Embed Size (px)
Citation preview

藥品生產驗證原則和實踐(一) - 設備確認及工藝驗證
1
如何提升香港藥品生產質量管理規範之培訓班
2015年7月14日下午 (龍志豪先生)

確認與驗證的關係
設備確認 定義 設備確認方法:V-model Approach 用戶需求說明 (URS) 功能說明 (FS) 設計說明 (DS) 設計確認 (DQ) 影響性評估 (IA) 及 風險評估 (RA) 安裝確認 (IQ) 運行確認 (OQ) 性能確認 (PQ)
工藝驗證 工藝驗證的定義 工藝驗證的目的 工藝驗證的類型 如何進行工藝驗證 工藝驗證文件要求 變更和再驗證 驗證總計劃 (VMP)
主要內容

確認 (Qualification) 和驗證 (Validation) 是製藥企業基本的品質活動,並且已經成為 GMP 法規要求。
確認 – 主要針對設備、人員和供應商;
驗證 – 將已確認的人員、設備、物料、軟體、程式等整合在一起,旨在證
明操作規程、系統、方法或生產程序,能夠達到既定目的;
確認與驗證的關係

確認與驗證的關係 確認
(Qualification)
是證明廠房、設施及設備,能夠正確運作並達到預期結果的一系列活動。
設備確認
設施確認 – 空調系統、壓縮空氣系統、純水系統
廠房確認 – 潔淨車間、物料貯存區域等
以硬件為主體
它強調的是結果的正確性
驗證 (Validation)
是證明任何操作規程、系統、方法或生產程序,能夠達到預期結果的一系列活動。
生產工藝 工藝驗證
清潔程序 清潔驗證
測驗方法 分析方法驗證
以軟件為主體
它強調的是過程及結果的同時正確性
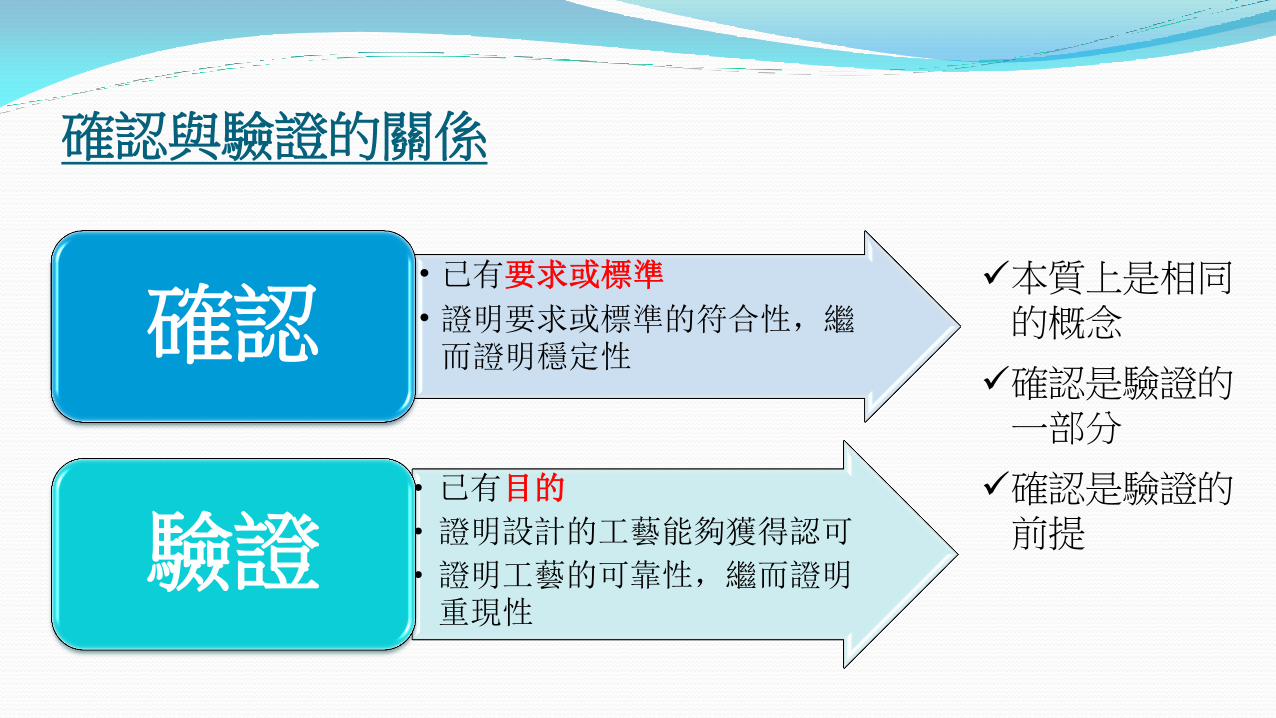
本質上是相同的概念
確認是驗證的一部分
確認是驗證的前提
• 已有要求或標準
• 證明要求或標準的符合性,繼而證明穩定性
確認
• 已有目的
• 證明設計的工藝能夠獲得認可
• 證明工藝的可靠性,繼而證明重現性
驗證
確認與驗證的關係

設備確認 (Equipment Qualification)

設備確認:V-model Approach
用戶需求說明
(URS)
功能說明 (FS)
設計說明 (DS)
設
計
確
認
(DQ)
系統建造
安裝確認 (IQ)
運行確認 (OQ)
性能確認 (PQ)
調試或試運行 (Commissioning) 建基於
建基於
建基於

確認步驟 - 設備、設施及廠房
文件證明到此設計符合用戶需求說明及要求 設計確認 (DQ)
工廠測試驗收 (FAT) / 現場測試驗收 (SAT)
安裝確認 (IQ)
操作確認 (OQ)
性能確認 (PQ)
對於新穎和複雜技術的設備或系統
FAT - 發貨前在製造場地對設備進行工廠接受度測試
SAT - 當設備到達後,就要進行現場接受度測試工作
文件證明項目是按照指定規格及要求 (例如設計及圖紙),供應及正確地安裝
文件證明項目是按照指定規格及要求,正確地運作
文件證明項目是與其他系統連接後,有效及重複地生產出符合預期質量的產品
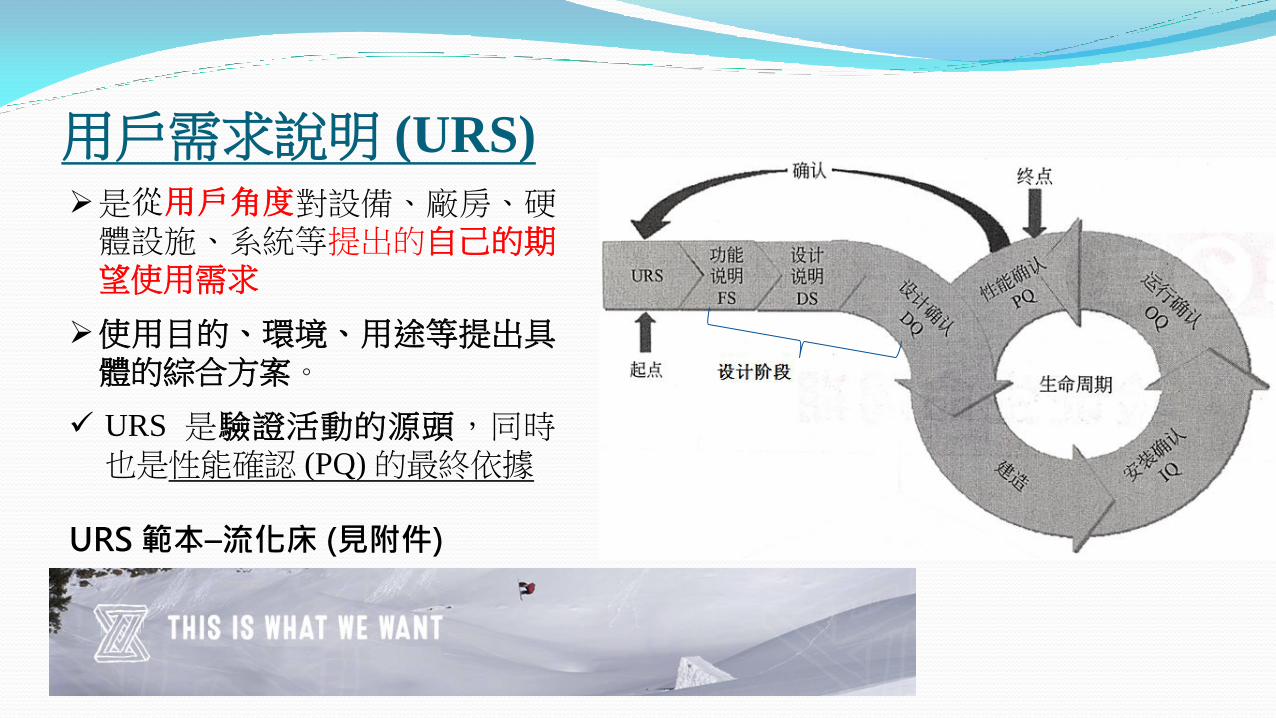
用戶需求說明 (URS)
是從用戶角度對設備、廠房、硬體設施、系統等提出的自己的期望使用需求
使用目的、環境、用途等提出具體的綜合方案。
URS 是驗證活動的源頭,同時也是性能確認 (PQ) 的最終依據
URS 範本–流化床 (見附件)

用戶需求說明 (URS) 應包括下列內容:
1.0 目的 用於描述編寫本使用者需求的目的
2.0 範圍 用於描述編寫本用戶需求的範圍
3.0 責任
4.0 法規需求
5.0 設備或系統描述
6.0 技術需求 6.1 生產工藝需求 6.7 安裝調試需求
6.2 外觀需求 6.8 能源需求
6.3 物料材質需求 6.9 清洗需求
6.4 程式需求 6.10 程式控制
6.5 功能需求 6.11 產能需求
6.6 運作需求 6.12 安全需求
7.0 通用需求 7.1 維修和維護需求 7.6 培訓需求
7.2 文件和證書需求 7.7 環境健康安全需求
7.3 FAT 及 SAT 需求 7.8 資料和安全需求
7.4 廠房設施及公用系統需求 7.9 包裝及運輸需求
7.5 備品零件需求
13.0 術語 用於描述用到的專業術語、縮略語進行解釋和說明
13.0 參考文件 描述適用和參考的法律、法規、公司指南等

功能說明 (FS)
功能說明 “URS” 所描述的功能在設備各組件的設計、材料的選用等方面做出詳細的描述, 例如: 過程控制 (所有操作過程階段/ 步驟/ 程式、狀態及相互功能轉換) 控制系統所具備的功能等
應由供應商完成及提供,再由用戶核實、複查及比對
如屬於非系統性或較小型的設備,供應商未必會提供一份獨立的功能說明

設計說明 (DS)
設計說明需說明如何滿足 “FS” 和 “URS” 的詳細的、具體的要求,需要詳細和準確
通過 DS,使用者能夠知道設備的正確安裝、測試和維護
應由供應商完成及提供,再由用戶核實、複查及比對
如對一些簡單或非系統性的設備,功能說明可以和設計說明合併成一個檔即功能設計說明 (FDS) 文件

用於新的廠房、公用設施、設備確認的第一步為設計確認 (DQ)
目的:
確保該項目的設計符合預定用途及相關法規要求制定的用戶要求
用戶要求、產品品質要求、法規要求、其他要求
經過批准的設計確認是後續確認活動的基礎 (如安裝確認、運行確認、性能確認)
進行設計確認工作,對下列文件進行複查 (verification) 及比對 (comparison):
用戶需求說明 (URS)
功能說明 (Functional Specifications)
設計說明 (Design Specifications)
可採用表格的方式將上述文件進行逐條的比對,並將對比的結果進行記錄。
建議對每一條需求和技術規格單獨編號。
設計確認 (DQ)

影響性評估(Impact Assessment) 及 風險評估 (Risk Assessment)
系統影響性評估 (System Impact Assessment)
用於確定設備的確認範圍和程度 (IQ, OQ, PQ)
針對每個設備進行評估,以判定其對整個系統的影響性:
直接影響 (Direct Impact)
間接影響 (Indirect Impact)
無影響 (Non-direct Impact)
部件關鍵性評估 (Component Criticality Assessment)
針對直接影響系統的設備進行部件關鍵性評估。以確定設備內各部件是:
關鍵 (Critical)
非關鍵 (Non-Critical)
風險評估 (Risk Assessment)
判斷出關鍵部件後,對關鍵部件繼續進行風險評估
採用風險評估工具(eg. FMEA),來找出關鍵部件的可能失效影響
參考資料:ISPE (Vol. 5) – Commissioning & Qualification

安裝確認 (IQ) 用於新的或發生改造 (Revamp) 之後的廠房、設施、設備等進行安裝確認;
證明所安裝或改造的廠房、系統和設備主要部件正確的安裝,以及和設計要求一致,符合已批准的設計、生產廠家建議;
設備、設施、管路的安裝以及所涉及的儀錶應對照工程技術圖紙 (Schematic Diagram) (eg. 管道及儀錶流程圖/P&ID; 竣工圖 (As-build Drawings) ; 線路圖 (Wiring Diagrams);
設備校正需求、操作手冊、預防性維護和清潔的要求等;
儀器應該經過校正
以表格形式每項核對及簽名確認
要有驗證報告(IQ Validation Report) 證明,該項目通過是次的安裝確認,確認效果是滿意及有效的。批准進行下一確認活動:運行確認 (OQ)。
適合於:
潔淨及非潔淨車間區域
實驗室 (特別留意:微生物實驗室)
主要公用設施 藥用壓縮空氣系統
工藝蒸汽(鍋爐)系統
製藥用水系統
關鍵生產設備
空氣淨化系統 (HVAC)

應包括以下的檢查項目但不局限於:
到貨的完整性
將到貨的實物與訂單、發貨單、DQ檔等進行對比
檢查 DQ 檔中所規定的檔(如操作說明、備件清單、圖紙等)是否齊全
安裝和連接情況
對照圖宅檢査安裝情況(機械安裝、電器安裝、控制回路等)
加工情況 (如焊接、排空能力、管路斜度等)
設備等的標識 (內部設備編號的標識、管路標識等)
檢查設備設施等與動力系統 (如供電) 的連接情況
檢查設備設施等與公用設施 (如壓縮空氣系統、冷水系統等) 的連接情況
安裝確認 (IQ) 材質和表面
檢查直接接觸產品: 設備材質類型 (SS304 / SS316L)
材質證明書 (Material Certificate)
設備表面的光滑程度 (Ra – Surface Roughness Value)
檢查可能對產品品質產生影響的其他物質(如潤滑劑、冷卻劑等)
清潔及消毒
校正
應對控制或測量用的儀錶等進行校正需求的評估
對需校正的儀錶等建立校正要求及方法
完成初始校正
文件
操作手冊
技術圖紙等的審核 管道及儀錶流程圖 (P&ID)
竣工圖 (As-build Drawings)
線路圖 (Wiring Diagrams)

安裝確認例子–瓶標籤機

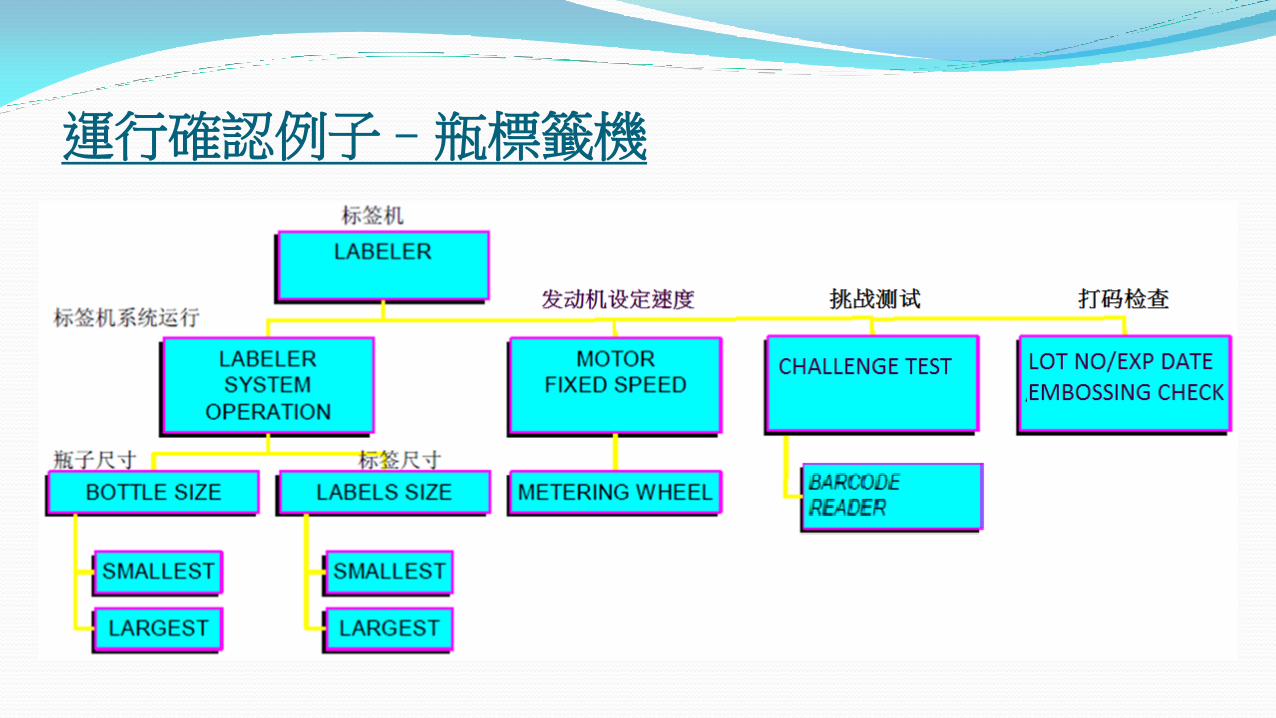
運行確認 (OQ) 應在安裝確認完成之後進行
測試應包括“最差條件”- 即指令引數的上下限度(例如最高和最低溫度)
測試應重複足夠的次數以確保結果可靠並且有意義
證明設備運行符合出廠技術參數,能滿足設備的 URS 和 DQ 中的功能技術指標,和證明各項技術參數能達到預先設定要求 (例如溫度、壓力、時間、轉速等)
設備相關的 SOPs,應該完成及審批
有負荷 (Wet Test Runs) 與無負荷試車 (Dry Test Runs) - 基於設備種類的考慮
混合機
口服液灌裝機
以表格形式每項核對及簽名確認
要有驗證報告(OQ Validation Report) 證明,該項目通過是次的運行確認,確認效果是滿意及有效的。批准進行下一確認活動:性能確認 (PQ)。

運行確認例子–瓶標籤機

性能確認 (PQ) 應在 IQ 和 OQ 成功完成之後執行
性能測試:
有些情況下,可以將 PQ 與 OQ 結合在一起進行
半自動數丸機 (Tablet/Capsule Counter)
瓶標籤機 (Bottle Labeler)
應該用實際產品作測試
經常包括一些“挑戰”測試 (Challenge Test)
主要涉及工藝相關的關鍵工藝參數 (CPP)。CPP 設定後,其實際生產運行時應在規定的範圍內波動。
輸出的品質均一及重現性:對輸出進行取樣檢測,各項品質指標均在預定標準之內。
根據PQ確認報告內容資料,並確認執行完成及偏差評估之後,需要對執行批准及驗收;
確認設備的性能確認已完成後,清潔驗證 (Cleaning Validation) 和工藝驗證 (Process Validation) 可以進行。
運行確認例子–瓶標籤機

設施 / 設備 描述
SIA CCA /
RA URS
DQ
FAT SAT
IQ
OQ
PQ
校正SOP
裝拆 SOP
操作SOP
清潔SOP
維護SOP
DI II NI
膠囊充填機 ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓
膠瓶壓蓋機 ✓ ✓ NA NA NA NA NA NA NA NA NA ✓
✓/ NA
✓/ NA
辦公室冷氣
系統 ✓ NA NA NA NA NA NA NA NA 內部使用手冊
純水系統 ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓ ✓
鍋爐
(非藥用蒸汽) ✓ NA NA NA NA NA NA NA NA 內部使用手冊
冷凍水系統 ✓ NA NA NA NA NA NA NA NA 內部使用手冊
設備確認計劃 (Validation Plan)

工藝驗證 (Process Validation)

《FDA行業指南-工藝驗證:一般原則和規範2011版》 收集並評估從 “工藝設計階段一直到商業化生產” 的資料,用這些資料確立科學證據,
證明該工藝能夠始終如一地生產出優質產品。工藝驗證涉及到了在產品生命週期及生產中所發生的一系列活動。
(EU GMP - 附錄:確認和驗證 2001) 為證明工藝在設定參數範圍內能有效及穩定地生產出符合預定質量標準和質量特性藥品
的驗證活動。
中國《藥品生產質量管理規範》(2010年修訂) 工藝驗證應當證明一個生產工藝按照規定的工藝參數能夠持續生產出符合預定用途和註
冊要求的產品。
(Annex 15 of PIC/S Guide – PE009/11)
工藝在已建立的參數內操作時,能有效且再現性地生產符合其預定規格及品質屬性的藥品的文件化證據。
工藝驗證的定義

《香港中成藥生產質量管理規範指引》對驗證工作的要求
第六章 「製造」 6.35 應驗證關鍵製造程序。
第七章「驗證」 - 原則 驗證是生產質量管理規範的基礎部分。 驗證是證明任何規程、製造程序、設備、物料、活動或系統確實能達到預期結果且有文件證明的一系列
活動。製造商應按照預定的方案進行驗證,並以書面報告形式記錄驗證結果和結論。 驗證結果和結論應妥善保存。製造程序和規程應在驗證的基礎上建立。製造程序和規程應定期接受再驗
證,以確保它們依然能達到預期的效果。 應特別注意製造程序、檢驗規程和清潔規程的驗證。
驗證應包括廠房、設施及設備安裝、運行和性能確認,以及產品驗證。
應對關鍵製造程序進行前瞻性或回顧性驗證。 採納新總處方或新製造方法前,應驗證其對常規製造的適用性,以確保當採用既定的製造程序、使用指
定的物料及設備,便能一致地製造符合規定品質的產品。 製造程序的重大變更,包括可影響產品品質和/或程序的重現性的設備或物料的任何變化,都應接受驗證。
工藝驗證的定義

評價生產方法及證明工藝的可靠性
證明產品均一/均勻性,即一批產品內部和批與批之間是均質的,始終生產出符合註冊要求的產品
為系統控制提供文件化證據
工藝驗證處於整個驗證活動的中心地位
廠房、設施、設備、清潔等驗證
為產品工藝驗證打下基礎
工藝驗證–目的


廠房與設施驗證
空調淨化系統確認 (驗證)
公用設施確認 (驗證)
(藥用壓縮空氣系統、工藝蒸汽(鍋爐)系統、
製藥用水系統)
生產設備確認(驗證)
工藝驗證
製藥企業相關的驗證活動及階段
測試儀器 校正
計算機系統驗證 (CSV)
分析方法 驗證

工藝驗證–先決條件
第三階段

工藝驗證的類型有三種:
前瞻性驗證 (Prospective Validation)
同步驗證 (Concurrent Validation)
回顧性驗證 (Retrospective Validation)
前瞻性驗證是首選的方法。
工藝驗證的類型
工藝驗證
前瞻性驗證
同步
驗證
回顧性
驗證

定義:
是指生產工藝投入使用前必須完成並達到設定要求的驗證。
適用條件:
1. 產品要求高,缺乏歷史資料,單靠生產過程監控及成品檢查不足以確保重現性及產品質量的生產工藝。
2. 引入新產品、新設備以及新的生產工藝時,或產品的某一生產過程發生了變更,且可能影響到產品特性時,在經過對變更程序 (Change Control) 進行評估並批准後,應採用前瞻性驗證的方式進行工藝的驗證。
工藝驗證的批數:前瞻性驗證的批數應足夠確定正常變化的範圍及趨勢,並有足夠的資料供評估。通常連續三批運行均在最終確定的參數範圍內,可認為工藝驗證符合要求。
工藝驗證的批次量:批次量應與正式生產批量相同
工藝驗證的類型–前瞻性驗證

前瞻性驗證方案:
職能部門和職責 工藝的簡短描述 應驗證的關鍵工藝步驟摘要 所要使用的設備/設施清單以及其校正狀態
各相關生產設備 監控設備 紀錄設備
成品放行的質量標準、相應的檢驗方法清單、在線控制及合格標準 分析方法驗證 (包括微生物檢驗方法) 取樣計劃 各相關紀錄 (包括批生產及包裝記錄、取樣記錄、檢驗記錄等) 評估結果的方法 (包括統計學分析方法) 驗收準則 (Acceptance Criteria)
工藝驗證的類型–前瞻性驗證

前瞻性驗證的一般流程
工藝驗證的類型–前瞻性驗證

定義:
在一些特殊的情況下,正常生產前沒有完成驗證,或該產品使用驗證過的但已經變更的工藝生產,無法從連續生產中得到資料可以考慮使用同步驗證的方式。
實際上是在不適合前驗證的情況下的退一步選擇。
適用條件:
1. 對所驗證的產品或工藝過程已有比較成熟的經驗與把握,如:該產品同原來已經驗證過的產品的規格不同,例如:藥片的形狀不同。
2. 已驗證的工藝進行週期性再驗證 (Periodic Revalidation)
3. 對進行同步驗證的決定必須證明其合理性、有文件紀錄並經過管理層批准
4. 同步驗證的文件要求與前瞻性驗證相同
工藝驗證的批數:通常三批運行均在最終確定的參數範圍內,可認為工藝驗證符合要求。
工藝驗證的批次量:批次量應與正式生產批量相同
工藝驗證的類型–同步驗證

定義:
當某一生產工藝有較長的穩定生產歷史,通過監控已積累了充分的歷史資料時,可採用回顧性驗證的方式,通過對歷史資料的回顧分析找出工藝受控、達到設定標準的文件依據。
適用條件:
1. 關 鍵 質 量 因 素 (Critical Quality Attitude) 和 關 鍵 工 藝 參 數 (Critical Process Parameters) 均已確定;
2. 已確立了合適的程序控制和認可標準;
3. 除人員操作失誤或設備故障外,從沒有出現過其它原因造成的重大產品不合格事件。
4. 當發生供應商、操作規程、檢驗方法、設備變更時,不可以使用回顧性驗證。
工藝驗證的批數: 回顧性驗證的樣本數應足夠大,除有合理的理由外,一般要 求 10~30 批連續批次的樣本。
工藝驗證的類型–回顧性驗證

回顧性驗證至少包括但不限於以下步驟:
驗證方案
選擇的批次
應具有回顧階段所生產批次的代表性,包括不合格批。
一般需總結連續10-30批產品連續生產過程的資料進行統計和分析
資料至少應包括:
批生產和批包裝紀錄、人員變更紀錄
廠房環境監控資料
設備維修保養紀錄、設備校正記錄
變更與偏差紀錄
中間品控制資料和成品檢驗資料、QC檢驗報告
所有批次的穩定性資料
評估結果的方法 (包括統計學分析方法)
驗收準則 (Acceptance Criteria)
工藝驗證的類型–回顧性驗證
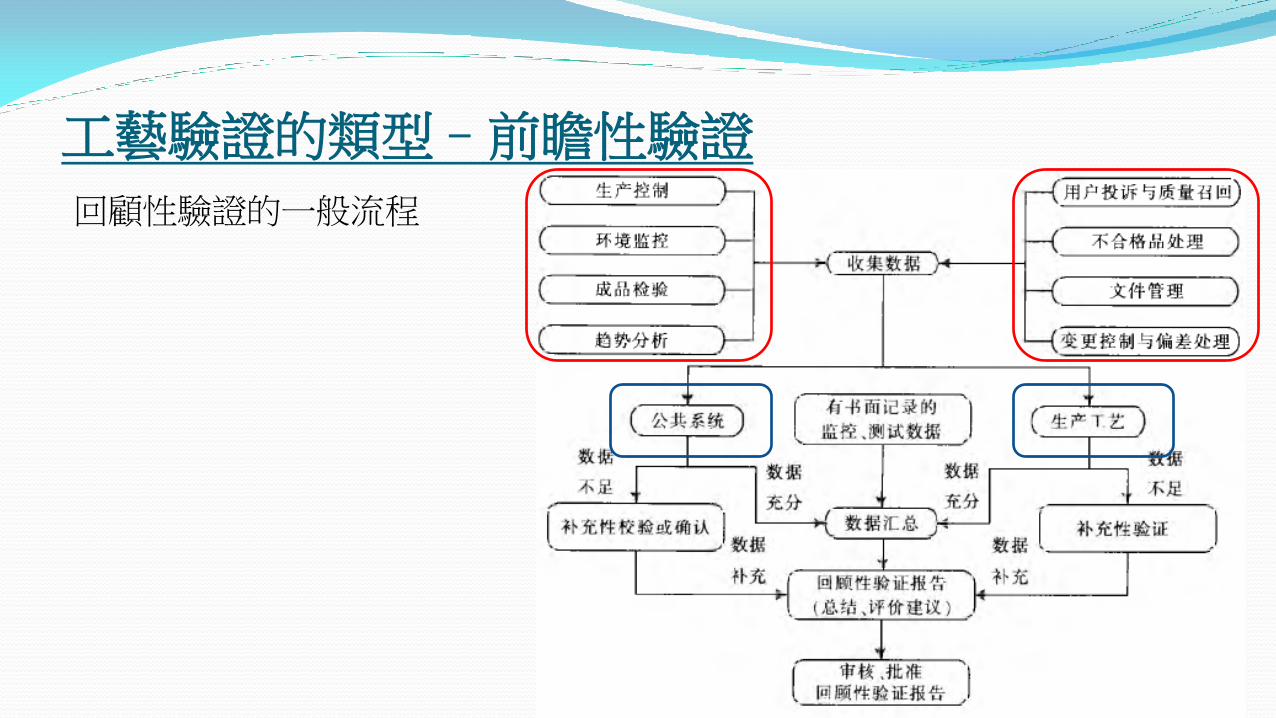
回顧性驗證的一般流程
工藝驗證的類型–前瞻性驗證
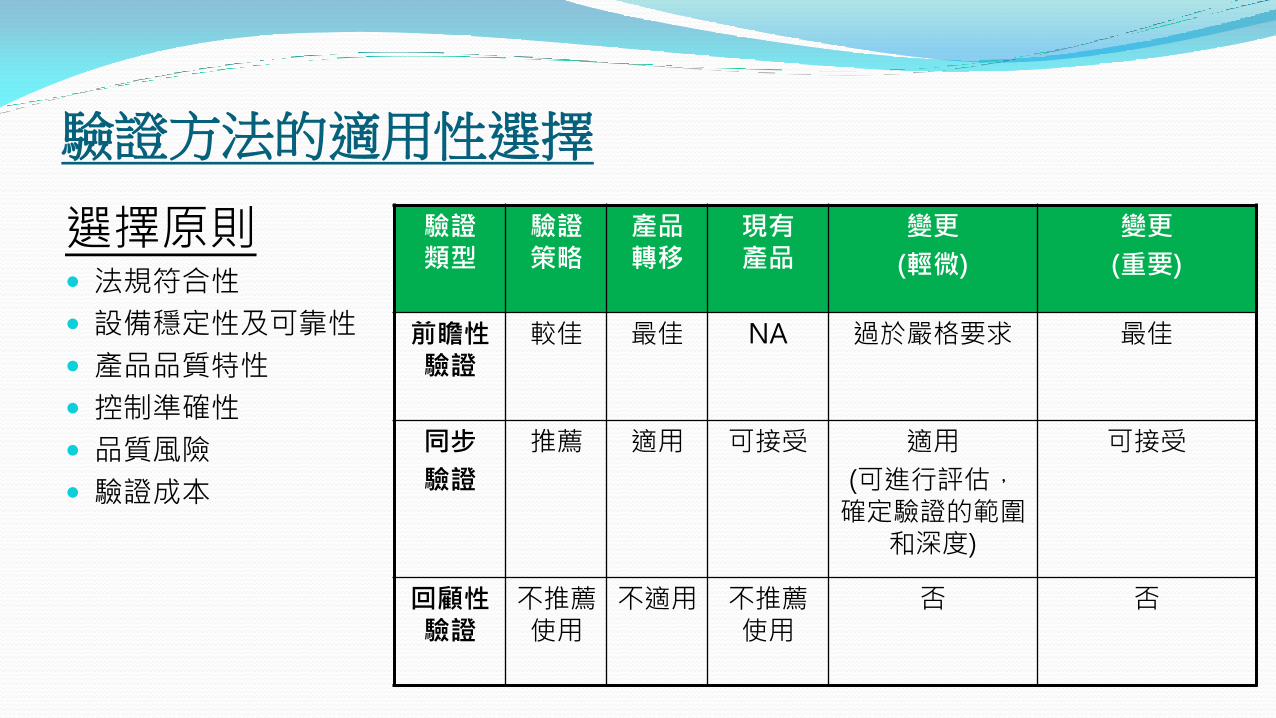
驗證方法的適用性選擇
選擇原則 法規符合性
設備穩定性及可靠性
產品品質特性
控制準確性
品質風險
驗證成本
驗證 類型
驗證 策略
產品 轉移
現有 產品
變更
(輕微)
變更
(重要)
前瞻性驗證
較佳 最佳 NA 過於嚴格要求 最佳
同步
驗證
推薦 適用 可接受
適用
(可進行評估,確定驗證的範圍
和深度)
可接受
回顧性驗證
不推薦 使用
不適用 不推薦 使用
否 否

如何進行工藝驗證
前提條件:
所有的前期確認均已完成,如生產設備確認、公用系統 (空調系統、工藝用水系統) 確認
各種批准的生產工藝文件都已具備,如經審批的批生產記錄、工藝驗證方案等
完成分析方法驗證 (包括微生物檢驗方法)
品質標準已經批准,包括成品、中間產品控制
取樣計劃:包括取樣位置及取樣量
確定驗證生產批次:通常進行連續三批
批准的產品穩定性試驗方案

工藝驗證的步驟
1. 編制批准批生產紀錄及檢驗規程 首先確認擬驗證的工藝編制,並批准了批生產紀錄及相應的產品質量標準和檢驗規程
2. 設備、公用設施確認 (Equipment & Utilities Qualification)
確認與驗證相關的生產設備、廠房環境及公用設施已完成了相關的確認,關鍵儀器儀錶均已校正,且都在有效期內
3. 工藝驗證的風險評估分析
4. 制訂工藝驗證方案 前期確認工作完成後應起草工藝驗證方案 一般工藝驗證最少應進行連續三批商業批量的驗證
5. 審批驗證方案 驗證方案應經驗證相關部門進行審核,並經質量負責人(獲授權人) 批准 應明確驗證的生產過程的 “關鍵工藝參數” 及 “關鍵質量因素”,證明其重現性 應明確進行驗證 (和分析) 的方法以及所要收集的資料 應對參與驗證的相關人員的培訓狀況、支持驗證的文件、相關的儀器儀錶、物料的適用性、生產設備
和公用系統的狀況以及產品質量標準及檢驗方法等進行預先確認

工藝驗證的步驟 6. 進行驗證
按照批准的驗證方案進行驗證
7. 驗證報告
按照方案規定匯總收集資料並進行分析,總結討論所有生產中的不符合項,如偏差、異常檢測結果,充分真實地描述對現有程序與控制方法所採取的任何糾正行動或變更,明確地陳述結論,說明資料是否表明了這一工藝與驗證方案中建立的條件相符合,以及工藝是否可視為處於足夠受控狀態
驗證報告應包括所有相關部門與質量部門的審核與批准
8. 驗證成功完成
驗證完成後,根據穩定性方案對留樣進行考察,並對穩定性資料進行評估,根據三批的穩定性資料確定產品的有效期;
針對新產品,其效期可執行生產批件上的暫定效期,待獲得足夠的穩定性資料支持後再確定其效期。
9. 驗證不成功
則應進行調查,只有在找出驗證不成功的原因後,才可以進行修訂驗證方案重新 進行驗證。

關鍵工藝參數 (Critical Process Parameter, CPP ):
有可能會對產品的標識、效力、品質、純度、效價、安全性和/ 或收率造成負面影響,所以必須對工藝相關變量或輸入進行緊密的控制。
關鍵工藝參數可能會對關鍵品質屬性或工藝的產出情況產生直接的影響。
對關鍵工藝參數有良好監控和控制就可以降低風險,但是並不會降低該工藝參數的關鍵性。
關鍵品質屬性(因素) (Critical Quality Attribute, CQA):
指的是物理、化學、生物學或微生物的性質或特征,其應在適當的限度、範圍或分佈內,以保證產品品質。
關鍵工藝參數 (CPP) 及 關鍵品質屬性 (CQA)

在開始工藝驗證活動前,應當在工藝驗證前確定產品的 CQA、影響產品關鍵品質屬性的 CPP、常規生產和工藝控制中的關鍵工藝參數範圍,通過驗證證明工藝操作的重現性。
對產品質量影響大的工藝參數稱為 CPP。其可接受範圍反映工藝參數的穩定性,在參數確認中影響產品質量的可改變參數範圍越寬,工藝越穩定。
CPP 和 CQA 通常在研發階段或根據歷史資料和數據確定。
要對準 “最差情況” 進行驗證。
關鍵工藝參數 (CPP) 及 關鍵品質屬性 (CQA)
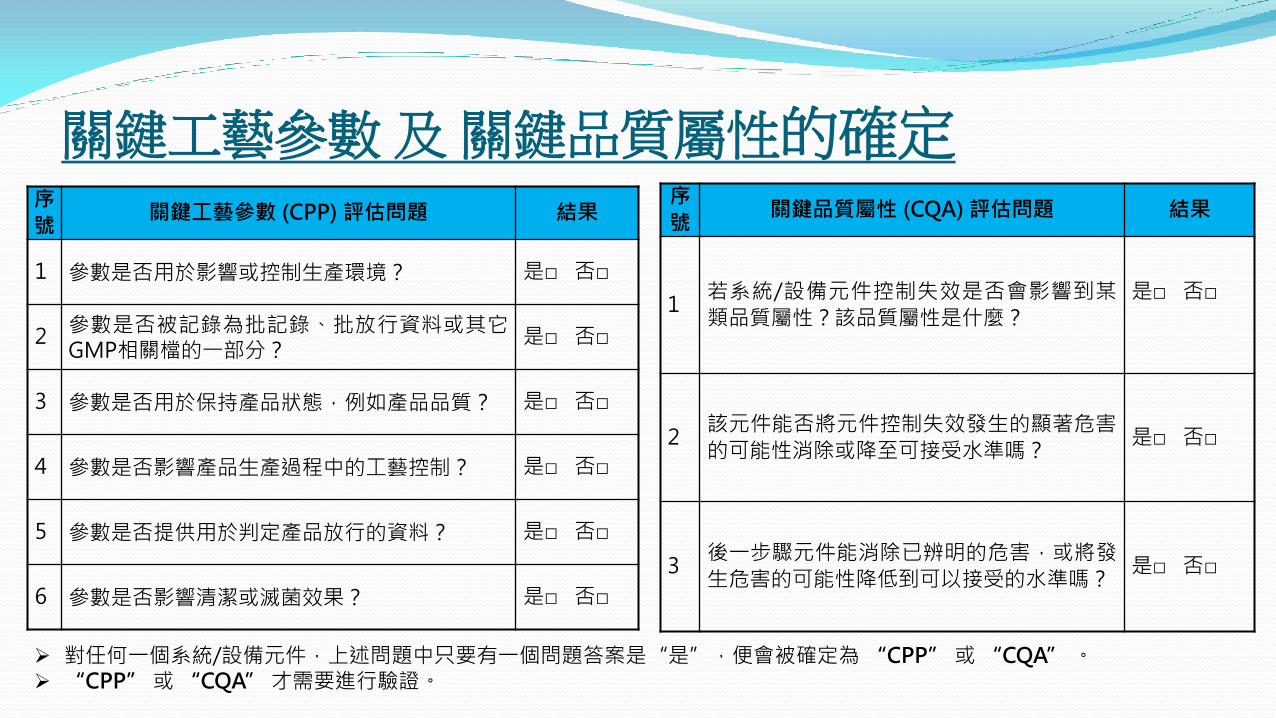
關鍵工藝參數 及 關鍵品質屬性的確定 序
號 關鍵品質屬性 (CQA) 評估問題 結果
1 若系統/設備元件控制失效是否會影響到某
類品質屬性?該品質屬性是什麼?
是□ 否□
2 該元件能否將元件控制失效發生的顯著危害
的可能性消除或降至可接受水準嗎? 是□ 否□
3 後一步驟元件能消除已辨明的危害,或將發
生危害的可能性降低到可以接受的水準嗎? 是□ 否□
序
號 關鍵工藝參數 (CPP) 評估問題 結果
1 參數是否用於影響或控制生產環境? 是□ 否□
2 參數是否被記錄為批記錄、批放行資料或其它GMP相關檔的一部分?
是□ 否□
3 參數是否用於保持產品狀態,例如產品品質? 是□ 否□
4 參數是否影響產品生產過程中的工藝控制? 是□ 否□
5 參數是否提供用於判定產品放行的資料? 是□ 否□
6 參數是否影響清潔或滅菌效果? 是□ 否□
對任何一個系統/設備元件,上述問題中只要有一個問題答案是“是”,便會被確定為 “CPP” 或 “CQA” 。 “CPP” 或 “CQA” 才需要進行驗證。

關鍵工藝參數 (CPP) 生產工序流程 關鍵質量屬性 (CQA)
粉粹及過篩的篩網目數 粉粹及過篩 粒度分佈 水分
批量 製粒機切刀及攪拌的速度 添加黏合劑的速度,溫度及方法 濕法整粒方式及篩網尺寸
濕法製粒 粒度分佈 水分
批量 進風溫度,濕度和風量和出風溫度 夥粒溫度,乾燥時間,水分
乾燥 水分
篩網尺寸,整粒速度,夥粒粒度分佈 整粒 粒度分佈 水分
批量,混合速度,混合時間 總混 混合均勻度 (含量)
充填機運行速度 膠囊充填 裝量差異,水分,崩解度,溶出度,含量均勻度
灌裝機運行速度 灌裝粒數
藥瓶灌裝 (數丸) 及入樽 灌裝粒數準確度
封口機運行速度 封口溫度
熱感應式鋁箔封口機 鋁箔封口密封度 (滲漏測試)

工藝驗證的關鍵步驟
通常應重點驗證的關鍵工藝步驟 (Critical Process
Steps):
A. 任何改變產品性狀的步驟;
B. 所有影響產品均一性的步驟;
C. 所有影響鑒別、純度或質量標準的步驟;
D. 延長儲存的步驟;
與品質無關的參數 (如與節能相關的參數),無需列入工藝驗證中。
關鍵工藝步驟
中藥提取及濃縮
口服液 (混合、過濾、灌裝、密封)
膠囊劑 (混合、充填、藥瓶灌裝、密封)
壓片 (制粒、混合、調配、壓片)

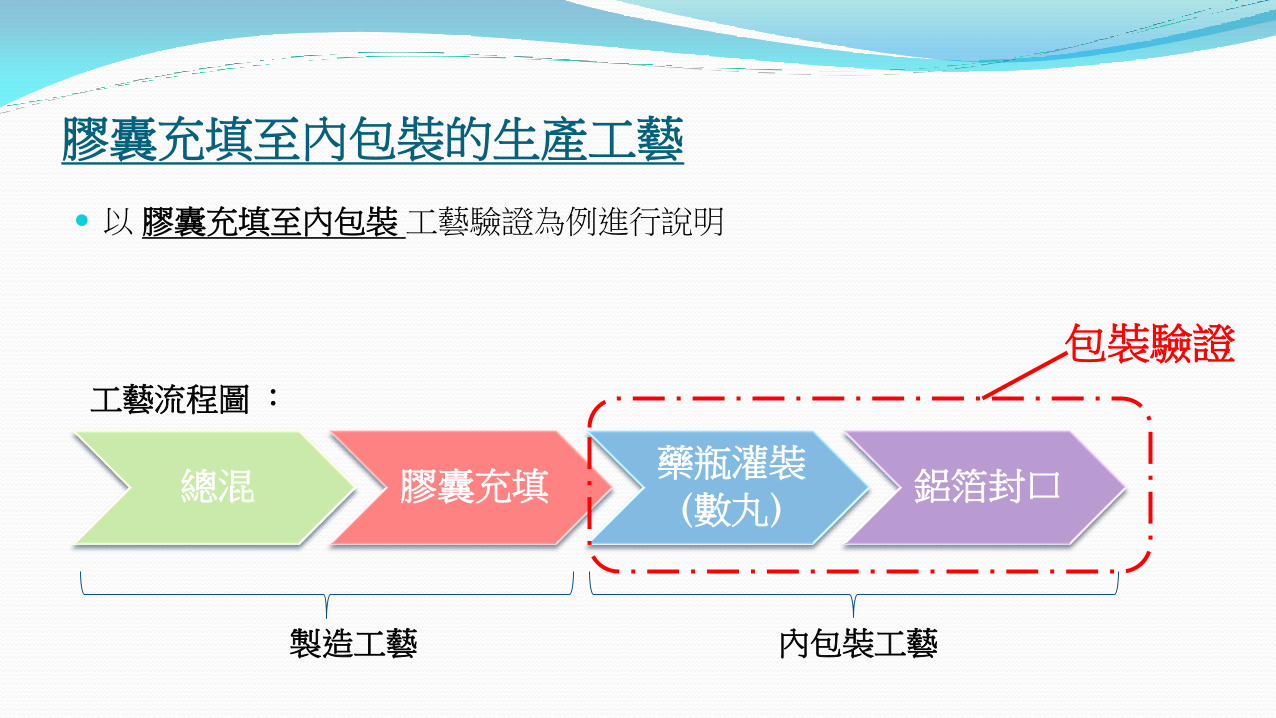
膠囊充填至內包裝的生產工藝
以 膠囊充填至內包裝 工藝驗證為例進行說明
總混 膠囊充填 藥瓶灌裝
(數丸) 鋁箔封口
工藝流程圖 :
製造工藝 內包裝工藝
包裝驗證

膠囊充填至內包裝的生產工藝
流程圖中,總混、膠囊充填、藥瓶灌裝及鋁箔封口過程為 “關鍵工藝步驟”,針對這四個步驟的驗證項目及驗證標準進行討論。
驗證項目
混合過程
分料過程
存放週期
膠囊充填過程
已充填膠囊存放時間
藥瓶灌裝過程
箔封口過程

膠囊生產工藝–總混過程
總混過程
在膠囊生產工藝中,混合是相當關鍵的步驟。對於小劑量藥物,混合顯的更為重要。如果混合不均勻,會對產品的安全性和有效性帶來風險。
在生產過程中有的時候是一步混合,有的是幾步混合。 一步混合是把過篩後的物料直接轉移至混合容器中,在指定的參數下進行混合。
幾步混合是指先把一部分物料轉移至混合容器中,在指定參數下進行混合,然後再加入某些物料 (例如:後下藥),以及潤滑劑、崩解劑等再在指定的參數下混合。
特別留意:幾何稀釋法 (Geometrical Dilution)
一步混合的時候,在混合完成時評估物料是否混合均勻。
幾步混合時,一般在最後一步混合後評估物料是否混合均勻。
非常著重投料的先後次序及物料的數量。

膠囊生產工藝–總混過程
驗證的目的: 確定混合參數
投料的先後次序及物料的數量
關鍵工藝參數: 批量、混合時間、混合轉速
驗證方法: 按生產指令依次加入物料,然後在規定轉速
下混合 XX 轉 (rpm) 或混合XX分鐘 (min)。
按計劃在混合容器中指定位置取樣檢驗
驗證的項目: 混合均勻度
檢測方法: 經驗證的測定混合均勻度的方法,如含量。

膠囊生產工藝–總混過程 驗證- 混合均勻度/一致性
目的: 評估混合顆粒均勻度
攪拌時間:
10mins
20mins
30mins
攪拌速度: 5 rpm (Low)
10rpm (Medium)
15rpm (High)
採樣數量: 10
接受標準: 含量一致及達標
CPP
關鍵工藝參數
CQA
關鍵質量因素

膠囊生產工藝–總混過程
總混過程
接受標準: 應符合質量標準規定的限度。 當第一份樣品檢測不合格時,應按照實驗室 “OOS管理程序” 進行調查,確定非實驗室偏差
後再進行其他樣品的檢驗,對所有檢驗樣品資料進行評估,確定是否符合要求。 取樣計劃:
取樣頻率應比正常生產高
取樣時間: 最終混合階段的各個驗證點
取樣點: 根據混合容器的構造,設計有代表性的取樣點 (特別留意:容器內 “死位”)
取樣量: 1 ~ 3 倍單位劑量,每個點取樣 3 份
無論使用什麼類型的混合容器,取樣都必須有代表性,一般取樣 6-10 個,每個取樣點重複取樣 3 份。

膠囊生產工藝–分料過程
分料過程
如果混合完成後,直接把整個混合容器轉移至膠囊工序,進行膠囊充填,這時候就不存在分料過程。
有的混合容器不能直接轉移至膠囊工序,需要把物料分卸到幾個小型的中轉容器中。由於顆粒或物料的大小,形狀和密度的不同,可能在分離流動或震盪的過程中將粗糙和精細的物料分開,導致物料分離或分層。因此需要對卸料過程進行驗證,證明物料不會分層。
驗證的目的: 評估卸料過程中物料是否分層
驗證方法: 卸料後在中轉容器中取樣
驗證的項目: 混合均勻度
檢測方法: 同混合階段
接受標準: 同混合階段
取樣計劃: 根據中轉容器的構造,設計有代表性的取樣點
取樣時間: 分料階段的各個驗證點
取樣量: 1-3倍單位劑量,每個點取樣3份

膠囊生產工藝–已總混物料存放週期
存放週期
在總混完成後,按照排產情況,進行 “已總混物料存放週期” 驗證。
例如,混合完成後,一般在 7 天內就進行膠囊充填,那麼在進行驗證時,可以進行為期7天或9天的存放週期驗證。
驗證的目的: 確定已總混物料在中間品暫存間正常條件下的最長存放週期
驗證方法: 取驗證批的一部分物料置於車間存放一定的週期
驗證項目: 如性狀、水分、含量、微生物限度等
取樣位置: 表面下有代表性的位置
接受標準: 無結塊,水分、含量、微生物限度符合要求

膠囊生產工藝–膠囊充填過程
膠囊充填過程
在正常的生產中,膠囊充填階段的中間控制 (IPC) 很多,這些中間控制都要在驗證中得到證實。
驗證的目的:
確定膠囊充填過程的工藝參數
關鍵工藝參數:
充填機運行速度 (轉速)
真空度
驗證方法:
按驗證參數進行膠囊充填
驗證項目:
裝量差異,水分,崩解度,溶出度,含量均勻度
接受標準:
按質量標準控制膠囊充填過程中膠囊的質量

膠囊生產工藝–已充填膠囊存放週期
存放週期
在膠囊充填完成後,按照排產情況,對已充填膠囊進行 “存放週期” 驗證。
例如,膠囊充填完成後,一般在 10 天內就進行內包裝,那麼在進行驗證時,可以進行為期10天或14天的存放週期驗證。
驗證的目的: 確定已充填膠囊在中間品暫存間正常條件下的最長存放週期
驗證方法: 取驗證批的一部分物料置於車間存放一定的週期
驗證項目: 如膠囊外觀、水分、崩解度、溶出度、含量均勻度、微生物限度等
取樣位置: 代表性取樣位置
接受標準: 無結塊,崩解度,溶出度、水分、含量、微生物限度符合要求

膠囊生產工藝–藥瓶灌裝 及 鋁箔封口 藥瓶灌裝 (數丸)
驗證的目的: 確定藥瓶灌裝過程的工藝參數
關鍵工藝參數: 灌裝機運行速度電磁感應式鋁箔封口機
震盤震動速度
驗證方法: 按驗證參數進行藥瓶灌裝
驗證項目: 藥瓶裝量差異
接受標準: 按質量標準控制藥瓶灌裝過程中膠囊的
數量
鋁箔封口
通過加熱線圈所產生的電磁場,利用電磁感應原理始通過磁場的物品感磁,在極短的時間內加熱至需要的溫度。
鋁箔能與磁場發生作用,鋁箔表面瞬間產生高熱並傳導至熱封膜,封膜與瓶口上緣接觸的部分迅即溶解並結合到瓶口上,如此達到封瓶的目的
驗證的目的:
確定藥瓶灌裝後,藥瓶通過電磁感應式鋁箔封口機,進行鋁箔封口過程的工藝參數
關鍵工藝參數: 鋁箔封口機磁場強度 運輸帶速度
驗證方法: 按驗證參數進行鋁箔封口
驗證項目: 鋁箔封口密封度
接受標準: 按質量標準控制藥瓶封口過程中,封口密封度

工藝驗證的文件要求
工藝驗證的文件要求
工藝驗證應有驗證方案 (Validation Protocol) 和驗證報告 (Validation Report)
驗證方案:應和驗證程序一致,包括格式、術語和起草 / 審核 / 批准職責,使用統一的範本
驗證報告:要以事實為依據,所有資料可追溯,如果有轉錄的數據,應加以覆核。同樣,驗證報告應由同一支團隊起草、審核及批准

工藝驗證方案
工藝驗證的方案至少包括下列內容:
驗證目的
驗證範圍
職責:明確工藝驗證由誰負責起草、批准、執行和記錄
參考法規和指南
產品和工藝描述:
產品總處方及工藝描述
關鍵工藝步驟及控制程式的總流程圖
關鍵工藝參數 (CPP) 及關鍵品質屬性(CQA)
確定驗證品種連續批號及所使用的起始物料:
批號
單位重量
驗證批量
所使用的起始物料名稱及批號

工藝驗證方案
生產環境確認: 工藝驗證中使用的廠房設施已經過充分的驗證或檢査確認 潔淨車間、空氣淨化系統
設備設施確認: 工藝驗證中使用的儀器儀錶、設施設備、輔助器具已經過充分的驗證或檢査確認 在開始進行工藝驗證測試樣品取樣之前,用於工藝驗證的所有實驗室設備和儀錶均已經過了充分的
確認或校正。 明確表明所有上述的編號及其校正確認列表
參與驗證的人員資格審查與方案培訓情況確認: 參與工藝驗證研究執行的所有扣關部門人員的培訓已經完成
驗證文件確認: 物料名稱、編碼、品質標準和檢驗SOP 關鍵工藝參數的中間控制檢驗、放行檢驗的檢驗方法和可接受標準 驗證使用的批生產紀錄、數據收集的表格和檢驗結果評估的SOP 取樣計劃:所有樣品須有明確的編碼

工藝驗證方案
驗證標準:
在驗證結束後,根據所擬訂的可接受標準 (Acceptance Criteria),對驗證結果進行評價和建議,做出驗證結論。
驗證執行:
必須按照已批准的驗證方案執行;
使用的記錄格式為經過批准的格式;
所記錄的驗證資料 – 應及時、真實、清晰、正確、完整;
必須按照已批准的的取樣計劃及方法;
及時收集編寫驗證報告所需的資料,測試完成後對測試資料進行搜集、整理和匯總生產工藝結束後,應對各樣本進行檢驗,檢驗結果應符合驗證方案內所擬定的可接受標準。
工藝驗證總結:
在驗證結束後,根據所擬訂的評估標準,對驗證結果進行評價和建議,做出驗證結論,及對工藝驗證結果的各步驟進行總結。
偏差報告
偏差處理按方案規定的偏差處理要求填寫偏差報告

工藝驗證報告
工藝驗證報告至少包括下列內容:
驗證的目的和參考的驗證方案
使用的原輔料及其批號
驗證中涉及的廠房、設施設備、儀器儀錶的編號及其校正情況表
所有中間控制、最終檢驗的原始記錄結果詳細匯總,結果須和確定的 “可接受標準” 比較
驗證結論 (接受或不接受的決定及其原因)
所有偏差 (方案偏差、工藝偏差和超限結果OOS) 的簡單描述、評估、糾正、預防措施和結論。任何偏差和分析結果OOS都需要有文字的調查,包括其對工藝和產品質量影響的探討和結論。

工藝驗證標準
驗證標准應嚴於放行標準或取產品品質指標範圍的中間值
對於生產工藝控制參數可取上限或下限值進行驗證,例如:
混 合 均 一 性 的 藥 典 標 準 為 RSD≤6% , 放 行 標 準 相 對 標 準 差 RSD
RSD≤5%,則驗證標準應不大於≤3%。;
溶液的配製溫度為50-60℃,則驗證時分別在50℃和60℃進行。
三個標準的比較:法定(註冊)標準 ≥ 放行 / 內控標準 > 驗證標準

變更和再驗證 工藝再驗證 (Revalidation) / 週期性再驗證 (Periodic Revalidation)
再驗證的目的,是為了保證工藝處於可控狀態,應在下列情況下進行再驗證: 工藝變更:
生產工藝變更、工藝技術參數變更和處方、批量變更(擴大或減小生產批量) 生產環境、設備或廠房變更:
生產設備種類變更 設施、設備位置 (Relocation) 變更 經嚴重維修的設備,應該進行再確認 生產區域或公用系統的變更 生產工藝從一個公司、工廠或建築轉移到其他公司、工廠或建築 設備上相同部件的替換 (Like for Like) 通常不需要進行再驗證,但可能影響產品品質的情況除外 (進行影響性評估)
法規環境變更: 分析方法或產品質量標準變更 法規指引要求變更
影響產品質量的起始物料的變更: 關鍵起始物料生產商的變更 關鍵起始物料的變更(可能影響產品品質的物理性質如密度、黏度或細微性分佈) 包裝材料的變更(例如,塑膠代替玻璃) 重複出現的產品質量問題,導致對驗證的狀態的懷疑
週期性再驗證

工藝驗證常見缺陷與分析 工藝驗證方案
1. 工藝驗證目的與目標不明確
2. 對工藝驗證批的生產、產品放行沒有控制要求
3. 工藝階段沒有界定
4. 每一階段工藝階段沒有設定關鍵參數和目標值範圍
5. 工藝驗證取樣沒有或不夠代表性
6. 驗證方案中未制定驗證過程中採用的紀錄、表格,或表格內容與方案內容不一致
7. 驗證紀錄內容填寫不完整,或者只有結論沒有紀錄支援
8. 缺少部分工序驗證,如方案設計不完整,混合工序只驗證攪拌時間,沒有對攪拌的轉速進行說明,導致結果存在疑問
9. 缺少人員培訓紀錄
10. 方案與報告格式不一致
11. 原始紀錄和原始資料未以檔形式歸檔,導致結果不具可追溯性,缺乏可信度
工藝驗證報告 1. 工藝驗證參數沒有回顧與評價
2. 工藝參數數理沒有統計分析
3. 驗證偏差處理沒有調查、分析、CAPA 處理
4. 驗證資料沒有進行匯總評價

驗證總計劃 (VMP)
管理層知道驗證項目所涉及的時間、人員和資
金
驗證團隊的所有成員知道他們各自的任務和
職責
所有成員能夠完全理解公司進行驗證的方法和進行
所有驗證活動所建立的組織

驗證總計劃 (VMP)
驗證總計劃 VMP
空調淨化 系統
VP
公用設施 VP
生產設備 VP
工藝驗證 VP
清潔驗證 VP
分析方法 VP
潔淨車間 VP
壓縮空氣系統
製藥用水系統

驗證總計劃 (VMP) 驗證總計劃應包括以下內容:
概述
公司的確認和驗證方針,對於驗證總計劃所包含的操作的一般性描述,位置和時間安排(包括優先順序別)等所生產和檢測的產品
各部門的職責和組織結構負責下列工作的部門或人員
驗證總計劃
起草確認和驗證方案、報告
確認和驗證的實施
批准確認和驗證檔
所有廠房、設施、設備、儀器等的清單以及確認的需求
應包含所有廠房、設施、設備、檢驗儀器等,以及對它們是否需確認的評估結論
確認的狀態
下一次再評估或週期性再確認的日期 (計劃)

驗證總計劃 (VMP) 所有工藝過程、分析方法和清潔程式的清單以及驗證的需求
應包含所有生產工藝、分析方法、清潔/消毒/滅菌程式、其他過程(如運輸),以及對它們是否需驗證的評估結論
驗證的狀態
下一次再評估或週期性再驗證的日期 (計劃)
所有電腦化系統的清單以及驗證的需求
應包括所有電腦化系統,以及對它們是否需驗證的評估結論
驗證的狀態
下一次再評估或週期性再驗證的日期 (計劃)
確認和驗證檔的格式:對確認和驗證的方案及報告的格式進行規定
計劃
制定上述確認和驗證活動的計劃,包括時間安排等


![Fujitsu ProcureMART - Interstage HTTP Server 2018 FUJITSU LIMITED 2. 新着情報の確認 [新着・確認済]から新着の対象案件を確認することが出来ます。[新着・確認済]をクリック](https://img.dokumen.tips/doc/110x75/5aff012f7f8b9a434e8fecf4/fujitsu-procuremart-interstage-http-server-2018-fujitsu-limited-2-.jpg)




























