Embed Size (px)
Citation preview

1
A DFT-PBC study of infinite single-walled carbon nanotubes withvarious tubular diameters
Bo-Cheng Wanga, Wen-Hao Chena, Houng-Wei Wang*,b and Michitoshi Hayashib
aDepartment of Chemistry, Tamkang University, Tamsui 251, Taiwan
bCenter for Condensed Matter Sciences, Taipei 106, Taiwan
Abstract
A density functional theory (DFT) calculation with Gaussian orbital and periodic
boundary condition (PBC) simulation model has been used to determine the electronic
and optimized geometrical structure of zigzag and armchair type SWNTs up to
infinite tubular length. We shed light on the electronic structures of zigzag and
armchair SWNTs with various tubular diameters ([n, 0] zigzag type SWNT for n = 6
~ 20; and [n, n] armchair type SWNT for n = 4 ~ 10 ). Calculated Eg (band gap
between HOCO and LUCO) of the zigzag SWNTs have oscillated with Δn = 3 (the
repeat section is n = 3m - 1, 3m and 3m + 1, m = 1, 2 ..). We conclude that [n, 0]
zigzag SWNTs have narrow Eg being a metal when n = 3m, otherwise, the zigzag
SWNT is a moderate Eg as a semiconductor. For the [n, n] armchair SWNT, the
calculated Eg are between 0.013 eV to 0.030 eV with metallic property; these results
also have oscillation forΔn = 2. The optimized structures of SWNTs being generated
by DFT-PBC method mentioned that the C-C bond length decreased with increase the
tubular diameter. Up to [10, 10] SWNT, it has equal bond length and present the π
bond delocalization. The calculated Eg of SWNT in this study are consistent with the
experimental data.

2
Introduction
Due to the unique physical properties (elasticity, stiffness and deformation) and
the applications in various materials (semiconducting, H2 storage and the probes) of
carbon nanotubes, they have attracted considerable attention.1-5 Almost twenty years
ago, Smalley et al. discovered the truncated-icosahedral C60 carbon cluster by laser
vaporization of graphite in a high-pressure supersonic nozzle.6 In 1991, Ijima detected
the multi-wall carbon nanotubes in a plasma arc discharge apparatus.7 Two years late,
the single walled nanotubes (SWNTs) have been achieved by Iijima and Bethune.
Later, the large-scale purification process and the SEM, TEM and STM
characterization of SWNTs have been obtained.8,9 Although, much scientific interest
was focused on the physical and electronic properties, and commercial applications of
these new materials, there have been no experimental structural data sufficiently
accurate to generate the C-C bond length of SWNTs. In order to investigate the
physical properties of SWNT, they need the theoretical analysis to determine the real
nature of SWNTs and specify their properties.
The geometrical structure of SWNT is a rolling up 2-D graphite sheet as a
hollow cylindrical shape or a one-by-one layering of cyclic carbon array shape as 1-D
tube axis infinity extension.10 The defect free SWNTs have various types of
cylindrical shapes with respect to the array of benzenoids in carbon nano-tube.
According to the geometrical analysis, there exist armchair, zigzag and chiral tubules
among SWNTs. Recently, the theoretical and experimental work predicted that the
infinity length SWNTs are π -bonded aromatic molecules that can be either
semiconducting or metallic depending upon the tubular diameter and helical angle.11,
13
Since the limitation of the CPU time, the quantum chemistry calculations cannot
simulate the real infinite length SWNT model. In 1992, Saito and Hamada used the

3
tight binding model to generate the band structure of SWNTs.14 Almost the same year;
Nakamura et al. predicted the infinite length [5, 5] and [6, 6] armchair SWNT using
DFT calculation.15 Recently, Brus et al. used the DFT calculation to generate the
HOMO-LUMO gap from C20H20 to C210H20 and simulate the infinite length of [5, 5]
armchair SWNT; they concluded that [5, 5] armchair SWNT with infinite length
contains narrow Eg having the metallic property.11 Our previous work used the
semiempirical PM3 method to determine the electronic and optimized structures of
zigzag and armchair SWNT with finite length and various tubular diameters.16
Although lots of calculations have been presented for the finite model (small segment)
of SWNT; their results cannot provide the sufficient data to support the real SWNT
model (infinite length). In order to investigate the infinite system with periodic unit,
the fast multipole method have been proposed.17 Late, the periodic boundary
condition (PBC) model has been presented that could solve the discrete MO model
into the continuous bands.18-20 Thus, one can generate the finite SWNT segment with
DFT calculation with Gaussian type molecular orbital and extend to infinite length
SWNTs model. Scuseria et al. used the DFT-PBC method to optimize the geometrical
structure and to generate the energies of [5,0] zigzag SWNTs.17 But they did not apply
PBC model to the SWNTs with various tubular diameters and to investigate the
diameter effect in these SWNTs.
In the present study, the DFT-PBE and DFT-VSXC methods with 3-21G* and
6-31G* basis sets and PBC function were used to generate the geometry-optimized
structure, band structures and Eg of SWNTs of the zigzag (from [6, 0] to [20, 0]) and
armchair (from [4, 4] to [10, 10]) types with different tubular diameters up to infinite
tubular length. Calculated Eg of SWNTs allow us to predict some physical properties
of SWNT. The calculation results reveal that the [n, 0] zigzag SWNTs have the
oscillation band gap with △n = 3. If n is a multiple of 3 (n = 6, 9, 12, 15), the zigzag

4
SWNTs may have narrow Eg being a metal; otherwise, the SWNT is a moderate-gap
and has a semiconductor. For armchair SWNTs, they have the narrow Eg being
oscillation also.
Calculations
The geometrical structure of SWNT can be described in terms of a role up a
section of a 2-D graphite sheet, which denotes the unit vectors of hexagonal
honeycomb lattice. The roll up vector is denoted by Ch = na1 + ma2, where n and m
are integers; the translation vector T is perpendicular to Ch and direct along the
tubular length of SWNT (Fig. 1). Conveniently, SWNT could be presented by a [n, m]
pair of number; [n, 0] and [n, n] zigzag and armchair types designate SWNTs,
respectively. The carbon atoms of the zigzag SWNT arrange as cis-polyenes with a
single circular of carbon atoms. On the other hand, the armchair SWNT is obtained by
rolling up hexagons as the σv symmetry plane that the carbon atoms arrange as
trans-polyenes with a single circular plane of carbon atoms. For the zigzag type
SWNT, n denotes the number of benzenoids in the circumference of the tube and the
translation axis is the trans-polyene rings along the tubular length. The tubular
diameter (dt) of [n, 0] zigzag SWNT could be determined: dt = [rcos(π/6)]/[sin(π
/2n)]/2, r is the length of C-C bond in the SWNT. For the [n, n] armchair SWNT, dt =
r/(sin2nπ /3). In this paper, we consider the zigzag and armchair SWNTs for
calculations only.
For the DFT-PBC calculation for SWNTs, we start the single layer for the unit
cell and extend along the tubular axis to infinite length. For example the unit cell of [5,
0] SWNT contains 20 carbon atoms for single circumference, thus we used this 20
carbon atoms for the start unit extending to the infinite tube in this calculation. The
optimized structure and band structure of SWNT are generated by using the DFT-PBC

5
model with 6-31G* basis set. The DFT-PBC method is implemented in the Gaussian
03 program package.21
The PBC model in Gaussian 03 package is based on Gaussian type orbitals,[] to
transform GTOs into “Crystalline orbitals”for calculating with periodic boundary
conditions is modulated by a phase factor eikl. Those functions are known as Bloch
functions:[]
ll
lieN
kk
1(1)
where k=(kx,ky,kz) is the reciprocal-lattice vector, which classifies periodic orbitals by
their irreducible representations of the infinite translation group, ψl is an orbital ψ
located in cell l, and i is the imaginary unit. Orbitals belong to different k do not
interact directly with each other and this allows one to solve self-consistent-field (SCF)
equations separately for each k point. The equations are similar as non-periodic case:
Fk Ck = Sk Ck Ek (2)
We note that Eq. (2) is valid both for HF and DFT methods. The exponent in the
Bloch orbital definition (1) introduces complex factors and therefore all matrices in
Eq. (2) are, in general, complex. Matrix elements between periodic orbitals defined in
Eq. (1) can be easily computed from matrix elements for localized GTOs,
l
lil
l
lil eAeAA kk
kk0
0 (3)
In this equation, lA0 is a matrix element of operator A between the Gaussian atomic
orbitals located in the central cell 0 and ψ located in cell l. The Kohn-Sham
Hamiltonian matrix elements (or Fock matrix elements in the HF case), lF 0, include
several contributions:
xclllll EJUTF ,00000 (4)
where lT 0 is the electronic kinetic energy term, lU 0
is the electron-nuclear

6
attraction term, lJ 0 is the electron-electron repulsion term, and xclE ,0
is the
contribution from the DFT exchange-correlation potential. lT 0 and lU 0
terms do
not depend on the density matrix, while lJ 0 and xclE ,0
do. An important feature of
the Kohn-Sham Hamiltonian matrix elements, lF 0, is their exponential decay with
respect to the increasing separation between the and GTOs. Such behavior arises
from the individual decay of the kinetic energy term, the exchange-correlation
potential term, and the exponential decay of the combined electrostatic terms. Overall,
all terms in Eq. (4) are quite similar to analogous terms in molecular calculations. The
electrostatic terms ( lU 0 and lJ 0
) include interactions of a given pair of basis
functions with charge distributions in the system. The number of such interactions is
infinite, and this is indeed different from the molecular case. The infinite sums can be
handled using the Ewald summation techniques[] or by the periodic fast multipole
method.[] The real-space density-matrix elements lP0 required for the construction
of the Coulomb, exchange, and correlation contributions can be obtained by
integrating the complex density kP in reciprocal space,
kkk
k
dePV
P lil
10 (5)
where Vk is the volume of the unit cell in k space. The matrix Pk is obtained from the
orbital coefficients Ck, which are solutions to the eigenvalue Eq. (2). The
transformation described by equation (5) is the only coupling of different k points
during the SCF procedure. In practice, the integration is replaced by a weighted sum
and the reader is referred to Ref. [] for detailed discussions on this topic. The energy
per unit cell can be computed as

7
0
0000
21
l lNRxc
llll EEJUTPE (6)
where Exc is the exchange-correlation energy and ENR is the nuclear repulsion energy.
In the following, triple sums like the one in Eq. (6) will be abbreviated by l. In
order to avoid convergence problems and to maximize accuracy, it is important that
electrostatic terms be grouped together into electronic Ee and nuclear EN terms,
lNR
llN
l
llle
EPUE
PJUE
00
000
21
21
(7)
Once the converged density is available, it is possible to compute gradients of the
total energy with respect to nuclear displacements (forces).
Results and discussions
To investigate the influence of the tubular diameter of SWNTs, the geometric
optimized structures, band structures and Eg of infinite length zigzag and armchair
SWNTs with various tubular diameters were calculated by DFT VSXC method with
PBC function and 6-31G* basis set.
Zigzag type SWNT
According to the theoretical analysis, the zigzag SWNT contains the number of
benzenoid (n) in the circumference of the tube. We used DFT VSXC with PBC
function and 6-31G* basis sets to generate the optimized geometry and the band
structures of zigzag SWNT with various tubular diameters from [6, 0] to [20, 0]. The
calculated Eg of infinite length [n, 0] zigzag SWNT exhibits oscillation properties
with the repeat unit having n = 3m - 1, 3m and 3m + 1, m is integer (Fig. 2). Table 1
shows the calculated Eg of zigzag SWNTs up to infinite tubular length. According to

8
this table, the highest band gap was obtained for the [n (=3m + 1), 0] SWNTs while
the [n (=3m), 0] SWNTs have the lowest in each △n = 3 section. [6, 0], [9, 0], [12, 0],
[15,0] and [18, 0] have 0.0296 eV, 0.1466 eV, 0.0677 eV, 0.0387 eV and 0.0308 eV
calculated Eg, respectively, these are the narrow Eg and have the metallic property. For
the [n (= 3m + 1), 0] zigzag SWNT, [7, 0], [10, 0], [13, 0], [16, 0] and [19, 0] zigzag
SWNTs have 0.2043 eV, 0.7643 eV, 0.6363 eV, 0.5327 eV and 0.4579 eV calculated
Eg, respectively, these values are semiconductor or metal. Thus, we conclude that [n, 0]
SWNT with n is a multiple of 3 may have the metallic property and others have a
semiconductor. We also conclude that [8, 0], [11, 0], [14, 0] and [17, 0] have the
highest band gap in each △n = 3 section. Comparing with other calculation results,
we obtained Eg = 0.2043 eV in the present study for [7, 0] SWNT. While Ito et al.
reported Eg = 0.1304 eV for GGA and 0.1943 eV for LDA. Very early, the calculated
Eg by tight-binding model was reported to be 1.04 eV for this SWNT. Our calculation
is very closed to that of LDA results. Hamada et al. used the tight-binding model to
determine the Eg = 0.697 eV for [13, 0] SWNT Our calculation for this SWNT is
0.6363 eV that is very closed to previous calculation data. Although tight-bind and
LDA methods can determine the larger scale carbon tube than other quantum
chemistry methods, they restrict the C-C bond length in the same distance for the
whole system. In present study, the DFT-PBC model optimized the tube structure up
to two different C-C bond lengths that may more close to the real carbon tube
structure than other methods.
The DFT-PBC calculated geometrical parameters and tubular diameter for the
infinite length zigzag SWNTs are presented in Table 2. The calculated tubular
diameters increased from 4.860 A ([6, 0] SWNT) to 15.796 A ([20, 0] SWNT); there
are nearly 0.8 A tubular diameter difference between any two neighboring SWNT.
Experimentally, the tubular diameters of SWNTs can vary ranging from 10 Å to 16 Å

9
with a peak maximum at 12 Å. We may predict that the most possible product is [15,
0] or [16, 0] SWNT in experiment.
Although several papers have been presented by using tight-bind model to
investigate the large scale of SWNTs, they assume that SWNT has the same C-C bond
lengths in the whole nano-tube system. In the present study, the C-C bond length of
SWNT has been optimized to two different magnitudes. According to Table 2, there
are two different calculated C-C bond lengths for [6, 0] SWNT 1.450 A and 1.416 A,
respectively. Up to [13, 0] SWNT, the two C-C bond lengths of the optimized
structure are very close (1.431 A and 1.428 A), then, these lengths keep the same
magnitude from [14, 0] to [20, 0] SWNTs. Concludly, increasing the tubular diameter
of infinite zigzag SWNTs may increase the π electron delocalization; the tubular
diameter may not affect the C-C bond length after [13, 0] SWNT. Thus, these SWNTs
are the nearly rolled up graphite sheet.
Fig. 3 shows the sketch of HOMO and LUMO with double layer unit cell of the
infinite tubular length tube of [6, 0] to [8, 0] zigzag SWNTs. Since there are △n = 3
oscillation period in the Eg of zigzag SWNT, the frontier orbital may have this
oscillation property, thus, the HOMO and LUMO of [6, 0] and [9, 0], [7, 0] and [10,
0], [8, 0] and [11,0] have very similar sketches, respectively. Due to the above
consideration, we determined the HOMO and LUMO for [6, 0] to [8, 0] zigzag
SWNTs only (Fig. 4). In this study, we simulate carbon nano-tube with infinite tubular
length, and ignored the influence of tubular length. So that, the influence of tubular
diameter of infinite length zigzag SWNT should be viewed as the armchair SWNT. Li
et al. were used DFT and the semiempirical PM3 computational techniques to
generate the electronic wave functions of same shortened [5, 5] and [6, 6] armchair
SWNTs.22 Our sketch of HOMO and LUMO are the same as those of Li’s results.
For the electronic band structure, we have systematically studied. The band

10
structures of [9. 0], [10, 0] and [11, 0] zigzag SWNTS are shown in Fig. 5. Bands
higher than EF (the Fermi level) are antibonding π* bands; π and σ (sp2)
bonding bands are located lower than the EF. Fig. 5 (a) shows the band structure of [9,
0] SWNT, the calculated HOCO and LUCO are –3.9825 eV and –3.8360 eV at Γ
point, respectively, its energy gap is 0.1466 eV. The calculated HOCO and LUCO
bands for [10, 0] SWNT are–4.3603 eV and–3.5960 eV and exhibit an energy gap is
0.7643 eV. The X point in these structures has a wave number near π/a, with a the
graphite lattice constant. Two bands stick together at the X point due to the screw
symmetry.
Armchair type SWNT
Recently, the electronic and geometrical structures of armchair of SWNTs using
computational methods have been proposed. The semiempirical calculation
mentioned that the HOMO/LUMO and Eg of the finite tubular length of armchair type
SWNTs have the oscillation properties with the repeat unit having 3m-1, 3m and 3m +
1 models (m is integer) in the carbon section along their latitudes. In this study, we
simulate the infinite tubular length of armchair type SWNTs, thus their tubular length
influence shall be ignored. The DFT-PBC calculated Eg and related geometrical
parameters of selected armchair SWNTs are shown in Tables 2 and 3, respectively.
Particularly, the calculated tubular diameters are from 5.545 A ([4, 4]) to 13.701 ([10,
10]), the distance between any two neighboring armchair SWNTs are not consistent in
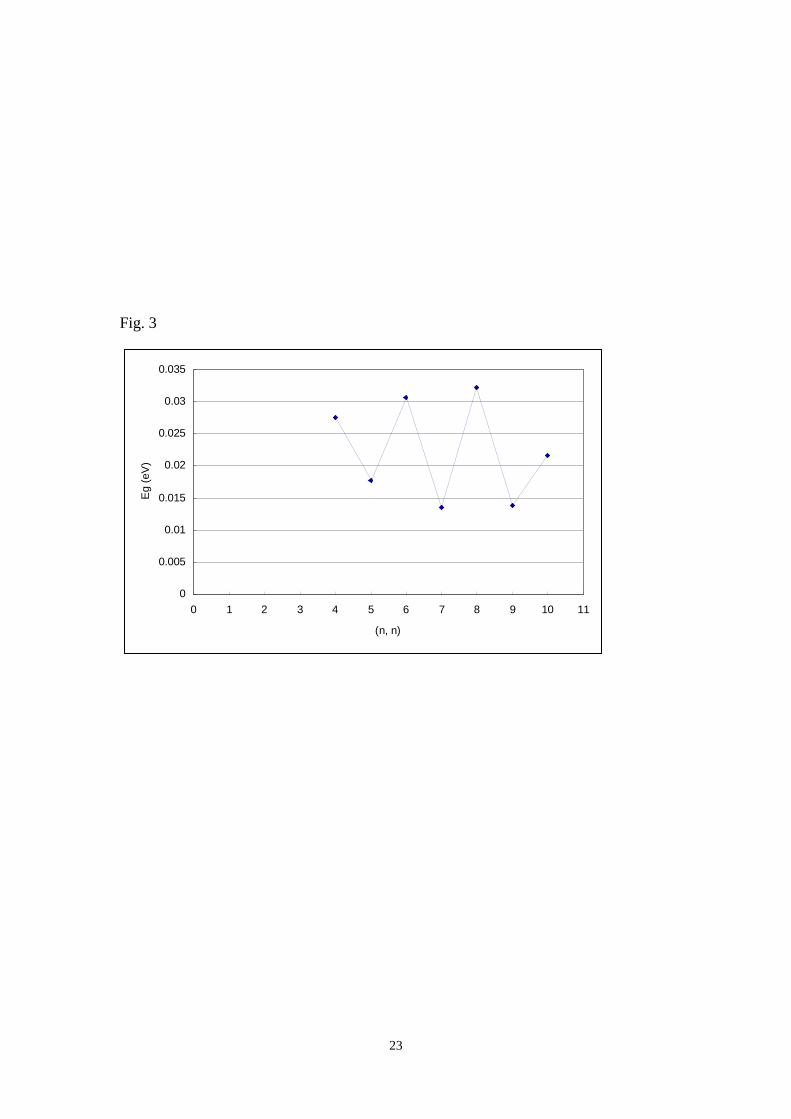
this series. All of the calculated Eg are between 0.01 to 0.03 eV for these SWNTs and
they should be a metal. Calculated Eg for [n, n] armchair SWNTs exhibit oscillations
with Δn = 2 (Fig. 3). This calculation result metioned that the calculated Eg of the
tube with even n is lower than that of tube with odd n. For example, the difference in
the calculated Eg for [5, 5] and [6, 6], [6,6] and [7,7] armchair SWNTs are 0.023 eV

11
and –0.017 eV, respectively. Recently, Nakamura et al. have presented the structures
and aromaticity of [5, 5] and [6, 6] armchair SWNTs by used DFT method. They
predict that [5, 5] zigzag SWNT possess higher HOMO and lower LUMO than that of
[6, 6] SWNT. Thus, [5, 5] SWNT contains higher Eg than that of [6, 6] SWNT. Our
calculations have the same trend as that of Nakamura’s data.
Table 4 shows the calculated optimized geometrical parameters of armchair
SWNT by DFT-PBC calculation. According to the calculation results, there are two
different C-C bond lengths in armchair SWNTs that is similar to that of zigzag
SWNTs. For [4, 4] armchair SWNT, it has 1.436 A and 1.432 A calculated C-C bond
length. Apparently, the C-C bond length decreases to 1.428 A for [10, 10] SWNT, that
is the only C-C bond length in this SWNT. This calculation results mentioned that the
C-C bond length decrease while the tubular diameter increase. Whereas, [n, n]
armchair SWNT. The structures and aromaticity of finite [5, 5] and [6, 6] armchair
SWNT have been generated by Nakamura et al.
The calculated sketch of HOMO and LUMO with double layer of the infinite
tubular length of [4, 4] and [5, 5] armchair SWNTs are shown in Fig. 4. The band
structures of [4, 4] and [5, 5] armchair SWNTs are presented in Fig. 6, HOCO and
LUCO are crossing in the K point, thus, both of [4, 4] and [5, 5] armchair SWNTs are
metal. This DFT-PBC calculation result is consistent with previous tight-binding
calculation.
Conclusion
In this study, DFT-PBC calculations were used to investigate the geometrical and
electronic structures of zigzag and armchair types of SWNT up to infinite tubular
length. The calculations reveal that the Eg of [n, 0] zigzag SWNTs have an oscillation
with △n = 3. The [n, 0] zigzag SWNTs with n is multiple by 3 that have narrow Eg

12
and have a metal, otherwise they have moderate Eg being the semiconducting bulk
materials. According to the calculated Eg for the [n, n] armchair SWNT, they have
narrow Eg and contain metal property. The calculated Eg in these SWNTs also have
oscillation with △n = 2, although there are very small Eg difference. For the
optimized structure of SWNTs, there are two different calculated C-C bond length,
those are decreasing with increasing the tubular diameter. This calculation provides
important information on the properties of SWNT needed for the design of new
nano-electronic devices.
Acknowledgment
We thank the National Science Council of ROC for supporting.

13
References
1. P. M. Aiayan, O. Stephan, C. Colliex, D. Trauth, Science, 265 (1994), 1212.
2. Y. Saito, K. Hamaguchi, K. Hata, K. Uchida, Y. Tasaka, F. Ikazaki, M. Yumura, A.
Kasuya, Y. Nishina, Nature, 389 (1997), 554.
3. W. A. de Heer, A. Chatelain, D. Ugarte, Science, 270 (1995), 1179.
4. P. G. Collins, A. Zettl, H. Bando, A. Theas, R. E. Smalley, Science, 278 (1997),
100.
5. M. B. Nardeli, B. I. Yokobson, J. Bernholc, Phys. Rev., 1357 (1998), R4277.
6. H. W. Kroto, J. R. Heaith, S. C. O’Brien, R. F. Curl, R. F. Smalley, Nature, 318
(1985), 162.
7. S. Iijima, Nature, 354 (1991), 56.
8. S. Iijima, T. Ichihashi, Nature, 363 (1993), 603.
9. D. S. Bethune, C. H. Kiang, M. S. de Vries, G. Gorman, R. Savoy, J. Vazquez. R.
Beyers, Nature, 363 (1993), 605
10. N. Hamada, S. Sawada and A. Oshiyama, Phys. Rev. Lett., 68 (1992), 1579.
11. Z. Zhou, M. Steigerwaid, M. Hybertsen, L. Brus and R. A. Friesner, J. Am. Chem.
Soc., 126 (2004), 3597.
12. T. Ito, K. Nishidate, Mamoru Baba and M. Hasegawa, Surface Science, 514
(2002), 222.
13. V. Barone, J. E. Peralta, M. Wert, J. Heyd and G. E. Scuseria, Nano Lett., 5
(2005), 1621.
14. (a). R. Saito, M. Fujita, G. Dresselhaus, M. S. Dresselhaus, Phy. Rev. B46 (1992),
1894. (b). N. Hamada, S. Sawada, A. Oshiyama, Phys. Rev. Lett., 68 (1993),
1579.
15. Y. Matsuo, K. Tahara and E. Nkamura, Organic Lett. 5 (2003), 3181.
16. B. C. Wang, H. W. Wang, I. C. Lin, Y. S. Lin, Y. M. Chou and H. L. Chiu, J.

14
Chienese Chemical Society, 50 (2003), 939.
17. K. N. Kudin and G. Scuseria, Chemical Physics Lett., 289 (1998), 611.
18. K. N. Kudin and G. Scuseria, Phys. Review B, 61 (2000), 16440.
19. S. Erkoc , International J. Mode. Phys., 11 (2000), 547.
20. H Cao, J. Ma, G. Zhang and Y. Jiang, Macromolecules, 38 (2005), 1123.
21. Gaussian
22. J. Li, Y. Zhang and M. Zhang, Chemical Physics Letters, 368 (20020, 328

15
Fig. 1
[1,0,0]
[2,1,0]Zigzag SWNT
Armchair SWNT
Chiral SWNT
m
n
m
n

16
Table 1 Calculated HOCO, LUCO and Eg of [n, 0] zigzag SWNTs by DFT-PBCmethod
[n, 0] HOCO LUCO Eg
n = 6 -4.0890 -4.0594 0.02967 -4.4539 -4.2496 0.20438 -4.5062 -3.8665 0.63979 -3.9826 -3.8360 0.1466
10 -4.3603 -3.5960 0.764311 -4.3824 -3.4191 0.963312 -3.9756 -3.9078 0.067713 -4.3028 -3.6665 0.636314 -4.3200 -3.5770 0.742915 -3.9893 -3.9505 0.038716 -4.2664 -3.7336 0.532717 -4.2704 -3.6668 0.603518 -4.0081 -3.9773 0.030819 -4.2333 -3.7753 0.457920 -4.2347 -3.7251 0.5096

17
Table 2 Calculated geometrical parameters for [n, 0] zigzag SWNTs
[n, 0] Diameter (Å) C1-C2 (Å) C2-C3 (Å)
n = 6 4.860 1.450 1.4167 5.662 1.441 1.4248 6.395 1.440 1.4229 7.210 1.437 1.425
10 7.947 1.435 1.42711 8.755 1.435 1.42512 9.514 1.433 1.42613 10.344 1.432 1.42814 11.083 1.432 1.42715 11.907 1.432 1.42716 12.649 1.431 1.42817 13.453 1.431 1.42718 14.222 1.431 1.42819 15.022 1.430 1.42820 15.796 1.431 1.428
tube axis

18
Table 3 Calculated HOCO, LUCO and Eg of [n, n] armchair SWNTs by DFT-PBCmethod
[n, n] HOCO LUCO Eg
n = 4 -3.9262 -3.8985 0.02765 -3.9396 -3.9217 0.01786 -3.9456 -3.9150 0.03067 -3.9545 -3.9409 0.0136
8 -3.9849 -3.9527 0.0322
9 -3.9707 -3.9569 0.013810 -3.9933 -3.9716 0.0216

19
Table 4 Calculated geometrical parameters for [n, n] armchair SWNTs
[n, n] Diameter (Å) C1-C2 (Å) C2-C3 (Å)
n = 4 5.545 1.436 1.4325-5 6.805 1.434 1.4306-6 8.249 1.432 1.4297-7 9.544 1.431 1.4298-8 10.962 1.430 1.4289-9 12.279 1.429 1.428
10-10 13.671 1.428 1.428
tube axis

20
tube
Figure captionsFig. 1 Hexagonal network of a single graphite sheet for zigzag, armchair and chiralSWNTFig. 2 Calculated Eg with oscillation property for [n, 0] zigzag SWNTs by DFT-PBCmethodFig. 3 Calculated Eg with oscillation property for [n, n] armchair SWNTs byDFT-PBC methodFig. 4 Sketch of HOMO and LUMO for [6, 0], [7, 0] and [8, 0] zigzag SWNTsFig. 5 Sketch of HOMO and LUMO for [4, 4] and [5, 5] armchair SWNTsFig. 6 Band structure of [9, 0], [10, 0] and [11, 0] zigzag SWNTsFig. 7 Band structure of [4, 4] and [5, 5] armchair SWNTs

21
Fig . 1
[1,0,0]
[2,1,0]Zigzag SWNT
Armchair SWNT
Chiral SWNT
m
n
m
n

22
Fig. 2
0
0.2
0.4
0.6
0.8
1
1.2
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
(n, 0)
Eg
(eV
)
23
Fig. 3
0
0.005
0.01
0.015
0.02
0.025
0.03
0.035
0 1 2 3 4 5 6 7 8 9 10 11
(n, n)
Eg
(eV
)

24
Fig. 4
HOMO LUMO
(6,0)
(7,0)

25
(8,0)
Fig. 5
HOMO LUMO
(4,4)

26
(5,5)
Fig. 6
-9-8
-7-6
-5-4
-3-2
-10
0 4 8 12 16 20 24 28 32 36 40 44 48 52 56 60 64 68 72 76
k-point
E(e
V)
HOCO-4 HOCO-3 HOCO-2 HOCO-1 HOCO
LUCO LUCO+1 LUCO+2 LUCO+3 LUCO+4

27
(9,0) SWNT
-8
-7
-6
-5
-4
-3
-2
-1
0
0 4 8 12 16 20 24 28 32 36 40 44 48 52 56 60 64 68 72 76
k-point
E(e
V)
HOCO-4 HOCO-3 HOCO-2 HOCO-1 HOCO
LUCO LUCO+1 LUCO+2 LUCO+3 LUCO+4
(10,0) SWNT
-8-7-6-5-4-3-2-10
0 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75
k-point
E(e
V)
HOCO-4 HOCO-3 HOCO-2 HOCO-1 HOCOLUCO LUCO+1 LUCO+2 LUCO+3 LUCO+4
(11,0) SWNTFig.5

28
-8
-7
-6
-5
-4
-3
-2
-1
0
0 3 6 9 12 15 18 21 24 27 30 33 36 39 42 45 48 51 54 57 60 63 66
k-point
E(e
V)
HOCO-4 HOCO-3 HOCO-2 HOCO-1 HOCO LUCO
LUCO+1 LUCO+2 LUCO+3 LUCO+4
(4,4) SWNT
-8
-7
-6
-5
-4
-3
-2
-1
0
0 3 6 9 12 15 18 21 24 27 30 33 36 39 42 45 48 51 54 57 60 63 66
k-point
E(e
V)
HOCO-4 HOCO-3 HOCO-2 HOCO-1 HOCO LUCO
LUCO+1 LUCO+2 LUCO+3 LUCO+4
(5,5) SWNT

![Double-walled carbon nanotubes: synthesis, structural ...077-088]-01.pdf · Double-walled carbon nanotubes: synthesis, structural characterization, and ... are seamless cylindrical](https://img.dokumen.tips/doc/110x75/5aa2b5537f8b9ac67a8d717c/double-walled-carbon-nanotubes-synthesis-structural-077-088-01pdfdouble-walled.jpg)



























