Embed Size (px)
Citation preview

Published Ahead of Print 4 August 2014. 2014, 196(20):3622. DOI: 10.1128/JB.02020-14. J. Bacteriol.
Delumeau, Libor Krásný and Peter J. LewisAndrew N. Keller, Xiao Yang, Jana Wiedermannová, Olivier in Gram-Positive Bacteria, a New Subunit of RNA Polymerase Foundε
http://jb.asm.org/content/196/20/3622Updated information and services can be found at:
These include:
SUPPLEMENTAL MATERIAL Supplemental material
REFERENCEShttp://jb.asm.org/content/196/20/3622#ref-list-1at:
This article cites 55 articles, 23 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on October 10, 2014 by T
he University of M
elbourne Librarieshttp://jb.asm
.org/D
ownloaded from
on O
ctober 10, 2014 by The U
niversity of Melbourne Libraries
http://jb.asm.org/
Dow
nloaded from

ε, a New Subunit of RNA Polymerase Found in Gram-PositiveBacteria
Andrew N. Keller,a Xiao Yang,a Jana Wiedermannová,b Olivier Delumeau,c Libor Krásný,b Peter J. Lewisa
School of Environmental and Life Sciences, University of Newcastle, Callghan, New South Wales, Australiaa; Institute of Microbiology, Academy of Sciences of the CzechRepublic, Prague, Czech Republicb; INRA, UMR-1319 Micalis, Jouy-en-Josas, Francec
RNA polymerase in bacteria is a multisubunit protein complex that is essential for gene expression. We have identified a newsubunit of RNA polymerase present in the high-A�T Firmicutes phylum of Gram-positive bacteria and have named it �. Previ-ously � had been identified as a small protein (�1) that copurified with RNA polymerase. We have solved the structure of � byX-ray crystallography and show that it is not an � subunit. Rather, � bears remarkable similarity to the Gp2 family of phage pro-teins involved in the inhibition of host cell transcription following infection. Deletion of � shows no phenotype and has no effecton the transcriptional profile of the cell. Determination of the location of � within the assembly of RNA polymerase core by sin-gle-particle analysis suggests that it binds toward the downstream side of the DNA binding cleft. Due to the structural similarityof � with Gp2 and the fact they bind similar regions of RNA polymerase, we hypothesize that � may serve a role in protectionfrom phage infection.
Transcription in bacteria is a tightly regulated and cyclic processcarried out by the highly conserved multisubunit enzyme
RNA polymerase (RNAP). The majority of work on bacterialRNAP has been performed in the Gram-negative bacterium Esch-erichia coli. The core E. coli RNAP complex consists of two �subunits and single �, �=, and � subunits (1). However, there aredifferences throughout the eubacteria in the subunit compositionof RNAP. In the model Gram-positive organism Bacillus subtilis,core RNAP has the subunit composition �2��=��1�2 (2). The �subunit is encoded by the gene rpoE and is involved in RNAPpromoter specificity and recycling (2–4). The � subunit appears tobe conserved across the Gram-positive bacteria, and despite a highcellular abundance and documented roles in gene expression, de-letion strains do not exhibit a strong phenotype (3–5). The occur-rence of two � subunits in Bacillus subtilis at approximately 11kDa (�1) and 9 kDa (�2) has been known for some time. The �2
subunit is encoded by rpoZ (synonym, yloH) and is similar to the� subunit present throughout eubacteria (6–8). The �1 subunithas been referred to as the second analogous � subunit simplybecause it has a size similar to that of the true � subunit (9–11).This annotation has been accepted despite there being no evidenceto suggest that these two proteins are related in function. UsingmTRAQ (mass differential tags for relative and absolute quantifi-cation) mass spectrometry, it has been shown that both of the �subunits are approximately equimolar with the � subunit (6). Pu-rification of RNAP shows that both � subunits remain tightlybound to RNAP even after anion-exchange chromatography (8).
We have determined the structure of �1, encoded by the ykzGgene, examined the phenotype generated on its deletion, and de-termined its location on RNAP. The ykzG gene is present in anoperon with the physiologically important RNase RNaseJ1 (RnjA)(12), and the two proteins are transcriptionally and translationallylinked. Surprisingly, there is no apparent linkage of the proteinswithin the cell with respect to any potential intermolecular inter-action, and subcellular localization of fluorescent protein fusionsindicates that they occupy different subcellular compartments.We propose that this small protein previously referred to as �1 isactually a small auxiliary subunit of B. subtilis RNAP, and we sub-
sequently named it ε and the gene encoding it rpoY. Structureanalysis indicated that ε is similar to phage T7 Gp2 which inhibitshost cell transcription through interaction with RNAP. Mutagen-esis studies indicated that both ε and Gp2 interact with RNAP viaa shared structural motif. Due to the lack of phenotype on its loss,location within an RNAP complex, and similarity to phage T7Gp2, we speculate that ε may help protect against phage infectionfrom Gp2-like proteins by occupying their binding sites.
MATERIALS AND METHODSStrains and growth conditions. Strains and plasmids used in this studyare presented in Table 1. Unless otherwise stated, all strains were grown inLB medium with appropriate antibiotics. Transformation of B. subtiliswas based on the methods outlined by Kunst and Rapoport (13). Briefly,B. subtilis strains to be transformed were grown on nutrient agar platesovernight. Colonies were removed from the plates and cultured in NuncF96 microwell cell culture plates containing 80 �l of MD medium (100mM K2HPO4-KH2PO4 at pH 7, 6 mM Na3 citrate, 300 �M MgSO4, 50�g/ml L-tryptophan, 2.5 mg/ml L-aspartate, 11 �g/ml ferric ammoniumcitrate) supplemented with 0.1% (wt/vol) casein hydrolysate at 37°C withshaking until cells entered stationary phase (A600 of 1.2 to 1.5). Eightymicroliters of prewarmed MD medium was added to the stationary-phaseculture, which was incubated for a further 1 h before 0.8 to 1 �g of DNAwas added. The culture was incubated for a further 20 min before 5 �l of20% (wt/vol) casein hydrolysate was added. The culture was incubated at37°C for a further 90 min before being plated on nutrient agar platescontaining appropriate antibiotics for the selection of transformants.
B. subtilis LK921 (Table 1) with a deletion of rpoY was constructed asfollows. The spectinomycin resistance cassette was obtained frompDG1728 (14) by BamHI-EcoRV cleavage and cloned into pGEX-5X-3
Received 23 June 2014 Accepted 31 July 2014
Published ahead of print 4 August 2014
Address correspondence to Peter J. Lewis, [email protected].
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02020-14.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JB.02020-14
3622 jb.asm.org Journal of Bacteriology p. 3622–3632 October 2014 Volume 196 Number 20
on October 10, 2014 by T
he University of M
elbourne Librarieshttp://jb.asm
.org/D
ownloaded from
(cleaved with BamHI-SmaI). Then, a multiple cloning site (HindIII,EcoRI, PstI; created by annealing oligonucleotides 5=-TCGACCGTGCGGGGCTTCAAATAAGAACTTC-3= and 5=-TCGAGAAGTTCTTATTTGAAGCCCCGCACGG-3=) was inserted at the BamHI site. The region up-
stream of rpoY was amplified by PCR with primers 5=-CGGAATTCGCCGTATGTTAGACTCGCTC-3= and 5=-CGGGATCCCTTGATTTTCAAAGACAACAAAC-3= and inserted between the BamHI-EcoRI sites of theplasmid. The region downstream of rpoY was amplified with primers 5=-
TABLE 1 Strains and plasmids used in this study
Strain or plasmid Genotypea Reference or source and/or description
E. coli strainsDH5� �� 80dlacZM15 (lacZYA-argF)U169 recA1 endA1 hsdR17(rK
� mK�)
supE44 thi-1 gyrA relA1Gibco BRL
BL21(DE3) F� ompT hsdSB(rB� mB
�) gal dcm (DE3) 53B834(DE3) F� ompT gal dcm met(lon) hsdSB(rB
� mB�) �DE3 Novagen
B. subtilis strainsBSB1 trpC2 chr::trp 15EU129 BSB1 chr::cat Pwt rpoC-gfp Pspac =rpoC 16EU164 BSB1 chr::erm spc Pspac rpoY-gfp Pxyl rpoC-mCherry cat This work; BSB1 transformed with pNG622 and pEU14LK921 BSB1 chr::spc rpoY This work; rpoY knockout with wild-type rpoY-rnjA
promoterBS489 BSB1 chr::tet rpoY-Pxyl-rnjA This work; Tetr cassette inserted by double crossover
resulting in deletion of rpoYBS491 BSB1 chr::erm Pwt rpoC-gfp Pspac =rpoC chr::rpoY tet Pxyl rnjA This work; BS489 transformed with pEU21EU337 BSB1 chr::rpoY tet-Pxyl-rnjA amyE::spc Pxyl rpoY-gfp mut1 This work; BS489 transformed with pNG1030EU344 BSB1 chr::rpoY tet-Pxyl-rnjA amyE::spc Pxyl rpoYF46A,T46A-gfp mut1 This work; BS489 transformed with pNG1037EU345 BSB1 chr::rpoY tet-Pxyl-rnjA amyE::spc Pxyl rpoY41–50A-gfp mut1 This work; BS489 transformed with pNG1039EU346 BSB1 chr::rpoY tet-Pxyl-rnjA amyE::spc Pxyl rpoY61–69A-gfp mut1 This work; BS489 transformed with pNG1040EU347 BSB1 chr::rpoY tet-Pxyl-rnjA amyE::spc Pxyl rpoYE34A-gfp mut1 This work; BS489 transformed with pNG1036BS509 trpC2 spo0A3 rpoC-His6 spc Pspac rpoY-gfp cam This work; BS200 (34) transformed with pEU14
PlasmidspETMCSIII bla P10-His6-T 54pGEX-5X-3 bla Ptac-GST GE HealthcarepSG1154 bla amyE3= spc Pxyl-=gfp mut1 amyE5= 55pDG1728 bla erm amyE5= spoVG-lacZ spc amyE3= 14pEU14 bla Pspac-=rpoY-gfp mut3-erm G. Doherty, University of Newcastle, unpublished datapEU21 bla Pspac-=rpoC-gfp mut3-erm 17pNG579 bla P10-rpoY-His9-T This work; PCR product of B. subtilis rpoY inserted into
NdeI and BamHI sites of pETMCSIIIpNG622 bla cat Pxyl-mCherry 17pNG689 bla P10-His6-rpoY=-T This work; G. stearothermophilus rpoY PCR product
containing a 9-aa N-terminal truncation ligated intoNdeI- and EcoRI-digested pETMCSIII
pNG729 bla P10-His6-rpoY=Leu23Met-T This work; G. stearothermophilus rpoY with Leu23mutated to Met by overlap extension PCR and a 9-aatruncation at C-terminus, ligated into NdeI- andEcoRI-digested pETMCSIII
pNG1030 bla amyE3= spc Pxyl-rpoY-gfp mut1 amyE5= This work; B. subtilis rpoY PCR product ligated intoKpnI- and EcoRI-digested pSG1154
pNG1036 bla P10-rpoYE34A-His6 T This work; B. subtilis rpoY overlap extension PCR productligated into KpnI- and EcoRI-digested pSG1154
pNG1037 bla amyE3= spc Pxyl-rpoYF46A,T48A-gfp mut1 amyE5= This work; B. subtilis rpoY overlap extension PCR productligated into KpnI- and EcoRI-digested pSG1154
pNG1039 bla amyE3= spc Pxyl-rpoY41–50A-gfp mut1 amyE5= This work; B. subtilis rpoY overlap extension PCR productamplified from pNG984 and ligated into KpnI- andEcoRI-digested pSG1154
pNG1040 bla amyE3= spc Pxyl-rpoY61–69A-gfp mut1 amyE5= This work; B. subtilis rpoY overlap extension PCR productamplified from pNG985 and ligated into KpnI- andEcoRI-digested pSG1154
pNG1041 bla P10-His6-rpoY41–50A-T This work; B. subtilis rpoY PCR product amplified frompNG984 ligated into NdeI- and EcoRI-digestedpETMCSIII
a bla, ampicillin resistance; Pwt, wild-type promoter; Pspac, IPTG-inducible promoter; Pxyl, xylose-inducible promoter; erm, erythromycin resistance; cat, chloramphenicolresistance; spc, spectinomycin resistance; GST, glutathione S-transferase. Gene subscripts using amino acids should be interpreted according to the following examples:rpoY=Leu23Met, the Leu-to-Met change at position 23 encoded by rpoY; rpoY41–50A, mutation of amino acids 41 to 50 to alanines encoded by the rpoY gene.
New Subunit of RNAP
October 2014 Volume 196 Number 20 jb.asm.org 3623
on October 10, 2014 by T
he University of M
elbourne Librarieshttp://jb.asm
.org/D
ownloaded from
CCGCTCGAGTTAAGGAGGATTTTAGAATGAAATTTGTAAAAAATG-3=(forward) and 5=-AAGGAAAAAAGCGGCCGCGATTTTCACTGTTTGTGCTG-3= (reverse) and inserted into the plasmid making use of theXhoI-NotI restriction sites. The forward primer (above) contained a shortsequence with a ribosome-binding site (from the tuf gene) to ensureproper translation of the downstream gene (rnjA) encoding RNAseJ1. Thewhole promoter region of the rpoY-rnjA operon was kept unchanged. Theresulting construct was named LK659. B. subtilis BSB1 (15) (Table 1) wastransformed with LK659 DNA and selected for spectinomycin resistanceto give B. subtilis LK921 (Table 1). The deletion was verified by PCR.
Wild-type B. subtilis rpoY was amplified using primers 5=-CACACACATATGATTTAT-3= and 5=-GAGGAAGAATTCTAACTCCAA-3=, di-gested with NdeI and BamHI, and inserted into pETMCSIII to givepNG579 (Table 1). Mutagenized B. subtilis rpoY with alanine substitu-tions was generated as follows. In the first step, the rpoY gene was ampli-fied by PCR (Expand; Roche) in two parts (separated by 10-amino-acid[aa] gaps that were subsequently substituted with alanines). Primers 5=-GGGCCGGCATATGATTTATAAGGTATTT-3= (forward) and 5=-CCGCTCGAGTAACTCCAATACTTTAAA-3= (reverse) were used in combina-tion with primers that were inside the rpoY gene. Internal primerscontained 30 nucleotides encoding 10 alanines to mutagenize the 10 re-spective amino acids at their 5= ends. In the second step, the two PCRproducts were mixed in 1�PCR buffer and 25 mM MgCl2, denatured,and, by decreasing the temperature by 1°C/min (95°C to 35°C), annealedvia the polyalanine region. In the third step, DNA polymerase (Expand;Roche) and deoxynucleoside triphosphates (dNTPs; 10 mM each) wereadded to create double-stranded DNA (dsDNA). The final product wasamplified by PCR with the primers 5=-CACACACATATGATTTAT-3=and 5=-GAGGAAGAATTCTAACTCCAA-3= and inserted into the NdeI/BamHI sites of pETMCSIII. Wild-type and mutant sequences were sub-sequently subcloned into pSG1154 (Table 1) prior to transformation intoB. subtilis. All clones were confirmed by DNA sequencing.
For the comparison of growth rates, strains were grown in 200 �l of LBmedium (minimum of five replicates for each strain) in Nunc F96 micro-well cell culture plates with shaking at 100 rpm in a PHERAstar FS (BMGLabtech) at 37°C. Cell density was quantified by measuring the A600 every10 to 12 min for 8 h. Maximum growth rate (doubling time) was deter-mined from the steepest part of the slope during exponential growth.
Fluorescence microscopy. Cells were grown and imaged as previouslydescribed (16, 17).
Phylogenetic work. All phylogenetic work was performed using in-formation obtained from the National Centre for Biotechnology Infor-mation databases (http://www.ncbi.nlm.nih.gov/). Alignments and phy-logenetic analysis were carried out using the Clustal W2 and MAFFTalgorithms (18, 19).
Protein overproduction and purification. Geobacillus stearothermo-philus rpoY (εGs) with a 9-amino-acid C-terminal truncation was ampli-fied from isolated genomic DNA using the primers 5=-GATCGGCATATGATTTTCAAAGTGTTTTACCA-3= and 5=-ATCGATGAATTCTCATGATGAAATCTCCAATA-3=, digested with NdeI and BamHI, and insertedinto pETMCSIII to give pNG689 (Table 1). In order to produce a proteinsuitable for structure solution by multiwavelength anomalous dispersion(MAD) through incorporation of Se-Met, the truncated rpoY gene frompNG689 was mutated using the primers 5=-CTTCGATGTACATTGTTTTCGTTTTTTC-3= and 5=-GAAAAAACGAAAACAATGTACATCGAAG-3= by overlap extension PCR (20), resulting in the replacement ofcodon Leu 23 with Met to give pNG729 (Table 1).
For the overproduction of εGs, pNG729 was transformed into the me-thionine auxotroph E. coli B834(DE3) (Table 1) and cultured in minimalM9 medium with amino acids and vitamin supplements as describedpreviously (21). Cells were grown to an A600 of 0.5 to 0.7, and proteinoverproduction was induced by the addition of isopropyl-�-D-thiogalac-topyranoside (IPTG) to 0.5 mM, followed by overnight culture at 18°C.Cells were harvested by centrifugation and stored at �80°C.
Cell pellets were resuspended in imidazole buffer A (20 mM KH2PO4,
pH 8.0, 500 mM NaCl, 20 mM imidazole), and cells were lysed by passagethrough an Avestin EmulsiFlex C5 homogenizer, followed by sonication.Following clarification by centrifugation, the lysate was then loaded ontoa 1-ml HisTrap HP column (GE Healthcare). Bound protein was washedwith 5 ml of imidazole buffer A buffer before being eluted with imidazolebuffer B (20 mM KH2PO4, pH 8.0, 500 mM NaCl, 500 mM imidazole) in0.5-ml fractions. If necessary, proteins were further purified by size exclu-sion chromatography using a HiLoad Superdex 75 16/60GL column (GEHealthcare) equilibrated with 20 mM Tris-HCl, pH 8.0, and 150 mMNaCl. Protein purity and yield were checked by SDS-PAGE. Fractionsshowing high purity and concentration were pooled and dialyzed into 20mM HEPES, pH 7.0, 150 mM NaCl, and 1 mM dithiothreitol (DTT).
RNAP with green fluorescent protein (GFP)-tagged ε for use in single-particle analysis was purified from B. subtilis BS509 (Table 1) as previouslydescribed (8). The purified sample was dialyzed in 20 mM Tris-HCl, pH7.8, 150 mM NaCl, 10 mM MgCl2, 5% (vol/vol) glycerol, and 1 mM DTT.
Protein crystallography and structure determination. Crystalliza-tion screening was carried out at 27°C by sitting-drop vapor diffusionusing MRC 96-well crystallization plates and PACT, JCSG, and structurescreens (Molecular Dimensions). εGs crystals grew in 100 mM sodiumacetate, pH 4.6, and 1.8 M ammonium sulfate.
Crystals were cryo-protected by replacing the lower well with 3.5 M
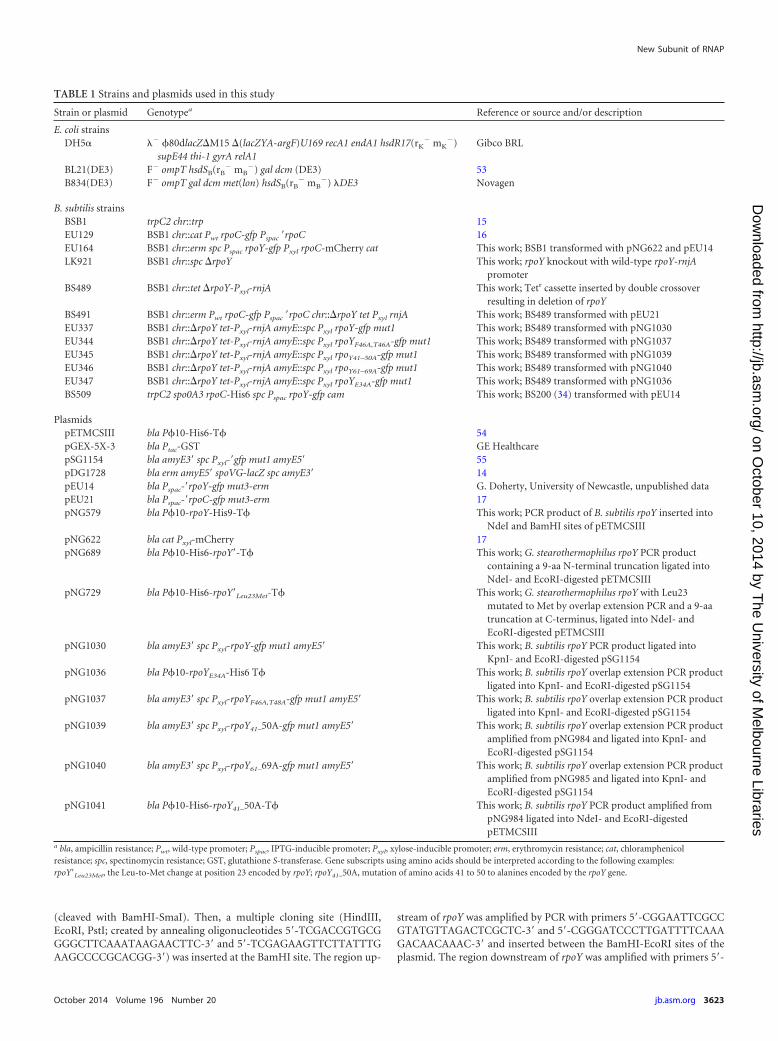
FIG 1 Colocalization of ε (YkzG) and RNAP. (A) Pseudocolored image ofDAPI-stained exponentially growing B. subtilis EU164 (rpoY-gfp rpoC-mCherry) to label nucleoid location and morphology. (B) Pseudocolored im-age of the RpoC-mCherry fusion illustration of the subcellular location ofRNAP. (C) Pseudocolored image of the ε (YkzG)-GFP fusion. (D) Overlay ofthe three fluorescence images shown in panels A to C. (E) Pseudocoloredimage of DAPI-stained nucleoids in B. subtilis BS491 (rpoC-gfp rpoY). (F)Pseudocolored image of the RpoC-GFP fusion. (G) Overlay of images frompanels E and F. Arrows indicate a transcription focus which represents the siteof rRNA synthesis, and a cartoon of a cell containing two nucleoids is shown inpanels D and G as a reference. Scale bar, 3 �m.
Keller et al.
3624 jb.asm.org Journal of Bacteriology
on October 10, 2014 by T
he University of M
elbourne Librarieshttp://jb.asm
.org/D
ownloaded from
ammonium sulfate and allowing the crystallization drop to equilibrateovernight. Crystals were mounted in cryo-loops and drop frozen in liquidnitrogen. A two-wavelength MAD data set was collected from a singlecrystal on the MX2 beamline at the Australian synchrotron using theBlu-Ice software (22). The collection wavelengths for peak anomaloussignal and a high-energy remote were determined from the anomalous f=and f� plots, which were calculated using CHOOCH (23). Reflections usedfor refinement were collected in-house at the University of New South
Wales (UNSW; Sydney, Australia), using a MAR345dtb image plate de-tector mounted on a Rigaku MicroMax HF007 rotating anode generatorwith Osmic confocal mirrors using Cu Ka radiation.
Diffraction data were indexed and integrated using iMOSFLM (24)and scaled in Scala (25). After integration, the space group was deter-mined to be P1 21 1 with the cell dimensions of 35.6 Å, 82.81 Å, 83.41 Å,90°, 92.7°, and 90°, and structure determination was performed using theCCP4 software suite (26). The calculation of phases and initial modelbuilding were achieved using the Crank automated structure solutionpipeline (27), which used Afro/Crunch2 (28) for substructure detection,Solomon (29) for hand determination and density modification, and,finally, Buccaneer (30) for model building. The initial model was thenextended using ARP/wARP and continued manually using COOT (31,32). Refinement was carried out using phenix.refine in conjunction withCOOT. The final model was assessed using the Phenix comprehensivevalidation tool (31, 33).
Electron microscopy and 3D reconstruction by single-particle anal-ysis. The purified RNAP-ε-GFP sample was diluted to 0.08 �M in 20 mMTris-HCl, pH 7.8, 150 mM NaCl, 10 mM MgCl2, and 1 mM DTT. Fourmicroliters of sample was applied to homemade continuous carbon gridsfor 30 s before being washed three times with the dilution buffer. Thespecimens were then stained with a 1% (wt/vol) uranyl formate aqueoussolution. The imaging conditions and particle selection were performed asdetailed by Yang et al. (34). A total of 260 images were taken, and 10,104particles were used in three-dimensional (3D) reconstructions.
The EMAN software package, version 1.8 (35), was used for imageprocessing. Individual particles were appended, center aligned, low-passfiltered to 15 Å, and boxed into 72- by 72-pixel images. The previouslypublished RNAP core negative stain electron density map (EMDataBankEMD-1577) (34) was filtered to 30 Å and employed as the initial model forrefinement. After 10 rounds of iteration, the model stably converged to 24-Å resolution as estimated with the EMAN e/o test.
Transcriptomics. B. subtilis BSB1 and LK921 were grown to A600 0.7in CH medium (36) and triplicate samples processed and analyzed asdetailed in (15).
Accession numbers. The final refined structure of ε was deposited inthe Protein Data Bank (PDB; http://www.pdb.org/pdb/home/home.do)under accession number 4NJC. The negatively stained molecular RNAPenvelope has been deposited in the EMDataBank (EMDB; http://www.emdatabank.org) under accession number EMD-2637.
RESULTS� (YkzG) is a core subunit of RNAP. In previous studies we con-firmed that the identity of the low-molecular-weight band copu-rifying with RNAP from B. subtilis that had been called �1 wasencoded by the ykzG gene (8). Due to the fact that YkzG could notbe separated from other core RNAP subunits using nondenatur-ing chromatography, we suggested that it was a subunit of theenzyme. These results were subsequently confirmed by Delumeau
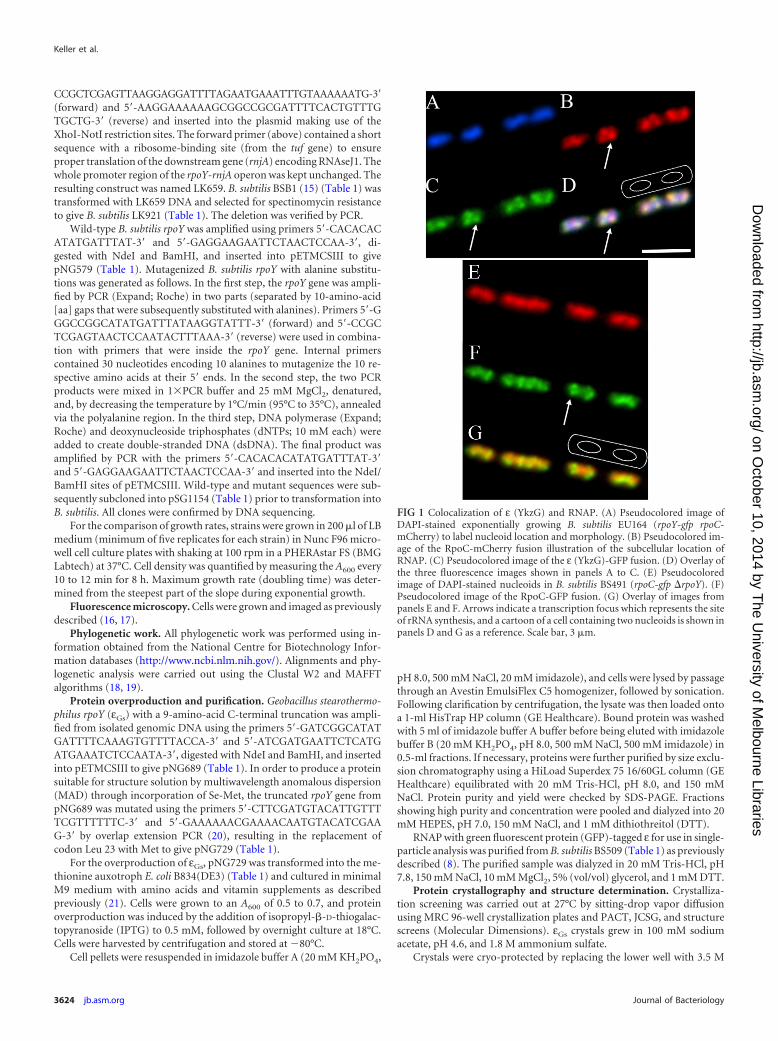
FIG 2 Transcription analysis of rpoY. (A) Levels of the rpoY-rnjA (ykzG-rnjA)transcript under 103 different growth conditions (15) taken from the B. subtilisExpression Data Browser (http://genome.jouy.inra.fr/cgi-bin/seb/index.py).A map of the region is shown below, along with an expanded portion showingthe location of the ribosome binding site (SD) for rnjA located at the end of theykzG (rpoY) gene. (B) Transcriptomics data for the rpoY-rnjA (ykzG-rnjA)region in wild-type (BSB1; black) and rpoY (LK921; magenta) strains. Topsection, coding strand; bottom, noncoding strand (with respect to rpoY-rnjA).The location and direction of transcription of genes are indicated in the middlesection.
TABLE 2 Gene transcript changes in the rpoY-deficient strain LK921
Expression groupand gene name Locus tag Description
Fold change inexpression (log2) P valuea
Increased expressionyhfH BSU10230 Hypothetical protein 1.53 0.036nadB BSU27870 L-Aspartate oxidase 1.00 0.008
Decreased expressionnarG BSU37280 Nitrate reductase (alpha subunit) �0.99 0.040adeC BSU14520 Adenine deaminase �1.12 0.019trpF BSU22650 Phosphoribosyl anthranilate isomerase �2.52 0.001rpoY BSU14540 RNAP ε subunit �8.13 0.006
a Two-tailed unequal variance Student’s t test.
New Subunit of RNAP
October 2014 Volume 196 Number 20 jb.asm.org 3625
on October 10, 2014 by T
he University of M
elbourne Librarieshttp://jb.asm
.org/D
ownloaded from

et al. (6), who isolated transcription complexes from B. subtiliscultures and showed that it was present at similar levels to RNAPcore subunits under a range of different growth conditions.
If YkzG is a subunit of core RNAP, it would be expected tocolocalize with it in the cell. In order to determine if this was thecase, we examined the localization patterns of fluorescent proteinfusions of YkzG and RNAP. Strain EU164 carrying chromosomalfusions of GFP to YkzG and mCherry to RpoC (the �= subunit ofRNAP) was grown to mid-exponential phase in LB medium, andfluorescence was observed as detailed in Materials and Methods.Due to the choice of fluorescent protein fusions used in this study,nucleoids could also be counterstained with the DNA stain 4=,6-diamidino-2-phenylindole (DAPI) so that fluorescent protein lo-calization could be correlated with nucleoid morphology as inprevious work (37).
As expected, both YkzG (Fig. 1C) and RNAP (Fig. 1B) colocal-
ized with the nucleoid (Fig. 1A and D). The arrows in Fig. 1 indi-cate the presence of a transcription focus which forms due to thehigh level of loading of RNAP onto rRNA operons in rapidlygrowing cells (36, 37). Since YkzG-GFP fluorescence exactly mir-rored that of the RpoC-mCherry fusion, this indicates that YkzG isassociated with RNAP involved in all classes of transcription(mRNA and stable RNA synthesis), further strengthening the hy-pothesis that it is an integral subunit of RNAP in B. subtilis. BLASTsearches indicated that YkzG was confined to the phylum Firmic-utes (see Fig. S1 in the supplemental material). There was also nosignificant sequence similarity between the true � subunit en-coded by yloH (rpoZ) and ykzG. Therefore, YkzG represents a newsubunit of RNAP and has been given the name ε, and the geneencoding it has been named rpoY. Core RNAP from B. subtilis andother firmicutes comprises at least seven subunits and should berepresented as �2��=��ε.
TABLE 3 Data collection and refinement statistics for the εGs crystal structure
Statistic In-house dataa Synchrotron dataa
Data collectionSpace group P 1 21 1 P 1 21 1Cell dimensions
a, b, c (Å) 35.65, 82.78, 83.43 36.54, 83.21, 83.43�, �, � (°) 90, 92.76, 90 90, 93.28, 90
Peak RemoteWavelength 1.54179 0.97941 0.95369Resolution (Å) 25.0–2.3 (2.42–2.3) 41.65–2.31 (2.44–2.31) 41.33–2.3 (2.42–2.3)Observations 158,061 (22,673) 138,779 (22,137) 139,240 (22,352)No. of unique reflections 21,590 (3,136) 19,757 (3,096) 19,727 (22,352)Rmerg 0.056 (0.237) 0.097 (0.749) 0.076 (0.489)Rmeas 0.060 (0.255) 0.135 (0.856) 0.098 (0.566)Mean I/�(I) 18.4 (5.9) 11.8 (2.9) 14.2 (3.6)Completeness (%) 99.92 (100.00) 90.1 (96.8) 91.5 (97.5)Multiplicity 7.3 (7.2) 7.0 (7.2) 7.1 (7.2)Anomalous completeness (%) 90.3 (97.0) 90.9 (97.0)Anomalous multiplicity (%) 3.6 (3.6) 3.6 (3.7)Wilson B-factors 46.36 35 34.2
RefinementResolution (Å) 23.07–2.30 (2.38–2.30)No. of reflections 21,575 (2,185)Rwork 0.214 (0.267)Rfree 0.256 (0.314)CC*b 0.819 (0.648)CCwork 0.890 (0.903)CCfree 0.873 (0.791)No. of atoms 6,955
Protein 3,506Ligand/ionWater 37
B-factorsOverall 61.7Protein 61.9Ligand/ionWater 42.1
RMS deviationsBond lengths (Å) 0.004Bond angles (°) 0.72
Ramachandran favored (%) 97Ramachandran outliers (%) 0.53
a Values for the highest-resolution shell are shown in parentheses.b CC*, true correlation coefficient, as defined by the phenix tool.
Keller et al.
3626 jb.asm.org Journal of Bacteriology
on October 10, 2014 by T
he University of M
elbourne Librarieshttp://jb.asm
.org/D
ownloaded from
Examination of sequence and transcriptome data revealed thatthe B. subtilis rpoY gene is located in a two-gene operon withanother gene called rnjA (15). The two genes are always cotrans-cribed at identical levels and are translationally linked (Fig. 2A).The gene rnjA encodes the RNase RNaseJ1 that is responsible forthe maturation of the 5= end of 16S rRNA and is crucial for properribosome assembly (38). Exhaustive searches of available anno-tated bacterial genomes in the NCBI failed to identify a case wherethe rpoY gene was not directly followed by rnjA although rnjA isdistributed more widely than rpoY and so is often present in itsabsence. The possibility that the close correlation of the rpoY andrnjA genes was related to the function of the translated proteinswas examined. Previously published work examining the localiza-tion of an RNaseJ1-GFP fusion showed that it is the same as ribo-somes, concentrating toward the poles of the cell and being absentfrom or in low concentration in the region occupied by the nucle-oid (39). This is a completely different localization pattern than weobserved for ε (Fig. 1), indicating that the two proteins occupydifferent subcellular regions and are unlikely to directly/stably in-teract with each other. In vitro studies also failed to identify anyinteraction between the two proteins (data not shown).
Phenotypic characterization of �. The rpoY gene was deletedfrom the B. subtilis chromosome to observe any phenotype asso-ciated with ε depletion in the cell. The rpoY gene was removedfrom BSB1 and replaced with a spectinomycin resistance gene togive strain LK921 (Materials and Methods), and the cellular levelsof RNaseJ1 were shown to be identical to those of the parent strainby quantitative Western blotting (C. Condon, personal commu-nication). Strain LK921 grew at growth rates identical to those ofthe wild type when cultured in a range of liquid media, and thecells appeared morphologically identical to the wild-type cellswhen examined by phase-contrast microscopy (data not shown).Competition growth experiments between LK921 and the wildtype showed that rpoY deletion did not detectably affect the fitnessof the knockout. Examination of knockout strain BS491 contain-ing an rpoC-gfp fusion indicated that deletion of ε had no visibleeffect on RNAP localization within the cell, with transcription fociclearly visible (Fig. 1F, arrow) and all RNAP fluorescence colocal-izing with nucleoid signal (Fig. 1E to G). Thus, ε does not appearto have a major influence on general bacterial growth, fitness, orRNAP distribution within the nucleoid.
To determine if the lack of ε in the cell caused changes in thetranscription profile of the cell, transcriptomics were carried outcomparing exponentially growing strain LK921 and wild-typeBSB1 cells (Table 1) as described in Materials and Methods. SincerpoY is expressed at high levels similar to other RNAP subunitsunder over 100 different growth conditions (15), transcriptomicswere performed on mid-exponentially growing cells grown in thedefined rich CH medium (36). Remarkably, no significant differ-ence in the transcription profiles between the two strains was ob-served, other than the loss of expression of rpoY (Fig. 2B). Thesmall number of genes showing a log2 change of �1.0 and a P of�0.05 are listed in Table 2. Of the two genes showing increasedlevels of expression, yhfH encodes a hypothetical protein, whereasnadB is the first gene in the nadBCA operon involved in NADbiosynthesis. Of the genes showing reduced levels of expression,narG is part of the narGHJI operon encoding nitrate reductase,and trpF is part of the trpEDCFBA operon involved in tryptophanbiosynthesis. None of the other genes in the trp operon showedaltered transcript levels between the wild type and knockout, and
previous studies have shown that trpF exhibits highly variabletranscript levels (15). Interestingly, adeC transcript levels werealso reduced moderately. This gene lies directly downstream fromthe rpoY-rnjA operon and is transcribed in the opposite direction.The reduced level of expression is not due to lack of termination oftranscription of rnjA in the knockout (Fig. 2B) and was observedin only two of the three transcriptome samples (Fig. 2B, magentalines), suggesting that it is unlikely to be highly significant withrespect to ε function.
Finally, we conducted a series of in vitro transcription assaysthat included multiple rounds of transcription (see the supple-mental material), promoter binding, open complex formation
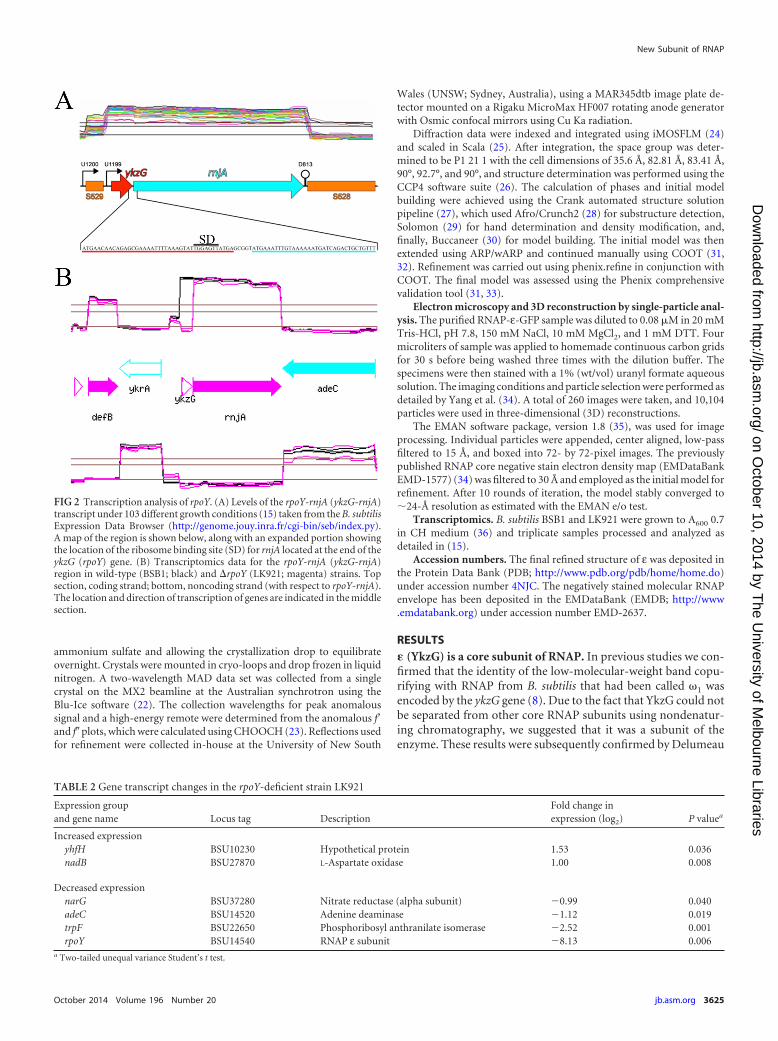
FIG 3 Structure of ε. (A) Electron density as a mesh focused on the loopbetween �1 and �2 from chain A of the asymmetric unit into which the loopwas built. (B) Cartoon representation of the structure of ε (PDB 4NJC) deter-mined by X-ray crystallography to a resolution of 2.3Å. (C) Structure of phageT7 protein Gp2 (PDB 2LMC, chain A) on the left side. The electrostatic po-tentials of ε and Gp2, scaled between 3 kT/e (blue) and �3 kT/e (red) (with kthe Boltzmann constant, T the temperature, and e the elementary charge), areshown mapped onto the protein surfaces on the right. Images were createdusing PyMol (version 1.3; Schrödinger, LLC) using the APBS plug-in (52).
New Subunit of RNAP
October 2014 Volume 196 Number 20 jb.asm.org 3627
on October 10, 2014 by T
he University of M
elbourne Librarieshttp://jb.asm
.org/D
ownloaded from
and decay, promoter escape, and sensitivity of RNAP to the con-centration of the initiating NTP (which also indicates the effectson the stability of open complexes). No effect by ε, either positiveor negative, in any of these assays could be detected. Overall, weconclude that ε has little effect on global transcript levels in vivo oron transcription initiation in vitro.
Determination of the structure of �. In a further attempt toestablish a functional role for ε, we undertook studies to deter-mine its structure by X-ray crystallography. In order to increasesolubility, a truncation of 9 amino acids at the carboxy-terminalend of the Geobacillus stearothermophilus protein was used in crys-tallographic studies (see Materials and Methods). In order to ob-tain experimental phases, leucine 23 was mutated to methioninein order to enable the production of seleno-methonine-substi-tuted protein (Materials and Methods) suitable for the generationof experimental phases that would allow structure solution bymultiwavelength anomalous dispersion (MAD) (Table 3), andthis protein was subsequently referred to as εGs. The asymmetricunit contained eight monomers, and initial model buildingproved difficult due to poorly defined density between the �1 and�2 strands in all but one of the monomers. However, one of themonomers (chain A) did contain a limited amount of density intowhich the backbone could be confidently built (Fig. 3A). Thisdensity was of insufficient quality to permit building and refine-ment of side chains for residues Asp19, Glu20, Arg24, and Asp25,
and so these side chains were assigned common rotamers; thepoor density fit is reflected in the inflated B-factors of these resi-dues. The resulting structure was a simple � sheet with an � helixrunning diagonally along the back of the structure forming a���� fold (Fig. 3B).
Searches for structural homologues using PDBeFOLD (http://www.ebi.ac.uk/msd-srv/ssm/ssmstart.html) found four struc-tures with a fold similar to that of εGs but no common functionalassignment. These four structures were the Gram-negative T7phage protein Gp2 (PDB 2WNM) (Fig. 3C), PaaB from Ralstoniaeutropha (PDB 3EGR), archaeal ribosomal protein LX (PDB3J21), and p56 from the Gram-positive bacteriophage 29 (PDB3ZOQ). PaaB is a small protein with unknown function in thephenyl-acetate catabolism pathway and has not been character-ized or linked directly to a cellular function. The LX protein is partof the 50S ribosomal subunit from Haloarcula marismortui, whereit interacts nonspecifically with the 23S rRNA (40). p56 is an in-hibitor of uracil DNA glycosylase that prevents host cell uracilexcision repair (41, 42).
Gp2, which had the highest structural similarity to εGs (rootmean square deviation [RMSD] of 1.85) (Fig. 3C), was of partic-ular interest as it is an RNAP binding protein that inhibits theformation of open complexes and initiation of transcription (43,44). Gp2 binds to a region of RNAP in the DNA binding cleftcalled the jaw (45, 46) and inhibits transcription initiation by pre-
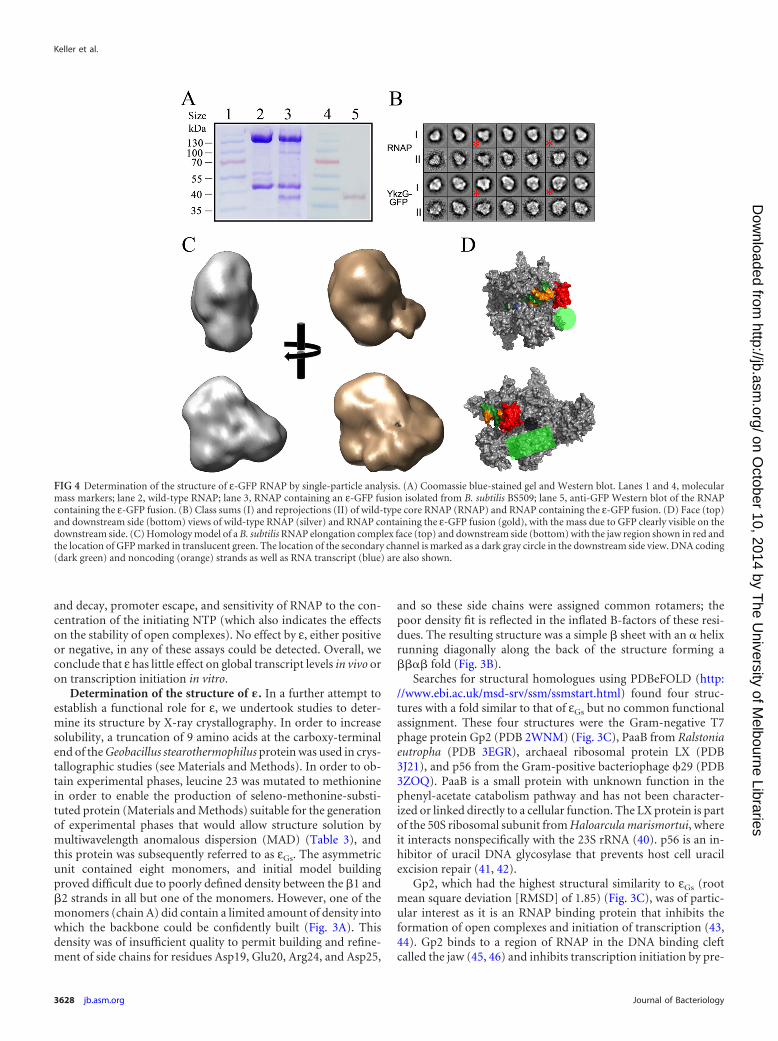
FIG 4 Determination of the structure of ε-GFP RNAP by single-particle analysis. (A) Coomassie blue-stained gel and Western blot. Lanes 1 and 4, molecularmass markers; lane 2, wild-type RNAP; lane 3, RNAP containing an ε-GFP fusion isolated from B. subtilis BS509; lane 5, anti-GFP Western blot of the RNAPcontaining the ε-GFP fusion. (B) Class sums (I) and reprojections (II) of wild-type core RNAP (RNAP) and RNAP containing the ε-GFP fusion. (D) Face (top)and downstream side (bottom) views of wild-type RNAP (silver) and RNAP containing the ε-GFP fusion (gold), with the mass due to GFP clearly visible on thedownstream side. (C) Homology model of a B. subtilis RNAP elongation complex face (top) and downstream side (bottom) with the jaw region shown in red andthe location of GFP marked in translucent green. The location of the secondary channel is marked as a dark gray circle in the downstream side view. DNA coding(dark green) and noncoding (orange) strands as well as RNA transcript (blue) are also shown.
Keller et al.
3628 jb.asm.org Journal of Bacteriology
on October 10, 2014 by T
he University of M
elbourne Librarieshttp://jb.asm
.org/D
ownloaded from
venting region 1.1 of �70 moving out of the DNA binding cleft toallow promoter DNA entry (45). The interaction between Gp2and RNAP is predominantly made between the �3 strand and theregion of amino acids 1045 to 1189 of �= (46). We also examinedthe electrostatic potential on the surface of ε and Gp2 as Gp2contains a negatively charged strip that is functionally importantin the inhibition of host-cell transcription (47). While the nega-tively charged strip was clearly visible in Gp2 (Fig. 3C, right-handside red surface), no such charge distribution was visible on thesurface of ε (Fig. 3B, right-hand side), supporting the conclusionsof the in vitro transcription assays that, unlike Gp2, ε does notinhibit transcription initiation.
Investigation of the � binding site. Due to the structural sim-ilarity of ε with Gp2, we considered the possibility that ε bound toa similar region of RNAP. A deletion of the jaw region of B. subtilisRNAP (�= subunit amino acids 963 to 1004) equivalent to thatfrom the E. coli enzyme (amino acids 1045 to 1198) (44) wasconstructed. Simultaneously, the same deletion was made in plas-mid pNG567 that is used for overproduction of recombinant B.subtilis RNAP containing ε (8). Despite multiple attempts, notransformants of jawless GFP-tagged RNAP could be obtained,and no soluble recombinant protein could be obtained, prevent-ing us from determining by either microscopic analysis of live cellsor mass spectrophotometric analysis of purified recombinant pro-tein (data not shown) whether ε binds RNAP in the absence of thejaw region.
As an alternative, we utilized negative-stain electron micros-copy with single-particle analysis (34) to generate a low-resolution3D structure in order to determine the approximate location of εon RNAP. Due to the small size of ε ( 8 kDa), it was not possibleto directly visualize ε on the core complex. To overcome the rela-tive size problem, strain BS509 (Table 1) was created which con-tained rpoC-His6 and rpoY-gfp fusions. The relatively large size ofGFP ( 27 kDa) would allow us to assign density to the approxi-mate region of RNAP to which ε binds. Coomassie blue-stainedgels and Western blotting with anti-GFP antibodies were used toshow that the ε-GFP fusion was present in the RNAP preparation(Fig. 4A, lanes 3 and 5). A total of 10,104 particles were used for a3D reconstruction of the GFP-tagged complex (Materials andMethods). Class sums (I) and reprojections (II) of RNAP core andthe purified RNAP ε-GFP complex are shown in Fig. 4B, and classsums that allowed identification of mass due to GFP are indicatedby an asterisk. The final reconstruction converged at a resolutionof 24Å, and the mass due to GFP can be clearly seen on thedownstream side of the enzyme compared with the equivalentRNAP core structure (Fig. 4C and D). The mass due to GFP (Fig.4D, green circle and rod) was close to the jaw (Fig. 4D, red) and thesecondary channel (Fig. 4D, dark gray circle). While this recon-struction does not permit unequivocal determination of the loca-tion of ε, it is consistent with ε being able to bind on the down-stream side of the DNA binding cleft in a similar region to Gp2(see below).
Since our results indicate that ε could bind to a similar regionof RNAP to Gp2, we explored the possibility that similar regions ofthe proteins were required for interaction with RNAP. Previousstudies have shown that E28, D37, E44, and, in particular, R56 andR58 are required for interaction of Gp2 with the �= jaw of E. coliRNAP (PDB 2LMC) (46, 48). There is no significant sequenceconservation between Gp2 and ε, so amino acids in (approxi-mately) equivalent positions were chosen for mutation. We mon-
itored the localization of ε-GFP fusions within live B. subtilis cellsto determine which regions/amino acids were important for in-teraction with RNAP (Materials and Methods). ε residue F46 andε T48 are in roughly equivalent positions to the Gp2 R56 and R58
FIG 5 Determination of the region of ε that interacts with RNAP. (A and B)Structures of Gp2 and εGs, respectively, with amino acids that have been mu-tated or shown to be involved in the interaction with RNAP highlighted in red.The region of ε mutated to 10 consecutive alanines (residues 41 to 50) is shownin purple. (C) Sequence and structural alignments of Gp2 and ε. Gp2 residuesinvolved in interaction with Escherichia coli RNAP are shown in green. ε resi-dues that were altered are represented the same as in panel B. �, alpha helix; �,� strand. (D to H) Localization of wild-type, εF46A,T48A (E), εR34A (F), ε41–50A
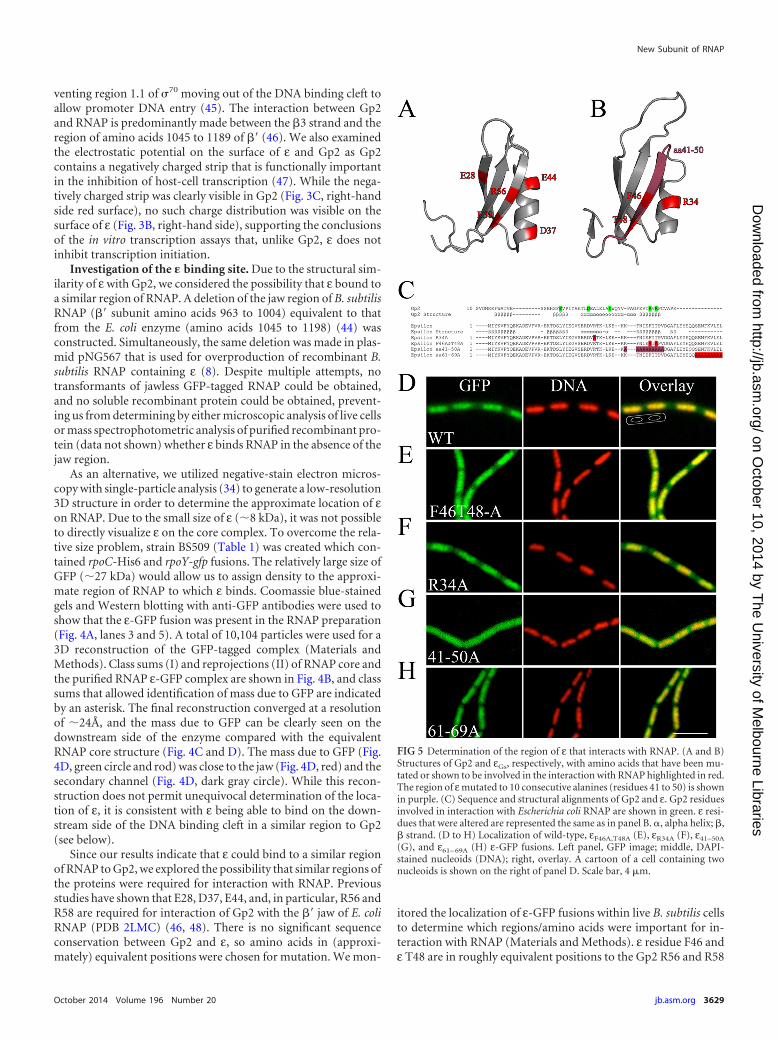
(G), and ε61– 69A (H) ε-GFP fusions. Left panel, GFP image; middle, DAPI-stained nucleoids (DNA); right, overlay. A cartoon of a cell containing twonucleoids is shown on the right of panel D. Scale bar, 4 �m.
New Subunit of RNAP
October 2014 Volume 196 Number 20 jb.asm.org 3629
on October 10, 2014 by T
he University of M
elbourne Librarieshttp://jb.asm
.org/D
ownloaded from
residues which are involved in binding to E. coli RNAP while εR34
is approximately equivalent to Gp2 D37 or E44 (Fig. 5A and B). Inaddition, the 10 amino acids spanning the �3 strand and the final10 amino acids were also mutated to alanines (ε residues 41 to 50mutated to alanines [ε41–50A] and ε61– 69A, respectively) (Fig. 5C).All of the mutants localized to the nucleoids with an identicalpattern to the wild-type ε except for ε41–50A (Fig. 5D to H), indi-cating that ε R34, F46, T48, and the final 9 amino acids were notessential for interaction with RNAP. Examination of the localiza-tion of the ε41–50A mutant showed that fluorescence was distrib-uted throughout the whole cell (Fig. 5G), suggesting that the 10amino acids spanning the �3 strand are important for interactionwith RNAP. It is possible that changing 10 amino acids to alanineresidues caused misfolding of the protein, but we do not believethis is the case as overproduced ε41–50A behaved identically towild-type ε during overproduction and purification. Additionalmutant proteins with 10 sequential alanines spanning the loopbetween �1 and �2 (amino acids 11 to 20) and across the �2 strand
(amino acids 21 to 30) aggregated in solution and were consideredunlikely to form functional GFP fusions in vivo (data not shown).We also created GFP fusions with alanine mutations in the mosthighly conserved residues (K4, E8, R17, E18, T20, and E45) thatcover other regions of ε, but all of them except the K4A mutantshowed identical localization to the wild-type protein (data notshown). The K4A mutant was not fluorescent on transformationof B. subtilis, which most likely indicates that the fusion was non-functional and was rapidly degraded. Therefore, our data are con-sistent with ε and Gp2 binding their cognate RNAPs at similarsites via the same regions.
DISCUSSION
We have identified a new subunit of RNAP that is restricted to themedically and industrially important group of high-A�T Gram-positive bacteria (the Firmicutes). While there are highly con-served subunits of RNAP across the kingdoms, within the Firmi-cutes we now know that there are at least two additional small
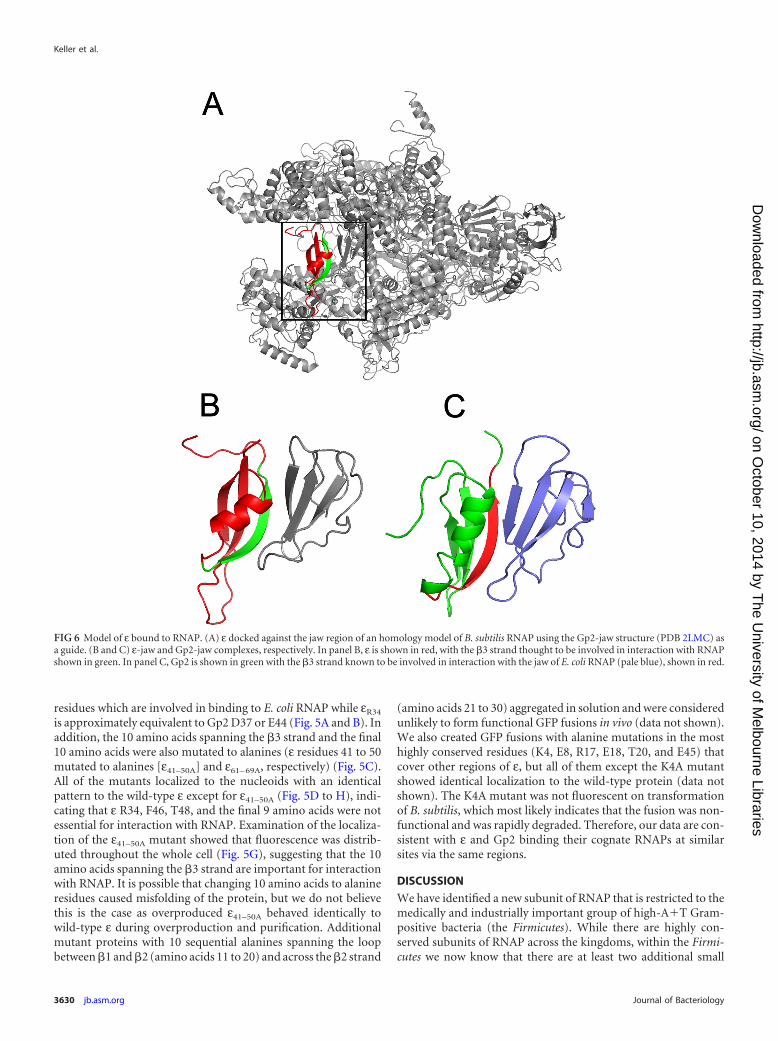
FIG 6 Model of ε bound to RNAP. (A) ε docked against the jaw region of an homology model of B. subtilis RNAP using the Gp2-jaw structure (PDB 2LMC) asa guide. (B and C) ε-jaw and Gp2-jaw complexes, respectively. In panel B, ε is shown in red, with the �3 strand thought to be involved in interaction with RNAPshown in green. In panel C, Gp2 is shown in green with the �3 strand known to be involved in interaction with the jaw of E. coli RNAP (pale blue), shown in red.
Keller et al.
3630 jb.asm.org Journal of Bacteriology
on October 10, 2014 by T
he University of M
elbourne Librarieshttp://jb.asm
.org/D
ownloaded from

subunits of RNAP, giving a subunit composition of �2��=�ε� inthese organisms. The �, ε, and � subunits all appear to be presentat levels similar to those of the other subunits of RNAP (6, 17),suggesting that they are all core subunit components.
As with � and �, ε is not essential for viability and, despitemuch work, the functional role of the � and � subunits is only nowbeing uncovered (2, 4, 49). No change in the transcriptome of B.subtilis could be detected on deletion of ε or any effect of ε in invitro transcription assays, and additional studies are needed tofully elucidate its functional role in transcription.
Determination of the structure of εGs by X-ray crystallographyrevealed it to have a ���� fold that is present in a diverse range ofsmall proteins of varied/uncharacterized function and that may beimportant in protein-protein interactions. 3D reconstruction bysingle-particle analysis indicated that ε is located on the down-stream side of RNAP, in the region of the �= jaw and secondarychannel. We do not believe it is likely that ε binds in/around thesecondary channel as it is structurally unrelated to proteins such asGreA/B and DksA that are known to bind in that region (50, 51),and we favor the idea that ε binds to the jaw in a similar fashion asGp2. GreA is present in Gram-positive bacteria, but DksA, whichhas been implicated in having a role in the stringent response, isnot. Transcriptomes of wild-type and ε strains following induc-tion of the stringent response were identical, indicating that ε hasno analogous role to DksA (data not shown). Therefore, we favorthe hypothesis that ε binds to/near the �= jaw in the DNA channelof RNAP in a location equivalent to Gp2 (Fig. 6A). While we couldnot unequivocally confirm binding to the �= jaw, interaction withRNAP is mediated via the �3 strand (Fig. 6A and B, green) that isalso important in Gp2 binding to RNAP (Fig. 6C, red) as mutationof this region abolished ε localization to the nucleoid.
At this stage the function of ε remains unclear, but one possi-bility is that it is required for phage protection by blocking accessof Gp2-like proteins to RNAP. Such proteins are widely distrib-uted among Gram-negative phage, but due to the small size andlevel of sequence divergence, it is not possible to ascertain whetherthey are also present in Gram-positive phage. A comprehensivesurvey of Bacillus phage infection in ε-positive and knockoutstrains in the future will help shed light whether ε plays a role inprotection from phage infection.
ACKNOWLEDGMENTS
We thank G. Doherty for fluorescent protein fusion strains, R. Rothnagelfor assistance with electron microscopy, and P. Curmi (UNSW) and theAustralian Synchrotron for access to X-ray diffraction facilities. O.D. ac-knowledges support from Philippe Noirot (INRA, Jouy en Josas, France).
This work was supported by grants 455646, LSHG-CT-2006-037469,and P305/12/G034 from the National Health and Medical ResearchCouncil, Australia (P.J.L.), European Union (P.J.L. and O.D.), and CzechScience Foundation (to L.K.).
REFERENCES1. Burgess RR. 1969. Separation and characterization of the subunits of
ribonucleic acid polymerase. J. Biol. Chem. 244:6168 – 6176.2. Wiedermannova J, Sudzinova P, Koval T, Rabatinova A, Sanderova H,
Ramaniuk O, Rittich S, Dohnalek J, Fu Z, Lewis P, Halada P, Krasny L.2014. Characterization of HelD, an interacting partner of RNA polymer-ase from Bacillus subtilis. Nucleic Acids Res. 42:5151–5163. http://dx.doi.org/10.1093/nar/gku113.
3. Juang YL, Helmann JD. 1994. The delta subunit of Bacillus subtilis RNApolymerase. An allosteric effector of the initiation and core-recyclingphases of transcription. J. Mol. Biol. 239:1–14.
4. Rabatinova A, Sanderova H, Jirat Matejckova J, Korelusova J, Sojka L,Barvik I, Papouskova V, Sklenar V, Zidek L, Krasny L. 2013. The deltasubunit of RNA polymerase is required for rapid changes in gene expres-sion and competitive fitness of the cell. J. Bacteriol. 195:2603–2611. http://dx.doi.org/10.1128/JB.00188-13.
5. Lopez de Saro FJ, Woody AY, Helmann JD. 1995. Structural analysis ofthe Bacillus subtilis delta factor: a protein polyanion which displaces RNAfrom RNA polymerase. J. Mol. Biol. 252:189 –202. http://dx.doi.org/10.1006/jmbi.1995.0487.
6. Delumeau O, Lecointe F, Muntel J, Guillot A, Guedon E, Monnet V,Hecker M, Becher D, Polard P, Noirot P. 2011. The dynamic proteinpartnership of RNA polymerase in Bacillus subtilis. Proteomics 11:2992–3001. http://dx.doi.org/10.1002/pmic.201000790.
7. Helmann JD. 2003. Purification of Bacillus subtilis RNA polymerase andassociated factors. Methods Enzymol. 370:10 –24. http://dx.doi.org/10.1016/S0076-6879(03)70002-0.
8. Yang X, Lewis PJ. 2008. Overproduction and purification of recombinantBacillus subtilis RNA polymerase. Protein Expr. Purif. 59:86 –93. http://dx.doi.org/10.1016/j.pep.2008.01.006.
9. Spiegelman GB, Hiatt WR, Whiteley HR. 1978. Role of the 21,000molecular weight polypeptide of Bacillus subtilis RNA polymerase in RNAsynthesis. J. Biol. Chem. 253:1756 –1765.
10. Achberger EC, Tahara M, Whiteley HR. 1982. Interchangeability of deltasubunits of RNA polymerase from different species of the genus Bacillus. J.Bacteriol. 150:977–980.
11. Gentry DR, Burgess RR. 1986. The cloning and sequence of the geneencoding the omega subunit of Escherichia coli RNA polymerase. Gene48:33– 40. http://dx.doi.org/10.1016/0378-1119(86)90349-5.
12. Figaro S, Durand S, Gilet L, Cayet N, Sachse M, Condon C. 2013.Bacillus subtilis mutants with knockouts of the genes encoding ribonu-cleases RNase Y and RNase J1 are viable, with major defects in cell mor-phology, sporulation, and competence. J. Bacteriol. 195:2340 –2348. http://dx.doi.org/10.1128/JB.00164-13.
13. Kunst F, Rapoport G. 1995. Salt stress is an environmental signal affect-ing degradative enzyme synthesis in Bacillus subtilis. J. Bacteriol. 177:2403–2407.
14. Guerout-Fleury AM, Frandsen N, Stragier P. 1996. Plasmids for ectopicintegration in Bacillus subtilis. Gene 180:57– 61. http://dx.doi.org/10.1016/S0378-1119(96)00404-0.
15. Nicolas P, Mader U, Dervyn E, Rochat T, Leduc A, Pigeonneau N,Bidnenko E, Marchadier E, Hoebeke M, Aymerich S, Becher D, Bisic-chia P, Botella E, Delumeau O, Doherty G, Denham EL, Fogg MJ,Fromion V, Goelzer A, Hansen A, Hartig E, Harwood CR, Homuth G,Jarmer H, Jules M, Klipp E, Le Chat L, Lecointe F, Lewis P, Lieber-meister W, March A, Mars RA, Nannapaneni P, Noone D, Pohl S, RinnB, Rugheimer F, Sappa PK, Samson F, Schaffer M, Schwikowski B, SteilL, Stulke J, Wiegert T, Devine KM, Wilkinson AJ, van Dijl JM, HeckerM, Volker U, Bessieres P, Noirot P. 2012. Condition-dependent tran-scriptome reveals high-level regulatory architecture in Bacillus subtilis.Science 335:1103–1106. http://dx.doi.org/10.1126/science.1206848.
16. Doherty GP, Meredith DH, Lewis PJ. 2006. Subcellular partitioning oftranscription factors in Bacillus subtilis. J. Bacteriol. 188:4101– 4110. http://dx.doi.org/10.1128/JB.01934-05.
17. Doherty GP, Fogg MJ, Wilkinson AJ, Lewis PJ. 2010. Small subunits ofRNA polymerase: localization, levels and implications for core enzymecomposition. Microbiology 156:3532–3543. http://dx.doi.org/10.1099/mic.0.041566-0.
18. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA,McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, ThompsonJD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0.Bioinformatics 23:2947–2948. http://dx.doi.org/10.1093/bioinformatics/btm404.
19. Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel methodfor rapid multiple sequence alignment based on fast Fourier transform.Nucleic Acids Res. 30:3059 –3066. http://dx.doi.org/10.1093/nar/gkf436.
20. Ling MM, Robinson BH. 1997. Approaches to DNA mutagenesis: anoverview. Anal. Biochem. 254:157–178. http://dx.doi.org/10.1006/abio.1997.2428.
21. Quin M, Newman J, Firbank S, Lewis RJ, Marles-Wright J. 2008.Crystallization and preliminary X-ray analysis of RsbS from Moorella ther-moacetica at 2.5 Å resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst.Commun. 64:196 –199. http://dx.doi.org/10.1107/S1744309108003849.
22. McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis
New Subunit of RNAP
October 2014 Volume 196 Number 20 jb.asm.org 3631
on October 10, 2014 by T
he University of M
elbourne Librarieshttp://jb.asm
.org/D
ownloaded from

PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM,Kuhn P. 2002. Blu-Ice and the Distributed Control System: software fordata acquisition and instrument control at macromolecular crystallogra-phy beamlines. J. Synchrotron Radiat. 9:401– 406. http://dx.doi.org/10.1107/S0909049502015170.
23. Evens G, Pettifer RF. 2001. CHOOCH: a program for deriving anoma-lous-scattering factors from X-ray fluorescence spectra. J. Appl. Crystal-logr. 34:82– 86. http://dx.doi.org/10.1107/S0021889800014655.
24. Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. 2011.iMOSFLM: a new graphical interface for diffraction-image processingwith MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67:271–281. http://dx.doi.org/10.1107/S0907444910048675.
25. Leslie AG. 2006. The integration of macromolecular diffraction data. ActaCrystallogr. D Biol. Crystallogr. 62:48 –57. http://dx.doi.org/10.1107/S0907444905039107.
26. CCP4. 1994. The CCP4 suite: programs for protein crystallography. ActaCrystallogr. D Biol. Crystallogr. 50:760 –763. http://dx.doi.org/10.1107/S0907444994003112.
27. Ness SR, de Graaff RA, Abrahams JP, Pannu NS. 2004. CRANK: newmethods for automated macromolecular crystal structure solution. Struc-ture 12:1753–1761. http://dx.doi.org/10.1016/j.str.2004.07.018.
28. de Graaff RA, Hilge M, van der Plas JL, Abrahams JP. 2001. Matrixmethods for solving protein substructures of chlorine and sulfur fromanomalous data. Acta Crystallogr. D Biol. Crystallogr. 57:1857–1862. http://dx.doi.org/10.1107/S0907444901016535.
29. Abrahams JP, Leslie AG. 1996. Methods used in the structure determi-nation of bovine mitochondrial F1 ATPase. Acta Crystallogr. D Biol. Crys-tallogr. 52:30 – 42. http://dx.doi.org/10.1107/S0907444995008754.
30. Cowtan K. 2006. The Buccaneer software for automated model building.1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62:1002–1011. http://dx.doi.org/10.1107/S0907444906022116.
31. Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and devel-opment of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486 –501. http://dx.doi.org/10.1107/S0907444910007493.
32. Langer G, Cohen SX, Lamzin VS, Perrakis A. 2008. Automated macro-molecular model building for X-ray crystallography using ARP/wARPversion 7. Nat. Protoc. 3:1171–1179. http://dx.doi.org/10.1038/nprot.2008.91.
33. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Echols N, Headd JJ,Hung LW, Jain S, Kapral GJ, Grosse Kunstleve RW, McCoy AJ, Mori-arty NW, Oeffner RD, Read RJ, Richardson DC, Richardson JS, Ter-williger TC, Zwart PH. 2011. The Phenix software for automated deter-mination of macromolecular structures. Methods 55:94 –106. http://dx.doi.org/10.1016/j.ymeth.2011.07.005.
34. Yang X, Molimau S, Doherty GP, Johnston EB, Marles-Wright J,Rothnagel R, Hankamer B, Lewis RJ, Lewis PJ. 2009. The structure ofbacterial RNA polymerase in complex with the essential transcriptionelongation factor NusA. EMBO Rep. 10:997–1002. http://dx.doi.org/10.1038/embor.2009.155.
35. Ludtke SJ, Baldwin PR, Chiu W. 1999. EMAN: semiautomated softwarefor high-resolution single-particle reconstructions. J. Struct. Biol. 128:82–97. http://dx.doi.org/10.1006/jsbi.1999.4174.
36. Davies KM, Lewis PJ. 2003. Localization of rRNA synthesis in Bacillus sub-tilis: characterization of loci involved in transcription focus formation. J.Bacteriol. 185:2346 –2353. http://dx.doi.org/10.1128/JB.185.7.2346-2353.2003.
37. Lewis PJ, Thaker SD, Errington J. 2000. Compartmentalization of tran-scription and translation in Bacillus subtilis. EMBO J. 19:710 –718. http://dx.doi.org/10.1093/emboj/19.4.710.
38. Britton RA, Wen T, Schaefer L, Pellegrini O, Uicker WC, Mathy N,Tobin C, Daou R, Szyk J, Condon C. 2007. Maturation of the 5= end ofBacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1.Mol. Microbiol. 63:127–138. http://dx.doi.org/10.1111/j.1365-2958.2006.05499.x.
39. Hunt A, Rawlins JP, Thomaides HB, Errington J. 2006. Functional
analysis of 11 putative essential genes in Bacillus subtilis. Microbiology152:2895–2907. http://dx.doi.org/10.1099/mic.0.29152-0.
40. Gabdulkhakov A, Nikonov S, Garber M. 2013. Revisiting the Haloarculamarismortui 50S ribosomal subunit model. Acta Crystallogr. D Biol. Crys-tallogr. 69:997–1004. http://dx.doi.org/10.1107/S0907444913004745.
41. Serrano-Heras G, Salas M, Bravo A. 2006. A uracil-DNA glycosylaseinhibitor encoded by a non-uracil containing viral DNA. J. Biol. Chem.281:7068 –7074. http://dx.doi.org/10.1074/jbc.M511152200.
42. Serrano-Heras G, Ruiz-Maso JA, del Solar G, Espinosa M, Bravo A,Salas M. 2007. Protein p56 from the Bacillus subtilis phage phi29 inhibitsDNA-binding ability of uracil-DNA glycosylase. Nucleic Acids Res. 35:5393–5401. http://dx.doi.org/10.1093/nar/gkm584.
43. Hesselbach BA, Nakada D. 1977. I protein: bacteriophage T7-codedinhibitor of Escherichia coli RNA polymerase. J. Virol. 24:746 –760.
44. Nechaev S, Severinov K. 1999. Inhibition of Escherichia coli RNA poly-merase by bacteriophage T7 gene 2 protein. J. Mol. Biol. 289:815– 826.http://dx.doi.org/10.1006/jmbi.1999.2782.
45. Bae B, Davis E, Brown D, Campbell EA, Wigneshweraraj S, Darst SA.2013. Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase in-volves misappropriation of sigma70 domain 1.1. Proc. Natl. Acad. Sci.U. S. A. 110:19772–19777. http://dx.doi.org/10.1073/pnas.1314576110.
46. James E, Liu M, Sheppard C, Mekler V, Camara B, Liu B, Simpson P,Cota E, Severinov K, Matthews S, Wigneshweraraj S. 2012. Structuraland mechanistic basis for the inhibition of Escherichia coli RNA polymer-ase by T7 Gp2. Mol. Cell 47:755–766. http://dx.doi.org/10.1016/j.molcel.2012.06.013.
47. Sheppard C, Camara B, Shadrin A, Akulenko N, Liu M, Baldwin G,Severinov K, Cota E, Matthews S, Wigneshweraraj SR. 2011. Inhibitionof Escherichia coli RNAp by T7 Gp2 protein: role of negatively chargedstrip of amino acid residues in Gp2. J. Mol. Biol. 407:623– 632. http://dx.doi.org/10.1016/j.jmb.2011.02.013.
48. Camara B, Liu M, Reynolds J, Shadrin A, Liu B, Kwok K, Simpson P,Weinzierl R, Severinov K, Cota E, Matthews S, Wigneshweraraj SR.2010. T7 phage protein Gp2 inhibits the Escherichia coli RNA polymeraseby antagonizing stable DNA strand separation near the transcription startsite. Proc. Natl. Acad. Sci. U. S. A. 107:2247–2252. http://dx.doi.org/10.1073/pnas.0907908107.
49. Vrentas CE, Gaal T, Ross W, Ebright RH, Gourse RL. 2005. Response ofRNA polymerase to ppGpp: requirement for the omega subunit and reliefof this requirement by DksA. Genes Dev. 19:2378 –2387. http://dx.doi.org/10.1101/gad.1340305.
50. Perederina A, Svetlov V, Vassylyeva MN, Tahirov TH, Yokoyama S,Artsimovitch I, Vassylyev DG. 2004. Regulation through the secondarychannel–structural framework for ppGpp-DksA synergism during tran-scription. Cell 118:297–309. http://dx.doi.org/10.1016/j.cell.2004.06.030.
51. Vassylyeva MN, Svetlov V, Dearborn AD, Klyuyev S, Artsimovitch I,Vassylyev DG. 2007. The carboxy-terminal coiled-coil of the RNA poly-merase �=-subunit is the main binding site for Gre factors. EMBO Rep.8:1038 –1043. http://dx.doi.org/10.1038/sj.embor.7401079.
52. Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. 2004. PDB2PQR:an automated pipeline for the setup of Poisson-Boltzmann electrostaticscalculations. Nucleic Acids Res. 32:W665–W667. http://dx.doi.org/10.1093/nar/gkh381.
53. Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymeraseto direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113–130. http://dx.doi.org/10.1016/0022-2836(86)90385-2.
54. Neylon C, Brown SE, Kralicek AV, Miles CS, Love CA, Dixon NE. 2000.Interaction of the Escherichia coli replication terminator protein (Tus)with DNA: a model derived from DNA-binding studies of mutant pro-teins by surface plasmon resonance. Biochemistry 39:11989 –11999. http://dx.doi.org/10.1021/bi001174w.
55. Lewis PJ, Marston AL. 1999. GFP vectors for controlled expression anddual labelling of protein fusions in Bacillus subtilis. Gene 227:101–110.http://dx.doi.org/10.1016/S0378-1119(98)00580-0.
Keller et al.
3632 jb.asm.org Journal of Bacteriology
on October 10, 2014 by T
he University of M
elbourne Librarieshttp://jb.asm
.org/D
ownloaded from












![Rif1 Supports the Function of the CST Complex in Yeast ... · cell cycle arrest [19–21]. Interestingly, Stn1 interacts with Pol12 [22], a subunit of the DNA polymerase a (pola)-primase](https://img.dokumen.tips/doc/110x75/608d5e2b94e36f65cb565cd0/rif1-supports-the-function-of-the-cst-complex-in-yeast-cell-cycle-arrest-19a21.jpg)
















