Embed Size (px)
Citation preview

Biochimica et Biophysica Acta 1834 (2013) 2736–2749
Contents lists available at ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r .com/ locate /bbapap
Probing the amino acids critical for protein oligomerisation and protein–nucleotide interaction in Mycobacterium tuberculosis PII protein throughintegration of computational and experimental approaches
Sriyans Jain a, Rahul Shubhra Mandal a, Swadha Anand b, Souvik Maiti a, Srinivasan Ramachandran a,⁎a CSIR—Institute of Genomics and Integrative Biology, Mall Road, New Delhi 110 007, Indiab National Institute of Immunology, Aruna Asaf Ali Marg, New Delhi 110 067, India
Abbreviations:MtbPII,Mycobacterium tuberculosis PII pCalorimetry; AMPPNP, Adenosine 5′-(β,γ-imido) triphospthio]-triphosphate; PIC, Protein Interaction Calculator sSurfaces and Assemblies server; 2-OG, 2-oxoglutarate⁎ Corresponding author at: Room No. 130, CSIR—Institu
Biology, Mathura Road, Near Sukhdev Vihar Bus DepTel./fax: +91 11 29879130.
E-mail addresses: [email protected], [email protected], ram(S. Ramachandran).
1570-9639/$ – see front matter © 2013 Elsevier B.V. All rhttp://dx.doi.org/10.1016/j.bbapap.2013.10.006
a b s t r a c t
a r t i c l e i n f oArticle history:Received 20 May 2013Received in revised form 5 October 2013Accepted 7 October 2013Available online 12 October 2013
Keywords:Protein stabilityMutagenesisIsothermal titration calorimetryMolecular dockingElectrostatic interactions
We investigated the interacting amino acids critical for the stability and ATP binding ofMycobacterium tuberculosisPII protein through a series of site specific mutagenesis experiments. We assessed the effect of mutants usingglutaraldehyde crosslinking and size exclusion chromatography and isothermal titration calorimetry. Mutationsin the amino acid pair R60–E62 affecting central electrostatic interaction resulted in insoluble proteins. Multiplesequence alignment of PII orthologs displayed a conserved pattern of charged residues at these positions.Mutationof amino acidD97 to a neutral residuewas toleratedwhereas positive chargewas not acceptable.Mutation of R107alone had no effect on trimer formation. However, the combination of neutral residues both at positions 97 and107 was not acceptable even with the pair at 60–62 intact. Reversal of charge polarity could partially restore theinteraction. The residues including K90, R101 and R103 with potential to form H-bonds to ATP are conservedthroughout across numerous orthologs of PII but when mutated to Alanine, they did not show significantdifferences in the total free energy change of the interaction as examined through isothermal titration calorimetry.The ATP binding pattern showed anti-cooperativity using three-site binding model. We observed compensatoryeffect in enthalpy and entropy changes and these may represent structural adjustments to accommodate ATP inthe cavity even in absence of some interactions to perform the requisite function. In this respect these smalldifferences between the PII orthologs may have evolved to suite species specific physiological niches.
© 2013 Elsevier B.V. All rights reserved.
1. Introduction
PII family of proteins are highly conserved and present in bacteria,archaea and in plastids of some plants. They play pivotal role in theregulation of nitrogen assimilation (reviewed in Refs. [1–7]). In nitrogenexcess conditions, PII activates the adenylylation activity of adenylyl-transferase (ATase/AR) enzyme, which in turn adenylylates glutaminesynthetase (GS) to inactive GS-AMP. Further, PII also interacts withmembrane bound ammonium ion transporter protein AmtB and blocksthe influx of ammonium ions [3]. Conversely under nitrogenlimiting conditions, PII proteins are post-translationally modifiedand interact with ATase/AR to allow deadenylylation of the GS-AMP to yield active GS. In some proteobacteria (Azospirilliumbrasilense, Rhodobacter capsulatus and Rhodospirillum rubrum),
rotein; ITC, Isothermal Titrationhate; ATP-γS, Adenosine 5′-[γ-erver; PISA, Protein Interfaces,
te of Genomics and Integrativeot, New Delhi 110 025, India.
ights reserved.
under nitrogen limiting conditions, PII protein also interactswith Dinitrogenase Reductase ADP-Ribosyl Glycohydrolase (DraG)to remove ADP-ribose from dinitrogenase reductase and activatesnitrogenase activity. When nitrogen is sufficient, PII interactswith Dinitrogenase Reductase ADP-Ribosyl Transferase (DraT) andpromotes ADP-ribosylation of dinitrogenase reductase to blockthe nitrogenase activity [8,9]. In cyanobacteria, PII interacts withN-acetyl-L-glutamate-kinase (NAGK) to increase its catalytic activityand in addition to strongly decrease its feedback inhibition by arginine[10]. In addition to controlling the activity of proteins involved inammonium assimilation, PII proteins also regulate the expression oftheir genes by interacting with a diverse variety of transcriptionalregulators. In Escherichia coli, PII proteins control the expression ofnitrogen regulatory genes by activating the two component regulatorysystemNtrBC [11–13]. In cyanobacteria, PII protein regulates the activityof transcriptional regulator NtcA by binding with its co-activatorPipX [14–16]. In Corynebacterium glutamicum, adenylylated PII (GlnK)interacts with transcriptional repressor AmtR and regulates thetranscription of 33 genes including those coding for transporters andenzymes for ammonium assimilation [17]. However, in actinobacteria,the activity of ATase and in turn the activity of GS is not controlled byPII (GlnK) [18,19]. In a recent study, Radchenko et al. demonstratedthat PII proteins from E. coli, A. brasilense and Arabidopsis thaliana also

2737S. Jain et al. / Biochimica et Biophysica Acta 1834 (2013) 2736–2749
have ATPase activity, which is inhibited by the addition of 2-oxoglutarate(2-OG) [20].
The crystal structures of GlnB/K subfamily proteins from variousspecies have been solved (reviewed in ref. [3]). Despite having a varietyof interacting partners in different organisms, these proteins havemaintained a conserved structure. Proteins of GlnB/K subfamily arehomotrimeric proteins of typically 112 amino acids in length. Eachsubunit of PII trimer comprises of two α-helices, four β-strands andthree loops namely, B-loop (a small loop between α2 and β4), C-loop (located at C-terminus) and T-loop (a large 19 residue longdisordered region dividing the protein in two β-α-β motifs). TheT-loop contains the site for post translational modification inresponse to nitrogen availability. In E. coli, A. brasilense, Rhizobiumleguminosarum, Herbaspirillum seropedicae and many other bacteria,this modification is uridylylation of Y51 under nitrogen limitationconditions [13,21–24]. In actinobacteria, PII proteins undergoadenylylation at Y51 whereas in cyanobacteria, PII proteins aremodified by phosphorylation at S49 [18,19,25–28]. In contrast to othercyanobacteria studied so far, Anabeana PII proteins are modified bynitration at Y51 [29].
ATP binding site has been shown to be present in all PII proteinsstudied so far. The B- and C-loop of adjacent subunits form a lateralcleft on the edge of the molecule and forms the binding site for ATP[30,31]. The crystal structure ofMethanococcus jannaschiiGlnK revealedthat ATP bindingwith PII results in a compact folded conformation of T-loop, which in turn creates binding site for 2-OG [32]. However, a recentstudy on X-ray structure of the A. brasilense PII protein GlnZ complexedwith Mg2+, ATP and 2-OG shows that 2-OG binds within the ATPbinding cleft and interacts with K58, G37, Q39 and G41. The ATP and2-OG binding coordinates the bound Mg2+ together with the oxygenatoms of phosphate tail of ATP and the side chain of the T-loop residueQ39 [33]. This observation was in complete agreement with the resultsobtained with structure of Synechococcus elongatus PII complexed withMg2+, ATP and 2-OG and that obtained with biochemical studies withE. coli and R. rubrum [34–36].
InMycobacterium tuberculosis, nitrogen assimilation is accomplishedonly by the glutamine synthetase/glutamine-2-OG-amidotransferase(GS/GOGAT) pathway due to absence of the glutamate dehydrogenase(GDH) [37]. Similar to other actinobacteria, M. tuberculosis codes for asingle gene Rv2919c coding for PII protein (MtbPII). MtbPII is annotatedas GlnB mainly due to its higher sequence identity with GlnB group ofproteins. On the basis of primary amino acid sequence and the geneticlinkage, Arcondeguy et al. classified prokaryotic PII proteins in threedistinct subgroups namely glnB, glnK, and nifI [2]. According to thisclassification, glnB genes are linked to glnA or nadE and their proteinsequences contain K3 and E5 or D5. The glnK genes are linked to amtB.GlnK protein sequences have amino acid L or I orM or F at third positionand I or T orMatfifth position. InM. tuberculosis, the geneRv2919c (glnB)is located in between the genes amtB and glnD as part of an operon.MtbPII has a L3 and T5. These features suggest that MtbPII is more likelyto be GlnK [2,38].
Although classified as non-essential, a transposon mutant ofRv2919c gene shows attenuated growth in macrophages preactivatedby interferon-gamma (IFN-γ) compared with naive macrophagesimplying that the gene is important for survival in the IFN-γ activatedmacrophages [39]. Similarly, a transposon mutant of M. bovis BCGhomolog of MtbPII showed attenuated growth in macrophageculture [40]. In another effort, Read et al. have reported that theywere unable to generate Rv2919c deletion mutant in contrastto the ease of constructing glnD mutant implying the perceivedessentiality of Rv2919c in M. tuberculosis [41]. These observationsshow the importance of MtbPII protein in cellular functioningand could be very important under conditions of stress thatM. tuberculosis faces during infection [42]. The supposed essentialityof MtbPII for survival of M. tuberculosis during infection, its absencein humans and the putative central role in regulation of nitrogen
assimilation makes MtbPII an attractive drug target against thispathogen [38].
In a previous study, we have shown that MtbPII exists as a trimerand binds to ATP with high affinity [43]. Subsequently, the crystalstructures of apo (PDB ID: 3BZQ) and ATP bound PII protein (PDBID: 3LF0) from M. tuberculosis have been solved confirming ourobservations [38]. The structural features observed in the crystal aretypical to PII family proteins. The root mean square deviation (RMSD)between Cα atoms of structures of Mtb apo PII and of MtbPII withbound ATP is 0.69 Å. The ATP bound MtbPII crystal structure showsbinding of ATP with protein without Mg2+ [38]. We have consistentlyobserved through both surface plasmon resonance (SPR) andisothermal titration calorimetry (ITC) experiments that MtbPII doesnot bind to ATP in absence of Mg2+. In addition, we observed that 2-OG does not alter the dissociation constant of MtbPII–ATP complexsignificantly but it contributes to enthalpy change [43].
Because MtbPII shows high conservation at the level of its aminoacid sequence and structure, we investigated its structural propertieswith respect to the amino acid residues, which are important for MtbPIItrimer formation and with potential to bind ATP.
2. Materials and methods
2.1. Computational identification of interacting residues for PII homotrimer
To decipher the interacting residues of partner chains, thecrystal structure co-ordinate files of MtbPII protein (PDB ID—3BZQand 3LF0) were analyzed using protein interaction calculator (PIC)server (http://crick.mbu.iisc.ernet.in/~PIC/) and Protein Interfaces,Surfaces and Assemblies (PISA) server [44,45]. The default settingswere used for analysis. To examine for the conservation of the predictedinteracting residues, sequences of PII and PII like proteins were retrievedusing BLASTP search against NCBI non-redundant database using MtbPIIas query sequence [46]. Redundant and partial sequenceswere removed.Resultant 1766 sequences were aligned using MUSCLE version 3.8.31[47].
2.2. Site directed mutagenesis
To over-express MtbPII in E. coli, M. tuberculosis gene Rv2919cencodingMtbPII was amplified and cloned in vector pET-23b (Novagen,USA) as described in our previous report [43]. To confirm the roleof various interacting residues in MtbPII oligomerisation, the plasmidpET-23b carrying Rv2919c was subjected to site directed mutagenesiswith Stratagene QuikChange Site Directed Mutagenesis kit (AgilentTechnologies Inc, Santa Clara, CA). Mutagenic primers were designedusing QuikChange primer design tool (Agilent Technologies Inc, SantaClara, CA). Mutations were confirmed by plasmid sequencing.
Plasmids carrying confirmed mutation were transformed into E. coliC41 (DE3) (Avidis, France), overexpressed and purified as describedpreviously [43]. Briefly, overnight grown culture of E. coliwas inoculatedin 1 l of Luria Bertani Broth (HiMedia Laboratories, India) containing100 μg/ml ampicillin at ratio of 1:100 and incubated at 37 °C withrigorous shaking. At an optical density.at 600nm of 0.6, the culture wasinduced by addition of 1 mM Isopropyl β-D-L-thiogalactopyranoside(IPTG; Sigma, USA) and further incubated at 37 °C for 3 h. For some ofthe mutants, over-expression was induced by addition of 0.1mM IPTGand incubation at 16 °C for 16 h. After induction cells were harvestedby centrifugation at 6800 ×g for 10 min at 4 °C. Harvested cell masswas suspended in sonication buffer (20 mM Tris–Cl pH 7.9, 500 mMNaCl, 5 mM imidazole, 10mM phenylmethanesulfonyl fluoride (PMSF;Sigma-Alrich, USA). To the suspension, 1 mg/ml lysozyme was addedand incubated for 1½ h on ice. Protein was extracted by sonication for5 min with 20 s pulse followed by centrifugation at 29000 ×g for30 min at 4 °C. The supernatant was separated and the pellet wasresuspended in equal volume of sonication buffer. A small sample was

2738 S. Jain et al. / Biochimica et Biophysica Acta 1834 (2013) 2736–2749
collected at every stage of induction and purification. To assess thefraction containing protein of interest, equal volume of samples, boiledwith SDS-PAGE running dye, were electrophoresed on SDS-PAGE (12%)in duplicates. One gel was stained with Coomassie brilliant blue R250and the other was subjected to western blot analysis using Penta-HisHRP conjugate antibody (Qiagen, Germany).
Supernatant containing extracted protein was incubated withnickel-nitrilotriacetate (Ni+-NTA) resin (Qiagen, Germany) at 4 °C for2h. After incubation, resin with supernatant was loaded on purificationcolumn. Non-specific proteins were removed by washing the columnwith 10 volumes of wash buffer containing gradient of imidazole inconcentration of 5 mM, 20 mM and 60 mM. His tagged protein waseluted with elution buffer containing 20 mM Tris–Cl pH 7.9, 500 mMNaCl and 150 mM imidazole. Purity of isolated protein was assessedby SDS-PAGE (12%) analysis. Identity of purified protein wasestablished by western blot using Penta-His HRP conjugate antibody(Qiagen, Germany). Protein estimation was carried out by Bradfordassay (Bio-Rad Laboratories, USA) using BSA (Sigma-Aldrich, USA) asstandard.
2.3. Glutaraldehyde crosslinking reaction
Purified proteins were dialysed against buffer containing 20 mMHEPES pH8.0 and 150mM NaCl. After dialysis, proteins were subjectedto crosslinking reaction using glutaraldehyde as crosslinking reagentas described previously [43]. Briefly, 5 μg protein suspended in 50 μlbuffer was incubated with varying concentrations of glutaraldehyde inthe range 0.5 – 2% v/v. The reaction mixture was incubated at 37 °C for1–2 h. The reaction was terminated by addition of 5 μl of 1 M Tris–ClpH 7.9. A fraction of reaction mixture containing approximately 1 μg ofcrosslinked protein was analysed by electrophoresis on 12% SDS-PAGEand by western blot analysis using Penta-His HRP conjugate antibody(Qiagen, Germany).
2.4. Size exclusion chromatography
Size exclusion chromatography was performed with Akta Purifier(GE healthcare, USA) coupledwith Superdex 75 column (GEHealthcare,USA). Purified proteins were dialyzed against a buffer containing 20mMTris–Cl pH7.9 and 150mMNaCl. Protein sample of 500μl at concentrationof 0.5 mg/ml was loaded on superdex 75 column pre-equilibrated withthe same buffer. The chromatogram was run by passing the buffer at aflow rate of 0.5 ml/min. The elution volume of protein was determinedas the volume of the eluent from the point of injection to the centreof the elution peak. The column was calibrated by passing standardproteins BSA (66kDa), Albumin (45kDa), carbonic anhydrase (29kDa),chymotrypsinogen A (25kDa) and cytochrome C (12.5kDa). A standardcurve between elution volume and log of the molecular weight of thestandard proteins was prepared to infer the molecular weight offractioning proteins (Supplementary Fig. 1).
2.5. MtbPII—nucleotide docking studies and in silico mutantprotein modelling
Molecular docking was performed in order to identify amino acidsinvolved in protein–ligand interactions. The MtbPII apo structure formwas used for dockingwith ATP, ADP and a non-hydrolysable ATP analogAdenosine 5′-(β,γ-imido) triphosphate (AMPPNP) with AutoDock Vina1.0.3 [48].
To examine probable effect and assess inter-residue bonding patternarising from different mutations on protein structure, we generatedin silico models of MtbPII mutants. Protein structure 3BZQ was used astemplate to model in silico MtbPII mutants with Discovery Studio v3.5(Accelrys Softwares Inc., San Diego). The mutant protein models weresubjected to energy minimisation to relax the steric hindrances, if any,
using UCSF Chimera version 1.6.1 with 1000 steps of steepest descentand 100 conjugate gradient steps [49].
2.6. PII-nucleotide binding study using ITC
Thermodynamic parameters of ATP interaction with various MtbPIImutants were determined using isothermal titration calorimetry (ITC)with VP-isothermal titration calorimeter (MicroCal Inc., USA). Purifiedproteins were extensively dialyzed against ITC buffer (20 mM Tris–ClpH 8.0 and 150mMNaCl). Ligand solutionswere prepared by dissolvingdifferent ligands at 1mMconcentration in the ITC buffer. Except in a fewexperiments, all ligand solutions contained 5 mM MgCl2. All solutionswere degassed extensively before the experiment. A solution containing20μM protein (on monomer basis) was titrated against ligand solution.The titrations were carried out with 25 additions of 9 μl ligand solutionwith 5 min interval between two successive injections. Each injectionlasted for 9 s. All titrations were performed at 30 °C with continuousstirring at 307 rpm. All experiments were repeated twice with sameinstrumental settings.
The binding constant Ka and the enthalpy change (ΔH) of theinteraction were derived by analysing the raw data obtained from theITC experiments with Origin 7.0 (MicroCal Inc., USA). The data wasfitted using a three sequential site binding model as well as single sitebinding model. The dissociation constant (Kd), change in Gibbs freeenergy (ΔG) and entropy change (ΔS) were subsequently calculatedusing thermodynamic relations Kd = 1/Ka, ΔG = −RT ln Ka andTΔS=ΔH−ΔG, respectively where T is reaction temperature (303 °K).Student's t-test was used for assessing the statistical significance ofdifferences of means of different thermodynamic quantities betweendifferent treatments and cases.
3. Results and discussion
3.1. Identification of residues interacting to form PII homotrimer
Analysis ofMtbPII apo structure formwith PISA predicted three pairsof amino acid forming interchain ionic bonds (K2–D97, R60–E62, andD75–R107) between the monomer units whereas analysis with PICserver suggested four pairs of amino acids forming interchain ionicbonds (K2–D97, R60–E62, D71–R107 and D75–R107). The additionalpair D71–R107 is predicted only by PIC server because it allows theprediction of ionic bonds up to a distance of 6 Å between the residues,which is about 1.5 times higher than usual ionic bond length of 4 Å[50]. Although the strength of the electrostatic interactions at 6 Åwould be 2.25 times weaker than the usual ionic bonds, we includedthis pair also for the experimental investigation of its role in theinteraction between monomer subunits.
Analysis of MtbPII–ATP complex structure with PIC and PISA serversresulted in somewhat different prediction of interchain ionic bonds(Table 1). In the crystal structure of MtbPII apo protein, subunits arepacked symmetrically and the interchain interactions are identicalbetween the subunits. However, in the ATP bound MtbPII, the subunitsare arranged asymmetrically within the unit cell and thereforethe interchain interactions between different subunits vary. The twopairs R60–E62 and K2–D97 were predicted to interact electrostaticallyamong all three chains as in the apo form. However no ionic interactionwas predicted between D75–R107 in MtbPII-ATP complex by PISA.Instead, PISA predicted a new interaction between D54 of chain C andK17 of chain A. PISA did not predict this interaction on the interfaceof other chain pairs A–B or B–C. However, PIC predicted electrostaticinteractions between D54 of chain B and K17 of chain C in addition.PIC also predicted electrostatic interactions between R38 of chain Bwith E106 of chain C and R38 of chain C with E106 of chain A. Thesedifferences are due to the rearrangement of monomer units in theMtbPII trimer upon binding to ATP. The rearrangement of monomerunits results in non-uniform electrostatic interactions among all three

Table 1Interchain ionic bonds between different subunits in crystal structure of MtbPII apo form (PDB ID: 3BZQ) and in crystal structure of MtbPII with ATP bound (PDB ID: 3LF0). + indicatesprediction of the interaction whereas – represents absence of the said interaction.
Position Residue ChainElectrostatic
conservation of residuein PII orthologsa
Position Residue ChainElectrostatic
conservationof residuein PII orthologsa
3BZQ 3LF0
PIC PISA PIC PISA
2 K A+ve = 97.9%–ve = 0.2%Neutral = 1.8%
97 D B+ve = 0.3%–ve = 80.2%Neutral = 19.4%
+ + + +
60 R A+ve = 98.3%–ve = 0.5%Neutral = 1.2%
62 E B+ve = 0.2%–ve = 99.3%Neutral = 0.5%
+ + + +
71 D A+ve = 1.2%–ve = 88.1%Neutral = 10.7%
107 R B+ve = 25.7%–ve = 13.4%Neutral = 60.9%
+
75 D A+ve = 2.7%–ve = 85.7%Neutral = 11.6%
107 R B+ve = 25.7%–ve = 13.4%Neutral = 60.9%
+ + +
2 K B 97 D C + + + +
38 R B+ve = 91.3%–ve = 0.3%Neutral = 8.4%
106 E C+ve = 0.2%–ve = 99.2%Neutral = 0.5%
+
54 D B+ve = 0.5%–ve = 80.1%Neutral = 19.4%
17 K C+ve = 98.1%–ve = 0Neutral = 1.9%
+
60 R B 62 E C + + + +
71 D B 107 R C + +
75 D B 107 R C + +
2 K C 97 D A + + + +
38 R C 106 E A +
54 D C 17 K A + +
60 R C 62 E A + + + +
71 D C 107 R A + +
75 D C 107 R A + +
– – –
–
–
–
–
–
– –
––
–
–
– –
–
––
–
–
–
–
Rows highlighted in grey are interactions preserved in all cases indicating their importance in trimer stabilisation.a Conservation of residues among orthologs is given once per residue only and iswith regards to electrostatic conservation rather than residual conservation.+ve=positively charged
amino acids (K or R);−ve=negatively charged amino acids (E or D); Neutral=uncharged amino acids.
2739S. Jain et al. / Biochimica et Biophysica Acta 1834 (2013) 2736–2749
chain pairs in the homotrimer complexedwith ATP. The amino acid K17is present on α-helix whereas residues R38 and D54 are present on Tloop and residue E106 on the C-loop. In the ATP bound MtbPII crystal,the T-loop of subunit B is stable which, according to authors, may bedue to crystal packing effects [38]. The electrostatic interaction betweenD71 and R107 was predicted to be maintained between the units B–Cand C–A but not in A–B where the interaction between D75 and R107was predicted to be preserved (Table 1). These results indicate thatthe electrostatic interactions between the pairs R60–E62 and K2–D97are preserved in both MtbPII apo form and MtbPII–ATP complexwhereas the interaction pairs D71–R107 and D75–R107 are mutuallyexclusive between the chain pairs in the trimer of MtbPII–ATP complex.Other interaction pairs in ATP bound MtbPII crystal may be the resultof loop readjustment on ATP binding.
In order to examine the evolutionary conservation of amino acidresidues predicted to interact electrostatically we carried out multiplesequence alignment of protein sequences of PII homologs from variousorganisms. A collection of 1766 non-redundant PII homologs fromdiverse organisms was analysed. In the interaction pair between theamino acid residues at positions 60 and 62 the following pattern wasobserved. We observed that in most of the cases, the amino acid atposition 60 is positively charged (R (13.1%) or K (85.2%)) whereas theamino acid at position 62 is negatively charged (E (92.5%) or D (6.8%)).A small subset of sequences has neutral (1.2%) or negatively charged(0.5%) residue at position 60. At position 62, neutral or positively chargedresidues occur in 0.5% or 0.2% sequences, respectively. At position 2,positively charged residue (K (96.9%) or R (1.0%)) shows an over-whelming prevalence. A small minority of 0.2% sequences has a
negatively charged amino acid at position 2 whereas other 1.8%sequences have a neutral amino acid at this position. The aminoacid at position 97 varies between negatively charged and neutralresidues in the proportion 0.802:0.194 with a small set of 0.3%sequences carrying positively charged residue at this position.This analysis shows conservation of electrostatic charge at thesepositions among various PII homologs. This type of evolutionaryconservation agrees well with the pattern of interaction seen in theMtbPII trimer in both apo form and in MtbPII complexed with ATPwherein the electrostatic interactions between these two pairs werepredicted among all three chain pairs in the homotrimer.
The amino acid residues at position 71 and at position 75 arepredominantly negatively charged residues with D (54.6%) or E (33.5%)at position 71 and D (31.5%) or E (54.2%) at position 75 with minorfraction of neutral amino acids (10.6% and 11.6% sequences at position71 and 75, respectively). The occurrence of positively charged aminoacids is very low (1.2% and 2.7% sequences at position 71 and 75,respectively). The amino acid residue at position 107 is highly variablewith occurrence of neutral (60.9%), positive (25.7%) and negatively(13.4%) charged amino acids. This pattern of variability of amino acidresidues observed in the pairs 71–107 and 75–107 points to possibleflexibility of these interactions between the apo and ATP bound forms.
With respect to the interaction pairs K17 and D54 in MtbPII–ATPcomplex, multiple sequence alignment of PII homologs showed thatamino acid at position 17 is predominantly positively charged residue(K (40.7%) or R (57.4%)). Only a small set (1.9%) of sequences havea neutral residue at this position. The amino acid at position 54 is anegatively charged (D (72.7%) or E (7.4%)), or neutral residue (19.4%)

Fig. 1. Western blot of native MtbPII after glutaraldehyde crosslinking experiment. Lanesare labelled after the% of glutaraldehyde in the reactionmixture. Themonomer and trimerform of protein are marked. Protein with no glutaraldehyde is negative control.
2740 S. Jain et al. / Biochimica et Biophysica Acta 1834 (2013) 2736–2749
with a few sequences (0.5%) having positively charged residue. In theinteraction pair R38 and E106, the amino acid at position 38 is primarilya positively charged amino acid (R (90.5%) or K (0.8%)) with a fewsequences having neutral residues (8.4%). At position 106, negativelycharged amino acid (E (96.5%) or D (2.7%)) is prevalent with a fewsequences having neutral or positively charged residue (0.5% and 0.2%,respectively). Even though theses interactions are temporarily engagedduring ATP binding to the trimer, the conservation of charges in therespective positions is maintained. However, their longer bond lengthsuggests weaker interactions.
3.2. Mutation studies on trimer formation
3.2.1. R60–E62 pairMutations were carried out either to neutralise or to reverse
the charge of one of the amino acid residue in this pair participating inthe ionic bond formation. The effect of mutations on the ability toform trimer was examined through glutaraldehyde crosslinking of theapo form. The results of various mutants are summarised in Table 2.Our previous study have shown that the recombinant MtbPII with C-terminal His tag purifies to near homogeneity with a 14.5 kDaband corresponding to monomer on SDS-PAGE. With glutaraldehydecrosslinking, it gives a slightly defused band with centre point at43.5kDa corresponds to the PII trimer (Fig. 1) [43]. The mutant proteinswith R60 to H60 (PII_R60H) or to E60 (PII_R60E) could not be purified asthese mutant proteins were found in insoluble fraction after sonication.We attempted to obtain solubilised forms of these mutant proteins byinducing the protein expression at 16 °C but without success (Fig. 2Aand Supplementary Fig. 2). Therefore, it appears that neutralizing orreversing the charge at position 60 is not acceptable because this pairof ionic interaction between the chains is conserved evolutionarily inboth the apo form and ATP bound form. If formation of electrostaticinteraction alone would suffice for the MtbPII to form a stable trimerand soluble protein then, in principle, complete reversal of polarityshould restore the native state. In order to examine this hypothesis, wegenerated anMtbPII double mutant PII_R60E/E62R. This mutant proteinwas found to be soluble only after expression at 16 °C in contrast to theexpression of nativeMtbPII at 37°C (Fig. 2B). The purified doublemutantprotein formed a stable trimer as observed through glutaraldehydecrosslinking (Fig. 2C). To assess the molecular weight of purified proteinsize exclusion chromatogram was run with Superdex 75 column withvoid volume of 8 ml. The wild type MtbPII was eluted as a peak at9.95ml corresponding to inferred molecular weight of 42.6kDa close tothe molecular weight of PII trimer (Fig. 3A). A small portion of proteinspassed through void volume indicated by peak appearing at elutionvolume of 7.71 ml which could be due to contaminating proteins or aminor proportion of the protein forming aggregates. The purified proteinPII_R60E/E62R showed two distinct peaks (Fig. 3B). The first peak
Table 2Summarised results for purification and crosslinking analysis of variousmutants of MtbPII.
Protein Expression Purification fraction Oligomerisation checked byglutaraldehyde crosslinking
PII_R60H + Pellet Could not be donePII_R60E + Pellet Could not be donePII_D97N + Soluble +PII_D97R + Pellet Could not be donePII_R107H + Soluble +PII_R107E + Soluble +PII_ R60H/D97N + Pellet Could not be donePII_R60E/D97R + Pellet Could not be donePII_D97N/R107H + Pellet Could not be donePII_ D97R/R107E + Pellet Could not be donePII_R60H/R107H + Pellet Could not be donePII_R60E/R107E + Pellet Could not be donePII_K2D/D97R + Soluble (16 °C) +PII_R60E/E62R + Soluble (16 °C) +
appeared at 7.71 ml corresponding to a protein passing through thevoid volume and the other peak was observed at 9.96ml correspondingto protein of molecular weight 42.6kDa. The result shows that althoughthe protein PII_R60E/E62R is soluble, a major proportion of it is formingaggregates indicating that swapping the charge at these sites does notrestore native characteristics completely.
Examination of double mutant model of structure of MtbPII apoform revealed that Oε1 atom of amino acid E60 of one subunit was ata distance of 2.81 Å from the atom Nη1 and at 2.69 Å from the atomNη2 of R62 from neighbouring subunits with potential to form strongionic bonds. The other oxygen atom (Oε2) of E60 was positioned at adistance of 4.83 Å from the atom Nη1 and at 4.35 Å from the atomNη2 of amino acid R62 fromneighbouring subunits. Themodel revealedthat the residues E60 and R62 have potential to interact electrostaticallybut the strength of this interaction is estimated to be weaker by about2.8 times than that in the native MtbPII. These changes may underliethe solubility of mutant protein PII_R60E/E62R during expression byinduction at 16 °C. These results elaborated the importance of theelectrostatic interaction in the 60–62 pair and are in agreementwith the evolutionary conservation pattern of the occurrence of apredominantly positively charged residue (R or K) at position 60and predominantly negatively charged residue (E or D) at position62 in overwhelming majority of PII homologs examined. In a smallproportion of PII sequences, charge reversal of both of these aminoacids is observed but it appears highly probable that the interatomicdistances in this pair reduces the possibility of occurrence of reversal ofpolarity in this pair.
These data taken together attest to the critical importance of theinteraction of 60–62 electrostatic pair in the trimer formation of MtbPIIand other PII orthologs.
3.2.2. K2-D97 pairThe importance of the electrostatic interaction in the K2–D97 pair
was examined first by neutralizing the charge at position 97 in theMtbPII mutant PII_D97N. This mutant was found to yield solubleprotein. It showed stable trimer formation after examination throughglutaraldehyde crosslinking representing intact folding and assemblyof this mutant protein. The size exclusion chromatography of theprotein PII_D97Nwas consistent with that of wild type MtbPII showing

Fig. 2. Mutational analysis of amino acid pair R60-E62: (A) Western blot of crude fractions of mutant protein PII_R60E. Lanes are labelled as M: Protein Molecular Weight Marker, Uni:Uninduced bacterial cell lysate, Ind: Bacterial cell lysate after induction at 16°Cwith addition of 0.1mM IPTG, Sup: Soluble fraction obtained after sonication, Plt: Insoluble cellmass (pellet)after sonication. The expressedmutant proteinwas present only in pellet fraction and therefore could not be purified. (B)Western blot of crude fractions ofmutant protein PII_R60E/E62R.Lanes are labelled as in (A). Significant amount of expressed protein was present in supernatant, which could be further purified. (C) Western blot of purified PII_R60E/E62R afterglutaraldehyde crosslinking. Lanes are labelled after the % of glutaraldehyde in the reactionmixture. Themonomer and trimer forms of protein aremarked. Proteinwith no glutaraldehydeis negative control.
2741S. Jain et al. / Biochimica et Biophysica Acta 1834 (2013) 2736–2749
a major peak at 9.95ml corresponding to inferred molecular weight of42.6 kDa close to the PII trimer (Fig. 3C). Reversing the charge as inD97R however rendered it expressed in inclusion bodies indicatingthat electric charge reversal of one of the amino acids in this pairwas not acceptable in this case. In the native MtbPII protein apo form,D97 interacts electrostatically with K2 with an interatomic distance of2.73Å. Evolutionary conservation analysis showed that the amino acidresidue at position 2 is predominantly a positively charged aminoacid whereas at position 97, although negatively charged amino acids(D or E) prevail with 80.2% sequences, other electrically neutral andhydrophobic amino acid residues also occur in minor proportion(19.4%). We sought to examine the reversibility of polarity of bothamino acid residues in this pair. We generated a double mutantPII mutant PII_K2D/D97R. Because Arginine and Lysine share similarphysiochemical properties and that we had already examined themutant protein PII_D97R, the mutant PII_K2D/D97R may be expectedto behave as native PII. We were able to obtain soluble form of thismutant protein only after expression at 16 °C. However, the majorfraction of protein was still insoluble (Fig. 4A). SDS-PAGE analysis ofpurified fractions of this protein showed multiple non-specific proteinswhich could not be removed by rigorous washing of the purificationcolumn. Glutaraldehyde crosslinking of the protein PII_K2D/D97R didnot show a clear band at trimer position, instead a band over 100 kDawas observed (Fig. 4B). The result from size exclusion chromatographyof this protein was in agreement with the glutaraldehyde crosslinkingresult. The size exclusion chromatogram of PII_K2D/D97R showed asingle peak of elution at volume of 7.93 ml indicating that the proteinpasses through the void volume (Fig. 3D). These result point to theformation of aggregates and may be the consequences of improperfolding. Among 1766 PII orthologs, position 2 is occupied overwhelminglyby a positively charged amino acid (K (96.9%) or R (1%)) whereas only asmall proportion of 0.2% of sequences had a negatively charged aminoacid at this position. This suggests an evolutionary preference for thepositively charged amino acid at this position. These data taken togetherpoint to the importance of preserving the electrostatic interaction alongwith sequence conservation at position 2, although at position 97 evena neutral polar residue like asparagine was acceptable.
Structure model of the double mutant protein PII_K2D/D97Rrevealed that the electrostatic interaction in the D2-R97 pair may not
be very strong because the interatomic distance exceeds 6 Å. This mayexplain its partial soluble form at 16 °C in contrast to the native MtbPII,which can be expressed in soluble form at 37 °C.
3.2.3. Other pairsOther electrostatically interacting pairs in MtbPII are D71–R107
and D75–R107. These pairs appear to be variable between the apo andthe ATP bound forms of MtbPII. In order to examine the importanceof these pairs on the formation of stable PII trimer we sought to firstneutralise and then to reverse the charge at position 107 because it isthe common amino acid residue in both interactions. We mutatedR107 to H107 (PII_R107H) (positive to electrically neutral) and toE107 (PII_R107E) (positive to electrically negative). In both thesecases, the proteins were soluble and also formed trimers (Fig. 5A).The size exclusion chromatograms also confirmed the results fromglutaraldehyde crosslinking of PII_R107H and PII_R107E (Fig. 3Eand F). These observations are in agreement with the acceptability ofhigh variability in the occurrence of other amino acid types at position107 across PII homologs.
We examined the double mutant protein PII_D97N/R107H, whichwas found in inclusion bodies (Fig. 5B) although the single mutantsPII_D97N and PII_R107H were soluble and formed trimer. These resultsshow that theminimal requirement for electrostatic interactions betweenmonomer units of PII for formation of stable trimer and soluble protein at37 °C involves the R60–E62 pair and either K2–D97 or D75/D71–R107intact between the monomer subunit chains. In MtbPII, amino acid pairsK2–D97 and D75/D71–R107 are present on the surface of the proteinwhereas the pair R60–E62 is present at the central region of the protein.Our results suggest that for proper assembly of MtbPII, the centralinteraction between R60 and E62 is critical but supporting interactionslocated at the surface are also important.
The MtbPII protein expressed in native form is largely soluble. Theprotein does not have additional sequence such as transmembranedomain or signal peptides regions. The protein is also not toxic. It wasobserved that the direct effect of structurally unfavourable mutationsis insolubility and this could be likely due to lack of proper folding andstructural integrity. The improperly folded mutant protein may betoxic and therefore they likely appear in the pellet and not in solublefraction.

Fig. 3. Size exclusion chromatograms of purifiedMtbPII and its mutants: Size exclusion chromatogram of purifiedMtbPII (A), and its mutants PII_R60E/E62R (B), PII_D97N (C), PII_K2D/D97R (D), PII_R107H (E), and PII_R107E (F). All proteins exceptPII_K2D/D97R showed two peaks: one appearing before elution volume of 8ml indicating a fraction of proteins passing through void volume of the column, the second peak appeared near elution volume of 9.95ml corresponding tomolecularweightof 42.6 kDa indicating trimeric MtbPII of its mutants. The columnwas calibrated by passing BSA (66kDa) eluted at 8.8ml, Albumin (45 kDa) eluted at 9.74ml, carbonic anhydrase (29 kDa) eluted at 10.96ml, chymotrypsinogen A (25kDa) eluted at11.64ml and cytochrome C (12.5 kDa) eluted at 12.77ml.
2742S.Jain
etal./Biochimica
etBiophysicaActa
1834(2013)
2736–2749

Fig. 4.Mutational analysis of amino acid pair K2-D97R: (A)Westernblot of crude fractions ofmutant protein PII_K2D/D97R. Lanes aremarked asM: ProteinMolecularWeightMarker, Uni:Uninduced bacterial cell lysate, Ind: Bacterial cell lysate after induction with IPTG, Sup: Soluble fraction obtained after sonication, Plt: Insoluble cell mass (pellet) after sonication. A smallamount of expressed protein was soluble. However, the mutant protein could not be purified to homogeneity. (B) Western blot of PII_K2D/D97R after glutaraldehyde crosslinking ofpurified protein. Lanes are labelled after the % of glutaraldehyde in the reactionmixture. Themonomer and trimer forms of protein aremarked. Protein with no glutaraldehyde is negativecontrol.
2743S. Jain et al. / Biochimica et Biophysica Acta 1834 (2013) 2736–2749
3.3. MtbPII—nucleotide docking studies
There are 47 crystal structures available for 19 PII proteins from 16organisms including M. tuberculosis PII [3]. E. coli has two orthologs ofPII proteins namely GlnB andGlnK. These proteins have been crystalisedin apo form and in presence of ATP or other ligands [31,51–54]. Crystalstructures of E. coli PII proteins (GlnB and GlnK) with bound ATP havebeen discussed in detail by Xu et al. [30]. Though PII proteins in E. coliGlnB and GlnK have similar sequence (67% sequence identity) andstructure, they differ in their interactionswith their receptors or ligands.The major contributor to these differences is conversion of Q82 in GlnBto Y82 in GlnK. This difference and its interaction with nearby residuesshifts the position of B loop to make ATP binding site wider in GlnKcompared with that in GlnB [30]. ATP bound with these proteins showdifferences in their conformation. These two proteins form similar H-bonds with sugar and base of ATP with differences that in GlnK atomNζ of K90 forms H-bond with N7 atom of adenine whereas in caseof GlnB the same residue interacts with oxygen atoms of phosphatetail of ATP. In addition, N atom of F36 of GlnB is hydrogen bondedwith ribose 3′-OH of ATP which is not the case in GlnK–ATP complex.The two complexes differ in the arrangement of phosphate tail of ATPand its interaction with protein despite the fact that ATP binding siteis conserved in both proteins. In GlnK–ATP complex, amide nitrogenof G89 is hydrogen bonded with oxygen atom (O1A) of α-phosphateof ATP, whereas in GlnB–ATP complex it is bonded with oxygen atomof β-phosphate of ATP. In contrast to GlnK, D88 of GlnB is H-bondedwith oxygen atom of β-phosphate of ATP [30].
The ATPboundMtbPII crystal structure 3LF0 shows that ATPbinds inthe cleft formed between neighbouring subunits (Fig. 6A) [38]. Thedetails of hydrogen bonds betweenATP and the protein are summarisedin Table 3. The structure is in close agreement to our docking resultspresented in our previous report [43]. ATP within the cleft appears tohave potential to form hydrogen bonds with R101 and R103 from onesubunit and R38, K40, I86, G87, D88, and G89 from the neighbouringsubunit. In this respect the structure appears highly similar to the E. coliPII–ATP complexes [30]. Multiple sequence alignment of PII homologsshowed that amino acids involved in ATP binding site are highlyconserved (N98.9% conservation) with the exception of R38 and I86which showed 90.5% and 91.9% conservation, respectively.
In ATP bound MtbPII, oxygen atoms of γ-phosphate of ATP couldform extensive H-bonds with Nε atom of R103 from A chain and
backbone N atoms of G87, D88 and G89 of B chain (Fig. 6B). Thephosphate tail of ATP is also stabilised by H-bonds between oxygenatoms of α-phosphate with hydrogen atoms of amino group of R103andbetween oxygen atomofβ-phosphate andhydrogen atomsof aminogroup of R101. Because Arginine is positively charged at physiologicalpH, it is possible that the phosphate chain of ATP forms electrostaticbonds with these residues to stabilise the complex. The head region ofATP appears stabilised with H-bonds between the adenine ring of ATPand carbonyl oxygen atoms of R38 and I86 and between the hydroxylgroup of ribose sugar of ATP and nitrogen atom of amino group of K40.High affinity of ATP with protein could be attributed to the extensiveH-bonds and to the possible electrostatic bonds between the proteinand phosphate tail of ATP. In addition, ATP molecules are H-bonded toa number of water molecules some of which bridge the interactionwith different amino acids, for example a water molecule bridges theinteraction between L112 of C chain and adenine ring of ATP moleculebound in the cleft between B and C subunits of the protein.
We sought to investigate the ATP binding pocket architecture andthe potential role of individual amino acids through modified analogsof ATP and through site specific mutagenesis.
In order to assess the binding pocket architecture and flexibility, weexamined binding of ADP and Adenosine 5′-(β,γ-imido) triphosphate(AMPPNP), a non-hydrolysable analog of ATP, with the protein bydockingusingAutoDockVina 1.0.3 [48]. Itwas observed that these ligandscould bind to the protein at the same site but their hydrogen bondingpattern differed significantly. In the structure of apo form of MtbPIIdockedwithADP (Fig. 6C), the phosphate chain of ADP could be stabilisedwith the H-bondswith G89 of subunit B and R101 and R103 of subunit A.R103 of subunit A also formed H-bonds with oxygen atom of hydroxylgroup of ribose sugar. In contrast to ATP, adenine ring of ADP formed H-bonds with H109, D110 and L112 of subunit A. The predicted bindingenergy of 3BZQ-ADP interaction was −5.45 kcal/mol which is lowerthan that for 3BZQ–ATP interactions (−7.98kcal/mol) indicating weakerbinding. In case of AMPPNP, the adenine ring formed H-bonds with E106from subunit A and the phosphate chain formed various H-bondswith amino acid residues K58, G87 and K90 of subunit B and R103of subunit A (Fig. 6D). 3BZQ–AMPPNP docking predicted bindingenergy of−3.88kcal/mol, which is further lower than that of interactionwith either ATP or ADP. It is noteworthy that although these ligandsATP, ADP and AMPPNP have structural similarities with respect toadenine ring and ribose sugar, the difference in the spatial arrangement
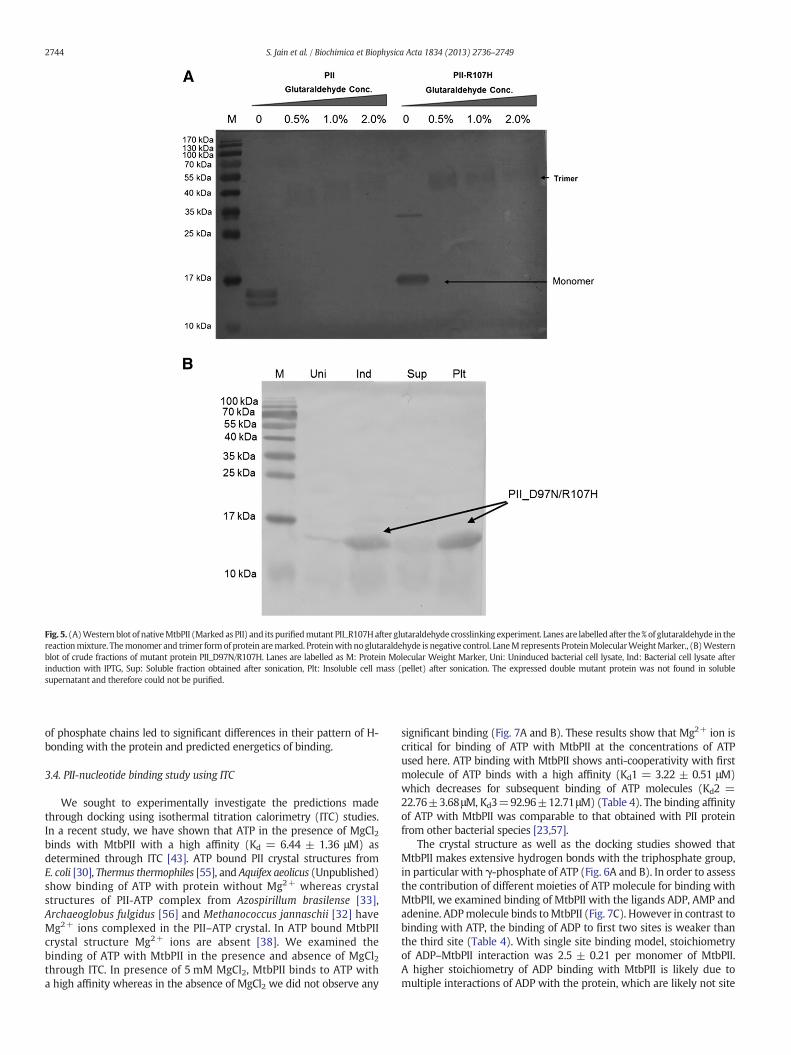
Fig. 5. (A)Western blot of nativeMtbPII (Marked as PII) and its purifiedmutant PII_R107Hafter glutaraldehyde crosslinking experiment. Lanes are labelled after the%of glutaraldehyde in thereactionmixture. Themonomer and trimer formof protein aremarked. Proteinwith noglutaraldehyde is negative control. LaneM represents ProteinMolecularWeightMarker., (B)Westernblot of crude fractions of mutant protein PII_D97N/R107H. Lanes are labelled as M: Protein Molecular Weight Marker, Uni: Uninduced bacterial cell lysate, Ind: Bacterial cell lysate afterinduction with IPTG, Sup: Soluble fraction obtained after sonication, Plt: Insoluble cell mass (pellet) after sonication. The expressed double mutant protein was not found in solublesupernatant and therefore could not be purified.
2744 S. Jain et al. / Biochimica et Biophysica Acta 1834 (2013) 2736–2749
of phosphate chains led to significant differences in their pattern of H-bonding with the protein and predicted energetics of binding.
3.4. PII-nucleotide binding study using ITC
We sought to experimentally investigate the predictions madethrough docking using isothermal titration calorimetry (ITC) studies.In a recent study, we have shown that ATP in the presence of MgCl2binds with MtbPII with a high affinity (Kd = 6.44 ± 1.36 μM) asdetermined through ITC [43]. ATP bound PII crystal structures fromE. coli [30], Thermus thermophiles [55], and Aquifex aeolicus (Unpublished)show binding of ATP with protein without Mg2+ whereas crystalstructures of PII-ATP complex from Azospirillum brasilense [33],Archaeoglobus fulgidus [56] and Methanococcus jannaschii [32] haveMg2+ ions complexed in the PII–ATP crystal. In ATP bound MtbPIIcrystal structure Mg2+ ions are absent [38]. We examined thebinding of ATP with MtbPII in the presence and absence of MgCl2through ITC. In presence of 5 mM MgCl2, MtbPII binds to ATP witha high affinity whereas in the absence of MgCl2 we did not observe any
significant binding (Fig. 7A and B). These results show that Mg2+ ion iscritical for binding of ATP with MtbPII at the concentrations of ATPused here. ATP binding with MtbPII shows anti-cooperativity with firstmolecule of ATP binds with a high affinity (Kd1 = 3.22 ± 0.51 μM)which decreases for subsequent binding of ATP molecules (Kd2 =22.76±3.68μM, Kd3=92.96±12.71μM) (Table 4). The binding affinityof ATP with MtbPII was comparable to that obtained with PII proteinfrom other bacterial species [23,57].
The crystal structure as well as the docking studies showed thatMtbPII makes extensive hydrogen bonds with the triphosphate group,in particular with γ-phosphate of ATP (Fig. 6A and B). In order to assessthe contribution of different moieties of ATP molecule for binding withMtbPII, we examined binding of MtbPII with the ligands ADP, AMP andadenine. ADPmolecule binds toMtbPII (Fig. 7C). However in contrast tobinding with ATP, the binding of ADP to first two sites is weaker thanthe third site (Table 4). With single site binding model, stoichiometryof ADP–MtbPII interaction was 2.5 ± 0.21 per monomer of MtbPII.A higher stoichiometry of ADP binding with MtbPII is likely due tomultiple interactions of ADP with the protein, which are likely not site
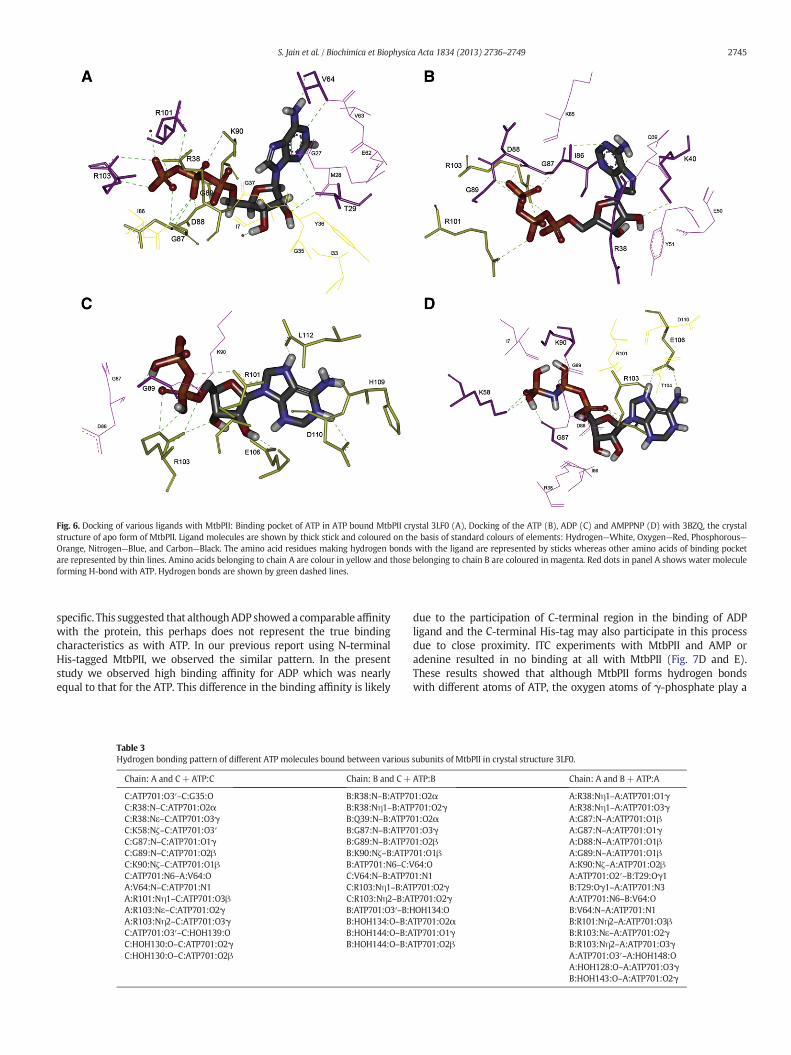
Fig. 6. Docking of various ligands with MtbPII: Binding pocket of ATP in ATP bound MtbPII crystal 3LF0 (A), Docking of the ATP (B), ADP (C) and AMPPNP (D) with 3BZQ, the crystalstructure of apo form of MtbPII. Ligand molecules are shown by thick stick and coloured on the basis of standard colours of elements: Hydrogen—White, Oxygen—Red, Phosphorous—Orange, Nitrogen—Blue, and Carbon—Black. The amino acid residues making hydrogen bonds with the ligand are represented by sticks whereas other amino acids of binding pocketare represented by thin lines. Amino acids belonging to chain A are colour in yellow and those belonging to chain B are coloured in magenta. Red dots in panel A shows water moleculeforming H-bond with ATP. Hydrogen bonds are shown by green dashed lines.
2745S. Jain et al. / Biochimica et Biophysica Acta 1834 (2013) 2736–2749
specific. This suggested that althoughADP showed a comparable affinitywith the protein, this perhaps does not represent the true bindingcharacteristics as with ATP. In our previous report using N-terminalHis-tagged MtbPII, we observed the similar pattern. In the presentstudy we observed high binding affinity for ADP which was nearlyequal to that for the ATP. This difference in the binding affinity is likely
Table 3Hydrogen bonding pattern of different ATP molecules bound between various
Chain: A and C+ATP:C Chain: B and C+
C:ATP701:O3′–C:G35:O B:R38:N–B:ATP70C:R38:N–C:ATP701:O2α B:R38:Nη1–B:ATC:R38:Nε–C:ATP701:O3γ B:Q39:N–B:ATP7C:K58:Nζ–C:ATP701:O3′ B:G87:N–B:ATP7C:G87:N–C:ATP701:O1γ B:G89:N–B:ATP7C:G89:N–C:ATP701:O2β B:K90:Nζ–B:ATP7C:K90:Nζ–C:ATP701:O1β B:ATP701:N6–C:VC:ATP701:N6–A:V64:O C:V64:N–B:ATP70A:V64:N–C:ATP701:N1 C:R103:Nη1–B:ATA:R101:Nη1–C:ATP701:O3β C:R103:Nη2–B:AA:R103:Nε–C:ATP701:O2γ B:ATP701:O3′–B:HA:R103:Nη2–C:ATP701:O3γ B:HOH134:O–B:AC:ATP701:O3′–C:HOH139:O B:HOH144:O–B:AC:HOH130:O–C:ATP701:O2γ B:HOH144:O–B:AC:HOH130:O–C:ATP701:O2β
due to the participation of C-terminal region in the binding of ADPligand and the C-terminal His-tag may also participate in this processdue to close proximity. ITC experiments with MtbPII and AMP oradenine resulted in no binding at all with MtbPII (Fig. 7D and E).These results showed that although MtbPII forms hydrogen bondswith different atoms of ATP, the oxygen atoms of γ-phosphate play a
subunits of MtbPII in crystal structure 3LF0.
ATP:B Chain: A and B+ATP:A
1:O2α A:R38:Nη1–A:ATP701:O1γP701:O2γ A:R38:Nη1–A:ATP701:O3γ01:O2α A:G87:N–A:ATP701:O1β01:O3γ A:G87:N–A:ATP701:O1γ01:O2β A:D88:N–A:ATP701:O1β01:O1β A:G89:N–A:ATP701:O1β64:O A:K90:Nζ–A:ATP701:O2β1:N1 A:ATP701:O2′–B:T29:Oγ1P701:O2γ B:T29:Oγ1–A:ATP701:N3TP701:O2γ A:ATP701:N6–B:V64:OOH134:O B:V64:N–A:ATP701:N1TP701:O2α B:R101:Nη2–A:ATP701:O3βTP701:O1γ B:R103:Nε–A:ATP701:O2γTP701:O2β B:R103:Nη2–A:ATP701:O3γ
A:ATP701:O3′–A:HOH148:OA:HOH128:O–A:ATP701:O3γB:HOH143:O–A:ATP701:O2γ
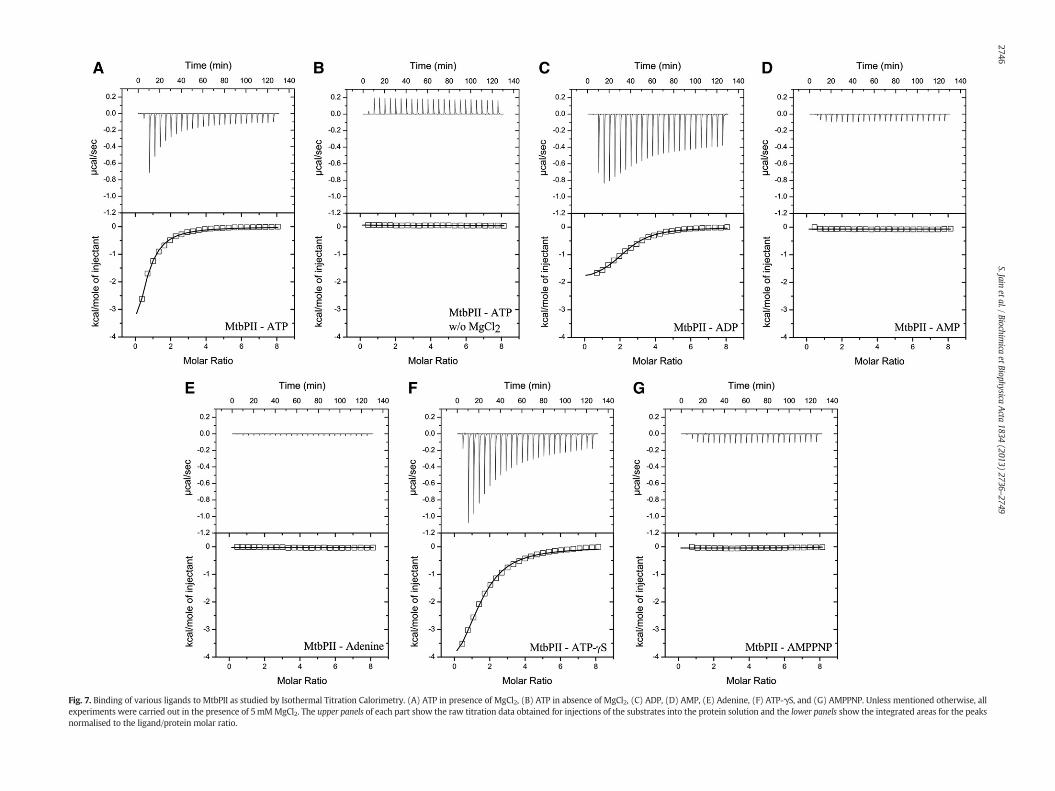
Fig. 7. Binding of various ligands to MtbPII as studied by Isothermal Titration Calorimetry. (A) ATP in presence of MgCl2, (B) ATP in absence of MgCl2, (C) ADP, (D) AMP, (E) Adenine, (F) ATP-γS, and (G) AMPPNP. Unless mentioned otherwise, allexperiments were carried out in the presence of 5mMMgCl2. The upper panels of each part show the raw titration data obtained for injections of the substrates into the protein solution and the lower panels show the integrated areas for the peaksnormalised to the ligand/protein molar ratio.
2746S.Jain
etal./Biochimica
etBiophysicaActa
1834(2013)
2736–2749

Table 4Dissociation constants of nucleotide binding with PII protein and its various mutants. All values represents average of two separate experiments ±Standard error of mean.
Protein Ligand Three sequential site binding model One site binding model
Kd1 (μM) Kd2 (μM) Kd3 (μM) Kd (μM) Stoichiometrya
PII (WT) ATP 3.22±0.51 22.76± 3.68 92.96±12.71 9.24±1.02 0.83±0.04ADP 68.29±21.50 109.90± 9.28 10.77±3.07 5.66±0.79 2.5± 0.21ATP-γS 15.35±6.01 33.77± 3.83 51.52±4.35 8.76±0.49 1.58±0.23
PII_R38A ATP 4.94±0.51 29.41± 3.73 84.53±23.36 9.44±1.40 0.84±0.01PII_Q39A ATP 5.64±2.90 27.58± 6.79 27.41±0.67 6.24±1.04 0.99±0.07PII_K90A ATP 6.31±1.38 38.30± 12.99 185.53±41.23 8.16±3.14 0.85±0.03PII_R101H ATP 4.68±0.37 19.09± 8.70 49.63±20.31 8.96±1.08 0.92±0.10PII_R103A ATP 6.19±3.07 25.71± 2.62 64.59±8.40 7.98±1.82 1.14±0.19PII_R103AR38A ATP 2.85±0.95 19.87± 6.31 32.36±23.19 7.08±2.25 0.88±0.06PII_R103AQ39A ATP 3.42±1.00 14.30± 7.11 57.26±37.99 7.56±2.24 0.79±0.03PII_R103AK90A ATP 4.31±0.18 19.77± 4.27 81.04±23.24 6.16±1.53 0.86±0.02
a Stoichiometry is given with respect to PII monomer.
2747S. Jain et al. / Biochimica et Biophysica Acta 1834 (2013) 2736–2749
definitive role. The data is in complete agreement with docking resultsof ATP with MtbPII where the oxygen atoms of γ-phosphate of ATPform extensive hydrogen bonds with the protein.
Next, we examined the binding of MtbPII with two non-hydrolysableATP analogs adenosine 5′-[γ-thio]-triphosphate (ATP-γS) and Adenosine5′-(β,γ-imido) triphosphate (AMPPNP). The binding of ATP-γS withMtbPII shows anti-cooperative behaviour as with ATP. However, thepattern of anti-cooperativity differs between the two ligands. The firstmolecule of ATP-γS binds with binding affinity 5-folds lesser than thatof ATP whereas the third molecule of ATP-γS binds with affinity higherthan that for ATP (Table 4) (Fig. 7F). AMPPNP displayed no bindingwith MtbPII (Fig. 7G). A sulphur atom in ATP-γS replaces one of theoxygen atoms at γ-phosphate of ATP whereas in AMPPNP the oxygenatom (O3B) connecting β- and γ-phosphates of ATP is replaced by anitrogen atom.
The results from ITC experimentswere comparablewith the dockingresults wherein ATP, ADP and AMPPNPwere estimated to bind to MtbPIIin decreasing order of binding energy. The lowest binding energy wasshown by AMPPNP, which has a twisted orientation at the γ-phosphatecompared to that in ATP. These results confirm that the γ-phosphateof ATP, positioned appropriately in space for interatomic H-bondinteractions and also for electrostatic interactions, has an importantrole in binding with MtbPII.
To determine the role of the amino acid residues predicted to beimportant for ATP binding, we generated a series of single and doublemutants of MtbPII using site directed mutagenesis. Mutants weregenerated by replacing the amino acid in question with Alanine(Histidine in case of R101) in order to shorten the side chain of aminoacid residues, thereby breaking the H-bonds and possible electrostaticinteractions between the respective amino acid and ATP [58]. Weobserved that unlike mutants studied for trimer stability, all thesemutant proteins showed good expression and were found in solublefraction after sonication. The mutant proteins were examined for ATPbinding using ITC. These results are displayed in Table 4.
Table 5Thermodynamicparameters for ATP bindingwith PII and its variousmutants from ITC datafitted with single site binding model. All values represents average of two separateexperiments± Standard error of mean.
Protein Enthalpy changeΔH (Kcal/mol)
Entropy changeTΔS (Kcal/mol)
Free energy changeΔG (Kcal/mol)
PII (WT) −4.21± 0.20 2.79±0.23 −7.00± 0.06PII_R38A −3.49± 0.34 3.48±0.43 −6.97± 0.09PII_Q39A −2.45± 0.04 4.79±0.06 −7.24± 0.10PII_K90A −2.68± 0.30 4.49±0.59 −7.17± 0.29PII_R101H −3.48± 0.44 3.53±0.49 −7.01± 0.07PII_R103A −2.30± 0.13 4.77±0.02 −7.07± 0.15PII_R103A_R38A −2.81± 0.21 4.36±0.42 −7.17± 0.21PII_R103A_Q39A −2.86± 0.88 4.27±1.06 −7.13± 0.12PII_R103A_K90A −2.74± 0.15 4.52±0.00 −7.25± 0.15
Mutated proteins with mutation of individual residues R38, Q39,K90, R101, and R103 showed same stoichiometry of binding to ATP,which was close to one per PII monomer as in native MtbPII. In mostcases anti-cooperativity was also observed as with the native protein.In the case of ATP binding with PII_Q39A, we observed loss of anti-cooperativity between the second and third site where the interactionshowed equivalent affinity (Table 4). As compared to ATP bindingwith native protein, MtbPII mutant PII_K90A resulted in approximatelytwo-folds reduction in the binding affinity at all three ATPs (Table 4).Analysis of ITC data with single site binding model showed that allmutant proteins on interaction with ATP resulted in reduced enthalpychange compared with native MtbPII protein but there was anequivalent increase in entropic change resulting in preserving thesame free energy change, upon binding of ATP to MtbPII (Table 5).
In order to identify any co-operative effect among the aminoacid predicted to interact, we also generated double mutants. Theamino acid R103 forms extensive H-bonds with γ-phosphate of ATP.Therefore, double mutants were designed by mutating above residuesin combination with R103. ITC analysis with these double mutants andATP showed no significant impact on the binding affinity (Table 4).Analysis of ITC data with single site binding model resulted in reducedenthalpic change, which was compensated by equivalent entropicchange indicating that the double mutants followed the samepattern as with single mutants. It is intriguing to us that none ofthe amino acids inferred to maintain H-bond interactions from bothcrystal structure and docking studies when mutated to Alanine had anyinfluence on the free energy change of binding of ATP with MtbPII.
The ITC data from PII mutants and ATP interaction showed that theenthalpy and entropy values were changed and appeared to display a“compensatory like” trend. These observations indicate that althoughthe overall binding constant remains same, the difference in enthalpychange is compensated through change in molecular order in thesystem. Ideally, when a charged or polar residue side chain group ischanged to non-polar type, we are likely to see a direct effect on theH-bond in terms of its disruption and if other variables remain same,this event is likely to influence in the free energy change. On the otherhand if the molecule may re-orient itself or if a water molecule couldoccupy the larger space created due to shortening of the sidechain, then a loss of H-bond could be compensated by adjustmentsin the molecular orientation. Thus, it appears that entropic contributionsignificantly aids in preserving the binding of ATP with MtbPII even inthe absence of the capacity of certain amino acids to form H-bonds.
These results indicate that the ATP binding pocket in MtbPII is veryrobust and mutating one or two amino acids among the participatingcandidates predicted to form H-bonds with the ATP does not affect theoverall ATP binding capacity despite the fact that these amino acidsshow a high degree of conservation among various orthologs of PII.
These observations are in contrast to those observed by Jiang et al. andRadchenko et al., who investigated the effect of amino acid mutations

2748 S. Jain et al. / Biochimica et Biophysica Acta 1834 (2013) 2736–2749
on binding of ATP with E. coli PII proteins [20,35]. Jiang et al. studiedbinding of ATP with E. coli GlnB and its mutants using ultrafiltrationmethod wherein the protein–ATP complexes, after short incubation,were centrifuged through Microcon-P10 filtration units to separate theunbound ligand and the effect of mutation was determined as the ratioof the amount of ATP bound to the mutant to that bound to the nativePII [35]. They showed that mutation K90R and G89A severely influencedthe ATP binding with the protein irrespective of presence or absence of2-OG. The E. coli GlnB mutant G89A completely abolished the binding ofATP with the protein whereas themutant K90R showed reduced bindingin absence of 2-OG which increased slightly in presence of 2-OG.The mutation R38H and Q39E had moderate effects on the ATP binding.The binding of ATP with mutation R38H was reduced to a fraction of0.36 relative to the wild type in absence of 2-OG but increased to afraction of 0.8 relative to that of wild type in presence of 2-OG. The ATPbindingwith E. coliGlnBmutant Q39E is reduce to a fraction of 0.3 relativeto the wild type irrespective of presence or absence of 2-OG. It isnoteworthy that the residue Q39 coordinates the binding of 2-OG inE. coli and therefore the reduced binding of ATP with this mutant couldbe due to insensitivity to 2-OG. Radchenko et al. examined the ATPbindingwith E. coliGlnKand itsmutantsK90A, R101AandR103A throughITC [20]. In E. coli GlnK, the mutation of these residues resulted insignificant reduction in the binding affinity. The dissociation constantfor wild type E. coli GlnK was observed to be 5 μM whereas dissociationconstants for the mutants K90A, R101A and R103A were found to be168μM, 260μMand 228μM, respectively. In our study, various mutantshad binding affinity equivalent to the wild type protein although inthe sequential binding site model we observed difference in thebinding in case of K90A and Q39A. In our previous study we haveshown that the binding affinity of ATP with MtbPII is not affectedby the presence of 2-OG, although it aids in the enthalpy change ofthe interaction. Therefore, although the role of the structurallyhomologous amino acids is likely to be very similar between E. coliGlnK and MtbPII, their extents may differ.
Despite maintaining structural conservation, PII proteins fromdifferent species have adapted to different response to the interactingproteins as reflected by different modes of posttranslational modifi-cations. They also have shown difference in their response to theeffector molecules. ITC experiments with E. coli and A. thaliana PIIproteins have shown enhanced binding of ATP in presence of 2-OGwhereas the binding affinity of ATP with MtbPII is not influenced by thepresence or absence of 2-OG [43,59,60]. In another study with B. subtilisPII, binding of ATP to the protein decreases by addition of Mg2+ butincreases with 2-OG addition whereas in many other species includingMtbPII, Mg2+ is required for this interaction [61]. These differences inthe interaction in effector molecule may reflect small differences tosuite the physiological niches of the respective species.
4. Conclusion
In this study we aimed to characterise MtbPII protein to identifyamino acid residues important for the stability of its trimeric structureand its binding with ATP. The amino acid pair R60–E62 present in thecentral core region of protein is critical in stabilizing trimeric structureof the protein. Among the amino acid pairs K2-D97 and D71/D75-R107 present at the surface of protein at least one of them must beintact. The pattern of ATP binding showed anti-cooperativity amongthe three binding sites as with other PII orthologs. The ATP bindingpocket of MtbPII is found to be robust preserving free energy changethrough enthalpy–entropy compensation with respect to mutation ofindividual amino acids predicted to form hydrogen bonds from crystalstructures. These differences between the PII orthologsmay have volvedto suite physiological niche of the species.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbapap.2013.10.006.
Acknowledgement
We thank Dr. Debasisa Mohanty, NII, Delhi for stimulatingdiscussions. Mr. Santosh Kumar (CSIR-IGIB, Delhi), Mr. Dilip Tiwari(CSIR-IGIB, Delhi) and Mr. Jaidev (INMAS, Delhi) are acknowledgedfor helping with ITC, size exclusion chromatography and dockingexperiment, respectively, SJ is recipient of research fellowship fromUniversity Grants Commission (UGC), India. This work was partiallysupported by funding from Council of Scientific and Industrial Research(CSIR) grants GAP0047 and BSC0121.
References
[1] A.J. Ninfa, M.R. Atkinson, PII signal transduction proteins, TrendsMicrobiol. 8 (2000)172–179.
[2] T. Arcondeguy, R. Jack, M. Merrick, P(II) signal transduction proteins, pivotal playersin microbial nitrogen control, Microbiol. Mol. Biol. Rev. 65 (2001) 80–105.
[3] L.F. Huergo, G. Chandra, M. Merrick, P(II) signal transduction proteins: nitrogenregulation and beyond, FEMS Microbiol. Rev. 37 (2013) 251–283.
[4] L.F. Huergo, F.O. Pedrosa, M. Muller-Santos, L.S. Chubatsu, R.A. Monteiro, M. Merrick,E.M. Souza, PII signal transduction proteins: pivotal players in post-translationalcontrol of nitrogenase activity, Microbiology 158 (2012) 176–190.
[5] K. Forchhammer, P(II) signal transducers: novel functional and structural insights,Trends Microbiol. 16 (2008) 65–72.
[6] K. Forchhammer, Global carbon/nitrogen control by PII signal transduction incyanobacteria: from signals to targets, FEMS Microbiol. Rev. 28 (2004) 319–333.
[7] J.A. Leigh, J.A. Dodsworth, Nitrogen regulation in bacteria and archaea, Annu. Rev.Microbiol. 61 (2007) 349–377.
[8] L.F. Huergo, L.S. Chubatsu, E.M. Souza, F.O. Pedrosa, M.B. Steffens, M. Merrick,Interactions between PII proteins and the nitrogenase regulatory enzymes DraTand DraG in Azospirillum brasilense, FEBS Lett. 580 (2006) 5232–5236.
[9] S. Nordlund, E.W. Triplett, Regulation of nitrogenase activity in phototrophic bacteriaby reversible covalent modification, Prokaryotic Nitrogen Fixation: a Model Systemfor the Analysis of a Biological Process, 2000. 149–164.
[10] M. Maheswaran, C. Urbanke, K. Forchhammer, Complex formation and catalyticactivation by the PII signaling protein of N-acetyl-L-glutamate kinase fromSynechococcus elongatus strain PCC 7942, J. Biol. Chem. 279 (2004) 55202–55210.
[11] L. Reitzer, Nitrogen assimilation and global regulation in Escherichia coli, Annu. Rev.Microbiol. 57 (2003) 155–176.
[12] A.A. Pioszak, P. Jiang, A.J. Ninfa, The Escherichia coli PII signal transduction proteinregulates the activities of the two-component system transmitter protein NRII bydirect interaction with the kinase domain of the transmitter module, Biochemistry39 (2000) 13450–13461.
[13] M.R. Atkinson, E.S. Kamberov, R.L. Weiss, A.J. Ninfa, Reversible uridylylation ofthe Escherichia coli PII signal transduction protein regulates its ability to stimulatethe dephosphorylation of the transcription factor nitrogen regulator I (NRI or NtrC),J. Biol. Chem. 269 (1994) 28288–28293.
[14] A.M. Fadi, J. Sauer, C. Spielhaupter, R. Schmid, K. Forchhammer, Signal transductionprotein P(II) is required for NtcA-regulated gene expression during nitrogendeprivation in the cyanobacterium Synechococcus elongatus strain PCC 7942,J. Bacteriol. 185 (2003) 2582–2591.
[15] J. Paz-Yepes, E. Flores, A. Herrero, Transcriptional effects of the signal transductionprotein P(II) (glnB gene product) on NtcA-dependent genes in Synechococcus sp.PCC 7942, FEBS Lett. 543 (2003) 42–46.
[16] J. Espinosa, K. Forchhammer, S. Burillo, A. Contreras, Interaction networkin cyanobacterial nitrogen regulation: PipX, a protein that interacts in a2-oxoglutarate dependent manner with PII and NtcA, Mol. Microbiol. 61(2006) 457–469.
[17] G. Beckers, J. Strosser, U. Hildebrandt, J. Kalinowski, M. Farwick, R. Kramer, A.Burkovski, Regulation of AmtR-controlled gene expression in Corynebacteriumglutamicum: mechanism and characterization of the AmtR regulon, Mol. Microbiol.58 (2005) 580–595.
[18] A. Hesketh, D. Fink, B. Gust, H.U. Rexer, B. Scheel, K. Chater, W.Wohlleben, A. Engels,The GlnD and GlnK homologues of Streptomyces coelicolor A3(2) are functionallydissimilar to their nitrogen regulatory system counterparts from enteric bacteria,Mol. Microbiol. 46 (2002) 319–330.
[19] J. Strosser, A. Ludke, S. Schaffer, R. Kramer, A. Burkovski, Regulation of GlnK activity:modification, membrane sequestration and proteolysis as regulatory principles inthe network of nitrogen control in Corynebacterium glutamicum, Mol. Microbiol.54 (2004) 132–147.
[20] M.V. Radchenko, J. Thornton, M. Merrick, PII signal transduction proteins areATPases whose activity is regulated by 2-oxoglutarate, Proc. Natl. Acad. Sci. U. S. A.110 (2013) 12948–12953.
[21] L.M. Araujo, L.F. Huergo, A.L. Invitti, C.I. Gimenes, A.C. Bonatto, R.A. Monteiro, E.M.Souza, F.O. Pedrosa, L.S. Chubatsu, Different responses of the GlnB and GlnZ proteinsupon in vitro uridylylation by the Azospirillum brasilense GlnD protein, Braz. J. Med.Biol. Res. 41 (2008) 289–294.
[22] S. Colonna-Romano, E.J. Patriarca, M. Amar, P. Bernard, G. Manco, A. Lamberti, M.Iaccarino, R. Defez, Uridylylation of the PII protein in Rhizobium leguminosarum,FEBS Lett. 330 (1993) 95–98.
[23] A.C. Bonatto, E.M. Souza, M.A. Oliveira, R.A. Monteiro, L.S. Chubatsu, L.F. Huergo, F.O.Pedrosa, Uridylylation of Herbaspirillum seropedicae GlnB and GlnK proteins is

2749S. Jain et al. / Biochimica et Biophysica Acta 1834 (2013) 2736–2749
differentially affected by ATP, ADP and 2-oxoglutarate in vitro, Arch. Microbiol. 194(2012) 643–652.
[24] A.C. Bonatto, G.H. Couto, E.M. Souza, L.M. Araujo, F.O. Pedrosa, L. Noindorf, E.M.Benelli, Purification and characterization of the bifunctional uridylyltransferaseand the signal transducing proteins GlnB and GlnK from Herbaspirillum seropedicae,Protein Expr. Purif. 55 (2007) 293–299.
[25] K.J. Williams, M.H. Bennett, G.R. Barton, V.A. Jenkins, B.D. Robertson, Adenylylationof mycobacterial Glnk (PII) protein is induced by nitrogen limitation, Tuberculosis(Edinb.) 93 (2013) 198–206.
[26] K. Forchhammer, M.N. Tandeau de, The PII protein in the cyanobacteriumSynechococcus sp. strain PCC 7942 ismodified by serine phosphorylation and signalsthe cellular N-status, J. Bacteriol. 176 (1994) 84–91.
[27] N. Kloft, G. Rasch, K. Forchhammer, Protein phosphatase PphA from Synechocystis sp.PCC 6803: the physiological framework of PII-P dephosphorylation, Microbiology 151(2005) 1275–1283.
[28] K. Forchhammer, A. Hedler, Phosphoprotein PII from cyanobacteria —analysis offunctional conservation with the PII signal-transduction protein from Escherichiacoli, Eur. J. Biochem. 244 (1997) 869–875.
[29] Y. Zhang, H. Pu, Q. Wang, S. Cheng, W. Zhao, Y. Zhang, J. Zhao, PII isimportant in regulation of nitrogen metabolism but not required for heterocystformation in the Cyanobacterium Anabaena sp. PCC 7120, J. Biol. Chem. 282 (2007)33641–33648.
[30] Y. Xu, P.D. Carr, T. Huber, S.G. Vasudevan, D.L. Ollis, The structure of the PII –ATPcomplex, Eur. J. Biochem. 268 (2001) 2028–2037.
[31] Y. Xu, E. Cheah, P.D. Carr, W.C. van Heeswijk, H.V. Westerhoff, S.G. Vasudevan, D.L.Ollis, GlnK, a PII-homologue: structure reveals ATP binding site and indicateshow the T-loops may be involved in molecular recognition, J. Mol. Biol. 282(1998) 149–165.
[32] O. Yildiz, C. Kalthoff, S. Raunser, W. Kuhlbrandt, Structure of GlnK1 with boundeffectors indicates regulatory mechanism for ammonia uptake, EMBO J. 26 (2007)589–599.
[33] D. Truan, L.F. Huergo, L.S. Chubatsu, M. Merrick, X.D. Li, F.K. Winkler, A new P(II)protein structure identifies the 2-oxoglutarate binding site, J. Mol. Biol. 400 (2010)531–539.
[34] O. Fokina, V.R. Chellamuthu, K. Forchhammer, K. Zeth, Mechanism of 2-oxoglutaratesignaling by the Synechococcus elongatus PII signal transduction protein, Proc. Natl.Acad. Sci. U. S. A. 107 (2010) 19760–19765.
[35] P. Jiang, P. Zucker, M.R. Atkinson, E.S. Kamberov, W. Tirasophon, P. Chandran, B.R.Schefke, A.J. Ninfa, Structure/function analysis of the PII signal transduction proteinof Escherichia coli: genetic separationof interactionswith protein receptors, J. Bacteriol.179 (1997) 4342–4353.
[36] A. Jonsson, P.F. Teixeira, S. Nordlund, The activity of adenylyltransferase inRhodospirillum rubrum is only affected by alpha-ketoglutarate and unmodified PIIproteins, but not by glutamine, in vitro, FEBS J. 274 (2007) 2449–2460.
[37] G. Harth, S. Maslesa-Galic, M.V. Tullius, M.A. Horwitz, All four Mycobacteriumtuberculosis glnA genes encode glutamine synthetase activities but only GlnA1 isabundantly expressed and essential for bacterial homeostasis, Mol. Microbiol. 58(2005) 1157–1172.
[38] N.D. Shetty, M.C. Reddy, S.K. Palaninathan, J.L. Owen, J.C. Sacchettini, Crystalstructures of the apo and ATP boundMycobacterium tuberculosis nitrogen regulatoryPII protein, Protein Sci. 19 (2010) 1513–1524.
[39] J. Rengarajan, B.R. Bloom, E.J. Rubin, Genome-wide requirements forMycobacteriumtuberculosis adaptation and survival in macrophages, Proc. Natl. Acad. Sci. U. S. A.102 (2005) 8327–8332.
[40] G.R. Stewart, J. Patel, B.D. Robertson, A. Rae, D.B. Young, Mycobacterial mutantswith defective control of phagosomal acidification, PLoS Pathog. 1 (2005)269–278.
[41] R. Read, C.A. Pashley, D. Smith, T. Parish, The role of GlnD in ammonia assimilation inMycobacterium tuberculosis, Tuberculosis (Edinb.) 87 (2007) 384–390.
[42] C.L. Stallings, M.S. Glickman, Is Mycobacterium tuberculosis stressed out? A criticalassessment of the genetic evidence, Microbes Infect. 12 (2010) 1091–1101.
[43] A. Bandyopadhyay, A. Arora, S. Jain, A. Laskar, C. Mandal, V.A. Ivanisenko, E.S. Fomin,S.S. Pintus, N.A. Kolchanov, S. Maiti, S. Ramachandran, Expression and molecularcharacterization of the Mycobacterium tuberculosis PII protein, J. Biochem. 147(2010) 279–289.
[44] K.G. Tina, R. Bhadra, N. Srinivasan, PIC: protein interactions calculator, Nucleic AcidsRes. 35 (2007) W473–W476.
[45] E. Krissinel, K. Henrick, Inference of macromolecular assemblies from crystallinestate, J. Mol. Biol. 372 (2007) 774–797.
[46] S.F. Altschul, T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang, W. Miller, D.J. Lipman,Gapped BLAST and PSI-BLAST: a new generation of protein database searchprograms, Nucleic Acids Res. 25 (1997) 3389–3402.
[47] R.C. Edgar, MUSCLE: multiple sequence alignment with high accuracy and highthroughput, Nucleic Acids Res. 32 (2004) 1792–1797.
[48] O. Trott, A.J. Olson, AutoDock Vina: improving the speed and accuracy of dockingwith a new scoring function, efficient optimization, and multithreading, J. Comput.Chem. 31 (2010) 455–461.
[49] E.F. Pettersen, T.D. Goddard, C.C. Huang, G.S. Couch, D.M. Greenblatt, E.C. Meng, T.E.Ferrin, UCSF Chimera —a visualization system for exploratory research and analysis,J. Comput. Chem. 25 (2004) 1605–1612.
[50] S. Kumar, R. Nussinov, Close-range electrostatic interactions in proteins, Chembiochem3 (2002) 604–617.
[51] M.J. Conroy, A. Durand, D. Lupo, X.D. Li, P.A. Bullough, F.K. Winkler, M. Merrick, Thecrystal structure of the Escherichia coli AmtB-GlnK complex reveals how GlnKregulates the ammonia channel, Proc. Natl. Acad. Sci. U. S. A. 104 (2007) 1213–1218.
[52] F. Gruswitz, J. O'Connell III, R.M. Stroud, Inhibitory complex of the transmembraneammonia channel, AmtB, and the cytosolic regulatory protein, GlnK, at 1.96 A,Proc. Natl. Acad. Sci. U. S. A. 104 (2007) 42–47.
[53] P.D. Carr, E. Cheah, P.M. Suffolk, S.G. Vasudevan, N.E. Dixon, D.L. Ollis, X-ray structureof the signal transduction protein from Escherichia coli at 1.9 A, Acta Crystallogr. DBiol. Crystallogr. 52 (1996) 93–104.
[54] E. Cheah, P.D. Carr, P.M. Suffolk, S.G. Vasudevan, N.E. Dixon, D.L. Ollis, Structure ofthe Escherichia coli signal transducing protein PII, Structure 2 (1994) 981–990.
[55] H. Sakai, H. Wang, C. Takemoto-Hori, T. Kaminishi, H. Yamaguchi, Y. Kamewari,T. Terada, S. Kuramitsu, M. Shirouzu, S. Yokoyama, Crystal structures of the signaltransducing protein GlnK from Thermus thermophilus HB8, J. Struct. Biol. 149 (2005)99–110.
[56] C. Litz, S. Helfmann, S. Gerhardt, S.L. Andrade, Structure of GlnK1, a signallingprotein from Archaeoglobus fulgidus, Acta Crystallogr. Sect. F Struct. Biol. Cryst.Commun. 67 (2011) 178–181.
[57] O. Fokina, V.R. Chellamuthu, K. Zeth, K. Forchhammer, A novel signal transductionprotein P(II) variant from Synechococcus elongatus PCC 7942 indicates a two-stepprocess for NAGK-P(II) complex formation, J. Mol. Biol. 399 (2010) 410–421.
[58] B.C. Cunningham, J.A. Wells, High-resolution epitope mapping of hGH-receptorinteractions by alanine-scanning mutagenesis, Science 244 (1989) 1081–1085.
[59] M.V. Radchenko, J. Thornton, M. Merrick, Control of AmtB –GlnK complex formationby intracellular levels of ATP, ADP, and 2-oxoglutarate, J. Biol. Chem. 285 (2010)31037–31045.
[60] C.S. Smith, A.M. Weljie, G.B. Moorhead, Molecular properties of the putativenitrogen sensor PII from Arabidopsis thaliana, Plant J. 33 (2003) 353–360.
[61] A. Heinrich, K. Woyda, K. Brauburger, G. Meiss, C. Detsch, J. Stulke, K.Forchhammer, Interaction of the membrane-bound GlnK–AmtB complex with themaster regulator of nitrogen metabolism TnrA in Bacillus subtilis, J. Biol. Chem. 281(2006) 34909–34917.





























