Embed Size (px)
Citation preview

イプラグリフロジン L-プロリン 2.7.6
個々の試験のまとめ
アステラス製薬
目次
2.7.6 個々の試験のまとめ
試験名[試験番号] 添付資料番号 頁2.7.6.1 食事の影響試験[CL-0071] 5.3.1.1-1 6
2.7.6.2 絶対バイオアベイラビリティ試験[CL-0057] 5.3.1.1-2 21
2.7.6.3 第 I 相試験[CL-0101] 5.3.3.1-1 37
2.7.6.4 海外第 I 相試験(単回・食事)[CL-0001] 5.3.3.1-2 73
2.7.6.5 海外第 I 相試験(単回・反復)[CL-0002] 5.3.3.1-3 96
2.7.6.6 マスバランス試験[CL-0055] 5.3.3.1-4 122
2.7.6.7 高齢者・性差試験[CL-0052] 5.3.3.3-1 134
2.7.6.8 腎機能低下患者 PK/PD 試験[CL-0073] 5.3.3.3-2 159
2.7.6.9 海外腎機能低下者試験[CL-0064] 5.3.3.3-3 187
2.7.6.10 海外肝機能低下者試験[CL-0063] 5.3.3.3-4 222
2.7.6.11 薬物相互作用試験(メトホルミン)[CL-0056] 5.3.3.4-1 247
2.7.6.12 薬物相互作用試験(グリメピリド)[CL-0059] 5.3.3.4-2 276
2.7.6.13 薬物相互作用試験(ピオグリタゾン)[CL-0060] 5.3.3.4-3 305
2.7.6.14 薬物相互作用試験(ミグリトール)[CL-0062] 5.3.3.4-4 332
2.7.6.15 薬物相互作用試験(シタグリプチン)[CL-0066] 5.3.3.4-5 350
2.7.6.16 薬物相互作用試験(ミチグリニド)[CL-0074] 5.3.3.4-6 375
2.7.6.17 薬物相互作用試験(フロセミド)[CL-0054] 5.3.3.4-7 398
2.7.6.18 QT/QTc 評価試験[CL-0058] 5.3.4.1-1 430
2.7.6.19 薬理作用機序検討試験[CL-0050] 5.3.4.1-2 460
2.7.6.20 血糖日内変動試験[CL-0070] 5.3.4.2-1 502
2.7.6.21 第 II 相用量設定試験[CL-0103] 5.3.5.1-1 530
2.7.6.22 第 III 相単独療法試験[CL-0105] 5.3.5.1-2 567
2.7.6.23 メトホルミン併用試験[CL-0106] 5.3.5.1-3 590
2.7.6.24 ピオグリタゾン併用試験[CL-0107] 5.3.5.1-4 628
2.7.6.25 スルホニルウレア剤併用試験[CL-0109] 5.3.5.1-5 665
2.7.6.26 腎機能低下患者試験[CL-0072] 5.3.5.1-6 713
2.7.6.27 海外第 IIa 相試験[CL-0016] 5.3.5.1-7 782
2.7.6.28 海外第 II 相用量設定試験[CL-0004] 5.3.5.1-8 805
2.7.6.29 海外第 II 相メトホルミン併用試験[CL-0005] 5.3.5.1-9 836
2.7.6.30 α-GI 併用試験[CL-0108] 5.3.5.2-1 861
2.7.6.31 DPP-4 阻害剤併用試験[CL-0110] 5.3.5.2-2 887
2.7.6.32 ナテグリニド併用試験[CL-0111] 5.3.5.2-3 913
2.7.6.33 長期投与試験[CL-0121] 5.3.5.2-4 941
2.7.6.34 長期投与試験[CL-0122] 5.3.5.2-5 978
1

2.7.6個々の試験のまとめ
2.7.6 個々の試験のまとめ
本項で使用した略号及び用語の定義一覧を表 2.7.6-1 に示す。
表 2.7.6-1 略号及び用語の定義一覧(その 1)略号及び用語 定義
25-OH ビタミン D 25-ヒドロキシビタミン DAe 尿中排泄量Ae% 尿中排泄率Aet 時間 0 から t までの尿中排泄量Aet% 時間 0 から t までの尿中排泄率Aeinf 時間 0 から無限時間まで外挿した尿中排泄量Aeinf % 時間 0 から無限時間まで外挿した尿中排泄率Aelast 時間 0 から定量可能最終時点までの尿中排泄量Aelast% 時間 0 から定量可能最終時点までの尿中排泄率Aetau 投与間隔ごとの尿中排泄量Aetau% 投与間隔ごとの尿中排泄率ALP アルカリホスファターゼALT (GPT) アラニン・アミノトランスフェラーゼASP1941 イプラグリフロジン L-プロリンの活性本体(フリー体)であるイプラグリ
フロジンの開発化合物番号AST (GOT) アスパラギン酸アミノトランスフェラーゼAUC 血漿中(全血中又は血清中)濃度-時間曲線下面積AUC%extra AUC 外挿部分が AUCinf に占める割合AUCt 時間 0 から t までの血漿中(全血中又は血清中)濃度-時間曲線下面積AUCinf 時間 0 から無限時間まで外挿した血漿中(全血中又は血清中)濃度-時間
曲線下面積AUCinf,u 非結合型の時間 0 から無限時間まで外挿した血漿中(全血中又は血清中)
濃度-時間曲線下面積AUClast 時間 0 から定量可能最終時点までの血漿中(全血中又は血清中)濃度-時
間曲線下面積AUClast,u 非結合型の時間 0 から定量可能最終時点までの血漿中(全血中又は血清中)
濃度-時間曲線下面積AUCtau 投与間隔ごとの血漿中(全血中又は血清中)濃度-時間曲線下面積AUCtau,u 非結合型の投与間隔ごとの血漿中(全血中又は血清中)濃度-時間曲線下
面積BAP 血清骨型アルカリホスファターゼBMI 体格指数[体重(kg)/身長(m)2]BQL 定量下限値未満BUN 血中尿素窒素CI 信頼区間CIOMS 国際医学団体協議会Cr Ae24h 時間 0 から治験薬投与後 24 時間までの尿中クレアチニン排泄量CL/F 経口クリアランスCLcr クレアチニンクリアランス
2

2.7.6個々の試験のまとめ
表 2.7.6-1 略号及び用語の定義一覧(その 2)略号及び用語 定義
CLu/F 非結合型の経口クリアランスCLr 腎クリアランスCLr,u 非結合型の腎クリアランスCmax 最高血漿中(全血中又は血清中)濃度Cmax,u 非結合型の最高血漿中(全血中又は血清中)濃度
CK(CPK) クレアチンキナーゼ(クレアチンホスホキナーゼ)Cre クレアチニンCRO 医薬品開発業務受託機関CRP C-反応性蛋白CT コンピューター断層撮影法CTx I 型コラーゲン架橋 C–テロペプチドCtrough トラフ濃度CV 変動係数DPP-4 ジペプチジルペプチダーゼ-4ED 有効量eGFR 推算糸球体濾過量EMA 欧州医薬品庁FAS 最大の解析対象集団FDA 米国食品医薬品局fu 非結合型分率γ-GTP γ-グルタミルトランスペプチターゼ(又は γ-グルタミルトランスフェラー
ゼ)GER グルコース排泄率GFR 糸球体濾過量GIP グルコース依存性インスリン分泌刺激ポリペプチドGLP-1 グルカゴン様ペプチド-1GMR 幾何平均比HAV A 型肝炎ウイルスHbA1c ヘモグロビン A1cHBs B 型肝炎ウイルス表面hCG ヒト絨毛性ゴナドトロピンHCV C 型肝炎ウイルスHDL 高密度リポ蛋白H-FABP 心臓由来脂肪酸結合蛋白HIPAA 医療保険の相互運用性と説明責任に関する法律HIV ヒト免疫不全ウイルスHPLC 高速液体クロマトグラフィーHRT ホルモン補充療法IgM 免疫グロブリン MIntact-PTH インタクト副甲状腺ホルモンJDS Japan Diabetes Societykel 消失速度定数LDH 乳酸脱水素酵素
3

2.7.6個々の試験のまとめ
表 2.7.6-1 略号及び用語の定義一覧(その 3)
略号及び用語 定義LDL 低密度リポ蛋白M1 イプラグリフロジン代謝物:ベンゾチオフェン環の 6-水酸化及びグルコー
ス環の 2’-O-β-グルクロン酸抱合体,AS2551864,H1M2 イプラグリフロジン代謝物:グルコース環の 2’-O-β-グルクロン酸抱合体,
AS2364093,H2,R2,Mk2M3 イプラグリフロジン代謝物:グルコース環の 6’-O-β-グルクロン酸抱合体,
AS2551840,H3,R3,Mk3M4 イプラグリフロジン代謝物:グルコース環の 3’-O-β-グルクロン酸抱合体,
AS2551844,H4,R4,Mk4M6 イプラグリフロジンの代謝物:ベンゾチオフェン環の 6-O-硫酸抱合体,
AS2551868,H6,R6,Mk6MAGE 平均血糖変動幅MDRD Modification of Diet in Renal DiseaseMedDRA ICH 国際医薬用語集MedDRA/J ICH 国際医薬用語集日本語版MID 最小不耐量MTD 最大耐量MRT 平均滞留時間MRTinf 無限時間まで外挿した平均滞留時間NA 該当せず/算出不能NC 算出せずNOAEL 最大無毒性量NGSP National Glycohemoglobin Standardization ProgramNT-proBNP N 末端プロ脳性ナトリウム利尿ペプチドNTX I 型コラーゲン架橋 N–テロペプチドNYHA ニューヨーク心臓協会OGTT 経口ブドウ糖負荷試験PD 薬力学PDAS 薬力学解析対象集団PK 薬物動態PKAS 薬物動態解析対象集団PPS 治験実施計画書に適合した対象集団PT MedDRA の基本語PTH 副甲状腺ホルモンPTR ピーク/トラフ比QTc 心拍数で補正した QT 間隔QTcB Bazett 法を用いて心拍数で補正した QT 間隔QTcF Fridericia 法を用いて心拍数で補正した QT 間隔QTcI 各被験者の心拍数で補正した QT 間隔Racc 累積比SAF 安全性解析対象集団SD 標準偏差SE 標準誤差
4

2.7.6個々の試験のまとめ
表 2.7.6-1 略号及び用語の定義一覧(その 4)
略号及び用語 定義SGLT Na+/グルコース共輸送担体SOC MedDRA の器官別大分類SU スルホニルウレアt1/2 消失半減期T-Bil 総ビリルビンT-Cho 総コレステロールTEAE 治療下で発現した有害事象tlag ラグタイムtmax 最高血漿中(全血中又は血清中)濃度到達時間TQT QT/QTc 評価TRACP-5b 酒石酸抵抗性酸ホスファターゼUA 尿酸ULN 正常上限値Vz/F みかけの分布容積Vz,u/F 非結合型のみかけの分布容積Ra グルコース出現率
なお,本項中のイプラグリフロジン L-プロリン錠の用量は,フリー体であるイプラグリフロジ
ン(ASP1941)としての用量を示す。
5

2.7.6個々の試験のまとめ
2.7.6.1 食事の影響試験[CL-0071](添付資料 5.3.1.1-1)
2.7.6.1.1 試験方法の概略
治験課題名:ASP1941 薬物動態試験 -ASP1941 の薬物動態に及ぼす食事の影響の検討-
治験実施医療機関:日本 1 施設
公表文献:なし
治験期間:
治験開始日:2010 年 10 月
治験終了日:2010 年 11 月
開発のフェーズ:第 I 相
目的:
非高齢健康成人男性を対象に,ASP1941 錠(以下,本剤)50 mg を単回経口投与したときの薬物
動態に及ぼす食事の影響を 6 群 3 用法 3 時期のクロスオーバー法にて検討する。また,薬力学に
及ぼす食事の影響及び安全性について検討する。
治験デザイン・治験方法:本試験は,非盲検無作為化,6 群 3 用法 3 時期のクロスオーバー試験として実施した。
非高齢健康成人男性(各群 5 例,計 30 例)を対象とし,本剤 50 mg を空腹時,食前又は食後に単回経
口投与した。第 1 期治験薬投与後,6 日以上の休薬期間(第 1 期治験薬投与日の翌日~第 2 期 Day −1)
を経過した後に,第 2 期治験薬投与を行った。更に,第 2 期治験薬投与後,6 日以上の休薬期間(第 2
期治験薬投与日の翌日~第 3 期 Day −1)を経過した後に,第 3 期治験薬投与を行った。
目標被験者数:30 例(各群 5 例)
【設定根拠】
これまで実施した本剤試験の結果より,本剤の最高血漿中濃度(Cmax)の個体内変動を変動係数
で 25%程度と想定し,本剤空腹時投与に対する本剤食前投与及び食後投与の真の幾何平均比
(GMR)を 0.95 と仮定して,GMR の 90%信頼区間が 80%の検出力で 0.8~1.25 の範囲にあるた
めに必要な最小総被験者数を算出した結果,28 例と算出された。この例数に脱落・中止例の生じ
る可能性を考慮して 30 例と設定した。
診断及び選択・除外基準:
以下の選択基準のすべてを満たし,かつ除外基準のいずれにも該当しない場合,本試験の対象
とした。
1.選択基準
(1) 性別:男性
(2) 年齢(同意取得時):20 歳以上 45 歳未満
(3) 体重(スクリーニング検査時):50.0 kg 以上 80.0 kg 未満
(4) BMI(スクリーニング検査時):17.6 kg/m2 以上 26.4 kg/m2 未満
(5) 治験担当医師が,スクリーニング検査から投与直前までに得られた診察(自覚症状,他覚所
見),すべての検査結果から健康であると判断した者
(6) 被験者本人から文書による同意が得られている者
2.除外基準
(1) スクリーニング検査前 120 日以内に,他の治験又は製造販売後臨床試験において投薬を受け
た者
(2) スクリーニング検査前 90 日以内に 400 mL の全血採血,30 日以内に 200 mL の全血採血又は
14 日以内に成分採血を実施した者
(3) 第 1 期入院日前 7 日以内に薬剤投与を受けた者又は薬剤投与の予定がある者
(4) スクリーニング検査時及び第 1 期入院時(Day −1)の血圧,脈拍数,体温,12 誘導心電図に
おいて以下の基準を逸脱する者
6

2.7.6個々の試験のまとめ
項目 基準範囲
臥位血圧 収縮期血圧:90 mmHg 以上,140 mmHg 以下
拡張期血圧:40 mmHg 以上,90 mmHg 以下
臥位脈拍数 40 回/分以上,100 回/分以下
腋窩体温 35.0°C 以上,37.0°C 以下
12 誘導心電図 正常あるいは臨床的に重要でない異常 a
a:例えば,洞性不整脈,非特異的 ST 上昇等
(5) スクリーニング検査時及び第 1 期入院時(Day −1)の臨床検査において以下の基準に抵触す
る者
なお,各種検査の基準範囲は実施医療機関・臨床検査施設の基準範囲とした。
[1] 血液学的検査:
基準範囲の上下限の±20%を逸脱する者
ただし,白血球数が基準範囲内であれば分画の個々の値は問わない。
[2] 血液生化学検査:
・AST,ALT,クレアチニン,Na,K,Cl 及び空腹時血糖値は,基準範囲を逸脱する者
・上記以外の項目は,基準範囲の上下限の±20%を逸脱する者
ただし,下限値の逸脱が臨床的に問題はないと考えられるもの(AST,ALT,ALP,γ-
グルタミルトランスペプチダーゼ,乳酸脱水素酵素,クレアチンキナーゼ,総ビリルビ
ン,総コレステロール,血中尿素窒素,尿酸,クレアチニン)は,下限を設定しない。
[3] 尿検査:
・尿中グルコース,尿中蛋白が(+)以上を認めた者
・尿中ウロビリノーゲンが(2+)以上を認めた者
[4] 尿中薬物検査:
ベンゾジアゼピン類,コカイン系麻薬,覚せい剤,大麻,バルビツール酸類,モルヒネ系
麻薬,フェンシクリジン類,三環系抗うつ剤のいずれかに陽性を認めた者
[5] 免疫学的検査:
HBs 抗原,HCV 抗体,HIV 抗原・抗体,梅毒のいずれかにおいて陽性を認めた者
(6) 薬物アレルギーの合併又は既往を有する者
(7) 第 1 期入院前 7 日以内に上部消化器症状(悪心,嘔吐,胃痛等)を発現した者
(8) 肝疾患(ウイルス性肝炎,薬物性肝障害等)の合併又は既往を有する者
(9) 心疾患(うっ血性心不全,狭心症,治療を要する不整脈等)の合併又は既往を有する者
(10) 呼吸器疾患(重篤な気管支喘息,慢性気管支炎等)の合併又は既往を有する者(小児期の非
重篤な喘息の既往は除く)
(11) 消化器系疾患(重篤な消化性潰瘍,逆流性食道炎等)の合併又は既往を有する者(虫垂炎の
既往は除く)
(12) 腎疾患(急性腎不全,糸球体腎炎,間質性腎炎等)の合併又は既往を有する者(結石の既往
は除く)
(13) 脳血管障害(脳梗塞等)の合併又は既往を有する者
(14) 悪性腫瘍の合併又は既往を有する者
(15) 日常的な飲酒量,喫煙量が過度である者
過度の目安;飲酒:アルコール量として平均 45 g/日以上(ビール大ビン 1 本で 25 g,日本酒
1 合で 22 g のアルコールを含有),喫煙:平均 20 本/日以上
(16) 本剤の投与歴のある者
(17) 治験依頼者,本試験に関係する CRO 又は実施医療機関に雇用されている者
(18) その他,治験担当医師により不適当と判断された者
7

2.7.6個々の試験のまとめ
治験薬,投与量及び投与方法:
1.治験薬及びロット番号
ASP1941 錠 50 mg:ASP1941 として 50 mg を含有するフィルムコート錠
ロット番号:10068A
2.投与量及び投与方法
被験者を A 群から F 群の 6 群に無作為に割り付けた。下表に従い,空腹時,食前又は食後条件下
で,各被験者へ投与日の 9 時(目安)に本剤を水 150 mL とともに単回経口投与した。
空腹時投与の場合には,Day −1 の 22 時以降絶食下に朝食を摂らずに本剤を投与した。食前投与
の場合には,Day −1 の 22 時以降絶食下に本剤投与後 5 分から食事を開始し,食事は 20 分以内に
終了した。食後投与の場合には,Day −1 の 22 時以降,朝食を摂取するまでは絶食とし,食事を 8
時 30 分(目安)より 20 分以内で摂取し,食後 10 分以内に本剤を投与した。
治験薬投与日の朝食(食前及び食後投与の被験者が摂取)は高脂肪食 aとし,第 1 期,第 2 期及
び第 3 期とも同一の食事内容とした。第 1 期と第 2 期の間(第 1 期治験薬投与日の翌日~第 2 期
Day −1),並びに第 2 期と第 3 期の間(第 2 期治験薬投与日の翌日~第 3 期 Day −1)に 6 日以上
の休薬期間を設けた。第 2 期及び第 3 期の投与の可否判定は,除外基準を参考に治験担当医師が
各期投与前までの検査結果等をもとに行った。
a:900 kcal 以上,かつ,総エネルギーに対する脂肪のエネルギーの占める割合は 35%以上(「後発医薬品の生物学
的同等性試験ガイドライン」による)
第 1 期 第 2 期 第 3 期
A 群(n=5)
ASP1941 錠
空腹時投与
ASP1941 錠
食前投与
ASP1941 錠
食後投与
B 群(n=5)
ASP1941 錠
空腹時投与
ASP1941 錠
食後投与
ASP1941 錠
食前投与
C 群(n=5)
ASP1941 錠
食前投与
ASP1941 錠
空腹時投与
ASP1941 錠
食後投与
D 群(n=5)
ASP1941 錠
食前投与
ASP1941 錠
食後投与
ASP1941 錠
空腹時投与
E 群(n=5)
ASP1941 錠
食後投与
ASP1941 錠
空腹時投与
ASP1941 錠
食前投与
F 群(n=5)
ASP1941 錠
食後投与
ASP1941 錠
食前投与
ASP1941 錠
空腹時投与
【設定根拠】
(1) 投与量の設定根拠
国内第 II 相試験[CL-0103]の結果から,本剤は 1 日 1 回 50 mg から 100 mg が臨床用法・用量と
想定され,国内第 III 相試験では,ASP1941 として 1 回 50 mg(50 mg 1 錠)を用量とし,増量基
準を満たす場合に 1 回 100 mg(50 mg 2 錠)を服用可能としている。また,国内第 I 相試験[CL-0101]
の結果より,本剤 50 mg 及び 100 mg を単回経口投与したときに,尿中グルコース排泄量では両投
与群間に大きな差はみられなかった。以上の試験成績を踏まえ,ASP1941 最終製剤の薬物動態及
び薬力学に及ぼす食事の影響を評価するために適切な用量として 50 mg に設定した。
(2) 投与間隔の設定根拠
休薬期間は,一般に消失半減期の 5 倍以上の時間をおけば十分と考えられる。国内の健康成人を
対象とした第 I 相試験[CL-0101]及び薬物相互作用試験(ミグリトール)[CL-0062]の結果より,
本剤(10~300 mg の用量範囲で単回経口投与したとき及び 100 mg を単独又はミグリトールと併
用で単回投与したとき)の消失半減期は平均 10.3~14.8 時間であった。以上より,休薬期間とし
8

2.7.6個々の試験のまとめ
て約 50~74 時間(約 2~3 日)程度が必要と考えられる。更に,頻回採血の影響等被験者保護の
観点も考慮して,6 日以上と設定した。
評価期間:
治験薬投与期間(治療期):各期 1 日(3 時期)
併用治療(薬剤及び療法):
第 1 期の治験薬投与から後観察まで,有害事象に対する治療を除き,併用治療(治験薬以外の薬
剤の使用及び治療)を禁止した。併用治療が行われた被験者の治験の継続は治験担当医師が判断
することとした。
評価項目,評価スケジュール及び評価基準:
1.薬物動態|薬力学
(1) 薬物動態
本剤の血漿中未変化体濃度から,以下の薬物動態パラメータをノンコンパートメント法を用いて
算出した。
・最高血漿中濃度(Cmax)
・時間 0 から無限時間まで外挿した血漿中濃度‐時間曲線下面積(AUCinf)
・最高血漿中濃度到達時間(tmax)
・時間 0 から定量可能最終時点までの血漿中濃度‐時間曲線下面積(AUClast)
・消失半減期(t1/2)
・消失速度定数(kel)
・経口クリアランス(CL/F)
・みかけの分布容積(VzF)
・無限時間まで外挿した平均滞留時間(MRTinf)
(2) 薬力学
尿中グルコース排泄量,尿中グルコース排泄速度,排尿量
(3) その他の評価項目
飲水量
2.安全性
有害事象,バイタルサイン(腋窩体温,臥位血圧,臥位脈拍数),臨床検査(血液学的検査,血液
生化学検査,尿検査),12 誘導心電図
治験薬投与後に認められた有害事象を調査対象とした。第 1 期治験薬投与開始時から第 2 期治験
薬投与直前までに発生した有害事象は第 1 期に発生した有害事象として,第 2 期治験薬投与開始
時から第 3 期治験薬投与直前までに発生した有害事象は第 2 期に発生した有害事象として,第 3
期治験薬投与開始時から後観察までに発生した有害事象は第 3 期に発生した有害事象として取り
扱った。有害事象と治験薬との関連性は以下の基準により判定し,「関連あるかもしれない」又は
「多分(おそらく)関連あり」のいずれかに該当したものを,「治験薬との関連性が否定できない
有害事象(副作用)」と定義した。
9

2.7.6個々の試験のまとめ
治験薬との関連性 関連性判定基準
否定できる 有害事象発現と薬剤投与との間に時間的関係から因果関係がありそうにないと思われ
る場合や,他の薬剤や合併症・基礎疾患等により説明ができる場合
関連あるかもしれ
ない
有害事象発現と薬剤投与との間に時間的に妥当な相関関係があるが,以下の項目のいず
れかに該当する場合
・要因として合併症・基礎疾患や他の薬剤による説明もできる場合
・投与中止に関する情報が不明確な場合
多分(おそらく)関
連あり
有害事象発現と薬剤投与との間に時間的に妥当な相関関係がある場合や,以下の項目に
該当する場合
・再投与により再発又は投与中止により消失又は軽快をしめす場合
・合併症・基礎疾患や他の薬剤では説明ができない,又はそれらによる可能性が考えに
くい場合
有害事象の程度は,平成 4 年 6 月 29 日薬安第 80 号添付「医薬品等の副作用の重篤度分類基準に
ついて」のグレードを参考に判定した。この基準に規定されていない項目については,以下に従っ
て判定した。
・軽度:日常の活動に支障がないもの
・中等度:日常の活動に支障があるもの
・重度:日常の活動が不可能になるもの
10

2.7.6個々の試験のまとめ
3.評価スケジュール
【試験全体のスケジュール】
X:実施。
a:第 1 例目の投薬は 9:00 を目安に実施した(2 例目以降順次実施した)。
b:治験期間中に被験者は自覚症状を発現した時点で各自「自覚症状」を治験担当医師又は治験協力者に申し出ることとした。
c:第 1 期,第 2 期及び第 3 期の Day −1 の午後に入院した。
d:入院時の詳細なスケジュールは次頁のとおり。
e:体重のみ測定。
f:投与−12 時間から投与前までをベースラインとした。
g:第 1 期治験薬投与前に被験者の入れ替えが生じた場合は再割り付けを実施することとした。
h:尿中蛋白及び尿中ウロビリノーゲンのみ実施した。
i:6 日以上の休薬期間(第 1[2]期治験薬投与日の翌日~第 2[3]期 Day −1)を経過した後に第 2[3]期治験薬投与を行った。
11

2.7.6個々の試験のまとめ
【入院期間中のスケジュール(第 1,2,3 期共通)】
X:実施。
a:第 1 例目の投薬は 9:00 を目安に実施した(2 例目以降順次実施した)。食前投与の被験者は,投与前採血→摂食 5 分前に治験薬投与→摂食(20 分以内)の順に実施した。
また,食後投与の被験者は,投与前採血→摂食(20 分以内)→摂食後 10 分以内の治験薬投与の順に実施した。
b:投与日を除く入院時の食事時刻は朝食 9:00,昼食 13:00,夕食 19:00 を目安とした。また,投与後 1 日,2 日及び 3 日(退院時)はすべての検査終了後,朝食を摂ることとした。
c:治験期間中に被験者は自覚症状を発現した時点で各自「自覚症状」を治験担当医師又は治験協力者に申し出ることとした。
d:蓄尿期間:投与−12 時間から投与前,投与後 0~2 時間,2~4 時間,4~6 時間,6~8 時間,8~10 時間,10~12 時間,12~24 時間,24~36 時間,36~48 時間,48~72 時間
e:第 2 期,第 3 期は有害事象として調査した。
f:第 1 期のみ実施。
g:体重のみ測定。第 1 期のみ実施。
h:第 1 期治験薬投与前に被験者の入れ替えが生じた場合は再割り付けを実施することとした。
i:治験薬を食前あるいは食後投与する被験者のみ朝食を摂取した。
j:13:30 摂食を目安とした(投与後 4 時間の血漿中未変化体濃度測定用採血終了後,摂食可)。
k:19:30 摂食を目安とした(投与後 10 時間の血漿中未変化体濃度測定用採血終了後,摂食可)。
l:尿中蛋白及び尿中ウロビリノーゲンのみ実施した。
12

2.7.6個々の試験のまとめ
統計解析:
1.解析対象集団
(1) 安全性解析対象集団[Safety analysis set(SAF)]
治験薬を服用したすべての被験者の集団を SAF とした。
(2) 薬物動態解析対象集団[Pharmacokinetics analysis set(PKAS)]
治験薬を服用し,薬物濃度測定用の試料が 1 時点以上で採取されている被験者を PKAS とした。
(3) 薬力学解析対象集団[Pharmacodynamics analysis set(PDAS)]
治験薬を服用し,治験薬投与後に薬力学評価測定用の試料が 1 時点以上で採取されている被験者
を PDAS とした。
2.被験者背景及びその他の基準値
SAF,PKAS 及び PDAS を対象に,計数値項目では度数集計を行い,計量値項目では要約統計量
を算出した。
3.薬物動態
PKAS を対象に,薬物動態パラメータの要約統計量を投与条件別に算出した。
Cmax及び AUClast を主要評価項目として空腹時投与に対する食前投与及び食後投与,並びに食前投
与に対する食後投与の幾何平均比及び 90%信頼区間をそれぞれ算出した。Cmax及び AUClast の幾何
平均比の 90%信頼区間が 0.8~1.25 の範囲内であることを目安として,空腹時投与に対する食前投
与及び食後投与,並びに食前投与に対する食後投与の薬物動態に及ぼす食事の影響の程度を検討
した。
4.薬力学|その他
PDAS を対象に,薬力学パラメータ及び飲水量の要約統計量を投与条件別に算出した。
5.安全性
SAF を対象に,以下の解析を投与条件別に行った。
有害事象:有害事象及び副作用の発現率を投与条件別に算出した。
臨床検査値:計量値項目では,測定時点ごとに実測値の要約統計量を算出した。計数値項目では,
Day −1 と各測定時点のクロス表を作成した。
バイタルサイン:測定時点ごとに実測値の要約統計量を算出した。
12 誘導心電図:心電図所見について,Day −1 と各測定時点のクロス表を作成した。
報告書の日付: 年 月 日
2.7.6.1.2 被験者の内訳及び解析対象集団
被験者の内訳を図 2.7.6.1-1 に示した。
計 30 例が登録され,A 群から F 群の 6 つの投与群に無作為に割り付けられた(各群 5 例)。30
例全例がすべての投与条件(空腹時,食前,食後)で治験薬を服用し,試験を完了した。治験薬
の投与を受けた 30 例すべての被験者を SAF,PKAS 及び PDAS とした。
13

2.7.6個々の試験のまとめ
図 2.7.6.1-1 被験者の内訳
同意取得 58
無作為化前脱落例 28
選択・除外基準関連 13
医療上好ましくない事象 1
同意の撤回 1
その他 a 13
登録 30
A 群 5 B 群 5 C 群 5 D 群 5 E 群 5 F 群 5
第 1 期 5 第 1 期 5 第 1 期 5 第 1 期 5 第 1 期 5 第 1 期 5
第 2 期 5 第 2 期 5 第 2 期 5 第 2 期 5 第 2 期 5 第 2 期 5
第 3 期 5 第 3 期 5 第 3 期 5 第 3 期 5 第 3 期 5 第 3 期 5
完了例 5 完了例 5 完了例 5 完了例 5 完了例 5 完了例 5
a:登録例数が予定数に達したため
Source:CL-0071 総括報告書(5.3.1.1-1)Table 12.1.1.1, Table 12.1.1.2, Table 12.1.1.3 and Table 12.1.1.4 and Appendix 13.2.1.1
2.7.6.1.3 人口統計学的及び他の基準値の特性
PKAS の被験者背景を表 2.7.6.1-1 に示した。
平均年齢は 28.9 歳,BMI の平均値は 21.09 kg/m2 であった。既往歴及び合併症を有していた被験
者はいなかった。
14

2.7.6個々の試験のまとめ
表 2.7.6.1-1 被験者背景(PKAS)
計N=30
年齢[同意取得時](歳) 平均値(標準偏差) 28.9(7.29)
最小値 – 最大値 20-44
中央値 27.0
身長(cm) 平均値(標準偏差) 171.55(5.321)
最小値 – 最大値 160.8-182.7
中央値 171.25
体重[スクリーニング時](kg) 平均値(標準偏差) 62.07(6.178)
最小値 – 最大値 50.3-73.4
中央値 61.15
BMI[スクリーニング時](kg/m2) 平均値(標準偏差) 21.09(1.929)
最小値 – 最大値 18.0-25.1
中央値 20.85
既往症 n(%) なし 30(100.0%)
あり 0
合併症 n(%) なし 30(100.0%)
あり 0
喫煙歴 n(%) なし 13(43.3%)
過去にあり 4(13.3%)
現在あり 13(43.3%)
飲酒歴 n(%) なし 16(53.3%)
過去にあり 0
現在あり 14(46.7%)
Source:CL-0071 総括報告書(5.3.1.1-1)Table 12.1.2.1.2 and Table 12.1.2.2.2
2.7.6.1.4 治験薬の曝露
30 例全例がすべての投与条件(空腹時,食前,食後)で治験薬を服用した。
2.7.6.1.5 薬物動態
2.7.6.1.5.1 血漿中未変化体濃度
血漿中未変化体濃度の平均値(標準偏差)の推移を図 2.7.6.1-2 に示した。また,空腹時,食前
及び食後投与時の未変化体の血漿中薬物動態パラメータを表 2.7.6.1-2 に示した。
Cmaxの平均値は食前投与で最も高く,食後投与で最も低かった。tmaxの平均値は,空腹時投与(1.25
時間)と比較して,食前投与では 0.65 時間に短縮し,食後投与では 2.17 時間に延長した。AUClast
の平均値は,3 つの投与条件下で同様であった。
15

2.7.6個々の試験のまとめ
図 2.7.6.1-2 血漿中未変化体濃度の平均値(標準偏差)の推移(PKAS)
Source:CL-0071 総括報告書(5.3.1.1-1)Figure 8-1
16

2.7.6個々の試験のまとめ
表 2.7.6.1-2 未変化体の血漿中薬物動態パラメータの要約統計量(PKAS)
N 平均値 標準
偏差
最小値 最大値 中央値 幾何
平均値
Na変動係
数(%)
Cmax
(ng/mL)
空腹時投与 30 1174.67 212.99 896.31 1758.82 1156.53 1156.82 30 18.13
食前投与 30 1464.17 340.67 878.67 2224.97 1503.09 1424.24 30 23.27
食後投与 30 976.32 236.00 627.80 1344.70 967.31 949.03 30 24.17
tmax
(h)
空腹時投与 30 1.25 0.41 0.50 2.00 1.25 1.18 30 32.80
食前投与 30 0.65 0.23 0.50 1.00 0.50 0.62 30 35.85
食後投与 30 2.17 0.81 0.50 4.00 2.00 2.00 30 37.52
AUClast
(ng·h/mL)
空腹時投与 30 5147.06 845.61 3239.30 6604.42 5036.17 5076.29 30 16.43
食前投与 30 5338.89 874.76 3176.33 7065.93 5281.62 5264.79 30 16.38
食後投与 30 5148.86 797.74 2898.56 6541.47 5197.73 5081.38 30 15.49
kel
(1/h)
空腹時投与 30 0.0616 0.0218 0.0152 0.1030 0.0617 0.0569 30 35.38
食前投与 30 0.0524 0.0219 0.0156 0.0958 0.0568 0.0470 30 41.73
食後投与 30 0.0493 0.0208 0.0180 0.0885 0.0474 0.0450 30 42.24
AUCinf
(ng·h/mL)
空腹時投与 30 5238.53 872.49 3252.51 6633.86 5223.03 5163.90 30 16.66
食前投与 30 5490.74 948.10 3199.28 7541.51 5471.45 5407.21 30 17.27
食後投与 30 5291.35 838.13 2940.04 6881.05 5479.31 5218.39 30 15.84
t1/2
(h)
空腹時投与 30 13.67 8.45 6.73 45.70 11.23 12.18 30 61.84
食前投与 30 16.90 9.97 7.24 44.51 12.20 14.74 30 58.97
食後投与 30 17.00 7.86 7.84 38.40 14.62 15.42 30 46.22
CL/F(L/h)
空腹時投与 30 9.83 1.84 7.54 15.37 9.57 9.68 30 18.71
食前投与 30 9.40 1.84 6.63 15.63 9.14 9.25 30 19.53
食後投与 30 9.74 1.97 7.27 17.01 9.13 9.58 30 20.22
Vz/F(L)
空腹時投与 30 188.20 108.00 78.58 629.07 156.80 170.14 30 57.38
食前投与 30 223.72 136.38 94.87 675.97 164.95 196.60 30 60.96
食後投与 30 232.15 100.54 115.35 508.58 206.75 213.10 30 43.31
MRTinf
(h)
空腹時投与 30 11.14 4.13 6.44 24.25 9.90 10.59 30 37.06
食前投与 30 12.53 4.65 5.45 24.64 10.57 11.78 30 37.08
食後投与 30 13.79 3.54 8.29 22.58 13.79 13.36 30 25.69
a:幾何平均値を算出した被験者数
Source:CL-0071 総括報告書(5.3.1.1-1)Table 8-1
2.7.6.1.5.2 食事の影響
本剤の薬物動態パラメータに及ぼす食事の影響を表 2.7.6.1-3 及び表 2.7.6.1-4 に示した。
空腹時投与に対する食前投与の Cmax及び AUClast の幾何平均比は,それぞれ 1.231(90% CI: 1.138,
1.332)及び 1.037(90% CI: 1.009, 1.066)であった。食前投与では空腹時投与と比較して,Cmaxが
約 23%高かったが,AUClast は同様であり,AUClast の幾何平均比の 90%信頼区間は,0.80~1.25 の
範囲に含まれていた。
空腹時投与に対する食後投与の Cmax及び AUClast の幾何平均比は,それぞれ 0.820(90% CI: 0.758,
0.888)及び 1.001(90% CI: 0.974, 1.029)であった。食後投与では空腹時投与と比較して,Cmaxは
約 18%低かったが,AUClast は同様であり,AUClast の幾何平均比の 90%信頼区間は,0.80~1.25 の
範囲に含まれていた。
食前投与に対する食後投与の Cmax及び AUClast の幾何平均比は,それぞれ 0.666(90% CI: 0.616,
0.721)及び 0.965(90% CI: 0.939, 0.992)であった。食後投与では食前投与と比較して,Cmaxは約
17

2.7.6個々の試験のまとめ
33%低かったが,AUClast は同様であり,AUClast の幾何平均比の 90%信頼区間は,0.80~1.25 の範
囲に含まれていた。
tmaxの最小二乗平均の変化率では,空腹時投与と比較して,tmaxは食前投与で短くなり,食後投
与で延長した。
表 2.7.6.1-3 ASP1941 の薬物動態パラメータ(Cmax及び AUClast)に及ぼす食事の影響
(PKAS)
比較 GMR GMR の 90%信頼区間
下限 上限
Cmax
食前投与/空腹時投与 1.231 1.138 1.332
食後投与/空腹時投与 0.820 0.758 0.888
食後投与/食前投与 0.666 0.616 0.721
AUClast
食前投与/空腹時投与 1.037 1.009 1.066
食後投与/空腹時投与 1.001 0.974 1.029
食後投与/食前投与 0.965 0.939 0.992
Source:CL-0071 総括報告書(5.3.1.1-1)Table 8-2
表 2.7.6.1-4 ASP1941 の薬物動態パラメータ(tmax)に及ぼす食事の影響(PKAS)
差 最小二乗平均の
変化率
変化率の 90%信頼区間
下限 上限
tmax
食前投与 − 空腹時投与 −0.480 −0.660 −0.300
食後投与 – 空腹時投与 0.733 0.553 0.914
食後投与 – 食前投与 2.333 1.987 2.680
Source:CL-0071 総括報告書(5.3.1.1-1)Table 8-4
2.7.6.1.6 薬力学|その他
尿中グルコース排泄速度の平均値(標準偏差)の推移を表 2.7.6.1-5 に示した。また,累積尿中
グルコース排泄量の平均値(標準偏差)の推移を表 2.7.6.1-6 に示した。
尿中グルコース排泄速度の平均値は,すべての投与条件で本剤投与後に速やかに増加し,投与
後 4~6 時間あるいは 6~8 時間でピークを示した。食前投与では,投与後 0~2 時間の尿中グルコー
ス排泄速度の平均値は,空腹時投与及び食後投与と比較して 2 倍以上に増加した。一方,食後投
与では投与後 2 時間以降に大きな増加がみられ,空腹時投与では投与後 4 時間まで他の投与条件
よりも低値で推移した。投与後 4 時間以降では,3 つの投与条件で,尿中グルコース排泄速度の
平均値に明らかな差はみられなかった。また,投与後 24 時間までの累積尿中グルコース排泄量の
平均値に,3 つの投与条件で明らかな差はみられなかった。
尿中グルコース排泄速度及び累積尿中グルコース排泄量のほか,薬力学パラメータとして排尿
量,並びにその他の評価項目として飲水量を測定した。その結果,排尿量及び飲水量ともに,3
つの投与条件で明らかな差は認められなかった。
18

2.7.6個々の試験のまとめ
表 2.7.6.1-5 尿中グルコース排泄速度(mg/h)の平均値(標準偏差)の推移(PDAS)
評価時点 a 空腹時投与(N=30)
食前投与(N=30)
食後投与(N=30)
−12~0 h 2.41(3.284) 1.80(0.710) 1.77(0.323)
0~2 h 1305.09(437.121) 3278.35(907.315) 1414.36(1157.190)
2~4 h 1987.70(473.322) 3282.13(607.865) 2578.36(428.282)
4~6 h 3975.89(917.878) 3463.59(768.129) 3661.62(989.774)
6~8 h 3751.87(940.173) 3493.40(661.592) 3546.32(824.957)
8~10 h 1765.85(371.963) 2306.54(456.493) 2346.04(628.737)
10~12 h 2872.94(1071.911) 2906.01(1133.393) 3059.72(886.918)
12~24 h 1058.09(275.366) 1203.99(363.620) 1336.27(427.913)
24~36 h 979.70(416.393) 1030.78(453.085) 1078.18(394.386)
36~48 h 122.36(154.650) 170.85(165.682) 182.48(174.710)
48~72 h 70.40(79.014) 114.79(126.573) 119.55(129.331)
平均値(標準偏差)
a:治験薬投与時点を 0 分とした。
Source:CL-0071 総括報告書(5.3.1.1-1)Table 12.3.1.1.1, Table 12.3.1.1.2 and Table 12.3.1.1.3
表 2.7.6.1-6 累積尿中グルコース排泄量(mg)の平均値(標準偏差)の推移(PDAS)
評価時点 a 空腹時投与(N=30)
食前投与(N=30)
食後投与(N=30)
0~2 h 2610.2(874.24) 6556.7(1814.63) 2828.7(2314.38)
0~4 h 6585.6(1586.51) 13121.0(2577.55) 7985.4(2547.01)
0~6 h 14537.4(3062.52) 20048.1(3723.35) 15308.7(3472.61)
0~8 h 22041.1(4601.61) 27034.9(4681.45) 22401.3(4614.61)
0~10 h 25572.8(4853.98) 31648.0(4930.34) 27093.4(5096.30)
0~12 h 31318.7(6475.91) 37460.0(6834.09) 33212.8(6397.06)
0~24 h 44015.8(8702.87) 51907.9(9010.28) 49248.1(9171.64)
0~36 h 55772.2(11969.01) 64277.3(12704.83) 62186.3(12509.02)
0~48 h 57240.5(12769.12) 66327.5(13915.26) 64376.0(13390.14)
0~72 h 58930.1(13798.98) 69082.5(16131.57) 67245.1(15522.11)
平均値(標準偏差)
a:治験薬投与時点を 0 分とした。
Source:CL-0071 総括報告書(5.3.1.1-1)Table 12.3.2.1.1, Table 12.3.2.1.2 and Table 12.3.2.1.3
2.7.6.1.7 安全性
2.7.6.1.7.1 有害事象
いずれの投与条件でも,有害事象は認められなかった。
2.7.6.1.7.2 臨床検査値
いずれの投与条件でも,臨床的に問題となる変化は認められなかった。
19

2.7.6個々の試験のまとめ
2.7.6.1.7.3 バイタルサイン
いずれの投与条件でも,腋窩体温,臥位血圧及び臥位脈拍数に明らかな変化は認められなかっ
た。
2.7.6.1.7.4 心電図
いずれの投与条件でも,心電図に異常所見が認められた被験者はいなかった。
2.7.6.1.8 結論
● 食前投与時の Cmaxは空腹時投与と比較して約 23%高かったが,AUClast は両投与条件間で同様
であり,AUClast の幾何平均比の 90%信頼区間は,0.80~1.25 の範囲に含まれていた。
● 食後投与時の Cmaxは空腹時投与と比較して約 18%低かったが,AUClast は両投与条件間で同様
であり,AUClast の幾何平均比の 90%信頼区間は,0.80~1.25 の範囲に含まれていた。
● 食後投与時の Cmaxは食前投与と比較して約 33%低かったが,AUClast は両投与条件間で同様で
あり,AUClast の幾何平均比の 90%信頼区間は,0.80~1.25 の範囲に含まれていた。
● tmaxの平均値は,空腹時投与の 1.25 時間と比較して,食前投与では 0.65 時間に短縮し,食後
投与では 2.17 時間に延長した。
● 尿中グルコース排泄速度の平均値は,すべての投与条件で本剤投与後に速やかに増加した。
空腹時投与では,尿中グルコース排泄速度の平均値は,投与後 4 時間までは他の投与条件と
比較して低値で推移した。投与後 24 時間までの累積尿中グルコース排泄量の平均値に,3 つ
の投与条件で明らかな差はみられなかった。
● いずれの投与条件でも有害事象は認められなかった。
20

2.7.6.2 絶対バイオアベイラビリティ試験[CL-0057](添付資料 5.3.1.1-2)
2.7.6.2.1 試験方法の概略
治験課題名:非高齢健康成人を対象に ASP1941 を単回投与したときの絶対バイオアベイラビリ
ティを検討する無作為化非盲検 2 期クロスオーバー試験
治験実施医療機関:英国 1 施設
公表文献:なし
治験期間:
治験開始日:2011 年 6 月
治験終了日:2011 年 7 月
開発のフェーズ:第 I 相
目的:
(1) 主要目的 ASP1941(以下,本剤)100 mg を単回経口投与したときの絶対バイオアベイラビリティを評価す
る。 (2) 副次目的 ・ 本剤 100 mg を単回経口投与したときと 25 mg を静脈内投与したときの,本剤及びその代謝物
M2 の薬物動態を評価する。 ・ 本剤 100 mg 単回経口投与の安全性及び忍容性を評価する。 ・ 本剤 25 mg 静脈内投与の安全性及び忍容性を評価する。 治験デザイン・治験方法:
本試験は,非高齢健康成人を対象とした単一施設,無作為化,非盲検,2 期クロスオーバー試験と
して実施した。 被験者は 2 期(各期 4 泊 5 日)とも治験実施医療機関に入院し,第 1 期 Day 1 と第 2 期 Day 1 の
間には 7 日間以上の休薬期間を設けた。健康被験者 14 例を,「第 1 期:経口投与 + 第 2 期:静脈
内投与」と「第 1 期:静脈内投与 + 第 2 期:経口投与」の 2 通りの投与順序に 7 例ずつ無作為に
割り付け,本剤を空腹時投与した。本剤及びその代謝物 M2 の薬物動態を評価するため,各期に
採血及び蓄尿を行った。第 2 期 Day 4 の退院(又は中止)から 7~14 日後に後観察を実施した。
目標被験者数:14 例(男女各 5 例以上)
【設定根拠】
EMA 及び FDA のバイオアベイラビリティ試験ガイドラインに従い,評価可能被験者が 12 例以上
となるよう,目標被験者数を 14 例と設定した。統計的手法による目標被験者数の算出は行ってい
ない。
診断及び選択・除外基準:
1.選択基準(以下の基準をすべて満たす者を対象とした)
(1) 年齢が 18 歳以上 55 歳以下の非高齢健康成人 (2) 女性は,妊娠の可能性がない者(閉経後,卵管結紮及び両側卵管切除等の不妊手術済み又は
子宮摘出の既往を有する)又はホルモン避妊薬以外の適切な(二重法による)避妊法を使用
する者。ホルモン避妊薬以外の適切な避妊法は以下のいずれかとした: ・ 治験薬投与開始 4 週間前から退院 3 カ月後までの禁欲(ライフスタイルとして選択する場
合であり,治験参加を目的とした禁欲は除く) ・ スクリーニング時から遡って 3 カ月以上前のパートナーの不妊手術 ・ スクリーニング時から退院 3 カ月後までのパートナーのコンドーム使用と他の適切な避妊
法(子宮内避妊器具,不妊手術,ペッサリー又は子宮頸管キャップ)の併用 男性は,不妊(不妊手術済み)の者又は適切な避妊法を使用する者。適切な避妊法は以下の
21
2.7.6個々の試験のまとめ

いずれかとした: ・ 治験薬投与開始 4 週間前から退院 3 カ月後までの禁欲(ライフスタイルとして選択する場
合であり,治験参加を目的とした禁欲は除く) ・ 治験薬投与開始日から退院 3 カ月後までのコンドーム使用とパートナーによる他の許容可
能な避妊法(経口又は注射ホルモン避妊薬,避妊パッチ,子宮内避妊器具,卵管結紮,膣
内ホルモンリング,ペッサリー又は子宮頸管キャップ)の併用 ・ パートナーに妊娠の可能性がない(閉経後,不妊手術済み又は子宮摘出の既往)
(3) BMI が 18.5 kg/m2 以上 30.0 kg/m2 未満の者 (4) 被験者本人から文書による同意が得られている者
2.除外基準(以下の基準のいずれかに該当する者は対象から除外した)
(1) スクリーニング時から遡って 6 カ月以内に妊娠していた女性,又はスクリーニング時から
遡って 3 カ月以内に授乳していた女性 (2) 本剤又は製剤の構成成分に対して過敏症を有する者,又は過敏症が疑われる者 (3) 基準値上限を超える肝機能検査値が認められた者(基準値上限を超えた場合,1 回まで再検
査を許容する) (4) 臨床的に重要な喘息,湿疹,アレルギー又は重度の薬物過敏症の既往を有する者(症状の出
ていない花粉症は除く) (5) 治験担当医師により臨床的に重要と判断された消化器系,心血管系,呼吸器系,腎機能,肝
機能,神経系,皮膚,精神又は代謝系の疾患又は障害の既往を有する者 (6) 本試験前の身体所見,心電図検査及び臨床検査の結果,治験担当医師により臨床的に重要な
異常があると判断された者 (7) 本試験前来院時に,脈拍数又は血圧に下記のいずれかの異常が認められた者: 脈拍数<40 bpm 若しくは>90 bpm,平均収縮期血圧>140 mmHg,平均拡張期血圧>90 mmHg(血
圧は臥位にて 5 分間安静後に 3 回測定する。脈拍数は自動測定とする。) (8) 430 msec(男性)又は 450 msec(女性)を超える QTc 延長が 2 回の測定で繰り返し認められ
た者,原因不明の失神,心停止,原因不明の不整脈若しくは Torsade de pointes,又は器質性心
疾患の既往を有する者,又は QT 延長症候群の家族歴を有する者 (9) 入院前 2 週間以内に処方薬又は一般用医薬品(ビタミン剤,経口避妊薬,ホルモン補充療法,
セイヨウオトギリソウ等の生薬又はハーブ製剤を含む)を使用した者(3 g/日以下のアセトア
ミノフェンの頓用を除く) (10) 入院前 3 カ月以内に肝代謝酵素誘導作用を有する薬剤(バルビツール酸類,リファンピシン
等)を常用した者 (11)入院 1 週間前から後観察までにグレープフルーツ(3 回 × 200 mL を超える)又はマーマレー
ド(3 回を超える)を摂取した者(被験者の報告に基づく) (12) 入院前 3 カ月以内に薬物乱用歴を有する者 (13) 入院前 3 カ月以内に 1 日 10 本を超える巻きタバコ(又は当量の刻みタバコ)を使用した者 (14) 入院前 3 カ月以内のアルコール摂取量が 1 週間あたり 21 単位(男性)又は 14 単位(女性)
[1 単位=アルコール 10 g=ビール(5%)250 mL,蒸留酒(35%)35 mL,ワイン(12%)100 mL]を超える者
(15) 入院前 3 カ月以内に献血(全血又は成分献血)を行った者 (16) HBs 抗原,HAV IgM 抗体,HCV 抗体又は HIV 抗体 1 及び 2 が陽性の者 (17) 何らかの理由のため本試験を完了できない可能性があると治験担当医師により判断された者 (18) 本試験登録予定日から遡って 3 カ月以内に他の治験に参加した者,又は 12 カ月以内に 4 件以
上の治験に参加した者,又は半減期の長い生物製剤を使用する治験に参加した者 (19) 治験担当医師により,本試験を安全に完了できない疾患を有すると判断された者 (20) アステラス製薬グループ又は本試験に関連する CRO に雇用されている者
22
2.7.6個々の試験のまとめ

治験薬,投与量及び投与方法:
1.治験薬及びロット番号:
(1) 経口投与 ASP1941 錠 50 mg:1 錠中に ASP1941 として 50 mg を含有するフィルムコート錠 ロット番号:10068E,BX1004035 (2) 静脈内投与 ASP1941 静注剤 25 mg:1 アンプルに ASP1941 として 25 mg を含有する静脈注射剤 ロット番号:BX1004036,W243(1APJ002) 2.投与量及び投与方法:
(1) 経口投与 各期 Day 1 に,100 mg(50 mg 錠 × 2)を 240 mL の水とともに空腹時に単回経口投与した。 (2) 静脈内投与 各期 Day 1 に,25 mg を空腹時に 1 時間かけて静脈内投与した。
【設定根拠】
本試験で単回経口投与した 100 mg は,本剤での有効性が期待される用量であった。 100 mg の静脈内投与は,本剤の溶解性に限度があることから 2000 mL の溶液が必要となり,1 時
間では投与不可能である。そのため,静脈内投与の用量を 1 時間以内で投与可能な 500 mL に溶け
うる 25 mg と設定した。本剤を経口投与した複数の試験の結果,本剤の薬物動態プロファイルは
用量によらず安定しているため,25 mg 静脈内投与による正確な経口バイオアベイラビリティ算
出は可能と考えられた。静脈内投与時間は,薬物動態シミュレーションにおいて本剤 25 mg 及び
100 mg 経口投与に類似した薬物動態プロファイルが得られた 1 時間と設定した。
評価期間:
治験薬投与期間:2 日間(各期 1 日)
併用治療(薬剤及び療法):
入院 2 週間前から後観察まで,すべての併用薬の使用を禁止した(ビタミン剤,ハーブ製剤及び
栄養補助食品を含む)。ただし,治験担当医師が認めた場合に限り,3 g/日以下のアセトアミノフェ
ンの使用は可とした。
23
2.7.6個々の試験のまとめ

評価項目,評価スケジュール及び評価基準:
1.評価スケジュール: スクリー
ニング 第 1 期及び第 2 期 f 後観察 h
Day −21~−2 −1 1 2~3 4 入院 X X X X 退院 X スクリーニング検査 a X 同意取得 X 身体所見 X X X 薬物・アルコール検査 X X X 妊娠検査(女性のみ) X X X 無作為化 X バイオバンキング用検体 投与後
(第 1 期のみ)
臨床検査 X X 投与後 24 時間 X バイタルサイン b (血圧,脈拍数)
X X 投与前及び 投与後 4 時間 投与後 24,48 時間 投与後 72 時間 X
12 誘導心電図(1 回測定) X X 投与前及び 投与後 4 時間 投与後 24,48 時間 投与後 72 時間 X
本剤投与 c 朝投与 食事 d
Day 1 の 投与前 10時間に投与前
最終摂取
投与後 4 時間
に摂取再開 通常どおり 通常どおり
薬物動態評価用採血 (静脈内投与後)e
投与前,投与後 0.25,0.5,1g,1.25,1.5,2,3,4,6,8,12,16,24,36,48,60,72 時間
薬物動態評価用採血 (経口投与後)
投与前,投与後 0.25,0.5,1,1.5,2,3,4,6,8,12,16,24,36,48,60,72 時間
薬物動態評価用蓄尿 投与前,投与後 0~4,4~8,8~12,12~16,16~24, 24~48,48~72 時間
併用薬,有害事象調査 X X X X X X
a: 同意取得,並びに選択・除外基準,被験者背景,既往歴,薬歴,血清学的検査,飲酒及び喫煙習慣の調査。
b: スクリーニング時は 3 回(脈拍数は 1 回),その他の来院では 1 回測定した。
c: 治験薬は,「第 1 期:経口投与 + 第 2 期:静脈内投与」又は「第 1 期:静脈内投与 + 第 2 期:経口投与」のいずれかで投
与した。
d: Day −1の投与前最終摂取及び Day 1 の投与後最初の摂取は eCRF に記録し,その他の食事は原資料のみに記録した。
e: 静脈内投与を実施した腕からは採血しないこととした。
f: 第 1 期 Day 1 と第 2 期 Day 1 の間は 7 日間以上おくこととした。
g: 静脈内投与開始 1 時間後の採血は,投与終了前 2 分間以内に実施した。
h: 後観察は退院(中止)7~14 日後に実施した。
2.薬物動態:
「1. 評価スケジュール」に示した時期に採血及び蓄尿を行った。各被験者の本剤未変化体及びそ
の代謝物 M2 の血漿中及び尿中濃度を測定し,ノンコンパートメント法を用いて以下の薬物動態
パラメータを算出した。
[血漿中未変化体] 主要評価項目
経口投与:無限時間まで外挿した血漿中濃度‐時間曲線下面積(AUCinf) 静脈内投与:AUCinf 絶対バイオアベイラビリティは以下の計算式を用いて算出した。 F =[AUCinf(経口)/投与量(経口)]/[AUCinf(静脈内)/投与量(静脈内)]
24
2.7.6個々の試験のまとめ

[血漿中未変化体] 副次評価項目
経口投与:
最高血漿中濃度到達時間(tmax),最高血漿中濃度(Cmax),定量可能最終時点までの血漿中濃度
‐時間曲線下面積(AUClast),消失半減期(t1/2),経口クリアランス(CL/F),みかけの分布容積
(Vz/F) 静脈内投与: tmax,Cmax,AUClast,t1/2,クリアランス(CL),分布容積(Vz),定常状態での分布容積(Vss)
[血漿中代謝物 M2] 経口投与:tmax,Cmax,AUClast,AUCinf,t1/2,AUCinf (M2)/AUCinf (未変化体)[未変化体に対す
る代謝物の比率(MPR)] 静脈内投与:tmax,Cmax,AUClast,AUCinf,t1/2,CL/F,Vz/F,MPR
[尿中未変化体及び代謝物 M2] 経口及び静脈内投与: 治験薬投与から定量可能最終時点までの尿中排泄量(Aelast),治験薬投与から定量可能最終時点
までの尿中排泄率(Aelast%),無限時間まで外挿した尿中排泄量(Aeinf),無限時間まで外挿した
尿中排泄率(Aeinf%),腎クリアランス(CLr)
3.安全性:
有害事象,臨床検査(血液学的検査,血液生化学検査,尿検査),バイタルサイン(収縮期及び拡
張期血圧,脈拍数),身体所見及び 12 誘導心電図
有害事象は,同意取得から後観察までに発現した事象を収集し,治験薬投与後から後観察までに
認められた有害事象を解析対象として集計した。有害事象と治験薬との関連性は以下の基準によ
り判定し,「関連あるかもしれない」又は「多分(おそらく)関連あり」のいずれかに該当したも
のを「治験薬との関連性が否定できない有害事象(副作用)」と定義した。 治験薬との関連性 関連性判定基準 否定できる 有害事象発現と薬剤投与との間に時間的関係から因果関係がありそうにないと思われる
場合や,他の薬剤や合併症・基礎疾患等により説明ができる場合 関連あるかもしれ
ない 有害事象発現と薬剤投与との間に時間的に妥当な相関関係があるが,要因として合併症・
基礎疾患や他の薬剤による説明もできる場合。投与中止に関する情報が不明確な場合 多分(おそらく)
関連あり 有害事象発現と薬剤投与との間に時間的に妥当な相関関係がある場合,合併症・基礎疾患
や他の薬剤では説明ができない,又はそれらによる可能性が考えにくい場合,再投与によ
り再発又は投与中止により消失又は軽快を示す場合 有害事象の程度は,以下に従って判定した。 ・ 軽度:日常の活動に支障がないもの ・ 中等度:日常の活動に支障があるもの ・ 重度:日常の活動が不可能になるもの 統計解析:
1.解析対象集団:
(1) 安全性解析対象集団(SAF) 治験薬を 1 回以上(経口又は静脈内)投与された被験者の集団を SAF とした。 (2) 薬物動態解析対象集団(PKAS) SAF に含まれる被験者のうち,1 つ以上の薬物動態パラメータ算出のために適切な薬物動態デー
タが得られた被験者の集団を PKAS とした。
25
2.7.6個々の試験のまとめ

2.被験者背景及びその他の基準値:
SAF を対象に,計量値項目では要約統計量(被験者数,平均値,標準偏差,最小値,中央値,最
大値)を算出し,計数値項目では度数集計を行った。
3.薬物動態:
PKAS を対象に,すべての薬物動態パラメータについて,投与法別に以下の要約統計量を算出し
た:被験者数,平均値,標準偏差,最小値,中央値,最大値,変動係数(連続量のみ),幾何平均
値(AUCinf及び Cmaxのみ)
未変化体の用量補正した AUCinfの対数変換値について,投与法,投与順序及び投与期を固定効果,
被験者を変量効果として分散分析を行った。 分散分析による推定値を通常のスケールに逆変換し,絶対バイオアベイラビリティ(経口投与 vs静脈内投与)の推定値として幾何平均比及びその 90%信頼区間を算出した。更に,被験者個々の
バイオアベイラビリティも算出した。
4.安全性:
安全性の解析では,SAF を解析対象とした。 有害事象:有害事象及び副作用の発現率を投与法別に算出した。 臨床検査値:評価時点ごとの実測値とベースライン(各期の治験薬投与開始前)からの変化量の
要約統計量を投与法別に算出した。ベースラインと各評価時点の測定値のクロス表を作成した。 バイタルサイン:評価時点ごとに実測値の要約統計量を投与法別に算出した。 12 誘導心電図:心電図所見の臨床的に重要な異常及び臨床的に重要でない異常の発現率を評価時
点ごとに算出した。 報告書の日付: 年 月 日
2.7.6.2.2 被験者の内訳及び解析対象集団
被験者の内訳を表 2.7.6.2-1 に示した。
14 例の被験者を 2 通りの投与順序に無作為に割り付けた。7 例が「第 1 期:本剤 100 mg経口投
与 + 第 2 期:本剤 25 mg静脈内投与」,7 例が「第 1 期:本剤 25 mg静脈内投与 + 第 2 期:本剤
100 mg経口投与」の投与順序で治験薬を投与された。14 例全例が試験を完了し,治験薬を投与さ
れた 14 例全例をSAF及びPKASとした。
表 2.7.6.2-1 被験者の内訳 第 1 期:経口投与 +
第 2 期:静脈内投与 第 1 期:静脈内投与 +
第 2 期:経口投与 計
無作為化例 7 7 14 治験薬投与例 7 7 14完了例 7 7 14中止例 0 0 0
Source:CL-0057 総括報告書(5.3.1.1-2)Table 12.1.1.1,12.1.1.2,12.1.1.3
26
2.7.6個々の試験のまとめ

2.7.6.2.3 人口統計学的及び他の基準値の特性
SAFの被験者背景を表 2.7.6.2-2 に示した。
全被験者のうち男性が 8 例,女性が 6 例であった。平均年齢は 41.6 歳,年齢の範囲は 20~54
歳であった。
表 2.7.6.2-2 被験者背景(SAF) 計
(N=14) 性別 n(%) 男性 8(57.1)
女性 6(42.9) 人種 n(%) 白人 12(85.7)
黒人 1(7.1) アジア人 0 その他 1(7.1)
年齢(歳) 平均値(標準偏差) 41.6(11.86) 最小値 – 最大値 20 – 54 中央値 47.0
体重(kg) 平均値(標準偏差) 76.74(10.427) 最小値 – 最大値 56.5 – 97.0 中央値 76.85
身長(cm) 平均値(標準偏差) 173.1(7.55) 最小値 – 最大値 161 – 184 中央値 174.0
BMI(kg/m2) 平均値(標準偏差) 25.614(3.0677) 最小値 – 最大値 20.75 – 29.74 中央値 25.745
Source:CL-0057 総括報告書(5.3.1.1-2)Table 12.1.2.1
2.7.6.2.4 治験薬の曝露
14 例全例が 2 通りの投与順序のいずれか(第 1 期:経口投与 + 第 2 期:静脈内投与,又は第 1
期:静脈内投与 + 第 2 期:経口投与)で治験薬を投与され,試験を完了した。
27
2.7.6個々の試験のまとめ

2.7.6.2.5 薬物動態
投与経路ごとの本剤の血漿中未変化体濃度の平均値の推移を図 2.7.6.2-1 に,本剤 100 mg経口投
与及び本剤 25 mg静脈内投与の血漿中未変化体の薬物動態パラメータを表 2.7.6.2-3 に示した。ま
た,本剤未変化体の絶対バイオアベイラビリティを表 2.7.6.2-4 に示した。
経口投与及び静脈内投与ともに,血漿中未変化体濃度はCmaxに達した後に急速に低下し,消失
相は緩やかに延長した。経口投与でのCmaxの平均値は 1406.423 ng/mL,tmaxの中央値は 1.000 時間
であり,静脈内投与でのCmaxの平均値は 612.182 ng/mL,tmaxの中央値は 0.970 時間であった。t1/2
の平均値は,経口投与と静脈内投与で同程度(それぞれ 16.32682 時間及び 16.78124 時間)であり,
血漿中未変化体濃度‐時間曲線の終末相は並行して推移した。経口投与のt1/2は,1 例(被験者 1009)
で 30.96 時間と顕著な高値が認められたが,これを除いた他の被験者では 10.08~21.77 時間であっ
た。絶対バイオアベイラビリティ(F)の平均値は 0.90180 であり,範囲は 0.8118~0.9855 であっ
た。静脈内投与のVzの平均値(260.28239 L)は,Vssの平均値(126.58781 L)の約 2 倍であった。
未変化体の用量補正後のAUC(AUCinf/Dose)の幾何平均値は,経口投与で 83.587 ng•h/mL/mg,
静脈内投与で 93.550 ng•h/mL/mgであった。静脈内投与に対する経口投与の幾何平均比は 0.8935
(90%信頼区間:0.8675,0.9203)であった。
図 2.7.6.2-1 投与経路ごとのASP1941の血漿中未変化体濃度の平均値の推移(片対数スケー
ル)(PKAS)
Source:CL-0057 総括報告書(5.3.1.1-2)Figure 12.4.1.1.2
28
2.7.6個々の試験のまとめ

表 2.7.6.2-3 ASP1941の100 mg経口投与及び25 mg静脈内投与の血漿中未変化体の薬物動
態パラメータ(PKAS) ASP1941 100 mg 経口投与 (N=14)
AUCinf (ng•h/mL)
AUClast (ng•h/mL)
CL/F (L/h)
Cmax (ng/mL)
Fc
t1/2 (h)
tmax (h)
Vss (L)
Vz/F (L)
N 14 14 14 14 14 14 14 − 14 平均値 8456.65937 8179.88020 12.10955 1406.423 0.90180 16.32682 1.500 − 281.96348 標準偏差 1320.096012 1250.038663 2.007799 338.1553 0.053326 5.494872 0.8771 − 97.760088 変動係数(%) 15.6 15.3 16.6 24.0 5.9 33.7 − − 34.7 中央値 8386.02740 8184.51270 11 92830 1427.950 0.90210 15.58955 1.000 − 262.62920 最小値 5925.7834 5887.4430 9.0112 859.39 0.8118 10.0812 0.50 − 178.6138 最大値 11097.3291 10494.6343 16.8754 1848.55 0.9855 30.9639 4.00 − 553.6867 幾何平均値 8358.71199 8089.12240 − 1365.038 − − − − −
ASP1941 25 mg 静脈内投与 (N=14) AUCinf
(ng•h/mL) AUClast
(ng•h/mL) CL/Fa (L/h)
Cmax (ng/mL)
F
t1/2 (h)
tmax (h)
Vss (L)
Vz/Fa (L)
N 14 14 14 13b − 14 13b 13b 14
平均値 2374.36326 2301.31054 10.93334 612.182 − 16.78124 0.972 126.58781 260.28239
標準偏差 428 572818 419.533491 1.969849 90.0090 − 4.994122 0.0038 33.724813 79.570795
変動係数(%) 18.1 18.2 18.0 14.7 − 29.8 − 26.6 30.6
中央値 2280.50900 2185.61560 11.01810 602.520 − 17.86985 0.970 122.67910 255.89390
最小値 1680 9982 1644.4138 8.0441 475.42 − 8.3946 0.97 77.0311 140.5737
最大値 3170.0077 3052.5020 14.8721 845.99 − 24.1338 0.98 215.8902 399.6003
幾何平均値 2338.75345 2266.14365 − 606.530 − − − − −
a:静脈内投与では,CL/F は CL,Vz/F は Vzの値を示した(F = 1)。 b:静脈内投与の中断のため被験者 1005 のパラメータは算出されなかった。 c:値は比率で示した。 −:該当なし Source:CL-0057 総括報告書(5.3.1.1-2)Table 12.4.4.1.1
表 2.7.6.2-4 ASP1941 の絶対バイオアベイラビリティ(PKAS)
ASP1941 100 mg 経口投与
(N=7)
ASP1941 25 mg静脈内投与
(N=7)
幾何平均比 (経口投与/静脈内投与)
幾何平均比の
90%信頼区間
幾何平均 幾何平均 AUCinf/Dose(ng•h/mL/mg) 83.587 93.550 0.8935 0.8675, 0.9203
Source:CL-0057 総括報告書(5.3.1.1-2)Table 12.4.6.1
29
2.7.6個々の試験のまとめ

投与経路ごとの血漿中M2 濃度の平均値の推移を図 2.7.6.2-2 に,本剤 100 mg経口投与及び本剤
25 mg静脈内投与の血漿中M2 の薬物動態パラメータを表 2.7.6.2-5 に示した。
経口投与及び静脈内投与ともに,血漿中M2 濃度はCmaxに達した後に急速に低下し,消失相は緩
やかに延長した。経口投与でのCmaxの平均値は 927.320 ng/mL,tmaxの中央値は 1.750時間であった。
静脈内投与でのCmaxの平均値は 207.307 ng/mL,tmaxの中央値は 1.480 時間であった。t1/2 の平均値
は,静脈内投与及び経口投与で同程度であり(それぞれ 16.84026 時間及び 16.28170 時間),血漿
中M2 濃度‐時間曲線の終末相は並行して推移した。MPRは,経口投与で 0.67273,静脈内投与で
0.56594 であった。 図 2.7.6.2-2 投与経路ごとの血漿中 M2 濃度の平均値の推移(片対数スケール)(PKAS)
Source:CL-0057 総括報告書(5.3.1.1-2)Figure 12.4.1.2.2
30
2.7.6個々の試験のまとめ

表 2.7.6.2-5 ASP1941 の 100 mg経口投与及び 25 mg静脈内投与の血漿中M2の薬物動態パ
ラメータ(PKAS) ASP1941 100 mg 経口投与 (N=14)
AUCinf(ng•h/mL)
AUClast (ng•h/mL)
CL/F (L/h)
Cmax (ng/mL)
MPRa
t1/2 (h)
tmax (h)
Vz/F (L)
N 14 14 − 14 14 14 14 − 平均値 5577.06956 5419.07802 − 927.320 0.67273 16.84026 2.036 − 標準偏差 1002.934034 958 549870 − 221.0358 0.145732 5 369481 0.8872 − 変動係数(%) 18.0 17.7 − 23.8 21.7 31.9 − − 中央値 5453.03300 5269 97170 − 896.450 0.69480 15.80630 1.750 − 最小値 4034.1019 3989.4957 − 495.72 0.4230 9.6390 1.00 − 最大値 7515.3015 7303 2038 − 1360.36 0.9396 28.2032 4.00 − 幾何平均値 5494.67055 5342 36574 − 901.807 − − − −
ASP1941 25 mg 静脈内投与 (N=14) AUCinf
(ng•h/mL) AUClast
(ng•h/mL) CL/F (L/h)
Cmax (ng/mL)
MPRa
t1/2 (h)
tmax (h)
Vz/F (L)
N 14 14 14 13b 14 14 13b 14 平均値 1309.22980 1264 25478 28.21886 207.307 0.56594 16.28170 1.383 654.50334 標準偏差 210.401962 200 545726 4.052598 31.9537 0.121469 4.023048 0.1283 157.428268 変動係数(%) 16.1 15.9 14.4 15.4 21.5 24.7 − 24.1 中央値 1296.60985 1236 17215 27.95775 205.750 0.60390 16.81940 1.480 660.99395 最小値 1040.1450 1008.8893 19.8993 158.68 0.3933 9.6169 1.25 364.6176 最大値 1804.0808 1722 9361 34.6106 271.86 0.7805 21.6047 1.50 946.5095 幾何平均値 1294.54764 1250 39409 − 205.072 − − − −
a:MPR = AUCinf (M2)/AUCinf (未変化体) b:静脈内投与の中断のため被験者 1005 のパラメータは算出されなかった。 −:該当なし Source:CL-0057 総括報告書(5.3.1.1-2)Table 12.4.4.2
本剤の尿中未変化体及び尿中M2 の薬物動態パラメータを,それぞれ表 2.7.6.2-6 及び表 2.7.6.2-7
に示した。
未変化体のAeinf%の平均値は,経口投与で 1.16589%,静脈内投与で 1.35746%であり,静脈内投
与でわずかに高かった。未変化体のCLrの平均値は,静脈内投与及び経口投与で同程度であった。
M2 のAeinf%の平均値は,経口投与で 43.08864%,静脈内投与で 44.40907%であり,両投与法で
同程度であった。M2 のCLrの平均値は,静脈内投与及び経口投与で同程度であった。
31
2.7.6個々の試験のまとめ

表 2.7.6.2-6 ASP1941の100 mg経口投与及び25 mg静脈内投与の尿中未変化体の薬物動態
パラメータ(PKAS) ASP1941 100 mg 経口投与 (N=14)
Aeinf (mg)
Aeinf% (%)
Aelast (mg)
Aelast% (%)
CLr (L/h)
N 14 14 14 14 14 平均値 1.16589 1.16589 1.12756 1.12756 0.13815 標準偏差 0.296985 0.296985 0.283544 0.283544 0.027135 変動係数(%) 25.5 25.5 25.1 25.1 19.6 中央値 1.05015 1.05015 1.02150 1.02150 0.13355 最小値 0.8212 0.8212 0.8159 0.8159 0.0955 最大値 1.8761 1.8761 1.7975 1.7975 0.1931 幾何平均値 1.13472 1.13472 1.09814 1.09814 0.13576
ASP1941 25 mg 静脈内投与 (N = 14) Aeinf (mg)
Aeinf% (%)
Aelast (mg)
Aelast% (%)
CLr (L/h)
N 14 14 14 14 14 平均値 0.34233 1.35746 0.33324 1.32349 0.14584 標準偏差 0.078843 0.316484 0.074767 0.301615 0.028848 変動係数(%) 23.0 23.3 22.4 22.8 19.8 中央値 0.32155 1.27410 0.31390 1.24910 0.14640 最小値 0.2363 0.9339 0.2372 0.9375 0.0946 最大値 0.5176 2.0703 0.5043 2.0251 0.2008 幾何平均値 0.33467 1.32659 0.32624 1.29503 0.14310
Source:CL-0057 総括報告書(5.3.1.1-2)Table 12.4.5.1.1
表 2.7.6.2-7 ASP1941 の 100 mg経口投与及び 25 mg静脈内投与の尿中M2の薬物動態パラ
メータ(PKAS) ASP1941 100 mg 経口投与 (N=14)
Aeinf (mg)
Aeinf% (%)
Aelast (mg)
Aelast% (%)
CLr (L/h)
N 14 14 14 14 14 平均値 62.04764 43.08864 60.42339 41.96069 11.33805 標準偏差 11.948402 8.297509 12.080965 8.389552 2.476046 変動係数(%) 19.3 19.3 20.0 20.0 21.8 中央値 60.73010 42.17370 59.84595 41.55970 11.28260 最小値 38.2541 26.5653 36.2596 25.1803 7.2908 最大値 85.2355 59.1913 82.8300 57.5208 17.1251 幾何平均値 60.92397 42.30832 59.23525 41.13560 11.08783
ASP1941 25 mg 静脈内投与 (N=14) Aeinf (mg)
Aeinf% (%)
Aelast (mg)
Aelast% (%)
CLr (L/h)
N 14 14 14 14 14 平均値 16.05389 44.40907 15.64019 43.26699 12.43707 標準偏差 3.036558 8.446189 2.946646 8.206376 2.414489 変動係数(%) 18.9 19.0 18.8 19.0 19.4 中央値 16.01520 44.98555 15.67640 43.69535 12.91265 最小値 10.7163 29.2795 10.3472 28.2710 7.7499 最大値 22.5782 62.8919 21.5627 60.0631 16.4491 幾何平均値 15.78602 43.65406 15.37612 42.52054 12.19423
Source:CL-0057 総括報告書(5.3.1.1-2)Table 12.4.5.2
32
2.7.6個々の試験のまとめ

2.7.6.2.6 安全性
2.7.6.2.6.1 有害事象
本試験で認められた有害事象の要約を表 2.7.6.2-8 に示した。
有害事象は,経口投与時に 14 例中 3 例(21.4%),静脈内投与時に 14 例中 7 例(50.0%)に認め
られた。
いずれの有害事象も程度は軽度であり,第 2 期の経口投与 9 日後に 1 例に認められたインフル
エンザを除き,他の事象はいずれも回復が確認された。
表 2.7.6.2-8 有害事象の要約(SAF) ASP1941
100 mg 経口投与 ASP1941
25 mg 静脈内投与 計
(N=14) (N=14) (N=14) 有害事象発現例数(発現率) 3(21.4%) 7(50.0%) 8(57.1%) 有害事象発現件数 8 15 23 副作用発現例数(発現率) 1(7.1%) 4(28.6%) 4(28.6%) 副作用発現件数 5 8 13 重篤な有害事象発現例数(発現率) 0 0 0 重篤な副作用発現例数(発現率) 0 0 0 中止に至った有害事象発現例数(発現率) 0 0 0 中止に至った副作用発現例数(発現率) 0 0 0
Source:CL-0057 総括報告書(5.3.1.1-2)Table 12.6.1.1.2,12.6.1.5
33
2.7.6個々の試験のまとめ

有害事象の内訳を表 2.7.6.2-9 に示した。
有害事象の器官別大分類で認められた主なものは,胃腸障害,全身障害および投与局所様態,
神経系障害であった。個別の有害事象では,浮動性めまいが静脈内投与時に 2 例(14.3%)に認め
られたが,他の有害事象はいずれの投与法でも 1 例(7.1%)にのみ認められた。
表 2.7.6.2-9 有害事象の内訳(SAF) MedDRA version 12.1 器官別大分類
基本語
ASP1941 100 mg 経口投与
(N=14)
ASP1941 25 mg 静脈内投与
(N=14) 発現例数(発現率) 発現件数 発現例数(発現率) 発現件数
すべての有害事象 3(21.4%) 8 7(50.0%) 15 胃腸障害 2(14.3%) 2 2(14.3%) 3 下痢 1(7.1%) 1 0 0 消化不良 0 0 1(7.1%) 1 悪心 1(7.1%) 1 1(7.1%) 2
全身障害および投与局所様態 1(7.1%) 3 2(14.3%) 4 無力症 1(7.1%) 1 0 0 カテーテル留置部位血腫 0 0 1(7.1%) 1 カテーテル留置部位疼痛 0 0 1(7.1%) 1 異常感 1(7.1%) 1 0 0 空腹 1(7.1%) 1 1(7.1%) 1 血管穿刺部位血腫 0 0 1(7.1%) 1
神経系障害 1(7.1%) 1 2(14.3%) 3 浮動性めまい 1(7.1%) 1 2(14.3%) 3
内分泌障害 0 0 1(7.1%) 1 早発初経 0 0 1(7.1%) 1
感染症および寄生虫症 1(7.1%) 1 0 0 インフルエンザ 1(7.1%) 1 0 0
傷害、中毒および処置合併症 0 0 1(7.1%) 1 挫傷 0 0 1(7.1%) 1
筋骨格系および結合組織障害 1(7.1%) 1 1(7.1%) 1 四肢痛 1(7.1%) 1 1(7.1%) 1
呼吸器、胸郭および縦隔障害 0 0 1(7.1%) 1 口腔咽頭痛 0 0 1(7.1%) 1
皮膚および皮下組織障害 0 0 1(7.1%) 1 薬疹 0 0 1(7.1%) 1
Source:CL-0057 総括報告書(5.3.1.1-2)Table 12.6.1.2,12.6.1.4
34
2.7.6個々の試験のまとめ

副作用の内訳を表 2.7.6.2-10 に示した。
副作用は,経口投与時の 14 例中 1 例(7.1%),静脈内投与時の 14 例中 4 例(28.6%)に認めら
れ,静脈内投与で多かった。経口投与時で 1 例に認められた有害事象(悪心,浮動性めまい,無
力症,異常感,空腹各 1 件)はいずれも,治験担当医師により「治験薬と関連あるかもしれない」
と判定された。静脈内投与時で 4 例に認められた有害事象(消化不良 1 件,悪心 2 件,浮動性め
まい 3 件,早発初経 1 件,空腹 1 件)はいずれも,治験担当医師により「治験薬と関連あるかも
しれない」と判定された。
表 2.7.6.2-10 副作用の内訳(SAF) MedDRA version 12.1 器官別大分類
基本語
ASP1941 100 mg 経口投与
(N=14)
ASP1941 25 mg 静脈内投与
(N=14) 発現例数(発現率) 発現件数 発現例数(発現率) 発現件数
すべての副作用 1(7.1%) 5 4(28.6%) 8 胃腸障害 1(7.1%) 1 2(14.3%) 3 消化不良 0 0 1(7.1%) 1 悪心 1(7.1%) 1 1(7.1%) 2
神経系障害 1(7.1%) 1 2(14.3%) 3 浮動性めまい 1(7.1%) 1 2(14.3%) 3
内分泌障害 0 0 1(7.1%) 1 早発初経 0 0 1(7.1%) 1
全身障害および投与局所様態 1(7.1%) 3 1(7.1%) 1 無力症 1(7.1%) 1 0 0 異常感 1(7.1%) 1 0 0 空腹 1(7.1%) 1 1(7.1%) 1
Source:CL-0057 総括報告書(5.3.1.1-2)Table 12.6.1.3,12.6.1.5
2.7.6.2.6.2 死亡,その他の重篤な有害事象
本試験では,死亡及びその他の重篤な有害事象は認められなかった。
2.7.6.2.6.3 中止に至った有害事象
本試験では,治験薬投与中止に至った有害事象は認められなかった。
2.7.6.2.6.4 臨床検査値
血液学的検査,血液生化学検査及び尿検査パラメータに,臨床的に重要と判定された変動は認
められなかった。
2.7.6.2.6.5 バイタルサイン
収縮期血圧,拡張期血圧及び脈拍数に,ベースラインからの明らかな変動は認められなかった。
35
2.7.6個々の試験のまとめ

2.7.6.2.6.6 心電図
治験担当医師により臨床的に重要と判定された心電図の異常所見は認められなかった。
2.7.6.2.7 結論
● 本剤 100 mg を空腹時に単回経口投与したときの絶対バイオアベイラビリティの平均値は
90.18%であり,範囲は 81.18%~98.55%であった。
● 経口投与及び静脈内投与ともに,血漿中未変化体濃度は Cmaxに達した後に急速に低下し,消
失相は緩やかに延長した。経口投与での Cmaxの平均値は 1406.423 ng/mL,tmax の中央値は 1.000
時間であり,静脈内投与での Cmaxの平均値は 612.182 ng/mL,tmaxの中央値は 0.970 時間であっ
た。t1/2 の平均値は,経口投与と静脈内投与で同程度(約 16 時間)であった。未変化体の
AUCinf/Dose の幾何平均値は,経口投与で 83.587 ng•h/mL/mg,静脈内投与で 93.550 ng•h/mL/mg
であり,静脈内投与に対する経口投与の幾何平均比は 0.8935(90%信頼区間:0.8675,0.9203)
であった。
● Cmax及び AUC に認められた個体間変動は経口投与及び静脈内投与で同程度であったことか
ら,本剤未変化体の吸収の個体間変動は低いことが示された。
● 静脈内投与での未変化体の Aeinf%の平均値は 1.35746%,M2 の Aeinf%の平均値は 44.40907%
であったことから,未変化体の消失には代謝など他の消失経路の寄与が大きいことが示され
た。更に,経口投与での未変化体の Aeinf%は 1.16589%であり,静脈内投与より低かったが,
その差(約 10%)は経口投与時のバイオアベイラビリティ(90.18%)により説明可能であり,
未変化体の消失経路は投与経路に依存しないことが示唆された。
● 未変化体及び M2 の CLr の平均値は,静脈内投与及び経口投与で同程度であった。
● 本剤 100 mg 単回経口投与及び 25 mg 単回静脈内投与の安全性及び忍容性は良好であった。
● 有害事象は,経口投与時に 14 例中 3 例(21.4%),静脈内投与時に 14 例中 7 例(50.0%)に認
められた。副作用は,経口投与時の 14 例中 1 例(7.1%),静脈内投与時の 14 例中 4 例(28.6%)
に認められた。重篤な有害事象及び治験薬投与中止に至った有害事象は認められなかった。
● 有害事象の器官別大分類で認められた主なものは,胃腸障害,全身障害および投与局所様態,
神経系障害であった。浮動性めまいが静脈内投与時で 2 例に認められたが,他の有害事象は
いずれの投与法でも 1 例にのみ認められた。
36
2.7.6個々の試験のまとめ

2.7.6.3 第 I 相試験[CL-0101](添付資料 5.3.3.1-1)
2.7.6.3.1 試験方法の概略
治験課題名:ASP1941 第Ⅰ相試験-健康成人男性を対象としたプラセボ対照単回及び反復経口
投与試験-
治験実施医療機関:日本 1施設
公表文献:なし
治験期間:
治験開始日:2006 年 12 月
治験終了日:2007 年 7 月
開発のフェーズ:第 I 相
目的:
(1) 主要目的
・健康成人男性を対象とし,ASP1941(以下,本剤)を単回経口及び反復経口投与したときの安
全性及び忍容性について検討する。
(2) 副次目的
・健康成人男性を対象とし,本剤を単回経口及び反復経口投与したときの薬物動態及び薬力学に
ついて検討する。また,100 mg において,安全性,薬物動態及び薬力学に与える食事の影響を予
備的に検討する。
治験デザイン・治験方法:
本試験は,第一部と第二部の 2 部構成で実施した。
第一部(単回経口投与)
治験薬投与前日の夕食以降絶食下の被験者に,本剤 1,3,10,30,100,300 mg あるいはプラセ
ボを単回経口投与した。各用量群の被験者数は,8 例(本剤群 6 例,プラセボ群 2 例)とし,本
剤の低用量から検討した。また,100 mg 用量群で空腹時単回経口投与を終了した被験者には,食
事の影響を検討するために食後に 100 mg あるいはプラセボを単回経口投与した。
第二部(反復経口投与)
治験薬投与日(Day 1)の朝食後,被験者に本剤 20,50,100 mg 又はプラセボを単回経口投与し,
1 日間の休薬後,Day 3 から 1 日 1 回朝食後,7 日間反復経口投与した。各用量群の被験者数は,
12 例(本剤群 8 例,プラセボ群 4 例)とし,本剤の低用量から検討した。
第一部,第二部とも,薬剤は無作為に割り付け,単盲検試験として実施した。
なお,本試験では,HbA1c 値の表記として Japan Diabetes Society(JDS)値を用いた。
目標被験者数:
第一部(単回経口投与):各用量 8 例(合計 48 例)
各用量群で,本剤を 6 例,プラセボを 2 例に割り付けた。100 mg の用量群では,更に同一被験者
にて食後の影響を検討した。
第二部(反復経口投与):各用量 12 例(合計 36 例)
各用量群で,本剤を 8 例,プラセボを 4 例に割り付けた。
【設定根拠】
実施可能性及びこれまで実施されてきた単回及び反復経口投与試験を考慮して被験者数を決定し
た。
診断及び選択・除外基準:
スクリーニング検査時及び投与前に以下の選択基準のすべてを満たし,かつ除外基準のいずれ
にも該当しない健康成人男性を対象とした。
1.選択基準
(1) 性別;男性
37
2.7.6個々の試験のまとめ

2.7.6個々の試験のまとめ
(2) 年齢(同意取得時);20 歳以上 45 歳未満
(3) 体重;50.0 kg 以上 85.0 kg 未満
(4) BMI;17.6 kg/m2 以上 26.4 kg/m2 未満
(5) 文書による同意が得られている
2.除外基準
(1) 以下の既往を有する者†
1) 肝疾患(ウィルス性肝炎,薬物性肝障害等)
2) 心疾患(うっ血性心不全,狭心症,治療を要する不整脈等)
3) 呼吸器疾患(重篤な気管支喘息,慢性気管支炎等)
4) 消化器系疾患(重篤な消化性潰瘍,逆流性食道炎,各種切除術を必要とする疾患等,な
お虫垂炎は除く)
5) 腎疾患(急性腎不全,糸球体腎炎,間質性腎炎等)
6) 脳血管障害(脳梗塞等)
7) 悪性腫瘍
8) 薬物アレルギー,花粉症以外のアレルギー性疾患
9) 薬物依存,アルコール依存
†第一部(単回経口投与)100 mg 群では,100 mg 空腹時投与後から食後投与前に発現した有害事
象も対象とすることとした。
(2) 何らかの疾患を合併する者
(3) 血圧・脈拍数,体温,12 誘導心電図において以下の基準を逸脱する者
項目 基準範囲
臥位血圧収縮期血圧:90 mmHg 以上,140 mmHg 未満
拡張期血圧:40 mmHg 以上,90 mmHg 未満
臥位脈拍数 40 回/分以上,100 回/分未満
腋窩体温 35.0℃以上,37.1℃未満
12 誘導心電図 正常あるいは臨床的に重要でない異常
(4) 臨床検査において以下の基準を逸脱する者
項目 許容範囲
血液学的検査:施設基準の上下限の±20%の範囲内
(ただし白血球数が上記範囲内であれば分画の個々の値については問わない。)
血液生化学検査,尿検査
(pH,比重,浸透圧,Ⅳ型コ
ラーゲン):
・AST,ALT,クレアチニン,Na,K;施設基準の範囲内
・-GTP,Cl,Ca,Glu;施設基準の上下限の±20%の範囲内
・上記以外の項目;治験責任医師(又は治験分担医師)が臨床的に重要な異常では
ないと判断する範囲内
(ただし,下限値の逸脱が臨床的に問題はないと考えられる AST,ALT,-GTP,
総ビリルビン,直接ビリルビン,中性脂肪,ALP,LDH,CK,CRP,総コレステ
ロール,尿中Ⅳ型コラーゲンについては,下限を設定しない。)
尿(潜血,蛋白,糖,ビリル
ビン,ケトン体),便検査:(+)未満
その他検査: 免疫学的検査,尿中薬物検査のいずれも陰性
注 1)±20%の計算値は,基準範囲のもつ有効数字まで四捨五入した。
注 2)血液生化学的検査項目のプロトロンビン時間,活性化部分トロンボプラスチン時間,血清浸透圧及び尿
浸透圧,尿中Ⅳ型コラーゲン,蓄尿の尿検査項目は,スクリーニング検査時のみ判断基準に用いた。上記尿検
査項目に関する基準に,尿沈渣を含まないこととした。
38

2.7.6個々の試験のまとめ
(5) 治験薬投与前 14 日以内に,薬剤投与を含む治療を受けた者
(6) スクリーニング検査前及び治験薬投与前 120 日以内に,他の治験又は製造販売後臨床試験に
おいて投薬を受けた者
(7) 本剤の治験において投与歴がある者‡
‡第一部(単回経口投与)100 mg 群では,100 mg 空腹時投与に引き続き,食後投与も受ける被験
者の食後投与前の確認については対象外とした。
(8) スクリーニング検査前及び治験薬投与前 90 日以内に 400 mL 以上の全血採血,30 日以内に
200 mL 以上の全血採血又は 14 日以内に成分献血を実施した者
(9) 日常的な飲酒量,喫煙量が過度である者(過度の目安; 飲酒:アルコール量として 45 g/日
(ビール大ビン1本で 25 g,日本酒1合で 22 g のアルコールを含有),喫煙:20 本/日)
(10) 治験実施計画書で規定されている被験者が遵守すべき事項を遵守できない者
(11) 治験依頼者又は治験実施医療機関に雇用されている者
(12) 治験担当医師により不適当と判断された者
(13) スクリーニング検査時及び投与前日(Day -1)の空腹時血糖値が 70 mg/dL 未満又は 110 mg/dL
以上,又はスクリーニング検査時の HbA1c 値が 5.8%以上の者
(14) スクリーニング検査時の心電図及び詳細 QT 計測(第二部のみ)において,以下の ECG の基
準を逸脱する者
項目 許容範囲
PQ 120 ms 以上 200 ms 未満
QRS 間隔 120 ms 未満
QTcB,QTcF 430 ms 未満
HR 40 bpm 以上
治験薬,投与量及び投与方法:
1.治験薬及びロット番号
(1) 被験薬
ASP1941 錠 1 mg:ASP1941 として 1 mg を含有するフィルムコート錠
ASP1941 錠 10 mg:ASP1941 として 10 mg を含有するフィルムコート錠
ASP1941 錠 100 mg:ASP1941 として 100 mg を含有するフィルムコート錠
ロット番号(共通):1941-CL-0101
(2) 対照薬
ASP1941 錠 1 mg プラセボ:ASP1941 を含有しない,ASP1941 錠 1 mg と外観上識別不能な錠
剤
ASP1941 錠 10 mg プラセボ:ASP1941 を含有しない,ASP1941 錠 10 mg と外観上識別不能な
錠剤
ASP1941 錠 100 mg プラセボ:ASP1941 を含有しない,ASP1941 錠 100 mg と外観上識別不能
な錠剤
ロット番号(共通):1941-CL-0101
2.投与量及び投与方法
第一部(単回経口投与)
治験薬投与前日の夕食以降絶食下の被験者に,本剤 1,3,10,30,100,300 mg あるいはプラセ
ボを水 150 mL とともに単回経口投与した。また,100 mg 空腹時単回経口投与を終了した被験者
に,空腹時単回経口投与前の割り付けに従って,食後に 100 mg あるいはプラセボを水 150 mL と
ともに単回経口投与した。
39

2.7.6個々の試験のまとめ
第二部(反復経口投与)
治験薬投与日(Day 1)の朝食後,被験者に本剤 20,50,100 mg 又はプラセボを水 150 mL ととも
に単回経口投与し,1 日間の休薬後,Day 3 から 1 日 1 回朝食後,7 日間反復経口投与した。
【設定根拠】
第一部(単回経口投与)
本剤の非臨床試験の結果,ラット及びサルの 2 週間反復経口投与毒性試験の無毒性量はそれぞれ
1 mg/kg/day 及び 100 mg/kg/day であった。従って,安全性試験成績からは,本試験の初回投与量
は,1 mg(1 mg/kg×1/60×60kg)と算出された。また,本剤の推定臨床用量は薬理試験成績(血糖
降下作用)を基に 5.7~7.4 mg/man/day と算定されていることから,この値の 1/10 を指標とした場
合,約 0.6~0.7 mg/man/day という用量が算出された。マウスの薬理試験成績からは正常血糖への
影響は糖負荷したモデルに比べて 100 倍以上の乖離(高用量が必要)があることが示されているこ
とから,健康成人を対象とした場合,初回用量から直ちに血糖への影響が現れるとは考えにくい。
これらのことを勘案した場合,健康成人では血糖降下にまではつながらないことが予想されたた
め,被験者の安全に関して特に問題はない用量として本剤の第Ⅰ相単回経口投与試験に用いる初
回用量を 1 mg とすることは妥当と判断した。
また,第一部では初回投与量に続き,3,10,30,100,300 mg の計 6 用量を計画しているが,低
用量から薬理効果並びに安全性評価を十分に行い,各投与群における安全性と忍容性を確認した
上で,公比 3 で慎重に増量していくことにより,安全に実施することが可能であると判断した。
予備的な食事の影響の検討は,第二部で実施可能性のある最高用量における影響を確認し,第二
部の用法用量設定の妥当性を確認するため 100 mg とした。
第二部(反復経口投与)
第一部で安全性と忍容性が確認された最高用量が 300 mg であった場合には 20,50,100 mg を,
100 mg であった場合には 10,20,50 mg と計画した。第二部の開始用量は,第一部で安全性と忍
容性が確認された最高用量の 10 分の 1 以下であることから,妥当な投与量と考えた。また,それ
以降の投与量については,低用量から薬理効果並びに安全性評価を十分に行い,前の投与量にお
ける安全性と忍容性を確認した上で,公比 3 未満で慎重に増量していくことにより,安全に実施
することが可能であると判断した。
評価期間:
第一部(単回経口投与)
治験薬投与期間:1 日間(100 mg を除く),2 日間(100 mg,空腹時及び食後)
評価期間:8 日間(100 mg を除く),16 日間(100 mg)
第二部(反復経口投与)
治験薬投与期間:1 日(単回)+7 日間(反復)
評価期間:16 日間
併用治療(薬剤及び療法):
有害事象に対する治療を除き,治験期間中の併用治療(治験薬以外の薬剤の使用,及び療法)を
原則禁止とした。
評価項目,評価スケジュール及び評価基準:
1.薬物動態|薬力学:
(1) 薬物動態
第一部と第二部[単回経口投与時(Day 1)及び反復経口投与時(Day 9)]に採血及び蓄尿を行
い,血漿中及び尿中の未変化体濃度を測定した。
血漿中及び尿中の未変化体濃度から,以下の薬物動態パラメータをノンコンパートメント法を用
いて算出した。
[血漿中パラメータ]
・最高血漿中濃度到達時間(tmax)
・最高血漿中濃度(Cmax)
40

2.7.6個々の試験のまとめ
・時間 0 から定量可能最終時点までの血漿中濃度-時間曲線下面積(AUClast)
・時間 0 から無限時間まで外挿した血漿中濃度-時間曲線下面積(AUCinf)
・経口クリアランス(CL/F)
・消失半減期(t1/2)
なお,第二部では,AUCinf及び AUClast は単回経口投与時(Day 1)のみ算出し,反復経口投与時
(Day 9)には AUC24hを算出した。
[尿中パラメータ]
・尿中排泄量(Ae)
・尿中排泄率(Ae%)
・腎クリアランス(CLr)
(2) 薬力学
血糖値,尿中グルコース排泄量,排尿量,尿中 Na,血中コルチゾール,飲水量及び体重・体脂肪
率
2.安全性:
バイタルサイン(腋窩体温,臥位血圧,臥位脈拍数),12 誘導心電図(標準及び詳細 QT 計測),
臨床検査(血液学的検査,血液生化学検査,尿検査,便検査),有害事象
有害事象は,治験薬投与後に認められた事象とした。第一部(単回経口投与)の 100 mg 群に参加
した被験者で認められた有害事象は,発現日時によって 1) 100 mg 空腹投与の投与後~100 mg 空
腹投与の後観察,2) 100 mg 空腹投与の後観察~100 mg 食後投与の投与前,又は 3) 100 mg 食後投
与の投与後~100 mg 食後投与の後観察の 3 つの期間のいずれかに分類した。
有害事象と治験薬との関連性は,「否定できる」,「関連あるかもしれない」,「多分(おそらく)関
連あり」の基準で判定し,「関連あるかもしれない」又は「多分(おそらく)関連あり」に該当し
た有害事象を「治験薬との関連性が否定できない有害事象(副作用)」と定義した。
治 薬との関連性 関連性判定基準
否定できる 有害事象発現と薬剤投与との間に時間的関係から因果関係がありそう
にないと思われる場合や,他の薬剤や合併症・基礎疾患等により説明
ができる場合
関連あるかもしれな
い
有害事象発現と薬剤投与との間に時間的に妥当な相関関係があるが,
以下の項目のいずれかに該当する場合
・要因として合併症・基礎疾患や他の薬剤による説明もできる場合
・投与中止に関する情報が不明確な場合
多分(おそらく)関連
あり
有害事象発現と薬剤投与との間に時間的に妥当な相関関係がある場合
や,以下の項目に該当する場合
・再投与により再発又は投与中止により消失又は軽快をしめす場合
・合併症・基礎疾患や他の薬剤では説明ができない,又はそれらによ
る可能性が考えにくい場合
有害事象の程度は,平成 4 年 6 月 29 日薬安第 80 号添付「医薬品等の副作用の重篤度分類基準に
ついて」のグレードを参考に判定した。この基準に規定されていない項目については,下記に従っ
て判定した。
・軽 度:日常の活動に支障がないもの
・中等度:日常の活動に支障があるもの
・重 度:日常の活動が不可能になるもの
41

2.7.6個々の試験のまとめ
3. Part 別の評価スケジュール及び評価項目
評価スケジュールを以下に示した。
第一部(単回経口投与)(100 mg 食後投与を除く)
a:治験薬の投与と重なった検査は,すべて投与前に実施した。
b:治験薬投与後は各時点の採血・採尿の後に食事をした。
c:ベースラインとした。ただし,空腹時血糖値では,Day 1 の朝食前血糖値をベースラインとした。
d:治験担当医師は,それぞれの時間で問診を実施したほか,被験者が自覚症状を発現した場合にはその時点における症状を記録した。
e:有害事象は発現ごとに記録した。
42

2.7.6個々の試験のまとめ
第一部(単回経口投与)(100 mg 食後投与)
本スケジュールは,100 mg 空腹時投与の後観察後のおおよそ 7~10 日後の入院から開始された。
a:治験薬の投与と重なった検査は,すべて投与前に実施した。
b:治験薬投与後は各時点の採血・採尿の後に食事をした。
c:ベースラインとした。ただし,空腹時血糖値では,Day 1 の朝食前血糖値をベースラインとした。
d:治験担当医師は,それぞれの時間で問診を実施したほか,被験者が自覚症状を発現した場合にはその時点における症状を記録した。
e:有害事象は発現ごとに記録した。
43

2.7.6個々の試験のまとめ
第二部(反復経口投与)前半
44

2.7.6個々の試験のまとめ
二部(反復経口投与)後半
a:治験薬の投与と重なった検査は,すべて投与前に実施した。
b:治験薬投与後は各時点の採血・採尿の後に食事をした。
c:ベースラインとした。ただし,空腹時血糖値では,Day 1 の朝食前血糖値をベースラインとした。
d:治験担当医師は,それぞれの時間で問診を実施したほか,被験者が自覚症状を発現した場合にはその時点における症状を記録した。
e:有害事象は発現ごとに記録した。
45

2.7.6個々の試験のまとめ
統計解析:
1.解析対象集団:
(1) 安全性解析対象集団(SAF)
治験薬を投与された全被験者を対象とした。
(2) 薬物動態解析対象集団(PKAS)
治験薬投与例のうち,本剤投与例でかつ血漿中未変化体濃度もしくは尿中未変化体濃度の検体が
採取されている被験者を対象とした。
(3) 薬力学解析対象集団(PDAS)
治験薬投与例のうち,薬力学の項目が治験薬投与後 1 時点以上で測定された被験者を薬力学解析
対象とした。
2.被験者背景及びその他の基準値:
SAF に対し,本剤群(第一部:1,3,10,30,100,300 mg 及び 100 mg 食後;第二部:20,50,
100 mg)及びプラセボ群別に解析を行い,プラセボ群は各投与群のプラセボ投与例を併合して解
析を行った。計数値項目は度数集計を行い,計量値項目は要約統計量を算出した。
3.薬物動態|薬力学:
(1)薬物動態
PKAS について解析を行った。血漿又は尿中未変化体濃度測定値を用いて薬物動態パラメータの
算出並びに用量ごとに要約統計量の集計を行った。測定値が定量下限値未満(BQL)の場合は,
t1/2 算出時には欠測,それ以外は薬物濃度を 0(ゼロ)として取り扱った。
また,薬物動態の用量依存性,食事の影響の検討,蓄積性の検討及び定常状態の検討を行った。
第一部(単回経口投与)
薬物動態の用量依存性の検討のため,パワーモデルによる解析,各用量群間の比較を行った。ま
た,食事の影響の検討のため,100 mg の空腹時と食後の場合について,Cmax及び AUCinfの空腹時
投与に対する食後投与の幾何平均比及びその 90%信頼区間を算出した。本剤の尿中排泄量が僅か
であったため,Ae%,CLr については食事の影響の評価を実施しなかった。
第二部(反復経口投与)
蓄積性の検討のため,Cmax及び AUC24hについて,単回経口投与時(Day 1)に対する反復経口投
与時(Day 9)の幾何平均比及びその 95%信頼区間を投与群別に算出した。また,定常状態の検討
のため,Day 1 から Day 9 の血漿中未変化体濃度について,トラフ濃度(治験薬投与後 24 時間の
濃度)の経時推移を投与群別にプロットした。また,単回経口投与時(Day 1)の AUCinfに対す
る反復経口投与時(Day 9)の AUC24hの幾何平均比及びその 95%信頼区間を投与群別に算出した。
(2)薬力学
PDAS について解析を行った。血糖値,尿中グルコース排泄量,排尿量,尿中 Na,血中コルチゾー
ル,飲水量及び体重・体脂肪率について各用量群ごとに,一覧表の作成,要約統計量の集計,経
時推移の図の作成,散布図の作成等を行った。
4.安全性:
安全性の解析では,SAF を解析対象とした。本剤群(第一部:1,3,10,30,100,300 mg 及び
100 mg 食後;第二部:20,50,100 mg)及びプラセボ群別に解析を行った。プラセボ群は各投与
群のプラセボ投与例を併合して解析を行った。
各安全性評価項目について一覧表を作成した。有害事象では,器官別大分類(SOC)別及び基本
語(PT)別に度数集計を行った。臨床検査値,バイタルサインについては,要約統計量の算出及
びケースプロットあるいはクロス表の作成を行った。12 誘導心電図については,一覧表を作成し
た。詳細 QT 計測(第二部のみ実施)については,要約統計量の算出及び度数集計を行った。
報告書の日付: 年 月 日
46

2.7.6個々の試験のまとめ
2.7.6.3.2 被験者の内訳及び解析対象集団
2.7.6.3.2.1 被験者の内訳
第一部(単回経口投与)
第一部(単回経口投与)の被験者の内訳を図 2.7.6.3-1 に示した。
本剤群 36 例,プラセボ群 12 例の合計 48 例に治験薬が投与され,本剤 300 mg 群の 1 例が本人
の都合により試験を中止した。
図 2.7.6.3-1 被験者の内訳:第一部(単回経口投与)
a:空腹時に治験薬を投与した。
b:100 mg 用量群(空腹時)と同じ被験者で実施した。
Source:CL-0101 総括報告書(5 3 3 1-1)図 6-1
第二部(反復経口投与)
第二部(反復経口投与)の被験者の内訳を図 2.7.6.3-2 に示した。
本剤群 24 例,プラセボ群 12 例の合計 36 例に治験薬が 1 回以上投与された。治験薬が投与され
たすべての被験者が試験を完了した。
47

2.7.6個々の試験のまとめ
図 2.7.6.3-2 被験者の内訳:第二部(反復経口投与)
Source:CL-0101 総括報告書(5 3 3 1-1)図 6-2
2.7.6.3.2.2 解析対象集団
第一部(単回経口投与)
治験薬を投与された全被験者 48 例を SAF とした。SAF に含まれる被験者のうち,本剤投与例
でかつ血漿中未変化体濃度若しくは尿中未変化体濃度の検体が採取されている被験者 36 例を
PKAS とした。また,SAF のうち,薬力学の項目が治験薬投与後 1 時点以上で測定された被験者
48 例を PDAS とした。
第二部(反復経口投与)
治験薬を投与された全被験者 36 例を SAF とした。SAF に含まれる被験者のうち,本剤投与例
でかつ血漿中未変化体濃度若しくは尿中未変化体濃度の検体が採取されている被験者 24 例を
PKAS とした。また,SAF のうち,薬力学の項目が治験薬投与後 1 時点以上で測定された被験者
36 例を PDAS とした。
2.7.6.3.3 人口統計学的及び他の基準値の特性
第一部(単回経口投与)
第一部(単回経口投与)の被験者背景を表 2.7.6.3-1 に示した。
平均年齢は本剤群で 22.5~26.7 歳,プラセボ群で 23.7 歳であった。平均体重は本剤群で 63.32
~69.87 kg,プラセボ群で 63.34 kg であり,BMI の平均値は,本剤群で 21.12~22.83 kg/m2,プラ
セボ群で 21.19 kg/m2 であった。
48

2.7.6個々の試験のまとめ
表 2.7.6.3-1 被験者背景:第一部(単回経口投与)(SAF)
項目ASP1941 プラセボ
1 mg 3 mg 10 mg 30 mg 100 mg 300 mgN=6 N=6 N=6 N=6 N=6 N=6 N=12
年齢(同意取得
時)[歳]23.8
(4.07)26.7
(6.92)25.2
(7.36)23.7
(4.93)22.5
(1.52)23.0
(1.79)23.7
(5.74)
体重(Day 1)[kg]
63.32(6.910)
66.37(4.408)
66.42(5.055)
64.07(4.713)
64.73(4.196)
69.87(4.323)
63.34(7.731)
身長(スクリー
ニング検査時)[cm]
172.00(5.109)
177.42(5.240)
170.80(5.528)
171.97(6.967)
169.48(4.367)
174.90(3.108)
172.57(4.953)
BMI(Day 1)[kg/m2]
21.35(1.647)
21.12(1.426)
22.78(1.499)
21.70(1.833)
22.60(2.287)
22.83(1.316)
21.19(1.630)
平均値(標準偏差)
Source:CL-0101 総括報告書(5.3.3.1-1)表 6-4
第二部(反復経口投与)
第二部(反復経口投与)の被験者背景を表 2.7.6.3-2 に示した。
平均年齢は本剤群で 25.6~30.0 歳,プラセボ群で 25.9 歳であった。平均体重は本剤群で 60.99
~63.31 kg,プラセボ群で 62.81 kg であり,BMI の平均値は,本剤群で 20.65~21.75 kg/m2,プラ
セボ群で 21.18 kg/m2 であった
表 2.7.6.3-2 被験者背景:第二部(反復経口投与)(SAF)
項目ASP1941 プラセボ
20 mg 50 mg 100 mgN=8 N=8 N=8 N=12
年齢(同意取得時)
[歳]
25.6(1.69)
30.0(5.58)
28.1(5.96)
25.9(4.52)
体重(Day 1)[kg]
63.19(4.341)
63.31(3.059)
60.99(2.589)
62.81(4.770)
身長(スクリーニング検査時)[cm]
171.35(3.862)
170.66(5.398)
171.89(4.019)
172.21(4.072)
BMI(Day 1)[kg/m2]
21.48(0.981)
21.75(1.316)
20.65(0.847)
21.18(1.438)
平均値(標準偏差)
Source:CL-0101 総括報告書(5.3.3.1-1)表 6-5
2.7.6.3.4 治験薬の曝露
本試験では,治験薬の投与を中止した被験者はいなかった。なお,第一部(単回経口投与)の
本剤 300 mg の被験者 1 例が,本人の都合により治験薬投与後に試験を中止した。
49

2.7.6個々の試験のまとめ
2.7.6.3.5 薬物動態|薬力学:
2.7.6.3.5.1 薬物動態の結果
第一部(単回経口投与)
本剤を空腹時に単回経口投与したときの未変化体の血漿中薬物動態パラメータの平均値(標準
偏差)を表 2.7.6.3-3 に示した。また,平均血漿中未変化体濃度–時間曲線を図 2.7.6.3-3 に示した。
健康成人男性に本剤 1~300 mg を空腹時に単回経口投与したとき,血漿中未変化体濃度は投与
後約 1~3 時間で Cmaxに達し,その後速やかに 2 相性で消失した。
Cmax及び AUCinfの平均値は投与量の増加に伴って上昇した。t1/2 の平均値は,消失相の血漿中濃
度が十分評価できなかったと推測される本剤 1 mg 群で 4.35 時間であったが,3 mg 群以上では
10.01~13.34 時間で,投与量に依存する傾向は認められなかった。CL/F の平均値は,1 mg 群を除
いて 10.49~12.49 L/h で,投与量に関わらずほぼ同程度の値であった。
本剤 100 mg を食後に単回経口投与したときの未変化体の血漿中薬物動態パラメータの平均値
(標準偏差)を表 2.7.6.3-4 に示した。また,平均血漿中未変化体濃度–時間曲線を図 2.7.6.3-4 に示
した。
本剤 100 mg を食後に単回経口投与時したときの血漿中薬物動態パラメータの平均値は,100 mg
空腹時単回経口投与時と比べて大きな差はないと考えられた。
表 2.7.6.3-3 未変化体の血漿中薬物動態パラメータの平均値(標準偏差):第一部(単回経口
投与,空腹時)(PKAS)
パラメータASP1941
1 mg 3 mg 10 mg 30 mg 100 mg 300 mgN=6 N=6 N=6 N=6 N=6 N=5
Cmax
(ng/mL)17.89(4.00)
53.72(15.55)
174.34(14.26)
523.63(103.36)
1392.39(422.99)
3421.41(690.45)
AUClast
(ng•h/mL)50.81(9.91)
212.48(25.76)
826.43(162.18)
2848.77(373.41)
9569.73(2169.51)
27121.21(4494.74)
AUCinf
(ng•h/mL)58.94
(11.24)244.64(35.02)
855.06(168.11)
2895.83(363.14)
9696.47(2242.15)
27298.54(4621.65)
tmax
(h)0.75
(0.27)0.92
(0.20)0.92
(0.20)1.58
(1.11)2.33
(1.21)2.60
(1.34)t1/2
(h)4.35
(1.05)10.01(2.28)
13.34(4.99)
12.43(5.05)
11.71(2.00)
10.34(1.59)
CL/F(L/h)
17.45(3.05)
12.49(1.92)
12.14(2.80)
10.49(1.27)
11.00(3.64)
11.26(2.00)
平均値(標準偏差)
300 mg 群では 1 例が同意撤回により中止した。当該被験者の薬物動態パラメータはデータが不十分なため算出せ
ず。
Source:CL-0101 総括報告書(5.3.3.1-1)表 8-1
50

2.7.6個々の試験のまとめ
図 2.7.6.3-3 平均血漿中未変化体濃度–時間曲線(平均値±標準偏差):第一部(単回経口投与,
空腹時)(PKAS)
1 mg
3 mg
10 mg
30 mg
100 mg
300 mg
0 1
1 0
10 0
100 0
1000 0
10000 0
0 6 12 18 24 30 36 42 48 54 60 66 72
Source:CL-0101 総括報告書(5.3.3.1-1)図 12-1
表 2.7.6.3-4 未変化体の血漿中薬物動態パラメータの要約統計量:第一部(単回経口投与,
100 mg 食後)(PKAS)
パラメータASP1941100 mg
N=6Cmax
(ng/mL)1593.68(379.98)
AUClast
(ng•h/mL)9355.89
(2164.68)AUCinf
(ng•h/mL)9711.38
(2327.61)tmax
(h)1.58
(0.20)t1/2
(h)16.81(7.97)
CL/F(L/h)
11.09(4.04)
平均値(標準偏差)
Source:CL-0101 総括報告書(5.3.3.1-1)表 8-2
時間(h)
血漿中未変化体濃度(
ng/m
L)
51

2.7.6個々の試験のまとめ
図 2.7.6.3-4 平均血漿中未変化体濃度–時間曲線(平均値±標準偏差):第一部(単回経口投与,
100 mg 空腹時及び食後)(PKAS)
100mg
0 1
1 0
10 0
100 0
1000 0
10000 0
0 6 12 18 24 30 36 42 48 54 60 66 72
Source:CL-0101 総括報告書(5.3.3.1-1)図 12-2
本剤単回投与時の未変化体の尿中薬物動態パラメータの平均値(標準偏差)を表 2.7.6.3-5 に示
した。
投与後 72 時間までの尿中未変化体排泄率(Ae%)は 0.65%~1.18%であり,いずれの投与量に
おいても尿中への未変化体の排泄率は小さかった。また,CLr は 0.09~0.13 L/h であり,いずれの
投与量においても同程度の値であった。
本剤 100 mg を食後に単回経口投与したときの未変化体の尿中薬物動態パラメータの平均値(標
準偏差)を表 2.7.6.3-6 に示した。
本剤 100 mg 食後単回経口投与時の尿中薬物動態パラメータの平均値は,100 mg 空腹時単回経
口投与時と比べて同様の値であった。
血漿
中未変化体濃度(
ng/m
L)
時間(h)
空腹時
食後
52

2.7.6個々の試験のまとめ
表 2.7.6.3-5 未変化体の尿中薬物動態パラメータの平均値(標準偏差):第一部(単回経口投
与,空腹時)(PKAS)
パラメー
タ
ASP19411 mg 3 mg 10 mg 30 mg 100 mg 300 mgN=6 N=6 N=6 N=6 N=6 N=5
Ae (μg)
6.47(1.94)
22.57(2.23)
74.94(13.73)
354.97(64.50)
1111.18(242.35)
3344.51(263.25)
Ae% (%dose)
0.65(0.19)
0.75(0.07)
0.75(0.14)
1.18(0.21)
1.11(0.24)
1.11(0.09)
CLr (L/h)
-(-)
0.10(0.01)
0.09(0.02)
0.12(0.01)
0.11(0.01)
0.13(0.01)
平均値(標準偏差)
1 mg 群の CLr は tlast が 24 時間未満のため算出せず。
300 mg 群では 1 例が同意撤回により中止した。当該被験者の薬物動態パラメータはデータが不十分なため算出せ
ず。
Source:CL-0101 総括報告書(5.3.3.1-1)表 8-3
表 2.7.6.3-6 未変化体の尿中薬物動態パラメータの要約統計量:第一部(単回経口投与,
100 mg 食後)(PKAS)
パラメータASP1941100 mg
N=6Ae
(μg)1157.11(260.07)
Ae%(%dose)
1.16(0.26)
CLr
(L/h)0.12
(0.01)
平均値(標準偏差)
Source:CL-0101 総括報告書(5.3.3.1-1)表 8-4
本試験の第一部(単回経口投与)では,本剤の用量反応性及び食事の影響を検討した。
本剤の用量反応性の検討では,対数変換された Cmax及び AUCinfに対してパワーモデル回帰を行
い,回帰直線の傾きとその 90%信頼区間を推定した。Cmax及び AUCinfの傾きはそれぞれ 0.927 及
び 1.072 であり,いずれもほぼ 1 に近かった。また,90%信頼区間はそれぞれ[0.892,0.961]及
び[1.040,1.103]であった。
食事の影響の検討では,本剤 100 mg の空腹時単回経口投与に対する食後単回経口投与の Cmax
の幾何平均比は 1.162 であり,食後単回経口投与の Cmaxの方が若干高いと考えられた。また,幾
何平均比の 90%信頼区間は[0.867,1.557]であった。AUClast 及び AUCinfの幾何平均比はそれぞ
れ 0.975 及び 0.998 であり,90%信頼区間はそれぞれ[0.727,1.308]及び[0.738,1.349]であっ
た。なお,本剤の尿中排泄量が僅かであったため,Ae%,CLr では食事の影響を検討しなかった。
53

2.7.6個々の試験のまとめ
第二部(反復経口投与)
本剤反復経口投与時の Day 1 及び Day 9 における未変化体の血漿中薬物動態パラメータの平均
値(標準偏差)を,表 2.7.6.3-7 及び表 2.7.6.3-8 に示した。また,Day 1 及び Day 9 の平均血漿中
未変化体濃度–時間曲線を図 2.7.6.3-5 及び図 2.7.6.3-6 に示した。
Day 1 投与後の血漿中未変化体濃度は,いずれの投与量においても投与後 1~2 時間で Cmaxに達
し,その後 2 相性で速やかに消失した。Cmax及び AUCinfは投与量の増加に伴って上昇した。t1/2
の平均値は 9.81~13.43 時間,CL/F の平均値は 11.32~11.68 L/h であり,いずれも投与量に依存す
る傾向は認められなかった。
Day 9 投与後の血漿中未変化体濃度は,いずれの投与量においても投与後約 1 時間で Cmaxに達
し,その後 2 相性で速やかに消失した。Cmax及び AUC24hは投与量の増加に伴って上昇した。t1/2
の平均値は 11.24~15.36 時間,CL/F の平均値は 10.82~11.17 L/h であり,いずれも投与量に依存
する傾向は認められなかった。
表 2.7.6.3-7 未変化体の血漿中薬物動態パラメータの平均値(標準偏差):第二部(反復経口
投与)Day 1(PKAS)
パラメータ
ASP1941
20 mg 50 mg 100 mg
N=8 N=8 N=8
Cmax
(ng/mL)338.99(58.04)
878.84(132.00)
1419.93(326.13)
AUClast
(ng•h/mL)1749.03(443.25)
4400.26(1057.35)
8476.24(1741.27)
AUCinf
(ng•h/mL)1863.02(495.33)
4534.84(1115.82)
8958.94(2022.73)
tmax
(h)1.25
(0.27)1.25
(0.27)1.81
(0.80)
t1/2
(h)13.43(2.95)
9.81(1.63)
12.55(4.12)
CL/F(L/h)
11.32(2.63)
11.50(2.28)
11.68(2.66)
平均値(標準偏差)
Source:CL-0101 総括報告書(5.3.3.1-1)表 8-7
54

2.7.6個々の試験のまとめ
表 2.7.6.3-8 未変化体の血漿中薬物動態パラメータの平均値(標準偏差):第二部(反復経口
投与)Day 9(PKAS)
パラメータ
ASP1941
20 mg 50 mg 100 mg
N=8 N=8 N=8
Cmax
(ng/mL)
398.05(60.14)
975.53(187.54)
1731.61(229.83)
AUC24h
(ng•h/mL)1894.04(323.89)
4693.11(1074.39)
9113.99(1326.31)
tmax
(h)1.25
(0.27)1.25
(0.38)1.38
(0.44)
t1/2
(h)13.93(3.52)
11.24(3.34)
15.36(5.03)
CL/F(L/h)
10.82(1.75)
11.05(2.04)
11.17(1.56)
平均値(標準偏差)
Source:CL-0101 総括報告書(5.3.3.1-1)表 8-8
図 2.7.6.3-5 平均血漿中未変化体濃度–時間曲線(平均値±標準偏差):第二部(反復経口投与)
Day 1(PKAS)
20 mg
50 mg
100 mg
0.1
1.0
10.0
00.0
00.0
10000.0
0 6 12 18 24 30 36 42 48
Source:CL-0101 総括報告書(5.3.3.1-1)図 12-4
血漿
中未変
化体濃
度(
ng/m
L)
時間(h)
55

2.7.6個々の試験のまとめ
図 2.7.6.3-6 平均血漿中未変化体濃度–時間曲線(平均値±標準偏差):第二部(反復経口投与)
Day 9(PKAS)
20 mg
50 mg
100 mg
0.1
1.0
10.0
100.0
1000.0
10000.0
0 6 12 18 24 30 36 42 48
Source:CL-0101 総括報告書(5.3.3.1-1)図 12-5
本剤反復経口投与時の Day 1 及び Day 9 における未変化体の尿中薬物動態パラメータの平均値
(標準偏差)を,表 2.7.6.3-9 及び表 2.7.6.3-10 に示した。
Day 1 における投与後 48 時間までの Ae%は 0.98%~1.24%であり,いずれの投与量においても
尿中への未変化体の排泄率は小さかった。また,CLr は 0.12~0.15 L/h であり,いずれの投与量に
おいても同程度の値であった。
Day 9 における投与後 24 時間までの Ae%は 1.29%~1.44%であり,いずれの投与量においても
尿中への未変化体の排泄率は小さかった。また,CLr は 0.14~0.15 L/h であり,いずれの投与量に
おいても同程度の値であった。
表 2.7.6.3-9 未変化体の尿中薬物動態パラメータの平均値(標準偏差):第二部(反復経口投
与)Day 1(PKAS)
パラメータ
ASP1941
20 mg 50 mg 100 mg
N=8 N=8 N=8Ae
(μg)248.36(33.22)
508.04(128.25)
982.58(208.26)
Ae%(%dose)
1.24(0.17)
1.02(0.26)
0.98(0.21)
CLr
(L/h)0.15
(0.02)0.12
(0.01)0.12
(0.01)
平均値(標準偏差)
Source:CL-0101 総括報告書(5.3.3.1-1)表 8-9
時間(h)
血漿中未変化体濃度(
ng/m
L)
56

2.7.6個々の試験のまとめ
表 2.7.6.3-10 未変化体の尿中薬物動態パラメータの平均値(標準偏差):第二部(反復経口投
与)Day 9(PKAS)
パラメータ
ASP1941
20 mg 50 mg 100 mg
N=8 N=8 N=8Ae
(μg)288.64(45.03)
677.21(139.44)
1286.83(254.34)
Ae%(%dose)
1.44(0.23)
1.35(0.28)
1.29(0.25)
CLr
(L/h)0.15
(0.02)0.15
(0.03)0.14
(0.02)
平均値(標準偏差)
Source:CL-0101 総括報告書(5.3.3.1-1)表 8-10
第二部(反復経口投与)では,本剤の蓄積性の検討及び定常状態の検討を行った。
本剤の蓄積性の検討では,Cmax及び AUC24hに対して,投与量ごとに Day 1 に対する Day 9 の幾
何平均比及びその 95%信頼区間を推定した。Cmaxの幾何平均比は,20 mg で 1.179,50 mg で 1.104,
100 mg で 1.235 であった。また,それぞれの 95%信頼区間は,50 mg では[0.990,1.230]と 1 を
含んでいたものの,20 mg では[1.051,1.322],100 mg では[1.013,1.505]と 1 を含まなかった。
AUC24hの幾何平均比は,20 mg で 1.257,50 mg で 1.190,100 mg で 1.231 であった。また,それ
ぞれの 95%信頼区間は,20 mg で[1.183,1.335],50 mg で[1.129,1.255],100 mg で[1.139,
1.330]と,いずれの投与量においても 1 を含まず蓄積性が認められた。しかしながら,いずれの
投与量においても Cmax及び AUC24hの幾何平均比は 1.2 程度であり,健康成人男性では ASP1941
を 7 日間食後反復経口投与したときの蓄積性は僅かであった。
定常状態の検討では,AUC(Day 1 では AUCinf,Day 9 では AUC24h)に対して,投与量ごとに
Day 1 に対する Day 9 の幾何平均比及びその 95%信頼区間を推定した。いずれの投与量においても
幾何平均比はほぼ 1 であり(20 mg:1.033,50 mg:1.038,100 mg:1.031),本剤は少なくとも 7
日間食後反復経口投与にて定常状態に達すると考えられた。
2.7.6.3.5.2 薬力学の結果
第一部(単回経口投与)
空腹時に本剤を単回経口投与したときの血糖値の経時的推移を図 2.7.6.3-7 に,食後に単回経口
投与したときの経時的推移を図 2.7.6.3-8 に示した。
空腹時の単回経口投与では,血糖値は食事により高値を示したと考えられたものの,経時的な
推移は,いずれの本剤群でもプラセボ群と大きな差はなかった。
食後の単回経口投与では,本剤 100 mg 群及びプラセボ群で血糖値は上昇し,また,空腹時投与
時と同様の推移を示した。
57

2.7.6個々の試験のまとめ
図 2.7.6.3-7 血糖値の経時的推移(平均値±標準偏差):第一部(単回経口投与,空腹時)
1 mg3 mg10 mg30 mg100 mg300 mgPlacebo
mg/dL
血糖値:Day1
血糖
値
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
220
230
Day1投与開始からの時間
0 hr 1 hr 2 hr 3 hr 4 hr 5 hr 6 hr 7 hr 8 hr 10 hr 12 hr 24 hr 36 hr 48 hr 72 hr
注)被験者数 本剤群:各群 6 例(ただし,300 mg 群では 6 時間以降 5 例),プラセボ群:12 例
Source:CL-0101 総括報告書(5.3.3.1-1)図 8-1
図 2.7.6.3-8 血糖値の経時的推移(平均値±標準偏差):第一部(単回経口投与,食後 100 mg)
100 mgPlacebo
mg/dL
血糖値:Day1
血糖値
70
80
90
100
110
120
130
140
150
160
170
180
190
200
210
220
230
Day1投与開始からの時間
-1 hr 1 hr 2 hr 3 hr 4 hr 5 hr 6 hr 7 hr 8 hr 10 hr 12 hr 24 hr 36 hr 48 hr 72 hr
注)被験者数 本剤群: 6 例,プラセボ群:2 例
Source:CL-0101 総括報告書(5.3.3.1-1)図 8-2
58

2.7.6個々の試験のまとめ
空腹時に単回経口投与したとき及び 100 mg を食後に単回経口投与したときの投与後 72 時間ま
での累積尿中グルコース排泄量の経時的推移を,図 2.7.6.3-9 及び図 2.7.6.3-10 に示した。
投与後 72 時間までの累積尿中グルコース排泄量は用量依存的に増加した。また,累積尿中グル
コース排泄量は,100 mg 空腹時単回経口投与時と食後単回投与時で同様の推移を示した。
図 2.7.6.3-9 累積尿中グルコース排泄量(平均値±標準偏差):第一部(単回経口投与,空腹
時)
1 mg3 mg10 mg30 mg100 mg300 mgPlacebo
mg
累積尿中グルコース排泄量:Day1
累積
尿中
グル
コー
ス排
泄量
0.00
20000.00
40000.00
60000.00
80000.00
100000.00
120000.00
140000.00
160000.00
180000.00
Day1投与開始からの時間
0 - 4hr 0 - 6hr 0 - 8hr 0 - 10hr 0 - 12hr 0 - 24hr 0 - 36hr 0 - 48hr 0 - 72hr
注)被験者数 30 mg 群を除く各群 6 例(300mg 群の 0-6 h 区間以降では 5 例),30 mg 群 5 例
プラセボ群:11 例
Source:CL-0101 総括報告書(5.3.3.1-1)図 8-5
59

2.7.6個々の試験のまとめ
図 2.7.6.3-10 累積尿中グルコース排泄量(平均値±標準偏差):第一部(単回経口投与,食後
100 mg)
100 mgPlacebo
mg
累積尿中グルコース排泄量:Day1
累積尿中
グルコ
ース排
泄量
0.00
20000.00
40000.00
60000.00
80000.00
100000.00
120000.00
140000.00
160000.00
180000.00
Day1投与開始からの時間
0 - 4hr 0 - 6hr 0 - 8hr 0 - 10hr 0 - 12hr 0 - 24hr 0 - 36hr 0 - 48hr 0 - 72hr
注)被験者数 本剤 100 mg 群 6 例,プラセボ群:2 例
Source:CL-0101 総括報告書(5.3.3.1-1)図 8-6
第二部(反復経口投与)
健康成人男性に本剤 20,50又は 100 mgを食後単回(Day 1)及び1日1回 7日間(Day 3~Day 9),
食後反復経口投与したときの Day 1 及び Day 9 の血糖値の経時的推移を,図 2.7.6.3-11 及び図
2.7.6.3-12 にそれぞれ示した。
Day 1,Day 9 いずれでも,血糖値は食事により高値を示したと考えられたものの,経時的な推
移はいずれの投与量でも,プラセボ群と大きな差はなかった。
60

2.7.6個々の試験のまとめ
図 2.7.6.3-11 血糖値の経時的推移(平均値±標準偏差):第二部(反復経口投与)Day 1
20 mg50 mg100 mgPlacebo
mg/dL
血糖値:Day1
血糖値
70
90
110
130
150
170
190
210
230
Day1投与開始からの時間
-1hr 0hr 1hr 2hr 3hr 4hr 5hr 6hr 7hr 8hr 10hr 12hr 23hr 24hr 36hr 47hr 48hr
注)被験者数 本剤群各群:8 例,プラセボ群:12 例
Source:CL-0101 総括報告書(5.3.3.1-1)図 8-19
図 2.7.6.3-12 血糖値の経時的推移(平均値±標準偏差):第二部(反復経口投与)Day 9
20 mg50 mg100 mgPlacebo
mg/dL
血糖値:Day9
血糖
値
70
90
110
130
150
170
190
210
230
Day9投与開始からの時間
-1hr 0hr 1hr 2hr 3hr 4hr 5hr 6hr 7hr 8hr 10hr 12hr 23hr 24hr 36hr 47hr 48hr
注)被験者数 本剤群各群:8 例,プラセボ群:12 例
Source:CL-0101 総括報告書(5.3.3.1-1)図 8-20
61

2.7.6個々の試験のまとめ
Day 1 投与後 48 時間までの累積尿中グルコース排泄量及びDay 9 投与後 96時間までの累積尿中
グルコース排泄量の経時的推移を,図 2.7.6.3-13 及び図 2.7.6.3-14 にそれぞれ示した。
Day 1 投与後 48 時間までの累積尿中グルコース排泄量及びDay 9 投与後 96時間までの累積尿中
グルコース排泄量ともに,用量依存的な増加が認められた。
図 2.7.6.3-13 累積尿中グルコース排泄量(平均値±標準偏差):第二部(反復経口投与)Day 1
20 mg50 mg100 mgPlacebo
mg
累積尿中グルコース排泄量:Day1
累積
尿中
グルコ
ース
排泄量
0.00
20000.00
40000.00
60000.00
80000.00
100000.0
120000.0
140000.0
160000.0
180000.0
Day1投与開始からの時間
Day1 0 - 4hr Day1 0 - 6hr Day1 0 - 8hr Day1 0 - 10hr Day1 0 - 12hr Day1 0 - 24hr Day1 0 - 36hr Day1 0 - 48hr
注)被験者数 本剤群各群:8 例,プラセボ群:12 例
Source:CL-0101 総括報告書(5.3.3.1-1)図 8-23
62

2.7.6個々の試験のまとめ
図 2.7.6.3-14 累積尿中グルコース排泄量(平均値±標準偏差):第二部(反復経口投与)Day 9
20 mg50 mg100 mgPlacebo
mg
累積尿中グルコース排泄量:Day9
累積尿中グルコース排泄量
0.00
20000.00
40000.00
60000.00
80000.00
100000.0
120000.0
140000.0
160000.0
180000.0
Day9投与開始からの時間
0 - 4hr 0 - 6hr 0 - 8hr 0 - 10hr 0 - 12hr 0 - 24hr 0 - 36hr 0 - 48hr 0 - 72hr 0 - 96hr
注)被験者数 本剤群各群:8 例,プラセボ群:12 例
Source:CL-0101 総括報告書(5.3.3.1-1)図 8-24
血糖値及び尿中グルコース排泄量の他,薬力学パラメータとして排尿量,尿中 Na,血中コルチ
ゾール,飲水量,体重及び体脂肪率を測定した。その結果,第一部(空腹時単回経口投与)では,
試験期間中の推移は本剤群とプラセボ群で大きな差は認められなかった。第二部(反復投経口投
与)では,いずれの投与群においても試験期間中ほぼ一定量で推移した。
2.7.6.3.6 安全性
2.7.6.3.6.1 有害事象
第一部(単回経口投与)
第一部(単回経口投与)の空腹時投与で認められた有害事象の要約を表 2.7.6.3-11 に,食後投与
で認められた有害事象の要約を表 2.7.6.3-12 に示した。
空腹時の単回経口投与ではすべての投与群で有害事象が認められ,本剤群で有害事象が最も多
く認められたのは 10 mg 群で 5 例(83.3%)であった。プラセボ群では有害事象は 4 例(33.3%)
に認められた。副作用は,本剤 3 mg 群,10 mg 群,100 mg 群及び 300 mg 群で認められた。用量
依存的な有害事象及び副作用の発現例数増加は認められなかった。
食後の単回経口投与では,本剤 100 mg 群の 2 例に有害事象が認められた。
63

2.7.6個々の試験のまとめ
また,空腹時の 100 mg 単回経口投与の後観察終了後から,食後の 100 mg 単回経口投与の入院
までに発現した有害事象はなかった。
第一部では重篤な有害事象及び低血糖関連の有害事象は認められなかった。
表 2.7.6.3-11 有害事象の要約:第一部(単回経口投与,空腹時)(SAF)
項目ASP1941 プラセボ 計
1 mg 3 mg 10 mg 30 mg 100 mg 300 mgN=6 N=6 N=6 N=6 N=6 N=6 N=12 N=48
有害事象発現例数
(発現率)1(16.7%) 3(50.0%) 5(83.3%) 1(16.7%) 3(50.0%) 3(50.0%) 4(33.3%) 20(41.7%)
有害事象発現件数 1 4 8 1 3 3 4 24
副作用発現例数
(発現率)0 2(33.3%) 5(83.3%) 0 2(33.3%) 3(50.0%) 0 12(25.0%)
副作用発現件数 0 3 8 0 2 3 0 16
低血糖関連の有害事象
発現例数(発現率)0 0 0 0 0 0 0 0
重篤な有害事象
発現例数(発現率)0 0 0 0 0 0 0 0
Source:CL-0101 総括報告書(5.3.3.1-1)表 9-1,付録 13.2.7.1,付録 13.2.7.2
表 2.7.6.3-12 有害事象の要約:第一部(単回経口投与,100 mg 食後)(SAF)
項目ASP1941 プラセボ 計100 mg
N=6 N=2 N=8
有害事象発現例数(発現率) 2(33.3%) 0 2(25.0%)
有害事象発現件数 3 0 3
副作用発現例数(発現率) 2(33.3%) 0 2(25.0%)
副作用発現件数 3 0 3
低血糖関連の有害事象発現例数(発現率) 0 0 0
重篤な有害事象発現例数(発現率) 0 0 0
Source:CL-0101 総括報告書(5.3.3.1-1)表 9-2,表 12-33,表 12-35
第一部(単回経口投与)の空腹時及び食後投与で認められた有害事象の内訳を表 2.7.6.3-13,
表 2.7.6.3-14,表 2.7.6.3-15 及び表 2.7.6.3-16 にそれぞれ示した。また副作用の内訳を表 2.7.6.3-17,
表 2.7.6.3-18,表 2.7.6.3-19 及び表 2.7.6.3-20 にそれぞれ示した。
空腹時の単回経口投与の本剤群で 2 例以上に認められた有害事象は,血中ビリルビン増加(4
例),活性化部分トロンボプラスチン時間延長(3 例),血中クレアチンホスホキナーゼ増加,尿
中白血球陽性(各 2 例)であった。食後の単回経口投与で認められた有害事象は,尿中 β2 ミクロ
グロブリン増加,C-反応性蛋白増加及び尿中白血球陽性(各 1 例)であり,いずれも本剤 100 mg
群に認められた。
空腹時の単回経口投与の本剤群で 2 例以上に認められた副作用は,血中ビリルビン増加(4 例),
活性化部分トロンボプラスチン時間延長(3 例),尿中白血球陽性(2 例)であり,いずれも本剤
群のみで認められた。また,食後の単回経口投与で 100 mg 群に認められた有害事象は,いずれも
治験薬との関連性が否定されなかった。
第一部(単回経口投与)で認められた有害事象はいずれも軽度と判定された。有害事象及び副
作用の発現率に用量依存性はみられなかった。
64

2.7.6個々の試験のまとめ
表 2.7.6.3-13 有害事象の内訳:第一部(単回経口投与,空腹時)(SAF)
MedDRA/J version 9.1器官別大分類
基本語
ASP1941 プラセボ
1 mg 3 mg 10 mg 30 mg 100 mg 300 mg
N=6 N=6 N=6 N=6 N=6 N=6 N=12
感染症および寄生虫症 0 0 0 1(16.7%) 1(16.7%) 0 1(8.3%)
鼻咽頭炎 0 0 0 0 1(16.7%) 0 1(8.3%)
咽頭炎 0 0 0 1(16.7%) 0 0 0
臨床検査 1(16.7%) 3(50.0%) 5(83.3%) 0 2(33.3%) 2(33.3%) 3(25.0%)
活性化部分トロンボプラスチン時
間延長
0 0 3(50.0%) 0 0 0 0
尿中 β2 ミクログロブリン増加 0 0 0 0 1(16.7%) 0 0
血中ビリルビン増加 0 0 2(33.3%) 0 1(16.7%) 1(16.7%) 1(8.3%)
血中クレアチンホスホキナーゼ増
加
1(16.7%) 1(16.7%) 0 0 0 0 0
血中カリウム増加 0 1(16.7%) 0 0 0 0 0
血中トリグリセリド増加 0 0 0 0 0 0 1(8.3%)
白血球数増加 0 1(16.7%) 0 0 0 0 0
尿中白血球陽性 0 0 2(33.3%) 0 0 0 1(8.3%)
血中アルカリホスファターゼ増加 0 1(16.7%) 0 0 0 0 0
潜血陽性 0 0 0 0 0 1(16.7%) 0
腎および尿路障害 0 0 1(16.7%) 0 0 0 0
頻尿 0 0 1(16.7%) 0 0 0 0
呼吸器、胸郭および縦隔障害 0 0 0 0 0 1(16.7%) 0
鼻出血 0 0 0 0 0 1(16.7%) 0
発現例数(発現率)
Source:CL-0101 総括報告書(5.3.3.1-1)表 9-3
表 2.7.6.3-14 有害事象の内訳(件数):第一部(単回経口投与,空腹時)(SAF)
MedDRA/J version 9.1器官別大分類
基本語
ASP1941 プラセボ1 mg 3 mg 10 mg 30 mg 100 mg 300 mg
感染症および寄生虫症 0 0 0 1 1 0 1
鼻咽頭炎 0 0 0 0 1 0 1
咽頭炎 0 0 0 1 0 0 0
臨床検査 1 4 7 0 2 2 3
活性化部分トロンボプラスチン時間延長 0 0 3 0 0 0 0
尿中 β2 ミクログロブリン増加 0 0 0 0 1 0 0
血中ビリルビン増加 0 0 2 0 1 1 1
血中クレアチンホスホキナーゼ増加 1 1 0 0 0 0 0
血中カリウム増加 0 1 0 0 0 0 0
血中トリグリセリド増加 0 0 0 0 0 0 1
白血球数増加 0 1 0 0 0 0 0
尿中白血球陽性 0 0 2 0 0 0 1
血中アルカリホスファターゼ増加 0 1 0 0 0 0 0
潜血陽性 0 0 0 0 0 1 0
腎および尿路障害 0 0 1 0 0 0 0
頻尿 0 0 1 0 0 0 0
呼吸器、胸郭および縦隔障害 0 0 0 0 0 1 0
鼻出血 0 0 0 0 0 1 0
Source:CL-0101 総括報告書(5.3.3.1-1)表 12-32
65

2.7.6個々の試験のまとめ
表 2.7.6.3-15 有害事象の内訳:第一部(単回経口投与,100 mg 食後)(SAF)
MedDRA/J version 9.1器官別大分類
基本語
ASP1941 プラセボ
100 mg
N=6 N=2
臨床検査 2(33.3%) 0
尿中 β2 ミクログロブリン増加 1(16.7%) 0
C-反応性蛋白増加 1(16.7%) 0
尿中白血球陽性 1(16.7%) 0
発現例数(発現率)
Source:CL-0101 総括報告書(5.3.3.1-1)表 9-4
表 2.7.6.3-16 有害事象の内訳(件数):第一部(単回経口投与,100 mg 食後)(SAF)
MedDRA/J version 9.1
器官別大分類
基本語
ASP1941 プラセボ100 mg
臨床検査 3 0
尿中 β2 ミクログロブリン増加 1 0
C-反応性蛋白増加 1 0
尿中白血球陽性 1 0
Source:CL-0101 総括報告書(5.3.3.1-1)表 12-33
表 2.7.6.3-17 副作用の内訳:第一部(単回経口投与,空腹時)(SAF)
MedDRA/J version 9.1
器官別大分類
基本語
ASP1941 プラセボ
1 mg 3 mg 10 mg 30 mg 100 mg 300 mg
N=6 N=6 N=6 N=6 N=6 N=6 N=12
臨床検査 0 2(33.3%) 5(83.3%) 0 2(33.3%) 2(33.3%) 0
活性化部分トロンボプラスチン時間延
長
0 0 3(50.0%) 0 0 0 0
尿中 β2 ミクログロブリン増加 0 0 0 0 1(16.7%) 0 0
血中ビリルビン増加 0 0 2(33.3%) 0 1(16.7%) 1(16.7%) 0
血中カリウム増加 0 1(16.7%) 0 0 0 0 0
白血球数増加 0 1(16.7%) 0 0 0 0 0
尿中白血球陽性 0 0 2(33.3%) 0 0 0 0
血中アルカリホスファターゼ増加 0 1(16.7%) 0 0 0 0 0
潜血陽性 0 0 0 0 0 1(16.7%) 0
腎および尿路障害 0 0 1(16.7%) 0 0 0 0
頻尿 0 0 1(16.7%) 0 0 0 0
呼吸器、胸郭および縦隔障害 0 0 0 0 0 1(16.7%) 0
鼻出血 0 0 0 0 0 1(16.7%) 0
発現例数(発現率)
Source:CL-0101 総括報告書(5.3.3.1-1)表 9-5
66

2.7.6個々の試験のまとめ
表 2.7.6.3-18 副作用の内訳(件数):第一部(単回経口投与,空腹時)(SAF)
MedDRA/J version 9.1器官別大分類
基本語
ASP1941 プラセボ1 mg 3 mg 10 mg 30 mg 100 mg 300 mg
臨床検査 0 3 7 0 2 2 0
活性化部分トロンボプラスチン時間延長 0 0 3 0 0 0 0
尿中 β2 ミクログロブリン増加 0 0 0 0 1 0 0
血中ビリルビン増加 0 0 2 0 1 1 0
血中カリウム増加 0 1 0 0 0 0 0
白血球数増加 0 1 0 0 0 0 0
尿中白血球陽性 0 0 2 0 0 0 0
血中アルカリホスファターゼ増加 0 1 0 0 0 0 0
潜血陽性 0 0 0 0 0 1 0
腎および尿路障害 0 0 1 0 0 0 0
頻尿 0 0 1 0 0 0 0
呼吸器、胸郭および縦隔障害 0 0 0 0 0 1 0
鼻出血 0 0 0 0 0 1 0
Source:CL-0101 総括報告書(5.3.3.1-1)表 12-34
表 2.7.6.3-19 副作用の内訳:第一部(単回経口投与,100 mg 食後)(SAF)
MedDRA/J version 9.1器官別大分類
基本語
ASP1941 プラセボ
N=2100 mg
N=6
臨床検査 2(33.3%) 0
尿中 β2 ミクログロブリン増加 1(16.7%) 0
C-反応性蛋白増加 1(16.7%) 0
尿中白血球陽性 1(16.7%) 0
発現例数(発現率)
Source:CL-0101 総括報告書(5.3.3.1-1)表 9-6
表 2.7.6.3-20 副作用の内訳(件数):第一部(単回経口投与,100 mg 食後)(SAF)
MedDRA/J version 9.1
器官別大分類
基本語
ASP1941 プラセボ100 mg
臨床検査 3 0
尿中 β2 ミクログロブリン増加 1 0
C-反応性蛋白増加 1 0
尿中白血球陽性 1 0
Source:CL-0101 総括報告書(5.3.3.1-1)表 12-35
第二部(反復経口投与)
第二部(反復経口投与)で認められた有害事象の要約を表 2.7.6.3-21 に示した。
プラセボ群を含むすべての投与群で有害事象が認められ,そのうち副作用は本剤群のみで認め
られた。有害事象は本剤 50 mg 群で 8 例と最も多く認められ,プラセボ群では 1 例に認められた。
用量依存的な有害事象及び副作用の発現例数増加は認められなかった。また,第二部でも重篤な
有害事象及び低血糖関連の有害事象は認められなかった。
67

2.7.6個々の試験のまとめ
表 2.7.6.3-21 有害事象の要約:第二部(反復経口投与)(SAF)
項目ASP1941 プラセボ 計
20 mg 50 mg 100 mgN=8 N=8 N=8 N=12 N=36
有害事象発現例数(発現率) 5(62.5%) 8(100.0%) 3(37.5%) 1(8.3%) 17(47.2%)
有害事象発現件数 11 11 3 1 26
副作用発現例数(発現率) 5(62.5%) 8(100.0%) 3(37.5%) 0 16(44.4%)
副作用発現件数 10 10 3 0 23
低血糖関連の有害事象発現例数(発現率) 0 0 0 0 0
重篤な有害事象発現例数(発現率) 0 0 0 0 0
Source:CL-0101 総括報告書(5.3.3.1-1)表 9-7,付録 13.2.7.3,付録 13.2.7.4
第二部(反復経口投与)で認められた有害事象及び副作用の内訳をそれぞれ表 2.7.6.3-22,表
2.7.6.3-23 表 2.7.6.3-24 及び表 2.7.6.3-25 に示した。
本剤群で 2 例以上に認められた有害事象は,β-N アセチル D グルコサミニダーゼ増加(10 例),
尿中 β2 ミクログロブリン増加(3 例),血中カリウム増加,尿中白血球陽性(各 2 例)であった。
本剤群で 2 例以上に認められた副作用は,β-N アセチル D グルコサミニダーゼ増加(10 例),
尿中 β2 ミクログロブリン増加(3 例),尿中白血球陽性(2 例)であり,いずれも本剤群のみで認
められた。
いずれの有害事象も軽度と判定された。有害事象及び副作用の発現率に用量依存性はみられな
かった。
表 2.7.6.3-22 有害事象の内訳:第二部(反復経口投与)(SAF)
MedDRA/J version 9.1器官別大分類
基本語
ASP1941 プラセボ
N=1220 mgN=8
50 mgN=8
100 mgN=8
全身障害および投与局所様態 1(12.5%) 1(12.5%) 0 0
異常感 1(12.5%) 0 0 0
発熱 0 1(12.5%) 0 0
臨床検査 4(50.0%) 8(100.0%) 3(37.5%) 0
尿中 β2 ミクログロブリン増加 2(25.0%) 1(12.5%) 0 0
β–NアセチルDグルコサミニダーゼ増加 1(12.5%) 6(75.0%) 3(37.5%) 0
血中カリウム増加 1(12.5%) 1(12.5%) 0 0
血中尿素減少 0 1(12.5%) 0 0
尿中白血球陽性 2(25.0%) 0 0 0
尿中蛋白陽性 1(12.5%) 0 0 0
尿検査異常 0 1(12.5%) 0 0
神経系障害 1(12.5%) 0 0 1(8.3%)
体位性めまい 1(12.5%) 0 0 1(8.3%)
発現例数(発現率)
Source:CL-0101 総括報告書(5.3.3.1-1)表 9-8
68

2.7.6個々の試験のまとめ
表 2.7.6.3-23 有害事象の内訳(件数):第二部(反復経口投与)(SAF)
MedDRA/J version 9.1器官別大分類
基本語
ASP1941 プラセボ20 mg 50 mg 100 mg
全身障害および投与局所様態 1 1 0 0
異常感 1 0 0 0
発熱 0 1 0 0
臨床検査 7 10 3 0
尿中 β2 ミクログロブリン増加 2 1 0 0
β–N アセチル D グルコサミニダーゼ増加 1 6 3 0
血中カリウム増加 1 1 0 0
血中尿素減少 0 1 0 0
尿中白血球陽性 2 0 0 0
尿中蛋白陽性 1 0 0 0
尿検査異常 0 1 0 0
神経系障害 3 0 0 1
体位性めまい 3 0 0 1
Source:CL-0101 総括報告書(5.3.3.1-1)表 12-36
表 2.7.6.3-24 副作用の内訳:第二部(反復経口投与)(SAF)
MedDRA/J version 9.1
器官別大分類
基本語
ASP1941 プラセボ
N=1220 mgN=8
50 mgN=8
100 mgN=8
全身障害および投与局所様態 1(12.5%) 1(12.5%) 0 0
異常感 1(12.5%) 0 0 0
発熱 0 1(12.5%) 0 0
臨床検査 4(50.0%) 8(100.0%) 3(37.5%) 0
尿中 β2 ミクログロブリン増加 2(25.0%) 1(12.5%) 0 0
β–NアセチルDグルコサミニダーゼ増加 1(12.5%) 6(75.0%) 3(37.5%) 0
血中尿素減少 0 1(12.5%) 0 0
尿中白血球陽性 2(25.0%) 0 0 0
尿中蛋白陽性 1(12.5%) 0 0 0
尿検査異常 0 1(12.5%) 0 0
神経系障害 1(12.5%) 0 0 0
体位性めまい 1(12.5%) 0 0 0
発現例数(発現率)
Source:CL-0101 総括報告書(5.3.3.1-1)表 9-9
69

2.7.6個々の試験のまとめ
表 2.7.6.3-25 副作用の内訳(件数):第二部(反復経口投与)(SAF)
MedDRA/J version 9.1器官別大分類
基本語
ASP1941 プラセボ20 mg 50 mg 100 mg
全身障害および投与局所様態 1 1 0 0
異常感 1 0 0 0
発熱 0 1 0 0
臨床検査 6 9 3 0
尿中 β2 ミクログロブリン増加 2 1 0 0
β–N アセチル D グルコサミニダーゼ増加 1 6 3 0
血中尿素減少 0 1 0 0
尿中白血球陽性 2 0 0 0
尿中蛋白陽性 1 0 0 0
尿検査異常 0 1 0 0
神経系障害 3 0 0 0
体位性めまい 3 0 0 0
Source:CL-0101 総括報告書(5.3.3.1-1)表 12-37
2.7.6.3.6.2 死亡,その他の重篤な有害事象
本試験では,第一部(単回経口投与)及び第二部(反復経口投与)いずれでも,死亡及びその
他の重篤な有害事象は認められなかった。
2.7.6.3.6.3 中止に至った有害事象
本試験では,第一部(単回経口投与)及び第二部(反復経口投与)いずれでも,中止に至った
有害事象は認められなかった。
2.7.6.3.6.4 臨床検査値
第一部(単回経口投与)
本剤の尿中グルコース排泄作用に起因すると考えられる有害事象として,尿中 β2 ミクログロブ
リン増加 1 例が本剤 100 mg 群の空腹時及び食後投与時に同一症例で認められた。本事象は軽度で
あり,いずれも治験薬との関連性は否定されなかった。なお,これらは後観察検査又は追跡検査
により基準値への回復が確認された。
第二部(反復経口投与)
本剤の尿中グルコース排泄作用に起因すると考えられる有害事象として,β-N アセチル D グル
コサミニダーゼ増加が本剤 20 mg 群 1 例,50 mg 群 6 例,100 mg 群 3 例に,尿中 β2 ミクログロブ
リン増加が 20 mg 群 2 例,50 mg 群 1 例に認められた。β-N アセチル D グルコサミニダーゼ増加,
尿中 β2 ミクログロブリン増加ともにプラセボ群では認められなかった。これらの事象はいずれも
軽度であり,後観察検査あるいは追跡検査により基準値への回復が確認された。また,用量依存
的な発現例数の増加や程度の悪化などは認められなかった。
70

2.7.6個々の試験のまとめ
2.7.6.3.6.5 バイタルサイン
本試験では,腋窩体温,臥位血圧及び臥位脈拍数に臨床上問題となる所見は認められなかった。
2.7.6.3.6.6 心電図
本試験では,12 誘導心電図に臨床上問題となる所見は認められなかった。
第二部(反復経口投与)のみで実施した詳細 QT 計測では,QTc の測定値及び変化量を Bazett,
Fridericia 及び Framingham 式により算出し,検討した。
500 ms 以上の QTc 測定値はいずれの算出式においても認められなかった。
ベースラインからの QTc 変化量については,Day 1 投与 8 時間後に Bazett 式で算出したときの
み 30~60 ms の延長が 20 mg 群の 1 例に認められ,Day 9 投与 8 時間後では,いずれの算出式にお
いても 30~60 ms の延長が 20 mg 群の 1 例に認められた。なお,60 ms 以上の QTc 変化量を示し
た被験者はいなかった。
2.7.6.3.7 結論
● 薬物動態の血漿中パラメータである t1/2 は,単回経口投与時では本剤 1 mg を除いて約 10~13
時間で,7 日間の食後反復経口投与時では約 11~15 時間であった。また,尿中未変化体排泄
率は,20~100 mg 反復投与時でも 1.29%~1.44%であった。
● 空腹時単回経口投与の検討では,本剤群及びプラセボ群に食事によるものと考えられる血糖
値の高値が認められたものの,経時的な推移は投与量によらず,プラセボ群と大きな差はな
かった。また,反復経口投与の検討でも,Day 1(食後単回経口投与)及び Day 9(7 日間食
後反復経口投与)ともに本剤群及びプラセボ群で食事によるものと考えられる血糖値の上昇
が認められた。いずれの時点でも,血糖値の上昇の程度は本剤群とプラセボ群で同程度であっ
た。
● 空腹時単回経口投与の検討では,投与後 72 時間までの累積尿中グルコース排泄量は用量依存
的に増加し,反復経口投与の検討でも,Day 1(食後単回経口投与)投与後 48 時間及び Day 9
(7 日間食後反復経口投与)投与後 96 時間までの累積尿中グルコース排泄量は用量依存的に
増加した。
● 空腹時単回経口投与の検討では,本剤 1~300 mg の用量範囲で良好な忍容性が認められ,反
復経口投与の検討でも 20~100 mg の用量範囲で良好な忍容性が認められた。また,本試験で
認められた有害事象はすべて軽度であり,安全性に大きな問題はないと考えられた。
● 反復経口投与の検討で,本剤の尿中グルコース排泄作用に起因すると考えられる有害事象と
して,β-N アセチル D グルコサミニダーゼ増加が本剤 20 mg 群 1 例,50 mg 群 6 例,100 mg
群 3 例に,尿中 β2 ミクログロブリン増加が 20 mg 群 2 例,50 mg 群 1 例に認められた。これ
らの事象はいずれも軽度であり,後観察検査あるいは追跡検査により基準値への回復が確認
された。また,用量依存的な発現例数の増加や程度の悪化などは認められなかった。
71

2.7.6個々の試験のまとめ
● 以上の結果から,本剤を 2 型糖尿病患者へ投与し,その有効性及び安全性を検討することに
問題はないと考えられた。
72

2.7.6個々の試験のまとめ
2.7.6.4 海外第 I 相試験(単回・食事)[CL-0001](添付資料 5.3.3.1-2)
2.7.6.4.1 試験方法の概略
治験課題名:健康成人男性を対象に,ASP1941 を単回経口投与したときの安全性,忍容性,薬物
動態,薬力学及び食事の影響の検討を目的とした二重盲検プラセボ対照用量漸増試験
治験実施医療機関:英国 1 施設
公表文献:なし
治験期間:
治験開始日:2006 年 11 月
治験終了日:2007 年 5 月
開発のフェーズ:第 I 相
目的:
Part A(用量漸増)
(1) 主要目的
ASP1941(以下,本剤)を単回で用量漸増投与したときの安全性及び忍容性を評価する。可能で
あれば,最大耐量(MTD)を決定する。
(2) 副次目的
本剤を単回で用量漸増投与したときの薬物動態及び薬力学を評価する。
Part B(食事の影響)
(1) 主要目的
本剤の薬物動態に及ぼす食事の影響を評価する。
(2) 副次目的
空腹時投与及び食後投与での本剤の安全性及び忍容性を評価する。
治験デザイン・治験方法:
本試験は,Part A(用量漸増)と Part B(食事の影響)の 2 部構成で実施した。
なお,本試験では,HbA1c 値の表記として National Glycohemoglobin Standardization Program(NGSP)値を用いた。
Part A(用量漸増)
健康成人男性を対象とした無作為化,二重盲検,逐次群,プラセボ対照,単回投与,用量漸増試
験として実施した。
本剤は 0.1 mg を開始用量とし,1,3,10,30,100,300 及び 600 mg へと増量するよう計画した。
次用量への移行は,治験担当医師が各用量段階での安全性及び忍容性を評価し,治験依頼者と協
議した上で決定した。各用量段階の被験者数は 8 例とし,6 例に本剤,2 例にプラセボを無作為に
割り付け,投与した。
各用量段階では,被験者は治験薬投与前日(Day 0)に入所し,Day 1 に割り付けられた治験薬を
空腹時に服用した。血中及び尿中の未変化体濃度,血糖値及び尿中グルコース濃度を測定するた
め,治験薬投与前から投与後 72 時間までの規定された評価時点で血液及び尿を採取した。安全性
に関するデータは試験期間を通じて収集した。被験者は Day 4 に退所し,退所から 5~9 日後(Day 8~12)に後観察を行った。
Part B(食事の影響)
健康成人男性を対象とした無作為化,非盲検,2 期クロスオーバー試験として実施した。
被験者を第 1 期空腹時投与及び第 2 期食後投与例,若しくは第 1 期食後投与及び第 2 期空腹時投
与例のいずれかに無作為に割り付けた。
被験者は各期の治験薬投与前日(Day 0)に入所し,Day 1 に本剤 100 mg を割り付けられた順に空
73

2.7.6個々の試験のまとめ
腹時又は食後に服用した。血中及び尿中の未変化体濃度,血糖値及び尿中グルコース濃度を測定
するため,各期の治験薬投与前から投与後 72 時間までの規定された評価時点で血液及び尿を採取
した。安全性に関するデータは試験期間を通じて収集した。被験者は Day 4 に退所した。第 2 期
の治験薬は,第 1 期の治験薬投与後,休薬期間を置いて投与した。休薬期間は,5 日以上かつ Part A で算出された本剤の消失半減期(t1/2)の最高値の 5 倍以上の期間とした。第 2 期の治験薬投与
8~12 日後に後観察を行った。
目標被験者数:
Part A(用量漸増):各用量 8 例(計 64 例)
Part B(食事の影響):12 例
【設定根拠】
Part A(用量漸増)
目標被験者数は,医学的見地に基づき設定した。本剤の安全性及び忍容性を十分に評価できる例
数としたが,本試験はヒト初回投与試験であることから,本剤に曝露される被験者を最小限に留
めた。
Part B(食事の影響)
統計学的な目標被験者数の算出は行っていない。Part B は本剤の薬物動態に及ぼす食事の影響を
評価する目的で計画したものであり,実施面から判断し,12 例と設定した。
診断及び選択・除外基準:
1.選択基準(以下の基準をすべて満たす者を対象とした)
(1) 18 歳以上 55 歳以下の健康男性
(2) 体重 60 kg 以上 100 kg 以下,かつ BMI 20 kg/m2 以上 30 kg/m2 以下の者
(3) 被験者本人から文書による同意が得られている者
2.除外基準(以下の基準のいずれかに該当する者は対象から除外した)
(1) 1 型又は 2 型糖尿病の既往を有する者
(2) 空腹時血糖値が 6.4 mmol/L を超える又は HbA1c 値が 6.2%を超える者
(3) 腎性糖尿又は蛋白尿が認められる者
(4) 本剤又は製剤の構成成分に対して過敏症を有する者又は過敏症が疑われる者
(5) 喘息,湿疹等のアレルギー疾患又は何らかの薬剤に対する重度の過敏症の既往といった臨床
的に重要な既往を有する者
(6) 治験実施医療機関入所前 4 週間以内に臨床的に重要な上部消化管症状(悪心,嘔吐,腹部不
快感又は腹部不調,胸やけ等)がみられた者
(7) その他,消化器系,心血管系,呼吸器系,腎機能,肝機能,神経系,皮膚,精神又は代謝系
に治験担当医師が臨床的に重要と判断した疾患又は障害の既往を有する者
(8) 本試験前の身体所見,心電図検査及び臨床検査の結果,治験担当医師が臨床的に重要な異常
があると判断した者
(9) 本試験前来院時に脈拍数又は血圧に下記の異常が認められた者:
脈拍数<40 bpm 又は>90 bpm,平均収縮期血圧<90 mmHg 又は>140 mmHg,平均拡張期血圧
<40 mmHg 又は>95 mmHg(血圧は臥位にて 5 分間安静にした後 3 回測定した。脈拍数は自動
測定とした。)
(10) 治験実施医療機関入所前 4 週間以内に処方薬又は市販薬(ビタミン剤,ハーブ製剤を含む)
を常用した者,若しくは治験実施医療機関入所前 2 週間以内にそれらの薬剤を使用した者
(11) 治験実施医療機関入所前 3 カ月以内に薬物乱用歴を有する者
(12) 治験実施医療機関入所前 3カ月以内に 1日 10本を超える巻きタバコ(又は当量の刻みタバコ)
74

2.7.6個々の試験のまとめ
を使用した者
(13) 治験実施医療機関入所前 3 カ月以内に 1 週間あたり 21 単位(1 単位=ビール 270 mL,蒸留酒
40 mL,ワイン グラス 1 杯)を超える飲酒をした者
(14) 治験実施医療機関入所前 3 カ月以内に献血(全血又は成分献血)を行った者
(15) 血清学的検査で HBs 抗原,HAV IgM 抗体,HCV 抗体又は HIV 抗体 1 及び 2 が陽性であった
者
(16) 何らかの理由のため本試験を完了できない可能性があると治験担当医師が判断した者
(17) 本試験登録予定日から遡って 3 カ月以内に他の臨床試験に参加した者,又は 12 カ月以内に 4件以上の臨床試験に参加した者
(18) 治験担当医師により,本試験を安全に完了できない疾患を有すると判断された者
(19) アステラス製薬グループ又は本試験の治験実施医療機関に雇用されている者
治験薬,投与量及び投与方法:
1.治験薬及びロット番号
(1) 被験薬
・経口液剤用の粉末剤(0.1 mg 用):ASP1941 として 0.1 mg を含有する粉末。水 20 mL に溶解し,
使用した。
[ロット番号]BN 06118000571
・ASP1941 錠(ASP1941 錠 1 mg,ASP1941 錠 10 mg,ASP1941 錠 100 mg):1 錠中に ASP1941 と
してそれぞれ 1,10,100 mg を含有するフィルムコート錠
[ロット番号]
ASP1941 錠 1 mg:BN 06118000572ASP1941 錠 10 mg:BN 06118000573ASP1941 錠 100 mg:BN 06118000574
(2) 対照薬
・粉末剤を含まない水:水 20 mL を用量 0.1 mg の対照として使用した。
・ASP1941 錠プラセボ(ASP1941 錠 1 mg プラセボ,ASP1941 錠 10 mg プラセボ,ASP1941 錠
100 mg プラセボ):1 錠中に ASP1941 を含有しないフィルムコート錠
[ロット番号]
ASP1941 錠 1 mg プラセボ:BN 06118000575ASP1941 錠 10 mg プラセボ:BN 06118000576ASP1941 錠 100 mg プラセボ:BN 06118000577
2.投与量及び投与方法
Part A(用量漸増)
本剤(0.1,1,3,10,30,100,300 又は 600 mg)又はプラセボを空腹時に単回経口投与した。
投与 4 時間後まで食事は摂取しないこととした。
Part B(食事の影響)
食後又は空腹時に本剤 100 mg を単回経口投与した。空腹時投与では,本剤を空腹時に服用し,投
与後 4 時間は絶食とした。食後投与では,標準的な高脂肪食を摂取し,治験薬服用 5 分前に完食
することとした。高脂肪食摂取開始の 30 分後に治験薬を服用した。
【設定根拠】
Part A(用量漸増)
非臨床試験で得られた最小の NOAEL に基づき,ヒトでの安全な開始用量は 1 mg と推測された。
1 型糖尿病罹患モデルラットを用いて行われた試験では,1 mg でも薬理学的活性が認められた。
75

2.7.6個々の試験のまとめ
薬理学的作用を生じない用量から投与を開始するため,開始用量を 0.1 mg とした。
Part B(食事の影響)
Part B で使用される用量は,治験実施計画書にて Part A の結果から決定することと規定し,100 mgとした。
評価期間:
Part A(用量漸増)
治験薬投与期間:1 日
Part B(食事の影響)
治験薬投与期間:各期 1 日
併用治療(薬剤及び療法):
治験実施医療機関入所 4 週間前から処方薬及び市販薬(ビタミン剤,ハーブ製剤を含む)の常用
を禁止した。入所 2 週間前から後観察までそれらの薬剤の使用を禁止した。
ただし,治験担当医師が認めた場合に限り,3 g/日以下のアセトアミノフェンの使用は可とした。
評価項目,評価スケジュール及び評価基準:
1.薬物動態|薬力学:
(1) 薬物動態
「3.Part 別の評価スケジュール及び評価項目」に示した時期に採血及び蓄尿を行い,血漿中及び
尿中の未変化体濃度を測定した。血漿中及び尿中の未変化体濃度から,以下の薬物動態パラメー
タをノンコンパートメント法を用いて算出した。
[血漿中パラメータ]
・最高血漿中未変化体濃度到達時間(tmax)
・最高血漿中未変化体濃度(Cmax)
・時間 0 から血漿中濃度定量可能最終時点までの血漿中濃度‐時間曲線下面積(AUClast)
・時間 0 から無限時間まで外挿した血漿中濃度‐時間曲線下面積(AUCinf)
・経口クリアランス(CL/F)・みかけの分布容積(Vz/F)・消失半減期(t1/2)
・ラグタイム(tlag)
[尿中パラメータ]
・時間 0 から定量可能最終時点までの尿中排泄量(Aelast)
・時間 0 から定量可能最終時点までの尿中排泄率(Aelast%)
・時間 0 から無限時間まで外挿した尿中排泄量(Aeinf)
・時間 0 から無限時間まで外挿した尿中排泄率(Aeinf%)
・腎クリアランス(CLr)
(2) 薬力学
「3.Part 別の評価スケジュール及び評価項目」に示した時期に採血及び蓄尿を行い,血糖値及び
尿中グルコース濃度を測定した。血糖値及び尿中グルコース濃度から以下のパラメータを算出し,
血糖値及び尿中グルコース排泄量に及ぼす本剤の影響を探索的に検討した。
[血糖値]
・時間 0 から治験薬投与後 24 時間までの濃度‐時間曲線下面積(AUC24h)
・時間 0 から定量可能最終時点までの濃度‐時間曲線下面積(AUClast)
[尿中グルコース]
・時間 0 から治験薬投与後 24 時間までの尿中排泄量(Ae24h)
・時間 0 から定量可能最終時点までの尿中排泄量(Aelast)
・腎クリアランス(CLr)
76

2.7.6個々の試験のまとめ
2.安全性:
有害事象は,治験薬投与後に認められた事象とした。Part B では,第 1 期治験薬投与時から第 2期治験薬投与直前までに発現した有害事象を第 1 期に発現した有害事象として,第 2 期治験薬投
与時から後観察までに発現した有害事象を第 2 期に発現した有害事象として取り扱うこととし
た。
有害事象と治験薬との関連性は,「否定できる」,「関連あるかもしれない」,「多分(おそらく)関
連あり」,「関連性不明」の基準で判定し,「関連あるかもしれない」,「多分(おそらく)関連あり」
又は「関連性不明」に該当した有害事象を「治験薬との関連性が否定できない有害事象(副作用)」
と定義した。有害事象の程度は,軽度,中等度,重度又は程度不明の 4 段階で判定した。
3.Part 別の評価スケジュール及び評価項目:
Part A(用量漸増)
評価スケジュールを以下に示した。
スクリー
ニング
後観察
入所前 3週間以内
Day 0 Day 1 Day 2~3 Day 4 退所又は
中止後
5~9 日
入所 X X X 10:00 まで
無作為化 X治験薬投与 9:00食事 22:00 投与 4 時間後 a,
18:009:00a,13:00,
18:009:00a
同意取得 X試験前スクリーニング X身体所見 X X X安全性評価用血糖値測定用採血 X b
血漿中未変化体濃度及び血糖値測定
用採血
Xc Xc Xc
尿中未変化体濃度及び尿中グルコー
ス濃度測定用蓄尿
Xd Xd Xd
薬物乱用及び飲酒調査 X X臨床検査(血液学的検査,血液生化学
検査,尿検査)
X X Xe Xe Xe X
バイタルサイン(血圧及び脈拍数) X X Xf Xf Xf X12 誘導心電図 X X Xg Xg Xg X併用薬及び有害事象の調査 X X X X X
a:同時刻に予定されていた採血及び評価終了後に食事の摂取を開始した。
b:採血時期:治験薬投与前,投与後 10 分,25 分,55 分,1 時間 25 分,1 時間 55 分,2 時間 25 分,2 時間 55 分,3 時間 25 分,
3 時間 55 分
c:採血時期:治験薬投与前,投与後 0.25 時間,0.5 時間,1 時間,1.5 時間,2 時間,3 時間,4 時間,5 時間,6 時間,7 時間,
8 時間,10 時間,12 時間,16 時間,24 時間,36 時間,48 時間,72 時間
d:蓄尿時期:治験薬投与前,投与後 0~2 時間,2~4 時間,4~6 時間,6~8 時間,8~10 時間,10~12 時間,12~16 時間,16~24 時間,24~36 時間,36~48 時間,48~72 時間
e:採血及び採尿時期:投与後 3 時間,24 時間,72 時間
f:測定時期:治験薬投与前,投与後 0.5 時間,1 時間,1.5 時間,2 時間,3 時間,4 時間,5 時間,6 時間,8 時間,12 時間,24時間,36 時間,48 時間,72 時間
g:測定時期:治験薬投与前,投与後 0.5 時間,1 時間,1.5 時間,2 時間,3 時間,4 時間,5 時間,6 時間,8 時間,12 時間,
24 時間,36 時間,48 時間,72 時間
Part A での評価項目は以下とした。
(1) 主要評価項目
[安全性]
77

2.7.6個々の試験のまとめ
・有害事象の種類,頻度及び程度
・身体所見及びバイタルサイン
・臨床検査(尿検査,血液学的検査,血液生化学検査)
・12 誘導心電図
(2) 副次評価項目
[薬物動態]
・血漿中パラメータ:AUCinf,AUClast,tmax,Cmax,t1/2,Vz/F,CL/F・尿中パラメータ:Aelast,Aeinf%,CLr
[薬力学]
・血糖値:AUClast,AUC24h
・尿中グルコース:Aelast,Ae24h,CLr
Part A では,安全性評価のために,腎機能マーカー(尿中の β2 ミクログロブリン,α1 ミクログロ
ブリン及び β-N-アセチル D グルコサミニダーゼ)を測定した。これらのマーカーは,本剤の薬理
作用(SGLT2 阻害)の結果として上昇する可能性がある。β-N-アセチル D グルコサミニダーゼ,
β2 ミクログロブリン及び α1 ミクログロブリンは腎毒性に特異的なマーカーではないが,腎臓に
及ぼす非特異的な影響の指標となる。
また,Part A では,各用量段階で以下のいずれかに該当する場合,増量を中止し当該用量を最小
不耐量(MID)とすることとした。MID の 1 段階低い用量を MTD とすることとした。
1. 中等度以上の副作用(「関連あるかもしれない」又は「多分(おそらく)関連あり」と判定さ
れた有害事象)が複数件認められた被験者の割合が,同一用量群で 50%以上となった場合
2. 1 回でも血糖値が 2.8 mmol/L を下回った場合
3. 重篤な副作用が発現した場合
78

2.7.6個々の試験のまとめ
Part B(食事の影響)
評価スケジュールを以下に示した。
スクリー
ニング
第 1 期及び第 2 期 後観察
第 1 期入
所前 3 週
間以内
Day 0 Day 1 Day 2~3 Day 4 第 2期退所
又は中止
後 5~9 日
入所 X X X 10:00 まで
無作為化 X治験薬投与 9:00食事 22:00 8:30~9:00(食後投与
のみ),投与 4 時間後a,18:00
9:00a,13:00,18:00
9:00a
同意取得 Xスクリーニング X身体所見 X X X血漿中未変化体濃度及び血糖
値測定用採血
Xb Xb Xb
尿中未変化体濃度及び尿中グ
ルコース濃度測定用蓄尿
Xc Xc Xc
薬物乱用及び飲酒調査 X X臨床検査(血液学的検査,血液
生化学検査,尿検査)
X X Xd Xd Xd X
バイタルサイン(血圧及び脈拍
数)
X X Xe Xe Xe X
12 誘導心電図 X X Xf Xf Xf X併用薬及び有害事象の調査 X X X X X
a:同時刻に予定されていた採血及び評価終了後に食事の摂取を開始した。
b:採血時期:治験薬投与前,投与後 0.25 時間,0.5 時間,1 時間,1.5 時間,2 時間,3 時間,4 時間,5 時間,6 時間,7 時間,
8 時間,10 時間,12 時間,16 時間,24 時間,36 時間,48 時間,72 時間
c:蓄尿時期:治験薬投与前,投与後 0~2 時間,2~4 時間,4~6 時間,6~8 時間,8~10 時間,10~12 時間,12~16 時間,16~24 時間,24~36 時間,36~48 時間,48~72 時間
d:採血及び採尿時期:投与後 3 時間,24 時間,72 時間
e:測定時期:治験薬投与前,投与後 0.5 時間,2 時間,4 時間,8 時間,12 時間,24 時間,36 時間,48 時間,72 時間
f:測定時期:治験薬投与前,投与後 2 時間,4 時間,6 時間,8 時間,12 時間,24 時間,72 時間
Part B での評価項目は以下とした。
(1) 主要評価項目
[薬物動態]
・血漿中パラメータ:Cmax 及び AUCinf(外挿部分が AUCinfの 20%を超える場合,AUClast を主
要評価項目とする。)
(2) 副次評価項目
[安全性評価項目]
・有害事象の種類,頻度及び程度
・身体所見及びバイタルサイン
・臨床検査(尿検査,血液学的検査,血液生化学検査)
・12 誘導心電図
[薬物動態]
・血漿中パラメータ:AUClast,tlag,tmax,t1/2,Vz/F,CL/F・尿中パラメータ:Aelast,Aeinf%,CLr
[薬力学]
・血糖値:AUClast,AUC24h
79

2.7.6個々の試験のまとめ
・尿中グルコース:Aelast,Ae24h,CLr
統計解析:
1.解析対象集団:
(1) 安全性解析対象集団(SAF)治験薬を 1 回以上服用した被験者の集団を SAF とした。
(2) 薬物動態解析対象集団(PKAS)SAF に含まれる被験者のうち,1 つ以上の薬物動態パラメータを算出できる血漿中濃度データが
得られた被験者の集団を PKAS とした。
2.被験者背景及びその他の基準値:
SAF を対象に,計数値項目では度数集計を行い,計量値項目では要約統計量を算出した。
3.薬物動態|薬力学:
薬物動態及び薬力学の解析では,PKAS を解析対象とした。
Part A(用量漸増)
薬物動態パラメータ及び薬力学パラメータの要約統計量を用量ごとに算出した。未変化体の AUC及び Cmaxの用量依存性を評価した。
Part B(食事の影響)
薬物動態パラメータ及び薬力学パラメータの要約統計量を投与条件ごとに算出した。
食事の影響を検討するため,未変化体の AUClast,AUCinf及び Cmaxについて,空腹時投与に対する
食後投与の比及び両側 90%信頼区間を算出した。AUClast,AUCinf及び Cmaxの空腹時投与に対する
食後投与の比の 90%信頼区間が 0.80~1.25 の範囲内にすべて含まれる場合,食事の影響なしと判
断することとした。
4.安全性:
安全性の解析では,SAF を解析対象とした。
Part A(用量漸増)
有害事象及び副作用の発現率を用量ごとに算出した。
臨床検査値に関しては,計量値項目では評価時点ごとに実測値の要約統計量を算出し,計数値項
目では治験薬服用前測定値と各評価時点の測定値のシフト表を作成した。
バイタルサインに関しては,評価時点ごとに実測値の要約統計量を算出した。
Part B(食事の影響)
有害事象及び副作用の発現率を投与条件ごとに算出した。
臨床検査値に関しては,計量値項目では評価時点ごとに実測値の要約統計量を算出し,計数値項
目では治験薬服用前測定値と各評価時点の測定値のシフト表を作成した。
バイタルサインに関しては,評価時点ごとに実測値の要約統計量を算出した。
報告書の日付: 年 月 日
80

2.7.6個々の試験のまとめ
2.7.6.4.2 被験者の内訳及び解析対象集団
2.7.6.4.2.1 被験者の内訳
Part A(用量漸増)
Part A の被験者の内訳を表 2.7.6.4-1 に示した。
計 62 例(本剤群 47 例,プラセボ群 15 例)に治験薬が投与され,すべての被験者が本試験を完
了した。
表 2.7.6.4-1 被験者の内訳:Part AASP1941プラセボ
0.1 mg 1 mg 3 mg 10 mg 30 mg 100 mg 300 mg 600 mg 計
無作為化/治
験薬投与例
15 6 6 6 6 6 5 6 6 47
完了例 15 6 6 6 6 6 5 6 6 47中止例 0 0 0 0 0 0 0 0 0 0
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.1.1.1
Part B(食事の影響)
Part B の被験者の内訳を表 2.7.6.4-2 に示した。
計 12 例に治験薬が 1 回以上投与された。2 例が第 1 期(食後)投与後第 2 期(空腹時)投与前
に本試験を中止した。その他の 10 例は 2 期 2 回の投与を完了した。
表 2.7.6.4-2 被験者の内訳:Part BASP1941 100 mg
第 1 期(食後投与)
第 2 期(空腹時投与)
第 1 期(空腹時投与)
第 2 期(食後投与)
計
無作為化/治験薬投与例 6 6 12完了例 4 6 10中止例 2a 0 2a
a:1 例が有害事象のため,1 例が追跡不能のため本試験を中止した。
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.2.1.1
2.7.6.4.2.2 解析対象集団
Part A(用量漸増)
治験薬を服用したすべての被験者 62 例(本剤群 47 例,プラセボ群 15 例)を SAF とした。
SAF に含まれる被験者のうち,1 つ以上の薬物動態パラメータを算出できる血漿中濃度データ
が得られた被験者 62 例(本剤群 47 例,プラセボ群 15 例)を PKAS とした。
Part B(食事の影響)
治験薬を服用したすべての被験者 12 例を SAF とした。
81

2.7.6個々の試験のまとめ
SAF に含まれる被験者のうち,1 つ以上の薬物動態パラメータを算出できる血漿中濃度データ
が得られた被験者(10 例)を PKAS とした。空腹時投与を受けなかった 2 例を PKAS から除外し
た。
2.7.6.4.3 人口統計学的及び他の基準値の特性
Part A(用量漸増)
Part A の被験者背景を表 2.7.6.4-3 に示した。
被験者の大部分が白人であった。平均年齢は本剤群で 32.9 歳,プラセボ群で 30.7 歳であった。
平均体重は本剤群で 78.45 kg,プラセボ群で 80.37 kgであり,BMIの平均値は,本剤群で 24.59 kg/m2,
プラセボ群で 24.34 kg/m2 であった。100 mg 群では,他群と比較して体重及び BMI が低い傾向が
みられ,平均体重の最も大きな差は 100 mg 群と 300 mg 群(それぞれ 67.60 kg 及び 86.50 kg)で
あった。その他,被験者背景に臨床的に重要な差はみられなかった。
表 2.7.6.4-3 被験者背景:Part A(SAF)ASP1941プラセボ
0.1 mg 1 mg 3 mg 10 mg 30 mg 100 mg 300 mg 600 mg 計N=15 N=6 N=6 N=6 N=6 N=6 N=5 N=6 N=6 N=47
年齢
(歳)
平均値(SD)
30.7(9.6)
35.7(13.9)
29.7(11.6)
36.7(10.7)
39.0(7.9)
29.5(8.4)
29.6(7.2)
31.3(10.7)
31.0(11.3)
32.9(10.3)
白人 15(100)
6(100)
6(100)
6(100)
5(83.3)
4(66.7)
5(100)
6(100)
5(83.3)
43(91.5)
黒人 0 0 0 0 1(16.7)
2(33.3)
0 0 0 3(6.4)
アジア
人
0 0 0 0 0 0 0 0 0 0
人種n(%)
その他 0 0 0 0 0 0 0 0 1(16.7)
1(2.1)
体重(kg)
平均値(SD)
80.37(10.12)
76.67(3.33)
76.33(3.31)
77.00(7.02)
81.58(8.26)
82.25(9.84)
67.60(4.62)
86.50(10.48)
77.83(7.28)
78.45(8.43)
身長(cm)
平均値(SD)
181.7(8.7)
178.3(4.8)
177.0(7.9)
180.0(4.9)
178.0(4.6)
180.2(4.8)
177.6(5.4)
180.7(6.3)
177.5(4.2)
178.7(5.2)
BMI(kg/m2)
平均値(SD)
24.34(2.46)
24.11(0.85)
24.46(2.09)
23.80(2.45)
25.82(3.12)
25.38(3.24)
21.43(1.15)
26.47(2.64)
24.74(2.68)
24.59(2.65)
SD:標準偏差
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.1.2.1
Part B(食事の影響)
SAF 12 例の平均年齢は 30.8 歳,平均体重は 75.75 kg,BMI の平均値は 25.02 kg/m2 であった。
2.7.6.4.4 治験薬の曝露
Part A(用量漸増)
被験者 62 例全例が治験薬(本剤又はプラセボ)を 1 回服用した。
82
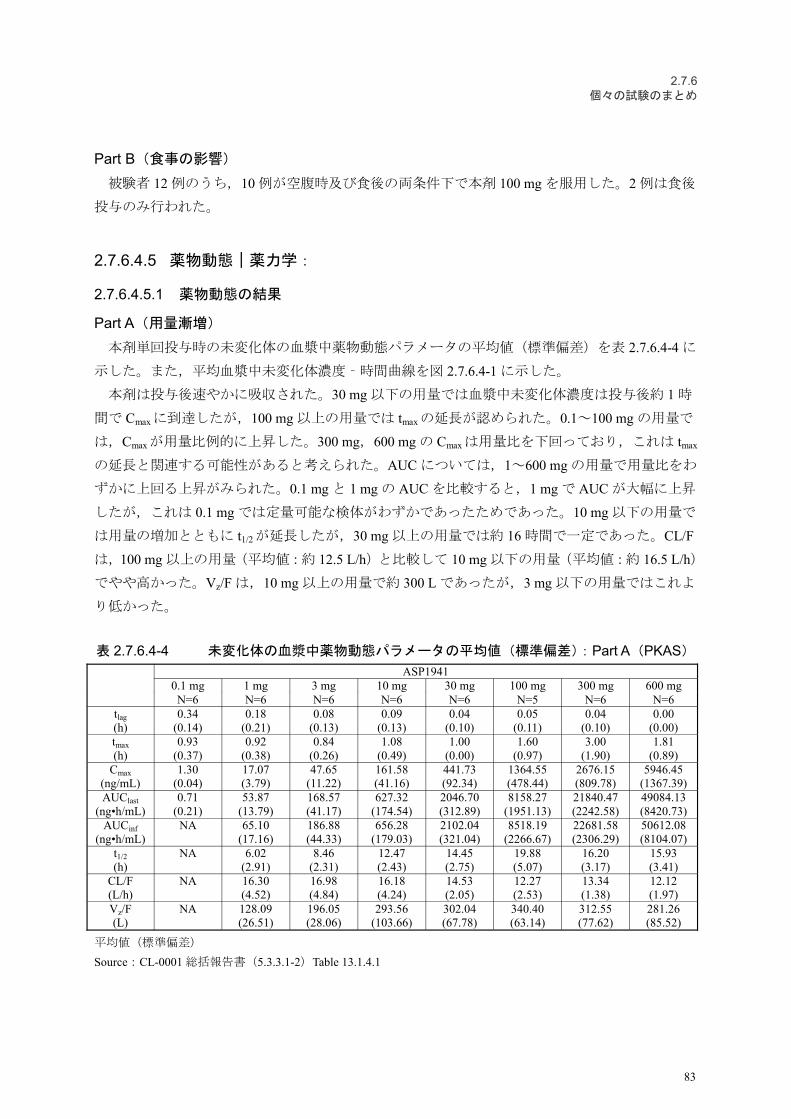
2.7.6個々の試験のまとめ
Part B(食事の影響)
被験者 12 例のうち,10 例が空腹時及び食後の両条件下で本剤 100 mg を服用した。2 例は食後
投与のみ行われた。
2.7.6.4.5 薬物動態|薬力学:
2.7.6.4.5.1 薬物動態の結果
Part A(用量漸増)
本剤単回投与時の未変化体の血漿中薬物動態パラメータの平均値(標準偏差)を表 2.7.6.4-4 に
示した。また,平均血漿中未変化体濃度‐時間曲線を図 2.7.6.4-1 に示した。
本剤は投与後速やかに吸収された。30 mg 以下の用量では血漿中未変化体濃度は投与後約 1 時
間で Cmaxに到達したが,100 mg 以上の用量では tmaxの延長が認められた。0.1~100 mg の用量で
は,Cmaxが用量比例的に上昇した。300 mg,600 mg の Cmaxは用量比を下回っており,これは tmax
の延長と関連する可能性があると考えられた。AUC については,1~600 mg の用量で用量比をわ
ずかに上回る上昇がみられた。0.1 mg と 1 mg の AUC を比較すると,1 mg で AUC が大幅に上昇
したが,これは 0.1 mg では定量可能な検体がわずかであったためであった。10 mg 以下の用量で
は用量の増加とともに t1/2が延長したが,30 mg 以上の用量では約 16 時間で一定であった。CL/F
は,100 mg 以上の用量(平均値:約 12.5 L/h)と比較して 10 mg 以下の用量(平均値:約 16.5 L/h)
でやや高かった。Vz/F は,10 mg 以上の用量で約 300 L であったが,3 mg 以下の用量ではこれよ
り低かった。
表 2.7.6.4-4 未変化体の血漿中薬物動態パラメータの平均値(標準偏差):Part A(PKAS)ASP1941
0.1 mg 1 mg 3 mg 10 mg 30 mg 100 mg 300 mg 600 mgN=6 N=6 N=6 N=6 N=6 N=5 N=6 N=6
tlag(h)
0.34(0.14)
0.18(0.21)
0.08(0.13)
0.09(0.13)
0.04(0.10)
0.05(0.11)
0.04(0.10)
0.00(0.00)
tmax(h)
0.93(0.37)
0.92(0.38)
0.84(0.26)
1.08(0.49)
1.00(0.00)
1.60(0.97)
3.00(1.90)
1.81(0.89)
Cmax(ng/mL)
1.30(0.04)
17.07(3.79)
47.65(11.22)
161.58(41.16)
441.73(92.34)
1364.55(478.44)
2676.15(809.78)
5946.45(1367.39)
AUClast(ng•h/mL)
0.71(0.21)
53.87(13.79)
168.57(41.17)
627.32(174.54)
2046.70(312.89)
8158.27(1951.13)
21840.47(2242.58)
49084.13(8420.73)
AUCinf(ng•h/mL)
NA 65.10(17.16)
186.88(44.33)
656.28(179.03)
2102.04(321.04)
8518.19(2266.67)
22681.58(2306.29)
50612.08(8104.07)
t1/2(h)
NA 6.02(2.91)
8.46(2.31)
12.47(2.43)
14.45(2.75)
19.88(5.07)
16.20(3.17)
15.93(3.41)
CL/F(L/h)
NA 16.30(4.52)
16.98(4.84)
16.18(4.24)
14.53(2.05)
12.27(2.53)
13.34(1.38)
12.12(1.97)
Vz/F(L)
NA 128.09(26.51)
196.05(28.06)
293.56(103.66)
302.04(67.78)
340.40(63.14)
312.55(77.62)
281.26(85.52)
平均値(標準偏差)
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.1.4.1
83

2.7.6個々の試験のまとめ
図 2.7.6.4-1 平均血漿中未変化体濃度‐時間曲線:Part A(PKAS)
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.1.3
本剤単回投与時の未変化体の尿中薬物動態パラメータの平均値(標準偏差)を表 2.7.6.4-5 に示
した。
最小用量の 0.1 mg では,尿中未変化体濃度はすべての検体で定量下限未満であり,薬物動態パ
ラメータは算出できなかった。
尿中の未変化体の量はわずかであった。尿中排泄量は用量に比例して増加し,排泄率はそれぞ
れの投与量の約 1%に相当した。CLr は各用量群とも約 0.14 L/h であり,ほぼ同程度であった。
表 2.7.6.4-5 未変化体の尿中薬物動態パラメータの平均値(標準偏差):Part A(PKAS)ASP1941
0.1 mg 1 mg 3 mg 10 mg 30 mg 100 mg 300 mg 600 mgN=6 N=6 N=6 N=6 N=6 N=5 N=5 N=5
Aelast(mg)
NA 0.01(0.00)
0.03(0.01)
0.10(0.02)
0.30(0.12)
1.04(0.19)
3.37(0.38)
6.25(0.70)
Aeinf(mg)
NA 0.01(0.00)
0.03(0.01)
0.10(0.02)
0.31(0.12)
1.09(0.23)
3.43(0.38)
6.48(0.70)
Aelast%(%)
NA 0.71(0.18)
0.92(0.33)
0.98(0.22)
1.01(0.39)
1.04(0.19)
1.12(0.13)
1.04(0.12)
Aeinf%(%)
NA 0.77(0.28)
0.93(0.35)
0.98(0.22)
1.04(0.39)
1.09(0.23)
1.14(0.13)
1.08(0.12)
CLr(L/h)
NA 0.12(0.03)
0.15(0.04)
0.15(0.02)
0.15(0.04)
0.13(0.01)
0.15(0.01)
0.13(0.02)
平均値(標準偏差)
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.1.4.5
時間(h)
血漿中未変化体濃度(
ng/m
L)
84

2.7.6個々の試験のまとめ
Part B(食事の影響)
空腹時投与時及び食後投与時の未変化体の血漿中薬物動態パラメータの平均値(標準偏差)を
表 2.7.6.4-6 に示し,Cmax及び AUCinfの空腹時投与時に対する食後投与時の比の平均値とその 90%
信頼区間をあわせて示した。また,平均血漿中未変化体濃度‐時間曲線を図 2.7.6.4-2 に示した。
Cmaxは,空腹時投与時と比較して食後投与時に 14%低下し,空腹時投与時に対する食後投与時
の比の 90%信頼区間下限が 0.80~1.25 の範囲を下回った。
AUClast 及び AUCinfに明らかな食事の影響はみられなかった。
tlagは空腹時投与時と比較して食後投与時に延長し,吸収開始の遅延が認められた。tmaxは空腹
時投与時と比較して食後投与時に延長し,食後投与時に吸収遅延がみられた。
t1/2,CL/F 及び Vz/F は両投与条件下で同程度であった。
表 2.7.6.4-6 未変化体の血漿中薬物動態パラメータの平均値(標準偏差):Part B(PKAS)空腹時投与 食後投与 比の平均値
N=10 N=10 [90%信頼区間]tlag(h)
0.03(0.08)
0.13(0.18)
-
tmax(h)
1.95(1.17)
3.31(1.70)
-
Cmax(ng/mL)
1142.33(175.65)
1002.98(332.86)
0.86[0.71 – 1.04]
AUClast(ng•h/mL)
8223.44(2666.29)
8942.80(2601.28)
1.09[1.05 – 1.14]
AUCinf(ng•h/mL)
8375.99(2763.85)
9077.05(2670.77)
1.09[1.05 – 1.14]
t1/2(h)
13.21(3.26)
12.33(3.21)
-
CL/F(L/h)
13.10(4.38)
11.79(3.19)
-
Vz/F(L)
235.74(50.95)
203.27(53.62)
-
平均値(標準偏差)
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.2.4.1.2 及び Table 13.2.4.2
85

2.7.6個々の試験のまとめ
図 2.7.6.4-2 平均血漿中未変化体濃度‐時間曲線:Part B(PKAS)
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.2.4.1.1
空腹時投与時及び食後投与時の未変化体の尿中薬物動態パラメータの平均値(標準偏差)を表
2.7.6.4-7 に示した。
尿中排泄率は,空腹時投与時及び食後投与時のいずれの投与条件下でも約 1%であった。CLr は
両投与条件下で同程度であった。
表 2.7.6.4-7 未変化体の尿中薬物動態パラメータの平均値(標準偏差):Part B(PKAS)空腹時投与 食後投与
N=10 N=10Aelast(mg)
1.080(0.280)
1.189(0.317)
Aeinf(mg)
1.097(0.291)
1.208(0.326)
Aelast%(%)
1.080(0.280)
1.189(0.317)
Aeinf%(%)
1.097(0.291)
1.208(0.326)
CLr(L/h)
0.134(0.029)
0.135(0.025)
平均値(標準偏差)
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.2.4.3.3
2.7.6.4.5.2 薬力学の結果
Part A(用量漸増)
血糖値の AUC の平均値(標準偏差)を表 2.7.6.4-8 に示した。
時間(h)
血漿中未変化体濃度(
ng/m
L)
空腹時投与
食後投与
86

2.7.6個々の試験のまとめ
血糖値に及ぼす本剤の影響はみられなかった。
表 2.7.6.4-8 血糖値の AUC の平均値(標準偏差):Part A(PKAS)ASP1941プラセボ
0.1 mg 1 mg 3 mg 10 mg 30 mg 100 mg 300 mg 600 mgN=15 N=6 N=6 N=6 N=6 N=6 N=5 N=6 N=6
AUC24h(h•mmol/L)
125.62(6.32)
127.58(5.36)
121.67(11.00)
135.01(11.11)
128.71(10.91)
125.49(8.32)
125.14(4.43)
125.07(9.91)
129.23(7.89)
AUClast(h•mmol/L)
372.51(14.75)
379.17(7.65)
358.04(27.35)
380.31(21.93)
377.58(22.80)
371.04(34.94)
360.34(15.60)
344.86(39.82)
376.55(27.56)
平均値(標準偏差)
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.1.4.7
尿中グルコースパラメータの平均値(標準偏差)を表 2.7.6.4-9 に示した。
尿中へのグルコース排泄量は,用量増加に伴って増加した。
表 2.7.6.4-9 尿中グルコースパラメータの平均値(標準偏差):Part A(PKAS)ASP1941プラセボ
0.1 mg 1 mg 3 mg 10 mg 30 mg 100 mg 300 mg 600 mgAe24h
(mmol)n=130.84
(0.19)
n=20.72
(0.17)
n=62.92
(5.17)
n=218.20(4.10)
n=654.96
(25.53)
n=591.34
(33.03)
n=5241.77(44.18)
n=5358.26(48.29)
n=5385.85(35.43)
Aelast(mmol)
n=152.61
(0.55)
n=62.55
(0.39)
n=66.66
(10.26)
n=517.41
(11.78)
n=659.00
(27.74)
n=6113.31(28.17)
n=5369.76(77.92)
n=5610.28(95.62)
n=5857.98
(263.16)CLr 24h(L/h)
n=130.01
(0.00)
n=20.01
(0.00)
n=60.02
(0.04)
n=20.15
(0.04)
n=60.42
(0.17)
n=50.75
(0.32)
n=51.93
(0.29)
n=52.87
(0.19)
n=52.98
(0.42)
平均値(標準偏差)
CLr 24h:24 時間の CLr
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.1.4.10
Part B(食事の影響)
血糖値の AUC の平均値(標準偏差)を表 2.7.6.4-10 に示した。
血糖値に及ぼす食事の影響はみられなかった。
表 2.7.6.4-10 血糖値の AUC の平均値(標準偏差):Part B(PKAS)空腹時投与 食後投与
N=10 N=10AUC24h (h•mmol/L) 128.03(6.64) 132.77(7.70)AUClast (h•mmol/L) 380.40(18.62) 386.40(19.59)
平均値(標準偏差)
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.2.4.4.2
尿中グルコースパラメータの平均値(標準偏差)を表 2.7.6.4-11 に示した。
尿中グルコースに及ぼす食事の影響はみられなかった。
87

2.7.6個々の試験のまとめ
表 2.7.6.4-11 尿中グルコースパラメータの平均値(標準偏差):Part B(PKAS)空腹時投与 食後投与
N=10 N=10Ae24h (mmol) 292.72(83.68) 300.02(107.17)Aelast (mmol) 423.18(109.52) 413.62(160.55)CLr 24h (L/h) 2.29(0.67) 2.27(0.84)
平均値(標準偏差)
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.2.4.5.3
2.7.6.4.6 安全性
2.7.6.4.6.1 有害事象
Part A(用量漸増)
Part A で認められた有害事象の要約を表 2.7.6.4-12 に示した。
有害事象発現率は,本剤群 23.4%(11/47 例),プラセボ群 13.3%(2/15 例)であった。本剤群は
すべての用量で忍容性は良好であった。副作用はプラセボ群では認められず,本剤群でのみ認め
られた。
表 2.7.6.4-12 有害事象の要約:Part A(SAF)ASP1941プラセボ
0.1 mg 1 mg 3 mg 10 mg 30 mg 100 mg 300 mg 600 mg 計N=15 N=6 N=6 N=6 N=6 N=6 N=5 N=6 N=6 N=47
有害事象発現例数(発現
率)
2(13.3%)
0 0 2(33.3%)
3(50.0%)
0 2(40.0%)
2(33.3%)
2(33.3%)
11(23.4%)
有害事象発現件数 2 0 0 3 6 0 2 5 6 22副作用発現例数(発現率) 0 0 0 2
(33.3%)3
(50.0%)0 1
(20.0%)2
(33.3%)2
(33.3%)10
(21.3%)副作用発現件数 0 0 0 2 6 0 1 4 3 16重篤な有害事象発現例数
(発現率)
0 0 0 0 0 0 0 0 0 0
重篤な有害事象発現件数 0 0 0 0 0 0 0 0 0 0中止に至った有害事象
発現例数(発現率)
0 0 0 0 0 0 0 0 0 0
程度別有害事象発現例数(発現率)軽度 2
(13.3%)0 0 2
(33.3%)3
(50.0%)0 2
(40.0%)1
(16.7%)2
(33.3%)10
(21.3%)中等度 0 0 0 0 0 0 0 1
(16.7%)0 1
(2.1%)重度 0 0 0 0 0 0 0 0 0 0程度不明 0 0 0 0 0 0 0 0 0 0
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.1.5.3
Part A で認められた有害事象及び副作用の内訳をそれぞれ表 2.7.6.4-13 及び表 2.7.6.4-14 に示し
た。
本剤群で 2 例以上に認められた有害事象は頭痛のみであり,6 例(12.8%)に認められた。300 mg
群の 1 例に発現した中等度の頭痛を除き,いずれの有害事象も軽度と判定された。有害事象発現
率に用量依存性はみられなかった。
88

2.7.6個々の試験のまとめ
低血糖は発現しなかった。また,腎機能について,β-N-アセチル D グルコサミニダーゼ増加及
び頻尿の副作用が各 1 例に認められたが,本剤投与による臨床的に重要な影響はみられなかった。
副作用発現率に明らかな用量依存性はみられなかった。
表 2.7.6.4-13 有害事象の内訳:Part A(SAF)ASP1941プラセボ
0.1 mg 1 mg 3 mg 10 mg 30 mg 100 mg 300 mg 600 mg 計
MedDRA version 9.1器官別大分類
基本語N=15 N=6 N=6 N=6 N=6 N=6 N=5 N=6 N=6 N=47
すべての
有害事象
2(13.3%)
0 0 2(33.3%)
3(50.0%)
0 2(40.0%)
2(33.3%)
2(33.3%)
11(23.4%)
眼障害 0 0 0 0 0 0 0 1(16.7%)
0 1(2.1%)
羞明 0 0 0 0 0 0 0 1(16.7%)
0 1(2.1%)
全身障害および
投与局所様態
0 0 0 1(16.7%)
1(16.7%)
0 1(20.0%)
0 1(16.7%)
4(8.5%)
冷感 0 0 0 0 1(16.7%)
0 0 0 0 1(2.1%)
空腹 0 0 0 0 0 0 1(20.0%)
0 0 1(2.1%)
注射部位血腫 0 0 0 0 0 0 0 0 1(16.7%)
1(2.1%)
血管穿刺部位
血腫
0 0 0 1(16.7%)
0 0 0 0 0 1(2.1%)
臨床検査 0 0 0 1(16.7%)
0 0 0 0 0 1(2.1%)
β-N アセチル
D グルコサミ
ニダーゼ増加
0 0 0 1(16.7%)
0 0 0 0 0 1(2.1%)
筋骨格系および
結合組織障害
1(6.7%)
0 0 0 0 0 1(20.0%)
0 0 1(2.1%)
背部痛 1(6.7%)
0 0 0 0 0 1(20.0%)
0 0 1(2.1%)
神経系障害 0 0 0 0 3(50.0%)
0 0 2(33.3%)
2(33.3%)
7(14.9%)
体位性めまい 0 0 0 0 0 0 0 0 1(16.7%)
1(2.1%)
味覚異常 0 0 0 0 0 0 0 0 1(16.7%)
1(2.1%)
頭痛 0 0 0 0 3(50.0%)
0 0 2(33.3%)
1(16.7%)
6(12.8%)
腎および
尿路障害
0 0 0 1(16.7%)
0 0 0 0 0 1(2.1%)
頻尿 0 0 0 1(16.7%)
0 0 0 0 0 1(2.1%)
呼吸器,胸郭
および縦隔障害
1(6.7%)
0 0 0 0 0 0 0 1(16.7%)
1(2.1%)
咳嗽 0 0 0 0 0 0 0 0 1(16.7%)
1(2.1%)
咽喉頭疼痛 1(6.7%)
0 0 0 0 0 0 0 0 0
皮膚および
皮下組織障害
0 0 0 0 0 0 0 1(16.7%)
1(16.7%)
2(4.3%)
ざ瘡 0 0 0 0 0 0 0 0 1(16.7%)
1(2.1%)
多汗症 0 0 0 0 0 0 0 1(16.7%)
0 1(2.1%)
発現例数(発現率)
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.1.5.4
89

2.7.6個々の試験のまとめ
表 2.7.6.4-14 副作用の内訳:Part A(SAF)ASP1941プラセボ
0.1 mg 1 mg 3 mg 10 mg 30 mg 100 mg 300 mg 600 mg 計
MedDRA version 9.1器官別大分類
基本語N=15 N=6 N=6 N=6 N=6 N=6 N=5 N=6 N=6 N=47
すべての
副作用
0 0 0 2(33.3%)
3(50.0%)
0 1(20.0%)
2(33.3%)
2(33.3%)
10(21.3%)
眼障害 0 0 0 0 0 0 0 1(16.7%)
0 1(2.1%)
羞明 0 0 0 0 0 0 0 1(16.7%)
0 1(2.1%)
全身障害および
投与局所様態
0 0 0 0 1(16.7%)
0 1(20.0%)
0 0 2(4.3%)
冷感 0 0 0 0 1(16.7%)
0 0 0 0 1(2.1%)
空腹 0 0 0 0 0 0 1(20.0%)
0 0 1(2.1%)
臨床検査 0 0 0 1(16.7%)
0 0 0 0 0 1(2.1%)
β-N アセチル
D グルコサミ
ニダーゼ増加
0 0 0 1(16.7%)
0 0 0 0 0 1(2.1%)
神経系障害 0 0 0 0 3(50.0%)
0 0 2(33.3%)
2(33.3%)
7(14.9%)
体位性めまい 0 0 0 0 0 0 0 0 1(16.7%)
1(2.1%)
味覚異常 0 0 0 0 0 0 0 0 1(16.7%)
1(2.1%)
頭痛 0 0 0 0 3(50.0%)
0 0 2(33.3%)
1(16.7%)
6(12.8%)
腎および
尿路障害
0 0 0 1(16.7%)
0 0 0 0 0 1(2.1%)
頻尿 0 0 0 1(16.7%)
0 0 0 0 0 1(2.1%)
皮膚および皮下
組織障害
0 0 0 0 0 0 0 1(16.7%)
0 1(2.1%)
多汗症 0 0 0 0 0 0 0 1(16.7%)
0 1(2.1%)
発現例数(発現率)
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.1.5.7
Part B(食事の影響)
Part B で認められた有害事象の要約を表 2.7.6.4-15 に示した。
有害事象発現率は空腹時投与で 60.0%(6/10 例),食後投与で 41.7%(5/12 例)であり,空腹時
投与と比較して食後投与で低かった。空腹時投与及び食後投与のいずれの投与条件下でも忍容性
は良好であった。
90

2.7.6個々の試験のまとめ
表 2.7.6.4-15 有害事象の要約:Part B(SAF)空腹時投与 食後投与 計
N=10 N=12 N=12有害事象発現例数(発現率) 6(60.0%) 5(41.7%) 8(66.7%)有害事象発現件数 21 7 28副作用発現例数(発現率) 4(40.0%) 3(25.0%) 5(41.7%)副作用発現件数 9 4 13重篤な有害事象発現例数(発現率) 0 0 0重篤な有害事象発現件数 0 0 0中止に至った有害事象発現例数(発現率) 0 1(8.3%) 1(8.3%)程度別有害事象発現例数(発現率)
軽度 5(50.0%) 4(33.3%) 6(50.0%)中等度 1(10.0%) 1(8.3%) 2(16.7%)重度 0 0 0程度不明 0 0 0
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.2.5.3
Part B で認められた有害事象及び副作用の内訳をそれぞれ表 2.7.6.4-16 及び表 2.7.6.4-17 に示し
た。
2 例以上に認められた有害事象は,空腹時投与で認められたカテーテル留置部位疼痛(2 例)の
みであった。食後投与で認められた疲労及び空腹時投与で認められた頚部痛(各 1 例,いずれも
中等度)を除き,いずれの有害事象も軽度と判定された。
腎機能に関連する有害事象及び低血糖は発現しなかった。
副作用発現件数は,空腹時投与(9 件)と比較して食後投与(4 件)で少なかった。
91

2.7.6個々の試験のまとめ
表 2.7.6.4-16 有害事象の内訳:Part B(SAF)空腹時投与 食後投与 計MedDRA version 9.1
器官別大分類
基本語N=10 N=12 N=12
すべての有害事象 6(60.0%) 5(41.7%) 8(66.7%)眼障害 0 1(8.3%) 1(8.3%)
瞳孔不同 0 1(8.3%) 1(8.3%)胃腸障害 1(10.0%) 1(8.3%) 2(16.7%)
下痢 0 1(8.3%) 1(8.3%)悪心 1(10.0%) 0 1(8.3%)
全身障害および投与局所様態 4(40.0%) 2(16.7%) 4(33.3%)カテーテル留置部位疼痛 2(20.0%) 1(8.3%) 2(16.7%)疲労 1(10.0%) 1(8.3%) 1(8.3%)局所腫脹 1(10.0%) 0 1(8.3%)穿刺部位疼痛 1(10.0%) 0 1(8.3%)
臨床検査 1(10.0%) 0 1(8.3%)肝酵素上昇 1(10.0%) 0 1(8.3%)筋酵素上昇 1(10.0%) 0 1(8.3%)
代謝および栄養障害 1(10.0%) 0 1(8.3%)脱水 1(10.0%) 0 1(8.3%)
筋骨格系および結合組織障害 2(20.0%) 2(16.7%) 4(33.3%)背部痛 0 1(8.3%) 1(8.3%)関節腫脹 0 1(8.3%) 1(8.3%)筋骨格硬直 1(10.0%) 0 1(8.3%)頚部痛 1(10.0%) 0 1(8.3%)
神経系障害 2(20.0%) 1(8.3%) 2(16.7%)浮動性めまい 1(10.0%) 0 1(8.3%)嗜眠 1(10.0%) 0 1(8.3%)傾眠 1(10.0%) 1(8.3%) 1(8.3%)
呼吸器,胸郭および縦隔障害 1(10.0%) 0 1(8.3%)咳嗽 1(10.0%) 0 1(8.3%)
皮膚および皮下組織障害 2(20.0%) 0 2(16.7%)アレルギー性皮膚炎 1(10.0%) 0 1(8.3%)皮膚反応 1(10.0%) 0 1(8.3%)
血管障害 1(10.0%) 0 1(8.3%)血腫 1(10.0%) 0 1(8.3%)
発現例数(発現率)
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.2.5.4
92

2.7.6個々の試験のまとめ
表 2.7.6.4-17 副作用の内訳:Part B(SAF)空腹時投与 食後投与 計MedDRA version 9.1
器官別大分類
基本語N=10 N=12 N=12
すべての副作用 4(40.0%) 3(25.0%) 5(41.7%)眼障害 0 1(8.3%) 1(8.3%)
瞳孔不同 0 1(8.3%) 1(8.3%)胃腸障害 1(10.0%) 1(8.3%) 2(16.7%)
下痢 0 1(8.3%) 1(8.3%)悪心 1(10.0%) 0 1(8.3%)
全身障害および投与局所様態 1(10.0%) 1(8.3%) 1(8.3%)疲労 1(10.0%) 1(8.3%) 1(8.3%)
代謝および栄養障害 1(10.0%) 0 1(8.3%)脱水 1(10.0%) 0 1(8.3%)
筋骨格系および結合組織障害 1(10.0%) 0 1(8.3%)筋骨格硬直 1(10.0%) 0 1(8.3%)
神経系障害 2(20.0%) 1(8.3%) 2(16.7%)浮動性めまい 1(10.0%) 0 1(8.3%)嗜眠 1(10.0%) 0 1(8.3%)傾眠 1(10.0%) 1(8.3%) 1(8.3%)
発現例数(発現率)
Source:CL-0001 総括報告書(5.3.3.1-2)Table 13.2.5.7
2.7.6.4.6.2 死亡,その他の重篤な有害事象
本試験では,Part A 及び Part B いずれでも,死亡及びその他の重篤な有害事象は認められなかっ
た。
2.7.6.4.6.3 中止に至った有害事象
Part A(用量漸増)
Part A では中止に至った有害事象は認められなかった。
Part B(食事の影響)
Part B では,1 例で食後投与後に中止に至った有害事象(瞳孔不同)が発現した。本事象は Day
11 に発現し,空腹時投与前に本試験を中止した。程度は軽度であり,治験薬との関連性は「関連
あるかもしれない」と判定された。神経学的検査では,ごく軽微な瞳孔不同を除いて正常と判定
された。神経科医は,試験継続に関して何らかの対策を行う必要性は低いと判断した。
2.7.6.4.6.4 臨床検査値
Part A(用量漸増)
臨床検査値について,治験薬投与に関連した又は用量依存的な臨床的に重要な傾向はみられな
かった。
93

2.7.6個々の試験のまとめ
腎機能マーカー(β-N-アセチル D グルコサミニダーゼ,β2 ミクログロブリン及び α1 ミクログ
ロブリン)に特記すべき影響は観察されなかった。一部の用量群で治験薬投与 3 時間後に上昇が
みられたが,一過性の変動であり,試験期間中に基準値内まで低下した。この変動に用量依存性
はみられなかった。
3 mg 群の 1 例で,治験薬投与 3 時間後に β-N-アセチル D グルコサミニダーゼの上昇
(158.7 μmol/h/mmol Cr;基準値上限 70.4 μmol/h/mmol Cr)が認められ,軽度の有害事象として報
告された。当該被験者のベースライン時の β-N-アセチル D グルコサミニダーゼは
61.9 μmol/h/mmol Cr であった。治験薬投与 24 時間後には β-N-アセチル D グルコサミニダーゼは
71.3 μmol/h/mmol Cr まで低下した。本事象と治験薬との関連性は「多分(おそらく)関連あり」
と判定された。本事象は無症候性であり,当該被験者では β2 ミクログロブリン及び α1 ミクログ
ロブリンはすべての評価時点で基準値内であった。
Part B(食事の影響)
中性脂肪の平均値が食後投与 3 時間後に一時的に軽度上昇したことを除き,臨床検査値に明ら
かな食事の影響はみられなかった。この中性脂肪の上昇は食事の摂取に関連するものであり,本
剤投与とは関連がないと考えられた。
2.7.6.4.6.5 バイタルサイン
Part A(用量漸増)
収縮期血圧,拡張期血圧及び脈拍数の平均値に臨床的に重要な変動は観察されなかった。
Part B(食事の影響)
食後投与で脈拍数の平均値に一過性の軽度の上昇がみられたことを除き,バイタルサインに明
らかな食事の影響はみられなかった。この脈拍数の上昇は食事の摂取に関連するものであり,本
剤の投与とは関連がないと考えられた。
2.7.6.4.6.6 心電図
Part A(用量漸増)
被験者の大部分に 1 測定時点以上で心電図異常(主に洞性徐脈及び洞性不整脈)が観察された
が,治験担当医師により臨床的に重要と判定されたものはなかった。心電図異常に一定の特徴は
みられなかった。
Part B(食事の影響)
被験者の大部分に複数の測定時点以上で心電図異常(主に洞性徐脈及び洞性不整脈)が観察さ
れたが,治験担当医師により臨床的に重要と判定されたものはなかった。心電図異常に一定の特
徴はみられなかった。
94

2.7.6個々の試験のまとめ
2.7.6.4.7 結論
● 本剤は速やかに吸収された。AUC は,1~600 mg の用量で用量比をわずかに上回る上昇がみ
られた。一方,300 mg 以上の用量の Cmaxは用量比を下回っていた。t1/2 は 30 mg 以上の用量
で約 16 時間であった。未変化体の尿中排泄率は低く,いずれの用量でも投与量の約 1%であっ
た。
● 本剤の吸収量に及ぼす食事の影響はみられなかった。一方,空腹時投与時と比較して食後投
与時に Cmaxが 14%とわずかに低下した。空腹時投与時に対する食後投与時の Cmaxの比の 90%
信頼区間の下限が 0.80~1.25 の範囲を下回ったことから,軽度ではあるものの食事の影響は
排除できないと考えられた。
● 尿中グルコース排泄量は用量増加に伴って増加した。血糖値に及ぼす本剤の影響はみられな
かった。
● 本剤を 600 mg までの用量で単回投与したとき,安全性及び忍容性は良好であった。検討した
用量範囲で MTD は見出せなかったが,600 mg を超える用量への増量は行わなかった。
● 本剤投与例で最も多く認められた有害事象は頭痛であった。有害事象発現率に明らかな用量
依存性はみられなかった。食後投与で認められた副作用の件数は,空腹時投与時と比較して
少なかった。しかし,被験者数が少ないことから,これは予備的な結果と判断すべきと考え
られた。また,低血糖は発現しなかった。
● 腎機能マーカーに特記すべき影響はみられなかった。
95

2.7.6個々の試験のまとめ
2.7.6.5 海外第 I 相試験(単回・反復)[CL-0002](添付資料 5.3.3.1-3)
2.7.6.5.1 試験方法の概略
治験課題名:健康成人を対象に,ASP1941 を反復経口投与したときの安全性,忍容性及び薬物動
態を評価し,ASP1941 の血糖値及び尿中グルコース濃度に及ぼす影響を探索的に検討する二重盲
検プラセボ対照用量漸増試験
治験実施医療機関:オランダ 1 施設
公表文献:なし
治験期間:
治験開始日:2007 年 5 月
治験終了日:2008 年 3 月
開発のフェーズ:第 I 相
目的:
(1) 主要目的
ASP1941(以下,本剤)を用量漸増反復投与したときの安全性及び忍容性を評価し,可能であれば,
最大耐量(MTD)を決定する。
(2) 副次目的
本剤を単回及び反復投与したときの薬物動態,薬力学及び薬物動態/薬力学関連性を評価する。
治験デザイン・治験方法:
本試験は,健康成人を対象とした単一施設,無作為化,二重盲験,逐次群,プラセボ対照,用量
漸増反復投与試験として実施した。
本剤は男性被験者を対象に 5 mg を開始用量とし,30,100,300 及び 600 mg へと増量するよう計
画した。次用量への移行は,治験担当医師が安全性及び忍容性データを慎重に評価し,治験依頼
者と協議した上で決定した。100 mg 群は女性被験者も対象とした。女性被験者への投与は,生殖
毒性試験(ICH M3)の終了を独立倫理委員会に通知し,男性被験者で 100 mg の忍容性が認められ
た後に開始した。各用量段階の被験者数は 8 例とし,6 例に本剤を,2 例にプラセボを無作為に割
り付けた。
各用量段階では,被験者は投与前日(Day −1)に入所し,割り付けられた治験薬を Day 1(単回投
与)及び Day 5~14(反復投与)に 1 日 1 回服用した。Day 1(単回投与)及び Day 14(反復投与
の最終投与日)の治験薬投与前から投与後 96 時間までの規定された評価時点で薬物動態及び薬力
学評価用の血液及び尿を採取した。安全性に関するデータは試験期間を通じて収集した。被験者
は Day 18 に退所し,退所から 7~14 日後(Day 25~31)に後観察を行った。
なお,本試験では,HbA1c 値の表記として National Glycohemoglobin Standardization Program(NGSP)値を用いた。
目標被験者数:48 例(男性 40 例,女性 8 例,各投与群 8 例とし男性若しくは女性のみで構成)
【設定根拠】
目標被験者数は,ヒトへの新規化学物質の曝露を最小限に留める必要性と各用量段階で化合物の
安全性及び忍容性を十分な被験者数で評価するための要件の両面を勘案し,医学的見地に基づき
設定した。被験者数の正式な統計学的算出は行わなかった。
96

2.7.6個々の試験のまとめ
診断及び選択・除外基準:
1. 選択基準(以下の基準をすべて満たす者を対象とした)
(1) 18 歳以上 55 歳以下の健康な男性又は女性
(2) 体重 60 kg 以上 100 kg 以下,かつ BMI 20 kg/m2 以上 30 kg/m2 以下の者
(3) 被験者本人から文書による同意が得られている者
2. 除外基準(以下の基準のいずれかに該当する者は対象から除外した)
(1) 1 型又は 2 型糖尿病の既往を有する者
(2) 適切な二重障壁避妊方法を施行していない妊娠可能な女性
(3) 妊娠している女性,又は授乳中の女性
(4) 空腹時血糖値が 6.4 mmol/L を超える又は HbA1c 値が 6.2%を超える者
(5) 腎性糖尿又は蛋白尿が認められる者
(6) 本剤又は製剤の構成成分に対して過敏症を有する又は過敏症が疑われる者
(7) 喘息,湿疹等のアレルギー疾患又は何らかの薬剤に対する重度の過敏症といった臨床的に重
要な既往を有する者
(8) 治験実施医療機関入所前 4 週間以内に臨床的に重要な上部消化管症状(悪心,嘔吐,腹部不
快感又は腹部不調,胸やけ等)がみられた者
(9) その他,消化器系,心血管系,呼吸器系,腎機能,肝機能,神経系,皮膚,精神又は代謝系
に治験担当医師が臨床的に重要と判断した疾患又は障害の既往を有する者
(10) 本試験前の身体所見,12 誘導心電図及び臨床検査の結果,治験担当医師が臨床的に重要な異
常があると判断した者
(11) 本試験前来院時の脈拍数及び/又は血圧に治験担当医師が異常と判断した所見が認められた
者
(12) 治験実施医療機関入所前 4 週間以内に処方薬又は市販薬(ビタミン剤,ハーブ製剤を含む)
を常用した者,若しくは治験実施医療機関入所前 2 週間以内にそれらの薬剤を使用した者
(13) 治験実施医療機関入所前 3 カ月以内に薬物乱用歴を有する者
(14) 治験実施医療機関入所前 3 カ月以内に 1 日 10 本を超える巻きタバコ(又は当量の刻みタバコ)
を使用した者
(15) 治験実施医療機関入所前 3 カ月以内に 1 週間あたり 21 単位(1 単位=ビール 270 mL,蒸留酒
40 mL,ワイン 175 mL)を超える飲酒をした者
(16) 治験実施医療機関入所前 3 カ月以内に献血(全血又は成分献血)を行った者
(17) 血清学的検査で HBs 抗原,HAV IgM 抗体,HCV 抗体又は HIV 抗体 1 及び 2 が陽性であった
者
(18) 何らかの理由のため本試験を完了できない可能性があると治験担当医師が判断した者
(19) 本試験登録予定日から遡って 3 カ月以内に他の臨床試験に参加した者,又は 12 カ月以内に 4件以上の臨床試験に参加した者
(20) 治験担当医師により,本試験を安全に完了できない疾患を有すると判断された者
(21) アステラス製薬グループ又は本試験に関連する医薬品開発業務受託機関に雇用されている者
97

2.7.6個々の試験のまとめ
治験薬,投与量及び投与方法:
1. 治験薬及びロット番号
(1) 被験薬
ASP1941 錠(ASP1941 錠 1 mg,ASP1941 錠 10 mg,ASP1941 錠 100 mg):1 錠中に ASP1941 としてそれぞれ 1,10,100 mg を含有するフィルムコート錠
[ロット番号]
ASP1941 錠 1 mg:BX1000212ASP1941 錠 10 mg:BX1000213ASP1941 錠 100 mg:BX1000214
(2) 対照薬
ASP1941 錠プラセボ(ASP1941 錠 1 mg プラセボ,ASP1941 錠 10 mg プラセボ,ASP1941 錠 100 mgプラセボ):
1 錠中に ASP1941 を含有しないフィルムコート錠
[ロット番号]
ASP1941 錠 1 mg プラセボ:BX1000209ASP1941 錠 10 mg プラセボ:BX1000210ASP1941 錠 100 mg プラセボ:BX1000211
2. 投与量及び投与方法
被験薬及び対照薬:
本剤(5,30,100,300 又は 600 mg)又はプラセボのいずれかを,Day 1 に単回経口投与し,Day 5~14 に 1 日 1 回反復経口投与した。なお,女性被験者には,本剤 100 mg 又はプラセボの投与のみ
を行った。Day 1 及び 14 では朝の空腹時に投与し,投与後 4 時間まで食事は摂取しないこととし
た。Day 5~13 では,ほぼ一定時刻の朝食の 30 分前に投与した。本試験期間中は標準食及び飲料
を被験者に提供した。
【設定根拠】
(1) 投与量の設定根拠
海外第 I 相試験(単回・食事)[CL-0001]では,本剤 3 mg で被験者の半数に,本剤 10 mg で全被
験者に明らかな薬理作用が確認されたことから,中間の用量として,本試験の最低用量 5 mg を設
定した。
性別による差を検出するため,100 mg の用量段階のみ女性被験者も対象とした。
(2) 投与方法の設定根拠
本剤の蓄積性を検討するため,単回投与時の薬物動態と反復投与時の薬物動態の比較を設定した。
また,同様の理由でトラフ値の測定を設定した。本剤の薬物動態が消失半減期(t1/2)の 5 倍で定
常状態に到達すると仮定して,反復投与期を 10 日間に設定した。t1/2の推定値(最高用量で最長
25 時間)に基づき,薬物動態データを Day 1 及び Day 14 の投与後 96 時間まで評価することとし
た。
評価期間:
治験薬投与期間:単回投与 1 日間,反復投与 10 日間
併用治療(薬剤及び療法):
治験実施医療機関入所 4 週間前から処方薬及び市販薬(ビタミン剤,ハーブ製剤を含む)の常用
を禁止した。入所 2 週間前から後観察までそれらの薬剤の使用を禁止した。ただし,治験担当医
師から了承が得られた場合に限り,一定限度のアセトアミノフェン(3 g/日以下)の使用は可とし
た。
98

2.7.6個々の試験のまとめ
評価項目,評価スケジュール及び評価基準:
1. 評価スケジュールスク
リーニ
ング
単回投与 反復投与 反復投与
の最終投
与日
反復投
与評価
期間
退所 後 察
Day –21~–1 –1 1 2~4 5a~13 14 15~17 18 25~31入所 X X X X X X 10:00
まで
無作為化 X治験薬投与 X Xb X食事 18:00
23:00投与後
4, 9 時間
9:00c
13:0018:00
投与後0.5, 4.5,9.5 時間
投与後
4, 9 時間
9:00c
13:0018:00
9:00c
同意取得 X試験前スクリーニング d X身体所見 X X X妊娠検査(女性) X X X血糖値測定 e X X X X X X尿中グルコース測定 f X X X X X X薬物動態評価用採血 e X X X X X X薬物動態評価用蓄尿 f X X X X X X薬物乱用及び飲酒調査 X X臨床検査(血液学的検査,
血液生化学検査,尿検査)
X X Xg Xg Xg Xg Xg X
バイタルサイン(血圧,
脈拍数)
X X Xh Xh Xh Xh Xh X X
12 誘導心電図 X X Xi Xi Xi Xi Xi X X併用薬/有害事象調査 X X X X X X X X
a:単回投与(Day 1)後の採血及び蓄尿の最終日かつ反復投与の初日
b:治験薬投与は朝食の 30 分前に実施
c:同時刻に予定されている採血及び評価終了後に食事の摂取を開始
d:被験者背景,病歴,治療歴,喫煙及び飲酒習慣の調査並びに血清学的検査
e:採血時期:Day 1 及び Day 14 の治験薬投与前,投与後 0.25,0.5,1,1.5,2,3,4,6,8,10,12,16,24,36,48,60,72,84,96 時間,Day 6~13 の投与前
f:蓄尿時期:Day 1 及び Day 14 の治験薬投与前,投与後 0~2,2~4,4~6,6~8,8~10,10~12,12~16,16~24,24~36,36~48,48~60,60~72,72~84,84~96 時間
g:測定時期:Day 1 の治験薬投与後 3,24 時間,Day 5,Day 8 及び Day 11 の投与前,Day 14 の治験薬投与後 3,24,72 時間
h:測定時期:Day 1 の治験薬投与前,投与後 3,24,48,72 時間,Day 5~13 の治験薬投与前,投与後 3 時間,Day 14 の治験薬
投与前,投与後 24,48,72 時間
i:測定時期:Day 1 の治験薬投与前,投与後 3,24,48,72 時間,Day 5,Day 8 及び Day 11 の治験薬投与前,投与後 3 時間,
Day 14 の治験薬投与前,投与後 3,24,48,72 時間
2. 薬物動態:
「1. 評価スケジュール」に示した時期に採血及び蓄尿を行い,血漿中及び尿中未変化体濃度を測
定した。血漿中及び尿中未変化体濃度から,以下の薬物動態パラメータをノンコンパートメント
法を用いて算出した。
[血漿中パラメータ]
・最高血漿中濃度到達時間(tmax)
・最高血漿中濃度(Cmax)
・時間 0 から定量可能最終時点までの血漿中濃度-時間曲線下面積(AUClast)(単回投与時のみ)
・時間 0 から無限時間まで外挿した血漿中濃度-時間曲線下面積(AUCinf)(単回投与時のみ)
・投与間隔ごとの血漿中濃度-時間曲線下面積(AUCtau)(単回投与時の投与後 24 時間までの血
漿中濃度-時間曲線下面積を含む)
・累積比(Racc)[反復投与時(最終投与後)のみ]
99

2.7.6個々の試験のまとめ
・経口クリアランス(CL/F)・みかけの分布容積(Vz/F)・消失半減期(t1/2)
・トラフ濃度(Ctrough)(Day 6~14 に毎日)
・ピーク/トラフ比(PTR)・ラグタイム(tlag)
[尿中パラメータ]
・時間 0 から定量可能最終時点までの尿中排泄量(Aelast)(単回投与時のみ)
・時間 0 から定量可能最終時点までの尿中排泄率(Aelast%)(単回投与時のみ)
・時間 0 から無限時間まで外挿した尿中排泄量(Aeinf)(単回投与時のみ)
・時間 0 から無限時間まで外挿した尿中排泄率(Aeinf%)(単回投与時のみ)
・投与間隔ごとの尿中排泄量(Aetau)[反復投与時(最終投与後)のみ]
・投与間隔ごとの尿中排泄率(Aetau%)[反復投与時(最終投与後)のみ]
・腎クリアランス(CLr)
3. 薬力学(血糖値及び尿中グルコース):
「1. 評価スケジュール」に示した時期に採血及び蓄尿を行い,血糖値及び尿中グルコース濃度を
測定した。血糖値及び尿中グルコース濃度から以下のパラメータを算出し,血糖値及び尿中グル
コース排泄量に及ぼす本剤の影響を探索した。
[血糖値]
・投与間隔ごとの血漿中濃度-時間曲線下面積(AUCtau)(単回投与時の投与後 24 時間までの血
漿中濃度-時間曲線下面積を含む)
・Ctrough
[尿中グルコース]
・Aelast(単回投与時のみ)
・投与間隔ごとの尿中排泄量(Aetau)(単回投与時の投与後 24 時間までの尿中排泄量を含む)
・蓄尿間隔ごと及び 24 時間の CLr
4. 安全性:
・有害事象
・身体所見,12 誘導心電図,バイタルサイン(脈拍数及び血圧)
・臨床検査値(血液学的検査,血液生化学検査,尿検査)
・尿中の β-N-アセチル D グルコサミニダーゼ,β2 ミクログロブリン及び α1 ミクログロブリン
・MTD の決定
有害事象は,治験薬投与開始後から後観察までに発現した事象を調査対象とした。有害事象と治
験薬との関連性は以下の基準により判定し,「関連あるかもしれない」又は「多分(おそらく)関
連あり」のいずれかに該当した有害事象を「治験薬との関連性が否定できない有害事象(副作用)」
と定義した。
治験薬との関連性 関連性判定基準
否定できる 有害事象発現と薬剤投与との間に時間的関係から関連性がありそうにないと思われる場
合や,他の薬剤や合併症・基礎疾患等により説明ができる場合
関連あるかもしれ
な 有害事象発現と薬剤投与との間に時間的に妥当な相関関係があるが,要因として合併症・
基礎疾患や他の薬剤による説明もできる場合。投与中止に関する情報が不明確な場合
多分(おそらく)
関連あり
有害事象発現と薬剤投与との間に時間的に妥当な相関関係がある場合,合併症・基礎疾患
や他の薬剤では説明ができない,又はそれらによる可能性が考えにくい場合,再投与によ
り再発又は投与中止により消失又は軽快を示す場合
100

2.7.6個々の試験のまとめ
有害事象の程度は,以下に従って判定した。
・軽度:日常の活動に支障がないもの
・中等度:日常の活動に支障があるもの
・重度:日常の活動が不可能になるもの
また,各用量群で以下のいずれかに該当する場合,増量を中止し当該用量を最小不耐量(MID)と
することとした。MID の 1 段階低い用量を MTD とすることとした。
1) 中等度以上の副作用(「関連あるかもしれない」又は「多分(おそらく)関連あり」と判定さ
れた有害事象)が複数件認められた被験者の割合が,同一用量群で 50%以上となった場合
2) 1 回でも血糖値が 2.8 mmol/L を下回った場合
3) 重篤な副作用が発現した場合
統計解析:
1. 解析対象集団:
(1) 安全性解析対象集団(SAF)治験薬を 1 回以上服用した被験者の集団を SAF とした。
(2) 薬物動態解析対象集団(PKAS)1つ以上の薬物動態パラメータを算出できる血漿中濃度データが得られた被験者の集団をPKASとした。
2. 被験者背景及びその他の基準値:
SAF を対象に,計数値項目では度数集計を行い,計量値項目では要約統計量を算出した。
3. 薬物動態:
PKAS を対象に,薬物動態パラメータの要約統計量を投与群ごとに算出した。
本剤の血漿中薬物動態に及ぼす性別の影響に関しては,血漿中未変化体濃度の対数変換 AUC(単
回投与時では AUCinf,定常状態では AUCtau)及び Cmaxについて,本剤 100 mg 群の男性被験者に
対する女性被験者の比及び 90%信頼区間を算出して検証した。AUC 及び Cmax共に 90%信頼区間が
0.8~1.25 の範囲内である場合,性別の影響は認められないと判断した。
血漿中未変化体濃度の用量比例性に関しては,単回投与時(Day 1)及び定常状態(Day 14)の AUC及び Cmaxの分散分析(パワーモデル)を行って検討した。
時間依存性に関しては,用量を分類変数として共変量に含めた混合モデルを使用し,Day 1 及び
Day 14 における AUC 及び Cmaxの比及び 95%信頼区間を算出して検討した。
4. 薬力学:
PKAS を対象に,薬力学パラメータの要約統計量を投与群ごとに算出した。
単回投与時(Day 1)及び定常状態(Day 14)の血糖値 AUCtau及び Ctroughの用量依存性に関しては,
用量を独立変数,性別及びベースラインの血糖値を共変量とする混合モデルを用いて評価した。
有意水準 0.10 で有意な場合に性別の影響が認められると判断することとした。
5. 安全性:
安全性の解析では,SAF を解析対象とした。
臨床検査値に関しては,計量値項目では評価時点ごとに実測値の要約統計量を算出し,治験薬服
用前測定値と各評価時点の測定値のシフト表を作成した。
バイタルサインに関しては,評価時点ごとに実測値の要約統計量を算出した。
報告書の日付: 年 月 日
101

2.7.6個々の試験のまとめ
2.7.6.5.2 被験者の内訳及び解析対象集団
2.7.6.5.2.1 被験者の内訳
被験者の内訳を表 2.7.6.5-1 に示した。
被験者 48 例が本剤群(36 例:男性被験者の 5,30,100,300,600 mg 群各 6 例及び女性被験
者の 100 mg 群 6 例)又はプラセボ群(12 例:各用量群に 2 例)のいずれかに割り付けられた。
100 mg 群の女性被験者 1 例を除き,全例が試験を完了した。
表 2.7.6.5-1 被験者の内訳
ASP1941プラセボ
(M, F)5 mg(M)
30 mg(M)
100 mg(M)
100 mg (F)
300 mg (M)
600 mg (M) 計
無作為化/治験薬投与例 12 6 6 6 6 6 6 36完了例 12 6 6 6 5 6 6 35中止例 0 0 0 0 1 0 0 1
M=男性被験者,F=女性被験者
Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.1.1
2.7.6.5.2.2 解析対象集団
被験者 48 例全例を SAF 及び PKAS の解析対象例とした。女性被験者 1 例(本剤 100 mg)が治
験薬の最終投与後に家族の都合により試験を中止したが,予定されていた投与は完了しており,
薬物動態及び薬力学用の検体採取は,Day 14 の最終投与後 36 時間(血液)又は 48 時間(尿)ま
で実施された。そのため,本被験者も SAF 及び PKAS の解析対象例とした。
2.7.6.5.3 人口統計学的及び他の基準値の特性
SAF の被験者背景を表 2.7.6.5-2 に示した。
被験者の大半(42/48 例,87.5%)が白人であった。平均年齢は本剤群では 29.9 歳(18~53 歳),
プラセボ群では 38.1 歳(20~54 歳),BMI の平均値は本剤群では 23.80 kg/m2,プラセボ群では
24.36 kg/m2 であった。本剤 100 mg の男性被験者と女性被験者を比べたところ,平均年齢では女性
被験者よりも男性被験者の方が高く,BMI の平均値では男性被験者と女性被験者で同程度であっ
た。
102

2.7.6個々の試験のまとめ
表 2.7.6.5-2 被験者背景(SAF)ASP1941プラセボ
(M=10, F=2)
(N=12)
5 mg (M)
(N=6)
30 mg (M)
(N=6)
100 mg (M)
(N=6)
100 mg (F)
(N=6)
300 mg (M)
(N=6)
600 mg (M)
(N=6)
計
(N=36)平均値 38.1 23.0 22.3 38.2 24.8 37.5 33.7 29.9標準偏差 14.2 6.8 1.8 10.6 5.0 11.6 12.8 10.8最小値 20 18 20 24 22 23 20 18
年齢
(歳)
最大値 54 34 25 46 35 52 53 53男性 10(83.3) 6(100.0) 6(100.0) 6(100.0) 0 6(100.0) 6(100.0) 30(83.3)性別
n(%) 女性 2(16.7) 0 0 0 6(100.0) 0 0 6(16.7)白人 11(91.7) 6(100.0) 6(100.0) 4(66.7) 5(83.3) 5(83.3) 5(83.3) 31(86.1)黒人 0 0 0 1(16.7) 0 1(16.7) 0 2(5.6)アジア人 1(8.3) 0 0 1(16.7) 1(16.7) 0 0 2(5.6)
人種n(%)
その他 0 0 0 0 0 0 1(16.7) 1(2.8)平均値 75.89 73.58 75.35 84.60 72.93 77.10 78.58 77.03標準偏差 9.11 5.46 4.90 14.26 10.40 8.91 10.87 9.80最小値 61.2 68.5 71.1 60.2 61.2 71.1 68.5 60.2
体重(kg)
最大値 94.6 82.3 84.9 97.3 90.8 94.4 92.4 97.3平均値 176.7 183.2 183.3 177.3 172.2 177.8 185.7 179.9標準偏差 8.3 3.4 4.4 8.5 9.0 6.4 6.6 7.8最小値 163 178 176 165 156 168 175 156
身長(cm)
最大値 191 188 188 187 183 186 193 193平均値 24.36 21.92 22.44 26.76 24.55 24.41 22.72 23.80標準偏差 2.85 1.26 1.68 3.16 2.26 2.78 2.01 2.69最小値 21.0 20.2 21.0 22.1 22.3 21.9 20.5 20.2
BMI(kg/m2)
最大値 29.2 23.3 25.4 30.0 27.2 29.5 25.6 30.0
M=男性被験者,F=女性被験者
Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.1.3
2.7.6.5.4 治験薬の曝露
被験者 48 例全例が,本剤又はプラセボを Day 1 に 1 回及び Day 5~14 に 1 日 1 回服用した。
2.7.6.5.5 薬物動態
血漿中薬物動態
本剤の単回投与時及び 1 日 1 回反復投与時(最終投与後)の血漿中未変化体の薬物動態パラメー
タを表 2.7.6.5-3 及び表 2.7.6.5-4 に,パワーモデルによる血漿中未変化体濃度の用量比例性の評価
結果を表 2.7.6.5-5 に示した。また,血漿中未変化体濃度の平均値の推移を図 2.7.6.5-1(単回投与
時)及び図 2.7.6.5-2[反復投与時(最終投与後)]に示した。
本剤は,すべての用量群で速やかに吸収され,治験薬投与後,単回投与時では 0.83333~2.00000
時間,反復投与時では 0.83333~2.35278 時間で Cmax に到達した。単回及び反復投与時いずれでも,
tmaxの平均値は最小用量の 5 mg 群で 0.83333 時間と短かったが,Cmaxに明らかな用量比例性はみ
られず(パワーモデルによる傾きの推定値の 95%信頼区間上限が 1 未満),5 mg から 600 mg への
120 倍の用量増量に対して,Cmaxは約 80 倍の上昇であった。単回及び反復投与時の Cmaxの平均値
103

2.7.6個々の試験のまとめ
は同程度であり,統計学的にも確認された。単回及び反復投与時の Cmax に及ぼす性別の影響は,
統計学的解析に基づくと完全には否定できなかったが,100 mg 群の男性被験者と女性被験者の差
はわずかであった。
t1/2 の平均値は,単回投与時 10.15587~13.51145 時間,反復投与時 11.16491~14.64702 時間であっ
た。また,t1/2の平均値は,男性被験者に比べ女性被験者でわずかに長かった。
PTR の平均値は用量増量に伴って低下し,600 mg 群では 5 mg 群の約 1/2 となり,血漿中濃度の
日内変動が用量増量に伴って小さくなることが示された。
AUCinfの平均値は,AUClast の平均値より高かったが,差は 10%以内であり,AUC の正確な推定
値を得られる十分な時間にわたって採血が実施されたことが示された。AUCinf及び AUCtauの平均
値はほぼ用量比例的に上昇し(いずれのパラメータもパワーモデルによる傾きの推定値の 95%信
頼区間が 1 を含む),5 mg から 600 mg への 120 倍の用量増量に伴って,AUCinf は約 150 倍,AUCtau
は約 140 倍に上昇した。AUCtau及び AUCinfの平均値は各用量群間で同程度であった。反復投与時
のAUCに一貫した変化はみられず,統計学的にも変化のないことが確認された。Raccの平均値は,
1.06651~1.19776 に達し,反復投与時の血漿中未変化体濃度が,単回投与時よりも平均して 7%~
20%上昇したことが示された。単回及び反復投与時の AUC に及ぼす性別の影響は,統計学的解析
に基づくと完全には否定できなかったが,100 mg 群の男性被験者と女性被験者の差はわずかで
あった。
CL/F の平均値は,単回投与時 13.10698~15.83315 L/h,反復投与時 13.13227~15.62589 L/h であ
り,各用量群間で同程度であった。反復投与による CL/F の変化はみられなかった。Vz/F の平均
値は,本剤の用量にかかわらず約 240 L に達し(単回投与時 224.25560~250.60640 L,反復投与時
231.33626~267.37403 L),定常状態では単回投与時よりもわずかに高い傾向がみられた。Ctroughよ
り,Day 7(反復投与開始後 2 日)で定常状態に到達することが示され,消失半減期が約 12 時間
であることと一致した。
104

2.7.6個々の試験のまとめ
表 2.7.6.5-3 ASP1941 単回投与時の血漿中未変化体の薬物動態パラメータ(PKAS)ASP1941パラメータ
5 mg (M)(N=6)
30 mg (M)(N=6)
100 mg (M)(N=6)
100 mg (F)(N=6)
300 mg (M)(N=6)
600 mg (M)(N=6)
tlag (h)平均値 0.0 0.0 0.0 0.0 0.0 0.0標準偏差 0.0 0.0 0.0 0.0 0.0 0.0変動係数 (%) NA NA NA NA NA NA最小値 0 0 0 0 0 0最大値 0 0 0 0 0 0中央値 0.0 0.0 0.0 0.0 0.0 0.0tmax (h)平均値 0.83333 1.50000 1.58333 1.66945 1.33333 2.00000標準偏差 0.25820 0.44721 0.37639 1.46933 0.81650 1.22474変動係数 (%) NA NA NA NA NA NA最小値 0.5000 1.0000 1.0000 0.5000 1.0000 1.0000最大値 1.0000 2.0000 2.0000 4.0000 3.0000 4.0000中央値 1.00000 1.50000 1.50000 1.00000 1.00000 1.50000Cmax (ng/mL)平均値 75.718 421.580 1277.048 1233.733 3325.498 5831.632標準偏差 13.095 55.734 211.232 353.793 887.535 1232.025変動係数 (%) 17.29 13.22 16.54 28.68 26.69 21.13最小値 58.26 322.66 1068.45 739.62 2456.65 4771.42最大値 92.46 476.65 1508.65 1574.44 4956.09 7785.95中央値 77.190 424.915 1268.640 1330.285 3120.235 5260.475AUClast (ng•h/mL)平均値 302.81089 2194.38175 7617.47094 7775.25894 20621.72666 48570.46314標準偏差 67.50092 515.75789 1292.27051 1381.65347 4409.76693 14792.45767変動係数 (%) 22.29 23.50 16.96 17.77 21.38 30.46最小値 226.22910 1635.44530 5669.12537 6093.77523 17222.38647 29390.25195最大値 386.45000 3007.53215 9161.07120 10205.77891 29138.77280 66875.69723中央値 298.54319 2171.78144 7606.11680 7620.91146 19375.06919 49051.01625AUCinf (ng•h/mL)平均値 326.03208 2220.17936 7650.10446 7820.95956 20656.58212 48780.71193標準偏差 63.39793 520.71499 1307.86595 1394.16048 4407.87606 14857.46157変動係数 (%) 19.45 23.45 17.10 17.83 21.34 30.46最小値 252.26520 1664.21046 5681.12039 6107.23344 17239.40254 29448.32901最大値 403.97535 3046.49158 9204.02102 10251.45170 29154.89539 67147.86601中央値 321.72515 2195.70125 7626.00803 7693.81605 19395.88437 49418.03417t1/2 (h)平均値 10.15587 11.79448 11.84553 13.51145 10.37221 12.62573標準偏差 1.46966 3.74429 2.89023 3.58610 2.10150 2.15865変動係数 (%) 14.47 31.75 24.40 26.54 20.26 17.10最小値 8.63908 7.43371 8.28320 8.63751 8.76882 11.06295最大値 12.65400 16.44306 15.05582 19.43159 13.96230 16.67799中央値 9.74798 10.89563 12.87665 13.65293 9.25097 11.99016CL/F (L/h)平均値 15.83315 14.12265 13.41965 13.10698 14.97876 13.41578標準偏差 3.08743 3.17437 2.47174 2.19809 2.58931 4.50671変動係数 (%) 19.50 22.48 18.42 16.77 17.29 33.59最小値 12.37699 9.84739 10.86481 9.75471 10.28986 8.93550最大値 19.82041 18.02656 17.60216 16.37402 17.40199 20.37467中央値 15.65085 13.73737 13.13162 13.02039 15.51118 12.15223Vz/F (L)平均値 231.31571 232.03250 224.25560 250.60640 224.55982 240.55007標準偏差 52.12802 64.76517 45.19731 59.76817 59.23420 71.94621変動係数 (%) 22.54 27.91 20.15 23.85 26.38 29.91最小値 164.32655 167.45042 151.01824 187.98524 137.10683 154.87690最大値 309.63364 349.85144 286.37976 349.69003 289.19970 326.87873中央値 229.15127 219.28128 226.17765 238.78409 223.08919 247.75177M=男性被験者,F=女性被験者,NA=評価できず
Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.4.2
105

2.7.6個々の試験のまとめ
表 2.7.6.5-4 ASP1941 反復投与時(最終投与後)の血漿中未変化体の薬物動態パラメータ
(PKAS)(その 1)ASP1941パラメータ
5 mg (M)(N=6)
30 mg (M)(N=6)
100 mg (M)(N=6)
100 mg (F)(N=6)
300 mg (M)(N=6)
600 mg (M)(N=6)
tmax (h)平均値 0.83333 1.00000 1.50278 2.35278 1.66667 1.25000 標準偏差 0.25820 0.00000 1.26229 1.17410 0.87560 0.41833 変動係数 (%) NA NA NA NA NA NA最小値 0.5000 1.0000 0.5167 1.0000 0.5000 1.0000 最大値 1.0000 1.0000 4.0000 4.0000 3.0000 2.0000 中央値 1.00000 1.00000 1.00000 2.27500 1.75000 1.00000 Cmax (ng/mL)平均値 75.352 446.340 1313.218 1341.238 3479.755 6431.100標準偏差 14.990 50.841 333.416 230.428 780.333 1228.190変動係数 (%) 19.89 11.39 25.39 17.18 22.42 19.10最小値 54.33 381.37 844.86 984.05 2388.34 4377.34最大値 98.61 505.46 1716.59 1652.01 4646.11 8068.19中央値 75.025 458.875 1316.385 1346.980 3503.495 6610.745AUCtau (ng•h/mL)平均値 328.31525 1998.70739 7496.04329 7980.34117 21325.18498 46500.72767標準偏差 56.01091 317.36413 1367.73904 1790.94374 3071.94205 11881.52927変動係数 (%) 17.06 15.88 18.25 22.44 14.41 25.55最小値 257.77188 1622.75107 6184.22133 5342.05968 17266.21200 33760.80604最大値 386.74118 2370.41730 9910.89298 10192.13184 26447.87692 60302.97630中央値 336.57974 2054.27366 7262.84368 8142.95302 20939.09603 44510.29302t1/2 (h)平均値 11.16491 11.31782 12.55477 14.64702 11.28508 11.91065標準偏差 3.14321 3.47536 1.65117 5.05546 2.78406 1.06049変動係数 (%) 28.15 30.71 13.15 34.52 24.67 8.90最小値 6.54799 7.57761 10.13281 9.17693 9.34133 10.96565最大値 14.57141 16.22108 14.60541 21.06916 16.81394 13.68275中央値 11.34331 11.51598 12.76099 14.22715 10.46938 11.68954CL/F (L/h)平均値 15.62589 15.34276 13.67704 13.13227 14.30569 13.62378標準偏差 2.80292 2.52656 2.23760 3.27611 2.00527 3.41232変動係数 (%) 17.94 16.47 16.36 24.95 14.02 25.05最小値 12.92854 12.65599 10.08990 9.81149 11.34306 9.94975最大値 19.39699 18.48712 16.17018 18.71937 17.37497 17.77208中央値 14.89034 14.66272 13.84075 12.42572 14.35046 13.68110Vz/F (L)平均値 244.47586 243.88675 251.13596 267.37403 231.33626 233.77024標準偏差 55.21966 64.08772 68.77410 80.45000 56.33342 59.49640変動係数 (%) 22.59 26.28 27.39 30.09 24.35 25.45最小値 183.23860 197.50468 163.41435 165.26293 167.02357 159.27422最大値 321.60869 364.90957 340.72457 382.19845 334.10822 302.80636中央値 234.67060 220.30800 254.57806 255.96312 221.02680 242.50247PTR平均値 21.84992 20.77711 15.04054 14.55526 13.58060 9.95871標準偏差 9.29070 5.00040 5.53941 4.22717 4.65884 3.72643変動係数 (%) 42.52 24.07 36.83 29.04 34.31 37.42最小値 12.59589 15.55810 9.86695 8.64103 6.68740 7.06761最大値 39.02941 27.55106 22.39087 20.58554 18.99815 16.07209中央値 18.93870 20.11414 13.08357 14.01137 13.15832 8.23223
M=男性被験者,F=女性被験者,NA=評価できず
106

2.7.6個々の試験のまとめ
表 2.7.6.5-4 ASP1941 反復投与時(最終投与後)の血漿中未変化体の薬物動態パラメータ
(PKAS)(その 2)ASP1941パラメータ
5 mg (M)(N=6)
30 mg (M)(N=6)
100 mg (M)(N=6)
100 mg (F)(N=6)
300 mg (M)(N=6)
600 mg (M)(N=6)
Racc
平均値 1.16144 1.06651 1.16339 1.19776 1.19133 1.17784標準偏差 0.14981 0.08547 0.09193 0.15237 0.16473 0.12041変動係数 (%) 12.90 8.01 7.90 12.72 13.83 10.22最小値 1.04416 0.93851 1.07391 0.99049 0.99079 1.02378最大値 1.45807 1.18499 1.28484 1.39349 1.43608 1.36499中央値 1.12560 1.07712 1.13005 1.22261 1.18746 1.18577Ctrough (ng/mL)平均値 3.860 22.835 91.677 100.917 272.550 693.970標準偏差 1.364 6.819 21.223 39.064 68.359 200.084変動係数 (%) 35.33 29.86 23.15 38.71 25.08 28.83最小値 1.70 14.10 65.98 54.82 190.00 454.56最大値 5.84 29.59 117.10 157.20 357.14 930.89中央値 4.005 24.640 92.585 102.445 265.595 694.900
M=男性被験者,F=女性被験者
Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.4.2
表 2.7.6.5-5 パワーモデルによる血漿中未変化体濃度の用量比例性の評価(PKAS)95%信頼区間
パラメータ 評価時点 傾きの推定値下限値 上限値
自由度
男性被験者 AUCinf Day 1 1.03 0.98 1.08 28AUClast Day 1 1.04 0.99 1.09 28AUCtau Day 14 1.03 0.99 1.07 28Cmax Day 1 0.91 0.87 0.95 28
Day 14 0.93 0.88 0.97 28AUCinf Day 1 1.03 0.98 1.08 34男性被験者及び
女性被験者 AUClast Day 1 1.04 1.00 1.09 34AUCtau Day 14 1.03 0.99 1.07 34Cmax Day 1 0.91 0.86 0.95 34
Day 14 0.93 0.88 0.97 34
Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.4.5
107

2.7.6個々の試験のまとめ
図 2.7.6.5-1 ASP1941 単回投与時の血漿中未変化体濃度の平均値の推移(PKAS)
Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.4.1
血漿中未変化体濃度(
ng/m
L)
時間(h)
女性
男性
108

2.7.6個々の試験のまとめ
図 2.7.6.5-2 ASP1941 反復投与時(最終投与後)の血漿中未変化体濃度の平均値の推移
(PKAS)
Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.4.1
尿中薬物動態
本剤の単回投与時及び 1 日 1 回反復投与時(最終投与後)の尿中未変化体の薬物動態パラメー
タを表 2.7.6.5-6 及び表 2.7.6.5-7 に示した。
単回投与時の Aelast%及び Aeinf%の平均値は,それぞれ 0.71423%~1.06690%及び 0.77820%~
1.07532%であり,未変化体の尿中排泄率は,本剤の用量にかかわらず投与量の 1%程度であった。
各群の反復投与時の Aetauの平均値は,単回投与時の Aeinfの平均値と同程度であった。CLr の平均
値は用量群間で同程度であり,単回投与時 0.12044~0.14722 L/h,反復投与時 0.12612~0.16874 L/h
であった。尿中排泄率及び CLr に関しては,単回投与時と反復投与時で差はみられなかった。
血漿中未変化体濃度(
ng/m
L)
時間(h)
女性
男性
109

2.7.6個々の試験のまとめ
表 2.7.6.5-6 ASP1941 単回投与時の尿中未変化体の薬物動態パラメータ(PKAS)ASP1941パラメータ
5 mg (M)(N=6)
30 mg (M)(N=6)
100 mg (M)(N=6)
100 mg (F)(N=6)
300 mg (M)(N=6)
600 mg (M)(N=6)
Aelast(mg)
0.03571(0.00733)
0.32007(0.06855)
0.98254(0.13028)
0.95032(0.18236)
2.94177(0.48854)
6.21261(1.18536)
Aelast%(%)
0.71423 (0.14669)
1.06690(0.22853)
0.98254(0.13028)
0.95032(0.18236)
0.98059(0.16284)
1.03543(0.19756)
Aeinf(mg)
0.03891 (0.00839)
0.32259 (0.06822)
0.98637 (0.13299)
0.95570(0.18703)
2.94589 (0.49070)
6.23808 (1.18581)
Aeinf%(%)
0.77820 (0.16798)
1.07532(0.22741)
0.98637(0.13299)
0.95570(0.18703)
0.98196(0.16356)
1.03968(0.19763)
CLr(L/h)
0.12044(0.02213)
0.14722(0.02433)
0.13106(0.02062)
0.12257(0.01577)
0.14691(0.03544)
0.13314(0.02653)
平均値(標準偏差)
M=男性被験者,F=女性被験者
Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.4.8
表 2.7.6.5-7 ASP1941 反復投与時(最終投与後)の尿中未変化体の薬物動態パラメータ
(PKAS)ASP1941パラメータ
5 mg (M)(N=6)
30 mg (M)(N=6)
100 mg (M)(N=6)
100 mg (F)(N=6)
300 mg (M)(N=6)
600 mg (M)(N=6)
Aetau(mg)
0.04587(0.00604)
0.33519(0.07542)
1.10303(0.25959)
0.99434(0.18244)
2.95516(0.52372)
6.55384(1.55535)
Aetau%(%)
0.91750(0.12096)
1.11733(0.25141)
1.10303(0.25959)
0.99434(0.18244)
0.98505(0.17457)
1.09230(0.25922)
CLr(L/h)
0.14135(0.01707)
0.16874(0.03170)
0.14749(0.02588)
0.12612(0.01233)
0.13905(0.01967)
0.14187(0.01121)
平均値(標準偏差)
M=男性被験者,F=女性被験者
Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.4.8
2.7.6.5.6 薬力学
血糖値
本剤の単回投与時及び 1 日 1 回反復投与時(最終投与後)の血糖値パラメータを表 2.7.6.5-8 に
示した。
単回及び反復投与時いずれでも,用量群間及び本剤群とプラセボ群間で血糖値パラメータに差
はみられず,用量反応性も認められなかった。本剤群とプラセボ群の統計的比較でも,反復投与
時の本剤 100 mg 群(男性被験者)の AUCtauを除き,有意差は認められなかった。反復投与時の
本剤 100 mg 群(男性被験者)の AUCtauは,プラセボ群と比較して有意に高かった(P<0.05)。な
お,探索的な解析のため多重性の調整は行わなかった。
女性被験者では,AUCtau及び Ctroughが男性被験者と比較してわずかに低く,AUCtauでは単回及
び反復投与時のいずれでも有意差が認められた(P<0.10)。
110

2.7.6個々の試験のまとめ
表 2.7.6.5-8 ASP1941 単回投与時及び反復投与時(最終投与後)の血糖値パラメータ(PKAS)ASP1941パラメータ プラセボ
(N=12)5 mg (M)
(N=6)30 mg (M)
(N=6)100 mg (M)
(N=6)100 mg (F)
(N=6)300 mg (M)
(N=6)600 mg (M)
(N=6)単回投与時
AUCtau(mmol•h/L)
125.25325 (6.13138)
128.16392 (7.87721)
122.61377 (4.49392)
124.06404(9.09450)
116.51814(3.94092)
125.30959(7.75291)
124.16413(5.42067)
Ctrough(mmol/L)
5.104(0.346)
5.055(0.124)
5.052(0.307)
5.163(0.558)
4.578(0.263)
5.113(0.451)
5.093(0.274)
反復投与時
AUCtau(mmol•h/L)
125.82486 (5.64412)
123.90449(5.89648)
124.04120 (5.54384)
133.06300(6.72533)
114.04638(3.80543)
126.03916(7.91139)
118.87377 (5.99977)
Ctrough(mmol/L)
4.999 (0.301)
5.057 (0.148)
4.970(0.245)
5.072(0.342)
4.445(0.100)
4.962(0.217)
4.887(0.314)
平均値(標準偏差)
M=男性被験者,F=女性被験者
Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.5.2
尿中グルコース
本剤の単回投与時及び 1 日 1 回反復投与時(最終投与後)の尿中グルコースパラメータ[Aetau
及び 24 時間の CLr(CLr 24h)]を表 2.7.6.5-9 に示した。また,本剤の単回投与時及び反復投与時(最
終投与後)の用量に対する 24 時間の尿中グルコース排泄量(Aetau)を図 2.7.6.5-3 に示した。
単回及び反復投与時いずれでも,Aetauの平均値は女性被験者で男性被験者と比較してわずかに
少なかったが,CLr 24hの平均値は女性及び男性被験者で同程度であった。
Aetauの平均値は,本剤 30 mg までは用量増量に伴って顕著に増加し,本剤 100 mg 以上では用
量の増加に対して増加量が小さくなり,300 mg 以上の用量では増加がみられなかった。血糖値が
用量群間で同程度であることを反映して,CLr 24h の平均値も同様の傾向を示した。
本剤 30 mg 群以上では,反復投与時の Aetau及び CLr 24hの平均値は,いずれも単回投与時よりも
低かった。
111

2.7.6個々の試験のまとめ
表 2.7.6.5-9 ASP1941 単回投与時及び反復投与時(最終投与後)の尿中グルコースパラメー
タ(PKAS)ASP1941パラメータ プラセボ
(N=12)5 mg (M)
(N=6)30 mg (M)
(N=6)100 mg (M)
(N=6)100 mg (F)
(N=6)300 mg (M)
(N=6)600 mg (M)
(N=6)単回投与時
Aetau(mmol)
0.62381 (0.28678)
17.10207a
(11.35743)172.74632(58.94355)
270.42386(48.49866)
228.60143(47.52354)
393.24410 (64.22058)
380.99921(73.37414)
CLr 24h(L/h)
0.00497(0.00226)
0.13122a
(0.08091)1.41494
(0.51274)2.17721
(0.36206)1.96457
(0.42518)3.14368
(0.52849)3.06390
(0.55056)反復投与時
Aetau(mmol)
0.60939(0.28116)
17.54734(3.93765)
135.24913(46.91364)
222.22878(66.95500)
192.51207(69.06577)
313.41566(62.49993)
326.70973(81.12828)
CLr 24h(L/h)
0.00482(0.00215)
0.14208(0.03360)
1.08783 (0.36315)
1.68748 (0.57636)
1.68571 (0.59921)
2.49612 (0.53215)
2.75138 (0.67091)
平均値(標準偏差)
M=男性被験者,F=女性被験者
a:N=5Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.5.9
図 2.7.6.5-3 ASP1941 単回投与時及び反復投与時(最終投与後)の用量に対する 24 時間の
尿中グルコース排泄量(Aetau)の平均値(男性被験者及び女性被験者の併合)
(PKAS)
Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.5.8
用量(mg)
単回投与時
反復投与時(最終投与後)
Ae t
au(
mm
ol)
112

2.7.6個々の試験のまとめ
2.7.6.5.7 薬物動態/薬力学関連性
本剤の単回投与時及び 1 日 1 回反復投与時(最終投与後)の未変化体の 24 時間の AUC(AUCtau)
に対する 24 時間の尿中グルコース排泄量(Aetau)を図 2.7.6.5-4 に示した。
未変化体の AUCtauに対する Aetauでは,単回及び反復投与時で類似する関連性がみられ,用量
増量に伴い Aetauが増加した。反復投与時の Aetauは,単回投与時よりも減少する傾向が示された。
反復投与による本剤の曝露量の減少はみられておらず,反復投与時の尿中グルコース排泄量の減
少傾向の原因は明らかではなかった。
図 2.7.6.5-4 ASP1941単回投与時及び反復投与時(最終投与後)の未変化体の24時間のAUC(AUCtau)に対する 24 時間の尿中グルコース排泄量(Aetau)(男性被験者及び
女性被験者の併合)(PKAS)
Source:CL-0002 総括報告書(5.3.3.1-3)Listing 13.2.6.1,Listing 13.2.6.1.1 及び Listing 13.2.6.9
2.7.6.5.8 安全性
2.7.6.5.8.1 有害事象
本試験で認められた有害事象の要約を表 2.7.6.5-10 に示した。
有害事象の発現率は,本剤群 63.9%(23/36 例),プラセボ群 66.7%(8/12 例)であり,同程度
であった。各用量群で有害事象発現率にやや差が認められたが,高用量において有害事象発現率
AUCtau(ng•h/mL)
単回投与時
反復投与時
(最終投与後)
Ae t
au(
mm
ol)
113

2.7.6個々の試験のまとめ
が高くなる傾向はみられなかった。また,本剤 100 mg を投与した男性群と女性群の有害事象発現
率にも明らかな差はみられなかった。
プラセボ群の男性被験者 1 例で発現した中等度の血管迷走神経性失神を除き,すべての有害事
象は軽度であった。
本試験では,死亡,その他の重篤な有害事象及び中止に至った有害事象は認められなかった。
表 2.7.6.5-10 有害事象の要約(SAF)ASP1941プラセボ
(M, F)(N=12)
5 mg (M)
(N=6)
30 mg (M)
(N=6)
100 mg (M)
(N=6)
100 mg (F)
(N=6)
300 mg (M)
(N=6)
600 mg (M)
(N=6)
計
(N=36)有害事象 8
(66.7%)4
(66.7%)2
(33.3%)5
(83.3%)4
(66.7%)4
(66.7%)4
(66.7%)23
(63.9%)副作用 5
(41.7%)3
(50.0%)0 3
(50.0%)4
(66.7%)3
(50.0%)1
(16.7%)14
(38.9%)重篤な有害事象 0 0 0 0 0 0 0 0中止に至った有害事象 0 0 0 0 0 0 0 0程度度別有害事象
軽度 7(58.3%)
4(66.7%)
2(33.3%)
5(83.3%)
4(66.7%)
4(66.7%)
4(66.7%)
23(63.9%)
中等度 1(8.3%)
0 0 0 0 0 0 0
重度 0 0 0 0 0 0 0 0
発現例数(発現率)
M=男性被験者,F=女性被験者
Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.6.1.2
本試験で認められた有害事象及び副作用の要約を,それぞれ表 2.7.6.5-11 及び表 2.7.6.5-12 に示
した。
プラセボ群及び本剤群において最も多く認められた有害事象は頭痛であった。発疹が本剤
100 mg 群の女性被験者 3 例(医師記載用語:いずれも皮膚発疹,顔面;治験薬と関連あるかもし
れない),300 mg 群の男性被験者 1 例(医師記載用語:心電図の電極に誘発された皮膚発疹;治
験薬との関連性は否定できる),600 mg 群の男性被験者 1 例(医師記載用語:頸上部皮膚発疹;
治験薬と関連あるかもしれない)及びプラセボ群の女性被験者 1 例(医師記載用語:皮膚発疹,
顔面及び腹部皮膚;治験薬と関連あるかもしれない)に認められた。これらの皮膚の異常は,600 mg
群で認められた発疹を除き,治験実施医療機関以外の外部の皮膚科医によって追跡調査された。
皮膚科医は,これらの皮膚の異常をざ瘡若しくは毛包炎の偶発的な発現,又は心電図の電極に誘
発された偶発的な事象であると判断し,治験薬との因果関係は否定された。
本試験では低血糖関連の有害事象は認められなかった。
腎機能関連の有害事象として,本剤 300 mg 群の男性被験者で頻尿(1 例)が,プラセボ群で頻
尿及び排尿困難(各 1 例)が認められた。本剤 300 mg 群の頻尿は,治験担当医師により「治験薬
と関連あるかもしれない」と判定された。
114

2.7.6個々の試験のまとめ
表 2.7.6.5-11 有害事象の内訳(SAF)ASP1941MedDRA version 10.1
器官別大分類
基本語
プラセボ(M, F)
(N=12)
5 mg(M)
(N=6)
30 mg(M)
(N=6)
100 mg(M)
(N=6)
100 mg(F)
(N=6)
300 mg(M)
(N=6)
600 mg(M)
(N=6)
計
(N=36)すべての有害事象 8(66.7%) 4(66.7%) 2(33.3%) 5(83.3%) 4(66.7%) 4(66.7%) 4(66.7%) 23(63.9%)耳および迷路障害 0 0 0 1(16.7%) 0 0 0 1(2.8%)聴力低下 0 0 0 1(16.7%) 0 0 0 1(2.8%)
眼障害 1(8.3%) 0 0 0 0 0 0 0霧視 1(8.3%) 0 0 0 0 0 0 0
胃腸障害 5(41.7%) 1(16.7%) 0 3(50.0%) 2(33.3%) 1(16.7%) 0 7(19.4%)腹部不快感 0 0 0 1(16.7%) 0 0 0 1(2.8%)腹部膨満 0 0 0 1(16.7%) 0 0 0 1(2.8%)腹痛 1(8.3%) 0 0 0 0 0 0 0便秘 1(8.3%) 0 0 0 0 0 0 0下痢 1(8.3%) 0 0 1(16.7%) 1(16.7%) 1(16.7%) 0 3(8.3%)口内乾燥 1(8.3%) 0 0 0 0 0 0 0鼓腸 2(16.7%) 0 0 2(33.3%) 0 0 0 2(5.6%)胃腸音異常 2(16.7%) 0 0 0 2(33.3%) 0 0 2(5.6%)胃酸過多 1(8.3%) 0 0 0 0 0 0 0口腔内潰瘍形成 0 1(16.7%) 0 0 0 0 0 1(2.8%)舌障害 1(8.3%) 0 0 0 0 0 0 0
全身障害および投与局所様態 2(16.7%) 1(16.7%) 1(16.7%) 2(33.3%) 0 1(16.7%) 0 5(13.9%)カテーテル留置部位疼痛 0 0 1(16.7%) 1(16.7%) 0 0 0 2(5.6%)カテーテル留置部位関連
反応0 0 0 1(16.7%) 0 1(16.7%) 0 2(5.6%)
悪寒 1(8.3%) 0 0 0 0 0 0 0疲労 0 1(16.7%) 0 0 0 0 0 1(2.8%)空腹 1(8.3%) 0 0 0 0 0 0 0
感染症および寄生虫症 1(8.3%) 2(33.3%) 1(16.7%) 0 0 0 0 3(8.3%)鼻咽頭炎 1(8.3%) 2(33.3%) 1(16.7%) 0 0 0 0 3(8.3%)
代謝および栄養障害 0 2(33.3%) 0 0 0 0 0 2(5.6%)食欲減退 0 2(33.3%) 0 0 0 0 0 2(5.6%)
筋骨格系および結合組織障害 1(8.3%) 0 0 0 0 1(16.7%) 2(33.3%) 3(8.3%)背部痛 0 0 0 0 0 0 1(16.7%) 1(2.8%)四肢不快感 1(8.3%) 0 0 0 0 0 0 0筋痛 0 0 0 0 0 1(16.7%) 1(16.7%) 2(5.6%)頚部痛 1(8.3%) 0 0 0 0 0 0 0四肢痛 1(8.3%) 0 0 0 0 0 0 0
神経系障害 5(41.7%) 1(16.7%) 1(16.7%) 1(16.7%) 2(33.3%) 1(16.7%) 3(50.0%) 9(25.0%)注意力障害 0 0 0 1(16.7%) 0 0 0 1(2.8%)浮動性めまい 1(8.3%) 0 0 0 1(16.7%) 0 0 1(2.8%)頭痛 3(25.0%) 1(16.7%) 0 1(16.7%) 1(16.7%) 1(16.7%) 3(50.0%) 7(19.4%)錯感覚 1(8.3%) 0 0 0 0 0 0 0血管迷走神経性失神 1(8.3%) 0 1(16.7%) 0 0 0 0 1(2.8%)
腎および尿路障害 2(16.7%) 0 0 0 0 1(16.7%) 0 1(2.8%)排尿困難 1(8.3%) 0 0 0 0 0 0 0頻尿 1(8.3%) 0 0 0 0 1(16.7%) 0 1(2.8%)
呼吸器,胸郭および縦隔障害 0 0 0 1(16.7%) 0 0 0 1(2.8%)咽喉頭疼痛 0 0 0 1(16.7%) 0 0 0 1(2.8%)
皮膚および皮下組織障害 2(16.7%) 0 0 0 3(50.0%) 1(16.7%) 1(16.7%) 5(13.9%)湿疹 1(8.3%) 0 0 0 0 0 0 0発疹 1(8.3%) 0 0 0 3(50.0%) 1(16.7%) 1(16.7%) 5(13.9%)
血管障害 1(8.3%) 0 0 0 0 0 0 0血腫 1(8.3%) 0 0 0 0 0 0 0
発現例数(発現率)
M=男性被験者,F=女性被験者
Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.6.1.3
115

2.7.6個々の試験のまとめ
表 2.7.6.5-12 副作用の内訳(SAF)ASP1941MedDRA version 10.1
器官別大分類
基本語
プラセボ
(M, F)(N=12)
5 mg(M)
(N=6)
30 mg(M)
(N=6)
100 mg(M)
(N=6)
100 mg(F)
(N=6)
300 mg(M)
(N=6)
600 mg(M)
(N=6)
計
(N=36)すべての副作用 5(41.7%) 3(50.0%) 0 3(50.0%) 4(66.7%) 3(50.0%) 1(16.7%) 14(38.9%)胃腸障害 4(33.3%) 1(16.7%) 0 3(50.0%) 2(33.3%) 1(16.7%) 0 7(19.4%)腹部不快感 0 0 0 1(16.7%) 0 0 0 1(2.8%)腹部膨満 0 0 0 1(16.7%) 0 0 0 1(2.8%)腹痛 1(8.3%) 0 0 0 0 0 0 0便秘 1(8.3%) 0 0 0 0 0 0 0下痢 1(8.3%) 0 0 1(16.7%) 1(16.7%) 1(16.7%) 0 3(8.3%)口内乾燥 1(8.3%) 0 0 0 0 0 0 0鼓腸 2(16.7%) 0 0 2(33.3%) 0 0 0 2(5.6%)胃腸音異常 2(16.7%) 0 0 0 2(33.3%) 0 0 2(5.6%)胃酸過多 1(8.3%) 0 0 0 0 0 0 0口腔内潰瘍形成 0 1(16.7%) 0 0 0 0 0 1(2.8%)
全身障害および投与局所様態 2(16.7%) 0 0 0 0 0 0 0悪寒 1(8.3%) 0 0 0 0 0 0 0空腹 1(8.3%) 0 0 0 0 0 0 0
代謝および栄養障害 0 1(16.7%) 0 0 0 0 0 1(2.8%)食欲減退 0 1(16.7%) 0 0 0 0 0 1(2.8%)
神経系障害 4(33.3%) 1(16.7%) 0 0 1(16.7%) 1(16.7%) 0 3(8.3%)頭痛 3(25.0%) 1(16.7%) 0 0 1(16.7%) 1(16.7%) 0 3(8.3%)錯感覚 1(8.3%) 0 0 0 0 0 0 0
腎および尿路障害 2(16.7%) 0 0 0 0 1(16.7%) 0 1(2.8%)排尿困難 1(8.3%) 0 0 0 0 0 0 0頻尿 1(8.3%) 0 0 0 0 1(16.7%) 0 1(2.8%)
皮膚および皮下組織障害 1(8.3%) 0 0 0 3(50.0%) 0 1(16.7%) 4(11.1%)発疹 1(8.3%) 0 0 0 3(50.0%) 0 1(16.7%) 4(11.1%)
発現例数(発現率)
M=男性被験者,F=女性被験者
Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.6.1.8
2.7.6.5.8.2 死亡,その他の重篤な有害事象
本試験では,死亡,及びその他の重篤な有害事象は認められなかった。
2.7.6.5.8.3 中止に至った有害事象
本試験では,試験中止に至った有害事象は認められなかった。
2.7.6.5.8.4 臨床検査値
各臨床検査項目の平均値に関しては,本剤の用量及び性別に関連した有意な変動傾向はみられ
なかった。
腎機能マーカーである尿中の β-N-アセチル D グルコサミニダーゼ,β2 ミクログロブリン及び
α1 ミクログロブリンの測定結果の要約を,それぞれ表 2.7.6.5-13,表 2.7.6.5-14 及び表 2.7.6.5-15
に示した。
116

2.7.6個々の試験のまとめ
各用量群の尿中の β-N-アセチル D グルコサミニダーゼは,ベースライン及び各測定時点のプラ
セボ群と比較して上昇し,男性被験者の本剤 100 mg 群及び 300 mg 群を除くすべての用量群で後
観察時にベースラインまで回復しなかった。しかし,これらの上昇には時間経過に伴う一貫性が
なく,本剤の用量及び性別との明らかな関連性は認められなかった。尿中の β2 ミクログロブリン
及び α1 ミクログロブリンが同時に上昇することはなかった。男性被験者では,本剤の各用量群で
尿中クレアチニンがわずかに低下したが,用量との明らかな関連性は認められなかった。その他
の腎機能関連の検査項目に変化はみられなかった。
表 2.7.6.5-13 尿中の β-N-アセチル D グルコサミニダーゼの要約(SAF)ASP1941β-N-アセチル D
グルコサミニ
ダーゼ(U/L)
プラセボ
(M, F)(N=12)
5 mg(M)
(N=6)
30 mg(M)
(N=6)
100 mg(M)
(N=6)
100 mg(F)
(N=6)
300 mg(M)
(N=6)
600 mg(M)
(N=6)
スクリーニング2.54
(1.51)1.83
(1.67)2.38
(1.11)4.47
(2.92)1.10
(0.33)4.87
(2.70)2.68
(0.73)
Day −1 2.01(1.23)
1.85(1.66)
1.93(1.35)
3.63(2.04)
0.88(0.55)
3.90(1.05)
2.82(1.76)
Day 1 2.46(0.78)
2.40(0.86)
1.95(0.85)
2.95(0.90)
1.30(0.37)
1.80(1.17)
1.73(0.57)
Day 2 2.26(0.91)
2.80(1.49)
4.50(1.49)
2.67(0.87)
3.10(2.13)
2.87(1.00)
3.05(0.72)
Day 5 2.10(0.92)
2.98(2.33)
4.18(2.95)
3.27(0.82)
2.68(0.95)
7.02(5.15)
7.83(5.40)
Day 8 2.61(0.82)
3.67(1.83)
8.02(3.00)
5.05(2.22)
4.87(2.67)
5.73(1.74)
4.30(1.14)
Day 11 2.18(0.88)
3.62(3.88)
11.12(3.23)
6.15(3.12)
4.40(2.29)
7.75(3.04)
4.28(1.35)
Day 14 1.27(0.48)
3.85(2.92)
1.88(0.84)
5.00(4.83)
2.20(0.91)
5.75(2.59)
4.45(2.43)
Day 15 1.96(1.17)
3.93(1.63)
8.17(2.23)
8.27(5.02)
4.28(2.67)
4.85(2.44)
5.00(1.52)
Day 17 2.31(1.64)
2.35(0.69)
5.88(4.82)
8.43(2.45)
4.70a
(0.52)8.27
(4.24)8.12
(6.06)
後観察3.48
(2.49)3.25
(1.89)3.53
(2.60)3.28
(2.16)2.83
(1.79)3.65
(2.65)3.95
(2.82)
平均値(標準偏差)
M=男性被験者,F=女性被験者
a:N=5Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.6.2.1
117

2.7.6個々の試験のまとめ
表 2.7.6.5-14 尿中の β2 ミクログロブリンの要約(SAF)ASP1941β2 ミクログロ
ブリン(µg/L)
プラセボ
(M, F)(N=12)
5 mg(M)
(N=6)
30 mg(M)
(N=6)
100 mg(M)
(N=6)
100 mg(F)
(N=6)
300 mg(M)
(N=6)
600 mg(M)
(N=6)
スクリーニング78.49
(40.61)70.97
(57.58)68.80
(52.30)98.12
(71.44)29.33
(16.17)102.70(43.06)
108.25(38.11)
Day −1 67.22(38.37)
44.75(50.45)
204.63(386.82)
88.47(61.77)
36.42(35.46)
87.05(27.63)
100.93(52.51)
Day 1 65.83(32.69)
59.47(18.51)
65.32(83.05)
47.07(16.69)
25.87(16.47)
27.55(10.46)
47.13(19.62)
Day 2 53.93(14.82)
62.62(28.67)
56.18(30.22)
34.37(16.01)
33.42(18.55)
61.77(19.48)
77.35(20.87)
Day 5 56.86(29.31)
58.77(36.15)
72.58(43.27)
33.60(14.81)
56.92(36.86)
61.72(42.19)
107.32(58.59)
Day 8 57.28(24.87)
74.28(20.59)
68.08(44.50)
42.90(20.50)
52.45(33.88)
56.72(25.00)
180.37(201.70)
Day 11 57.59(23.27)
77.17(61.95)
72.95(24.50)
79.75(32.28)
34.53(27.27)
85.65(36.10)
57.97(32.76)
Day 14 35.07(27.89)
45.97(30.94)
35.20(20.76)
67.62(30.92)
47.27(13.83)
86.77(47.64)
129.70(119.29)
Day 15 50.46(34.96)
63.35(29.06)
37.18(47.74)
48.30(49.37)
25.87(26.99)
56.35(27.01)
108.62(98.51)
Day 17 43.80(30.59)
52.97(28.11)
62.88(47.82)
49.95(34.34)
42.92a
(33.82)42.25
(33.76)117.27(73.31)
後観察77.38
(46.36)75.25
(20.69)147.60
(132.28)134.72
(146.34)92.45
(66.10)89.02
(43.49)118.22(76.55)
平均値(標準偏差)
M=男性被験者,F=女性被験者
a:N=5Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.6.2.1
118

2.7.6個々の試験のまとめ
表 2.7.6.5-15 尿中の α1 ミクログロブリンの要約(SAF)ASP1941α1 ミクログロ
ブリン(mg/dL)
プラセボ
(M, F)(N=12)
5 mg(M)
(N=6)
30 mg(M)
(N=6)
100 mg(M)
(N=6)
100 mg(F)
(N=6)
300 mg(M)
(N=6)
600 mg(M)
(N=6)
スクリーニング0.871
(0.402)0.802
(0.528)0.707
(0.454)1.100
(0.476)0.390
(0)1.152
(0.722)0.875
(0.349)
Day −1 0.825(0.499)
0.530(0.225)
0.822(0.864)
1.122(0.673)
0.397(0.016)
1.080(0.619)
0.915(0.620)
Day 1 1.876(3.086)
0.562(0.228)
0.472(0.146)
0.473(0.115)
0.390(0)
0.430(0.098)
8.685(2.266)
Day 2 0.518(0.273)
0.473(0.127)
0.623(0.213)
0.535(0.229)
0.435(0.110)
0.587(0.107)
0.463(0.127)
Day 5 0.464(0.083)
0.492(0.235)
0.532(0.188)
0.507(0.116)
0.390(0)
0.540(0.258)
0.745(0.399)
Day 8 0.471(0.086)
0.557(0.241)
0.767(0.114)
0.665(0.264)
0.443(0.112)
0.713(0.296)
0.928(0.585)
Day 11 0.502(0.191)
0.578(0.189)
0.777(0.210)
0.667(0.246)
0.425(0.072)
1.002(0.622)
0.713(0.311)
Day 14 0.473(0.237)
0.433(0.106)
0.427(0.068)
0.968(0.691)
0.400(0.024)
1.022(0.755)
1.008(0.637)
Day 15 0.454(0.120)
0.497(0.196)
0.758(0.168)
1.130(0.226)
0.503(0.240)
0.707(0.206)
0.930(0.369)
Day 17 0.478(0.104)
0.480(0.187)
0.632(0.326)
0.943(0.430)
0.432a
(0.073)0.922
(0.489)1.062
(0.862)
後観察1.059
(0.706)0.817
(0.624)1.427
(0.698)1.398
(1.055)0.877
(0.986)1.148
(0.765)1.472
(0.904)
平均値(標準偏差)
M=男性被験者,F=女性被験者
a:N=5Source:CL-0002 総括報告書(5.3.3.1-3)Table 12.6.2.1
2.7.6.5.8.5 バイタルサイン
収縮期血圧,拡張期血圧及び脈拍数に関しては,本剤の用量及び性別による臨床的に意義のあ
る変化はみられなかった。
2.7.6.5.8.6 心電図
多くの男性及び女性被験者では,本試験期間中の 1 測定時点以上で,心拍数の異常所見が観察
されたが,治験担当医師により臨床的に重要と判定された事象はなかった。また,異常所見に一
貫した傾向はみられず,これらは,通常観察される所見であり,プラセボ群でも認められた。
2.7.6.5.8.7 身体所見
退所時の身体所見に,スクリーニング時からの臨床的に重要な変化は認められなかった。
119

2.7.6個々の試験のまとめ
2.7.6.5.8.8 最大耐量(MTD)
本試験では,健康男性被験者に本剤を最大 600 mg まで単回投与及び 1 日 1 回 10 日間反復投与
した。最高用量群を含む各投与群の安全性に問題はなく,忍容性は良好であり,MTD には達しな
かった。
2.7.6.5.9 結論
薬物動態及び薬力学
● 本剤は速やかに吸収された。Cmaxに明らかな用量比例性はみられなかったが,AUC は,ほぼ
用量比例的に上昇した。本剤の t1/2は単回投与時 10.15587~13.51145時間,反復投与時 11.16491
~14.64702 時間であった。血漿中未変化体濃度の平均値は,反復投与により 7%~20%上昇し
た。血漿中未変化体濃度の日内変動は用量増量に伴い小さくなった。未変化体の尿中排泄率
は,投与量の 1%程度であった。本剤の吸収,クリアランス及び分布に関する薬物動態パラメー
タには,反復投与による変化はみられなかった。
● 血糖値に及ぼす本剤の単回投与及び 1 日 1 回 10 日間の反復投与の影響はみられなかった。尿
中グルコース排泄量は,本剤投与により増加し,本剤 5 mg 投与例でもプラセボ投与例と比較
して高かった。本剤投与量の増加に伴う尿中グルコース排泄量の増加は,投与量 300 mg まで
認められた。血糖値及び尿中グルコース濃度は,女性被験者で男性被験者よりも低い傾向が
みられた。尿中グルコースの Aetau及び CLr 24hの平均値は,本剤 30 mg 群以上では,反復投与
時の方が単回投与時よりも低かった。
● 未変化体の AUCtauに対する尿中グルコースの Aetauに関しては,単回及び反復投与時で類似
する関連性がみられ,用量増量に伴い Aetauが増加した。本剤の薬物動態は,単回投与時及び
1 日 1 回 10 日間反復投与時で類似していたが,Aetauは,反復投与時に単回投与時よりも減少
する傾向が示された。
安全性
● 本剤 600 mg までの 1 日 1 回反復投与では安全性及び忍容性が良好であり,MTD には達しな
かった。最も多く認められた有害事象は頭痛であったが,プラセボ投与例でも認められた。
頭痛の発現率に本剤の投与量との関連性はみられなかった。
● 腎機能マーカーでは,本剤投与により尿中の β-N-アセチル D グルコサミニダーゼが上昇した
が,用量及び性別との関連性は認められなかった。尿中の β2 ミクログロブリン及び α1 ミク
ログロブリンが同時に上昇することはなかった。その他の腎機能マーカーには変化はみられ
なかった。
● 発疹が複数例で認められた。治験実施医療機関以外の外部の皮膚科医の判断では,ほとんど
の発疹の治験薬との因果関係はなしであった。
120

2.7.6個々の試験のまとめ
性別の影響
本剤の薬物動態,薬力学及び安全性の成績に対して,性別は臨床的に重要な影響を及ぼさなかっ
た。
121

2.7.6個々の試験のまとめ
2.7.6.6 マスバランス試験[CL-0055](添付資料 5.3.3.1-4)
2.7.6.6.1 試験方法の概略
治験課題名:健康成人男性に 14C 標識 ASP1941 を単回経口投与したときの ASP1941 の薬物動態を
評価する非盲検試験
治験実施医療機関:オランダ 1 施設
公表文献:なし
治験期間:
治験開始日:2008 年 5 月
治験終了日:2008 年 10 月
開発のフェーズ:第 I 相
目的:
(1) 主要目的
ASP1941(以下,本薬)の 14C 標識体 100 mg を単回経口投与したときの本薬の薬物動態,特に代
謝・排泄の経路及び寄与度を評価する。
(2) 副次目的
1) 本薬の 14C 標識体 100 mg を単回経口投与したときのヒトの血漿,尿及び糞中における本薬
の代謝プロフィールを評価する。
2) 本薬の 14C 標識体 100 mg を単回経口投与したときの安全性及び忍容性を評価する。
治験デザイン・治験方法:
本試験は,健康成人男性を対象とした単一施設,非盲検試験として実施した。
被験者は治験薬投与前日(Day −1)の午後に入所し,Day 1 に本薬の 14C 標識体 100 mg 経口液剤
を空腹時に服用した。本試験は吸収,分布,代謝及び排泄(ADME)試験であり,14C 放射能の分
析及び本薬の代謝プロフィールの評価のため,採血(全血及び血漿),蓄尿,蓄便及び呼気採取
を投与後少なくとも 144 時間まで行った。安全性に関するデータは試験期間を通じて収集した。
被験者は,14C 放射能の累積排泄量に基づく以下の条件に従って Day 7(投与後約 144 時間)又は
Day 10 に退所することとした。
1 入所期間の延長を要する医学的事由がなく,かつ以下のいずれかに該当する
2a 投与された放射能が十分に回収された(簡易測定による計数で投与量の 95%以上)
2b 尿及び糞中放射能が許容限度未満[簡易測定による計数で連続 2 日間(24 時間間隔の 2 倍)
の排泄量が投与量の 1%未満]
Day 10 に上記の条件に該当しなかった場合,被験者は 2a 又は 2b に該当するまで最長 Day 13 まで
入所期間を延長することとした。Day 13 に 2a 及び 2b のいずれにも該当しなかった被験者は,2a
又は 2b に該当するまで自宅で蓄尿及び/又は蓄便を継続することとした。
退所後,Day 7~14 に後観察を行った。
目標被験者数:6 例
【設定根拠】
統計学的な目標被験者数の算出は行っていない。未変化体及び代謝物の排泄経路の推定及びマス
バランスの評価を目的とした試験に適切な被験者数として 6 例を設定した。この被験者数は,マ
スバランス試験で一般的に許容されている。
122

2.7.6個々の試験のまとめ
診断及び選択・除外基準:
1. 選択基準(以下の基準をすべて満たす者を対象とした)
(1) 18 歳以上 55 歳以下の健康男性
(2) 体重 60 kg 以上 100 kg 未満,かつ BMI 18.5 kg/m2 以上 30 kg/m2 未満の者
(3) 被験者本人から文書による同意が得られている者
2. 除外基準(以下の基準のいずれかに該当する者は対象から除外した)
(1) 本薬又は製剤の構成成分に対して過敏症を有する又は過敏症が疑われる者
(2) 喘息,湿疹等のアレルギー疾患又は何らかの薬剤に対する重度の過敏症といった臨床的に重
要な既往を有する者
(3) 薬剤の吸収の妨げになる又は排便異常を引き起こす恐れのある臨床的に重要な上部消化管症
状を有する者
(4) 心血管系の疾患又は障害の既往を有する又は現在診断されている者
(5) 臨床的に重要な心電図異常所見の既往を有する者
(6) その他,消化器系,呼吸器系,腎機能,肝機能,神経系,皮膚,精神又は代謝系に臨床的に
意義のある疾患又は障害の既往を有する者
(7) 試験前の身体所見,心電図及び臨床検査の結果を治験担当医師が確認し,臨床的に重要な異
常が認められた者
(8) 治験担当医師により試験を完了できないと判断された疾患を有する者
(9) スクリーニング時に心拍数又は血圧に下記の異常が認められた者:
心拍数<40 bpm 又は>90 bpm,平均収縮期血圧>140 mmHg,平均拡張期血圧>90 mmHg(血圧は
臥位にて 5 分間安静にした後 3 回測定した)
(10)治験実施医療機関入所前 2 週間以内に処方薬又は市販薬(ビタミン剤,自然療法薬,ハーブ製
剤を含む)を使用した者(3 g/日以下のアセトアミノフェンを除く)
(11)治験実施医療機関入所前 3 カ月以内に薬物乱用歴を有する者
(12)本試験登録予定日から遡って 3 カ月以内に他の臨床試験に参加した者,又は 12 カ月以内に 3
件以上の臨床試験に参加した者
(13)治験実施医療機関入所前 3 カ月以内に 1 週間あたり 21 単位(1 単位=ビール 270 mL,蒸留酒
40 mL,ワイン 125 mL)を超える飲酒をした者
(14)治験実施医療機関入所前 3 カ月以内に 1 日 10 本を超える巻きタバコ(又は当量の刻みタバコ
)を使用した者
(15)血清学的検査で HBs 抗原,HAV IgM 抗体,HCV 抗体又は HIV 抗体 1 及び 2 が陽性であった者
(16)治験実施医療機関への入所前 3 カ月以内に献血(全血>400 mL 又は成分献血)を行った者,又
は 4 週間以内に血漿交換を行った者
(17)排便頻度が不規則(2 日に 1 回未満)の者
(18)何らかの理由のため試験を完了できない可能性があると治験担当医師が判断した者
(19)アステラス製薬グループ又は本試験の治験実施医療機関に雇用されている者
(20)過去 1 年間に仕事中又は臨床試験参加中に診断目的で放射線照射を受けた者(歯科 X 線検査
,胸部 X 線検査及び脊椎骨以外の体幹骨の X 線検査を除く)
123

2.7.6個々の試験のまとめ
治験薬,投与量及び投与方法:
1. 治験薬及びロット番号
ASP1941 経口液剤:1.8 MBq ± 10%の 14C-ASP1941 を含む ASP1941 100 mg/水 100 mL[1%
hydroxypropyl-β-cyclodextrin(HP-β-CD)]
[ロット番号]
ASP1941:BX100093714C-ASP1941:CP-3355-G
2. 投与量及び投与方法
本薬の 14C 標識体(1.8 MBq ± 10%)を含む 100 mg 経口液剤を空腹時に投与した。投与直後に,使
用した薬剤容器を水 50 mL で 2 回洗浄し,洗浄後の液も摂取させた。投与後 4 時間まで食事は摂
取しないこととした。
【設定根拠】
本薬 100 mg は臨床用量範囲内と推測され,本薬の代謝及び様々な排泄経路を評価する上で本用量
は適切であると考えられた。
約 1.8 MBq の放射能量は,ヒトに対する放射能の負荷を制限し,かつ本試験の目的を最大限達成
できるよう設定した。
評価期間:
治験薬投与期間:単回投与 1 日
併用治療(薬剤及び療法):
治験実施医療機関入所 2 週間前から処方薬及び市販薬(ビタミン剤,自然療法薬,ハーブ製剤を
含む)の使用を禁止した。ただし,治験担当医師から了承が得られた場合に限り,一定限度のア
セトアミノフェン(3 g/日以下)の使用は可とした。試験期間中に,その他の薬剤を必要とする医
学的事由が被験者の健康状態に生じた場合,当該被験者は試験を中止することとした(治験担当
医師と治験依頼者との協議の結果,薬剤の使用が試験の結果に影響を及ぼさないと判定された場
合を除く)。
124

2.7.6個々の試験のまとめ
評価項目,評価スケジュール及び評価基準:
1. 評価スケジュールスクリー
ニング
退所 a (退所)a 後観察
Day –21~–1 –1 1 2~6 7 (8~9) (10) (11~13) 7~14
入所 X X X 17:00 まで (X) (17:00 まで) (X)
同意取得 X
試験前スクリーニング b X
身体所見 X X X X
薬物乱用及び飲酒調査 X X
血清学的検査 X
選択基準/除外基準 X X X
臨床検査(血液学的検査,
血液生化学検査,尿検査)
X X Xc Xc Xc (X)c (X)c X
バイタルサイン X X Xd Xd Xd (X)d (X)d X
12 誘導心電図 e X X Xd Xd Xd (X)d (X)d X
治験薬投与 X
食事 X Xf X X (X) (X) (X)
採血 g X X X (X) (X) (X)
蓄尿 h X X X (X) (X) (X)
蓄便 i X X X X (X) (X) (X)
呼気採集 j X X X (X) (X) (X)
併用薬/有害事象調査 X X X X X (X) (X) (X) X
a: 被験者は,所定の条件に従って Day 7 又は Day 10 に退所することとした。また,Day 10 に所定の条件を満たさなかった場合,
最大 3 日間(Day 13 まで)退所を延期することとした。
b: 被験者背景,病歴,薬歴,喫煙及び飲酒習慣の調査
c: 測定時期:Day 1 の治験薬投与後 3,24,96,144 時間(Day 7 の退所が延期された場合は Day 1 の投与後 216 時間,Day 11
~13 の退所時にも測定)
d: 測定時期:Day 1 の治験薬投与前,投与後 2,24,96,144 時間(Day 7 の退所が延期された場合は Day 1 の治験薬投与後 216
時間,Day 11~13 の退所時にも測定)
e: 測定時点ごとに,臥位にて 10 分間安静にした後,5 分以内に 10 秒間ずつ 3 回測定
f: 同時刻に予定されていた採血及び評価終了後に食事の摂取を開始
g: 血漿中薬物動態,血漿及び全血中 14C 放射能評価用の採血時期:Day 1 の治験薬投与前,投与後 0.25,0 5,1,1.5,2,3,4,
6,8,10,12,16,24,36,48,60,72,84,96,120,144 時間[Day 7 の退所が延期された場合は採血を継続(Day 1 の治
験薬投与後 168,192,216 時間,Day 11~13 の 24 時間ごと)]
血球中 14C 放射能計数のためのヘマトクリット測定用の採血時期:Day 1 の治験薬投与前,投与後 24,48,72,96,120,144
時間[Day 7 の退所が延期された場合は採血を継続(退所まで 24 時間ごと)]14C 標識代謝物評価用の採血時期:Day 1 の治験薬投与前,投与後 0.5,1,2,4,6,8,10,12,24,48,72 時間
h: 尿中薬物動態,尿中 14C 放射能,14C 標識代謝物の 14C 標識体評価及び簡易測定用の蓄尿時期:Day 1 の治験薬投与前(–12~
0 時間),投与後 0~4,4~8,8~12,12~24,24~36,36~48,48~60,60~72,72~84,84~96,96~120,120~144 時
間[Day 7 の退所が延期された場合は蓄尿を継続(Day 1 の治験薬投与後 144~168,168~192,192~216 時間,Day 11~13
の各 24 時間)]
i: 糞中 14C 放射能,14C 標識代謝物の 14C 標識体評価及び簡易測定用の蓄便時期:入所時(Day –1)から Day 1 の投与直前まで,
Day 1~7 の各 24 時間[Day 7 の退所が延期された場合は蓄便を継続(Day 8~10 の各 24 時間,Day 11~13 の排便ごと)]
j: 14C 標識二酸化炭素評価用の呼気採集時期:Day 1 の治験薬投与前,投与後 1,2,3,4,6,8,12,24,48,72,96,120,
144 時間[Day 7 の退所が延期された場合は呼気採集を継続(Day 1 の治験薬投与後 168,192,216 時間,Day 11~13 の 24
時間ごと)]
2. 薬物動態:
「1. 評価スケジュール」に示した時期に採血,蓄尿,蓄便及び呼気採集を行った。血漿中及び尿
中未変化体濃度を測定し,以下の薬物動態パラメータをノンコンパートメント法を用いて算出し
た。また,全血,血漿,尿,糞及び呼気中の総放射能(並びに尿及び糞中簡易測定)を測定し,
以下の 14C 放射能の薬物動態パラメータを算出した。
125

2.7.6個々の試験のまとめ
[血漿中未変化体濃度並びに血漿及び全血中 14C 放射能]
・最高血漿中(又は全血中)濃度(Cmax)
・最高血漿中(又は全血中)濃度到達時間(tmax)
・ラグタイム(tlag)
・消失速度定数(kel)
・消失半減期(t1/2)
・時間 0 から定量可能最終時点までの血漿中(又は全血中)濃度‐時間曲線下面積(AUClast)
・無限時間まで外挿した血漿中(又は全血中)濃度‐時間曲線下面積(AUCinf)
・最終評価時点から無限外挿した部分の AUCinfに対する割合(%AUCextra)
・経口クリアランス(CL/F)(血漿中未変化体濃度のみ)
・みかけの分布容積(Vz/F)(血漿中未変化体濃度のみ)
・全血中放射能/血漿中放射能比
・赤血球中放射能
[尿中未変化体濃度](蓄尿間隔ごと及び最終蓄尿時間まで)
・時間 0 から定量可能最終時点までの累積尿中排泄量(Aelast)
・時間 0 から無限時間まで外挿した累積尿中排泄量(Aeinf)
・1 時間あたりの尿中排泄率(dU/dt)
・腎クリアランス(CLr)
[尿中 14C 放射能]
・蓄尿時間ごとの尿中放射能排泄量(Aeurine,14C,t1-t2)
・最終蓄尿時間までの累積尿中放射能排泄量(Aeurine,14C)
・1 時間あたりの尿中放射能排泄率(dU14C,t1-t2/dt)
[糞中 14C 放射能]
・蓄便時間ごとの糞中放射能排泄量(Aefeces,14C,t1-t2)
・最終蓄便時間までの累積糞中放射能排泄量(Aefeces,14C)
・1 時間あたりの糞中放射能排泄率(dF14C,t1-t2/dt)
[呼気中 14C 放射能]
・呼気採集時間ごとの呼気中放射能排泄量(Aeexpired air,14C,t1)
・最終呼気採集時間までの累積呼気中放射能排泄量(Aelastexpired air,14C)
・呼気中放射能排泄率(dA14C,t1-t2/dt)
[14C 放射能総排泄率]
・体外への総累積放射能排泄量(Aetotal,14C)
[代謝プロフィール]
・血漿,尿,糞中の代謝物を特定及び定量することとした(本報告書には含まない)。
3. 安全性:
・有害事象
・臨床検査値
・バイタルサイン
・心電図
・身体所見
有害事象は,治験薬投与開始後から後観察までに発現した事象を調査対象とした。
有害事象と治験薬との関連性は以下の基準により判定し,「関連あるかもしれない」又は「多分
(おそらく)関連あり」のいずれかに該当したものを,「治験薬との関連性が否定できない有害
事象(副作用)」と定義した。
126

2.7.6個々の試験のまとめ
治験薬との関連性 関連性判定基準
否定できる 有害事象発現と薬剤投与との間に時間的関係から因果関係がありそうにないと思われる
場合や,他の薬剤や合併症・基礎疾患等により説明ができる場合
関連あるかもしれ
ない
有害事象発現と薬剤投与との間に時間的に妥当な相関関係があるが,要因として合併症・
基礎疾患や他の薬剤による説明もできる場合。投与中止に関する情報が不明確な場合
多分(おそらく)
関連あり
有害事象発現と薬剤投与との間に時間的に妥当な相関関係がある場合,合併症・基礎疾患
や他の薬剤では説明ができない,又はそれらによる可能性が考えにくい場合,再投与によ
り再発又は投与中止により消失又は軽快を示す場合
有害事象の程度は,以下に従って判定した。
・軽度:日常の活動に支障がないもの
・中等度:日常の活動に支障があるもの
・重度:日常の活動が不可能になるもの
統計解析:
1. 解析対象集団:
(1) 安全性解析対象集団(SAF)
治験薬を服用した被験者の集団を SAF とした。
(2) 薬物動態解析対象集団(PKAS)
SAF に含まれる被験者のうち,1 つ以上の薬物動態パラメータを算出できる血漿中濃度データが
得られた被験者の集団を PKAS とした。
2. 被験者背景及びその他の基準値:
SAF を対象に,計数値項目では度数集計を行い,計量値項目では要約統計量を算出した。
3. 薬物動態:
薬物動態の解析では,PKAS を対象に,薬物動態パラメータの要約統計量を算出した。
4. 安全性:
安全性の解析では,SAF を解析対象とした。
有害事象及び副作用の発現率を算出した。
バイタルサインに関しては,評価時点ごとに実測値の要約統計量を算出した。
報告書の日付: 年 月 日
2.7.6.6.2 被験者の内訳及び解析対象集団
2.7.6.6.2.1 被験者の内訳
被験者の内訳を表 2.7.6.6-1 に示した。
計 6 例に治験薬が投与され,すべての被験者が本試験を完了した。
表 2.7.6.6-1 被験者の内訳
被験者数
登録例 12
治験薬投与例 6
完了例 6
中止例 0
Source:CL-0055 総括報告書(5.3.3.1-4)Table 12.1.1
127

2.7.6個々の試験のまとめ
2.7.6.6.2.2 解析対象集団
被験者 6 例全例が治験薬を服用し,すべての薬物動態パラメータを算出できる血漿中濃度デー
タが得られた。したがって,6 例全例を SAF 及び PKAS の解析対象例とした。
2.7.6.6.3 人口統計学的及び他の基準値の特性
SAF の被験者背景を表 2.7.6.6-2 に示した。
被験者全例が白人であった。平均年齢は 33.7 歳であり,BMI の平均値は 24.02 kg/m2であった。
表 2.7.6.6-2 被験者背景(SAF)
N=6
年齢 (歳) 平均値 33.7
標準偏差 13.1
最小値 – 最大値 21 – 55
性別 n(%) 男性 6(100.0)
女性 0
人種 n(%) 白人 6(100.0)
体重 (kg) 平均値 75.37
標準偏差 9.33
最小値 – 最大値 65.5 – 89.3
身長 (cm) 平均値 177.3
標準偏差 4.8
最小値 – 最大値 173 – 183
BMI (kg/m2) 平均値 24.02
標準偏差 3.33
最小値 – 最大値 19.8 – 29.8
Source:CL-0055 総括報告書(5.3.3.1-4)Table 12.1.2 及び Appendix 13.2.4.1
2.7.6.6.4 治験薬の曝露
被験者 6 例全例が,本薬の 14C 標識体 100 mg を空腹時に 1 回服用した。本試験で投与された本
薬の 14C 標識体の放射能は,1.78 MBq であった。
2.7.6.6.5 薬物動態
血漿及び全血中 14C 放射能並びに血漿中未変化体濃度の主な薬物動態パラメータの要約を表
2.7.6.6-3 に示した。
本薬の 14C 標識体(1.78 MBq)100 mg を投与したとき,tmaxの平均値は,血漿及び全血中放射
能ではいずれも 1.00 時間,血漿中未変化体濃度では 0.75 時間であった。tlag の平均値は,血漿及
び全血中放射能並びに血漿中未変化体濃度のいずれでも 0.00 時間であった。t1/2 の平均値は,血漿
及び全血中放射能では,それぞれ 12.6 時間及び 10.0 時間,血漿中未変化体濃度では 11.1 時間で
128

2.7.6個々の試験のまとめ
あった。全血中放射能/血漿中放射能比(平均値)は,Cmaxでは 0.685,AUClast では 0.560 及び
AUCinfでは 0.625 であった。血漿中未変化体濃度/血漿中放射能比(平均値)は,Cmaxでは 0.619,
AUClast では 0.485 及び AUCinfでは 0.472 であったことから,本薬の代謝物が 1 種類以上生成され
たことが示された。
全血中放射能/血漿中放射能比(平均値)は,本薬の 14C 標識体投与後 24 時間までの間に 0.543
~0.700 と比較的一定であり,赤血球への放射能の分布は最小限であることが示された。赤血球中
放射能の最高値(平均値)743 ng eq/mL は,血漿中放射能の最高値(平均値)2724 ng eq/mL の約
27%であった。赤血球中放射能(平均値)は投与後 1 時間で最高値に達し,その後低下したこと
から,投与後 24 時間までは 14C 放射能の赤血球への蓄積はなかったことが示唆された。
表 2.7.6.6-3 血漿及び全血中 14C 放射能並びに血漿中未変化体濃度の薬物動態パラメータの
要約(PKAS)
Cmax
(ng eq/mL)tmax
(h)tlag
(h)AUClast
(ng eq•h/mL)AUCinf
(ng eq•h/mL)t1/2
(h)
血漿中放射能 (N=6)
平均値 2724 1.00 0.00 14880 15326 12.6
標準偏差 133 0.00 0.00 1654 1586 4.55
全血中放射能 (N=6)
平均値 1863 1.00 0.00 8313 9577 10.0
標準偏差 58.4 0.00 0.00 795 1436 2.81
全血中放射能/血漿中放射能比 a
(N=6)平均値 0.685 ― ― 0.560 0.625 ―
標準偏差 0.0216 ― ― 0.0368 0.0690 ―
Cmax
(ng/mL)tmax
(h)tlag
(h)AUClast
(ng•h/mL)AUCinf
(ng•h/mL)t1/2
(h)
血漿中未変化体濃度 (N=6)
平均値 1688 0.75 0.00 7229 7251 11.1
標準偏差 185 0.27 0.00 1365 1367 3.57
血漿中未変化体濃度/
血漿中放射能比 a (N=6)
平均値 0.619 ― ― 0.485 0.472 ―
標準偏差 0.0556 ― ― 0.0633 0.0608 ―
a:単位なし
Source:CL-0055 総括報告書(5.3.3.1-4)Table 12.4.3,Table 12.4.8 及び Table 12.4.10
Day 7(治験薬投与後 144 時間)までの尿,糞及び呼気中の 14C 放射能の累積排泄率及び累積排
泄量の要約統計量を表 2.7.6.6-4 に示した。
Day 7 までは,尿及び糞中放射能の累積排泄率(平均値)は,投与された放射能(1.78 MBq)
の 100.6%であった。被験者ごとの放射能の累積排泄率は,97.9%~102.9%であった。14C 放射能の
主要な排泄経路は尿であり(尿中放射能の累積排泄率の平均値は 67.9%),残りは糞中に排泄され
た(糞中放射能の累積排泄率の平均値は 32.7%)。被験者ごとの放射能の累積排泄率は,尿中 58.5%
~72.6%,糞中 25.3%~44.4%であった。
129

2.7.6個々の試験のまとめ
表 2.7.6.6-4 Day 7 までの尿,糞及び呼気中の 14C 放射能の累積排泄率及び累積排泄量
(PKAS)
尿中 (%) 糞中 (%) 呼気中 (%) 計 (%)
平均値 (N=6) 67.9 32.7 0a 100.6
標準偏差 5.2 6.6 0a 1.8
中央値 69.4 31.4 0a 100.9
最小値 58.5 25.3 0a 97.9
最大値 72.6 44.4 0a 102.9
尿中 (kBq) 糞中 (kBq) 呼気中 (kBq) 計 (kBq)
平均値 (N=6) 1209 581 0a 1790
標準偏差 93.0 118 0a 31.2
中央値 1234 559 0a 1794
最小値 1041 450 0a 1742
最大値 1292 790 0a 1832
a:定量下限値未満(呼気中放射能は検出されなかった)
Source:CL-0055 総括報告書(5.3.3.1-4)Table 12.4.3 及び Table 12.4.5
尿中未変化体濃度の薬物動態パラメータの要約を表 2.7.6.6-5 に示した。
Day 7(治験薬投与後 144 時間)までの未変化体の累積 Aelast(平均値)は,投与された本薬の14C 標識体 100 mg のうち 0.955 mg(1.0%)であった。未変化体の CLr(平均値)は 0.133 L/h であっ
た。
表 2.7.6.6-5 尿中未変化体濃度の薬物動態パラメータの要約(PKAS)
累積 Aelasta (mg) 累積 Aelast
a (%) CLr (L/h)
平均値 (N=6) 0.955 1.0 0.133
標準偏差 0.254 0.3 0.0293
変動係数 (%) 26.6 26.6 22.1
中央値 0.883 0.9 0.126
最小値 0.713 0.7 0.104
最大値 1.33 1.3 0.169
a:最終の蓄尿間隔(投与後 120~144 時間)までの累積データ
Source:CL-0055 総括報告書(5.3.3.1-4)Table 12.4.9
2.7.6.6.6 安全性
2.7.6.6.6.1 有害事象
本試験で認められた有害事象の要約を表 2.7.6.6-6 に示した。
有害事象発現率は 83%(5/6 例)であり,有害事象の程度はすべて軽度であった。副作用発現率
は 67%(4/6 例)であった。
本試験では,死亡,その他の重篤な有害事象及び中止に至った有害事象は認められなかった。
130

2.7.6個々の試験のまとめ
表 2.7.6.6-6 有害事象の要約(SAF)
計N=6
有害事象発現例数(発現率) 5(83%)
有害事象発現件数 11
副作用発現例数(発現率) 4(67%)
副作用発現件数 8
重篤な有害事象発現例数(発現率) 0
中止に至った有害事象発現例数(発現率) 0
程度別有害事象発現例数(発現率)
軽度 5(83%)
中等度 0
重度 0
Source:CL-0055 総括報告書(5.3.3.1-4)Table 12.6.1.2,Appendix 13.2.7.1,Appendix 13.2.7.2 及び Appendix 13.2.7.3
本試験で認められた有害事象及び副作用の内訳を表 2.7.6.6-7 に示した。
有害事象は 6 例中 5 例に計 11 件認められた。すべての事象が一過性で後遺症なく回復した。副
作用は 6 例中 4 例に計 8 件認められ,いずれも治験薬と「関連あるかもしれない」と判定された。
表 2.7.6.6-7 有害事象及び副作用の内訳(SAF)
MedDRA version 11.0器官別大分類
基本語
有害事象発現例数
(発現率)N=6
有害事象発現件数N=6
副作用発現例数
(発現率)N=6
副作用発現件数N=6
すべての有害事象/
副作用5(83%) 11 4(67%) 8
胃腸障害 2(33%) 3 2(33%) 2
排便回数増加 1(17%) 1 1(17%) 1
胃腸音異常 1(17%) 1 1(17%) 1
肛門周囲痛 1(17%) 1 0 0
神経系障害 2(33%) 3 1(17%) 1
味覚異常 1(17%) 1 1(17%) 1
頭痛 2(33%) 2 0 0
腎および尿路障害 2(33%) 2 2(33%) 2
多尿 2(33%) 2 2(33%) 2
呼吸器,胸郭および縦
隔障害2(33%) 2 2(33%) 2
鼻出血 1(17%) 1 1(17%) 1
口腔咽頭痛 1(17%) 1 1(17%) 1
皮膚および皮下組織
障害1(17%) 1 1(17%) 1
皮膚乾燥 1(17%) 1 1(17%) 1
Source:CL-0055 総括報告書(5.3.3.1-4)Table 12.6.1.2 及び Appendix 13.2.7.1
2.7.6.6.6.2 死亡,その他の重篤な有害事象
本試験では,死亡,その他の重篤な有害事象は認められなかった。
131

2.7.6個々の試験のまとめ
2.7.6.6.6.3 中止に至った有害事象
本試験では,中止に至った有害事象は認められなかった。
2.7.6.6.6.4 臨床検査値
臨床検査値(腎機能マーカーである尿中の β-N-アセチル D グルコサミニダーゼ,β2 ミクログロ
ブリン及び α1 ミクログロブリンを含む)に関しては,基準値外の検査値がごく少数みられたが,
臨床的に重要な異常又は一定の傾向は観察されなかった。
本薬の作用機序から予測されたとおり,初回及び 2 回目の評価時点(本薬投与後 3 時間及び 24
時間)で全被験者の尿中グルコース排泄量が基準値を超えて増加した。血糖値に対する本薬の明
らかな影響はみられなかった。
2.7.6.6.6.5 バイタルサイン
血圧及び脈拍数に関して,臨床的に重要な異常又は一定の傾向は観察されなかった。
2.7.6.6.6.6 心電図
心拍数に関して,臨床的に重要な異常又は一定の傾向は観察されなかった。また,臨床的に重
要な心電図の異常所見はみられなかった。
2.7.6.6.6.7 身体所見
身体所見に関して,臨床的に重要な異常はみられなかった。
2.7.6.6.7 結論
● 本薬の 14C 標識体を含む 100 mg 経口液剤の単回投与後,本薬は速やかに吸収された。血漿及
び全血中放射能並びに血漿中未変化体濃度の tmax の平均値は,0.75~1.00 時間であった。血漿
中未変化体濃度/血漿中放射能比(平均値)は,Cmaxでは 0.619,AUClast では 0.485 及び AUCinf
では 0.472 であり,本薬の代謝物が 1 種類以上生成されたことが示された。血漿中未変化体
濃度,血漿及び全血中放射能の t1/2 の平均値は,10.0~12.6 時間であり,いずれも同程度の速
度で排泄された。赤血球への放射能の分布は最小限であることが示された。Day 7 までに,投
与された放射能の 100.6%(平均値)が排泄され,そのうち 67.9%は尿中に,32.7%は糞中に
排泄された。呼気中に放射能は検出されなかった。未変化体の累積尿中排泄率の平均値は投
与量の 1.0%であり,放射能の累積尿中排泄率の平均値は投与された放射能の 67.9%であった
ことから,本薬の代謝物が 1 種類以上生成されて尿中に排泄されたと考えられた。
132

2.7.6個々の試験のまとめ
● 臨床検査値,バイタルサイン,心電図及び身体所見に関して,臨床的に重要な異常又は一定
の傾向はみられなかった。健康男性被験者 6 例に対する本薬の 14C 標識体(1.78 MBq)100 mg
の単回経口投与は,安全であり,忍容性は良好であった。
133

2.7.6個々の試験のまとめ
2.7.6.7 高齢者・性差試験[CL-0052](添付資料 5.3.3.3-1)
2.7.6.7.1 試験方法の概略
治験課題名:健康成人男女(18 歳以上 45 歳以下及び 65 歳以上)を対象に,ASP1941 を反復経口
投与したときの薬物動態,安全性及び忍容性を検討し,ASP1941 の血糖値及び尿中グルコース排
泄に及ぼす影響を探索する第 I 相無作為化二重盲検プラセボ対照試験
治験実施医療機関:米国 1 施設
公表文献:なし
治験期間:
治験開始日:2007 年 11 月
治験終了日:2008 年 1 月
開発のフェーズ:第 I 相
目的:
(1) 主要目的
非高齢及び高齢健康成人男女を対象に ASP1941(以下,本剤)100 mg を単回及び反復投与したと
きの,本剤の薬物動態,並びに血糖値及び尿中グルコース排泄に対する薬力学的作用を評価する。
(2) 副次目的
非高齢及び高齢健康成人男女を対象に本剤 100 mg/日を 14 日間朝食前に反復経口投与したとき
の,安全性及び忍容性を評価する。
治験デザイン・治験方法:
本試験は,非高齢(18 歳以上 45 歳以下)及び高齢(65 歳以上)の健康成人男女を対象とした単
一施設,無作為化,二重盲検,並行群間,プラセボ対照,反復投与試験として実施した。
同意取得後,治験薬投与開始(Day 1)前 42 日以内にスクリーニング検査を行った。適格と判断
された被験者は Day −2 に治験実施医療機関に入所した。被験者数は各年齢層で 32 例(男女 16 例
ずつ)と計画し,年齢及び性別で層別したのちに,同一性別内で本剤群とプラセボ群の割合が 3:1
になるよう無作為に割り付けた。治験薬の初回投与は Day 1 の朝に行い,投与後 96 時間まで薬物
動態及び薬力学評価用の採血及び蓄尿を実施した。治験薬の反復投与は Day 5 から Day 18 まで行
い,未変化体のトラフ濃度測定のため Day 6,Day 8,Day 10,Day 12,Day 15,Day 18 の投与前
に採血を行った。Day 18 には,薬物動態及び薬力学評価用の採血及び蓄尿を投与前及び投与後 96
時間まで実施した。退所(Day 22)の 7~10 日後に後観察を実施した。
なお,本試験では,HbA1c 値の表記として National Glycohemoglobin Standardization Program(NGSP)
値を用いた。
目標被験者数:64 例
非高齢者(18 歳以上 45 歳以下):男女各 16 例(それぞれ本剤群 12 例,プラセボ群 4 例)
高齢者(65 歳以上):男女各 16 例(それぞれ本剤群 12 例,プラセボ群 4 例)
【設定根拠】
目標被験者数は他の類似の第 I 相試験を参考に設定し,統計的手法は用いていない。
診断及び選択・除外基準:
1. 選択基準(以下の基準をすべて満たす者を対象とした)
(1) 本試験に関連する行為(併用禁止薬の中止を含む)を開始する前に,治験審査委員会/独立
倫理委員会が承認した同意説明文書及び各国のプライバシー規則(米国では HIPAA 法)に
則って,本人又はその代諾者から文書による同意を得た者
(2) 非高齢(18 歳以上 45 歳以下)又は高齢(65 歳以上)の健康男女
(3) 非高齢女性(18 歳以上 45 歳以下)では,妊婦又は授乳中でない女性,スクリーニング時及
び治験実施医療機関入所時の妊娠検査†が陰性の女性,適切な避妊法をスクリーニングから試
験終了まで実施することに同意する女性(注:65 歳以上の女性は妊娠検査の対象外とした)
134

2.7.6個々の試験のまとめ
(4) スクリーニング時及び Day −2 の体重が 60 kg 以上 100 kg 以下,BMI が 20 kg/m2 以上 30 kg/m2
以下の者
(5) 治験実施医療機関入所前 48 時間以内のアルコール摂取禁止を含む食事制限に同意する者
(6) 治験実施医療機関入所期間中,本試験の標準食及び飲料の規定時間での摂取に同意する者
(7) スクリーニング時及び治験実施医療機関入所時の尿中薬物検査(アルコール,コチニンを含
む)が陰性である者(注:受動喫煙によりコチニン陽性となった被験者は,一酸化炭素呼気
検査が陰性であれば組入れ可能とした)
(8) 既往歴,身体所見,12 誘導心電図,バイタルサイン(起立性低血圧の評価を含む血圧,脈拍,
体温及び呼吸),スクリーニング時及び治験実施医療機関入所時の臨床検査[血液学的検査,
血液生化学検査,尿検査,血清学的(肝炎)検査]に基づき健康と判断された者
†:妊娠可能な女性被験者の妊娠検査に用いる尿検体は,スクリーニング時,治験実施医療機関入
所時(Day −2),退所時(又は中止時)に採取した。
2. 除外基準(以下の基準のいずれかに該当する者は対象から除外した)
(1) 1 型又は 2 型糖尿病の既往を有する者
(2) 空腹時血糖値が 6.4 mmol/L を超える又は HbA1c 値が 6.2%を超える者
(3) 腎性糖尿又は蛋白尿を有する者
(4) 臨床的に重要な喘息,湿疹等のアレルギー疾患の既往を有する者
(5) 治験実施医療機関入所前 4 週間以内に臨床的に重要な上部消化管症状(胸やけ,悪心,嘔吐,
腹部不快感,腹部不調)がみられた者
(6) 臨床的に重要な神経系,消化器系,腎機能,肝機能,肺,代謝系,心血管系,精神,内分泌
系,血液の疾患若しくは障害の既往,又は治験担当医師が本試験に参加不適切と判断した医
学的状態を有する者
(7) 治験薬投与開始前 4 週間以内に臨床的に重要な疾患又は異常がみられた者
(8) 複数の薬剤に対するアレルギーの既往を有する者,治験薬,治験薬に由来する誘導体,製剤
の構成成分に対するアレルギー又は過敏症を有する者,あるいはアレルギー又は過敏症が疑
われる者
(9) 肝炎を有する者,又はスクリーニング時の血清学的検査で HAV IgM 抗体,HBs 抗原又は HCV
抗体が陽性である者
(10) HIV 抗体が陽性である者
(11) 治験薬投与開始前 60 日以内に 1 単位(450 mL)以上の全血又は成分献血を行った者
(12) 過去に平均アルコール摂取量が 2 オンス/日を超えていた者,過去 3 年以内にアルコール依存
症又は薬物乱用の既往を有する者
(13) 治験薬投与開始前 120 日以内にタバコ又はニコチン含有製品を使用した者
(14) 治験薬投与開始前 21 日以内に全身又は局所投与の処方薬を使用した者(避妊薬を除く)
(15) 治験薬投与開始前 10 日以内に全身又は局所投与の市販薬,又はビタミン剤,ハーブ製剤,栄
養補助食品等の補完代替医療を使用した者(アセトアミノフェンを除く)
(16) 治験薬投与開始前 30 日以内に他の治験薬の投与を受けた者
(17) 治験実施計画書に規定された治験薬の服用,試験手順又は本試験の要求事項を遵守できない
と治験担当医師が判断した者
治験薬,投与量及び投与方法:
1. 治験薬及びロット番号
(1) 被験薬
ASP1941 錠 100 mg:1 錠中に ASP1941 として 100 mg を含有するフィルムコート錠
ロット番号:BX1000211
(2) 対照薬
ASP1941 錠 100 mg プラセボ:1 錠中に ASP1941 を含有しないフィルムコート錠
135

2.7.6個々の試験のまとめ
ロット番号:BX1000214
2. 投与量及び投与方法
ASP1941 錠 100 mg 又はプラセボを 1 日 1 回午前 8 時~10 時の朝食 5 分前以内に経口投与した。
治験薬の初回投与は Day 1 に行い,投与後 96 時間まで薬物動態及び薬力学の検体を採取した。治
験薬の反復投与は Day 5 から 14 日間行い,Day 18 の最終投与後 96 時間まで薬物動態及び薬力学
の検体を採取した。
【設定根拠】
尿中グルコース排泄に対する薬力学的作用を検討したこれまでの臨床試験の結果から,本試験の
用量は本剤での有効性が期待される 100 mg と設定した。また,0.1~600 mg の単回投与を検討し
た海外第 I 相試験(単回・食事)[CL-0001]の結果から,本剤の安全性及び忍容性は確認されて
いる。100 mg 単回投与時の最高血漿中未変化体濃度(Cmax)及び血漿中未変化体濃度‐時間曲線
下面積(AUC)は,サルでの 13 週間投与時の Cmax及び AUC と比較して低かった。ラットにおけ
る 13 週間毒性試験では,NOAEL での曝露量はヒトでの 100 mg 単回投与時の曝露量と比較して
低かったが,NOAEL を超える用量でみられた所見は,血中尿素窒素,尿中 β-N-アセチル D グル
コサミニダーゼ及び β2 ミクログロブリンの上昇,並びに腎臓の軽微な病理組織学的変化のみで
あった。
評価期間:
治験薬投与期間:15 日間(Day 1 及び Day 5~18)
併用治療(薬剤及び療法):
治験薬投与開始から試験終了まで,避妊薬及びアセトアミノフェンを除く処方薬,市販薬,補完
代替医療(ビタミン剤,ハーブ製剤,栄養補助食品等)の併用を禁止した。
136

2.7.6個々の試験のまとめ
評価項目,評価スケジュール及び評価基準:
1. 評価スケジュール:スク
リーニ
ング
入
所
選択・除
外基準
再確認
単回
投与
PK/PD評価
反復
投与
最終
投与
PK/PD評価
退所
(中止時)
後
観
察
Day−42~
−3−2 −1 1 2~4 5~17 18 19~22 22 29
同意取得 X
既往歴 X
身長 X
体重 X X X Xa Xa X X X
妊娠検査 X X X
尿中薬物検査,
コチニン・アルコール検査X X
一酸化炭素呼気検査 b X
血清学的検査(肝炎) X
HbA1c 値 X
無作為化 X
治験薬投与 X X X
前治療薬調査(治験薬投与前) X X X X
身体所見 X Xc X X
臨床検査(血液学的検査,
血液生化学検査及び尿検査)X Xc Xd Xd Xd X X
α1 酸性糖蛋白質 X X
安全性評価用蓄尿(24 時間)e Xf Xf
安全性評価用採血
(24 時間蓄尿の中央時点)g Xh Xh
安全性評価用蓄尿(2 時間)f Xi Xi
安全性評価用採血
(2 時間蓄尿の中央時点)g Xj Xj
血糖値測定用指穿刺採血 k X X X X X X X
尿中未変化体濃度及び尿中
グルコース濃度測定用蓄尿 k X X X X X X
血漿中未変化体濃度測定用採血 j X X X X X X
便潜血(腸管自発運動) X Xc Xl X
バイタルサイン X X X Xm Xm Xm Xm Xm X X
12 誘導心電図 X Xc Xn Xn Xn X
連続 12 誘導心電図 o X X X X X
併用薬及び有害事象調査 X X X X X X X
a: Day 2,Day 4,Day 6,Day 8,Day 10,Day 12,Day 14 及び Day 16
b:受動喫煙によりコチニン陽性となった被験者は,一酸化炭素呼気検査が陰性であれば組入れ可能とした。
c:入所前 72 時間以内にスクリーニング検査を実施した場合は不要とした。
d: 投与期間中は,Day 2(初回投与後 24 時間),Day 5 及び Day 12 の投与前,Day 18 の投与前及び投与後 3 時間に実施した。
入所から退所(中止時)まで,毎朝食前に指穿刺採血による血糖値測定もあわせて実施した(下表を参照)。
e:測定項目:総蛋白,電解質(ナトリウム,カリウム及びクロール)及びクレアチニン
f: 初回投与(Day 1)前 24 時間及び Day 17 の投与前~Day 18 の投与前
g:測定項目:電解質(ナトリウム,カリウム及びクロール)及びクレアチニン
h:初回投与(Day 1)前 12 時間及び Day 17 の投与後 12 時間
i: Day 1 及び Day 18 の投与後 0~2,2~4,4~6 時間
j: Day 1 及び Day 18 の投与後 1,3,5 時間
k:詳細は下表に示した。
l: Day 6 のみ
m:Day 1 の投与前及び投与後 1.25 時間,Day 2(初回投与後 24 時間),Day 3(初回投与後 48 時間),Day 5~18 の投与前,
Day 19(最終投与後 24 時間)及び Day 21(最終投与後 72 時間)
n:Day 1 の投与前及び投与後 1.5 時間,Day 10 及び Day 18 の投与前
o: Day −1,Day 1,Day 18 に 23.5 時間以上連続測定した。評価時点は以下の通り。Day −1(初回投与 24,23,21,16 時間前)
,
Day 1(初回投与前及び投与後 1,3,8 時間),Day 2(初回投与後 23.5 時間),Day 18(最終投与後 1,3,8 時間)及び
Day 19(最終投与後 23.5 時間)
137

2.7.6個々の試験のまとめ
血糖値,尿中未変化体及びグルコース濃度並びに血漿中未変化体濃度測定用の検体採取スケ
ジュールDay −1 1 2~4 5~17 18 19~22 22
血糖値
測定用
指穿刺
採血(PD/
安全性)
初回投与
24a,23.75,
23.5,23,
22.5,22,21,
20,18,16,
12 時間前
初回投与 8 時
間前,投与前 a 朝食前 a 投与前 a
投与前 a,投与
後 0.25,0.5,1,
1.5,2,3,4,
6,8,12 時間
最終投与後
16,24a,36a,
48a,72a時間
最終投
与後 96
時間 a
尿中
未変化体,
グルコー
ス濃度測
定用蓄尿
(PD/PK)
投与前,投与
後 0~2,2~4,
4~6,6~8,8
~12 時間
初回投与後
12~24,24~
36,36~48,
48~72 時間
初回投与後 72
~96 時間
(Day 5 の投
与前まで)
投与前,投与後
0~2,2~4,4
~6,6~8,8
~12 時間
最終投与後12
~24,24~36,
36~48,48~
72 時間
最終投
与後 72
~96 時
間
血漿中
未変化体
濃度測定
用採血
(PK)
投与前,投与
後 0.25,0.5,1,
1.5,2,3,4,
6,8,12 時間
初回投与後
16,24,36,
48,72 時間
初回投与後 96
時間(Day 5 投
与前)
トラフ濃度;
Day 6,8,10,
12,15
投与前,投与後
0.25,0.5,1,
1.5,2,3,4,
6,8,12 時間
最終投与後
16,24,36,
48,72 時間
最終投
与後 96
時間
a:空腹時
2. 薬物動態:
「1. 評価スケジュール」に示した時期に採血及び蓄尿を行った。血漿中及び尿中未変化体濃度を
測定し,以下の薬物動態パラメータをノンコンパートメント法(血管外投与モデル 200)を用い
て算出した。
[血漿中パラメータ]
・ Day 1
治験薬投与から定量可能最終時点までの血漿中濃度‐時間曲線下面積(AUClast)
治験薬投与から無限時間まで外挿した血漿中濃度‐時間曲線下面積(AUCinf)
経口クリアランス(CL/F)
最高血漿中濃度(Cmax)
最高血漿中濃度到達時間(tmax)
消失半減期(t1/2)
みかけの分布容積(Vz/F)
・ Day 6,Day 8,Day 10,Day 12,Day 15 及び Day 18
トラフ濃度(Ctrough)
・ Day 18投与間隔ごとの血漿中濃度‐時間曲線下面積(AUCtau)CL/FCmax
tmax
t1/2
Vz/F
ピーク/トラフ比(PTR)
[尿中パラメータ]
・ Day 1最終評価時点までの累積尿中排泄量(Ae)
最終評価時点までの累積尿中排泄率(Ae%)
定量可能最終時点までの累積尿中排泄量(Aelast)
138

2.7.6個々の試験のまとめ
定量可能最終時点までの累積尿中排泄率(Aelast%)
腎クリアランス(CLr)
・ Day 18投与間隔ごとの累積尿中排泄量(Aetau)
投与間隔ごとの累積尿中排泄率(Aetau%)CLr
3. 薬力学:
「1. 評価スケジュール」に示した時期に採血及び蓄尿を行った。血糖値及び尿中グルコース濃度
を測定し,以下の薬力学パラメータを算出した。
[血糖値]
・ Day −1 及び Day 18:最高血漿中濃度(Cmax),投与間隔ごとの血漿中濃度‐時間曲線下面積
(AUCtau)
[尿中グルコース]
・ Day 1:治験薬投与後 24 時間の尿中グルコース排泄量(Ae24h),定量可能最終時点までの累
積尿中グルコース排泄量(Aelast)
・ Day 18:投与間隔ごとの累積尿中グルコース排泄量(Aetau),Aelast
[尿中グルコース排泄率]
・ Day 1 及び Day 18 の蓄尿間隔のグルコース排泄率(GER)
・ Day 1 及び Day 18 の治験薬投与後 24 時間のグルコース排泄率(GER24h)
4. 安全性:
・ 有害事象の種類,頻度及び程度
・ バイタルサイン(血圧,脈拍数,呼吸数及び体温)。体位変換時の血圧及び脈拍数の評価を含
む。
・ 12 誘導心電図
・ 身体所見
・ 臨床検査(血液学的検査,血液生化学検査,尿検査)。血清クレアチニン及び指穿刺採血によ
る血糖値測定を含む。
・ 低血糖の発現率[低血糖は血糖値 70 mg/dL(3.9 mmol/L)未満と規定した]
・ 尿中の β-N-アセチル D グルコサミニダーゼ,α1 ミクログロブリン,β2 ミクログロブリン
・ 血清中の α1 酸性糖蛋白質,電解質(ナトリウム,カリウム及びクロール),クレアチニン
・ 尿中の総蛋白,電解質(ナトリウム,カリウム及びクロール),クレアチニン
・ クレアチニンクリアランス
有害事象は治験薬投与開始日から後観察までに発現した事象を収集し,初回投与日から最終投与
5 日後までに認められた事象(TEAE)を集計した。有害事象と治験薬との関連性は以下の基準で
判定し,「関連あるかもしれない」又は「多分(おそらく)関連あり」に該当した有害事象を「治
験薬との関連性が否定できない有害事象(副作用)」と定義した。治験薬との関連性 関連性判定基準
否定できる 有害事象発現と薬剤投与との間に時間的関係から因果関係がありそうにないと思われる
場合や,他の薬剤や合併症・基礎疾患等により説明ができる場合
関連あるかもしれ
ない
有害事象発現と薬剤投与との間に時間的に妥当な相関関係があるが,要因として合併症・
基礎疾患や他の薬剤による説明もできる場合。投与中止に関する情報が不明確な場合
多分(おそらく)
関連あり
有害事象発現と薬剤投与との間に時間的に妥当な相関関係がある場合,合併症・基礎疾患
や他の薬剤では説明ができない,又はそれらによる可能性が考えにくい場合,再投与によ
り再発又は投与中止により消失又は軽快を示す場合
有害事象の程度は,以下に従って判定した。
・ 軽度:日常の活動に支障がないもの
・ 中等度:日常の活動に支障があるもの
139

2.7.6個々の試験のまとめ
・ 重度:日常の活動が不可能になるもの
5. 有効性:
本試験では有効性の評価は行わなかった。
統計解析:
1. 解析対象集団:
(1) 安全性解析対象集団(SAF)
無作為化後,治験薬が少なくとも 1 回投与され,初回投与後に何らかのデータが得られたすべて
の被験者の集団を SAF とした。
(2) 薬物動態解析対象集団(PKAS)
主要な薬物動態パラメータを算出するために必要な薬物動態測定用検体が得られたすべての被験
者の集団を PKAS とした。
(3) 薬力学解析対象集団(PDAS)
主要な薬物動態パラメータを算出するために必要な薬力学測定用検体が得られたことが開鍵前ま
でに確認されたすべての被験者の集団を PDAS とした。
2. 被験者背景及びその他の基準値:
SAF,PKAS 及び PDAS を対象に,投与群別,年齢層別(非高齢,高齢)に,計数値項目では度
数集計を行い,計量値項目では要約統計量(平均値,標準偏差,最小値,中央値,最大値)を算
出した。
3. 薬物動態|薬力学:
PKAS 及び PDAS を対象に,薬物動態及び薬力学パラメータを測定日ごとの血漿中及び尿中濃度
から算出した。主要パラメータは以下の通りとした。
[薬物動態]
Day 1:AUCinf,AUClast,Cmax
Day 18:AUCtau,Cmax
[薬力学]
血糖値:AUCtau,Cmax(Day −1 及び Day 18)
尿中グルコース:Ae24h(Day 1),Aetau(Day 18)
薬物動態及び薬力学の主要パラメータについて年齢(非高齢,高齢)及び性別(男性,女性)の
影響を評価するため,2 つの片側 t 検定を行い,Day 1 及び Day 18 の各パラメータについて幾何平
均比とその 90%信頼区間(CI)を算出した。年齢,性別,及び年齢と性別との交互作用を固定効
果としたモデルを用いた。年齢と性別との交互作用が有意水準 15%で統計学的に有意な場合
(P≤0.15)は,各年齢層での性別の影響及び各性別での年齢の影響を評価することとした。
4. 安全性:
SAF を対象に,投与群別,年齢層別(非高齢,高齢)に,計数値項目では度数集計を行い,計量
値項目では要約統計量(平均値,標準偏差,最小値,中央値,最大値)を算出した。
報告書の日付: 年 月 日
2.7.6.7.2 被験者の内訳及び解析対象集団
被験者の内訳を表 2.7.6.7-1 に示した。
計 65 例が年齢及び性別で層別され,本剤群又はプラセボ群に無作為に割り付けられた。中止例
は本剤群の 4 例(非高齢者 2 例及び高齢者 2 例)であった。中止理由の内訳は,追跡不能 2 例(非
高齢者 1 例及び高齢者 1 例),有害事象 1 例(非高齢者),その他 1 例(高齢者)であった。
140

2.7.6個々の試験のまとめ
被験者 65 例全例(本剤群 49 例,プラセボ群 16 例)を SAF 及び PDAS の解析対象例とし,ま
た,本剤の投与を受けた被験者 49 例全例を PKAS の解析対象例とした。
表 2.7.6.7-1 被験者の内訳
非高齢 高齢計
プラセボ ASP1941 プラセボ ASP1941
無作為化例 8 24 8 25 65
投与完了例 8 22 8 23 61
中止例 0 2 0 2 4
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.1.1.1 及び Table 12.1.2.1
2.7.6.7.3 人口統計学的及び他の基準値の特性
被験者背景を表 2.7.6.7-2 に示した。
非高齢者及び高齢者いずれの層別集団でも,投与群間で平均年齢,体重,BMI に大きな差はみ
られなかった。非高齢者の男女の割合は各投与群で男性 50.0%,女性 50.0%と同じであり,高齢者
では本剤群で男性 48.0%,女性 52.0%,プラセボ群で男性 50.0%,女性 50.0%とほぼ同じであった。
141

2.7.6個々の試験のまとめ
表 2.7.6.7-2 被験者背景(SAF)
非高齢 高齢
プラセボ ASP1941 プラセボ ASP1941
項目 分類 (N=8) (N=24) (N=8) (N=25)
性別 n(%) 男性 4(50.0) 12(50.0) 4(50.0) 12(48.0)
女性 4(50.0) 12(50.0) 4(50.0) 13(52.0)
人種 n(%) 白人 7(87.5) 22(91.7) 7(87.5) 24(96.0)
黒人 1(12.5) 2(8.3) 1(12.5) 1(4.0)
民族 n(%) ヒスパニック系/ラテン系 7(87.5) 24(100.0) 8(100.0) 23(92.0)
非ヒスパニック系/ラテン系 1(12.5) 0 0 2(8.0)
年齢(歳) N 8 24 8 25
平均値 33.75 34.13 72.25 70.88
標準偏差 7.498 7.415 5.651 5.028
最小値 24.0 21.0 66.0 65.0
中央値 36.50 34.50 72.50 68.00
最大値 41.0 44.0 80.0 81.0
体重(kg) N 8 24 8 25
平均値 71.71 73.27 72.69 71.23
標準偏差 8.455 8.838 6.709 7.689
最小値 60.9 60.5 65.0 60.0
中央値 72.20 74.00 73.00 72.30
最大値 84.5 95.0 85.0 89.0
BMI(kg/m2) N 8 24 8 25
平均値 26.27 26.64 28.37 28.19
標準偏差 2.538 2.169 2.578 1.758
最小値 22.8 23.0 24.2 24.9
中央値 26.23 26.82 29.07 28.88
最大値 29.4 30.5 31.4 31.2
N:年齢層内での投与群別被験者数
n(%):年齢層内での投与群別分類別被験者数及び割合
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.1.3.1
2.7.6.7.4 治験薬の曝露
治験薬の投与量を表 2.7.6.7-3 に示した。
本剤の投与期間及び平均 1 日投与量は,非高齢者と高齢者で大きな違いはなかった。各年齢層
の本剤投与被験者全例が解析対象例となったため,本剤の投与期間及び平均 1 日投与量は,SAF,
PDAS 及び PKAS の解析対象集団間で同じであった。
142

2.7.6個々の試験のまとめ
表 2.7.6.7-3 治験薬の投与量(SAF)
非高齢 高齢
項目 (N=24) (N=25)
投与期間(日) 平均値 14.42 14.92
標準偏差 2.858 0.400
最小値 1.0 13.0
中央値 15.00 15.00
最大値 15.0 15.0
平均 1 日投与量(mg/kg) 平均値 1.38 1.42
標準偏差 0.162 0.150
最小値 1.1 1.1
中央値 1.35 1.38
最大値 1.7 1.7
N:本剤の投与を受け,データが得られた被験者数
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.2.1.1
2.7.6.7.5 薬物動態
2.7.6.7.5.1 血漿中薬物動態
単回投与時の血漿中未変化体濃度の推移(平均値 ± 標準偏差)を図 2.7.6.7-1 に,血漿中未変化
体の薬物動態パラメータの平均値(標準偏差)を表 2.7.6.7-4 に示した。
本剤 100 mg は単回経口投与後,速やかに吸収され,血漿中未変化体濃度は投与後約 2 時間で
Cmaxに到達した。tmaxは,高齢男性,高齢女性,非高齢男性,非高齢女性で同程度であった。曝露
量は,非高齢者より高齢者,男性より女性で高かった。
143

2.7.6個々の試験のまとめ
図 2.7.6.7-1 ASP1941 100 mg 単回投与時の血漿中未変化体濃度の推移(平均値 ± 標準偏
差)(PKAS)
Source:CL-0052 総括報告書(5.3.3.3-1)Figure 12.4.1.5
時間(h)
血漿
中未変化体濃度(
ng/m
L)
非高齢男性
非高齢女性
高齢男性
高齢女性
Day 1
144

2.7.6個々の試験のまとめ
表 2.7.6.7-4 ASP1941 100 mg 単回投与時の血漿中未変化体の薬物動態パラメータ:Day 1
(PKAS)
tmax Cmax AUClast AUCinf t1/2 CL/F Vz/F年齢性別層 (h) (ng/mL) (h•ng/mL) (h•ng/mL) (h) (L/h) (L)
非高齢男性N 12 12 12 12 12 12 12平均値 2.083 1260.674 8451.889 8519.799 15.046 12.025 259.652
標準偏差 0.8528 367.9539 1422.6066 1441.1076 2.8772 1.8957 59.4455
非高齢女性N 12 12 12 12 12 12 12平均値 1.876 1690.752 9698.288 9844.079 17.930 10.758 276.330
標準偏差 0.7211 594.7038 2390.5414 2460.9571 3.6679 2.8094 93.6361
高齢男性N 12 12 12 12 12 12 12
平均値 1.894 1491.293 10215.509 10325.042 15.234 10.017 217.894
標準偏差 0.9204 346.3221 2010.9129 2040.5144 4.7889 1.8555 71.2123
高齢女性N 13 13 13 13 13 13 13
平均値 2.122 1975.718 13640.997 13772.018 15.568 7.515 165.954
標準偏差 1.0096 441.6300 2414.8059 2447.3279 4.5213 1.5753 49.4773
N:本剤の投与を受け,データが得られた被験者数
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.4.2.9
反復投与時の血漿中未変化体濃度の推移(平均値 ± 標準偏差)を図 2.7.6.7-2 に,血漿中未変化
体の薬物動態パラメータの平均値(標準偏差)を表 2.7.6.7-5 に示した。
本剤 100 mg の 1 日 1 回 14 日間反復投与時の tmaxは,単回投与時と同程度であった。累積係数
(AI)は非高齢男性 1.592,高齢男性 1.595,非高齢女性 1.988,高齢女性 1.795 であり,同一性別
内では同程度であった。これら 4 集団の反復投与時の経口クリアランスは,単回投与時と同程度
であった。反復投与時の t1/2 は,単回投与時と比較して高齢男性で 10.3%,高齢女性で 30.3%,非
高齢男性で 11.1%,非高齢女性で 31.9%延長した。
145

2.7.6個々の試験のまとめ
図 2.7.6.7-2 ASP1941 100 mg 反復投与時の血漿中未変化体濃度の推移(平均値 ± 標準偏
差)(PKAS)
Source:CL-0052 総括報告書(5.3.3.3-1)Figure 12.4.1.5
時間(h)
血漿
中未変化体濃度(
ng/m
L)
非高齢男性
非高齢女性
高齢男性
高齢女性
Day 18
146

2.7.6個々の試験のまとめ
表 2.7.6.7-5 ASP1941 100 mg 反復投与時の血漿中未変化体の薬物動態パラメータ:Day 18
(PKAS)
tmax Cmax AUCtau t1/2 CLss/F Vz/F PTR AI年齢性別層 (h) (ng/mL) (h•ng/mL) (h) (L/h) (L)
非高齢男性N 12 12 12 12 12 12 12 12平均値 1.625 1592.925 8231.224 16.717 12.367 298.483 15.99 1.592
標準偏差 0.6557 363.8184 1169.7507 5.0494 1.7060 99.5728 4.624 0.2840
非高齢女性N 10 10 10 10 10 10 10 10平均値 1.983 1676.536 9582.693 23.654 10.751 364.412 14.93 1.988
標準偏差 0.9661 321.7939 1731.1535 9.2468 1.9651 148.2436 4.351 0.5326
高齢男性N 12 12 12 12 12 12 12 12
平均値 1.764 1584.996 10050.118 16.798 10.351 246.559 12.09 1.595
標準偏差 0.6492 400.3300 2143.4169 4.4408 2.0775 63.0501 2.755 0.2492
高齢女性N 12 12 12 12 12 12 12 12
平均値 1.812 2124.083 14027.041 20.292 7.461 211.744 12.53 1.795
標準偏差 0.5150 519.7452 2941.7786 9.6795 1.7552 93.1055 4.050 0.5642
N:本剤の投与を受け,データが得られた被験者数
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.4.2.10
血漿中未変化体の薬物動態パラメータに及ぼす年齢の影響を表 2.7.6.7-6 に,性別の影響を表
2.7.6.7-7 に示した。
統計解析の結果,反復投与時の AUCtau及び Cmaxに対する年齢と性別との交互作用は統計学的に
有意であった(P<0.15)が,単回投与時の AUCinf 及び Cmaxに対する交互作用は統計学的に有意で
はなかった。
反復投与時の年齢の影響は男性より女性で顕著であった。高齢女性の非高齢女性に対する
AUCtau及びCmaxの比は,それぞれ 145.35%,125.40%であり,高齢男性の非高齢男性に対するAUCtau
の比は,120.74%であったが,Cmaxは 2 つの年齢層で同程度であった。
反復投与時の性別の影響は,非高齢者より高齢者で顕著であった。高齢者では,女性の男性に
対する AUCtau及び Cmaxの比は,それぞれ 139.33%,134.66%であり,非高齢者では,女性の男性
に対する AUCtau及び Cmax の比は,それぞれ 115.74%,105.92%であった。
147

2.7.6個々の試験のまとめ
表 2.7.6.7-6 血漿中未変化体の薬物動態パラメータに及ぼす年齢の影響(PKAS)
性別 Day パラメータ
非高齢 高齢 比
(高齢/非高齢)b 比の 90% CIbN LSMa N LSMa
女性 1 AUCinf 12 9569.78 13 13550.25 141.59 123.65, 162.14
AUClast 12 9433.03 13 13424.34 142.31 124.40, 162.81Cmax 12 1588.33 13 1924.78 121.18 99.57, 147.48
18 AUCtau 10 9441.60 12 13723.56 145.35 126.64, 166.82Cmax 10 1649.00 12 2067.93 125.40 105.90, 148.50
男性 1 AUCinf 12 8415.00 12 10149.00 120.61 105.04, 138.48AUClast 12 8349.00 12 10043.44 120.30 104.87, 137.99
Cmax 12 1214.43 12 1453.53 119.69 97.96, 146.2318 AUCtau 12 8157.46 12 9849.39 120.74 105.88, 137.69
Cmax 12 1556.83 12 1535.63 98.64 83.96, 115.89
N:データが得られた被験者数
a:自然対数変換値の最小二乗幾何平均
b:幾何平均比及び信頼区間をパーセンテージで示した。
外れ値は解析の対象外とした。
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.4.3.5
表 2.7.6.7-7 血漿中未変化体の薬物動態パラメータに及ぼす性別の影響(PKAS)
年齢層 Day パラメータ
男性 女性 比
(女性/男性)b 比の 90% CIbN LSMa N LSMa
高齢 1 AUCinf 12 10149.00 13 13550.25 133.51 116.59, 152.89
AUClast 12 10043.44 13 13424.34 133.66 116.84, 152.91Cmax 12 1453.53 13 1924.78 132.42 108.81, 161.16
18 AUCtau 12 9849.39 12 13723.56 139.33 122.18, 158.89Cmax 12 1535.63 12 2067.93 134.66 114.62, 158.21
非高齢 1 AUCinf 12 8415.00 12 9569.78 113.72 99.04, 130.58
AUClast 12 8349.00 12 9433.03 112.98 98.50, 129.60Cmax 12 1214.43 12 1588.33 130.79 107.05, 159.79
18 AUCtau 12 8157.46 10 9441.60 115.74 100.85, 132.84Cmax 12 1556.83 10 1649.00 105.92 89.45, 125.42
N:データが得られた被験者数
a:自然対数変換値の最小二乗幾何平均
b:幾何平均比及び信頼区間をパーセンテージで示した。
外れ値は解析の対象外とした。
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.4.3.6
2.7.6.7.5.2 尿中薬物動態
Day 1 及び Day 18 の尿中未変化体の薬物動態パラメータの平均値(標準偏差)を表 2.7.6.7-8 に
示した。
未変化体の尿中への排泄率は 2%未満と低く,単回投与時の Aelast は非高齢男性 1.3171 mg,非高
齢女性 1.2469 mg,高齢男性 1.1938 mg,高齢女性 1.1566 mg であった。反復投与時では尿中排泄
量がわずかに増加し,Aetauは非高齢男性 1.5161 mg,非高齢女性 1.5780 mg,高齢男性 1.3629 mg,
高齢女性 1.3414 mg であった。高齢女性の腎クリアランス(CLr)は,非高齢男性と比較して単回
投与時で 46%,反復投与時で 47%低かった。腎クリアランスが全身クリアランスに占める割合は
148

2.7.6個々の試験のまとめ
2%未満と低いため,高齢女性における腎クリアランスの低さは臨床的に重要ではないと考えられ
た。
表 2.7.6.7-8 ASP1941 100 mg 投与時の尿中未変化体の薬物動態パラメータ:Day 1 及び
Day 18(PKAS)
Day 1 Day 18Aelast Aelast% CLr Aetau Aetau% CLr
年齢性別層 (mg) (L/h) (mg) (L/h)
非高齢男性N 12 12 12 12 12 12平均値 1.3171 1.3171 0.1589 1.5161 1.5161 0.1860
標準偏差 0.19193 0.19193 0.02967 0.27005 0.27005 0.03341
非高齢女性N 12 12 12 10 10 10
平均値 1.2469 1.2469 0.1334 1.5780 1.5780 0.1683
標準偏差 0.18608 0.18608 0.02815 0.29926 0.29926 0.04115
高齢男性N 12 12 12 12 12 12
平均値 1.1938 1.1938 0.1219 1.3629 1.3629 0.1431
標準偏差 0.30872 0.30872 0.04475 0.50733 0.50733 0.07036
高齢女性N 13 13 13 12 12 12平均値 1.1566 1.1566 0.0858 1.3414 1.3414 0.0977
標準偏差 0.24665 0.24665 0.01767 0.26319 0.26319 0.01947
N:本剤の投与を受け,データが得られた被験者数
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.4.2.11 及び Table 12.4.2.12
2.7.6.7.6 薬力学
Day −1(初回投与前 24 時間)及び Day 18(最終投与後 24 時間)の血糖値(平均値±標準偏差)
を図 2.7.6.7-3 に示した。
本剤 100 mg 又はプラセボの 14 日間反復投与時の変動は,各投与群ともに層別集団のいずれも
比較的小さく,年齢又は性別による差は認められなかった。
図 2.7.6.7-3 Day −1 及び Day 18 の血糖値(平均値 ± 標準偏差)(PDAS)
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.5.1.1,Table 12.5.1.2,Table 12.5.1.6 及び Table 12.5.1.7
プラセボ プラセボ プラセボ プラセボ
非高齢者 高齢者 男性 女性
149

2.7.6個々の試験のまとめ
Day −1 及び Day 18 の血糖値パラメータの平均値(標準偏差)を表 2.7.6.7-9 に,Day 1 及び Day
18 の尿中グルコースパラメータの平均値(標準偏差)を表 2.7.6.7-10 に示した。
本剤群では,反復投与時に血糖値の AUC24hが 6%~7%低下し,Cmaxは 7%~8%低下した。
また,本剤群では,プラセボ群と比較して単回及び反復投与時ともに尿中へのグルコース排泄
量が顕著に増加した。この所見は,SGLT2(SGLT:Na+/グルコース共輸送担体)阻害薬としての
本剤の作用機序と合致する。SGLT2 は腎近位尿細管でのグルコース及びナトリウムの再吸収を
担っており,本剤は尿へのグルコース排泄を促進することが期待される。
本剤 100 mg 単回投与時の男性の尿中グルコース排泄量(Ae24h)は 244.98 mmol であり,女性の
184.84 mmol と比較して多かった。反復投与時の Aetauも,男性 245.33 mmol,女性 170.88 mmol で
あり,女性より男性で多かった。
本剤100 mg単回投与時の非高齢者及び高齢者のAe24hは,それぞれ250.04 mmol及び179.99 mmol
であった。反復投与時の非高齢者の Aetauは単回投与時と同程度であったが,高齢者では反復投与
時に 169.02 mmol に減少した。
150

2.7.6個々の試験のまとめ
表 2.7.6.7-9 血糖値パラメータ:年齢層別及び性別(PDAS)
Day −1 Day 18
年齢層/ AUC24h Cmax AUCtau Cmax
投与群 性別 (h•mmol/L) (mmol/L) (h•mmol/L) (mmol/L)
プラセボ 非高齢N 8 8 8 8平均値 135.997 7.5771 129.376 6.9318
標準偏差 10.5168 0.97782 11.0968 0.78191
高齢N 8 8 8 8平均値 149.351 7.5632 142.001 7.7645
標準偏差 14.6893 1.25946 10.9744 1.34489
ASP1941 非高齢N 24 24 23 23平均値 136.032 7.3851 127.646 6.8615
標準偏差 8.5711 0.82732 8.4754 0.79650
高齢N 25 25 24 24
平均値 147.305 8.0401 138.281 7.3805
標準偏差 10.3701 0.96476 10.6405 0.96973
プラセボ 男性N 8 8 8 8平均値 140.004 7.5632 137.873 7.5910
標準偏差 18.7930 1.39385 14.6617 1.57420
女性N 8 8 8 8平均値 145.343 7.5771 133.504 7.1053
標準偏差 7.6687 0.77431 10.4298 0.45095
ASP1941 男性N 24 24 24 24平均値 143.877 7.9009 135.641 7.2325
標準偏差 11.1078 1.01207 12.2488 0.98888
女性N 25 25 23 23平均値 139.773 7.5449 130.401 7.0160
標準偏差 10.7679 0.87137 8.8917 0.84497
N:データが得られた被験者数
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.5.2.1,Table 12.5.2.2,Table 12.5.2.5 及び Table 12.5.2.6
151

2.7.6個々の試験のまとめ
表 2.7.6.7-10 尿中グルコースパラメータ:年齢層別及び性別(PDAS)
Day 1 Day 18
年齢層/ Ae24h GER24h Aelast Aetau GER24h Aelast
投与群 性別 (mmol) (mmol/h) (mmol) (mmol) (mmol/h) (mmol)
プラセボ 非高齢N 8 8 8 8 8 8平均値 0.43 0.017 3.00 0.61 0.025 1.21
標準偏差 0.317 0.0130 2.397 0.501 0.0206 0.853
高齢N 8 8 8 8 8 8平均値 0.32 0.013 2.01 1.34 0.055 2.17
標準偏差 0.217 0.0089 2.956 1.558 0.0640 1.816
ASP1941 非高齢N 24 24 24 22 22 22平均値 250.04 10.278 385.47 254.14 10.443 440.29
標準偏差 58.640 2.4148 121.519 81.142 3.3343 181.400
高齢N 25 25 25 24 24 24
平均値 179.99 7.400 275.77 169.02 6.949 316.68
標準偏差 41.686 1.7201 88.538 52.335 2.1533 129.843
プラセボ 男性N 8 8 8 8 8 8平均値 0.51 0.02 3.35 0.98 0.04 1.84
標準偏差 0.302 0.012 3.152 0.857 0.035 1.350
女性N 8 8 8 8 8 8平均値 0.24 0.01 1.66 0.97 0.04 1.53
標準偏差 0.146 0.006 1.861 1.500 0.062 1.635
ASP1941 男性N 24 24 24 24 24 24平均値 244.98 10.07 390.65 245.33 10.08 440.11
標準偏差 52.256 2.148 105.179 78.896 3.242 183.390
女性N 25 25 25 22 22 22平均値 184.84 7.59 270.81 170.88 7.02 305.64
標準偏差 55.447 2.283 100.998 60.801 2.497 114.037
N:データが得られた被験者数
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.5.2.3,Table 12.5.2.4,Table 12.5.2.7 及び Table 12.5.2.8
尿中グルコースパラメータに及ぼす年齢及び性別の影響を表 2.7.6.7-11 に示した。
尿中へのグルコース排泄に対し,年齢及び性別による影響が認められた。年齢の影響に関して,
非高齢者の方が高齢者より尿中へのグルコース排泄が多かった。単回投与時で,高齢者の非高齢
者に対する Ae24h及び GER24 の比は,それぞれ 72.77%,72.79%であり,反復投与時での高齢者の
非高齢者に対する Aetau及び GER24 の比は,それぞれ 68.50%,68.49%であった。
性別の影響に関して,男性の方が女性より尿中へのグルコース排泄が多かった。単回投与時で,
女性の男性に対する Ae24h 及び GER24の比は,それぞれ 74.52%,74.42%であり,反復投与時での
女性の男性に対する Aetau及び GER24の比は,それぞれ 70.52%,70.55%であった。
152

2.7.6個々の試験のまとめ
表 2.7.6.7-11 尿中グルコースパラメータに及ぼす年齢及び性別の影響(PDAS)
非高齢 高齢 比Day パラメータ N LSMa N LSMa
(高齢/非高齢)b 比の 90% CIb
1 Ae24h 24 242.42 25 176.42 72.77 66.05, 80.19Aelast 24 364.89 25 263.68 72.26 62.92, 83.00
GER24h 24 9.96 25 7.25 72.79 66.04, 80.2318 Aelast 22 396.37 24 293.93 74.16 60.86, 90.35
Aetau 22 235.44 24 161.27 68.50 58.83, 79.75GER24h 22 9.68 24 6.63 68.49 58.85, 79.79
男性 女性 比Day パラメータ N LSMa N LSMa
(女性/男性)b 比の 90% CIb
1 Ae24h 24 239.56 25 178.53 74.52 67.64, 82.12Aelast 24 377.04 25 255.19 67.68 58.93, 77.74
GER24h 24 9.85 25 7.33 74.42 67.55, 82.0518 Aelast 24 400.95 22 290.57 72.47 58.48, 88.30
Aetau 24 232.04 22 163.63 70.52 60.57, 82.11GER24h 24 9.54 22 6.73 70.55 60.56, 82.11
N:データが得られた被験者数
a:自然対数変換値の最小二乗幾何平均
b:幾何平均比及び信頼区間をパーセンテージで示した。
外れ値は解析の対象外とした。
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.5.3.4 及び Table 12.5.3.5
薬力学パラメータにおける年齢と性別との交互作用は統計学的に有意ではなかった。高齢者及
び女性では曝露量が高かったが,薬力学パラメータには影響しなかった。尿中へのグルコース排
泄量はむしろ高齢者及び女性でより少なかったことから,薬力学的作用には曝露量よりも腎機能
が重要な役割を果たしていることが示唆された。
2.7.6.7.7 安全性
2.7.6.7.7.1 有害事象
本試験で認められた有害事象(TEAE)の要約を表 2.7.6.7-12 に,有害事象の内訳を表 2.7.6.7-13
及び表 2.7.6.7-14 に,副作用の内訳を表 2.7.6.7-15 及び表 2.7.6.7-16 に示した。
有害事象は,プラセボ群の 1 例(非高齢者)及び本剤群の 9 例(非高齢者 5 例,高齢者 4 例)
に認められた。本試験で認められたすべての有害事象は軽度又は中等度であり,回復が確認され
た。治験担当医師により治験薬との関連性が否定されなかった有害事象は,本剤群の 49 例中 6 例
に認められた。その内訳は,便秘 3 例[非高齢男性 1 例(Day 18 に発現),高齢女性 2 例(Day 11
に発現)],頭痛 2 例[非高齢女性 1 例(Day 1 に発現),非高齢男性 1 例(Day 18 に発現)],全身
性皮疹,そう痒症及び眼窩周囲浮腫[非高齢女性 1 例(いずれも投与中止に至った有害事象で Day
2 に発現)]であった。本試験では,低血糖及び泌尿生殖器感染,重篤な有害事象は認められなかっ
た。
153

2.7.6個々の試験のまとめ
表 2.7.6.7-12 有害事象の要約(SAF)
非高齢 高齢計
(N=65)プラセボ
(N=8)ASP1941 (N=24)
プラセボ(N=8)
ASP1941 (N=25)
有害事象発現例数(発現率) 1(12.5%) 5(20.8%) 0 4(16.0%) 10(15.4%)
有害事象発現件数 1 9 0 5 15
副作用発現例数(発現率) 1(12.5%) 4(16.7%) 0 2(8.0%) 7(10.8%)
副作用発現件数 1 6 0 2 9
重篤な有害事象発現例数(発現率) 0 0 0 0 0
中止に至った有害事象発現例数(発現率) 0 1(4.2%) 0 0 1(1.5%)
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.6.1.1,Table 12.6.1.2,Table 12.6.1.3,Table 12.6.1.4 及び Appendix 13.2.7
表 2.7.6.7-13 有害事象の内訳(SAF)
MedDRA version 9.1 非高齢 高齢計
(N=65)器官別大分類 プラセボ
(N=8)ASP1941 (N=24)
プラセボ(N=8)
ASP1941 (N=25)基本語
すべての有害事象 1(12.5%) 5(20.8%) 0 4(16.0%) 10(15.4%)
皮膚および皮下組織障害 0 2(8.3%) 0 2(8.0%) 4(6.2%)
そう痒症 0 2(8.3%) 0 1(4.0%) 3(4.6%)
接触性皮膚炎 0 0 0 2(8.0%) 2(3.1%)
眼窩周囲浮腫 0 1(4.2%) 0 0 1(1.5%)
全身性皮疹 0 1(4.2%) 0 0 1(1.5%)
胃腸障害 0 1(4.2%) 0 2(8.0%) 3(4.6%)
便秘 0 1(4.2%) 0 2(8.0%) 3(4.6%)
傷害,中毒および処置合併症 0 2(8.3%) 0 0 2(3.1%)
処置後局所反応 0 2(8.3%) 0 0 2(3.1%)
神経系障害 0 2(8.3%) 0 0 2(3.1%)
頭痛 0 2(8.3%) 0 0 2(3.1%)
臨床検査 1(12.5%) 0 0 0 1(1.5%)
血中クレアチンホスホキナーゼ増加 1(12.5%) 0 0 0 1(1.5%)
発現例数(発現率)
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.6.1.1
154

2.7.6個々の試験のまとめ
表 2.7.6.7-14 有害事象の内訳(件数)(SAF)
MedDRA version 9.1器官別大分類
基本語
非高齢 高齢 計
プラセボ ASP1941 プラセボ ASP1941
すべての有害事象 1 9 0 5 15
皮膚および皮下組織障害 0 4 0 3 7
そう痒症 0 2 0 1 3
接触性皮膚炎 0 0 0 2 2
眼窩周囲浮腫 0 1 0 0 1
全身性皮疹 0 1 0 0 1
胃腸障害 0 1 0 2 3
便秘 0 1 0 2 3
傷害,中毒および処置合併症 0 2 0 0 2
処置後局所反応 0 2 0 0 2
神経系障害 0 2 0 0 2
頭痛 0 2 0 0 2
臨床検査 1 0 0 0 1
血中クレアチンホスホキナーゼ増加 1 0 0 0 1
Source:CL-0052 総括報告書(5.3.3.3-1)Appendix 13.2.7
表 2.7.6.7-15 副作用の内訳(SAF)
MedDRA version 9.1 非高齢 高齢計
(N=65)器官別大分類 プラセボ
(N=8)ASP1941 (N=24)
プラセボ(N=8)
ASP1941 (N=25)基本語
すべての副作用 1(12.5%) 4(16.7%) 0 2(8.0%) 7(10.8%)
胃腸障害 0 1(4.2%) 0 2(8.0%) 3(4.6%)
便秘 0 1(4.2%) 0 2(8.0%) 3(4.6%)
神経系障害 0 2(8.3%) 0 0 2(3.1%)
頭痛 0 2(8.3%) 0 0 2(3.1%)
臨床検査 1(12.5%) 0 0 0 1(1.5%)
血中クレアチンホスホキナーゼ増加 1(12.5%) 0 0 0 1(1.5%)
皮膚および皮下組織障害 0 1(4.2%) 0 0 1(1.5%)
そう痒症 0 1(4.2%) 0 0 1(1.5%)
眼窩周囲浮腫 0 1(4.2%) 0 0 1(1.5%)
全身性皮疹 0 1(4.2%) 0 0 1(1.5%)
発現例数(発現率)
Source:CL-0052 総括報告書(5.3.3.3-1)Table 12.6.1.2
155

2.7.6個々の試験のまとめ
表 2.7.6.7-16 副作用の内訳(件数)(SAF)
MedDRA version 9.1器官別大分類
基本語
非高齢 高齢 計
プラセボ ASP1941 プラセボ ASP1941
すべての副作用 1 6 0 2 9
胃腸障害 0 1 0 2 3
便秘 0 1 0 2 3
神経系障害 0 2 0 0 2
頭痛 0 2 0 0 2
臨床検査 1 0 0 0 1
血中クレアチンホスホキナーゼ増加 1 0 0 0 1
皮膚および皮下組織障害 0 3 0 0 3
そう痒症 0 1 0 0 1
眼窩周囲浮腫 0 1 0 0 1
全身性皮疹 0 1 0 0 1
Source:CL-0052 総括報告書(5.3.3.3-1)Appendix 13.2.7
2.7.6.7.7.2 死亡,その他の重篤な有害事象
本試験では死亡及びその他の重篤な有害事象は認められなかった。
2.7.6.7.7.3 中止に至った有害事象
投与中止に至った有害事象は,本剤群の非高齢女性 1 例(全身性発疹,そう痒症及び眼窩周囲
浮腫)に認められた。全身性発疹,そう痒症及び眼窩周囲浮腫はいずれも Day 11 に回復し,治験
担当医師は治験薬との関連性をそれぞれ「多分(おそらく)関連あり」,「関連あるかもしれない」,
「関連あるかもしれない」と判定した。
2.7.6.7.7.4 臨床検査値
年齢層別各投与群の血液学的及び血液生化学検査値(平均値及び中央値)は,すべての項目で
基準値内であった。各投与群のいずれの年齢層でも,検査項目の平均値又は中央値に異常な変動
はみられず,治験薬投与による影響はみられなかった。
事前に規定した臨床検査項目(血糖値,総ビリルビン,肝酵素,血清クレアチニン,電解質,
血小板数,白血球数)に関しては,試験期間中に有害事象と判断されるような異常を示す変動は
みられなかった。臨床検査値関連の有害事象としては,クレアチンホスホキナーゼの上昇が Day 12
にプラセボ群の非高齢者 1 例に認められ,血中クレアチンホスホキナーゼ増加として報告された。
本事象の治験薬との関連性は「多分(おそらく)関連あり」と判定された。更に,Day 29 以降に
本剤群の 4 例(非高齢者 2 例及び高齢者 2 例)に有害事象として血中クレアチンホスホキナーゼ
増加が認められ,非高齢者 1 例及び高齢者 1 例(いずれも Day 29 に発現)は治験薬との関連性が
「関連あるかもしれない」と判定された。
156

2.7.6個々の試験のまとめ
血清中 α1 酸性糖蛋白質(平均値±標準偏差,以下同様)は,ベースライン時では非高齢者で 20.79
± 2.783~20.96 ± 7.429 μmol/L,高齢者で 19.68 ± 3.177~20.46 ± 4.822 μmol/L と同程度であった。
Day 18 では年齢層別の各投与群で同程度の低下がみられ,ベースラインからの変化量は非高齢者
で−3.00 ± 1.333 μmol/L(本剤群)~−3.80 ± 2.239 μmol/L(プラセボ群),高齢者で−1.06 ± 2.689 μmol/L
(本剤群)~−1.17 ± 3.195 μmol/L(プラセボ群)であった。
腎機能マーカーに関しては,本試験中に本剤群の非高齢者 15 例及び高齢者 21 例で β-N-アセチ
ル D グルコサミニダーゼ(基準値上限:5 U/L)が上昇した。プラセボ群での β-N-アセチル D グ
ルコサミニダーゼ上昇は高齢者 2 例のみであり,このうち 1 例はベースライン時ですでに高値を
示していた。本試験中に α1 ミクログロブリン上昇は,本剤群の非高齢者 6 例及び高齢者 11 例,
プラセボ群の高齢者 4 例にみられ,測定値は 1.3~3.1 mg/dL(基準値上限:1.2 mg/dL)であった。
β2 ミクログロブリン上昇は本剤群の非高齢者 3 例及び高齢者 2 例にみられ,プラセボ群ではみら
れなかった。これら 5 例のうち 4 例では α1 ミクログロブリンも同時に上昇していた。β2 ミクロ
グロブリンの測定値は 329~892 μg/L(基準値上限:300 μg/L)であった。
2.7.6.7.7.5 バイタルサイン
本剤群の 3 例及びプラセボ群の 1 例で起立時の収縮期血圧に 20%以上の低下がみられたが,有
害事象として報告された事象はなく,症候性の起立性低血圧は発現しなかった。
起立時の収縮期血圧,拡張期血圧及び脈拍数は,年齢層別の各投与群で投与期間を通してわず
かな上昇と低下が混在してみられた。本剤群とプラセボ群との比較の結果,起立時のバイタルサ
インに及ぼす本剤の影響は認められなかった。
2.7.6.7.7.6 心電図
臨床的に重要な心電図の異常は,いずれの投与群でもみられなかった。
2.7.6.7.8 結論
非高齢及び高齢健康男女を対象に本剤を単回及び反復投与したとき,本剤の曝露量は非高齢者
より高齢者,男性より女性で高かった。統計解析の結果,反復投与時の年齢と性別との交互作用
は統計学的に有意であった(P<0.15)。
反復投与時の主要薬物動態パラメータに及ぼす年齢の影響は,男性より女性で顕著であった。
高齢女性の非高齢女性に対する AUCtau及び Cmaxの比は,それぞれ 145.35%,125.40%であり,高
齢男性の非高齢男性に対する AUCtauの比は,120.74%であった。
反復投与時の主要薬物動態パラメータに及ぼす性別の影響は,非高齢者より高齢者で顕著で
あった。高齢者では,女性の男性に対する AUCtau及び Cmaxの比は,それぞれ 139.33%,134.66%
であり,非高齢者では,女性の男性に対する AUCtau及び Cmaxの比は,それぞれ 115.74%,105.92%
であった。
157

2.7.6個々の試験のまとめ
本剤の未変化体の尿中排泄量は少なく,腎クリアランスが全身クリアランスに占める割合は 2%
未満と低かった。
高齢者の非高齢者に対する尿中グルコース排泄量(Ae24h及びAetau)の比は,単回投与時で 72.77%
であり,反復投与時で 68.50%であった。また,女性の男性に対する尿中グルコース排泄量(Ae24h
及び Aetau)の比は,単回投与時で 74.52%,反復投与時で 70.52%であった。薬力学的作用では,
年齢と性別との交互作用はみられなかった。
本剤 100 mg の 1 日 1 回 14 日間反復投与は,健康高齢者及び健康非高齢者いずれの集団でも安
全であることが確認された。本試験での有害事象の発現はごく少数であり,低血糖及び泌尿生殖
器感染,重篤な有害事象は発現しなかった。
158

2.7.6個々の試験のまとめ
2.7.6.8 腎機能低下患者 PK/PD 試験[CL-0073](添付資料 5.3.3.3-2)
2.7.6.8.1 試験方法の概略
治験課題名:ASP1941 臨床薬理試験-腎機能低下を伴う 2 型糖尿病患者における薬物動態・薬力
学の検討-
治験実施医療機関:日本 3 施設
公表文献:なし
治験期間:
治験開始日:2010 年 2 月
治験終了日:2010 年 6 月
開発のフェーズ:第 I 相
目的:
正常な腎機能を有する 2 型糖尿病患者,及び軽度又は中等度腎機能低下を伴う 2 型糖尿病患者に
ASP1941(以下,本剤)50 mg を単回経口投与し,薬物動態,薬力学(尿中グルコース排泄量)及
び安全性に及ぼす腎機能の影響を検討する。
治験デザイン・治験方法:
本試験は,多施設共同,非盲検試験として実施した。
患者本人から文書同意を取得した後,スクリーニングを実施し,被験者の適格性を確認した。適格
性に問題がなかった被験者は,治験薬投与予定日の前々日(Day −2)の午後に入院し,前観察期検
査・観察を実施した。前観察期の検査・観察終了後,治験担当医師が治験薬を投与することに問題
ないと判断した被験者に,本剤 50 mg を朝食前(Day 1)に単回経口投与した。被験者は評価期の終
了(Day 4)まで入院を継続し,評価期終了後には 1~2 週間の後観察期を設けた。
なお,本試験では HbA1c 値の表記として Japan Diabetes Society(JDS)値を用いた。
目標被験者数:
18 例(腎機能低下の重症度分類ごとに 6 例ずつ)
腎機能低下の重症度は,スクリーニング時の日本人用糸球体濾過量(GFR)推算式†から推定される
GFR 値(eGFR)に基づき,分類した。
正常な腎機能:90 mL/min/1.73 m2 以上
軽度腎機能低下:60 mL/min/1.73 m2 以上 90 mL/min/1.73 m2 未満
中等度腎機能低下:30 mL/min/1.73 m2 以上 60 mL/min/1.73 m2 未満
† 日本人用 GFR 推算式:eGFR(mL/min/1.73 m2)=194×血清クレアチニン−1 094×年齢−0 287
(女性の場合:194×血清クレアチニン−1 094×年齢−0 287×0.739)
【設定根拠】
本剤投与時の薬物動態及び薬力学の評価が可能と思われる例数として,実施可能性も考慮して設定
した。
診断及び選択・除外基準:
1.選択基準(以下の基準をすべて満たす者を対象とした)
(1) 同意取得時の年齢が 20 歳以上 80 歳未満で,治験実施計画書に定められた期間,治験実施医療
機関に入院することが可能な患者。性別は問わない
(2) スクリーニング開始以前に本人から文書による同意が得られている患者
(3) 同意取得時点で 2 型糖尿病と診断されてから 12 週(84 日)以上経過している患者
(4) スクリーニング時の空腹時血糖値が 240 mg/dL 未満の患者
(5) 食事・運動療法のみで治療されている患者。若しくはスクリーニング開始時に,以下の血糖降
下薬†を単剤かつ同一の用法・用量で 4 週(27 日)以上服用している患者で,治験担当医師が,
評価期終了まで用法・用量を変更しないことが可能と判断した患者
† スルホニルウレア剤,ボグリボース,アカルボース,ピオグリタゾン塩酸塩,メトホルミン
159

2.7.6個々の試験のまとめ
塩酸塩
(6) 治験担当医師が,スクリーニング開始から評価期終了まで,合併症に対する治療内容(食事療
法を含む)を変更しないことが可能と判断した患者(感冒等の一過性の疾患に対する治療は除
く)
(7) スクリーニング時の日本人用 GFR 推算式から推定される eGFR が 30 mL/min/1.73 m2 以上であ
り,かつ透析を行っていない患者
(8) スクリーニング時の BMI が 20.0 kg/m2 以上 35.0 kg/m2 以下の患者
2.除外基準(以下の基準のいずれかに該当する者は対象から除外した)
(1) 1 型糖尿病の患者
(2) スクリーニング開始日から遡って 12 週(84 日)以内にインスリンの投与を受けた患者
(3) 糖尿病性ケトアシドーシスの患者
(4) 副腎皮質ステロイド製剤,免疫抑制剤等の継続的投与(内服,注射,吸入)が必要とされる慢
性疾患を有する患者
(5) スクリーニング開始から評価期終了までに利尿薬の使用が必要な患者
(6) 手術前後,重症感染症又は重篤な外傷のある患者
(7) スクリーニング開始前 4 週(27 日)以内に,臨床所見や臨床検査値から急速に腎機能が低下し
たと判断された患者
(8) 腎血管閉塞性疾患,腎摘出,腎移植等,医学的に重要な腎疾患の既往歴がある患者
(9) ファンコニー症候群,間質性腎炎等の尿細管機能障害を有する患者
(10) 神経因性膀胱や前立腺肥大症等により,明らかな排尿障害を合併している患者
(11) 自覚症状を有する尿路感染症,性器感染症を有する患者
(12) スクリーニング時の AST 値及び ALT 値が基準値上限の 3 倍を超える患者,又は重篤な肝疾患
を合併している患者
(13) 重篤な消化管障害を合併している患者,又は重篤な消化管障害の手術歴を有する患者
(14) スクリーニング開始日から遡って 12 週(84 日)以内に,脳血管発作,不安定狭心症,心筋梗
塞,血管形成術,重篤な心疾患(NYHA Class III–IV)の既往歴がある患者,又は本剤の安全性
の評価に影響を与えると治験担当医師が判断するような心・脳血管疾患を合併している患者
(15) 血圧のコントロールが不良の患者(スクリーニング時の 5 分間安静後の臥位の収縮期血圧が
170 mmHg を超える,又は拡張期血圧が 95 mmHg を超える患者)
(16) 悪性腫瘍を合併している患者
(17) 不安定な精神医学的疾患を有する患者
(18) 薬物中毒,又はアルコール依存症の患者
(19) 試験期間中に適切な避妊方法を施行できない男性又は閉経前の女性。妊娠を希望している女性,
妊娠している女性,又は授乳中の女性
(20) スクリーニング時の妊娠検査(尿)で陽性を示した閉経前の女性
(21) 過去に本剤やその類薬(SGLT 阻害作用を有する治験薬)に対してアレルギー反応を示した患
者
(22) 過去に本剤の投与(プラセボを除く)を受けた患者
(23) 文書同意取得日から遡って 12 週(84 日)以内に,他の医療用医薬品・医療機器の治験又は製
造販売後臨床試験に参加した患者,又は現在参加している患者
(24) 本試験で規定した来院や服薬ルール等,被験者が遵守すべき事項をひとつでも遵守できない患
者,又はこれに同意しない患者
(25) スクリーニング検査前 90 日以内に 400 mL 以上の全血採血,30 日以内に 200 mL 以上の全血採
血又は 14 日以内に成分献血を実施した患者
(26) その他,治験担当医師が,試験対象として不適当と判断した患者
160

2.7.6個々の試験のまとめ
治験薬,投与量及び投与方法:
1.被験薬及びロット番号
ASP1941 錠 50 mg:1 錠中に ASP1941 として 50 mg を含有するフィルムコート錠
ロット番号:08078G
2.投与量及び投与方法
本剤 50 mg を朝食前に水 150 mL とともに単回経口投与した。
【設定根拠】
(1) 投与量の設定根拠
第 II 相用量設定試験[CL-0103]で,日本人 2 型糖尿病患者に本剤 12.5,25,50 又は 100 mg を 12
週間投与したとき,100 mg 群まで安全性に大きな問題はなく,主要評価項目である HbA1c 値の変
化量は 50 mg 群と 100 mg 群でほぼ同等であったことから,50 mg/日が臨床推奨用量になると考えら
れた。本試験では,臨床推奨用量になると考えられる 50 mg を投与量として設定した。
(2) 投与期間の設定根拠
第 I 相試験[CL-0101]で,日本人健康成人男性に本剤 20,50 又は 100 mg を単回経口投与し(Day 1),
1 日間の休薬後,Day 3 から 7 日間反復経口投与したとき,Day 1 と Day 9 における薬物動態パラメ
ータ及び 1 日尿中グルコース排泄量に大きな差は認められなかった。この結果から,本剤は 50 mg
の単回投与で薬物動態,薬力学に及ぼす腎機能の影響を十分に評価することが可能と考え,単回投
与とした。
(3) 投与方法の設定根拠
日本人健康成人男性を対象とした第 I 相試験[CL-0101]において食事の影響を検討したところ,空
腹時と食後で血漿中未変化体濃度及び尿中グルコース排泄量に大きな差がみられなかったことか
ら,空腹時,朝食前,朝食後のいずれでも投与が可能であると考えられた。本試験では,被験者ご
との投与時期を統一するため,朝食前投与とした。
評価期間:
治験薬投与期間:1 日
併用治療(薬剤及び療法):
1.併用禁止薬
(1) スクリーニング開始 4 週(27 日)前から評価期終了まで,単剤で使用する以下の薬剤†を除き,
血糖降下薬の使用を禁止した。
† スルホニルウレア剤,ボグリボース,アカルボース,ピオグリタゾン塩酸塩,メトホルミン
塩酸塩
(2) スクリーニング開始 12 週(83 日)前から評価期終了までインスリンの使用を禁止した。
(3) スクリーニング開始から評価期終了まで利尿薬の使用を禁止した。
2.条件付併用可能薬
(1) スクリーニング開始 4 週(27 日)前から評価期終了まで,単剤で使用する以下の血糖降下薬‡
の用法・用量の変更を禁止した。
‡ スルホニルウレア剤,ボグリボース,アカルボース,ピオグリタゾン塩酸塩,メトホルミン
塩酸塩
(2) スクリーニング開始から評価期終了まで,副腎皮質ステロイド製剤及び免疫抑制剤の投与(内
服,注射,吸入)を禁止した。ただし,塗布等の局所使用の場合や,スクリーニング期の一時
的な投与の場合は使用を認めた。
(3) スクリーニング開始から評価期終了まで,血糖値に直接影響を与えると考えられるブドウ糖及
びグルカゴンの投与(点滴,注射)を禁止した。ただし,スクリーニング期の一時的な投与の
161

2.7.6個々の試験のまとめ
場合は使用を認めた。
(4) スクリーニング開始から評価期終了まで,感冒等の一過性の疾患を除く合併症に対する薬剤の
用法・用量の変更及び新たな使用の開始を原則禁止した。なお,前観察期の好ましくない医療
上のできごと又は評価期の有害事象に対する治療は除く。
3.併用禁止療法
スクリーニング開始から評価期終了まで,食事療法の内容変更及び血糖管理を目的とした新たな入
院療法を禁止した。
162

2.7.6個々の試験のまとめ
評価項目,評価スケジュール及び評価基準:
1.評価スケジュール:スクリー
ニング期前観察期 評価期
後観
察期中止時 o
Day - −2 −1 1 2 3 4 - -
許容範囲(日目)a −14~–7 −2 −1 1 2 3 4 11~18
同意取得 b X
選択・除外基準確認,患者背景調査 X X
入院 Xk X X X X Xn
X外来 Xj Xj
治験薬投与 Xm
服薬状況の確認 X
食事内容の確認 X X X X X
併用薬調査
体重・身長 c X X Xl X X X X X
バイタルサイン(血圧・脈拍数)d X X Xl X X X X X
自覚症状・他覚所見 X X X X X X X X X
12 誘導心電図 X Xl X X X X
妊娠検査 e X X X
血液学的検査,血液生化学検査,
尿検査X Xl X X X X
HbA1c 値 X
血糖値 f X X Xl X X X X X
薬物動態(血漿中・尿中未変化体
濃度)g X
薬力学(尿中グルコース排泄量・
排泄速度,排尿量),飲水量 h Xl X
CLcri Xl X
好ましくない医療上のできごと
有害事象
a:治験薬投与日を Day 1 とし,その前日を Day −1 とした。
b:スクリーニング開始以前までに同意取得した。
c:身長はスクリーニング時のみ測定した。
d:バイタルサインは臥位にて測定した。また,Day 1 は複数回,以下の時点に測定した。
評価時点:治験薬投与時(午前 9 時目安)を 0 時間として,投与前(朝食前,ベースライン),投与後 2 時間,4 時間(昼食前
),10 時間(夕食前)
e:閉経前の女性のみ実施した。
f:Day 1 は複数回,以下の時点に測定した(Day 1 及び中止時以外は空腹時血糖値を測定)。
評価時点:治験薬投与時(午前 9 時目安)を 0 時間として,投与前(朝食前,ベースライン),投与後 4 時間(昼食前),10
時間(夕食前)
g:血漿中未変化体濃度:Day 1 治験薬投与時(午前 9 時目安)を 0 時間として,投与前(朝食前),投与後 0.5 時間,1 時間,1.5
時間,2 時間,3 時間,4 時間(昼食前),6 時間,8 時間,12 時間,24 時間(Day 2 朝食前),36 時間,48 時間(Day 3 朝食
前),72 時間(Day 4 朝食前)
尿中未変化体濃度:Day 1 治験薬投与時(午前 9 時目安)を 0 時間として,投与後 0~4 時間(昼食前),4~10 時間(夕食前
),10~24 時間(Day 2 朝食前),24~36 時間,36~48 時間(Day 3 朝食前),48~72 時間(Day 4 朝食前)。
h:前観察期:Day −1 蓄尿開始時刻(午前 9 時目安)を 0 時間として,蓄尿開始後 0~4 時間(昼食前),4~10 時間(夕食前),
10~24 時間(Day 1 投与前)
評価期:Day 1 治験薬投与時(午前 9 時目安)を 0 時間として,投与後 0~4 時間(昼食前),4~10 時間(夕食前),10~24
時間(Day 2 朝食前),24~36 時間,36~48 時間(Day 3 朝食前),48~72 時間(Day 4 朝食前)
i:CLcr(実測クレアチニンクリアランス):24 時間蓄尿データ及び蓄尿終了時(24 時間後)の血清クレアチニンを利用した。
j:朝食を摂らず,また血糖降下薬を服用せずに午前中に来院した。
k:午後入院した。
l:ベースラインとした。
m:すべてのベースラインの検査終了後(午前 9 時目安)に投与した。
n:すべての検査・観察を実施した後に退院した。
o:治験薬投与後の中止例について実施することとした。
163

2.7.6個々の試験のまとめ
2.薬物動態|薬力学:
(1)薬物動態
「1.評価スケジュール」に示した時期に採血及び蓄尿を行い,血漿中及び尿中の未変化体濃度を測
定した。血漿中及び尿中未変化体濃度から,以下の薬物動態パラメータをノンコンパートメント法
を用いて算出した。
[血漿中パラメータ]
・最高血漿中濃度(Cmax)
・最高血漿中濃度到達時間(tmax)
・時間 0 から定量可能最終時点までの血漿中濃度‐時間曲線下面積(AUClast)
・時間 0 から無限時間まで外挿した血漿中濃度‐時間曲線下面積(AUCinf)
・時間 0 から治験薬投与後 24 時間までの血漿中濃度‐時間曲線下面積(AUC24h)
・消失速度定数(kel)
・消失半減期(t1/2)
・経口クリアランス(CL/F)
・ラグタイム(tlag)
・みかけの分布容積(Vz/F)
・無限時間まで外挿した平均滞留時間(MRTinf)
[尿中パラメータ]
・尿中排泄量(Ae)
・時間 0 から治験薬投与後 24 時間までの尿中排泄量(Ae24h)
・時間 0 から定量可能最終時点までの尿中排泄量(Aelast)
・尿中排泄率(Ae%)
・時間 0 から定量可能最終時点までの尿中排泄率(Aelast%)
・腎クリアランス(CLr)
(2)薬力学
「1.評価スケジュール」に示した時期に採血,蓄尿及び測定を行い,以下の薬力学パラメータを算
出した。
・尿中グルコース排泄量
・尿中グルコース排泄速度
・排尿量
・体重
・血糖値
3.安全性:
・有害事象
・臨床検査(血液学的検査,血液生化学検査,尿検査)
・バイタルサイン(臥位血圧,臥位脈拍数)
・12 誘導心電図
有害事象は,治験薬投与から後観察期終了までに新たに発現した事象とした。
有害事象と治験薬との関連性は以下の基準により判定し,「関連あるかもしれない」又は「多分(お
そらく)関連あり」のいずれかに該当したものを,「治験薬との関連性が否定できない有害事象(副
作用)」と定義した。
164

2.7.6個々の試験のまとめ
治験薬との関連性 関連性判定基準
否定できる 有害事象発現と薬剤投与との間に時間的関係から因果関係がありそうにないと思われる
場合や,他の薬剤や合併症・基礎疾患等により説明ができる場合
関連あるかもしれ
ない
有害事象発現と薬剤投与との間に時間的に妥当な相関関係があるが,要因として合併症・
基礎疾患や他の薬剤による説明もできる場合。投与中止に関する情報が不明確な場合
多分(おそらく)
関連あり
有害事象発現と薬剤投与との間に時間的に妥当な相関関係がある場合,合併症・基礎疾患
や他の薬剤では説明ができない,又はそれらによる可能性が考えにくい場合,再投与によ
り再発又は投与中止により消失又は軽快を示す場合
有害事象の程度は,平成 4 年 6 月 29 日薬安第 80 号添付「医薬品等の副作用の重篤度分類基準につ
いて」のグレードを参考に判定した。この基準に規定されていない項目については,以下に従って
判定した。
·軽度:日常の活動に支障がないもの
·中等度:日常の活動に支障があるもの
·重度:日常の活動が不可能になるもの
4.その他:
「1.評価スケジュール」に示した時期に測定を行い,以下の項目を評価した。
・CLcr
下記計算式にて実測 CLcr を算出した。
実測 CLcr(mL/min)=尿中クレアチニン(mg/dL)×排尿量(mL)/[血清クレアチニン(mg/dL)
×1440(min)]
体表面積による補正[実測 CLcr×1.73/体表面積(m2)]も行った。
・飲水量
・eGFR
統計解析:
1.解析対象集団:
(1) 安全性解析対象集団(SAF)
治験薬を服用した被験者の集団を SAF とした。
(2) 薬物動態解析対象集団(PKAS)
治験薬を服用し,血漿中又は尿中未変化体濃度が 1 時点以上測定された被験者の集団を PKAS とし
た。
(3) 薬力学解析対象集団(PDAS)
治験薬を服用し,治験薬服用後の尿中グルコース排泄量が評価可能な被験者の集団を PDAS とした。
2.被験者背景及びその他の基準値:
SAF,PKAS,PDAS を解析対象とした。腎機能低下の重症度分類別に,計数値項目では度数集計を
行い,計量値項目では要約統計量を算出した。
3.薬物動態:
薬物動態の解析では,PKAS を解析対象とした。
(1) 血漿中パラメータ
血漿中パラメータの要約統計量を腎機能低下の重症度分類別に算出した。
(2) 尿中パラメータ
尿中パラメータの要約統計量を腎機能低下の重症度分類別に算出した。
(3) 腎機能低下を伴う被験者と正常な腎機能を有する被験者の比較
Cmax及び AUCinfを主要パラメータ,t1/2,AUClast,CL/F 及び CLr を副次パラメータとして,腎機能
低下を伴う被験者と正常な腎機能を有する被験者の比較を行った。
165

2.7.6個々の試験のまとめ
Cmax,AUCinf,t1/2,AUClast,CL/F 及び CLr について,正常な腎機能を有する被験者と軽度又は中等
度の腎機能低下を伴う被験者の最小二乗平均の差及びその 90%信頼区間を真数に逆変換し,正常な
腎機能を有する被験者に対する軽度又は中等度の腎機能低下を伴う被験者の幾何平均比及びその
90%信頼区間を算出した。
また,体重で補正した薬物動態パラメータ(Cmax×体重,AUCinf×体重,CL/F/体重)も同様に,正常
な腎機能を有する被験者に対する軽度又は中等度の腎機能低下を伴う被験者の幾何平均比及びその
90%信頼区間を算出した。
(4) eGFR と CL/F 又は CLr との相関
CL/F と CLr について,スクリーニング時の eGFR と CL/F,及びスクリーニング時の eGFR と CLr
の相関を示す図をそれぞれ作成し,視覚的に評価した。
4.薬力学:
薬力学の解析では,PDAS を解析対象とした。
各薬力学評価項目の要約統計量を評価時点ごとに算出した。また,ベースラインからの変化量につ
いても要約統計量を算出した。スクリーニング時の eGFR と尿中グルコース排泄量のベースライン
(Day −1)からの変化量との相関を示す図を作成した。また,スクリーニング時の eGFR と血糖値
の投与後 24,48,72 時間のベースライン(投与前)からの変化量との相関を示す図も作成した。
5.安全性:
安全性の解析では,SAF を解析対象とした。
(1) 有害事象
有害事象及び副作用の発現率を腎機能低下の重症度分類別に算出した。また,低血糖に関連する事
象を注目すべき有害事象として集計した。
(2) 臨床検査値
計量値項目は評価時点ごとの要約統計量を腎機能低下の重症度分類別に算出した。計数値項目はベ
ースライン(治験薬投与前)と各評価時点(投与後 24,72 時間,及び後観察)の測定値のクロス表
を腎機能低下の重症度分類別に作成した。
(3) バイタルサイン
評価時点ごとの測定値の要約統計量を腎機能低下の重症度分類別に算出した。また,ベースライン
(治験薬投与前)からの変化量を集計した。
(4) 12 誘導心電図
心電図異常所見についてベースライン(治験薬投与前)と各評価時点(投与後 24,72 時間,及び後
観察)の判定結果のクロス表を腎機能低下の重症度分類別に作成した。
報告書の日付: 年 月 日
2.7.6.8.2 被験者の内訳及び解析対象集団
2.7.6.8.2.1 被験者の内訳
被験者の内訳を表 2.7.6.8-1 に示した。
計 25 例の 2 型糖尿病患者が登録された。その内訳は,正常な腎機能を有する被験者が 8 例,
軽度の腎機能低下を伴う被験者が 9 例,中等度の腎機能低下を伴う被験者が 8 例であった。全 25
例が本剤 50 mg の単回経口投与を受け,本試験を完了した。
166

2.7.6個々の試験のまとめ
表 2.7.6.8-1 被験者の内訳
腎機能低下の重症度計
正常 軽度 中等度
登録例 8 9 8 25
治験薬投与例 8 9 8 25
完了例 8 9 8 25
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.1.1.1
2.7.6.8.2.2 解析対象集団
25 例全例を SAF,PKAS 及び PDAS の解析対象例とした。
2.7.6.8.3 人口統計学的及び他の基準値の特性
被験者背景を表 2.7.6.8-2 に示した。
eGFR の平均値は,正常な腎機能を有する被験者で 111.00 mL/min/1.73 m2,軽度腎機能低下を伴
う被験者で 71.92 mL/min/1.73 m2,中等度腎機能低下を伴う被験者で 46.80 mL/min/1.73 m2 であっ
た。HbA1c 値の平均値は,正常な腎機能を有する被験者(8.28%)では,腎機能低下を伴う被験
者(軽度 6.57%,中等度 6.74%)と比べて高かった。2 型糖尿病の平均罹病期間は,正常な腎機能
を有する被験者(141.5 カ月)では,腎機能低下を伴う被験者(軽度 87.4 カ月,中等度 91.0 カ月)
と比較して長かった。平均年齢は,腎機能の低下に伴い高くなる傾向がみられた。
167

2.7.6個々の試験のまとめ
表 2.7.6.8-2 被験者背景(PKAS)
腎機能低下の重症度計
正常 軽度 中等度
N=8 N=9 N=8 N=25
年齢[同意取得時](歳) 平均値(標準偏差) 54.5(11.71) 60.8(10.69) 64.1(9.69) 59.8(11.02)
最小値 – 最大値 38 – 72 45 – 74 47 – 75 38 – 75
65 歳未満 n(%) 6(75.0) 4(44.4) 4(50.0) 14(56.0)
65 歳以上 n(%) 2(25.0) 5(55.6) 4(50.0) 11(44.0)
性別 n(%) 男性 5(62.5) 7(77.8) 6(75.0) 18(72.0)
女性 3(37.5) 2(22.2) 2(25.0) 7(28.0)
身長[スクリーニング
時](cm)
平均値(標準偏差) 164.06(9.356) 162.78(10.353) 162.23(7.062) 163.01(8.741)
最小値 – 最大値 150.3 – 181.0 145.9 – 180.4 153.3 – 174.0 145.9 – 181.0
体重[スクリーニング
時](kg)
平均値(標準偏差) 73.84(10.259) 66.98(13.154) 63.50(10.876) 68.06(11.890)
最小値 – 最大値 51.6 – 85.8 45.6 – 80.7 50.2 – 81.3 45.6 – 85.8
eGFR[スクリーニング
時](mL/min/1.73 m2)
平均値(標準偏差) 111.00(18.901) 71.92(9.538) 46.80(9.662) 76.39(29.332)
最小値 – 最大値 93.6 – 142.6 61.2 – 86.7 31.8 – 57.4 31.8 – 142.6
BMI[スクリーニング
時](kg/m2)
平均値(標準偏差) 27.31(2.126) 25.14(3.802) 24.01(2.853) 25.48(3.224)
最小値 – 最大値 22.8 – 29.7 20.0 – 31.2 21.4 – 29.4 20.0 – 31.2
合併症 n(%) なし 0 0 0 0
あり 8(100.0) 9(100.0) 8(100.0) 25(100.0)
飲酒歴 n(%) なし 3(37.5) 2(22.2) 4(50.0) 9(36.0)
過去にあり 0 2(22.2) 1(12.5) 3(12.0)
現在あり 5(62.5) 5(55.6) 3(37.5) 13(52.0)
飲酒量 a n(%) なし 3(37.5) 4(44.4) 5(62.5) 12(48.0)
LEVEL 1 2(25.0) 2(22.2) 2(25.0) 6(24.0)LEVEL 2 3(37.5) 3(33.3) 1(12.5) 7(28.0)LEVEL 3 0 0 0 0
罹病期間[同意取得時]
(月)
平均値(標準偏差) 141.5(105.84) 87.4(55.39) 91.0(68.24) 105.9(79.20)
最小値 – 最大値 50 – 375 35 – 207 25 – 245 25 – 375
HbA1c 値[スクリーニ
ング時](%)
平均値(標準偏差) 8.28(2.445) 6.57(0.487) 6.74(1.444) 7.17(1.743)
最小値 – 最大値 5.5 – 13.5 6.0 – 7.4 5.4 – 9.7 5.4 – 13.5
a:飲酒量は以下の 3 レベルに分類した。
LEVEL 1=ビール中ビン 1 本未満/清酒 1 合未満/ウイスキー・ブランデー60 mL 未満/焼酎(35 度)90 mL 未満
/ワイン 240 mL 未満
LEVEL 2=ビール中ビン 1 本以上 3 本未満/清酒 1 合以上 3 合未満/ウイスキー・ブランデー60 mL 以上 180 mL
未満/焼酎(35 度)90 mL 以上 270 mL 未満/ワイン 240 mL 以上 720 mL 未満
LEVEL 3=ビール中ビン 3 本以上/清酒 3 合以上/ウイスキー・ブランデー180 mL 以上/焼酎(35 度)270 mL 以
上/ワイン 720 mL 以上
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.1.2.1-2,Table 12.1.2.2.1-2 及び Table 12.1.2.2.2-2
2.7.6.8.4 治験薬の曝露
全 25 例が Day 1 に本剤 50 mg の経口単回投与を受けた。
168

2.7.6個々の試験のまとめ
2.7.6.8.5 薬物動態|薬力学
2.7.6.8.5.1 薬物動態の結果
(1) 血漿中未変化体濃度
血漿中未変化体濃度の平均値の推移を図 2.7.6.8-1 に,未変化体の血漿中薬物動態パラメータの
要約統計量を表 2.7.6.8-3 に示した。
本剤は速やかに吸収された。投与後約 1 時間で Cmaxに到達し,血漿中未変化体濃度はいずれの
群でも二相性に消失した。
Cmax及び AUCinfの平均値はそれぞれ,正常な腎機能を有する被験者で 1044.74 ng/mL 及び
4821.35 ng•h/mL,軽度腎機能低下を伴う被験者で 1088.77 ng/mL 及び 4482.28 ng•h/mL,中等度腎
機能低下を伴う被験者で 1160.97 ng/mL 及び 5947.60 ng•h/mL であった。
t1/2 の平均値は 14.97~17.08 時間,CL/F の平均値は 9.64~12.16 L/h であった。
図 2.7.6.8-1 血漿中未変化体濃度の平均値(標準偏差)の推移(PKAS)
Source:CL-0073 総括報告書(5.3.3.3-2)Figure 12.2.2-2
血漿中未変化体濃度(
ng
/mL)
時間(h)
正常な腎機能を有する被験者
軽度の腎機能低下を伴う被験者
中等度の腎機能低下を伴う被験者
169

2.7.6個々の試験のまとめ
表 2.7.6.8-3 未変化体の血漿中薬物動態パラメータの要約統計量(PKAS)
パラメータ
腎機能
低下の
重症度
N 平均値標準
偏差最小値 最大値 中央値
幾何
平均値Na
変動
係数(%)
Cmax
(ng/mL)
正常 8 1044.74 347.55 265.12 1329.52 1132.44 954.47 8 33.27
軽度 9 1088.77 223.40 829.05 1581.45 1057.54 1070.48 9 20.52
中等度 8 1160.97 357.94 682.82 1845.17 1127.08 1114.89 8 30.83
AUCinf
(ng•h/mL)
正常 8 4821.35 1558.49 2276.30 7279.98 4921.07 4570.89 8 32.32
軽度 9 4482.28 1587.80 3089.74 8079.33 3816.66 4276.95 9 35.42
中等度 8 5947.60 2461.76 3117.51 10411.06 5105.65 5543.07 8 41.39
tmax
(h)
正常 8 1.43 1.86 0.50 6.00 0.98 0.96 8 129.85
軽度 9 0.84 0.26 0.50 1.08 1.00 0.80 9 30.47
中等度 8 0.95 0.32 0.50 1.50 1.00 0.90 8 33.30
t1/2
(h)
正常 8 14.97 4.58 8.00 23.07 14.62 14.34 8 30.60
軽度 9 15.37 8.46 7.42 34.29 14.19 13.70 9 55.01
中等度 8 17.08 7.79 9.76 31.72 14.66 15.72 8 45.63
CL/F(L/h)
正常 8 11.65 4.85 6.87 21.97 10.21 10.94 8 41.61
軽度 9 12.16 3.35 6.19 16.18 13.10 11.69 9 27.52
中等度 8 9.64 3.63 4.80 16.04 9.82 9.02 8 37.71
a:幾何平均値を算出した被験者数
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.2.3-1,Table 12.2.3-2 及び Table 12.2.3-3
(2) 尿中未変化体濃度
未変化体の尿中薬物動態パラメータの要約統計量を表 2.7.6.8-4 に示した。
Aelast%の平均値は 0.84%~1.05%,CLr の平均値は 0.07~0.12 L/h であり,いずれの群でも著し
く低かった。
表 2.7.6.8-4 未変化体の尿中薬物動態パラメータの要約統計量(PKAS)
パラメータ
腎機能
低下の
重症度
N 平均値標準
偏差最小値 最大値 中央値
幾何
平均値Na
変動
係数(%)
Aelast
(μg)
正常 8 525.23 126.56 368.84 750.66 545.37 512.12 8 24.10
軽度 9 428.15 74.34 348.37 527.36 408.70 422.56 9 17.36
中等度 8 418.44 201.86 231.13 878.94 377.54 386.07 8 48.24
Aelast%(% dose)
正常 8 1.05 0.25 0.74 1.50 1.09 1.02 8 24.10
軽度 9 0.86 0.15 0.70 1.05 0.82 0.85 9 17.36
中等度 8 0.84 0.40 0.46 1.76 0.76 0.77 8 48.24
CLr
(L/h)
正常 8 0.12 0.03 0.07 0.17 0.11 0.11 8 27.24
軽度 9 0.10 0.02 0.06 0.12 0.11 0.10 9 17.96
中等度 8 0.07 0.02 0.05 0.11 0.08 0.07 8 28.70
a:幾何平均値を算出した被験者数
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.2.4
(3) 腎機能の低下を伴う被験者と正常な腎機能を有する被験者の比較
本剤の薬物動態パラメータに及ぼす腎機能低下の影響を表 2.7.6.8-5 に示した。
Cmax及び AUCinfの幾何平均比は,軽度の腎機能低下を伴う被験者では正常な腎機能を有する被
験者に比べ Cmaxは約 12%高く,AUCinfは約 6%低かった。また,中等度の腎機能低下を伴う被験
170

2.7.6個々の試験のまとめ
者では正常な腎機能を有する被験者に比べ Cmax及び AUCinfがそれぞれ約 17%及び約 21%高かっ
た。幾何平均比の 90%信頼区間はいずれも 1 を含んでおり,有意な差は認められなかった。
t1/2 及び CL/F には,腎機能低下による影響は認められなかった。
体重で補正した薬物動態パラメータ(Cmax,AUCinf,CL/F)についても,上述と同様の結果で
あった。
腎機能の低下に伴い CLr に明らかな低下がみられたが,未変化体の尿中排泄量は極めてわずか
であり,CLr の低下は本剤の曝露量増加とは関連しないと考えられた。
表 2.7.6.8-5 ASP1941 の薬物動態パラメータに及ぼす腎機能低下の影響(PKAS)
パラメータ 比較 幾何平均比90%信頼区間
下限 上限
Cmax軽度/正常 1.122 0.827 1.522
中等度/正常 1.168 0.853 1.599
AUCinf軽度/正常 0.936 0.694 1.261
中等度/正常 1.213 0.892 1.649
t1/2軽度/正常 0.955 0.671 1.360
中等度/正常 1.096 0.762 1.576
CL/F軽度/正常 1.069 0.793 1.441
中等度/正常 0.825 0.606 1.121
AUClast軽度/正常 0.932 0.693 1.252
中等度/正常 1.207 0.891 1.636
CLr軽度/正常 0.885 0.712 1.099
中等度/正常 0.623 0.498 0.778
腎機能低下の 2 つの重症度間(正常な腎機能を有する被験者と軽度又は中等度の腎機能低下を伴う被験者)で,
対数変換した薬物動態パラメータの最小二乗平均の差及びその 90%信頼区間を真数に逆変換し,幾何平均比及び
その 90%信頼区間を算出した。
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.2.5 及び Table 12.2.6
(4) eGFR と未変化体の CL/F 又は CLr との相関
スクリーニング時の eGFR と CL/F の相関を図 2.7.6.8-2 に,スクリーニング時の eGFR と CLr
の相関を図 2.7.6.8-3 に示した。
CL/F と eGFR には,明らかな相関が認められなかった(r2=0.09)。一方,スクリーニング時の
eGFR が高いほど,CLr は上昇する傾向を示した(r2=0.53)。
171

2.7.6個々の試験のまとめ
図 2.7.6.8-2 スクリーニング時の eGFR と CL/F の相関(PKAS)
Source:CL-0073 総括報告書(5.3.3.3-2)Figure 12.2.3
図 2.7.6.8-3 スクリーニング時の eGFR と CLrの相関(PKAS)
Source:CL-0073 総括報告書(5.3.3.3-2)Figure 12.2.4
172

2.7.6個々の試験のまとめ
2.7.6.8.5.2 薬力学の結果
(1) 尿中グルコース排泄量
1 日尿中グルコース排泄量の要約統計量を表 2.7.6.8-6 に,ベースラインからの変化量の要約統
計量を表 2.7.6.8-7 に示した。
ベースライン(Day −1)の 1 日尿中グルコース排泄量は,正常な腎機能を有する被験者より腎
機能低下を伴う被験者で少なかった。本剤の単回投与後,Day 1 の 1 日尿中グルコース排泄量の
平均値はいずれの群でも増加した。Day 1 のベースラインからの平均変化量は,正常な腎機能を
有する被験者で 71106.5 mg,軽度の腎機能低下を伴う被験者で 61196.8 mg,中等度の腎機能低下
を伴う被験者で 38460.0 mg であった。ベースラインからの平均変化量は,正常な腎機能を有する
被験者と軽度の腎機能低下を伴う被験者では同程度であったが,中等度の腎機能低下を伴う被験
者では正常な腎機能を有する被験者より小さかった。
表 2.7.6.8-6 1 日尿中グルコース排泄量(mg)の要約統計量(PDAS)
腎機能低下の重症度 評価時点 N 平均値 標準偏差 最小値 最大値 中央値第 1
四分位点
第 3
四分位点
正常
Day −1 (0-24 h) 8 56744.7 82414.26 29 231399 6782.4 5556.3 98926.3
Day 1 (0-24 h) 8 127851.1 85489.06 33032 298855 110534.0 63908.5 171018.5
Day 2 (24-48 h) 8 75213.3 63471.34 15913 220257 56021.0 39285.3 87462.0
Day 3 (48-72 h) 8 50359.3 58431.74 1532 184457 27319.4 18204.7 62918.7
軽度
Day −1 (0-24 h) 9 6872.6 7008.34 88 18216 6114.7 582.8 10440.1
Day 1 (0-24 h) 9 68069.4 29170.20 22738 123752 63411.0 57617.0 82624.5
Day 2 (24-48 h) 9 28801.3 20358.07 2788 67396 29700.0 11606.0 40239.0
Day 3 (48-72 h) 9 13810.3 14022.20 198 44997 11246.6 2044.4 20410.2
中等度
Day −1 (0-24 h) 8 4819.8 11252.30 49 32531 646.5 226.2 2116.4
Day 1 (0-24 h) 8 43279.8 16950.89 25418 71616 40336.0 29093.0 55173.0
Day 2 (24-48 h) 8 20150.4 17120.58 2495 54334 14262.4 8998.0 28927.1
Day 3 (48-72 h) 8 7708.7 7591.59 98 20428 6414.0 584.8 13573.4
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.3.3.1
表 2.7.6.8-7 1 日尿中グルコース排泄量(mg)のベースラインからの変化量の要約統計量
(PDAS)
腎機能低下の重症度 評価時点 N 平均値 標準偏差 最小値 最大値 中央値第 1
四分位点
第 3
四分位点
正常
Day 1 (0-24 h) 8 71106.5 25011.85 33003 117490 68961.8 58281.0 81936.5
Day 2 (24-48 h) 8 18468.6 27127.64 −18277 51164 23521.5 −7896.5 41806.3
Day 3 (48-72 h) 8 −6385.4 28745.85 −46942 22265 4864.1 −36007.6 17940.4
軽度
Day 1 (0-24 h) 9 61196.8 25186.79 22650 105536 62828.2 41971.1 72184.4
Day 2 (24-48 h) 9 21928.7 15542.01 2700 49180 17321.7 11496.6 30010.3
Day 3 (48-72 h) 9 6937.6 8765.72 89 26781 2832.2 1956.7 9483.9
中等度
Day 1 (0-24 h) 8 38460.0 12111.60 24763 58332 35364.5 28749.9 48178.3
Day 2 (24-48 h) 8 15330.7 10153.07 2446 35348 13573.6 8650.8 20201.1
Day 3 (48-72 h) 8 2888.9 8162.55 −12104 15030 2539.5 −236.3 7789.1
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.3.3.2
173

2.7.6個々の試験のまとめ
スクリーニング時の eGFR と Day 1 の尿中グルコース排泄量のベースラインからの変化量を図
2.7.6.8-4 に示した。
スクリーニング時の eGFR が高いほど,Day 1 の尿中グルコース排泄量のベースラインからの
変化量は増加する傾向がみられた。
図 2.7.6.8-4 eGFR と Day 1 の尿中グルコース排泄量のベースラインからの変化量(PDAS)
Source:CL-0073 総括報告書(5.3.3.3-2)Figure 12.3.3.4
(2) 尿中グルコース排泄速度
尿中グルコース排泄速度の要約統計量を表 2.7.6.8-8 に示した。
ベースライン(Day −1)の尿中グルコース排泄速度は,正常な腎機能を有する被験者より腎機
能低下を伴う被験者で遅かった。Day 1 の尿中グルコース排泄速度の平均値は,いずれの群もす
べての評価時点でベースラインと比較して増加した。尿中グルコース排泄速度のベースラインか
らの平均変化量は,軽度の腎機能低下を伴う被験者では正常な腎機能を有する被験者と同程度で
あったが,中等度の腎機能低下を伴う被験者では正常な腎機能を有する被験者と比較して小さか
った。
Da
y1
(0-2
4 h
)の尿中グルコース排泄量のベ
ースラインからの変化量
eGFR[スクリーニング時]
174

2.7.6個々の試験のまとめ
表 2.7.6.8-8 尿中グルコース排泄速度(mg/h)の要約統計量(PDAS)
腎機能低下の重症度 評価時点 N 平均値 標準偏差 最小値 最大値 中央値第 1
四分位点
第 3
四分位点
正常
Day −1: 0-4 h 8 3784.21 4890.968 3.7 12138.8 699.00 534.24 7832.38
Day −1: 4-10 h 8 3569.80 4888.673 1.7 13161.8 622.30 221.69 6853.50
Day −1: 10-24 h 8 1443.92 2546.556 0.4 7401.9 130.92 41.19 1902.45
Day 1: 0-4 h 8 7489.08 5320.579 1676.9 17674.8 5783.68 4044.71 10452.09
Day 1: 4-10 h 8 7470.88 4880.593 3250.0 17834.7 5841.29 3870.08 9629.83
Day 1: 10-24 h 8 3788.29 2661.634 478.9 8695.5 3921.12 1588.96 5055.85
Day 2: 24-36 h 8 4237.22 2918.498 1076.7 10395.8 3251.18 2464.19 5497.31
Day 2: 36-48 h 8 2012.31 2469.530 237.7 7959.0 1363.47 801.06 1786.34
Day 3: 48-72 h 8 2100.48 2435.276 64.1 7685.7 1137.29 759.45 2630.29
軽度
Day −1: 0-4 h 9 915.54 912.082 5.3 2378.5 849.71 45.05 1405.63
Day −1: 4-10 h 9 289.06 355.784 3.3 979.2 59.79 9.00 532.58
Day −1: 10-24 h 9 108.39 123.797 1.7 316.8 35.22 25.05 203.14
Day 1: 0-4 h 9 4790.34 1658.422 1969.2 7558.5 5087.34 4404.50 5638.24
Day 1: 4-10 h 9 3677.69 1504.784 1459.4 6362.3 3471.78 2835.20 4371.71
Day 1: 10-24 h 9 1894.14 1059.066 420.0 3946.0 1637.36 1251.43 2546.06
Day 2: 24-36 h 9 1742.83 1197.656 185.1 3963.3 1642.50 711.65 2521.00
Day 2: 36-48 h 9 653.26 517.817 17.7 1653.1 819.47 247.83 893.75
Day 3: 48-72 h 9 576.27 584.331 8.2 1874.9 473.54 85.24 850.43
中等度
Day −1: 0-4 h 8 203.90 320.612 1.1 882.0 70.11 3.22 300.73
Day −1: 4-10 h 8 422.20 1130.578 2.6 3219.3 11.07 3.18 63.57
Day −1: 10-24 h 8 104.89 238.324 1.6 690.3 14.03 2.99 56.57
Day 1: 0-4 h 8 2839.00 1125.285 1347.2 4421.9 2594.96 2006.12 3870.37
Day 1: 4-10 h 8 2361.73 1066.709 1362.3 4625.0 2092.65 1630.30 2730.32
Day 1: 10-24 h 8 1258.90 511.817 572.2 1909.3 1198.71 855.12 1741.02
Day 2: 24-36 h 8 1173.24 747.707 183.6 2399.8 1026.15 645.94 1729.13
Day 2: 36-48 h 8 506.57 721.293 24.0 2128.0 165.17 103.67 681.47
Day 3: 48-72 h 8 320.48 315.972 4.1 851.2 266.87 24.38 563.04
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.3.1.1
(3) 排尿量
1 日排尿量の要約統計量を表 2.7.6.8-9 に,ベースラインからの変化量を表 2.7.6.8-10 に示した。
本剤の単回投与後,Day 1 の 1 日排尿量の平均値は,いずれの群もベースライン(Day −1)と
比較して増加した。Day 1 のベースラインからの平均変化量は各群で同程度であった。
175

2.7.6個々の試験のまとめ
表 2.7.6.8-9 1 日排尿量(mL)の要約統計量(PDAS)
腎機能低下の重症度 評価時点 N 平均値標準
偏差最小値 最大値 中央値
第 1
四分位点
第 3
四分位点
正常
Day −1 (0-24 h) 8 2677.5 1317.31 955 4595 2467.5 1772.5 3695.0
Day 1 (0-24 h) 8 3153.1 1047.87 1700 4860 3075.0 2460.0 3797.5
Day 2 (24-48 h) 8 2483.8 886.91 1135 3750 2590.0 1785.0 3117.5
Day 3 (48-72 h) 8 2126.0 802.59 896 3366 1931.0 1661.0 2781.0
軽度
Day −1 (0-24 h) 9 1915.9 794.44 945 3033 1675.0 1355.0 2345.0
Day 1 (0-24 h) 9 2488.9 846.36 1035 3775 2630.0 1900.0 3080.0
Day 2 (24-48 h) 9 1736.7 786.19 550 2740 1785.0 1220.0 2400.0
Day 3 (48-72 h) 9 1798.8 896.16 551 3261 1991.0 1061.0 2481.0
中等度
Day −1 (0-24 h) 8 2183.3 920.66 1345 4250 1965.0 1600.0 2370.5
Day 1 (0-24 h) 8 2629.6 817.41 1920 4265 2256.0 2077.5 3092.5
Day 2 (24-48 h) 8 2067.3 629.59 1330 3060 1985.0 1535.0 2554.0
Day 3 (48-72 h) 8 1867.1 504.58 1126 2751 1916.0 1526.0 2088.0
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.3.3.1
表 2.7.6.8-10 1 日排尿量(mL)のベースラインからの変化量の要約統計量(PDAS)
腎機能低下の重症度 評価時点 N 平均値 標準偏差 最小値 最大値 中央値第 1
四分位点
第 3
四分位点
正常
Day 1 (0-24 h) 8 475.6 620.34 −185 1665 300.0 12.5 850.0
Day 2 (24-48 h) 8 −193.8 846.76 −1365 1525 −332.5 −640.0 117.5
Day 3 (48-72 h) 8 −551.5 661.69 −1714 391 −521.5 −916.5 −106.5
軽度
Day 1 (0-24 h) 9 573.0 541.83 −215 1545 580.0 80.0 855.0
Day 2 (24-48 h) 9 −179.2 283.60 −610 345 −225.0 −315.0 −95.0
Day 3 (48-72 h) 9 −117.1 499.07 −1042 336 221.0 −404.0 266.0
中等度
Day 1 (0-24 h) 8 446.4 471.38 −120 1285 426.0 50.0 727.0
Day 2 (24-48 h) 8 −116.0 690.07 −1462 670 42.5 −477.5 367.0
Day 3 (48-72 h) 8 −316.1 708.68 −1499 536 −237.0 −844.5 298.5
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.3.3.2
(4) 体重
体重の要約統計量を表 2.7.6.8-11 に示した。
試験期間中,いずれの群でも体重にベースライン(投与前)からの顕著な変化は認められなか
った。
176

2.7.6個々の試験のまとめ
表 2.7.6.8-11 体重(kg)の要約統計量(PDAS)
腎機能低下の重症度 評価時点 N 平均値 標準偏差 最小値 最大値 中央値第 1
四分位点
第 3
四分位点
正常
Day −1 8 73.16 9.963 51.0 82.7 74.55 71.75 79.50
投与前 a 8 72.86 9.487 51.3 81.5 74.10 72.05 78.90
24 h 8 72.38 9.356 51.1 80.6 73.80 71.40 78.45
48 h 8 72.16 9.317 50.8 80.6 73.80 71.35 77.80
72 h 8 72.31 9.624 50.6 81.2 73.70 71.20 78.45
後観察 8 73.61 10.179 51.7 84.1 74.50 71.75 80.30
軽度
Day −1 9 67.16 12.919 46.4 81.2 71.40 58.70 77.80
投与前 a 9 66.83 12.492 46.2 80.3 71.00 58.30 77.50
24 h 9 66.20 12.282 46.6 79.8 70.40 57.90 76.70
48 h 9 66.31 12.410 46.2 79.6 70.00 58.20 76.90
72 h 9 66.18 12.339 45.8 79.2 70.80 57.80 76.70
後観察 9 66.90 12.938 45.3 81.6 70.60 59.50 78.20
中等度
Day −1 8 62.84 11.047 49.4 81.2 59.35 54.80 71.90
投与前 a 8 62.66 10.738 49.2 80.0 59.30 55.00 71.75
24 h 8 62.40 10.720 49.0 80.0 59.00 54.90 71.20
48 h 8 62.31 10.688 48.7 79.6 58.95 54.90 71.25
72 h 8 62.41 10.669 48.9 79.4 58.95 55.00 71.55
後観察 8 62.86 11.240 48.6 80.8 58.80 55.20 72.75
a:ベースライン
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.3.4.1
(5) 血糖値
血糖値の要約統計量を表 2.7.6.8-12 に,ベースラインからの変化量を表 2.7.6.8-13 に示した。
ベースライン(投与前)の血糖値は,正常な腎機能を有する被験者より腎機能低下を伴う被験
者で低かった。投与後 24 時間の血糖値の平均値はいずれの群でもベースラインと比較して低下
した。投与後 24 時間の血糖値のベースラインからの変化量の平均値は,正常な腎機能を有する
被験者で−24.9 mg/dL,軽度の腎機能低下を伴う被験者で−11.9 mg/dL,中等度の腎機能低下を伴う
被験者で−4.1 mg/dL であり,腎機能が低下するほどベースラインからの低下量は減少した。
177

2.7.6個々の試験のまとめ
表 2.7.6.8-12 血糖値(mg/dL)の要約統計量(PDAS)
腎機能低下の重症度 評価時点 N 平均値 標準偏差 最小値 最大値 中央値第 1
四分位点
第 3
四分位点
正常
Day −1a 8 169.9 63.79 90 253 168.5 113.0 226.5
投与前 a 8 159.9 57.49 102 255 143.5 109.5 208.0
4 h 8 165.0 68.07 71 277 165.0 112.0 209.0
10 h 8 143.1 52.89 91 242 135.5 95.5 175.0
24 ha 8 135.0 35.78 96 194 130.5 102.0 162.5
48 ha 8 145.3 40.28 98 205 143.5 106.5 179.5
72 ha 8 145.3 44.04 97 226 144.5 106.0 169.0
後観察 a 8 155.4 54.26 98 228 138.0 108.5 212.0
軽度
Day −1a 9 137.0 31.73 86 189 138.0 120.0 164.0
投与前 a 9 133.3 29.81 84 168 136.0 116.0 159.0
4 h 9 135.8 35.20 102 198 122.0 106.0 153.0
10 h 9 104.6 19.84 80 127 93.0 91.0 124.0
24 ha 9 121.4 22.73 80 153 127.0 109.0 137.0
48 ha 9 126.7 24.18 82 158 135.0 111.0 140.0
72 ha 9 127.4 26.76 81 169 136.0 110.0 146.0
後観察 a 9 139.2 27.15 95 176 148.0 120.0 160.0
中等度
Day −1a 8 114.0 15.78 96 143 112.0 101.0 123.5
投与前 a 8 121.5 29.35 93 188 111.5 106.0 128.0
4 h 8 139.6 81.82 89 338 114.0 100.0 131.0
10 h 8 115.5 62.30 63 264 103.5 84.5 110.5
24 ha 8 117.4 36.68 86 200 104.0 96.0 126.5
48 ha 8 117.4 24.31 96 164 106.5 101.5 131.5
72 ha 8 114.0 23.89 92 166 105.5 99.0 122.5
後観察 a 8 125.8 34.32 104 207 115.5 105.5 126.5
a:空腹時
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.3.5.1
表 2.7.6.8-13 血糖値(mg/dL)のベースラインからの変化量の要約統計量(PDAS)
腎機能低下の重症度 評価時点 N 平均値 標準偏差 最小値 最大値 中央値第 1
四分位点
第 3
四分位点
正常
24 h 8 −24.9 22.67 −61 −1 −14.5 −45.5 −8.5
48 h 8 −14.6 19.67 −50 4 −4.5 −28.5 −2.5
72 h 8 −14.6 18.47 −42 6 −8.0 −32.5 0.0
後観察 8 −4.5 11.74 −28 8 −2.0 −10.5 4.5
軽度
24 h 9 −11.9 11.10 −31 −1 −7.0 −22.0 −4.0
48 h 9 −6.7 12.78 −33 5 −2.0 −7.0 3.0
72 h 9 −5.9 13.18 −31 14 −5.0 −13.0 0.0
後観察 9 5.9 8.40 −8 17 6.0 2.0 12.0
中等度
24 h 8 −4.1 10.44 −24 12 −5.5 −7.0 2.0
48 h 8 −4.1 11.10 −24 15 −5.5 −8.0 1.5
72 h 8 −7.5 8.12 −22 1 −5.0 −13.0 −1.5
後観察 8 4.3 13.02 −7 30 −1.0 −3.0 9.5
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.3.5.2
178

2.7.6個々の試験のまとめ
スクリーニング時の eGFR と投与後 24 時間の血糖値のベースラインからの変化量を図 2.7.6.8-5
に示した。
スクリーニング時の eGFR が高いほど,投与後 24 時間の血糖値のベースラインからの低下量は
増加する傾向がみられた。
図 2.7.6.8-5 eGFR と投与後 24 時間の血糖値のベースラインからの変化量(PDAS)
Source:CL-0073 総括報告書(5.3.3.3-2)Figure 12.3.5.4
2.7.6.8.5.3 その他の評価項目の結果
(1) eGFR
eGFR の要約統計量を表 2.7.6.8-14 に示した。
試験期間中,いずれの群も eGFR にベースライン(投与前)からの顕著な変化は認められなか
った。
投与後
24時間の血糖値のベースラインからの変化量
eGFR[スクリーニング時]
179

2.7.6個々の試験のまとめ
表 2.7.6.8-14 eGFR(mL/min/1.73 m2)の要約統計量(PDAS)
腎機能低下の重症度 評価時点 N 平均値 標準偏差 最小値 最大値 中央値第 1
四分位点
第 3
四分位点
正常
投与前 8 111.54 19.559 92.6 152.4 106.39 97.49 119.75
24 h 8 100.12 19.716 80.3 142.7 97.74 86.06 105.19
72 h 8 106.07 17.192 89.7 139.7 100.47 95.39 113.75
後観察 8 111.70 24.298 91.6 167.7 106.79 96.12 114.26
軽度
投与前 9 76.88 11.830 64.1 100.4 72.89 69.94 77.07
24 h 9 70.33 11.891 58.5 90.7 65.67 62.79 73.21
72 h 9 75.76 10.319 63.4 92.5 74.17 68.19 80.17
後観察 9 76.50 10.430 64.1 92.5 71.70 67.92 84.33
中等度
投与前 8 45.84 13.119 27.4 63.4 46.66 35.55 55.76
24 h 8 41.20 9.864 25.2 52.2 44.09 33.56 48.42
72 h 8 43.93 10.459 27.1 57.3 45.49 36.68 51.37
後観察 8 44.98 8.261 29.4 55.0 45.65 40.87 51.22
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.4.5.1
(2) CLcr
CLcr の要約統計量を表 2.7.6.8-15 に示した。
いずれの群でも Day −1 と Day 3 の CLcr に顕著な差は認められなかった。
表 2.7.6.8-15 CLcr(mL/min)の要約統計量(PDAS)
腎機能低下の重症度 評価時点 N 平均値 標準偏差 最小値 最大値 中央値第 1
四分位点
第 3
四分位点
正常Day −1 8 147.278 44.8815 68.94 196.65 156.733 121.433 178.154
Day 3 8 142.144 39.5441 63.62 198.36 141.178 129.113 167.297
軽度Day −1 9 117.880 41.6129 60.42 163.01 133.864 72.576 147.937
Day 3 9 99.874 35.1647 31.87 132.86 103.952 76.181 130.721
中等度Day −1 8 67.481 21.6475 38.65 104.80 64.905 51.507 81.788
Day 3 8 65.216 20.0146 39.25 101.71 63.440 51.614 75.330
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.4.1.1
(3) 飲水量
1日飲水量の要約統計量を表 2.7.6.8-16に,ベースラインからの変化量を表 2.7.6.8-17に示した。
ベースライン(Day −1)の 1 日飲水量の平均値は,正常な腎機能を有する被験者よりも腎機能
低下を伴う被験者で少なかった。本剤投与後,Day 1 の平均 1 日飲水量は増加し,ベースライン
からの平均変化量は各群で同程度であった。
180

2.7.6個々の試験のまとめ
表 2.7.6.8-16 1 日飲水量(mL)の要約統計量(PDAS)
腎機能低下の重症度 評価時点 N 平均値 標準偏差 最小値 最大値 中央値第 1
四分位点
第 3
四分位点
正常
Day −1 (0-24 h) 8 2117.8 948.66 1050 3960 2007.5 1430.0 2528.5
Day 1 (0-24 h) 8 2366.3 731.04 1520 3480 2240.0 1835.0 2890.0
Day 2 (24-48 h) 8 1843.8 898.08 730 3640 1685.0 1325.0 2180.0
Day 3 (48-72 h) 8 1926.3 770.05 1150 3420 1755.0 1325.0 2340.0
軽度
Day −1 (0-24 h) 9 1440.6 637.33 760 2350 1490.0 790.0 1895.0
Day 1 (0-24 h) 9 1705.6 580.35 900 2820 1740.0 1400.0 1970.0
Day 2 (24-48 h) 9 1507.8 683.44 640 2960 1510.0 1150.0 1750.0
Day 3 (48-72 h) 9 1357.8 527.47 740 2350 1350.0 1000.0 1440.0
中等度
Day −1 (0-24 h) 8 1610.3 637.20 1190 3120 1400.0 1255.0 1631.0
Day 1 (0-24 h) 8 1890.0 717.00 1260 3440 1605.0 1415.0 2190.0
Day 2 (24-48 h) 8 1777.5 748.15 1180 3110 1380.0 1280.0 2305.0
Day 3 (48-72 h) 8 1670.0 865.73 870 3600 1340.0 1170.0 1935.0
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.4.4.1
表 2.7.6.8-17 1 日飲水量(mL)のベースラインからの変化量の要約統計量(PDAS)
腎機能低下の重症度 評価時点 N 平均値 標準偏差 最小値 最大値 中央値第 1
四分位点
第 3
四分位点
正常
Day 1 (0-24 h) 8 248.5 424.53 −520 920 259.0 60.0 475.0
Day 2 (24-48 h) 8 −274.0 570.31 −1240 543 −302.5 −560.0 115.0
Day 3 (48-72 h) 8 −191.5 382.47 −600 590 −255.0 −470.0 −36.0
軽度
Day 1 (0-24 h) 9 265.0 270.58 −270 610 260.0 120.0 510.0
Day 2 (24-48 h) 9 67.2 369.82 −660 610 40.0 −55.0 260.0
Day 3 (48-72 h) 9 −82.8 578.29 −1310 650 −40.0 −110.0 140.0
中等度
Day 1 (0-24 h) 8 279.8 338.74 −240 870 290.0 60.0 454.0
Day 2 (24-48 h) 8 167.3 536.39 −360 1348 80.0 −175.0 270.0
Day 3 (48-72 h) 8 59.8 327.39 −400 480 125.0 −245.0 319.0
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.4.4.2
2.7.6.8.6 安全性
2.7.6.8.6.1 有害事象
本試験で認められた有害事象の要約を表 2.7.6.8-18 に示した。
有害事象はいずれの群も 3 例(正常な腎機能を有する被験者の 37.5%,軽度の腎機能低下を伴
う被験者の 33.3%,中等度の腎機能低下を伴う被験者の 37.5%)に認められ,有害事象の発現率
は各群で同程度であった。副作用は,正常な腎機能を有する被験者に 3 例(37.5%),軽度の腎機
能低下を伴う被験者に 2 例(22.2%),中等度の腎機能低下を伴う被験者に 1 例(12.5%)認めら
れた。
181

2.7.6個々の試験のまとめ
表 2.7.6.8-18 有害事象の要約(SAF)
腎機能低下の重症度計
正常 軽度 中等度N=8 N=9 N=8 N=25
有害事象発現例数(発現率) 3(37.5%) 3(33.3%) 3(37.5%) 9(36.0%)
有害事象発現件数 5 5 4 14
重篤な有害事象発現例数(発現率) 0 0 0 0
低血糖関連の有害事象発現例数
(発現率)
0 1(11.1%) 0 1(4.0%)
副作用発現例数(発現率) 3(37.5%) 2(22.2%) 1(12.5%) 6(24.0%)
副作用発現件数 4 2 1 7
重篤な副作用発現例数(発現率) 0 0 0 0
低血糖関連の副作用発現例数
(発現率)
0 0 0 0
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.5.1.1,Table 12.5.1.2.1 及び Table 12.5.1.2.2
本試験で認められた有害事象の内訳を表 2.7.6.8-19 及び表 2.7.6.8-20 に示した。
正常な腎機能を有する被験者では,血中クレアチンホスホキナーゼ増加,血中カリウム増加,
尿中細菌検出及び湿疹が各 1 例,軽度の腎機能低下を伴う被験者では,回転性めまい,尿中細菌
検出,冷汗及び接触性皮膚炎が各 1 例,中等度の腎機能低下を伴う被験者では,鼻咽頭炎,血中
カリウム増加,糖尿病性自律神経ニューロパチー及び頻尿が各 1 例に認められた。いずれの有害
事象も軽度と判定された。腎機能低下の重症度と有害事象の発現率に明らかな関連はみられなか
った。
182

2.7.6個々の試験のまとめ
表 2.7.6.8-19 有害事象の内訳(SAF)
MedDRA/J version 12.1器官別大分類
基本語
腎機能低下の重症度計
正常 軽度 中等度N=8 N=9 N=8 N=25
すべての有害事象 3(37.5%) 3(33.3%) 3(37.5%) 9(36.0%)
耳および迷路障害 0 1(11.1%) 0 1(4.0%)
回転性めまい 0 1(11.1%) 0 1(4.0%)
感染症および寄生虫症 0 0 1(12.5%) 1(4.0%)
鼻咽頭炎 0 0 1(12.5%) 1(4.0%)
臨床検査 2(25.0%) 1(11.1%) 1(12.5%) 4(16.0%)
血中クレアチンホスホキナーゼ増加 1(12.5%) 0 0 1(4.0%)
血中カリウム増加 1(12.5%) 0 1(12.5%) 2(8.0%)
尿中細菌検出 1(12.5%) 1(11.1%) 0 2(8.0%)
神経系障害 0 0 1(12.5%) 1(4.0%)
糖尿病性自律神経ニューロパチー 0 0 1(12.5%) 1(4.0%)
腎および尿路障害 0 0 1(12.5%) 1(4.0%)
頻尿 0 0 1(12.5%) 1(4.0%)
皮膚および皮下組織障害 1(12.5%) 2(22.2%) 0 3(12.0%)
冷汗 0 1(11.1%) 0 1(4.0%)
接触性皮膚炎 0 1(11.1%) 0 1(4.0%)
湿疹 1(12.5%) 0 0 1(4.0%)
発現例数(発現率)
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.5.1.3.1
表 2.7.6.8-20 有害事象の内訳(件数)(SAF)
MedDRA/J version 12.1器官別大分類
基本語
腎機能低下の重症度計
正常 軽度 中等度
すべての有害事象 5 5 4 14
耳および迷路障害 0 1 0 1
回転性めまい 0 1 0 1
感染症および寄生虫症 0 0 1 1
鼻咽頭炎 0 0 1 1
臨床検査 4 1 1 6
血中クレアチンホスホキナーゼ増加 1 0 0 1
血中カリウム増加 1 0 1 2
尿中細菌検出 2 1 0 3
神経系障害 0 0 1 1
糖尿病性自律神経ニューロパチー 0 0 1 1
腎および尿路障害 0 0 1 1
頻尿 0 0 1 1
皮膚および皮下組織障害 1 3 0 4
冷汗 0 2 0 2
接触性皮膚炎 0 1 0 1
湿疹 1 0 0 1
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.5.1.2.1,Table 12.5.1.4.1
183

2.7.6個々の試験のまとめ
本試験で認められた副作用の内訳を表 2.7.6.8-21 及び表 2.7.6.8-22 に示した。
副作用として,正常な腎機能を有する被験者では血中カリウム増加,尿中細菌検出及び湿疹が
各 1 例,軽度の腎機能低下を伴う被験者では回転性めまい及び尿中細菌検出が各 1 例,中等度の
腎機能低下を伴う被験者では頻尿が 1 例に認められた。
表 2.7.6.8-21 副作用の内訳(SAF)
MedDRA/J version 12.1器官別大分類
基本語
腎機能低下の重症度計
正常 軽度 中等度N=8 N=9 N=8 N=25
すべての副作用 3(37.5%) 2(22.2%) 1(12.5%) 6(24.0%)
耳および迷路障害 0 1(11.1%) 0 1(4.0%)
回転性めまい 0 1(11.1%) 0 1(4.0%)
臨床検査 2(25.0%) 1(11.1%) 0 3(12.0%)
血中カリウム増加 1(12.5%) 0 0 1(4.0%)
尿中細菌検出 1(12.5%) 1(11.1%) 0 2(8.0%)
腎および尿路障害 0 0 1(12.5%) 1(4.0%)
頻尿 0 0 1(12.5%) 1(4.0%)
皮膚および皮下組織障害 1(12.5%) 0 0 1(4.0%)
湿疹 1(12.5%) 0 0 1(4.0%)
発現例数(発現率)
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.5.1.3.2
表 2.7.6.8-22 副作用の内訳(件数)(SAF)
MedDRA/J version 12.1
器官別大分類
基本語
腎機能低下の重症度計
正常 軽度 中等度
すべての副作用 4 2 1 7
耳および迷路障害 0 1 0 1
回転性めまい 0 1 0 1
臨床検査 3 1 0 4
血中カリウム増加 1 0 0 1
尿中細菌検出 2 1 0 3
腎および尿路障害 0 0 1 1
頻尿 0 0 1 1
皮膚および皮下組織障害 1 0 0 1
湿疹 1 0 0 1
Source:CL-0073 総括報告書(5.3.3.3-2)Table 12.5.1.2.2,Table 12.5.1.4.2
2.7.6.8.6.2 死亡,その他の重篤な有害事象
本試験では,死亡及びその他の重篤な有害事象は認められなかった。
2.7.6.8.6.3 中止に至った有害事象
本試験では,中止に至った有害事象は認められなかった。
184

2.7.6個々の試験のまとめ
2.7.6.8.6.4 注目すべき有害事象
本試験では,低血糖関連の有害事象を注目すべき有害事象として扱った。
後観察期の Day 13 及び Day 15 に,冷汗(程度:軽度)が軽度の腎機能低下を伴う被験者 1 例
(71 歳男性)に認められた。本事象は併用薬(グリメピリド)の使用によるものと考えられ,2
件とも治験薬との関連性は「関連なし」と判定された。被験者は自己判断でジュースと飴を摂取
し,発現日の当日にいずれも回復した。本試験では他に低血糖関連の有害事象は認められなかっ
た。
2.7.6.8.6.5 臨床検査値
血液学的検査,血液生化学検査,尿検査に,臨床的に問題となるベースラインからの変化はみ
られなかった。
2.7.6.8.6.6 バイタルサイン
血圧及び脈拍数に,ベースラインからの顕著な変化はみられなかった。
2.7.6.8.6.7 心電図
心電図に臨床的に重要な異常が認められた被験者はいなかった。
2.7.6.8.7 結論
● 本剤の単回投与後,軽度の腎機能低下を伴う被験者では,正常な腎機能を有する被験者と比
較して,血漿中薬物動態パラメータにわずかな影響が認められた。中等度の腎機能低下を伴
う被験者では,Cmax及び AUCinfの幾何平均比は,正常な腎機能を有する被験者と比較してそ
れぞれ約 17%及び約 21%高かったが,有意な差は認められなかった。t1/2 には腎機能低下に
よる影響はみられなかった。腎機能の低下に伴い CLr に明らかな低下がみられたが,未変化
体の尿中排泄量は極めてわずかであり,CLr の低下は本剤の曝露量増加とは関連しないと考
えられた。
● 本剤の単回投与後,1 日尿中グルコース排泄量の平均値はいずれの群でも増加した。1 日尿
中グルコース排泄量と対応して,投与後 24 時間の血糖値の平均値はいずれの群でも低下し
た。Day1 の 1 日尿中グルコース排泄量のベースラインからの平均変化量は,正常な腎機能を
有する被験者と軽度の腎機能低下を伴う被験者では同程度であったが,中等度の腎機能低下
を伴う被験者では正常な腎機能を有する被験者より少なかった。また,血糖値のベースライ
ンからの平均変化量は,腎機能の低下に伴い減少した。
185

2.7.6個々の試験のまとめ
● 本剤の投与後,Day 1 の 1 日排尿量の平均値は,いずれの群でもベースライン(Day −1)と
比較して増加した。1 日排尿量と対応して,Day 1 の 1 日飲水量の平均値はいずれの群でもベ
ースラインと比較して増加した。Day 1 の 1 日排尿量及び 1 日飲水量のベースラインからの
平均変化量は,各群で同程度であった。
● 正常な腎機能を有する 2 型糖尿病患者,軽度又は中等度の腎機能低下を伴う 2 型糖尿病患者
に,本剤 50 mg を単回投与したときの安全性及び忍容性は良好であった。
186

2.7.6個々の試験のまとめ
2.7.6.9 海外腎機能低下者試験[CL-0064](添付資料 5.3.3.3-3)
2.7.6.9.1 試験方法の概略
治験課題名:程度が異なる腎機能障害を有する 2 型糖尿病患者での ASP1941 の薬物動態,薬力学
並びに安全性及び忍容性に及ぼす腎機能障害の影響を,腎機能が正常の 2 型糖尿病患者及び健康
被験者と比較し,検討する非盲検並行群間比較試験
治験実施医療機関:欧州 5 施設
公表文献:なし
治験期間:
治験開始日:2010 年 1 月
治験終了日:2010 年 6 月
開発のフェーズ:第 I 相
目的:
(1) 主要目的
・健康被験者,腎機能が正常の 2 型糖尿病患者及び軽度,中等度又は重度の腎機能障害を有する
2 型糖尿病患者を対象として,ASP1941(以下,本剤)100 mg 単回投与時の本剤の薬物動態を
比較する。
(2) 副次目的
・健康被験者,腎機能が正常の 2 型糖尿病患者及び軽度,中等度又は重度の腎機能障害を有する
2 型糖尿病患者を対象として,本剤 100 mg 単回投与時の本剤の薬力学(尿中グルコース排泄)
を比較する。
・健康被験者,腎機能が正常の 2 型糖尿病患者及び軽度,中等度又は重度の腎機能障害を有する
2 型糖尿病患者を対象として,本剤 100 mg 単回投与時の本剤の安全性及び忍容性を比較する。
治験デザイン・治験方法:
本試験は,健康被験者,腎機能が正常の 2 型糖尿病患者及び軽度,中等度又は重度の腎機能障害
を有する 2 型糖尿病患者を対象とした非盲検,並行群間比較試験として実施した。
本試験では,modification of diet in renal disease(MDRD)式†により血清クレアチニンを用いて推
定糸球体濾過量(eGFR)を算出し,被験者の腎機能を eGFR に基づき以下の 4 段階に分類した:
・腎機能正常:90 mL/min/1.73 m2 以上
・軽度腎機能障害:60 mL/min/1.73 m2 以上 90 mL/min/1.73 m2 未満
・中等度腎機能障害:30 mL/min/1.73 m2 以上 60 mL/min/1.73 m2 未満
・重度腎機能障害:15 mL/min/1.73 m2 以上 30 mL/min/1.73 m2 未満
† MDRD 式:eGFR(mL/min/1.73 m2)=32788×血清クレアチニン−1 154×年齢−0 203×[1.210(黒人の
場合]×[0.742(女性の場合)]
上記基準に基づきスクリーニング時に各被験者の腎機能を評価し,健康被験者(腎機能正常)8
例,腎機能が正常の 2 型糖尿病患者(以下,腎機能正常患者)8 例,軽度腎機能障害を有する 2
型糖尿病患者(以下,軽度腎機能低下患者)8 例,中等度腎機能障害を有する 2 型糖尿病患者(以
下,中等度腎機能低下患者)8 例及び重度腎機能障害を有する 2 型糖尿病患者(以下,重度腎機
能低下患者)8 例を,適格性確認後,本試験に組み入れた。各群には,男女それぞれ 3 例以上組
み入れることとした。被験者は治験薬投与前日(Day −1)に入所し,Day 1 に本剤 100 mg を 1
回服用した。未変化体及び代謝物の血漿中及び尿中濃度,血糖値,尿中グルコース及び尿中クレ
アチニンを測定するため,治験薬投与前から投与後 120 時間までの規定された評価時点で血液及
び尿を採取した。被験者は Day 6 に治験実施計画書で規定した評価が終了した後,退所した。
目標被験者数:
計 40 例(各群 8 例)
187

2.7.6個々の試験のまとめ
【設定根拠】
目標被験者数は医学的見地に基づき設定したものであり,統計学的な目標被験者数の算出は行っ
ていない。
診断及び選択・除外基準:
1.選択基準(以下の基準をすべて満たす者を対象とした)
(1) 45 歳以上 80 歳以下の安定した 2 型糖尿病患者又は健康被験者
(2) 男性の場合,スクリーニング時から治験薬投与後3カ月まで禁欲する,又は有効な避妊法を使
用することに同意した者。女性の場合,妊娠の可能性がない(閉経後,卵管結紮等の不妊手
術済み,子宮摘出の既往を有する),又はホルモン避妊薬以外の適切な避妊法を使用してい
る者。ホルモン避妊薬以外の適切な避妊法は,以下とする:
○ 入所1カ月前から退所後3カ月までの禁欲
○ スクリーニング時から遡って3カ月以上前のパートナーの不妊手術
○ 以下の避妊法のうち,2種類以上の併用:
・ 殺精子剤入りペッサリー
・ 子宮内避妊器具
・ パートナーが殺精子剤入りクリームとコンドームを併用する
(3) スクリーニング時の eGFR(MDRD 式で算出)が以下の者:
○ 腎機能正常被験者:90 mL/min/1.73 m2以上
○ 軽度腎機能障害患者:60 mL/min/1.73 m2以上90 mL/min/1.73 m2未満
○ 中等度腎機能障害患者:30 mL/min/1.73 m2以上60 mL/min/1.73 m2未満
○ 重度腎機能障害患者:15 mL/min/1.73 m2以上30 mL/min/1.73 m2未満
(4) BMIが25.0 kg/m2以上40.0 kg/m2以下の者
(5) 空腹時血糖値が,2 型糖尿病患者では 11.5 mmol/L 以下,健康被験者では 5.6 mmol/L 未満の
者
(6) 被験者本人から文書による同意が得られている者
2.除外基準(以下の基準のいずれかに該当する者は対象から除外した)
すべての被験者
(1) 1 型糖尿病(C-ペプチドで判定)の患者
(2) スクリーニング前 6 カ月以内に妊娠していた又は授乳中であった者
(3) 血液透析が必要である,又は腎移植の既往を有する者
(4) 本試験前の身体所見,心電図及び臨床検査の結果,治験担当医師が臨床的に重要な異常があ
ると判断した者
(5) 試験前来院時に脈拍数又は血圧に下記の異常が認められた者:
脈拍数が 40 bpm 未満又は 90 bpm を超える,平均収縮期血圧が 160 mmHg を超える,平均拡
張期血圧が 100 mmHg を超える(血圧は臥位にて 5 分間安静にした後 3 回測定した。脈拍数
は自動測定とした。)
(6) 腎機能障害患者では,疾患のコントロール目的で試験前から使用していた薬剤の用法・用量
を 1 カ月(又はその薬剤の半減期の 5 倍のいずれか長い方の期間)以内に変更した者。健康
被験者では,アセトアミノフェン(3 g/日以下)の頓用を除き,入所前 2 週間以内に処方薬又
は市販薬(経口避妊薬,ビタミン剤,セイヨウオトギリソウ等の生薬又はハーブ製剤を含む)
を使用した者
(7) 血清学的検査で HBs 抗原,HAV IgM 抗体,HCV 抗体又は HIV 抗体 1 及び 2 が陽性であった
者
(8) 腎機能正常被験者では,肝機能パラメータ(ALT,AST,ビリルビン及びアルカリホスファ
ターゼ)のいずれかが連続して基準値上限を超えた者。2 型糖尿病患者では,肝機能パラメー
タが連続して以下に当てはまる者:
188

2.7.6個々の試験のまとめ
(9) a. ALT/AST:基準値上限の 2 倍未満
b. ビリルビン:基準値上限の 1.5 倍未満
c. アルカリホスファターゼ:基準値上限の 1.5 倍未満
肝機能パラメータがこれらの範囲を超えた場合,治験担当医師が被験者の組入れの可否を治
験依頼者と相談の上,決定することとした。
(10) 治験実施医療機関入所前 3 カ月以内に薬物乱用歴を有する者
(11) 本剤又は製剤の構成成分に対して過敏症を有する又は過敏症が疑われる者
(12) 治験実施医療機関入所前 3カ月以内に 1日 10本を超える巻きタバコ(又は当量の刻みタバコ)
を使用した者
(13) 治験実施医療機関入所前 3 カ月以内に 1 週間あたり 21 単位(1 単位=アルコール 10 g=ビール
[アルコール 5%]250 mL,蒸留酒[アルコール 35%]35 mL,ワイン[アルコール 12%]100
mL)を超える飲酒をした者,又は 14 単位を超える飲酒をした女性
(14) 本試験登録予定日から遡って 3 カ月以内に他の臨床試験に参加した者,又は 12 カ月以内に 4
件以上の臨床試験に参加した者,又は半減期の長い生物製剤を使用する臨床試験に参加した
者
(15) 本試験を安全に完了できない疾患を有すると治験担当医師が判断した者
(16) 治験実施医療機関入所前 3 カ月以内に献血(全血又は成分献血)を行った者
(17) アステラス製薬グループ又は本試験に関係する CRO に雇用されている者
腎機能障害を有する 2 型糖尿病患者
(18) スクリーニング前 3 カ月以内に,治験担当医師が試験参加に支障があると判断した臨床的に
重要な疾患(腎疾患,2 型糖尿病及びそれらに関連する症状を除く)の既往,病態又は臨床
検査値異常を有する患者
(19) Day 1 から遡って 2 週間(又はその薬剤の半減期の 5 倍のいずれか長い方の期間)以内に併
用している薬剤の用量を変更した患者,若しくは試験期間中に用量の変更が必要となる可能
性がある患者
(20) 閉塞性尿路疾患を有する患者,若しくは実質性腎疾患や腎臓病と関連がないその他の腎機能
低下の要因を有する患者
(21) 悪性腫瘍に続発する腎疾患を有する患者
(22) スクリーニング期に臨床所見若しくは臨床検査値又は腎機能障害の大幅な変化又は悪化が認
められ,試験前 4 週間以内の腎機能の変動又は急激な悪化が示唆された患者
2 型糖尿病患者
(23) 食事療法のみによる治療を受けている患者
(24) 試験期間中に新たな薬剤が必要となる,又はその可能性がある患者
(25) 試験前(例えば 6 カ月以内)に,重度の低血糖(例えば,血糖値が 3 mmol/L[55 mg/dL]未
満が認められた,低血糖から回復するために他者の助けを必要とした,又は入院を要した)
が認められた患者
(26) ヘモグロビンが 5.6 mmol/L(9 g/dL)未満である患者
正常腎機能被験者
(27) 治験担当医師が試験参加に支障があると判断した臨床的に重要な疾患の既往又は病態を有す
る者
(28) スクリーニング前 3 カ月以内に,治験担当医師が試験参加に支障があると判断した臨床検査
値の異常が認められた者
189

2.7.6個々の試験のまとめ
治験薬,投与量及び投与方法:
1.被験薬及びロット番号
ASP1941 錠 50 mg:1 錠中に ASP1941 として 50 mg を含有するフィルムコート錠
ロット番号:BX1002810
2.投与量及び投与方法
本剤 100 mgを空腹時に単回経口投与した。治験薬投与前 8時間から投与後 2時間まで絶食とした。
【設定根拠】
100 mg/日は,2 型糖尿病患者での本剤の有効用量の範囲(最大の有効性の 75%~90%の有効性が
認められる用量)に含まれると予想された。本剤の尿中排泄率は 1%であるため,腎機能障害患者
で血漿中未変化体濃度は上昇しないと予想されたが,本薬の代謝物(主にグルクロニド)の血漿
中濃度は,腎機能障害患者で上昇する可能性があった。健康被験者を対象とした単回投与試験
[CL-0001]で 600 mg まで忍容性が確認されており,2 型糖尿病患者を対象とした 4 週間投与試
験[CL-0016]で 300 mg まで忍容性が確認されていることから,本剤 100 mg(単回投与)は,腎
機能障害患者に対して安全に投与できると予想された。
評価期間:
治験薬投与期間:1 日
併用治療(薬剤及び療法):
健康被験者では,入所 2 週間前からすべての薬剤の併用を禁止した。ただし,治験担当医師から
了承が得られた場合に限り,一定限度のアセトアミノフェン(3 g/日以下)の使用は可とした。
2 型糖尿病患者では,糖尿病治療薬の使用は可としたが,試験開始 1 カ月(又はその薬剤の半減
期の 5 倍のいずれか長い方の期間)前から用量の変更を禁止した。試験期間中のアセトアミノフェ
ン(3 g/日以下)の頓用は可とした。治験担当医師は,Day 1 に糖尿病治療に関連する併用薬の中
止の可否を治験依頼者と相談の上,決定した。
190

2.7.6個々の試験のまとめ
評価項目,評価スケジュール及び評価基準:
1.評価スケジュール:スクリー
ニング
入所期間 後観察
Day −21~−2
Day −1 Day 1 Day 2~5 Day 6 Day 13~20
適格性確認 a X Xb
入所 X X X X
同意取得 X
身体所見 X X X
薬物・アルコール検査 X X
妊娠検査 X X
治験薬投与 X
食事 f X X X X
クレアチニンクリアラ
ンス
投与前 20~0
時間
薬物動態及び薬力学評
価用採尿
投与前
20~16,16
~12,12
~8 時間 e
投与前 8~0 時
間,投与後 0~
4,4~8,8~12
時間
投与後 12~16,
16~24,24~36,
36~48,48~60,
60~72,72~96
時間
投与後
96~120
時間
薬物動態評価用採血 投与前,投与後
0.5,1,1.5,2,
3,4,6,12
時間
投与後 16,24,
36,48,72,96
時間
投与後
120 時間
臨床検査 X X 投与後 24 時間 X
eGFR(MDRD) Xc X X
空腹時血糖値 X 投与前 20,
16,12,8
時間 d, e
投与前,投与後
4,8,12 時間 d
投与後 16,24,
36,48,60,72,
96 時間 d
投与後
120 時間
バイタルサイン
(血圧,脈拍数)
X X 投与前,投与後
2,3,4,6,12
時間
投与後 24,48,
72,96 時間
投与後
120 時間
X
12 誘導心電図 X X 投与前 投与後 72 時間 投与後
120 時間
X
併用薬及び有害事象の
調査
X X X X X X
a:適格性は,同意の有無,選択・除外基準,被験者背景,既往歴,喫煙習慣,飲酒習慣,薬歴,血清学的検査結
果を確認し,判断した。
b:選択・除外基準のみ確認した。
c:スクリーニング時に eGFR を算出し,腎機能障害の程度を判定した。
d:治験薬投与前の検体は空腹時に採取した。その他の Day −1 の投与前 20,16,12 及び 8 時間,Day 1 の投与後
4,8 及び 12 時間,Day 2 の投与後 16 及び 36 時間,Day 3 の投与後 60 時間の検体は,空腹時以外に採取した。
e:検体採取時期は Day 1 の治験薬投与予定時刻と対応する。
f:入所期間中は標準食(朝食,昼食及び夕食)を摂取した。
2.薬物動態|薬力学:
(1) 薬物動態
「1.評価スケジュール」に示した時期に採血及び蓄尿を行い,未変化体及び代謝物の血漿中及び
尿中濃度を測定した。各被験者の未変化体及び代謝物の血漿中及び尿中濃度から,ノンコンパー
191

2.7.6個々の試験のまとめ
トメント法を用いて以下の薬物動態パラメータを算出した。
血漿中パラメータ
[未変化体]
•最高血漿中濃度(Cmax)
•時間 0 から無限時間まで外挿した血漿中濃度‐時間曲線下面積(AUCinf)
•時間 0 から定量可能最終時点までの血漿中濃度‐時間曲線下面積(AUClast)
•経口クリアランス(CL/F)
•ラグタイム(tlag)
•最高血漿中濃度到達時間(tmax)
•消失半減期(t1/2)
•みかけの分布容積(Vz/F)
•非結合型の最高血漿中濃度(Cmax,u)
•非結合型の時間 0 から無限時間まで外挿した血漿中濃度‐時間曲線下面積(AUCinf,u)
•非結合型の定量可能最終時点までの血漿中濃度‐時間曲線下面積(AUClast,u)
•非結合型の経口クリアランス(CLu/F)
•非結合型の Vz/F(Vz,u/F)
•非結合型分率(fu)
[代謝物]
•AUCinf
•AUClast
•Cmax
•CL/F
•tlag
•tmax
•t1/2
尿中パラメータ
[未変化体]
•腎クリアランス(CLr)
•非結合型未変化体の CLr(CLr,u)
•時間 0 から治験薬投与後 24 時間までの尿中排泄量(Ae24h)
•時間 0 から定量可能最終時点までの尿中排泄量(Aelast)
•時間 0 から定量可能最終時点までの尿中排泄率(Aelast%)
•時間 0 から無限時間まで外挿した尿中排泄量(Aeinf)
•時間 0 から無限時間まで外挿した尿中排泄率(Aeinf%)
[代謝物]
•Ae24h
•CLr
•Aelast
•Aelast%
•Aeinf
•Aeinf%
(2) 薬力学
「1.評価スケジュール」に示した時期に蓄尿及び採血を行った。尿中グルコース濃度,尿中クレ
192

2.7.6個々の試験のまとめ
アチニン濃度及び血糖値を測定し,以下の薬力学パラメータを算出した。
血糖値
•治験薬投与前 20 時間から時間 0 までの血漿中濃度‐時間曲線下面積(AUC−20-0h)(Day −1)
•治験薬投与後 4 時間から投与後 24 時間までの血漿中濃度‐時間曲線下面積(AUC4-24h)(Day 1)
尿中グルコース
•治験薬投与前 20 時間から時間 0 までの尿中排泄量(Ae−20-0h)(Day −1)
•治験薬投与後 4 時間から投与後 24 時間までの尿中排泄量(Ae4-24h)(Day 1)
•時間 0 から治験薬投与後 120 時間までの尿中排泄量(Ae0-120h)(Day 1~Day 6)
腎クリアランス(CLr)
•治験薬投与前 20 時間から時間 0 までの腎クリアランス(CLr −20-0h)(Day −1)
•治験薬投与後 4 時間から投与後 24 時間までの腎クリアランス(CLr 4-24h)(Day 1)
3.安全性:
• 有害事象
• バイタルサイン
• 臨床検査値
• 身体所見
• 12 誘導心電図
有害事象は,治験薬投与時以降,試験完了までに発現した事象を収集した。
有害事象と治験薬との関連性は以下の基準により判定し,「関連あるかもしれない」又は「多分(お
そらく)関連あり」のいずれかに該当したものを,「治験薬との関連性が否定できない有害事象(副
作用)」と定義した。治験薬との関連性 関連性判定基準
否定できる 有害事象発現と薬剤投与との間に時間的関係から関連性がありそうにないと思われる場
合や,他の薬剤や合併症・基礎疾患等により説明ができる場合
関連あるかもしれ
ない
有害事象発現と薬剤投与との間に時間的に妥当な相関関係があるが,要因として合併症・
基礎疾患や他の薬剤による説明もできる場合。投与中止に関する情報が不明確な場合
多分(おそらく)
関連あり
有害事象発現と薬剤投与との間に時間的に妥当な相関関係がある場合,合併症・基礎疾患
や他の薬剤では説明ができない,又はそれらによる可能性が考えにくい場合,再投与によ
り再発又は投与中止により消失又は軽快を示す場合
有害事象の程度は,以下に従って判定した。
・軽度:日常の活動に支障がないもの
・中等度:日常の活動に支障があるもの
・重度:日常の活動が不可能になるもの
統計解析:
1.解析対象集団:
(1) 安全性解析対象集団(SAF)
治験薬を服用したすべての被験者の集団を SAF とした。
(2) 薬物動態解析対象集団(PKAS)
治験薬を服用し,血漿中又は尿中の未変化体濃度測定用の血液又は尿が 1 時点以上で採取され,
検体の採取時刻及び治験薬服用時刻が記録されている被験者の集団
(3) 薬力学解析対象集団(PDAS)
治験薬を服用し,血糖値又は尿中グルコース測定用の血液又は尿が 1 時点以上で採取され,検体
の採取時刻及び治験薬服用時刻が記録されている被験者の集団
193

2.7.6個々の試験のまとめ
2.被験者背景及びその他の基準値:
SAF,PKAS 及び PDAS を対象に,計数値項目では度数集計を行い,計量値項目では要約統計量
を算出した。
3.薬物動態|薬力学:
(1) 薬物動態
PKAS を対象に,未変化体及び代謝物の薬物動態パラメータの要約統計量を投与群別に算出した。
薬物動態の主要パラメータは,未変化体の AUCinf 及び Cmaxとした。
腎機能障害が本剤の薬物動態に及ぼす影響を検討するため,主要な薬物動態パラメータ(AUCinf
及び Cmax)の自然対数変換値について分散分析を行い,腎機能正常患者群又は健康被験者群を対
照群として,対照群に対する各群の比及びその 90%信頼区間を算出した。比の 90%信頼区間が 80%
~125%の範囲内であった場合,臨床的に腎機能障害の影響はないと判断することとした。
同様の比較を,未変化体の CLr 及び CLr,uについても行った。また,代謝物の AUCinf及び Cmaxに
ついても腎機能正常患者群を対照として同様に各群と比較した。
(2) 薬力学
PDAS を対象に,薬力学パラメータの要約統計量を投与群別に算出した。
腎機能と尿中グルコース排泄量の関連性を検討するため,Day −1(ベースライン)及び Day 1(治
験薬投与後)の同時刻に尿中グルコース排泄量(それぞれ Ae−20-0h及び Ae4-24h)を測定し,その変
化量(Ae4-24h − Ae−20-0h)を算出した。尿中グルコース排泄量の変化量について,投与群を固定効
果とした分散分析モデルを用いて,腎機能低下患者各群と腎機能正常患者群を比較した。同様の
比較を,CLr についても行った。
4.安全性:
安全性の解析では,SAF を解析対象とした。
(1) 有害事象
有害事象及び副作用の発現率を投与群別に算出した。
(2) 臨床検査値
計量値項目は,評価時点ごとに実測値の要約統計量を算出した。計数値項目は,治験薬投与日前
日と評価時点ごとのクロス表を作成した。
(3) バイタルサイン
評価時点ごとに実測値の要約統計量を算出した。
(4) 12 誘導心電図
心電図所見の臨床的に重要な異常及び臨床的に重要でない異常の発現率を評価時点ごとに算出し
た。
報告書の日付: 年 月 日
194

2.7.6個々の試験のまとめ
2.7.6.9.2 被験者の内訳及び解析対象集団
被験者の内訳を表 2.7.6.9-1 に示した。
計 40 例が登録され,全例に本剤 100 mg が単回投与された。内訳は,健康被験者 8 例,腎機能
正常患者 8 例,軽度腎機能低下患者 8 例,中等度腎機能低下患者 8 例,重度腎機能低下患者 8 例
であった。試験を中止した被験者はいなかった。
全 40 例を SAF,PKAS 及び PDAS の解析対象例とした。
表 2.7.6.9-1 被験者の内訳
健康被験者 2 型糖尿病患者
腎機能正常軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下
治験薬投与例 8 8 8 8 8
完了例 8 8 8 8 8
中止例 0 0 0 0 0
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.1.1.3
2.7.6.9.3 人口統計学的及び他の基準値の特性
被験者背景を表 2.7.6.9-2 に示した。
腎機能障害を有する 2 型糖尿病患者群では,健康被験者群と比較して平均年齢が高かった。す
べての被験者が白人であり,軽度腎機能低下患者群を除き,男性の割合が高かった。
195

2.7.6個々の試験のまとめ
表 2.7.6.9-2 被験者背景(SAF,PKAS 及び PDAS)
健康被験者 2 型糖尿病患者
腎機能正常軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下(N=8) (N=8) (N=8) (N=8) (N=8)
年齢 (歳)
平均値 (標準偏差) 54.9(7.66) 57.1(8.11) 64.6(2.20) 68.9(6.83) 66.0(5.40)
最小値 – 最大値 47 – 66 46 – 70 62 – 69 61 – 79 57 – 72
性別 n(%)
男性 5(62.5) 5(62.5) 3(37.5) 5(62.5) 5(62.5)
女性 3(37.5) 3(37.5) 5(62.5) 3(37.5) 3(37.5)
人種 n(%)
白人 8(100) 8(100) 8(100) 8(100) 8(100)
黒人 0 0 0 0 0
アジア人 0 0 0 0 0
その他 0 0 0 0 0
体重 (kg)
平均値 (標準偏差) 77.9(10.47) 95.9(11.63) 79.1(13.60) 94.1(15.00) 85.6(19.96)
最小値 – 最大値 55 – 88 70 – 105 63 – 100 61 – 105 54 – 118
BMI (kg/m2)平均値 (標準偏差) 27.3(1.75) 33.3(4.00) 28.8(3.26) 33.2(2.48) 31.1(5.63)
最小値 – 最大値 25 – 30 26 – 38 25 – 34 28 – 36 25 – 39
身長 (cm)
平均値 (標準偏差) 168.8(10.26) 169.9(6.29) 165.4(9.93) 167.6(9.66) 165.5(12.86)
最小値 – 最大値 148 – 184 165 – 184 151 – 182 148 – 178 147 – 180
eGFRa (mL/min/1.73 m2)平均値 (標準偏差) 98.6(4.96) 107.1(13.83) 74.5(8.90) 43.8(10.32) 25.0(4.14)
最小値 – 最大値 93 – 106 94 – 131 63 – 85 32 – 57 18 – 30
a:スクリーニング時
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.1.2.1.1,Table 12.1.2.1.2 及び Table 12.4.1.1
2.7.6.9.4 治験薬の曝露
全被験者 40 例が,本剤 100 mg を 1 回服用した。
2.7.6.9.5 薬物動態|薬力学
2.7.6.9.5.1 薬物動態の結果
未変化体の薬物動態
(1) 血漿中薬物動態
本剤単回投与時の未変化体の血漿中薬物動態パラメータを表 2.7.6.9-3 に示した。また,腎機能
障害が未変化体の血漿中薬物動態に及ぼす影響を評価するため,未変化体の AUCinf及び Cmaxを比
較した[表 2.7.6.9-4(腎機能正常患者群を対照とした検討)及び表 2.7.6.9-5(健康被験者群を対照
とした検討)]。
2 型糖尿病患者群では,AUCinfは,腎機能正常患者群と比較すると,軽度腎機能低下患者群で
は 1.255 倍,中等度腎機能低下患者群では 1.401 倍,重度腎機能低下患者群では 1.466 倍であり,
196

2.7.6個々の試験のまとめ
腎機能の低下に伴い上昇した。Cmaxに一定の傾向は認められず,腎機能障害の影響はみられなかっ
た。腎機能が正常の被験者群間で比較すると,健康被験者群と比較して,AUCinf及び Cmaxはいず
れも腎機能正常患者群で高かった。
t1/2 の平均値は,2 型糖尿病患者群と比較して健康被験者群で短かったが,健康被験者群では変
動係数が大きかった(50.7%)。2 型糖尿病患者群では,t1/2 に一定の傾向は認められず,腎機能障
害の影響はみられなかった。
腎機能正常患者群と健康被験者群で tmaxに差はなかった。2 型糖尿病患者群では,tmaxの変動に
一定の傾向は認められなかった。CL/F の平均値は,2 型糖尿病患者群では腎機能の低下に伴い低
下した。2 型糖尿病患者群と健康被験者群で Vz/F に差はなかったが,Vz/F の変動係数は,2 型糖
尿病患者群(22.0%~40.7%)と比較して健康被験者群(61.9%)で大きかった。Vz/F の平均値は
腎機能の低下に伴い低下した。
197

2.7.6個々の試験のまとめ
表 2.7.6.9-3 未変化体の血漿中薬物動態パラメータ(PKAS)
健康被験者 2 型糖尿病患者
腎機能正常軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下AUCinf N 8 8 8 7 8
(ng•h/mL) 平均値 7325.784 8240.802 10506.144 12103.727 12687.138
標準偏差 2037.1308 1812.0386 3165.2302 4906.4535 4840.1949
変動係数 (%) 27.8 22.0 30.1 40.5 38.2
最小値 – 最大値 4989.10 –10050.35
6395.89 –12045.28
7147.96 –17238.07
6971.34 –21000.49
6455.17 –20665.20
中央値 6779.552 8106.767 10028.125 11990.443 12894.511
CL/F N 8 8 8 7 8
(L/h) 平均値 14.584 12.589 10.172 9.393 9.079
標準偏差 3.8945 2.4215 2.5962 3.4308 3.7973
変動係数 (%) 26.7 19.2 25.5 36.5 41.8
最小値 – 最大値 9.95 – 20.04 8.30 – 15.64 5.80 – 13.99 4.76 – 14.34 4.84 – 15.49
中央値 14.805 12.336 9.975 8.340 7.758
Cmax N 8 8 8 8 8
(ng/mL) 平均値 1277.194 1456.045 1625.634 1448.140 1576.083
標準偏差 360.1587 155.8559 417.2681 419.6489 404.2510
変動係数 (%) 28.2 10.7 25.7 29.0 25.6
最小値 – 最大値 775.36 –1925.98
1276.56 –1692.38
1017.43 –2473.78
798.24 –2001.82
990.05 –2044.19
中央値 1202.135 1443.330 1603.040 1552.570 1697.950t1/2 N 8 8 8 7 8
(h) 平均値 15.184 19.156 20.561 20.763 21.218
標準偏差 7.6936 6.1590 8.0842 9.7629 9.2240
変動係数 (%) 50.7 32.2 39.3 47.0 43.5
最小値 – 最大値 8.22 – 28.53 13.53 – 29.45 11.29 – 33.43 10.07 – 39.20 13.41 – 41.37
中央値 12.498 16.593 20.307 17.429 18.954tmax N 8 8 8 8 8
(h) 平均値 1.563 1.195 1.063 1.313 1.500
標準偏差 0.6781 0.4500 0.3204 0.7039 0.8018
変動係数 (%) NA NA NA NA NA
最小値 – 最大値 1.00– 3.00 0.53 – 2.00 0.50 – 1.50 1.00– 3.00 0.50– 3.00
中央値 1.500 1.015 1.000 1.000 1.250Vz/F N 8 8 8 7 8
(L) 平均値 318.417 335.461 289.163 257.199 254.226
標準偏差 196.9787 73.7622 106.0580 104.7623 79.4472
変動係数 (%) 61.9 22.0 36.7 40.7 31.3
最小値 – 最大値 164.73 –749.71
217.26 –456.09
155.70 –473.13
181.50 –471.70
127.70 –345.98
中央値 248.758 341.703 275.896 208.459 274.987
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.4.4.1
198

2.7.6個々の試験のまとめ
表 2.7.6.9-4 腎機能障害が未変化体の血漿中薬物動態に及ぼす影響[腎機能正常患者群を対
照とした検討](PKAS)
投与群 最小二乗平均比
比較群/対照群 a 90%信頼区間
AUCinf
(ng•h/mL)
腎機能正常患者 8084.03 - -
健康被験者 7084.19 0.876 [0.670, 1.146]
軽度腎機能低下患者 10143.92 1.255 [0.960, 1.640]
中等度腎機能低下患者 11326.66 1.401 [1.062, 1.849]
重度腎機能低下患者 11848.37 1.466 [1.121, 1.916]
Cmax
(ng/mL)
腎機能正常患者 1448.90 - -
健康被験者 1233.69 0.851 [0.685, 1.059]
軽度腎機能低下患者 1580.92 1.091 [0.878, 1.357]
中等度腎機能低下患者 1388.83 0.959 [0.771, 1.192]
重度腎機能低下患者 1527.25 1.054 [0.848, 1.311]
a:対照群:腎機能正常患者群
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.4.6.1.1
表 2.7.6.9-5 腎機能障害が未変化体の血漿中薬物動態に及ぼす影響[健康被験者群を対照と
した検討](PKAS)
投与群 最小二乗平均比
比較群/対照群 a 90%信頼区間
AUCinf
(ng•h/mL)
健康被験者 7084.19 - -
腎機能正常患者 8084.03 1.141 [0.873, 1.492]
軽度腎機能低下患者 10143.92 1.432 [1.095, 1.872]
中等度腎機能低下患者 11326.66 1.599 [1.212, 2.110]
重度腎機能低下患者 11848.37 1.673 [1.279, 2.186]
Cmax
(ng/mL)
健康被験者 1233.69 - -
腎機能正常患者 1448.90 1.174 [0.945, 1.460]
軽度腎機能低下患者 1580.92 1.281 [1.031, 1.593]
中等度腎機能低下患者 1388.83 1.126 [0.905, 1.400]
重度腎機能低下患者 1527.25 1.238 [0.996, 1.539]
a:対照群:健康被験者群
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.4.6.1.2
本剤単回投与時の非結合型未変化体の血漿中薬物動態パラメータを表 2.7.6.9-6 に示した。
fuの変動に,腎機能の低下に伴う一定の傾向は認められなかった。また,健康被験者群と腎機
能正常患者群で違いは認められなかった。
199

2.7.6個々の試験のまとめ
表 2.7.6.9-6 非結合型未変化体の血漿中薬物動態パラメータ(PKAS)
健康被験者 2 型糖尿病患者
腎機能正常軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下AUCinf,u N 8 8 8 7 8
(ng•h/mL) 平均値 227.968 257.711 307.297 414.855 405.484
標準偏差 61.1332 50.4378 78.4017 168.0634 171.5297
変動係数 (%) 26.8 19.6 25.5 40.5 42.3
最小値 – 最大値 134.56–312.46
178.97 –328.45
216.75 –447.82
246.83 –675.57
206.32 –736.33
中央値 221.548 249.390 294.789 321.272 408.437
CLu/F N 8 8 8 7 8
(L/h) 平均値 471.522 401.961 343.850 275.566 289.612
標準偏差 144.5289 82.9901 84.8548 102.1071 126.5061
変動係数 (%) 30.7 20.6 24.7 37.1 43.7
最小値 – 最大値 320.04 –743.15
304.46 –558.76
223.30 –461.37
148.02–405.13
135.81 –484.68
中央値 451.532 401.678 339.601 311.263 244.837
Cmax,u N 8 8 8 8 8
(ng/mL) 平均値 39.962 45.844 47.676 50.829 50.176
標準偏差 11.7870 6.7581 10.6982 16.0492 14.5755
変動係数 (%) 29.5 14.7 22.4 31.6 29.0
最小値 – 最大値 20.91 – 60.46 35.13 – 54.95 30.85 – 64.27 30.95 – 74.47 31.64 – 72.48
中央値 37.723 46.310 46.804 49.169 51.469fu (2h) N 8 8 8 8 8
平均値 0.030 0.028 0.028 0.034 0.030
標準偏差 0.0040 0.0042 0.0028 0.0072 0.0034
変動係数 (%) 13.4 14.9 10.2 21.4 11.5
最小値 – 最大値 0.03 – 0.04 0.02 – 0.03 0.02 – 0.03 0.02 – 0.05 0.03 – 0.04
中央値 0.030 0.028 0.027 0.032 0.030fu (6h) N 8 8 8 8 8
平均値 0.033 0.035 0.032 0.038 0.034
標準偏差 0.0031 0.0033 0.0044 0.0054 0.0028
変動係数 (%) 9.6 9.4 13.8 14.2 8.4
最小値 – 最大値 0.03 – 0.04 0.03 – 0.04 0.02 – 0.04 0.03 – 0.04 0.03 – 0.04
中央値 0.033 0.036 0.033 0.039 0.033Vz,u/F N 8 8 8 7 8
(L) 平均値 10573.614 10797.548 9661.191 7338.531 7995.894
標準偏差 7560.8170 2623.2109 3156.8317 2102.3801 2430.9998
変動係数 (%) 71.5 24.3 32.7 28.6 30.4
最小値 – 最大値 5251.77 –27796.55
5944.44 –13880.41
5993.28 –15834.06
4879.21 –11245.53
4540.78 –10841.86
中央値 7899.917 11436.954 9384.222 7227.426 8039.985
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.4.4.1
(2) 尿中薬物動態
本剤単回投与時の未変化体の尿中薬物動態パラメータと,非結合型未変化体の腎クリアランス
(CLr,u)をあわせて表 2.7.6.9-7 に示した。また,腎機能障害が未変化体の尿中薬物動態に及ぼす
影響を評価するため,未変化体の CLr 及び CLr,uを比較した[表 2.7.6.9-8(腎機能正常患者群を対
照とした検討)及び表 2.7.6.9-9(健康被験者群を対照とした検討)]。
200

2.7.6個々の試験のまとめ
未変化体の尿中排泄量に腎機能の低下に伴う一定の傾向は認められなかったが,尿中排泄量の
平均値は重度腎機能低下患者群で最も少なかった。未変化体の尿中排泄率は,軽度腎機能低下患
者群で 1.108455%,重度腎機能低下患者群で 0.694629%であった。未変化体及び非結合型未変化体
の腎クリアランスは,いずれも腎機能の低下に伴い低下した。腎機能が正常の被験者群間で比較
すると,健康被験者群及び腎機能正常患者群間で,腎クリアランスに差はみられなかった。
201

2.7.6個々の試験のまとめ
表 2.7.6.9-7 未変化体の尿中薬物動態パラメータ(PKAS)
健康被験者 2 型糖尿病患者
腎機能正常軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下Ae24h N 8 8 8 8 8
(mg) 平均値 0.727828 0.739377 0.876781 0.767610 0.502963
標準偏差 0.1186461 0.1550958 0.5099496 0.2656759 0.1459597
変動係数 (%) 16.3 21.0 58.2 34.6 29.0
最小値 – 最大値 0.52480 –0.91394
0.47842 –0.95616
0.55458 –2.06071
0.49683 –1.26962
0.24026 –0.72497
中央値 0.717440 0.731795 0.674305 0.783505 0.513535
Aeinf N 8 8 8 7 8
(mg) 平均値 0.924396 1.040354 1.108455 1.045070 0.694629
標準偏差 0.1230365 0.2626096 0.5855547 0.4266980 0.2156706
変動係数 (%) 13.3 25.2 52.8 40.8 31.0
最小値 – 最大値 0.70292 –1.08549
0.71401 –1.44019
0.76606 –2.47132
0.63074 –1.90030
0.34371 –1.03203
中央値 0.966795 0.992870 0.856345 1.000190 0.725055
Aeinf% N 8 8 8 7 8
(%) 平均値 0.924396 1.040354 1.108455 1.045070 0.694629
標準偏差 0.1230365 0.2626096 0.5855547 0.4266980 0.2156706
変動係数 (%) 13.3 25.2 52.8 40.8 31.0
最小値 – 最大値 0.70292 –1.08549
0.71401 –1.44019
0.76606 –2.47132
0.63074 –1.90030
0.34371 –1.03203
中央値 0.966795 0.992870 0.856345 1.000190 0.725055
Aelast N 8 8 8 8 8(mg) 平均値 0.923021 1.030765 1.092416 1.050611 0.681766
標準偏差 0.1215868 0.2566560 0.5725241 0.3884721 0.2123273
変動係数 (%) 13.2 24.9 52.4 37.0 31.1
最小値 – 最大値0.69701 –1.08896
0.70946 –1.40670
0.75745 –2.42367
0.61342 –1.83763
0.34168 –1.01507
中央値 0.962745 0.990500 0.854465 1.010065 0.710895
Aelast% N 8 8 8 8 8
(%) 平均値 0.923021 1.030765 1.092416 1.050611 0.681766
標準偏差 0.1215868 0.2566560 0.5725241 0.3884721 0.2123273
変動係数 (%) 13.2 24.9 52.4 37.0 31.1
最小値 – 最大値 0.69701 –1.08896
0.70946 –1.40670
0.75745 –2.42367
0.61342 –1.83763
0.34168 –1.01507
中央値 0.962745 0.990500 0.854465 1.010065 0.710895
CLr N 8 8 8 8 8
(L/h) 平均値 0.131456 0.130496 0.106263 0.095978 0.062414
標準偏差 0.0239810 0.0369892 0.0448138 0.0324732 0.0279245
変動係数 (%) 18.2 28.3 42.2 33.8 44.7
最小値 – 最大値0.09939 –0.15453
0.06856 –0.17178
0.07767 –0.20995
0.06048 –0.14347
0.01992 –0.10564
中央値 0.142635 0.130825 0.087470 0.084685 0.062135CLr,u N 8 8 8 8 8
(L/h) 平均値 4.231519 4.108166 3.572636 2.717290 1.970456
標準偏差 0.8142283 0.9902662 1.3508909 0.7915054 0.8833947
変動係数 (%) 19.2 24.1 37.8 29.1 44.8
最小値 – 最大値 3.16853 –5.25989
2.60796 –5.10292
2.63903 –6.65060
1.44199 –4.05212
0.70830 –3.31029
中央値 4.162420 4.246645 2.951725 2.664465 1.938770
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.4.5.1
202

2.7.6個々の試験のまとめ
表 2.7.6.9-8 腎機能障害が未変化体の尿中薬物動態に及ぼす影響[腎機能正常患者群を対照
とした検討](PKAS)
投与群 最小二乗平均 比
比較群/対照群 a
90%信頼区間
CLr
(L/h)腎機能正常患者 0.13 - -
健康被験者 0.13 1.034 [0.760, 1.407]
軽度腎機能低下患者 0.10 0.801 [0.589, 1.090]
中等度腎機能低下患者 0.09 0.730 [0.536, 0.993]
重度腎機能低下患者 0.06 0.446 [0.328, 0.607]
CLr,u
(L/h)腎機能正常患者 3.99 - -
健康被験者 4.16 1.042 [0.781, 1.391]
軽度腎機能低下患者 3.40 0.852 [0.638, 1.137]
中等度腎機能低下患者 2.61 0.653 [0.489, 0.872]
重度腎機能低下患者 1.77 0.442 [0.331, 0.590]
a:対照群:腎機能正常患者群
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.4.6.1.1
表 2.7.6.9-9 腎機能障害が未変化体の尿中薬物動態に及ぼす影響[健康被験者群を対照とし
た検討](PKAS)
投与群 最小二乗平均 比
比較群/対照群 a
90%信頼区間
CLr
(L/h)健康被験者 0.13 - -
腎機能正常患者 0.13 0.967 [0.711, 1.316]
軽度腎機能低下患者 0.10 0.775 [0.569, 1.054]
中等度腎機能低下患者 0.09 0.706 [0.519, 0.960]
重度腎機能低下患者 0.06 0.432 [0.317, 0.587]CLr,u
(L/h)健康被験者 4.16 - -
腎機能正常患者 3.99 0.960 [0.719, 1.281]
軽度腎機能低下患者 3.40 0.817 [0.612, 1.091]
中等度腎機能低下患者 2.61 0.627 [0.470, 0.837]
重度腎機能低下患者 1.77 0.424 [0.318, 0.567]
a:対照群:健康被験者群
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.4.6.1.2
代謝物の薬物動態
(1) 血漿中薬物動態
本剤単回投与時の本薬の代謝物[M1(AS2551864),M2(AS2364093),M3(AS2551840),M4
(AS2551844)及び M6(AS2551868)]の血漿中薬物動態パラメータを表 2.7.6.9-10 に示した。ま
た,腎機能障害が代謝物の血漿中薬物動態に及ぼす影響を評価するため,代謝物のAUCinf及び Cmax
を比較した[表 2.7.6.9-11(腎機能正常患者群を対照とした検討)]。
すべての代謝物の AUCinf が腎機能の低下に伴い上昇したが,M6 では他の代謝物と比較して上
昇の程度が小さかった。2 型糖尿病患者群間の比較では,腎機能正常患者群と比較して,中等度
腎機能低下患者群,重度腎機能低下患者群で AUCinfの上昇が認められた。腎機能正常患者群の
AUCinfの平均値は,健康被験者群と比較して M2,M3,M4 及び M6 で高く,M1 で低かった。
203

2.7.6個々の試験のまとめ
M1,M2,M3 及び M4 の Cmaxは腎機能の低下に伴い上昇した。M6 の Cmax に腎機能の低下に伴
う一定の傾向は認められなかった。M2,M3,M4 及び M6 の Cmaxの平均値は,腎機能が正常の被
験者群間で比較したところ,健康被験者群と比較して,腎機能正常患者群で高かった。対照的に,
M1 の Cmaxは,腎機能正常患者群と比較して,健康被験者群で高かった。
t1/2 は,腎機能障害の程度にかかわらず,すべての代謝物で健康被験者群と比較して 2 型糖尿病
患者群で長くなったが,腎機能の低下に伴う一定の傾向は認められなかった。tmaxは,すべての代
謝物で腎機能正常患者群が最も短かった。また,すべての代謝物で腎機能の低下に伴い tmaxは延
長した。
表 2.7.6.9-10 代謝物の血漿中薬物動態パラメータ(PKAS)(その 1)
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下
M1(AS2551864)AUCinf N 7 7 8 6 8
(ng•h/mL) 平均値 524.031 353.503 582.242 1075.209 2110.726
標準偏差 184.1187 157.1556 464.4589 584.6102 1260.9725
変動係数 (%) 35.1 44.5 79.8 54.4 59.7
最小値 – 最大値 301.35 –808.82
209.03 –637.15
222.58 –1527.17
323.20 –1740.17
363.08 –4427.25
中央値 555.024 268.385 389.785 1238.233 2096.287
Cmax N 8 8 8 8 8
(ng/mL) 平均値 62.438 37.346 53.031 56.660 99.376
標準偏差 17.2876 14.3588 32.4524 23.0839 51.1537
変動係数 (%) 27.7 38.4 61.2 40.7 51.5
最小値 – 最大値 40.48 – 88.16 24.89 – 62.1421.05 –110.94
24.06 – 86.3930.43 –152.93
中央値 60.815 33.025 46.340 60.940 99.105
t1/2 N 7 7 8 6 8(h) 平均値 14.658 23.460 22.715 22.860 21.225
標準偏差 5.1081 12.6997 15.1049 11.8103 7.9391
変動係数 (%) 34.8 54.1 66.5 51.7 37.4
最小値 – 最大値 9.21 – 24.19 9.69 – 43.60 7.80 – 47.69 10.68 – 45.37 11.66 – 35.80
中央値 13.120 25.953 17.973 20.714 20.720
tmax N 8 8 8 8 8(h) 平均値 2.438 1.885 2.563 3.563 6.625
標準偏差 0.8210 0.2140 0.9797 1.2939 4.4701
変動係数 (%) NA NA NA NA NA
最小値 – 最大値 1.50 – 4.00 1.50 – 2.00 1.50 – 4.00 1.50 – 6.00 3.00 – 12.00
中央値 2.000 2.000 2.000 3.500 4.000
204

2.7.6個々の試験のまとめ
表 2.7.6.9-10 代謝物の血漿中薬物動態パラメータ(PKAS)(その 2)
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下
M2(AS2364093)AUCinf N 8 8 8 8 8
(ng•h/mL) 平均値 5065.207 5593.528 8107.285 14992.566 27504.430
標準偏差 1430.0553 1433.8908 3574.8301 5334.7161 9610.9321
変動係数 (%) 28.2 25.6 44.1 35.6 34.9
最小値 – 最大値 2707.97 –7270.88
3277.75 –7153.39
5014.28 –14776.90
8034.16 –21982.60
16559.01 –48198.56
中央値 4789.517 6002.619 6356.160 15267.199 25450.691
Cmax N 8 8 8 8 8
(ng/mL) 平均値 857.709 994.691 1114.923 1258.144 1953.140
標準偏差 262.6935 275.5752 327.5426 310.6905 474.9321
変動係数 (%) 30.6 27.7 29.4 24.7 24.3
最小値 – 最大値 431.09 –1226.00
580.93 –1356.47
757.35 –1621.64
753.52 –1670.20
1219.83 –2645.44
中央値 936.260 962.810 1058.500 1262.015 1921.250
t1/2 N 8 8 8 8 8(h) 平均値 17.132 20.801 22.116 20.020 20.141
標準偏差 9.0920 5.4400 11.1720 10.2312 5.6481
変動係数 (%) 53.1 26.2 50.5 51.1 28.0
最小値 – 最大値 7.83 – 31.76 14.83 – 29.52 11.89 – 45.51 10.66 – 39.46 13.67 – 28.15
中央値 14.096 18.445 19.341 17.059 20.405
tmax N 8 8 8 8 8
(h) 平均値 2.188 1.760 1.823 2.250 2.750
標準偏差 0.9234 0.2579 0.5307 0.4629 1.3887
変動係数 (%) NA NA NA NA NA
最小値 – 最大値 1.00 – 4.00 1.50 – 2.00 1.50 – 3.00 2.00 – 3.00 2.00 – 6.00
中央値 2.000 1.790 1.515 2.000 2.000
205

2.7.6個々の試験のまとめ
表 2.7.6.9-10 代謝物の血漿中薬物動態パラメータ(PKAS)(その 3)
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下
M3(AS2551840)AUCinf N 8 8 8 7 8
(ng•h/mL) 平均値 1404.335 1601.336 2015.736 4622.515 9094.693
標準偏差 550.6940 574.3382 868.7633 1135.3936 5174.1072
変動係数 (%) 39.2 35.9 43.1 24.6 56.9
最小値 – 最大値 656.21 –2249.84
613.04 –2611.52
963.16 –3289.79
3174.08 –6143.45
4067.48 –19944.21
中央値 1349.996 1611.134 1808.901 4693.991 7498.370
Cmax N 8 8 8 8 8
(ng/mL) 平均値 164.768 177.233 179.496 205.128 337.553
標準偏差 45.7205 53.6900 54.4044 53.5775 248.9196
変動係数 (%) 27.7 30.3 30.3 26.1 73.7
最小値 – 最大値 82.74 –217.62
71.47 –235.48
102.16 –270.88
107.90 –271.61
193.65 –941.84
中央値 185.555 179.515 189.330 197.770 247.735
t1/2 N 8 8 8 7 8(h) 平均値 16.169 21.241 20.776 20.795 20.473
標準偏差 9.1774 7.7935 10.9805 9.3932 4.4671
変動係数 (%) 56.8 36.7 52.9 45.2 21.8
最小値 – 最大値 8.58 – 32.25 8.46 – 29.65 7.34 – 41.69 10.05 – 38.15 13.88 – 27.05
中央値 12.438 21.418 19.791 17.758 21.578
tmax N 8 8 8 8 8
(h) 平均値 2.500 2.010 2.375 3.000 6.750
標準偏差 0.7559 0.4513 0.7440 1.4142 5.6252
変動係数 (%) NA NA NA NA NA
最小値 – 最大値 2.00 – 4.00 1.50 – 3.00 2.00 – 4.00 2.00 – 6.00 2.00 – 16.00
中央値 2.000 2.000 2.000 2.500 3.500
206

2.7.6個々の試験のまとめ
表 2.7.6.9-10 代謝物の血漿中薬物動態パラメータ(PKAS)(その 4)
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下
M4(AS2551844)AUCinf N 8 8 8 7 8
(ng•h/mL) 平均値 1063.771 1422.534 1679.322 2977.443 4979.150
標準偏差 52.9598 462.5122 705.2593 1093.2382 2918.6537
変動係数 (%) 5.0 32.5 42.0 36.7 58.6
最小値 – 最大値 1002.71 –1133.66
738.34 –1962.52
1022.57 –2816.06
1606.24 –4612.02
1784.65 –11579.47
中央値 1053.981 1394.447 1408.699 3100.437 4119.224
Cmax N 8 8 8 8 8
(ng/mL) 平均値 167.896 229.161 232.501 279.216 384.008
標準偏差 44.2819 76.6028 74.0812 91.4461 125.4112
変動係数 (%) 26.4 33.4 31.9 32.8 32.7
最小値 – 最大値 125.02 –249.30
107.66 –349.76
142.47 –360.19
157.58 –412.96
192.26 –641.28
中央値 154.000 241.980 234.780 281.700 366.540
t1/2 N 8 8 8 7 8(h) 平均値 15.292 19.722 19.466 22.271 21.055
標準偏差 8.0731 7.2052 7.9038 8.8969 6.7188
変動係数 (%) 52.8 36.5 40.6 39.9 31.9
最小値 – 最大値 8.32 – 33.26 10.19 – 30.06 9.41 – 30.59 9.14 – 34.97 13.75 – 31.49
中央値 12.580 18.940 18.757 19.169 20.594
tmax N 8 8 8 8 8
(h) 平均値 2.188 1.698 1.823 2.063 2.313
標準偏差 0.9234 0.2519 0.5307 0.8210 1.6022
変動係数 (%) NA NA NA NA NA
最小値 – 最大値 1.00 – 4.00 1.50 – 2.00 1.50 – 3.00 1.50 – 4.00 1.00 – 6.00
中央値 2.000 1.540 1.515 2.000 1.750
207

2.7.6個々の試験のまとめ
表 2.7.6.9-10 代謝物の血漿中薬物動態パラメータ(PKAS)(その 5)
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下
M6(AS2551868)AUCinf N 8 8 8 7 8
(ng•h/mL) 平均値 677.573 862.888 1140.298 1133.302 1079.828
標準偏差 265.4408 456.4230 745.2005 666.3183 472.9215
変動係数 (%) 39.2 52.9 65.4 58.8 43.8
最小値 – 最大値 331.51 –1044.88
286.43 –1620.06
273.63 –2590.57
392.19 –2174.59
276.90 –1770.94
中央値 660.540 753.943 852.940 942.238 1075.706
Cmax N 8 8 8 8 8
(ng/mL) 平均値 48.850 55.301 66.558 45.130 40.874
標準偏差 25.0819 34.4156 46.9921 33.8740 15.3760
変動係数 (%) 51.3 62.2 70.6 75.1 37.6
最小値 – 最大値 22.21 – 83.2923.56 –129.80
23.88 –166.78
19.64 –117.24
15.66 – 55.90
中央値 45.880 43.645 51.360 34.650 47.145
t1/2 N 8 8 8 7 8(h) 平均値 19.309 27.667 27.033 30.370 27.046
標準偏差 9.1180 10.8535 13.9276 14.9356 6.7560
変動係数 (%) 47.2 39.2 51.5 49.2 25.0
最小値 – 最大値 10.44 – 37.65 11.10 – 41.56 7.32 – 48.16 11.95 – 56.42 15.40 – 35.74
中央値 16.534 29.291 25.915 25.524 28.487
tmax N 8 8 8 8 8
(h) 平均値 3.000 2.198 2.688 2.750 5.125
標準偏差 1.3093 0.5161 0.5939 0.8864 4.2908
変動係数 (%) NA NA NA NA NA
最小値 – 最大値 2.00 – 6.00 1.58 – 3.00 1.50 – 3.00 2.00 – 4.00 2.00 – 12.00
中央値 3.000 2.000 3.000 2.500 3.000
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.4.4.1
208

2.7.6個々の試験のまとめ
表 2.7.6.9-11 腎機能障害が代謝物の血漿中薬物動態に及ぼす影響[腎機能正常患者群を対照
とした検討](PKAS)(その 1)
投与群 最小二乗平均 比
比較群/対照群 a
90%信頼区間
M1 (AS2551864)AUCinf
(ng•h/mL)腎機能正常患者 327.13 - -
健康被験者 494.76 1.512 [0.860, 2.660]
軽度腎機能低下患者 460.49 1.408 [0.815, 2.432]
中等度腎機能低下患者 899.35 2.749 [1.527, 4.948]
重度腎機能低下患者 1716.06 5.246 [3.036, 9.062]
Cmax
(ng/mL)腎機能正常患者 35.21 - -
健康被験者 60.36 1.714 [1.140, 2.578]
軽度腎機能低下患者 45.27 1.286 [0.855, 1.933]
中等度腎機能低下患者 51.99 1.477 [0.982, 2.220]
重度腎機能低下患者 85.72 2.435 [1.619, 3.661]
M2 (AS2364093)AUCinf
(ng•h/mL)腎機能正常患者 5409.17 - -
健康被験者 4873.87 0.901 [0.676, 1.202]
軽度腎機能低下患者 7519.26 1.390 [1.042, 1.854]
中等度腎機能低下患者 14127.01 2.612 [1.958, 3.483]
重度腎機能低下患者 26248.10 4.853 [3.638, 6.472]
Cmax
(ng/mL)腎機能正常患者 959.17 - -
健康被験者 817.33 0.852 [0.665, 1.091]
軽度腎機能低下患者 1073.79 1.120 [0.874, 1.434]
中等度腎機能低下患者 1221.52 1.274 [0.994, 1.631]
重度腎機能低下患者 1900.56 1.981 [1.547, 2.538]
M3 (AS2551840)AUCinf
(ng•h/mL)腎機能正常患者 1495.68 - -
健康被験者 1306.04 0.873 [0.609, 1.252]
軽度腎機能低下患者 1855.83 1.241 [0.865, 1.779]
中等度腎機能低下患者 4497.78 3.007 [2.070, 4.368]
重度腎機能低下患者 8007.08 5.353 [3.733, 7.678]
Cmax
(ng/mL)腎機能正常患者 167.81 - -
健康被験者 158.09 0.942 [0.687, 1.293]
軽度腎機能低下患者 171.95 1.025 [0.747, 1.406]
中等度腎機能低下患者 198.11 1.181 [0.860, 1.620]
重度腎機能低下患者 291.83 1.739 [1.267, 2.386]
M4 (AS2551844)AUCinf
(ng•h/mL)腎機能正常患者 1350.95 - -
健康被験者 1062.62 0.787 [0.573, 1.080]
軽度腎機能低下患者 1569.26 1.162 [0.846, 1.595]
中等度腎機能低下患者 2795.32 2.069 [1.490, 2.873]
重度腎機能低下患者 4390.57 3.250 [2.367, 4.462]
Cmax
(ng/mL)腎機能正常患者 216.50 - -
健康被験者 163.27 0.754 [0.571, 0.996]
軽度腎機能低下患者 222.28 1.027 [0.777, 1.356]
中等度腎機能低下患者 265.19 1.225 [0.927, 1.618]
重度腎機能低下患者 366.42 1.692 [1.281, 2.235]
a:対照群:腎機能正常患者群
209

2.7.6個々の試験のまとめ
表 2.7.6.9-11 腎機能障害が代謝物の血漿中薬物動態に及ぼす影響[腎機能正常患者群を対照
とした検討](PKAS)(その 2)
投与群 最小二乗平均 比
比較群/対照群 a
90%信頼区間
M6 (AS2551868)AUCinf
(ng•h/mL)腎機能正常患者 754.11 - -
健康被験者 629.51 0.835 [0.509, 1.370]
軽度腎機能低下患者 940.64 1.247 [0.760, 2.048]
中等度腎機能低下患者 960.31 1.273 [0.762, 2.127]
重度腎機能低下患者 959.61 1.273 [0.775, 2.089]
Cmax
(ng/mL)腎機能正常患者 48.22 - -
健康被験者 42.92 0.890 [0.547, 1.449]
軽度腎機能低下患者 55.31 1.147 [0.705, 1.867]
中等度腎機能低下患者 36.57 0.758 [0.466, 1.234]
重度腎機能低下患者 37.69 0.782 [0.480, 1.272]
a:対照群:腎機能正常患者群
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.4.6.2
(2) 尿中薬物動態
本薬の代謝物の尿中薬物動態パラメータを表 2.7.6.9-12 に示した。
M1,M2,M3 及び M6 の尿中排泄量に腎機能の低下に伴う一定の傾向はみられなかったが,M4
の尿中排泄量は腎機能の低下に伴い減少した。代謝物の CLr は,腎機能の低下に伴い低下し,ま
た健康被験者群と比較して腎機能正常患者群で低かった。
210

2.7.6個々の試験のまとめ
表 2.7.6.9-12 代謝物の尿中薬物動態パラメータ(PKAS)(その 1)
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下
M1(AS2551864)Ae24h N 8 8 8 8 8
(mg) 平均値 5.008644 2.401496 3.726084 2.860389 2.344175
標準偏差 1.0536459 0.5801238 2.1934904 1.1408253 1.1285000
変動係数 (%) 21.0 24.2 58.9 39.9 48.1
最小値 – 最大値 3.41408 –6.50490
1.43790 –3.13545
1.41592 –7.83197
1.42848 –4.52256
0.83018 –4.16697
中央値 5.055525 2.647315 2.892165 2.719720 2.184825
Aeinf N 7 7 8 6 8
(mg) 平均値 6.240507 3.208811 4.867631 4.036240 3.479378
標準偏差 1.0250425 0.6523469 2.9162013 1.3475528 1.8806198
変動係数 (%) 16.4 20.3 59.9 33.4 54.1
最小値 – 最大値 4.78082 –7.79703
2.50088 –4.34016
2.00823 –9.81374
2.23572 –6.10378
1.10412 –6.44228
中央値 6.296070 3.092240 3.603440 3.993515 2.915185
Aeinf% N 7 7 8 6 8(%) 平均値 4.216561 2.168113 3.288940 2.727188 2.350930
標準偏差 0.6925967 0.4407768 1.9704065 0.9105069 1.2706898
変動係数 (%) 16.4 20.3 59.9 33.4 54.1
最小値 – 最大値 3.23029 –5.26827
1.68978 –2.93254
1.35691 –6.63091
1.51062 –4.12417
0.74603 –4.35289
中央値 4.254100 2.089350 2.434760 2.698320 1.969715
Aelast N 8 8 8 8 8
(mg) 平均値 5.893903 3.077070 4.695001 3.961566 3.382675
標準偏差 1.1514283 0.5455997 2.7871173 1.4967149 1.8529314
変動係数 (%) 19.5 17.7 59.4 37.8 54.8
最小値 – 最大値 4.11839 –7.50013
2.23770 –3.98717
1.99683 –9.77745
2.21458 –5.97970
1.09655 –6.27657
中央値 6.025295 3.145905 3.605130 3.736735 2.802200
Aelast% N 8 8 8 8 8
(%) 平均値 3.982369 2.079104 3.172298 2.676735 2.285594
標準偏差 0.7779934 0.3686503 1.8831865 1.0112924 1.2519815
変動係数 (%) 19.5 17.7 59.4 37.8 54.8
最小値 – 最大値 2.78270 –5.06766
1.51196 –2.69404
1.34921 –6.60638
1.49634 –4.04034
0.74091 –4.24093
中央値 4.071145 2.125615 2.435900 2.524820 1.893380CLr N 8 8 8 8 8
(L/h) 平均値 12.846555 11.056996 9.220338 4.409704 1.898553
標準偏差 3.9867878 3.9815731 1.9279405 1.8589834 0.7250956
変動係数 (%) 31.0 36.0 20.9 42.2 38.2
最小値 – 最大値 8.34251 –19.40528
3.92512 –15.49178
6.42610 –11.97261
2.12417 –7.52557
0.94871 –3.04099
中央値 11.728180 11.990700 8.883520 3.973485 1.935100
211

2.7.6個々の試験のまとめ
表 2.7.6.9-12 代謝物の尿中薬物動態パラメータ(PKAS)(その 2)
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下
M2(AS2364093)Ae24h N 8 8 8 8 8
(mg) 平均値 48.591860 41.348976 52.466136 41.863379 36.954035
標準偏差 7.2320285 10.6630862 23.9302286 14.5256542 11.9882907
変動係数 (%) 14.9 25.8 45.6 34.7 32.4
最小値 – 最大値 37.52395 –59.70562
20.08561 –57.46625
32.44330 –106.67348
22.06813 –66.78414
13.37521 –51.76757
中央値 47.160965 41.205975 44.083915 40.223155 39.166795
Aeinf N 8 8 8 8 8
(mg) 平均値 57.125655 52.621751 64.205714 55.887333 50.679905
標準偏差 8.0358426 12.7325896 25.5157207 15.8864194 15.4720155
変動係数 (%) 14.1 24.2 39.7 28.4 30.5
最小値 – 最大値 45.97638 –70.86811
31.12958 –71.59054
44.46717 –122.81833
35.96420 –85.19066
19.93470 –65.34692
中央値 58.620540 51.581740 55.439410 54.984970 52.512900
Aeinf% N 8 8 8 8 8(%) 平均値 39.670595 36.542885 44.587301 38.810648 35.194379
標準偏差 5.5804480 8.8420748 17.7192516 11.0322369 10.7444567
変動係数 (%) 14.1 24.2 39.7 28.4 30.5
最小値 – 最大値 31.92804 –49.21397
21.61777 –49.71566
30.87998 –85.29051
24.97514 –59.16018
13.84354 –45.37981
中央値 40.708710 35.820655 38.499590 38.184010 36.467290
Aelast N 8 8 8 8 8
(mg) 平均値 56.767536 52.090390 63.376963 55.015294 49.918905
標準偏差 7.9853917 12.7117841 25.0868459 15.8136686 15.0673917
変動係数 (%) 14.1 24.4 39.6 28.7 30.2
最小値 – 最大値 45.04233 –70.62698
30.14365 –71.22160
43.42380 –120.58218
35.62564 –84.41992
19.74374 –64.08133
中央値 58.299455 51.326175 53.938160 53.372035 51.540220
Aelast% N 8 8 8 8 8
(%) 平均値 39.421903 36.173879 44.011780 38.205065 34.665905
標準偏差 5.5454113 8.8276262 17.4214220 10.9817141 10.4634663
変動係数 (%) 14.1 24.4 39.6 28.7 30.2
最小値 – 最大値 31.27940 –49.04652
20.93309 –49.45944
30.15542 –83.73763
24.74003 –58.62494
13.71093 –44.50092
中央値 40.485735 35.643175 37.457055 37.063915 35.791820CLr N 8 8 8 8 8
(L/h) 平均値 12.153141 10.117528 8.378164 3.914731 1.948303
標準偏差 3.8790602 3.7933829 2.1996720 0.9332343 0.6525827
変動係数 (%) 31.9 37.5 26.3 23.8 33.5
最小値 – 最大値 6.70158 –19.09441
4.35173 –17.21930
4.73199 –11.53484
2.51663 –5.66074
0.78823 –2.68997
中央値 11.415110 10.030440 8.625285 3.728545 2.141230
212

2.7.6個々の試験のまとめ
表 2.7.6.9-12 代謝物の尿中薬物動態パラメータ(PKAS)(その 3)
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下
M3(AS2551840)Ae24h N 8 8 8 8 8
(mg) 平均値 4.483549 3.484053 4.175030 3.546488 2.786348
標準偏差 0.5333329 1.1486218 2.3915432 1.0270254 1.1878864
変動係数 (%) 11.9 33.0 57.3 29.0 42.6
最小値 – 最大値 3.55681 –5.09661
1.70202 –5.20721
1.68811 –9.78260
1.84944 –4.99047
1.15340 –5.15703
中央値 4.427345 3.314595 3.775665 3.318460 2.568480
Aeinf N 8 8 8 7 8
(mg) 平均値 5.352061 4.581971 5.319264 5.738139 4.686029
標準偏差 0.7409314 1.3003707 2.6006180 0.9600225 1.8602740
変動係数 (%) 13.8 28.4 48.9 16.7 39.7
最小値 – 最大値 4.02492 –6.11100
2.81087 –6.62760
2.45836 –11.26656
4.70218 –7.15271
1.85985 –8.14551
中央値 5.413650 4.742125 5.078340 5.378860 4.732430
Aeinf% N 8 8 8 7 8(%) 平均値 3.716710 3.181924 3.693934 3.984819 3.254185
標準偏差 0.5145353 0.9030370 1.8059839 0.6666802 1.2918560
変動係数 (%) 13.8 28.4 48.9 16.7 39.7
最小値 – 最大値 2.79509 –4.24375
1.95199 –4.60250
1.70720 –7.82400
3.26541 –4.96716
1.29156 –5.65660
中央値 3.759480 3.293140 3.526625 3.735320 3.286405
Aelast N 8 8 8 8 8
(mg) 平均値 5.270416 4.442085 5.138973 5.203904 4.516475
標準偏差 0.7140299 1.2415158 2.6237785 1.3181800 1.8206232
変動係数 (%) 13.5 27.9 51.1 25.3 40.3
最小値 – 最大値 3.94900 –5.98232
2.66860 –6.49730
2.12356 –11.09700
2.75477 –6.88112
1.79126 –7.92239
中央値 5.411265 4.499025 4.956395 5.174865 4.514465
Aelast% N 8 8 8 8 8
(%) 平均値 3.660011 3.084780 3.568731 3.613821 3.136443
標準偏差 0.4958560 0.8621637 1.8220674 0.9154053 1.2643221
変動係数 (%) 13.5 27.9 51.1 25.3 40.3
最小値 – 最大値 2.74236 –4.15439
1.85319 –4.51201
1.47470 –7.70625
1.91303 –4.77856
1.24393 –5.50166
中央値 3.757820 3.124325 3.441940 3.593655 3.135045CLr N 8 8 8 8 8
(L/h) 平均値 4.265645 3.226939 2.699215 1.254275 0.581138
標準偏差 1.4111232 1.2992937 0.6269870 0.2762124 0.2302383
変動係数 (%) 33.1 40.3 23.2 22.0 39.6
最小値 – 最大値 2.54827 –6.23598
1.07634 –5.45050
1.63995 –3.65192
0.77002 –1.66627
0.27231 –1.00748
中央値 4.036660 3.202225 2.626345 1.261980 0.550755
213

2.7.6個々の試験のまとめ
表 2.7.6.9-12 代謝物の尿中薬物動態パラメータ(PKAS)(その 4)
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下
M4(AS2551844)Ae24h N 8 8 8 8 8
(mg) 平均値 12.998706 13.350201 13.564178 9.651818 6.156528
標準偏差 2.9379189 4.0832638 5.1238791 2.4156924 2.2491667
変動係数 (%) 22.6 30.6 37.8 25.0 36.5
最小値 – 最大値 8.57704 –17.05243
7.31871 –18.22681
5.68354 –21.80267
6.89128 –13.09843
1.70695 –9.18036
中央値 13.398415 13.917635 13.340630 9.532860 6.751720
Aeinf N 8 8 8 7 8
(mg) 平均値 15.547230 16.868891 16.562061 13.736727 8.219405
標準偏差 3.1919672 4.6118402 5.4687849 3.4290955 3.0199011
変動係数 (%) 20.5 27.3 33.0 25.0 36.7
最小値 – 最大値 10.31665 –19.30361
10.87620 –22.86460
7.81790 –25.15146
9.62390 –20.75333
2.63369 –13.27615
中央値 16.842150 17.858900 17.484555 12.792040 8.925230
Aeinf% N 8 8 8 7 8(%) 平均値 10.796688 11.714508 11.501431 9.539394 5.707920
標準偏差 2.2166429 3.2026651 3.7977672 2.3813191 2.0971544
変動係数 (%) 20.5 27.3 33.0 25.0 36.7
最小値 – 最大値 7.16434 –13.40528
7.55292 –15.87819
5.42910 –17.46629
6.68326 –14.41204
1.82895 –9.21955
中央値 11.695940 12.402010 12.142055 8.883360 6.198080
Aelast N 8 8 8 8 8
(mg) 平均値 15.439983 16.789138 16.264439 12.860086 8.004068
標準偏差 3.1096308 4.8426632 5.3841085 3.4607192 2.9378601
変動係数 (%) 20.1 28.8 33.1 26.9 36.7
最小値 – 最大値 10.43955 –19.36457
10.67415 –23.83259
7.79237 –24.69506
8.92547 –19.99866
2.56720 –12.86556
中央値 16.608510 17.261285 17.278740 12.525315 8.728170
Aelast% N 8 8 8 8 8
(%) 平均値 10.722210 11.659121 11.294749 8.930615 5.558381
標準偏差 2.1594651 3.3629610 3.7389636 2.4032766 2.0401802
変動係数 (%) 20.1 28.8 33.1 26.9 36.7
最小値 – 最大値 7.24969 –13.44762
7.41260 –16.55041
5.41137 –17.14935
6.19825 –13.88796
1.78278 –8.93442
中央値 11.533685 11.987000 11.999120 8.698130 6.061230CLr N 8 8 8 8 8
(L/h) 平均値 14.676853 13.496843 10.777331 4.992479 1.957906
標準偏差 3.1997026 7.6933237 3.8447605 2.0113619 0.9549672
変動係数 (%) 21.8 57.0 35.7 40.3 48.8
最小値 – 最大値 9.10029 –18.81273
5.62158 –30.96772
3.72405 –15.33155
2.48202 –7.75186
0.70095 –3.65490
中央値 15.793445 11.374540 11.454370 4.561915 1.985410
214

2.7.6個々の試験のまとめ
表 2.7.6.9-12 代謝物の尿中薬物動態パラメータ(PKAS)(その 5)
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下
M6(AS2551868)Ae24h N 3 1 1 - -
(mg) 平均値 0.822737 1.152000 1.140770 - -
標準偏差 0.0393973 - - - -
変動係数 (%) 4.8 - - - -
最小値 – 最大値 0.79207 –0.86717
1.15200 –1.15200
1.14077 –1.14077
- -
中央値 0.808970 1.152000 1.140770 - -
Aeinf N 3 1 1 - -
(mg) 平均値 1.189923 1.899630 2.073190 - -
標準偏差 0.1498344 - - - -
変動係数 (%) 12.6 - - - -
最小値 – 最大値 1.02376 –1.31475
1.89963 –1.89963
2.07319 –2.07319
- -
中央値 1.231260 1.899630 2.073190 - -
Aeinf% N 3 1 1 - -(%) 平均値 0.959617 1.531960 1.671930 - -
標準偏差 0.1208388 - - - -
変動係数 (%) 12.6 - - - -
最小値 – 最大値 0.82561 –1.06029
1.53196 –1.53196
1.67193 –1.67193
- -
中央値 0.992950 1.531960 1.671930 - -
Aelast N 3 1 1 - -
(mg) 平均値 1.163237 1.638690 1.870220 - -
標準偏差 0.1736067 - - - -
変動係数 (%) 14.9 - - - -
最小値 – 最大値 0.97277 –1.31261
1.63869 –1.63869
1.87022 –1.87022
- -
中央値 1.204330 1.638690 1.870220 - -
Aelast% N 3 1 1 - -
(%) 平均値 0.938090 1.321530 1.508240 - -
標準偏差 0.1400033 - - - -
変動係数 (%) 14.9 - - - -
最小値 – 最大値 0.78449 –1.05855
1.32153 –1.32153
1.50824 –1.50824
- -
中央値 0.971230 1.321530 1.508240 - -CLr N 3 1 1 - -
(L/h) 平均値 1.528113 1.414710 0.800290 - -
標準偏差 0.3439962 - - - -
変動係数 (%) 22.5 - - - -
最小値 – 最大値 1.17838 –1.86607
1.41471 –1.41471
0.80029 –0.80029
- -
中央値 1.539890 1.414710 0.800290 - -
注:M6 濃度は,大部分の尿検体で定量限界未満であったため,濃度データが得られなかった。
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.4.5.1
215

2.7.6個々の試験のまとめ
2.7.6.9.5.2 薬力学の結果
血糖値
血糖値パラメータを表 2.7.6.9-13 に示した。
Day 1 の血糖値の AUC(AUC4-24h)は Day −1(ベースライン,AUC−20-0h)と比較して変化がな
かった。Day −1 及び Day 1 のいずれでも,2 型糖尿病患者群では健康被験者群と比較して血糖値
の AUC の平均値が高かった。2 型糖尿病患者群での血糖値の AUC の変動係数(Day −1 の変動係
数:18.0%~36.0%)は,健康被験者群(Day −1 の変動係数:6.9%)と比較して大きかった。血糖
値の AUC の変化に腎機能の低下に伴う一定の傾向はみられなかった。
表 2.7.6.9-13 血糖値パラメータ(PDAS)
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下AUC−20-0h
(h•mmol/L)N 8 8 8 8 8
平均値 109.1775 142.4400 157.8636 158.0600 150.0425
標準偏差 7.48529 25.61387 38.78554 55.19743 54.01450
変動係数 (%) 6.9 18.0 24.6 34.9 36.0
最小値 – 最大値 99.560 –119.320
111.800 –176.920
122.320 –233.120
88.940 –235.000
94.720 –229.440
中央値 109.6500 132.6400 141.3000 143.6100 134.5600
AUC4-24h
(h•mmol/L)N 8 8 8 8 8
平均値 110.0050 132.8741 153.6550 163.4301 151.5875
標準偏差 9.45671 12.63190 33.24504 43.17668 28.24882
変動係数 (%) 8.6 9.5 21.6 26.4 18.6
最小値 – 最大値 93.760 –119.980
117.760 –155.060
118.080 –203.240
107.980 –226.540
120.200 –198.720
中央値 113.1500 127.8500 146.4200 155.4500 143.1500
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.5.2
尿中グルコース
尿中グルコースパラメータを表 2.7.6.9-14 に示した。また,腎機能障害が尿中グルコース排泄に
及ぼす影響を評価するため,尿中グルコースパラメータの投与前後の変化を腎機能正常患者群と
腎機能障害を有する 2 型糖尿病患者群間で比較した(表 2.7.6.9-15)。
腎機能障害の程度にかかわらず,Day −1(ベースライン)の尿中グルコース排泄量(Ae−20-0h)
は健康被験者群と比較して 2 型糖尿病患者群で多かった。Day 1(治験薬投与後)の尿中グルコー
ス排泄量(Ae4-24h)は,すべての投与群で Day −1 と比較して増加した。ベースラインからの Day 1
時点の尿中グルコース排泄量の変化量を腎機能正常患者群と比較したところ,軽度腎機能低下患
者群で大きかった。中等度腎機能低下患者群及び重度腎機能低下患者群では,腎機能正常患者群
と比較して,ベースラインからの変化量は小さかった。尿中グルコース排泄量の増加量は腎機能
の低下に伴い減少し,腎臓の濾過能の低下と一致していた。
ベースラインのグルコースの CLr は,腎機能障害の程度にかかわらず,健康被験者群と比較し
て 2 型糖尿病患者群で高かった。グルコースの CLr は,腎機能障害の程度にかかわらず,すべて
216

2.7.6個々の試験のまとめ
の投与群で治験薬投与後に増加したが,中等度腎機能低下患者群及び重度腎機能低下患者群では,
ベースラインからの変化量が腎機能正常患者群と比較して小さかった。本剤投与後のグルコース
の CLr は,健康被験者群,腎機能正常患者群及び軽度腎機能低下患者群で同程度であった。グル
コースの CLr の変化量は,重度腎機能低下患者群,中等度腎機能低下患者群,腎機能正常患者群,
軽度腎機能低下患者群,健康被験者群の順に小さかった。
表 2.7.6.9-14 尿中グルコースパラメータ(PDAS)
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下Ae−20-0h
(mmol)N 7 8 8 8 8
平均値 0.86963 6.15360 19.79820 5.86891 8.96974
標準偏差 1.39283 15.11480 43.57834 9.07062 16.42366
最小値 – 最大値 0.03757 –3.99477
0.16980 –43.51920
0.15235 –125.11435
0.08905 –24.38750
0.09984 –42.00846
中央値 0.44094 0.77266 0.60738 0.43771 0.47454
Ae4-24h
(mmol)N 8 8 8 7 8
平均値 243.14701 271.13316 361.20265 124.43988 66.83415
標準偏差 100.27000 108.39793 199.71519 93.90385 48.63903
最小値 – 最大値 112.31388 –407.39040
90.71481 –388.49440
159.81470 –733.82807
42.25110 –312.37615
18.97797 –163.90824
中央値 230.59235 297.23274 358.94895 89.14985 54.50866
変化 a 253.29127 264.97956 341.40445 117.77785 57.86441
CLr −20-0h
(L/h)N 7 8 8 8 8
平均値 0.00746 0.03730 0.08927 0.02668 0.04053
標準偏差 0.01160 0.09011 0.18690 0.03877 0.07196
最小値 – 最大値 0.00037 –0.03348
0.00133 –0.26010
0.00125 –0.53670
0.00070 –0.10543
0.00087 –0.18646
中央値 0.00392 0.00525 0.00461 0.00319 0.00388
CLr 4-24h
(L/h)N 8 8 8 7 8
平均値 2.20276 2.02050 2.34209 0.71525 0.41537
標準偏差 0.86439 0.75461 1.37655 0.51609 0.22616
最小値 – 最大値 0.98039 –3.76030
0.73098 –3.01263
1.30291 –5.47143
0.32186 –1.83190
0.13885 –0.82482
中央値 2.19502 2.25481 2.00445 0.59257 0.38495
変化 b 2.30364 1.98319 2.25281 0.68515 0.37483
a:変化(Ae4-24h − Ae−20-0h)の平均値
b:変化(CLr 4-24h − CLr −20-0h)の平均値
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.5.4
217

2.7.6個々の試験のまとめ
表 2.7.6.9-15 腎機能障害が尿中グルコースパラメータに及ぼす影響(PDAS)
投与群 最小二乗
平均
差
比較群−対照群 a
90%信頼区間
Ae4-24h − Ae−20-0h 腎機能正常患者 264.98 - -
軽度腎機能低下患者 341.40 76.42 [−26.39, 179.24]
中等度腎機能低下患者 117.78 −147.20 [−253.63, −40.78]
重度腎機能低下患者 57.86 −207.12 [−309.93, −104.30]
CLr 4-24h − CLr −20-0h 腎機能正常患者 1.98 - -
軽度腎機能低下患者 2.25 0.27 [−0.44, 0.98]
中等度腎機能低下患者 0.69 −1.30 [−2.03, −0.56]
重度腎機能低下患者 0.37 −1.61 [−2.32, −0.90]
a:対照群:腎機能正常患者群
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.5.5.1.1
2.7.6.9.6 安全性
2.7.6.9.6.1 有害事象
本試験で認められた有害事象の要約を表 2.7.6.9-16 に示した。
有害事象は,健康被験者群 1 例(12.5%),中等度腎機能低下患者群 2 例(25.0%)に発現した。
副作用は認められなかった。死亡,重篤な有害事象及び投与中止に至った有害事象は認められな
かった。
表 2.7.6.9-16 有害事象の要約(SAF)
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下(N=8) (N=8) (N=8) (N=8) (N=8)
有害事象発現例数(発現率) 1(12.5%) 0 0 2(25.0%) 0
有害事象発現件数 3 0 0 4 0
副作用発現例数(発現率) 0 0 0 0 0
副作用発現件数 0 0 0 0 0
死亡発現例数(発現率) 0 0 0 0 0
重篤な有害事象発現例数(発現率) 0 0 0 0 0
重篤な有害事象発現件数 0 0 0 0 0
重篤な副作用発現例数(発現率) 0 0 0 0 0
重篤な副作用発現件数 0 0 0 0 0
中止に至った有害事象発現例数
(発現率)0 0 0 0 0
中止に至った有害事象発現件数 0 0 0 0 0
中止に至った副作用発現例数(発
現率)0 0 0 0 0
中止に至った副作用発現件数 0 0 0 0 0
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.6.1.1
有害事象の内訳を表 2.7.6.9-17 及び表 2.7.6.9-18 に示した。
218

2.7.6個々の試験のまとめ
健康被験者群 1 例に頭痛が 2 件,嘔吐が 1 件認められた。いずれの事象も軽度であり,治験薬
との関連性は否定された。頭痛では薬物治療が行われ,また,嘔吐では無処置で回復が確認され
た。その他の投与群では,中等度腎機能低下患者群 2 例に有害事象が認められた。1 例では,頭
痛が 2 件,高血圧が 1 件認められた。いずれの事象も軽度であり,治験薬との関連性は否定され
た。いずれも薬物治療が行われ,回復が確認された。他の 1 例では,下痢が 1 件認められた。軽
度であり,治験薬との関連性は否定された。無処置にて回復が確認された。
表 2.7.6.9-17 有害事象の内訳(SAF)
MedDRA version 12.0器官別大分類
基本語
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下(N=8) (N=8) (N=8) (N=8) (N=8)
すべての有害事象 1(12.5%) 0 0 2(25.0%) 0
胃腸障害 1(12.5%) 0 0 1(12.5%) 0
下痢 0 0 0 1(12.5%) 0
嘔吐 1(12.5%) 0 0 0 0
神経系障害 1(12.5%) 0 0 1(12.5%) 0
頭痛 1(12.5%) 0 0 1(12.5%) 0
血管障害 0 0 0 1(12.5%) 0
高血圧 0 0 0 1(12.5%) 0
発現例数(発現率)
Source:CL-0064 総括報告書(5.3.3.3-3)Table 12.6.1.2
表 2.7.6.9-18 有害事象の内訳(件数)(SAF)
MedDRA version 12.0
器官別大分類
基本語
健康被験者 2 型糖尿病患者
腎機能正常 軽度
腎機能低下
中等度
腎機能低下
重度
腎機能低下
すべての有害事象 3 0 0 4 0
胃腸障害 1 0 0 1 0
下痢 0 0 0 1 0
嘔吐 1 0 0 0 0
神経系障害 2 0 0 2 0
頭痛 2 0 0 2 0
血管障害 0 0 0 1 0
高血圧 0 0 0 1 0
Source:CL-0064 総括報告書(5.3.3.3-3)Appendix13.2.7.1
2.7.6.9.6.2 死亡,その他の重篤な有害事象
本試験で,死亡及びその他の重篤な有害事象は認められなかった。
2.7.6.9.6.3 中止に至った有害事象
本試験で,中止に至った有害事象は認められなかった。
219

2.7.6個々の試験のまとめ
2.7.6.9.6.4 注目すべき有害事象
低血糖の発現を示唆する有害事象は認められなかった。
2.7.6.9.6.5 臨床検査値
臨床検査では,臨床的に重要な変化は観察されなかった。
2.7.6.9.6.6 バイタルサイン
バイタルサインに臨床的に問題となるような変動は観察されなかった。
2.7.6.9.6.7 心電図
健康被験者群の 2 例(25%),腎機能正常患者群の 5 例(62.5%),軽度腎機能低下患者群の 6 例
(75%),中等度腎機能低下患者群の 5 例(62.5%),重度腎機能低下患者群の 7 例(87.5%)に 1
評価時点以上で心電図異常(主に洞性徐脈及び脚ブロック)が観察されたが,治験担当医師によ
り臨床的に重要と判定されたものはなかった。
2.7.6.9.7 結論
(1) 薬物動態
未変化体の薬物動態
腎機能が正常の被験者群間(健康被験者群と腎機能正常患者群)で比較したところ,健康被験
者群と比較して,AUCinf 及び Cmaxはいずれも腎機能正常患者群で高かった。tmax及び Vz/F に差は
なく,t1/2(平均値)は腎機能正常患者群の方が長かった。CLr(平均値)に差はなかった。
2 型糖尿病患者群間(腎機能正常患者群と腎機能低下患者群)で比較したところ,AUCinf(平均
値)は腎機能の低下に伴い上昇した。t1/2,Cmax及び tmaxに一定の傾向は認められなかった。CL/F,
CLr 及び非結合型未変化体の CLr(平均値)は,腎機能の低下に伴い減少した。
代謝物(M1,M2,M3,M4 及び M6)の薬物動態
腎機能が正常の被験者群間(健康被験者群と腎機能正常患者群)で比較したところ,腎機能正
常患者群では,健康被験者群と比較して M2,M3,M4 及び M6 の AUCinf は高かったが,M1 の
AUCinfは低かった。M2,M3,M4 及び M6 の Cmax は腎機能正常患者群で高かったが,M1 の Cmax
は腎機能正常患者群で低かった。t1/2 はすべての代謝物で腎機能正常患者群の方が長かった。CLr
は腎機能正常患者群の方が低かった。
2 型糖尿病患者群間(腎機能正常患者群と腎機能低下患者群)で比較したところ,すべての代
謝物の AUCinf及び tmaxが,腎機能の低下に伴いそれぞれ増加及び延長した。M1,M2,M3 及び
220

2.7.6個々の試験のまとめ
M4 の Cmaxは,腎機能の低下に伴い上昇したが,M6 の Cmaxに,腎機能の低下に伴う一定の傾向
はみられなかった。t1/2 に,腎機能の低下に伴う一定の傾向はみられなかった。
(2) 薬力学
血糖値
血糖値の AUC の変化に,腎機能の低下に伴う一定の傾向はみられなかった。
尿中グルコース
尿中グルコース排泄量(Ae)の Day −1 の値は,腎機能障害の程度にかかわらず,健康被験者群
と比較して 2 型糖尿病患者群で多かった。すべての投与群で治験薬投与後に Ae が増加した。中等
度腎機能低下患者群及び重度腎機能低下患者群では,尿中グルコース排泄量のベースラインから
の変化が,腎機能正常患者群と比較して小さかった。尿中グルコース排泄量の増加量は腎機能の
低下に伴い減少し,腎臓の濾過能の低下と一致していた。
グルコースの CLr は,腎機能にかかわらず,健康被験者群と比較して 2 型糖尿病患者群で高かっ
た。グルコースの CLr は,腎機能にかかわらず,すべての投与群で治験薬投与後に増加した。中
等度腎機能低下患者群及び重度腎機能低下患者群では,腎機能正常患者群と比較して,ベースラ
インからの変化量が小さかった。本剤投与後のグルコースの CLr は,健康被験者群,腎機能正常
患者群及び軽度腎機能低下患者群で同程度であった。グルコースの CLr の変化量は,重度腎機能
低下患者群,中等度腎機能低下患者群,腎機能正常患者群,軽度腎機能低下患者群,健康被験者
群の順に小さかった。
(3) 安全性
有害事象発現例は少数であった。健康被験者群 1 例(頭痛,嘔吐),中等度腎機能低下患者群 2
例(頭痛,下痢,高血圧)に認められたが,いずれの事象も軽度であった。死亡及び重篤な有害
事象は認められなかった。投与中止に至った有害事象は認められなかった。低血糖の発現を示唆
する有害事象は認められなかった。臨床検査値,バイタルサイン及び心電図に,臨床的に問題と
なるような変化は観察されなかった。
221





























