Embed Size (px)
Citation preview

www.actamat-journals.com
Acta Materialia 54 (2006) 3953–3960
A thick-interface model for diffusive and massive phasetransformation in substitutional alloys
J. Svoboda a,*, J. Vala b, E. Gamsjager c, F.D. Fischer c
a Institute of Physics of Materials, Academy of Sciences of the Czech Republic, Zizkova 22, CZ-61662 Brno, Czech Republicb Faculty of Civil Engineering, Brno University of Technology, Zizkova 17, CZ-60200 Brno, Czech Republic
c Institute of Mechanics, Montanuniversitat Leoben, Franz-Josef-Straße 18, A-8700 Leoben, Austria
Received 24 February 2006; received in revised form 20 April 2006; accepted 22 April 2006Available online 7 July 2006
Abstract
Based on the application of the thermodynamic extremal principle, a new model for the diffusive and massive phase transformationin multicomponent substitutional alloys is developed. Interfacial reactions such as the rearrangement of the lattice, solute drag and trans-interface diffusion are automatically considered by assigning a finite thickness and a finite mobility to the interface region. As an appli-cation of the steady-state solution of the derived evolution equations, the kinetics of the massive c! a transformation in the Fe-richFe–Cr–Ni system is simulated. The thermodynamic properties of the interface may influence significantly the contact conditions atthe interface as well as the conditions for the occurrence of the massive transformation and its kinetics. The model is also used forthe simulation of the diffusion-induced grain boundary migration in the same system. By application of the model a realistic valuefor the Gibbs energy per unit interface area is obtained.� 2006 Acta Materialia Inc. Published by Elsevier Ltd. All rights reserved.
Keywords: Phase transformation; Diffusion; Modelling; Interface kinetics; Diffusion-induced grain boundary migration
1. Introduction
To simulate diffusional phase transformations, it is nec-essary to solve the coupled problem of diffusion and inter-face migration. Usually, the interface is assumed to besharp, and ortho-equilibrium (continuous chemical poten-tials of all components across the interface) or para-equilib-rium (continuous chemical potentials of interstitialcomponents and continuous site fractions of substitutionalcomponents across the interface) contact conditions areapplied at the interface. A more general set of contact con-ditions at a sharp interface with a finite mobility restrictsthe continuity in chemical potentials to the interstitial ele-ments only and allows identical jumps in chemical poten-tials for all substitutional elements [1]. In the case ofsubstitutional alloys, assuming a sharp migrating interface
1359-6454/$30.00 � 2006 Acta Materialia Inc. Published by Elsevier Ltd. All
doi:10.1016/j.actamat.2006.04.027
* Corresponding author. Tel.: + 420 5 412 18657.E-mail address: [email protected] (J. Svoboda).
of finite mobility, the contact condition represented byidentical jumps of the chemical potentials of the substitu-tional elements is also derived by application of the ther-modynamic extremal principle [2].
A real migrating interface of finite thickness h may,however, drag segregated impurity atoms forming concen-tration profiles across the interface. Such a local diffusionprocess may reduce the migration velocity v of the interfacedue to the Gibbs energy dissipated by this local diffusionprocess. This decelerating effect is called solute drag. Prom-inent approaches by Lucke and Stuwe [3] and Cahn et al.[4] are reported in the literature – for other references seee.g. Ref. [5]. In contrast to the results of the classic workson solute drag, Svoboda et al. [5] allow differences of thechemical potential for the solvent as well as for the intersti-tial solute component in binary systems. The diffusion ofthe solute across the interface is driven by the gradient ofthe chemical potential of the solute, and the interfacemigration is driven by the difference of the chemical
rights reserved.

3954 J. Svoboda et al. / Acta Materialia 54 (2006) 3953–3960
potential of the solvent across the interface (a finite inter-face mobility is supposed). A similar approach, based onthe balance for the total Gibbs energy in the interface,was later used by Odquist et al. [6] for multicomponentsystems.
The solute drag effect does not only lead to the occur-rence of jumps of the chemical potentials of interstitial ele-ments segregating in the interface, but may also changesignificantly the jumps of the chemical potentials of substi-tutional components, which eventually breaks the condi-tion to be identical for all substitutional components aspredicted in Refs. [1,2]. As a consequence this may leadto a situation in which the massive transformation mayoccur in the two-phase region as has been experimentallyproven for several systems (e.g. Ref. [7]) and discussed indetail in Ref. [8].
The goal of this paper is to present a thick-interfacemodel for the diffusive transformation in multicomponentsystems consisting of substitutional components that isbased on the application of the thermodynamic extremalprinciple (e.g. see Refs. [2,9,10]). The model takes intoaccount solute drag and trans-interface diffusion as wellas a finite interface mobility. The steady-state solution ofthe derived equations is then applied to the Fe-rich Fe–Cr–Ni system to simulate the massive c! a transforma-tion. The influence of thermodynamic properties of theinterface on kinetics and conditions for the massive trans-formation are discussed in detail. The model is also usedfor simulation of the diffusion-induced grain boundarymigration (DIGM) in the same system.
2. The model
A one-dimensional, two-phase system of unit cross-sec-tional area with r substitutional components is considered,in which the c! a transformation occurs (see Fig. 1). Thelocal chemical composition is described by mole fractions~x ¼ ðx1ðz; tÞ; . . . ; xr�1ðz; tÞÞ, with z being the coordinateand t being the time. The mole fraction of the componentr is given by
xrðz; tÞ ¼ 1�Xr�1
k¼1
xkðz; tÞ: ð1Þ
Fig. 1. System geometry.
The system moves with the velocity v(t), being the velocityof interface migration to the left, relative to a fixed coordi-nate system. Thus, the position of the interface is fixed inthe coordinate system, and one can choose the origin ofthe coordinate system to coincide with the left-hand surfaceof the interface of thickness h (see Fig. 1). The left and rightends of the system have the coordinates zL and zR, respec-tively, both of which depend on time. Furthermore, weassume that non-zero diffusive fluxes ja�
k and jc�k can be im-
posed at both ends of the specimen.Let a be a scalar quantity a = a(z, t). Then its material
(or total time) derivative in the actual configuration con-nected with the moving system _a ¼ da=dt and the partialtime derivative oa/ot are related by
_a ¼ oaot� v
oaoz: ð2Þ
Furthermore, the time derivative of an integral quantity isgiven by
d
dt
Z zR
zL
a dz ¼Z zR
zL
_adz: ð3Þ
if ddt ðdzÞ ¼ 0 is assumed (see Section 2.1 of Ref. [11]). The
chemical potential lk of component k is supposed to be afunction of ~x and of z. The dependence on z determinesto which phase or to which position in the interface thematerial point belongs. Consequently, as ~x is a functionof z and t, we can formally write lk as a certain functionof z and t.
We suppose that the dependencies of the chemicalpotentials on ~x are known for both the a and c phases aswell as for the interface I between both phases supposedto be a third phase denoted by b (see Fig. 1); i.e. la
kð~xÞ,lb
k ð~xÞ and lckð~xÞ are known functions. For the chemical
potentials of phases a, b or c the well-known Gibbs–Duhem relation (see Appendix in Ref. [12] for details)holdsXr
k¼1
xk dlfk ¼
Xr
k¼1
xkolf
k
ozdzþ olf
k
otdt
!¼ 0: ð4Þ
where f represents the phase a, b or c. If we choose dt = 0,the Gibbs–Duhem relation readsXr
k¼1
xkolf
k
oz¼ 0: ð5Þ
Dividing Eq. (4) by the time increment dt yieldsXr
k¼1
xk _lfk ¼ 0: ð6Þ
Alternatively Eq. (6) can be obtained using dz = �vdt andEq. (2).
The chemical potential lk can be calculated at any pointof the specimen as
lkðz;~xÞ ¼X
f2fa;b;cgwf ðzÞlf
k ð~xÞ: ð7Þ

J. Svoboda et al. / Acta Materialia 54 (2006) 3953–3960 3955
The functions wf(z) are supposed to be reasonably contin-uous weight functions, having the properties
waðzÞ ¼ 1 for z 6 0; waðzÞ ¼ 0 for z P h=2;
wcðzÞ ¼ 0 for z 6 h=2; wcðzÞ ¼ 1 for z P h: ð8Þ
and wb(z) = 1 � wa(z) � wc(z) for every z between zL andzR. Note that the Gibbs–Duhem Eqs. (5) and (6) for lk in-stead of lf
k are not fulfilled in the region 0 6 z 6 h (in theinterface I).
For the application of the thermodynamic extremalprinciple, it is necessary to calculate the rate _G of the totalGibbs energy G of the system and the total dissipation Q inthe system.
2.1. Total Gibbs energy and its rate
The total Gibbs energy G of the system is given by
G ¼ 1
X
Xr
k¼1
Z zR
zL
xklk dz; ð9Þ
where X is the molar volume. By assuming that the im-posed fluxes ja�
k � jkðzLÞ and jc�k � jkðzRÞ (see Fig. 1) are
zero, the time derivative of G can be calculated using Eq.(3) as
_G ¼ 1
X
Xr
k¼1
Z zR
zL
ð _xklk þ xk _lkÞdz: ð10Þ
Using Eqs. (2), (7) and (8) together with the Gibbs–Duhemrelation in the form of Eq. (4), one gets
_G ¼ 1
X
Xr
k¼1
Z zR
zL
_xklk dz� vX
f2fa;b;cg
Z h
0
xkdwf
dzlf
k dz
!:
ð11ÞAccording to the Gibbs–Duhem relation in the form of Eq.(5) and using Eq. (7), Eq. (11) can be rewritten as
_G ¼ 1
X
Xr
k¼1
Z zR
zL
_xklk dz� vZ h
0
xkolk
ozdz
� �: ð12Þ
The mass conservation law for the component k with thediffusive flux jk reads
_xk ¼ �Xojk
oz: ð13Þ
Insertion of Eq. (13) into Eq. (12) and integration of thefirst term in Eq. (12) by parts yield
_G ¼Xr
k¼1
Z zR
zL
jk
olk
ozdz� 1
XvZ h
0
xkolk
ozdz: ð14Þ
2.2. Rate of the total Gibbs energy dissipation
The rate of the total Gibbs energy dissipation due tobulk diffusion and interface migration is given by [2,10]
Q ¼Xr
k¼1
Z zR
zL
j2k
Akdzþ v2
M; Ak ¼
xkDk
XRT: ð15Þ
The quantity Dk is the tracer diffusion coefficient of compo-nent k, R is the gas constant, T is the absolute temperatureand M is the interface mobility.
We assume negligible vacancy fluxes in the system,which together with the vacancy mechanism of diffusionleads to the constraintXr
k¼1
jk � 0: ð16Þ
Using Eqs. (13) and (16) leads toXr
k¼1
_xk ¼ 0: ð17Þ
Thus Eq. (1) is automatically fulfilled during the whole sys-tem evolution, if the starting conditions obey Eq. (1). Theconstraint of Eq. (16) can be taken into account in the for-mulation of the thermodynamic extremal principle bymeans of the Lagrange multiplier method.
2.3. Application of the thermodynamic extremal principle
We use the principle of maximum dissipation rate as itwas first applied by Onsager [9] in 1945 for a diffusionalprocess. According to this thermodynamic extremal princi-ple the kinetics of the system corresponds to the variation[10]
d _Gþ Q2þZ zR
zL
kXr
k¼1
jk dz
!ðj1; . . . ; jr; vÞ ¼ 0: ð18Þ
where k is the Lagrange multiplier to meet Eq. (16).Performing the variations
d _GþQ2þZ zR
zL
kXr
k¼1
jk dz
!ð~j1; . . . ;~jrÞ
¼Xr
k¼1
Z zR
zL
ejkolk
ozdzþ
Xr
k¼1
Z zR
zL
jkejk
Akdzþ
Xr
k¼1
Z zR
zL
~jkkdz¼ 0;
d _GþQ2þZ zR
zL
kXr
k¼1
jk dz
!ðevÞ
¼ � evX
Xr
k¼1
Z h
0
xkolk
ozdzþ vev
M¼ 0; ð19Þ
we obtain for k = 1,. . ., r
olk
ozþ jk
Akþ k ¼ 0 ð20Þ
and
v ¼ MX
Xr
k¼1
Z h
0
xkolk
ozdz: ð21Þ
Multiplication of Eqs. (20) by Ak, their summation andapplication of Eq. (17) allow one to calculate k as

Fig. 2. Weight functions used in the calculations.
3956 J. Svoboda et al. / Acta Materialia 54 (2006) 3953–3960
k ¼ �Pr
k¼1AkolkozPr
l¼1Al: ð22Þ
The fluxes jk follow from Eq. (20) as
jk ¼ Ak �olk
ozþPr
i¼1AioliozPr
l¼1Al
!: ð23Þ
The diffusional law Eq. (23) is the same as that derivedin Ref. [12] for constrained fluxes (see Eq. (16)). Eq.(21) for the interface motion can be considered as anew result.
Combination of Eqs. (21) and (23), together with givenexpressions for the chemical potentials and the conserva-tion laws (Eq. (13)), yields the system evolution equations.Integrating Eq. (20) with respect to z from 0 to h, oneobtains
lkðhÞ � lkð0Þ ¼ �Z h
0
kdz�Z h
0
jk
Akdz: ð24Þ
which provides the contact conditions at the migratinginterface depending on the interface properties and thekinetics of the system. Assuming that jk is bounded forany k, then the second integral tends to zero for h! 0and/or for all Ak !1 in the interface I. As the first inte-gral in Eq. (24) is independent of k, identical jumps of thechemical potentials for all components are guaranteed for asharp interface. This agrees with results presented in Refs.[1,2]. Identical jumps of chemical potentials are also ob-tained for infinite diffusivities Dk in the interface I of allcomponents assuming xk > 0.
3. Numerical solution of the steady-state problem
During massive transformation the concentration pro-files are limited to the interface and its nearest neighbour-hood, and the transformation occurs practically as asteady-state process with a constant velocity v and oxk/ot
= 0. For the sake of simplicity we assume that both – zL
and zR tend to infinity and the imposed fluxes ja�k and jc�
k
(see Fig. 1) are zero. Thus, we can set xkðzLÞ ¼ x1k withx1k being the (a priori known) mole fractions in the a phasefar from the interface. Integration of Eq. (13), in combina-tion with Eq. (2), yields
jk ¼vXðxk � x1k Þ: ð25Þ
Let us now introduce the decomposition
lfk ð~xÞ ¼ lf
0k þ RT ln xk þ ufk ð~xÞ: ð26Þ
for all f 2 {a,b,c}; lf0k are constants for a given tempera-
ture and ufk are functions of ~x (not dominant, but rather
complicated in practice). By inserting Ak from Eq. (15)into Eq. (23) and by applying Eqs. (17) and (25) one ob-tains after time-consuming calculations a matrix equationfor ~x
ðIþ BÞ d~xþ ðKþ vNÞ~x ¼ vN~x1; ð27Þ
dzwhere I is an identity matrix, B is a square matrix and K
and N are two square diagonal matrices, all of the orderr � 1. Their forms are given in Appendix A. The matrix ele-ments of B and K depend on~x. The matrix N depends onlocal diffusional properties only. The Crank–Nicholsonscheme is used in the numerical method to obtain the stea-dy-state mole fraction profiles xk(z). The value of v can beobtained from Eq. (21) for a known ~xðzÞ in the interface.The code was developed within the framework of MAT-LAB environment (http://www.mathworks.com/).
In particular, K is a zero matrix in both phases a and c.Thus,~x ¼~x1 for z 6 0 (in the whole phase a). The profilesfor the mole fractions xk for z P h (in the whole phase c)are similar to exponential curves but, as B is not a zeromatrix, not exactly identical with them. For z tending toinfinity we again have~x!~x1.
4. Results of simulations based on the model
Usually the diffusion coefficients Dak and Dc
k for thephases a and c are known. Also the diffusion coefficientsDb
k for the centre of the interface I represented by the bphase can be estimated. The diffusion coefficients Dk insidethe interface (0 < z < h) can be interpolated by applying theformula
ln DkðzÞ ¼X
f2fa;b;cgwf ðzÞ ln Df
k ð28Þ
with the same weight functions wa(z), wb(z) and wc(z) givenby Eq. (8). In the simulations the cubic Hermite interpola-tion splines are used as the weight functions given bywa(0) = wc(h) = 1, wa(h/2) = wa(h) = wc(h/2) = wc(0) = 0and dwa/dz = dwc/dz = 0 for all nodes z 2 {0,h/2,h}. Theweight functions are plotted in Fig. 2.
4.1. Massive phase transformation in Fe–Cr–Ni system
We assume that the thermodynamic properties in thecentre of the interface I (b phase) can be approximatedby the thermodynamic properties of the liquid phase. Thechemical potentials in the a and c phases as well as in theliquid are calculated based on a thermodynamic assessment
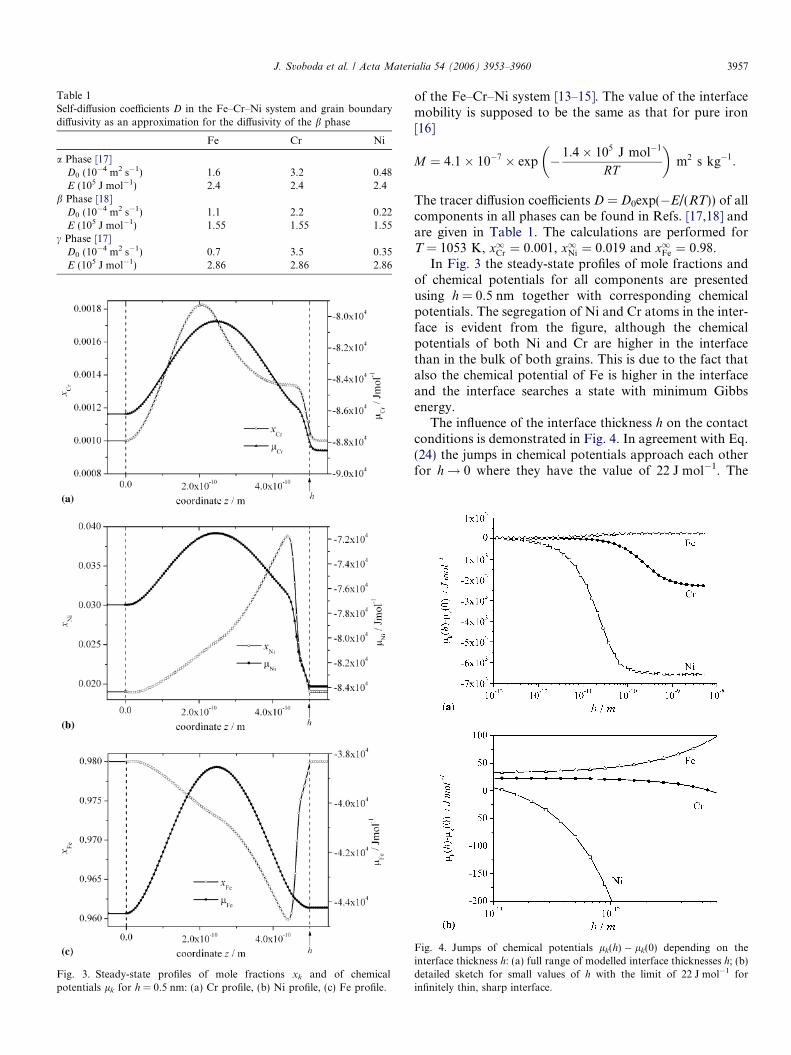
Table 1Self-diffusion coefficients D in the Fe–Cr–Ni system and grain boundarydiffusivity as an approximation for the diffusivity of the b phase
Fe Cr Ni
a Phase [17]D0 (10�4 m2 s�1) 1.6 3.2 0.48E (105 J mol�1) 2.4 2.4 2.4
b Phase [18]D0 (10�4 m2 s�1) 1.1 2.2 0.22E (105 J mol�1) 1.55 1.55 1.55
c Phase [17]D0 (10�4 m2 s�1) 0.7 3.5 0.35E (105 J mol�1) 2.86 2.86 2.86
Fig. 3. Steady-state profiles of mole fractions xk and of chemicalpotentials lk for h = 0.5 nm: (a) Cr profile, (b) Ni profile, (c) Fe profile.
J. Svoboda et al. / Acta Materialia 54 (2006) 3953–3960 3957
of the Fe–Cr–Ni system [13–15]. The value of the interfacemobility is supposed to be the same as that for pure iron[16]
M ¼ 4:1� 10�7 � exp � 1:4� 105 J mol�1
RT
� �m2 s kg�1:
The tracer diffusion coefficients D = D0exp(�E/(RT)) of allcomponents in all phases can be found in Refs. [17,18] andare given in Table 1. The calculations are performed forT = 1053 K, x1Cr ¼ 0:001, x1Ni ¼ 0:019 and x1Fe ¼ 0:98.
In Fig. 3 the steady-state profiles of mole fractions andof chemical potentials for all components are presentedusing h = 0.5 nm together with corresponding chemicalpotentials. The segregation of Ni and Cr atoms in the inter-face is evident from the figure, although the chemicalpotentials of both Ni and Cr are higher in the interfacethan in the bulk of both grains. This is due to the fact thatalso the chemical potential of Fe is higher in the interfaceand the interface searches a state with minimum Gibbsenergy.
The influence of the interface thickness h on the contactconditions is demonstrated in Fig. 4. In agreement with Eq.(24) the jumps in chemical potentials approach each otherfor h! 0 where they have the value of 22 J mol�1. The
Fig. 4. Jumps of chemical potentials lk(h) � lk(0) depending on theinterface thickness h: (a) full range of modelled interface thicknesses h; (b)detailed sketch for small values of h with the limit of 22 J mol�1 forinfinitely thin, sharp interface.
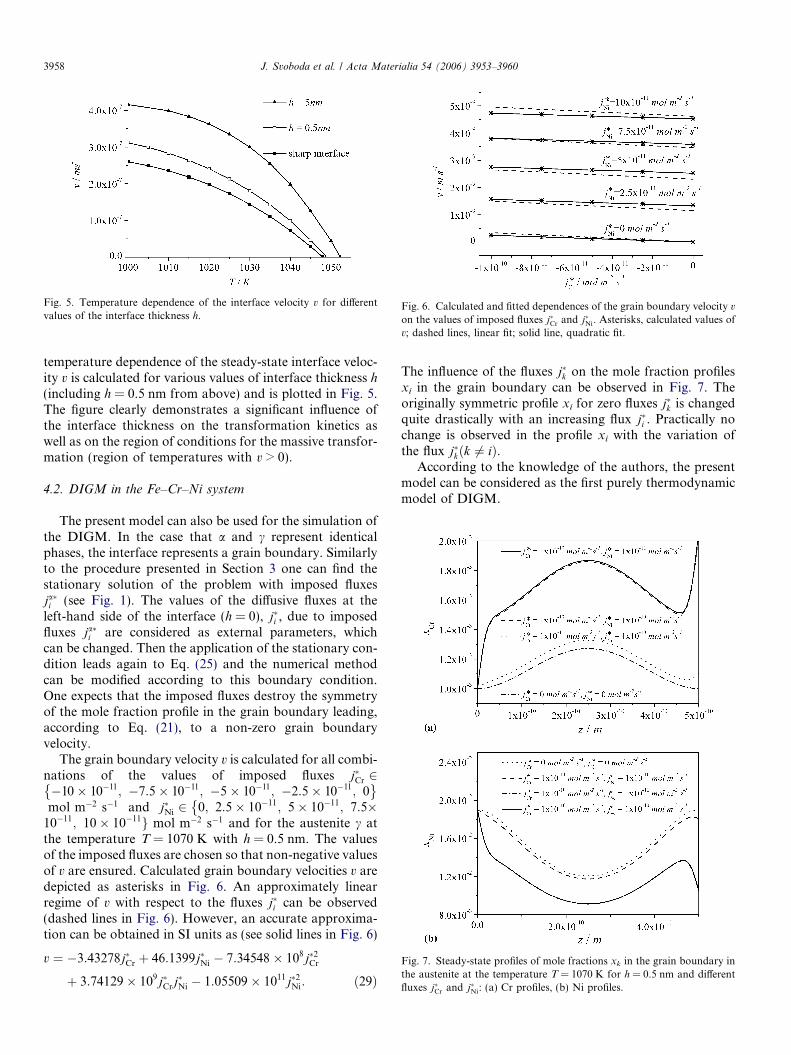
Fig. 6. Calculated and fitted dependences of the grain boundary velocity v
on the values of imposed fluxes j�Cr and j�Ni. Asterisks, calculated values ofv; dashed lines, linear fit; solid line, quadratic fit.
Fig. 5. Temperature dependence of the interface velocity v for differentvalues of the interface thickness h.
Fig. 7. Steady-state profiles of mole fractions xk in the grain boundary inthe austenite at the temperature T = 1070 K for h = 0.5 nm and differentfluxes j�Cr and j�Ni: (a) Cr profiles, (b) Ni profiles.
3958 J. Svoboda et al. / Acta Materialia 54 (2006) 3953–3960
temperature dependence of the steady-state interface veloc-ity v is calculated for various values of interface thickness h
(including h = 0.5 nm from above) and is plotted in Fig. 5.The figure clearly demonstrates a significant influence ofthe interface thickness on the transformation kinetics aswell as on the region of conditions for the massive transfor-mation (region of temperatures with v > 0).
4.2. DIGM in the Fe–Cr–Ni system
The present model can also be used for the simulation ofthe DIGM. In the case that a and c represent identicalphases, the interface represents a grain boundary. Similarlyto the procedure presented in Section 3 one can find thestationary solution of the problem with imposed fluxesja�
i (see Fig. 1). The values of the diffusive fluxes at theleft-hand side of the interface (h = 0), j�i , due to imposedfluxes ja�
i are considered as external parameters, whichcan be changed. Then the application of the stationary con-dition leads again to Eq. (25) and the numerical methodcan be modified according to this boundary condition.One expects that the imposed fluxes destroy the symmetryof the mole fraction profile in the grain boundary leading,according to Eq. (21), to a non-zero grain boundaryvelocity.
The grain boundary velocity v is calculated for all combi-nations of the values of imposed fluxes j�Cr 2�10� 10�11; �7:5� 10�11; �5� 10�11; �2:5� 10�11; 0� �mol m�2 s�1 and j�Ni 2 0; 2:5� 10�11; 5� 10�11; 7:5�
�10�11; 10� 10�11g mol m�2 s�1 and for the austenite c atthe temperature T = 1070 K with h = 0.5 nm. The valuesof the imposed fluxes are chosen so that non-negative valuesof v are ensured. Calculated grain boundary velocities v aredepicted as asterisks in Fig. 6. An approximately linearregime of v with respect to the fluxes j�i can be observed(dashed lines in Fig. 6). However, an accurate approxima-tion can be obtained in SI units as (see solid lines in Fig. 6)
v ¼ �3:43278j�Cr þ 46:1399j�Ni � 7:34548 � 108j�2Cr
þ 3:74129� 109j�Crj�Ni � 1:05509� 1011j�2Ni: ð29Þ
The influence of the fluxes j�k on the mole fraction profilesxi in the grain boundary can be observed in Fig. 7. Theoriginally symmetric profile xi for zero fluxes j�k is changedquite drastically with an increasing flux j�i . Practically nochange is observed in the profile xi with the variation ofthe flux j�kðk 6¼ iÞ.
According to the knowledge of the authors, the presentmodel can be considered as the first purely thermodynamicmodel of DIGM.

J. Svoboda et al. / Acta Materialia 54 (2006) 3953–3960 3959
4.3. Calculation of the interface and grain boundary Gibbs
energy densities in the Fe–Cr–Ni system
The calculation of interface and grain boundary Gibbsenergies per unit interface or grain boundary area may beconsidered as a check of the reasonability of the model.The Gibbs energy density r of the c/a interface is calculatedaccording to the formula
r ¼ 1
X
Xr
k¼1
Z h
0
xklk dz� h2ðxkð0Þlkð0Þ þ xkðhÞlkðhÞÞ
� �ð30Þ
representing the difference of the total Gibbs energies givenby Eq. (9) for the thick interface and for the sharp interfacein position h/2. For temperatures ranging from T = 1030 to1070 K the Gibbs energy densities r are calculated for thec/a interface as well as for grain boundaries in a and c. Forh = 0.5 nm the values of the Gibbs energy densities r rangefrom 0.186 to 0.226 J m�2 which can be considered as real-istic values supporting the reasonability of the model.
5. Summary
A new thick-interface thermodynamic model of the sol-ute drag and trans-interface diffusion during diffusivetransformation and of DIGM is presented. The steady-state solution of the model equations is found, whichdescribes the massive transformation. The model is appliedto the Fe–Cr–Ni system. The simulations of the massivec! a transformation show that the existence of the solutedrag in the interface influences the contact conditions at theinterface allowing the massive transformation to occur alsoin the two-phase region. By choosing a and c as identicalphases and by imposing fluxes to the interface (grainboundary), DIGM is simulated. The interface and grainboundary Gibbs energy densities are calculated from themodel, and their realistic values support the reasonabilityof the model.
Acknowledgements
Financial support by the Osterreichische For-schungsforderungsgesellschaft mbH, the Province of Sty-ria, the Steirische Wirtschaftsforderungsgesellschaft mbHand the Municipality of Leoben under the frame of theAustrian Kplus Programme is gratefully acknowledged.The work has also been supported by the Grant Agencyof the Academy of Sciences of the Czech Republic (GrantNo. A200410601) and by the program Kontakt–Austrian–Czech Scientific Cooperation (No. 05/2006).
Appendix A
The detailed form of the matrices B, K and N from Eq.(27) originates in Eq. (7): differentiating Eq. (7), oneobtains
olk
oz¼
Xf2fa;b;cg
owf
ozlf
k þ wfolf
k
oz
!: ðA:1Þ
Inserting lfk from Eq. (26) together with
olfk
oz¼ RT
xk
oxk
ozþXr
l¼1
oufk
oxl
oxl
oz; ðA:2Þ
taking into account that wa + wb + wc = 1 and conse-quently alsoXf2fa;b;cg
owf
oz¼ 0; ðA:3Þ
we obtain
olk
oz¼
Xf2fa;b;cg
owf
ozðlf
0k þ ufk Þ þ
RTxk
oxk
oz
þX
f2fa;b;cgwf
Xr
l¼1
oufk
oxl
oxl
oz: ðA:4Þ
The notation
blk ¼X
f2fa;b;cg
owf
ozðlf
0k þ ufk Þ; bukl ¼
Xf2fa;b;cg
wf
Xr
l¼1
oufk
oxl
ðA:5Þconverts Eq. (A.4) into its brief form
olk
oz¼ blk þ
RTxk
oxk
ozþXr
l¼1
bukloxl
oz: ðA:6Þ
By means of Dk from Eq. (15) the diffusive fluxes from Eq.(23) can be expressed as
jk ¼ �xkDk
RT
Xr
l¼1
xlDlolk
oz� oll
oz
� � !, Xr
m¼1
xmDm
!ðA:7Þ
(Dk appears here instead of Ak). The additional notation
fk ¼Dk
D; g ¼
Xr
l¼1
flxl ðA:8Þ
with some (non-zero) reference value D of the diffusioncoefficient enables us to simplify Eq. (A.7) into the form
jk ¼ �xkfkD
RT
Xr
l¼1
flxl
golk
oz� oll
oz
� �: ðA:9Þ
According to Eq. (A.4) and dividing Eq. (A.9) by fkD, weobtain
� jk
fkD¼ xk
RTblk þ
RTxk
oxk
ozþXr
l¼1
bukloxl
oz
!
� xk
RT
Xr
l¼1
flxl
gbllþ
RTxl
flxl
goxl
ozþ flxl
g
Xr
m¼1
bulmoxm
oz
!ðA:10Þ

3960 J. Svoboda et al. / Acta Materialia 54 (2006) 3953–3960
and using the new notation
lk ¼ blk �Xr
l¼1
flxl
gbll; ukl ¼ bukl �
Xr
m¼1
fmxm
gbuml ðA:11Þ
finally
� jk
fkD¼ oxk
oz� xkfl
goxl
ozþ xklk
RTþ xk
Xr
l¼1
ukl
RToxl
oz: ðA:12Þ
From Eq. (25) it is known
jk
fkDþ v
fkDXðxk � x1k Þ ¼ 0; ðA:13Þ
this together with Eq. (A.12) yields
oxk
ozþ xk
Xr
l¼1
� fl
gþ ukl
RT
� �oxl
ozþ lk
RTþ v
fkDX
� �xk ¼
vfkDX
x1k :
ðA:14Þ
Clearly x1 + . . . + xr = 1, thus
oxr
oz¼ � ox1
oz� . . .� oxr�1
ozðA:15Þ
and also
g ¼Xr�1
l¼1
flxl þ fr 1�Xr�1
l¼1
xl
!¼ fr þ
Xr�1
l¼1
ðfl � frÞxl:
ðA:16Þ
Consequently the contribution of the summation with theindex r in Eq. (A.9) can be eliminated with the result
oxk
ozþ xk
Xr�1
l¼1
� fl
gþ ukl
RT
� �oxl
oz� xk
Xr�1
l¼1
� fr
gþ ukr
RT
� �oxl
oz
þ lk
RTþ v
fkDX
� �xk ¼
vfkDX
x1k ðA:17Þ
for k 2 {1, . . . , r � 1}; this can be rewritten as
oxk
ozþ xk
Xr�1
l¼1
Bkloxl
ozþ ðKk þ vN kÞxk ¼ vN kx1k ðA:18Þ
where
Bkl ¼xkðfr � flÞ
gþ ukl � ukr
RT; Kkk ¼
lk
RT; Nkk ¼
1
fkDX
ðA:19Þ
are exactly the elements of matrices B, K and N needed inEq. (27).
In particular, outside the interface all second additiveterms in the relations for Bkl by Eq. (A.19) vanish, Kkk=0 and Nkk are constants. With respect to Eq. (A.16) onecan also see that Bkl = 0 just as in the case of the same val-ues of diffusion coefficients for all components(f1 = . . . = fr). However, even for non-zero elements of B,if xkðzÞ ¼ x1k holds for some z and each k 2 {1, . . . , r � 1}then xk ¼ x1k everywhere. Inside the interface such simpleconsiderations are not available. The practical structureof both B and K is rather complicated.
References
[1] Fischer FD, Simha NK, Svoboda J. ASME J Eng Mater Technol2003;125:266.
[2] Svoboda J, Gamsjager E, Fischer FD, Fratzl P. Acta Mater2004;52:959.
[3] Lucke K, Stuwe H-P. Acta Metall 1971;19:1087.[4] Cahn JW, Fife P, Penrose O. Acta Mater 1997;45:4397.[5] Svoboda J, Fischer FD, Gamsjager E. Acta Mater 2002;50:967.[6] Odquist J, Sundman B, Agren J. Acta Mater 2003;51:1035.[7] Veeraraghavan D, Wang P, Vasudevan VK. Acta Mater
2003;51:1721.[8] Svoboda J, Gamsjager E, Fischer FD. Metall Mater Trans A
2006;37A:125.[9] Onsager L. Ann New York Acad Sci 1945;46:241.
[10] Svoboda J, Turek I, Fischer FD. Philos Mag 2005;85:3699.[11] Fischer FD, Simha NK. Acta Mech 2004;171:213.[12] Svoboda J, Fischer FD, Fratzl P, Kroupa A. Acta Mater
2002;50:1369.[13] Lee B-J. CALPHAD 1992;16:121.[14] Hillert M, Qiu C. Metall Trans A 1991;22A:2187.[15] Hillert M, Qiu C. Metall Trans A 1992;23A:1593.[16] Krielaart G. Ph.D. thesis, TU-Delft; 1995.[17] Lee B-J, Oh KH. Z Metallkd 1996;87:195.[18] Fridberg J, Torndahl L-E, Hillert M. Jernkont Ann 1969;153:263.







![On Energy Gaps in a New Type of Analytically Solvable ... · various generalizations of the Saxon-Hutner conjecture [3] (concerning binary substitutional alloys) for alloys consisting](https://img.dokumen.tips/doc/110x75/5e9fab59eb1e6729824be3b1/on-energy-gaps-in-a-new-type-of-analytically-solvable-various-generalizations.jpg)





















