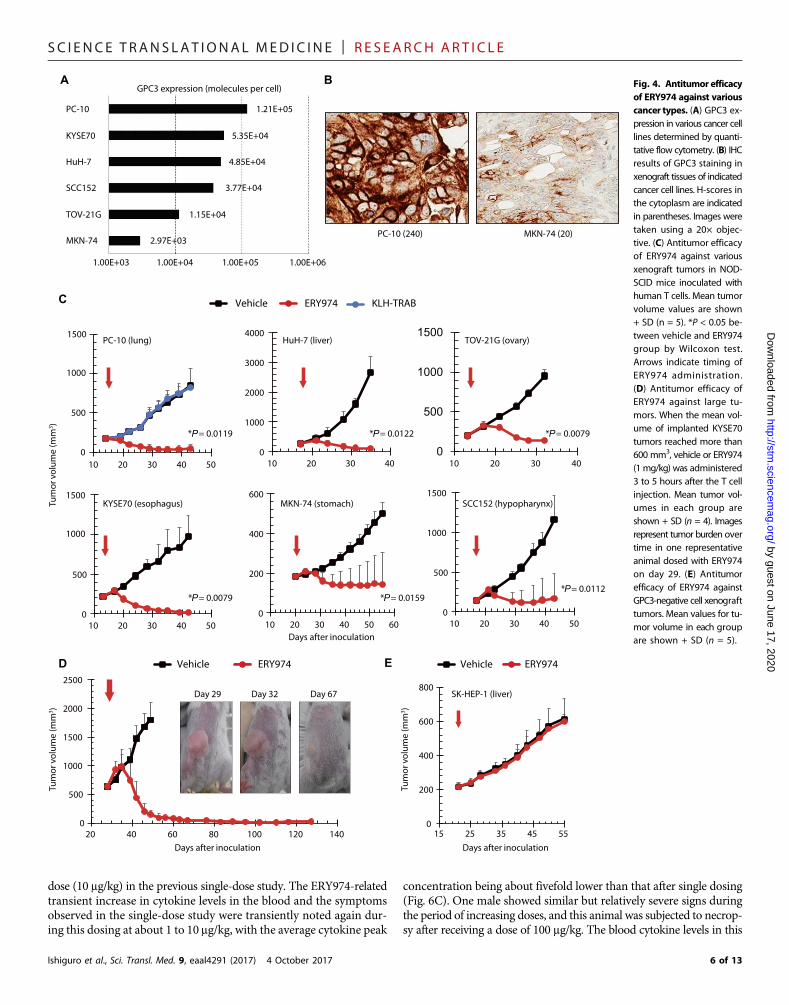
In vivo antitumor efficacy of ERY974 against various typesof tumorsWe then examined the in vivo antitumor efficacy of ERY974 againstGPC3-positive cell lines derived from various types of cancer (MKN-74 from gastric adenocarcinoma, PC-10 from lung squamous cell car-cinoma, TOV-21G from ovarian clear cell carcinoma, HuH-7 fromhepatocellular carcinoma, KYSE70 from esophageal squamous cell car-cinoma, and SCC152 from hypopharynx squamous cell carcinoma).These six cell lines were shown to have various levels of GPC3 expres-sion by quantitative flow cytometry; PC-10 had the highest (1.21 × 105
molecules per cell) and MKN-74 had the lowest (2.97 × 103 moleculesper cell) expression (Fig. 4A). To compare the expression of GPC3 inthese two cell lines in in vivo setting with those in clinical tumorsamples, we inoculated these cell lines into nonobese diabetic/severecombined immunodeficiency (NOD-SCID)mice and stained xenografttissues using the mGC33 antibody under the same conditions used forTMAanalysis as described above. Staining intensity was consistent withthe levels observed by flow cytometry using in vitro cultured cells; stron-ger GPC3 staining was observed in PC-10 xenograft tissue (H-score incytoplasm = 240), and staining inMKN-74 xenograft tissue was weaker(H-score in cytoplasm = 20) (Fig. 4B). The staining intensity in thesetwo xenograft tissues was comparable to the range observed in clinicaltumors shown in Fig. 2.
For evaluation of the in vivo antitumor efficacy of ERY974, the sixaforementioned cell lines were inoculated into NOD-SCID mice. Afterthe tumor volume reached around 200mm3, themice were treatedwithERY974. Because ERY974 has no cross-reactivity withmouse CD3, hu-man T cells were intraperitoneally injected into mice as effector cells onthe sameday of ERY974 administration (humanT cell–injectedmodel).With a single administration of ERY974, marked tumor growth inhibi-tion (TGI) was observed (Fig. 4C). The TGI (%) values of PC-10,KYSE70, HuH-7, SCC152, TOV-21G, and MKN-74 cells were 119,127, 106, 97, 107, and 112%, respectively, indicating tumor shrinkage.Using ERY974, marked tumor regression was also observed against re-latively large tumors (Fig. 4D). In contrast, antitumor efficacy was notobserved against GPC3-negative SK-HEP-1 cells (Fig. 4E).
Antitumor efficacy of ERY974 in immunocompetent humanCD3 transgenic miceWe then explored the antitumor efficacy of ERY974 in an immuno-competent mouse model. Because ERY974 is not cross-reactive to mu-rine Cd3e, we created a human CD3 transgenic mouse in which allthree components of the Cd3 complex, Cd3e, Cd3d, and Cd3g, werereplaced by their human counterparts, CD3E, CD3D, and CD3G(41). As for target tumor cells to be used for immunocompetent mousemodels, we introduced the human GPC3 gene into two mouse tumorcell lines, Hepa1-6 (hepatoma) and LLC1 (Lewis lung carcinoma), andestablished cell lines that stably express humanGPC3 (Hepa1-6/hGPC3and LLC1/hGPC3, respectively).
We inoculated human GPC3–expressing mouse tumor cells intohuman CD3 transgenic mice and established syngeneic tumor models.We then evaluated the immunogenicity of the tumors, that is, the abilityto induce an adaptive immune response, by examining immuno-histochemically the levels of tumor-infiltrating T cells and PD-L1 ex-pression in the syngeneic tumor tissues (Fig. 5A and table S9). IHCanalysis in Hepa1-6/hGPC3 tumors (at day 10) revealed high immuno-genicity, specifically moderate infiltration of CD3+ T cells and positivePD-L1 expression. In contrast, LLC1/hGPC3 tumors (at day 14) showedlow immunogenicity. The levels of tumor-infiltrating T cells were low,
No bindingability to FcγRs
Conservedbinding ability
to FcRn
Commonlight chain
Anti-glypican 3 Anti-CD3
Fc hetero-dimerization
A
B
FITC
FITCFITC
FITC
Coun
tsCo
unts
Coun
tsCo
unts
SK-HEP-1 SK-HEP-1/hGPC3
BAF3 BAF3/CD3ε
ERY974Control
Fig. 1. Generation of ERY974. (A) Schematic illustration of ERY974 structure andthe introduced mutations. The two Fab arms share a common light chain, depictedin green, with a variable domain. (B) Flow cytometry results showing specific bindingof ERY974 to GPC3 and CD3E in SK-HEP-1 (a parental human liver/ascites adenocar-cinoma cell line), SK-HEP-1/hGPC3, (SK-HEP-1 cells that overexpress human GPC3),BAF3 (a parental mouse pro–B cell line), and BAF3/CD3e (BAF3 cells that overexpresshuman CD3E). The blue lines indicate staining with a control IgG4 antibody, and thered lines indicate staining with ERY974. FITC, fluorescein isothiocyanate.
3 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on June 17, 2020http://stm
.sciencemag.org/
Dow
nloaded from
and PD-L1 was negative in tumor tissues. We further examined the de-gree of immunogenicity in these syngeneic tumors with RNA sequencing(RNA-seq)–based gene expression analysis. A group of immune-relatedgenes was selected on the basis of a previous report that investigated theimmunogenicity of implanted mouse syngeneic tumors (42). The geneset included markers of immune cell infiltration, indicators of dendriticcell and T cell activation, mediators of immune suppression, vasculature-related factors, and immune function–related transcription factors, asshown in Fig. 5B. Higher expression of these genes suggests higher im-munogenicity of the tumor. The expression of these genes in Hepa1-6/hGPC3 tumors was higher than that in LLC1/hGPC3 tumors. Thissupports the aforementioned IHC results, in which Hepa1-6/hGPC3tumors exhibited high immunogenicity, whereas LLC1/hGPC3 tumorsdisplayed low immunogenicity.
Next, the antitumor efficacy of four antibodies, ERY974, anti-mouse CTLA-4, anti-mouse PD-1, and anti-mouse PD-L1, was exam-ined using the syngeneic tumor models with human CD3 transgenicmice. In Hepa1-6/hGPC3 tumors, which are predicted to be immuno-genic, ERY974 showedmarked efficacy (P = 0.0068) because completeremission of tumors was observed in most mice. Treatment with theanti–PD-1 antibody also showed significant efficacy (P = 0.0208), andanti–CTLA-4 and anti–PD-L1 antibodies showed signs of efficacy(Fig. 5C). In contrast, in the nonimmunogenic LLC1/hGPC3 tumors,only ERY974 showed statistically significant antitumor efficacy (P =0.0003); anti–CTLA-4, anti–PD-1, and anti–PD-L1 antibodies did notshow efficacy (Fig. 5C).
Ishiguro et al., Sci. Transl. Med. 9, eaal4291 (2017) 4 October 2017
To examine the mechanism by which ERY974 changed the tumormicroenvironment, we analyzed the histopathological features and geneexpression of the aforementioned immune-related gene set in the tumors.As shown in Fig. 5A and table S9, in both Hepa1-6/hGPC3 and LLC1/hGPC3 tumors, a higher degree of immune cell infiltration, such as that ofT cells and granulocytes, was observed by histopathological analysis intumors treated with ERY974 compared to that in tumors treated withvehicle. In addition, a higher degree of PD-L1 expression was observedin tumors treated with ERY974. GPC3 expression and the degree ofFoxP3+ cell infiltration were not changed by ERY974 treatment.Consistently, treatment with ERY974 resulted in further up-regulationof immune-related genes in both tumor types when compared with ve-hicle treatment (Fig. 5B).
Toxicology and PK of ERY974 in cynomolgus monkeysTo extrapolate the PK and safety of ERY974 to humans, the cynomol-gus monkey was selected on the basis of similar binding affinities toGPC3 and CD3 (Fig. 6A) and similar median effective concentrationvalues forTDCCactivity (humanPBMCs, 0.054nM; cynomolgusmon-key PBMCs, 0.11 nM).We first conducted a single-dose study with lowdoses and slow intravenous infusion (0.1, 1, and 10 mg/kg over 30min;n = 3 per sex per dose), and the PK parameters were calculated afterexcluding the data of anti-ERY974–positive animals (6 of 18 animals;Table 1 and fig. S3). ERY974 exhibited biphasic disposition and alinear PK profile in the range of 0.1 to 10 mg/kg. The total body clear-ance (CLtotal) was independent of dose, and the elimination half-life
Hepatocellular carcinoma (240) Hepatocellular carcinoma (100)
Lung squamous cell carcinoma (190) Lung small cell carcinoma (160) Lung squamous cell carcinoma (5) Head and neck squamous cell carcinoma (26)
Normal placenta (300)Esophagus squamous cell carcinoma (114)
Gastric carcinoma (102) Gastric carcinoma (25)
Cardia adenocarcinoma (128) Normal endometrium (30)
Fig. 2. GPC3 expression in normal and tumor tissues. IHC analysis of an anti-GPC3 mouse antibody (mGC33) in a multi-tissue array. Representative photomicrographs ofGPC3 staining in tumor and normal tissues are shown. Cytoplasmic H-scores are indicated in parentheses. H-scores were calculated as values between 0 and 300, defined as[1 × (percentage of cells staining at 1+ intensity) + 2 × (percentage of cells staining at 2+ intensity) + 3 × (percentage of cells staining at 3+ intensity) =H-score]. Photoswere takenusing a 20× objective lens.
4 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on June 17, 2020http://stm
.sciencemag.org/
Dow
nloaded from
(t1/2) was 2.89 to 3.87 days. The maximum plasmaconcentration (Cmax) and the area under the plas-ma concentration–time curve from zero to infinity(AUCinf) exhibited a dose-proportional increasefrom 0.1 to 10 mg/kg. The steady-state volume ofdistribution (Vd,ss) was 104 to 123 ml/kg, which isless than the volume of extracellular fluid inmonkeys,suggesting that ERY974 has low tissue penetration(43). On the basis of these properties, by a single-animal species allometric scaling method with a fixedexponent of 0.85 for clearance and 1.0 for volume ofdistribution (44), t1/2 in humans was predicted to be5.1 days.
Here, a transient increase in blood cytokines wasobserved from1mg/kg andbecamemore pronouncedat a 10-fold higher dose, with IL-6 being the mostprominent cytokine (Fig. 6B). Signs of a deteriorat-ing general condition such as red skin, reduced foodconsumption, and body weight loss were also notedin a dose-dependentmanner. The relevant effects onclinical pathology such as elevation of C-reactiveprotein, decreased red blood cell count, and in-creased white blood cell numbers were mostlylimited to the highest dose. Histopathological find-ings such as decreased lymphocytes in the thymusand increased immune cell infiltration in multipletissues were also seen at the highest dose and wereconsistentwith an inflammatory response.However,clear cytotoxic changes were not detected, and thefindings were transient, with the animals exhibitingrapid recovery.
In the second study, we evaluated the safetyprofile at a higher dose. Animals (n = 3 per sex perdose) were treated with a dose of 0.1 mg/kg on thefirst day, and then the dosage was increased aboutthreefold per day up to 1500 mg/kg (Fig. 6C). In thisperiod, the plasma concentration of ERY974 in-creased with dosage, and Cmax reached a value thatwas about 100-fold higher than that at the highest
Ishiguro et al., Sci. Transl. Med. 9, eaal4291 (2017) 4 October
TDCC
(%)
TDCC
( %)
10−6 10−5 10−4 10−3 10−2 10−1
10−6 10−5 10−4 10−3 10−2 10−1 10−6 10−5 10−4 10−3 10−2 10−1
10−6 10−5 10−4 10−3 10−2 10−110−6 10−5 10−4 10−3 10−2 10−1
100−10
0
10
20
30
Antibody concentration (μg/ml)
TD
CC (%
) ERY974_SK-HEP-1
KLH-TRAB_SK-HEP-1/hGPC3
ERY974_SK-HEP-1/hGPC3
KLH-TRAB_SK-HEP-1
A
Antibody concentration (μg/ml)100
SK-HEP-1
SK-HEP-1/hGPC3
B
100
Antibody concentration (μg/ml)
CD69
+ /CD
3+ (%)
C
100−20
0
20
40
60
80
CD4
CD8
CD25
+ /CD
3+ (%)
Antibody concentration (μg/ml) Antibody concentration (μg/ml)
Donor A
CD4
CD8
Donor B
D
0
20,000
40,000
60,000
80,000
tnuoc llec lat oT
CD4-positive T cells
Day 0Day 3
Day 0Day 3
SK-HEP-1 SK-HEP-1/hGPC3CD8-positive T cells
SK-HEP-1 SK-HEP-1/hGPC3
0
20,000
40,000
60,000
80,000
tnuoc llec latoT
Day 0Day 3
Day 0Day 3
ERY974KLH-TRAB
E
CellTrac
Cell
coun
tC e
ll c o
unt
CellTrac
CellTrac CellTrac
Cell
coun
tC e
ll c o
unt
SK-HEP-1/hGPC3
SK-HEP-1
CD4-positive T cells CD8-positive T cells
0
20
40
60
80
100
0
20
40
60
80
100SK-HEP-1
SK-HEP-1/hGPC3
10
100
−10
0
10
20
30
40
KLH-TRABERY974
Fig. 3. GPC3-dependent TDCC and polyclonal T cell ac-tivation induced by ERY974. (A) Target cells and PBMCswere incubated with various concentrations of ERY974 for24 hours. A keyhole limpet hemocyanin (KLH)–TRAB antibodywas used as a negative control. TDCC was measured as de-scribed in Materials and Methods. Data represent means ±SD (n = 3). (B) T cell activation was assessed by measuringCD25 and CD69 levels on CD3+ T cells by flow cytometry. Datarepresent means ± SD (n = 3). (C) TDCC of ERY974 elicited byCD4+ or CD8+ T cells was measured. Data represent means ±SD (n = 3). (D) Target cells and CFSE-labeled PBMCs were in-cubated with ERY974 (1 mg/ml) or KLH-TRAB for 3 days. Totalnumbers of viable CD4+ or CD8+ T cells at day 0 (immediatelyafter reaction started) and day 3 were counted by flow cytom-etry. Data represent means ± SD (n = 3). (E) Red lines repre-sent the CFSE profiles in the presence of ERY974, and bluelines represent the profile in the presence of KLH-TRAB. Thehistograms show one representative result of triplicate assays.
2017 5 of 13
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on June 17, 2020http://stm
.sciencemag.org/
Dow
nloaded from
dose (10 mg/kg) in the previous single-dose study. The ERY974-relatedtransient increase in cytokine levels in the blood and the symptomsobserved in the single-dose study were transiently noted again dur-ing this dosing at about 1 to 10 mg/kg, with the average cytokine peak
Ishiguro et al., Sci. Transl. Med. 9, eaal4291 (2017) 4 October 2017
concentration being about fivefold lower than that after single dosing(Fig. 6C). One male showed similar but relatively severe signs duringthe period of increasing doses, and this animal was subjected to necrop-sy after receiving a dose of 100 mg/kg. The blood cytokine levels in this
AGPC3 expression (molecules per cell)
1.00E+03 1.00E+04
1.21E+05
1.00E+05 1.00E+06
5.35E+04
4.85E+04
3.77E+04
1.15E+04
2.97E+03
PC-10
KYSE70
HuH-7
SCC152
TOV-21G
MKN-74
B
PC-10 (240) MKN-74 (20)
0
500
1000
1500
10 20 30 40 50
0
1000
2000
3000
4000
10 20 30 40
0
500
1000
1500
10 20 30 40 500
200
400
600
10 20 30 40 50 60
0
500
1000
1500
10 20 30 40
Days after inoculation
Tum
or v
olum
e (m
m3 )
0
1000
1500
10 20 30 40 50
PC-10 (lung)
* P = 0.0119
C Vehicle ERY974 KLH-TRAB
0
500
1000
1500
2000
2500
20 40 60 80 100 120 140Days after inoculation
D Vehicle ERY974
Tum
or v
olum
e (m
m3 )
Day 32 Day 67Day 29
E
HuH-7 (liver) TOV-21G (ovary)
KYSE70 (esophagus) MKN-74 (stomach) SCC152 (hypopharynx)
* P = 0.0122 * P = 0.0079
* P = 0.0079 * P = 0.0159* P = 0.0112
0
200
400
600
800
15 25 35 45 55
SK-HEP-1 (liver)
Vehicle ERY974
500Tu
mor
vol
ume
(mm
3 )
Days after inoculation
Fig. 4. Antitumor efficacyof ERY974 against variouscancer types. (A) GPC3 ex-pression in various cancer celllines determined by quanti-tative flow cytometry. (B) IHCresults of GPC3 staining inxenograft tissues of indicatedcancer cell lines. H-scores inthe cytoplasm are indicatedin parentheses. Images weretaken using a 20× objec-tive. (C) Antitumor efficacyof ERY974 against variousxenograft tumors in NOD-SCID mice inoculated withhuman T cells. Mean tumorvolume values are shown+ SD (n = 5). *P < 0.05 be-tween vehicle and ERY974group by Wilcoxon test.Arrows indicate timing ofERY974 administration.(D) Antitumor efficacy ofERY974 against large tu-mors. When the mean vol-ume of implanted KYSE70tumors reached more than600 mm3, vehicle or ERY974(1 mg/kg) was administered3 to 5 hours after the T cellinjection. Mean tumor vol-umes in each group areshown + SD (n = 4). Imagesrepresent tumor burden overtime in one representativeanimal dosed with ERY974on day 29. (E) Antitumorefficacy of ERY974 againstGPC3-negative cell xenografttumors. Mean values for tu-mor volume in each groupare shown + SD (n = 5).
6 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on June 17, 2020http://stm
.sciencemag.org/
Dow
nloaded from
individual were similar to those in others, and clear cytotoxicchanges and organ toxicity were not detected. In the other fiveanimals, dosing was tolerated until the target dose of 1500 mg/kg.The main pathological findings were similar to those in thesingle-dose study, and no clear cytotoxic changes or organ toxi-cities were evident.
Effect of steroid premedication on antitumor efficacyand cytokine releasePremedication with corticosteroids, as represented by dexa-methasone (DEX), is a common practice in the clinic to preventinfusion reactions including cytokine release syndrome (CRS)(45). Thus, we examined the influence of DEX premedicationon cytokine release caused by ERY974 treatment and its anti-tumor efficacy using a humanized NOD/Shi-scid, IL-2Rgnull(huNOG)mousemodel, in which CD34+ human hematopoieticstem cells (HSCs) were transplanted to reconstitute human im-mune cells. About 100 days after HSC transplantation, each hu-manizedmouse was examined for the presence of human CD45+
leukocytes, humanCD3+ T cells, andmurine CD45+ leukocytesin the mouse peripheral blood by flow cytometry. Human-ization was successfully achieved; the percentage of humanleukocytes among all leukocytes was 53.9 to 93.8%, and thepercentage of human T cells among human leukocytes was4.4 to 20.7%. Next, we compared ERY974 dose dependencyfor antitumor efficacy and that for cytokine release (Fig. 7,A and B). ERY974 was administered as a single dose of 1, 0.2,0.04, or 0.008 mg/kg 4 weeks after inoculation of PC-10 cancercells. Substantial antitumor efficacy was observed at all dosages.Among the cytokines tested (IL-2, IL-4, IL-6, IL-10, tumor ne-crosis factor, and interferon-g), transient induction of IL-6 andIL-2 was notable, similar to that observed in cynomolgusmonkeys. Both antitumor efficacy and cytokine release weredose-dependent and showed similar dose responses. We nextexamined the effect of premedication by administering DEX at18 hours and 1 hour before the first administration of ERY974.DEX premedication almost completely inhibited cytokine re-lease, whereas it did not show any suppressive effect on the anti-tumor efficacy of ERY974 (Fig. 7, C and D). After the secondadministration of ERY974 on day 7, cytokine elevation wasnot observed, althoughDEXwas not given before that time point(Fig. 7D). These results were the same at a lower ERY974 dosewith submaximal efficacy (fig. S4).
Ishiguro et al., Sci. Transl. Med. 9, eaal4291 (2017) 4 October 2017
0
500
1000
1500
2000
2500
10 15 20 25 30
H&E
LLC1/hGPC3Hepa1-6/hGPC3
Vehicle ERY974
CD3
FoxP3
PD-L1
GPC3
Vehicle ERY974
(Scale bars, 100 μm)
A
LLC1/hGPC3Hepa1-6/hGPC3
Vehicle ERY974 Vehicle ERY974
B
CD45CD11cCD11bCD4CD8CD19F4/80CD49bCD127IL-6RCD62LCD25CD80CD86CD83CD137LCD40OX40LIFNgCD40LCTLA-4ARG-1iNOSIDOTGF-bIL-10PD-L1PD-L2B7H4COX2IL-6FoxP3VEGFPLGFVEGFR1VEGFR2TbetGATA3STAT3HIF1aC/EBPb
Cell
popu
latio
nsD
endr
itic
cell
and
T ce
ll ac
tivat
ion
Supp
ress
ion
mol
ecul
esTM
ETr
ansc
riptio
nal
fact
ors
Row scale
2
1
0
−1
−2
C
0
500
1000
1500
2000
5 10 15 20 25 30Days after inoculation Days after inoculation
Tum
or v
olum
e (m
m3 )
Hepa1-6/hGPC3 LLC1/hGPC3
VehicleERY974 (*P = 0.0068) Anti−CTLA-4Anti−PD-1 (*P = 0.0208) Anti−PD-L1
VehicleERY974 (*P = 0.0003) Anti−CTLA-4Anti−PD-1Anti−PD-L1
Fig. 5. Antitumor efficacy of ERY974 in immunocompetent humanCD3 transgenic mice. (A) Histopathological analysis of Hepa1-6/hGPC3and LLC1/hGPC3 tumors. Tumor tissue samples taken 3 days afteradministering vehicle or ERY974 (5 mg/kg) were stained as indicated.H&E, hematoxylin and eosin. (B) Gene expression analysis in Hepa1-6/hGPC3and LLC1/hGPC3 tumors. RNA from tumors treated with vehicle or ERY974was used for RNA-seq. Each group was tested in triplicate (n = 3). Z scoreswere calculated using log2-transformed fragments per kilobase of exon permillion mapped fragments values for all target genes. (C) Antitumor efficacyof ERY974 and immune checkpoint inhibitors. Values represent means + SD(n = 5). *P < 0.05 between vehicle group and the antibody treatment groupat day 25 determined by Dunn’s multiple comparisons test. Arrows indicatethe timing of antibody administration.
7 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on June 17, 2020http://stm
.sciencemag.org/
Dow
nloaded from
DISCUSSIONHere, we generated ERY974, a bispecific antibody that redirects the cy-tolytic activity of T cells against tumor cells expressing the highly tumor-specific antigen GPC3. ERY974 has a whole IgG-like structure with twodistinct heavy chains and one common light chain. The presence of a
Ishiguro et al., Sci. Transl. Med. 9, eaal4291 (2017) 4 October 2017
common light chain completely circumventsmismatched pairing of theheavy chain and light chain, which is a general issue formanufacturing abispecific antibody consisting of two distinct heavy and light chains. Inaddition to the common light chain, another set of mutations that fa-cilitate the heterodimerization of the two heavy chains and modify thepI to allow straightforward separation of mispaired homodimers by ionchromatography was also introduced to ERY974. Thesemutations helpensure a robust and efficient manufacturing process for a whole IgG-like bispecific antibody. This set of antibody engineering technologies,termed ART-Ig technology, has been applied to our previous bispecificantibody drug named ACE910/emicizumab (37), and similar to thatcase, large-scale production of good manufacturing practice-gradeERY974 has been successfully accomplished.
ERY974 has a whole IgG-like structure with intact neonatal Fc re-ceptor (FcRn) binding properties of the Fc portion, which allows it tohave a longer plasma half-life than non-IgG formats. From the cyno-molgus monkey PK study, the plasma half-life of ERY974 in humanswas predicted to be 5.1 days; this enables a weekly or biweekly dose regi-men in the clinic. This is one of the advantages of ERY974 having awhole IgG-like structure because this is not feasible for bispecific mole-cules such as BiTE that lack FcRn binding ability and as a result havepoor PK. As an alternative approach to a bispecific antibody, severalchimeric antigen receptor T cell–based therapies targeting GPC3 arebeing developed (46, 47). However, considering that a T cell–based ther-apy requires ex vivo immune cell manipulation, ERY974 is expected tohave an advantage over such approaches, especially with respect tomanufacturing and access.
A
Peptide SpeciesK D
Mean
GPC3Human
Cyno. 1.35 × 10−9
CD3Human 2.07 × 10−7
Cyno. 1.55 × 10−7
B
C
(M)
1.46 × 10−9
Days after first dosing
IL-6
(pg/
ml)
Vehicle 0.1 μg/kg 1 μg/kg 10 μg/kg
−5 −4 −3 −2 −1 0 1 2 3 4 5 6 7 8 9 100.01
0.1
1
10
100
1000
10,000
0
200
400
600
800
1000
1200
IL-6
(pg/
ml)
Days after first dosing
Dose (μg/kg)
Vehicle ERY974
0
500
1000
1500
2000
2500
3000
3500
4000
4500
–1 0 1 2 3 4 5 6 7
Fig. 6. Safety assessment in cynomolgus monkey. (A) Binding affinity of ERY974 to human or cynomolgus monkey epitope peptides derived from GPC3 or CD3 wasmeasured by surface plasmon resonance analysis. KD, dissociation constant. (B) Change in serum interleukin (IL)–6 concentration over time in a single-dose study. Five animalsper sex per group received a single administration of ERY974. Necropsieswere performedonday 22 after the dose. (C) Serum IL-6 concentration (red andblack lines) over time in arepeated-dose study. Three animals per sex received doses (blue bars) that increased daily by about threefold to 1.5 mg/kg. Necropsies were performed on day 10.
Table 1. PK parameters of ERY974 in cynomolgus monkeys after asingle intravenous infusion of ERY974. Animals that developed anti-drug antibodies (ADA) were excluded from the analysis. The mean ofthree animals is shown. MRT, mean residence time.
Dose
Sex t1/2 AUCinf CLtotal Vd,ss Cmax MRT(day)
(ng/dayper ml)(ml/dayper kg)
(ml/kg)
(ng/ml) (day)0.1 mg/kg
Male* 3.87 4.4 22.8 111 1.86 4.88Female
2.89 3.75 26.8 104 1.67 3.891 mg/kg
Male* 3.72 37.8 26.5 116 16.9 4.38Female*
3.62 34.6 29.3 123 15.9 4.2910 mg/kg
Male† 3.68 366 27.3 109 217 3.97Female*
3.52 379 26.6 108 189 4.08*Data from one ADA-positive animal were excluded. †Data from twoADA-positive animals were excluded.
8 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on June 17, 2020http://stm
.sciencemag.org/
Dow
nloaded from
Because TRABs induce cytotoxicity against target-expressing cells,one issue that limits their clinical use is undesired cytotoxicity againstnormal tissues that express the target molecule. It is crucial to select ahighly tumor-specific antigen as the target to avoid such effects; howev-er, only a few antigens are currently known to meet the strict criteriarequired forTRABuse in solid tumors, and this is hampering the clinicalsuccess of TRABs. GPC3 is a molecule reported to have highly tumor-specific expression, and this was further confirmed by IHC in this studyusing a large set of tumor and normal tissues. High expression of GPC3was observed in multiple solid tumors, whereas only weak expressionwas observed in a limited range of normal tissues, such as the endome-
Ishiguro et al., Sci. Transl. Med. 9, eaal4291 (2017) 4 October 2017
trium and placenta. Maeda et al. (32) observed expression of GPC3in the endometrium in 8% (1 of 12) of secretory phase samples but notin the proliferative phase (0 of 12) or menstrual phase samples (0 of 7).In accordance with that report, our examined TMA core of the endo-metrium, which was GPC3-positive, suggests features of the secretoryphase: stromal edema, prominent spiral arterioles, and predecidualiza-tionwith endometrial gland lumens. Although the positive ratewas low,administration of ERY974 to premenopausal females requires carefulattention. Strong GPC3 expression in the placenta is a well-known ob-servation (25). Therefore, administration of ERY974 to pregnant wom-en should be avoided. Because only a limited number of normal tissues
A
0
500
1000
1500
2000
2500
3000
25 35 45 55 65
Tum
or v
olum
e (m
m3 )
Days after inoculation
Vehicle
0.008 mg/kg (*P = 0.0385)
0.04 mg/kg (*P = 0.0022)
0.2 mg/kg (*P < 0.0001)
1 mg/kg (*P < 0.0001)
B
0
200
400
600
800
1000
1200
1400
1600
1800
25 30 35 40 45 50
Days after inoculation
Tum
or v
olum
e (m
m3 )
VehicleDEXERY974 (1 mg/kg) (*P < 0.0001)ERY974 (1 mg/kg) + DEX (*P < 0.0001)
C
0
100
200
300
400
500
600
−17 2 6 240
200
400
600
800
1000
−17 2 6 24
IL-6
(pg/
ml)
IL-2
(pg/
ml)
Vehicle
0.008 mg/kg
0.04 mg/kg
0.2 mg/kg
1 mg/kg
Vehicle
0.008 mg/kg
0.04 mg/kg
0.2 mg/kg
1 mg/kg
Time after ERY974 treatment (hour)
D
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
0
50
100
150
200
250
300
Time after f irst ERY974 treatment (hour)
IL-6
(pg/
ml)
IL-2
(pg/
ml)
−19 6 24 149(−19)
174(6)
−19 6 24 149(−19)
174(6)
VehicleDEXERY974 (1 mg/kg)ERY974 (1 mg/kg) + DEX
VehicleDEXERY974 (1 mg/kg)ERY974 (1 mg/kg) + DEX
Fig. 7. Effect of premedication on antitumor efficacy and cytokine release. (A) Antitumor efficacy of ERY974 in huNOG mice. Mean tumor volume values are shown+ SD (n = 4 or 5). *P <0.05 between vehicle and ERY974 groups by Dunnett’s multiple comparisons test. Arrows indicate ERY974 administration. (B) IL-6 and IL-2induction. Blood was collected 17 hours before and 2, 6, and 24 hours after ERY974 administration. Plasma concentrations of IL-6 and IL-2 are shown. Values representmeans + SD (n = 5). (C) Influence of DEX premedication on antitumor efficacy. huNOG mice bearing tumors were administered vehicle, DEX premedication alone, orERY974 (1 mg/kg) with or without DEX premedication. Mean tumor volume values are shown + SD (n = 4 or 5). *P < 0.05 between the vehicle and ERY974 groups byDunnett’s multiple comparisons test. (D) Effect of DEX on induction of IL-6 and IL-2. Blood was collected 19 hours before and 6 and 24 hours after the first dose ofERY974, and 19 hours before and 6 hours after the second dose (149 and 174 hours after the first dose, respectively) of ERY974. Values represent means + SD (n = 5).
9 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on June 17, 2020http://stm
.sciencemag.org/
Dow
nloaded from
showed GPC3 expression, this highly tumor-specific expression profilesuggests acceptable toxicity for the TRAB, as supported by the results incynomolgus monkeys including female animals to assess toxicity inendometrium in this study. Administration of ERY974 at a singledose (10 mg/kg) did not cause any cytotoxic changes in normal tis-sues. Furthermore, even aftermultiple rounds of administration withincreasing doses, to a final dose (1500 mg/kg), cytotoxic changes werenot detected in normal tissues, and all other abnormalities were tran-sient and reversible.
In studies on cynomolgusmonkeys, themost prominent clinical ob-servation was CRS. As observed with other TRABs such as blinatumo-mab (48), CRS is thought to be a common side effect induced byTRABs. Thus, the development of agents or clinical regimens to mini-mize CRS is desirable for expanding the therapeutic index of TRABs.Here, we successfully demonstrated that cytokine release can be man-aged by corticosteroid premedication. Corticosteroids are widely usedduring clinical cancer treatment, especially at first dosing, to suppressCRS, which occurs through the use of various cytotoxic and biologicalagents. However, corticosteroids inhibit T cell activation and prolifera-tion, which results in the concern that premedication with corticoste-roidsmight dampen the antitumor efficacy of TRABs. Therefore, in thisstudy, we examined the effects of corticosteroids on the pharmaco-logical action of ERY974 in the huNOG mouse model, in which bothcytokine release and antitumor efficacy can be evaluated in the samesystem. We found that corticosteroid premedication effectively sup-pressed cytokine release but did not affect antitumor efficacy. This sug-gests that a corticosteroid, which has a relatively short half-life, couldeffectively suppress early and transient cytokine release, without a sig-nificant effect on ERY974-mediated cytotoxic activity against tumors.An in vitro study to examine the effect of corticosteroids on cytokinerelease and pharmacological activity of blinatumomab has also beenpreviously reported (49). This study also demonstrated the selective ac-tivity of corticosteroids in significantly reducing cytokine productionwhile maintaining the cytotoxic activity of T effector cells.
Here, we examined the antitumor efficacy of ERY974 and check-point inhibitors by using two mouse tumor models, namely LLC1/hGPC3 andHepa1-6/hGPC3, which present nonimmunogenic or im-munogenic features, respectively. A previous report suggested thatLLC1 tumors have low major histocompatibility complex (MHC)class I expression (42), and as expected, we found that checkpoint in-hibitors have marginal antitumor efficacy against LLC1/hGPC3 tu-mors. In contrast, ERY974 treatment resulted in marked TGI.Because ERY974 can activate T cells in a polyclonal manner and re-direct them to GPC3-expressing tumors regardless of the presence ofneoantigens inMHCclass Imolecules, this therapeutic could efficient-ly elicit an antitumor effect even in tumors with nonimmunogenicfeatures. In contrast, in immunogenic Hepa1-6/hGPC3 tumors, treat-ment with anti–PD-1 was effective, suggesting that an immuno-suppressive environment is created in the tumor tissue by immunecheckpoint molecules. Even in the immunosuppressive environmentof an immunogenic tumor, ERY974 alone was shown to inducecomplete tumor regression. In contrast, multiple studies have sug-gested that the pharmacological action of TRABs is inhibited by im-munosuppressive activity induced by the PD-1/PD-L1 axis (50). Forexample, Junttila et al. (51) reported that PD-L1 expression in tumorslimited anti-HER2 TRAB (HER2-TDB) activity, and this resistancecould be reversed by anti–PD-L1 treatment. Here, ERY974 treatmentincreased the number of PD-L1+ cells in both Hepa1-6/hGPC3 andLLC1/hGPC3 tumors. This suggests that combining the treatment
Ishiguro et al., Sci. Transl. Med. 9, eaal4291 (2017) 4 October 2017
with administration of immune checkpoint inhibitors could furtheramplify the antitumor efficacy of ERY974 in either immunogenic ornonimmunogenic clinical tumors, which represents an attractive ap-proach for future clinical studies.
The current study thus provides preclinical safety profiles of ERY974and its potent antitumor efficacy not only in immunogenic but also innonimmunogenic tumors, which are difficult to treat with immunecheckpoint inhibitors. This strongly supports the clinical testing ofERY974 for the treatment of GPC3-positive solid tumors. ERY974phase 1 clinical trials are currently ongoing (NCT02748837).
MATERIALS AND METHODSStudy designThe main objective of our study was to evaluate the antitumor efficacyand safety of ERY974 bispecific antibody targeting GPC3 and CD3E.The in vivo antitumor efficacy was assessed in three different tumor-grafted mouse platforms: NOD-SCID mouse inoculated with humanT cells, immunocompetent human CD3E/D/G transgenic mouse, andhuNOGmouse. Sample size (n = 4 or 5 per group) was determined onthe basis of the consistency of tumor growth observed in preliminaryexperiments as the one that would give statistically significant differ-ences in tumor size between the various treatment groups. Animalswere randomly assigned to groups on the basis of tumor size so thateach group had the same average size. Tumor volumes were calculatedaccording to the following formula: [(length × width2)/2]. All tumorvolume data (mean tumor volume with SD) were plotted except thedata of dead animals (two in Fig. 7A and one in Fig. 7C). Animals weresacrificed at the end of the study. Human PBMCs and HSCs were usedin those studies upon approval by an Institutional Review Board. Thetoxicity of ERY974 was assessed using cynomolgus monkeys. Animalsjudged unsuitable for toxicological evaluation of ERY974 were excludedfrom the study before grouping. Animals were assigned to each groupusing a computerized procedure designed to balance bodyweight equal-ly among groups. The number of animals per group (n = 3 per sex pergroup) was chosen according to the ICH-S4A guideline as one thatwould enable a scientific conclusion on the general safety items withthe minimum use of animals. For PK analysis, the data from animalsthat showed ADA production were excluded, regardless of the titerlevels. Primary data are located in table S10.
In vitro TDCC and T cell activation assaysHuman PBMCswere purified from fresh blood of healthy donors usinga conventional Ficoll-Paque PLUS gradient (GE Healthcare). Adherenttarget cellswere detachedwithAccutase (InnovativeCell Technologies),and 10,000 cells per well were seeded in 96-well U-bottom plates.ERY974 and human PBMCs were added (E/T ratio of 20:1). Target cellkilling was assessed after 24 hours at 37°C and 5% CO2 through thequantification of lactate dehydrogenase (LDH) release into cell super-natants by dead cells (LDH cytotoxicity detection kit; Takara Bio). Allsamples were assessed in triplicate. Maximal lysis of target cells (100%)was achieved by incubation with Triton X-100. Minimal lysis (0%) re-fers to target cells incubatedwith effector cells but without ERY974. Thepercentage of TDCC was calculated as (sample release − spontaneousrelease)/(maximum release − spontaneous release) × 100. For TDCCassays using purified CD4+ and CD8+ T cells as effectors, CD4+ andCD8+ T cells were purified from PBMCs from two donors usingCD4 and CD8microbead separation systems (Miltenyi Biotec), respec-tively. All procedures were performed in the samemanner as described
10 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on June 17, 2020http://stm
.sciencemag.org/
Dow
nloaded from
previously, except for an alteration to the effector/target cell ratio (E/Tratio of 10). A KLH-TRAB antibody (bispecific to CD3 and keyholelimpet hemocyanin) was used as a negative control. After the TDCCassay, the remaining cells were collected and used to measure T cell ac-tivation by examining the expression of CD69 and CD25 in the CD3+
population using FACSVerse. Anti-CD3 (allophycocyanin-conjugated;clone SK7, BDBiosciences), anti-CD25 (phycoerythrin-conjugated; cloneM-A251, BD Biosciences), and anti-CD69 (FITC-conjugated; cloneFN50, BD Biosciences) antibodies were used.
In vivo human T cell–injected mouse modelAllmouse studies were performed in accordance with the policies of theInstitutional Animal Care and Use Committee (IACUC) at ChugaiPharmaceutical Co., Ltd. NOD-SCIDmice (CLEA Japan Inc..) receivedsubcutaneous implants of human cancer cells (1 × 107 cells per mouse).After palpable tumors were established, randomization was performedon the basis of tumor volume and body weight. Human T cells wereselectively amplified by culturing human PBMCs using Dynabead Hu-man T-Activator CD3/CD28 (Life Technologies) and were intra-peritoneally injected into mice (3 × 107 cells per mouse) as effectorcells. ERY974 (1 mg/kg), KLH-TRAB (1 mg/kg), or vehicle was intra-venously administered 3 to 5hours after T cell injection. Tumor sizewasmeasured twice per week. TGI was calculated using the followingformula: TGI (%) = [1 − (T − T0)/(C − C0)] × 100, where T and T0are the mean tumor volumes on a specific experimental day and onthe randomization day, respectively, for the experimental groups, andC and C0 are the corresponding mean tumor volumes for the controlgroup. Values > 100% represent tumor shrinkage.
Cynomolgus monkey studiesAll cynomolgus monkey studies were conducted at Covance Labora-tories Inc. according to the guidelines of the IACUC, using purpose-bred, naïve, cynomolgusmonkeys of Chinese origin. For the single-dosestudy, three cynomolgus monkeys per sex per group were administereda single intravenous infusion of ERY974 (0.1, 1, and 10 mg/kg) for30 min. For the second study, three cynomolgus monkeys per sexwere administered an intravenous infusiondose of ERY974 (1500mg/kg)after a daily increasing dose regimen (0.1 to 300 mg/kg per dose).Whole-blood samples or tissues were collected at selected time points for clinicalpathology and histopathology. Plasma ERY974 concentration wasdetermined by an electrochemiluminescence immunoassay (ECLIA).This method is based on an indirect immunoassay using GPC3 asa solid-phase antigen, with anti-ERY974 antibody binding to CDRfor CD3 antigen as a detection antibody. Anti-ERY974 antibodies incynomolgus monkey plasma were analyzed by ECLIA using biotin-labeled ERY974 and ruthenium-labeled ERY974. PK analysis wasperformed using WinNonlin Professional Edition computer soft-ware, version 6.1 (Pharsight).
Statistical analysisData are presented as means ± SD, means + SD, or means only asstated in the figure legends. Statistically significant differences weretested using specific tests as indicated in the figure legends. P < 0.05was considered statistically significant.
SUPPLEMENTARY MATERIALSwww.sciencetranslationalmedicine.org/cgi/content/full/9/410/eaal4291/DC1Materials and Methods
Ishiguro et al., Sci. Transl. Med. 9, eaal4291 (2017) 4 October 2017
Fig. S1. Cancer cell growth inhibition induced by ERY974.Fig. S2. TDCC and polyclonal T cell activation induced by ERY974 targeting cancer cell lines.Fig. S3. Plasma concentration–time profiles of ERY974 after a single intravenousadministration.Fig. S4. Influence of DEX pretreatment on the antitumor efficacy of ERY974 at a dose withsubmaximal efficacy (0.04 mg/kg).Table S1. H-scores for 31 hepatocellular carcinoma TMA cores staining for GPC3.Table S2. H-scores for 40 lung squamous cell carcinoma TMA cores staining for GPC3.Table S3. H-scores for 69 lung small cell carcinoma TMA cores staining for GPC3.Table S4. H-scores for 87 esophagus squamous cell carcinoma TMA cores staining for GPC3.Table S5. H-scores for 30 cardiac adenocarcinoma TMA cores staining for GPC3.Table S6. H-scores for 40 gastric cancer TMA cores staining for GPC3.Table S7. H-scores for 68 head and neck cancer TMA cores staining for GPC3.Table S8. H-scores for 30 different normal TMA cores staining for GPC3.Table S9. Histopathological analysis of Hepa1-6/hGPC3 and LLC1/hGPC3 tumors.Table S10. Primary dataReferences (52, 53)
REFERENCES AND NOTES1. M. A. Postow, M. K. Callahan, J. D. Wolchok, Immune checkpoint blockade in cancer
therapy. J. Clin. Oncol. 33, 1974–1982 (2015).
2. I. Márquez-Rodas, P. Cerezuela, A. Soria, A. Berrocal, A. Riso, M. González-Cao,S. Martín-Algarra, Immune checkpoint inhibitors: Therapeutic advances in melanoma.Ann. Transl. Med. 3, 267 (2015).
3. F. S. Hodi, S. J. O’Day, D. F. McDermott, R. W. Weber, J. A. Sosman, J. B. Haanen,R. Gonzalez, C. Robert, D. Schadendorf, J. C. Hassel, W. Akerley, A. J. M. van den Eertwegh,J. Lutzky, P. Lorigan, J. M. Vaubel, G. P. Linette, D. Hogg, C. H. Ottensmeier, C. Lebbé,C. Peschel, I. Quirt, J. I. Clark, J. D. Wolchok, J. S. Weber, J. Tian, M. J. Yellin, G. M. Nichol,A. Hoos, W. J. Urba, Improved survival with ipilimumab in patients with metastaticmelanoma. N. Engl. J. Med. 363, 711–723 (2010).
4. S. L. Topalian, F. S. Hodi, J. R. Brahmer, S. N. Gettinger, D. C. Smith, D. F. McDermott,J. D. Powderly, R. D. Carvajal, J. A. Sosman, M. B. Atkins, P. D. Leming, D. R. Spigel,S. J. Antonia, L. Horn, C. G. Drake, D. M. Pardoll, L. Chen, W. H. Sharfman,R. A. Anders, J. M. Taube, T. L. McMiller, H. Xu, A. J. Korman, M. Jure-Kunkel,S. Agrawal, D. McDonald, G. D. Kollia, A. Gupta, J. M. Wigginton, M. Sznol, Safety,activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 366,2443–2454 (2012).
5. T. N. Schumacher, R. D. Schreiber, Neoantigens in cancer immunotherapy. Science 348,69–74 (2015).
6. N. A. Rizvi, M. D. Hellmann, A. Snyder, P. Kvistborg, V. Makarov, J. J. Havel, W. Lee,J. Yuan, P. Wong, T. S. Ho, M. L. Miller, N. Rekhtman, A. L. Moreira, F. Ibrahim,C. Bruggeman, B. Gasmi, R. Zappasodi, Y. Maeda, C. Sander, E. B. Garon, T. Merghoub,J. D. Wolchok, T. N. Schumacher, T. A. Chan, Mutational landscape determinessensitivity to PD-1 blockade in non–small cell lung cancer. Science 348, 124–128(2015).
7. P. Sharma, J. P. Allison, The future of immune checkpoint therapy. Science 348, 56–61(2015).
8. G. Riethmüller, Symmetry breaking: Bispecific antibodies, the beginnings, and 50 yearson. Cancer Immun. 12, 12 (2012).
9. S. R. Frankel, P. A. Baeuerle, Targeting T cells to tumor cells using bispecific antibodies.Curr. Opin. Chem. Biol. 17, 385–392 (2013).
10. P. A. Baeuerle, C. Reinhardt, Bispecific T-cell engaging antibodies for cancer therapy.Cancer Res. 69, 4941–4944 (2009).
11. B. D. Choi, P. C. Gedeon, J. E. Herndon II, G. E. Archer, E. A. Reap, L. Sanchez-Perez,D. A. Mitchell, D. D. Bigner, J. H. Sampson, Human regulatory T cells kill tumor cellsthrough granzyme-dependent cytotoxicity upon retargeting with a bispecific antibody.Cancer Immunol. Res. 1, 163 (2013).
12. R. Bargou, E. Leo, G. Zugmaier, M. Klinger, M. Goebeler, S. Knop, R. Noppeney, A. Viardot,G. Hess, M. Schuler, H. Einsele, C. Brandl, A. Wolf, P. Kirchinger, P. Klappers, M. Schmidt,G. Riethmüller, C. Reinhardt, P. A. Baeuerle, P. Kufer, Tumor regression in cancer patients byvery low doses of a T cell–engaging antibody. Science 321, 974–977 (2008).
13. M. S. Topp, N. Gökbuget, A. S. Stein, G. Zugmaier, S. O’Brien, R. C. Bargou, H. Dombret,A. K. Fielding, L. Heffner, R. A. Larson, S. Neumann, R. Foà, M. Litzow, J.-M. Ribera,A. Rambaldi, G. Schiller, M. Brüggemann, H. A. Horst, C. Holland, C. Jia, T. Maniar, B. Huber,D. Nagorsen, S. J. Forman, H. M. Kantarjian, Safety and activity of blinatumomab foradult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia:A multicentre, single-arm, phase 2 study. Lancet Oncol. 16, 57–66 (2015).
14. L. L. Sun, D. Ellerman, M. Mathieu, M. Hristopoulos, X. Chen, Y. Li, X. Yan, R. Clark,A. Reyes, E. Stefanich, E. Mai, J. Young, C. Johnson, M. Huseni, X. Wang, Y. Chen,P. Wang, H. Wang, N. Dybdal, Y.-W. Chu, N. Chiorazzi, J. M. Scheer, T. Junttila, K. Totpal,
11 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on June 17, 2020http://stm
.sciencemag.org/
Dow
nloaded from
M. S. Dennis, A. J. Ebens, Anti-CD20/CD3 T cell–dependent bispecific antibody forthe treatment of B cell malignancies. Sci. Transl. Med. 7, 287ra70 (2015).
15. G. R. Chichili, L. Huang, H. Li, S. Burke, L. He, Q. Tang, L. Jin, S. Gorlatov, V. Ciccarone,F. Chen, S. Koenig, M. Shannon, R. Alderson, P. A. Moore, S. Johnson, E. Bonvini, ACD3xCD123 bispecific DART for redirecting host T cells to myelogenous leukemia:Preclinical activity and safety in nonhuman primates. Sci. Transl. Med. 7, 289ra82 (2015).
16. K. Brischwein, B. Schlereth, B. Guller, C. Steiger, A. Wolf, R. Lutterbuese, S. Offner,M. Locher, T. Urbig, T. Raum, P. Kleindienst, P. Wimberger, R. Kimmig, I. Fichtner, P. Kufer,R. Hofmeister, A. J. da Silva, P. A. Baeuerle, MT110: A novel bispecific single-chainantibody construct with high efficacy in eradicating established tumors. Mol. Immunol.43, 1129–1143 (2006).
17. R. Lutterbuese, T. Raum, R. Kischel, P. Lutterbuese, B. Schlereth, E. Schaller,S. Mangold, D. Rau, P. Meier, P. A. Kiener, K. Mulgrew, M. D. Oberst, S. A. Hammond,P. A. Baeuerle, P. Kufer, Potent control of tumor growth by CEA/CD3-bispecificsingle-chain antibody constructs that are not competitively inhibited by soluble CEA.J. Immunother. 32, 341–352 (2009).
18. M. Friedrich, T. Raum, R. Lutterbuese, M. Voelkel, P. Deegen, D. Rau, R. Kischel,P. Hoffmann, C. Brandl, J. Schuhmacher, P. Mueller, R. Finnern, M. Fuergut, D. Zopf,J. W. Slootstra, P. A. Baeuerle, B. Rattel, P. Kufer, Regression of human prostatecancer xenografts in mice by AMG 212/BAY2010112, a novel PSMA/CD3-BispecificBiTE antibody cross-reactive with non-human primate antigens. Mol. Cancer Ther.11, 2664–2673 (2012).
19. M. Klinger, J. Benjamin, R. Kischel, S. Stienen, G. Zugmaier, Harnessing T cells to fightcancer with BiTE® antibody constructs—Past developments and future directions.Immunol. Rev. 270, 193–208 (2016).
20. R. Lutterbuese, T. Raum, R. Kischel, P. Hoffmann, S. Mangold, B. Rattel, M. Friedrich,O. Thomas, G. Lorenczewski, D. Rau, E. Schaller, I. Herrmann, A. Wolf, T. Urbig,P. A. Baeuerle, P. Kufer, T cell-engaging BiTE antibodies specific for EGFR potentlyeliminate KRAS- and BRAF-mutated colorectal cancer cells. Proc. Natl. Acad. Sci. U.S.A.107, 12605–12610 (2010).
21. J. Filmus, S. B. Selleck, Glypicans: Proteoglycans with a surprise. J. Clin. Invest. 108,497–501 (2001).
22. A. D. Gonzalez, M. Kaya, W. Shi, H. Song, J. R. Testa, L. Z. Penn, J. Filmus, OCI-5/GPC3, aglypican encoded by a gene that is mutated in the Simpson-Golabi-Behmel overgrowthsyndrome, induces apoptosis in a cell line–specific manner. J. Cell Biol. 141, 1407–1414 (1998).
23. S. Paine-Saunders, B. L. Viviano, J. Zupicich, W. C. Skarnes, S. Saunders, glypican-3controls cellular responses to Bmp4 in limb patterning and skeletal development.Dev. Biol. 225, 179–187 (2000).
24. S. Grisaru, D. Cano-Gauci, J. Tee, J. Filmus, N. D. Rosenblum, Glypican-3 modulatesBMP- and FGF-mediated effects during renal branching morphogenesis. Dev. Biol. 231,31–46 (2001).
25. S. Khan, M. Blackburnz, D. L. Mao, R. Hube, D. Schlessinger, M. Fant, Glypican-3 (GPC3)expression in human placenta: Localization to the differentiated syncytiotrophoblast.Histol. Histopathol. 16, 71–78 (2001).
26. B. V. Iglesias, G. Centeno, H. Pascuccelli, F. Ward, M. G. Peters, J. Filmus, L. Puricelli,E. B. de Kier Joffé, Expression pattern of glypican-3 (GPC3) during human embryonic andfetal development. Histol. Histopathol. 23, 1333–1340 (2008).
27. T. Nakatsura, Y. Nishimura, Usefulness of the novel oncofetal antigen glypican-3 fordiagnosis of hepatocellular carcinoma and melanoma. BioDrugs 19, 71–77 (2005).
28. Z.-W. Zhu, H. Friess, L. Wang, M. Abou-Shady, A. Zimmermann, A. D. Lander,M. Korc, J. Kleeff, M. W. Büchler, Enhanced glypican-3 expression differentiates themajority of hepatocellular carcinomas from benign hepatic disorders. Gut 48,558–564 (2001).
29. N. Yamauchi, A. Watanabe, M. Hishinuma, K.-i. Ohashi, Y. Midorikawa, Y. Morishita, T. Niki,J. Shibahara, M. Mori, M. Makuuchi, Y. Hippo, T. Kodama, H. Iwanari, H. Aburatani,M. Fukayama, The glypican 3 oncofetal protein is a promising diagnostic marker forhepatocellular carcinoma. Mod. Pathol. 18, 1591–1598 (2005).
30. Q. Lin, L.-W. Xiong, X.-F. Pan, J.-F. Gen, G.-L. Bao, H.-F. Sha, J.-X. Feng, C.-Y. Ji, M. Chen,Expression of GPC3 protein and its significance in lung squamous cell carcinoma.Med. Oncol. 29, 663–669 (2012).
31. T. Ushiku, H. Uozaki, A. Shinozaki, S. Ota, K. Matsuzaka, S. Nomura, M. Kaminishi,H. Aburatani, T. Kodama, M. Fukayama, Glypican 3-expressing gastric carcinoma: Distinctsubgroup unifying hepatoid, clear-cell, and alpha-fetoprotein-producing gastriccarcinomas. Cancer Sci. 100, 626–632 (2009).
32. D. Maeda, S. Ota, Y. Takazawa, H. Aburatani, S. Nakagawa, T. Yano, Y. Taketani, T. Kodama,M. Fukayama, Glypican-3 expression in clear cell adenocarcinoma of the ovary.Mod. Pathol. 22, 824–832 (2009).
33. T. Umezu, K. Shibata, H. Kajiyama, E. Yamamoto, A. Nawa, F. Kikkawa, Glypican-3expression predicts poor clinical outcome of patients with early-stage clear cellcarcinoma of the ovary. J. Clin. Pathol. 63, 962–966 (2010).
34. T. Mounajjed, L. Zhang, T. Wu, Glypican-3 expression in gastrointestinal and pancreaticepithelial neoplasms. Hum. Pathol. 44, 542–550 (2013).
Ishiguro et al., Sci. Transl. Med. 9, eaal4291 (2017) 4 October 2017
35. D. Baumhoer, L. Tornillo, S. Stadlmann, M. Roncalli, E. K. Diamantis, L. M. Terracciano,Glypican 3 expression in human nonneoplastic, preneoplastic, and neoplastic tissues: Atissue microarray analysis of 4,387 tissue samples. Am. J. Clin. Pathol. 129, 899–906(2008).
36. T. Ishiguro, M. Sugimoto, Y. Kinoshita, Y. Miyazaki, K. Nakano, H. Tsunoda, I. Sugo,I. Ohizumi, H. Aburatani, T. Hamakubo, T. Kodama, M. Tsuchiya, H. Yamada-Okabe, Anti–glypican 3 antibody as a potential antitumor agent for human liver cancer. Cancer Res.68, 9832–9838 (2008).
37. Z. Sampei, T. Igawa, T. Soeda, Y. Okuyama-Nishida, C. Moriyama, T. Wakabayashi,E. Tanaka, A. Muto, T. Kojima, T. Kitazawa, K. Yoshihashi, A. Harada, M. Funaki,K. Haraya, T. Tachibana, S. Suzuki, K. Esaki, Y. Nabuchi, K. Hattori, Identification andmultidimensional optimization of an asymmetric bispecific IgG antibody mimicking thefunction of factor VIII cofactor activity. PLOS ONE 8, e57479 (2013).
38. K. Nakano, T. Orita, J. Nezu, T. Yoshino, I. Ohizumi, M. Sugimoto, K. Furugaki, Y. Kinoshita,T. Ishiguro, T. Hamakubo, T. Kodama, H. Aburatani, H. Yamada-Okabe, M. Tsuchiya,Anti-glypican 3 antibodies cause ADCC against human hepatocellular carcinoma cells.Biochem. Biophys. Res. Commun. 378, 279–284 (2009).
39. T. Kuramochi, T. Igawa, H. Tsunoda, K. Hattori, Humanization and simultaneousoptimization of monoclonal antibody. Methods Mol. Biol. 1060, 123–137 (2014).
40. K. Gunasekaran, M. Pentony, M. Shen, L. Garrett, C. Forte, A. Woodward, S. B. Ng, T. Born,M. Retter, K. Manchulenko, H. Sweet, I. N. Foltz, M. Wittekind, W. Yan, Enhancingantibody Fc heterodimer formation through electrostatic steering effects:Applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 285,19637–19646 (2010).
41. O. Ueda, N. A. Wada, Y. Kinoshita, H. Hino, M. Kakefuda, T. Ito, E. Fujii, M. Noguchi, K. Sato,M. Morita, H. Tateishi, K. Matsumoto, C. Goto, Y. Kawase, A. Kato, K. Hattori, J. Nezu,T. Ishiguro, K.-i. Jishage, Entire CD3e, d, and g humanized mouse to evaluate humanCD3-mediated therapeutics. Sci. Rep. 7, 45839 (2017).
42. M. G. Lechner, S. S. Karimi, K. Barry-Holson, T. E. Angell, K. A. Murphy, C. H. Church,J. R. Ohlfest, P. Hu, A. L. Epstein, Immunogenicity of murine solid tumor models as adefining feature of in vivo behavior and response to immunotherapy. J. Immunother. 36,477–489 (2013).
43. B. Davies, T. Morris, Physiological parameters in laboratory animals and humans. Pharm.Res. 10, 1093–1095 (1993).
44. R. Deng, S. Iyer, F.-P. Theil, D. L. Mortensen, P. J. Fielder, S. Prabhu, Projecting humanpharmacokinetics of therapeutic antibodies from nonclinical data: What have welearned? MAbs 3, 61–66 (2011).
45. C. H. Chung, Managing premedications and the risk for reactions to infusionalmonoclonal antibody therapy. Oncologist 13, 725–732 (2008).
46. H. Gao, K. Li, H. Tu, X. Pan, H. Jiang, B. Shi, J. Kong, H. Wang, S. Yang, J. Gu, Z. Li,Development of T cells redirected to glypican-3 for the treatment of hepatocellularcarcinoma. Clin. Cancer Res. 20, 6418–6428 (2014).
47. S. S. Hoseini, N.-K. V. Cheung, Immunotherapy of hepatocellular carcinoma using chimericantigen receptors and bispecific antibodies. Cancer Lett. 399, 44–52 (2017).
48. M.-E. Goebeler, R. Bargou, Blinatumomab: A CD19/CD3 bispecific T cell engager (BiTE)with unique anti-tumor efficacy. Leuk. Lymphoma 57, 1021–1032 (2016).
49. C. Brandl, C. Haas, S. d’Argouges, T. Fisch, P. Kufer, K. Brischwein, N. Prang, R. Bargou,J. Suzich, P. A. Baeuerle, R. Hofmeister, The effect of dexamethasone on polyclonal T cellactivation and redirected target cell lysis as induced by a CD19/CD3-bispecificsingle-chain antibody construct. Cancer Immunol. Immunother. 56, 1551–1563 (2007).
50. J. Feucht, S. Kayser, D. Gorodezki, M. Hamieh, M. Döring, F. Blaeschke, P. Schlegel,H. Bösmüller, L. Quintanilla-Fend, M. Ebinger, P. Lang, R. Handgretinger, T. Feuchtinger,T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated bybispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 onleukemic blasts. Oncotarget 22, 6902–76919 (2016).
51. T. T. Junttila, J. Li, J. Johnston, M. Hristopoulos, R. Clark, D. Ellerman, B.-E. Wang, Y. Li,M. Mathieu, G. Li, J. Young, E. Luis, G. L. Phillips, E. Stefanich, C. Spiess, A. Polson,B. Irving, J. M. Scheer, M. R. Junttila, M. S. Dennis, R. Kelley, K. Totpal, A. Ebens,Antitumor efficacy of a bispecific antibody that targets HER2 and activates T cells.Cancer Res. 74, 5561–5571 (2014).
52. Y. Sato, K. Mukai, S. Watanabe, M. Goto, Y. Shimosato, The AMeX method. A simplifiedtechnique of tissue processing and paraffin embedding with improved preservationof antigens for immunostaining. Am. J. Pathol. 125, 431–435 (1986).
53. M. Suzuki, K. Katsuyama, K. Adachi, Y. Ogawa, K. Yorozu, E. Fujii, Y. Misawa, T. Sugimoto,Combination of fixation using PLP fixative and embedding in paraffin by the AMeXmethod is useful for histochemical studies in assessment of immunotoxicity. J. Toxicol.Sci. 27, 165–172 (2002).
Acknowledgments:We thank the donors and patients who consented to the use of their cellsfor the studies. We also thank N. Hironiwa, T. Kuramochi, and K. Esaki for their help withantibody generation and characterization; N. Ikeda and A. Kato for pharmacological
12 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
evaluation; O. Kondoh for facilitating the pharmacology studies; O. Ueda, N. A. Wada, H. Hino,and K. Jishage for generating the human CD3 transgenic mice; A. Shioda and S. Akai forthe safety evaluation; and S. Matsuura for review of the manuscript. Funding: This study wasfunded and supported by Chugai Pharmaceutical Co., Ltd. Author contributions: T. Ishiguro,Y. Sano, S.i.-K., and J.N. wrote the manuscript; T. Ishiguro, Y. Sano, Y. Kinoshita, Y.A., T. Tsunenari,N.O., Y. Kayukawa, Y. Sonobe, K.S., T.F., Y.M., M.N., M.E., E.F., H.M., Y.N., M.T., Y. Kawabe, andM.A. executed the pharmacological studies and analyzed the assay data; S.i.-K., A.K., A.H., W.F.,E.N., M.I., and S.C. executed the toxicological and PK studies and analyzed the assay data;L.S. and E.E. executed the GPC3 IHC studies and analyzed the assay data; M.K.-S., H.S., A.N., A.S.,T.W., H.K., H.S., T. Tsushima, T. Igawa, K.H., and J.N. designed, produced, and characterized theantibodies; and T. Ishiguro, Y. Sano, S.i.-K., A.K., H.S., and M.K.-S. designed and supervised thestudies. Competing interests: Chugai Pharmaceutical Co., Ltd. is developing ERY974 as a clinicalcompound. T. Ishiguro, Y. Sano, S.i.-K., M.K.-S., A.K., Y. Kinoshita, H.S., Y.A., T. Tsunenari, N.O.,Y. Kayukawa, Y. Sonobe, K.S., T.F., Y.M., M.N., M.E., Y. Kawabe, A.H., W.F., E.F., E.N., A.N., A.S.,M.T., T. Tsushima, T.W., H.K., H.S., H.M., Y.N., T. Igawa, M.I., S.C., M.A., K.H., and J.N. areemployees of Chugai Pharmaceutical Co., Ltd., and L.S. and E.E. are employees of VentanaMedical Systems Inc..; Chugai Pharmaceutical Co., Ltd. has filed patent applications related tothis work for ERY974 and TRAB. T. Ishiguro, M.K., H.S., Y.A., A.N., T. Igawa, and J.N. are inventors
Ishiguro et al., Sci. Transl. Med. 9, eaal4291 (2017) 4 October 2017
on patent application (WO/2016/047722) submitted by Chugai Pharmaceutical Co., Ltd. thatcovers the ERY794 molecule. Data and materials availability: Materials are available fromChugai Pharmaceutical Co., Ltd. under a material transfer agreement.
Submitted 26 November 2016Resubmitted 26 May 2017Accepted 23 August 2017Published 4 October 201710.1126/scitranslmed.aal4291
Citation: T. Ishiguro, Y. Sano, S.-i. Komatsu, M. Kamata-Sakurai, A. Kaneko, Y. Kinoshita, H. Shiraiwa,Y. Azuma, T. Tsunenari, Y. Kayukawa, Y. Sonobe, N. Ono, K. Sakata, T. Fujii, Y. Miyazaki, M. Noguchi,M. Endo, A. Harada, W. Frings, E. Fujii, E. Nanba, A. Narita, A. Sakamoto, T. Wakabayashi, H. Konishi,H. Segawa, T. Igawa, T. Tsushima, H. Mutoh, Y. Nishito, M. Takahashi, L. Stewart, E. ElGabry,Y. Kawabe, M. Ishigai, S. Chiba, M. Aoki, K. Hattori, J. Nezu, An anti–glypican 3/CD3bispecific T cell–redirecting antibody for treatment of solid tumors. Sci. Transl. Med. 9,eaal4291 (2017).
13 of 13
by guest on June 17, 2020http://stm
.sciencemag.org/
Dow
nloaded from

tumorsredirecting antibody for treatment of solid−glypican 3/CD3 bispecific T cell−An anti
Ishigai, Shuichi Chiba, Masahiro Aoki, Kunihiro Hattori and Junichi NezuTsushima, Hironori Mutoh, Yukari Nishito, Mina Takahashi, Lorraine Stewart, Ehab ElGabry, Yoshiki Kawabe, MasakiAtsushi Narita, Akihisa Sakamoto, Tetsuya Wakabayashi, Hiroko Konishi, Hiroaki Segawa, Tomoyuki Igawa, Takashi Toshihiko Fujii, Yoko Miyazaki, Mizuho Noguchi, Mika Endo, Asako Harada, Werner Frings, Etsuko Fujii, Eitaro Nanba,Shiraiwa, Yumiko Azuma, Toshiaki Tsunenari, Yoko Kayukawa, Yukiko Sonobe, Natsuki Ono, Kiyoaki Sakata, Takahiro Ishiguro, Yuji Sano, Shun-ichiro Komatsu, Mika Kamata-Sakurai, Akihisa Kaneko, Yasuko Kinoshita, Hirotake
DOI: 10.1126/scitranslmed.aal4291, eaal4291.9Sci Transl Med
further development of this antibody for therapeutic use in multiple cancer types.was quite large. Treatment also appeared to be safe when administered to monkeys. These results suggestantibody was effective in a variety of mouse cancer models, even when treatment was initiated after the tumor
bispecifictesting of a bispecific antibody recognizing CD3 and glypican 3, a common antigen on solid tumors. This . describe the development and preclinicalet alcell therapy, or in vivo, such as with bispecific antibodies. Ishiguro
designed to redirect T cells to tumor cells. This can be done by engineering the cells ex vivo, such as in CAR T Because the endogenous immune response is not enough to clear a patient's cancer, therapies are being
Double trouble for solid tumors
ARTICLE TOOLS http://stm.sciencemag.org/content/9/410/eaal4291
MATERIALSSUPPLEMENTARY http://stm.sciencemag.org/content/suppl/2017/10/02/9.410.eaal4291.DC1
CONTENTRELATED
http://stm.sciencemag.org/content/scitransmed/12/534/eaax1315.fullhttp://stm.sciencemag.org/content/scitransmed/10/424/eaan5488.fullhttp://stm.sciencemag.org/content/scitransmed/9/399/eaaa0984.fullhttp://stm.sciencemag.org/content/scitransmed/9/376/eaak9537.fullhttp://stm.sciencemag.org/content/scitransmed/8/320/320ra4.full
REFERENCES
http://stm.sciencemag.org/content/9/410/eaal4291#BIBLThis article cites 53 articles, 20 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
registered trademark of AAAS. is aScience Translational MedicineScience, 1200 New York Avenue NW, Washington, DC 20005. The title
(ISSN 1946-6242) is published by the American Association for the Advancement ofScience Translational Medicine
of Science. No claim to original U.S. Government WorksCopyright © 2017 The Authors, some rights reserved; exclusive licensee American Association for the Advancement
by guest on June 17, 2020http://stm
.sciencemag.org/
Dow
nloaded from





























