Embed Size (px)
Citation preview

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 1
1. Metabolism of energy in Cells To do mechanical work, organisms need energy. This energy is used for the
active transport of molecules, ions and synthesis of macromolecules and other
biomolecules. The free energy used in these processes comes from the
environment. However, energy from the nutrients cannot be utilized directly
by the cell. It has to be converted to chemical energy and stored in the form of
useful energy. This energy conversion occurs in mitochondria and is known
as oxidative phosphorylation (Figure 1.1). Under aerobic conditions, the
glucose metabolism starts with glycolysis in the cytosol and the overall
reaction results in conversion of glucose into two pyruvate molecules. A net
energy yield of two adenosine triphosphate (ATP) molecules is generated by
this reaction and also involves the reduction of two nicotinamide adenine
dinucleotide molecules (NADH). Under aerobic conditions, the NADH and
pyruvate are transferred to mitochondria. When pyruvate enters the
mitochondrial matrix, it reacts with coenzyme A to form acetyl-CoA, CO2 and
reduced NADH, the acetyl-CoA being further oxidized in the Krebs cycle. The
net energy yield of the overall oxidation of two pyruvate molecules from one
glucose molecule is conserved in ten NADH, two flavin adenine dinucleotide
(FADH2) and two ATP molecules. Under anaerobic conditions pyruvate is
converted into lactate by the enzyme lactate dehydrogenase in a reversible
reaction that enables glycolysis to proceed transiently in active tissues such
as contracting muscle. The lactate formed in these active tissues is secreted
into the bloodstream and then oxidized back to pyruvate in the liver (Berg et
al. 2002)
The acetyl-CoA can also be generated for Krebs cycle by the oxidation of fatty
acids in the mitochondrial matrix. Fatty acids are coupled to coenzyme A to
form acyl-CoA in the outer mitochondrial membrane. The long-chain acyl-
CoA esters are unable to traverse through mitochondrial membrane and are
carried across as carnitine esters. On the matrix side, the regenerated acyl-

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 2
CoA molecules are shortened by two carbon atoms in a cyclic process called β-
oxidation. This continues until all the carbon atoms in the acyl-CoA have
been converted into two carbon acetyl-CoA molecules, which enter the Krebs
cycle. One molecule of NADH and one of FADH2 and two of acetyl-CoA are
produced in every cycle (Berg et al. 2002).
Figure 1.1: Energy metabolism in eukaryotic cells. Oxidation of nutrients in the cytosol and in the
mitochondria leads to adenosine triphosphate (ATP) synthesis by the oxidative phorphorylation system.
The ATP generated by this process is then used to drive biosynthetic reactions and other processes in
the cell that require energy.
2. Mitochondria Mitochondria are membrane-bound organelles present in almost all
eukaryotic cells except a few (Henze and Martin 2003). These organelles are
sometimes described as "cellular power plants", because they generate most
of the cell’s ATP supply that is used as a source of chemical energy. The
number of mitochondria in a cell varies by organism, tissue type or cell type.
Many cells possess only a single mitochondrion, while others can have several
million mitochondria (Alberts et al. 1994; Voet et al. 2006). A mitochondrion

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 3
contains inner and outer membranes made up of phospholipid bilayers and
proteins. The two membranes are very much different in their properties.
Due to this double-membrane organization, mitochondrion is divided into five
compartments; the outer membrane, inner membrane, cristae (infoldings of
inner membrane) and the matrix. The size of mitochondrion ranges from 1 to 10 micrometers (μm).
The outer membrane encloses the entire organelle and has a protein-to-
phospholipid ratio similar to plasma membrane of eukaryotic cells (about 1:1
by weight). It contains many integral proteins called porins, which contain a
large internal channel (about 2-3 nm) that is permeable to all molecules of
about 5000 daltons or less (Henze and Martin 2003). Larger molecules can
only traverse the outer membrane by active transport through mitochondrial
membrane transport proteins. The outer membrane also contains enzymes
involved in activities such as elongation of fatty acids, oxidation of
epinephrine (adrenaline), and the degradation of tryptophan.
The space between outer and inner membrane is known as inter-membrane
space (IMS). The main function of IMS is nucleotide phosphorylation.
Channel proteins (porins) in the outer membrane allow free movement of ions
and small molecules into the IMS. The contents of the intermembrane space
are similar to that of the contents of cytoplasm. Enzymes destined for the
mitochondrial matrix can pass through this space via transport through
translocators. These are known as translocase of the outer membrane (TOM)
and transloacase of the inner mitochondrial membrane (TIM). The IMS tends
to have a low pH because of the proton gradient, which results when protons
are pumped from mitochondrial matrix into the intermembrane space during
the electron transport.
The inner mitochondrial membrane contains proteins with four types of
functions (Alberts et al. 1994): (a) Proteins carrying out the oxidation
reactions of the respiratory chain (b) ATP synthase, which makes ATP in the
matrix. (c) Specific transport proteins that regulate the passage of

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 4
metabolites into and out of the matrix and (d) Protein import machinery. The
inner membrane contains more than 100 different polypeptides, and has a
very high protein to phospholipid ratio (more than 3:1 by weight, which is
about 1 protein for 15 phospholipids). It is rich in unusual phospholipids
known as cardiolipin, which was first isolated from beef hearts. Unlike the
outer membrane, the inner membrane does not contain porins, and is highly
impermeable. Most of the ions and molecules require special membrane
transporters to enter or exit the matrix. In addition, there is a membrane
potential across the inner membrane. The inner mitochondrial membrane is
compartmentalized into numerous cristae, which expand the surface area of
the inner mitochondrial membrane, enhancing its ability to generate ATP.
Mitochondria of cells, which have greater demand for ATP, such as muscle
and nerve cells, contain more cristae than typical liver mitochondria.
The matrix is the space enclosed by the inner membrane. The matrix is
important in the production of ATP with the help of the ATP synthase located
in the inner membrane. The matrix contains a mixture of hundreds of
enzymes, in addition to the special mitochondrial ribosomes, tRNAs, and
several copies of the mitochondrial genome. Of the enzymes, the major
functions include oxidation of pyruvate and fatty acids, and the citric acid
cycle (Alberts et al. 1994).

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 5
Figure 1.2: Simplified structure of mitochondrion. Mitochondria are bounded by a double-membrane
system, consisting of inner and outer membranes. Folds of the inner membrane (cristae) extend into the
matrix.
2.1. The Respiratory Chain The respiratory chain in the inner mitochondrial membrane contains three
respiratory enzyme complexes through which electrons pass on their way
from NADH to O2. Although the respiratory chain usually proceeds in the
forward direction (towards the formation of water) due to the exergonic
nature of the reaction cascade, except for the final reaction, all of these steps
are fully reversible. In order to be reversed, sufficient energy must be
provided to drive the reaction in this direction. For example, the reducing
equivalents derived from succinate are usually carried by FAD (as FADH+H+).
These can be transferred to NAD (as NADH++H+) with the concomitant
hydrolysis of ATP. Electron transport across the other two phosphorylation
sites can also be reversed, again, only if sufficient energy is provided. The
respiratory chain consists of four major enzyme complexes (Figure 1.3)
located in the inner mitochondrial membrane. The enzymes of the respiratory

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 6
chain are arranged so as to transport hydrogen ions from the matrix across
the inner membrane. When this occurs, a proton gradient develops in close
proximity to the F1F0 ATPase (ATP synthase) complex and provides sufficient
energy to drive ATP synthesis by causing a dehydration of ADP and Pi.
Reducing equivalents transported into the mitochondrial compartment by the
various substrate shuttle systems or generated in the compartment are
passed down the respiratory chain in carefully regulated steps. Reducing
equivalents enter the chain through the NAD-dehydrogenase complex
(complex I) via mitochondrial shuttles or via the FAD-ubiquinone complex
(complex II). With respect to the latter complex, reducing equivalents collect
via three pathways:
Succinate contributes its reducing equivalents to a flavoprotein with
an iron–sulfur center.
Glycerol 3-phosphate also uses FAD flavoprotein with an iron–sulfur
center, but it is a different protein.
The products of fatty acid oxidation (a mitochondrial process) are
picked up by a FAD-flavoprotein, transferred to an electron-
transferring flavoprotein, again with an iron–sulfur center, and then
transferred to ubiquinone.
In mammals, complex I consist of about 42 subunits (Weiss et al. 1981). Of
these, seven are encoded by the mitochondrial genome and the rest by the
nuclear genome synthesized on the ribosomes in the cytoplasm and imported
into the mitochondrial compartment. It is the largest of the four respiratory
chain complexes. Complex I is known as NADH-coenzyme Q reductase or
NADH dehydrogenase. As the name implies, this complex transfers a pair of
electrons from NADH to coenzyme Q, a lipid-soluble compound embedded in
the inner membrane. Complex I has a molecule of flavin mononucleotide
(FMN) and two binuclear iron-sulfur centers and four tetranuclear iron-
sulfur centers (Ohnishi 1993). Because of its FMN, it is called a flavoprotein.

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 7
The complex catalyzes the transfer of electrons to complex III via ubiquinone
and this transfer is coupled with the vectorial transfer of protons across the
mitochondrial membrane. There are two distinct species of tightly bound
ubiquinones in complex I that differ in spin relaxation and redox properties. The transfer of electrons leads to the formation of a proton gradient (∆μH+)
that, in turn, drives ATP production. The stoichiometry of proton transfer for
complex I is 4H+/2e–. This distinguishes this complex from those that follow it
in the respiratory chain. The other two H+ translocating complexes (III and
IV) have a stoichiometry of 2 H+/2e–.
Complex II, succinate:quinone reductase (succinate dehydrogenase), is the
smallest of the respiratory chain complexes (Hederstedt and Ohnishi 1992).
None of the subunits of complex II are encoded by the mitochondrial genome.
The complex consists of four subunits with several different redox prosthetic
groups: a covalently bound FAD, three iron-sulfur clusters, and a cytochrome
b. The head of the complex protrudes out into the matrix where its FAD can
accept succinate-donated electrons from the citric acid cycle. Actually, this
enzyme is a component of the respiratory chain and the citric acid cycle. It too
is a flavoprotein because of its FAD content. The FAD is bound to a histidine
residue (Paudel 1994). When succinate is converted to fumarate in the citric
acid cycle, a concomitant reduction of FAD to FADH2 occurs. This FADH2
transfers its electrons to the iron-sulfur cluster, which, in turn, passes them
on to ubiquinone. Because of insufficient energy to elicit a proton gradient,
reducing equivalents and associated electrons entering the respiratory chain
via complex II yield only two ATPs via OXPHOS rather than the three ATPs
generated when entry occurs via complex I.
Once reducing equivalents enter the chain via site 1 or site 2 (the sites
correspond to entry via complex I or II), they are passed to complex III, the
ubiquinone-cytochrome bc1 reductase. This complex takes the electrons
passed to it from ubiquinone and then passes them to complex IV, cytochrome
c oxidase. This passage uses a unique redox pathway called the Q cycle

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 8
(Slater 1985). Three different cytochromes (bc, b, c1) are involved as well as
an iron-sulfur protein. The iron is in the middle of a porphorin ring much like
that of hemoglobin and oscillates between the reduced and oxidized states
(ferrous to ferric).
The Q cycle begins when a molecule of reduced ubiquinone diffuses to the Qp
site on complex III near the outer face of the inner mitochondrial membrane.
An electron from the reduced ubiquinone is transferred to a mobile protein
called the Rieske protein. The electrons are then transferred to cytochrome c1.
This releases two H+ and leaves UQ–, a semiquinone anion form of
ubiquinone, at the Qp site. The second electron is then transferred to the bL
heme, converting UQ– to ubiquinone. The Rieske protein and cytochrome c1
are similar in structure; each has a globular domain and each is anchored to
the inner membrane by a hydophobic segment. The segments differ: the
Rieske protein has an N-terminal and the cytochrome c1 has a C-terminal.
The electron on the bL heme is passed to the bH heme against a membrane
potential of 0.15 V and is driven by the loss of redox potential as the electron
moves from bL to bH. The electron is then passed from bH to ubiquinone at the
second binding site, converting the ubiquinone to UQ–. The resulting UQ–
remains firmly bound to the Qn site. This completes the first half of the Q
cycle. The second half is similar in that a second molecule of reduced
ubiquinone is oxidized at the Qp site. One electron is passed to cytochrome c1
and the other is passed to heme bH. The bH electron is transferred to the
semiquinone anion UQ– at the Qn site. With the addition of two H+, this
produces UQH2. The UQH2 is released and returns to the coenzyme Q pool
and the Q cycle is complete. Actually, the Q cycle is an unbalanced proton
pump. Cytochrome c is a mobile electron carrier, as is ubiquinone. Electrons
travel from c to the water-soluble c1. The c1 associates loosely with the inner
mitochondrial membrane to acquire electrons from the iron-sulfur centers.
The c1 of complex III then migrates along the membrane in a reduced state so
as to give these electrons to complex IV.

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 9
Complex IV, cytochrome-c oxidase, contains two heme centers (a and a3) as
well as two copper centers. The copper oscillates between the reduced
(cuprous) and oxidized (cupric) states. Complexes III and IV elicit a proton
gradient and thus ATP is formed at each of these sites. Complex IV accepts
the electrons from cytochrome c and directs them to molecular oxygen to form
water. The water thus formed quickly passes out of the compartment into the
cytoplasm.
2.2. Origin of mitochondria Mitochondria are only formed by the division of other mitochondria and
contain ribosomes and transfer RNAs that are similar to bacteria. They
contain their own DNA, which is circular as is true with bacteria, along with
their own transcriptional and translational machinery. Hence, it is generally
accepted that they were originally derived from endosymbiont prokaryotes.
Studies of mitochondrial DNA, which uses a variant genetic code, show that
the ancestor proto-mitochondrion was a member of the phylum
Proteobacteria (Futuyma and Douglas 2005). In particular, the pre-
mitochondrion was probably related to the rickettsias, group of eubacterial
obligate intracellular parasites (Gray et al. 1999). Infact, the most
mitochondrion-like bacterial genome found to date is that of Proteobacteria
Rickettsia Prowasekii (Andersson et al. 1998). The endosymbiotic hypothesis
suggests that mitochondria descended from specialized bacteria (probably
purple non-sulfur bacteria) that survived endocytosis by other cell types, and
became incorporated into the cytoplasm. The ability of symbiont bacteria to
conduct cellular respiration in host cells that had relied on glycolysis and
fermentation would have provided a considerable evolutionary advantage.
Similarly, host cells with symbiotic bacteria capable of photosynthesis would
also have an advantage. In both cases, the number of environments in which
the cells could survive would have been greatly expanded.

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 10
The endosymbiotic relationship developed a few years ago and mitochondria
still show the signs of their ancient origin. Mitochondrial ribosomes in
mammals are the 70S type like bacteria, in contrast to the 80S ribosomes
found elsewhere in the cell (O’Brien 2003). One mitochondrion can contain 2-
10 copies of its genome (Wiesner et al. 1992). As in prokaryotes, there is a
very high proportion of coding DNA, and an absence of repeats, mitochondrial
genes are transcribed as multigenic transcripts, which are cleaved, and
polyadenylated to yield mature mRNAs. In humans, mitochondrial genes lack
introns (Anderson et al. 1981), conforming to the bacterial pattern. Further,
there are codon differences in mitochondria (Fernandez-Silva et al. 2003). In
the mitochondria, the UGA codon specifies tryptophan; AGA and AGG are
stop codons; and AUA, AUC, and AUU are each allowable start codons.
2.3. Functions of mitochondria Mitochondria not only produce most of the energy in eukaryotic cells but are
also involved in a number of other processes such as: lipid metabolism, Krebs
cycle, apoptosis (programmed cell death), cellular proliferation, regulation of
the cellular redox state, heme synthesis and steroid synthesis. Some
mitochondrial functions are performed only in specific types of cells. For
example, mitochondria in liver cells contain enzymes that allow them to
detoxify ammonia, a waste product of protein metabolism. A mutation in the
genes regulating any of these functions can result in mitochondrial diseases.
2.3.1. Heat production Under certain circumstances, protons re-enter the mitochondrial matrix
without contributing to ATP synthesis. This process is known as proton leak
or mitochondrial uncoupling and is due to the facilitated diffusion of protons
into the matrix. This process results in the unharnessed potential energy of
the proton electrochemical gradient being released as heat. The process is
mediated by a proton channel called thermogenin, or UCP1 (Mozo et al. 2005).

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 11
Thermogenin is a 33kDa protein (Nicholls and Lindberg 1973) primarily
found in brown adipose tissue, or brown fat, and is responsible for non-
shivering thermogenesis. Brown adipose tissue is found in mammals and is
at its highest levels in early life and in hibernating animals. In humans,
brown adipose tissue is present at birth and decreases with age (Mozo et al.
2005).
2.3.2. Calcium storage The concentration of free calcium in the cell can regulate a number of
reactions and is important for signal transduction. Mitochondria store
calcium, thus maintains calcium homeostasis. The release of this calcium
back into the cells interior can initiate calcium spikes or waves. These events
coordinate processes such as neurotransmitter release in nerve cells and
release of hormones in endocrine cells.
2.3.3. Citric acid cycle Pyruvate molecules produced by glycolysis are actively transported across the
inner mitochondrial membrane, and into the matrix where these are oxidized
and combined with coenzyme A to form CO2, acetyl-CoA and NADH. The
acetyl-CoA is the primary substrate to enter the citric acid cycle. The
enzymes of the citric acid cycle are present in the mitochondrial matrix
except succinate dehydrogenase, which is bound to the inner mitochondrial
membrane. The citric acid cycle oxidizes the acetyl-CoA to carbon dioxide and
in the process produces reduced cofactors (three molecules of NADH and one
molecule of FADH2), which are a source of electrons for the electron transport
chain, and a molecule of GTP (that is converted to an ATP).
2.3.4. Oxidative phosphorylation Oxidative phosphorylation (OXPHOS) is the synthesis of ATP from ADP and
inorganic phosphate. It is carried out by five multi-subunit enzyme complexes

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 12
(Figure 1.3). Electrons from NADH and FADH2 are transported to oxygen by
respiratory chain complexes; I, II, III and IV. The final step, the production of
ATP from ADP and inorganic phosphate is carried out by ATP synthase
(complex V). Rather than being isolated complexes, these enzymes seem to
occur together, forming super-complexes (Schagger and Pfeiffer 2000,
Schagger and Pfeiffer 2001), with varying stoichiometries depending on the
use of different solubilization detergents (Schagger 2002, Schafer et al. 2006).
The functional significance of this super-complex formation is thought to
involve catalytic enhancement by channeling of the substrates and
prevention of competition from other enzymes, prevention of the reactive
oxygen species (ROS) and stabilization of the individual OXPHOS enzyme
complexes (Schagger 2002, Acin-Perez et al. 2004). Two processes
characterize the operation of the respiratory chain, electron flow within the
enzyme complexes and the transport of protons across the inner
mitochondrial membrane. NADH is oxidized by the first and largest enzyme
of the respiratory chain, complex I. Electrons are passed through the enzyme
via prosthetic groups flavin mononucleotide (FMN) and seven iron-sulphur
clusters, to ubiquinone (UQ). FADH2 donates the electrons to complex II. The
enzyme operates both in the respiratory chain and in the Krebs cycle, where
it oxidizes succinate to fumarate, yielding FADH2. Electrons are carried
through complex II via flavin adenine dinucleotide (FAD) and three iron-
sulphur clusters to ubiquinone. Complex II also contains one b heme, the
functional role of which is currently unclear. The reaction of complex II is
reversible and the direction of electron flow through the enzyme is dictated
by the relative concentrations of the reactants and products. Thus, in
addition to being an entry point for electrons into the respiratory chain,
complex II also participates in regulation of the Krebs cycle. Electrons from
complexes I and II reduce ubiquinone (UQ) to ubiquinol (QH2), which is
hydrophobic and shuttles within the inner mitochondrial membrane,
transferring the electrons further to complex III. The electrons pass through

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 13
the complex III dimer via cytochrome b, a binuclear iron-sulphur cluster and
cytochrome c1, which donates them to ferricytochrome c. Complex IV,
catalyzes the final step of the respiratory chain, the transfer of the electrons
from ferrocytochrome c to dioxygen, to produce water. The electron
transportation occurs via four redox centres, a binuclear copper centre (CuA),
a mononuclear copper centre (CuB) and two hemes (a & a3). In addition to
electron transport, complexes I, III and IV serve as proton pumps. The
enzymes use energy from the electron transfer to translocate protons from
the matrix side into the intermembrane space, producing an electrochemical
gradient across the inner mitochondrial membrane. The free energy released
from the flow of protons back across the membrane is then used by complex V
for chemical work to produce ATP from ADP and inorganic phosphate
(Schultz and Chan 2001, Berg et al. 2002).
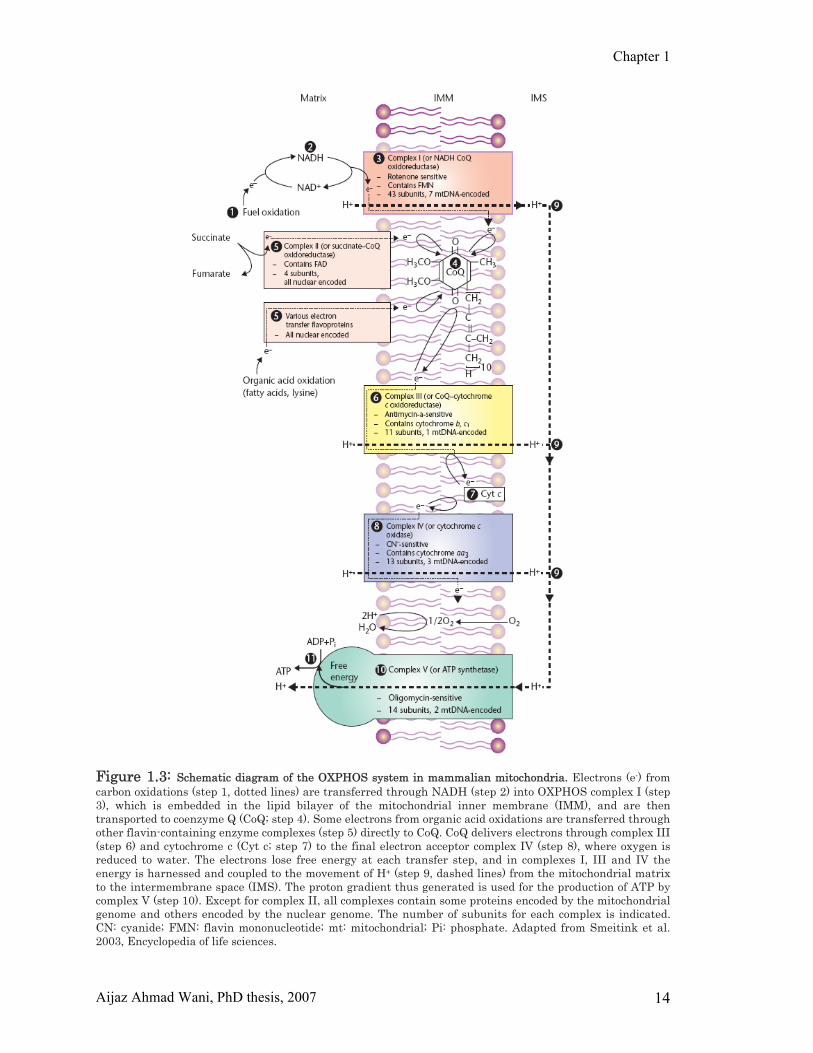
Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 14
Figure 1.3: Schematic diagram of the OXPHOS system in mammalian mitochondria. Electrons (e-) from carbon oxidations (step 1, dotted lines) are transferred through NADH (step 2) into OXPHOS complex I (step 3), which is embedded in the lipid bilayer of the mitochondrial inner membrane (IMM), and are then transported to coenzyme Q (CoQ; step 4). Some electrons from organic acid oxidations are transferred through other flavin-containing enzyme complexes (step 5) directly to CoQ. CoQ delivers electrons through complex III (step 6) and cytochrome c (Cyt c; step 7) to the final electron acceptor complex IV (step 8), where oxygen is reduced to water. The electrons lose free energy at each transfer step, and in complexes I, III and IV the energy is harnessed and coupled to the movement of H+ (step 9, dashed lines) from the mitochondrial matrix to the intermembrane space (IMS). The proton gradient thus generated is used for the production of ATP by complex V (step 10). Except for complex II, all complexes contain some proteins encoded by the mitochondrial genome and others encoded by the nuclear genome. The number of subunits for each complex is indicated. CN: cyanide; FMN: flavin mononucleotide; mt: mitochondrial; Pi: phosphate. Adapted from Smeitink et al. 2003, Encyclopedia of life sciences.

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 15
2.3.5. Generation of ROS by mitochondria The respiratory chain is also one of the main sources of ROS formation. It is
estimated that 1% to 3% of O2 reduced in mitochondria is in the form of
superoxide free radical (O2•–) (Turrens 2003). Superoxide (O2•–) is formed
when electrons passing through respiratory chain, leak and react with
molecular oxygen. O2•– is rapidly converted to hydrogen peroxide (H2O2) by
mitochondrial superoxide dismutase (MnSOD). In the presence of reduced
metals such as Fe2+, H2O2 can be converted to hydroxyl radical (OH•). These
three free radicals (O2•–, H2O2 and OH•) are collectively called as ROS and
can damage cellular macromolecules including DNA, proteins and lipids
(Andersen 2004). ROS production increases when respiratory flux is
depressed by a high ATP/ADP ratio, high electronegativity of auto oxidizable
redox carriers in complex I and complex III or a rise in oxygen tension (state
4 respiration). Defects in respiratory chain complexes and normal ageing also
lead to increased mitochondrial ROS production (Esposito et al. 1999;
Cadenas and Davies 2000). Although within a certain local concentration
range, ROS play important roles in regulating many cellular functions and
acting as a secondary messenger to activate specific transcription factors
such as NF-kB and AP-1 (Dalton et al. 1999), an excess production of ROS is
harmful to cells. ROS are extremely reactive molecules and oxidative damage
is believed to be involved in many neurodegenerative diseases,
mitochondriopathies and normal ageing (Shigenaga et al. 1994). A recent
study indicates that mitochondrial ROS homeostasis plays a key role in life
and death of eukaryotic cells (Fleury et al. 2002) as mitochondria not only
respond to ROS but also releases ROS in response to a number of pro-
apoptotic stimuli. However, mitochondria are not the sole source of ROS
within the cell; it is also formed in peroxisomes and cytosol.
Because of their potential harmful effects, excessive ROS must be promptly
eliminated from the cells by a variety of antioxidant defense mechanisms
including important enzymes such as superoxide dismutase (SOD), catalase

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 16
and various peroxidases. The cytosolic copper/zinc-containing SOD
(Cu/ZnSOD, or SOD1) and the mitochondrial manganese-containing SOD
(MnSOD, or SOD2) are two essential enzymes responsible for catalyzing the
conversion of O2•– to H2O2, which is further eliminated by catalase and
peroxidases (Halliwell et al. 1999). Since mitochondrial respiratory chain is a
major site of O2•– generation in the cells, MnSOD plays an important role in
maintaining cellular ROS balance. Vicious cycle theories of aging and
oxidative stress propose that ROS produced by the electron transport chain
damages the mitochondrial DNA leading exponentially to more ROS
production and mitochondrial damage. Although this theory is widely
discussed in the field of research on aging and oxidative stress, there is little
supporting data available.

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 17
Figure 1.4: A schematic representation of ROS generation through respiratory chain and other
sources, its consequences upon cell function. A fraction of oxygen is incompletely reduced by one
electron-transfer to generate ROS and organic free radicals, which are disposed by coordinated action of
antioxidant enzymes. If ROS escapes the antioxidant defense, they may cause oxidative damage and
mutate the mtDNA. This may finally result in defective electron transport chain (ETC). The defective
ETC not only works less efficiently in ATP synthesis but also generates more ROS, which will further
enhance the oxidative damage to various biomolecules in mitochondria or the whole cell.
2.3.6. Mitochondria and apoptosis Mitochondria play a central role in activating apoptotic cell death in response
to cellular dysfunction. Under certain conditions (e.g. high Ca2+, oxidative
stress and low ATP), mitochondrial permeability transition pore (PTP) opens
allowing diffusion of lower molecular weight solutes across the mitochondrial
inner membrane (Crompton 1999). This further results in mitochondrial
swelling and rupturing of outer membrane, leading to the release of proteins
ROS
Oxidative stress
Damage to lipids and proteins
Damage to mtDNA
Permeability transition pore (PTP) activation
Release of cytochrome and AIF
Apoptosis Energy deficit
Defective electron transport chain
mtDNA mutations
Defective mtDNA encoded subunits
ROS scavenging enzymes and antioxidants
Environmental insults, Peroxisomes
Vicious cycle
Cellular dysfunction/cell loss
Mitochondrial ETC
Damage to lipids and proteins
ROS scavenging enzymes and antioxidants
Environmental insults, Peroxisomes
Cellular dysfunction/cell loss
Redox imbalance

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 18
present in the intermebrane space such as cytochrome c, SMAC/Diablo,
apoptosis inducing factor (AIF) and endonuclease G into the cytoplasm. These
proteins then activate the downstream pathways of cell death (Bernardi et al.
2001). The protein components of PTP are not known and it still unclear how
the PTP recognizes apoptotic signals and causes release of proapoptotic
proteins. The pore complex probably involves both inner membrane and outer
membrane proteins and its susceptibility to induction is believed to be
regulated by both pro and anti-apoptotic members of the Bcl-2 family
proteins (Harris and Thompson 2000; Adams and Cory 2001). The bcl-2 is a
family of proteins that are involved in the response to apoptosis. Some of
these proteins (bcl-2 and bcl-XL) are anti-apoptotic, while others (Bad, Bax or
Bid) are pro-apoptotic. The sensitivity of cells to apoptotic stimuli can depend
on the balance of pro and anti-apoptotic bcl-2 proteins. When there is an
excess of pro-apoptotic proteins the cells are more sensitive to apoptosis,
when there is an excess of anti-apoptotic proteins, the cells will tend to be
more resistant. An excess of pro-apoptotic bcl-2 proteins at the surface of the
mitochondria is thought to be important in the formation of the PTP.
The pro-apoptotic bcl-2 proteins are often found in the cytosol where they act
as sensors of cellular damage or stress. Following cellular stress they relocate
to the surface of the mitochondria where the anti-apoptotic proteins are
located. This interaction between pro- and anti-apoptotic proteins disrupts
the normal function of the anti-apoptotic bcl-2 proteins and can lead to the
formation of pores in the mitochondria and the release of cytochrome c and
other pro-apoptotic molecules from the intermembrane space. This in turn
leads to the formation of the apoptosome and the activation of the caspase
cascade. The release of cytochrome c from the mitochondria is particularly an
important event in the induction of apoptosis. Once cytochrome c has been
released into the cytosol it is able to interact with a protein called Apaf-1.
This leads to the recruitment of pro-caspase 9 into a multi-protein complex
with cytochrome c and Apaf-1 called the apoptosome. Formation of the

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 19
apoptosome leads to activation of caspase 9 and the induction of apoptosis.
The role of mitochondria in the induction of apoptosis is summarized in figure
1.5. Mitochondria play a role in integrating many of the signaling pathways
that sense cellular dysfunction and decide whether to commit the cell to
apoptosis, it is tempting to speculate that mitochondrial dysfunction caused
by mitochondrial (mtDNA) or nuclear DNA (nDNA) mutations would promote
cell death. Supporting this hypothesis, progressive degeneration and loss of
neural tissue is a common feature of mitochondrial diseases, but it is unclear
whether the cell loss is apoptotic or necrotic and the degree of cell loss varies
considerably with brain region (Sparaco et al. 1993). Cell death may be
triggered by a range of distinct mechanisms including increases in ROS,
oxidation of the mitochondrial glutathione pool, elevation of free Ca2+, ATP
depletion or changes in intracellular pH, all of which can be affected by
mutations that cause mitochondrial dysfunction. ROS are an important
proapoptotic signal in a number of biological systems in response to cell
damage and in some cases may even be induced to signal cell death (Hwang
et al. 2001).
Understanding how and when cells die in response to mitochondrial
dysfunction is critical for understanding the pathophysiology of mitochondria
related diseases and to design therapies. Also it is important for
understanding what role mitochondrial dysfunction plays in ageing and other
neurodegenerative disorders.

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 20
Figure 1.5: Illustration of the main apoptotic signaling pathways involving mitochondria
3. Mitochondrial DNA Mitochondria contain their own genome known as mitochondrial DNA
(mtDNA). Each cell contains hundreds to thousands of mtDNA molecules; the
phenomenon called polyplasmy and a single mitochondrion has 2-10 copies of
DNA. The mtDNA is maternally inherited, although a few mitochondria from
the sperm cell may enter the oozyte during fertilization, they are eliminated
by a ubiquitin-dependent mechanism (Sutovsky et al. 2000). Interestingly,
data related to human disease has shown that the paternal mtDNA can
escape this elimination and be transmitted to the muscle tissue of the
offspring (Schwartz and Vissing 2002). MtDNA is double-stranded circular
structure (Heavy and Light strand) and contains 16569 base pair nucleotide
sequence (Anderson et al. 1981; Andrews et al. 1999) (Figure 1.6). The two
strands of mtDNA have different base compositions; the heavy (H) strand is

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 21
rich in guanine residues and light (L) strand is rich in thymine residues.
MtDNA encodes 37 genes, of which 28 are encoded by the H-strand and 9 by
the L-strand. Majority of the genes encode an RNA product. There are 22
mitochondrial tRNAs and two rRNAs (12S and 16S). The remaining 13 genes
encode proteins of the OXPHOS system. The mitochondrial genome is
compact, the genes lack introns and are tightly packed, overlapping each
other, or separated by only one or few non-coding bases. There are only two
non-coding regions in mtDNA, a displacement (D) loop of about 1 kilo baspair
(kb) between tRNA-phenyalanine and tRNA-proline. The other non-coding
region of approximately 30 nucleotides is located inside a tRNA cluster about
two-thirds of the way from the D-loop.
Figure 1.6: Structure of mitochondrial DNA. mtDNA encodes 37 genes, 2 rRNAs (12S and 16S), 22
tRNAs and 13 subunits of OXPHOS enzyme complexes: ND1-ND6 and ND4L for complex I, cytochrome
b gene for complex III, COX 1-3 genes for complex IV and ATPase 6 and 8 genes for complex V. D loop =
displacement loop, HSP and LSP = heavy and light strand promoters for transcription, OH and OL =
heavy and light strand replication origins.

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 22
3.1. Replication and transcription of mtDNA The mitochondrial genome is replicated and transcribed within the organelle.
Its replication is under relaxed control and none of the mechanism assures
that each molecule is replicated once and only once during cell cycle. The cis
elements, which are located within the D-loop region of mtDNA, are
responsible for the regulation of both replication and transcription of mtDNA.
However, the trans-acting factors e.g. mtRNA polymerase, mtDNA
polymerase and other regulatory factors are encoded by nDNA (Garesse and
Vallejo 2001.)
The mechanism of mtDNA replication is currently under debate. For many
years the most accepted model was asymmetric (strand–asynchronous) model
of mtDNA replication (Clayton 1982). This model proposes two sites of
initiation of DNA synthesis, one for each strand, which lie far a part. The
synthesis of leading H-strand starts at a point in the major non-coding region
of mtDNA denoted as OH and OL. This model has been challenged by strand-
synchronous (symmetrical) model of mtDNA replication (Holt et al. 2000) in
which the origin of replication of both the leading and lagging strands is
located downstream from the original OH. The replication fork is thought to
move bidirectionally from the origin of replication along the parental mtDNA
strand until the OH is reached (Bowmaker et al. 2003). Differences between
mtDNA replication in cultured cells and in solid tissues have been observed
and it remains to be resolved whether or not both of these replication
mechanisms exist. The two mechanisms are perhaps regulated by different
physiological conditions in the cell.
The mtDNA contains 37 genes that are distributed on both H and L strand
and are expressed as three polycistronic transcription units. The replication
and transcription are linked, because the same RNA primer is used in both
transcription of the L-strand and replication of the mtDNA. The
mitochondrial heavy strand promoters (HSP) and light strand promoters
(LSP) are located about 150 bp apart on regulatory D-loop region.

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 23
Transcription of the L-strand begins at the initiation point (L), located in the
LSP area of the D-loop (Figure 1.6). The L-strand is transcribed as a single
polycistronic precursor RNA containing eight tRNAs and the ND6 mRNA
(Attardi & Schatz 1988). Two models exist for the transcription of the H-
strand. Montoya et al. (1983) suggested that the RNA synthesis starts at two
transcription initiation points, H1 and H2, located in the HSP area of the D-
loop, while another model proposes the existence of only one major
transcription initiation point, H1 (Clayton 1992). The transcription of both
strands of mtDNA leads to a polycistronic primary RNA molecule in which
both of the rRNA genes and almost all the protein genes are flanked by tRNA
genes. This unique genetic organization has led to a proposal that the
cloverleaf secondary structures of the tRNA sequences may act as a signal for
the processing enzymes. Precise endonucleolytic excision of the tRNAs from
the polycistronic transcripts would then yield correctly processed rRNAs and
mRNAs (Ojala et al. 1981). In some cases, where there are no tRNAs flanking
the mRNA, there may be secondary structures resembling tRNA cloverleafs
that are recognized by the processing enzymes.
3.2. Mitochondrial DNA inheritance MtDNA is known to inherit maternally. No mtDNA from sperm enters the
fertilized ovum at the time of conception; the embryo is thus developing with
maternal mtDNA alone. However, a single report regarding the paternal
inheritance of a deletion mutation in mtDNA complex I gene indicated that
paternal inheritance of mtDNA is also possible (Schwartz et al. 2002).
Analysis of infants born after intracytoplasmic sperm injection failed to
identify paternal mtDNA with methods capable of detecting low levels as low
as 0.001% (Marchington et al. 2002; Danan et al. 1997; Houshmand et al.
1997). This implied that paternal mtDNA replication was either suppressed
or diluted beyond the limits of detection and its contribution may not be
necessary.

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 24
Evidence for selective targeting of sperm mitochondria for degradation by the
ovum is also available (Sutovsky et al. 1999; Sutovsky et al. 2004). Failure of
ovum to eliminate paternal mtDNA could result in loss of the embryo at the
blastocyst stage (St. John et al. 2000). Investigators have also failed to detect
paternal mtDNA in patients with sporadic mitochondrial myopathies (Filosto
et al. 2000; Taylor et al. 2003). Thus, if paternal transmission does occur, it is
rare and might depend on the presence of particular paternal mutations that
allow the sperm’s mtDNA to escape destruction as well as to permit
replication. In any event, the dogma that human mtDNA is exclusively
maternally inherited remains as a basis for genetic counselling.
Figure 1.7: Mitochondrial Inheritance. As mitochondria are inherited almost exclusively from the
mother, defects in mtDNA will be passed on from the mother to her children.
3.3. Homoplasmy and heteroplasmy A single cell contains thousands of mtDNA molecules. In normal cases, the
sequence of all these DNA molecules will be identical. However, somatic
mtDNA mutations arise and accumulate with ageing, and could have a role
in the senescence of tissues. If the mutation is present in all the DNA
molecules of mitochondria, then it is known as homoplasmy. The most
common source of somatic mutation of mtDNA is the free radicals generated
by the respiratory chain itself. Most of the mitochondrial diseases are

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 25
characterized by the coexistence of wild type and mutant mtDNA in various
proportions, called heteroplasmy. When the amount of wild-type DNA drops
below a certain level (40-20%), the functioning of the respiratory chain is
distributed and clinical symptoms emerge, a phenomenon called threshold
effect. The level of threshold effect is tissue specific, being lower in tissues
with high-energy demand e.g. heart, Brain, retina, kidney and skeletal
muscle. This means that these tissues are more vulnerable to energy defects.
Mitochondria are randomly segregated at cell division including oogenesis.
During oogenesis, wild-type and mutant mtDNA molecules are randomly
passed to oocytes creating a spectrum of heteroplasmy across the oocyte
population. Oocyte maturation is associated with the rapid replication of this
mtDNA population. This restricted-amplification event can lead to random
shift of mtDNA mutational load between generations and is responsible for
the mutated mtDNA observed in affected offsprings from mothers with
pathogenic mtDNA mutations.
3.4. Mitochondrial DNA mutations Defects in mtDNA can be either point mutations, deletions, duplications or
rearrangements. Point mutations are usually maternally inherited whereas
deletions or large-scale rearrangements are sporadic. As there are no introns,
no splice site mutations are found. MtDNA mutates 10-20 times faster than
nDNA, probably because mtDNA is less protected, especially from ROS
generated in its vicinity, and its repair mechanisms are less efficient
(Fernandez-Silva et al. 2003).
Deletions in mtDNA were the first described mutations to associate with
disease (Holt et al. 1988). The deletion can range from a single base or many
bases (about 6kb) and can be located on any part of mtDNA. The commonest
deletion is 5kb long spanning the region between cytochrome b and
cytochrome c oxidase subunit II (COX II), thus encompassing tRNA and
protein coding genes. The large-scale mtDNA deletions are commonly

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 26
associated with diseases such as chronic progressive external
ophthalmoplegia (CPEO), Kearns-Sayre syndrome, and Pearson’s syndrome.
However, the pathological expression of deletion mutation is not only related
to these phenotypes but has also been described in association with all
mitochondrial syndromes. The prevalence of single deletion disorders is
estimated at 1.2 per 100 000 (Schapira 2006).
Deletions exist in heteroplasmic form, the proportion of deleted molecules
varies between tissues, and the degree of heteroplasmy can shift over time.
Single deletions arise as a primary mtDNA mutation, probably within the
oocyte, and are transmitted to offspring, which may then develop clinical
features. Some patients have duplications of mtDNA. Duplications might not
be pathogenic themselves, but could be an intermediate step in the
generation of deletions. Small size deletions have been described in
cytochrome oxidase, cytochrome b and complex I genes, which are associated
with a variety of clinical presentations.
Approximately, 100 point mutations have been described to be associated
with human disease (www.mitomap.org), but pathogenicity has not been
confirmed for all. These occur in protein coding, tRNA, and rRNA genes.
Evolution has produced related sets of mtDNA sequences, called haplogroups
that can be recognized by certain sequence changes. Since the mtDNA
haplogroups are associated with specific populations on different continents,
a great deal has been learned about the evolution of modern humans and
about human migrations by studying the haplogroup distributions in
different populations (Herrnstadt & Howell 2004). It has also been suggested
that haplogroup-specific polymorphisms may play a role in the pathogenesis
of mitochondrial diseases. The higher mutation rate of mtDNA makes it
difficult to distinguish pathogenic mutations from polymorphisms (Mitchell et
al. 2006). Furthermore, there is also no genotype-phenotype correlation, as
the same mutation can cause different phenotypes and the same clinical
features can be caused by different mutations. The G11778A mutation in

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 27
ND4 was the first known mtDNA point mutation to be associated with
human disease (Wallace et al. 1988). Although, majority of pathogenic
mtDNA mutations are heteroplasmic, those causing Leber’s hereditary optic
neuropathy (LHON) are mostly homoplasmic. The mtDNA genes encoding
subunits of complex I are among the most frequently encountered carriers of
pathogenic mutations, and base pair changes causing variable clinical
features have been identified in all of them. The majority of the pathogenic
mutations reported to affect complex I seem to reside in the ND6 and ND1
genes, which have therefore been called hot spots for mutations causing
especially LHON (Chinnery et al. 2001, Valentino et al. 2004). In addition,
mutations in mitochondrial tRNA genes can lead to the absence or mis-
incorporation of certain amino acids during translation and can thereby cause
complex I deficiency (DiMauro and Hirano 2005). The effect of tRNA
mutation on the functioning of the OXPHOS enzymes varies depending on
codon usage, the subunits that are most dependent on the mutant tRNA for
protein elongation being more seriously affected (Triepels et al. 2001). In
addition, a mutation in a tRNA gene may affect the adjacent subunit gene
through changes in the processing of the polycistronic transcript (Bindoff et
al. 1993).
Criteria for a pathologically relevant mtDNA mutation are:
(i) Co-segregation of mutation with the particular clinical phenotype
(ii) Heteroplasmy
(iii) Absence of the particular mutation in more than 100 normal controls
(iv) Functional impairment in one or more respiratory chain complexes
(v) Haplogroup divergence of identical mutations in different index patients
(vi) Evolutionary conservation of each affected nucleotide
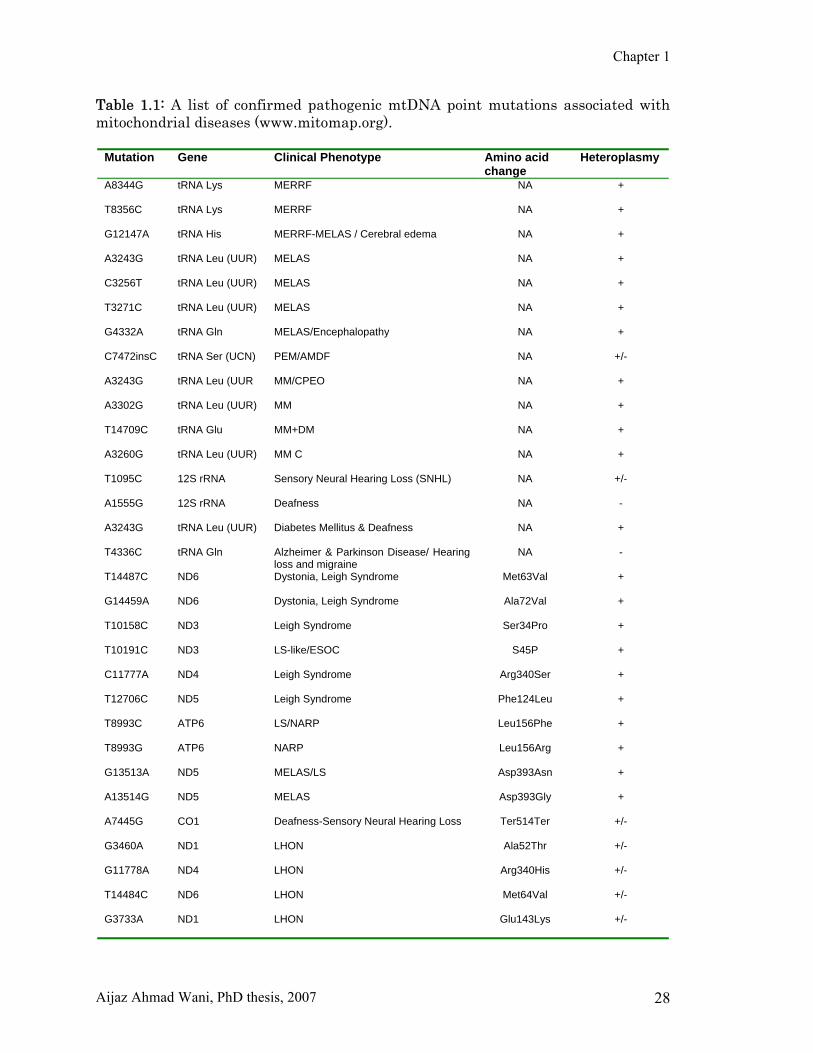
Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 28
Table 1.1: A list of confirmed pathogenic mtDNA point mutations associated with mitochondrial diseases (www.mitomap.org).
Mutation Gene Clinical Phenotype Amino acid change
Heteroplasmy
A8344G
tRNA Lys MERRF NA +
T8356C
tRNA Lys MERRF NA +
G12147A
tRNA His MERRF-MELAS / Cerebral edema NA +
A3243G
tRNA Leu (UUR) MELAS NA +
C3256T
tRNA Leu (UUR) MELAS NA +
T3271C
tRNA Leu (UUR) MELAS NA +
G4332A
tRNA Gln MELAS/Encephalopathy NA +
C7472insC
tRNA Ser (UCN) PEM/AMDF NA +/-
A3243G
tRNA Leu (UUR MM/CPEO NA +
A3302G
tRNA Leu (UUR) MM NA +
T14709C
tRNA Glu MM+DM NA +
A3260G
tRNA Leu (UUR) MM C NA +
T1095C
12S rRNA Sensory Neural Hearing Loss (SNHL) NA +/-
A1555G
12S rRNA Deafness NA -
A3243G
tRNA Leu (UUR) Diabetes Mellitus & Deafness NA +
T4336C tRNA Gln Alzheimer & Parkinson Disease/ Hearing loss and migraine
NA -
T14487C
ND6 Dystonia, Leigh Syndrome Met63Val +
G14459A
ND6 Dystonia, Leigh Syndrome Ala72Val +
T10158C
ND3 Leigh Syndrome Ser34Pro +
T10191C
ND3 LS-like/ESOC S45P +
C11777A
ND4 Leigh Syndrome Arg340Ser +
T12706C
ND5 Leigh Syndrome Phe124Leu +
T8993C
ATP6 LS/NARP Leu156Phe +
T8993G
ATP6 NARP Leu156Arg +
G13513A
ND5 MELAS/LS Asp393Asn +
A13514G
ND5 MELAS Asp393Gly +
A7445G
CO1 Deafness-Sensory Neural Hearing Loss Ter514Ter +/-
G3460A
ND1 LHON Ala52Thr +/-
G11778A
ND4 LHON Arg340His +/-
T14484C
ND6 LHON Met64Val +/-
G3733A
ND1 LHON Glu143Lys +/-

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 29
Table 1.1 continued:
Besides this, a number of secondary or provisionally pathogenic variants are also reported, but their
pathogenicity has not yet been established. MM= mitochondrial myopathy, MICM= Maternally
Inherited Cardiomyopathy, NIDDM= Non-Insulin Dependent Diabetes Mellitus, MMC= mitochondrial
myopathy/cytopathy, MERRF= myoclonic epilepsy with ragged red fibresa, MELAS= mitochondrial
encephalopathy with stroke like episodes, NA= not applicable
4. Nuclear genes encoding respiratory chain proteins In contrast to the wealth of information gained on mtDNA and its disease-
related mutations, the number of nuclear OXPHOS-related genes that have
been proven to be associated with mitochondrial syndromes is still rather
small. However, a clinical genetic classification can now be proposed for these
defects (Zeviani et al. 2003) as follows:
4.1. Genes coding structural subunits of the respiratory chain
complexes Approximately 30% of the mitochondrial disorders involve deficiency of
complex I (Benit et al. 2001). The primary genetic defect may either be at the
mtDNA or at the nDNA level. Majority of complex I deficient cases seem to
follow an autosomal recessive mode of inheritance, suggesting a defect of
nuclear origin (Loeffen et al. 2000). In the past few years, several disease-
associated mutations have been discovered in some 36 nuclear-encoded
subunits of complex I (Triepels et al. 2001a; Petruzzella et al. 2002). These
include substitutions of functionally important amino acids, frame shifts and
C4171A
ND1 LHON L289M +/-
T10663C
ND4 LHON Val65Ala -
G14459A
ND6 LHON and dystonia, Leigh syndrome Ala72Val +
C14482G
ND6 LHON Met64Ile +
C14482A
ND6 LHON Met64Ile +/-
A14495G
ND6 LHON Leu60Ser +
C14568T
ND6 LHON Gly36Ser -

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 30
premature stop codon mutations and also large-scale deletions (Triepels et al.
2000; Ogilvie et al. 2005). The NDUFV1 and NDUFS4 genes seem to be hot
spots for mutations in nuclear complex I genes. In most of these cases, the
clinical presentation is that of an early-onset progressive neurological
disorder with lactic acidosis, most often Leigh syndrome, occasionally
complicated by cardiomyopathy, or multisystem involvement. However, no
mutation in structural genes has been found in many cases of complex I
deficiency, suggesting that still unknown assembly factors for complex I
(Janssen et al. 2002) or other gene products involved in its formation and
activity may be responsible for these forms. Currently, in about 40% of
isolated complex I deficiencies, the OXPHOS defect can be explained by
mutations in structural nuclear genes (Table 1.2).
Complex II is a FAD-dependent enzyme at a cross-point between OXPHOS
and Krebs-cycle pathways. It comprises four protein subunits, all encoded by
nuclear genes (SDH-A, B, C, and D). Mutations in SDHA, the largest subunit
of complex II, are a rare cause of Leigh syndrome or late onset
neurodegenerative disease (Rustin & Rotig 2002). However, the most
interesting discovery concerning defects of complex II is their association
with inherited paragangliomas (Maher and Eng 2002; Ackrell 2002; Baysal
2002; Dannenberg et al. 2002). In 10-15% of the cases, these neuro-
ectodermal tumors are inherited in an autosomal dominant fashion with
incomplete penetrance. It now appears that mutations in SDHB, SDHC, and
SDHD are responsible for the majority of familial paragangliomas (Baysel et
al. 2002) and also for a significant fraction of non-familial tumors, including
phaeochromocytomas (tumors of the adrenal medulla) (Cascon et al. 2002,
Neumann et al. 2002).
Complex III catalyzes electron transfer from succinate and NADH-linked
dehydrogenases to cytochrome c. Complex III is made up of 11 subunits, of
which 10 are encoded by nDNA and one (cytochrome b) is coded by mtDNA.
Although pathogenic mutations in the gene encoding mitochondrial

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 31
cytochrome b have been described, mutations in the nDNA encoded subunits
were never reported.
Coenzyme Q10 deficiency Coezyme Q10 (CoQ10), or ubiquinone, is a lipophilic component of the
electron-transport chain and transfers electrons from various dehydrogenases
to complex III and acts as a lipid-soluble antioxidant and as a membrane
stablizer. The first syndrome of CoQ10 deficiency was characterized by the
triad of recurrent myoglobinuria, brain involvement (seizures, ataxia, and
mental retardation) and ragged-red fibers/lipid storage in muscle. Coenzyme
Q10 is mainly synthesized intracellularly and requires many enzymatic steps.
The mevalonate pathway is a sequence of reactions that leads to farnesyl
pyrophosphate- the common substrate for synthesis of ubiquinone, cholesterol,
dolichol, dolicholphosphate, as well as for prenylation of proteins. Irrespective
of the genetic causes of the defect, which are presently unknown, early
recognition of CoQ deficiency is important, as its supplementation can lead to
clinical improvement.
4.2. Genes involved in the assembly of respiratory chain complexes This group comprises of genes encoding assembly factors for complex I, III
and IV. To date, only two complex I assembly factors (B17.2 and CIA30) have
been identified and how each functions is not clear. Mutations in B17.2L
chaperone are associated with progressive encephalopathy (Ogilvie et al.
2005) and that of CIA30 are reported to associate with
cardioencephalomyopathy (Dunning et al. 2007).
Human cytochrome oxidase (COX) is composed of 13 subunits: the three
largest ones are encoded by mtDNA genes, while the remaining subunits are
encoded by nuclear genes. The most frequent manifestation of isolated COX
deficiency in infancy is Leigh syndrome. Other phenotypes such as severe
cardiomyopathy or complex encephalocardiomyopathies are also known to

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 32
associate with COX deficiency (Shoubridge 2001). No mutation in nDNA
encoded subunits of COX are reported, whereas all of the nuclear-gene
defects of COX identified till date are caused by mutations in assembly
factors of the enzyme, including SURF1, SCO1, SCO2, COX10 and COX15.
SURF1 is a 30kDa hydrophobic protein located in inner membrane of
mitochondria. Mutations in SURF1 are relatively frequent, accounting for the
majority of the Leigh syndrome cases.
Mutations in other COX assembly genes are rare and have been reported in
only a few families or singleton cases. Human SCO1 and SCO2 are nuclear-
encoded copper ion binding proteins, presumed to be responsible for the
insertion of copper into the COX holoenzyme. Whereas mutations in SCO1
were found in only one family, mutations in SCO2 are more frequent. Copper
supplementation can restore COX activity in cells harboring mutations in
genes involving copper transport, including SCO2 (Jaksch et al. 2001;
Salviati et al. 2002). Similar to COX10, COX15 (heme A farnesyl-transferase)
is involved in the synthesis of heme A, the prosthetic group for COX. The first
deleterious mutations in COX15 have been identified in a patient with fatal,
infantile hypertrophic cardiomyopathy (Antonicka et al. 2003a).
BCS1L, a mitochondrial inner-membrane protein, is a chaperone necessary
for the assembly of mitochondrial respiratory chain complex III. Mutations in
BCS1L have been shown in infantile cases of complex III deficiency
associated with neonatal proximal tubulopathy, hepatic involvement and
encephalopathy (de Lonlay et al. 2001) and in GRACILE (growth retardation,
aminoaciduria, cholestasis, iron overload, lactacidosis, and early death)
syndrome (Visapaa et al. 2002). More recently, BCS1L mutations were
associated with isolated encephalopathy (Fernandez-Vizarra et al. 2007) and
Bjornstad syndrome (Hinson et al. 2007)

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 33
4.3. Genes involved in mtDNA maintenance MtDNA is dependent upon nDNA for the production of a number of proteins
involved in its replication, transcription, translation, repair, and
maintenance. Mutations of these genes can result in multiple mtDNA
deletions or depletion of mtDNA. Adenine nucleotide translocator-1 (ANT-1)
is an isoform specific to muscle, heart, and brain. It regulates the adenine
nucleotide pool within mitochondria. ANT1 mutations cause adult onset
autosomal dominant chronic progressive external opthalmoplegia (CPEO)
with ragged red fibers and multiple mtDNA deletions in skeletal muscle
(Kaukonen et al. 2000). Twinkle is a hexomeric 5′-3′ DNA helicase protein
encoded by the C10orf2 gene, which is responsible for unwinding the mtDNA
replication fork (Spelbrink et al. 2001; Korhonen et al. 2003). Inhibition of
twinkle in cultured cells results in rapid mtDNA depletion, whereas over-
expression of the gene leads to mtDNA accumulation, confirming its
importance in regulating copy number (Tyynismaa et al. 2004). Twinkle is
highly expressed in human skeletal muscle and in a specific splice variant in
testes, which is of interest since mtDNA replication is down regulated during
spermatogenesis. Twinkle co-localizes with mitochondrial transcription factor
A and mitochondrial single-stranded DNA-binding protein, and together they
are thought to stabilize mtDNA. Several mutations causing autosomal
dominant progressive external ophthalmoplegia (PEO) are located at or near
putative subunit interaction sites in the holoenzyme. The clinical
manifestations of C10orf2 (twinkle) mutations typically include PEO. In some
cases, this can be of late onset (above 50 years of age) and be associated with
myopathy and cardiomyopathy in addition to axonal neuropathy, diabetes,
deafness, and osteoporosis (Kiechl et al. 2004). MtDNA polymerase γ (POLG) is a heterodimer comprising a 140kDa alpha
subunit and a 41kDa beta subunit. It is located within the inner
mitochondrial membrane and is essential for mtDNA replication. The alpha
subunit is catalytic and contains both polymerase and exonuclease activities,

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 34
the beta subunit facilitate DNA binding and promote DNA synthesis (Filosto
et al. 2003). Mutations of POLG have been associated with a range of clinical
phenotypes including PEO. The human POLG gene contains a 10-CAG repeat
length encoding a polyglutamine tract. A variation in this microsatellite has
been associated with male subfertility (Rovio et al. 2001; Jensen et al. 2004).
POLG mutations also cause Alpers syndrome, an autosomal recessive
disorder characterized by epilepsy, cortical blindness, micronodular hepatic
cirrhosis, and episodic psychomotor regression (Naviaux and Nguyen 2004;
Ferrari et al. 2005). POLG mutations have been identified in patients with
PEO and Parkinsonism.
Mitochondrial DNA depletion syndrome (MDS) is a clinically heterogeneous
group of disorders characterized by a reduction in mtDNA copy number. MDS
has been linked to mutations in two genes involved in deoxyribonucleotide
metabolism: thymidine kinase 2 (TK2) and deoxyguanosine kinase, which are
responsible for the myopathic form and the hepatoencephalopathic form of
MDS, respectively (Hirano et al. 2001). Both deoxyguanosine kinase and TK2
genes are involved in the formation of the mitochondrial nucleotide pool, as is
thymidine phosphorylase, whose deficiency causes a syndrome known as
mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). MNGIE
is associated with both depletion and multiple deletions of mtDNA.

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 35
Table 1.2: Mutations in nuclear OXPHOS-related disease associated genes
Location Clinical phenotype Inheritance Accession no.
Respiratory-enzyme subunits
NDUFS1 (Complex I) 2q33-q34 Lactic acidosis, mitochondrial complex I defficiency
AR NM_005006
NDUFS2 (Complex I) 1q23 Cardiomyopathy and encephalomyopathy AR NM_004500
NDUFV1(Complex I) 11q13 Leigh syndrome
AR NM_007103
NDUFS4 (Complex I) 5q11.1 Fatal multisystem complex I deficiency AR NM_002495
NDUFS7 (Complex I) 19p13 Leigh syndrome
AR NM_024407
NDUFS8 (Complex I) 11q13 Leigh syndrome
AR NM_002496
SDHA (Complex II) 5p15/3q29* Leigh syndrome AR NM_004168
SDHB (Complex II) 1p36.1-p35 Phaeochromocytoma and cervical paraganglioma
AD NM_003000
SDHC (Complex II) 1q21 Familial paraganglioma - PGL3
AD NM_003001
SDHD (Complex II) 11q23 Familial paraganglioma - PGL1
AD NM_003002
Krebs cycle enzymes
FH (Fumarate hydratase) 1q42.3-q43 Multiple cutaneous and uterine leiomyomatosis
AD NM_000143
Assembly factors
B17.2L (Complex I) 5q12.1 Encephalopathy AD NM_174889
CIA30 (Complex I) 15q13.3 Cardioencephalomyopathy
AD NM_01601
SURF1 (COX assembly) 9q34 Leigh syndrome
AR NM_003172
SCO1 (COX assembly) 17p13-p12 Ketacidotic coma and hepatopathy
AR NM_004589
SCO2 (COX assembly) 22q13 Hypertrophic cardiomyopathy
AR NM_005138
COX10 (COX assembly) 17p13.1-q11.1 Tubulopathy and Leukodystrophy
AR NM_001303
COX15 (COX assembly) 10q24 Hypertrophic cardiomyopathy
AR NM_078470
BCS1L (Complex III assembly)
2q33-37
Tubulopathy, encephalopathy, and liver failure, complex III Deficiency, GRACILE
AD
NM_004328
MtDNA maintenance
TP (thymidine phosphorylase)
22q13.32-qter Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)
AR NM_001953
ANT1 (adenine nucleotide translocator 1)
4q34 Autosomal dominant progressive external ophthalmoplegia (adPEO)
AD J04982
C10 ORF2 (Twinkle) 10q24 Autosomal dominant progressive external ophthalmoplegia (adPEO)
AD AF292005
POLG1 (Polymerase gamma γ)
15q25 Autosomal dominant, autosomal recessive progressive external ophthalmoplegia (adPEO, arPEO)
AD, AR NM_002693
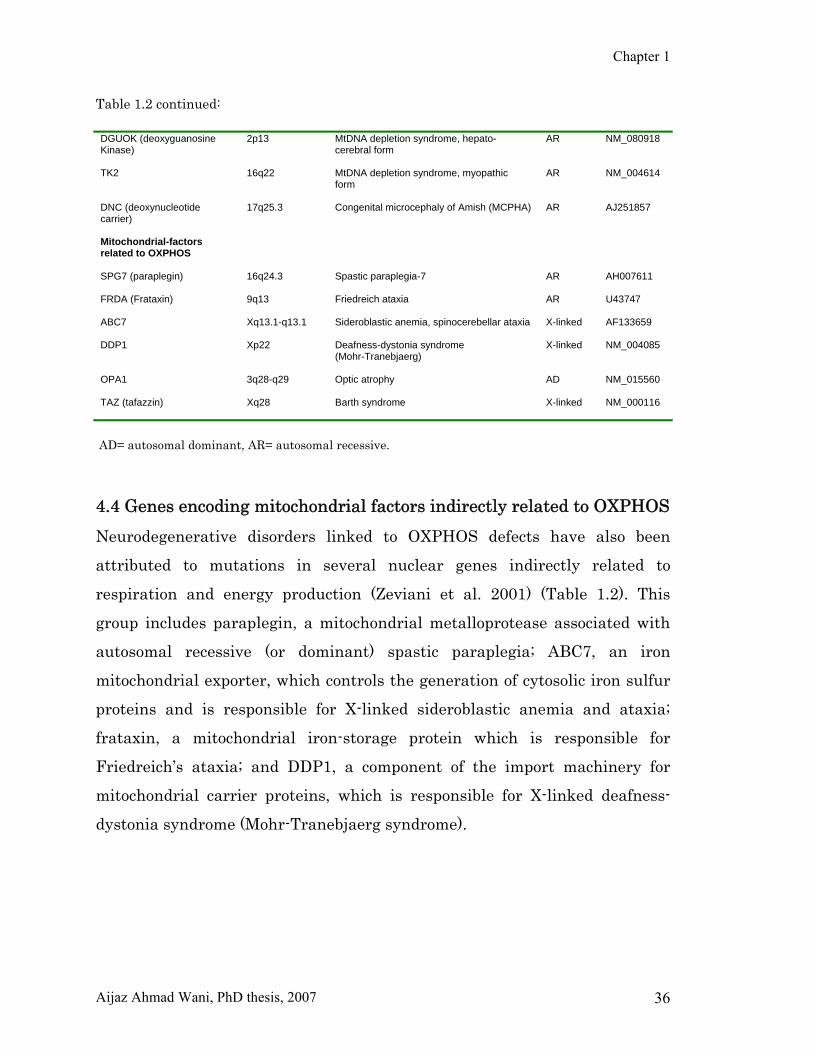
Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 36
Table 1.2 continued: DGUOK (deoxyguanosine Kinase)
2p13 MtDNA depletion syndrome, hepato-cerebral form
AR NM_080918
TK2 16q22 MtDNA depletion syndrome, myopathic form
AR NM_004614
DNC (deoxynucleotide carrier)
17q25.3 Congenital microcephaly of Amish (MCPHA) AR AJ251857
Mitochondrial-factors related to OXPHOS
SPG7 (paraplegin) 16q24.3 Spastic paraplegia-7
AR AH007611
FRDA (Frataxin) 9q13 Friedreich ataxia
AR U43747
ABC7
Xq13.1-q13.1 Sideroblastic anemia, spinocerebellar ataxia
X-linked AF133659
DDP1 Xp22 Deafness-dystonia syndrome (Mohr-Tranebjaerg)
X-linked NM_004085
OPA1 3q28-q29 Optic atrophy
AD NM_015560
TAZ (tafazzin) Xq28 Barth syndrome
X-linked NM_000116
AD= autosomal dominant, AR= autosomal recessive. 4.4 Genes encoding mitochondrial factors indirectly related to OXPHOS Neurodegenerative disorders linked to OXPHOS defects have also been
attributed to mutations in several nuclear genes indirectly related to
respiration and energy production (Zeviani et al. 2001) (Table 1.2). This
group includes paraplegin, a mitochondrial metalloprotease associated with
autosomal recessive (or dominant) spastic paraplegia; ABC7, an iron
mitochondrial exporter, which controls the generation of cytosolic iron sulfur
proteins and is responsible for X-linked sideroblastic anemia and ataxia;
frataxin, a mitochondrial iron-storage protein which is responsible for
Friedreich’s ataxia; and DDP1, a component of the import machinery for
mitochondrial carrier proteins, which is responsible for X-linked deafness-
dystonia syndrome (Mohr-Tranebjaerg syndrome).

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 37
5. Mitochondrial encephalomyopathies Human diseases caused by impaired respiratory chain function have been
increasingly recognized in recent years. Although neurological diseases are
the commonest consequence of respiratory chain dysfunction, it is now
apparent that virtually any tissue in the body can be affected. The
neurological diseases associated with respiratory chain impairment are
collectively known as mitochondrial encephalomyopathies (MCPs), a term
which reflects the common involvement of both the central nervous system
and skeletal muscle in these patients. Organs such as the brain, heart and
skeletal muscle are highly energy dependent and thus vulnerable to defects
in energy metabolism. Skeletal muscle involvement presents with exercise
intolerance, weakness and myalgias, in association with involvement of other
organs, most commonly with encephalopathy or cardiomyopathy (Scaglia et
al. 2004). Lactic acidosis with an increased lactate/pyruvate ratio is
frequently observed. MCPs are caused by mutations in both nuclear and
mitocondrial DNA (DiMauro and Gurgel-Giannetti 2005).
Proteins most frequently affected by mutations are those of the respiratory
chain and oxidative phosphorylation. That is why mitochondrial
encephalomyopathies are sometimes also called as respiratory chain
disorders. However, MCPs may also be due to defects in pathways and
components of the mitochondrion other than respiratory chain. Proteins of
the respiratory chain that are encoded by nDNA; after being synthesized in
the cytoplasm, are imported into mitochondria, where they assemble together
with their mtDNA encoded counterparts to form holoenzymes in the
mitochondrial inner membrane. MCPs present with a wide spectrum of
disease and their clinical features overlap (Leonard and Schapira 2000a & b).
Concerning mtDNA, a single mutation or different mutations in the same
gene may present with different clinical manifestations, while as the same
clinical phenotype may be caused by different mutations, called genetic

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 38
heterogeneity (Dimauro 1996). MtDNA mutations were first reported in 1988
(Holt et al. 1988; Wallace et al. 1988). Subsequently, the first nuclear
mutation leading to MCP was identified (Bourgeois et al. 1992).
5.1. Classical mitochondrial syndromes The onset of MCPs ranges from early embryogenesis to late adulthood. The
MCPs can present at any age (Wolf and Smeitink 2002). The frequency is
approximately 1 per 5000 individuals. The clinical phenotypes associated
with classical mitochondrial disorders are:
5.1.1. Chronic progressive external opthalmoplegia Chronic progressive external ophthalmoplegia (CPEO) is the commonest
manifestation of a mtDNA mutation and is characterized by ophthalmoplegia
and ptosis (Holt et al. 1989). Later on cataracts, retinitis pigmentosa,
deafness, fatigue, ataxia, limb weakness, neuropathy, cardiomyopathy and
renal insufficiency may develop (Schapira and Cock 1999; McFarland et al.
2002). The particular susceptibility of the extra-ocular muscles is explained
by three to four times greater mitochondrial volume compared with limb
muscles (Schapira and Cock 1999). The clinical course is usually benign in
that additional tissue or organ failure is rare with low risk of serious
disability. CPEO is due to mtDNA deletions (40% of the cases), nDNA
mutations and point mutations in mtDNA encoding tRNAs (Zeviani et al.
1989).
5.1.2. Kearns-sayre syndrome Kearns-Sayre syndrome (KSS) is a subtype of CPEO with pigmentary
retinopathy, cardiac conduction defects, cerebellar ataxia, raised CSF protein
and onset is above 20 years (Kearns and Sayre 1958). Its expression is
systemic, but most common expressions are in the eyes, with
ophthalmoplegia and retinal degeneration, specifically retinitis pigmentosa.

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 39
Other characteristic features are dysphagia, proximal weakness, hearing loss,
cerebellar ataxia and cardiac conduction defects. The prognosis of KSS is
worse than that of CPEO and patients rarely survive beyond the age of 30
years. KSS is due to sporadic, single large deletions ranging from 1.3 to 8.8
kb (90% of the cases) or duplications. The origins of replication and
transcription within the mtDNA are usually spared (Schapira and Cock 1999).
5.1.3. Pearson syndrome Pearson syndrome is characterized by sideroblastic anemia and exocrine
pancreas dysfunction. Bone marrow biopsy is characterized by normal
cellularity, but vacuolization of the precursor cells (Pearson et al. 1979;
Schapira and Cock 1999). Additional features are failure to thrive, and
chronic diarrhoea with villous atrophy. With disease progression,
hepatomegaly, raised transaminases, hyperbilirubinaemia, coagulopathy and
tubular dysfunction with aminoaciduria and glucosuria (Fanconi’s syndrome)
may occur (Schapira and Cock 1999). The syndrome is usually fatal in
infancy. In patients who survive beyond infancy, the syndrome evolves into
KSS. It is caused by a deletion in mtDNA with a heteroplasmy rate of up to
90% in blood (McFarland et al. 2002).
5.1.4. Mitochondrial encephalomyopathy with lactic acidosis and
stroke-like episodes Mitochondrial encephalomyopathy with lactic acidosis and stroke-like
episodes (MELAS) syndrome is characterized by migraine-like headache,
recurrent vomiting, seizures, short stature, normal early development, lactic
acidosis and ragged-red fibers in muscle (Pavlakis et al. 1984). The classical
MELAS phenotype affects children at 5-15 years of age. However, stroke-like
episodes usually occur in early infancy whereas several atypical
manifestations, like delayed motor development and failure to thrive are
observed (Sue et al. 1999; Okhuijsen-Kroes et al. 2001). In adults, common

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 40
clinical manifestations include sensorineural hearing impairment, diabetes,
myopathy, cardiomyopathy and cognitive decline. Complex I is frequently the
most affected respiratory chain enzyme detected in MELAS (Ciafaloni et al.
1992). About 80% of the MELAS patients have the heteroplasmic missense
mutation A3243G in the tRNA Leu(UUR) gene (Goto et al. 1990). The
genotype-phenotype correlation of A3243G mutation is rather loose, since the
observed clinical manifestations are not restricted solely on MELAS.
Identification of mutations in mitochondrial ND genes associated with
MELAS or with MELAS/LHON overlap syndrome further address the link
between complex I defect and MELAS phenotype (Corona et al. 2001).
5.1.5. Myoclonic epilepsy with ragged-red fibers Myoclonic epilepsy with ragged-red fibers (MERRF) is a neuromuscular
disorder characterized by myoclonus, epilepsy, muscle weakness, cerebellar
ataxia, deafness and dementia (Fukuhara et al. 1980; Silvestri et al. 1993).
Complex IV deficiency is the most prominent biochemical finding in patient’s
muscle with MERRF. In some cases, complex I is also affected. Most patients
with MERRF do harbor a heteroplasmic A8344G mtDNA mutation in tRNA-
lysine gene (Shoffner et al. 1990). Clinical, biochemical and molecular studies
on large pedigrees with A8344G mutation have shown a positive correlation
between the severity of the disease, age at onset, mtDNA heteroplasmy and
reduced activity of respiratory chain enzymes in skeletal muscle (Zeviani and
Di Donato 2004).
5.1.6. Neuropathy, ataxia retinitis pigmentosa (NARP) The neuropathy, ataxia, retinitis pigmentosa (NARP) syndrome, first
described in 1990 (Holt et al. 1990), is characterized by weakness due to
motor neuropathy, sensory disturbances, cerebellar ataxia and retinitis
pigmentosa. Additional features may be developmental delay, mental
retardation, dementia, ataxia, cardiomyopathy and epilepsy. There is no

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 41
clinical or histological evidence of myopathy in NARP. The disease is caused
due to mutations in ATPase 6 gene of mtDNA (McFarland et al. 2002).
5.1.7. Leigh syndrome Leigh syndrome is the most common pediatric phenotype of isolated complex
I deficiency (Loeffen et al. 2000). The disease is a progressive
neurodegenerative disorder involving encephalopathy with lactic acidosis,
occasionally complicated by cardiomyopathy or a multisystemic presentation
(van Erven et al. 1987; Robinson 1998). The onset is usually in the first year
of life, and the children present with developmental delay and failure to
thrive. Motor and intellectual retardation, ataxia, dystonia, hypotonia, and
optic atrophy are frequently encountered. Neuroimaging shows symmetrical
lesions in the basal ganglia, midbrain and brainstem. On pathological
examination, a bilateral symmetrical focus of spongy necrosis is detected with
myelin degeneration, vascular proliferation and gliosis in the thalami, brain
stem and spinal cord. Patients with similar symptoms but with a typical or
unknown neuropathology are referred to as Leigh-like cases (LLS) (Rahman
et al. 1996; Morris et al. 1996). Besides complex I deficiency, LS can have
several other biochemical causes, and it has been described in association
with defects of all OXPHOS system enzymes (Dahl 1998). Mutations in both
mtDNA and nDNA may lead to LS, but a severe form of the disease, called
maternally inherited Leigh syndrome (MILS), is caused by mutations in
mtDNA. The T8993G mutation in the ATPase 6 gene is a common cause of
MILS (Holt et al. 1990). The degree of heteroplasmy of the T8993G mutation
correlates well with the severity of the disease. High levels of mutated
mtDNA (>95%) lead to Leigh’s phenotype, whereas low levels of mutation
load can cause NARP syndrome (Tatuch et al. 1992).

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 42
5.1.8. Leber hereditary optic neuropathy Leber’s hereditary optic neuropathy is the commonest cause of maternally
inherited blindness particularly in young men. The unique anatomical and
physiological features of the optic nerve seem to make it especially vulnerable
to complex I deficiency (Bristow et al. 2002). The main phenotype of LHON is
a tissue-specific, subacute, painless visual loss characterized by central
scotomas, abnormal color vision and optic atrophy. Vision deteriorates over a
period of days or weeks in one eye followed by the other eye. LHON typically
affects young adults, the average age being 20 years. Men are affected three
to four times more often than women (Simon and Johns 1999; Man et al.
2002; Zeviani and Di Donato 2004). Approximately 90% of LHON cases carry
one of the three mtDNA mutations, G3460A (Howell et al. 1991), G11778A
(Wallace et al. 1988) or T14484C (Chinnery et al. 2001). All these mutations
are residing on genes encoding complex I subunits and are usually
homoplasmic. Interestingly, only 50% of men and 10% of women harboring a
pathogenic mtDNA mutation develop the optic neuropathy. This marked
incomplete penetrance and gender bias imply that additional genetic or
environmental factors modulate the phenotypic expression of LHON (Hudson
et al. 2005; Man et al. 2002).
5.1.9. Myoneurogastrointestinal encephalopathy (MNGIE) Myoneurogastrointestinal encephalopathy (MNGIE) is a multisystem
disorder (Ionasescu 1983) characterized by gastrointestinal dysmotility,
manifesting before age of 20 years as episodic nausea, vomiting,
gastroparesis, progressive intestinal pseudo-obstruction, abdominal pain,
dilation and dysmotility of oesophagus, stomach and small intestine,
diarrhoea, and malabsorption with progressive malnutrition, leading to death
around 40 years of age. Additional features include generalized myopathy
with CPEO, cognitive decline due to leucencephalopathy, retinitis pigmentosa,
deafness, hoarseness, dysarthria and polyneuropathy. Post-mortem changes

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 43
include visceral neuropathy (loss of neurons and fibrosis in the coeliac,
mesenteric and Auerbach plexuses) and scleroderma-like changes (Schapira
and Cock 1999). MNGIE has been shown to result from mutations in the
nDNA encoded gene for the thymidine-phosphorylase (Nishino et al. 2001;
DiMauro and Schon 2003) (Table 1.2).
6. Treatment of mitochondrial disorders Treatment options for mitochondrial encephalomyopathies are very limited
and there is no cure available. Some effects such as cardiac arrhythmia,
seizure disorders, renal bicarbonate loss and hypoglycemia can be treated.
Seizures usually respond to conventional anticonvulsants. However, valproic
acid should be used with caution and in association with L-carnitine, because
it inhibits carnitine uptake (Tein et al. 1993). In patients with CPEO,
improvement in severe ptosis is possible by surgery; congenital cataracts are
also treated surgically. A specific lactic acid lowering agent is dichloroacetate
(DCA), which acts by inhibiting pyruvate dehydrogenase (PDH) kinase, thus
keeping PDH in the dephosphorylated, active form and favoring pyruvate
metabolism and lactate oxidation (Stacpoole 1989). DCA should not be used
long term in patients with mitochondrial disorders, who are already prone to
development of peripheral neuropathy by virtue of their mitochondrial
dysfunction. Since defects of the respiratory chain result in the increased
production of free radicals, the use of antioxidants do play a significant role.
N-acetylcysteine and coenzyme Q10, both antioxidants, improved OXPHOS
function and reduced free radical production in cybrid cells carrying the
T8993G mutation that causes NARP or MILS (Mattiazzi et al. 2004).
However, the use of antioxidants in mtDNA disease has yet to be tested in a
clinical trial. In vitro studies have revealed a potentially useful therapeutic
approach to a fatal infantile form of encephalocardiomyopathy associated
with COX deficiency due to mutations in the SCO2 gene. When copper was

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 44
added to the medium of cultured COX-deficient myoblasts harboring SCO2
mutations, COX activity was restored (Jaksch et al. 2001; Salviati et al. 2002).
This suggests that copper supplementation should be tried in infants with
cardiopathy and SCO2 mutations.
Various strategies are being assessed to modify the mtDNA mutant load in
cells and tissues of patients. An obvious target would be the preferential
expansion of wildtype mtDNA or the suppression of mutant mtDNA
expansion. A few approaches are:
Inhibiting Replication of Mutant Genomes Selective hybridization of nucleic acid derivatives to mutant mtDNA would
inhibit their replication while allowing propagation of wild-type genomes,
thus causing the proportion of mutant genomes to fall below the pathogenic
threshold. Some success has ben obtained in decreasing the ratio of A8344G
MERRF mutants in vitro (Taylor et al. 1997), but there were problems with
the delivery of peptide nucleic acids to human mitochondria (Chinnery et al.
1999). Importing RNAs into Mitochondria Normal yeast tRNAs can be imported from the cytoplasm to compensate for
mutant mitochondrial tRNAs, and human mitochondria can internalize yeast
tRNA derivatives in the presence of a specific yeast transport factor
(Kolesnikova et al. 2000). Interestingly, the same yeast tRNA-lysine
derivatives expressed in cybrid human cell lines harboring the MERRF
mutation (A8344G in tRNA-lysine) were imported into mitochondria and
were able to partially restore mitochondrial function (Kolesnikova et al. 2004).
Recently, import of cytosolic tRNAs by Leishmania RNA import complexes
into mitochondria restored mitochondrial function in MERRF cybrids
harboring a mutant mitochondrial tRNA lysine (Mahata et al. 2006).

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 45
Importing Polypeptides into Mitochondria This therapeutic strategy is based on the phenomenon of allotropic
expression and xenotopic expression, and import of restriction endonucleases.
Allotopic expression refers to a strategy aimed at reducing the load of mutant
proteins by importing a normal version of a mutant mtDNA-encoded protein
from a gene ‘‘snuck’’ into the nucleus. For example, the ATPase 6 gene of
mtDNA can be converted from the mitochondrial into the nuclear genetic
code. To be sure that the novel nuclear protein encoded by the converted gene
is recognized by, and transported into mitochondria, it has to be provided
with a leader peptide, whose genetic sequence can be ‘‘borrowed’’ from
another mtDNA encoded protein. This approach has been realized in vitro to
correct the biochemical defect in cybrid cells harboring the T8993G
NARP/MILS mutation (Manfredi et al. 2002) and in cybrids harboring the
G11778A LHON mutation (Guy et al. 2002).
Still another molecular ‘‘trick’’ is to correct a respiratory chain defect due to a
mtDNA mutation by transfecting affected mammalian cells with either
mitochondrial or nuclear genes from other organisms but encoding cognate
proteins (‘‘xenotopic expression’’) (Seo et al. 1998; Bai et al. 2001; Ojaimi et al.
2002). A more direct molecular approach is to import specific restriction
endonucleases as ‘‘magic bullets’’ to selectively destroy mutant mtDNAs. This
approach has proven successful in cybrid cell lines harboring the T8993G
NARP/MILS mutation in the ATPase 6 gene, which creates a unique SmaI
site in human mtDNA. The gene for SmaI was fused to mitochondrial
targeting sequences and transiently expressed in heteroplasmic cybrids,
which lost mutant mtDNAs (Tanaka et al. 2002). Genetic counselling Prenatal diagnosis for tRNA point mutations, including the more common
ones associated with MELAS and MERRF is practically impossible by two
concerns. First, the mutation load in amniocytes or chorionic villi does not

Chapter 1
Aijaz Ahmad Wani, PhD thesis, 2007 46
necessarily correspond to that of other fetal tissues. Second, mutation load
measured in prenatal samples may shift in utero or after birth due to mitotic
segregation. At the other end of the spectrum, large-scale deletions of mtDNA
as a rule are neither inherited nor transmitted and either arise de novo in
oogenesis or in early embryogenesis. Mitochondrial diseases caused by
nuclear gene mutations will be transmitted by Mendelian inheritance.
Genetic counselling in mitochondrial disorders is a considerable challenge
given the diversity of the clinical manifestations and the poor link between
phenotype and genotype. Practical advice can include the possibility of in vitro fertilization with a donor egg, but another possibility for the future is of
nuclear transfer from a maternal egg and fertilization in a donor cytoplasm
using paternal sperm. The recurrence risks for the relative of an individual
with LHON are 30% for brothers, 8% for sisters, 46% for nephews, 10% for
nieces, and 31% and 6% for male and female cousins, respectively (Harding et
al. 1995).
Advances in molecular and diagnostic technology will help us better
understand these complex diseases and help medical practitioners in better
managing their patients.




![CURRICULUM VITAE - Aligarh Muslim UniversityCURRICULUM VITAE [ Prof. Aijaz Ahmed Khan ] [Jan. 2020] PERSONAL DETAILS Name DR. AIJAZ AHMED KHAN Date of Birth 10th January, 1960 Sex](https://img.dokumen.tips/doc/110x75/5ecb53828a7fdc41d3644d6f/curriculum-vitae-aligarh-muslim-university-curriculum-vitae-prof-aijaz-ahmed.jpg)
























