Embed Size (px)
Citation preview

Nemoci spojené s expanzí mikrosatelitních repetitivních sekvencí – myotonická dystrofie typu 1
Kongenitální myotonie a mutace v genu CLCN1

Half of the human genome consists of repetitive DNA, significant proportion is organized in tandem arrays (copy number variation).
• Repeat unit sizes 1- 4 nucleotides and spanning less than 100 bp are typically defined as microsatellite repeats. • Repeat unit sizes 10 - 40 nucleotides covering several hundreds of bp are referred to as minisatellite repeats. • The term midisatellite repeats has been proposed for loci containing repeat units of 40 - 100 nucleotides that can extend over distances of 250–500 kb. • Macrosatellite repeats are the largest class of repeat arrays with unit sizes of >100 nucleotides but which are typically much larger and can span hundreds of kb of DNA.
Repeat sequences in the human genome
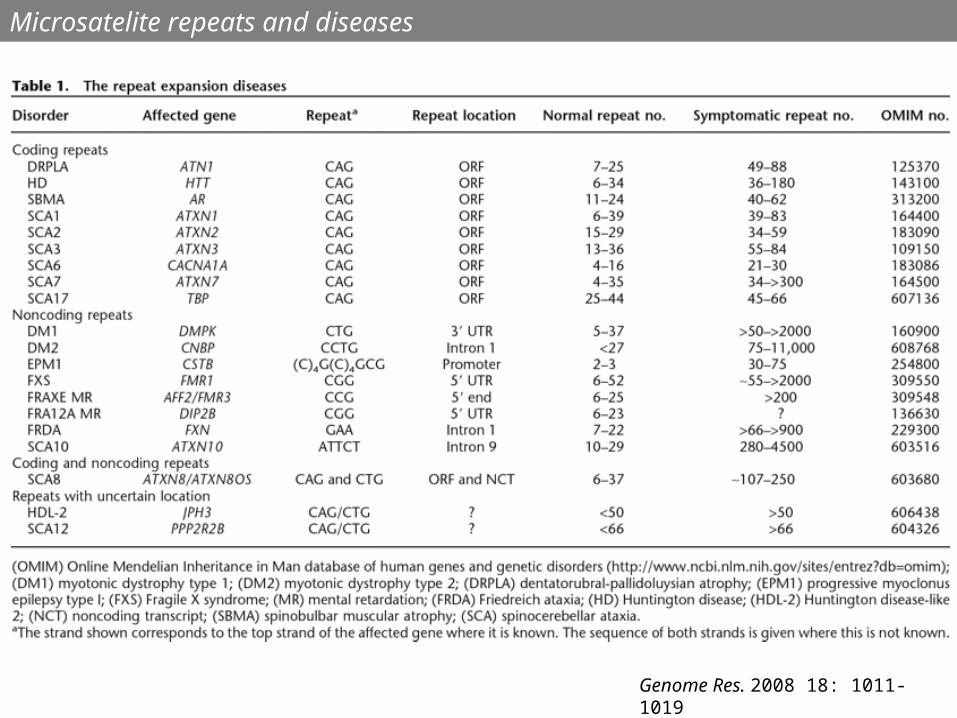
Genome Res. 2008 18: 1011-1019
Microsatelite repeats and diseases

• Repeat expansions can occur in 5′UTRs, coding regions, introns, 3′UTRs.
• Repeat lengths in introns, 3′UTRs, and 5′UTRs can become much larger
than in coding regions.
• Premutation alleles do not show usually disease symptoms, but can expand
in the next generation.
• As repeats get longer, symptoms are seen at an earlier age and are more
severe.
Spinocerebellar ataxias (SCA12), Spinal Bulbar Muscular Atrophy (SBMA), myotonic dystrophy (DM)
FMR1, 5´UTR, CGG repeats• FXS: >200 • FXTAS: 55-200• Normal: 5-54
Microsatelite repeats and diseases

DM1 is caused by an expansion
of the CTG repeat in the 3’UTR of
the dystrophia myotonica protein
kinase gene (DMPK, 19q13.3),
AD inheritance
• Individuals with 5 to 37 repeats are unaffected. • Individuals with 38-50 repeats carry the premutation. These individuals
are asymptomatic. However, these repeats are unstable and can expand
during meiosis. As a result, such individuals are at risk of having affected
children. • ~ 50 to 150 repeats are consistent with the mild adult-onset form of DM1,
~100 to 1000 repeats are consistent with the classic adult or childhood
onset form of DM1,
> 1000 repeats are consistent with the congenital form of DM1 and often
result in severe neonatal complications.
Myotonic dystrophy type 1; DM1

• Patients with 50–150 CTG repeats
(mild adult onset form of DM1) may
develop cataract, diabetes, myotonia,
mild muscle weakness. • Patients with 100–1000 CTG
repeats (the classic form of DM1)
are affected earlier and more
severely. • CTG repeat size above 1000 is
associated with the congenital form
of DM1, which may be fatal due to
respiratory failure. Feeding difficulties,
muscle weakness, foot deformity, and
cognitive impairments are present in
surviving infants.
Myotonic dystrophy type 1; DM1

• The expanded CTG repeat -
‘dynamic’ mutation - the
number of repeats tends to
increase in size over
generations.
• Expansion of the CTG repeats
commonly occurs during
meiosis. As a result, children of
affected individuals tend to have
severe symptoms and earlier
onset than their parents.
Myotonic dystrophy type 1; DM1

DM2 (also known as proximal myotonic myopathy [PROMM]) is caused by an expansion of the CCTG repeat in the first intron of the zinc finger 9 gene (ZNF9, 3q21), AD inheritance.
• Unaffected individuals have less than 24 repeats. • Affected individuals have between 75 and 11000 repeats. • The repeat structure in DM2 is more complex than the triplet repeat seen in DM1. The
normal repeat structure is (TG)12-26(TCTG)7-12(CCTG)3-9(g/tCTG)0-4(CCTG)4-15. • Individuals with 22-33 uninterrupted CCTG repeats carry a premutation. These individuals are asymptomatic. However, these repeats are unstable and very likely to expand during meiosis (risk of having affected children).• The minimum pathogenic length of the expanded region appears to be 75 uninterrupted CCTG repeats. Repeat counts can increase to over 11000 in affected individuals, with a mean repeat length of ~5000 repeats. The expanded region has been shown to display an even greater instability than the DM1 mutation. • Unlike DM1, the length of the DM2 expansion does not appear to correlate significantly with the age of onset or severity of disease symptoms.
Myotonic dystrophy type 2; DM2

• Nuclei of cells of DM1/DM2 patients → expression of genes containing CTG/CCTG repeat expansions
→ nuclear foci containing RNA with expanded CUG/CCUG repeats → capture of RNA binding proteins (proteins regulating mRNA splicing) – muscleblind (MBNL) protein and others → misregulation of splicing of certain genes: chloride channel 1 (CLCN1), insulin receptor (IR), cardiac troponin T, skeletal troponin T, and others. • Insulin receptor and chloride ion channel pre-mRNAs are misspliced in DM patients → the symptoms of insulin resistance and myotonia.
In these two images of the same muscle precursor cell, the top image shows the location of the Mbnl1 splicing factor (green) and the bottom image shows the location of RNA repeats (red) inside the cell nucleus (blue). The white arrows point to two large foci in the cell nucleus where Mbnl1 is sequestered with RNA.
Pathogenesis of DM1 and DM2

Model of CLCN1 splicing regulation. Exon splicing enhancer (ESE) is located at the 5′ end of exon 7A. MBNL1 represses exon 7A inclusion through inhibiting ESE. RNAs carrying expanded CUG/CCUG repeats deplete MBNL1 protein, resulting in the facilitation of exon 7A inclusion.
Pathogenesis of DM1 and DM2
Y. Kino, Nucleic Acids Res 2009

Proper CLCN1 pre-mRNA splicing in normal skeletal muscle is regulated by MBNL1 protein. Depletion of MBNL1 proteins results in inclusion of additional exons (e.g., exon 7a) containing premature termination codons. Aberrantly spliced CLCN1 transcripts are exported from the nucleus, degraded through the nonsense-mediated decay pathway, and/or produce truncated proteins. These effects result in a dramatic reduction in the number of functional ClC-1 channel.
J Gen Physiol 2007;129:79-94


Toxic RNAs have myriad downstream effects on cellular metabolism.
•Expression of repeat-containing RNAs can induce hypermethylation and heterochromatinization of the neighboring DNA (FRAXA) (1). •The double-stranded hairpin structure formed by the repeat RNAs can sequester RNA-binding proteins such as MBNL1 (2). This leads to altered splicing of MBNL1 target RNAs (3). •In addition, in some cases kinase pathways are activated through unknown mechanisms, leading to aberrant phosphorylation and localization of CUGBP1 (4). This also has impact on splice site choice and perhaps on decay and translation of CUGBP1 target messenger RNAs (mRNAs) (5 and 6). •The toxic RNA can be cleaved by the Dicer protein to generate siRNAs that may inhibit expression of genes containing complementary repeats (7). This siRNA pathway can also induce silencing of the toxic repeat-containing gene (8). •Finally, when CAG repeats lie within a coding region they can encode polyglutamine, which also has toxic effects on the cell (9).
A.M. Dickson 2010

• MBNL1 is a RNA-binding protein (binding to hairpins formed by CAG, CUG, CCUG), sequestration of MBNL1 is strongly implicated in disease presentation.
• MBNL1 localization to the RNA foci sequesters MBNL1 away from its normal targets and this leads to disease symptoms. Several mRNAs whose splicing is regulated by MBNL1 exhibit aberrant splicing in DM.
• MBNL1 knockout mouse model reproduces much of the pathology as seen in DM mouse models. Overexpression of MBNL1 in a DM mouse model is sufficient to ameliorate both splicing defects and disease symptoms.
Muscleblind (MBNL1) sequestration

• In DM1 cells, CUG-containing RNAs form hairpin structures, which can be cleaved by the RNA interference (RNAi) machinery to generate small interference RNAs (siRNAs).
• These siRNAs are capable of binding complementary sequences in target mRNAs, possibly interfering with their expression and contributing to disease pathogenesis.
RNA interference

G. Sicot, Human Molecular Genetics, 2011
- The expanded gene is transcribed into sense and anti-sense transcripts. - Sense and anti-sense transcripts might be connected with repeat-associated non-ATG (RAN) translation in all possible reading frames generating homopolymers- iRNA pathways might be activated by the processing of dsRNA structures, which can result from the folding of CUG-containing transcripts into hairpin structures, or from the hybridization of complementary sense and anti-sense transcripts.


Three disease mechanisms for microsatellite expansion disorders. Expanded microsatellite repeats have been traditionally classified as either coding disorders or non-coding disorders that give rise to protein gain-of-function or loss-of-function or RNA toxicity mechanisms. For traditional ‘coding’ disorders, the repeat expansion is translated as part of a larger open-reading frame (ORF) and results in the expression of a mutant protein that disrupts normal cellular function and induces toxicity. For example Huntington’s disease (HD), a late-onset neurodegenerative disorder, is caused by a CAG expansion within the first exon of huntingtin gene that is translated as a polyglutamine tract in the huntingtin protein, HTT. For traditional ‘non-coding’ disorders (blue), the repeat expansion remains in the RNA transcript, accumulates as RNA foci that sequester RNA binding proteins and lead to a loss of their normal function. For example, in myotonic dystrophy, CUG(G) expanded RNA transcripts sequester MBNL proteins from their normal splicing targets leading to a MBNL loss-of-function and alternative splicing dysregulation. The recent discovery of repeat associated non-ATG (RAN) translation adds a third pathway for disease. RNA transcripts from both ‘non-coding’ and ‘coding’ disorders may undergo RAN translation. Once in the cytoplasm, these transcripts are capable of producing proteins in all three reading frames, which may contribute to cellular toxicity/stress. Depending upon the flanking sequences, each of these RAN proteins will have a distinct expanded peptide repeats (colored boxes) and unique different C-terminal regions (f1, f2 and f3). If the repeat is also within an ATG-initiated open-reading frame, this ATG-initiated protein will share the expanded peptide repeat and C-terminal region with one of the RAN proteins but will have an additional N-terminal region. Further complexity is added by fact that many expansion mutations are bidirectionally transcribed, which doubles the number of distinct RAN proteins that may be produced. J.D. Cleary, Current Opinion in Genetics & Development 2014

• The expanded DMPK gene is transcribed into sense and anti-sense transcripts.
• CUG-containing DMPK transcripts form alternative RNA structures and accumulate in the nucleus of DM1 cells, resulting in reduced DMPK protein levels.
• Loss of function of MBNL1, through sequestration into RNA foci, deregulates alternative splicing.
• CELF1 (CUGBP/Elav-like family member 1) hyperphosphorylation and upregulation affect alternative splicing and translation efficiency.
• Leaching of transcription factors (TF) by expanded DMPK transcripts may mediate changes in gene expression.
• iRNA pathways might be activated by the processing of dsRNA structures, which can result from the folding of CUG-containing transcripts into hairpin structures, or from the hybridization of complementary sense and anti-sense DMPK transcripts.
• Both sense and anti-sense DMPK transcripts might be connected with repeat-associated non-ATG (RAN) translation in all possible reading frames generating homopolymers, which might be deleterious to the cell.
Molecular pathogenesis of DM1: mechanisms of RNA toxicity, spliceopathy, deregulation of gene expression and proteotoxicity.

• Repeat-associated non-ATG (RAN) translation occurs independently of an ATG initiation codon in all reading frames. RAN translation occurs across long, hairpin-forming repeats which affect translational initiation.
• RNA translation on sense CUG-containing transcript s produce polyleucine (CUG), polycysteine (UGC) and polyalanine (GCU) tracts.
• RAN translation on antisense CAG-containing transcripts produce polyglutamine (CAG), polyserine (AGC) and polyalanine (GCA) homopolymeric peptides.
• These toxic homopolymeric proteins are considered in pathogenic models of microsatellite disorders.
Repeat-associated non-ATG (RAN) translation

Warner JP, J Med Genet 1996
Repeat primed PCR.Stippled box represents(CAG)n repeat. F shows 5' fluoresceinated primer. (A) For large alleles exceeding 100 CAG the PCR using flanking primers P1 and P2 fails to give a product. (B) In the early amplification cycles primer P4 (the repeat specific 3' terminus) binds at multiple sites within CAG alleles giving rise to a mixture of products. Specificity is dictated by P1. A 10:1 molar ratio of P3 to P4 ensures that primer P4 is exhausted in the early amplification cycles. (C) The primer P3 amplifiesfrom the end of products from previous amplification rounds. A long extension time is usedto allow complete extension of the larger sized products within the PCR product mixture.

Warner JP, J Med Genet 1996
Electrophoreogram of PCR (primers P1 + P2)The axis shows migration time in minutes. CAG allele sizes shown with the arrows. (A) Trace obtained from a heterozygous normal subject. (B) Trace obtained from a heterozygoussubject with a small expansion. (C) Trace obtained from a patient with myotonic dystrophy and an expanded allele size of >4 kb as determined by Southern blot analysis.The larger allele fails to amplify.

Warner JP, J Med Genet 1996
Electrophoreogram of repeat-primed PCR (P1+P3+P4) The axis shows migration time in minutes. CAG allele sizes shown with the arrows. The same people typed in fig 2 were retyped. Note the characteristic ladder with a 3 bp periodicity. (A) Both alleles give peaks and all the intermediate priming sites give peaks.There is a slight continuation of the pattern beyond the maximum allele size owing to mispriming at the end of the repeat. This usually extends for less than 30 bp.(B) Both alleles give peaks as in (A). (C) The ladder shows the presence of a large CAG allele undetectable using flanking primers.

Close up of traces shown in fig 3. The axis shows migration time in minutes.CAG allele sizes shown with the arrows. (A) Detail from trace shown in fig 3B. (B) Detail from trace shown in fig 3C.
Warner JP, J Med Genet 1996

• Restrikční štěpení DNA.
• Elektroforetické rozdělení naštěpené DNA v agarózovém gelu
Southern blot:
• 0,25 M HCl - depurinace DNA
• 0,5M NaOH - denaturace DNA, rozštěpení cukr-fosfátové vazby v místě depurinace (účinnější přenos DNA z gelu na membránu).
• Alkalický přenos DNA na membránu v 0.5 M NaOH (vazba negativně nabité DNA k pozitivně nabité membráně), různé možnosti.

Myotonia congenita (MC)
Mutations in the genes coding skeletal muscle chloride channel 1 (CLCN1) and alpha subunit of voltage-gated sodium channel 4 (SCN4A)
CLCN1:• chromosome 7q35• 23 exons• mutations associated with recesive and/or dominant inheritance
Rayan et al. (2013)
SCN4A:• chromosome 17q23• 24 exons• mutations associated with dominant inheritance
Sato et al. (2001) and Yu et al. (2004).

CLCN1 exists as homodimer with each individual subunit forming Cl pore.
Myotonia congenita (MC); CLCN1 protein
• The heterozygous situation - mixture of 25% WT channels, 25% channels carrying two mutant subunits, and 50% channels carrying one WT and one mutant subunit.
• Dominant MC is due to dominant-negative effects of the mutant subunit on the WT subunit.

• PTC mutations - recessive MC• Missense mutations -
recessive or dominant MC.• p.(Glu291Lys) is inherited
recessively; reversing the charge is expected to have drastic effects – misfolding and degradation of the protein, inability to create the dimer structure.
• p.(Glu291Asp) is inherited dominantly; conserving the negative charge by replacing glutamate with aspartate – subtle effect on the protein structure.
Myotonia congenita (MC); CLCN1 mutations
Glutamate
Aspartate
Lysine

Myotonia congenita (MC); CLCN1 mutations
Réblová K. (2013)
Cederholm et al. (2010)
• A homology model of the human dimeric ClC-1 channel on the basis of known crystallographic structure.• Identification of AAs which form the dimer interface or the Cl- ion pathway. • A search for mutations of AAs forming the dimer interface or the Cl- ion pathway.• A assesment of the correlation between the localisation of a mutation and the type of MC (recessive/dominant).• In case of mutations localised in the dimer interface, the correlation between the localisation of a mutation and the dominant MC was observed.



















