Embed Size (px)
Citation preview

4
Accelerated titration designs
Janet DanceyInvestigational Drug Branch, CTEp, DCTD, NCI, Bethesda,Maryland, USA
Boris Freidlin and Larry RubinsteinBiometric Research Branch, DCTD, NCI, Bethesda, Maryland, USA
4.1 Introduction
A decade ago, investigators in oncology had a clear interest in modifications to thestandard phase I design to make it more effcient, to treat fewer patients at nontoxicdose levels (which may be less effcacious) and to increase the precision of phase IIdose recommendations. This was the conclusion of the 1996 Joint Meeting of theUS National Cancer Institute and the European Organization for the Research andTreatment of Cancer (1,2). At approximately the same time, a review of the recentphase I oncology literature revealed that few investigators were makng use of theinnovative phase I tral designs developed over the previous decade (3), which weremeant to accomplish these very objectives.
Approximately five years previous to this, Sheiner and coworkers published a se-ries of papers in which they argued for the use of dose-response models in the analysisof phase I trals (4-). Standard practice in oncology trals, among other fields, was to
analyze the dose-toxicity relationship only in terms of the population as a whole, andto analyze it separately for each dose. Rarely were attempts made to fit a dose-toxicitymodel to the phase I data that accounted for interpatient and intrapatient varabilty
Statistical Methods/or Dose-Finding Experiments Edited by s. Chevret(Q 2006 John Wiley & Sons, Ltd
91

92 ACCELERATED TITRATION DESIGNS
separately, acc0!l0dated the possibility of cumulative toxicity and allowed for theconstrction of dose-toxicity curves for the sensitive as well as the typical patients.In addition, Sheiner argued for the use of intrapatient dose escalation, to maximizethe possibilty of individual patients receiving effcacious doses and to increase theaccuracy of the analysis of the phase I data. This was not commonly practised inoncology phase I trals.
In response to the above, Simon et at. (7) developed a family of 'accelerated titra-tion designs' and proposed the use of an accompanying dose-toxicity model, basedon the work of Sheiner and coworkers (4, 5). The main distinguishing features ofthese designs are (a) a rapid initial escalation phase, (b) intrapatient dose escalationand (c) the ability to analyze tral results using a dose-toxicity model that incorporatesparameters for intrapatient and interpatient varation in toxicity and cumulative toxi-city. The distinguishing features of the model are its simplicity and the incorporationof separate varables for interpatient and intrapatient varability, as well as for thepossibility of cumulative toxicity.
4.2 Design
Simon et at. (7) proposed a family of accelerated titration dose-escalation designs.In their formulation all designs use 40 % dose-escalation steps. The dose-escalation!de-escalation rules are based on definitions of dose-limiting toxicity (DLT) and of'moderate' toxicity. These definitions may be protocol specific. For example, Simonet at. used any grade 2 toxicity that was considered to be treatment related as moderatetoxicity. For purposes of comparson, they designated the standard phase I design(with 40 % escalation steps in place of the standard modified Fibonacci escalation)as 'design 1'. They then introduced accelerated designs designated as 'design 2','design 3' or 'design 4' .
Design 1 dictates that patients are dose escalated in cohorts of three until DLT isobserved. One instace of DLT leads to treatment of three additional patients at thecurrent dose level (with escalation continuing if no additional DLT is observed). Twoinstances ofDLT, at a dose level, leads to a halt in dose escalation, with the prior doselevel declared the MTD so long as six patients have been treated at that level, withone instance of DLT (de-escalation continues until such a dose level is determned).
Design 2 stars with an accelerated phase that uses single-patient cohorts per doseleveL. When the first instance of first-course DLT is observed or the second instanceof first-course moderate toxicity is observed, the cohort for the current dose level isexpanded to thee patients and the tral reverts to use of design 1 for fuer cohorts.
Design 3 is similar to design 2 except that double-dose steps are used during theaccelerated phase. Two 40 % dose steps correspond to approximately a doubling ofthe actual dose. The accelerated phase ends, as with design 2, when the first instance offirst-course DLT or the second instance of first-course moderate toxicity is observed.After that, design 1 is used for fuher patients.
I
I',i.,
I .
Il
ll
!

ACCELERATED TITRATION DESIGNS93
Design 4 is similar to design 3, except for the criterion that is used for trggeringthe end of the accelerated phase. With designs 2 and 3, the accelerated phase ends withthe first-course instance of DLT or second instance of first-course moderate toxicity.With design 4, the trgger is the first instance of any-course DLT or the second instanceof any-course intermediate toxicity. In addition, when the first instace of moderatetoxicity is observed, two additional patients must have been treated at that dose, or ahigher dose (during any course), without experiencing moderate or worse toxicity, inorder that the accelerated phase continues. This may require the treatment of one ortwo additional patients at that dose. Hence, design 4 may stop the accelerated phaseearlier than design 3.
Table 4.1 summarzes the characteristics of the four dose escalation designs.
Table 4.1 Summar of the four dose-escalation designs and the two intrapatientdose-escalation options.
Design Description
Cohorts of 3 new patients per dose leveL. If i of 3 patientsexperiences DLT in first course, expand to 6 patientsCohorts of i patient per dose leveL. When first instance offirst-course DLT is observed, or second instance of first-coursegrade 2 toxicity of any type, expand cohort for current doselevel and revert to use of design 1 for all furher cohortsSame as design 2, except that double-dose steps are usedduring initial accelerated stage of trial (both forbetween-patient and within-patient escalation)Cohort of 1 new patient per dose level and double-dose stepsare used durng the initial accelerated stage of the tral. Whenthe first instance of DLT is observed at any course, or thesecond instance of any-course grade 2 toxicity of any type,expand cohort for current dose level and revert to use of design1 for all further cohorts. When the first instance of moderatetoxicity is observed, two additional patients must have beentreated at that dose, or a higher dose (during any course),without experiencing moderate or worse toxicity, in order thatthe accelerated phase continues
Description
No withn-patient dose escalation. De-escalate if grade 3 orworse toxicity at previous courseEscalate if grade 0-1 toxicity at previous course. De-escalateif grade 3 or worse toxicity at previous course
1
2
3
4
Escalation
A
B
-
-,

94 ACCELERATED TITRATION DESIGNS4.2.1 Intrapatient dose-escalation
In order to maximize each patient's chance to be treated at a potentially active dose,the accelerated titration design allows intrapatient dose-escalation for a patient whoremains on study and has no evidence of toxicity at the current dose. Specifically,the dose for the next course is escalated if less than moderate toxicity was observedfor the patient during the current course. If moderate toxicity occured, then the dosestays the same for the next course for that patient. If DLT occurred, then the patientgenerally goes off-study, but if not, then the dose is reduced. For design 2, single-dosesteps are used for intrapatient dose changes. For designs 3 and 4, double-dose stepsare used for intrapatient dose changes durng the accelerated stage, and single-dosesteps subsequently.
All four designs may be used with and without intrapatient dose-escalation. Simonet at. (7) compared the performance of the four designs, with and without intrapatientdose-escalation, in terms of toxicity, potential effcacy (reduction of treatment at dosesbelow the MTD) and tral length. Table 4.1 also summarzes the two intrapatient dose-escalation options.
4.3 Evaluation of performance
Simon et at. (7) fit the above model to data from 20 phase I trals (involving 9 distinctagents). Only thee ofthe trals showed any evidence of cumulative toxicity (a :: 0).The estimates of a for the other trals were zero or very close to zero. The trials varedsubstantially in the other parameters and thus provide a broad range of experience forevaluation of the accelerated titration designs.
Simon et at. (7) evaluated the performance of the accelerated titration designs bysimulating phase I data based on the 20 sets of parameters estimated from the 20 realtrals that they studied. For each of the 20 sets of parameters, they generated data for1000 phase I trals and applied each of their designs to the simulated data. Figure 4.1shows the average number of patients per tral utilzed by each of the designs. Foreach design, the average is taken over the same 20000 simulated datasets generatedfrom the sets of parameters derived from the 20 actual trals analyzed. Results foreight designs are shown. Designs 1 to 4 are as described above. The designs labeledwith B utilize intrapatient dose-escalation if the toxicity in the previous course is lessthan intermediate. Designs labeled with A do not permt intrapatient dose-escalation.
Design lA corresponds to the standard design, although it does not use Fibonaccidose steps. Design lB is the standard design augmented to permt intrapatient dose-escalation. As can be seen in Figure 4.1, the average number of patients is much greaterfor the standard design lA or lB than for any of the accelerated titration designs. Theaverage number of patients is somewhat less for designs 3 and 4 that use double-dose steps compared to design 2. Although the average differences are not great, thedifferences for individual trals can be; i.e. for a tral in which the starng dose isvery low relative to the dose at which intermediate toxicity is expected, designs 2and 3 wil require substantially fewer patients.

ACCELERATED TITRATION DESIGNS
Patients and Cohorts Distribution for8 Designs
40
35
lJ 30ñi"¡:
25l-eC\
Q)20
~ 15Q)
~10
5
0
Totalpatients
Cohorts
95
c: Design 1 A
. Design 1 8
o Design 2A
c: Design 28
. Design 3A
. Design 38
D Design 4A
. Design 48
Figure 4.1 Average number of patients and number of cohorts for the eight designs.
Figure 4.1 also shows the average number of patient cohorts utilized by eachdesign. The average is lowest for designs 3 and 4, which use double-dose steps.Although the difference in average number of cohorts is not large, the difference inaverage time to complete the trals wil be much shorter for designs 2 to 4 if patientsare not instantaneously available, since the accelerated phase of those designs requiresonly one patient per cohort.
Figure 4.2 shows the average number of patients experiencing each level of toxicityas their worst toxicity durg their treatment on the tral. With the standard design, anaverage of 23 patients experience less than intermediate toxicity (labeled 'no toxicity'in the figure). These patients are underteated. For design 2B the average number ofunderteated patients is about eight and for designs 3B and 4B the number is less thanfive. Ths major reduction in the number of underteated patients is achieved with verysmall increases in the average number of patients experiencing DLT or unacceptabletoxicity with the accelerated titration designs. Figure 4.3 shows the average percentageof patients experiencing each level of toxicity as their worst toxicity during theirtreatment on the tral.
The accelerated titration designs without intrapatient dose-escalation, 2A, 3A and4A, performed quite well with regard to reduction in average number of patients andreduction of number of underteated patients. However, they do not provide patientsaccrued early in the tral a full opportnity to be treated at a therapeutic dose. Theyare also less effective in situations where interpatient varabilty in susceptibility totoxicity is large.
These designs may be attactive, however, when there is concern about cumulativetoxicity. It is worth noting in ths regard that analysis of the 20 phase I trals used

96
25
en 20+-CQ)'"§
15a.-0..Q) 10.0E::Z
5
0
ACCELERATED TITRATION DESIGNS
Toxicity Distribution for 8 Designs
;: ;: ~ ..+- +-'õ 'õ a. ;:
Cl Q) +-'x 'x (. 'õ0 0 (. 'x+- +- ct 00 ~ c +-Z :2
:J
c: Design 1 A
.. Design 18
o Design 2A
D Design 28.. Design 3A
.. Design 38
o Design 4A
II Design 48
Figure 4.2 Average number of patients with worst toxicity at each toxicity level, for
the eight designs.
Toxicity Distribution for 8 Designs
0.7
0.6en+-
0.5cQ)'"§
a. 0.4-0+- 0.3cQ)(...
0.2Q)a.
0.1
0 ;: ;: ~ ..+- +-'õ 'õ a. ;:
Cl Q) +-'x 'x (. 'õ0 0 (. 'x+- +- ct 00 :Q c +-Z ~ :J
o Design 1 A
.. Design 18
o Design 2A
o Design 28
.. Design 3A
.. Design 38
o Design 4A
II Design 48
Figure 4.3 Average percentage of patients with worst toxicity at each toxicity level,for the eight designs.
-- l. '-'."_.'''' ~.

.--
ACCELERATED TITRATION DESIGNS 97
Comparison of Design 1 A vs Design 48
181614
ëi 12;:j 10
~ 8
8 642
o
- Design 1 A
- Design 48
LO CJ C' I" ,. LO 0) C' I" ,. LO CJ C' I",. ,. C\ C\ C\ C' C' 'V 'V 'V LO LO
Number of Patients
Figure 4.4 Diagram of the comparative performances of design 1A and design 4B,in terms of patients required to reach each dose level and to define the MID.
for evaluation of these designs revealed no evidence of il effects from intrapatientdose-escalation and lead the investigators to conclude that 'cumulative toxicity doesnot appear to be a valid reason to prohibit intra-patient dose escalation, as it occursrarely' (1).
To ilustràte furter the effciency of the accelerated designs in comparson withthe standard, we give in Figure 4.4 a simulated comparson of the performance of de-sign 4B versus design lA for a paricular dose-toxicity modeL. The accelerated designcompletes the tral with less than half the number of patients required by the stan-dard. More dramatically, due to the single-patient cohorts and two-step escalations,it requires only one patient for every six of the stadard design to escalate throughthe portion of the dose-toxicity curve where DLT is unlikely. Of course, if the initialdose of the tral is not defined so conservatively, the comparson is not so extreme.
Hr::
j',i~:l-i'
r¡:
~;~l
4.4 Model-based analysis
By using a model for the statistical distrbution of toxicity, based on current andprevious doses, a graded toxicity scale, based on the unobserved continuous varableassociated with toxicity and multi course treatment results, the accelerated titrationdesigns allow for an effcient approach to analysis of phase I data. The model used inSimon et at. (7) was based on measurng the worst toxicity experience for each patientdurng each course of treatment; i.e. the model does not consider separate toxicity foreach organ system, but takes the maximum over all organ systems' and records thatworst toxicity separately for each course of treatment for each patient. The modelwas designed to represent different levels of worst toxicity. The toxicity experiencedin a paricular course was determned by the curent dose administered and the total
-

98 ACCELERATED TITRATION DESIGNS
Table 4.2 Summar of the dose-toxicity modelfor both the unobserved continuous toxicityvarable and the observed toxicity grade leveL.
Model relating toxicity to dose
Yij = log(dij + aDi) + ßi + êijdij = dose for the ith patient in course ja = cumulative toxicity parameter
Dij = cumulative dose up to course jßi = interpatient random effect, N (0, aÐêij = intrapatient random effect, N(Q, a;)
Yij -: K 1 grade 0-1 toxicityKi -: Yij -: K2 grade 2 toxicityK 2 -: Yij -: K 3 grade 3 toxicity
Yij ~ K3 grade 4 toxicity
dose administered in the previous courses. The model incorporated parameters forboth intrapatient and interpatient varabilty, and for cumulative toxicity.
Suppose that the ith patient receives dose dij during dose j and received a totaldose Dij for courses prior to j. Let a represent the effect of cumulative toxicity
(a = 0 indicates no effect of cumulative toxicity). The random varable ßi representsinterpatient varability in the toxic effects; ßi is taken to be normally distrbutedwith mean zero and varance aJ. The random varable Bij represents intrapatientvarabilty in the toxic response; B¡j is taen to be normally distrbuted with meanzero and varance a;. These terms and random varables determne the unobservedmagnitude Yij of the worst toxicity for patient i in course j, according to the formula
Yij = log(d¡j + aDij) + ßi + Bij.
In addition to the thee parameters a, aJ and a;, there are also several parameters
for converting value Yij into a graded level of toxicity. Values of Yij less than K 1
correspond to less then moderate toxicity values between K 1 and K2 correspond tomoderate toxicity, values between K2 and K3 correspond to dose-limiting toxicity andvalues greater than K3 correspond to life-theatening toxicity: If one does not wishto distinguish DLT from life-theatening toxicity, then only K 1 and K2 are needed.
Therefore there are five or six parameters to be estimated from the data. Table 4.2summarzes the characteristics of the modeL. Ths model is a generalization of the K maxmodel of Sheiner, Beal and Sambol (5) and of the model of Chou and Talalay (8,9).
Given the data of the grade of toxicity (worst over organ systems) for eachcourse of each patient, the method of maxmum likelihood is used to estimatethe model parameters. Splus software for fitting the parameters is available athttp://inus.nci.nih.govlbrb. That website also contains an Excel macro for managing
. i.Il. j .
. . ~-l.1
j¡

ACCELERATED TITRATION DESIGNS 99
Probabilties of Grade 3+ Toxicity
(Chloroquinoxaline Sulfonamide)
1
0.90.8
~ 0.7
:E 0.6
B 0.5e 0.4D. 0.3
0.20.1
o OC\""COC:OC\,,.,.,.,.,.,C\C\C\
-Gr 3+ (16%tile)
- Gr 3+ (50%tie)
.. Gr 3+ (84%tie)
Dose Level
Figure 4.5 Probabilities of grade 3+ toxicity at varous dose levels for the meanpatient and the patients one standard deviation above and below the mean.
dose assignments to patients durng accelerated titration design trals. The macroassists investigators in quality controlling the dose assignment and provides a conve-nient way of recording dose assignments in a systematic manner that makes the dataavailable for subsequent analysis.
Figure 4.5 ilustrates the power of the model-based analysis to constrct a dose-toxicity curve, not only for the typical patient (50th percentile) but also for the patientwho falls one standard deviation below the typical in terms of increased susceptibilityto toxicity (16th percentile). The standard approach to defining the MID is based onthe probabilty of toxicity at a given dose for the population as a whole, which oftenroughly corresponds to the probabilty of toxicity for the typical patient. With thisapproach, the initial phase II dose would be set, for Figure 4.5, at dose level 16 or17, to keep the probabilty of DLT below 30 %. However, the model-based analysisreveals that such a dose level results in at least a 40-0 % likelihood of DLT for anontrvial subgroup of the patient population (those at the 16th percentile or below).This might suggest that a more prudent approach would be to define a lower initialphase II dose, to accommodate the susceptibilty of this subgroup.
Figure 4.6 ilustrates the power of the model-based analysis to constrct com-parative dose-toxicity cures for the different levels of toxicity. For example, theanalysis suggests that the dose-toxicity cures for grade 2 versus grade 3 toxicity areseparated by approximately four dose levels. This indicates that for a given patient,as well as for the population as a whole, DLT is likely to occur approximately fourdose levels beyond moderate toxicity, suggesting that accelerated dose-escalation islikely to be safe, both for the population and for a given individual. Even though thedose-toxicity curve for DLT is relatively steep, it is well separated from the curve formoderate toxicity.
~

100
1
0.90.8
;: 0.7ŠO.6.g 0.5.c20.4D. 0.3
0.20.1
o
ACCELERATED TITRATION DESIGNS
Probabilties of Toxicity (50%tie Patient)
(Chloroquinoxaline Sulfonamide)
- Gr 2+ toxicity
- Gr 3+ toxicity
- Gr 4+ toxicity
10 12 14 16 18 20 22 24Dose Level
Figure 4.6 Probabilities of grade 2+, grade 3+ and grade 4+ toxicity at varous
dose levels for the mean patient.
Sheiner and coworkers (4,5) proposed the use of dose-toxicity models for phase Itrals a decade ago. They are stil rarely used, despite their potential for facilitatingthe definition of a phase II staring dose.
4.5 Clinical applications
First-in-man phase I tral designs of oncology agents share the following charac-
teristics: selection of a 'safe' starng dose, sequential dose-escalation in cohorts ofpatients and determnation of a recommended dose based on a prespecified primarendpoint, usually the occurrence of unacceptable toxicity in a defined number of pa-tients treated at a given dose leveL. Optimal phase I designs result in the identificationof a dose for furer evaluation in a manner that is both safe and effcient. Higher
staring doses, fewer patients per dose level and large escalation steps require fewer
patients overall. However, safety is enhanced by lower staing doses, more patientsper dose level to ensure safety of the dose and smaller dosing increments. Phase Idesigns must strke a balance between these elements. Accelerated titration designsproposed by Simon et aI. (7) use the following modifications to enhance effciency:as few as one patient per level are enrolled and initial dose-escalation steps are larger
(e.g. 100 % increments in the absence of toxicity). The number of patients per doselevel increases to thee, once toxicity of a minimum degree (e.g. second instace of
grade 2; first instance of DLT) has been seen in at least one patient. Thereafter aminimum of thee patients in each cohort are recruited, expanding to six in the eventthat one of thee has a DLT in the protocol prescribed observation period (usuallyone cycle or 4-8 weeks of chronic therapy) and the dose-escalation increments arereduced.
t.
L-"";",:..,,...~

ACCELERATED TITRATION DESIGNS 101
To assess the use and utilty of the accelerated titration designs in the evaluationof novel oncology therapeutics, we conducted a literature search using the ISI Webof Knowledge Database (Thomson ISI, Thomson Corporation, Philadelphia, Penn-sylvania) in May and August 2005. All aricles in the database that cited the originalpaper by Simon et at. (7) were retreved and reviewed. In tota, 106 publications wereidentified. Aricles that focused on statistical methodology of phase I studies (10),were not of phase I studies (4), that evaluated combinations of agents (12), werereview aricles (34), or of phase I studies that did not use the Simon et at. (7) acceler-ated titration designs (10), were not included in our review. In total, 36 publicationsof phase I trals of novel cancer therapeutics were identified. From the tral publi-
cations, the following details were abstracted: agent/class, schedule, type of design,study-specific modifications to the design (e.g. patients/cohort and dose-escalationincrements during the accelerated phase, rules for termnating the accelerated phase,dose-escalation increments following termnation of the accelerated phase, dose lev-els evaluated and the number of dose levels evaluated during the accelerated phaseand subsequently). A summar of these trals are provided in Tables 4.3 and 4.4.
From the results of our review a number of observations can be made regarding theutilzation of the designs. Firstly, the classes of agents selected for evaluation using anaccelerated titration design favor agents that belong to chemical classes that have beenpreviously studied or to biological agents not associated with significant risk of severe,irreversible organ toxicity. This is not surprising, as agents with these characteristicswould engender a level of comfort regarding the safety of using an accelerated titrationdesign. Secondly, designs 3 and 4, which utilize single-patient cohorts and 100 % dose-escalations, are the most commonly used. Thirdly, almost half of the studies do notutilze intrapatient dose-escalation. Fourtly, the most common modifications to thedesigns are those determning the dose-escalation increments following termnationof the accelerated phase and/or modifications to rules for termnating the acceleratedphase. Most trals with modifications in the dose increments following termnationof the accelerated phase stipulated dose increments of 15-30 % rather than 40 %.A few utilzed higher-dose increments (50-67 %) and a few reverted to a modifiedFibonacci escalation schema. Rules for termnating the accelerated phase included
the first occurrence of any toxicity, or the achievement of a prespecified dose (e.g.mouse equivalent MTD). Given the frequency and nature of these modifications,it appears that investigators retain concerns regarding the safety of the acceleratedtitration design dose-escalation increments and termnation rules.
To assess the effciency and safety of the accelerated designs, the numbers ofpatients and dose levels, overall and during the accelerated phase, were evaluatedacross the 36 studies. Patients treated above the ultimately recommended phase IIdose (RP2D) were identified. If a patient was treated at a dose level prescribed bythe accelerated phase dose increase and that dose exceeded the RP2D, then the doselevel was considered to have exceeded the recommended dose due to the acceleratedphase rules. Similarly, patients that died on the study due to obvious or suspectedtreatment-related toxicity were identified. Those fatalities which occured at dosesprescribed during the accelerated phase of the study were classified as deaths during
~

f
Tab
le 4
.3Ph
ase
I tr
ials
usi
ng a
ccel
erat
ed ti
trat
ion
desi
gn.
.. 0 N
Intr
a pa
tient
Aut
hor/
dose
-R
ecom
men
ded
refe
renc
eA
gent
Sche
dule
Des
ign
esca
latio
ndo
se
LoR
usso
5-Fl
uoro
-pyr
imid
inon
eO
rally
dai
ly f
or 5
day
s4B
Yes
625
mg/
m2/
day
oral
ly f
or
eta!. (10)
ever
y 4
wee
ks5
days
eve
ry 4
wee
ks
Goe
tz17 -(Allylamino )-17-
iv weekly x 3 every
2BY
es30
8 m
g/m
2 w
eekl
y x
3
et a
t. (1
1)de
met
hoxy
geld
anam
ycin
4 w
eeks
ever
y 4
wee
ks
Gre
m17 -(Allylamino )-17-
1 ho
ur is
iv in
fusi
on2B
Yes
40 m
g/m
2 da
ily x
5 e
very
et a
i. (1
2)de
met
hoxy
geld
anam
ycin
daily
for
5 d
ays
ever
y3
wee
ks
3 w
eeks
Sess
aB
BR
3464
, cat
ioni
civ daily x 5 every
4BY
es0.
12 m
g/m
2/da
y x
5 ev
ery
et a
t. (1
3)tr
ipla
tinum
com
plex
3 w
eeks
3 w
eeks
Mro
ssB
BR
3576
,iv infusion every
4AN
o15
0 m
g/m
2 ev
ery
4 w
eeks
f)
et a
t. (1
4)az
a-an
thra
pyra
zole
4 w
eeks
n
Plum
mer
BM
S-18
4476
, tax
ane
anal
ogiv weekly x 3 every
4BY
es50
mg/
m2/
wee
k x
2 ev
ery
~ tret
at.
(15)
4 w
eeks
, lat
er21
day
s~
amen
ded
tow
eekl
y x
2 ev
ery
~21
day
s"" "" ""
Man
iB
MS-
2475
50, d
eriv
ativ
e of
1 ho
ur iv
infu
sion
eve
ryM
odif
ied
4BY
es40
mg/
m2
ever
y 3
wee
ks~
et a
t. (1
6)E
poth
ilone
B3
wee
ks~ ""
Abr
aham
BM
S-24
7550
, der
ivat
ive
of1
hour
iv in
fusi
onM
odif
ied
3BY
es6
mg/
m2/
day
x 5
ever
y0 Z
et a
i. (1
7)E
poth
ilone
Bdaily x 5 every
21 d
ays
ei21
day
str cr
Gad
geel
BM
S-24
7550
, der
ivat
ive
of1
hour
iv in
fusi
wye
ryM
odif
ied
2BY
es40
mg/
m2
ever
y 21
day
s"" ci
et a
t. (1
8)E
poth
ilone
B21
day
sZ cr
.1:
\~¿"
.~C
':,.'~
; .'
,,;.:'
.1,-
~~\O
¡.t~
r''':'
''~t''
'''~~~
~~t..
.~~~
~....
~~t~
~.
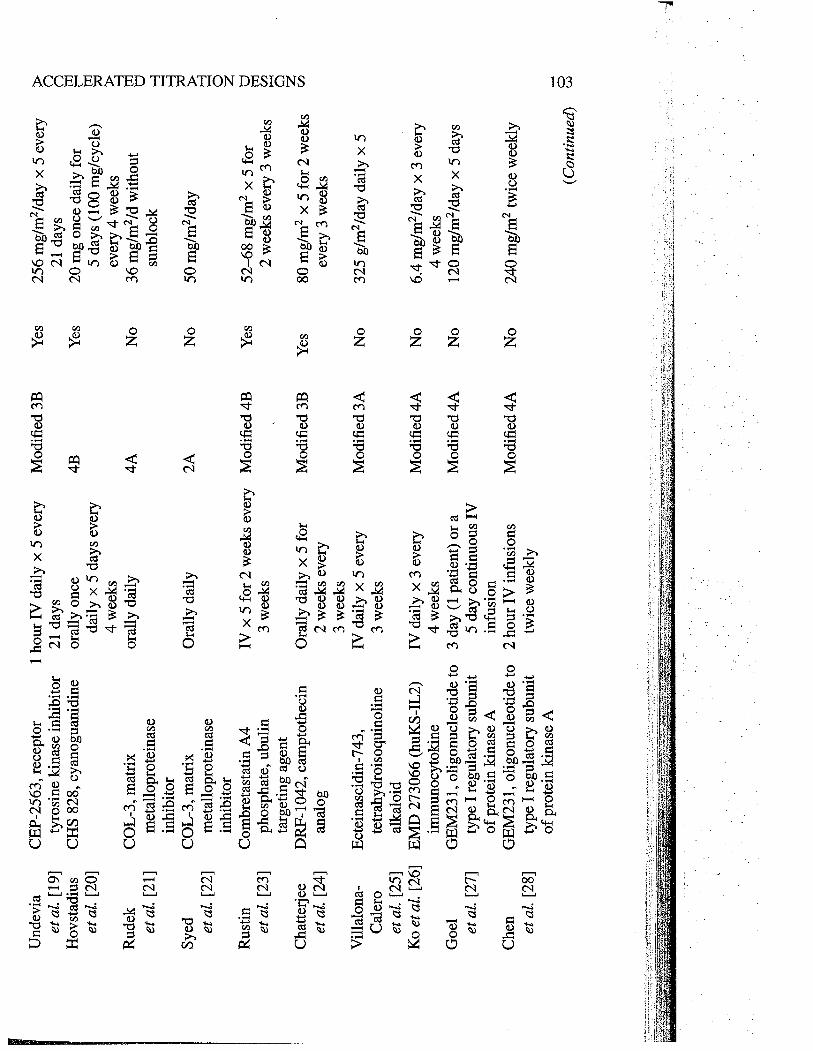
Und
evia
CE
P-2
563,
rec
epto
r1 hour iv daily x 5 every
Mod
ifie
d 3B
Yes
256
mg/
m2/
day
x 5
ever
y~
et a
t. (1
9)ty
rosi
ne k
inas
e in
hibi
tor
21 d
ays
21 d
ays
nH
ovst
adiu
sC
HS
828,
cya
nogu
anid
ine
oral
ly o
nce
4BY
es20
mg
once
dai
ly f
ortr r;
et a
t. (2
0)da
ily x
5 d
ays
ever
y5
days
(10
0 m
g/cy
cle)
ìO
4 w
eeks
ever
y 4
wee
ks~
Rud
ekC
OL-
3, m
atrix
oral
ly d
aily
4AN
o36
mg/
m2/
d w
ithou
tm 0
et a
t. (2
1)m
etal
lopr
otei
nase
sunb
lock
din
hibi
tor
i-Sy
edC
OL-
3, m
atrix
Ora
lly d
aily
2AN
o50
mg/
m2/
day
~et
at.
(22)
met
allo
prot
eina
se¡. 0
inhi
bito
rZ
Rus
tinC
ombr
etas
tatin
A4
iv x 5 for 2 weeks every
Mod
ifie
d 4B
Yes
52-6
8 m
g/m
2 x
5 fo
r0 tr
et a
t. (2
3)phosphate, ubulin
3 w
eeks
2 w
eeks
eve
ry 3
wee
ksen ¡.
targ
etin
g ag
ent
a ZC
hatte
rjee
DR
F-10
42, c
ampt
othe
cin
Ora
lly d
aily
x 5
for
Mod
ifie
d 3B
Yes
80 m
g/m
2 x
5 fo
r 2
wee
ksen
et a
t. (2
4)an
alog
2 w
eeks
eve
ryev
ery
3 w
eeks
3 w
eeks
Vila
lona
-E
ctei
nasc
idin
-743
,iv daily x 5 every
Mod
ified
3A
No
325
g/m
2/da
y da
ily x
5C
aler
ote
trah
ydro
isoq
uino
line
3 w
eeks
et a
t. (2
5)al
kalo
idK
o et
at.
(26)
EM
D 2
7306
6 (h
uKS-
IL2)
iv daily x 3 every
Mod
ifie
d 4A
No
6.4
mg/
m2/
day
x 3
ever
yim
mun
ocyt
okin
e4
wee
ks4
wee
ksG
oel
GE
M23
1
, olig
onuc
leot
ide
to3
day
(1 p
atie
nt)
or a
Mod
ifie
d 4A
No
120
mg/
m2/
day
x 5
days
et a
t. (2
7)ty
pe i
regu
lato
ry s
ubun
it5 day continuous iv
of p
rote
in k
inas
e A
infu
sion
Che
nG
EM
23
1 , o
ligon
ucle
otid
e to
2 hour iv infusions
Mod
ifie
d 4A
No
240
mg/
m2
twic
e w
eekl
yet
at.
(28)
type
I r
egul
ator
y su
buni
ttw
ice
wee
kly
of p
rote
in k
inas
e A
..(
Con
tinue
d)0 U
J
i

r~~
..""
. -"
.-~
---~
-~~
.~_.
...-
Tab
le 4
.3(
Con
tinue
d)
0 .t
Intr
a pa
tient
Aut
hor/
dose
-R
ecom
men
ded
refe
renc
eA
gent
Sche
dule
Des
ign
esca
latio
ndo
se
Bor
chm
ann
H22
xKi-
4, b
ispe
cifi
cIV days 1,3,5,
4BY
es80
mg/
m2
per
cycl
e
et a
l. (2
9)an
ti-C
D30
and
CD
647-21 days
mon
oclo
nal a
ntib
ody
Gad
geel
KR
N55
00, s
pica
myc
in1
hour
iV d
aily
x 5
2BY
es4.
3 m
g/m
2/da
y x
5 ev
ery
et a
l. (3
0)de
riva
tive
ever
y 3
wee
ks3
wee
ks
Scho
emak
erM
AG
-CP
T, p
olym
erIV
infu
sion
ove
r 3
days
Mod
ifie
d 2A
No
68 m
g/m
2/da
y fo
r 3
days
et a
t. (3
1)co
njug
ate
ofev
ery
4 w
eeks
ever
y 4
wee
ks
cam
ptot
heci
nM
atsu
mur
aM
CC
-465
, dox
orub
icin
1 ho
ur I
V in
fusi
on e
very
AT
DN
o32
.5 m
g/m
2 ev
ery
3 w
eeks
~et
al.
(32)
PEG
imm
unol
ipos
ome
3 w
eeks
plan
ned
n
Cliv
eA
ntag
onis
t G s
ubst
ance
PIV
eve
ry 3
wee
ks u
ntil
Mod
ifie
d 4A
No
400
mg/
m2
wee
kly
6 ho
ur~
et a
l. (3
3)an
alog
the
targ
et m
axim
umIV
infu
sion
~pl
asm
a co
ncen
trat
ion
of 1
0 p,
M th
en w
eekl
y~
Mat
sum
ura
NK
9l1,
mic
elle
IV e
very
3 w
eeks
Mod
ifie
d 3A
No
50 m
g/m
2 ev
ery
3 w
eeks
i-
et a
t. (3
4)en
caps
!lla
ted
doxo
rubi
cin
- i-
de J
onge
PN
U -
1595
48, a
lky1
cycl
ine
IV e
very
3 w
eeks
Mod
ifie
d 4B
Yes
12 a
nd 1
4 m
g/m
2 IV
eve
ry~
et a
l. (3
5)(2
stu
dies
)3
wee
ks in
HP
and
MP
- 0pa
tient
sZ
Loc
khar
PNU
-16
6196
A, b
rost
allc
in,
IV w
eekl
y x
3 ev
ery
Mod
ifie
d 3B
Yes
2.4
mg/
m2/
wee
kci tI
et a
l. (3
6)a
nona
lkyl
atin
g D
NA
4 w
eeks
C/ ..
min
or g
roov
e bi
nder
0 Z C/
';;.::
,:,¡:
\~;i:
:;\,~
,.:"
.~~
#~,~
ii~1¡
¥.~'
i'Niim
l~.~
~.,\'
..i!~
,.~~J
!~~~
,~'"
,~~~
~,""
".~'
7'~~
,"=
-.,-
,.~,.~
...-,
..,,.,
./~."
.,....
.",..
_...,
,_.,,
-.

j

106ACCELERATED TITRATION DESIGNS
Table 4.4 Summar of phase I studies using accelerated titration design.
Trials (n)
Patients (n)t
¡
1i
I
I
r!
il
i
Il
Design
Intrapatientdose-escalation
Modifications
Types of
modifications
Patients/study(n)
Dose levels (n)Fold dose rangePatients treatedAbove RP2DDose levelsAbove RP2D
Deaths due totoxicity
Type1
23
4Unknown
YesNoYesNo
Dose-escalation duringthe accelerated phase
Patients/cohort duringthe accelerated phase
Rules for terminationof accelerated phase
Dose-escalationfollowingaccelerated phase
During accelerated phase
Median5
Range0-15
36911
Trials (n)1
610
18
1
2016
15
11
3
2
8
19
For study
Median Range
22 7-74
6 3-15
16 2-320
6 0-28
1 0-3
o
(13 patients acrossall studies dieddue to toxicities)
0-3
3 0-12
o 0-
o 0-1
(4 of 36 studies had adose level thatexceeded the RP2Ddurng the ATD)
0-1(1 patient across all
studies died due totoxicities)
o,I'
"
.,1-i
I
1i
" '.~;.-,'".-
d

ACCELERATED TITRATION DESIGNS 107
the accelerated phase. As summarzed in Table 4.4, the accelerated titration designs,as used in these studies, rarely resulted in dose escalation beyond the recommendedphase II dose. Only 4 of 36 exceeded the recommended phase II dose during theaccelerated titration phase and only 1 death from toxicity occurred during the accel-erated titration phase, among the 91 I patients enrolled in these studies. (It should benoted, however, that the use of acceleration may have contrbuted, in some trials, toexceeding the RP2D by a greater number of doses, or for a greater number of patients,than would otherwise have happened. Thus, the use of acceleration in these trials mayhave increased the overall number of patients treated above the RP2D, even beyondthe acceleration phase itself, and thus contrbuted to a greater death rate from toxic-ity.) Based on its utilization in these selected studies, the accelerated titration designappears to provide an enhanced effciency with acceptable safety. However, thereare a number of issues investigators might consider prior to selecting an acceleratedtitration design to evaluate a novel agent in a first-in-man phase I clinical tral.
The use of minimum one-patient cohorts and larger dose-escalation steps may beadvantageous under the following circumstances: (a) the agent is of a chemical classthat has been widely studied; (b) tle agent is predicted to have minimal interpatientvarability in pharacokinetics; (c) the agent's anticipated toxicity is unlikely to besevere or ireversible and is amenable to close monitoring and supportive interven-tions. Examples of agents most amenable to evaluation using a phase I acceleratedtitration design might be the following: a new formulation of a previously studiedagent (e.g.liposomal formulation ofpaclitaxel), a biological agent with minimal tox-icity based on animal models (e.g. antibody or small molecule inhibitor of a receptortyrosine kinase inhbitor) or an agent for which significant interspecies varabilty in
preclinical toxicology has led to a very conservative staring dose in a human phase Istudy. Under these circu:Qstances, the increased effciency and presumed safety of anaccelerated design might make it preferable.
Conversely, there are situations where an accelerated titration design may notprovide the optimal balance between safety and effciency as either larger numbersof patients/dose cohort and/or smaller dose increments would be preferable. Agentsassociated with steep dose-response curves for toxicity, severe ireversible toxicity,unexplained mortalty in anmal toxicology studies or large varabilty in doses orplasma drg levels eliciting effects may require alternative designs to balance safetyand effciency optimally. For example, larger patient numbers/dose cohorts may bepreferred if there is anticipated wide interpatient varabilty in toxic effects due topharacokinetic or pharacogenomic differences between patients. For this circum-stance, larger patient numbers per dose level is appropriate since decisions about thesafety of a given dose may require more than a single patient's experience. Similarly,when a pharacokinetic or a pharacodynamc endpoint, rather than toxicity, is theprimar endpoint, larger numbers of patients per dose level are recommended due toanticipated interpatient varability in these endpoints. With either situation, the useof an accelerated titration design with single-patient cohorts may not be optimal.
There are also situations where small dose-escalation increments may be advis-able. For example, if the agent is predicted to have severe, ireversible or potentially
-

l~:r~:f!
108ACCELERATED TITRATION DESIGNS
"".,".~.;
fatal organ toxicity based on animal toxicology, paricularly if associated with a steepdose-response curve for toxicity, relatively small changes in dose/concentration maylead from minimal toxicity to severe toxicity, and thus smaller dose-escalation incre-ments are preferable to ensure safety.
Approaches to enhancing the proportion of patients in a phase I tral receiving'therapeutic' dose levels includes not only limiting enrolment on lower dose levels butalso allowing dose-escalation within individual patients. Intrapatient dose-escalationprovides two advantages: it improves the lielihood of benefit from the agent for the,individual patient and it increases the experience at higher dose levels. Acceleratedtitration designs proposed by Simon et at. (7) allow intrapatient dose escalation if notoxicity :: grade 1 was seen in the first cycle at the assigned dose leveL. Whle it didnot appreciably shorten the study duration, it did allow more patients to be treated ator near the recommended phase II dose and increased the number of cycles evaluatedat the higher dose levels.
Although the rationale supportng intrapatient dose-escalation is appealing, itdoes not seem to be widely applied. Thus, despite the appeal of escalating patientsto higher doses than they were assigned initially, should safety criteria be met, someissues remain with its routine application in phase I protocols. Many phase I pro-tocols continue to be written prohibiting intrapatient escalation since it is believedto have a minimal impact on tral effciency while bringing with it concerns aboutpractical issues regarding the 'rules' for implementing dose-escalation and safety.Studies that have allowed intrapatient escalation withn their protocols have gener-ally allowed dose-escalation to occur after the patient has been evaluated at the currentdose level for the duration of the observation period and where the patient has hadminimal/no toxicity. Less commonly used are rules that require not only that the pa-tient has not had significant toxicity but also that the next higher dose level has beenevaluated in one or more new patients - a more strngent and cumbersome criterionthat may be favored to enhance safety and also to distinguish between acute ver-sus cumulative toxic events. Although experience with intrapatient dose-escalationwithn phase I studies is limited, to date its use within phase I studies using an ac-celerated titration design did not appear to compromise patient safety or complicatethe interpretation of the study results. Of note, withn a given protocol, it is impor-tant to require a minimum number of newly recruited patients at each dose leveland to base furter dose-escalation decisions upon the behavior of the drg in these
individuals.
'0.1
; ~ f"~~ ;~.~ ;:.;;.Î'31 !
J.-r"
~~ I
~ ¡'/11
4.6 Conclusions
Accelerated titration designs can dramatically reduce the number of patients accruedto a phase I tral, in comparson to the standard phase I design. They can also sub-stantially shorten the duration of the phase I tral. With intrapatient dose-escalationand application of a dose-toxicity model, they provide much greater information thanthe stadard design and analysis with regard to cumulative toxicity, interpatient and
t...
1
~

ACCELERATED TITRATION DESIGNS 109
intrapatient varability, steepness of the dose-toxicity curve and separation of thedose-toxicity curves for the varing toxicity levels. They also provide all patientsentered in the tral a maximum opportunity to be treated at a therapeutic dose.
Despite this, we find that the designs are not widely used, which is likely tobe due to the conservativeness of investigators. Even when they are used, theyare often used with an initial dose set much more conservatively than would bedone for the standard design and without use of intrapatient dose-escalation, therebyreducing their effectiveness. A recent comprehensive review of the risk-benefit rela-tionship for phase I trals conducted over the past decade reveals an overall tox-
icity death rate of only O.OOS (46). An accompanying editorial (47) argues thatsuch a low toxicity death rate, in the context of treatment for an often rapidly fa-tal disease, suggests that phase I trals may be conducted in an overly cautiousfashion. Appropriate utilization of designs such as the accelerated titration designsmight increase the potential for benefit in phase I trals, with little increase inrisk.
References1. S.G. Arbuck (1996) Workshop on phase I study design. Ninth NCIÆORTC New Drug
Development Symposium, Amsterdam, March 12, 1996. Ann. Oncology, 7(6), 567-73.
2. E.A. Eisenhauer, P.J. O'Dwyer, M. Christian and J.S. Humphrey (2000). Phase I clinicaltral design in cancer drug development. J. Clin. Oncology, 18(3),684-92.
3. S.P. Dent and E.A Eisenhauer (1996) Phase I tral design: are new methodologies beingput into practice? Ann. Oncology, 7(6), 561-6.
4. L.B. Sheiner (1990) Implications of an alternative approach to dose-response trals. J.Acquired Immune Deficiency Syndrome, 3(Suppl. 2), 20-.
5. L.B. Sheiner, S.L. Beal and N.e. Sambol (1989) Study designs for dose-ranging. Clin.Pharmacology Theory, 46(1), 63-77.
6. L.B. Sheiner, Y. Hashimoto and S.L. Beal (1991) A simulation study comparng designsfor dose ranging. Statistics in Medicine, 10(3), 303-21 March.
7. R. Simon, B. Freidlin, L. Rubinstein, S.G. Arbuck, J. Collns and M.e. Chrstian (1997)Accelerated titration designs for phase I clinical trals in oncology. J. Natl Cancer Inst.,89(15),1138-47.
8. T.e. Chou and P. Talalay (1981) Generalized equations for the analysis of inhibitions ofMichaelis-Menten and higher-order kinetic systems with two or more mutually exclusiveand nonexclusive inhibitors. Eur. J. Biochemistry, 115(1), 207-16.
9. T.e. Chou and P. Talalay (1984) Quantitative analysis of dose-effect relationships: thecombined effects of multiple drgs or enzyme inhbitors. Adv. Enzyme Regulation, 22,
27-55.10. P.M. LoRusso, S. Prakash, A. Wozniak, L. Flaherty, M. Zalupski, A Shields, H.
Sands, R. Parchment and B. Jasti (2002) Phase I clinical tral of 5-fluoropyrimidinone(5FP), an oral prodrg of 5-fluorouracil (5FU). Investigational New Drugs, 20(1), 63-71.
11. M.P. Goetz, D. Toft, J. Reid, M. Ames, B. Stensgard, S. Safgren, AA. Adjei, J. Sloan, P.Atherton, V. Vasile, S. Salazaar, A Adjei, G. Croghan and e. Erlichman (2005) Phase I
--

~'Y
110 ACCELERATED TITRATION DESIGNS
tral of 17 -allylamino-17 -demethoxygeldanamycin in patients with advanced cancer J.Clin. Oncology, 23(6), 1078-87.
12. J.L. Grem, G. Momson, X.D. Guo, E. Agnew, C.H. Takmoto, R Thomas, E. Szabo,L. Grochow, E GroUman, J .M. Hamlton, L. Neckers and RH. Wilson (2005) Phase Iand pharacologic study of 17-( allylamno )-17 -demethoxyge1danamycin in adult patientswith solid tumors. J. Clin. Oncology,
23(9), 1885-93.
13. C. Sessa, G. Capri, L. Gianni, E Peccatori, G. Grassell, J. Bauer, M. Zucchett, L. Viganò,A Gatti, C. Minoia, P. Liati, S. Van den Bosch, A Bemareggi, G. Camboni and S.Marsoni (2000) Clinical and pharacological phase I study with accelerated titrationdesign of a daily times five schedule of BBR3464, a novel cationic trplatinum complex.Ann. Oncology, 11(8),977-83.
14. K. Mross, M.E. Scheulen, T. Licht, C. Unger, H. RicWy, AC. Stem, K. Kutz, M.G.Camboni, P. Barbieri, E. Verdi, B. Vincenzi and A Bemareggi (2004) Phase I clinicaland pharacokinetic study of BBR 3576, a novel aza-anthapyrazole, administered Lv.every 4 weeks in patients with advanced solid tumors: a phase I study group tral of
the Central European Society of Anticancer-Drug Research (CESAR). Anticancer Drugs,15(1), 15-22.
15. R Plummer, M. Ghielmini, P. Calvert, M. Voi, J. Renard, G. Gallant, E. Gupta, H. Calvertand C. Sessa (2002) Phase I and pharacokinetic study of the new taxane analog BMS-184476 given weekly in patients with advanced malignancies. Clin. Cancer Res., 8(9),2788-97.
16. S. Mani, H. McDaid, A Hamlton, H. Hochster, M.B. Cohen, D. Khabelle, T. Griffn,D.E. Lebwohl, L. Liebes, E Muggia and S.B. Horwitz (2004) Phase I clinical and phar-macokinetic study of BMS-247550, a novel derivative of epothilone B, in solid tumors.Clin. Cancer Res., 10(4), 1289-98.
17. J. Abraham, M. Agrawal, S. Bake, A Rutt, M. Edgerly, EM. Balis, B. Widemann, L.Davis, B. Damle, D. Sonnichsen, D. Lebwohl, S. Bates, H. Kotz and T. Fojo (2003) PhaseI tral and pharacokinetic study of BMS-247550, an epothilone B analog, administered
intravenously on a daily schedule for five days. J. Clin. Oncology, 21(9), 1866-73.18. S.M. Gadgeel, A. Wozniak, RR Boinpally, R. Wiegand, L.K. Heilbrun, V. Jain, R
Parchment, D. Colevas, M.B. Cohen and P.M. LoRusso (2005) Phase I clinical trial ofBMS-247550, a derivative of epothilone B, using accelerated titration 2B design. Clin.Cancer Res., 11(17), 6233-9.
19. S.D. Undevia, N.J. Vogelzang, AM. Mauer, L. Janisch, S. Mani and M.J. Ratain (2004)Phase I clinical tral of CEP-2563 dihydrochloride, a receptor tyrosine kinase inhibitor, inpatients with refractory solid tumors. Invest. New Drugs, 22(4),449-58.
20. P. Hovstadius, R Larsson, E. Jonsson, T. Skov, AM. Kissmeyer, K. Krasilnikoff, J. Bergh,M.O. Karlsson, A Lönnebo and J. Ahlgren (2002) A phase I study of CHS 828 in patientswith solid tumor malignancy. Clin. Cancer Res., 8(9), 2843-50. -
21. M.A Rudek, W.D. Figg, V. Dyer, W. Dahut, M.L. Turner, S.M. Steinberg, DJ. Liewehr,D.R. KoWer, J.M. Pluda and E. Reed (2001) Phase I clinical trial of oral COL-3, a matrxmetalloproteinase inhibitor, in patients with refractory metastatic cancer. J. Clin. Oncology,19(2), 584-92.
22. S. Syed, C. Takmoto, M. Hidalgo, J. Rizzo, J.G. Kuhn, L.A Hammond, G. Schwar,A. To1cher, A. Patnaik, S.G. Eckhardt and E.K. Rowinsky (2004) A phase I and phar-macokinetic study of Col-3 (Metastat), an oral tetracycline derivative with potent matrxmetalloproteinase and antitumor properties. Clin. Cancer Res., 10(19),6512-21.

ACCELERATED TITRATION DESIGNS 111
23. G.J. Rustin, S.M. Galbraith, H. Anderson, M. Stratford, L.K. Folkes, L. Sena, L. Gumbrelland P.M. Price (2003) Phase I clinical tral of weekly combretastatin A4 phosphate: clinicaland pharacokinetic results. J. Clin. Oncology, 21(15), 2815-22.
24. A. Chatterjee, R Digumari, RN. Mamdi, K. Katneni, V.V. Upreti, A. Surath, M.L.Srinivas, S. Uppalapati, S. Jiwatani, S. Subramanam and N.R Srinivas (2004) Safety,tolerabilty, pharacokinetics, and pharacodynamics of an orally active novel camp-tothecIn analog, DRF-1042, in refractory cancer patients in a phase I dose escalationstudy. J. CUn. Pharmacology, 44(7), 723-36.
25. M.A. Vilalona-Calero, S.G. Eckhardt, G. Weiss, M. Hidalgo, J.H. Beijnen, C. vanKesteren, H. Rosing, E. Campbell, M. Kraynak, L. Lopez-Lazaro, C. Guzman, D.D.Yon Hoff, J. Jimeno and E.K. Rowinsky (2002) A phase I and pharacokinetic studyof ecteinascidin-743 on a daily x 5 schedule in patients with solid malignancies. Clin.Cancer Res., 8(1), 75-85.
26. Y.J. Ko, G.J. Bubley, R Weber, C. Redfern, D.P. Gold, L. Finke, A. Kovar, T. Dahl and
S.D. Gilies (2004) Safety, pharacokinetics, and biological pharacodynamics of theimmunocytokine EMD 273066 (huKS-IL2): results of a phase I tral in patients withprostate cancer. 1. Immunotherapy, 27(3), 232-9.
27. S. Goel, K. Desai, A. Bulgar, A. Fields, G. Goldberg, S. Agrawal, R Marin, M. Grndeland S. Mani (2003) A safety study of a mixed-backbone oligonucleotide (GEM231) tar-geting the type I regulatory subunit alpha of protein kinase A using a continuous infusionschedule in patients with refractory solid tumors. CUn. Cancer Res., 9(11), 4069-76.
28. H.X. Chen, J.L. Marshall, E. Ness, RR Marin, B. Dvorchik, N. Rizvi, J. Marquis, M.McKinlay, W. Dahut and M.J. Hawkins (2000) A safety and pharacokinetic study ofa mixed-backbone oligonucleotide (GEM231) targeting the type I protein kinase A bytwo-hour infusions in patients with refractory solid tumors. CUn. Cancer Res., 6(4), 1259-66.
29. P. Borchmann, R Schnell, L Fuss, O. Manzke, T. Davis, L.D. Lewis, D. Behnke, C. Wick-enhauser, P. Schiler, V. Diehl and A. Engert (2002) Phase I tral of the novel bispecificmolecule H22 x Ki-4 in patients with refractory Hodgkin lymphoma. Blood, 100(9),3101-7.
30. S.M. Gadgeel, R.R Boinpally, L.K. Heilbrun, A. Wozniak, V. Jain, B. Redman, M. Zalup-ski, R Wiegand, R Parchment and P.M. LoRusso (2003) A phase I clinical tral of spi-camycin derivative KR5500 (NSC 650426) using a phase I accelerated titration '2B'design. Investigational New Drugs, 21(1), 63-74.
31. N.E. Schoemaker, C. van Kesteren, H. Rosing, S. Jansen, M. Swar, J. Lieverst, D. Fraier,M. Breda, C. Pellzzoni, R Spinell, M. Grazia Porro, J.H. Beijnen, J.H. Schellens andW.W. ten Bokkel Huinink (2002) A phase I and pharacokinetic study of MAG-CPT, awater-soluble polymer conjugate of camptothecin. Br. 1. Cancer, 87(6),608-14.
32. Y. Matsumura, M. Gotoh, K. Muro, Y. Yamada, K. Shirao, Y. Shimada, M. Okuwa,S. Matsumoto, Y. Miyata, H. Ohkura, K. Chin, S. Baba, T. Yamao, A. Kannam, Y.Takamatsu, K. Ito and K. Takahashi (2004) Phase I and pharacokinetic study of
MCC-465, a doxorubicin (DXR) encapsulated in PEG iinunoliposome, in patients withmetastatic stomach cancer. Ann. Oncology, 15(3),517-25.
33. S. Clive, D.J. Webb, A. MacLellan, A. Young, B. Byrne, L. Robson, J.E Smyth and D.LJodrell (2001) Forear blood flow and local responses to peptide vasodilators: a novelpharacodynamc measure in the phase I tral of antagonist G, a neuropeptide growthfactor antagonist. CUn. Cancer Res., 7(10), 3071-8.
ii

112 ACCELERATED TITRATION DESIGNS
34. Y. Matsumura, T. Hamaguchi, T. Ura, K Muro, Y. Yamada, Y. Shimada, K Shirao, T.Okusaka, H. Ueno, M. Ikeda and N. Watanabe (2004) Phase I clinical trial and phara-cokinetic evaluation ofNK911, a micelle-encapsulated doxorubicin. Br. J. Cancer,
91(10),
1775-81.35. MJ. de Jonge, J. Verweij, A van der Gaast, O. Valota, O. Mora, AS. Planting, M.A
Mantel, S.V. Bosch, MJ. Lechuga, F. Fiorentini, D. Hess and C. Sessa (2002) Phase I andpharacokinetic studies of PNU-159548, a novel alycycline, administered intravenouslyto patients with advanced solid tumours. Eur. J. Cancer, 38(18), 2407-15.
36. AC. Lockhar, M. Howard, KR. Hande, BJ. Roth, J.D. Berlin, F. Vreeland, A Campbell,E. Fontana, F. Fiorentini, C. Fowst, V.A. Paty, O. Lanord and M.L. Rothenberg (2004)A phase I dose-escalation and pharacokinetic study of brostallicin (PNU-166196A),a novel DNA minor groove binder, in adult patients with advanced solid tumors. CUn.Cancer Res., 10(2),468-75.
37. AJ. Ten Tije, 1. Verweij, A Spareboom, A Van Der Gaast, C. Fowst, F. Fiorentini, J.Tursi, A Antonellni, M. Mantel, C.M. Harman, G. Stoter, A.S. Planting and MJ. DeJonge (2003) Phase I and pharacokinetic study of brostallicin (PNU-166196), a newDNA minor-groove binder, administered intravenously every 3 weeks to adult patientswith metastatic cancer. CUn. Cancer Res., 9(8), 2957-6.
38. J. Dupont, B. Bienvenu, C. Aghajanian, S. Pezzull, P. Sabbatini, P. Vongphrachan, C.Chang, C. Perkell, K Ng, S. Passe, L. Breimer, J. Zhi, M. DeMaro, D. Spriggs and S.L.Soignet (2004) Phase I and pharacokinetic study of the novel oral cell-cycle inhibitorRo 31-7453 in patients with advanced solid tumors. J. CUn. Oncology, 22(16), 3366-74.
39. R. Salazar, D. Bissett, C. Twelves, L. Breimer, M. DeMaro, S. Campbell, J. Zhi, S. Ritlandand J. Cassidy (2004) A phase I clinical and pharacokinetic study of Ro 31-7453 givenas a 7- or 14-day oral twice daily schedule every 4 weeks in patients with solid tumors.Clin. Cancer Res., 10(13),4374-82.
40. S. Wadler, D. Makower, C. Clairmont, P. Lambert, K Fehn and M. Sznol (2004) PhaseI and pharacokinetic study of the ribonucleotide reductase inhbitor, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone, administered by 96-hour intravenous continuousinfusion. J. CUn. Oncology, 22(9), 1553-63.
41. 1. Murren, M. Modiano, C. Clairmont, P. Lambert, N. Savaraj, T. Doyle and M. Sznol(2003) Phase I and pharacokinetic study of trapine, a potent ribonucleotide reductaseinhibitor, administered daily for five days in patients with advanced solid tumors. CUn.Cancer Res., 9(11), 4092-100.
42. P.H. Jones, R.D. Burnett, i. Fainar, P. Nadolny, P. Walker, Z. Yu, D. Tang-Liu, T.S.
Ganesan, D.C. Talbot, AL. Hars and GJ. Rustin (2003) A phase 1 study of tazarotenein adults with advanced cancer. Br. J. Cancer, 89(5), 808-15.
43. S.E. AI-Batran, A Atmaca, F. Bert, D. Jäger, C. Frisch, A. Neumann, J. Ort, A Knuthand E. Jäger (2004) Dose escalation study for defining the maximum tolerated dose ofcontinuous oral trofosfamide in pretreated patients with metastatic lung cancer. Onkologie,27(6), 534-8.
44. E.C. Dees, S.D. Baker, S. O'Reily, M.A Rudek, S.B. Davidson, C. Ayleswort, K Elza-Brown, M.A Carducci and R.C. Donehower (2004) A phase I and phanacokinetic studyof short infusions ofUCN-Ol in patients with refractory solid tumors. Clin. Cancer Res.,11(2 Pt 1), 664-71.
-

T¡
¡¡
i
It
I
¡
l¡
t
i
I
t¡rl
I,
r~
rl
ii
Ir
tr
Fe
la
I
li~~~~~~,fiIL
ACCELERATED TITRATION DESIGNS 113
45. B.C. Goh, M.J. Ratain, D. Bertucci, R Smith, S. Mani, N.J. Vogelzang, R.L. Schilsky,
M. Hutchison, M. Smith, S. Averbuch and E. Douglass (2001) Phase I study of ZD9331on short daily intravenous bolus infusion for 5 days every 3 weeks with fixed dosingrecommendations. J. Clin. Oncology, 19(5), 1476-84.
46. E. Horstmann, M.S. McCabe, L. Grochow, S. Yamamoto, L. Rubinstein, T. Budd, D.Shoemaker, E.J. Emanuel and C. Grady (2005) Risks and benefits of phase 1 oncologytrals, 1991 through 2002. N. Engl. J. Medicine, 352(9),895-904.
47. R Kurzrock and RS. Benjamin (2005) Risks and benefits of phase I oncology trals,revisited. N. Engl. 1. Medicine, 352(9), 930-2.





























