Embed Size (px)
Citation preview

A Novel Recombinant Fusion Protein Encoding a 20-AminoAcid Residue of the Third Extracellular (E3) Domain ofCCR2 Neutralizes the Biological Activity of CCL21
Liat Izhak,* Gizi Wildbaum,* Yaniv Zohar,* Rachel Anunu,* Leah Klapper,† Adi Elkeles,†
Jane Seagal,* Eitan Yefenof,‡ Michal Ayalon-Soffer,† and Nathan Karin2*
CCL2 is a key CC chemokine that has been implicated in a variety of inflammatory autoimmune diseases and in tumor progressionand it is therefore an important target for therapeutic intervention in these diseases. Soluble receptor-based therapy is a knownapproach for neutralizing the in vivo functions of soluble mediators. Owing to the complexity of seven-transmembrane G protein-coupled receptors, efforts to generate neutralizing soluble chemokine receptors have so far failed. We developed a strategy thatis based on the generation of short recombinant proteins encoding different segments of a G protein-coupled receptor, and testedthe ability of each of them to bind and neutralize its target chemokine. We show that a fusion protein comprised of as few as 20aa of the third extracellular (E3) domain of the CCL2 receptor, stabilized by the IgG H chain Fc domain (E3-IgG or BL-2030),selectively binds CCL2 and CCL16 and effectively neutralizes their biological activities. More importantly, E3-IgG (BL-2030)could effectively suppress the in vivo biological activity of CCL2, attenuating ongoing experimental autoimmune encephalomy-elitis, as well as the development of human prostate tumor in SCID mice. The Journal of Immunology, 2009, 183: 732–739.
C hemokines are small (!8–14 kDa), structurally relatedproteins that regulate cell trafficking via interactions witha subset of seven-transmembrane G-coupled protein re-
ceptor (1–4). CCL2 (MCP-1) is a key CC chemokine that partic-ipates in the regulation of inflammatory responses. CCL2 and itspredominant receptor, CCR2, have been implicated in a varietyof autoimmune diseases including multiple sclerosis (MS),3
rheumatoid arthritis, atherosclerosis, myocarditis, and others(5– 8). Its major role involves the recruitment of macrophagesto sites of inflammation, initiating a cascade of events that leadsto tissue destruction in chronic inflammatory diseases. We havepreviously shown that during experimental autoimmune en-cephalomyelitis (EAE), the murine model of MS, a T cell-me-diated autoimmune disease of the CNS, autoantibodies to CCL2that are capable of suppressing the disease are being produced,supporting the rational of developing CCL2 inhibitors for MStreatment (7).
Many studies have shown the importance of chemokines in tu-mor progression. Various chemokines are being produced by tu-
mor cells, which also express their cognate receptors (for a generalreview see Ref. 9). Functionally, they are likely to direct tumorprogression, angiogenesis, and survival. Cancer of the prostate isthe most common cancer in American males, and the second lead-ing cause of cancer-related mortality (10, 11). The three majorchemokines produced by human prostate cancer cells that also ex-press their target receptors are CXCL12 (SDF-1!), CXCL8 (IL-8),and CCL2 (MCP-1) (12–18). These chemokines are likely to pro-mote tumor development and angiogenesis (12–18). Thus far, mostof the attention has focused on the role of CXCL12, which is likelyto be the key chemokine that attracts these cells to the bones (19–24). Interestingly, CCL2 became a major target of interest in a fewrecent reports showing that 1) in humans, CCR2 expression wascorrelated with the Gleason score and clinical pathologic stages,whereas lower levels of CCR2 were expressed in normal prostatetissues (25), 2) CCL2 is a potent regulator of prostate cancer cellmigration and proliferation (12), 3) CCL2 acts as a paracrine andan autocrine factor for prostate cancer growth and invasion (13),and 4) CCL2 is involved in recruiting macrophages that assisttumor development (12, 14). These areas of interest have moti-vated us to select the third extracellular (E3) domain of the CCL2receptor, stabilized by the IgG H chain Fc domain (E3-IgG) as apotential candidate for therapy of this disease.
Its pivotal role in cancer and autoimmunity has made CCL2 anattractive target for therapy. We have focused on developing asoluble receptor-based approach for inhibiting CCL2. Previousstudies have shown that CCR2 binds its ligand CCL2 via the N-terminal region alone (26), or the N-terminal region combined withthe third extracellular loop (E3 domain) element (27). We show inthis study that a fusion protein, encoding 20 aa of the E3 domain,selectively binds and neutralizes CCL2 as effectively as a recom-binant protein composed of the E3 domain fused to the N-terminalregion. We then explored, for the first time, how in vivo neutral-ization of CCL2 with this compound affects the development ofcancer of the prostate, as well as the manifestation of ongoing EAEin C57BL/6 mice.
*Department of Immunology, Rappaport Family Institute for Research in the MedicalSciences, Haifa, †BioLine Innovations Jerusalem, Jerusalem, and ‡Lautenberg Centerfor General and Tumor Immunology, The Hebrew University Hadassah Medical Cen-ter, Jerusalem, Israel
Received for publication August 20, 2008. Accepted for publication May 4, 2009.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This study was supported by grants from the Israel Science Foundation, the IsraelMinistry of Health Chief Scientist, and a sponsored research grant from the IsraelMinistry of Industry, together with BiolineRx.2 Address correspondence and reprint requests to Dr. Nathan Karin, Bruce Rap-paport Faculty of Medicine, Technion, Rappaport Family Institute for Research inthe Medical Sciences, PO Box 9697, Haifa 31096, Israel. E-mail address: [email protected] Abbreviations used in this paper: MS, multiple sclerosis; EAE, experimental auto-immune encephalomyelitis.
Copyright © 2009 by The American Association of Immunologists, Inc. 0022-1767/09/$2.00
The Journal of Immunology
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0802746

Materials and MethodsConstruction of soluble receptors
Human and mouse cDNA encoding the constant region (Fc, Hinge-CH2-CH3) IgG1 were constructed as follows: human Fc was generated by RT-PCR on RNA extracted from human PBMC that was cultured for 4 dayswith LPS and IL-4. The primers used for this reaction were 5"-ctcgagCCCAAATCTTGTGACAAAAC and 3"-gggcccTTTACCCGGGGACAGGGAGA (AF237583). The mouse Fc was extracted from Con A-stimu-lated spleen cells and the primers were 5"-ccgCCGCTCGAGGTGCCCAGGGATTGTGGTTG and 3"-gaacaaTTGTTCGGGCCCTTTACCAGGAGAGTGGGAGA (L35037).
The PCR products were digested with XhoI and ApaI and ligated intomammalian expression/secretion vector pSecTag2/Hygro B (InvitrogenLife Technologies). A different set of primers, 5" (sense) and 5" (antisense),were used to generate cDNA encoding the different human (NM000647)(28) and mouse (NM009915) (29) domains of CCR2 from the PC-3 and theTRAMP mouse C-1 cell line (30), respectively. The cDNA encoding thehuman CXCR4 (NM001008540) (31) was obtained from the PC-3 line.Primers were designed as follows: human CCR2 N terminus 5"-CCCAAGCTTATGCTGTCCACATCTCGTTCTCGGTT and 3"-CCGCTCGAG TGGGCCCCAATTTGCTTCACGTCAA; human CCR2-E1 5"-CCCAAGCTTTCTGCTGCAAATGAGTGGGT and 3"-CCGCTCGAGTTTGCACATTGCATTCCCAA; human CCR2-E2 5"-CCCAAGCTTACTAAATGCCAGAAAGAAGA and 3"-CCGCTCGAGTGTGTGGAAATTATTCCATC;human CCR2-E3 5"-CCCAAGCTTAACACCTTCCAGGAATTCTTCGGCCT and 3"-CCGCTCGAGTTGGTCCAGTTGACTGGTGCTTTCAC;human CXCR4 N terminus 5"-CCCAAGCTTTGGAGGGGATCAGTATATACACTTC and 3"-CCGCTCGAGATTTTATTGAAATTAGCATTTTCTTC; mouse CCR2 N terminus 5"-CCAAGCTTATGGAAGACAATAATATGTTAC and 3"-CCGCTCGAGCACACTGGTTTTATGACAAGGC;andmouseCCR2-E35"-CCCAAGCTTGAATCCTTGGGAATGAGTAACTGT and 3"-CCGCTCGAGTCCAAGAGTCTCTGTCACCTGCAT.
The constant region of the human Fc, (Hinge-CH2-CH3) alone (Ig) wasalso cloned and used later as a control for BiaCore analysis (see Fig. 3A).Each PCR product was digested with HindIII and XhoI and subcloned intothe vector containing the human/mouse (h/m) IgG1 fragment. Each fusedfragments was sequenced by dideoxynucleotide sequencing at our facility(Sequenase version 2; Upstate Biotechnology).
Expression and purification of fusion proteins
Expression and purification of the various fusion proteins was done usingCHO dhfr#/# (DG44) cells provided by Dr. L. Chasin (Columbia Univer-sity, New York, NY) according to the method described in detail (32). Thefusion protein was expressed as a disulfide-linked homodimer similar toIgG1, and it was purified from the culture medium by the High-Trap pro-tein G affinity column (BD Biosciences).
Evaluation of the affinity of E3-IgG to CCL2 by surfaceplasmon resonance
E3-IgG affinity to CCL2 was determined by BIAcore analysis, using thebiosensor BIAcore 3000. CCR2-IgG or Fc, as a negative control, wereimmobilized directly to CM5 chip (100 "g/ml in 10 mM acetate (pH $ 4))using amine coupling kit (BIAcore) to reach up to 2500 response unitsbinding level. All subsequent binding experiments were performed in HBS(0.01 M HEPES (pH $ 7.4), 0.15 M NaCl, 3.4 mM EDTA, and 0.005%Tween 20 at 25°C). CCL2 (R&D Systems) was used in concentrationsranging from 94 to 1500 nM and a flow rate of 30 "l/min. The surface wasregenerated back to baseline level by injecting 10 "l of 100 mM NaOH.For binding kinetics, BIA-evaluation (version 4.1 software; BIAcore) wasapplied, and the 1:1 Langmuir binding model was chosen.
Production of anti-human CCL2 mAb
BALB/c mice were administered 50 "g of human (h)CCL2 in CFA, fol-lowed by six injections every 3 wk, of 50 "g of hCCL2 in IFA. Spleno-cytes were isolated and fused with NSO myeloma cells by standard pro-cedures (33). Hybridoma supernatants were first screened for their abilityto bind hCCL2 by using an ELISA immunoassay. Selected clones werethen tested for their binding specificity by Western blotting using differentchemokines (PeproTech), and for their in vitro ability to inhibit CCL2-induced migration of the human monocyte cell line THP-1 in a Transwellsystem (Corning; Costar). Our anti-CCL2 mAb selectively binds and neu-tralizes human CCL2 with low cross-reactivity to its mouse countermolecule.
Evaluation of the affinity of our anti-hCCL2 mAb to CCL2
The evaluation of the affinity of our anti-hCCL2 mAb to CCL2 was con-ducted by indirect competitive ELISA (34). Briefly, microtiter plates werecoated with recombinant CCL2 (dilution 1/10,000, 200 "l/well). Afterovernight incubation at 4°C the plates were washed three times with wash-ing buffer, blocked with 1% BSA in PBS (w/v, 250 "l/well), and incubatedfor 20 min. To titrate mAb affinity, a mixture of 100-2000 ng/100 "l ofunlabelled mAbs and 100,000 cpm of 125I-labeled mAb was added to eachwell for 90 min at room temperature. Wells were washed three times andradioactivity was counted in a gamma counter.
Cytokine/chemokine detection by ELISA
The specificity binding of the soluble receptors was detected by an ELISAas follows: each well was coated with 10 ng of either mouse or humanproteins, respectively—CCL2, CCL4, CCL5, CCL7, CCL7, CCL8,CCL13, CCL11, CCL16, CCL19, CCL20, CXCL8, CXCL9, CXCL10,CXCL11, CXCL12, XCL1, IL-1#, CD40L (PeproTech) using coatingbuffer (PBS X1), and incubated at 4°C overnight. Wells were incubatedwith 200 "l of 1% BSA/PBS blocking buffer for 1 h at room temperature.Soluble chemokine receptors were added (50 "g/ml) in 1% BSA/PBSbuffer (50 "l per well) and incubated overnight at 4°C and washed fourtimes with PBS/Tween 20 (0.05%). Then 50 "l of goat anti-human IgG-HRP (Jackson ImmunoResearch) was added at 1/10,000 in 1% BSA/PBSfor 1 h and washed four times with PBS/Tween 20 (0.05%). The substratesolution (tetramethylbenzidine) was then added at 50 "l/well. When a bluecolor appeared, the reaction was terminated by adding 50 "l of H2SO4 (1M). OD was determined at 450 nm with the reference filter set to 620 nm.
Chemokine detection in sera by ELISA
Detection of mouse CCL2 was done by ELISA development kit (MouseCCL2/JE ELISA kit; R&D Systems) and conducted according to the man-ufacturer’s instruction.
In vitro chemotaxis assays
Chemotaxis assays in which the chemokine-induced migration of mono-cyte cell lines (THP-1 and RAW 264.7 cells, 1 % 106) were determined,were performed in a Transwell system (5-"m pore size, Corning; Costar).Chemotaxis assays in which the chemokine-induced migration of PC-3cells (1 % 106) were conducted using the CytoSelect cell migration assay(8-"m pore size; Cell Biolabs. Briefly, cells were loaded into the upperchamber of the two systems. The lower chambers were loaded with 20ng/ml chemokines (h/m CCL2, h/m CCL7, h/m CCL8, h/m CCL11,hCCL16, hCCL13, mCCL12; PeproTech) and 50 "g/ml soluble receptors(h/m E3-Ig) or Abs. Cells were let to migrate for 2 h under a humidified7.5% CO2 atmosphere at 37°C. The content of the lower chambers wascollected and counted using the FACSCalibur (BD Biosciences). The che-motaxis index was then calculated by dividing the number of migratingcells in the presence of chemoattractant by the number of cells migrated inits absence.
Evaluation of IC50
Chemokine-induced cell migration was assessed using 5-"m pore Trans-well filter membranes (Costar). A total of 1 % 106 THP-1, or RAW 264.7cells, were loaded into each Transwell filter. Soluble receptors (h/m E3-Ig)or mAbs (our anti-hCCL2 mAb and commercial anti-mCCL2 mAb; R&DSystems) were added in the lower chamber at different concentrations(0.01–100 "g/ml) with 20 ng/ml CCL2. Plates were then incubated at 37°Cfor 2 h. The migrated cells were collected and counted by FACSCalibur(BD Biosciences). The IC50 calculation was performed by Origin8 soft-ware (OriginLab).
In vitro proliferation assay
A total of 1 % 104 PC-3 cells were cultured in 96-well flat-bottom tissueculture plates in their respective medium. After 24 h, wells were supple-mented with fresh medium, 200 "g/ml neutralizing Abs, 200 "g/ml solublereceptors, or 200 "g/ml total IgG from preimmune mice. Cells were cul-tured in the presence and absence of these supplements for 5 days. Eachwell was pulsed with 2 "Ci of thymidine (specific activity 10 Ci/mmol) forthe final 16 h. The cultures were then harvested on fiberglass filters. Resultsare shown as the mean cpm of six replicates & SE from three independentexperiments, divided by the mean cpm of control cells.
Animal models
An animal model of EAE used C57BL/6 female mice purchased fromHarlan Laboratories and maintained in independent ventilated cages under
733The Journal of Immunology
pathogen-free conditions. At 6 wk of age, mice were subjected to activedisease induction by a single administration of MOG33–55 emulsified inCFA as described elsewhere (35). At the onset of disease only mice withan apparent clinical manifestation of disease were selected and separatedinto equally sick groups for therapy. Animals were then monitored forclinical signs daily by an observer blind to the treatment protocol. EAE wasscored as follows: 0, clinically normal; 1, flaccid tail; 2, hind limb paral-ysis; 3, total hind limb paralysis, accompanied by an apparent front limbparalysis; and 4, total hind limb and front limb paralysis.
The animal xenograft model of prostate cancer used SCID/Beige malemice purchased from Harlan Laboratories and maintained in independentventilated cages under pathogen-free conditions. At 6 wk of age, mice weres.c. injected between the two flanks with 5 % 106 PC-3 cell line (AmericanType Culture Collection). Mice were monitored daily for tumor volume.Tumor diameters were measured using a caliper. Tumor volume was cal-culated using the formula $/6 % a % b2, where a is the longest dimension,and b is the width.
Histology
Spinal cords were subjected to histology analysis. Briefly, tissue sampleswere fixed overnight in 4% paraformaldehyde in PBS, then dehydrated,paraffin-embedded, and sectioned into 5-"m sections. Sections were thendeparaffinized and stained with H&E and with Luxol fast blue.
Statistical analysis
The significant difference was examined using Student’s t test. Values forp ' 0.05 were considered statistically significant.
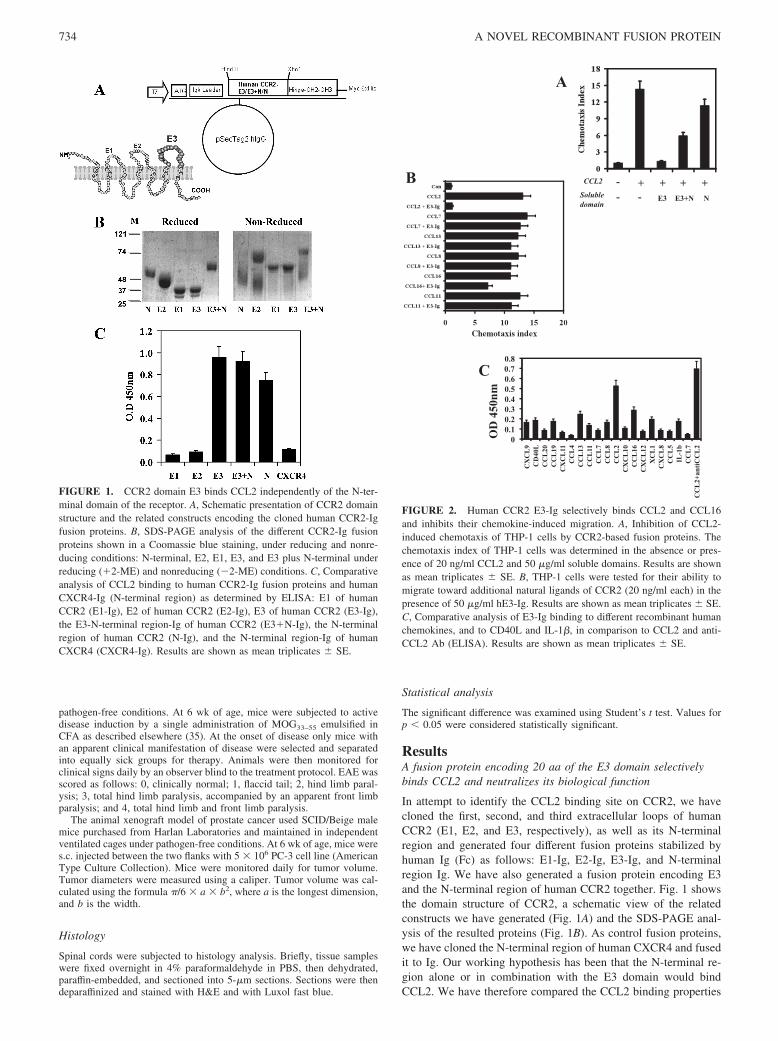
ResultsA fusion protein encoding 20 aa of the E3 domain selectivelybinds CCL2 and neutralizes its biological function
In attempt to identify the CCL2 binding site on CCR2, we havecloned the first, second, and third extracellular loops of humanCCR2 (E1, E2, and E3, respectively), as well as its N-terminalregion and generated four different fusion proteins stabilized byhuman Ig (Fc) as follows: E1-Ig, E2-Ig, E3-Ig, and N-terminalregion Ig. We have also generated a fusion protein encoding E3and the N-terminal region of human CCR2 together. Fig. 1 showsthe domain structure of CCR2, a schematic view of the relatedconstructs we have generated (Fig. 1A) and the SDS-PAGE anal-ysis of the resulted proteins (Fig. 1B). As control fusion proteins,we have cloned the N-terminal region of human CXCR4 and fusedit to Ig. Our working hypothesis has been that the N-terminal re-gion alone or in combination with the E3 domain would bindCCL2. We have therefore compared the CCL2 binding properties
FIGURE 1. CCR2 domain E3 binds CCL2 independently of the N-ter-minal domain of the receptor. A, Schematic presentation of CCR2 domainstructure and the related constructs encoding the cloned human CCR2-Igfusion proteins. B, SDS-PAGE analysis of the different CCR2-Ig fusionproteins shown in a Coomassie blue staining, under reducing and nonre-ducing conditions: N-terminal, E2, E1, E3, and E3 plus N-terminal underreducing ((2-ME) and nonreducing (#2-ME) conditions. C, Comparativeanalysis of CCL2 binding to human CCR2-Ig fusion proteins and humanCXCR4-Ig (N-terminal region) as determined by ELISA: E1 of humanCCR2 (E1-Ig), E2 of human CCR2 (E2-Ig), E3 of human CCR2 (E3-Ig),the E3-N-terminal region-Ig of human CCR2 (E3(N-Ig), the N-terminalregion of human CCR2 (N-Ig), and the N-terminal region-Ig of humanCXCR4 (CXCR4-Ig). Results are shown as mean triplicates & SE.
FIGURE 2. Human CCR2 E3-Ig selectively binds CCL2 and CCL16and inhibits their chemokine-induced migration. A, Inhibition of CCL2-induced chemotaxis of THP-1 cells by CCR2-based fusion proteins. Thechemotaxis index of THP-1 cells was determined in the absence or pres-ence of 20 ng/ml CCL2 and 50 "g/ml soluble domains. Results are shownas mean triplicates & SE. B, THP-1 cells were tested for their ability tomigrate toward additional natural ligands of CCR2 (20 ng/ml each) in thepresence of 50 "g/ml hE3-Ig. Results are shown as mean triplicates & SE.C, Comparative analysis of E3-Ig binding to different recombinant humanchemokines, and to CD40L and IL-1#, in comparison to CCL2 and anti-CCL2 Ab (ELISA). Results are shown as mean triplicates & SE.
734 A NOVEL RECOMBINANT FUSION PROTEIN
of each of our fusion proteins. Fig. 1C shows that of all fusionproteins the E3 domain, the N-terminal domain, or their fusion(E3-N-terminal) bind CCL2, with the first two exhibiting a slightlyhigher binding (OD450 nm 0.96 & 0.1 and 0.92 & 0.08, comparedwith 0.74 & 0.06, p ' 0.001).
Fig. 2A shows the ability of each domain to inhibit the CCL2-induced migration of THP-1 cells. In these recombinant fusionproteins, the E3 domain alone (E3-Ig or BL-2030) not only couldsignificantly inhibit CCL2-induced migration of THP-1 cells (che-motaxis index of 1.32 & 0.2 compared with 14.34 & 1.5, p '0.001) but could also do so better than each of the other fusionproteins, including the E3-N-terminal fusion (chemotaxis index of1.32 & 0.2 vs 5.9 & 0.6, p ' 0.01). Human CCR2 binds multiplechemokines, including CCL2, CCL8, CCL16, CCL7, CCL11, andCCL13. We have determined the ability of human E3-Ig to inhibitthe migration of THP-1 cells induced by each of these chemokines,as well as its direct binding to each of them (ELISA). Our obser-vations clearly show that of these chemokines, E3-Ig (BL-2030)selectively inhibits THP-1-induced migration of two only of them:CCL2 and CCL16. It also exclusively binds both chemokines (Fig.2C). Its ability to block CCL2-induced migration is significantlyhigher than to inhibit CCL16-induced chemotaxis under the sameconditions (Fig. 2B, p ' 0.01).
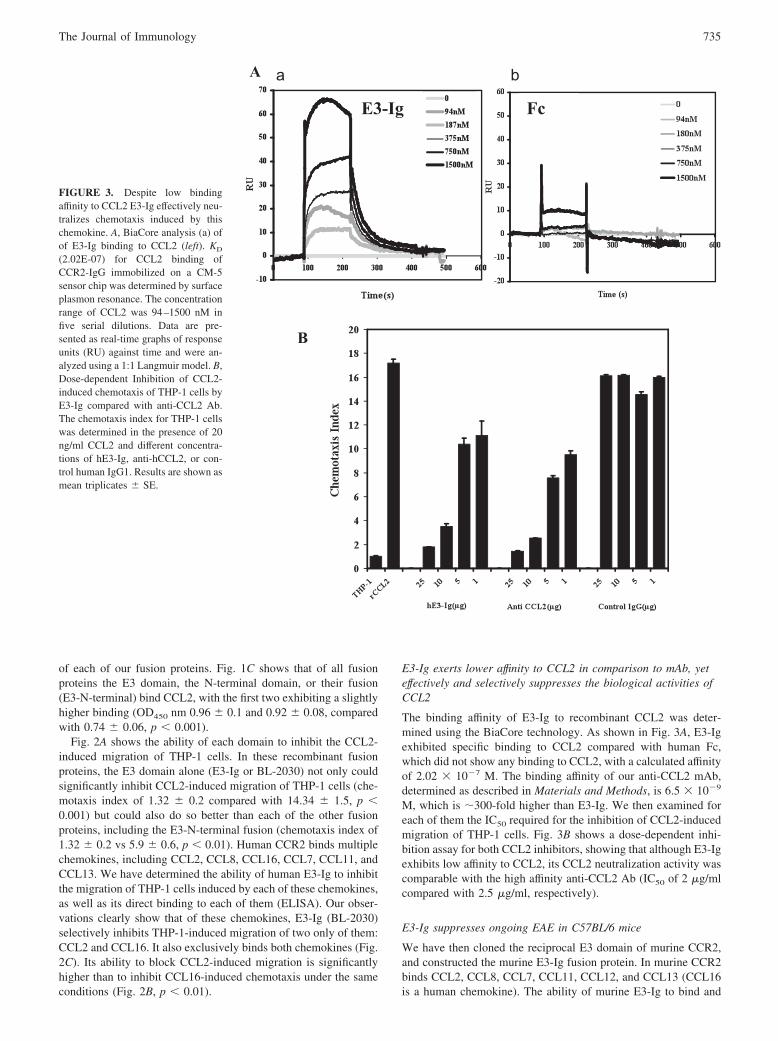
E3-Ig exerts lower affinity to CCL2 in comparison to mAb, yeteffectively and selectively suppresses the biological activities ofCCL2
The binding affinity of E3-Ig to recombinant CCL2 was deter-mined using the BiaCore technology. As shown in Fig. 3A, E3-Igexhibited specific binding to CCL2 compared with human Fc,which did not show any binding to CCL2, with a calculated affinityof 2.02 % 10#7 M. The binding affinity of our anti-CCL2 mAb,determined as described in Materials and Methods, is 6.5 % 10#9
M, which is !300-fold higher than E3-Ig. We then examined foreach of them the IC50 required for the inhibition of CCL2-inducedmigration of THP-1 cells. Fig. 3B shows a dose-dependent inhi-bition assay for both CCL2 inhibitors, showing that although E3-Igexhibits low affinity to CCL2, its CCL2 neutralization activity wascomparable with the high affinity anti-CCL2 Ab (IC50 of 2 "g/mlcompared with 2.5 "g/ml, respectively).
E3-Ig suppresses ongoing EAE in C57BL/6 mice
We have then cloned the reciprocal E3 domain of murine CCR2,and constructed the murine E3-Ig fusion protein. In murine CCR2binds CCL2, CCL8, CCL7, CCL11, CCL12, and CCL13 (CCL16is a human chemokine). The ability of murine E3-Ig to bind and
FIGURE 3. Despite low bindingaffinity to CCL2 E3-Ig effectively neu-tralizes chemotaxis induced by thischemokine. A, BiaCore analysis (a) ofof E3-Ig binding to CCL2 (left). KD
(2.02E-07) for CCL2 binding ofCCR2-IgG immobilized on a CM-5sensor chip was determined by surfaceplasmon resonance. The concentrationrange of CCL2 was 94–1500 nM infive serial dilutions. Data are pre-sented as real-time graphs of responseunits (RU) against time and were an-alyzed using a 1:1 Langmuir model. B,Dose-dependent Inhibition of CCL2-induced chemotaxis of THP-1 cells byE3-Ig compared with anti-CCL2 Ab.The chemotaxis index for THP-1 cellswas determined in the presence of 20ng/ml CCL2 and different concentra-tions of hE3-Ig, anti-hCCL2, or con-trol human IgG1. Results are shown asmean triplicates & SE.
735The Journal of Immunology

inhibit chemokine-induced migration of the mouse monocytic cellline (RAW 264.7) in response to each of these chemokines wasdetermined. Our observations clearly show that of these chemo-kines mE3-Ig exclusively inhibits the CCL2-induced migration ofRAW 264.7 cells (Fig. 4A). It also solely binds this chemokine(Fig. 4B). We then examined the IC50 required for the inhibition ofmurine CCL2-induced migration of RAW 264.7, according theprotocol used in Fig. 2B. Our results show that the IC50 of murineE3-Ig (5.2 "g/ml) is comparable to the commercially availablecontrol mAb (clone 123616; R&D Systems) (Fig. 4C), which is4.7 "g/ml.
Although mice lacking CCL2 displayed markedly reduced EAE(5), there is a dispute whether its targeted neutralization in wild-type rodents suppresses the disease (6, 7, 36). Based on its highspecificity in binding and neutralization of CCL2 we have ex-plored the ability of E3-Ig to interfere in the regulation of ongoingEAE in C57BL/6 mice.
These mice were subjected to active induction of EAE and, fol-lowing the onset of disease (day 15), treated (every 3 days) with200 "g/mouse of E3-Ig, control mouse IgG1, or PBS and moni-tored for disease progression by an observer blind to the experi-mental protocol (Fig. 4D). We show that administration of E3-Ig(BL-2030) rapidly suppresses EAE (mean maximal score of 1.3 &0.2, compared with 3 & 1.2 and 3.3 & 0.9 in PBS or control IgGtreated mice, respectively, p ' 0.01). At the peak of disease, rep-resentative mice were sacrificed, and lumbar spinal cords weresubjected to histological analysis. Fig. 4D shows the results ofrepresentative sections (of 18 sections per group), showing amarked decrease in leukocyte infiltrate around HEV of micetreated with E3-Ig but not with control IgG or PBS. A comple-mentary Luxol fast blue staining was conducted to verify that re-covery is associated with reduction in demyelination that occursduring EAE in C57BL/6 mice (Fig. 4E). Thus, E3-Ig (BL-2030)effectively suppresses an ongoing EAE disease. Finally, We have
FIGURE 4. E3-Ig selectively bindsand neutralizes murine CCL2 and sup-presses ongoing EAE in C57BL/6mice. A, The monocytic cell line (RAW264.7) was tested for its ability to mi-grate toward additional natural ligandsof CCR2 (20 ng/ml each) in the pres-ence of 50 "g/ml mE3-Ig. Results areshown as mean triplicates & SE. B,Murine E3-Ig was determined for itsbinding specificities to various chemo-kines. Results of triplicates are shownas mean OD at 450 nm & SE. C, Dose-dependent inhibition of CCL2-inducedchemotaxis of RAW 264.7 cells bymE3-Ig compared with anti-CCL2mAb. The chemotaxis index for RAW264.7 cells was determined in the pres-ence of 20 ng/ml CCL2 and differentconcentrations of either mE3-Ig or anti-mCCL2. Results are shown as meantriplicates & SE. D, C57BL/6 micewere subjected to active induction ofEAE. Following disease onset (day 15)mice were treated (every 3 days) with200 "g/mouse of E3-Ig (f), controlmouse IgG1 (E), or PBS (Œ) and mon-itored for disease progression by an ob-server blind to the experimental proto-col. Results of six mice/group areshown as mean EAE score & SE. E,H&E (a, b, and c), or Luxol fast blue (d,e, and f) staining of spinal cord samplesfrom mice treated with PBS (a and d),control IgG (b and e), and E3-Ig (c andf) at a magnification of %20.
736 A NOVEL RECOMBINANT FUSION PROTEIN

measured the levels of CCL2 in circulating blood 1 day after thesecond mE3-Ig administration during EAE (Fig. 4D). We identi-fied an 1.8-fold increase in CCL2 levels in the circulation oftreated mice (57.15 & 6.1 ng/ml in E3-Ig-treated mice comparedwith 30.1 & 4.2 ng/ml in PBS-treated mice.
E3-Ig inhibits the proliferation and CCL-2 induced migration ofPC-3 cells in vitro
Three human prostate cancer cell lines are commonly used inxenograft experiments: LNCaP, an androgen-dependent cellline derived from a patient’s lymph node, and DU-145 andPC-3, both androgen-independent cell lines, derived from brainand bone metastases, respectively. Of these lines, PC-3 is themost aggressive (37) and has therefore been selected for our invivo xenograft studies. After verification of CCR2 expression(FACS analysis, data not shown), we tested whether hE3-Igwould affect CCL2-induced migration of PC-3 cells in vitro(Fig. 5A). We show that hE3-Ig effectively blocks CCL2-in-duced chemotaxis of PC-3 cells (chemotaxis index of 3.16 &0.2 vs 10.3 & 0.09, p ' 0.01), and that the migration of these
cells is directed via the CCL2-CCR2 interaction (Fig. 5A). Ananti-CCR2 mAb (38) provided by Dr. C. Martinez-A (CentroNacional de Biotecnologia, Universidad Autonoma de Madrid,Madrid, Spain) also effectively suppressed CCL2-induced mi-gration of these cells (chemotaxis index of 2.6 & 0.2 vs 10.3 &1.4, p ' 0.01) (Fig. 5A). We then determined the ability of thissoluble receptor to inhibit the proliferation/growth of PC-3cells. As shown in Fig. 5B, addition of E3-Ig to cultured PC-3cells significantly reduced the proliferation of these cells (41 &2.4 % 103 cpm vs 100 & 8.7 % 103 cpm, p ' 0.01). Similarresults were obtained using the anti-CCR2 mAb (28 & 1.9 %103 cpm vs 100 & 8.5 % 103 cpm, p ' 0.01).
FIGURE 5. Human E3-Ig inhibits the proliferation and CCL2-in-duced migration of PC3 cells. A, Human E3-Ig inhibits CCL2-inducedmigration of PC-3 cells as determined by the CytoSelect cell migrationassay. Results are shown as mean triplicates & SE. Add amount ofCCL2. B, Human E3-Ig inhibits PC-3 cell proliferation. The 96-welltissue culture plates were loaded with 1 % 104 PC-3 cells and culturedin the presence of culture medium, control IgG, anti-CCR2 mAb, andhE3-Ig. All gene products including control IgG, all mAbs, and E3-Igwere isotype matched (IgG1). Results are shown as mean of [3H] uptakeof triplicates & SE.
FIGURE 6. Human E3-Ig inhibits the development of primary tumor inPC3 prostate cancer xenograft model. A, SCID/Beige mice (six mice/group) were implanted with 5 % 106 PC-3 cells and were treated twice aweek, starting from day 7, with either PBS (Œ), isotype-matched IgG1 (f),50 "g/mouse (E), 100 "g/mouse (‚), or 200 "g/mouse (F) of hE3-Ig.Mice were monitored for the progression of the primary tumor until sac-rificed on day 40. In the 100 and 200 "g/mouse groups, treatment wasterminated on day 34 and mice were then monitored for tumor growth untiltumors reached the volume of 1000 mm3 (day 54 for 100 "g/mouse groupand day 68 for 200 "g/mouse group). Data presented represent one of threeindependent experiments with similar data. Results are shown as meantumor volume & SE. B, SCID/Beige mice (six mice/group) were implantedwith 5 % 106 PC-3 cells and were treated twice a week, starting from day7, with either PBS (f), isotype-matched IgG1 (!), 200 "g/mouse anti-CCL2 (‚), or 200 "g/mouse E3-Ig (F). Mice were monitored for theprogression of the primary tumor using a caliper. Data presented representone of three independent experiments with similar data. Results are shownas mean tumor volume & SE.
737The Journal of Immunology

E3-Ig inhibits tumor growth in a prostate cancer xenograftmodel
Next, we explored the ability of hE3-Ig to suppress the growth ofPC-3 in SCID/Beige mice. Briefly, mice were implanted with 5 %106 PC-3 cells and beginning on day 7, when tumors were clearlyidentified in all animals, were treated (twice a week) with PBS,isotype-matched IgG1, or 50, 100, or 200 "g/mouse of hE3-Ig andmonitored for tumor growth (Fig. 6). Our results clearly show adose-dependent blockade of primary tumor growth in E3-Ig-treated mice (tumor size at day 40, 1550 mm3 & 123.7 and 2020mm3 & 134.8 in control groups, compared with 90.5 mm3 & 26.1and 505 mm3 & 91.1 in those treated with 200 and 100 "g ofE3-Ig, respectively, p ' 0.001). Most importantly, E3-Ig inhibi-tory effect was still observed 15–35 days following treatment ter-mination, hinting to its therapeutic potential as anticancer agent.
After determining the dissociation constant (Kd) and neutraliz-ing activity (IC50) of our anti-human CCL2 mAb in comparison tohE3-Ig (Fig. 3), we have compared their ability to suppress thegrowth of human prostate cancer cell line (PC-3) in SCID mice. Ithas been previously shown that in this xenograft model eventhough both the murine and tumor-derived CCL2 contribute totumor development, targeted neutralization of the human-derivedchemokine markedly suppresses tumor development (14). Our re-sults clearly show that under the same experimental conditions,both equally very effectively suppressed tumor development (Fig.6B) despite the major difference in anti-human CCL2 mAb in com-parison to hE3-Ig binding affinity to CCL2 (!300-fold higher forthe Ab).
DiscussionIn this study, we describe a novel recombinant soluble form of theCCL2 receptor, E3-Ig (BL-2030). We have shown that E3-Ig se-lectively binds CCL2 and neutralizes its biological activity, mainlythe chemotaxis of human monocytes in vitro. In addition, it inhib-its the proliferation and migration of the human prostate cancercell line PC-3 that expresses CCL2 and its receptor. The in vivoactivity of E3-Ig was demonstrated in two separate disease modelsin which the CCL2-CCR2 pathway was shown to play a role, theEAE model of MS and the PC-3 prostate cancer model. E3-Ig(BL-2030) significantly suppressed ongoing EAE disease and ex-hibited dose-dependant inhibition of PC-3 tumor growth. The in-hibition of PC-3 cell proliferation and migration in vitro togetherwith the suppression of tumor growth in vivo indicate an anti-tumorigenic and anti-metastatic potential of E3-Ig. In support ofour results, a previous study has demonstrated that an anti-CCL2-neutralizing Ab attenuates tumor burden in the PC-3 prostate can-cer model (14).
Chemokine receptors belong to the superfamily of seven-trans-membrane G protein-coupled receptors (2) that span the plasmamembrane seven times generating three loops (domains). Theirthree-dimensional structure is dependent on plasma-membrane sta-bilization, and therefore the generation of soluble chemokine re-ceptors that are highly effective in neutralizing their target chemo-kines is still a major challenge. In an attempt to overcome thisobstacle, we generated short recombinant proteins encoding dif-ferent segments of the G protein-coupled receptor CCR2, andtested their ability to bind and neutralize its target chemokineCCL2. We show that although both the E3 domain and the Nterminus of CCR2 bind CCL2, only the E3 domain alone, or incombination with the N-terminal region, neutralizes the basic bi-ological activity of CCL2; chemoattraction of monocytes. Surpris-ingly, the E3 domain alone comprising only 20 aa of CCR2 (fused
to Ig) was the most effective inhibitor of CCL2-induced chemo-taxis in vitro.
Neutralizing mAbs and soluble receptor-based fusion proteinswere shown to be effective in the treatment of autoimmune dis-eases and cancer. Use of anti-TNF-! mAbs and recombinant sol-uble TNF-! receptor fusion proteins is a classical example of suc-cessful treatment of rheumatoid arthritis and other relatedautoimmune diseases (39–41). One of the major disadvantages ofmAb-based therapies is their tendency to elicit anti-idiotypic neu-tralizing Abs, as a part of a natural regulatory network (42). Fromthis perspective, soluble receptors have a therapeutic advantage.The CCL2/CCR2 axis has been implicated in the pathophysiologyof a wide range of both acute and chronic inflammatory conditions,such as rheumatoid arthritis, MS, atherosclerosis, uveitis, asthma,psoriasis, diabetes, inflammatory bowel disease, lupus nephritis,transplant rejection, and several CCL2/CCR2 antagonists are cur-rently under clinical development.
The role of CCL2 in EAE has been studied by either usingdeficient mice, or by administering anti-CCL2 polyclonal Abs toEAE in mice or rats. Izikson et al. (43) showed that mice lackingCCR2 are EAE-resistant. Nevertheless, at least six different che-mokines bind CCR2, therefore other chemokines, aside of CCL2,might contribute to EAE resistance in CCR2-deficient mice asdemonstrated in that study. A complementary publication ofHuang et al. (5) showing that mice lacking CCL2 display a mark-edly reduced form of disease, further emphasizes the pivotal roleof CCL2 in the pathogenesis of EAE. Thus far the evidence thattargeting CCL2 alone, in wild-type rodents suppresses EAEemerged from studies that used polyclonal Ab-based therapies,with their limitations. There is a dispute in these studies whethertargeted neutralization of CCL2 in wild-type rodents suppressesthe disease (6, 7, 36). We show in this study that in C57BL/6 EAEmice injection of E3-Ig, that specifically and exclusively binds andneutralizes CCL2 (Fig. 4, A–C), effectively suppressed an ongoingdisease (Fig. 4, D and E) further implies that this chemokine playsa pivotal role in EAE, as previously demonstrated in mice lackingCCL2 (5).
It has been reported that administration of anti-CCL2 mAbs topatients suffering from rheumatoid arthritis led to a 2000-fold in-crease in their circulating CCL2 (44), which may explain, in part,why therapy was ineffective. In the current study we show thatrepeated administration of E3-Ig only moderately increased circu-lating blood levels CCL2 (1.8-fold increase). It is possible that inhuman therapy with E3-Ig would also result in a moderated in-crease in circulating CCL2. This effect would be a major advan-tage to the recipients, though this feature has yet to be carefullydetected along clinical trails.
In addition of being a key regulator of monocytes infiltration tosites of inflammation, CCL2 was shown to possess protumorigenicand proangiogenic functions. The mechanisms by which CCL2promotes tumor progression are still unclear. CCL2 is primarilyresponsible for the recruitment of tumor infiltrating macrophagesinto the tumor site, stimulating angiogenesis and metastasis. Inaddition, CCL2 has been shown to have direct effects on the tumorcells in several neoplasms including breast, lung, cervix, ovary,sarcoma, and prostate, inducing cancer cell proliferation, migra-tion, and survival.
We clearly show that E3-Ig well suppresses the establishment ofhuman tumor cells in SCID mice. Our human E3-Ig (BL-2030)binds and neutralizes both CCL2 and CCL16 (Fig. 2). Humancancer cells produce both chemokines (9). CCL16 is also likely tocontribute to tumor invasion and angiogenesis (45). Therefore itcould be that its neutralization by BL-2030 also contributes totumor suppression in a xenograft model of cancer (Fig. 6).
738 A NOVEL RECOMBINANT FUSION PROTEIN

E3-Ig (BL-2030) represents a novel approach for inhibitingCCL2. Its potent anti-tumorigenic effect, together with its anti-inflammatory activity, suggests a therapeutic potential for the treat-ment of autoimmune disease, as well as various malignancies. Fur-ther studies exploring the therapeutic potential of E3-Ig inadditional inflammatory disease models and various cancer modelsare required.
DisclosuresN.K., L.I., G.W. and Y.Z. hold a pending patent on therapy of inflamma-tory autoimmunity and cancer using CCL2 E3-Ig that has been licensed outto BiolineRx. L.K., M.A.-S., and A.E. are research scientists at BiolineRXand preformed part of the in vitro analysis within the manuscripts and alsoparticipate in discussing the results.
References1. Rollins, B. J. 1997. Chemokines. Blood 90: 909–928.2. Zlotnic, A., and O. Yoshei. 2000. Chemokines: a new clasification system and
their role in immunity. Immunity 12: 121–127.3. Mackay, C. R. 2001. Chemokines: immunology’s high impact factors. Nat. Im-
munol. 2: 95–101.4. Proudfoot, A. E. 2002. Chemokine receptors: a multifaceted therapeutic targets.
Nat. Rev. Immunol 2: 106–115.5. Huang, D. R., J. Wang, P. Kivisakk, B. J. Rollins, and R. M. Ransohoff. 2001.
Absence of monocyte chemoattractant protein 1 in mice leads to decreased localmacrophage recruitment and antigen-specific T helper cell type 1 immune re-sponse in experimental autoimmune encephalomyelitis. J. Exp. Med. 193:713–726.
6. Kennedy, K. J., R. M. Strieter, S. L. Kunkel, N. W. Lukacs, and W. J. Karpus.1998. Acute and relapsing experimental autoimmune encephalomyelitis are reg-ulated by differential expression of the CC chemokines macrophage inflammatoryprotein-1! and monocyte chemotactic protein-1. J. Neuroimmunol. 92: 98–108.
7. Youssef, S., G. Wildbaum, G. Maor, N. Lanir, A. Gour-Lavie, N. Grabie, andN. Karin. 1998. Long-lasting protective immunity to experimental autoimmuneencephalomyelitis following vaccination with naked DNA encoding C-C chemo-kines. J. Immunol. 161: 3870–3879.
8. Goser, S., R. Ottl, A. Brodner, T. J. Dengler, J. Torzewski, K. Egashira,N. R. Rose, H. A. Katus, and Z. Kaya. 2005. Critical role for monocyte che-moattractant protein-1 and macrophage inflammatory protein-1! in induction ofexperimental autoimmune myocarditis and effective anti-monocyte chemoattrac-tant protein-1 gene therapy. Circulation 112: 3400–3407.
9. Homey, B., A. Muller, and A. Zlotnik. 2002. Chemokines: agents for the immu-notherapy of cancer? Nat. Rev. Immunol. 2: 175–184.
10. Garnick, M. B. 1993. Prostate cancer: screening, diagnosis, and management.Ann. Intern. Med. 118: 804–818.
11. Pienta, K. J., and P. S. Esper. 1993. Risk factors for prostate cancer. Ann. Intern.Med. 118: 793–803.
12. Loberg, R. D., L. L. Day, J. Harwood, C. Ying, L. N. St. John, R. Giles,C. K. Neeley, and K. J. Pienta. 2006. CCL2 is a potent regulator of prostatecancer cell migration and proliferation. Neoplasia 8: 578–586.
13. Lu, Y., Z. Cai, D. L. Galson, G. Xiao, Y. Liu, D. E. George, M. F. Melhem,Z. Yao, and J. Zhang. 2006. Monocyte chemotactic protein-1 (MCP-1) acts as aparacrine and autocrine factor for prostate cancer growth and invasion. Prostate66: 1311–1318.
14. Loberg, R. D., C. Ying, M. Craig, L. L. Day, E. Sargent, C. Neeley, K. Wojno,L. A. Snyder, L. Yan, and K. J. Pienta. 2007. Targeting CCL2 with systemicdelivery of neutralizing antibodies induces prostate cancer tumor regression invivo. Cancer Res. 67: 9417–9424.
15. Caruso, D. J., A. J. Carmack, V. B. Lokeshwar, R. C. Duncan, M. S. Soloway,and B. L. Lokeshwar. 2008. Osteopontin and interleukin-8 expression is inde-pendently associated with prostate cancer recurrence. Clin. Cancer Res. 14:4111–4118.
16. Waugh, D. J., and C. Wilson. 2008. The interleukin-8 pathway in cancer. Clin.Cancer Res. 14: 6735–6741.
17. Akashi, T., K. Koizumi, O. Nagakawa, H. Fuse, and I. Saiki. 2006. Androgenreceptor negatively influences the expression of chemokine receptors (CXCR4,CCR1) and ligand-mediated migration in prostate cancer DU-145. Oncol. Rep.16: 831–836.
18. Mydlo, J. H., M. I. Gerstein, C. F. Harris, and A. S. Braverman. 2003. Immunefunction, mitogenicity, and angiogenic growth factor concentrations in lean andobese rodent sera: implications in obesity-related prostate tumor biology. Pros-tate Cancer Prostatic Dis. 6: 286–289.
19. Darash-Yahana, M., E. Pikarsky, R. Abramovitch, E. Zeira, B. Pal, R. Karplus,K. Beider, S. Avniel, S. Kasem, E. Galun, and A. Peled. 2004. Role of highexpression levels of CXCR4 in tumor growth, vascularization, and metastasis.FASEB J. 18: 1240–1242.
20. Engl, T., B. Relja, D. Marian, C. Blumenberg, I. Muller, W. D. Beecken, J. Jones,E. M. Ringel, J. Bereiter-Hahn, D. Jonas, and R. A. Blaheta. 2006. CXCR4chemokine receptor mediates prostate tumor cell adhesion through !5 and #3integrins. Neoplasia 8: 290–301.
21. Kukreja, P., A. B. Abdel-Mageed, D. Mondal, K. Liu, and K. C. Agrawal. 2005.Up-regulation of CXCR4 expression in PC-3 cells by stromal-derived factor-
1alpha (CXCL12) increases endothelial adhesion and transendothelial migration:role of MEK/ERK signaling pathway-dependent NF-%B activation. Cancer Res.65: 9891–9898.
22. Sun, Y. X., J. Wang, C. E. Shelburne, D. E. Lopatin, A. M. Chinnaiyan,M. A. Rubin, K. J. Pienta, and R. S. Taichman. 2003. Expression of CXCR4 andCXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J. Cell Biochem. 89:462–473.
23. Taichman, R. S., C. Cooper, E. T. Keller, K. J. Pienta, N. S. Taichman, andL. K. McCauley. 2002. Use of the stromal cell-derived factor-1/CXCR4 pathwayin prostate cancer metastasis to bone. Cancer Res. 62: 1832–1837.
24. Vaday, G. G., S. B. Hua, D. M. Peehl, M. H. Pauling, Y. H. Lin, L. Zhu,D. M. Lawrence, H. D. Foda, and S. Zucker. 2004. CXCR4 and CXCL12(SDF-1) in prostate cancer: inhibitory effects of human single chain Fv antibod-ies. Clin. Cancer Res. 10: 5630–5639.
25. Lu, Y., Z. Cai, G. Xiao, E. T. Keller, A. Mizokami, Z. Yao, G. D. Roodman, andJ. Zhang. 2007. Monocyte chemotactic protein-1 mediates prostate cancer-in-duced bone resorption. Cancer Res. 67: 3646–3653.
26. Monteclaro, F. S., and I. F. Charo. 1997. The amino-terminal domain of CCR2is both necessary and sufficient for high affinity binding of monocyte chemoat-tractant protein 1: receptor activation by a pseudo-tethered ligand. J. Biol. Chem.272: 23186–23190.
27. Datta-Mannan, A., and M. J. Stone. 2004. Chemokine-binding specificity of sol-uble chemokine-receptor analogues: identification of interacting elements by chi-mera complementation. Biochemistry 43: 14602–14611.
28. Charo, I. F., S. J. Myers, A. Herman, C. Franci, A. J. Connolly, andS. R. Coughlin. 1994. Molecular cloning and functional expression of two mono-cyte chemoattractant protein 1 receptors reveals alternative splicing of the car-boxyl-terminal tails. Proc. Natl. Acad. Sci. USA 91: 2752–2756.
29. Kurihara, T., and R. Bravo. 1996. Cloning and functional expression of mCCR2,a murine receptor for the C-C chemokines JE and FIC. J. Biol. Chem. 271:11603–11607.
30. Gingrich, J. R., R. J. Barrios, B. A. Foster, and N. M. Greenberg. 1999. Patho-logic progression of autochthonous prostate cancer in the TRAMP model. Pros-tate Cancer Prostatic Dis. 2: 70–75.
31. Loetscher, M., T. Geiser, T. O’Reilly, R. Zwahlen, M. Baggiolini, and B. Moser.1994. Cloning of a human seven-transmembrane domain receptor, LESTR, thatis highly expressed in leukocytes. J. Biol. Chem. 269: 232–237.
32. Carothers, A. M., G. Urlaub, J. Mucha, D. Grunberger, and L. A. Chasin. 1989.Point mutation analysis in a mammalian gene: rapid preparation of total RNA,PCR amplification of cDNA, and Taq sequencing by a novel method. BioTech-niques 7: 494–499.
33. Harlow, E., and D. Lane. Antibodies, A Laboratory Manual. Cold Spring HarborLaboratory, New York; 1988.
34. Friguet, B., A. F. Chaffotte, L. Djavadi-Ohaniance, and M. E. Goldberg. 1985.Measurements of the true affinity constant in solution of antigen-antibody com-plexes by enzyme-linked immunosorbent assay. J. Immunol. Methods 77:305–319.
35. Mendel, I., N. Kerlero de Rosbo, and A. Ben-Nun. 1995. A myelin oligodendro-cyte glycoprotein peptide induces typical chronic experimental autoimmune en-cephalomyelitis in H-2b mice: fine specificity and T cell receptor V# expressionof encephalitogenic T cells. Eur. J. Immunol. 25: 1951–1959.
36. Karpus, W. J., and K. J. Kennedy. 1997. MIP-1! and MCP-1 differentially reg-ulate acute and relapsing autoimmune encephalomyelitis as well as Th1/Th2 lym-phocyte differentiation. J. Leukocyte Biol. 62: 681–687.
37. Rubio, N., M. M. Villacampa, N. El Hilali, and J. Blanco. 2000. Metastaticburden in nude mice organs measured using prostate tumor PC-3 cells expressingthe luciferase gene as a quantifiable tumor cell marker. Prostate 44: 133–143.
38. Frade, J. M., M. Mellado, G. del Real, J. C. Gutierrez-Ramos, P. Lind, andA. C. Martinez. 1997. Characterization of the CCR2 chemokine receptor: func-tional CCR2 receptor expression in B cells. J. Immunol. 159: 5576–5584.
39. Feldmann, M., F. M. Brennan, and R. N. Maini. 1996. Role of cytokines inrheumatoid arthritis. Annu. Rev. Immunol. 14: 397–440.
40. Moreland, L. W., G. Margolies, L. W. Heck, Jr., A. Saway, C. Blosch, R. Hanna,and W. J. Koopman. 1996. Recombinant soluble tumor necrosis factor receptor(p80) fusion protein: toxicity and dose finding trial in refractory rheumatoid ar-thritis. J. Rheumatol. 23: 1849–1855.
41. Moreland, L. W., S. W. Baumgartner, M. H. Schiff, E. A. Tindall,R. M. Fleischmann, A. L. Weaver, R. E. Ettlinger, S. Cohen, W. J. Koopman,K. Mohler, et al. 1997. Treatment of rheumatoid arthritis with a recombinanthuman tumor necrosis factor receptor (p75)-Fc fusion protein. N. Engl. J. Med.337: 141–147.
42. Hebert, J., D. Bernier, Y. Boutin, M. Jobin, and W. Mourad. 1990. Generation ofanti-idiotypic and anti-anti-idiotypic monoclonal antibodies in the same fusion:support of Jerne’s Network Theory. J. Immunol. 144: 4256–4261.
43. Izikson, L., R. S. Klein, I. F. Charo, H. L. Weiner, and A. D. Luster. 2000.Resistance to experimental autoimmune encephalomyelitis in mice lacking theCC chemokine receptor (CCR)2. J. Exp. Med. 192: 1075–1080.
44. Haringman, J. J., D. M. Gerlag, T. J. Smeets, D. Baeten, F. van den Bosch,B. Bresnihan, F. C. Breedveld, H. J. Dinant, F. Legay, H. Gram, et al. 2006. Arandomized controlled trial with an anti-CCL2 (anti-monocyte chemotactic pro-tein 1) monoclonal antibody in patients with rheumatoid arthritis. ArthritisRheum. 54: 2387–2392.
45. Strasly, M., G. Doronzo, P. Cappello, D. Valdembri, M. Arese, S. Mitola,P. Moore, G. Alessandri, M. Giovarelli, and F. Bussolino. 2004. CCL16 activatesan angiogenic program in vascular endothelial cells. Blood 103: 40–49.
739The Journal of Immunology




![Supporting Information - UvA · 3.1. Cloning and expression The gene encoding for GOx[1] connected to a C‐terminal triple Strep‐tag was cloned into pET42a. For recombinant expression](https://img.dokumen.tips/doc/110x75/5f2afce1a1126542ed405d85/supporting-information-uva-31-cloning-and-expression-the-gene-encoding-for-gox1.jpg)























