Embed Size (px)
Citation preview

Listen to this manuscript’s
audio summary by
JACC Editor-in-Chief
Dr. Valentin Fuster.
J O U R N A L O F T H E A M E R I C A N C O L L E G E O F C A R D I O L O G Y V O L . 6 9 , N O . 6 , 2 0 1 7
ª 2 0 1 7 T H E A U T H O R . P U B L I S H E D B Y E L S E V I E R O N B E H A L F O F T H E A M E R I C A N
C O L L E G E O F C A R D I O L O G Y F O U N DA T I O N . T H I S I S A N O P E N A C C E S S A R T I C L E U N D E R
T H E C C B Y - N C - N D L I C E N S E ( h t t p : / / c r e a t i v e c o mm o n s . o r g / l i c e n s e s / b y - n c - n d / 4 . 0 / ) .
I S S N 0 7 3 5 - 1 0 9 7
h t t p : / / d x . d o i . o r g / 1 0 . 1 0 1 6 / j . j a c c . 2 0 1 6 . 1 1 . 0 4 2
REVIEW TOPIC OF THE WEEK
A Test in Context: Lipoprotein(a)Diagnosis, Prognosis, Controversies, and Emerging Therapies
Sotirios Tsimikas, MD
ABSTRACT
Fro
for
Ca
ceu
dio
Ma
Evidence that elevated lipoprotein(a) (Lp[a]) levels contribute to cardiovascular disease (CVD) and calcific aortic valve
stenosis (CAVS) is substantial. Development of isoform-independent assays, in concert with genetic, epidemiological,
translational, and pathophysiological insights, have established Lp(a) as an independent, genetic, and likely causal risk
factor for CVD and CAVS. These observations are consistent across a broad spectrum of patients, risk factors, and
concomitant therapies, including patients with low-density lipoprotein cholesterol <70 mg/dl. Statins tend to increase
Lp(a) levels, possibly contributing to the “residual risk” noted in outcomes trials and at the bedside. Recently approved
proprotein convertase subtilisin/kexin-type 9 inhibitors and mipomersen lower Lp(a) 20% to 30%, and emerging
RNA-targeted therapies lower Lp(a) >80%. These approaches will allow testing of the “Lp(a) hypothesis” in clinical
trials. This review summarizes the current landscape of Lp(a), discusses controversies, and reviews emerging therapies to
reduce plasma Lp(a) levels to decrease risk of CVD and CAVS. (J Am Coll Cardiol 2017;69:692–711) © 2017 The Author.
Published by Elsevier on behalf of the American College of Cardiology Foundation. This is an open access article under the
CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
D espite significant advances in the diagnosisand therapy of cardiovascular disease(CVD), patients continue to experience
myocardial infarction, stroke, peripheral arterialdisease, and need for revascularization. The ad-vances in identifying modifiable risk factors forCVD, including smoking, hypertension, dyslipide-mias, diabetes mellitus, and obesity, have allowedthe development of practice patterns andevidence-based guidelines in medical therapy andrevascularization that have contributed to thereduction of CVD mortality. However, despite theseadvances, w40% of all deaths can be attributed toCVD (1). Furthermore, in the short time window inwhich clinical trials evaluate therapies, only 20% to30% of patients benefit, and more events developin patients on active therapy than are prevented.These observations suggest that the presence of
m the Division of Cardiovascular Medicine, Sulpizio Cardiovascular Cent
nia. Dr. Tsimikas is a coinventor of and receives royalties from patents
lifornia San Diego (UCSD) on oxidation-specific antibodies; and currently
ticals, Inc., during a partial leave of absence in which he is leading the c
vascular diseases.
nuscript received September 6, 2016; revised manuscript received Novem
additional modifiable risk factors contributes toCVD risk.
Lipid disorders can be broadly divided into 4“clinical” categories: elevated low-density lipopro-tein cholesterol (LDL-C), low high-density lipopro-tein cholesterol (HDL-C), elevated triglycerides,and elevated lipoprotein(a) (Lp[a]). In the currentgenetic era, it has become apparent that onlyelevated levels of apolipoprotein B-100 (apoB)�containing lipoproteins (very low-density lipopro-tein, intermediate-density lipoprotein, LDL, Lp[a]) arecausally associated with increased cardiovascularrisk. In contrast, variations in genes associated withhigher HDL-C or drugs that raise HDL-C are not asso-ciated with improvement in cardiovascular risk (2,3).
At the clinical level, elevated Lp(a) has been theleast studied of these 4 lipid disorders. However, thisis rapidly changing with the awareness of its role in
er, University of California San Diego, La Jolla, Cali-
or patent applications owned by the University of
has a dual appointment at UCSD and Ionis Pharma-
linical development of antisense molecules for car-
ber 10, 2016, accepted November 21, 2016.

AB BR E V I A T I O N S
AND ACRONYM S
apo(a) = apolipoprotein(a)
apoB = apolipoprotein B-100
ASO = antisense
oligonucleotides
CAD = coronary artery disease
CAVS = calcific aortic valve
stenosis
CVD = cardiovascular disease
EAS = European
Atherosclerosis Society
HDL-C = high-density
lipoprotein cholesterol
LDL-C = low-density
lipoprotein cholesterol
LDLR = low-density
lipoprotein receptor
Lp(a) = lipoprotein(a)
MACE = major adverse cardiac
events
OxPL = oxidized
phospholipid(s)
PCSK9 = proprotein
convertase subtilisin/
kexin-type 9
SNP = single nucleotide
orphism
J A C C V O L . 6 9 , N O . 6 , 2 0 1 7 TsimikasF E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1 Lipoprotein(a)
693
CVD and calcific aortic valve stenosis (CAVS), and thepotential of novel therapies to substantially lowerLp(a). This review summarizes the Lp(a) field from atranslational and clinical perspective with its rela-tionship with CVD and CAVS.
HOW IS Lp(a) DIFFERENT FROM LDL AND
PLASMINOGEN?
Lp(a) is composed of an LDL-like particle in whichapoB is covalently bound by a single disulfide bond toapolipoprotein(a) (apo[a]), the pathognomoniccomponent of Lp(a) (Figure 1). For unknown etiolog-ical and physiological reasons, apo(a) has evolvedfrom the plasminogen gene through duplication andremodeling over the millennia (4). Plasminogen con-tains 5 kringles (KI to KV) and a protease domain.Plasminogen is a proenzyme, which is converted tothe fibrinolytic enzyme plasmin by plasminogen ac-tivators, such as urokinase and tissue plasminogenactivator, either endogenously or iatrogenically.Apo(a) does not contain KI to KIII of plasminogen, butinstead contains 10 subtypes of KIV (with KIV1 andKIV3–10 present in 1 copy, and KIV2 present in 1 to >40copies), 1 copy of KV, and an inactive proteasedomain. Unlike apoB, apo(a) does not contain lipiddomains or transport lipid, but instead, it is hydro-philic, and can bind to denuded and exposed lysine-rich vascular endothelium, similar to and also incompetition with plasminogen. However, except forhigh Lp(a) levels, on a molar basis, plasminogen isalmost always in excess of apo(a), which calls intoquestion its in vivo potency in inhibiting plasmin-ogen activity in most patients.
Within populations, there is extensive apo(a) pro-tein size heterogeneity, with >40 different isoforms,and thus, >40 different sizes of Lp(a) particles, whichis a unique occurrence unlike other circulating pro-teins, which usually have 1 defined mass (5). Withinindividuals, >80% carry 2 different-sized apo(a) iso-forms, each inherited from 1 parent. For example, anindividual may carry 2 small isoforms, a small and alarge one, or 2 large isoforms, with plasma Lp(a)levels determined by the net production of apo(a) ineach isoform, with the major contribution generallydriven by the small isoform (Figure 1).
METABOLISM OF Lp(a)
Circulating Lp(a) levels are primarily determined bythe LPA gene locus, without significant dietary orenvironmental influences (6). Apo(a) is synthesizedalmost exclusively in the liver, but the site of as-sembly of Lp(a) has not been confirmed, and may bewithin the hepatocyte, the space of Disse, or the
plasma compartment (7). The steps of as-sembly include apo(a) docking to LDL, andthen formation of a covalent disulfide bondbetween KIV-9 of apo(a) and apoB of LDL. Itappears that the LDL component is derivedfrom a newly synthesized apoB-100 and isnot derived from a very low-density lipo-protein precursor. Lp(a) has a longer plasmaresidence time than LDL. This may due tothe fact that the apo(a) component, which iscovalently attached near the LDL receptor(LDLR) binding site of apoB and which canbe larger than apoB, interferes with dockingto the LDLR, reduces clearance through theLDLR, and also requires clearance throughalternative pathways. The size of the apo(a)isoform correlates modestly and inverselywith plasma Lp(a) levels due to the consti-tutive hepatocyte production of apo(a), andbecause small isoforms can be made inhigher molar quantities per unit time versuslarge isoforms. The mechanisms throughwhich Lp(a) is cleared from plasma remaincontroversial. The LDLR likely has a modestrole, as evidenced by the fact that statinsraise LDLR numbers, yet do not lower Lp(a),and proprotein convertase subtilisin/kexin-
type 9 (PCSK9) inhibitors increase LDLR numbers,yet reduce Lp(a). However, animal and humanturnover studies with labeled Lp(a) suggest minimal(if any) effect, but cell culture studies and clinicalphenotypes of LDLR deficiency, such as those inpatients with familial hypercholesterolemia due toLDLR mutations, show higher Lp(a) levels than un-affected siblings. The kidney, as well as other un-defined clearance mechanisms, such as scavengerreceptor B1 and plasminogen receptors, and pro-teolytic cleavage of apo(a), may also have a role incatabolism.WHAT ARE THE MECHANISMS THROUGH
WHICH Lp(a) MEDIATES CVD?
Lp(a) contributes to CVD risk via multiple, nonre-dundant mechanisms. Lp(a) quantitatively carries allof the atherogenic risk of LDL particles, includingtheir propensity to oxidize after entry into the vesselwall, creating highly immunogenic and proin-flammatory oxidized LDL (8). However, on an equi-molar basis, Lp(a) is more atherogenic than LDLbecause, by definition, it not only contains all theproatherogenic components of LDL, but also ofapo(a). Apo(a) potentiates atherothrombosis throughadditional mechanisms, including inflammation
polym

FIGURE 1 Composition of Lp(a)
Lipoprotein [Lp(a)] is composed of apolipoprotein B-100 (apoB-100) covalently bound to apolipoprotein (a) [apo(a)], which is derived from kringle IV (KIV) and KV, and
the protease domain of plasminogen. Plasminogen has 1 copy each of KI to KV and an active protease domain. Apo(a) contains 10 subtypes of KIV repeats, composed of
1 copy each of KIV1, multiple copies of KIV2, and 1 copy of KIV3�10, KV, and an inactive protease-like (P) domain. In these examples, apo(a) isoforms of 4, 8, 24, and 40
KIV2 repeats are shown, representing 13, 17, 33, and 49 total KIV repeats. Oxidized phospholipids (OxPL), represented here by 1-palmitoyl-2-oxovaleroyl-sn-glycero-3-
phosphocholine (POVPC), are present covalently bound to apo(a), and also dissolved in the lipid phase of apoB-100.
Tsimikas J A C C V O L . 6 9 , N O . 6 , 2 0 1 7
Lipoprotein(a) F E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1
694

FIGURE 2 Pathogenic Mechanisms of Lp(a)
The atherogenicity of Lp(a) can be broadly classified in 3 categories: proatherogenic, proinflammatory, and potentially antifibrinolytic. The
major individual mechanisms within each category are listed. EC ¼ endothelial cell; IL ¼ interleukin; MCP ¼ monocyte chemoattractant
protein; PAI ¼ plasminogen activator inhibitor; SMC ¼ smooth muscle cell; TFPI ¼ tissue factor pathway inhibitor; other abbreviation as
in Figure 1.
J A C C V O L . 6 9 , N O . 6 , 2 0 1 7 TsimikasF E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1 Lipoprotein(a)
695
through its content of oxidized phospholipids(OxPL), the presence of lysine binding sites thatallow accumulation in the arterial wall, and potentialantifibrinolytic effects by inhibiting plasminogenactivation (Figure 2) (reviewed in Spence andKoschinsky [9]). However, Lp(a)-mediated CVD riskshould be considered in a quantitative mannerbecause most patients (70% to 80%) at risk for CVDhave low Lp(a) levels, such that LDL-C is present insignificant excess to Lp(a), and therefore, most ofthe apoB-driven risk is due to a higher number ofLDL particles. However, as Lp(a) levels increase to>25 to 30 mg/dl, which represents w30% of thepopulation, the risk of Lp(a) rises in a linear fashionaccording to absolute circulating Lp(a) mass.
The OxPL components of Lp(a), which can bepresent in the lipid phase of Lp(a) and also becovalently bound to apo(a) (10–12), are proin-flammatory and impart many of the proatherogenicproperties of Lp(a) (Figure 2). This has been shown inclinical studies by measuring levels of OxPL on
apoB-containing particles (OxPL-apoB), which pri-marily reflect the OxPL content of Lp(a) (10,11),which demonstrates that elevated OxPL-apoB levelsare either similar or superior predictors to Lp(a) inthe diagnosis or prognosis of CVD (13–18) and CAVS(19,20). In a recent study that validated thesemechanisms in vivo, patients with elevated Lp(a)had increased arterial inflammation detected byincreased accumulation of 18-fluorodeoxyglucose inthe carotid arteries and aorta. In addition, theirmonocytes demonstrated increased production ofproinflammatory cytokines upon stimulation and anenhanced capacity to transmigrate in an in vitrosystem, which reflected vascular penetration ofmonocytes through an endothelial cell barrier. Theseproinflammatory effects were associated withenhanced in vivo monocyte trafficking to the arterialwall using autologous radiolabeled monocytes, andwere abrogated by inactivating OxPL with a specificantibody or using recombinant apo(a) constructslacking OxPL, which suggested that these effects

FIGURE 3 Presence of SMCs, Macrophages, ApoB, Apo(a), and OxPL in Ruptured Human Coronary Plaques and in Distal Protection Devices Following
Percutaneous Coronary Intervention
2 4 6 8 10Time, min
OxPL(POVPC)
12
100%
75%
50%
Rela
tive
Inte
nsity
(%)
25%
0%14 16
H
O
OO
O
O CH
OH
m/z=594
OH2C
CH2
CH3
CH3
CH3
H2
H2O
CC
NO
P
18 20
HH
Continued on the next page
Tsimikas J A C C V O L . 6 9 , N O . 6 , 2 0 1 7
Lipoprotein(a) F E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1
696

J A C C V O L . 6 9 , N O . 6 , 2 0 1 7 TsimikasF E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1 Lipoprotein(a)
697
were largely driven by the presence of OxPL onLp(a) (12). In separate studies, OxPL on Lp(a) alsoup-regulated inflammatory genes, and inducedrelease of interleukin-8 (21) and monocyte chemo-attractant protein-1 (22). Monocyte chemoattractantprotein-1 is physically present on Lp(a), and thus,may enhance its entry into the vessel wall. Finally,apo(a) contains lysine-binding sites that allows it tobind tightly to exposed surfaces on denuded endo-thelium, enter, and accumulate into subintimalspaces or aortic valve leaflets, which leads toinflammation. Additional mechanisms are shown inFigure 2.
Human coronary lesions from subjects with sud-den death or carotid endarterectomy specimensshowed a progressive increase in the presence ofLp(a) and OxPL as lesions progressed, with thehighest presence of these epitopes in ruptured pla-ques (Figures 3A to 3D) (23). These findings werelater confirmed in patients who underwent coro-nary, carotid, renal, and peripheral interventions, inwhich similar strong immunostaining for Lp(a) andOxPL, and the direct presence of OxPL in debrisfrom distal protection devices by liquidchromatography-tandem mass spectrometry, wasdocumented (Figures 3E and 3F) (24).
WHAT IS THE CLINICAL EVIDENCE THAT
ELEVATED Lp(a) MEDIATES CVD?
Elevated Lp(a) mediates myocardial infarction,stroke, and peripheral arterial disease. Over the lastdecade, data in subjects without previous CVD fromepidemiological studies and meta-analyses (25),Mendelian randomization studies (26), and genome-wide association studies (27,28) have conclusivelyshown that elevated Lp(a) levels are associated with ahigher risk of CVD. In 63,746 coronary artery disease(CAD) cases and 130,681 control subjects from theCardiogramPlus4CD Consortium, 46 loci and 104 in-dependent variants that reached genome-wide
FIGURE 3 Continued
(A) Human coronary lesion with fibrous cap disruption (arrow), empty p
SMCs in the fibrous cap. (C) Diffuse and strong presence of macrophages
rupture site. Apo(a) is not present at sites of apoB staining, which sugg
colocalize at sites of apo(a), but are present more diffusely, similar to m
oxidized low-density lipoprotein (OxLDL) in macrophages and cell memb
intervention, with captured atheroma debris. (H) The OxPL POVPC deriv
trometry (LC-MS/MS). Arrows in G and H denote the journey of OxPL f
Magnification �200. I ¼ intima; M ¼media; MAC ¼macrophage; NC ¼ ne
from van Dijk et al. (23) and Ravandi et al. (24).
significance for susceptibility for CAD were identi-fied, with the most potent variants clustered in net-works linked to lipid metabolism and inflammation.Analyzing individual variants revealed that the mostpotent genetic association with CAD was the LPA lo-cus, which was numerically more potent than LDL-,PCSK9-, and 9p21-related variants. This observationthat the LPA gene is 1 of the strongest (if not thestrongest) monogenetic risk factors for CAD is un-derappreciated, and points to the potential of tar-geting Lp(a) lowering to reduce CVD risk with specifictherapies (29).
In meta-analyses of clinical outcomes, the risk ofLp(a) for CVD is curvilinear (25). In contrast, geneticstudies, which reflect life-long elevated Lp(a) andless confounding by the multiple heterogeneouslimitations in meta-analyses, show a more potentand linear risk, with odds ratios as high as 4 rela-tive to subjects with low levels (27,30). Figure 4represents the salient studies that show such asso-ciations, but the overall data in published reportsare consistent in their preponderance of evidence ofa causal association. Finally, there is evidence thatsome individuals have alleles that do not expressapo(a); therefore, they have either very low or ab-sent circulating Lp(a) levels, have a reduced risk ofCVD, but they are not at increased risk for non-CVDevents, which is consistent with the causality ofLp(a) for vascular disease (31,32).
WHAT IS THE CLINICAL EVIDENCE THAT Lp(a)
MEDIATES CAVS? Although it has long been sus-pected that Lp(a) is a risk factor for CAVS; onlyrecently has Lp(a) become appreciated as a potentrisk factor (33). Recent studies have shown that of>2.5 million single-nucleotide polymorphisms (SNPs)analyzed, the LPA SNP rs10455872, which is associ-ated with markedly elevated Lp(a) levels, was theonly monogenetic risk factor for aortic valve calcifi-cation and CAVS in multiple racial groups (34–37).Similar to the studies with myocardial infarction, a
laque cavity with embolized lipid gruel, and superimposed luminal thrombus. (B) A paucity of
. (D) Scant presence of apoB epitopes. (E) Apo(a) staining, primarily in the fibrous cap at the
ests that as Lp(a) enters the cell, apo(a) is retained, but apoB is degraded. (F) OxPL, which
acrophage staining. This suggests that additional sources of OxPL are present, such as from
ranes of apoptotic cells. (G) A filter distal protection device used in a saphenous vein graft
ed from the atheroma in G, and identified by liquid chromatography-tandem mass spec-
rom a ruptured plaque to a distal protection device to definitive identification by LC-MS/MS.
crotic core. Other abbreviations as in Figures 1 and 2. Modified and reprinted with permission

FIGURE 4 Evidence Base for Lp(a) as an Independent, Causal, Genetic Risk Factor for CVD
1.8
1.6
1.4
1.2
1.0
0.9
0.83 6 12
Usual Lp(a), Geometric Mean, mg/dL
Meta-analysis Mendelian Randomization Genome-Wide Association
Risk
Rat
io (9
5% C
I)
Odds
Rat
io fo
r Cor
onar
y Di
seas
e
Hazard Ratio (95% CI)
Multivariable adjusted 0 variantalleles
1 variantallele
2 variantalleles
Geometric Mean Lp(a), mg/dL
Adjustment for age and sex onlyNonfatal Ml and coronary death
24 48 96 192 0.8 1 2 4
P<.001
0
8.0
4.0
2.0
1.0
0.025 50 75 100 125
Lp(a)
mg/dL
>117
77-117
30-76
5-29
<5
Epidemiological, meta-analyses, Mendelian randomization, and genome-wide association studies demonstrate that genetically elevated lipoprotein(a)
[Lp(a)] leads to higher risk for cardiovascular disease (CVD) events, particularly acute myocardial infarction. Reprinted with permission from the Emerging
Risk Factors Collaboration (25), Clarke et al. (27), and Kamstrup et al. (30). CI ¼ confidence interval; MI ¼ myocardial infarction.
Tsimikas J A C C V O L . 6 9 , N O . 6 , 2 0 1 7
Lipoprotein(a) F E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1
698
strong case for causality can be made because thesestudies linked a genetic trait (LPA SNPs) that couldnot be altered by environment or diet to a quantita-tive and substantial (5- to 20-fold) increase in plasmaLp(a) levels, and to a clinical phenotype of aorticvalve calcification and CAVS.
However, the genetic readouts are often binary(i.e., risk of disease; yes/no), and additional insightsinto natural history would be helpful to clinicianswho manage such patients over time. In that regard,recent data from the ASTRONOMER Trial (AorticStenosis Progression Observation: Measuring Effectsof Rosuvastatin) (38) showed that patients with pre-existing mild-to-moderate aortic stenosis andelevated levels of Lp(a) and OxPL-apoB (third tertileor >58.5 mg/dl and >5.5 nM, respectively) had a fasterprogression rate, with annual changes in peak aorticjet velocities of 0.26 � 0.26 m/s per year versus 0.17 �0.21 m/s per year in the highest versus the lowesttertiles (Figures 5A and 5B), and a need for aortic valvereplacement (Figures 5C and 5D), which occurred in20.4% of the entire group (19). Similar rates of pro-gression were noted in patients with tricuspid valvesversus bicuspid valves, which represented 48% of thestudy patients. Interestingly, younger patients(median: younger than 57 years of age) had nearlydouble the progression rate and the highest need foraortic valve replacement, which is consistent with theprimary driver of genetically elevated Lp(a). Inter-estingly, rosuvastatin was shown not to affect
progression of CAVS, and it actually increased Lp(a)levels by 20% and OxPL-apoB levels by 46%(Figures 5E and 5F). However, it has been acknowl-edged that the beneficial effects of lowering LDL-Cmay confound the appropriate interpretation of theeffect of statins on CAVS in these trials. There areseveral potential explanations for these findings. Forexample, an adverse effect of raising Lp(a) with astatin might be offset by a beneficial effect oflowering LDL-C. Other possibilities include that mostpatients had low Lp(a) on entry, only patients with aninitially high Lp(a) level would have been adverselyaffected, and that the Lp(a)-raising effect of statins ismodest.
In more recent work, the enzyme autotaxin hasbeen identified as an important contributor moleculein CAVS (39). Oxidation of phospholipids generateslysophosphatidylcholine, which is present in calcifiedaortic valves in mouse models and explanted calcifiedhuman valves (39). Autotaxin is a lysophospholipasethat converts lysophosphatidylcholine into lyso-phosphatidic acid, which promotes inflammation,fibrosis, and cell motility. In a case–control study in150 patients with CAVS plus CAD and 150 matchedpatients with CAD without CAVS, patients with CAVShad elevated autotaxin mass and activity, Lp(a)(>50 mg/dl) and OxPL-apoB (>2.02 nM), and auto-taxin activity in combination with either Lp(a) orOxPL-apoB, which significantly increased the risk ofCAVS, with odds ratios of 3.46 and 5.48, respectively,

J A C C V O L . 6 9 , N O . 6 , 2 0 1 7 TsimikasF E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1 Lipoprotein(a)
699
compared with patients with lower levels (Figures 5Gand 5H) (20).
Therefore, a unifying hypothesis for the develop-ment and progression of CAVS is that Lp(a) carriesboth autotaxin and OxPL into aortic valve leaflets,and initiates inflammation and calcification viaOxPL metabolites that induce up-regulation of pro-calcifying genes, which ultimately results in hemo-dynamically significant disease and clinical sequelae(Figure 5I) (33).
AREA OF CONTROVERSY I: WHAT ARE THE
AVAILABLE Lp(a) ASSAYS, AND IS THE
MEASUREMENT OF Lp(a) ADEQUATELY
STANDARDIZED FOR ROUTINE CLINICAL CARE?
Lp(a) levels are generally expressed as milligrams perdeciliter of the mass of the entire particle, which in-cludes the protein content of apoB-100 and apo(a),and their associated lipids (cholesterol, cholesterylesters, phospholipids, and triglycerides), as well ascarbohydrates attached to apo(a), or in nanomoles perliter as a particle number of apo(a). Lp(a) mass assayshave an inherent limitation due to the heterogeneityof Lp(a) particle sizes, making it difficult to stan-dardize assays with appropriate calibrators. In addi-tion, because most antibodies used in assays arepolyclonal and cross-react with multiple KIV2 repeats,these assays may overestimate Lp(a) levels in patientswith large isoforms and underestimate levels inpatients with small isoforms. Most clinical labora-tories have overcome this limitation by using appro-priate calibrators, along with linking the results tothe World Health Organization/International Federa-tion of Clinical Chemistry and Laboratory MedicineInternational Reference Reagent, making the assaysrelatively isoform independent. Lp(a) cholesterolcontent can also be estimated with gel electrophoresistechniques, but these techniques are not validated interms of accuracy or in predictive value. This topicwas recently reviewed by Marcovina and Albers (40),which provided full details of assay methodologiesand limitations. For routine clinical care, currentlyavailable assays, except for Lp(a) cholesterol assays,can be considered fairly accurate for separating low-risk patients from high-risk patients, except perhapswhen patients are near the assay thresholds for what isconsidered elevated, which is generally >30 mg/dl or>75 nmol/l in the United States. Currently availableassays linked to the World Health Organization/In-ternational Federation of Clinical Chemistry andLaboratory Medicine standard are able to detect high-risk patients (i.e., levels >50 mg/dl or >125 nmol/l)with acceptable accuracy. Future efforts should focus
on performing comparison studies of different assays(41), and fully standardizing assays across all plat-forms and instruments.
AREA OF CONTROVERSY II: WHAT ARE
CONSIDERED ELEVATED Lp(a) LEVELS AND
WHAT ARE APPROPRIATE RISK CUTOFFS
FOR CVD AND CAVS?
Previous data that showed an inflection for risk ofmyocardial infarction at Lp(a) >30 mg/dl (42) wererecently confirmed by a large meta-analysis of126,634 participants and 1.3 million person-years offollow-up, in which the risk was curvilinear, butstarted to accelerate at >24 mg/dl (25). Genome-wideassociation studies and Mendelian randomizationstudies were also consistent with these estimates(26,27). In general, the higher the Lp(a) level, thegreater the average risk for CVD. Detailed data onCAVS are not available for risk according to baselineLp(a) levels, except that risk was significantly higherat >40 to 60 mg/dl (19,20,35,36). The EuropeanAtherosclerosis Society (EAS) proposed <50 mg/dl(approximately <100 to 125 nmol/l) as an optimallevel, which represents 20% of the population withhigher levels. However, this recommendation missesrisk in patients with levels between 25 and 50 mg/dl,as shown by the Copenhagen data (43) and recentLp(a) analyses of randomized trials (44,45). The2016 Canadian Cardiovascular Society Guidelines forthe Management of Dyslipidemia consider Lp(a)>30 mg/dl to be a risk factor, and suggest measuringLp(a) to inform decision-making, particularly in pa-tients at intermediate risk and those with a familyhistory of premature CAD, and in younger patientswho may not meet treatment risk criteria (46).
Unlike normally distributed LDL-C or other labo-ratory values, Lp(a) levels are skewed leftward, andmost individuals (w70%) have values in the normalrange of <30 mg/dl. However, it must be emphasizedthat 30% of the global population amounts tow2 billion people with Lp(a) levels in the atherogenicrange. Both the German and U.K. apheresis guidelinesuse Lp(a) >60 mg/dl to specifically allow reimburse-ment for patients with both isolated Lp(a) elevationand recurrent CVD events, or in conjunction withuncontrolled elevated LDL-C.
AREA OF CONTROVERSY III:
DOES MEASURING Lp(a) ENHANCE
RISK PREDICTION AT THE BEDSIDE?
Although genetic and epidemiological data stronglysupport the prognostic causality of Lp(a), the

FIGURE 5 Relationship of Lp(a), OxPL, and Autotaxin to the Progression of CAVS
0.30
0.25
0.20
0.15
0.10
0.05
0Tertiles 1 & 2
Lp(a) ≤58.5mg/dL
Prog
ress
ion
Rate
of
V peak
(m/s
/yr)
Tertile 3Lp(a) >58.5mg/dL
p = 0.005
0.26 ± 0.03(n = 73)
0.17 ± 0.02(n = 147)
0.30
0.25
0.20
0.15
0.10
0.05
0Tertiles 1 & 2
OxPL-apoB ≤5.50nM
Prog
ress
ion
Rate
of
V peak
(m/s
/yr)
Tertile 3OxPL-apoB >5.50nM
p = 0.01
0.26 ± 0.03(n = 73)
0.17 ± 0.02(n = 147)
A B
100
80
60
40
20
00 1 2 3 4 5
266
5930
10656
Follow-Up (Years)
Adju
sted
Even
t-Fr
ee S
urvi
val (
%)
12266
*HR = 2.0 (1.1-3.7); p = 0.02
Tertiles 1 & 2 of Lp(a)
Tertile 3 of Lp(a)
13971
14773
100
80
60
40
20
00 1 2 3 4 5
266
6326
10557
Follow-Up (Years)
Adju
sted
Even
t-Fr
ee S
urvi
val (
%)
12266
*HR = 1.9 (1.0-3.4); p = 0.04
Tertiles 1 & 2 of OxPL-apoB
Tertile 3 of OxPL-apoB
13971
14773
C D
StatinPlacebo
p <0.001
*60
55
50
45
40Baseline
Lp(a
) Pla
sma
Leve
ls (m
g/dL
)
1 year
p = 0.007
*†10
9
8
7
6
5
Baseline
OxPL
-apo
B Pl
asm
a Le
vels
(nM
)
1 year
E F
(A) Elevated Lp(a) and (B) OxPL-apoB predict the progression of pre-existing mild-to-moderate calcific aortic valve stenosis (CAVS) on echocardiography in meters/
second/year in the ASTRONOMER trial. (C) Elevated Lp(a) or (D) OxPL-apoB predict a higher rate of aortic valve replacement (AVR) compared with those with low
Lp(a) levels. Rosuvastatin raises both (E) Lp(a) and (F) OxPL-apoB levels. The synergistic effect of elevated autotaxin along with either (G) Lp(a) or (H) OxPL-apoB in
predicting the presence of CAVS. (I) A unifying hypothesis of the Lp(a)-autotaxin-OxPL axis in the progression of CAVS. ATX ¼ autotaxin; OR ¼ odds ratio; other
abbreviations as in Figure 1. Reprinted with permission from Capoulade et al. (19) and Nsaibia et al. (20).
Continued on the next page
Tsimikas J A C C V O L . 6 9 , N O . 6 , 2 0 1 7
Lipoprotein(a) F E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1
700
clinician and patient are most interested in beingable to recategorize the potential risk into a lower orhigher risk category, and to change therapy on thebasis of the value. This was addressed by the Bru-neck study (47) in 826 subjects from the general
community with a 15-year prospective follow-up. Insubjects at intermediate risk based on Framinghamrisk score and Reynolds risk score variables, whichare the categories of highest need of risk stratifica-tion, the addition of Lp(a) to these risk scores
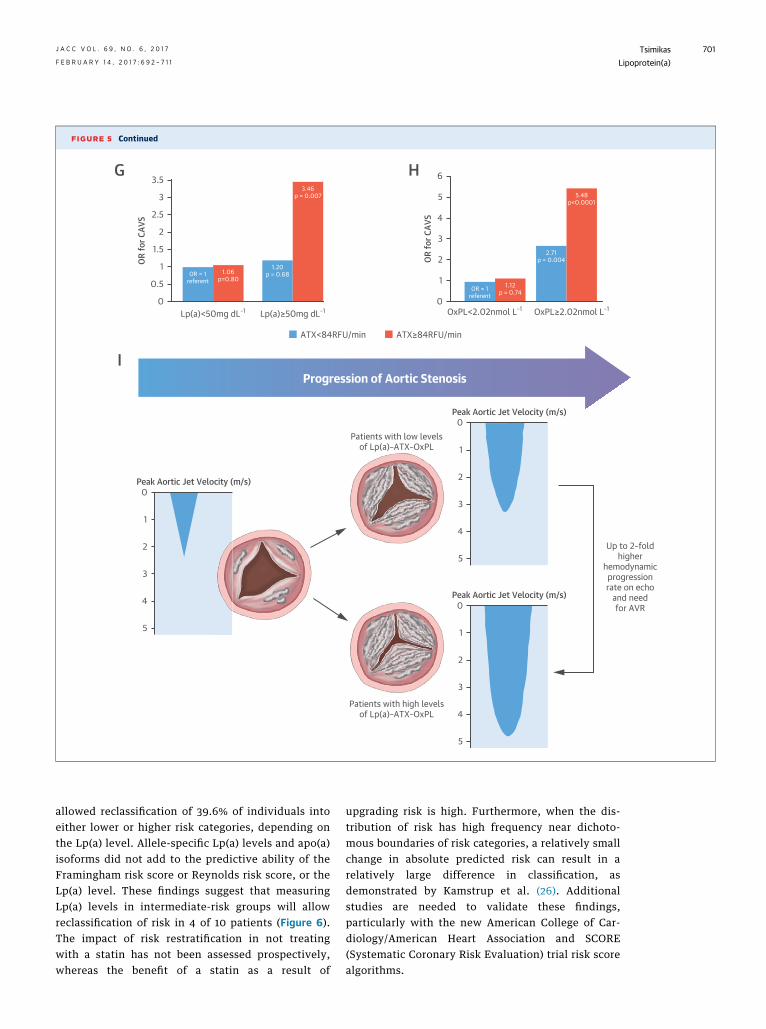
FIGURE 5 Continued
1.20p = 0.68
3.46p = 0.007
1.06p=0.80
OR = 1referent
3.5
3
2.5
2
1.5
1
0.5
0Lp(a)<50mg dL–1 Lp(a)≥50mg dL–1
OR fo
r CAV
S
6
5
4
3
2
1
0OxPL<2.02nmol L–1 OxPL≥2.02nmol L–1
Up to 2–foldhigher
hemodynamicprogressionrate on echo
and needfor AVR
Patients with low levelsof Lp(a)–ATX–OxPL
0
1
2
3
4
5
Patients with high levelsof Lp(a)–ATX–OxPL
Peak Aortic Jet Velocity (m/s)
Progression of Aortic Stenosis
0
1
2
3
4
5
Peak Aortic Jet Velocity (m/s)
0
1
2
3
4
5
Peak Aortic Jet Velocity (m/s)
1.12p = 0.74
2.71p = 0.004
5.48p<0.0001
OR = 1referent
OR fo
r CAV
S
G
I
H
ATX≥84RFU/minATX<84RFU/min
J A C C V O L . 6 9 , N O . 6 , 2 0 1 7 TsimikasF E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1 Lipoprotein(a)
701
allowed reclassification of 39.6% of individuals intoeither lower or higher risk categories, depending onthe Lp(a) level. Allele-specific Lp(a) levels and apo(a)isoforms did not add to the predictive ability of theFramingham risk score or Reynolds risk score, or theLp(a) level. These findings suggest that measuringLp(a) levels in intermediate-risk groups will allowreclassification of risk in 4 of 10 patients (Figure 6).The impact of risk restratification in not treatingwith a statin has not been assessed prospectively,whereas the benefit of a statin as a result of
upgrading risk is high. Furthermore, when the dis-tribution of risk has high frequency near dichoto-mous boundaries of risk categories, a relatively smallchange in absolute predicted risk can result in arelatively large difference in classification, asdemonstrated by Kamstrup et al. (26). Additionalstudies are needed to validate these findings,particularly with the new American College of Car-diology/American Heart Association and SCORE(Systematic Coronary Risk Evaluation) trial risk scorealgorithms.

FIGURE 6 Reclassification of Individuals Predicted to Be at Intermediate 15-Year CVD Risk by Additional Assessment of Lp(a)
120 40 / 67 / 13
4 / 26 / 1141
Intermediaterisk group
Non-cases
CVD cases
Reclassified tolower risk group
Reclassified tohigher risk group
Intermediate (15% to <30%) risk groupby Reynolds Risk Score*
Additional Lp(a)assessment
Individuals predicted to be at intermediate 15-year CVD risk by additional assessment of Lp(a) predicted risk groups were defined as: lower risk
group, <15%; intermediate risk group, 15% to <30%; and higher risk group, 30%. This reclassification table compares a model on the basis
of the Reynolds Risk Score only with a model considering the Reynolds Risk Score plus Lp(a) level. Net reclassification improvement (NRI)
denotes the classic retrospective categorical net NRI with calculations on the basis of 148 subjects with events and 502 subjects without
events and a complete follow-up over the 15-year period. *The Reynolds Risk Score contains information on age, sex, diabetes, smoking,
systolic blood pressure, total cholesterol, high-density lipoprotein cholesterol, parental history of premature myocardial infarction, and logeC-reactive protein. Abbreviations as in Figures 1 and 4. Reprinted with permission from Willeit et al. (47).
Tsimikas J A C C V O L . 6 9 , N O . 6 , 2 0 1 7
Lipoprotein(a) F E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1
702
AREA OF CONTROVERSY IV: IS IT POSSIBLE
TO ONLY MEASURE Lp(a) ONCE IN A
PERSON’S LIFETIME TO ASCERTAIN RISK,
AND WHICH PATIENTS SHOULD HAVE A
Lp(a) LEVEL MEASURED?
Because >90% of circulating Lp(a) levels are geneti-cally determined, and the levels are quantitativelyrelated to the LPA gene, with little influence from dietand environment, and because plasma levels do notfluctuate significantly around a pre-set baseline over alifetime, this test can be considered similar tomeasuring a SNP in a gene. However, unlikemeasuringa particular SNP, there are tens (if not hundreds) of LPASNPs that can influence Lp(a) levels; thus, an Lp(a)level is the most appropriate measurement (19,25–27).Most laboratories charge $50 to $100 for an Lp(a) level;therefore, this is likely to be a cost-effective testbecause it only has to be done once for screening ordiagnostic purposes. Because most patients are notaware of their Lp(a)-mediated risk, there is a rationaleto add an Lp(a) measurement to the lipid panel of apatient in whom lipids are measured for the first time.If Lp(a) is in the normal range, then subsequent
measurements are not needed, irrespective of anychange in the patient’s medical therapy.
In 2010, the EAS recommended Lp(a) measurementonce in all patients with premature CVD, and in pa-tients with intermediate or high CVD risk, familialhypercholesterolemia, family history of prematureCVD and/or elevated Lp(a), recurrent CVD despitestatin treatment, or a 3% 10-year risk of fatal and/or10% 10-year risk of fatal and nonfatal coronary heartdisease (44). The U.S. National Lipid Associationprovided similar recommendations on testing (48).The 2016 European Society of Cardiology/EAS guide-lines recommended measurement of Lp(a) in selectedcases at high risk, in patients with a family history ofpremature CVD, and for reclassification in subjectswith borderline risk, with a Class IIa, Level ofEvidence: C (49).
AREA OF CONTROVERSY V: IS THERE A
DIFFERENCE IN Lp(a)-MEDIATED CVD RISK
IN DIFFERENT RACIAL GROUPS?
It is well appreciated that racial differences exist inLp(a) levels, apo(a) isoforms, and LPA SNPs

FIGURE 7 Apo(a) Isoform Differences, Lp(a) Values, and CVD Events in Different
Racial Groups
200Black
White
Hispanic
12-20
African American
Caucasian
African American
Caucasian
African American
Caucasian
0.8 0.9 1.0 1.1 1.2 1.3 1.4
21-26 27-41
Black White Hispanic
150
100
50
0100
7550250
50403020100
100
70
50
30
20
10
7 9 11 13 15 19 17 21 23Number of Kringles
Number of Kringles
Lp(a
), nm
ol/L
Hazard Ratio per Standard DeviationIncrease in Log-Transformed Lp(a) Levels
CVD
CHD
Stroke
Num
ber o
f Sub
ject
s25 27 29 31 33 35 37 39 41
A
B
C
(A) Differences in apo(a) isoform size in black, white, and Hispanic subjects from the Dallas
Heart Study. Number of kringles represents total KIV repeats. A very small number of
samples were inappropriately categorized as <10 repeats due to sample degradation.
(B) Corresponding Lp(a) levels. Note that despite similar isoforms, black subjects had
significantly elevated Lp(a). (C) Similar CVD outcomes in black and white subjects from
the ARIC study using log2 transformed Lp(a) data. Modified and reprinted with permission
from Tsimikas et al. (51) and Virani et al. (53). ARIC ¼ Atherosclerosis Risk In Communities;
J A C C V O L . 6 9 , N O . 6 , 2 0 1 7 TsimikasF E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1 Lipoprotein(a)
703
(Figures 7A and 7B) (26,27,50–52). For example, in-dividuals of African descent have the highest levels ofthese, and are generally followed by South Asians,Caucasians, Hispanics, and East Asians. The differ-ences likely reflect the geographic migration of theLPA gene out of Africa, with additional changes ingenetic architecture over the past w40 to 60 millionyears because the LPA gene duplicated itself from theplasminogen gene and expanded in humans over thelast 3 million years or so. It is now evident thatelevated Lp(a) is an independent CVD risk factor in allracial groups studied to date (51,53–57). For example,in the ARIC (Atherosclerosis Risk In Communities)study, with 20 years of follow-up in 3,467 black and9,851 white subjects, Lp(a) levels were positively andsimilarly associated with CVD events in both groups,despite a larger range of distribution in black subjectscompared with white subjects (Figure 7C). However,because the prevalence of elevated Lp(a) is differentamong racial groups, the overall clinical expression ofdisease and incidence ascribed to Lp(a) relative toother risk factors may be variable.
AREA OF CONTROVERSY VI: IS Lp(a) A RISK
FACTOR WHEN LDL-C IS CONTROLLED?
Previous data from angiographic progression studiessuggested that Lp(a) was no longer a risk factorwhen LDL-C was controlled. For this reason, manyclinicians have practiced with the assumption thatwhen an elevated Lp(a) level is discovered, the mostappropriate course of action was to treat the LDL-Cand not the Lp(a). Recent studies have suggestedthat this is a false assumption, and that elevatedLp(a) remains a risk factor even in patients whoachieve LDL-C <70 mg/dl (44,45,58). Furthermore,the concept of diminishing returns is now apparentin outcomes trials of LDL-C lowering, in which thestarting LDL-C is now often <100 mg/dl, but theabsolute risk reduction is small. For example, inIMPROVE-IT (Improved Reduction of Outcomes:Vytorin Efficacy International Trial) (59), after amedian of 6 years of follow-up, the major adversecardiac event (MACE) rate was 32.7% in the simva-statin/ezetimibe group, which achieved LDL-C of54 mg/dl, and 34.7% in the simvastatin-alone arm,which achieved LDL-C of 70 mg/dl. Although thiswas a laudatory achievement, a 32.7% recurrent hardMACE rate in the setting of an LDL-C of 54 mg/dlsuggests that LDL-C–directed risk reduction mightnot reduce events optimally, even with PCSK9 in-hibitors. Recent reports from the AIM-HIGH (Athe-rothrombosis Intervention in Metabolic Syndromewith Low HDL/High Triglyceride and Impact on
CHD ¼ coronary heart disease; other abbreviations as in Figures 1 and 4.

FIGURE 8 Demonstration of Increased Risk of Events in Patients With Elevated Lp(a) and on Statin Therapy in the AIM-HIGH, LIPID, and JUPITER trials
Study Name NBaseline 4th Quartile Lp(a)
(mg/dL)Achieved LDL-C
(mg/dL)Odds Ratio(95% CI) P-Value Odds ratio (95% CI) Weight
AIM HIGH - Niacin group 1427 ~>50 65.2 1.90 (1.33 - 2.72) 0.001 10.84%
LIPID - Pravastatin group 7863 >73.7 112.5 1.23 (1.09 - 1.40) <0.001 58.72%
JUPITER - Rosuvastatin group 3877 ~>21 55 1.71 (0.99 - 2.95) 0.06 29.44%
OVERALL 13167 54.9(weighted value)
89.1(weighted value)
1.61(weighted value) 100%
Hazard Ratio (95% CI) for CV Events0.5 1 1.5 2 2.5 3
This forest plot shows a study-level analysis of baseline low-density lipoprotein cholesterol (LDL-C) levels and Lp(a) levels greater than the fourth quartile and the
associated hazard ratio compared with the lowest quartile. For AIM-HIGH, quartile 3 was>33.8 to 125.2 nmol/l and quartile 4 was >125.2 nmol/l; quartiles 1 and 2 were
not given. For LIPID, quartile 1 was #13.9 mg/dl, quartile 2 was 13.9 to 44.1 mg/dl, quartile 3 was 44.1 to 73.7 mg/dl, and quartile 4 was >73.7 mg/dl. For JUPITER,
quartile 1 was #10 nmol/l, quartile 2 was 11 to 23 nmol/l, quartile 3 was 24 to 49 nmol/l, and quartile 4 was $50 nmol/l. Conversion of milligrams per deciliter to
nanomoles is roughly made by multiplying milligrams per deciliter by 2.5. AIM-HIGH ¼ Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High
Triglyceride and Impact on Global Health Outcomes; JUPITER ¼ Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin; LIPID ¼Long-Term Intervention with Pravastatin in Ischaemic Disease; other abbreviations as in Figures 1 and 4.
Tsimikas J A C C V O L . 6 9 , N O . 6 , 2 0 1 7
Lipoprotein(a) F E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1
704
Global Health Outcomes) (45), JUPITER (Justificationfor the Use of Statins in Prevention: an InterventionTrial Evaluating Rosuvastatin) (44), and LIPID (Long-Term Intervention with Pravastatin in IschaemicDisease) (58) trials have suggested that a portion ofthis “residual risk” is due to highly elevated Lp(a) inthe setting of controlled LDL-C. For example, inAIM-HIGH, patients who achieved LDL-C levels of65.2 mg/dl and had Lp(a) >125 nmol/l (w50 mg/dl),which was $75th percentile of Lp(a) levels, had an89% higher risk of MACEs compared with those whohad a similar LDL-C level, but low Lp(a). In JUPITER,patients who achieved LDL-C of 55.0 mg/dl and Lp(a)>54 nmol/l (w21 mg/dl) had a 71% higher risk ofMACEs. In LIPID, in patients who achieved LDL-C ofw112 mg/dl and Lp(a) >73.7 mg/dl, a 23% increase inMACEs was present (Figure 8). The overall dataencompassing 13,167 statin-treated patients shows aweighed hazard ratio of 1.61 in the setting of LDL-Cof 89.1 mg/dl and Lp(a) of 54.9 mg/dl. These datastrongly support the independent role of Lp(a) inmediating CVD events that may explain some of theresidual risk in patients on statin therapy. It shouldalso be acknowledged that clinical benefit has notbeen seen in cholesterol ester transfer protein in-hibitor trials, despite a modest 20% to 30% reductionin Lp(a), along with LDL-C reductions and HDL-Cincreases. These data, together with the AIM-HIGHstudy, suggest that a potential benefit of Lp(a)
lowering may not be seen unless >50% mean re-ductions in Lp(a) can be achieved with no detri-mental off-target cardiovascular effects.
AREA OF CONTROVERSY VII:
DO STATINS INCREASE Lp(a) LEVELS?
The knowledge on the effect of statins on Lp(a) isevolving. It has been believed that statins do notaffect Lp(a) levels because the LDLR is thought toplay either no role or a minor role in Lp(a) clearance.Although definitive data do not currently exist, aclose assessment of published reports has suggestedthat statins tend to raise Lp(a) by 10% to 20%,particularly when the data are evaluated as pre- andpost-statin levels in individual patients rather thanmean levels in groups. For example, in a recentanalysis of 3,896 patients in whom Lp(a) and OxPL-apoB were measured pre- and post-statin therapy,including atorvastatin, pravastatin, rosuvastatin,pitavastatin, and simvastatin/ezetimibe, the meanpatient-level Lp(a) increased by 11% (and up to 50% insome studies), and OxPL-apoB increased by 24%(Figure 9) (60). This might also explain why somepatients do not respond well to LDL-C lowering bystatins, because most of their cholesterol is on Lp(a),rather than LDL particles (61), and Lp(a) can increasewith statin therapy. This should be a clue to physi-cians that lack of response to statin therapy could be

FIGURE 9 Effect of Statin Therapy on OxPL-ApoB and Lp(a)
(n=29)(n=21)
(24%) (11%)
-10 500 -10 500Mean % Change with Statins/Ezetimibe
Statin Study OxPL-apoB Lp(a)
Atorvastatin 10mg
Atorvastatin 80mg
Pravastatin 40mg
Pitivastatin 2mg
Rosuvastatin 40mg
Simvastatin/Ezetimibe
Capoulade et al (ASTRONOMER) 2015
2016 (n=162)
(n=1850)
(n=134)
Rodenberg et alChoi et al (REVERSAL)
Ky et al
Yoshida et al
Yeang et al
2012
200620082008
Tsimikas et al (MIRACL)Ky et al
Choi et al (REVERSAL)
200420082008
(n=1151)(n=26)
(n=108)
(n=90)(n=106)
(n=24)
(n=21)
20082012
Ky et al Yoshida et al
A forest plot showing the mean percent change in OxPL-apoB and Lp(a) in patients treated with a variety of and different doses of statins, where a baseline and
follow-up OxPL-apoB and Lp(a) level were available. Reprinted with permission from Yeang et al. (60). ASTRONOMER ¼ Aortic Stenosis Progression Observation:
Measuring Effects of Rosuvastatin; MIRACL ¼ Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering; REVERSAL ¼ Reversal of Atherosclerosis with
Aggressive Lipid Lowering Therapy; other abbreviations as in Figure 1.
J A C C V O L . 6 9 , N O . 6 , 2 0 1 7 TsimikasF E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1 Lipoprotein(a)
705
due to highly elevated Lp(a). Thus, although statintherapy results in overall benefit, it is possible thatpatients who have increased Lp(a) post-statin therapydo not obtain the full benefit of the statin. A formalpatient-level meta-analysis that assesses the role ofbaseline and on-treatment Lp(a), and the change inLp(a) post-statin therapy would be helpful to confirmthe potential change in Lp(a) and its relationship toCVD outcomes. The mechanism(s) by which statinsraise Lp(a) and OxPL-apoB on Lp(a) are undefined,and require further investigation.
AREA OF CONTROVERSY VIII:
ARE CURRENT THERAPIES TO LOWER Lp(a)
EFFECTIVE AND HAVE THEY REALLY FAILED
IN CLINICAL TRIALS?
First, it must be emphasized that there are noapproved medications to specifically lower Lp(a),and until the recent trials with the antisense oligo-nucleotides (ASOs), there have never been anyrandomized trials of Lp(a) lowering (29,62). Thecurrent pharmaceutical armamentarium of approveddrugs to lower Lp(a) levels in broad populations isessentially limited to niacin, with PCSK9 inhibitors,mipomersen, and estrogen in more limited pop-ulations. When apheresis has been performed tolower Lp(a), a reduction in cardiovascular risk hasbeen suggested (63,64). However, a firm conclusionof benefit of apheresis or an estimation of the
magnitude of benefit is hindered by the fact thatstudies performed to date have had no simulta-neous control group; rather, the outcomes of pa-tients who underwent apheresis were comparedwith historical controls (63). In addition, most sub-studies of Lp(a) risk that have evaluated othertherapies have not recruited patients with elevatedLp(a) levels, and all are post hoc analyses withmost patients having low levels; therefore, aproper interpretation is difficult. For example, theAIM-HIGH trial showed that niacin added to excel-lent LDL-C control did not change CVD outcomes.The baseline mean Lp(a) level was w13.5 mg/dl, andniacin reduced Lp(a) 19% to w11 mg/dl (43), changeswhich would be unlikely to result in clinical benefit.However, niacin also did not reduce the event ratein the subgroup of patients in the fourth quartile ofLp(a) (>125 nmol/l or approximately >50 mg/dl),despite achieving a mean Lp(a) reduction of 39%.However, this analysis was underpowered, withonly w700 patients and an event rate of w16%.Estrogen with progestin lowered Lp(a) by w15% to20% in HERS (Heart and Estrogen/ProgestinReplacement Study) and, in a post hoc analysis,post-menopausal women with elevated Lp(a) in thefourth quartile (55 to 236 mg/dl) had the mostbenefit (65), but this is not generally an option inwomen at risk of atherothrombosis. Mipomersenand PCSK9 inhibitors may reduce Lp(a) by 20% to30%, but are currently only indicated for rare

FIGURE 10 Mechanism of ASO Targeted to Apo(a) to Reduce Lp(a) Levels
Because only apo(a) is targeted, the liver can continue to make very low-density lipoprotein and LDL, and export them to the circulation. Antisense oligonucleotide
(ASO) to apo(a) inhibits both alleles of apo(a). Abbreviations as in Figures 1 and 3.
Tsimikas J A C C V O L . 6 9 , N O . 6 , 2 0 1 7
Lipoprotein(a) F E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1
706
patient populations (66–69). Thus, it can beconcluded that the Lp(a) hypothesis has not beentested with any currently available therapy.
AREA OF CONTROVERSY IX: IS Lp(a)
PROATHEROGENIC AND PROTHROMBOTIC,
OR ONLY PROATHEROGENIC?
Because of its high homology with plasminogen andpotential for interfering with plasminogen activa-tion, Lp(a) may not necessarily be prothrombotic apriori, but may tilt the balance towards thrombosisdue to the potential loss of fibrinolytic activity.Basic investigations studying plasminogen proper-ties in vitro have documented loss of plasminogenactivation to plasmin in the presence of apo(a) or
Lp(a) (70). However, a relationship of Lp(a) kringlerepeats or LPA SNPs with elevated Lp(a) levels inclinical venous thrombosis studies could not beshown, but associations with factor V Leiden werepresent (71,72). Although further study is needed,this does suggest that elevated Lp(a) may needa second underlying propensity to propa-gate thrombus, and that most patients with elevatedLp(a) are at risk primarily for atherosclerosis.Interestingly, in the Women’s Health Study,although the overall trial was negative, the onesubgroup that benefited from aspirin use werewomen with elevated Lp(a), which suggested riskcould be modified by antiplatelet therapy (73).Anecdotal evidence exists for use of vitamin K an-tagonists in patients with recurrent thromboses and

FIGURE 11 Mean Percentage Change in Lp(a) Concentration in the IONIS-APO(a)-LRx
Trial, a Multiple-Ascending-Dose Randomized Trial
1
IONIS-APO(a)-LRx 10 mgPlacebo
treatment period
IONIS-APO(a)-LRx 20 mg IONIS-APO(a)-LRx 40 mg
5
0
-20
-40
-60
-80
-1008 15 22 29/ET 36 50 64 85
Study Day
Mea
n (+
/- S
EM) %
Cha
nge
from
Bas
elin
eFa
stin
g Lp
(a) (
nmol
/L)
113
***
***
***
The shaded area represents the dosing window and arrows indicate dosing. p values are
only shown at day 36 for the multiple-ascending-dose phase, as determined by the exact
Wilcoxon rank-sum test comparing IONIS-APO(a)-LRx versus placebo. ***p < 0.001.
Lp(a) ¼ lipoprotein(a). Reprinted with permission from Viney et al. (62).
J A C C V O L . 6 9 , N O . 6 , 2 0 1 7 TsimikasF E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1 Lipoprotein(a)
707
high Lp(a), but formal studies have not been re-ported. To test the fibrinolysis side of the hypothe-sis, future studies with Lp(a)-lowering agents coulddirectly assess comprehensive coagulation parame-ters in patients after lowering of Lp(a).
EMERGING THERAPIES FOR LOWERING Lp(a)
Small molecules and antibodies that inhibit enzymeactivity and/or receptors or inactivate proteins,respectively, are the mainstay of current pharma-ceuticals. Because Lp(a) has no enzyme activity, andthe circulating mass levels are relatively high,neither of these approaches may reduce Lp(a) levels.Because apo(a) is synthesized by hepatocytes, ther-apies directed at the hepatocyte are likely to be mostefficacious. In this regard, Figure 10 demonstratesthe mechanism of ASOs in inhibiting apo(a) synthe-sis, and thus, Lp(a) secretion. Liver-targeted ASOsare injected subcutaneously, bind to plasma pro-teins, and enter the liver, where they accumulateintracellularly. They then bind to their target mRNAmainly in the nucleus but also in cytoplasm if mRNAis present in this compartment. Once a double-stranded complex is formed, ribonuclease H1 thencleaves the sense strand to prevent protein synthe-sis, but the antisense strand (i.e., the ASO) can thenbind to additional mRNA targets (74). In the case ofASO to apo(a), the hepatocytes can continue tosynthesize LDL particles and export them; therefore,steatosis should not occur, but both apo(a) alleleswill be inhibited, Lp(a) assembly prevented, andplasma Lp(a) levels reduced.
Initial studies suggested that mipomersen, an ASOthat inhibited apoB mRNA, could lower Lp(a) andOxPL-apoB in transgenic Lp(a) (75). Interestingly, themechanism was a marked reduction of apoB produc-tion, such that apoB was limiting Lp(a) assembly.Mipomersen did not affect production of apo(a),which continued to be secreted into the circulation as“free” apo(a). These preclinical findings were subse-quently confirmed in 4 randomized trials of LDL-Clowering, with mipomersen showing a 25% reduc-tion in Lp(a) (76). This observation confirmed thatlowering Lp(a) could be achieved with ASOs, andsubsequently, it was reported that an ASO specific toapo(a) reduced circulating apo(a) by 86% withoutaffecting apoB in apo(a) transgenic mice (77).Recently, optimized ASOs to apo(a) have been re-ported for human trials (78), and 3 randomized,controlled trials (29,62) have reported dose-dependent reductions in mean Lp(a) lowering of>80%. In addition, a significant reduction was notedin proinflammatory OxPL and in the inflammatory
effects of monocytes, which are cells that initiateand accelerate CVD, as well as plasma LDL-C (62).A subsequent advance in potency of ASOs wasmade with IONIS-APO(a)-LRx, which contains anN-acetyl-galactosamine (GalNac3)�conjugated mole-cule designed to be highly and selectively taken upby hepatocytes, with mean reductions of 66% to 92%and up to 99% reduction of Lp(a) in some patients(Figure 11), together with a reduction in OxPL.
CONCLUSIONS
The Central Illustration depicts the current thresholdsfor what is considered Lp(a)-driven risk, and theeffects of approved and emerging therapies. Lp(a)levels <30 mg/dl are considered optimal, with negli-gible Lp(a)-mediated risk. Lp(a) levels <50 mg/dl arerecommended by the EAS as optimal, and Lp(a) levels>60 mg/dl are used as a cutoff for the reimbursementof apheresis in Germany and the United Kingdom. In atheoretical patient with a Lp(a) level of 150 mg/dl, useof statins tends to increase Lp(a) by 10% to 20%. Incontrast, use of niacin, PCSK9 inhibitors, cholesterol

CENTRAL ILLUSTRATION Lp(a) Cutoffs Signifying Increased CVD Risk and Effect of TherapeuticAgents in Achieving These Targets
Tsimikas, S. J Am Coll Cardiol. 2017;69(6):692–711.
The effect of various therapies on lipoprotein(a) [Lp(a)] in a hypothetical patient with Lp(a) of 150 mg/dl, which is >98th percentile for Lp(a)
levels. Statins tend to increase the level by 10% to 20%, to 165 to 180 mg/dl. Niacin, proprotein convertase subtilisin/kexin-type 9 (PCSK9)
antibodies, cholesterol ester transfer protein inhibitors (CETPi), and mipomersen decrease levels by 20% to 30%, to 105 to 120 mg/dl.
Apheresis results in a time-averaged reduction of 30% to 35%, reducing levels to 98 to 105 mg/dl. Antisense oligonucleotides reduce Lp(a)
by 80% to 99%, reaching levels of 1.5 to 30 mg/dl. CAVS ¼ calcific aortic valve stenosis; CVD ¼ cardiovascular disease; EAS ¼ European
Atherosclerosis Society.
Tsimikas J A C C V O L . 6 9 , N O . 6 , 2 0 1 7
Lipoprotein(a) F E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1
708

J A C C V O L . 6 9 , N O . 6 , 2 0 1 7 TsimikasF E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1 Lipoprotein(a)
709
ester transfer protein inhibitors, and mipomersenleads to a 20% to 30% reduction. Similarly, apheresisleads to a time-averaged reduction of 30% to 35%. Useof ASO, which may reduce Lp(a) >80% and up to 99%,may reach levels in which the risk due to Lp(a) is quitelow in most patients. Such potent and specific thera-pies are currently being developed clinically in pa-tients with elevated Lp(a), and will be able to finallytest the hypothesis that pharmacologically reducinggenetically elevated Lp(a) levels may reduce the riskof CVD and CAVS in the setting of what is consideredoptimal medical therapy at present.
ACKNOWLEDGMENTS The author thanks his men-tors, colleagues, collaborators, and trainees in theLp(a) arena for their contributions in bringing thisfield to the clinical frontier. He also thanks TracyReigle of Ionis Pharmaceuticals for expert assistancewith the illustrations.
ADDRESS FOR CORRESPONDENCE: Dr. Sotirios Tsi-mikas, Vascular Medicine Program, Department ofMedicine, Sulpizio Cardiovascular Center, Universityof California, San Diego, La Jolla, California 92093-0682. E-mail: [email protected].
RE F E RENCE S
1. MozaffarianD,BenjaminEJ,GoAS, etal.,AmericanHeart Association Statistics Committee and StrokeStatistics Subcommittee. Heart disease and strokestatistics–2015 update: a report from the AmericanHeart Association. Circulation 2015;131:e29–322.
2. AIM-HIGH-Investigators. Niacin in patients withlow HDL cholesterol levels receiving intensivestatin therapy. N Engl J Med 2011;365:2255–67.
3. Barter PJ, Rye KA. New era of lipid-loweringdrugs. Pharmacol Rev 2016;68:458–75.
4. Schmidt K, Noureen A, Kronenberg F, et al.Structure, function, and genetics of lipoprotein(a). J Lipid Res 2016;57:1339–59.
5. Marcovina SM, Albers JJ, Gabel B, et al. Effectof the number of apolipoprotein(a) kringle 4domains on immunochemical measurements oflipoprotein(a). Clin Chem 1995;41:246–55.
6. Kronenberg F. Human genetics and the causalrole of lipoprotein(a) for various diseases. Car-diovasc Drugs Ther 2016;30:87–100.
7. Kronenberg F, Utermann G. Lipoprotein(a):resurrected by genetics. J Intern Med 2013;273:6–30.
8. Steinberg D, Witztum JL. Oxidized low-densitylipoprotein and atherosclerosis. ArteriosclerThromb Vasc Biol 2010;30:2311–6.
9. Spence JD, Koschinsky ML. Mechanisms of lip-oprotein(a) pathogenicity. Arterioscler ThrombVasc Biol 2012;32:1550–1.
10. Bergmark C, Dewan A, Orsoni A, et al. A novelfunction of lipoprotein [a] as a preferential carrierof oxidized phospholipids in human plasma. J LipidRes 2008;49:2230–9.
11. Leibundgut G, Scipione C, Yin H, et al. De-terminants of binding of oxidized phospholipids onapolipoprotein (a) and lipoprotein (a). J Lipid Res2013;54:2815–30.
12. van der Valk FM, Bekkering S, Kroon J, et al.Oxidized phospholipids on lipoprotein(a) elicitarterial wall inflammation and an inflammatorymonocyte response in humans. Circulation 2016;134:611–24.
13. Kiechl S, Willeit J, Mayr M, et al. Oxidized phos-pholipids, lipoprotein(a), lipoprotein-associatedphospholipase A2 activity, and 10-year cardiovas-cular outcomes: prospective results from the
Bruneck study. Arterioscler Thromb Vasc Biol 2007;27:1788–95.
14. Tsimikas S, Mallat Z, Talmud PJ, et al. Oxida-tion-specific biomarkers, lipoprotein(a), and risk offatal and nonfatal coronary events. J Am CollCardiol 2010;56:946–55.
15. Tsimikas S, Willeit P, Willeit J, et al. Oxida-tion-specific biomarkers, prospective 15-yearcardiovascular and stroke outcomes, and netreclassification of cardiovascular events. J AmColl Cardiol 2012;60:2218–29.
16. Bertoia ML, Pai JK, Lee JH, et al. Oxidation-specific biomarkers and risk of peripheral arterydisease. J Am Coll Cardiol 2013;61:2169–79.
17. Tsimikas S, Duff GW, Berger PB, et al. Pro-in-flammatory interleukin-1 genotypes potentiatethe risk of coronary artery disease and cardiovas-cular events mediated by oxidized phospholipidsand lipoprotein(a). J Am Coll Cardiol 2014;63:1724–34.
18. Byun YS, Lee JH, Arsenault BJ, et al., TNTTrial Investigators. Relationship of oxidizedphospholipids on apolipoprotein B-100 to car-diovascular outcomes in patients treated withintensive versus moderate atorvastatin therapy:the TNT trial. J Am Coll Cardiol 2015;65:1286–95.
19. Capoulade R, Chan KL, Yeang C, et al. Oxidizedphospholipids, lipoprotein(a), and progression ofcalcific aortic valve stenosis. J Am Coll Cardiol2015;66:1236–46.
20. Nsaibia MJ, Mahmut A, Boulanger MC, et al.Autotaxin interacts with lipoprotein(a) andoxidized phospholipids in predicting the risk ofcalcific aortic valve stenosis in patients with cor-onary artery disease. J Intern Med 2016;280:509–17.
21. Scipione CA, Sayegh SE, Romagnuolo R, et al.Mechanistic insights into Lp(a)-induced IL-8expression: a role for oxidized phospholipidmodification of apo(a). J Lipid Res 2015;56:2273–85.
22. Wiesner P, Tafelmeier M, Chittka D, et al.MCP-1 binds to oxidized LDL and is carried bylipoprotein(a) in human plasma. J Lipid Res 2013;54:1877–83.
23. van Dijk RA, Kolodgie F, Ravandi A, et al. Dif-ferential expression of oxidation-specific epitopesand apolipoprotein(a) in progressing and rupturedhuman coronary and carotid atherosclerotic le-sions. J Lipid Res 2012;53:2773–90.
24. RavandiA,LeibundgutG,HungMY,etal. Releaseand capture of bioactive oxidized phospholipids andoxidized cholesteryl esters during percutaneouscoronary and peripheral arterial interventions inhumans. J Am Coll Cardiol 2014;63:1961–71.
25. Emerging Risk Factors Collaboration. Lip-oprotein(a) concentration and the risk of coronaryheart disease, stroke, and nonvascular mortality.JAMA 2009;302:412–23.
26. Kamstrup PR, Tybjærg-Hansen A,Nordestgaard BG. Extreme lipoprotein(a) levelsand improved cardiovascular risk prediction. J AmColl Cardiol 2013;61:1146–56.
27. Clarke R, Peden JF, Hopewell JC, et al.,PROCARDIS Consortium. Genetic variants associ-ated with Lp(a) lipoprotein level and coronarydisease. N Engl J Med 2009;361:2518–28.
28. CARDIoGRAMplusC4D Consortium, Deloukas P,Kanoni S, et al. Large-scale association analysisidentifies new risk loci for coronary artery disease.Nat Genet 2013;45:25–33.
29. Tsimikas S, Viney NJ, Hughes SG, et al. Anti-sense therapy targeting apolipoprotein(a): arandomised, double-blind, placebo-controlledphase 1 study. Lancet 2015;386:1472–83.
30. Kamstrup PR, Tybjærg-Hansen A,Steffensen R, et al. Genetically elevated lip-oprotein(a) and increased risk of myocardialinfarction. JAMA 2009;301:2331–9.
31. Kyriakou T, Seedorf U, Goel A, et al., PRO-CARDIS Consortium. A common LPA null alleleassociates with lower lipoprotein(a) levels andcoronary artery disease risk. Arterioscler ThrombVasc Biol 2014;34:2095–9.
32. Lim ET, Würtz P, Havulinna AS, et al.Sequencing Initiative Suomi (SISu) Project distri-bution and medical impact of loss-of-functionvariants in the Finnish founder population. PLoSGenet 2014;10:e1004494.
33. Yeang C, Wilkinson MJ, Tsimikas S. Lip-oprotein(a) and oxidized phospholipids in calcific

Tsimikas J A C C V O L . 6 9 , N O . 6 , 2 0 1 7
Lipoprotein(a) F E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1
710
aortic valve stenosis. Curr Opin Cardiol 2016;31:440–50.
34. Thanassoulis G, Campbell CY, Owens DS,et al., CHARGE Extracoronary Calcium WorkingGroup. Genetic associations with valvular calcifi-cation and aortic stenosis. N Engl J Med 2013;368:503–12.
35. Kamstrup PR, Tybjærg-Hansen A,Nordestgaard BG. Elevated lipoprotein(a) and riskof aortic valve stenosis in the general population.J Am Coll Cardiol 2014;63:470–7.
36. Arsenault BJ, Boekholdt SM, Dubé MP, et al.Lipoprotein(a) levels, genotype, and incidentaortic valve stenosis: a prospective Mendelianrandomization study and replication in a case-control cohort. Circ Cardiovasc Genet 2014;7:304–10.
37. Vongpromek R, Bos S, Ten Kate GJ, et al.Lipoprotein(a) levels are associated with aorticvalve calcification in asymptomatic patients withfamilial hypercholesterolaemia. J Intern Med 2015;278:166–73.
38. Chan KL, Teo K, Dumesnil JG, et al.,ASTRONOMER Investigators. Effect of lipidlowering with rosuvastatin on progression ofaortic stenosis: results of the Aortic Stenosis Pro-gression Observation: Measuring Effects of Rosu-vastatin (ASTRONOMER) trial. Circulation 2010;121:306–14.
39. Bouchareb R, Mahmut A, Nsaibia MJ, et al.Autotaxin derived from lipoprotein(a) and valveinterstitial cells promotes inflammation andmineralization of the aortic valve. Circulation2015;132:677–90.
40. Marcovina SM, Albers JJ. Lipoprotein (a)measurements for clinical application. J Lipid Res2016;57:526–37.
41. Verbeek R, Boekholdt SM, Stoekenbroek RM,et al. Population and assay thresholds for thepredictive value of lipoprotein (a) for coronaryartery disease: the EPIC-Norfolk ProspectivePopulation Study. J Lipid Res 2016;57:697–705.
42. Kostner GM, Avogaro P, Cazzolato G, et al.Lipoprotein Lp(a) and the risk for myocardialinfarction. Atherosclerosis 1981;38:51–61.
43. Nordestgaard BG, Chapman MJ, Ray K, et al.Lipoprotein(a) as a cardiovascular risk factor: cur-rent status. Eur Heart J 2010;31:2844–53.
44. Khera AV, Everett BM, Caulfield MP, et al.Lipoprotein(a) concentrations, rosuvastatin ther-apy, and residual vascular risk: an analysis fromthe JUPITER Trial (Justification for the Use ofStatins in Prevention: an Intervention Trial Evalu-ating Rosuvastatin). Circulation 2014;129:635–42.
45. Albers JJ, Slee A, O’Brien KD, et al. Relation-ship of apolipoproteins A-1 and B, andlipoprotein(a) to cardiovascular outcomes: theAIM-HIGH trial (Atherothrombosis Intervention inMetabolic Syndrome with Low HDL/High Triglyc-eride and Impact on Global Health Outcomes).J Am Coll Cardiol 2013;62:1575–9.
46. Anderson TJ, Grégoire J, Pearson GJ, et al.2016 Canadian Cardiovascular Society guidelinesfor the management of dyslipidemia for the pre-vention of cardiovascular disease in the adult. CanJ Cardiol 2016;32:1263–82.
47. Willeit P, Kiechl S, Kronenberg F, et al.Discrimination and net reclassification of cardio-vascular risk with lipoprotein(a): prospective 15-year outcomes in the Bruneck Study. J Am CollCardiol 2014;64:851–60; erratum J Am Coll Car-diol 2016;67:737.
48. Davidson MH, Ballantyne CM, Jacobson TA,et al. Clinical utility of inflammatory markers andadvanced lipoprotein testing: advice from anexpert panel of lipid specialists. J Clin Lipidol 2011;5:338–67.
49. Catapano AL, Graham I, De Backer G, et al.2016 ESC/EAS Guidelines for the Managementof Dyslipidaemias. Eur Heart J 2016;37:2999–3058.
50. Deo RC, Wilson JG, Xing C, et al. Single-nucleotide polymorphisms in LPA explain most ofthe ancestry-specific variation in Lp(a) levels inAfrican Americans. PLoS One 2011;6:e14581.
51. Tsimikas S, Clopton P, Brilakis ES, et al. Rela-tionship of oxidized phospholipids on apolipopro-tein B-100 particles to race/ethnicity,apolipoprotein(a) isoform size, and cardiovascularrisk factors: results from the Dallas Heart Study.Circulation 2009;119:1711–9.
52. Nikpay M, Goel A, Won HH, et al., CARDIo-GRAMplusC4D Consortium. A comprehensive1,000 genomes-based genome-wide associationmeta-analysis of coronary artery disease. NatGenet 2015;47:1121–30.
53. Virani SS, Brautbar A, Davis BC, et al. Associ-ations between lipoprotein(a) levels and cardio-vascular outcomes in black and white subjects: theAtherosclerosis Risk in Communities (ARIC) study.Circulation 2012;125:241–9.
54. Gambhir JK, Kaur H, Prabhu KM, et al. Associ-ation between lipoprotein(a) levels, apo(a) iso-forms and family history of premature CAD in youngAsian Indians. Clin Biochem 2008;41:453–8.
55. Cai DP, He YM, Yang XJ, et al. Lipoprotein (a) isa risk factor for coronary artery disease in ChineseHan ethnic population modified by some tradi-tional risk factors: a cross-sectional study of 3462cases and 6125 controls. Clin Chim Acta 2015;451:278–86.
56. Lanktree MB, Anand SS, Yusuf S, et al., SHAREInvestigators. Comprehensive analysis of genomicvariation in the LPA locus and its relationship toplasma lipoprotein(a) in South Asians, Chinese,and European Caucasians. Circ Cardiovasc Genet2010;3:39–46.
57. Guan W, Cao J, Steffen BT, et al. Race is a keyvariable in assigning lipoprotein(a) cutoff valuesfor coronary heart disease risk assessment: theMulti-Ethnic Study of Atherosclerosis. ArteriosclerThromb Vasc Biol 2015;35:996–1001.
58. Nestel PJ, Barnes EH, Tonkin AM, et al. Plasmalipoprotein(a) concentration predicts future coro-nary and cardiovascular events in patients withstable coronary heart disease. Arterioscler ThrombVasc Biol 2013;33:2902–8.
59. Cannon CP, Blazing MA, Giugliano RP, et al.,IMPROVE-IT Investigators. Ezetimibe added tostatin therapy after acute coronary syndromes.N Engl J Med 2015;372:2387–97.
60. Yeang C, Hung MY, Byun YS, et al. Effect oftherapeutic interventions on oxidized phospholipidson apolipoprotein B100 and lipoprotein(a). J ClinLipidol 2016;10:594–603.
61. Yeang C, Witztum JL, Tsimikas S. ’LDL-C’ ¼LDL-C þ Lp(a)-C: implications of achieved ultra-low LDL-C levels in the proprotein convertasesubtilisin/kexin type 9 era of potent LDL-Clowering. Curr Opin Lipidol 2015;26:169–78.
62. Viney NJ, van Capelleveen JC, Geary RS, et al.Antisense oligonucleotides targeting apolipopro-tein(a) in people with raised lipoprotein(a): tworandomised, double-blind, placebo-controlled,dose-ranging trials. Lancet 2016;388:2239–53.
63. Moriarty PM, Hemphill L. Lipoprotein apher-esis. Endocrinol Metab Clin North Am 2016;45:39–54.
64. Roeseler E, Julius U, Heigl F, et al. Lipoproteinapheresis for lipoprotein(a)-associated cardiovas-cular disease: prospective 5 years of follow-up andapolipoprotein(a) characterization. ArteriosclerThromb Vasc Biol 2016;36:2019–27.
65. Shlipak MG, Simon JA, Vittinghoff E, et al.Estrogen and progestin, lipoprotein(a), and therisk of recurrent coronary heart disease eventsafter menopause. JAMA 2000;283:1845–52.
66. Duell PB, Santos RD, Kirwan BA, et al. Long-term mipomersen treatment is associated with areduction in cardiovascular events in patients withfamilial hypercholesterolemia. J Clin Lipidol 2016;10:1011–21.
67. Hossne NA, Cruz E, Buffolo E, et al. Long-termand sustained therapeutic results of a specificpromonocyte cell formulation in refractory angina:ReACT (Refractory Angina Cell Therapy) clinicalupdate and cost-effective analysis. Cell Transplant2015;24:955–70.
68. Raal FJ, Giugliano RP, Sabatine MS, et al.Reduction in lipoprotein(a) with PCSK9 mono-clonal antibody evolocumab (AMG 145): a pooledanalysis of more than 1,300 patients in 4 phase IItrials. J Am Coll Cardiol 2014;63:1278–88.
69. Raal FJ, Stein EA, Dufour R, et al.,RUTHERFORD-2 Investigators. PCSK9 inhibitionwith evolocumab (AMG 145) in heterozygousfamilial hypercholesterolaemia (RUTHERFORD-2):a randomised, double-blind, placebo-controlledtrial. Lancet 2015;385:331–40.
70. Boffa MB, Koschinsky ML. Lipoprotein (a):truly a direct prothrombotic factor in cardiovas-cular disease? J Lipid Res 2016;57:745–57.
71. Kamstrup PR, Tybjærg-Hansen A,Nordestgaard BG. Genetic evidence that lip-oprotein(a) associates with atherosclerotic steno-sis rather than venous thrombosis. ArteriosclerThromb Vasc Biol 2012;32:1732–41.
72. Helgadottir A, Gretarsdottir S, Thorleifsson G,et al. Apolipoprotein(a) genetic sequence variantsassociated with systemic atherosclerosis and cor-onary atherosclerotic burden but not with venousthromboembolism. J Am Coll Cardiol 2012;60:722–9.
73. Chasman DI, Shiffman D, Zee RY, et al. Poly-morphism in the apolipoprotein(a) gene, plasmalipoprotein(a), cardiovascular disease, and low-

J A C C V O L . 6 9 , N O . 6 , 2 0 1 7 TsimikasF E B R U A R Y 1 4 , 2 0 1 7 : 6 9 2 – 7 1 1 Lipoprotein(a)
711
dose aspirin therapy. Atherosclerosis 2009;203:371–6.
74. Crooke ST, Geary RS. Clinical pharmacologicalproperties of mipomersen (Kynamro), a secondgeneration antisense inhibitor of apolipoprotein B.Br J Clin Pharmacol 2013;76:269–76.
75. Merki E, Graham MJ, Mullick AE, et al. Anti-sense oligonucleotide directed to human apolipo-protein B-100 reduces lipoprotein(a) levels andoxidized phospholipids on human apolipoprotein
B-100 particles in lipoprotein(a) transgenic mice.Circulation 2008;118:743–53.
76. Santos RD, Raal FJ, Catapano AL, et al.Mipomersen, an antisense oligonucleotide toapolipoprotein B-100, reduces lipoprotein(a) invarious populations with hypercholesterolemia:results of 4 phase III trials. Arterioscler ThrombVasc Biol 2015;35:689–99.
77. Merki E, Graham M, Taleb A, et al. Antisenseoligonucleotide lowers plasma levels of
apolipoprotein (a) and lipoprotein (a) in transgenicmice. J Am Coll Cardiol 2011;57:1611–21.
78. Graham MJ, Viney N, Crooke RM, et al. Anti-sense inhibition of apolipoprotein (a) to lowerplasma lipoprotein (a) levels in humans. J LipidRes 2016;57:340–51.
KEY WORDS aortic stenosis, cardiovasculardisease, genome-wide association studies,therapy

![Cyclosporin A-Induced Hyperlipidemia · 2012. 9. 30. · Cyclosporin A-Induced Hyperlipidemia 341 2.4. Plasma lipoprotein (a) Lipoprotein (a) [Lp(a)] is a LDL-like lipoprotein consisting](https://img.dokumen.tips/doc/110x75/60b482bc2d15520abb15cefc/cyclosporin-a-induced-hyperlipidemia-2012-9-30-cyclosporin-a-induced-hyperlipidemia.jpg)






![Lipoprotein(a) and Oxidized Phospholipids Promote Valve ... · BACKGROUND Lipoprotein(a) [Lp(a)], a major carrier of oxidized phospholipids (OxPL), is associated with an increased](https://img.dokumen.tips/doc/110x75/5e5f5b29282d4a2232338b34/lipoproteina-and-oxidized-phospholipids-promote-valve-background-lipoproteina.jpg)




![Clinical Significances of Lipoprotein Metabolism · 2017. 11. 30. · Lipoprotein(a) [Lp(a)] was originally described as a new serum lipoprotein particle by Kare Berg in 1963. Lp(a)](https://img.dokumen.tips/doc/110x75/608de5358d44ff6179489354/clinical-significances-of-lipoprotein-metabolism-2017-11-30-lipoproteina.jpg)















