Embed Size (px)
Citation preview

BIOSIMILARSTRENDS IN2016
REPORTOUR NEXT CHAPTER IN HEALTHCARE

TABLE OF CONTENTS
INTRODUCTION………………………………….………….01What are Biologics and Biosimilars?, Biosimilars are not Generics, Message from Amgen, Message from Editorial Council
BIOSIMILARS LANDSCAPE………………………..….05U.S. Biosimilar Timeline, International Experience
UNCOVERING THE UNKNOWNS…………….……..13U.S. Approval Pathway, Interchangeability & Substitution, Naming, Coding & Payment Guidance
GAME CHANGERS………………………………………....20Coverage Policies, Provider & Patient Education, Logistics, Manufacturer Reliability & Contracting, Reimbursement, Safety & Pharmacovigilance
LOOKING AHEAD……………………………………….....47
GLOSSARY………………………………………………….....50
REFERENCES……………………………………………......54

1
INTRODUCTION

2
WHAT ARE BIOLOGICS AND BIOSIMILARS?
Biologics are therapeutic proteins, such as monoclonal antibodies (mAbs), that are manufactured from natural sources, including living “host” systems, such as human and animal cells, yeast, and bacteria.1 These living systems are genetically engineered to generate proteins, typically by inserting the genetic sequence, encoding a therapeutic protein, into the DNA of the host.2 Because they are small microscopic organisms, the hosts are typically further engineered to increase the amount of protein they produce, ensuring that therapeutically useful quantities of protein can be cultivated.2
“Biosimilar” is a U.S. Food and Drug Administration (FDA) designation used to describe a biological product that is
“highly similar” to an approved biologic (known as a reference product) already being used to treat patients.1 To be classified as a biosimilar, the protein must have no clinically meaningful differences in terms of safety and effectiveness from the reference product; only minor differences in clinically inactive components are allowed.3
BIOSIMILARS ARE NOT GENERICS
Biosimilars are reverse-engineered approximations of marketed biologics, meaning that biosimilars are highly similar to their reference products.2 However, due to the incredible complexity of proteins and the fact that they are literally grown in living cells, biosimilars are also unique and can never be identical to the reference product.2 This is in contrast to traditional small molecule medicines that can be directly copied as generic compounds in a laboratory.2 Therefore, it can be clearly stated that biosimilars are not generics and should be considered differently.
For regulatory and clinical purposes, the inevitable differences between a biosimilar and the reference product will vary based on a number of factors, including the manufacturing process itself, as well as the ability of a biosimilar manufacturer to reverse engineer, and scientifically document, a highly similar biologic to the reference product.4 As a result, no two biosimilars are identical to each other or to the reference product, although they share many attributes.5
Because biologics and their reference products are never identical, the Biologics Price Competition and Innovation Act was passed in 2010 to create a separate approval pathway for biosimilars.3,7 Unlike the abbreviated pathway for small molecules,8 the FDA requires an additional level of evidence for biosimilars than for generics.9 To be considered a biosimilar, it must be shown that any subtle structural or functional differences between the candidate protein and the reference product do not impact the clinical profile.9 Given all these differences between biosimilars and generics, it is no wonder that payers, prescribers, and other decision makers have additional considerations when evaluating a biosimilar.
INTRODUCTION

3
MESSAGE FROM AMGEN
Dear Colleagues,
This year promises to bring greater clarity to the biosimilar landscape. In 2015, we witnessed some significant biosimilar milestones, including the finalization of some of the long-standing draft biosimilar guidances from the U.S. Food and Drug Administration, as well as the approval and launch of the first biosimilar in the U.S. With the evolution of this marketplace, education and informed choice are important components of biosimilar adoption and utilization. Payers, employers, patients, pharmacists, physicians, and other key stakeholders all play a key role as the biosimilars market develops. As a pioneer in the development and commercialization of biologics medicines, Amgen has the opportunity to support this education.
At Amgen, we know that there are still unknowns in the market and there is much to learn from one other as we take this next step in biologic medicine together.
We have taken that approach for this year’s report. Working alongside an expert Editorial Council comprised of leading medical and pharmacy directors, as well as employers representing a broad mix of the U.S. reimbursement community, we are proud to share the third edition of the Trends in Biosimilars Report. This report is intended to give stakeholders a guide to the latest topics, trends, and issues pertinent to biosimilar introduction and adoption in the U.S.
We look forward to working together with you, and we hope the 2016 Trends in Biosimilars Report is a valuable resource for you and your colleagues as you manage the ongoing introduction, adoption, and management of biosimilars.
DUANE H. BARNES
Vice President & General ManagerU.S. Value and Access, Amgen
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
GA
ME
CH
AN
GE
RS
LOO
KIN
G A
HE
AD
UN
CO
VE
RIN
G T
HE
U
NK
NO
WN
S

4
INTRODUCTION
This report offers insights from our Editorial Council, comprised of highly experienced medical and pharmacy directors representing the broad mix of managed care organizations throughout the U.S., as well as employer and benefit design consultants.
ROBERT ADAMSON, PharmD
Chief Pharmacy Officer, Barnabas Health
MICHAEL BOSKELLO
Clinical Strategy, Viking Healthcare Solutions
CHERYL LARSON
Vice President, Midwest Business Group on Health
SANDRA MORRIS, RN, MSN, CHC
Former U.S. Benefits Design Senior Manager,
Procter & Gamble Company
PETER NEUMANN, ScD
Director, Evaluation of Value and Risk in Health,
Tufts Medical Center
ELAN RUBINSTEIN, PharmD, MPH
Principal Consultant, EB Rubinstein Associates
KENNETH L. SCHAECHER, MD
Medical Director, SelectHealth
DEAR COLLEAGUES,
The first commercial launch of a biosimilar in the U.S. represented a significant new development in the biologic market, and it is expected that biosimilars will play an increasingly prominent role in our value-based healthcare system. To more fully leverage this trend, we believe that all stakeholders, including payers and healthcare providers, will benefit from a better understanding of regulatory, policy, and manufacturing considerations for biosimilar introduction and adoption. Payers and healthcare providers alike will likely be making specific evaluations and critical decisions around new biosimilar entrants, in order to determine whether biosimilars are aligned with a value-based healthcare model’s focus on managing costs, driving quality care, and achieving intended patient outcomes. The community of health plans, hospital systems, Integrated Delivery Networks (IDNs), pharmacy benefit managers (PBMs), employers, and government officials, who determine reimbursement is already working to evaluate biosimilar therapies, their benefits and their emerging role in formulary structures and medical coverage policies. Providers and other stakeholders are also assessing the financial and organizational implications of biosimilars adoption and acceptance within their institutions, including the educational and new structural costs (i.e., electronic medical record programming) associated with biosimilar implementation.
We are proud to contribute to the 2016 Trends in Biosimilars Report, which provides timely and useful information on some of the crucial biosimilar issues that the broader healthcare community is grappling with today. It is our hope that this report will serve as a unique resource for all of those seeking a more complete understanding of these issues, and to this end we have also included varying stakeholder perspectives to encourage informed dialogue within the healthcare community. The following pages first provide an overview of biologics and biosimilars, and then delve more deeply into a market overview of biosimilars (including an international perspective), pathways for approval, payment guidance, and potential drivers of success for biosimilars in the current market.
Editorial Council members are participating
independently and their views may not reflect the
interests of their respective companies.
MESSAGE FROM THE EDITORIAL COUNCIL

5
BIOSIMILARS LANDSCAPE

6
BIOSIMILARS LANDSCAPEU.S. TIMELINE
2008 2009 2010 2011 2012 2013 2014 2015
No guidelines on biosimilarsin the U.S.3
December 2009 Implementation of the Biologics Price Competition and Innovation Act3
March 2010Pathway passes as part of the A�ordable Care Act3
February 2012 FDA issues draft guidance to assist industry
with development of biosimilars10
December 20143 active FDA filings for biosimilars pending
review as of December 31, 201411,12,13
July 2014FDA accepts application for first biosimilar in the U.S.11
March 2015FDA approves the first biosimilar in the U.S.14
CMS issues guidance documents on the treatment of biosimilar products under Medicare Part B, Medicare Part D, and Medicaid Drug Rebate Program15,16,17
April 2015FDA issues three final biosimilars guidance documents18
July 2015CMS issues proposed payment provisions for biosimilars under Medicare Part B19
August 2015FDA issues draft guidance on biosimilar naming20
September 2015Launch of first biosimilar in the U.S.21
October 2015CMS finalizes provisions for biosimilars22
November 2015FDA issues final guidance on formal meetings
between FDA and biosimilar product applicants18
December 20156 active FDA filings for biosimilars pending review
as of December 31, 201524-29

7
2008 2009 2010 2011 2012 2013 2014 2015
No guidelines on biosimilarsin the U.S.3
December 2009 Implementation of the Biologics Price Competition and Innovation Act3
March 2010Pathway passes as part of the A�ordable Care Act3
February 2012 FDA issues draft guidance to assist industry
with development of biosimilars10
December 20143 active FDA filings for biosimilars pending
review as of December 31, 201411,12,13
July 2014FDA accepts application for first biosimilar in the U.S.11
March 2015FDA approves the first biosimilar in the U.S.14
CMS issues guidance documents on the treatment of biosimilar products under Medicare Part B, Medicare Part D, and Medicaid Drug Rebate Program15,16,17
April 2015FDA issues three final biosimilars guidance documents18
July 2015CMS issues proposed payment provisions for biosimilars under Medicare Part B19
August 2015FDA issues draft guidance on biosimilar naming20
September 2015Launch of first biosimilar in the U.S.21
October 2015CMS finalizes provisions for biosimilars22
November 2015FDA issues final guidance on formal meetings
between FDA and biosimilar product applicants18
December 20156 active FDA filings for biosimilars pending review
as of December 31, 201524-29
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
GA
ME
CH
AN
GE
RS
LOO
KIN
G A
HE
AD
UN
CO
VE
RIN
G T
HE
U
NK
NO
WN
S

8
BIOSIMILARS LANDSCAPEINTERNATIONAL EXPERIENCE
Biosimilars, also referred to as follow-on biologics, have limited exposure to date in the U.S. compared with the rest of the world, given that the first U.S. biosimilar was approved in March 2015 and launched in September that year.14, 21 Ten years earlier, the European Medicines Agency (EMA) was the frontrunner in biosimilar regulation after providing an overarching regulatory approval framework for biosimilars30 and subsequently approving its first biosimilar the following year in 2006.31 Since then, the European Union (EU) has led the world in biosimilar development, having approved 22 different biosimilar products as of March 2016.31 In addition to the U.S. and EU, other countries with leading global pharmaceutical markets, such as, Japan, Australia and Canada have followed suit, even adapting the framework provided by the EMA for their respective markets: 11 biosimilars have been approved in Australia since 2010,32 8 biosimilars have been approved in Japan33 and 3 in Canada since 2009.34 Moreover, as many of the leading biologic products face patent expiration in mature markets around the world, it is estimated that the global biosimilar market may reach as high as $20 billion by 2020.35
Despite this upward trend in approval of biosimilars, uptake of these agents has varied across national markets.36 Given the variability of healthcare system market structures outside the U.S., translating these experiences to the U.S. may present its own set of challenges. Nonetheless, key observations from these markets may provide some insights on how the U.S. biosimilars market will evolve.
EUROPEAN UNION
The increased availability of biosimilars following the expiry of patents or other forms of market exclusivity has helped to advance the progress of biosimilar uptake across EU countries, although considerable variances in uptake by geography and therapeutic area persist.37 In 2013, the range of market penetration for biosimilar human growth hormone (HGH) products ranged from a low of 2 percent in Norway to a high of 99 percent in Poland.37 Erythropoietin (EPO) biosimilar penetration has varied in a slightly narrower range, from 1 percent in Croatia to 62 percent in Bulgaria. Granulocyte-colony stimulating factor (G-CSF) biosimilar penetration is lowest in Belgium, with 2 percent market share, and highest in Croatia, Czech Republic, Hungary and Romania, capturing nearly 100 percent share of the accessible market in these countries.37 This speaks to the heterogeneity of the EU market and further explains why the EU experience may not necessarily offer comparable insights on the trajectory of the U.S. market.
In the EU countries with the largest economies, often referred to as the EU5, uptake has increased over time, although it remains measured and shows significant variations (see Figure 1).37, 38
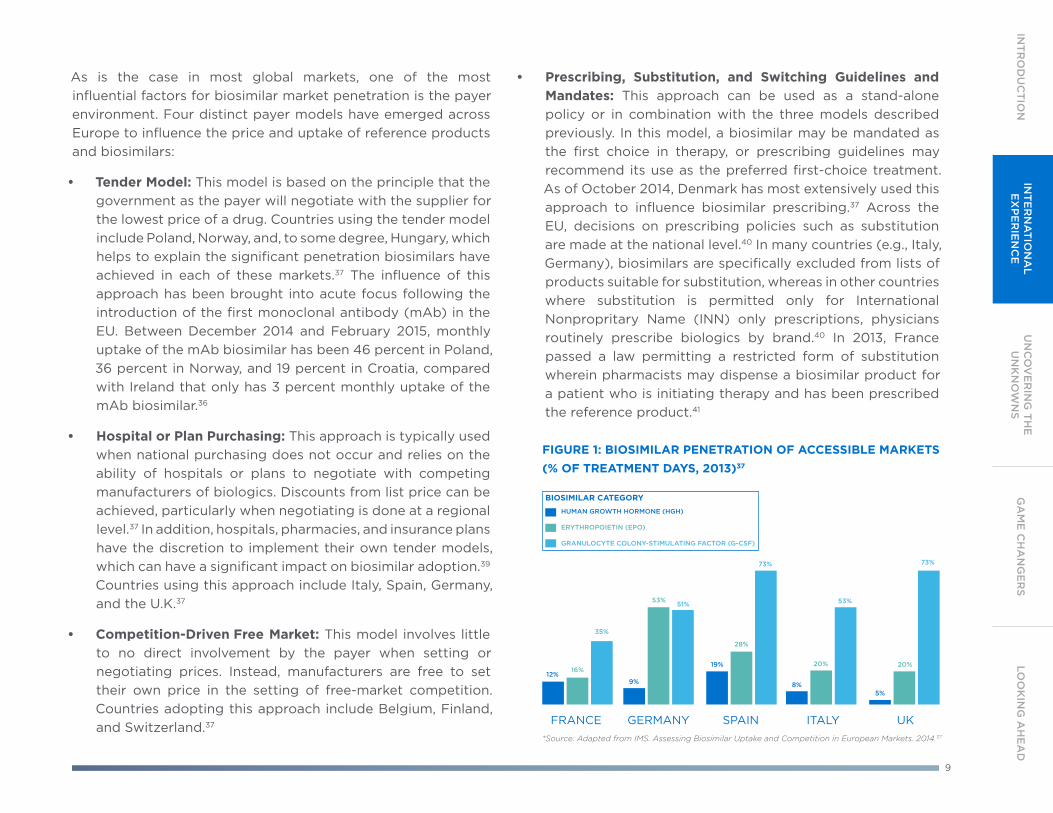
9
As is the case in most global markets, one of the most influential factors for biosimilar market penetration is the payer environment. Four distinct payer models have emerged across Europe to influence the price and uptake of reference products and biosimilars:
• Tender Model: This model is based on the principle that the government as the payer will negotiate with the supplier for the lowest price of a drug. Countries using the tender model include Poland, Norway, and, to some degree, Hungary, which helps to explain the significant penetration biosimilars have achieved in each of these markets.37 The influence of this approach has been brought into acute focus following the introduction of the first monoclonal antibody (mAb) in the EU. Between December 2014 and February 2015, monthly uptake of the mAb biosimilar has been 46 percent in Poland, 36 percent in Norway, and 19 percent in Croatia, compared with Ireland that only has 3 percent monthly uptake of the mAb biosimilar.36
• Hospital or Plan Purchasing: This approach is typically used when national purchasing does not occur and relies on the ability of hospitals or plans to negotiate with competing manufacturers of biologics. Discounts from list price can be achieved, particularly when negotiating is done at a regional level.37 In addition, hospitals, pharmacies, and insurance plans have the discretion to implement their own tender models, which can have a significant impact on biosimilar adoption.39 Countries using this approach include Italy, Spain, Germany, and the U.K.37
• Competition-Driven Free Market: This model involves little to no direct involvement by the payer when setting or negotiating prices. Instead, manufacturers are free to set their own price in the setting of free-market competition. Countries adopting this approach include Belgium, Finland, and Switzerland.37
• Prescribing, Substitution, and Switching Guidelines and Mandates: This approach can be used as a stand-alone policy or in combination with the three models described previously. In this model, a biosimilar may be mandated as the first choice in therapy, or prescribing guidelines may recommend its use as the preferred first-choice treatment. As of October 2014, Denmark has most extensively used this approach to influence biosimilar prescribing.37 Across the EU, decisions on prescribing policies such as substitution are made at the national level.40 In many countries (e.g., Italy, Germany), biosimilars are specifically excluded from lists of products suitable for substitution, whereas in other countries where substitution is permitted only for International Nonpropritary Name (INN) only prescriptions, physicians routinely prescribe biologics by brand.40 In 2013, France passed a law permitting a restricted form of substitution wherein pharmacists may dispense a biosimilar product for a patient who is initiating therapy and has been prescribed the reference product.41
FIGURE 1: BIOSIMILAR PENETRATION OF ACCESSIBLE MARKETS (% OF TREATMENT DAYS, 2013)37
BIOSIMILAR CATEGORY
*Source: Adapted from IMS. Assessing Biosimilar Uptake and Competition in European Markets. 2014.37
HUMAN GROWTH HORMONE (HGH)
ERYTHROPOIETIN (EPO)
GRANULOCYTE COLONY-STIMULATING FACTOR (G-CSF)
12%9%
19%
8%5%
16%
53%
28%
20% 20%
35%
51%
73%
53%
73%
INT
RO
DU
CT
ION
IN
TE
RN
AT
ION
AL
EX
PE
RIE
NC
EG
AM
E C
HA
NG
ER
SLO
OK
ING
AH
EA
DU
NC
OV
ER
ING
TH
E
UN
KN
OW
NS

10
BIOSIMILARS LANDSCAPEAUSTRALIA
Australia has modeled its approach to biosimilars largely from the experience in the EU,32 including policies and guidelines to facilitate the adoption of biosimilars.42 The Australian health authority recently signed legislation that gives the payer body exclusive authority to determine substitution of biosimilars at the pharmacy level.43
The Australian government has publicly pledged to invest $20 million Australian dollars in launching a national education campaign to foster biosimilar adoption and uptake.43
In August 2015, Australia’s Therapeutic Goods Administration (TGA) approved the country’s first biosimilar mAb, infliximab.32
The following December, this biosimilar mAb was officially added to the Pharmaceutical Benefits Scheme (PBS) with the recommendation that the biosimilar be substituted for the reference product at the pharmacy level.45 This means that pharmacists may offer patients the option of receiving the reference product or the lower, cost-saving biosimilar. There is no automatic or uncontrolled substitution of PBS medicines in clinical practice in Australia, but a pharmacist can substitute a medicine if: 46
• The prescriber has not ticked the ‘brand substitution not permitted’ box; and
• The pharmacist consults the patient before any substitution occurs; and
• The patient agrees to the substitution.
CANADA
Canada independently developed a regulatory framework in 2010 that, similar to the EMA framework, requires head-to-head analytical and clinical comparisons of the biosimilar and its reference product.30 Biosimilars are still in early development in Canada, and providers have been slow to adopt biosimilars, perhaps because of a relative unfamiliarity with the products compared with providers in more established markets.47 However, the Canadian healthcare market is undergoing significant change, with several trends that may affect the future of the country’s biosimilar market.47 Concurrent with the first introduction of biosimilars in Canada in 2009, a number of factors that may influence adoption of biosimilars include: 47
• Transition to a pan-Canadian process for reimbursement price negotiations;
• Generally decreased access to physicians, accompanied by the increasing importance of healthcare provider groups;
• Movement toward greater reliance on health economic evidence;
• Reevaluation of generics pricing regulations; and
• Resistance to high-cost biologics and drugs for rare diseases.
Within this landscape, provincial payers are emerging as critical decision-makers in biosimilar uptake, and are evaluating new therapies based on their clinical and economic value.47 Importantly, however, since biosimilars are not considered to be therapeutically or pharmaceutically equivalent to the reference biologic drug, Health Canada does not support substitution of biosimilars.47,48

11
2008
2008
2008
2008
2009
2009
2009
2009
2010
2010
2010
2010
BIOSIMILARS *DOES NOT INCLUDE LUXEMBOURGINNOVATOR
2011
2011
2011
2011
2012
2012
2012
2012
2013
2013
2013
2013
2014
2014
2014
2014
ESAs
ESAs
G-CSFs
G-CSFs
JAP
AN
EU
27*
% T
RE
AT
ME
NT
DA
YS
% T
RE
AT
ME
NT
DA
YS
PERCENTAGE OF TREATMENT DAYS EXAMPLE
3% 6% 9% 15%
19% 25% 29%
1% 8%
21% 36% 50% 60% 67%
0% 0% 7%
36% 57% 69% 75%
0% 0% 0% 0% 0% 3% 15%
FIGURE 2. COMPARISON OF INNOVATOR (REFERENCE PRODUCTS) AND BIOSIMILAR USE IN THE EU27* AND JAPAN51
Source. Data on File, Amgen. IMS Health MIDAS 2014.
JAPAN
Japan has a clearly established approval pathway for biosimilars that is based on EU processes and was published in 2009 by its regulatory approval agency (Pharmaceuticals and Medical Devices Agency) and its regulatory body (Ministry for Health, Labour and Welfare).30, 33 Japan’s first biosimilar was approved in 2009 and seven subsequent biosimilars were approved as of
February 2016.33 Figure 2 shows how biosimilar use in Japan compares with that in the EU for erythropoiesis-stimulating agents (ESAs) and granulocyte-colony stimulating factors (G-CSFs). Japan, along with the U.S. and EU, holds a large portion of the healthcare cost burden as it comprises the majority of value size and growth of the global biologic markets.50
INT
RO
DU
CT
ION
IN
TE
RN
AT
ION
AL
EX
PE
RIE
NC
E
GA
ME
CH
AN
GE
RS
LOO
KIN
G A
HE
AD
UN
CO
VE
RIN
G T
HE
U
NK
NO
WN
S

12
BIOSIMILARS LANDSCAPE
AUSTRALIA• New legislation gives the payer body exclusive
authority to determine substitution of biosimilars at the pharmacy level43
• Substitution of biosimilars recommended as its default policy43
EUROPE• Uptake varies over time, between countries and across therapeutic areas36
• Four distinct payer models have emerged across Europe to influence the price and uptake of reference product and biosimilars37
• Pharmacy-level substitution for biologic drugs is not widely practiced in any EU country40
U.S.• The U.S. is the only country
that has established a legislative standard and definition in order for an interchangeability designation to be granted52
CANADA• Government doesn’t provide direction
regarding therapeutic equivalence with originator products48
• Health Canada does not support biosimilar substitution47
JAPAN• Holds a large portion
of the healthcare cost burden as it comprises the majority of value size and growth of the global biologic markets50

13
UNCOVERING THE UNKNOWNS

14
UNCOVERING THE UNKNOWNSU.S. FDA APPROVAL PATHWAY
In recognition of the unique and complex biological properties of biologics, the FDA has established a distinct pathway for the approval of biosimilars.7 This process differs significantly from that used for approval of small-molecule generics and carries a higher threshold of clinical evidence.8,9 To receive FDA approval, generics must be shown to be bioequivalent and have the same active ingredient, strength, dosage form, route of administration, and condition of use as the original product.53 Conversely, biosimilars require head-to-head analytical, and nonclinical and clinical comparisons with a reference product.9 Within this framework, the clinical evidence must be sufficient to demonstrate that the biosimilar product is highly similar to the reference product and safe, pure, and potent for one or more approved conditions of use.9 While the FDA’s approval pathway
is aligned with U.S. law, and therefore it is independent from the EU framework, both frameworks share a requirement for head-to-head analytical, nonclinical, and clinical testing.9, 54
It is important to note that, while a biosimilar may require fewer clinical trials than its reference product in order to attain approval, it may also require a greater preponderance of analytical characterization, and nonclinical and clinical pharmacology data (see Figure 3).9, 55 Importantly, the FDA does not require comparative analytical or clinical studies between two biosimilars.9 This means that biosimilar candidates will be evaluated only against their reference products and not against other biosimilars.5
ANALYTICAL CHARACTERIZATION(structure & function assessment)
NONCLINICAL
CLINICAL PHARM.(pharmacokinetics/pharmacodynamics)
SAFETY &EFFICACY
ANALYTICAL CHARACTERIZATION(structure & function assessment)
NONCLINICAL
CLINICAL PHARM.(pharmacokinetics/pharmacodynamics)
SAFETY &EFFICACY
REFERENCE PRODUCT DEVELOPMENT351(A) Biologics License Application
BIOSIMILAR DEVELOPMENT351(K) Biologics License Application
Demonstrate safety,purity, and potency
Clinical StudiesOne study to inform immunogenicity and will likely need at least one clinical studyin a sensitive population to confirm safety and e cacy
Demonstrate biosimilarity to the reference product
FIGURE 3. CLINICAL DEVELOPMENT OF REFERENCE PRODUCTS AND BIOSIMILARS9,55
Source. Graphic was adapted from Kozlowski S. Presented at: Biotechnology Technology Summit; June 13, 2014; Rockville, MD.

15
Determination of Interchangeability
When the FDA approves a sponsored product as a biosimilar, it is not automatically considered interchangeable with its reference product, and it should be noted that only the FDA can grant this designation of interchangeability, which requires additional evidence beyond that required for FDA approval.3 To be deemed interchangeable, the product must be approved as a biosimilar, and the sponsor must provide evidence of an expectation of the same clinical result as the reference product in any given patient.7 For a product that is administered more than once, the sponsor must also prove that there are no additional risks to safety or diminished efficacy as a result of switching the reference product for a biosimilar.7
Biosimilar Substitution
The laws regarding substitution for biosimilars are still coming into focus, and will vary at the state and federal levels (see Figure 4).56 However, it is important to keep in mind that state laws governing substitution will only apply to biosimilars designated by the FDA as interchangeable—and that has not been achieved by any biosimilar candidates as of the printing of this report.56 FDA policies on approval standards for biosimilars do not address substitution.1, 3 Within multiple state jurisdictions, there is ongoing legislative activity regarding substitution of interchangeable biologics with the reference product.56
Interchangeability
RUNNERS SLIDE / IMAGE
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
GA
ME
CH
AN
GE
RS
LOO
KIN
G A
HE
AD
AP
PR
OV
AL
PAT
HW
AY
Prior to 2013, no state laws specifically addressed the substitution of biologics.56 See Figure 4 on the next page to see how U.S. state legislation related to biosimilar substitution has changed since then.

16
UNCOVERING THE UNKNOWNSAs of February 2016, several states have passed legislation with varying stipulations regarding substitution of interchangeable biologics, even though there are currently no interchangeable biologics on the market.56
While the approval of the first biosimilar has helped to clarify some aspects of the regulatory, naming, and reimbursement pathways in the U.S., it is too early to determine its ultimate impact on the U.S. healthcare system or on the future of the biosimilar marketplace.
As noted previously, the FDA has stated that biosimilar candidates will be evaluated only against their reference products, and not against other biosimilars.5, 9 Therefore, it cannot be assumed that biosimilars are comparable to one another.5
FIGURE 4. STATE LEGISLATION REGARDING BIOSIMILAR SUBSTITUTION IN THE U.S. AS OF FEBRUARY 8, 201656
MAP BREAKDOWN
WA
OR
CA
NVUT
CO
ND
SD
MN
WI
IA
AK
MT
WY
NMOK
KS MO
TN
LA
AL
SC
NC
KY
WV
OH
NY
ME
NE
ID
AZ
MS
IL IN
GA
FL
VA
PA
VT NH
MI
TX
AR
MA
RI
CT
NJ
DE
MD
DC
HI VI PR
Source: Adapted from NCSL, 2016
Key
Enacted law
No decision has been made
Filed; failed/-adjourned
Bill pending

17
NAMING, CODING, & PAYMENT GUIDANCE FOR BIOSIMILARS
Developments in 2015 offered some clarity on how key regulatory agencies plan to accommodate naming and reimbursement for biosimilars. While many questions still remain, both the FDA and the Centers for Medicare and Medicaid Services (CMS) issued guidance in late 2015 that provided more direction on the future of biosimilars in the U.S.20, 22
FDA Draft Naming Guidance
Appropriate naming of a biosimilar may seem, at first glance, to be a relatively minor concern, but it constitutes a necessary step for post-market monitoring of biosimilar efficacy and safety, relative to each other and to the reference product. There are a number of perspectives on how a biosimilar should be named.
Many stakeholders believe that most biosimilars will be marketed with unique brand names, at least in the initial development of the biosimilars market.57 However, brand names are not required, and prescribers as well as other healthcare providers are not required to use them. In contrast, non-proprietary names (sometimes referred to as the United States Adopted Name (USAN), International Nonproprietary Name (INN), or “active ingredient name”) are required for all drugs and biologics, and are often preferentially used in prescribing and health records. The question has been raised about whether biosimilars should
have unique non-proprietary names to ensure that they are not treated like multisource generic drugs for purposes of prescription ordering, health records, and pharmacovigilance.58, 59
Pharmacovigilance and ongoing monitoring efforts could be impacted by the FDA’s final decision on biosimilar naming. While not all biologic manufacturers and healthcare providers may have the same perspective, a great number have argued that assigning biosimilars unique, non-proprietary names is an important consideration in guarding against inappropriate substitution and ensuring accurate safety monitoring.58
Potential variables and considerations for naming biosimilars include: 20,58
• The reference product and all corresponding biosimilars should have the exact same name
• All biosimilars should have distinguishable names—represented by prefixes to the name of the shared core drug name of the reference product
• All biosimilars should have distinguishable names—represented by suffixes to the name of the shared core drug name of the reference product
• The number of letters in the prefix or suffix should be specified, and could either have meaning or be random
In August 2015, the FDA issued draft guidance on biosimilar naming.20 In this report, the FDA stated that shared non-proprietary names are not appropriate for all biologic products, and proposed an alternate naming pathway that would use four-letter suffixes to differentiate between reference products and their biosimilar counterparts.20
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
GA
ME
CH
AN
GE
RS
LOO
KIN
G A
HE
AD
NA
MIN
G, C
OD
ING
&
PAY
ME
NT

18
UNCOVERING THE UNKNOWNSNAMING, CODING, & PAYMENT GUIDANCE FOR BIOSIMILARS
Key components of the draft guidance include:
• A proposal that reference products and biosimilars have non-proprietary names that share a core drug substance name and an FDA-designated suffix that would allow for better identification of each product.20 This approach would require manufacturers of reference biologics to retroactively modify the current name of the reference product to which biosimilars have been approved. The challenges to manufacturers, other stakeholders, and systems to implement a new name for currently marketed biologics are unclear.
• Outstanding questions regarding how the FDA plans to approach naming for reference products and biosimilars that have been designated as interchangeable.20 For interchangeable biologic products, the FDA requested feedback from the public to determine whether the non-proprietary name for an interchangeable biologic should include a distinguishable suffix, or whether it should share the same suffix as the reference product.20 In the latter scenario, should the
FDA designate a previously marketed non-interchangeable biologic as interchangeable with its reference product, the name of the biosimilar would then need to be changed to match that of the reference product.
• Verification that an identical name would not be required to facilitate substitution of interchangeable biologics. The FDA intends to convey interchangeability designations via the
“Purple Book.”60 As of February 2016, state pharmacy laws permitting substitution of interchangeable biologics refer to the FDA interchangeability designation and do not require the products to use the same name.56
In a web-based survey of 401 U.S. pharmacists conducted in October 2015 by Industry Standard Research on behalf of the Alliance for Safe Biologic Medicines, 77 percent of respondents indicated they preferred a suffix that connects the biosimilar to the product’s manufacturer rather than a suffix with no meaning.61
77% OF RESPONDENTS INDICATED THEY PREFER A SUFFIX THAT CONNECTS THE BIOSIMILAR TO THE PRODUCT’S MANUFACTURER61

19
Final CMS Reimbursement Provisions for Biosimilars
In October 2015, CMS issued its final ruling on Payment for Medicare Part B Covered Drugs and Biologics for 2016, including policies for biosimilars.22 Per the guidance, CMS will maintain its policy standard of reimbursing products according to the current calculation-based methodology of average sales price (ASP) plus 6 percent* reimbursement for Medicare Part B covered drugs and biologics for coverage year 2016.22
In the payment rule, CMS finalized its proposal for reimbursement of biosimilar products, confirming that all biosimilar products of the same reference product will be pooled and combined into a common Healthcare Common Procedure Coding System (HCPCS) code.22 The combined HCPCS code will use a weighted average to determine the biosimilar ASP. The reference product will continue to have its own HCPCS code and payment amount and will not be included in the biosimilar ASP calculation.22
For coverage year 2016, all biosimilar products associated with a particular reference product will be paid under a single billing code and receive a payment equal to 100 percent of the weighted average ASP for the biosimilar products, plus a constant dollar add-on equal to 6 percent* of the reference product’s ASP.22 The reference product would remain in its own billing code and continue to be paid 106 percent* of its own ASP.23 Prior to establishment of an ASP for a biosimilar, CMS will pay 106 percent* of the Wholesale Acquisition Cost (WAC) of the biosimilar, not of the reference product.22
These policies went into effect on January 1, 2016.22 CMS generally sets Hospital Outpatient Prospective Payment System payment policy for biosimilars in alignment with the provisions for payment in the physician setting.62
*The current federal spending sequester requires all Medicare government payments to be reduced by 2 percent.63
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
GA
ME
CH
AN
GE
RS
LOO
KIN
G A
HE
AD
NA
MIN
G, C
OD
ING
&
PAY
ME
NT
ONE HCPCS CODE
BIOSIMILAR ONE BIOSIMILAR TWO BIOSIMILAR THREE
ONE HCPCS CODE
ORIGINAL BIOLOGIC

20
GAME CHANGERS

21
Many of the factors that inform coverage and preference decisions for biologic medicines, including biosimilars—and which may ultimately justify a change in an organization’s
preferred biologic when more biosimilars are approved—are shown in the figure below and discussed throughout this report.
GAME CHANGERS
COVERAGE POLICIES
LOGISTICS
REIMBURSEMENT
EDUCATION
RELIABILITY
BIOSIMILAR
SAFETY
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
GA
ME
CH
AN
GE
RS
LOO
KIN
G A
HE
AD
UN
CO
VE
RIN
G T
HE
U
NK
NO
WN
S

22
GAME CHANGERSCOVERAGE POLICIES
Pharmacy vs. Medical Benefit Coverage
The coverage and appropriate utilization of biosimilars may be influenced by whether a biosimilar is managed through the pharmacy benefit or through the medical benefit.64 To the extent that biosimilars and reference products are covered through a medical benefit, payers will likely face the same issues regarding utilization management and tracking in the medical benefit specialty category for biosimilars as they do today for all biologics. For biosimilars, tracking of safety over time, or pharmacovigilance, is particularly important in order to better understand the benefits and risks of these products.4
A prescription drug covered through the medical benefit is typically administered by a healthcare professional, while a prescription drug covered through the pharmacy benefit is typically self-administered by the patient.65 The pharmacy benefit can include specialty, mail-order, and retail pharmacy, while drugs covered through the medical benefit may be administered in a variety of settings (see Figure 5).66
Source: Artemetrx. Specialty Drug Management. 2015
MEDICAL BENEFIT
DRUG SPEND
PHARMACY BENEFIT
SPECIALTY PHARMACY
MAIL-ORDER PHARMACY
RETAIL PHARMACY
OUTPATIENT HOSPITAL
PHYSICIAN OFFICE
HOME INFUSION
AMBULATORY INFUSIONCENTERS
INPATIENT HOSPITAL
FIGURE 5. COVERAGE OF PHARMACY VS. MEDICAL BENEFITS66

23
Payers often use specialty pharmacy dispensing for prescription drugs with particular characteristics—such as high cost, special handling requirements, special storage/preparation, or complex administration. Biologics, including biosimilars, are typically considered specialty drugs.67
Payers may stipulate that specialty drugs be predominately dispensed through contracted specialty or local retail pharmacies.64 Prescription drugs dispensed through retail, mail order, or specialty pharmacies are typically adjudicated online. However, this is not the case for prescription drugs covered through the medical benefit, which are adjudicated retroactively and paid to the medical office, clinic, or other provider on a buy-and-bill basis.64
To help manage the utilization of drugs covered under the medical benefit, payers may develop coverage and reimbursement policies and implement them prior to authorization.64
In addition, while pharmacy benefit and specialty pharmacy claims are adjudicated using the National Drug Code (NDC), drug claims covered under the medical benefit submitted by medical providers are typically adjudicated using a less specific Healthcare Common Procedure Coding System (HCPCS) Level II J-Codes.68 Therefore, tracking use and safety of medical benefit therapies can be more difficult than tracking pharmacy benefit therapies. It is reasonable to assume that this challenge will continue to be true for all biologics, including biosimilars. Given that CMS has issued final guidance designating that all biosimilars in a category have a blended code, further issues could result with tracking use and safety for biosimilars through the medical benefit channel.
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
CO
VE
RA
GE
PO
LICIE
S LO
OK
ING
AH
EA
DU
NC
OV
ER
ING
TH
E
UN
KN
OW
NS
24
GAME CHANGERSPatient Population Size
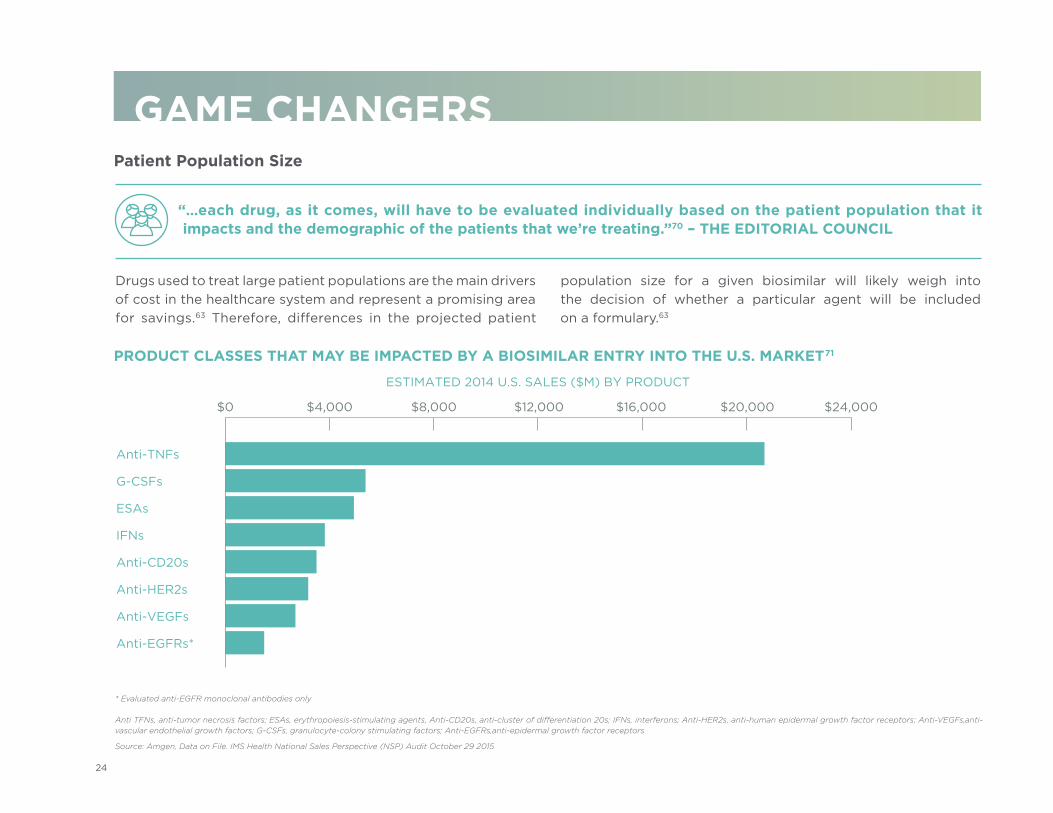
PRODUCT CLASSES THAT MAY BE IMPACTED BY A BIOSIMILAR ENTRY INTO THE U.S. MARKET71
Source: Amgen, Data on File. IMS Health National Sales Perspective (NSP) Audit October 29 2015
* Evaluated anti-EGFR monoclonal antibodies only
Anti TFNs, anti-tumor necrosis factors; ESAs, erythropoiesis-stimulating agents, Anti-CD20s, anti-cluster of differentiation 20s; IFNs, interferons; Anti-HER2s, anti-human epidermal growth factor receptors; Anti-VEGFs, anti-vascular endothelial growth factors; G-CSFs, granulocyte-colony stimulating factors; Anti-EGFRs, anti-epidermal growth factor receptors
“…each drug, as it comes, will have to be evaluated individually based on the patient population that it impacts and the demographic of the patients that we’re treating.”70 – THE EDITORIAL COUNCIL
Anti-TNFs
G-CSFs
ESAs
IFNs
Anti-CD20s
Anti-HER2s
Anti-VEGFs
Anti-EGFRs*
ESTIMATED 2014 U.S. SALES ($M) BY PRODUCT
$0 $4,000 $8,000 $12,000 $16,000 $20,000 $24,000
Drugs used to treat large patient populations are the main drivers of cost in the healthcare system and represent a promising area for savings.63 Therefore, differences in the projected patient
population size for a given biosimilar will likely weigh into the decision of whether a particular agent will be included on a formulary.63

25
Formulary: Drug Formulary Assessment, Fit, & Policies
Realizing the savings, however, involves a complex set of considerations. For example, assessment of a biosimilar’s fit within a formulary design requires not only an analysis of potential savings, but also of current clinical guidelines and practices, societal factors and behaviors, operating costs, logistical implications, physician and patient acceptance, and the likelihood of clinical use and adoption.72 When faced with these multiple considerations, stakeholders may feel that the potential benefits of adding a biosimilar to a formulary may not justify the required effort and resources to enact the change.
As a result, appropriate utilization of biosimilars, both overall and within particular therapeutic categories, will be determined by multifaceted decisions by payers, patients, integrated delivery networks (IDNs), hospitals, and employers. For example, some payers may rank potential savings higher than consumer choice when it comes to assessing biosimilars.73 On the other hand, some self-insured employers may be more concerned about the impact on their company’s reputation and employee morale if employees feel they are being “forced” into lower-cost treatments.
An organization’s formulary is developed and maintained by its Pharmacy & Therapeutics (P&T) committee, which appraises, evaluates, and selects drugs for the formulary. The vast majority of coverage plan sponsors specify a two-, three-, four-, or higher-tiered formulary, and then structure their coverage options in accordance with these tiers.74 For drugs covered under the pharmacy benefit, traditional tiering is based on several drug characteristics, including whether a drug is available from multiple sources or only from a single source, the drug’s net cost, and its specialty designation.75 This structure may now need to be adapted to accommodate biosimilars.
As biosimilars may be treated as specialty drugs (as some biologics are),67 there may be challenges incorporating them into the current tiering system. Questions may arise as to whether or not to place biosimilars on a currently existing tier, or to create a new tier for a specialty drug that is also a biosimilar (see Figure 6).76
NON-PREFERRED BRAND TIER ($$$)
PREFERRED BRAND TIER ($$)
GENERIC TIER ($)
Typically the highest cost sharing. Non-preferred brands have generally not been found to be any more cost e�ective than available generics, preferred brands, or over-the-counter drugs.
Typically a slightly higher cost sharing than generics. Preferred brands have been proven to be safe and e�ective, and are favorably priced compared to other brand drugs that treat the same condition.
Typically the most a�ordable cost sharing. The active ingredient in a generic drug is chemically identical to the active ingredient of the corresponding brand. PA
TIE
NT
CO
ST
SH
AR
ING
($
)
FIGURE 6. FORMULARY TIERS FOR DRUG DISPENSING77
Source: Avalere. Value Based Formulary Design. 2015
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
CO
VE
RA
GE
PO
LICIE
S LO
OK
ING
AH
EA
DU
NC
OV
ER
ING
TH
E
UN
KN
OW
NS

26
GAME CHANGERSFormulary decision makers, including P&T committees, may consider the following when evaluating a biosimilar: 72, 78, 80
PATIENT: Acceptability (related to factors such as therapeutic category, therapeutic vs. supportive care, acute vs. chronic therapy, and new vs. continuing therapy); disease severity and comorbidities; education; cost share and savings; adherence.
PROVIDER: Clinical studies and evidence-based review; electronic medical record and medication system; educational requirements for staff, providers, and patients; administration-related costs; economic considerations, including 340B; costs for pharmacovigilance; costs associated with potential for drug shortages; costs for monitoring the response to biosimilar treatment
PRODUCT: Clinical data; evidence-based reviews and guidelines; indications, labeling, immunogenicity, interchangeability, pharmacovigilance requirements; preparation, administration, and storage requirements
MANUFACTURER: Supply reliability and history of shortages; supply chain security and anti-counterfeit measures; patient assistance and reimbursement support
EMPLOYER / PURCHASER: Employer purchasers may provide guidance, or potentially overrule coverage decisions.
COMMERCIAL & GOVERNMENT PAYERS: Financial impact; sponsor (i.e. commercial, Medicare, Medicaid); coverage and reimbursement policies needed to support, transition from a reference product to a biosimilar; information and claims system; provider acceptability; support programs, including education, for providers and patients; costs associated with potential for drug shortages, for monitoring the response to biosimilar treatment, for monitoring of therapeutic interchange, and cost impact of patient-assistance programs; negotiation and influence of bundled contracting approaches; cost impact of patient-assistance programs
WHAT TO EXPECT NEXT
As we look to the future, it is unclear how payers will manage biosimilars, whether through the pharmacy benefit or through the medical benefit. Furthermore, we may see more direct contracting
by employer purchasers with IDNs.79 We expect to see biosimilars within formularies, but the placement within the formulary, whether to an existing tier or creating a new tier, is undetermined.

27
EDUCATION
As of March 2016, only one biosimilar has been marketed for use in the U.S.14 This limited exposure to the functional and operational aspects of biosimilars may contribute to why patients, physicians, pharmacists, and allied health workers
have various levels of awareness and familiarity with biosimilars today.72 Several surveys show that stakeholders have one thing in common: the belief that education about biosimilars is essential.61, 81
Informed Choice for Physicians
“If you speak to physicians, you’ll hear the term ‘financial toxicity’49 when the physician has identified the appropriate therapy for their patient, the problem is the patient may not be able to afford it or have access to it.”70 – THE EDITORIAL COUNCIL
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
ED
UC
AT
ION
LOO
KIN
G A
HE
AD
UN
CO
VE
RIN
G T
HE
U
NK
NO
WN
S

28
GAME CHANGERSThe use of any biologic in medical practice could be impacted by the treatment setting (medical office/clinic, home, hospital, or long-term care facility), chronic versus acute use, and its indication. Introducing a biosimilar option adds to the
complexity of this choice. In addition to considering the potential savings that a biosimilar may (or may not) present to the patient, a provider may have other questions or concerns related to biosimilars:
• Does a biosimilar have all the indications of the reference product?;72
• Were the products’ indications approved through extrapolation or through clinical trials?;72
• Is the biosimilar as safe and efficacious as the reference product?;82
• Is the dosing and administrative device equivalent for the biosimilar and the reference product?;80, 83
• Has the biosimilar been designated as interchangeable by the FDA?;80
• Are formulary barriers in my therapeutic choice for my patients?;72
• Are there reimbursement support and other programs available for my patients?;80
• How does the site of care or payment model impact reimbursement (e.g., is it retail or buy-and-bill)?;64
• Is my patient drug-naive to treatment options for the molecular target?;72
• How do I track and trace which version of the molecule will be dispensed?;72
• How comfortable is my patient with the use of a biosimilar?;84
• Will providers need to educate the patient about drug administration devices or managing injection site discomfort?72, 85
QUESTIONS

29
Medical history is also a factor. In many cases, patients with chronic disease may be stabilized on a particular product and the provider may choose to maintain the patient on that product. This, too, could influence a provider’s decision to prescribe a biosimilar.84 Finally, one can add a manufacturer’s known or
expected reliability to deliver a consistent supply of the selected biologic to the list.72 Navigating this long list of considerations may require a significant amount of education for a physician to make a truly informed choice.72
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
ED
UC
AT
ION
LOO
KIN
G A
HE
AD
UN
CO
VE
RIN
G T
HE
U
NK
NO
WN
S

30
GAME CHANGERS
Providers
Integrated Delivery Networks [IDNs] have been spending the last three years prepping everybody for biosimilars because we knew physician engagement was going to be the key driver to making this [utilization of biosimilars] successful. And if we start educating after it [biosimilar] has been released, it’s too late.”70 – THE EDITORIAL COUNCIL
According to recent studies, pharmacists and physicians want additional and in-depth education about biosimilars.61, 82 Based on the key roles both pharmacists and physicians play in the treatment and care of patients, ensuring these key stakeholders are educated on the myriad of topics related to biosimilars may be one of the pivotal factors to support appropriate utilization of a biosimilar.61, 82
In March 2015, QuantiaMD conducted a survey of nearly 300 primary care physicians and specialists in the therapeutic areas where biosimilars are most concentrated —including endocrinology, gastroenterology, hematology, infectious disease, oncology, nephrology, neurology, and rheumatology—and found that prescribing specialists have a general level of awareness about biosimilars.82 However, the awareness level drops sharply as they are asked about specifics as seen in Figure 7.82
“

31
0% 25% 50% 75% 100%
78%
60%
53%
38%
33%
ARE FAMILIAR WITH THE TERM “BIOSIMILAR”;
ALREADY PRESCRIBE BIOLOGICS;
ARE AWARE THAT THE FIRST BIOSIMILAR WAS APPROVED BY THE FDA;
ARE AWARE OF A BIOSIMILAR UNDER CONSIDERATION FOR APPROVAL THAT WOULD BE RELEVANT TO THEIR PATIENT POPULATION; AND
CAN NAME A BIOSIMILAR UNDER CONSIDERATION FOR APPROVAL.
FIGURE 7. PHYSICIAN INTEREST IN LEARNING ABOUT BIOSIMILARS82
Source: QuantiaMD. Physician Biosimilar Survey. 2015
Similar results were observed in a separate survey of 401 pharmacists, sponsored by the Alliance for Safe Biologic Medicines in October 2015: only 35 percent of respondents said that they had a complete understanding of biosimilars.61
Of particular note, hospital pharmacists were more likely to be “very familiar” with biosimilars than were retail pharmacists (44 percent vs. 23 percent, respectively).61
It is possible that knowledge of biosimilars will increase since these surveys were completed, given that the first biosimilar was launched in September 201521 and that the FDA provided some new draft guidances as well as finalized other previous draft guidances that year.18
35%OF RESPONDENTS SAID THAT THEY HAD A COMPLETE UNDERSTANDING OF BIOSIMILARS61
ONLY
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
ED
UC
AT
ION
LOO
KIN
G A
HE
AD
UN
CO
VE
RIN
G T
HE
U
NK
NO
WN
S

32
• Should the labeling disclose data from the clinical trials that compared the biosimilar with the reference product?
• Should the labeling briefly describe the FDA’s basis of approval for a biosimilar to a specific reference product?
• Should the labeling disclose whether or not the FDA has designated the product as interchangeable with the reference product?
• Should the labeling disclose if any indications or conditions of use of the reference product are not approved for the biosimilar?
• Should a clinician be made aware that the data presented for extrapolated indications were not actually based on clinical testing
of the biosimilar?
Labeling for the first biosimilar product approved by the FDA discloses none of the above information.88 While formulary directors may be able to obtain some desired details not disclosed in a biosimilar’s label from FDA’s approval history or
from an Academy of Managed Care Pharmacy (AMCP) dossier submitted by the manufacturer, that information may not be as accessible to patients or providers who may want to learn more about a product to make more informed treatment decisions.89
In February 2016, in an effort to help educate providers on biosimilars, the FDA introduced a free Continuing Education Course for healthcare professionals—titled, FDA Overview of Biosimilar Products—to help strengthen providers’ knowledge and understanding of biosimilars and interchangeable products.87
However, there are still opportunities to gain further clarity on some biosimilar topics. As of March 2016, the FDA has yet to issue final guidance on the exact amount and type of information required in a biosimilar’s labeling, which may raise several questions:69
“For [P&T committees] trying to understand the clinically meaningful differences between the biosimilar and its reference product, it would be helpful to have that information available on the product label. This is where I would like to see the clinical trials section or others replicated into formal biosimilars labeling structures.”70– THE EDITORIAL COUNCIL
GAME CHANGERS
QUESTIONS

33
Patients
FIGURE 8. PATIENT PREFERENCE ON MEDICATION CHANGES84
One should expect that patients want to make completely informed decisions regarding the choice of which biologic to use - a biosimilar, or its reference product. Patients may receive their information regarding healthcare from a number of sources, but physicians often remain their primary point of reference. This has marked implications for patient education and utilization of biosimilars, as patients may mirror or adopt their physician’s point of view.
The primacy of the patient/physician relationship is reflected in a 2015 survey by the Global Healthy Living Foundation.84 Of the 177 arthritis patients surveyed, a majority of respondents stated that the decision to prescribe a biologic should remain in the hands of the patient and his or her physician, while only 2 percent of respondents felt payers should make this decision (see Figure 8.).84
Physicians
90%
7%
2% Pharmacists
Payers
Source: Global Healthy Living Foundation. Patient Perspectives on Medication Switching for Non-Medical Reasons. 2015WHAT TO EXPECT NEXT
Evidence shows that there is need for additional patient education around the appropriate use and considerations associated with biosimilars. It will be important for physicians to have sufficient knowledge and experience with biosimilars to speak comfortably with their patients about these agents. To this end, we anticipate there may be potential friction between
payers and providers as more payers have increasing influence in financial decisions in regard to biosimilars. To help alleviate this, it will be important for payers and manufacturers to help facilitate the additional education of physicians, pharmacists, and patients on the science supporting the use of biosimilars.72
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
ED
UC
AT
ION
LOO
KIN
G A
HE
AD
UN
CO
VE
RIN
G T
HE
U
NK
NO
WN
S

34
GAME CHANGERS“I really just cannot stress enough how difficult it can be to make changes within our electronic medical record [EMR] infrastructure. One small change when you’re looking at a drug formulary and [you have to weigh] the EMR infrastructure changes across your healthcare organization…And if we’re going to save a relatively small amount, it’s not worth it, at all.”70 – THE EDITORIAL COUNCIL
Payer and provider organizations may need to address logistical considerations resulting from the addition of a biosimilar to their inventory and data management systems.
Of particular importance is the need to reprogram and adapt existing information technology (IT) systems to accommodate the inclusion of biosimilars into hospital/Integrated Delivery Network (IDN) formularies. This endeavor will likely involve associated changes in the electronic medication-administration systems (eMARs), adapting Electronic Medical Records (EMRs), reprogramming to support preference for a certain product, and making the changes needed to support various coding and pricing schemes.72 Evaluation of the potential costs associated with making needed system changes may be compared to potential savings to help determine if a change will be beneficial in the short and long term.72
An additional logistical consideration is managing different pricing schedules for inpatient and outpatient pharmacies within the same health system. This may be a challenging problem for health systems that use the same EMRs for 340B hospitals as well as for other pharmacy fulfillment mechanisms that are not
supported by federal discount programs, such as commercial direct mail, purchases by pharmacy benefit managers, HMOs, mail-order pharmacies, insurers, hospitals, and clinics.90 Many health systems today include hospitals, home health agencies, hospices, stand-alone clinics, and physician practices in order to provide a continuum of care as an IDN.91 An IDN’s operations may be divided between inpatient and several types of outpatient treatment settings as well as commercial and government operations.91 As a result of this complexity, IT standardization and integration across settings may be challenging.
From the financial perspective, sites within a single healthcare system that qualify as 340B covered entities can obtain federally mandated “ceiling price” discounts for covered outpatient drugs, while other sites that are not 340B-eligible usually pay a higher net price.92 Keeping track of pricing differences for a single product could be a significant burden in a complex healthcare system which may have ongoing data integration efforts. Since biosimilars may present another price point to an already established treatment option, differential pricing could be even more complex if biosimilars were added to a formulary.
LOGISTICS

35
“340B pricing may hamper the adoption of biosimilars with 340B organizations. This delay will stem from the fact that there would be no historical discount set for biosimilar products to base a 340B or Medicaid price off of.”70 – THE EDITORIAL COUNCIL
340B Health is a membership organization of more than 1,100 hospitals and health systems, both public and private nonprofits, throughout the U.S. that participate in the 340B Drug Pricing Program.93 It is managed by the Health Resources and Services Administration (HRSA).
The 340B program requires drug manufacturers to sell outpatient drugs at a discount to safety-net providers serving high numbers of low-income Medicare, Medicaid, and Supplemental Security Insurance patients. Congress created the program to allow these providers to “stretch scarce federal resources as far as possible, reaching more eligible patients and providing more comprehensive services.” Savings from the program help fund free and low-cost medications as well as HIV/AIDS, diabetes, cancer, dental, and primary care clinics that serve our most vulnerable citizens.93
Quick facts about the 340B Program:
• In August 2015, HRSA issued proposed guidance for governing policies related to the 340B Drug Pricing Program.44
• Covered entities include six categories of hospitals: disproportionate share hospitals, children’s hospitals, and cancer hospitals exempt from the Medicare prospective payment system, sole community hospitals, rural referral centers, and critical access hospitals.90
• 340B is a growing sector: Covered entities and their affiliated sites spent more than $7 billion to purchase 340B drugs in 2013, three times the amount spent in 2005.90
• Approximately 40 percent of all U.S. hospitals participated in 340B in 2015.94
• Ceiling prices for 340B drugs are not publicly disclosed. However, it has been estimated that 340B hospitals receive discounts on the average sales prices of medications ranging from 15 to 60 percent.90, 95
• Medicare Part B pays for certain 340B treatments, such as biologic drugs used to treat cancer and rheumatoid arthritis, at the same rate as 340B hospitals and non-340B hospitals. It does this despite the fact that 340B hospitals are able to obtain those treatments at a discount.90
THE 340B DRUG PRICING PROGRAM
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
LOG
ISTIC
SLO
OK
ING
AH
EA
DU
NC
OV
ER
ING
TH
E
UN
KN
OW
NS
40% OF HOSPITALS IN THE U.S. ARE PART OF THE 340B PROGRAM94

36
GAME CHANGERSWHAT TO EXPECT NEXT
Support for biosimilar utilization may pose challenges to existing IT systems and processes across the healthcare industry, including the need for customization of EMRs and eMARs.72
In addition, there is the potential need to develop a standardized approach to pricing schedule, especially for health systems participating in the federal 340B Drug Pricing Program.
Drug shortages can have a dramatic fiscal impact on hospitals because shortages could increase the cost of delivering patient care. For example, when a drug shortage occurs, the cost of acquiring alternative drug supplies might incur costs if re-contracting is required. 96 Drug shortages can also lead to a substantial number of treatment changes. Physicians report that in some cases, when a patient’s treatment is modified due to supply problems, the available alternatives have a weaker evidence base than the medications in shortage.97 Concerns about reliability of supply are well-founded based on the generic manufacturing industry’s history with the production of sterile injectables, which is a segment of the pharmaceutical market that has suffered numerous shortages over the years—largely due to quality control issues (see Figure 9).98 An economic analysis suggests that shortages of injectable drugs may also be associated with inadequate reimbursement for multisource drugs predominantly covered by Medicare,99 a risk factor that also might apply in the future for certain classes of biosimilar products.
As more manufacturers plan to enter the biosimilar market, it will likely become increasingly important for stakeholders to have an understanding of each company’s manufacturing capabilities, quality assurance process, reputation for consistent supply, and plans for avoiding shortages.72 In choosing one product over
another, a P&T committee may evaluate a manufacturer’s reliability, safety, and quality control history as well as its safeguards to ensure manufacturing capacity and dependability.80 It may also be important to evaluate a manufacturer’s ability to institute procedures to ensure a steady product supply, including backup or multisite manufacturing capabilities to accommodate unexpected disruptions at the primary manufacturing site.80
RELIABILITY
Productdiscontinuation
Active pharmaceutical ingredient (API) or non-API component issue
Loss of manufacturing site or site change
Quality problems
Manufacturing delays and capacity issues
Increased demand4
0%
30%
3%
9%
12%
6%
Source: United States Government Accountability Office. Drug Shortages. 2014
FIGURE 9. REPORTED CAUSES OF DRUG SHORTAGES, 2011-201398

37
There are a lot of challenges to consider before jumping to adopt a biosimilar. We have been so heavily impacted by drug shortages at our organizations that if we have a good stable supply of a very necessary product to treat our really sick patients, we’re going to stick with that until we can truly ascertain whether or not the biosimilar is going to be supplied in the quantities that we need it.” 70 – THE EDITORIAL COUNCIL
MEASURING THE VALUE OF BIOSIMILARS70
Achieving better value is the central focus of American health policy debates. By definition, biosimilars should help in the pursuit. They promise similar health outcomes and cost savings. However, whether biosimilars deliver on the promise hinges on several considerations.
Broadly speaking, estimating the value of interventions means judging the costs and health outcomes they generate compared to a relevant alternative. In practice, it means weighing multiple dimensions, including the quality of clinical data, the magnitude of treatment effects, the likelihood of adverse events, and a product’s overall cost-effectiveness and budget impact. There are uncertainties in each estimation step. Moreover, judgments about how to weight various dimensions vary within and among interested parties, including patients, physicians, employers, and payers.
In considering the value of biosimilars, there may be residual questions at time of launch that can only be answered through additional experience in clinical practice. The biosimilar will have been shown to have no clinically meaningful differences in its approved indications, but it may not be licensed for
all of the reference product’s indications. Biosimilars may require a slightly different dose (for titrated products), or may differ in terms of certain patients’ ability to stay on the therapy long term. Considerations of value must consider whether a lower price is worth the potential differential in these attributes.
Going forward, there is a need for more evaluation. Cost effectiveness analysis in which costs and health outcomes are measured and compared to alternative strategies is one tool to help. Research would also be useful on the impact of various coverage and formulary policies that incorporate biosimilars. Finally, there will be an important role for education – of patients, physicians, pharmacists – around all of these issues.
PETER NEUMANN, SCD
Director, Evaluation of Value and Risk in Health, Tufts Medical Center
Member of the Editorial Council
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
RE
LIAB
ILITY
&
ME
ASU
RIN
G V
ALU
ELO
OK
ING
AH
EA
DU
NC
OV
ER
ING
TH
E
UN
KN
OW
NS
“

38
GAME CHANGERS
The availability of biosimilars may impact contracts among payers and IDNs, wholesalers, group purchasing organizations (GPOs), and manufacturers by presenting several new considerations to evaluate.
Stakeholders may negotiate discounts with biosimilar manufacturers in a similar manner as they do with the manufacturers of reference products. Having biosimilars on the market that are FDA-designated as interchangeable may afford payers greater leverage when negotiating with manufacturers that have biosimilars that have not been
designated as interchangeable with the same reference product. Furthermore, a manufacturer may earn additional negotiating power should it have the first interchangeable biosimilar in a multi-biosimilar marketplace.7 In fact, by statute, an exclusivity period will be granted should a biosimilar receive an interchangeable designation by the FDA, and if it is the first to receive that designation, no other biosimilar to the same reference product may receive an interchangeable designation by the FDA.7 As of March 2016, there are no interchangeable biosimilars in the U.S. market.
CONTRACTING
“One consideration when contracting for a biosimilar, is that the reference product may have a high level of support from healthcare providers, which may make it more difficult to drive adoption, especially for patients already on established therapies.” 70 – THE EDITORIAL COUNCIL

39
In recent years, we have witnessed an increase in collaborations, mergers, and acquisitions among pharmaceutical companies looking to expand their portfolios into biosimilars.100, 101, 102 As the makeup of biosimilar manufacturers changes with manufacturers
both marketing biologics and developing biosimilars, the contracting environment is expected to become more complex as new biosimilars come to market.103
WHAT TO EXPECT NEXT
The biologics marketplace has become sensitive to shortages in recent years. Payers, IDNs, and providers may make their degree of confidence in a manufacturer’s ability to avoid shortages a key consideration when evaluating their preferences among multiple biosimilars as well as the reference product.80 The contracting process will likely evolve as the complexion of
biosimilar manufacturers continues to shape through mergers and collaborations as well as the potential negotiating power both manufacturers and formulary decision makers may have should the FDA begin to approve biosimilars with an interchangeable designation.
REBATES
BUNDLED PAYMENTS
CONTRACTING
LANDSCAPEPRICING CAPS
340B PRICING/CONTRACTING
PAYER MERGERS
BIOSIMILAR MERGERS
MULTI-STATE IDNS
DIFFERENTIATED BIOSIMILARS (Extrapolation, Number of indications, Interchangeability, Varying clinical study designs)
VALUE-BASED CONTRACTING/REIMBURSEMENT
THE PACE OF ENTRANTS AND NUMBER OF BIOSIMILAR COMPETITORS
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
CO
NT
RA
CT
ING
LOO
KIN
G A
HE
AD
UN
CO
VE
RIN
G T
HE
U
NK
NO
WN
S

40
GAME CHANGERS
Biosimilars bring the opportunity for savings to a healthcare system that is constantly searching for efficiencies while simultaneously aware of the need to preserve incentives that will reward future innovation. Across Medicare program Parts A (inpatient hospital), B (physician office and outpatient hospital),
C (Medicare Advantage), and D (prescription drug benefit), prescription drugs totaled 19 percent of overall Medicare programming spend in the U.S. in 2013.104 Part D represented 57 percent of Medicare drug and pharmacy spending (see Figure 10).104
The reimbursement landscape continues to change, especially in light of CMS’ finalized Medicare program reimbursement provisions22 and the Part B drug payment model demonstration
program that was proposed in March 2016.105 These guidelines may influence not only reimbursement incentives, but also appropriate use and understanding of biosimilars.
REIMBURSEMENT
FIGURE 10. MEDICARE PROGRAM SPENDING, 2013 104
MEDICARE PROGRAM SPENDING=$574 BILLION
COMPONENTS OF MEDICARE DRUG AND PHARMACY SPENDING
Drugs and pharmacy
All else
19%
81%
Inpatient hospital
Physicianand supplier
Outpatienthospital
Part D
Part A and B services deliveredin Medicare Advantage
Other
11%
12%
5%
5%
57%
10%
Source: Schmidt R, et al. Medicare Drug Spending. 2015

41
6%* OF WAC OF BIOSIMILAR
6%* OF ASP OF REFERENCE PRODUCT
The Medicare program pays for most Part B drugs at a prospective rate of ASP plus 6 percent which, since 2013, has been subject to a 2 percent reduction due to sequestration.22, 63 In order to incentivize physicians to prescribe biosimilars, CMS stipulated that once ASP is established for a biosimilar, CMS reimbursement
will be paid at 100 percent of the biosimilar’s own ASP, plus 6 percent* of the ASP for the reference product.22 Prior to ASP establishment for a biosimilar, CMS will pay 106 percent* of WAC of the biosimilar (not of the reference product) until ASP can be used for payment.19
CMS issued guidelines in 2015 with respect to the coverage of biosimilars and clarified that the payment amount for a biosimilar biological product is based on the ASP of all biosimilar products included within the same billing and payment code.22
This decision has proven to be controversial. During a February 2016 hearing of the House Energy & Commerce Subcommittee on Health Biosimilars, House and Senate Members voiced their concerns and opposition to CMS’ policy to group and issue multiple biosimilars under the same J-code (the most commonly used HCPCS codes) for Medicare reimbursement purposes.106
Many argued this proposed payment policy would not provide the right incentives for manufacturers to invest in further trials for additional indications, subsequently reducing the number of biosimilar products available in the U.S. market.106
In March 2016, CMS announced a proposal for a two-phase payment model for Part B drugs that would test whether alternative drug payment designs will lead to a reduction in Medicare expenditures.105
Part D plans must cover generic and brand name prescription drugs, including biologicals licensed under section 351 of the Public Health Services Act, that are generally needed by Medicare recipients for treatment of medically accepted indications.107 Part D drug plan formularies must include at least two drugs from each drug category or class unless only one drug is available for a particular category or class, or only two drugs are available but one drug is clinically superior to the other.107
*The current federal spending sequester requires all Medicare government payments to be reduced by 2 percent.63
CMS Guidelines for Biosimilar Coverage
WAC OF BIOSIMILAR
BEFORE ASP IS ESTABLISHED FOR BIOSIMILARS ONCE ASP IS ESTABLISHED FOR BIOSIMILARS
ASP OF BIOSIMILAR
Payment for a biosimilar is based on ASP of all biosimilar products included within the same HCPCS code.22
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
RE
IMB
UR
SEM
EN
TLO
OK
ING
AH
EA
DU
NC
OV
ER
ING
TH
E
UN
KN
OW
NS
+ +

42
GAME CHANGERS
The CMS guidance notes that biosimilars “may provide Part D sponsors with new products that create formulary design options to help control costs while still ensuring beneficiaries have access to the medications they need.”17
The guidelines include:17
• “Biosimilars may be added to plan formularies at any time as a formulary enhancement.”
• “For the purposes of Part D transition supply and notice requirements, biosimilars and the reference biological product should be treated like different products.”
• “Because biosimilars are not interchangeable with the reference biological product, CMS expects that Part D sponsors’ Pharmacy and Therapeutics (P&T) committees will review newly approved biosimilars in accordance with section 30.1.5 of Chapter 6 of the Medicare Prescription Drug Benefit Manual.”
• “Biosimilars do not meet the CMS definition of a generic drug …or the… definition of a multiple source drug.”
• “…biosimilars are non-applicable drugs for purposes of establishing coverage gap cost sharing under the basic Part D benefit, and are not discounted or otherwise subject to (Coverage Gap) Discount Program requirements.”
A 2011 Amgen-sponsored report from Milliman observed “If CMS does not require the biosimilar manufacturer to comply with the 50 percent discount (i.e., the Coverage Gap Discount Program), since it applies only to brand-name drugs, then the [reference]
biologic price would be more competitive with the biosimilar for the Medicare-eligible population in the coverage gap. Thus, the senior population and Medicare Part D may indirectly impact the biosimilar pricing in the commercial sector.” 108
CMS Guidelines for Biosimilar Coverage

43
REFERENCE PRODUCTS
% C
ON
TR
IBU
TIO
N O
F T
OT
AL
DR
UG
CO
ST
S
100%
0%2017
10%
40%
50%
2016
5%
45%
50%
2018
15%
35%
50%
2019
20%
30%
50%
2020
25%
25%
50%
GENERICS / BIOSIMILARS
% C
ON
TR
IBU
TIO
N O
F T
OT
AL
DR
UG
CO
ST
S
100%
0%2016
58%
42%
2017
51%
49%
2018
44%
56%
2019
37%
63%
2020
25%
75%
BENEFICIARY PLAN MANUFACTURER
Compared to Medicare, CMS’ approach to how they classify biosimilars is different when it comes to Medicaid services. According to a Medicaid Drug Rebate Program Notice issued to participating drug manufacturers in March 2015, State Medicaid programs were instructed that biosimilars should fall within the definition of single source drugs in the Medicaid Drug Rebate
program.16 Furthermore, states may consider the total rebates for reference biological products as well as those that have been determined to be biosimilar to, or interchangeable with, reference biological products in their determination of preferred drugs lists consistent with the requirements for prior authorization programs.16
WHAT TO EXPECT NEXT
Reimbursement for biosimilars will likely be shaped by payers and providers as is the case with branded, generic, and specialty medications. Stakeholders are expected to strive to maximize biosimilars’ cost effectiveness and value, while remaining sensitive to patient and provider perceptions and needs.63
Over time, payers will actively learn, both clinically and economically, where the opportunity of biosimilars can meet the ongoing reality of the U.S. healthcare system, and will likely continue to adjust reimbursement mechanisms accordingly.
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
RE
IMB
UR
SEM
EN
TLO
OK
ING
AH
EA
DU
NC
OV
ER
ING
TH
E
UN
KN
OW
NS
FIGURE 11. CLOSING THE COVERAGE GAP 109
Source: Adapted from Medicare Rights Center. Closing the Doughnut Hole. 2014

44
GAME CHANGERS
With more biosimilars expected to launch in the U.S. market in the next several years, experts have stressed the importance of tracking the safety of biosimilars through pharmacovigilance.110,111
A core principle of pharmacovigilance is traceability, the ability to identify each medical product and link it back to its manufacturer.112 It is necessary to differentiate between the biosimilar(s) and the reference product to ensure that adverse event reports associated with a specific biologic are not inappropriately associated with another biologic112—either the reference product and/or among one or more of its biosimilars.112, 113 In the U.S., it has been found that medication orders captured in patients’ EMRs can help validate previously
reported Adverse Event Reports (AERs), and detect new ones.114 Consequently, it is important that the healthcare professionals prescribing, dispensing, and administering biologic medicines, including biosimilars, have a clear understanding of which products are reference products and which are biosimilars, and—if there is more than one biosimilar available for a reference product—which biosimilar is being given to a specific patient.
During the February 2016 hearing of the House Energy & Commerce Subcommittee on Health Biosimilars, a CMS representative discussed processes they have put in place to address this issue: 106
SAFETY
“We [CMS] implemented a requirement for claims for biosimilars to include a modifier that identifies the manufacturer of the specific product...This will allow us and others to track which specific biosimilars a beneficiary receives.”
- Sean Cavanaugh, Deputy Administrator and Director of the Center for Medicare, CMS

45
ENOXAPARIN ACTIVE SURVEILLANCE
• Administrative claims databases• Useful for answering specific questions
FINDINGS: Pharmacy Benefit
• 33% of unit volume• Using pharmacy claims to identify• Products readily identifiable via NDC
Medical Benefit• 67% of unit volume• Using institutional and medical claims to identify• Products have a shared code and cannot be uniquely
identified
ENOXAPARIN CASE STUDY: EXAMPLE OF INADEQUACIES OF CURRENT SYSTEMS FOR ACCURATE PHARMACOVIGILANCE113
ADVERSE EVENT REPORTSUNIT VOLUME SHARE
69%
5%
26%
4%
Not identifiable
Other
Sandoz
Winthrop
SanofiIdentifiable to a specific generic
Identifiable to Sanofi
50%
6%
40%
Source: Adapted from Grampp et al. ASHP. 2014
* Includes both Pharmacy and Medical Benefit
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
SAF
ET
YLO
OK
ING
AH
EA
DU
NC
OV
ER
ING
TH
E
UN
KN
OW
NS
ENOXAPARIN PASSIVE SURVEILLANCE*
• Reports submitted to the FDA or manufacturers• Used to detect new safety signals
FINDINGS: • Using FDA AE database to identify• 26% ambiguous AE reports (generic name only)• Identification of AE reports to brand or generic
manufacturers did not track to unit volume share

46
GAME CHANGERS
WHAT TO EXPECT NEXT
Availability of biosimilars in the U.S. market brings with it the need to monitor patient safety related to use of these new products. With biosimilars, it is imperative that regulatory and industry come together to create the most appropriate pharmacovigilance and safety monitoring process, inclusive of
agreed-to naming guidance, to understand patient experience and to be able to proactively, quickly, and accurately identify potential safety signals across categories/medications. All constituents participating in the healthcare system are vested in the safe and appropriate use of biosimilars.
“By differentiating among biological products that have not been determined to be interchangeable, the goal of this naming convention is to help minimize inadvertent substitution. Inadvertent substitution may lead to unintended alternating or switching of biological products that have not been determined by FDA to be interchangeable. This naming convention may also facilitate pharmacovigilance for multiple biological products containing related drug substances when other means to track a specific dispensed product are not readily accessible…”
- U.S. FDA. Guidance for Industry: Nonproprietary Naming of Biological Products. Draft Guidance. 2015
Some experts believe that International Nonproprietary Names (INNs), while useful for tracing the safety of multisource drug classes, are insufficient for pharmacovigilance of biosimilars without the addition of a brand name or the manufacturer’s name.112 Naming of biosimilars continues to be a controversial topic, but is an important factor in pharmacovigilance.
Without distinguishable nonproprietary naming, product traceability can be degraded and safety signals can be attributed to the wrong product and inappropriately deemed class effects (e.g., effects found throughout the class of biologics).115
Distinguishable nonproprietary naming also increases the level of accountability of the manufacturer.
This is vital for both pharmacovigilance purposes as well as to ensure that non-interchangeable biologics are not inappropriately
substituted for reference products. As the FDA states in its Draft Guidance on biosimilar naming in August 2015:20

47
LOOKING AHEAD

48
Our current healthcare system places a premium on value, but this emphasis reflects more than just cost savings. Equally important are innovation and competition in the medical marketplace, which set the stage for development of alternatives to currently available medications. Biosimilars are uniquely poised to increase value in the healthcare system, both now and in the future. The ultimate impact of biosimilars will be driven by the answers to several key questions:
How will payers respond to the launch of biosimilars? Payer responses are likely to evolve on several issues, such as the relative speed in which formulary, coverage, and policy decisions will be addressed after the initial launch of a biosimilar. It also remains to be seen whether payers will take differing approaches to biosimilars based on the structural complexity of the compounds.72
How will employer purchasers influence uptake in the biosimilar market? There will be a combination of factors that determine uptake/utilization. Employers may not always follow national formularies,78 because they are willing to pay more to have PBMs administer a formulary that offers more choice to their employers and families.116
Will biosimilar market uptake reflect factors beyond opportunity for savings? Driving down costs of biologics is an ongoing initiative across the healthcare market. However, without a track record for U.S. market differentiation, it is unclear which key features and benefits—aside from cost-savings projections—will be most important in driving biosimilar uptake for particular stakeholder categories. For example, each biosimilar will be reviewed and approved on a case-by-case basis;9 which could potentially be influenced by a range of factors such as indication extrapolation, administration devices, and interchangeability designation.1 In addition, each manufacturer of a biosimilar may also influence market differentiation through experience, reliability, and support services.80
LOOKING AHEAD

49
How will appropriate use in therapy evolve as various stakeholders become more familiar with biosimilars? Biosimilars are likely to gain increased acceptance over time.72 The increasing influence of evidence-based practice guidelines on payer coverage policy117 suggests that inclusion of biosimilars in such guidelines will help stakeholders appreciate their appropriate role in therapy. Physician and patient acceptance are important for market uptake in the U.S.118
How will naming of biosimilars be determined, and what are the ramifications? The process of naming a biosimilar is not as straightforward as it may seem. For example, a distinguishable nonproprietary name could potentially reduce the likelihood of unintended switching or substitution, but might also negatively impact the prescribing and dispensing of the biosimilar.106
There are also questions about whether the nonproprietary names should be changed when manufacturers merge or when products change hands, or when biosimilars are designated
as interchangeable.106 As one can assume, the outcome of this debate will have wide impact, including to ePrescribing, EMRs, claims systems, historical databases, drug inventories, inventory management, and drug labels.
How will stakeholders value factors related to reimbursement? CMS’ final rule provides that, for reimbursement purposes, all biosimilars of a single reference product will be pooled within the same HCPCS code on the basis of weighted average sales.22
This pooling methodology does not distinguish the relative value of biosimilars with more or fewer approved indications.106 It is also blind to the market experience of individual biosimilars, and to market history of these biosimilars’ manufacturers.106
Each stakeholder, in evaluating and acting upon these and many other, future considerations, plays a critical role in developing a long-term, sustainable market for biosimilars in the U.S.
INT
RO
DU
CT
ION
B
IOSIM
ILAR
S LA
ND
SCA
PE
GA
ME
CH
AN
GE
RS
LOO
KIN
G A
HE
AD
UN
CO
VE
RIN
G T
HE
U
NK
NO
WN
S

50
GLOSSARY

51
AE An adverse event (AE) is an unexpected medical occurrence in a patient that happens during treatment with a drug or other therapy.6
ASP The average sales price (ASP) is a calculation of the weighted average of manufacturer’s sales price for a drug for all purchasers, net of price adjustments.119
Biologic A substance derived from a living organism or its products that is used in the diagnosis, prevention, or treatment of disease. Examples of biologic medicines include recombinant proteins, allergy shots, vaccines, and hematopoietic growth factors.6
Biosimilar A biological product that is highly similar to a U.S.-licensed reference biological product notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product.9
CD20 Cluster of differentiation 20 (CD20) is a protein found on B cells (a type of white blood cell). It may be found in higher than normal amounts in patients with certain types of B-cell lymphomas and leukemia.6
CDC The Centers for Disease Control and Prevention (CDC) is a U.S. federal government agency whose mission is to protect public health by preventing and controlling disease, injury, and disability.6
EGFR Epidermal growth factor receptor (EGFR) is a protein on the surface of cells that binds with epidermal growth factor and is therefore involved in cell division.6
EMA The European Medicines Agency (EMA) is a decentralized agency of the EU that is responsible for the scientific evaluation of medicines developed by pharmaceutical companies for use in the EU.120
ESA Erythropoiesis-stimulating agent (ESA) is a substance that stimulates the bone marrow to make more red blood cells.6
Extrapolation The process by which a proposed biosimilar product may be licensed in one or more additional conditions of use for which the reference product is licensed, if appropriate scientific justification is provided and the patent landscape allows for it. In practical terms, this means that the biosimilar can receive approval for multiple indications without undergoing clinical testing in those conditions as long as the reference product itself was approved in those conditions.9
FDA The U.S. Food and Drug Administration (FDA) is an agency in the U.S. federal government whose mission is to protect public health by assuring the safety, efficacy, and security of human and veterinary drugs, biological products, medical devices, the nation’s food supply, cosmetics, and nutritional supplements.6
G-CSF Granulocyte-colony stimulating factor (G-CSF) is a protein that stimulates bone marrow to produce granulocytes and stem cells and release them into the bloodstream.6
Generic Medicine A prescription drug that has the same active-ingredient formula as a brand-name drug. Generic drugs usually cost less than brand-name drugs and are rated by the FDA to be as safe and effective as brand-name drugs.121
HCPCS The Healthcare Common Procedure Coding System (HCPCS) is a collection of standardized codes that represent medical procedures, supplies, products, and services. The codes are used to facilitate the processing of health insurance claims.121
HER2 Human epidermal growth factor receptor 2 (HER2) is a protein involved in normal cell growth. It is found on some types of cancer cells, including breast and ovarian.6
GLO
SSAR
YR
EF
ER
EN
CE
S

52
HGH Human growth hormone (HGH) is a hormone secreted by the anterior pituitary gland that stimulates body growth generally and the lengthening of long bones in particular.6
IDN Integrated Delivery Network (IDN) is a network of healthcare organizations under a parent holding company.91
Interchangeability An “interchangeable” biological product is biosimilar to the reference product and can be expected to produce the same clinical result as the reference product in any given patient. If administered more than once to an individual (as many biological products are), the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product will not be greater than the risk of using the reference product without such alternation or switch. A biosimilar should only be considered interchangeable with the reference product if the FDA has approved it as a biosimilar and designated it as interchangeable, once additional criteria have been met.1
INN Allocated by the World Health Organization, an International Nonproprietary Name (INN) identifies pharmaceutical substances or active pharmaceutical ingredients. Each INN is a unique name that is globally recognized and is public property. A non-proprietary name is also known as a generic name.122
INF Interferon (INF) is a biological response modifier (a substance that can improve the body’s natural response to infections and other diseases). Interferons interfere with the division of cancer cells and can slow tumor growth.6
J-Code A subset of the most commonly used Healthcare Common Procedure Coding System (HCPCS) Level II Codes with a high-order value of “J” (See HCPCS).121
mAb A monoclonal antibody (mAb) is a type of protein made in the laboratory that can bind to substances in the body, including cancer cells.6
PBM Pharmacy benefit manager (PBM) is most often a third-party organization that provides prescription drug programs and services to help maximize drug effectiveness and contain drug expenditures by influencing the behaviors of prescribing physicians, pharmacists, and members. PBMs can be a service inside of an integrated healthcare system (See IDN).123
P&T Committee A Pharmacy and Therapeutics Committee is an advisory committee responsible for developing and maintaining a formulary and establishing and implementing policies on the use of drug products.124
Pharmacovigilance Procedures that monitor the safety of medicines to detect, assess, understand, and prevent adverse events or any other safety-related issues.122
Reference Product A reference product (aka, originator product) is a previously licensed product used as the comparator for head-to-head comparability studies with the similar biotherapeutic product in order to show similarity in terms of quality, safety, and efficacy. Is sometimes also referred to as the innovator or originator product that the biosimilar product is intended to copy.9
GLOSSARY

53
Switching A practice wherein a prescriber (or the prescriber’s delegate, under direct supervision of the prescriber) may change the prescription from one biologic medicine to another biologic medicine.
Substitution A practice allowed by law wherein a pharmacist may dispense an alternative biologic medicine for a prescribed biologic medicine without the prior approval of the prescriber. In some U.S. states, there is ongoing dialogue regarding post-dispensing notification and documentation. Private organization management of substitution may vary based on formulary decisions and other factors.
VEGF Vascular endothelial growth factor (VEGF) is a signal protein produced by cells that stimulates vasculogenesis and angiogenesis.6
WAC Wholesale acquisition cost (WAC) is an estimate of the manufacturer’s list price for a drug to wholesalers or direct purchasers, not including discounts or rebates.119
WHO The World Health Organization (WHO) is the directing and coordinating authority for health within the United Nations system. The WHO sets standards for disease control, health care, and medicines; conducts education and research programs; and publishes scientific papers and reports.125
GLO
SSAR
YR
EF
ER
EN
CE
S

54
REFERENCES

55
1. U.S. Food and Drug Administration. Information for Consumers
(Biosimilars). August 2015. Available at: http://www.fda.gov/Drugs/
DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/
ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/
ucm241718.htm. Accessed March 1, 2016.
2. Camacho LH, Frost CP, Abella E, et al. Biosimilars 101: considerations for
U.S. oncologists in clinical practice. Cancer Medicine 2014; 3(4): 889–899
3. Patient Protection and Affordable Care Act. December
2009. Available at: http://www.fda.gov/downloads/Drugs/
GuidanceComplianceRegulatoryInformation/UCM216146.pdf. Accessed on
March 1, 2016.
4. Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars. Ann
Oncol. 2008;19:411-419.
5. Declerck, PJ. Biologicals and biosimilars: a review of the science and its
implications. GaBI Journal. 2012;1(1):13-6.
6. National Cancer Institute. Dictionary of Cancer Terms. Available at: http://
www.cancer.gov/publications/dictionaries. Accessed on March 1, 2016.
7. U.S. Food and Drug Administration. Guidance for Industry: Biosimilars:
Additional Questions and Answers Regarding Implementation of the
Biologics Price Competition and Innovation Act of 2009. Draft Guidance.
May 2015. Available at: http://www.fda.gov/downloads/Drugs/.../
Guidances/UCM273001.pdf. Accessed on March 1, 2016.
8. U.S. Food and Drug Administration. Drug Price Competition and Patent
Term Restoration Act of 1984 (Hatch-Waxman Amendments). August 2003.
Available at: http://www.fda.gov/NewsEvents/Testimony/ucm115033.htm.
Accessed on March 1, 2016.
9. U.S. Food and Drug Administration. Guidance for Industry: Scientific
Considerations in Demonstrating Biosimilarity to a Reference Product.
Final Guidance. April 2015. Available at: http://www.fda.gov/downloads/
DrugsGuidanceComplianceRegulatoryInformation/Guidances/UCM291128.
pdf. Accessed on March 1, 2016.
10. U.S. Food and Drug Administration. FDA issues draft guidance on biosimilar
product development. News Release. February 9, 2012.Available at: http://
www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm291232.
htm. Accessed on March 1, 2016.
11. Sandoz. FDA accepts Sandoz application for biosimilar filgrastim. News
release. July 24, 2014. Available at: http://www.sandoz-biosimilars.com/en/
mediacenter/press_releases/140724_FDA_accepts_Sandoz_application_
for_biosimilar_filgrastim.shtml. Accessed on March 1, 2016.
12. Celltrion. Celltrion files for US FDA approval of Remsima. News Release.
August 11, 2014.Available at: http://www.celltrion.com/en/company/notice_
view.asp?idx=456&code=ennews&intNowPage=1&menu_num=&align_
year=all. Accessed on March 1, 2016.
13. Apotex. Apotex Announces FDA Has Accepted For Filing its Biosimilar
Application for Pegfilgrastim. News release. December 17, 2014. Available
at: http://www.apotex.com/global/about/press/20141217.asp. Accessed on
March 1, 2016.
14. U.S. Food and Drug Administration. FDA approves first biosimilar product
Zarxio. News Release. March 6, 2015. Available at: http://www.fda.gov/
NewsEvents/Newsroom/PressAnnouncements/ucm436648.htm. Accessed
on March 1, 2016.
15. Centers for Medicare and Medicaid Services. Food and Drug Administration
Approval of First Biosimilar Product. MLN Matters. Available at: https://
www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/
MLNMattersArticles/Downloads/SE1509.pdf. Accessed March 1, 2016.
16. Centers for Medicare and Medicaid Services. Medicaid Drug Rebate
Program Notice for Participating Drug Manufacturers. March 30, 2015.
Available at: http://www.medicaid.gov/Medicaid-CHIP-Program-
Information/By-Topics/Benefits/Prescription-Drugs/Downloads/Rx-
Releases/MFR-Releases/mfr-rel-092.pdf. Accessed March 1, 2016.
17. Centers for Medicare and Medicaid Services. Part D Requirements for
Biosimilar Follow-On Biological Products. March 30, 2015. Available at:
http://www.amcp.org/WorkArea/DownloadAsset.aspx?id=19358.
Accessed March 1, 2016.
GLO
SSAR
YR
EF
ER
EN
CE
S

56
REFERENCES
18. U.S. Food and Drug Administration. Guidances (Drugs)—Biosimilars.
November 2015. Available at: http://www.fda.gov/Drugs/
GuidanceComplianceRegulatoryInformation/Guidances/ucm290967.htm.
Accessed March 1, 2016.
19. Centers for Medicare and Medicaid Services. Medicare Program; Revisions
to Payment Policies under the Physician Fee Schedule and Other Revisions
to Part B for CY 2016. Available at: https://www.cms.gov/Medicare/
Medicare-Fee-for-Service-Payment/PhysicianFeeSched/PFS-Federal-
Regulation-Notices-Items/CMS-1631-P.html. Accessed March 1, 2016.
20. U.S. Food and Drug Administration. Guidance for Industry.
Nonproprietary Naming of Biological Products. Draft Guidance.
August 2015. Available at: http://www.fda.gov/downloads/drugs/
guidancecomplianceregulatoryinformation/guidances/ucm459987.pdf.
Accessed March 1, 2016.
21. Sandoz. Court ruling paves the way for launch of Sandoz’s Zarxio as first
U.S. biosimilar. News Release. July 22, 2015. Available at: http://www.sandoz.
com/media_center/press_releases_news/global_news/2015-07-22-court-
ruling-paves-the-way-for-launch-of-sandozs-zarxio-as-first-us-biosimilar.
shtml. Accessed March 1, 2016.
22. Centers for Medicare and Medicaid Services. Proposed policy, payment,
and quality provisions changes to the Medicare Physician Fee Schedule
for Calendar Year 2016. October 30, 2015. Available at: https://www.cms.
gov/Newsroom/MediaReleaseDatabase/Fact-sheets/2015-Fact-sheets-
items/2015-10-30-2.html. Accessed March 1, 2016.
23. Slavitt A. MedPAC. File Code CMS-1631-P. Letter. September 8, 2016.
Available at: http://www.medpac.gov/documents/comment-letters/medpac-
comment-on-cms’s-proposed-rule-on-the-physician-fee-schedule-and-
other-revisions-to-part-b.pdf?sfvrsn=0. Accessed April 5, 2016.
24. Hospira. Hospira Submits New Biologics License Application to U.S. FDA
for Proposed Epoetin Alfa Biosimilar. News Release. January 12, 2015.
Available at: http://phx.corporate-ir.net/phoenix.zhtml?c=175550&p=irol-
newsArticle&ID=2006860. Accessed March 1, 2016.
25. Apotex. Apotex Announces FDA Has Accepted For Filing its Biosimilar
Application for Filgrastim. News Release. February 17, 2015. Available at:
http://www.apotex.com/global/about/press/20150217-2.asp. Accessed
March 1, 2016.
26. Amgen. Data on File. News Release. 25 November 2015.
27. Sandoz. FDA accepts Sandoz regulatory submission for a proposed
biosimilar etanercept. News Release. October 2, 2015. Available at: https://
www.novartis.com/news/media-releases/fda-accepts-sandoz-regulatory-
submission-proposed-biosimilar-etanercept. Accessed March 1, 2016.
28. U.S. Food and Drug Administration. 2015-2016 Advisory Committee
Tentative Meetings. November 2015.Available at: http://www.fda.gov/
AdvisoryCommittees/ucm414515.htm. Accessed March 1, 2016.
29. Amgen. Data on File. 2016 Potential Approvals: Apotex. November 13, 2015.
30. Wang J, Chow SC. On the Regulatory Approval Pathway of Biosimilar
Products. Pharmaceuticals 2012, 5, 353-368.
31. European Medicines Agency. European public assessment reports. February
7, 2016. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/
medicines/landing/epar_search.jsp&mid=WC0b01ac058001d125. Accessed
on March 1, 2016.
32. GABI. Biosimilars approved in Australia. February 5, 2016. Available at:
http://www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-
Australia. Accessed on March 1, 2016.
33. GABI. Biosimilars approved in Japan. February 19, 2016. Available at: http://
www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Japan.
Accessed on March 1, 2016.

57
34. Health Canada. Summary Basis of Decision Documents: Drugs. February 4,
2016. Available at: http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/
drug-med/index-eng.php. Accessed on March 1, 2016.
35. Dalgaard K. et al. Biosimilars seven years on: Where are we and what’s next.
[White Paper] February 2013. Available at: http://www.mckinsey.com/~/
media/mckinsey/dotcom/client_service/pharma%20and%20medical%20
products/pmp%20new/pdfs/biosimilars%20seven%20years%20on_
white%20paper.ashx. Accessed on March 1, 2016.
36. Long, D. Impact of Biosimilars on the Pharmaceutical Industry. September
2015. Presented at Magellan Rx Specialty Summit; September 1-3, 2015;
New York, NY.
37. IMS. Assessing biosimilar uptake and competition in European markets.
October 2014. Available at: https://www.imshealth.com/files/web/
IMSH%20Institute/Healthcare%20Briefs/Assessing_biosimilar_uptake_and_
competition_in_European_markets.pdf. Accessed on March 1, 2016.
38. Cber. World Economic League Table 2015 Highlights.December 26, 2014.
Available at: http://www.cebr.com/reports/world-economic-league-
table-2015/. Accessed on March 1, 2016.
39. Bacharidis N. Tendering & Contracting for generic & branded medicines
across Europe—a comparison and benchmarking. Presented in November
2011; Birmingham, UK. Available at: http://www.ghp.org.uk/ContentFiles/
PDIG%20Nico%20Bacharidis%20Presentation%20-%20Nov%202011.pdf.
Accessed on March 1, 2016.
40. Neiderwieser D, Schmitz S. Biosimilar agents in oncology⁄haematology:
from approval to practice. Eur Jrnl Hematol. 2010. 277-288.
41. EuropaBio. EuropaBio’s Statement on French Substitution Policy. January
24, 2014. Available at: http://www.europabio.org/news/europabios-
statement-french-substitution-policy. Accessed on March 1, 2016.
42. GABI. Australian guidelines for biosimilars. August 7, 2015. Available at:
http://gabionline.net/Guidelines/Australian-guidelines-for-biosimilars.
Accessed on March 1, 2016.
43. Commonwealth of Australia. Economics Legislation Committee - National
Health Amendment (Pharmaceutical Benefits) Bill 2015 [Provisions].
June 2015.
44. Health and Human Services. 340B Drug Pricing Program Omnibus
Guidance. August 28, 2015. Available at: https://www.gpo.gov/fdsys/pkg/
FR-2015-08-28/pdf/2015-21246.pdf. Accessed March 1, 2016.
45. Pharmaceutical Benefits Scheme. Letter to pharmacists - Biosimilar
infliximab on the PBS. 1 December 2015. Available at: http://www.pbs.gov.
au/info/publication/factsheets/biosimilars/letter-to-pharmacist-biosimilar-
infliximab-on-the-pbs-2015-12. Accessed on February 7, 2016.
46. Pharmaceutical Benefits Scheme. Biosimilar Medicines—Factsheet for
Healthcare Professionals. Available at: http://www.pbs.gov.au/publication/
factsheets/biosimilars/biosimilars-factsheet-healthcare-professionals.pdf.
Accessed on February 4, 2016.
47. Canada Market Access Briefing. [White Paper] 2015. Available at: http://
www.precisionadvisors.com/wp-content/uploads/2015/03/Canada-White-
Paper.pdf. Accessed on February 7, 2016.
48. Health Canada. Interchangeability/Substitutability of Subsequent Entry
Biologics (SEBs). [Letter] July 29, 2010.
49. Zafar SY, Peppercorn JM, Schrag D, et al. The financial toxicity of cancer
treatment: a pilot study assessing out-of-pocket expenses and the insured
cancer patient’s experience. Oncologist 2013;18:381-90.
50. Rickwood S, Di Biase S. Searching for Terra Firma in the Biosimilars and
Non-Original Biologics Market. [White Paper] 2013.
51. Data on File, Amgen. IMS Health MIDAS 2014. August 20, 2015.
52. Data on File, Amgen. Comparison of Biosimilar Rules and Guidelines.
December 4, 2015.
53. U.S. Food and Drug Administration. News events: NDA, ANDA, IND & RDRC.
Available at: www.fda.gov/downloads/Drugs/NewsEvents/UCM245363.pdf.
Accessed on February 7, 2016.
54. European Medicines Agency. Guideline on Similar Biological Medicinal
Products. October 23, 2014. Available at: http://www.ema.europa.eu/docs/
en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf.
Accessed on March 1, 2016.
55. Kozlowski S. U.S. FDA Perspectives on Biosimilar Biologic Products. Presented
at: Biotechnology Technology Summit. June 13, 2014; Rockville, MD.
GLO
SSAR
YR
EF
ER
EN
CE
S

58
REFERENCES
56. National Conference of State Legislatures. State Laws and Legislation
Related to Biologic Medications and Substitution of Biosimilars. February
8, 2016. Available at: http://www.ncsl.org/research/health/state-laws-and-
legislation-related-to-biologic-medications-and-substitutionof-biosimilars.
aspx. Accessed on March 1, 2016.
57. Neas RG, Gaugh DR. What’s in a name?—the identification of biologic
products. Journal of Generic Medicines: 2013 vol. 10 no. 3-4 148-158.
58. Casadevall N, Felix T, Strober B, Warnock D. Similar Names for Similar
Biologics. BioDrugs. 2014:Oct;28(5):439-44.
59. Grampp G, Ramanan S. The Diversity of Biosimilar Design and Development:
Implications for Policies and Stakeholders. BioDrugs. 2015 Nov 18.
60. U.S. Food and Drug Administration. Purple Book: Lists of Licensed
Biological Products with Reference Product Exclusivity and
Biosimilarity or Interchangeability Evaluations. 3 September 2015.
Available at: http://www.fda.gov/drugs/developmentapprovalprocess/
howdrugsaredevelopedandapproved/approvalapplications/
therapeuticbiologicapplications/biosimilars/ucm411418.htm. Accessed on
February 7, 2016.
61. Olson K. Biosimilar naming and labeling: a study of U.S. pharmacists.
October 2015. Available at: http://docs.house.gov/meetings/IF/
IF14/20160204/104408/HHRG-114-IF14-20160204-SD009.pdf. Accessed
March 1, 2016.
62. Centers for Medicare & Medicaid Services. CMS Finalizes Hospital
Outpatient and Ambulatory Surgical Center Policy and Payment Changes,
Including Changes to the Two-Midnight Rule and Quality Reporting for 2016.
October 30, 2015.
63. Mulcahy AW, Predmore Z and Mattke S. The Cost Savings Potential
of Biosimilar Drugs in the United States. Santa Monica, CA: RAND
Corporation, 2014.
64. Collins S. Specialty Pharmacy Management Will Become More Intense.
Managed Care Magazine. October 2010. Available at: http://www.
managedcaremag.com/archives/2010/10/specialty-pharmacy-management-
will-become-more-intense. Accessed on March 1, 2016.
65. Milliman. Commercial Specialty Medication Research: 2016 Benchmark
Projections. Research Report. 28 December 2015. Available at: http://
us.milliman.com/uploadedFiles/insight/2016/commercial-specialty-
medication-research.pdf. Accessed on March 1, 2016.
66. Artemetrx. Specialty Drug Management. 22 April 2015. Available at: http://
businesshealthcaregroup.org/wp-content/uploads/2009/11/Wisconsin-
External-Slides-April-2015-FINAL.pdf. Accessed on March 1, 2016..
67. UnitedHealth Center for Health Reform & Modernization. The Growth of
Specialty Pharmacy: Current trends and future opportunities. Issue Brief.
April 2014. Available at: http://www.unitedhealthgroup.com/~/media/uhg/
pdf/2014/unh-the-growth-of-specialty-pharmacy.ashx. Accessed on
March 1, 2016.
68. Stern D, Reissman, D. Specialty Pharmacy Cost Management Strategies of
Private Health Care Payers. J Manag Care Pharm. 2006;12(9):736-44
69. Olson K. ASBM Labeling Survey. February 2015. ISR Report. Available at:
http://docs.house.gov/meetings/IF/IF14/20160204/104408/HHRG-114-IF14-
20160204-SD010.pdf. Accessed on March 1, 2016.
70. Amgen, Data on File. IMS Health National Sales Perspective (NSP) Audit
October 29 2015.
71. Amgen, Data on File. 2015 Biosimilars Roundtable Advisory Board.
September 26, 2015. Dallas, TX.
72. Ventola CS. Evaluation of Biosimilars for Formulary Inclusion - Factors for
Consideration by P&T Committees. P T. 2015 Oct; 40(10) 680–689

59
73. Milliman. Understanding biosimilars and projecting the cost savings
to employers. December 2011. Available at: http://us.milliman.com/
uploadedFiles/insight/health-published/understanding-biosimilars.pdf.
Accessed on March 1, 2016.
74. Centers for Medicare and Medicaid Services. How Medicare Prescription
Drug Plans & Medicare Advantage Plans with Prescription Drug Coverage
(MA-PDs) Use Pharmacies, Formularies, & Common Coverage Rules.
October 2015. Available at: https://www.medicare.gov/Pubs/pdf/11136.pdf.
Accessed: March 1, 2016.
75. ExpressScripts. Plan Design Review Guide. December 2010. Available at:
https://www.express-scripts.com/art/corporate/pdf/plan_design_review_
guide.pdf. Accessed on March 1, 2016.
76. Academy of Managed Care Pharmacy. Specialty Pharmacy and Biosimilars.
[Presentation] February 2015. Available at: http://www.amcp.org/
WorkArea/DownloadAsset.aspx?id=19133. Accessed on March 1, 2016.
77. Avalere. Value Based Formulary Design: Is Premera A Voice Crying
in the Wilderness? Presented at ISPOR Annual Meeting; May 16-
20, 2015; Philadelphia. Available at: http://www.ispor.org/Event/
GetReleasedPresentation/327. Accessed on March 1, 2016.
78. Teton County Navitus. PHARMACY BENEFIT MANAGEMENT SERVICES
AGREEMENT (10202015). January 1, 2016. Available at: http://www.tetonwyo.
org/cc/docs/StaffReports/2015-BCC/10-20/1020-15-NavitusContract.pdf.
Accessed on March 1, 2016.
79. Fitch Ratings. 2015 Outlook: U.S. Nonprofit Hospitals and Healthcare
Systems. December 9, 2014. Available at: http://www.calhospital.org/sites/
main/files/file-attachments/fitch_2015_healthcare_outlook.pdf. Accessed
on March 1, 2016.
80. Griffith N, McBride A, Stevenson JG, Green L. Formulary Selection Criteria
for Biosimilars Considerations for US Health-System Pharmacists. Hospital
Pharmacy. 2014;49(9) 813-825.
81. Jacobs IA, Singh E, Swell KL et al. Patient Understanding and Attitudes
About Biosimilars: An International Cross-Sectional Survey. Value in Health.
2015 (18) A335 – A766.
82. QuantiaMD. Reading the Signs: A Roadmap for Engaging Physicians in the
Biosimilars Discussion. Web-based survey. March 2016.
83. Minutolo R, Borzumati M, Sposini S, et al. Am J Kidney Dis. 2016. Feb 13.
84. Global Healthy Living Foundation. Patient Perspectives on Medication
Switching for Non-Medical Reasons: A survey of stabilized patients by
the Global Health Living Foundation. April 23, 2015. Available at: http://
www.50statenetwork.org/wp-content/uploads/2015/04/GHLF-Switching-
Stable-Patients-Survey_Summary.pdf. Accessed on March 1, 2016.
85. Floodmark et al; Switching from originator to biosimilar human growth
hormone using dialogue teamwork: single-center experience from Sweden.
Biol Ther (2013) 3: 35-43.
86. Panaccione R, Ghosh S. Optimal use of biologics in the management of
Crohn’s disease. Therapeutic Advances in Gastroenterology. 2010;3(3):179-
189.
87. U.S. Food and Drug Administration Voice Blog. FDA Offers Free, Continuing
Education Course to Help Health Care Providers Understand ‘Biosimilars’.
February 18, 2016. Available at: http://blogs.fda.gov/fdavoice/index.
php/2016/02/fda-offers-free-continuing-education-course-to-help-health-
care-providers-understand-biosimilars/. Accessed on March 1, 2016.
88. Sandoz. ZARXIO™ (filgrastim-sndz). [Prescribing Information] March
2015. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/
label/2015/125553lbl.pdf. Accessed on March 1, 2016.
89. Academy of Managed Care Pharmacy (AMCP). AMCP Format for Formulary
Submissions. 2015. Available at: http://www.amcp.org/Tertiary.aspx?id=8435.
Accessed on March 1, 2016.
90. Medicare Payment Advisory Commission (MedPAC). Overview of the 340B
Drug Pricing Program, May 2015. Available at: http://www.medpac.gov/
documents/reports/may-2015-report-to-the-congress-overview-of-the-
340b-drug-pricing-program.pdf?sfvrsn=0. Accessed on March 1, 2016.
91. The Advisory Board Company. Post-Acute Care Cheat Sheet: Integrated
Delivery Networks. Available at: https://www.advisory.com/research/post-
acute-care-collaborative/members/resources/cheat-sheets/integrated-
delivery-networks. Accessed on March 1, 2016.
GLO
SSAR
YR
EF
ER
EN
CE
S

60
REFERENCES
92. Health Resources and Services Administration. 42 CFR Part 10: 340B Drug
Pricing Program Ceiling Price and Manufacturer Civil Monetary Penalties
Regulation. June 17, 2015. Available at: https://www.gpo.gov/fdsys/pkg/FR-
2015-06-17/pdf/2015-14648.pdf. Accessed March 1, 2016.
93. 340B Health Organization. About. Available at: http://www.340bhealth.org/
about. Accessed on January 3, 2016. Accessed on March 1, 2016.
94. Government Accountability Office. Medicare Part B Drugs. Report to
Congressional Requesters. June 2015. Available at: http://gao.gov/
assets/680/670676.pdf. Accessed on March 1, 2016.
95. National Association of Community Health Centers (NACHC).
Understanding the 340B Program: A primer for health centers. May 2011.
Available at: http://www.nachc.org/client/documents/5.11%20340%20
Manual%20Primer%20for%20Health%20Centers1.pdf. Accessed on March 1,
2016.
96. ASHP Expert Panel on Drug Product Shortages. ASHP Guidelines on
Managing Drug Product Shortages in Hospitals and Health Systems. Am J
Health Syst Pharm. 2009 Aug 1;66(15):1399-406 at 1399
97. Becker DJ, Talwar S, Levy BP, et al. Impact of Oncology Drug Shortages
on Patient Therapy: Unplanned Treatment Changes. Journal of Oncology
Practice. 2013;9(4):e122-e128 at e125-6
98. Government Accountability Office. Drug Shortages: Public Health Threat
Continues, Despite Efforts to Help Ensure Product Availability. February
2014. Available at: http://www.gao.gov/assets/670/660785.pdf. Accessed
on March 1, 2016.
99. Yurukoglu, Liebman and Ridley. The role of government reimbursement in
drug shortages. Nat. Bur. Econ.Res. 2016.
100. Pfizer. Pfizer Completes Acquisition of Hospira. News Release. September 3,
2015. Available at: http://www.pfizer.com/news/press-release/press-release-
detail/pfizer_completes_acquisition_of_hospira. Accessed on March 1, 2016.
101. Epirus. Epirus and Zalicus Announce Merger Agreement. News Release.
April 16, 2014. Available at: http://www.businesswire.com/news/
home/20140416005356/en/Epirus-Zalicus-Announce-Merger-Agreement.
Accessed on March 1, 2016.
102. Amgen. Amgen and Watson Announce Collaboration to Develop and
Commercialize Oncology Biosimilars. News Release. December 19, 2011.
Available at: http://www.prnewswire.com/news-releases/amgen-and-
watson-announce-collaboration-to-develop-and-commercialize-oncology-
biosimilars-135880803.html. Accessed on March 1, 2016.
103. Johnson SJ. Pfizer-Allergan deal could reduce biosimilar cost savings.
Modern Healthcare. December 5, 2015. http://www.modernhealthcare.com/
article/20151205/MAGAZINE/312059970?template=print. Accessed on
March 1, 2016.
104. Schmidt R, Suzuki S, Neuman K. Medicare Drug Spending. 11 September
2015. Available at: http://www.medpac.gov/documents/september-2015-
meeting-presentation-medicare-drug-spending.pdf?sfvrsn=0. Accessed on
March 1, 2016.
105. Centers for Medicare and Medicaid Services. Medicare Program; Part B Drug
Payment Model. March 8, 2016. Available at: https://s3.amazonaws.com/
public-inspection.federalregister.gov/2016-05459.pdf. Accessed on March
10, 2016.
106. House of Representatives Subcommittee on Health Committee on
Energy and Commerce. Examining Implementation of the Biologics
Price Competition and Innovation Act. [Transcript] February 4, 2016;
Washington, D.C. Available at: http://docs.house.gov/meetings/IF/
IF14/20160204/104408/HHRG-114-IF14-Transcript-20160204.pdf. Accessed
on March 1, 2016.

61
107. Centers for Medicare and Medicaid Services. Medicare Prescription Drug
Benefit Manual: Chapter 6 – Part D Drugs and Formulary Requirements.
Available at: https://www.cms.gov/Medicare/Prescription-Drug-Coverage/
PrescriptionDrugCovContra/Downloads/Chapter6.pdf. Accessed on
January 19, 2016
108. Kopenski F. Understanding biosimilars and projecting the cost savings
to employers. December 2011. Available at: http://us.milliman.com/
uploadedFiles/insight/health-published/understanding-biosimilars.pdf.
Accessed on March 1, 2016.
109. Medicare Rights. Closing the Doughnut Hole. January 1, 2014. Available at:
https://www.medicare.gov/Pubs/pdf/11493.pdf. Accessed on March 1, 2015.
110. Lucio SD, Stevenson JG, Hoffman JM. Biosimilars Primer for the Health-
System Pharmacist. Am J Health Syst Pharm. 2013;70(22)2004-2017.
111. Schellekens H. Biosimilar therapeutics-what do we need to consider? NDT
Plus. 2009;2(Suppl_1):i27-i36.
112. Zuñiga L, Calvo B. Biosimilars: pharmacovigilance and risk management.
Pharmacoepidemiol Drug Saf. 2010 Jul;19(7):661-9 at 663
113. Grampp G, Machaon B, Felix T, et al. Active and passive surveillance of
enoxaparin generics: a case study relevant to biosimilars. Expert Opin Biol
Ther. 2014;14(3):1-12.
114. Liu M, McPeek Hinz ER, Matheny ME, et al. Comparative analysis of
pharmacovigilance methods in the detection of adverse drug reactions
using electronic medical records. JAMIA. 2013;20(3):420-426.
115. Casadevall N, Edwards R, Felix T, et al. Pharmacovigilance and biosimilars:
considerations, needs and challenges Expert Opin Biol Ther. 2013(13):1039-
1047.
116. Miller S. Restrictive Formularies and Drug Exclusions: In Whose Interest?
November 20, 2014. Available at: https://www.shrm.org/hrdisciplines/
benefits/articles/pages/drug-formularies-exclusions.aspx. accessed on
March 1, 2016.
117. McCauley JL. Guidelines and Value-Based Decision Making: An Evolving
Role for Payers. N C Med J. 2015;76(4):243-246.
118. European Commission (EC). Pharmaceutical Industry: A strategic sector for
the European Economy. November 2014.
119. Mattingly J. Understanding Drug Pricing. US Pharm. 2012;37(6)(Generic
Drug Review suppl):4045.
120. European Medicines Agency. About Us. Available at: http://www.ema.
europa.eu/ema/index.jsp%3Fcurl=pages/about_us/general/general_
content_000235.jsp%26mid=. Accessed on March 1, 2016.
121. Centers for Medicaid and Medicare Services. Glossary. Available at: https://
www.cms.gov/apps/glossary/. Accessed on March 1, 2016.
122. World Health Organization. Essential medicines and health products: Terms.
Available at: http://www.who.int/medicines/services/inn/en/ Accessed on
March 1, 2015.
123. Kingery. The Basics of Pharmacy Benefits Management. Anthem Virginia
CE Forum. 2009. Available at: https://www.anthem.com/shared/va/f5/s1/t0/
pw_b135247.pdf?refer=ahpagent&state=va. Accessed on March 14, 2016.
124. Tyler LS. ASHP Guidelines on the Pharmacy and Therapeutics Committee
and the Formulary System. Am J Health-Syst Pharm. 2008; 65:1272-83
125. World Health Organization. About Who. Available at: http://www.who.int/
about/en/. Accessed on March 1, 2016.
GLO
SSAR
YR
EF
ER
EN
CE
S

62
Amgen Inc. One Amgen Center Drive
Thousand Oaks, CA 913210-1799www.amgen.com
© 2016 Amgen. All rights reserved. USA-BIO-122466
KEY QUESTIONS TO CONSIDER WHEN EVALUATING BIOSIMILARS
With the evolution of the biosimilar marketplace and remaining unknowns, there are still many considerations that have short and long-term implications for building a sustainable market for biosimilars in the U.S. Below is a foundation of key elements to help
guide you as you consider the appropriate utilization of a biosimilar for you and your organization:
HOW WILL…
…an interchangeability designation, or lack thereof, impact decisions made around biosimilars?
…formulary decision makers manage biosimilars?
…patient and provider understanding of biosimilars impact uptake?
…contracting change as more biosimilars are introduced?
…biosimilar utilization pose challenges to existing information technology systems and processes?
…reimbursement change as more biosimilars enter the marketplace?
…the naming process impact the prescribing and dispensing of biosimilars?





























