Embed Size (px)
Citation preview

D. X. Hu Burns GroupTowards Catalytic Enantioselective Halogenation of Alkenes
Catalytic Enantioselective Halogenation
Literature Review
Organic Synthesis
October 6th, 2012
Relevant Problems:
Challenges in Enantioselective
Halofunctionalization
Strategies for Catalytic
Enantioselective Halofunctionalization
Research Proposal
Tens of thousands of chiral halogenated compounds
have been isolated from nature, with most of them
from marine sources. Given that the sources of these
natural products are difficult to trace due to the
difficulty in culturing marine bacteria, how can
scientists recreate these on large scale in the
laboratory for further research on their functions?
10, 20, 50 Years from Now…?
Today it is possible to couple unactivated secondary alkyl
halides using transition-metal methodology. Could the
development of simple methods for enantioselective alkyl
halide synthesis help the development of stereoretentive alkyl
halide cross-coupling methodology?
Could the development of stereoretentive alkyl halide cross-
coupling methodology combined with a “chiral halide pool”
simplify the synthesis of “3D” molecules in the way palladium-
catalyzed cross-coupling trivialized the synthesis of many
“2D” molecules?

D. X. Hu Burns GroupLiterature Review: Catalytic Enantioselective Halogenation
Challenges in Enantioselective Halogenation:Facing Reality:
How many methods can you think of for secondary alkyl
halide synthesis?
How many mechanisms of halogenation can you think of?
Why are there so few?
Inter-alkene halonium transfer is fast, resulting in rapid loss of
optical activity:
For a bis-adamantyl olefin with Br2 and I2, the second-order
rate constants for inter-alkene transfer were found to be ~2 x
106 and ~7.6 x 106 M-1 s-1 at -80°C(!!) with most of the rate
suppression due to a high entropy of activation (-21 eu). The
incredibly low enthalpic barrier (~1.8 kcal/mol) results in the
rate of reaction being dictated primarily by steric factors and
the rate of diffusion!
Halogens rarely form more than one covalent bond, resulting
in fewer orbital geometries for reactivity. More on this later.

D. X. Hu Burns GroupLiterature Review: Catalytic Enantioselective Halogenation
Challenges in ES Halogenation (cont’d):Challenges in ES Halogenation (cont’d):
Limited range of mechanisms to work with to due the
following requirements:
Process requires a non-stereotopic substrate (i.e.
true reagent control) or the formation of a non-
stereotopic intermediate (i.e. dynamic kinetic
resolution): this means we must invoke planar starting
materials or intermediates (alkenes, carbocations, or
rapidly interconverting radicals)
Halonium ions generated by solvolysis can be trapped
enantiospecifically in the absence of olefins, but addition of
olefin results in erosion of enantiospecificity (es):
This is detrimental to catalytic processes in which there is
always an excess of alkene relative to halonium. This effect
has been observed to be concentration dependent:
The rate of halonium exchange may also be dependent on a
number of other factors, such as counterion coordination
ability, solvent nucleophilicity, and the presence of added
Lewis bases.
Neutral halogens are non-basic compared to
chalcogens or pnictogens. This limits concerted
“halene”-type reactivity.

D. X. Hu Burns GroupLiterature Review: Catalytic Enantioselective Halogenation
Challenges in ES Halogenation (cont’d):Challenges in ES Halogenation (cont’d):
As a consequence of a lack of bonding modes and extreme
energies of oxidation state >X(I), there are few orbital
geometries available for reactivity, making transfer of
stereochemistry difficult (i.e. linear σ* maximizes distance
from covalent delivery partner).
Due to their valence saturation, halides cannot carry a
“leaving group,” further restricting concerted “halene”-type
reactivity.
As a consequence of the lack of a stable “halene,” any
enantioselective C-Br bond formation must be accompanied
by a second C-X functionalization process. This results in an
inevitable problem of regioselectivity except with C2-
symmetric substrates.
As a corollary, any stoichiometric halogenation agent
leaves behind a radical or anionic partner that must be
accounted for.
Due to their incredible reactivity, non-enantioselective
background reaction rates tend to be high.
Is there any hope?
With conjugated substrates (styrenyl or cinnamyl-type),
diastereomers often form through the intermediacy of an open
bromocarbocation.

D. X. Hu Burns GroupLiterature Review: Catalytic Enantioselective Halogenation
Type I: Chiral Lewis Base CatalysisCurrent Catalytic ES Halogenation Methods:
Current strategies for catalysis of halofunctionalization
revolve around four paradigms:
Early studies focused on the use of preformed halogen/amine
complexes:
For an enantioselective process, tactics are needed to
maintain a chiral environment in the product-determining
step:
Such studies demonstrated the importance of both the
activator and the counterion:

D. X. Hu Burns GroupLiterature Review: Catalytic Enantioselective Halogenation
Type II: Chiral Ion Pairing CatalysisType I: Chiral Lewis Base Catalysis (cont’d)
Electron-rich phosphines were found to be catalytically active
with NIS and NBS for enantioselective halogenative polyene
cyclizations with greater catalytic activity in DCM and toluene.
Conversely, stoichiometric quantities of chiral phosphine were
necessary for high enantioselectivity with better performance
in toluene than in DCM.
Phosphate bases have been shown to catalyze
haloetherification, with hypo-halogen species being
suggested as possible intermediates. It is also possible that
interaction with the alcohol accelerates the ring-closure step
and/or that anion exchange is faster than cyclization of an
intermediate bromiranium.
The authors propose a stereochemical model based on
preferential approach to the phosphoramidite assuming that
halogen delivery is the product-determing step. Denmark and
co-workers, however, suggest that the product-determining
step is not delivery of the halogen but the actual C-C-bond-
forming cyclization.

D. X. Hu Burns GroupLiterature Review: Catalytic Enantioselective Halogenation
Type II: Chiral Ion Pairing Catalysis (cont’d)Type II: Chiral Ion Pairing Catalysis (cont’d)
The same catalyst under slightly different conditions provided
the same result. Denmark and Shi provide different
mechanistic proposals for stereoinduction – it is likely that
one or both are wrong.
An intermolecular coupling using this strategy has been
attempted, and while product was formed in fair ee the
product was formed in poor yield due to trapping of the
bromiranium by the catalyst.
Denmark’s proposal:
Shi’s proposal:

D. X. Hu Burns GroupLiterature Review: Catalytic Enantioselective Halogenation
Type II: Chiral Ion Pairing Catalysis (cont’d)Type II: Chiral Ion Pairing Catalysis (cont’d)
A subset of chiral ion pairing catalysis is phase-transfer
catalysis. This strategy has been used effectively in
enantioselective fluorination to form oxazoline compounds.
The use of an insoluble halogenation reagent is one strategy
for preventing non-catalyzed background reaction. Tailoring
the delivery agent also allowed bromocyclization and
iodocyclizations to take place. While phase-transfer
fluorination almost certainly involves fluoronium delivery as
the product-determining step, this is not necessarily the case
for bromination or iodination.

D. X. Hu Burns GroupLiterature Review: Catalytic Enantioselective Halogenation
Type II: Chiral Ion Pairing Catalysis (cont’d)Type II: Chiral Ion Pairing Catalysis (cont’d)
It has been shown that a chiral hydrogen bond donor can
catalyze a reaction through activation of a halogenating agent
while also inducing product selectivity by leaving a chiral
anion after halogen delivery.
Templation of carboxylic acid substrates with a chiral base
has been employed successfully for lactonization reactions.
Alcohols do not cyclize selectively.
Suggested transition state:

D. X. Hu Burns GroupLiterature Review: Catalytic Enantioselective Halogenation
Type III: Hydrogen-Bonding Catalysts (cont’d)Type III: Hydrogen-Bonding Catalysts
Most Lewis-base catalysts also need hydrogen-bonding
coordination or activation for successful stereochemical
transfer.
In many cases the source of selectivity-induction is not well
understood.

D. X. Hu Burns GroupLiterature Review: Catalytic Enantioselective Halogenation
Type III: Hydrogen-Bonding Catalysts (cont’d)Type III: Hydrogen-Bonding Catalysts (cont’d)
Many Lewis-basic catalysts are susceptible to decay by the
stoichiometric oxidants. Incubation of quinuclidine catalysts
with NBS for a few hours prior to introduction of substrate
results in dramatically diminished ee.
There is experimental evidence that the stoichiometric
oxidant’s counterion is associated with the quinuclidine
complexes during the product-determining step.
D. X. Hu Burns GroupLiterature Review: Catalytic Enantioselective Halogenation
Type III: Hydrogen-Bonding Catalysts (cont’d)Type III: Hydrogen-Bonding Catalysts (cont’d)
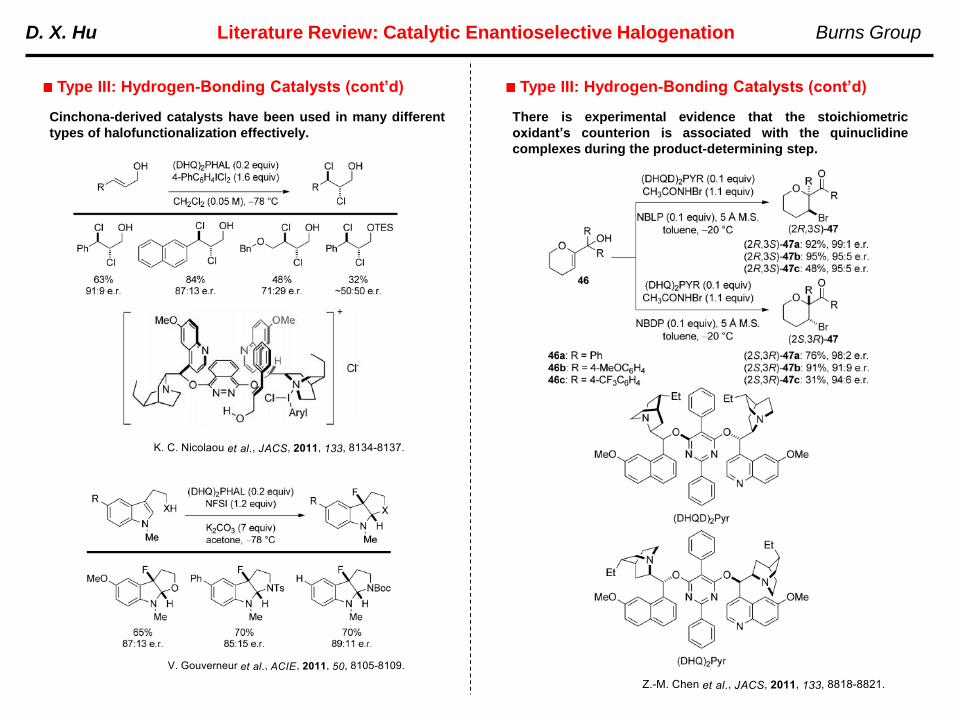
Cinchona-derived catalysts have been used in many different
types of halofunctionalization effectively.
There is experimental evidence that the stoichiometric
oxidant’s counterion is associated with the quinuclidine
complexes during the product-determining step.
D. X. Hu Burns GroupLiterature Review: Catalytic Enantioselective Halogenation
Type IV: Lewis-Acid MediatedType III: Hydrogen-Bonding Catalysts (cont’d)
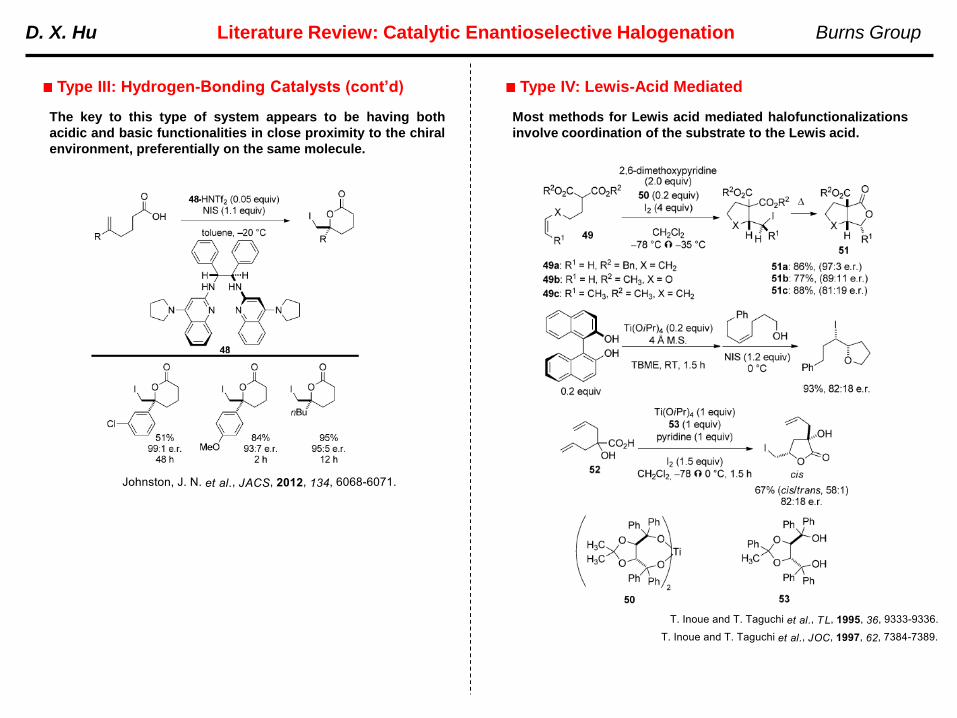
The key to this type of system appears to be having both
acidic and basic functionalities in close proximity to the chiral
environment, preferentially on the same molecule.
Most methods for Lewis acid mediated halofunctionalizations
involve coordination of the substrate to the Lewis acid.

D. X. Hu Burns GroupLiterature Review: Catalytic Enantioselective Halogenation
Type IV: Lewis-Acid MediatedType IV: Lewis-Acid Mediated (cont’d)
A key point is that Lewis acid mediated processes must
generate a chiral counterion.
In more specific cases, Lewis acid activation probably
proceeds by activating carbonyl groups in the substrate rather
than the halogen-donor.

D. X. Hu Burns GroupLiterature Review: Catalytic Enantioselective Halogenation
Other Important Precedents (cont’d):Other Important Precedents:
Alpha-halogenation of carbonyls is another popular field of
research. Organocatalysts and metal catalysts have been
employed to great effect.