Embed Size (px)
Citation preview

A N A D V A N C E D
L A B O R A T O R Y M A N U A L
o r
O R G A N I C C H E M I S T R Y
BY
MICHAEL H E I D E L B E R G E R , B.S., A.M., P H . D .ASSOCIATE IX CItEMIBTnV, IIOCKEFEIJJ-n INSTITUTE
TOIt MEDICAL IlEflEARCH
BOOK DEPABTMENT
The CHEMICAL CATALOG COMPANY, Inc.19 EAST 24TH STREET, NEW YORK, U. S. A.
1923

COPYRIGHT, 1923, BY
The CHEMICAL CATALOG COMPANY, Ino.
All Bighta Reserved
Press ofJ, J Uttte & Ivos Company
HOT York, U. S. L.

TO
N. T . H.


P R E F A C E .
In the field of organic chemistry there are a numberof elementary laboratory manuals, any one of whichmay be used to the student's advantage. When itcomes to the choice of a guide for an advanced course,however, there is a vast amount of material availablefrom which a selection in the form of a laboratorymanual has never been made. Hence the student isoften permitted to follow some line in which he isinterested, regardless of its practicability or its valuefrom the standpoint of training, or else the planningof the experiments devolves entirely upon the instructor.
With the object of providing a brief advanced coursein manipulative organic chemistry embodying experi-ments scattered as widely as possible over the importanttypes of substances and reactions, the author desiresto present this little book in the hope of renderingsimpler the task both of the advanced student and hisinstructor.
It has been the writer's aim to select experiments ofgreater difficulty than those ordinarily included m ele-mentary manuals, but to avoid preparations of so diffi-cult or involved a nature as to become a source of dis-couragement rather than a stimulus to the student. Inthis connection a word of apology may be necessaryfor including as much as has been done of the workof Dr. Walter A. Jacobs of the Rockefeller Instituteand the writer, but it is very strongly felt that the valueof a volume such as the present one depends largely onthe personal experience of its author, and for this rea-
5

6 -PREFACE
son the writer has drawn freely on his own work anthat -of his colleagues. It has been attempted also 1preserve as just a balance as possible between the chenistry of aliphatic and aromatic compounds, and to uelude products of technical and biological, as well \theoretical importance, in order to provide as broadfoundation as possible for the student in his futuiwork. In the selection of experiments, care has beetaken to exclude those involving great expense, andfurther economy is effected by the use of many of tlinitial products as steps in the synthesis of others.
Finally, the author takes great pleasure in acknowedging his indebtedness to his colleagues at the Rockfeller Institute for Medical Research for the use <their records in individual experiments, to Prof Maston T Bogert of Columbia University for some vepertinent suggestions, and to Prof John 'M Nelson <Columbia University, whose encouragement and helful advice stimulated the writer to the preparation <this manual
MICHAEL HEIDELBERGER.
New York City, December, 1922.

TABLE OF CONTENTS
PAGE
PREFACE 5
INTRODUCTORY WARNING n
CHAPTER I. NITRATION AND NITROSATION . 13
A. c^Nitraniline 13B. />-Nitroso-0-cresol 16
CHAPTER II. HALOGENATION 18
A. Chloroacetone 18B. dZ-a-Bromopropionic acid . . . 20C. 5-Iodo-2-toluidine 21
CHAPTER III . SUBSTITUTIONS 23
A. Ethylene cyanohydrin 23(3-Chloropropionic acid 23P-Bromopropionic acid 24
B. Benzylamine 24C. w-Aminophenol 28D. 0-Nitrophenylarsonic acid . 29
CHAPTER IV. ESTERIFICATION, ETHERIFICA-TION, DE-AUCYLATION, AND RELATED REAC-TIONS 32
A. Methyl Anthranilate 32B. p-Nitrophenoxyacetic acid . . . . 33
7

8 TABLE OF CONTENTS
PAGEC. w-Phenetidine 34D. Allyl Phenyl Ether and Its Molecular
Rearrangements . . . . 36
1. Allyl bromide . . 362. Allyl phenyl ether . . . 373. o-Allylphenol . . - 3 74. 0-Propenylpheftol . . • 395 a-Methylcoumarane . . . . 39
E. Dihydrocupreine . . . . . . 41
CHAPTER V REDUCTION . . . . - 4 5
A. With stannous chloride- p-Ammodi-methylamline . . . . . . 45
B. With ferrous sulfate and ammonia-/>-Aminophenoxyacetic acid . . 46
C. With sodium amalgam dl-1 -Phenyl-1-hydroxy-2-aminoethane . . 4 8
D. With palladium black. Dihydroqumine 50
CHAPTER VI. OXIDATION . . . . , . 54
A. With potassium f erricyanide: jt»-Nitro-o-cresol . 54
B. With nitrites: Isonitrosoc&gaphor andCampj^rqumone . . . . 55
C. With aft^ospherk oxygen: Camphoric
acid 58
D. With bromine • Calcium gluconate . . 59
E. With hydrogen peroxide: rf-Arabinose 61

TABLE OF CONTENTS' g
PJ.QHCHAPTER VII. FORMATION OF HETEROCYCLES
AND DYES . . 64
A. Diethylmalonic ester 64Diethylbarbitunc acid, "Veronal," "Bar-
bital" 65B. 9-Methylacndme . 66C Quinicine (quinotoxine) hydrochloride 6jD Meldola's blue 70E Rosindulme . . . . . . 71
Rosindone 73
CHAPTER VIII. SUGARS, PROTEINS, AND AMINO-
ACIDS 74
A. [3-Glucose . . . 74(3-Glucose penta-acetate . . . 75
B. Hydrolysis of a Biose- Galactose from
lactose . . . . . 76
C. Preparation of d/-alanine . . . 78
D. Separation of dZ-alanine into its opticalisomers . . 791. Benzoylation of alanine . . . 802. /-Benzoylalanine . . . . 803. Z-Alanine 814. d-Alanine 82
E. Crystalline egg albumin 83
CHAPTER IX. PREPARATION AND REACTIONS OF
ORGANOMETALLIC COMPOUNDS . . . . 88
A, Direct arsenatipn of phenol. p-Hydroxy-pftenylarsonic acid 88

i o TABLE OF CONTENTS
PAGEB. Synthesis of Salvarsan (Arsphena-
mme), 3,3'-diammo-4,4'-dihydroxyar-senobenzene dihydrochlonde . . . 92
1. 3-Nitro-4-hydroxyphenylarsomcacid 92
2 Reduction of the nitro acid . . 93C. Mercuric compounds of aniline . . 96
1. />-Aminophenylmercunc acetate gt2 />-Mercuri-&«-anilme . . . 9^

INTRODUCTORY WARNING.
The student will remember from his elementarycourse that many organic reactions, harmless under con-trolled conditions, may gather speed and violence if notcarefully watched. Every experiment should thereforebe considered as a whole from the point of view of itspotential sources of danger, and a plan of proceduremapped out accordingly.
If the reaction is accompanied by a rise of tempera-ture, even if no minimum is specified m the directions,accidents may often be prevented by keeping a pot ofice water or freezing mixture at hand, into which thevessel may be plunged in time to prevent boiling over ordecomposition. If gases such as hydrobromic acid, forexample, are evolved, the reaction should be carried outunder the hood, and the vapors led into a flask of waterby a tube terminating above the surface.
Many a weary repetition may also be avoided bykeeping in pots as much as possible large flasks orbeakers containing material on which much time hasbeen expended.
It should also be kept in mind that most organiccompounds are more or less toxic and many are ex-tremely dangerous. Distillations other than underdiminished pressure should therefore be carried outunder the hood, and care should be taken to avoid con-tact of the substances handled with the skin, or inhala-tion of their vapors or dusts. The writer still has vivid
ii

12 INTRODUCTORY WARNING
recollections of several extremely uncomfortable daysmany years ago as a result of getting minute traces of
/ \ C H B r 2
co-tetrabromo-o-xylene, [ j , on his finger tips\ / C H B r 2
and thence indirectly on his face and into his eyes.When working therefore with substances which areknown to be irritating the student will find it advisableto wear rubber gloves. Dried crystalline material orpowders should also be transferred under the hood, aprecaution which it is particularly unsafe to overlook inthe case of the alkaloid, arsenic, and mercury deriva-tives of which the preparation is described in the fol-lowing pages.
The student will also frequently handle highly in-flammable solvents, and must therefore remember thatmany painful and even fatal accidents have resultedfrom working with these near a flame or electric switch.
While there need be no occasion for timidity, it mustbe borne m mind that constant vigilance and concen-tration are the price that must be paid for the joys, thesatisfaction, and the thrills that come to those who workin organic chemistry.

A N A D V A N C E D L A B O R A T O R Y
M A N U A L O F O R G A N I C
C H E M I S T R Y
I. NITRATION AND NITROSATION.
(See also p. 92)
A. Nitration.
o-Nitraniline, o-O2NCaH4NHfl.In the nitration of benzene, it will be remembered,
only one mononitro compound was capable of forma-tion, and the by-product occurring in the reaction wasw-dinitrobenzene:
NO2 NO 2
If one substituent ia already present in the benzenenucleus,1 however, the case becomes more complicated,and using aniline or its acetyl derivative as an example,three fsotineric monotiitro derivatives are theoreticallypossible c
13

14 ADVANCED LABORATORY MANUAL
NHCOCH3
|NO2
o-Nitro-acetamlide.
NHCOCH3
w-Nitro-acetamlide.JNO,
NHCOCHa
p-Nitro-acetanilide.
NO2
The relative amounts of the position isomers formedvary within wide limits according to the conditionsused, for if one nitrates aniline itself in cold, concen-trated sulfunc acid a relatively large proportion ofmetQr and para- mtranilines are the chief products,while if acetanilide is nitrated in the same solvent thepara- nitro derivative is obtained almost exclusively.If the nitration is carried out in an excess of fumingnitric acid the main product is again the para- com-pound, with only 6 to 8 per cent of o-nitramline. Wittand Utermann1 found, however, that by carrying outthe nitration in glacial acetic acid in the presence ofacetic anhydride as dehydrating agent, about 75 percent of the acetanilide nitrated was converted into theortho- compound, the remainder being p-nitro-acetan-llide. By this method large amounts of o-nitran-
1Ber. 39, 3901 (1906), 41, 3090 (1908).

OF ORGANIC CHEMISTRY 15
ihne can readily be prepared. Batches of the sizegiven below may be conveniently handled in the labora-tory.
Nitration.90 g. of acetanihde are dissolved by warming
gently in a mixture of 80 g of acetic anhydride and44 g. of glacial acetic acid, and the solution is thenchilled in ice-water. 50 g. of fummg nitric acid(d 1.52) are mixed with 46 g. of glacial acetic acid,also cooled, and added in small portions to the chilledacetanilide solution, from which a portion of theacetanihde may separate. The mixture is stirred wellwith a thermometer, adding the nitric acid solutionslowly enough to prevent the temperature from risingabove that of the room, and controlling the speed of thereaction by immersing in ice-water when necessary.When the tendency of the temperature to rise becomesvery slight the reaction mixture should be removedfrom the ice-water and allowed to stand 24 hours atroom temperature. Care should be taken by coolingoccasionally if necessary during the first hour or twothat the reaction does not become too vigorous. Thenext day the mixture is poured on to ice, stirred well,and the crude crystalline o-nitro-acetamhde filtered off,washed well with ice-cold water, and sucked as dry aspossible.
Separation of Isomers.In the meantime a mixture of one volume, of 50 per
cent aqueous potassium hydroxide, 4 volumes of water,and one volume of alcohol is prepared, cooled to o°,and the nitration product thoroughly rubbed up (inportions) in a chilled mortar with about 600 cc. of thesolution. The onitro-acetanilide dissolves, while thebare*- compound remains insoluble in the cold mixtureind is sucked off and washed with a little of the cold

16 ADVANCED LABORATORY MANUAL
solution, then with a little ice-cold water. After re-crystalhzation from water the yield is 20 g , melting at207 °.Saponification.
The filtrate and washings from the crude paro nitroderivative are now allowed to come to room tempera-ture, and on letting stand for 24 hours saponificationoccurs and pure o-nitraniline separates in long, orangered needles, the reaction being as follows:
o-CH8CONHC6H4NO2 -f H2O > CH8COOH +H N C H N O
just as acetamide, CH8CONH2, when warmed withdilute alkali, splits into acetic acid and ammonia. Afterwashing with ice-cold water the yield of o-nitraniline is30-40 g., melting at 71 50 .
B. Nitrosation.
OH
p-Nitroso-o-cresol, 1 j CHa.
NO
While the nitrosation of phenols occurs at least asreadily as that of tertiary aromatic amines -such asdimethylanilme, the conditions must be very carefullycontrolled owing to the ease with which most phenolsoxidize. For this reason a higher proportion of tarryby-products is formed and the yields are smaller thanin the case of the dialkylanilines. Taking o-cresol asa typical example of a phenol with an unsubstitutedpara- position, the main product of the reaction is^-nitroso-o-cresol«

OF ORGANIC CHEMISTRY 17
54 g. of o-cresol are dissolved in 4 liters of ice-waterin a battery jar provided with an adequate mechanicalstirrer, and 34 5 g of 100 per cent sodium nitrite (or anequivalent amount of a less pure salt) are then added.Since nitrosation is effected by means of free nitrousacid and not its salts no reaction takes place at thispoint A solution of 18.5 cc. of concentrated sulfuricacid in 500 cc. of water is then added through adropping funnel during one-half to three-quarters ofan hour, keeping the temperature between 50 and io°by means of additional ice, and stirring continually.In this way the nitrous acid reacts with the cresol asfast as liberated to yield the ^-nitroso compound, andsince any local excess of nitrous acid is largely avoidedby the slow addition of the sulfuric acid and the efficientstirring, the formation of tarry by-products is reducedto a minimum While the nitroso compound mayseparate oily at first, it soon crystallizes. After stand-ing in the cold for one to two hours after the additionof the sulfuric acid the mixture is filtered on a largeBuchner funnel and washed with ice-cold water. Theresulting brown solid is purified by dissolving in 10per cent sodium carbonate solution, stirring with bone-black to collect insoluble tar, and filtering into an excessof dilute sulfuric acid. 40-45 g. of the nitroso com->pound should be obtained in this way as glistening,orange-brown scales melting at 1340.

18 ADVANCED LABORATORY MANUAL
I I . HALOGENATION.
A. Chlorination.
Chloroacetone, C1CH2COCH8If acetone be treated with a chlorinating agent such
as phosphorus pentachloride, the keto group is at-tacked and 2,2-dichloro-propane, CH8CClaCH8, results.If, however, elementary chlorine is used, the hydrogenatoms of the methyl groups are successively replaced.Not only is a mixture of mono- and poly-chlorinatedacetones formed, but the hydrochloric acid liberatedcondenses the acetone to products of higher molecularweight, of which mesityl oxide may be taken as anexample.
CH8 CH8> C O + H8CCOCH8 => > C .CHCOCHa.
CH8 CH8
Thus, unless some substance is at hand to bind thehydrochloric acid as fast as formed, exceedingly com-plex mixtures are obtained from which it is virtuallyimpossible to isolate pure products; mesityl oxide, forinstance, boiling at practically the same point as mono-chloroacetone. Fritsch1 found that small pieces ofmarble were very satisfactory, as these reacted at oncewith the hydrochloric acid liberated, and his methodis accordingly the basis of that given below.
M«« 279, 313 (1894).

OF ORGANIC CHEMISTRY 19
Chlorination.21 g. of marble, broken into small pieces, and 84 g.
of acetone are placed in a flask provided with an inlettube, dropping funnel, and reflux condenser, andwarmed to 400 in a water bath. A slow stream ofchlorine is then passed in and enough water (a totalof 50 to 60 cc. slowly dripped in to keep in solu-tion the calcium chloride formed by interaction ofthe marble and hydrochloric acid This is also aidedby frequent agitation of the flask. The reaction mustbe very carefully watched, for if a yellow color develops(and according to Khng2 this usually happens at thelower reaction temperature originally given by Fritsch)it indicates the formation of hypochlorous acid, andthis, if it accumulates, may react explosively with theacetone In the event, then, that the solution turnsyellow the stream of chlorine is at once interrupteduntil the coloration disappears. When only a littlemarble is left, the reaction is discontinued, for althougha large excess of acetone is present the main productwould be the symmetrical dichloro derivative,C1CH2COCH2C1, if this excess were not maintained.The mixture is allowed to stand at 400 until the evolu-tion of carbon dioxide ceases, making sure that an ex-cess of marble is present, and is then poured off from themarble into a separatory funnel. The two layers formedare separated and the lower, consisting of a strongaqueous solution of calcium chloride, is discarded.Fractionation.
The upper layer of acetone and its chlorinationproducts is fractionated with the aid of a good distill-ing column, the monochloroacetone boiling at 118-200.The yield is 16.8 g., plus an additional 5 g. on refrac-tionation of the lower and upper fractions.
'Bull, soc chvm. [3] 33, 32a (1905).

20 ADVANCED LABORATORY MANUAL
The chloro-acetones are extremely irritating, both invapor form and if dropped on the skin. Gloves shouldbe worn and all operations conducted under the hood.This applies to the next experiment as well.
B . Bromination.
di-a-Bromopropionic Acid, CH8CHBrCO2H.The method used depends upon the fact that although
acetic acid and its homologs react with difficulty withbromine, the anhydrides and acid bromides readilyyield bromo substitution products.8
Acid Bromide.To 50 g. of propionic acid and 7.6 g. of dry amor-
phous phosphorus are added, drop by drop, 66 7 g. ofbromine, at about which point evolution of hydro-bromic acid ceases. Through the intermediate forma-tion of phosphorus bromides the acid is converted intothe bromide according to the equation (Zehnsky):
4CH8CH2COaH + P + 5Br >4CH8CHsCOBr + P O ( O H ) 8 + HBr.
Bromo Acid Bromide.In order now that substitution should take place it is
unnecessary to isolate the acid bromide—-the mixtureis simply warmed to 40-50 ° on the water bath, usinga reflux condenser, and an additional ip$ & ef bromineis added drop by drop, the reaction b e | % :CHaCH3COBr + BrB — » CH8CHBrCGBr + HBr.Bromination proceeds rapidly and may be consideredcomplete two Ijoursi after all of the bromine has beenadded, Bromoprogiony! 'bromide boils at 154° under
, Ber. 14, api (1881); VbiSiard, Arm 242, 141 (1887);, Ber. 20. 2036 (18S7) ; We&fe, Ann. 280, 247 (1894).

OF ORGANIC CHEMISTRY 2\
atmospheric pressure, but distillation of the mixture isthen difficult, and it is accordingly purified by distilla-tion in a moderate vacuum. The yield is 75-80 percent of the theory.Bromo Acid.
If an ester of a-bromopropionic acid were desiredthe appropriate alcohol would now be used, but as theacid itself is required, the bromide is decomposed byaddition of one and one-third equivalents of water, themixture being shaken under a reflux condenser, with apot of ice close at hand, until homogeneous, and finallywarmed for one-half hour on the water-bath. Thesolution is then cooled, treated with several volumesof ether, dried over sodium sulfate, and concentrated.When fractionated in vacuo the residue boils mainly at124 ° under a pressure of 18-19 mm. and solidifies ina freezing mixture, then melting at about 25 °. Theacid should be protected from the moisture of the air,as it is quite hygroscopic. It is also a powerful skinirritant. The yield should be 60 per cent of the bro-mide used.
C. Iodination.
5-Iodo-2-toluidine,CH8
NH <0In general the introduction of iodine into the aro-
matic nucleus is not as readily effected as in the case ofchlonne or bromine, butarornaticamines unsubstitutedinthe paro- position nevertheless react easily with iodme.
* Wheeler and Liddle, Am. Chem. I. 4a, 501 (igog).

22 ADVANCED LABORATORY MANUAL
An equimolecular amount of iodine is dissolved ino-toluidine, substitution taking place with evolution ofheat and conversion of one-half of the base into thehydnodide, according to the equation:
2CH8CaH4NH2 + I2 >I(CH8)C aH8NH2 + CH 8 C 6 H,NH a .HI .
In order to utilize the remainder of the o-toluidine andiodine the reaction is completed by heating the mixtureunder a reflux condenser with an equal volume ofwater, 2 molecular equivalents of powdered calciumcarbonate, and 2 volumes of ether to take up the iodobase formed, the entire reaction being represented by
2 + 2l2 + CaCO8 >2l(CH3)C eH8NH2 + Cala + CO2 + H a O.
After one hour's heating the ether is allowed to boil offand the mixture is distilled with steam, the iodo com-pound passing over slowly m a yield of 75 per cent ofthe theory. The practically pure product is dried andrecrystallized from ligroin, forming prisms which meltat 90-1 °.

OF ORGANIC CHEMISTRY 23
I I I . S U B S T I T U T I O N S .
A. (3-Chloropropionic Acid.
1. Ethylene Cyanohydrin, a a32 g of ethylene chlorohydnn, C1CH2CH2OH, are
dissolved in 160 cc of absolute alcohol and boiled undera reflux condenser To the boiling liquid is added,drop by drop, a solution of 27.2 g. of potassium cyanidein 42 cc of water, and the boiling continued for 8-10hours. A precipitate of potassium chloride forms, andat the end this is filtered off, washed with a little alcohol,and the filtrate concentrated to a syrup and fractionatedm vacuo. The yield of cyanohydrin should be 20 g.,boiling at u o ° under 15 mm pressure.
2. p-Chloropropionic Acid, C1CH2CH2CO2H.10 g. of ethylene cyanohydrin are heated in sealed
tubes at ioo° with 75 cc. of concentrated hydrochloricacid for three hours If the cyanohydrin is boiled withdilute sodium hydroxide2 or warmed with acid indilute alcohol,8 hydrolysis of the mtnle or CN groupoccurs,
HOCHaCH8CN -f 3H2O »HOCH2CH2COOH + NH<OH,
and the so-called hydracrylic acid is formed, amongother products. If, on the other hand, the cyanohydrin
aMoureu, Bull, soc chim [3] g, 426 (1893); Jacobs andHeidelberger, J. Am. Chem Soc 39, 1465 (1917),
•Wislicenus, Ann 128, 6 (1863)•Erlenmeyer, Ann. 191, 268 (1878),

24 ADVANCED LABORATORY MANUAL
is heated in a sealed tube with concentrated hydro-chloric acid, not only is the nitrile group saponified tocarboxyl, but chlorine replaces the hydroxyl group,*the end result being
HOCH2CH2CN + 2HCI + H2O >C1CH2CH2COOH + NH<C1.
There is no evidence at hand as to whether the inter-mediate product is hydracrylic acid or ClCHjCH2CN,or both, although it is known that [3-chloropropionicacid can be prepared from the former."
The contents of the tubes are diluted with justenough water to dissolve the ammonium chloridewhich has separated and extracted repeatedly withether, thorough extraction being necessary, as thechloropropionic acid is also quite soluble in water.After drying the ethereal extracts over anhydroussodium sulfate and concentrating, a syrupy residue isleft, which crystallizes readily on cooling and rubbingwith a rod The yield should be 10.5 g. Recrystallizedfrom ligroin, it melts at 38.5-9 50 (corr.).
Similarly 10 g. of ethylene cyanohydrin, boiled threehours with 100 cc. of hydrobromic acid (d 1.49) andworked up in the same way, yield 17 g. of (3-bromo-propionic acid, melting at 60-1 ° (corr.) after recrys-tallization from ligroin.
B. Benzylamine, C6H5CH2NH8.
Benzylhexamethylenetetraminium Chloride.70 g. of benzyl chloride are added to a suspension
of 70 g. of finely powdered hexamethylenetetr«»mine in4 parts of chloroform, heated to boiling under ^ refluxcondenser on the water bath, and removed if necessary
* Jacobs and Heidelberger, loc. cit."Beckurts and Otto, Ber, 18, 236 (1885).

OF ORGANIC CHEMISTRY 25
until the initially often vigorous reaction is over. Themixture is then heated one-half hour longer, placingthe flask on a cork or rubber ring to protect it fromthe bumping which usually occurs
This method, of course, is an indirect one for replac-ing the halogen of benzyl chloride by ammonia. Ifammonia itself is used, the usual mixture of primary,secondary, and tertiary bases is formed, and the yieldof primary benzylamine is poor. Dele"pine6 found,however, that the primary amme was the main productif benzyl chloride was first combined with hexameth-ylenetetramine and the resulting compound suitablydecomposed As the method is of wide application forthe preparation of primary amines it is given here.
As is well known, formaldehyde and ammonia com-bine to yield hexamethylenetetramine, C0H iaN4. Dudenand Scharff7 found that most of the reactions of thissubstance could be explained on the basis of the threedimensional formula.
Among its other properties, hexamethylenetetraminecombines with one molecular equivalent of an aliphatic
8 Del6piri«, Bull soc. chim. [3] 17, 393 (1897).TPydc8 and Scharff, Am. a88, 218 (1895).

26 ADVANCED LABORATORY MANUAL
halide, RX, such as methyl iodide or benzyl chloride,to yield quaternary ammonium salts of the typeCeH12N4.RX,8 just as trimethylamine forms quaternarysalts of the type (CH 8) 8N.RX. The chief objectionto the Duden and Scharff formula is that the reactionstops when one equivalent of halide has reacted, whileaccording to the formula, which postulates four tertiarynitrogen atoms, four molecules of halide should com-bine with one of hexamethylenetetramme. However,only one enters into reaction, and for our present pur-pose the equation is: ^TT ̂ „
C6H8CHaCl + CaH l aN4 > CeH12N4 <
Isolation of the Salt C 1
The reaction mixture is cooled, filtered, and thesalt washed with a little chloroform and sucked dryThe yield should be 90 per cent of the theory. Thesalt darkens above 1800 and melts at 1920
If some of the salt is dissolved in a little water andboiled, formaldehyde is evolved, and the solution sud-denly becomes turbid, owing to the decomposition ofthe quaternary salt to yield methylene-benzylamine,CaHBCH2N CHa, and as this product is fairly stable,vigorous treatment is necessary for its hydrolysis, aswill be seen below.Decomposition.
The salt is transferred to a distilling flask providedwith a condenser and treated with the theoreticalamounts of 95 per cent alcohol and concentrated hydro-chloric acid according to the equation:
CflHiaN4.CeHBCH2Cl + 3HC1 + i2C2H8OH >6CH2(OCaHB)3 + 3NH.C1 + C6HBCHaNHa.HCl,
"An indication of the extent to which RX may be variedmay be obtained by glancing through / Bxol Chetn, 20, 659,685; ax, 103, 145, 439, 455, 4^5 (1915).

OF ORGANIC CHEMISTRY 27
the formaldehyde split off combining at once with thealcohol to form methylal." The flask is rotatedgently until the salt is dissolved and then warmed care-fully until crystals of ammonium chloride begin toseparate, removing the source of heat until certain thatthe reaction does not become violent. The liquid sepa-rates into two layers, of which the upper is mainlymethylal. When this no longer increases it is distilledoff, and the residue in the flask cooled and filtered.The mixture of ammonium chloride and benzyl-amine hydrochloride on the filter is washed with someof the hydrochloric acid-alcohol mixture, the filtratereturned to the flask, and treated with one-third of theoriginal amount of acid-alcohol mixture. A volumeof liquid equal to that added is now distilled off, andthe process again repeated, by which time the distillateshould be free from methylal. As stated above, amethylene compound is initially formed, the equationbeing:
CflHiaN4.CeHBCH2Cl + 3HCI + ioCaH6OH >5CH a(OC2H6) a + 3NH4C1 + CeH8CH8N:CH2 .HCl,
and the prolonged treatment described is necessary inorder to decompose this completely.
Benzylaminc.The final residue in the flask and the crystals already
filtered off are dissolved in water, chilled, and the solu-tion made alkaline with strong sodium hydroxide solu-tion or solid sodium carbonate. The amine layer isseparated, dried over a few sticks of sodium hydroxide,and distilled.
A representative run started with 70 g. of hexa-methylenetetramine and 70 g. of benzyl chloride gave129 g. of the quaternary salt, which in turn yielded 45 g.of pure benzylamine, boiling at 1840.

28 ADVANCED LABORATORY MANUAL
OH
C. rn-Aminophenol,* INH.
Of the methods for the preparation of this substancetwo would seem best suited for laboratory use Inthe first place, one can start with w-nitranilme, diazo-tize this, and decompose the diazo compound by boil-ing with dilute acid, forming m-nitrophenol, but as theyield is not very good and the nitrophenol must sub-sequently be reduced to the ammo compound, themethod is scarcely suited for the preparation of con-siderable amounts of this material. On the other hand,the method involving the direct replacement of one ofthe hydroxyl groups of resorcinol, w-C eH4(OH) a byammonia, was found to be very satisfactory.
The direct replacement of a phenolic hydroxyl groupby the ammo group is relatively difficult in the benzeneseries, requiring high temperatures, although it takesplace somewhat more readily m the case of the naph-thols. If, for example, resorcinol and ammonia aloneare heated under pressure, a high reaction temperatureis required, and the yield of m-ammophenol is poor.If, however, ammonium chloride is present,9 the reac-tion may be carried out at a lower temperature andthe yield is more satisfactory.
200 g. of resorcinol, 120 g. of ammonium chloride,and 400 cc of 10 per cent aqueous ammonia are heatedin an autoclave in a bath the temperature of which is2200, the heating being continued for 14 hours afterthe pressure reaches a constant value The yields aresmaller if the heating is earned out at a lower tem-perature or for a shorter period and are not improved
"Ger pat 49,060.

OF ORGANIC CHEMISTRY 29
by longer heating. After the autoclave and contentshave cooled, the mixture is concentrated to dryness invacuo, taken up in 650-750 cc. of hot water, and allowedto cool. 90-100 g. of crude m-aminophenol separateon standing in the cold. The filtrate is acidified stronglywith concentrated hydrochloric acid and shaken outseveral times with ether to remove unchanged resor-cinol. The aqueous liquor is then treated with anexcess of ammonia and again shaken out with ether,an additional 30-35 g. of aminophenol being recovered.The crude product is recrystallized from 2-3 parts ofwater, about 115 g. separating as sandy crystals melt-ing at 1230.
AsO8Ha
D. o-Nitrophenylarsonic Acid, ] J NO2.
The student is already familiar with the replacementof the aromatic ammo group by the cyano- and halogenresidues by means of the Sandmeyer reaction, and itsconversion into the phenol group through the diazoreaction has been touched upon in the discussion ac-companying the preceding preparation. With theseexamples the usefulness of the diazo group as an inter-mediary between the amino and other groups is by nomeans exhausted, and the so-called Bart reaction,10
given here, affords a typical example of the extensiveapplication of the fundamental reaction in question.One of the examples in Bart's patent is o-nitrophenyl-arsonic acid, but his description of the substance isinaccurate and the variation of the method here givenis believed to be more convenient.11
MGer pat 250,264.n Jacobs, Heidelberger and Rolf, /. Am. Chem. Soc 40, 1582
(1918)

30 ADVANCED LABORATORY MANUAL
50 g. of o-nitraniline (p. 13) are ground finely under250 cc. of 1:1 hydrochloric acid, and the mixture ischilled to io° and slowly diazotized with a strong solu-tion of 275 g. of sodium nitrite. After 10 to 15minutes' stirring the solution is filtered from traces ofundissolved nitraniline and poured slowly with vigor-ous rotation into 275 cc. of thoroughly chilled 25 percent sodium hydroxide solution, keeping the tempera-ture below o° by immersion in a freezing mixture. Theresulting alkaline solution is added to a cold solution of67.5 g. of sodium arsemte in 625 cc. of water, using asuffiaently large flask to allow for the frothing whichoccurs during the subsequent decomposition of the diazocompound. The mixture is then heated on the waterbath to 60-700 for 1.5 to 2 hours, during which a slowbut steady nitrogen evolution occurs. Overheating isparticularly to be avoided. The reaction may be con-sidered to take place according to the following equa-tion : _ .
ONa
O2NC6H<N :N — OH + As — ONa >
OH
O N a "
O2NCeH4N2 —As — O N a >
H O OH
O
O2NCeH4As — ONa + N2 + H 2 0 .
ONa"Cf. Schmidt, Ann 431, 159 (1920).

OF ORGANIC CHEMISTRY 31
Owing to the fact that sodium arsenite is a reducingagent, the reaction does not proceed quantitatively,some of the diazo compound being reduced with forma-tion of nitrobenzene:
ONa
O2NC6H4N :N — OH + As — ONa >
OH
O
CaHBNO2 + Na + As —ONa.
ONaPurification.
After the evolution of nitrogen ceases the hot mix-ture is made faintly add with acetic acid, shaken withboneblack to remove the tarry by-products, and filtered.0-Nitrophenylarsonic acid is a strong acid, and is notliberated by a slight excess of acetic acid, so that inorder to obtain the free acid the deep yellow filtratemust be treated with hydrochloric or sulfuric aciduntil strongly acid to Congo red. After chilling thor-oughly and letting stand 55 g. of o-nitrophenyl-arsonic acid are obtained as a heavy, pale yellow powder.Recrystallized'from water it forms pale yellow, glisten-ing, hexagonal plates containing one molecule 01 waterof crystallization. After this is driven off %n vacuo atioo° the acid melts and decomposes at 235-40°, withpreliminary softening.18
M Instead of Expt, V B the nitro acid may be reduced withferrous sulfate and alkali according to I. Am. Chem. Soc. 40,1583 (191S), yielding o-arsanilic acid.

32 ADVANCED LABORATORY MANUAL
IV. E S T E R I F I C A T I O N , E T H E R I F I C A T I O N ,DE-ALKYLATION, AND R E L A T E D
R E A C T I O N S .
A. Esterification; Preparation of the Perfume,Methyl Anthranilate.1
COOCH3
NH9
Either anthranilic acid itself, or acetanthranilic acid,the intermediate product in the preparation of an-thranihc acid from. o-acet-toluidide, may be used forthe esterification, since alcoholic hydrochloric acidhydrolyzes, or rather alcoholyzes, the acetamino group.
7 g of anthranilic acid or 9 g. of the acetyl deriva-tive are dissolved or suspended in 50 cc of methylalcohol which has previously been dried over potassiumcarbonate. Dry hydrochloric acid is passed m untilthe solution, which becomes hot, is saturated. It isthen boiled for one hour under a reflux condenser sur-mounted by a calcium chloride tube. When the solu-tion is cooled methyl anthranilate hydrochloride crys-
*Cf Erdmann, Ger. pats 110,386, 113,942.

OF ORGANIC CHEMISTRY 33
talhzes. The mixture is diluted with about 200 cc.of water and made alkaline with sodium carbonate.The oily ester is shaken out with ether and the etherealsolution washed first with 5 per cent sodium carbonatesolution, and finally with water. The ethereal layer isdried over sodium sulfate and evaporated to smallbulk, after which the ester is distilled in vacuo. Itboils at 1350 under a pressure of 15 mm of mercury,and forms a crystalline mass when cooled and rubbed,melting at 24.5°.
Methyl anthranilate occurs in oil of orange blossoms(neroh oil), oil of orange peel, and in the essential oilof jasmine flowers, and is extensively used in per-fumery.
OCHflCOOH
B. p-Nitrophenoxyacetic Acid,
The reaction of phenols with alkyl halides or dimethylsulfate in the presence of alkali to combine with the acidliberated is, of course, well known. The followingpreparation2 is selected as an example of the varietyof alkyl compounds that enter into this reaction, itbeing, of course, possible to consider chloroacetic acid,ClCHaCOaH, as a methyl halide in which one of thehydrogens has been replaced by a carboxyl group.
35 g- °f jfr-nitrophenol, 40 g. of 50 per cent sodiumhydroxide solution (the additional molecular equivalentbeing to neutralize the chloroacetic acid), 24 g. ofchloroacetic acid, and 200 cc. of water are boiled gently
"Jacobs and Heidelberger, / . Am Chem, Soc. 39, 1437(1917).

34 ADVANCED LABORATORY MANUAL
in an open flask until the solution is no longer alkalineto litmus. As the ethenfication is not quantitativeowing to the action of free hydroxyl ions on thechloroacetic acid according to the equation:
ClCH2COONa + OH" > HOCH2COONa + Cl",
one-half of the above quantities of alkali and chloro-acetic acid and 50 cc. of water are added, and the solu-tion is boiled again until neutral. It is then acidifiedstrongly with hydrochloric acid and cooled, precipitatingthe nitrophenoxyacetic acid The crude product maybe purified either by recrystalhzation from alcohol, 01by dissolving it m dilute sodium hydroxide solution andreprecipitating with hydrochloric acid. The yield shouldbe 25-30 g. The acid forms glistening platelets whichmelt at 183°, as recorded in the literature
OC2H5
C. m-Phenetidine,1NH,
Acetylation.w-Aminophenol may be conveniently acetylated by
dissolving it in a little more than the minimum amountof warm 50 per cent acetic acid, cooling quickly to roomtemperature, and adding 1.1 molecular equivalents ofacetic anhydride. After one-half hour the solution isdiluted with an equal volume of water and chilled,rubbing the walls of the vessel to complete the crystal-lization. The product so obtained is filtered off,washed with ice water, and dried. It should melt at148-90 a and is then sufficiently pure for preparativepurposes.
•Ikuta, Am. Chem. J. 15, 41 (1893)

OF ORGANIC CHEMISTRY 35
Ethylation.27 g. of m-acetaminophenol are dissolved in 180 cc.
of normal potassium hydroxide solution, warmed to50-600, and shaken with 23 cc of diethyl sulfate,*added in small portions, keeping the temperaturewithin the limits indicated. It is of course necessaryto protect the ammo group, otherwise this, too, will bealkylated, and acetylation is a very convenient methodfor this purpose, as the acetylamino group is unaffectedby the conditions used in the reaction, and may bereadily removed by saponification at a later stage. Thetemperature must be raised owing to the fact that diethylsulfate is more inert than dimethyl sulfate, with whichmethylatjons can be carried out in the cold. It is thusalso less sensitive than dimethyl sulfate to the hydroxylions present m the alkaline reaction mixture, and goodyields of ethyl ethers may therefore be obtained with itsaid in spite of the saponification of the reagent intoalcohol and sulfuric acid, which does occur to an appre-ciable extent, as in the case of dimethyl sulfate
In order to allow for this side reaction the mixture,from which a portion of the acet-w-phenetidine hascrystallized, is again subjected to similar treatment withone-half the initial quantities of alkali and diethyl sul-fate. It is then allowed to stand overnight after add-ing 100 cc. of concentrated aqueous ammonia to assistm the decomposition of any remaining excess of diethylsulfate, the ammonia being alkylated just as any otheramine.
Saponification.The crude acetyl product is filtered off, washed with
a little ice-cold water, sucked dry, and boiled one-halfhour with 150 cc. of 1: 1 hydrochloric acid, the acetyl-
* Heidelberger and Jacobs, /. Am. Chem. Soc. 41, 1452(1910).

S6 ADVANCED LABORATORY MANUAL
amino group thus splitting into acetic acid and theamine hydrochlonde, which separates on cooling.Enough water is added to dissolve this salt and thesolution is then made strongly alkaline with sodiumhydroxide and the liberated base is {shaken out withether. The ethereal layer is dried over sodium sulfateor crushed sodium hydroxide, concentrated, and theresidue distilled in vacuo. 15 g. of m-phenetidineshould l?e obtained, boiling at 142-4 50 under 20 mm.pressure. According to Reverdm and Lokietek5 asomewhat better yield is obtained using ethyl bromideas the alkylatmg agent.
D. Allyl Phenyl Ether and its MolecularRearrangements.
1. Allyl Bromide, CH2 : CHCH2Br.The method is essentially that of Merlmg and Jacobi.6
140 g of allyl alcohol, CH8 . CHCH2OH, are cooledin ice-water. A stream of hydrobromic acid, whichmay be conveniently generated by dropping bromine onnaphthalene and passing the vapors through severalbottles containing naphthalene, is then passed m untilthe liquid is saturated, after which it is boiled under areflux condenser for one hour, the reaction being:
CH2 : CHCHaOH + HBr >CHa :CHCHBBr + H2O.
Under the conditions given very little addition ofhydrobromic acid takes place at the double bond, al-though this would undoubtedly occur on long standingof the saturated solution in the cold.
The mixture is then poured into water and the crudebromide separated and washed first with normal sodium
'Reverdin and Lokietek, Bull soc chim [4] 17, 407 (1915).•Merlmg and Jacobi, Ann. 278, 11 (1894)

OF ORGANIC CHEMISTRY 37
hydroxide solution and then with water. After dryingover calcium chloride and distilling, the yield shouldbe 85 per cent of the theory, boiling at 70-1 °.
2. Allyl Phenyl Ether, C0HnOCH2CH. CH2.7
94 g of phenol, 121 g. of allyl bromide, 140 g. of drypotassium carbonate, and 150 g of acetone are boiledfor eight hours on the water bath under a reflux con-denser In this case potassium carbonate is used tocombine with the acid liberated in the alkylation, andthe reaction is carried on in a non-aqueous solventPotassium bromide soon separates and the mixturethickens, to a paste After cooling, water is added andthen ether to take up the allyl phenyl ether. The ethe-real layer is shaken out twice with 10 per cent sodiumhydroxide solution to remove unchanged phenol, washedwith a little water, and then dried over potassium car-bonate and distilled in vacua The yield should be115-30 g., boiling at 850 under a pressure of 19 mm.The purification must be earned out under diminishedpressure, as will be seen from the next experiment.
3. o-Allylphenol (o-Chavicol).OH
CH aCH:CH9 .8
Allyl phenyl ether is boiled under an air condenseruntil the temperature no longer rises, a process requir-ing four to six hours. From an initial temperature ofabout 1900 the thermometer finally rises to about 2200.The gradual increase m boiling point is due to a re-
TClaisen and Eisleb, Ann 401, 21 (1913); 418, 78 (1918).'Claisen, loc. cit; Jacobs and Heidelberger, /. Am Ckem.
Soc, 39, 3202 (1917).

38 ADVANCED LABORATORY MANUAL
markable rearrangement, somewhat analogous to thattaking place when the alkyl anilines (in the form ofsalts) are heated to high temperatures, and consisting inthe wandering of the allyl group to the o-position inthe nucleus. While this reaction takes place compara-tively readily in the case of the alkyl aniline salts, thealkyl phenol ethers exhibit no such phenomenon. Ap-parently the allyl group is the only one possessing suchlability in the phenol series.
Purification.After the boiling has been completed the product
is cooled, dissolved in 20 per cent aqueous sodiumhydroxide, and the solution shaken with petroleumether to remove traces of unchanged allyl phenyl etherand small amounts of methylcoumarane, formed ac-cording to the following scheme •
OH O CH.CHa
C H C H . C H * 1 I —CH 9
The o-allylphenol is then liberated from the alkalinesolution by acidification with sulfuric acid, and is takenup in ether, dried over sodium sulfate, and concen-trated. 'Distillation in vacuo yields an oil with aguaiacol-like odor, boiling at 109-100 under a pressureof 22 mm and solidifying in a freezing mixture to amass of crystals which melt at - ^ 6 ° . The yield isalmost quantitative
An interesting property of o-allylphenol is that, byrepetition of the ethenfication and heating, it can be

OF ORGANIC CHEMISTRY 39
converted successively into 0, o'-diallylphenol, and0, 0'', />-tnallylphenol.
OH
4. o-Propenylphenol (o-Anol),[ |CH:CH.CH 8 .
o-Allylphenol is dissolved in three parts of the strong-est possible methyl alcoholic potassium hydroxide andthe solution boiled until one part of the methyl alcoholhas boiled off, after which a reflux condenser is at-tached and the mixture boiled in an oil bath for twohours. A similar shifting of the double bond underthe influence of alkali is observed in many other cases,and reactions of this type must be carefully avoidedin determining the constitution of an unknown sub-stance if the position of any unsaturated linking pres-ent is to be fixed with certainty.
The mixture is diluted with water, acidified, and thephenol isolated as in the preceding case. It boils at230-1 ° under atmospheric pressure, while according toPauly and Buttlar0 it boils at 112-30 under a pressureof 12 mm, solidifying when chilled and rubbed Itmay then be recrystalhzed from ligroin, when it meltsat 34-50-
O — CHCH8
5. a-Methylcoumarane, ( | — CHa.
The formation of traces of this substance fromj-allylphenol was encountered in the preparation of the
9 Pauly and Buttlar, Ann. 383, 380 (1911).

40 ADVANCED LABORATORY MANUAL
latter, and this remarkable intramolecular reairange-ment may be made the chief reaction by using a catalyst,such as pyndine hydrochlonde. This salt may readilybe obtained by passing dry hydrochloric acid gasthrough a wide tube into a chilled solution of pyridmein several volumes of benzene.
20 g of o-allylphenol are boiled with 2 g. of drypyridine hydrochlonde until the boiling point sinks to aminimum10 The solution is diluted with ether andwashed with dilute acid to remove the pyndine salt,with dilute sodium hydroxide to remove any unchangedallylphenol, and then with water. After the ethereallayer is dried over sodium sulfate and concentrated theresidue is distilled, the a-methylcoumarane boiling at197-80.
The following scheme will serve to summarize thegroup of reactions occurring between phenol and ally]bromide
OH
|+2C8H f lBr + K2CO8 >
0CH2CH CH2
+ 2KBr- f C O a + H a O .
0CH 2CH • CH2
1 Cf, Ger. pat 279,864

OF ORGANIC CHEMISTRY
OH
OCH.CHCH,
|CH2CH.CH a , O —CHCH a
\ I l - C H f l
E. Dihydrocupreine, "> i a .CH
\ C H C H 2 C H aCHa
HO
MA summary of the classic researches leading to the deter-mination of the constitution of the cinchona alkaloids may befound m Schmidt and Grafe's book on "Alkaloide" in Abder-halden's collection "Handbuch der biologischen Arbeits-methoden" (Part I, Section IX), or better, the original articlesof Koenigs, Ann. 347, 143 (1906); and Rabe, Ann. 365, 353(1909), 373, 85 (1910). These and Rabe's later articles areonly one of the many fascinating and inspiring records af-forded by organic chemistry of year-long patient and brilliantinvestigation, overcoming obstacles one by one, revising ideasoriginally thought correct, and finally arriving at the true solu-tion.
^Heidelberger and Jacobs, /, Am, Cketrh $oc. 41, 821(1919).

42 ADVANCED LABORATORY MANUAL
Demethylation.50 g of dihydroquinine (p. 50) are boiled with
200 cc. of aqueous hydrobromic acid (d. 1.49), allow-ing the water boiling off to escape until the temperaturehas again reached that of the constant-boiling hydrate,when an air-condenser is attached and the boiling con-tinued for four hours The methyl bromide liberatedmay be collected if desired by leading tubes from theair-condenser into a test tube immersed in a freezingmixture.
Phenolic ethers, of which dihydroquinine is an ex-ample, may be dealkylated by heating with acids Hy-drochloric aad is often used, but the reaction rarelytakes place at the boiling point of the constant-boilinghydrate and is therefore generally carried out in sealedtubes The pressure developed by the methyl chlorideliberated often results in the breakage of tubes, butcan be avoided to some extent by frequently openingand resealmg the tubes Concentrated hydriodic aadis also a useful dealkylating agent, but is relativelyexpensive, and hydrobromic aad may therefore beused to advantage in many cases especially when largeamounts of material are m question. While a solutionof the acid in glaaal acetic acid is often used, the con-stant-boiling hydrate (d 1 49) which boils at 126 ° at760 mm. pressure, and contains 47.8 per cent of hydro-bromic aad, has been found very satisfactory for casessuch as the present one.
Filtration of the Dihydrobromide.The dark brown solution is cooled and allowed to
stand overnight m the ice box The heavy, crystallinedihydrocupreine dihydrobromide which separates is fil-tered off on cloth on a Buchner funnel and washedwith a little of the hydrobromic acid. The filtrate isthen distilled to about one-third of its original volume,which, besides concentrating the mother liquors, effects

OF ORGANIC CHEMISTRY 43
the conversion of any unchanged dihydroquinine pres-ent. The crop of crystals collected after cooling theconcentrated liquid is filtered off as above and addedto the first fraction, while the filtrate may be added tothe hydrobromic acid used for another preparation ofthe same substance, in case this is undertaken.Conversion to Base.
The combined fractions of the dihydrobromide aredissolved in about 2 liters of warm water, cooled, andcautiously treated with 10 per cent sodium hydroxidesolution, with vigorous stirring, until the localized pre-cipitate first formed begins to dissolve slowly. Addi-tion of sodium hydroxide is then continued rapidlywith vigorous stirring until the copious precipitate ofthe base just redissolves m the excess of alkali, solu-tion taking place, of course, by virtue of the phenolicgroup exposed by the demethylation. In this way theseparation of the dihydrocupreine as a gum may beavoided, as this dissolves in excess alkali only with thegreatest difficulty. In case any gummy material isformed in spite of all precautions, it may be removed,dissolved in dilute hydrochloric acid, and the aboveprocess repeated, adding the alkaline solution to themam portion. Bone black is next added to collect atrace of gelatinous material, and the solution is filteredthrough large folded filters, for if the manipulationshave been properly carried out the alkalinity of thesolution should not be so great as to attack the paper.Since alkaline phenolic solutions are subject to oxida-tion, this part of the preparation should be carried outas rapidly as possible. The base is precipitated fromthe clear yellow filtrate by the addition of not too largean excess of saturated ammonium chloride solution.The amorphous base is filtered off on a large Buchnerfunnel, washed with water, sucked as dry as possible,

44 ADVANCED LABORATORY MANUAL
and added to 190 cc. of boiling 95 per cent alcoholMost of the material dissolves before crystals begin toseparate At this point the solution is rapidly decantedfrom the undissolved substance, which is then dis-solved in the minimum amount of boiling alcohol andadded to the rest of the solution 25 cc. of waterare added and the solution is allowed to cool and standover night m the ice box, when 23 g. of quite puredihydrocupreme are obtained On concentration ofthe mother liquors and washings to about one-halfvolume and addition of about one-quarter volume ofwarm water, a further crop of 5 g. of slightly lesspure alkaloid is obtained, while about 2 g additionalmay be recovered by shaking out the ammoniacal fil-trate from the crude amorphous base with chloroform
The main fraction, recrystalhzed from 85 per centalcohol, yields the pure alkaloid as compact aggregatesof thick, minute plates which swell and evolve gas at185-900, with preliminary softening, forming a glassymass which adheres to the walls of the capillary tubeand only liquefies completely at 230°, with simultaneous
darkening. [O]T-V in absolute alcohol is —148.70 ,
c = i 13The interest attached to this alkaloid lies in the fact
that its ethyl ether, the so-called "optochin," is a re-markable and specific bactericide for the pn^umococcus,while its homologous ethers are highly bactericidal forother micro-organisms 18
"Morgenroth and Levy, Berl Mm. Wochschr. 48, 1560(1911), and later articles

OF ORGANIC CHEMISTRY 45
V. REDUCTION.
(See also p. 93)
A. Reduction with Stannous Chloride.
p-Aminodimethylaniline,N(CH 8 ) 2
NH2.
This technically important intermediate may be pre-pared as follows 1 50 g. of />-nitrosodimethylanilmeor a corresponding amount of its hydrochloride, pre-pared according to the directions in any elementarylaboratory manual, are added m small amounts to awarm solution of 225 g. of stannous chloride in 450 ccof concentrated hydrochloric acid, cooling occasionallyto prevent the mixture from getting hot during the ad-dition of the amine The reduction is completed bywarming for one-half hour on the water bath, afterwhich the mixture is cooled Part of the double tinsalt of the aminodimethylaniline is deposited at thispoint, and the precipitation is completed by saturatingthe mixture at o° with hydrochloric acid gas. The saltis then sucked off through a cloth filter and dissolvedin water. As the base is readily oxidized by the air
* Jacobs and Hddelberger, /. Biol. Chem. 21, 113 (1915).

46 ADVANCED LABORATORY MANUAL
the acid solution is covered by a layer of ether, ice isadded, and the compound only then liberated by makingvery strongly alkaline with 50 per cent sodium hy-droxide, being sure that enough ice is present to keepthe mixture cold. After shaking cautiously the ether isremoved and the alkaline solution again shaken outseveral times with ether. The combined ethereal ex-tracts are dried over sodium sulfate and sodium hy-droxide and concentrated, and the residue is distilledin vacuo. The yield of aminodimethylaniline shouldbe 36 g., boiling at 146-80 under a pressure of 24 mm.and solidifying in the receiver to a crystalline masswhich melts at 38-41 °. Owing to its sensitiveness tothe oxygen of the air the base is best preserved invacuum ampoules (which may be made of thick-walledtest tubes), or in glass-stoppered bottles.
B. Reduction with Ferrous Sulfate and Ammonia.
OCH2COaH
Ap-Aminophenoxyacetic Acid,
NHa20 g. of p-nitrophenoxyacetic acid (p. 33) are dis-solved in a slight excess of dilute ammonia and thesolution is poured in a thin stream into a vigorously ro-tated, boiling solution of 7 molecular equivalents (onem excess) of ferrous sulfate (FeSO4>7HaO) in 2 to2.5 parts of water. Small portions of concentratedaqueous ammonia are then immediately added, rotatingthe solution vigorously after each addition and continu-1 Jacobs and Heidelberger, 7. Am. Chetn. Soc. 39, 1436(1917).

OF ORGANIC CHEMISTRY 4.7
ing the addition of ammonia until the solution remainsdefinitely alkaline to litmus after shaking and boiling".The mixture is boiled 5 minutes, making certain thatthe reaction remains alkaline, and is filtered hotthrough a large Buchner funnel which, with the suc-tion flask, has previously been warmed to preventcracking. The precipitate of iron hydroxides is washedwith a little hot dilute ammonia solution and the filtrateconcentrated in vacuo until the ammonium salt filtrateof the amino acid begins to separate owing to the salt-ing-out action of the ammonium sulfate present. Themixture is then heated until the salt dissolves andtreated with an excess of acetic acid, whereupon 15 7 g.of />-aminophenoxyacetic acid precipitate almost atonce. The acid crystallizes with one molecule of waterof crystallization. Contrary to the statements ofothers8 both the air-dry and anhydrous acids melt withgas evolution and resolidification at about 2200, leav-ing a residue which does not melt below 285 °.
The reduction with ferrous sulfate and ammoniatakes place according to the equation:
OaNCnH4OCH2CO2NH4 + 6FeSO, + I2NH.OH +6H2O > HaNCeH4OCHaCOaNH4 +
6(NH 4 ) 2 SO 4 + 6Fe(OH) s + 2 H 2 O.The method is of great value in dealing, with sub-
stances which are sensitive fo strong acids or bases, suchas o-nitro- and ammo-benzaldehydes, nitro- and amino-benzamides and benzoylureas, and is also of service inthe preparation of other ammo actds.
In the reduction of o-nitrophenylarsonic acid withthe aid of ferrous sulfate, ammonia is not a strongenough base to decompose the iron salt of the aminoarsonic acid, and sodium hydroxide must be used.
"Howard, Bar. 30, 547 (1897); Kyra, J. prakt. Cham, [2]55, 113 (1897).

48 ADVANCED LABORATORY MANUAL
After preparing the o-nitrophenylarsonic acid (p 29)this modification may be tried instead of, or in additionto, the above preparation. In the case of arsonic acidsthe method is of particular utility in that it resultsonly in the reduction of the nitro group, while strongerreduang agents reduce the arsonic acid group as well.
C. Reduction with Sodium Amalgam.
<i/-Phenylethanolamine? l-phenyl-l-hydroxy-2-aminoethane, OH
— CH,NH a .
The benzaldehyde cyanohydrin required for thispreparation is very conveniently made according toGer pat 85,230 as follows Benzaldehyde is thoroughlyshaken, best m a shaking machine for one-half hour,with an excess of concentrated sodium bisulfite solu-tion The crystalline bisulfite addition product,
OHC0HBCH< , is filtered off, washed with al-
OSO2Na
cohol, and stirred to a thin paste with water. A con-centrated solution of 1 1 molecular equivalents of potas-sium cyanide is then added all at once, with vigorousstirring The bisulfite compound quickly dissolves, and
OHthe oily cyanohydrin, Cf lHBCH< , precipitates in al-
CN

OF ORGANIC CHEMISTRY 49
moat quantitative yield. It is separated from the solu-tion as quickly as possible and used directly for thereduction.
35 g. of the cyanohydrm are dissolved in 550 cc. of50 per cent alcohol, chilled in a freezing mixture, andreduced with continual chilling and mechanical stir-ring, with 1400 g. of 4 per cent sodium amalgam, onto all of which the solution should be poured.* Dur-ing the reduction, which requires several hours and pro-ceeds according to the equation:
OHQ # 0 C H < + 4Na + 4 H 2 O >
CNOH
C0H aCH< + 4NaOH,CH2NH2
the reaction is kept just acid to litmus by slowly drippingin first 50 per cent acetic acid, then the glacial acid,regulating the speed of addition by careful tests withlitmus paper, as the reaction should neither be allowedto remain alkaline nor become strongly acid. Whenall of the sodium has been used up the solution ispoured from the mercury and concentrated to aboutone-half volume. The solid mass of salts obtained oncooling is ground up in a mortar, thinned with a littledilute hydrochloric acid, filtered off, and washed withmore of the dilute acid The filtrate is shaken out withether to remove non-basic impurities and is then cov-ered with a layer of ether, made very strongly alkalinewith sodium hydroxide, shaken out, and the extractionwith ether repeated several times. The ethereal solu-tion is dried over crushed sodium hydroxide and con-centrated to small bulk.
The resulting oily base does not crystallize, but* Cf. Ger. pat 183,634.

SO ADVANCED LABORATORY MANUAL
forms a characteristic N-benzoyl derivative It is dis-solved m a mixture of 5 parts each (calculating cc perg of base) of glacial acetic acid and saturated sodiumacetate solution and shaken in the cold with 1 1 molecu-lar equivalents of benzoyl chloride.8 The benzoylderivative separates as an oil which crystallizes afterbeing rubbed Recrystallized from the minimumamount of alcohol it forms nacreous scales which meltat 148.5-9.00.6
D. Reduction with Palladium Black.
Dihydroquinine,CH
H H a C /
CHBO
One of the most important methods for the identi-fication of unsaturated linkages, and a method whichhas had important consequences in the study of ringsystems in general and alkaloids in particular, is thePaal-Skita method of reduction with colloidal metals,in which finely divided palladium or platinum acts as
1 For this general method of acylation see Jacobs and Heidel-berger, /. Am. Chem. Soc 39, 1439 (1917)
"Kolshorn, Ber. 37, 2483 (1904), and Rosenmund, Ber. 46,1046 (1913), give 147°.

OF ORGANIC CHEMISTRY 51
carrier for the hydrogen either in the presence or ab-sence of a "protective colloid." 7 Many of the modi-fications of the method have been applied on an enor-mous scale industrially, as m the hardening of fats withthe aid of reduced nickel.
In the cinchona series 8 the method is particularlyeasy of application and has been used for the produc-tion of the dihydro alkaloids, whose* biological prbp-erties were first extensively studied by Morgenroth andhis collaborators.9
40 g. of quinine
CHflO
CHCH:CH 2
are dissolved in 180 g. of 10 per cent aqueous sulfuricacid, filtered, and treated with 8 to 10 cc. of a 2 percent solution of palladious chloride10 prepared by dis-solving the salt m a little hot 1: 1 hydrochloric acid,concentrating as far as possible on the water bath, anddiluting to volume with water. The precipitate whichforms in the quinine solution dissolves readily on stir-
7 For a detailed discussion see Skita's monograph.8 See footnote, p. 41.0 Loc. cit.10Ger. pat 252,136; Heidelberger and Jacobs, 7. Am. Chem.
Soc. 41, 819 (1919).

52 ADVANCED LABORATORY MANUAL
ring, and the clear solution is then rinsed into a shak-ing apparatus (that shown in the cut is a convenientform), the air is displaced by hydrogen, and the wholeshaken with free access of hydrogen under pressure ofa column of water varying from about 25 to 60 cm.The reaction proceeds slowly until the palladium is
entirely reduced, after which the absorption of hydro-gen takes place rapidly until almost the calculatedamount has been absorbed, slowing down towards theend. The mixture is poured out of the shaker andeither centrifuged or allowed to stand until the pal-ladium has settled, after which the liquid is carefullypoured off and the palladium washed either by centrif u-gation or decantation, after which it may be used forfour or five additional reductions The solution andwashings are diluted with a large volume of waterand rapidly made alkaline with 10 per cent aqueoussodium hydroxide, for if the addition of alkali is tooslow the sparingly soluble neutral sulfate of dihydro-quinine, (C20H28O2Na)2.H!jSO4, may crystallize outand contaminate the base The amorphous precipitateis filtered off on a large Buchner funnel, washed wellwith water, pressed out and sucked as dry as possible,and then spread out to dry in the air. It is recrystal-lized by adding to hot dry acetone or toluene, and whendried melts at 171-20 as recorded in the literature forthe natural alkaloid which occurs in small amountsassociated with quinine.

OF ORGANIC CHEMISTRY 53
While quinine in dilute sulfuric acid immediately re-duces a one per cent solution of potassium permangan-ate, the permanganate color persists for some momentsin a similar solution of pure dihydroquinine, and thistest should be performed as a control of the purity ofthe product obtained.

54 ADVANCED LABORATORY MANUAL
VI. OXIDATION.
A. Oxidation with Potassium Ferricyanide.
OH
p-Nitro-o-cresol, | C H s '
In the direct nitration of o-cresol a mixture of thep- and o~ derivatives is formed, from which relativelylittle of the para- isomer can be isolated.1 Bettermethods for the preparation of the />ara-compound areby diazotization of 5-nitro-o-tolmdme and replacementof the diazo group by hydroxyl, or by simply boiling thenitrotoluidme with strong aqueous alkali.8
Another very convenient method which gives ahomogeneous product in good yield involves the oxida-tion of ^-nitroso-o-cresol to the />-nitro derivative, asgiven by Borsche and Berkhout,8 except that almostquantitative yields are obtained in the cold instead of onboiling as recommended by these authors
15 g. of £-nitroso-0-cresol (p 16) are dissolved in asolution of 150 g. of sodium hydroxide and 150 g. ofpotassium ferricyanide in 3 liters of water, and allowed
1 Hirsch, Ber 18, 1512 (1885)a Neville and Winther, Ber 15, 2978 (1882)'Borsche and Berkhout, Ann 330, 95 (1904).

OF ORGANIC CHEMISTRY 55
to stand at room temperature for two days. The oxida-tion may be considered to take place as follows:
RNO + 2K8Fe(CN)0 + sNaOH >RNO a + 2K3NaFe(CN)0 + H aO,
the ferricyanide being reduced to ferrocyanide. Thelarge excess of ferncyanide is necessary in order toinsure the completion of the reaction.
The solution is finally acidified with sulf uric acid andextracted several times with ether. The ethereal layeris in turn shaken out with 5 per cent sodium hydroxidesolution and this warmed on the water bath to drive outthe dissolved ether. On cooling and acidifying withsulfuric acid the nitrocresol separates as faintly yellowneedles melting at 93-5 ° after drying in vacuo oversulfuric acid. The yield should be 15 g.
B. Oxidation with Nitrites: Isonitrosocamphorand Camphorquinone.4
CH3 CH8
CH9 C C:O CHa C C:O
i.CH8.C.CH8CHa CH C:NOH CHaIsonitrosocamphor CamphorquinoneC:O
56 g. Of camphor (natural) are dissolved in 250 cc. ofdry ether in a large flask and 7.6 g. of sodium wireadded. The mixture is cooled with ice water, afterwhich 39 g. of isoamyl nitrite are added in small por-tions with shaking and continued cooling, the addition
*Cf. Gaisen and M&nasse, Ann. 974, 73 (1893),

56 ADVANCED LABORATORY MANUAL
of the nitrite taking 10-15 minutes It should also beremembered that isoamyl nitrite is quite volatile andextremely toxic The reaction may be considered totake place as follows
CO2C8H14< I + Na2 + CBH nONO »
CHaCO CHOH
C8H14< I + C8H14 < I + C 6 H u ONa B
C:NONa CH2
Punfication.After several hours ice water is added (if there is
no sodium left), giving a reddish solution of thesodium salt of the isonitroso compound Unchangedcamphor and borneol,
CH3
•CHOH
CH n .C .CH,.C.C
CH-
formed by reduction are removed by shaking out cau-tiously several times with benzene, and the aqueous layeris filtered through a wet paper to remove traces of sol-vent. Dilute acetic acid is then added until no furtherprecipitation occurs The yield of crude product,which is, of course, a mixture of isomers, should beabout 22 g.
' Ci Baubigny, Ann chivt phys. [4] 19, 221 (1870), Beck-mann, Ber aa, 912 (1889), for a discussion of the action ofsodium on camphor,

OF ORGANIC CHEMISTRY 57
Isomerization.Owing to the presence of a low-melting isomer,
Claisen and Manasse had much difficulty recrystalhz-ing the compound to constant melting point, butForster ° found that the low-melting isomer is con-verted into the high-melting form when the dried mix-ture is heated at 150° for a few minutes After re-crystallizing the melt from boibng ligrom the isonitroso-camphor melts at 152-30.
Fission.12 5 g of isonitrosocamphor are dissolved in 22 5 cc.
of glacial acetic acid and treated slowly with 50 g. offinely powdered, hydrated sodium sulfite, while thesolution is shaken vigorously and heated over a freeflame T When all of the sulfite has been added themixture is boiled for one hour under a reflux con-denser, using a sand bath. According to von Pech-mann 8 the sodium bisulfite initially formed reacts withthe isonitroso compound as follows:
R . C O . C . N O H + NaHSO a >
R . C O . C ' N , O S O 8 N a
50 cc. of concentrated hydrochloric acid are then runinto the boiling solution, and the boiling is continuedfor one-half hour, von Pechmann formulates the re-action as :
J Forster, /. Chtm. Soc. 83, 535 C1903).Lapworth and Chapmau, J.Ckem, Soc. 79, 380 (1901).8voi» P$cJimai}a, $frf 30, ^504 (1887).

58 ADVANCED LABORATORY MANUAL
R.CO.C:NOSO 2 Na
R'R. CO. CO. R ' + NH4NaSO4
The solution is finally diluted with water, cooled, andthe crude camphorquinone filtered off and purified bydistillation with steam. The yield should be about 7 g.The qumone forms yellow prisms and needles whichmelt at 1980. It sublimes rapidly above ioo°, and, likecamphor itself, is quite volatile at room temperature
C. Oxidation with Atmospheric Oxygen:Camphoric Acid.
CH8
CH2 CH CO2H
CH8 — C — CH8
CH2 C COaH.
Camphorquinone is extremely sensitive to oxidizingagents and yields camphoric acid when merely boiledin alkaline solution in the presence of air e Thediketone is boiled during one day with 3-4 molecularequivalents of alcoholic potassium hydroxide Thealcohol is then evaporated off, and the residue takenup m water. After filtering, the camphoric acid isprecipitated with dilute sulfuric acid and recrystallized
from water, melting at 176-80 and giving [a] \\ + 46°
in absolute alcohol (c = 1).0 Gaisen and Manasse, loc. cit.

OF ORGANIC CUBMtSTRY 59
D. Oxidation with B r o m i n e : Gluconic AcidCalcium Sal t ,
H H H O H H
H o - c - d : - c - - c — c -
•k <!)H O H i OH
To a solution of 200 g. of anhydrous glucose in oneliter of water add gradually 200 g. of bromine, withfrequent shaking. About 50 g. of the bromine may beadded at once and then the res t in 25-50 g. portions asthe fluid bromine disappears into solution. Kiliani'soriginal method calls for double the amount of bromine,but Ruff " found that an equal weight was sufficient.It is well, however, to control ^ the progress of theoxidation by the gradual diminution in reducing power(Fehhng's solution) of the solution, and to use a littlemore bromine if the reducing powder has not fallen toa minimum after an equal weight of bromine has re-acted. The oxidation may be considered to take placeaccording to the equation:
RCHO + Br2 + H ,O > RCOOH + 2HBr.
After the reaction is over t h e excess of bromine isboiled off, stirring to prevent local overheating, and thegolden yellow solution is cooled and the volume meas-ured. Bromine ion is then determined in an aliquotportion, after which the calculated amount of lead car-bonate, ground to a thin paste wi th water, is added insmall portions, with stirring. A n appreciable excessof lead carbonate is to be avoided, as lead gluconate is
"Kiliatii, Ber 17, 1298 (1884).uRuff, Ber. 39, 2273 (1899).

60 ADVANCED LABORATORY MANUAL
then formed and this prevents the lead bromide fromcrystallizing out, presumably owing to the formationof a double salt. The resulting mixture is concen-trated, best in vacuo, to SCO cc , chilled, and allowed tostand in the ice box for 24 hours, after which the leadbromide is filtered off and washed with a little ice-coldwater. Traces of bromine ion in the filtrate are re-moved by adding a little freshly precipitated silveroxide or a suspension of silver carbonate, and afterfiltration hydrogen sulfide is passed in to remove minuteamounts of lead and silver ions in solution. Anysulfides formed are filtered off and the filtrate, whichnow consists of a solution of glucomc acid, is boiledwith an excess of calcium carbonate After cooling,and filtering off the excess of carbonate, the filtrate isconcentrated in vacuo to a thin syrup. The calciumsalt of gluconic acid separates on cooling and lettingstand, with occasional rubbing, and is filtered off andrecrystallized from the minimum amount of boilingwater or dilute alcohol. The yield of crude calciumsalt should be 140-50 g.
The free gluconic acid is a syrup and passes over
H H H OH H
into the lactone, HO—C — C — C — C — C — CO -r
H OH A i :>H
O
from which crystals have been obtained on long stand-
"Kiliani, loc. at., footnote, p 1300
m g "

O F ORGANIC CHEMISTRY 61
E. Oxidation with Hydrogen Peroxide:d-Arabinose,18
H H H OHl | I I OH
H O — C — C — C — C — C < (a- andI I I / H
125 g. of calcium gluconate are dissolved in 375 cc.of hot water, cooled to 35 °, and treated with an amountof hydrogen peroxide solution corresponding to 1.5atoms of active oxygen, and 25 cc. of a basic ferricacetate solution containing 5 per cent of iron (preparedby adding with vigorous stirring, a solution of 18 g. offerric sulfate in 500 cc. of water to 42.5 cc of concen-trated aqueous ammonia diluted with 150 cc. of water.After allowing to settle, sucking off and washing freefrom ammonia (Nessler test) with hot water, the pre-cipitate, which should be pressed down on the funneluntil it weighs less than 35 g., is added to 13 g. ofacetic acid, stirred until dissolved, and diluted to 50cc ) . The evolution of carbon dioxide which accom-panies the reaction, according to the equation:
H H H OH H
H O — C — C — C — C r - C —COOH + H aO a — >
H OH OH H OH
) 35,

62 ADVANCED LABORATORY MANUAL
H H H OH
1 1 1 1 O H
HO — C — C — C — C — C <H
is over in about 6 hours, and all the hydrogen peroxideis then used up The calcium acetate and ferric hy-droxide are filtered off and the solution is concentratedto a thick syrup w vacuo This is kneaded thoroughlywith 500 cc. of alcohol until the undissolved calciumsalts form a crumbly mass. The solution is poured offand the salts, which occlude appreciable amounts ofarabinose, are dissolved in the minimum amount of hotwater and again kneaded with a smaller amount ofalcohol, repeating the solution and reprecipitation ofthe salts until a test portion of the alcoholic solution,when concentrated, no longer appreciably reducesFehlmg's solution. The alcoholic extracts are com-bined and concentrated again in vacuo to a thick syrup.This is boiled out repeatedly with 90 per cent alcohol,seeding the chilled filtrates with arabinose if necessaryin order to start crystallization The yield of fairlypure sugar so obtained should be 18-21 g. An addi-tional 5 g. may be recovered by taking the calcium saltsremaimng from the alcoholic extraction, which containunchanged calcium gluconate, and putting them throughthe entire process again, using one-fourth of the initialamount of hydrogen peroxide and ferric acetate 2 5 g.more may also be isolated from the mother liquorsof the crystalline arabinose by concentrating as far as
"A ketonic acid is possibly the intermediate stage Ruff,£er 31, 1574 (1898)

OF ORGANIC CHEMISTRY 63
possible in vacuo, taking up in 95 per cent alcohol, add-ing one-quarter volume of ether, and filtering off theprecipitated calcium salts quickly with the aid of bone-black before the arabmose begins to separate.
The sugar may be freed from traces of ash by re-crystallization from one-half its weight of water, withthe aid of bone-black. It should then form rhombicprisms which melt at 158.5-9.5° when anhydrous and
show [<*]?) = —105.1 ° on coming to equilibrium
(c = 9.4253).

64 ADVANCED LABORATORY MANUAL
VII . F O R M A T I O N O F H E T E R O C Y C L E SAND DYES.
A. Diethylbarbituric Acid, "Veronal," "Barbital,"
H N — C = O l
1 1
o = c c<I I 2
H N — C = OI I C2HB
N — C = O
1. Diethylmalonic Ester, (CaHB)2C(COaC2H0)a .The essential requirement m the preparation of
diethylmalonic ester and "veronal" is that all reagentsmust be absolutely anhydrous, otherwise the yields willbe vanishingly small. All reagents and solutions musttherefore be protected from the moisture of the air afterdrying.
Required are ioo g. of carefully dried and freshlydistilled malomc ester, prepared according to the direc-tions in the elementary text books, 28 8 g. of sodium(2 molecular equivalents), 380 cc of absolute alcohol(dried by adding to ordinary absolute alcohol enoughsodium to combine with 1 per cent of water and distill-ing), and 195 g of carefully dried and freshly distilledethyl iodide (2 molecular equivalents). One-half of thesodium is dissolved m one-half of the alcohol under areflux condenser surmounted by a calcium chloridetube, the malonic ester added with stirring, and then
1 The writer is indebted to Dr F A. Taylor of the RockefellerInstitute for Medical Research for placing at his disposalthe results of working over the method of Fischer and Dilthey,Ann. 335, 338 (1904)-

OF ORGANIC CHEMISTRY 65
slowly through a dropping funnel 1 equivalent of theethyl iodide. The mixture is finally boiled, and whenneutral to moistened red litmus the second half of thesodium is dissolved in the remainder of the alcohol,the neutral reaction mixture added, and the balance ofthe ethyl iodide slowly dripped in, after which the mix-ture is boiled over night, when it should again be neu-tral. As much alcohol as possible is distilled off, etherand water are added, the two layers separated, and theethereal solution is dried over the sodium sulfate.112 g. of ester boiling at 220-20 should be obtained, or83 per cent of the theory, and on redistillation, 102.5 g.boiling at 221-2 °.
2. Diethylbarbituric acid,16 g. of sodium are dissolved in 300 g. of absolute
alcohol (dried as in 1), the solution cooled to roomtemperature, and 50 g of diethylmalonic ester added.20 g. of pulverized and carefully dried urea are thendissolved in the mixture by gentle warming and thewhole is heated in an autoclave at 1080 for 5 hours(internal temperature) The precipitated sodium saltof diethylbarbituric acid,
NaCO — N <
(C aH 6 ) 9C< CO,CO — N <
H
is filtered off, washed with alcohol, dissolved in water,and converted into the free aad by adding concentratedhydrochloric acid. After recrystallization from waterthe yield is 27.5 g. melting at 183-50, while Fischerand Dilthey give 10,1 ° (corr.) for the pure product
The acid so obtained is not quite pure, and is recrya-tallized from 95 per cent alcohol. About one-half of
66 ADVANCED LABORATORY MANUAL
the quantity recrystallized is obtained in the first crop,and the purity of this should be controlled by melting-point determinations and analyses for carbon, hydrogen,and nitrogen (Kjeldahl). The fractions obtained byconcentrating the mother liquors do not give such satis-factory analyses
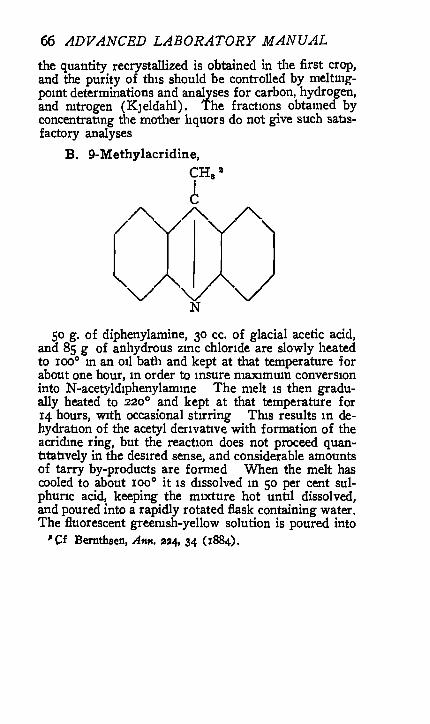
B. 9-Methylacridine,
\ \
\
\
N
50 g. of diphenylamine, 30 cc. of glacial acetic acid,and 85 g of anhydrous zinc chloride are slowly heatedto ioo° in an oil bath and kept at that temperature forabout one hour, in order to insure maximum conversioninto N-acetyldiphenylamme The melt is then gradu-ally heated to 2200 and kept at that temperature for14 hours, with occasional stirring This results in de-hydration of the acetyl derivative with formation of theacridine ring, but the reaction does not proceed quan-titatively in the desired sense, and considerable amountsof tarry by-products are formed When the melt hascooled to about ioo° it is dissolved in 50 per cent sul-phuric acid, keeping the mixture hot until dissolved,and poured into a rapidly rotated flask containing water.The fluorescent greenish-yellow solution is poured into
'Cf Bernthsen, Ann. 224, 34 (1884).

OF ORGANIC CHEMISTRY
a large vessel and the resinous residue extracted 4-6times with hot, very dilute sulphuric acid, adding thesolution to the original aqueous extract. The coldacid solution is finally filtered with the aid of bone-black and the 9-methylacndine precipitated by addingammonia. It is apt to separate amorphous at first, butcrystallizes on standing, and is then filtered off, washedwith water, and recrystallized from 85 per cent alcohol.It forms greenish brown needles which melt at 1140,and the yield should be about 25-30 g Furtheramounts may be obtained by diluting the alcoholicmother liquors and recrystallizing the impure productwhich separates.
In order to obtain the base in a state of absolutepurity Koenigs 8 dissolves 10 g. in 50 cc. of alcohol andadds a solution of 10 g of tartanc add in 100 cc. ofalcohol. The tartrate which separates is washed withalcohol, dissolved in water, and converted into the basewith ammonia. The acndme should then form green-ish yellow needles which melt at 117-80.
C. Quinicine (Quinotoxine) Hydrochloride,
O H
CHftO
HBCUCHCH:CHaCHaN* Koenigs, Ber. 32, 3607 (1899).
H

68 ADVANCED LABORATORY MANUAL
50 g. of quinine or quinidine (calculated to the an-hydrous basis) are dissolved in ioo cc. of 50 per centacetic acid and 600 cc of water and boiled for 32 hoursunder a reflux condenser.
Rabe * has found that quinine and quinidine
H
C H C H r C H
CHflO
are stereoisomers differing in the spatial relationshipsabout the asymmetric carbon atom (3) and identicalas far as asymmetric carbon atoms (1) and (2) areconcerned.6 Therefore, in the remarkable intramolecu-lar change into quinicine effected as above by boilingwith weak acids, or by heating the acid sulfate of thebase, it would be expected that both quinine andquinidine would yield the same quinicine, as the asym-metry about carbon atoms (3) and (4) disappears.This is actually found to be the case; and it is accord-ingly a matter of indifference whether quinine orquinidine be used as starting material.
*Rabe, Ann. 373, 91 Os^)"For a possible elucidation of the asymmetry at (4), see
King and Palmer, / . Chem. Soc. tax, 4577 (ipes).

OF ORGANIC CHEMISTRY 69
The solution, which is now colored a deep brownishorange, is chilled, made alkaline with sodium hydroxide,and the base extracted with ether. As cjuinicine itselfhas never been obtained crystalline it is isolated as thehydrochloride.8 The ethereal solution is accordingly,carefully dried over crushed sodium hydroxide, con-centrated, and the viscous residue taken up in about3 volumes of absolute alcohol. It is then neutralized,with cooling, by carefully adding absolute alcohol satu-rated at o° with dry hydrochloric acid gas, until a testdrop proves neutral to wet litmus paper. It is a goodplan to keep about one-tenth of the solution separate,and if the endpoint is accidentally slightly overrun, anexcess of the base can be restored by adding the portionwithdrawn. The neutral solution should be protectedfrom the moisture of the air and allowed to stand inthe ice-box with occasional rubbing. In case the saltdoes not crystallize under these conditions crystals forseeding may be obtained by adding dry ether to a littleof the solution until just turbid, and letting stand in awell-stoppered test tube.
The yield should be 37 g. Recrystallized from abso-lute alcohol the hydrochloride forms aggregates ofminute leaflets which melt with decomposition at 180-2 °
and give [ a ] ^ = + 13.70 in water, c = 1.861, while
anhydrous quinine hydrochloride has a specific rotationof — 149 8° and anhydrous quinidine hydrochloride oneof + 2 0 0 8° in water.
9 Heidelberger and Jacobs, / . Am. Cham. Sac, 41,83a (1919).

70 ADVANCED LABORATORY MANUAL
D. Meldola's Blue,
This representative of the oxazine dyes is prepared asfollows: 15 g. of (3-naphthol are dissolved on the waterbath under a reflux condenser in 50 g. of 95 per centalcohol. To the boiling solution is gradually added asolution of 15 g. of /j-nitroso-dimethylanihne hydro-chloride in 100 g. of hot 50 per cent alcohol, care beingtaken that boiling does not cease. As soon as themixture becomes a pure violet in color (about 20minutes) it is removed from the bath and allowed tostand over night at room temperature Longer heat-ing does not improve the yield. The oxazonium chlo-ride separates as black needles with a brassy reflex,and is sucked off and washed with a little alcohol.To the 7 g of impure dye so obtained 3 g. may beadded by cautious addition of an equal volume ofether to the filtrate. The dye is analytically pure onlyafter two recrystallizations from alcohol, and thenforms narrow, black platelets with a greenish metallicluster. It then sinters slightly, but does not melt,

OF 0RGAM1C CHEMISTRY 71when heated to 265 °. The aqueous solution is violetin color. £. Rosinduline,
//\
NNHa
XCBHB aThis representative of the large group of phenazinedyes is prepared essentially according to Fischer andHeppT as follows:28 g. of aniline are diazotized in the cold with 75 cc.of water, 75 cc. of concentrated hydrochloric acid, and21 g of sodium nitrite in a little water, and the solutionis added slowly, with stirring, to a lukewarm solution°f 43 S °f ct-naphthylamine in 500 cc. of 90 per centalcohol. In order to insure complete conversion of theresulting pasty mass of the dye into the hydrochloride200 cc. of 1:1 hydrochloric acid are added and thestirring continued for one-half hour.One part of the dried phenylazo-a-naphthylaminehydrochloride, 2 parts of aniline, and 4 of alcohol areheated for 6-8 hours at 160-70 ° in sealed tubes, thecondensation probably taking place as follows:1 Fischer and Hepp, Ann. 956, 236 (1890).

ADVANCED LABORATORY MANUAL
+ CflH8NHa
NH..HC1
> Rosinduline + NH a + 2H,
the hydrogen not appearing as such, but instead reduc-ing a portion of the azo dye.
The contents of the tubes are distilled with steam toremove aniline and alcohol, and the residue is boiled outrepeatedly with water. The intensely colored solutionso obtained is allowed to stand for several days, pouredoff from the tar which settles, acidified with hydro-chloric acid, and the dye precipitated by the carefuladdition of salt. It forms long red needles with agreen reflex, and contains 3 5 molecules of water ofcrystallization. To be obtained analytically pure itmust be repeatedly purified by solution in hot water,and crystallized by carefully adding saturated saltsolution to incipient turbidity, and seeding, washingthe crystals so obtained with small amounts of ice-coldwater.

OP ORGANIC CHEMISTRY 73
A by-product, rosindone,
which is also formed in the reaction and is insolublein water may be isolated from the insoluble tarryresidue by extracting with alcohol and recrystallizmgthe product from a mixture of alcohol and toluene. Itforms red hexagonal plates with a slight green reflex,melting at 259 °.

74 ADVANCED LABORATORY MANUAL
VIII. SUGARS, PROTEINS, A N DAMINO ACIDS.
A. Separation of Stereoisomers— (5-Glucosefrom Glucose.1
The pyridine required for this preparation must beabsolutely anhydrous, and may be obtained by boil-ing pyridine with an excess of calcium oxide for 5 or6 hours and distilling immediately before use
12 g. of anhydrous, crystalline glucose, which con-sists of an equilibrium mixture of the a- and (3- forms,
H H H OH H
H O —C—C—C—C—C—C<
U 1 'OH
HO
H
a-Glucose
H H H OH H
— C — C — C — C — C — C < 2
H k H
^-GlucoseO
H
xCf. Behrend, Ann 353, 106 (1907). Thanks are due Dr.G. M Meyer of the Rockefeller Institute for Medical Researchfor several improvements on the original methods in this andsection B.
'Cf. Boeseken, Ber 46, 2622 (1913); Pictet, HelveticaChim. Ada 3, 649 (1920).

OF ORGANIC CHEMISTRY 75
are dissolved in 30 g. of hot, dry pyridine, boiled for10 minutes under a reflux condenser surmounted bya tube containing calcium oxide, cooled to body tem-perature, and shaken in a shaking machine in a coolroom until crystallization has taken place. Afterhaving stood over night the crystalline deposit of(5-glucose is filtered off and washed first with a littledry pyridme, then with absolute alcohol, and finallywith dry ether. After drying in a desiccator toremove the pyridine of crystallization, the yieldshould be about 4 g. of sugar melting at 148-50°.When dissolved in water, |3-glucose exhibits mutarota-tion, that is, it slowly reverts to the original equilibriummixture, and this change may be followed by observingthe optical rotation of an aqueous (for example, 4per cent) solution from time to time until the constantequilibrium value has been attained. For instance,Behrend read the rotation after 8, 18, 28, 38, etc.,minutes, and found that equilibrium was attainedwithm 23 hours at a value of [a] j) = + 52.70. From
the curve plotted from his observations, the value ofpure P-glucose by extrapolation to o time was foundto be + 20.70. The student should repeat these ob-servations and check up the values so obtained withthose given in Behrend's article.
^-Glucose Penta-acetate.8
H H H OAc HI I I I I
AcO — C — C — C — C — C — C <
J.A XX XX \-/J.l.\* 4.XI I I I I OAc
_ C — C — C — C — C — 1k Ac [ t O
•Cf. Behrend, he. cit.

76 ADVANCED LABORATORY MANUAL
1.8 g. of finely powdered (3-glucose are added to anice-cold mixture of 7 g. of acetic anhydride and io g.of dry pyndine The mixture is frequently shakenwith occasional cooling until all the sugar is dissolved,after which it is allowed to stand at .room temperaturefor 2 days. It is then poured into ioo g. of a mixtureof ice and water and stirred vigorously until thegummy precipitate crystallizes The acetate is suckedoff after 2 hours, dried in a desiccator, and recrystal-lized from 40 cc. of 95 per cent alcohol. The yieldshould be about 2.9 g., melting at 130-10. According
20to Hudson and Dale * [a] ~ of a 7 per cent solution inchloroform is + 3.80.
The corresponding a- compound obtained similarlyfrom a-glucose5 melts at 111°, and gives a value of
20-f-101.60 for [a]-Q m chloroform.
B. Hydrolysis of a Biose— Galactose fromLactose.
200 g. of milk sugar are dissolved in 500 cc. of warmwater. 5.5 cc. of concentrated sulfuric acid are added,and the solution is boiled under a reflux condenser for2 hours, hydrolysis occurring according to the scheme:
4 Hudson and Dale, / Am. Chem Soc. 37, 1265•Behrend and Roth, Ann 331, 379 (1904).

OF ORGANIC CHEMISTRY 77
H OH
V I -
H — C —OHHO — C — HH-AH-A_
O
o
H —C —H
OH
Lactose
+ HaO —H OH
H — C OHHO —C —H
H - A
H — C — OH
H — C — H
OHGlucose
6
H
- C
HO
H —C — OH
- A - H
C —H
H — C —OH
— C —H H
H OH
H-L OHHO —C —H
A-HC —OHH
H-cLHHG&lactose

78 ADVANCED LABORATORY MANUAL
The mixture is boiled a few moments longer withbone black to remove most of the color developed, andfiltered hot. The sulfunc acid is then quantitativelyremoved by means of barium hydroxide, of whichabout 30 g. of the dried base are necessary. Thesolid compound can be added in the beginning, finish-ing with a hot, concentrated solution and avoiding anexcess of alkali, to which the hexoses are very sensitive.6 cc. of glacial acetic acid are then added, the mixtureis again decolorized with bone black, filtered, and con-centrated in vacua to a thick syrup, of about 120 cc.in volume, immersing the flask in a water bath, thetemperature of which does not exceed 60-700. Beforethe syrup cools, one to one and one-half volumes ofglacial acetic aad are added, and the mixture is stirredwell, cooled, and rubbed, or better, seeded with a crys-tal of galactose. After three hours the sugar is suckedoff on a Buchner funnel and washed with a little coldacetic acid, then with a very little alcohol, and finallywith ether. The yield should be 50 g.
The purity of the galactose obtained should be con-trolled by a determination of the optical rotatory
power, [ a ] 2 ^ of a 10 per cent solution of the pure
sugar at equilibrium in water is + 81.50.
C. Preparation of d/-Alanine,CH2CHNH2COaH.f l
While the alanine obtained by hydrolysis of proteinsis the dextro- isomer, synthetic methods, of course,yield the dl- mixture, from which the d- isomer maybe separated as given below.
50 g. of a-bromopropionic acid (p. 20) are slowly•Acknowledgment is due Dr L. A. Mikeska of the Rocke-
feller Institute for Medical Research for details of this methodand of slight modifications of Fischer's procedure in the fol-lowing preparations.

OF ORGANIC CHEMISTRY 79
stirred into two volumes of concentrated aqueousammonia cooled by means of a freezing mixture. Thesolution is then transferred to an autoclave and heatedat ioo° for three hours, after which it is concentratedto dryness in vacua. The ammonium bromide m theiesidue is extracted by heating repeatedly with smallportions of 95 per cent alcohol and decanting the hotsolution, until a test shows that very little more is beingremoved. The residue of crude alanine is dissolved mthe minimum amount of hot water and about fivevolumes of alcohol are then stirred in, causing thealanine to crystallize quickly. The product is filteredoff, dried, and the purity controlled by a nitrogen de-termination (Kjeldahl) and a test for bromine ion,as well as a determination of amino nitrogen by theVan Slyke or Soerensen method The yield shouldbe about 50 per cent of the theory.
D. Separation of <f/-Alanine into its OpticalIsomers.7
Of the methods for the separation of racemic mix-tures into their components one of the most useful isthe formation of salts with optically active acids orbases. Early attempts to apply this method to theamino acids failed, since these compounds are not onlyweak acids, and therefore form rather unstable saltswith bases, but are also weak bases, and form unstablesalts with acids. Fischer overcame this difficulty byconverting the amino acids into their benzoyl deriva-tives, which he found to be stronger acids and soformed stable salts with optically active bases such as thealkaloids strychnine and brucme. Ordinary methodsof benzoylation failed in this instance, but good yieldswere obtained in the presence of sodium bicarbonate.
TE, Fischer, Ber. 3a, 2451 (1899).

80 ADVANCED LABORATORY MANUAL
1. Benzoylation of Alanine.To a solution of 45 g. of J/-alanine in 450 cc. of
water 330 g. of sodium bicarbonate are added, andthen 218 g (3 mols ) of benzoyl chloride in aboutten portions The reaction mixture is shaken vigor-ously and frequently cooled with ice-water. Whenall of the chloride has been added the mixture isallowed to stand at room temperature for 4 to 5hours, with frequent shaking, and is then filtered Anybenzoyl chloride in the nitrate is shaken out with ether,after which the aqueous layer is acidified to Congo redwith 1.1 hydrochloric acid, causing the separation of amixture of benzoic acid and benzoylalanine. Afterletting stand over night in the cold the crystals arecollected, dried, and boiled out repeatedly with ligrointo remove the benzoic acid. The yield of crude productshould be over 90 g. After recrystallization fromwater, 70 g. of pure benzoylalanine, melting at 165-60
(corr.), are obtained.
2. /-Benzoylalanine.65 g. of the d/-benzoyl compound and 157 g of
brucine (hydrate) are dissolved in 240 cc. of hotwater After 15 hours at o° the brucine salt whichhas separated is filtered off and purified by recrystal-lizing twice from 100 cc. of water, filtering each cropof crystals only after 12 hours in the ice box. Thepurified salt forms radiating masses of long, pointedplates, and should be obtained in a yield of about 87 g.
Recovery of Brucine.801 g. of the brucine salt are converted into Z-benzoyl-
alanine by dissolving in 240 cc of hot water andstirring in 140 cc. of normal sodium hydroxide solu-tion. The mixture is cooled to o° in order that thebrucine may be as completely removed as possible, and

OF ORGANIC CHEMISTRY 81
the alkaloid is filtered off and washed with a little ice-cold water. The filtrate is then acidified with 140 cc.of normal hydrochloric acid solution, concentrated tosmall volume in vacuo at 40-500, and cooled. 19 g.of J-benzoyl derivative should separate as an oil whichsoon crystallizes. Recrystallized from 5 parts of waterit forms glistening plates which melt at 150-1° (corr.),considerably lower than the racemic compound. If thecrystallizations have been carefully carried out andthe benzoyl compound is free from the rf-isomer,
20[a] - , of a solution of 0.75 g. in 3.9 cc. of normal
potassium hydroxide solution and 2.9 cc. of watershould be — 37-3°-
3. /-Alanine.5 g of the benzoyl derivative are hydrolyzed by
hearing with 25 cc. of 20 per cent aqueous hydrochloncacid in a boiling water bath for 5 hours. The ben-zoic acid and unchanged benzoylalanine which crys-tallize pn cooling are shaken out with ether, afterwhich 1 g. of benzoylalanine may be rocevered fromthe ethereal residue as in the original preparation ofthis substance. The aqueous acid solution containingthe hydrolyzed ammo acid is evaporated dry, leavinga crystalline residue of /-alanine hydrochlonde. Thisis purified by solution in a small amount of warmabsolute alcohol, addition of a few drops of alcoholichydrochloric acid, and gradual addition of ether. Theresulting delicate needles are dissolved in 30 parts ofwater and boiled 15 minutes with an excess of leadoxide prepared by pouring lead acetate solution intowarm barium hydroxide solution in excess, filtering,and washing. After the alanine salt-lead oxide mix-ture has been boiled and a filtered test portion showsonly traces of chlorine ion the mixture is cooled and

82 ADVANCED LABORATORY MANUAL
filtered. The filtrate is freed from lead salts withhydrogen sulfide, and evaporated to dryness Thealmost pure Z-alanine so obtained is dissolved in 5parts of water, warmed on the water bath, and treatedwith alcohol until crystallization begins. The ammoacid separates as rods or prisms on cooling. Whenrapidly heated it melts at 297 ° with vigorous gasevolution. An 8 8 per cent solution in water rotatesthe plane of polarized light 0 2 1 0 to the left.
4. d-Alanine.The mother-liquor from the first crop of the crude
brucme salts of Z-benzoylalanme contains the d-com-pound together with smaller amounts of the Z-com-pound which render purification as the brucine saltextremely difficult The following procedure is ac-cordingly adopted: The original mother-liquor is dilutedwith two volumes of water, made alkaline with 250 cc.of normal sodium hydroxide solution, and cooled.The alkaloid is filtered off and the filtrate again acidi-fied with 250 cc. of normal hydrochloric acid solution.After one day 9 g of iZ-benzoylalanine separate, whichare of no use as far as the preparation of the d-com-pound is concerned. The filtrate is concentrated tosmall volume in vacuo, yielding about 18 g of a crys-talline mixture consisting of about 90 per cent ofd-benzoylalanine and 10 per cent of the Z-compound.
This is best separated by means of the strychninesalts. 13.3 g. of the crystals and 23 g of strychnineare dissolved in 300 cc. of hot water and allowed tostand over night in the ice box. The strychnine saltof d-benzoylalanine separates as thick platelets, butmust be recrystallized four times from the same volumeof water before the optical rotation reaches a constantvalue and all traces of the Z-compound are eliminated.The yield should then be about 20 g.

OF ORGANIC CHEMISTRY 83
d-B enzoylalanine.A solution of the pure strychnine salt in 600 cc. of
water is made alkaline with 40 cc. of normal sodiumhydroxide solution and chilled The strychnine isfiltered off and the filtrate acidified with 40 cc. of nor-mal acid and concentrated to small bulk in vacuo. 5 g.of pure c?-benzoylalanine separate on cooling. The
30melting point should be 150-1° (corr.), while [ a ] ^of a 10 per cent solution containing one equivalent ofpotassium hydroxide is + 3 7 - i ° .
(/-Alanine.Hydrolysis of the tf-benzoyl derivative is accom-
plished as in the case of the Z-compound, and theS-alanine is isolated in the same way. It is very similarin its properties to the Z-compound except that theslight rotation of polarized light is in the oppositedirection. The synthetic tf-alanine is identical in everyrespect with the d-alanme isolated from the hydrolyticproducts of silk and other proteins.
E. Crystalline E g g Albumin.8
The initial requirement for the success of this prepa-ration, which may, in any event, require several weeksfor completion, is that the eggs used be not more thantwo or three days old. The albumin originally presentin the egg rapidly undergoes a modification which soon
'Hopkins, 7. Physxol, 25, 306 (1809-1900). Soerensen,Compt. rend. lab. Carlsberg, 12 (1917) ; Chem. Abstrs. 12,2575 (1918).
Acknowledgment is gratefully made to Dr. Lillian E. Bakerof the Rockefeller Institute for Medical Research for the useof her notes on this preparation. Hopkins' original method,modified In several details, and made more precise, was foundto give crystalline preparations more consistently than Soeren-sen s modification.

84 ADVANCED LABORATORY MANUAL
makes it increasingly difficult and finally impossiblto obtain crystals of this sensitive protein. As few afour fresh eggs may be used, but the conditions ccrystallization are somewhat more easily realized whemore are taken.
Removal of Globulins.The whites of the eggs are carefully separated froi
all traces of yolk and the volume is measured. Aequal amount of saturated ammonium sulfate solutiois slowly added while the mixture is beaten with aegg-beater in order to break up the membranes enclosing the substance of the egg-white. To ensure conplete precipitation of the globulins the mixture iallowed to stand over night in the cold, and is thefiltered through large folded filters, or better, certnfuged, as the precipitated globulin rapidly clogs trfilters. The volume of the clear yellowish filtratemeasured and recorded.
Crystallization.io per cent acetic acid is next added slowly froi
a burette, with vigorous stirring, until a permanenwell-defined precipitate fills the liquid in amount suJficient to make it actually milky in appearance, ncmerely opalescent. When this point has been reachei cc of io per cent acetic acid for each ioo cc. recordeabove is slowly stirred in, giving rise to a heavamorphous precipitate of albumin which crystallizemore or less rapidly. The process is accelerated binoculation with a crystal of egg albumin if availaband letting the mixture stand in the ice-box witoccasional stirring.
If the eggs were very fresh and the conditions cprecipitation just right, crystallization may be qompleiwithin a few hpurs, but several days may be require<

OF ORGANIC CHEMISTRY 85
In case microscopic examination fails to reveal crystals,aliquot portions of the mixture are removed, one ofwhich is treated with a small measured amount of acid,and another with alkali in order to find more favorableconditions for crystallization if possible. If these arefound, the acidity of the entire mixture is adjustedaccordingly, otherwise it is better to begin again.
Theoretically, the precipitation of a protein, which isbuilt up of ammo-acids and functions as a typical atn-photenc electrolyte, should best be accomplished atthe isoelectric point, that is, at the hydrogen ion con-centration at which the acidic and basic functions ofthe protein are equal and at a minimum.9 Accordingto Soerensen, the iso-electric point of egg albumin isat pH 4.8,10 and while precipitation would undoubtedlybe most complete at this point, it is not necessarilythe most favorable for crystallization, as the egg al-bumin crystals are not isoelectric protein, but a com-pound of this with the sulfate ion, and perhaps theammonium ion as well.11
The mixture should be allowed to stand at least 24hours in the cold after all amorphous material hasbecome crystalline, in order to ensure a maximum yield.A little toluene may also be added to retard putre-faction.RecrystalUzation.
The crystals are finally filtered or centrifuged off,drained as well as possible from mother-liquor, and
0 For a full discussion of the consequences of this viewpointsee Loeb, Protetns and the Theory of Colloidal Behavior,McGraw-Hill, 1922; MichaeHs, Wasserstoifionen-komentra-tton. Berlin, 1914.M It is assumed that the student is familiar with the meaningof this expression. If not, he is referred to W. M. Clark'sbook, The Determination of Hydrogen Ions, Williams andWilkins, 1920.u This point is fully discussed by Soerensen.

86 ADVANCED LABORATORY MANUAL
dissolved in as small a volume of water as possibliThe amount required is surprisingly small, so thjonly a little is added at a time to the crystals, witconstant stirring, and addition of water is stoppebefore the solution is entirely clear It will be founthat a small portion of the albumin is usually "d<natured," or rendered insoluble by the crystalhzatioprocess, and this must be filtered off The pH of trslightly opalescent filtrate should then be adjusted t5.2 to 5.412 Success in recrystallization of the albumidepends entirely on the hydrogen ion concentratioiand if the reaction is too acid a gelatinous mass pr<cipitates from which it is impossible to obtain crystalIf the reaction is too alkaline, on the other hand, verlittle of the albumin comes out
After the acidity of the concentrated albumin sohtion has been adjusted, saturated ammonium sulfaisolution is stirred in, drop by drop, until enough albtmin remains precipitated to make the solution mill*in appearance Again the amorphous albumin gradially changes over into the crystalline form, and ttprecipitate increases in amount when the solutionkept in the cold and stirred from time to time Itbest, at this point also, to have a little toluene preset
Three recrystallizations are sufficient to remove sbut traces of the amorphous "conalbumin" and oth<
** Young, Proc Roy Soc [B] 93, 15 (1921-2) gives 4but good crystallization could not fee obtained at this pH.is possible, however, that eggs from different sources vaiso that the student may have to determine the optimum comtions for himself.
A standard of pH 52 may be prepared by mixing 4.2 <of 0.2 N acetic acid with 158 cc. of 02 N sodium acetawhile the 5.4 standard contains 29 cc. of the add to 1;cc of sodium acetate solution Bromo-cresol purple mayused as indicator The range 5.2 to 5.4 was determined witout correcting for the protein or salt error. For detailspH determinations the student is referred to Clark's book.

OF ORGANIC CHEMISTRY 87
impurities originally present, but each time a smallamount of the crystalline albumin is usually trans-formed into an insoluble modification and must beremoved. The crystals are stable only in the presenceof the mother-liquor from which they are deposited,but an aqueous solution free from salts may be ob-tained by dialysis against water in the cold with theaid of a parchment or collodion membrane. Excel-lent collodion membranes for the dialysis of proteinsolutions may be prepared with the aid of one of themixtures suggested by Eggerth,18 e g-, 7 g of "Par-lodion" to 60 cc. of ether, 30 cc. of alcohol, and 10 cc.of glacial acetic acid. This mixture yields very toughmembranes which are easily removed, after the addi-tion of water, from the vessel in which they areformed. If a concentrated salt-free albumin solutionis desired, the bag may be prepared m a 50 cc. test-tube,loosely filled with the albumin solution, and clampedat the open end with a rubber-faced screw pmchcock.14
The pressure developed when the solution is dialyzedin this way prevents undue dilution. Complete re-moval of the salts may be tested for in the dialysis waterby the Nessler method At least 4 or 5 daily changesof water are necessary before the test becomes negativeIn winter the dialysis may be completed more rapidlyby immersing the bag in running tap water and finish-ing after 48 hours with one or two changes of distilledwater.
20[•a] D of a 10 per cent aqueous solution of pure
crystalline tgg albumin is about —30.50 .M Eggerth, / B\ol. Chem. 48, 203 (1921).MThis method of dialysis under pressure was suggested by
Adair, Barcroft, and Bock, / Physxol 55, 333 (1921).

88 ADVANCED LABORATORY MANUAL
IX. P R E P A R A T I O N AND R E A C T I O N S OFORGANOMETALLIC COMPOUNDS.
(See also p. 2gi)
A. The Direct Arsenation of Phenol.p-Hydroxyphenylarsonic Acid.
AsOflH,
OH
Just as phenol can "be nitrated or sulfonated, it isalso possible to introduce the arsonic acid residue intcthe molecule by treatment of phenol with arsenic acidThe following conditions have been found very satisfactory on a laboratory scale: x
Arsenation.480 g. of 80 per cent aqueous arsenic acid are boilec
in an open flask, allowing the water to escape untithe temperature reaches 1500. 200 g. of molterphenol are then quickly poured in and an air-condenser is attached, after which the flask is placed iran oil bath kept at 155-60°. The contents of the flasl
*Ger. pat 205,616, Jacobs and Heidelberger, / . Am. ChemSoc. 41, 1440 (1917)

OF ORGANIC CHEMISTRY 89
boil steadily (a few fragments of porous tile should ofcourse be added), rendering mechanical stirring un-necessary. The air condenser (a long glass tube ofabout 1% cm. bore) acts as an efficient reflux, prevent-ing undue loss of phenol. The mixture is boiled for 7hours, taking care, of course, that it does not boil sohard as to escape from the top of the condenser. Ini-tially two layers are formed, but the mixture finallybecomes homogeneous and darkens somewhat.
Purification.It is diluted with about 2 liters of water, resulting
in the precipitation of a small amount of tar, and,without filtering, treated, with vigorous stirring, witha hot, strong solution of barium hydroxide until justneutral to litmus. It is important to avoid an un-necessary excess of barium hydroxide, for althoughthe neutral barium salt of p-hydroxyphenylarsonicacid is soluble under these conditions, an excess maylead to the formation of basic salts which would beprecipitated with the barium arsenate. The clear,almost colorless filtrate is then treated with just enoughsulphuric acid to remove the barium ion present, a pre-liminary heating greatly facilitating the subsequentfiltration of the barium sulfate. The precipitationshould be followed during the addition of the sulfuncacid by tests on filtered samples, and it is a simplematter to strike the point at which the nitrate no longerreacts for either barium or sulfate ion.
The filtrate is next concentrated to about one liter,preferably in vacuo, and the further manipulations willdepend on whether the student will proceed to thesynthesis of "salvarsan (arsphenamine)," in which casetwo-thirds of the solution should be worked up for thesodium salt according to method 2, or whether he willstop at the isolation of the ^-bydroxyphenylarsonic acid,
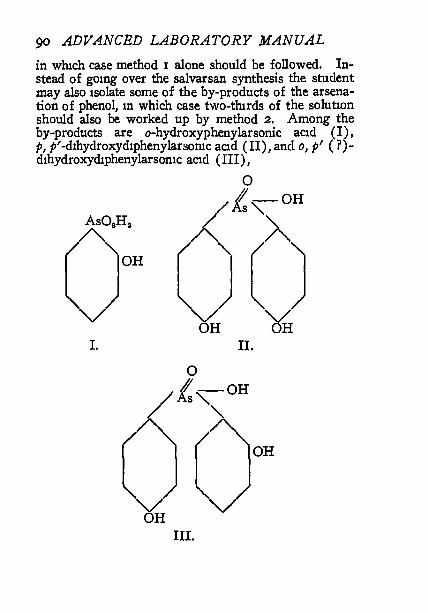
90 ADVANCED LABORATORY MANUAL
in which case method I alone should be followed. In-stead of going over the salvarsan synthesis the studentmay also isolate some of the by-products of the arsena-tion of phenol, in which case two-thirds of the solutionshould also be worked up by method 2. Among theby-products are o-hydroxyphenylarsonic acid ( I ) ,p, p'-dihydroxydiphenylarsonic acid ( I I ) , and 0, p' ( ?)-dihydroxydiphenylarsonic acid ( I I I ) ,
OH
AsOaH
OHIII .

OF ORGANIC CHEMISTRY 9*
and a full theoretical discussion of the subject and direc-tions for isolating the by-products will be found in theoriginal article referred to above.
Isolation.1. The solution (or one-third of it if the alter-
natives given above are followed) is further concen-trated in vacuo to a thick syrup consisting of a mixtureof free arsonic acids. This is dissolved in about 3volumes of hot glacial acetic acid, yielding a faintlycolored solution which gradually sets to a thickpaste of colorless crystals of /j-hydroxyphenylarsonicacid on chilling and rubbing. After standing 24 hoursin the ice-box the acid is filtered off and washed withsmall portions of cold glacial acetic acid. The yieldfrom 200 g. of phenol averages 40 g, or considerablyless than if the acid is isolated as the sodium salt. Thefree acid melts at 170-30, with preliminary softening.
2 Two-thirds of the concentrated solution are neu-tralized to litmus with sodium hydroxide and the solu-tion concentrated further in vacuo until partial crys-tallization of the sodium ^-hydroxyphenylarsonate oc-curs The mixture is then heated on the water bathuntil the salt redissolves, with the addition of a verysmall amount of hot water if necessary, and then treatedwhile still hot with several volumes of alcohol until aslight permanent turbidity is observed. The sodiumsalt quickly separates, and after several hours of thor-ough chilling m a freezing mixture it is filtered off andwashed with small portions of chilled 85 per cent al-cohol. The yield from two-thirds of the original solu-tion should be about 80 g., and if the manipulations havebeen carefully carried out the salt 19 pure, giving notest for barium, arsetiate/or sulfate ions.
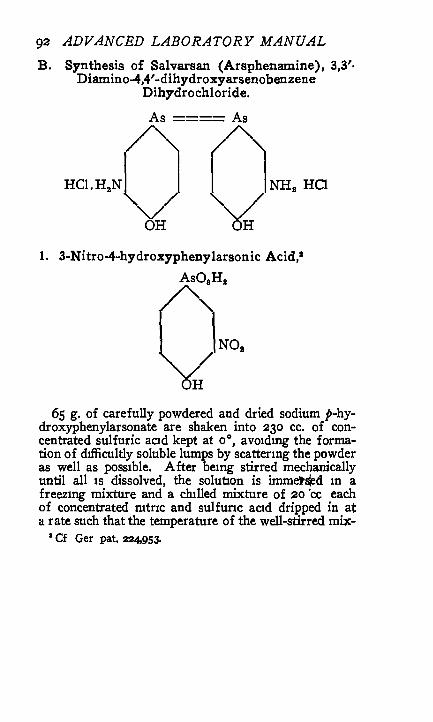
92
B.
ADVANCED LABORATORY MANUAL
Synthesis of Salvarsan (Arsphenamine), 3,3'-Diamino-4,4/-dihydroxyarsenobenzene
Dihydrochloride.
As = = = = = As
HC1.H2N\
OH
NH0 HC1
OH
1. 3-Nitro-4-hydroxyphenylarsonic Acid,2
AsO8H2
NO,
H
65 g. of carefully powdered and dried sodium />-hy-droxyphenylarsonate are shaken into 230 cc. of con-centrated sulfuric acid kept at o°, avoiding the forma-tion of difficultly soluble lumps by scattering the powderas well as possible. After being stirred mechanicallyuntil all is dissolved, the solution is immefrsjed m afreezing mixture and a chilled mixture of 20 'cc eachof concentrated nitric and sulfuric acid dripped in ata rate such that the temperature of the well-stirred mix-
1 Cf Ger pat. 224,953.

OF ORGANIC CHEMISTRY 93
ture varies between — 5 0 and o° When the mamreaction is over the vessel is removed from the freez-ing mixture and the stirring continued until the tem-perature reaches 10 °. The solution is then pouredinto 700 g. of ice and water, stirred well, and allowedto stand over night in the ice-box The bright yellowarsonic acid is filtered off, washed with small portionsof ice-cold water, and dried. The yield should beabout 38 g
2. Reduction of 3-Nitro-4-hydroxyphenylarsdnicacid to 3,3'-Diamino-4,4'-dihydroxyarsenoben-zene.
In the reduction of an arsonic acid, the end productdepends largely on the reducing agent used. For in-stance, sulfur dioxide and tiydnodic acid together con-vert the arsonic radical into the arsinoxide group,— As = O, while stronger reducing agents such ashypophosphorous acid (also in the presence of hydriodicacid) or sodium hyposulfite (hydrosulfite) effect reduc-tion to a still lower stage, that of the arseno- compounds— As As—, in which two molecules of the arsonicacid are linked through the arsenic atoms after reduc-tion. Zinc dust and hydrochloric acid reduce arsonicacids even more powerfully, giving arsines, RAsH2.
When the reduction of a nitrophenylarsonic acid isin question the state of affairs is still more complicated,for it becomes possible to reduce either the nitro- orthe arsonic-aad group without affecting the other, or toreduce both simultaneously. For example, ferrous sul-fate and alkali give ammophenylarsonic acids, while sul-fur dioxide and hydriodic acid together permit the prepa-ration of certain nitrophenylarsinoxides. Sodium hypo-sulfite (hydrosulfite) in excess, on the other hand, re-duces the nitro- to the amino- group, with simultaneousf ormatign of arseno compounds, This rea^ge^t was use«i

94 ADVANCED LABORATORY MANUAL
by Ehrlich and Bertheima for the direct preparation of"Salvarsan," and while other methods are said to yielda product of lower toxicity,4 the method is given hereas well for its historical interest as because it avoidsthe isolation of an intermediate product and gives satis-factory yields when carefully earned out.
Reduction.A solution of 25 7 g. of magnesium chloride crys-
tals B in 650 cc. of water is poured into a 1.5 literbeaker suspended in a water bath A thermometerand mechanical stdrrer are adjusted in the beaker, afterwhich 148 g. of commercial sodium hyposulfite(hydrosulfite) (80 per cent) are stirred in, avoidingthe formation of lumps, and followed immediatelyby a cold solution of 9.9 g. of 3-nitro-4-hydroxy-phenylarsonic acid (0.038 mols.) in 225 cc. of water and6 8 cc. of 10 N sodium hydroxide The water bath isthen heated until a temperature of 55-60° is maintainedwithin the beaker A yellowish, amorphous (contraryto Ehrlich and Bertheim) precipitate soon begins toseparate, and gradually increases in amount as the reac-tion proceeds After one and a half to two hours'stirring a filtered test portion should remain clear orgive only a slight turbidity on heating to the boilingpoint. The mixture is then filtered on a Buchnerfunnel, washed well with water, and pressed downthoroughly.
Purification.The crude, very impure, water-insoluble, easily
oxidizable arseno-compound is then converted into itsdihydrochlonde (Salvarsan, Arsphenamine). The
"Ehrlich and Bertheim, Ber. 45, 756 (1912).*Cf Christiansen, / Am Chem Soc 43,2402 (1920)8 The presence of this salt in some way prevents the forma-
tion of undesirable by-products.

OF ORGANIC CHEMISTRY 95
calculated amount of strong methyl alcoholic hydro-chloric acid (0 038 mols HC1) is added to 85 cc. ofmethyl alcohol and the crude product stirred withthis until as much as possible dissolves The insolu-ble material is collected with the aid of decoloriz-ing carbon and the mixture filtered The filtrate isadded, with stirring, to one liter of chilled ether (Ehr-hch and Bertheim) or acetone and the amorphous (con-trary to Ehrlich and Bertheim) yellow precipitate iswashed with acetone or ether and dried m vacuo oversulfunc acid and paraffin, after which it still retainssolvent corresponding roughly to one molecule ofmethyl alcohol. The yield should be about 80 per centof the theory, or 7 g.
The entire reduction and purification should be car-ried out in as short a time as possible, and with theminimum exposure of the product to the air, for notonly is the arsenic of the arseno group easily oxidizable,but the orf/zo-aminophenol grouping in addition makesthe compound extremely sensitive.
The product obtained in this way is not an absolutelypure chemical individual, and usually contains an ap-preciable amount of sulfur (2 to 3 per cent), at leasta portion of which appears to be present as a sulfonicacid.8 The purity of the product may be controlledby an arsenic determination, or better, by titration withiodine after dissolving in water and acidifying slightlywith hydrochloric acid, one molecule of the arseno com-pound being1 oxidized to two of 3-amino-4-hydroxy-phenylarsonic acid under these conditions according tothe equation:
R A s : A s R + 4l a + 6HaO > 2RAsO8Ha + 8HI
If the arseno compound is to be preserved it mustbe kept in vacuum ampoules, for it undergoes oxida-
"King, 7. Chem. Soc, 1x9, 1416 (1921).

96 ADVANCED LABORATORY MANUAL
tion even in the dry slate and as the hydrochloride.Practically, the resultant alteration involves seriousdangers, as the toxicity of the drug is thereby greatlyincreased
C. Mercuric Compounds of Aniline.
1. p-Aminophenylmercuric Acetate,
NHH
The "mercuration" of aromatic compounds is of suf-ficiently general applicability to be classed with suchprocesses as bromination, nitration, and sulfonation,and is particularly easily carried out in the case ofphenols and amines.
To a solution of 63.6 g. of pulverized mercuricacetate m 320 cc. of water containing a few drops ofacetic acid to suppress hydrolysis, add 37.2 g. of aniline,shake until dissolved, and filter rapidly if the solutionis not clear. Entrance of the mercury into the nucleustakes place with extreme ease, giving mainly thep- compound, which soon begins to crystallize. Sincedeposition of the isomenc 0- acetate, which is formedin relatively small amount, also takes place after sev-eral hours, the crystals are filtered off after three hours,thus avoiding contamination, with the mixture of the
TCf, Dimroth, Ber. 35, 2038 (190a),

OF ORGANIC CHEMISTRY 97
two compounds which would separate later A smallportion of the />-aminophenylmercunc acetate should berecrystallized from chloroform for analysis, the meltingpoint being 166-70. The remainder is sufficiently purefor the preparation of />-mercuri-&w-aniline.
The isomeric o-aminophenylmercunc acetate is diffi-cult to separate from the larger amount of the p- com-pound which would separate with it if the filtrate fromthe initial crop of the p- acetate were allowed to stand.Dimroth found, however, that there was a considerabledifference m the solubilities of the chlorides, so thefiltrate is treated with an excess of sodium chloridesolution, precipitating p-aminophenylmercunc chloridein the amorphous state, while the 0-amino chlorideseparates in crystalline form. The mixture is suckedoff, dried, and the 0- derivative extracted with not toomuch warm alcohol, separating as leaflets after thesolution is chilled and allowed to stand The amor-phous residue of p- chloride may be boiled with alcoholor benzene, and separates from the cold solution asleaflets melting at 188°, with decomposition.
In all of these compounds the mercury retains itssalt-forming character, and mercuric ions are split offwith relative ease. For instance, boiling the salts withsodium or ammonium sulfide solution results in thedeposition of mercuric sulfide, while alkaline stannouschloride solution immediately gives a dark gray pre-cipitate of mercury. The latter test is, in fact, a veryconvenient one for identifying mercury compounds ofthis, the so-called "half complex" class, in which onevalence of the mercury retains its salt-forming inor-ganic character, as distinguished from the "full com-plex" class, in which the mercury is fully bound be-tween two organic radicals, and is more completelymasked. The preparation of a typical member of the"full complex" group is given below.

98 ADVANCED LABORATORY MANUAL
2. p-Mercuri-Ws-aniline,
NH9 NH a
18 g. of />-aminophenylmercunc acetate are finelypulverized, suspended m 200 cc. of warm water, stirrecmechanically on the water bath, and treated with aqueous ammonia until the substance is entirely dissolvedJust as inorganic mercuric salts form complex ionswith ammonia, so do the aminophenylmercuric saltsand in this case the ammonia compound is easiljsoluble.
A solution of 14 g. (1.1 equivalents) of crystallinesodium sulfi.de in a little water is next added, and thestirring and heating continued for two and one-haUhours Mercuric sulfi.de and crystalline />-mercuri-&W"-aniline are precipitated apparently according to thescheme.
N H , NH a
-{-NaaS
HgOCOCHa
NH a
Hg — S — Hg8 Prom unpublished' experiments of Dr. W. A. Jacobs and
the writer.

OF ORGANIC CHEMISTRY
NH99
NH,
+ 2CH8COaNa > HgS
The precipitate is then filtered from the hot solu-tion, washed with a little hot water, sucked dry, andextracted with pyndine on the water bath, as thissolvent dissolves much larger quantities of the mer-cun compound than chloroform, which is usually pro-posed. After concentrating to small bulk in vacuo thesolution is carefully diluted with hot water and rubbed,whereupon the fas-compound gradually separates asglistening needles which melt at 1740 with decomposi-tion The yield should be 6-7 g. In contradistinctionto the starting material, the &«-compound does notblacken when treated m the cold with alkaline stan-nous chloride solution.


INDEX
PAGEAcylation 34, 30, 66, 76, 80rf-Alanine . 82, 83rf/-Alanine . . .J-Alanine . . . . . . . .Albumin, crystalline eggAlkylation . . . .Allyl bromide . . . .o-AllylphenolAllyl phenyl ether
.. . 83
33, 35. 373637
•• 36, 37^-Aminodimethylanihne £m-Aminophenol 28^-Aminophenoxyacetic acid 46o-Aminophenylmercurjc chloride 97/>-Aminophenylmercuric acetate p6Anthranilic acid, methyl ester 32rf-Arabinose 61Arsenation 29, 88"Arsphenamine" 92
"Barbital" 64rf-Benzoylalanine 83ii-Benzoylalanine, strychnine salt 82rf/-Benzoylalanine 80J-Benzoylalanine 80/-Benzoylalanine, brucine salt 80Benzylamine 24, 27Benzylhexamethylenetetraminium chloride 24Bromination 20d/-a-Bromopropic>nic acid 20/5-Bromopropionic acid , 24
Camphoric acid 58Camphorquinone 55. 58
Chlorination 18Chloroacetone 18jS-Chloropropionic acid 03
101

102 INDEX
PAGEDe-alkylation 423,3'-Diamino-4,4'-dihydroxyarsenobenzene and hydrochlo-
nde . . . . 92Dxazotization . . . . 30, 71Diethylbarbituric acid . 64, 65Diethylmalonic ester . . . 64Dihydrocupreine . . 41Dihydroqmnine 50
Ethylene cyanohydrin 23Egg albumin, crystalline 83Galactose . . . 76Gluconic acid, calcium salt 59/3-Glucose 74/S-Glucose penta-acetate 75
^-Hydroxyphenylarsomc acid • . 8 8
Iodination / 21S-Iodo-2-toluidine ' ••*• 2*Isomtrosocamphor /, ...'/.. .... . . . . SSMeldola's blue 70Mercuration 96/)-Mercuri-fcw-aniline 989-Miethylacndine .. 66Methyl anthramlate 32a-Methylcoumarane 39
o-Nitraniline 13, 16Nitration 13. 93/>-Nitroacetanilide IS, 16/i-Njtro-o-cresol 543-Nitro-4-hydroxyphenylarsonic acid . . . 92^-Nltrophenoxyacetic acid 33o-Nitrophenylarsonic acid 29^-Nitrosoo-cresol 16
m-Phenetidine 34Phenylazo-a-naphthylamme hydrochlorlde . . . . . . . . . . 71i-Phenyl-i-benzoyloxy-2-aminoethane . . . 50i-Phenyl-i-hydroxy-2-aminoethane 480-Propcnylphenol 39

INDEX 103
PAGEQuinidne (quinotoxine) hydrochloride 67
Rosindone 73
Rosinduhne 71
"Salvarsan" 92
"Veronal" 64





























