Embed Size (px)
Citation preview

© 2010 John T. Whitteck

INVESTIGATION OF PHOSPHONATE BIOSYNTHESIS: I. STRUCTURE OF DEHYDROPHOS
II. MECHANISM OF HYDROXYETHYLPHOSPHONATE DIOXYGENASE
BY
JOHN THOMAS WHITTECK
DISSERTATION
Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry
in the Graduate College of the University of Illinois at Urbana-Champaign, 2010
Urbana, Illinois
Doctoral Committee:
Professor Wilfred A. van der Donk, Chair Professor John Katzenellenbogen Professor Martin Burke Professor William Metcalf

ii
ABSTRACT
Natural product phosphonates are used extensively in the clinic as antibacterials and in
commercial agriculture as herbicides. In an effort to efficiently discover new natural product
phosphonates, a multidisciplinary, collaborative program has been established at the Institute for
Genomic Biology at the University of Illinois at Urbana-Champaign to mine genomes for novel
phosphonate structures and biosynthetic enzymes. Detailed herein are my contributions to this
effort through assigning the structure of dehydrophos and through investigations into the
mechanism of hydroxyethylphosphonate dioxygenase.
Dehydrophos was discovered as a secondary metabolite of Streptomyces luridus and was
shown to have broad spectrum activity against both Gram-negative and Gram-positive bacteria.
Chemical synthesis of the originally proposed structure showed it to be inconsistent with the
isolated material. Labeling studies with extensive NMR spectroscopic analysis led to
reassignment of the structure as a tripeptide containing an aminophosphonate analogue of
dehydroalanine. This structure was confirmed through organic synthesis.
Hydroxyethylphosphonate dioxygenase (HEPD) catalyzes a biochemically unprecedented
carbon-carbon bond cleavage reaction as part of the early steps of phosphinothricin biosynthesis.
Characterization of HEPD has shown it to be a non-heme iron dependent dioxygenase that is
dependent on only ferrous iron and molecular oxygen for activity. Studies with substrate
isotopologues and substrate analogues have given insight into the mechanism and suggest a
hydroperoxylation mechanism for the early steps.

iii
For My Wife

iv
ACKNOWLEDGEMENTS
The success of these projects has only been possible through the support of many people
over the course of my graduate career. First, and foremost, I would like to thank my advisor,
Prof. Wilfred van der Donk. It was Wilfred’s generous guidance and financial support through
the years that helped refine me into the scientist that I am. I am very grateful for his careful
analysis and insight.
I would like to thank the various people and institutions that provided funding for my
graduate career. I would like to thank the NIH and Howard Hughes Medical Institute for
providing funds for my salary and for equipment and reagents. I would like to thank Dr. Seemon
Pines for his generous contributions to the department that funded the Seemon Pines Award and
the Seemon Pines Travel Award that helped to enrich my graduate experience. I would also like
to thank those who contributed to the Fuson Travel Award that funded my travel to the
Bioorganic Gordon Research Conference.
I would also like to thank the members of my committee: Prof. Martin Burke, Prof. John
Katzenellenbogen and Prof. William Metcalf. I feel that each have gone above and beyond their
required duties and have provided insight, advice and encouragement. Particularly, I would like
to thank William Metcalf as the lead PI for the Mining Microbial Genomes (MMG) theme at the
Institute for Genomic Biology (IGB). In many respects he has served as an unofficial part-time
co-advisor.
I would like to thank the professors at the University of Missouri – Columbia that
inspired me to pursue chemistry as a career. I would like to thank Prof. John Adams as my
undergraduate advisor. I would like to thank Prof. John McCormick who ignited the spark of

v
interest as my organic chemistry professor. I would particularly like to thank Prof. Kent Gates
who was my undergraduate research advisor. His knowledge, advice, perspective and humor
helped me greatly as an undergraduate.
I would like to thank the current and former members of the MMG theme. I have been
fortunate to work with such a talented and scientifically diverse group. I feel that working so
closely with different scientists towards a common goal has enriched my graduate experience
and prepared me for the next step of my career. In particular I would like to thank: Dr. Svetlana
Borisova, Dr. Stephanie Bumpus, Benjamin Circello, Joel Cioni, Dr. Heather Cooke, Dr.
Benjamin Griffin and Juan Velasquez.
I would like to thank the current and former members of the van der Donk laboratory for
their help and camaraderie over the years. Early in my graduate career it was great to be able to
draw on the experiences of such an intelligent group of scientists. Later in my graduate career it
was great to see the evolution of the younger students into great scientists and colleagues. In
particular I would like to thank: Noah Bindman, Kevin Clark, Dr. Lisa Cooper, Dr. Emily Fogle,
Dr. Yuki Goto, Dr. Leigh Anne Ihnken, Dr. Cyril Jacquot, Christopher Kerwood, Patrick Knerr,
Dr. Mathew Levengood, Trent Oman and Dr. Xingang Zhang.
I would also like to thank the administrative assistants that made my time here much
easier by helping me navigate through the system. I would like to thank Patti Silver, Stacy Olson
and Susan Lighty as members of the organic division team that helped me. I would also like to
thank Martha Freeland who, beyond helping me get things done on the paperwork side of things,
was and continues to be a confidant and consistent source of entertainment.
There are two people I worked with in particular that have had a huge impact on who I
am as a scientist and as a person. Dr. Heather McGinley was very influential when I first started

vi
in the group. She taught me a great deal about persistence, hard work and perspective. The
second person I would like to thank is Dr. Robert Cicchillo (aka “Dude” aka “That’s a joke” aka
“Chickenloaf” aka “You’re breaking the mass spec.”) Under Rob’s mentorship I was able to
learn more about biochemistry and mechanistic enzymology than I thought possible in a
relatively short time period. Rob was also a great colleague and friend in the lab.
Beyond the van der Donk lab, there are other people I have met at the University of
Illinois that have been great friends over the years. I would like to thank Dave Connors (aka “my
good friend Dave Connors), Raymond Hart (though not a chemist), Jared Delcamp, Dustin
Covell, Josh Ritchey, Nicolaas Vermeulen for their friendship.
Beyond interactions in the lab there are many other people I would like to thank. I would
like to thank my mom, Lanetta Chapin and step-father, Nathan Chapin, for the support and
caring they have provided over the years. Particularly, for my mom, I have only recently come to
realize the love and sacrifice that you have given to provide me with the life I enjoy today and
would like to thank you. I would like to thank my uncle Freeman Pryer for his care and support
throughout the years. I would also like to thank other members of the Chapin clan that have
welcomed me into their family. There are too many to list, but I would like to thank: Robert and
Teresa, Raymond and Donna and family, Linda and Gary and family, Laura and Dale and
family, Amy, and Alan and Jenny and family. I would also like to thank the Browns and Elliotts
as recent additions to the family for their support. I would like to say “thank you” to Bill and
Trish Elliott, Kelly Elliott, Neil and Janet Brown and Cecil (aka “bad ass Jack”) and Joy Elliott.
Finally, and most importantly, I would like to thank my wife Erin. Her love, support,
guidance, sacrifice and generosity have enriched my life immensely and both sustained and

vii
uplifted me when I’ve been down down. Already we have experienced so many good times and
made it through some difficult times together and I look forward to our next adventure together.

viii
TABLE OF CONTENTS
LIST OF FIGURES ....................................................................................................................x
LIST OF SCHEMES.................................................................................................................xii
LIST OF TABLES...................................................................................................................xiv
CHAPTER 1: MINING MICROBIAL GENOMES FOR NOVEL PHOSPHONATE
CONTAINING ANTIBIOTICS AND ENZYME TRANSFORMATIONS .................................1
1.1 Traditional and Genomic Approaches to Antibiotic Discovery.........................................1
1.2 Phosphonate Biosynthesis ...............................................................................................2
1.3 Structural Diversity and Activity of Phosphonates ...........................................................3
1.4 Unusual Enzyme Catalyzed Transformations During Phosphonate Biosynthesis..............6
1.5 Summary.......................................................................................................................12
1.6 References.....................................................................................................................12
CHAPTER 2: STRUCTURE REASSIGNMENT AND SYNTHESIS OF DEHYDROPHOS
(A53868) ..................................................................................................................................18
2.1 Background...................................................................................................................18
2.2 Structural Reassignment of A53868 via NMR Spectroscopy..........................................19
2.3 Synthesis of Dehydrophos .............................................................................................21
2.4 Future Work..................................................................................................................23
2.5 Experimental .................................................................................................................25
2.6 References.....................................................................................................................30
CHAPTER 3: MECHANISTIC STUDIES ON THE CONVERSION OF 2-HEP TO HMP BY
HEPD .......................................................................................................................................33
3.1 Background...................................................................................................................33
3.2 Fate of the Carbon Atoms of 2-HEP upon Conversion to the Products...........................34
3.3 Tracking the Hydrogen Atoms from 2-HEP to HMP and Formate .................................35
3.4 Kinetic Parameters for HEPD........................................................................................37
3.5 Molecular Oxygen and Water are Incorporated into HMP and Formate .........................38
3.6 X-Ray Crystallographic Studies.....................................................................................39
3.7 Proposed Mechanisms ...................................................................................................42
3.8 Tracking the Hydroxyl Group from 2-HEP into HMP and Formate ...............................44
3.9 O-Formylhydroxymethylphosphonate is Not Accepted as a Substrate............................48

ix
3.10 Stereochemistry of HMP from Sterospecifically Labeled 1-1H-2-HEP........................49
3.11 Revised Mechanisms ...................................................................................................52
3.12 Experimental ...............................................................................................................54
3.13 References...................................................................................................................69
CHAPTER 4: CHEMISTRY OF SUBSTRATE ANALOGUES WITH HEPD .........................73
4.1 Background...................................................................................................................73
4.2 HMP is Converted to Phosphate and Formate by HEPD................................................74
4.3 1-Hydroxyethylphosphonates are Substrates..................................................................75
4.4 2-Hydroxypropylphosphonate is Oxidized by HEPD.....................................................77
4.5 Studies with Additional Substrate Analogues.................................................................83
4.6 Chemistry of Substrate Analogues Show HEPD is Capable of Hydroperoxylation.........84
4.7 Experimental .................................................................................................................88
4.8 References.....................................................................................................................94
APPENDIX A: CHEMISTRY OF 4-PHOSPHONOCARBOXYLIC ACIDS WITH HEPD......97
A.1 Glyphosate and 4-Phosphonobutyric Acid are Substrates for HEPD .............................97
A.2 Experimental .............................................................................................................. 100
A.3 References.................................................................................................................. 100
AUTHOR’S BIOGRAPHY .................................................................................................... 102

x
LIST OF FIGURES
Figure Page
1.1 Structural diversity of known phosphonate containing natural products ........................... 4
1.2 FR900098 and phosphinothricin, their targets and mimicked primary metabolites ........... 5
2.1 Proposed structures for A53868 ....................................................................................... 18
2.2 13C NMR spectrum of 15N13C A53868: (a) non-vinyl carbon atom and (b) quaternary carbon
atom. Tree diagrams are shown to show couplings. (c) Vinyl region of the 1H NMR
spectrum of 15N A53868 .................................................................................................. 20
3.1 (a) 31P NMR spectrum of an HEPD assay with 1-13C-2-HEP as the substrate, (b) 13C NMR
spectrum of an HEPD assay with 2-13C-2-HEP as the substrate........................................ 35
3.2 LC-MS analysis of derivatized formate produced from oxidation of (a) 1,1-2H2-2-HEP and
(b) 2,2-2H2-HEP............................................................................................................... 36
3.3 LC-MS analysis of derivatized formate produced from oxidation of (a) (2S)-2-2H-2-HEP
and (b) (2R)-2-2H-2-HEP................................................................................................. 37
3.4 Michaelis Menten plot for (a) unlabeled HEP and (b) 2,2-2H2-2-HEP .............................. 38
3.5 (a) Derivatized formate from 2-13C-2-HEP and 18O2 as analyzed by GC-MS. The m/z 106
corresponds to 13C,18O-formate and 104 corresponds to 13C-formate. The m/z 103 (asterisk)
is spurious formate. (b) LC-MS analysis showing 18O2 incorporation into HMP as evidenced
by LC-MS. The black line corresponds to an EIC of m/z 115 (18O-HMP) and the red line
corresponds to an EIC of m/z 113 (HMP). The mass spectrum at 5.9 min is shown in the
inset. (c) LC-MS analysis showing incorporation of 18OH2 into HMP. The solid line
corresponds to HMP and the dotted line corresponds to 18O-HMP ................................... 39
3.6 a), b) Orthogonal views of Cd(II)–HEPD showing the tandem arrangement of the HppE fold
including cupin domains (blue and purple), α-helical domains (red and cyan), cadmium
(white sphere) and metal ligands (green). C) Stereoview of electron density maps using
model phases (Fo – Fc). The first map (contoured at 3σ in blue) was calculated by omitting
metal-bound solvents (red sphere) before one round of refinement. The second map,
contoured at 5σ (purple mesh) and 14σ (yellow mesh), was calculated by omitting the
cadmium (gold sphere) before one round of refinement. d) Stereoview of electron density
maps (Fo – Fc) calculated using HEPD–HEP model phases. The map was calculated by

xi
omitting non-protein metal-bound ligands before one round of refinement (contoured at 3σ
in blue mesh and 6σ in red). HEP carbons are shown in green ......................................... 41
3.7 (a) Extracted ion chromatograms for HMP (black line) and 18O-HMP (gray line) from
reaction of 2-18OH-HEP. (b) Mass spectrum of 18O-formate (105 m/z), the peak at 103 m/z is
spurious formate .............................................................................................................. 46
3.8 31P NMR spectrum of (a) a standard of OFHMP and HMP in buffered solution, (b) the same
sample after incubation with HEPD for 2 h ...................................................................... 49
4.1 HEPD catalyzes oxidation of HMP to afford (a) Phosphate (Pi) and (b) 2H-Formate from 2H2-HMP ......................................................................................................................... 75
4.2 31P NMR spectra after incubation of HEPD with (a) 1-HEP, (b) 1-HEP-CF3, Pi = inorganic
phosphate (AP = acetyl phosphate) .................................................................................. 77
4.3 31P NMR spectrum after incubation of HEPD with (2R)-HPP, demonstrating formation of
OPP as well as inorganic phosphate (Pi)........................................................................... 83
4.4 Additional analogues tested as substrates for HEPD......................................................... 84

xii
LIST OF SCHEMES
Figure Page
1.1 Isomerization of PEP to PnPy by PEPM .......................................................................... 2
1.2 Steps that are thought to be common to phosphonate biosynthesis ................................... 3
1.3 Proposed biosynthetic pathway for fosfomycin ................................................................ 7
1.4 Proposed mechanism for Fom3 catalyzed C-H insertion of a methyl group...................... 8
1.5 Proposed mechanism for HppE catalyzed epoxidation ..................................................... 10
1.6 Early steps in the revised pathway in phosphinothricin biosynthesis ................................ 11
2.1 Initial retrosynthetic analysis for 3 ................................................................................... 22
2.2 Synthesis of dehydrophos ................................................................................................ 23
2.3 Potential modes of action for dehydrophos either via (a) proteolysis or (b) covalent
inhibition ......................................................................................................................... 24
3.1 2-HEP is converted to HMP by the gene product of phpD (HEPD) during phosphinothricin
biosynthesis ..................................................................................................................... 33
3.2 Analysis of HMP and formate produced from oxidation of (a) 1,1-2H2-2-HEP and (b) 2,2-2H2-HEP show both hydrogen atoms at C1 are retained in HMP and one hydrogen atom at
C2 is present in formate. Deuterium atoms are labeled in blue ......................................... 36
3.3 Two proposed mechanisms for HEPD............................................................................. 43
3.4 Potential mechanism for exchange of a ferryl oxygen in HEPD with solvent.................... 44
3.5 An alternative hydroperoxylation mechanism for HEPD. Blue circles indicate oxygen from
substrate, orange circles indicate oxygen from O2, and half filled circles represent oxygen
atoms with potential solvent wash-out/wash-in ................................................................ 45
3.6 Alternative mechanism that would result in retention of 18O label from 2-18OH-HEP in
HMP.a Blue circles indicate oxygen from substrate, orange circles indicate oxygen from O2,
and half filled circles represent oxygen atoms with potential solvent wash-out/wash-in.
Products from one round of exchange are shown in shaded boxes with products with two
rounds of exchange shown in regular boxes. Exchange is defined as incorporation of oxygen
from water into HMP; incorporation from 2-HEP is defined as incorporation of the hydroxyl
group of 2-HEP into HMP ............................................................................................... 48
3.7 Proposed stereochemical outcomes for the HEPD mechanisms ........................................ 50

xiii
3.8 When incubated stoichiometrically, 2-HEP is oxidized by HEPD to HMP, which is then
converted to phosphate .................................................................................................... 51
3.9 Both (1S) and (1R)-2H-2-HEP (reactions a and b, respectively) were converted to racemic 2H-HMP by HEPD as evidenced by conversion of the 2H-HMP product to dimethyl 2H-
HMP and Moshers ester analysis ..................................................................................... 52
3.10 Revised mechanisms that can account for production of racemic HMP from
stereoselectively labeled 1-2H-2-HEP .............................................................................. 54
3.11 Synthesis of 1,1-2H2-2-HEP............................................................................................. 61
3.12 Synthesis of 2,2-2H2-2-HEP............................................................................................. 62
3.13 Synthesis of (2R)- and (2S)-2H-2-HEP ............................................................................. 64
4.1 Two proposed mechanisms for HEPD.............................................................................. 74
4.2 Two proposed mechanisms that can account for inactivation of HEPD during oxidation of 2-
HPP ................................................................................................................................. 79
4.3 Reaction of (2R)-HPP partitions between formation of OPP and phosphate (presumably via
HMP) .............................................................................................................................. 83
4.4 Mechanism for reaction of 1-hydroxyphosphonates with HEPD ...................................... 84
4.5 Published example of formation of acylphosphates via Baeyer-Villiger oxidation of
acylphosphonates............................................................................................................. 85
4.6 Mechanism for oxidation of (2R)-HPP to OPP as well as acetate, formate and phosphate 87
A.1 Potential products from glyphosate and PBA oxidation by HEPD under single turnover
conditions ........................................................................................................................ 97
A.2 Oxidation of 4-phosphonocarboxylic acids could proceed via (a) hydroxylation or (b)
hydroperoxylation............................................................................................................ 99

xiv
LIST OF TABLES
Table ................................................................................................................................... Page
3.1 Ratio of 1H-:2H-formate from oxidation of various 2-HEP isotopologues to phosphate by
HEPD.............................................................................................................................. 51
3.2 (R)-Mosher ester analysis of (1R)- and (1S)-2-Benzyloxy-2H-ethanol. The chemical shifts
and area for each diasterotopic proton are given in addition to the calculated ee............... 68
4.1 Hydrogen peroxide assays for the reaction 2-HEP and 2-HPP with HEPD....................... 81

1
CHAPTER 1: MINING MICROBIAL GENOMES FOR NOVEL PHOSPHONATE
CONTAINING ANTIBIOTICS AND ENZYME TRANSFORMATIONS*
1.1 Traditional and Genomic Approaches to Antibiotic Discovery
The discovery and subsequent clinical development of natural products as antibiotics has
revolutionized modern medicine.1 Beginning in the 1930s with penicillin, a dramatic increase in
the discovery of both the number and types of new natural product antibiotics ensued (mostly as
secondary metabolites of actinobacteria).2-4 Emphasis on natural product discovery has since
declined for commercial reasons. Increasing regulatory costs and the resource intensive nature of
the traditional approach to natural product discovery has resulted in low profitability.5-7
For soil bacteria in particular, one possibility to increase the commercial attractiveness of natural
product discovery is to replace expensive traditional methods with a genomics-guided
approach.8-10 In the traditional approach a soil sample is cultured for bacteria, screened for
activity, the active compound purified via phenotype guided bioassay and its structure solved
with NMR spectroscopy and mass spectrometry.11 While initially successful, recent problems
have been identified that include: low culturability of soil bacteria,12, 13 redundant metabolite
isolation14 and difficulty accessing “cryptic” metabolic gene clusters (i.e. gene clusters that we
know are present via genomics but are not metabolically active under culture conditions).15, 16 In
a genomics guided approach, isolated DNA is sequenced for genes involved in natural product
biosynthesis, which are then moved into a genetically amenable heterologous host for
* Parts of this chapter have been reproduced from 1) Metcalf, W.W., van der Donk, W.A., Ann. Rev. Biochem. 2009, 78, 65-94 and 2) Cicchillo, R.M., Zhang, H., Blodgett, J.A.V., Whitteck, J.T., Li, G., Nair, S.K., van der Donk, W.A. Metaclf, W.W., Nature, 2009, 459, 871-874 with permission from Annual Reviews and Nature Publishing Group, respectively

2
production, isolation and characterization of the natural product. This approach helps to
circumvent the aforementioned problems since the producing organism does not need to be
cultured, redundant metabolic gene clusters are ignored, and sequencing can find cryptic
biosynthetic gene clusters. A potential disadvantage is that a genomic screen may result in many
new compounds but none with bioactivity resulting in wasted resources and labor.
1.2 Phosphonate Biosynthesis
An attractive feature of phosphonate containing natural products is that they seem amenable to a
genomics guided approach to discovery.17 Analysis of the biosynthetic pathways of
phosphinothricin,18-21 FR900098,22 fosfomycin,23, 24 the rhizocticins25 and dehydrophos25
suggests that the first step of phosphonate biosynthesis is the isomerization of
phosphoenolpyruvate (PEP) to phosphonopyruvate (PnPy) by phosphoenolpyruvate mutase
(PEPM, Scheme 1.1). Studies on K-26 biosynthesis suggest it is the only known example of
phosphonate biosynthesis without PEPM.26 Therefore, isolated DNA that contains a PEPM gene
is likely to have genes that code for phosphonate biosynthesis and PEPM serves as an effective
screen to quickly identify new gene clusters. In addition, degenerate primers for PEPM can be
used to extract phosphonate biosynthetic gene clusters from DNA isolates that have been
identified as containing PEPM.
Scheme 1.1 Isomerization of PEP to PnPy by PEPM.

3
In addition to PEPM, other transformations are common in phosphonate biosynthesis. Since the
equilibrium of PEPM lies heavily in favor of PEP, an additional, irreversible step is necessary to
drive biosynthesis forward.27, 28 One way in which this is accomplished is by decarboxylation of
PnPy by PnPy decarboxylase (PPD) to afford phosphonoacetaldehyde (PnAA, Scheme 1.2).29 In
the cases of fosfomycin,30 phosphinothricin31 and dehydrophos,32 PnAA undergoes reduction by
an alcohol dehydrogenase to afford 2-hydroxyethylphosphonate (2-HEP, Scheme 1.2).32 In
addition to decarboxylation, aldol addition to either PnPy or PnAA is another way to drive
phosphonate biosynthesis forward. Genetic evidence suggests PnPy undergoes a homocitrate
synthase-like condensation during FR900098 biosynthesis (Scheme 1.2).22 In vitro studies
demonstrate that PnAA undergoes aldol condensation with oxaloacetate during biosynthesis of
the rhizocticins and plumbemycins.25 Knowledge of these steps then serves as a secondary probe
of DNA isolates containing PEPM to confirm a phosphonate biosynthetic gene cluster.
Scheme 1.2 Steps that are thought to be common to phosphonate biosynthesis.
1.3 Structural Diversity and Activity of Phosphonates
Phosphonates represent a structurally diverse class of compounds as shown in Figure 1.1.17
Correspondingly, phosphonates demonstrate a variety of biological activities including:

4
antibacterial (fosfomycin, dehydrophos, plumbemycin), antifungal (rhizocticins), antimalarial
(FR900098 and fosmidomycin), herbicidal (phosphinothricin, phosphonothrixin) and
antihypertensive (K-4 and K-26) activities.
Figure 1.1 Structural diversity of known phosphonate containing natural products.17
Phosphonates generally exert their bioactivity by similar mechanisms of mimicking either
phosphoryl or carboxyl moieties of primary metabolic intermediates. Figure 1.2 shows two
examples of phosphonates, their targets and the mimicked metabolite. In the case of FR900098,
the phosphonate mimics the phosphate group of deoxy-xyulose phosphate (DXP) and inhibits

5
DXP reductoisomerase.33, 34 This enzyme is an important target since it is involved in the non-
mevalonate pathway of isoprenoid biosynthesis. This pathway is present in plants and many
pathogens but absent in humans. As a second example, the commercial herbicide
phosphinothricin (a phosphinate since it contains two phosphorus carbon bonds) mimics the
tetrahedral intermediate during conversion of glutamate to glutamine thereby inhibiting
glutamine synthase, resulting in a toxic buildup of ammonia and glutamine starvation in the
plant.35
Figure 1.2 FR900098 and phosphinothricin, their targets and mimicked primary metabolites.
The diverse structures and activities of phosphonate antibiotics are a second attractive feature for
this class of compounds. Potentially innumerable processes can be targeted as the vast majority
of known primary metabolic pathways contain intermediates with a phosphoryl or carboxyl
moiety. Additionally, the general activity of phosphonates may help alleviate potential problems
with a genomic approach to antibiotic discovery since the effort required for genotyping should
result in a compound with some bioactivity.

6
1.4 Unusual Enzyme Catalyzed Transformations During Phosphonate Biosynthesis
Given the structural diversity of phosphonates it is not surprising that many unusual and
interesting enzyme catalyzed transformations take place during their biosynthesis. Study of these
enzymes may lead to their development in commercial applications or commercial production of
therapeutics and high-value products.36 Moreover, these enzymes may serve as inspiration for
biomimetic chemistry, which may help streamline organic syntheses.37 Further work into
phosphonate discovery and biosynthesis will likely afford more interesting transformations.
Given below are some examples of unique enzyme reactions during the well-studied
biosyntheses of fosfomycin and phosphinothricin.
1.4.1 Fosfomycin
Fosfomycin [(1R, 2S)-epoxypropylphosphonic acid] is a clinical antibiotic used to treat urinary
tract infections.38 The minimal fosfomycin biosynthetic gene cluster from Streptomyces
wedmorensis has been identified by heterologous production.24 The proposed biosynthetic
pathway is shown in Scheme 1.3. The first three steps have already been mentioned but are:
isomerization of PEP to PnPy by PEPM, decarboxylation by PPD to afford PnAA and reduction
by FomC to afford 2-HEP. The final two steps of fosfomycin biosynthesis are C-H insertion of a
methyl group by Fom3 and epoxidation by hydroxypropylphosphonate epoxidase (HppE), both
of which are chemically and biochemically unique.

7
Scheme 1.3 Proposed biosynthetic pathway for fosfomycin.
Bioinformatic analysis of Fom3 shows that it has two conserved domains: an N-terminal vitamin
B12-like (methyl cob(III)alamin, MeCbl) binding domain and a C-terminal domain homologous
to the [4Fe-4S]+ containing radical-SAM family.30 Mechanistic insight has come from in vivo
feeding studies using deuterated 2-HEP. Fosfomycin produced from 2,2-2H2-2-HEP and (2S)-2H-
2-HEP retains some deuterium label whereas fosfomycin from (2R)-2H-2-HEP contains none and
suggests the pro-R hydrogen atom is abstracted.39, 40 A mechanism has been proposed based on
these data and is shown in Scheme 1.4. The reduced [4Fe-4S]+ cluster reductively cleaves SAM
to form a 5’-adenosyl radical intermediate that has been proposed for other radical-SAM
proteins.41, 42 The 5’-adenosyl radical then abstracts the pro-R hydrogen atom at C2 of 2-HEP to
afford 5’-deoxyadenosine and a carbon-centered radical. The carbon radical recombines with the
methyl group of MeCbl to afford 2-hydroxypropylphosphonate (2-HPP) and cob(II)alamin.
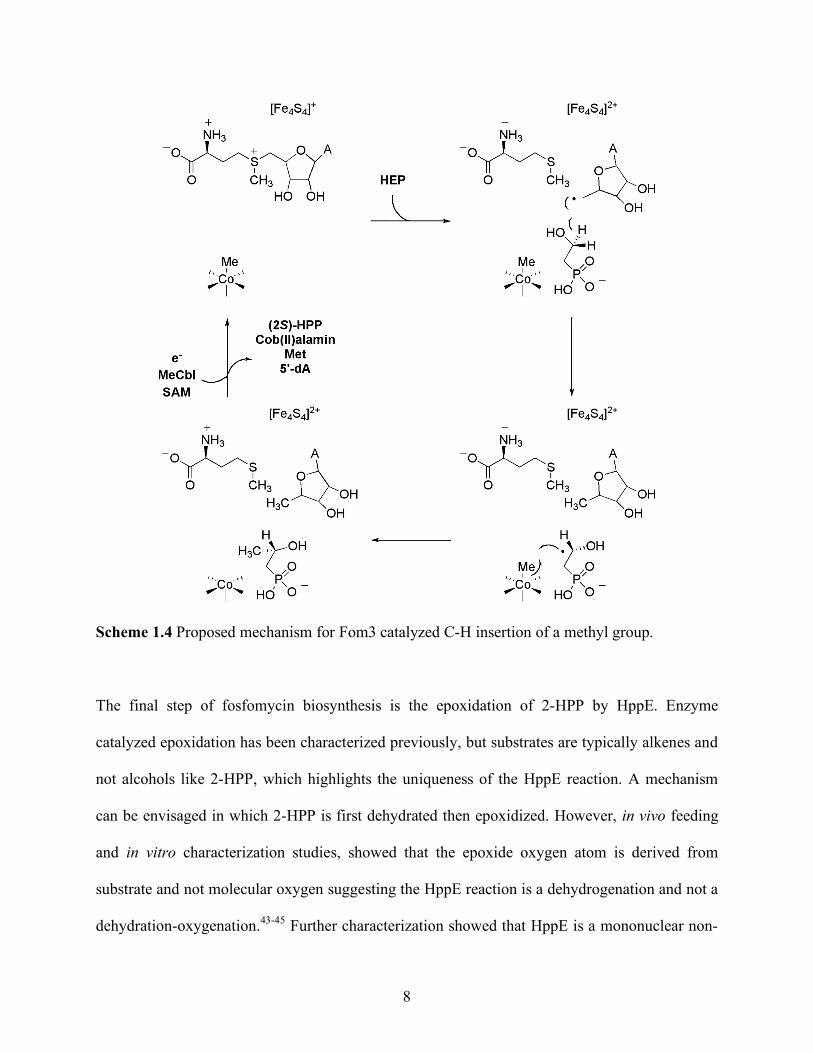
8
Scheme 1.4 Proposed mechanism for Fom3 catalyzed C-H insertion of a methyl group.
The final step of fosfomycin biosynthesis is the epoxidation of 2-HPP by HppE. Enzyme
catalyzed epoxidation has been characterized previously, but substrates are typically alkenes and
not alcohols like 2-HPP, which highlights the uniqueness of the HppE reaction. A mechanism
can be envisaged in which 2-HPP is first dehydrated then epoxidized. However, in vivo feeding
and in vitro characterization studies, showed that the epoxide oxygen atom is derived from
substrate and not molecular oxygen suggesting the HppE reaction is a dehydrogenation and not a
dehydration-oxygenation.43-45 Further characterization showed that HppE is a mononuclear non-

9
heme iron-containing enzyme whose activity is not dependent on α-ketoglutarate but is
dependent on exogenous reducing equivalents and molecular oxygen for activity.45-47
Mechanistic studies using spectroscopic techniques, substrate analogues and X-ray
crystallographic analysis has led to two proposed mechanisms for this transformation (Scheme
1.5).48-51 Both mechanisms begin with bidentate binding of 2-HPP and molecular oxygen to
ferrous iron. In addition, both mechanisms have abstraction of the pro-R hydrogen atom,
agreeing with in vivo feeding studies and in vitro assays.39 The mechanisms differ in the timing
of electron input and whether the hydrogen atom abstracting species is an Fe(III)-OO or a
Fe(IV)=O intermediate.
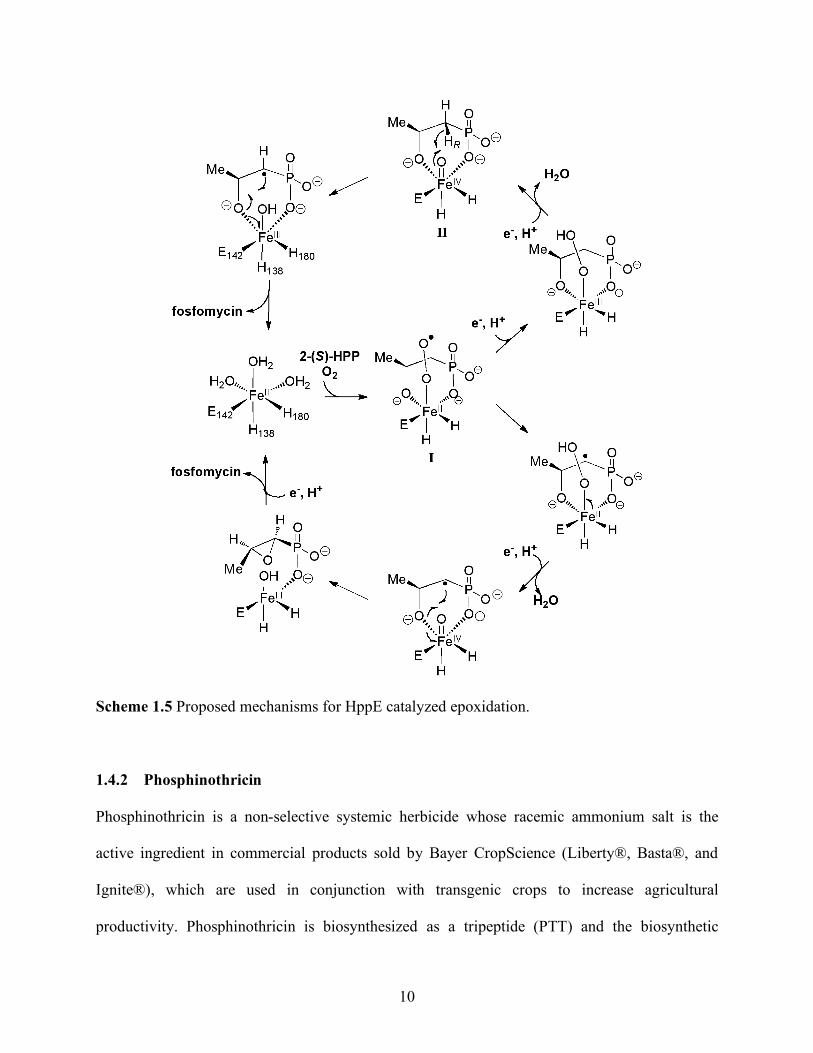
10
Scheme 1.5 Proposed mechanisms for HppE catalyzed epoxidation.
1.4.2 Phosphinothricin
Phosphinothricin is a non-selective systemic herbicide whose racemic ammonium salt is the
active ingredient in commercial products sold by Bayer CropScience (Liberty®, Basta®, and
Ignite®), which are used in conjunction with transgenic crops to increase agricultural
productivity. Phosphinothricin is biosynthesized as a tripeptide (PTT) and the biosynthetic
11
pathway has been studied for decades. These studies were pioneered by Seto and coworkers and
have used a combination of in vivo feeding studies, in vitro biochemistry, and genetics.52, 53
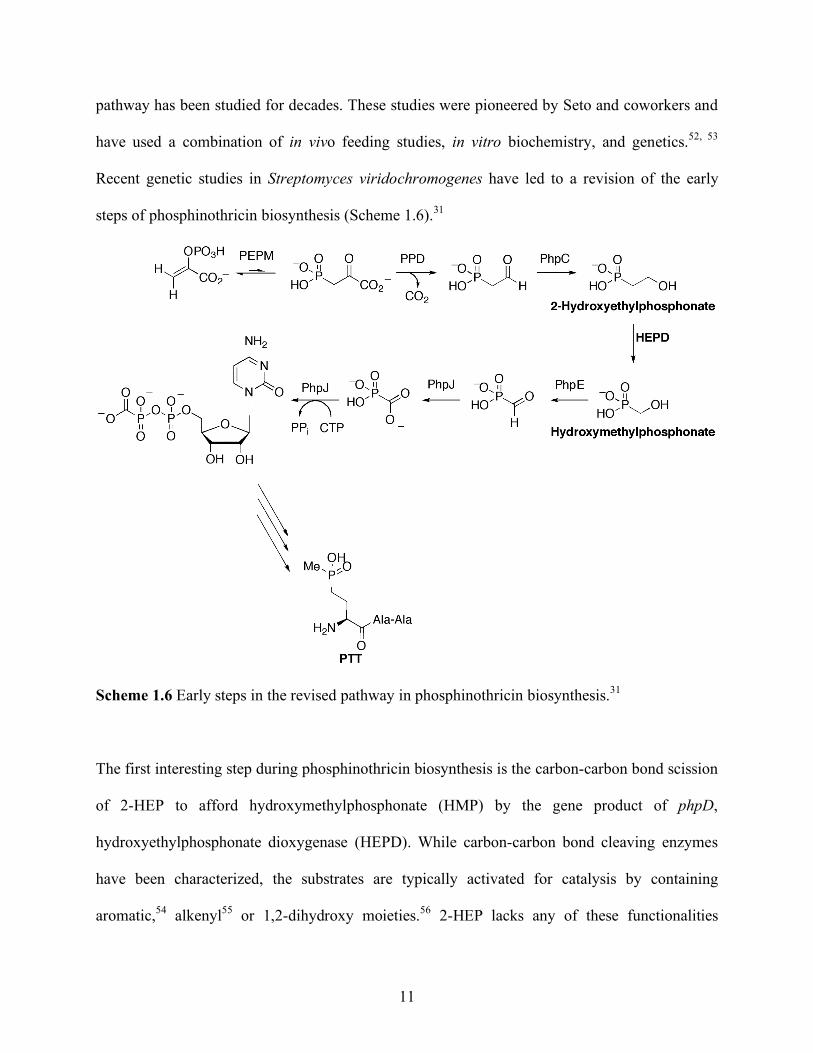
Recent genetic studies in Streptomyces viridochromogenes have led to a revision of the early
steps of phosphinothricin biosynthesis (Scheme 1.6).31
Scheme 1.6 Early steps in the revised pathway in phosphinothricin biosynthesis.31
The first interesting step during phosphinothricin biosynthesis is the carbon-carbon bond scission
of 2-HEP to afford hydroxymethylphosphonate (HMP) by the gene product of phpD,
hydroxyethylphosphonate dioxygenase (HEPD). While carbon-carbon bond cleaving enzymes
have been characterized, the substrates are typically activated for catalysis by containing
aromatic,54 alkenyl55 or 1,2-dihydroxy moieties.56 2-HEP lacks any of these functionalities

12
making the HEPD catalyzed reaction biochemically unprecedented. Initial characterization has
shown that HEPD is dependent on only ferrous iron and molecular oxygen for activity.57
Therefore HEPD does not require exogenous electron input or utilize organic cofactors, unlike
HppE and other iron dependent enzymes, further highlighting its uniqueness.54, 58
1.5 Summary
To continue the medical success of natural product antibiotics, new approaches must be
identified for their discovery. Phosphonate natural products seem amenable to a genomics-
guided approach in which isolates are screened by genotyping instead of phenotyping.
Additionally, previously discovered phosphonates demonstrate that this class of compounds
seem generally bioactive and have diverse structures and activities. In this thesis, Chapter 2 will
detail my contributions to exploring structurally unique phosphonates by reassigning the
structure and completing the first total synthesis of dehydrophos (A53868).
Studies into the biosynthesis of phosphonates have shown that these diverse structures come
about through novel biosynthetic chemistries as typified by Fom3, HppE and HEPD. Further
study into phosphonate discovery and biosynthesis will no doubt reveal further structural and
enzymatic diversity. In this thesis, Chapters 3 and 4 will detail my contribution to elucidating
biochemically unprecedented enzyme activities through mechanistic study of HEPD.
1.6 References
1. McDermott, W.; Rogers, D. E., John Hopkins Med. J. 1982, 151, (6), 302-312. Social ramifications of control of microbial disease. 2. Fischbach, M. A.; Walsh, C. T., Science 2009, 325, (5944), 1089-1093. Antibiotics for Emerging Pathogens.

13
3. Madigan, M.; Martinko, J., Brock Biology of Microorganisms. Prentice Hall: Upper Saddle River, NJ, 2005. 4. Kieser, T.; Bibb, M. J.; Buttner, M. J.; Chater, K. F.; Hopwood, D. A., Practical Streptomyces Genetics. 2nd ed.; John Innes Foundation: Norwich, England, 2000. 5. Projan, S. J., Curr. Opin. Microbiol. 2003, 6, (5), 427-430. Why is big Pharma getting out of antibacterial drug discovery? 6. Koehn, F. E.; Carter, G. T., Nat. Rev. Drug. Discov. 2005, 4, (3), 206-220. The evolving role of natural products in drug discovery. 7. Paterson, I.; Anderson, E. A., Science 2005, 310, (5747), 451-453. The Renaissance of Natural Products as Drug Candidates. 8. Challis, G. L., J. Med. Chem. 2008, 51, (9), 2618-2628. Genome Mining for Novel Natural Product Discovery. 9. Malek, Z.; Gregory, L. C., ChemBioChem 2009, 10, (4), 625-633. Strategies for the Discovery of New Natural Products by Genome Mining. 10. Zazopoulos, E.; Huang, K.; Staffa, A.; Liu, W.; Bachmann, B. O.; Nonaka, K.; Ahlert, J.; Thorson, J. S.; Shen, B.; Farnet, C. M., Nat. Biotech. 2003, 21, (2), 187-190. A genomics-guided approach for discovering and expressing cryptic metabolic pathways. 11. Li, J. W. H.; Vederas, J. C., Science 2009, 325, (5937), 161-165. Drug Discovery and Natural Products: End of an Era or an Endless Frontier? 12. Daniel, R., Curr. Opin. Biotech. 2004, 15, (3), 199-204. The soil metagenome - a rich resource for the discovery of novel natural products. 13. Van Lanen, S. G.; Shen, B., Curr. Opin. Microbiol. 2006, 9, (3), 252-260. Microbial genomics for the improvement of natural product discovery. 14. Tulp, M.; Bohlin, L., Bioorg. Med. Chem. 2005, 13, (17), 5274-5282. Rediscovery of known natural compounds: Nuisance or goldmine? 15. Bentley, S. D.; Chater, K. F.; Cerdeno-Tarraga, A. M.; Challis, G. L.; Thomson, N. R.; James, K. D.; Harris, D. E.; Quail, M. A.; Kieser, H.; Harper, D.; Bateman, A.; Brown, S.; Chandra, G.; Chen, C. W.; Collins, M.; Cronin, A.; Fraser, A.; Goble, A.; Hidalgo, J.; Hornsby, T.; Howarth, S.; Huang, C. H.; Kieser, T.; Larke, L.; Murphy, L.; Oliver, K.; O'Neil, S.; Rabbinowitsch, E.; Rajandream, M. A.; Rutherford, K.; Rutter, S.; Seeger, K.; Saunders, D.; Sharp, S.; Squares, R.; Squares, S.; Taylor, K.; Warren, T.; Wietzorrek, A.; Woodward, J.; Barrell, B. G.; Parkhill, J.; Hopwood, D. A., Nature 2002, 417, (6885), 141-147. Complete genome sequence of the model actinomycete Streptomyces coelicolor.

14
16. Omura, S.; Ikeda, H.; Ishikawa, J.; Hanamoto, A.; Takahashi, C.; Shinose, M.; Takahashi, Y.; Horikawa, H.; Nakazawa, H.; Osonoe, T.; Kikuchi, H.; Shiba, T.; Sakaki, Y.; Hattori, M., Proc. Natl. Acad. Sci. U.S.A. 2001, 98, (21), 12215-12220. Genome sequence of an industrial microorganism Streptomyces avermitilis: Deducing the ability of producing secondary metabolites. 17. Metcalf, W. W.; van der Donk, W. A., Ann. Rev. Biochem. 2009, 78, (1), 65-94. Biosynthesis of Phosphonic and Phosphinic Acid Natural Products. 18. Seto, H.; Imai, S.; Tsuruoka, T.; Satoh, A.; Kojima, M., J. Antibiot. 1982, 35, (12), 1719-1721. Studies on the Biosynthesis of Bialaphos (Sf-1293) .1. Incorporation of C-13-Labeled and H-2-Labeled Precursors into Bialaphos. 19. Schwartz, D.; Berger, S.; Heinzelmann, E.; Muschko, K.; Welzel, K.; Wohlleben, W., Appl. Environ. Microbiol. 2004, 70, (12), 7093-7102. Biosynthetic Gene Cluster of the Herbicide Phosphinothricin Tripeptide from Streptomyces viridochromogenes Tu494. 20. Blodgett, J. A. V.; Zhang, J. K.; Metcalf, W. W., Antimicrob. Agents Chemother. 2005, 49, (1), 230-240. Molecular Cloning, Sequence Analysis, and Heterologous Expression of the Phosphinothricin Tripeptide Biosynthetic Gene Cluster from Streptomyces viridochromogenes DSM 40736. 21. Seto, H.; Sasaki, T.; Imai, S.; Tsuruoka, T.; Ogawa, H.; Satoh, A.; Inouye, S.; Niida, T.; Otake, N., J. Antibiot. 1983, 36, (1), 96-98. Studies on the Biosynthesis of Bialaphos (Sf-1293) .2. Isolation of the 1st Natural-Products with a C-P-H Bond and Their Involvement in the C-P-C Bond Formation. 22. Eliot, A. C.; Griffin, B. M.; Thomas, P. M.; Johannes, T. W.; Kelleher, N. L.; Zhao, H.; Metcalf, W. W., Chem. Biol. 2008, 15, (8), 765-770. Cloning, Expression, and Biochemical Characterization of Streptomyces rubellomurinus Genes Required for Biosynthesis of Antimalarial Compound FR900098. 23. Rogers, T. O.; Birnbaum, J., Antimicrob. Agents Chemother. 1974, 5, (2), 121-132. Biosynthesis of Fosfomycin by Streptomyces fradiae. 24. Woodyer, R. D.; Shao, Z.; Thomas, P. M.; Kelleher, N. L.; Blodgett, J. A. V.; Metcalf, W. W.; van der Donk, W. A.; Zhao, H., Chem. Biol. 2006, 13, (11), 1171-1182. Heterologous Production of Fosfomycin and Identification of the Minimal Biosynthetic Gene Cluster. 25. Circello, B. T.; Eliot, A. C.; Lee, J.-H.; van der Donk, W. A.; Metcalf, W. W., Chem. Biol. 2010, 17, (4), 402-411. Molecular Cloning and Heterologous Expression of the Dehydrophos Biosynthetic Gene Cluster. 26. Ntai, I.; Manier, M. L.; Hachey, D. L.; Bachmann, B. O., Org. Lett. 2005, 7, (13), 2763-2765. Biosynthetic Origins of C-P Bond Containing Tripeptide K-26.

15
27. Seidel, H. M.; Freeman, S.; Seto, H.; Knowles, J. R., Nature 1988, 335, (6189), 457-458. Phosphonate biosynthesis: isolation of the enzyme responsible for the formation of a carbon-phosphorus bond. 28. Bowman, E.; McQueney, M.; Barry, R. J.; Dunaway-Mariano, D., J. Am. Chem. Soc. 1988, 110, (16), 5575-5576. Catalysis and thermodynamics of the phosphoenolpyruvate/phosphonopyruvate rearrangement. Entry into the phosphonate class of naturally occurring organophosphorus compounds. 29. Barry, R. J.; Bowman, E.; McQueney, M.; Dunaway-Mariano, D., Biochem. Biophys. Res. Comm. 1988, 153, (1), 177-182. Elucidation of the 2-aminoethylphosphonate biosynthetic pathway in Tetrahymena pyriformis. 30. Woodyer, R. D.; Li, G.; Zhao, H.; van der Donk, W. A., Chem. Comm. 2007, (4), 359-361. New insight into the mechanism of methyl transfer during the biosynthesis of fosfomycin. 31. Blodgett, J. A. V.; Thomas, P. M.; Li, G.; Velasquez, J. E.; van der Donk, W. A.; Kelleher, N. L.; Metcalf, W. W., Nat. Chem. Biol. 2007, 3, (8), 480-485. Unusual transformations in the biosynthesis of the antibiotic phosphinothricin tripeptide. 32. Shao, Z.; Blodgett, J. A. V.; Circello, B. T.; Eliot, A. C.; Woodyer, R.; Li, G.; van der Donk, W. A.; Metcalf, W. W.; Zhao, H., J. Biol. Chem. 2008, 283, (34), 23161-23168. Biosynthesis of 2-Hydroxyethylphosphonate, an Unexpected Intermediate Common to Multiple Phosphonate Biosynthetic Pathways. 33. Jomaa, H.; Wiesner, J.; Sanderbrand, S.; Altincicek, B.; Weidemeyer, C.; Hintz, M.; uuml; rbachova, I.; Eberl, M.; Zeidler, J.; Lichtenthaler, H. K.; Soldati, D.; Beck, E., Science 1999, 285, (5433), 1573-1576. Inhibitors of the Nonmevalonate Pathway of Isoprenoid Biosynthesis as Antimalarial Drugs. 34. Koppisch, A. T.; Fox, D. T.; Blagg, B. S. J.; Poulter, C. D., Biochemistry 2001, 41, (1), 236-243. E. coli MEP Synthase: Steady-State Kinetic Analysis and Substrate Binding. 35. Manderscheid, R.; Wild, A., J. Plant Phys. 1986, 123, (2), 135-142. Studies on the mechanism of inhibition by phosphinothricin of glutamine synthase isolated from Triticum aestivum L. 36. Yang, S.-T., Bioprocessing for Value-Added Products from Renewable Resources: New Technologies and Applications. 1st ed.; Elsevier: 2007. 37. Breslow, R., J. Biol. Chem. 2009, 284, (3), 1337-1342. Biomimetic Chemistry: Biology as an Inspiration. 38. Allerberger, F.; Klare, I., J. Antimicrob. Chemother. 1999, 43, (2), 211-217. In-vitro activity of fosfomycin against vancomycin-resistant enterococci.

16
39. Hammerschmidt, F.; Kaehlig, H., J. Org. Chem. 1991, 56, (7), 2364-2370. Biosynthesis of natural products with a phosphorus-carbon bond. 7. Synthesis of [1,1-2H2]-, [2,2-2H2]-, (R)- and (S)-[1-2H1](2-hydroxyethyl)phosphonic acid and (R,S)-[1-2H1](1,2-dihydroxyethyl)phosphonic acid and incorporation studies into fosfomycin in Streptomyces fradiae. 40. Hammerschmidt, F., Liebigs Ann. Chem. 1992, 1992, (6), 553-557. Biosynthese von Naturstoffen mit einer P-C-Bindung, IX. Synthese und Einbau von (S)- und (R)-2-Hydroxy-[2-2H-]ethylphosphonsure in Fosfomycin durch Streptomyces fradiae. 41. Booker, S. J.; Cicchillo, R. M.; Grove, T. L., Curr. Opin. Chem. Biol. 2007, 11, (5), 543-552. Self-sacrifice in radical S-adenosylmethionine proteins. 42. Booker, S. J., Curr. Opin. Chem. Biol. 2009, 13, (1), 58-73. Anaerobic functionalization of unactivated C-H bonds. 43. Hammerschmidt, F.; Bovermann, G.; Bayer, K., Liebigs Ann. Chem. 1990, 1990, (11), 1055-1061. Biosynthese von Naturstoffen mit einer P-C-Bindung, V. Das Oxiran-Sauerstoff-Atom des Fosfomycins entstammt nicht dem Luft-Sauerstoff. 44. Hammerschmidt, F., J. Chem. Soc. - Perkin Trans. 1 1991, (8), 1993-1996. Biosynthesis of Natural-Products with a P-C Bond .8. On the Origin of the Oxirane Oxygen Atom of Fosfomycin in Streptomyces Fradiae. 45. Liu, P.; Murakami, K.; Seki, T.; He, X.; Yeung, S.-M.; Kuzuyama, T.; Seto, H.; Liu, H.-w., Journal of the American Chemical Society 2001, 123, (19), 4619-4620. Protein Purification and Function Assignment of the Epoxidase Catalyzing the Formation of Fosfomycin. 46. Liu, P.; Liu, A.; Yan, F.; Wolfe, M. D.; Lipscomb, J. D.; Liu, H.-w., Biochemistry 2003, 42, (40), 11577-11586. Biochemical and Spectroscopic Studies on (S)-2-Hydroxypropylphosphonic Acid Epoxidase: A Novel Mononuclear Non-heme Iron Enzyme. 47. Liu, P. H.; Murakami, K.; Seki, T.; He, X. M.; Yeung, S. M.; Kuzuyama, T.; Seto, H.; Liu, H. W., J. Am. Chem. Soc. 2001, 123, (19), 4619-4620. Protein purification and function assignment of the epoxidase catalyzing the formation of fosfomycin. 48. Zhao, Z. B.; Liu, P. H.; Murakami, K.; Kuzuyama, T.; Seto, H.; Liu, H. W., Angew. Chem., Int. Ed. Engl. 2002, 41, (23), 4529-4530. Mechanistic studies of HPP epoxidase: Configuration of the substrate governs its enzymatic fate. 49. Liu, P.; Liu, A.; Yan, F.; Wolfe, M. D.; Lipscomb, J. D.; Liu, H.-w., Biochemistry 2003, 42, (40), 11577-11586. Biochemical and Spectroscopic Studies on (S)-2-Hydroxypropylphosphonic Acid Epoxidase: A Novel Mononuclear Non-heme Iron Enzyme. 50. Yan, F.; Moon, S.-J.; Liu, P.; Zhao, Z.; Lipscomb, J. D.; Liu, A.; Liu, H.-w., Biochemistry 2007, 46, (44), 12628-12638. Determination of the Substrate Binding Mode to the Active Site

17
Iron of (S)-2-Hydroxypropylphosphonic Acid Epoxidase Using 17O-Enriched Substrates and Substrate Analogues. 51. Higgins, L. J.; Yan, F.; Liu, P.; Liu, H.-w.; Drennan, C. L., Nature 2005, 437, (7060), 838-844. Structural insight into antibiotic fosfomycin biosynthesis by a mononuclear iron enzyme. 52. Seto, H.; Kuzuyama, T., Nat. Prod. Rep. 1999, 16, (5), 589-596. Bioactive natural products with carbon-phosphorus bonds and their biosynthesis. 53. Thompson, C. J.; Seto, H., Bialaphos. Butterworth-Heinemann: Newton, MA, 1995. 54. Costas, M.; Mehn, M. P.; Jensen, M. P.; Que, L., Chem. Rev. 2004, 104, (2), 939-986. Dioxygen activation at mononuclear nonheme iron active sites: Enzymes, models, and intermediates. 55. Grogan, G., Biochem. J. 2005, 388, 721-730. Emergent mechanistic diversity of enzyme-catalysed beta-diketone cleavage. 56. Xing, G.; Diao, Y.; Hoffart, L. M.; Barr, E. W.; Prabhu, K. S.; Arner, R. J.; Reddy, C. C.; Krebs, C.; Bollinger, J. M., Jr., Proc. Natl. Acad. Sci. U.S.A. 2006, 103, (16), 6130-6135. Evidence for C-H cleavage by an iron-superoxide complex in the glycol cleavage reaction catalyzed by myo-inositol oxygenase. 57. Cicchillo, R. M.; Zhang, H.; Blodgett, J. A. V.; Whitteck, J. T.; Li, G.; Nair, S. K.; van der Donk, W. A.; Metcalf, W. W., Nature 2009, 459, (7248), 871-874. An unusual carbon-carbon bond cleavage reaction during phosphinothricin biosynthesis. 58. Kovaleva, E. G.; Lipscomb, J. D., Nat. Chem. Biol. 2008, 4, (3), 186-193. Versatility of biological non-heme Fe(II) centers in oxygen activation reactions.

18
CHAPTER 2: STRUCTURE REASSIGNMENT AND SYNTHESIS OF DEHYDROPHOS
(A53868)*
2.1 Background
In the 1980s, Eli Lilly and Company isolated a phosphonate antibiotic from the spent medium of
Streptomyces luridus NRRL 15101 and found it to have antibacterial activity against both Gram-
negative and Gram-positive bacteria.1 The compound was designated A53868 and structure 1
was assigned on the basis of 1H NMR spectroscopy and mass spectrometric analysis (Figure
2.1).1 The defining structural features of A53868 are a 3-carbon amino phosphonic acid
containing a vinyl moiety that is attached via amide linkage to a leucine-glycine dipeptide (Gly-
Leu). Later, scientists at the same company revised the structure of A53868 to 2 on the basis of
proton coupled 13C NMR spectroscopy (Figure 2.1).2 Structure 2 is a constitutional isomer of 1
and contains the defining features of A53868 and differs only in that the phosphonate moiety is
vinylic instead of allylic.
Figure 2.1 Proposed structures for A53868.
Dr. Weijuan Ni, a former postdoctoral researcher in the van der Donk laboratory, synthesized
* Parts of this chapter have been reproduced from 1) Whitteck, J.T., Griffin, B.M., Eliot, A.C., Thomas, P.M., Kelleher, N.L., Metcalf, W.W., van der Donk, W.A., Angew. Chem. Int. Ed. Engl. 2007, 46, 9089-9092 with permission from John Wiley and Sons Inc.

19
structure 2. Surprisingly, the recorded 31P NMR spectrum showed that 2 displays a different
chemical shift than isolated A53868, which was confirmed by spiking a sample of 2 with the
material isolated from S. luridus. A crystal structure of synthetic 2 was obtained and showed
unambiguously that the structure of synthetic 2 was correct and hence that the proposed structure
for A53868 was incorrect.
2.2 Structural Reassignment of A53868 via NMR Spectroscopy
In collaboration with Dr. Benjamin Griffin, a former postdoctoral researcher in the William
Metcalf laboratory in the Department of Microbiology at UIUC, Streptomyces luridus was grown
on media containing either (15NH4)2SO4 or (15NH4)2SO4 and 13C6-D-glucose as the sole sources of
nitrogen or carbon, resulting in material with complete incorporation of either 15N or both 15N
and 13C label, respectively. The A53868 isotopologues were purified from the spent media and
multiple NMR spectroscopic analyses were performed that led to a new proposed structure (3,
Figure 2.1).
Most telling in the structural reassignment was analysis of the 13C NMR spectrum of the 15N and
13C double-labeled compound. The resonances for the Gly-Leu dipeptide and secondary vinyl
carbon atom had splitting patterns and couplings consistent with 2. However, the peak
corresponding to the non-vinyl carbon appears as a doublet just as is observed for unlabeled
material despite uniform labeling of carbon and nitrogen (Figure 2.2a). The lack of additional
splitting suggests that this carbon atom is not connected to either carbon or nitrogen, which is
inconsistent with structure 2. Furthermore, although the splitting pattern of the quaternary vinyl
carbon is the expected doublet of doublet of doublets (Figure 2.2b), the coupling constants (190
Hz, 73 Hz and 11 Hz) correspond to splitting from phosphorus, the secondary vinyl carbon

20
(confirmed by 13C-13C COSY) and a nitrogen atom, respectively. This observation suggests that
this carbon atom is bonded to phosphorus, the vinylic carbon, and a nitrogen but not to the non-
vinyl carbon atom.
Figure 2.2 13C NMR spectrum of 15N13C A53868: (a) non-vinyl carbon atom and (b) quaternary
carbon atom. Tree diagrams are shown to show couplings. (c) Vinyl region of the 1H NMR
spectrum of 15N A53868.
The 1H NMR spectrum of the 15N single-labeled isotopologue also supports structure 3 (Figure
2.2c). The signal corresponding to the non-vinyl hydrogen atoms is a doublet that is also seen in
unlabeled material. This finding confirms that the carbon carrying these protons is not bonded to
nitrogen and that structure 2 is inconsistent with data. Additionally, the signals for the vinyl
protons are consistent with the proposed double bond connectivity in 3. The peaks are doublet of
doublets which show cis and trans splitting for 15N and 31P (3J(P,Hcis) = 15.7 Hz; 3J(P,Htrans) =
36.3 Hz; 3J(N,Hcis) = 2.8 Hz; 3J(N,Htrans) = 6.1 Hz) which suggest an α,β-unsaturated-α-
aminophosphonic acid moeity.

21
To help distinguish between the proposed structures, from here on A53868 will refer to structure
2 and the S. luridus metabolite has been named dehydrophos. The major revision from 2 to 3 is
that the non-vinyl carbon has been reassigned from an allylic carbon to a phosphonate methyl
ester carbon. The misassignment of methyl protons as a methylene group may be due to the low
integration under standard instrument settings, which is closer to 2 than 3. Only upon increasing
the recycle delay to twice the relaxation time (T1) did the peak integrate to 3. Dehydrophos is
then a tripeptide containing a unique phosphonate analogue of dehydroalanine, an important
structural feature of many natural products such as the lantibiotics,3 microcystins,4 and
thiostrepton.5
2.3 Synthesis of Dehydrophos
Since dehydrophos contains a dehydroalanine-like moiety, our retrosynthetic analysis has drawn
heavily on methods by which dehydroalanine containing peptides have been synthesized. Many
methods have been developed for the synthesis of dehydroalanine containing peptides since, in
addition to being an important structural feature, these structures have also been important
intermediates in chemical and biosyntheses due to their electrophilicity.6-10 Typically,
dehydroalanine is synthesized via activation and elimination of serine derivatives,11 Hoffmann
elimination of 2,3-diaminopropionic acid,12 or oxidative elimination of S-aryl or Se-aryl
protected cysteine-like derivatives.10, 13
Initial synthetic efforts focused on activation and elimination strategies since the synthesis of the
phosphonate analogue of serine has been reported.14 A retrosynthesis following this strategy is
shown in Scheme 2.1. Protected dehydrophos (4) could be synthesized from tripeptide 5
following activation and elimination of the hydroxyl group of the serine-like phosphonate. The

22
tripeptide 5 would then be synthesized by iterative coupling of Leu and Gly onto serine analogue
6. Though 6 was synthesized, it was never successfully coupled to N-protected leucine or an N-
terminal protected Gly-Leu dipeptide under multiple different peptide coupling conditions. One
reason for this difficulty could be the strongly electron withdrawing nature of the phosphonate,
which decreases the nucleophilicity of the α-amine.
Scheme 2.1 Initial retrosynthetic analysis for 3.
Alternatively, a more convergent method was envisaged in which an α-oxo phosphonate is
condensed with a terminal amide. Such methodology has been reported for synthesis of C-
terminal dehydroalanine containing peptides15 and for synthesis of diethyl N-acetyl-α,β-
unsaturated-α-amino phosphonic acids.16 Following this strategy, dimethyl 1-
oxoethylphosponate (7) and CBZ protected Gly-Leu dipeptide (8) underwent acid catalyzed
condensation to afford protected dehydrophos (9, Scheme 2.2). The yield of this reaction was
very low and it is necessary to purge the solvent of oxygen beforehand and have p-
hydroxyanisole as an oxygen scavenger to obtain any product. The low yield is believed to be
due to oxygen initiated oligomerization at the reactive vinyl group. The N-terminus was then
deprotected first by palladium-catalyzed transesterification of the benzylcarbamate to a
triethylsilylcarbamate, which was susceptible to base hydrolysis to afford the amine. It should be
noted that normal deprotection of the CBZ protecting group cannot be utilized since the
hydrogenolysis conditions would also reduce the alkene. The phosphonate was then selectively

23
hydrolyzed to the monomethyl ester17 to afford 3. Spectroscopic characterization (1H, 13C and 31P
NMR spectroscopy and HRMS) of synthetic 3 showed that it is identical to the phosphonate
produced by S. luridus, thus confirming the structure of dehydrophos. Though the overall yield
using this scheme is low, it has the advantages of being a highly convergent and rapid route to 3.
Scheme 2.2 Synthesis of dehydrophos.
2.4 Future Work
The unique structure of dehydrophos brings up interesting questions concerning its biosynthesis
and mode of action. Other antibiotics including phosphonate containing antibiotics such as
phosphinothricin tripeptide, alafosfalin, the rhizocticins and plumbemycins are biosynthesized as

24
short peptides to promote uptake by the target organism.18 Upon uptake and translocation these
peptides are hypothesized to undergo hydrolysis by a non-specific protease to afford an active
phosphonate containing amino acid.19 Hydrolysis of dehydrophos at the C-terminus would afford
an enamine, which would subsequently isomerize and hydrolyze to afford a mimic of pyruvate,
methyl acetylphosphonate (10, Scheme 2.3a), which has been shown to inhibit pyruvate
dehydrogenase in vitro.20 Alternatively, dehydrophos could derive activity without hydrolysis in
analogy to the phosphonate K-26, which directly inhibits angiotensin-converting enzyme.21 In
this mechanism the highly electrophilic dehydroalanine-like moiety could undergo Michael-type
addition by a nucleophile on the target leading to covalent inhibition (Scheme 2.3b). Studies into
the mode of action of dehydrophos are currently ongoing. Recent work by Ben Circello in the
William Metcalf laboratory on the biosynthesis of dehydrophos has identified the minimal
biosynthetic gene cluster and afforded much insight into the biosynthetic steps.22
Scheme 2.3 Potential modes of action for dehydrophos either via (a) proteolysis or (b) covalent
inhibition.

25
2.5 Experimental
2.5.1 Production of A53868 by Streptomyces luridis
As reported by Benjamin Griffin, S. luridis (NRRL 15101) was obtained from the Agricultural
Research Service Culture Collection (Peoria, IL). S. luridis was plated on ISP medium 4 (Difco,
Sparks, MD) with the following composition (g/L): soluble starch (10), dipotassium phosphate
(1.0), magnesium sulfate (1.0), sodium chloride (1.0), ammonium sulfate (2.0), calcium
carbonate (2.0), agar (20), ferrous sulfate (0.001), manganous chloride (0.001), and zinc sulfate
(0.001) at pH 7.2. After incubating at 30 °C for three days, the agar-solidified medium was
liquefied by repeated freezing and subsequent thawing. The resulting supernatant was separated
from the residual agar by filtration, before being concentrated ten-fold via rotary evaporation.
15N-labeled A53868 was produced in a similar manner using ISP medium 4 plates that contained
15N-ammonium sulfate (98+ atom% 15N, Isotec, Miamisburg, OH) as the sole nitrogen source.
Double-labeled 15N13C-A53868 was produced by S. luridis in broth culture using a modified ISP
medium 4 that did not contain agar or soluble starch. The carbon source was 13C6-D-glucose (10
g/L, 99 atom% 13C, Isotec, Miamisburg, OH), and the nitrogen source was 15N-ammonium
sulfate (2 g/L). The liquid cultures were incubated for three weeks at 30 °C on a rotary shaker at
225 rpm. Biomass was removed by filtration through a 0.22 µ filter and the supernatant was
concentrated ten-fold via rotary evaporation.
2.5.2 Purification of Bacterial Cell Free Broth
As detailed in Notebook I on pages 48-50, cell free broth was purified via reverse phase HPLC
(C18) using the following conditions: Solvent A = 79.9% acetonitrile, 19.9% deionized H2O,

26
0.09% trifluoroacetic acid. Solvent B: 99.9% deinoized H2O, 0.1% trifluoroacetic acid. A linear
gradient of 2-25% of A in B over 45 min at 8 mL/min was used. The retention time for A53868
was 21 min. Acetonitrile and trifluoroacetic acid were removed under reduced pressure, and then
the sample was flash frozen in liquid N2. Water was removed by lyophilization on a
LABCONCO (model Freezone 4.5) at -49 oC.
2.5.2.1 Spectral Characterization of Unlabeled A53868
1H NMR (400 MHz, D2O) δ 0.73 (d, J = 6.2 Hz, 3H), 0.78 (d, J = 6.2 Hz, 3H), 1.46 (m, 3H), 3.34
(d, J = 11 Hz, 3H), 3.72 (s, 2H), 4.25 (m, 1H), 5.52 (d, J = 15 Hz, 1H), 6.01 (d, J = 36 Hz, 1H);
31P NMR (162 MHz, D2O) 10.5; HRMS (FTMS+) calcd m/z (C11H22N3O5P+H+)+ 308.1365,
found 308.1367.
2.5.2.2 Spectral Characterization of 15N-Labeled A53868
1H NMR (400 MHz, D2O) 0.73 (d, J = 6.2 Hz, 3H), 0.78 (d, J = 6.2 Hz, 3H), 1.46 (m, 3H), 3.34
(d, J = 11 Hz, 3H), 3.72 (s, 2H), 4.25 (m, 1H), 5.52 (dd, J = 6, 16 Hz, 1H), 6.01 (dd, J = 2, 36 Hz,
1H); 31P NMR (162 MHz, D2O) 10.5 (d, J = 8.5 Hz).
2.5.2.3 Spectral Characterization of 15N,13C-Labeled A53868
13C NMR (100 MHz, D2O) 21.7 (d, J = 34 Hz), 22.9 (d, J = 34.9 Hz), 25.2 (m), 40.6 (m), 41.2
(m), 52.9 (d, J = 9 Hz), 54.2 (m), 117.1 (dd, J = 12, 73 Hz), 135.7 (ddd, J = 11, 73, 190 Hz),
167.9 (dd, J = 17, 51 Hz), 174.2 (dd, J = 7.4, 14.4, 52.8 Hz).

27
2.5.3 Synthesis
2.5.3.1 N-Carboxybenzyl-glycinyl-leucine methyl ester
As detailed in Notebook IV on page 45, in a 500 mL round bottom flask L-leucine methyl ester
hydrochloride (10.4 g, 50 mmol, 1 eq.), N-carboxybenzyl-glycine and O-(benzotriazol-1-yl)-
N,N,N′,N′-tetramethyluronium hexafluorophosphate (20.8 g, 55 mmol, 1.1 eq) was dissolved in
250 mL of DMF. While stirring, N-methylmorpholine (12 mL, 110 mmol, 2.2 eq) was added.
The yellow solution was stirred overnight at room temperature. The solution was poured into a 1
L separatory funnel, diluted with 500 mL of EtOAc, washed with aqueous 5% citric acid (1x200
mL), saturated aqueous NaHCO3 (1x300 mL), and brine (1x200 mL). The organic layer was
saved and dried over Na2SO4, filtered, and concentrated via rotary evaporation. Purification by
silica gel flash chromatography (1:1 EtOAc:hexanes) afforded the product (15.2 g, 45.2 mmol,
90%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 0.91 (d, J = 4.9, 6H, CH3), 1.61 (m, 3H,
CH, CH2), 3.71 (s, 3H, OCH3), 3.91 (m, 2H, N-CH2), 4.63 (m, 1H, N-CH), 5.12 (s, 2H, Ph-
CH2), 5.54 (bs, 1H, NH), 6.53 (bs, 1H, NH), 7.34 (m, 5H, Ph); 13C NMR (100 MHz, CDCl3) δ
22.0, 23.0, 25.0, 41.6, 44.6, 50.9, 52.6, 67.4, 128.3, 128.4, 128.7, 136.3, 156.8, 169.0, 173.5;
HRMS (ESI+) calcd m/z (C17H24N2O5+H+)+ 337.1763, found: 337.1755
2.5.3.2 N-Carboxybenzyl-glycinyl-leucinamide (8)
As detailed in Notebook IV on page 47, in a 500 mL round bottom flask with a magnetic stir bar
CBZ-Gly-Leu-OMe (11 g, 33 mmol) was dissolved in 100 mL of methanol. To this colorless
solution was then added 100 mL of NH4OH (28-30% ammonia) and the reaction mixture was
stirred for 2 d at room temperature. The yellow solution was poured into a 1 L separatory funnel

28
and diluted with 200 mL of brine. The product was extracted with EtOAc (3x300 mL). The
organic layers were combined and washed with 100 mL of brine, dried over Na2SO4, filtered and
concentrated to dryness under reduced pressure to afford 8.45 g of 8 (26.2 mmol, 80%) as a
white foam. 1H NMR (400 MHz CD3CN) δ 0.89 (d, J = 6.2 Hz, 3H, CH3), 0.92 (d, J = 6.3 Hz,
3H, CH3), 1.583 (m, 3H, CH + CH2), 3.75 (d, J = 6.0 Hz, 2H, N-CH2), 4.32 (m, 1H, N-CH), 5.10
(d, J = 1.5, 2H, Ph-CH2), 6.01 (bs, 1H, CO-NH), 6.20 (t, J = 5.8 Hz, 1H, NHCH2), 6.36 (bs, 1H,
CO-NH), 7.04 (d, J = 7.5, 1H, NH-CH) 7.39 (m, 5H, Ph); 13C NMR (100 MHz, CD3CN) δ 21.0,
22.7, 40.8, 44.3, 51.7, 66.6, 128.1, 128.2, 128.7, 137.2, 157.2, 169.8, 175.0 HRMS (ESI+); calcd
m/z (C16H23N3O4+H+) 322.1767, found 322.1763
2.5.3.3 Acetylphosphonate dimethyl ester (7)
As detailed in Notebook IV on page 38, in a dry three-necked round bottom flask with a
magnetic stir bar under positive N2 pressure, acetyl chloride (3.5 mL, 50 mmol, 1 eq) was cooled
to 0 oC in an ice-water bath. Then, trimethyl phosphite (5.9 mL, 50 mmol, 1 eq) was slowly
added over a period of 1 h, which resulted in evolution of MeCl. After full addition of trimethyl
phosphite, the colorless solution was allowed to warm to room temperature and stirred until
evolution of gas ceased (about 1 h). Unreacted materials were removed under vacuum to afford 7
(7.3 g, 48 mmol, 96%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 2.47 (d, J = 5.28 Hz, 3H,
CH3), 3.85 (d, J = 10.71, 6H, OCH3); 13C NMR (100 MHz, CDCl3) 30.90 (d, J = 59.5 Hz), 54.07
(d, J = 7.00 Hz), 208.40 (d, J = 170.4 Hz); 31P NMR (162 MHz, CDCl3) δ 0.02 HRMS (EI-) calcd
m/z (C4H9O4P) 152.0328, found 152.0326.

29
2.5.3.4 N-Carboxybenzyl-glycinyl-leucinyl-1-amidoethylene-1-phosphonate dimethyl ester
(9)
As detailed in Notebook IV on page 49, in a 250 mL round bottom flask equipped with a Dean-
Stark apparatus, Ar was bubbled through 75 mL of toluene for 1 h. 4-Hydroxyanisole (253 mg,
2.04 mmol, 20 mol%) was added with stirring. After all 4-hydroxyanisole was dissolved, 8 (3.28
g, 10.2 mmol, 1 eq) and 7 (1.55 g, 10.2 mmol, 1 eq) and p-toluenesulfonic acid monohydrate
(194 mg, 1.02 mmol, 10% mol) were added and the suspension was heated under reflux
overnight resulting in a yellow solution. The reaction mixture was allowed to cool to room
temperature and toluene was removed via rotary evaporation. The resulting red foam was taken
up in 500 mL of EtOAc, washed with saturated aqueous NaHCO3 (1x200 mL), and brine (1x150
mL). The organic layer was dried over Na2SO4, filtered, and concentrated via rotary evaporation.
Purification by silica gel flash chromatography (30:1 DCM:MeOH) yielded 9 (519 mg, 1.14
mmol, 11.2%) as a white foam. 1H NMR (400 MHz, CDCl3) δ 0.86 (d, J = 5.5 Hz, 3H, CH2),
0.89 (d, J = 5.4 Hz, 3H, CH3), 1.55 (m, 3H, CH + CH2), 3.69 (d, J = 11.2 Hz , 6H, OCH3), 3.89
(d, J = 5.38 Hz, 2H, NCH2), 4.62 (m, 1H, N-CH), 5.08 (s, 2H, Ph-CH2), 5.58 (d, J = 19.1 Hz, 1H,
C=CH), 5.95 (m, 1H, NH), 6.61 (d, J = 41.7 Hz, 1H, C=CH), 7.05 (d, J = 8.7 Hz, 1H, NH), 7.30
(m, 5H, Ph), 8.29 (d, J = 7.8 Hz, 1H, NH); 13C NMR (100 MHz, CDCl3) 22.09, 23.08, 24.92,
40.90, 44.50, 52.63, 53.58 (d, J = 5.5 Hz), 67.35, 116.29 (d, J = 10.0 Hz), 128.30, 128.45,
128.76, 130.22 (d, J = 201 Hz), 136.37, 156.97, 170.00, 172.07 (d, J = 10.0 Hz); 31P NMR (162
MHz, CDCl3) 15.7; HRMS (ESI+) calcd m/z (C20H30N3O7P+H+)+ 456.1900 found 456.1901.

30
2.5.3.5 Glycinyl-leucinyl-1-amidoethylene-1-phosphonate monomethyl ester (3)
As detailed in Notebook IV on page 50, in a clean 25 mL round bottom flask equipped with a
magnetic stir bar, 9 (433 mg, 0.95 mmol, 1 eq) was dissolved in 5 mL of dry DCM. Then,
triethylsilane (303 µL, 1.9 mmol, 2 eq), palladium(II) chloride (16 mg, 0.09 mmol, 10% mol),
and triethylamine (50 µL, 0.66 mmol, 70%) were added while stirring. The black suspension was
stirred at room temperature for 2 h, filtered over a column of Celite 545®, and concentrated to
dryness under reduced pressure to yield a yellow oil. This oil was suspended in 3 mL of an
aqueous solution of 10% NaOH and stirred vigorously overnight at room temperature. The
resulting yellow solution was diluted to 10 mL with H2O, filtered through a 0.22 µm filter unit,
and purified by HPLC (C18) using the same method to isolate A53868 from cell free broth to
yield 175 mg of (3) (0.57 mmol, 60%) as a white solid. 1H NMR (400 MHz, D2O) δ 0.68 (d, J =
5.5 Hz, 3H, CH3), 0.72 (d, J = 5.7 Hz, 3H, CH3), 1.45 (m, 3H, CH2, CH), 3.28 (d, J = 11 Hz. 3H,
OCH3), 3.65 (s, 2H, N-CH2), 4.21 (m, 1H, N-CH), 5.53 (d, J = 16 Hz, 1H, C=CH), 5.95 (d, J =
36 Hz, 1H, C=CH); 13C NMR (100 MHz, D2O) 20.69, 22.14, 24.4, 39.7, 40.3 52.1 (d, J = 5.2),
53.4, 117.6 (d, J = 11.7), 134.5 (d, J = 190.4), 167.2, 173.8; 31P NMR (162 MHz, D2O) 10.5;
HRMS (FTMS+) calcd m/z (C11H22N3O5P+H+)+ 308.1365, found 308.1367.
2.6 References
1. Johnson, R. D.; Gordee, R. S.; Kastner, R. M.; Larsen, S. H.; Ose, E. E. (Eli Lilly). UK 2,127,413, 1984, [ Chem. Abstr. 1984, 101, 88837r]. 2. Hunt, A.; Elzey, T., J. Antibiot. 1988, 41, 802. 3. Chatterjee, C.; Paul, M.; Xie, L.; van der Donk, W. A., Chem. Rev. 2005, 105, (2), 633-684. Biosynthesis and Mode of Action of Lantibiotics.

31
4. Rinehart, K. L.; Harada, K.; Namikoshi, M.; Chen, C.; Harvis, C. A.; Munro, M. H. G.; Blunt, J. W.; Mulligan, P. E.; Beasley, V. R.; Dahlem, A. M.; Carmichael, W. W., J. Am. Chem. Soc. 1988, 110, (25), 8557-8558. Nodularin, microcystin, and the configuration of Adda. 5. Anderson, B.; Hodgkin, D. C.; Viswamitra, M. A., Nature 1970, 225, (5229), 233-235. The Structure of Thiostrepton. 6. Schmidt, U.; Lieberknecht, A.; Wild, J., Synthesis 1988, 1988, (03), 159-172. Didehydroamino Acids (DDAA) and Didehydropeptides (DDP). 7. Okeley, N. M.; Zhu, Y.; van der Donk, W. A., Org. Lett. 2000, 2, (23), 3603-3606. Facile Chemoselective Synthesis of Dehydroalanine-Containing Peptides. 8. Zhu, Y.; van der Donk, W. A., Org. Lett. 2001, 3, (8), 1189-1192. Convergent Synthesis of Peptide Conjugates Using Dehydroalanines for Chemoselective Ligations. 9. Galonic, D. P.; van der Donk, W. A.; Gin, D. Y., Chem. Eur. J. 2003, 9, (24), 5997-6006. Oligosaccharide-Peptide Ligation of Glycosyl Thiolates with Dehydropeptides: Synthesis of S-Linked Mucin-Related Glycopeptide Conjugates. 10. Levengood, M. R.; van der Donk, W. A., Nat. Protocols 2007, 1, (6), 3001-3010. Dehydroalanine-containing peptides: preparation from phenylselenocysteine and utility in convergent ligation strategies. 11. Ranganathan, D.; Shah, K.; Valsh, N., J. Chem. Soc. Chem. Comm. 1992, 1145-1147. 12. Blettner, C.; Bradley, M., Tetrahedron Lett. 1994, 35, (3), 467-470. Asparagine as a masked dehydroalanine residue in solid phase peptide synthesis. 13. Sarah, B.; Tony, R.; Glyn, W.; Jonathan, W. E.; Carl, A.; Marianne, C.; Vinay, S.; Mark, B., Chem. Eur. J. 2000, 6, (8), 1455-1466. Biomimetic Synthesis of Lantibiotics. 14. Corcoran, R. C.; Green, J. M., Tetrahedron Lett. 1990, 31, (47), 6827-6830. Conversion of [alpha]-aminocarboxylic acids to [alpha]-aminophosphonic acids. 15. Maciej, M.; Barbara, R.; Zbigniew, K.; Grzegorz, P.; nacute; nski, Liebigs Ann. Chem. 1985, 1985, (5), 893-900. Synthesis of Peptides with alpha,beta-Dehydroamino Acids, II. Synthesis of tert-Butyloxycarbonyldipeptides of Dehydroalanine and Dehydrophenylalanine. 16. Zon, J., Synthesis 1981, 1981, (04), 324-324. A Simple Preparation of Diethyl 1-Acylamino-1-ethenephosphonates. 17. Rabinowitz, R., J. Am. Chem. Soc. 1960, 82, (17), 4564-4567. Synthesis of Monoesters of Phosphonic Acids.

32
18. Elizabeth, M. N.; Christopher, T. W., ChemBioChem 2009, 10, (1), 34-53. How Nature Morphs Peptide Scaffolds into Antibiotics. 19. Metcalf, W. W.; van der Donk, W. A., Ann. Rev. Biochem. 2009, 78, (1), 65-94. Biosynthesis of Phosphonic and Phosphinic Acid Natural Products. 20. Kluger, R.; Pike, D. C., J. Am. Chem. Soc. 1977, 99, (13), 4504-4506. Active site generated analogs of reactive intermediates in enzymic reactions. Potent inhibition of pyruvate dehydrogenase by a phosphonate analog of pyruvate. 21. Yamato, M.; Koguchi, R.; Okachi, K.; Yamada, K.; Nakayama, H.; Kase, A.; Karasawa, K.; Shuto, J., J. Antibiot. 1986, 39, 44. 22. Circello, B. T.; Eliot, A. C.; Lee, J.-H.; van der Donk, W. A.; Metcalf, W. W., Chem. Biol. 2010, 17, (4), 402-411. Molecular Cloning and Heterologous Expression of the Dehydrophos Biosynthetic Gene Cluster.

33
CHAPTER 3: MECHANISTIC STUDIES ON THE CONVERSION OF 2-HEP TO HMP BY
HEPD*
3.1 Background
As described in chapter 1.4.2, genetic studies have suggested that during the biosynthesis of
phosphinothricin, 2-hydroxyethylphosphonate (2-HEP) is converted to hydroxymethylphosphonate
(HMP) by the product of the phpD gene (Scheme 3.1). Dr. Robert Cicchillo, a former postdoctoral
researcher in the van der Donk laboratory, was successful in expressing and purifying the enzyme
(labeled HEPD) in E. coli as an N-terminal His6 fusion protein. He was also able to show that the
enzyme is dependent only on molecular oxygen and one equivalent of ferrous iron for activity.1
Scheme 3.1 2-HEP is converted to HMP by the gene product of phpD (HEPD) during phosphinothricin
biosynthesis.
In order to further study this biochemically unique transformation, synthetic stable isotopologues
(defined in this thesis as synthetic compounds that are enriched in a naturally low abundance, non-
radioactive isotope) have been synthesized. Stable isotopologues have been used extensively to study
primary and secondary metabolism in addition to organic and enzymatic mechanisms. Enriching in
NMR-active isotopes (such as 13C, natural abundance = 1.1%) can increase the sensitivity and selectivity
* Parts of this chapter have been reproduced from 1) Whitteck, J.T., Cicchillo, R.M., van der Donk, W.A., J. Am. Chem. Soc. 2009, 131, 16225-16232 and 2) Cicchillo, R.M., Zhang, H., Blodgett, J.A.V., Whitteck, J.T., Li, G., Nair, S.K., van der Donk, W.A., Metaclf, W.W., Nature, 2009, 459, 871-874 with permission from the American Chemical Society and Nature Publishing Group, respectively.

34
of assays to aid in identification of products. More generally, enriching in other isotopes can be used in
conjunction with mass spectrometry to follow atoms from the substrate to the products. In the context of
these experiments, isotopologues of 2-HEP have been synthesized and presented to HEPD as substrates.
Characterization of the products has given insight into the mechanism of this biochemically unique
transformation.
3.2 Fate of the Carbon Atoms of 2-HEP upon Conversion to the Products
In experiments performed by Dr. Robert Cicchillo, to unambiguously identify the phosphorus-
containing product of the HEPD reaction, purified His6-HEPD (10 µM) was incubated with 1-13C-2-
HEP in oxygenated HEPES buffer (pH = 7.5) for 2 h. The 31P NMR spectrum was then recorded and
showed two doublets with splitting from the 13C nucleus (Figure 3.1a). One doublet corresponded to
unoxidized 1-13C-2-HEP. The chemical shift of the other doublet (δ = 18 ppm) corresponded to HMP.
The assay was again performed using unlabeled 2-HEP as a substrate and the 31P NMR spectrum was
recorded. Two peaks were observed with chemicals shifts that were identical to the previous assay.
Spiking the sample with synthetic HMP confirmed the identity of the phosphorus-containing product.
Together with the 13C labeling experiments HMP was thus shown unambiguously to be a product of 2-
HEP oxidation by HEPD.
In experiments performed by Dr. Robert Cicchillo, to identify the product of the excised carbon atom,
HEPD (10 µM) was incubated with 2-13C-2-HEP in phosphate buffer (pH = 7.5) for 2 h. It was
necessary to use phosphate buffer to avoid peaks in the 13C NMR spectrum from buffer (though the
substrate is labeled, buffer is used in a 25 fold concentration and would likely produce peaks in the
spectrum). The recorded 13C NMR spectrum showed two peaks: one corresponding to 2-13C-2-HEP and
a second peak at δ = 171.1 ppm, which could correspond to either formate or formaldehyde (Figure
3.1b). Spiking with commercially available 13C-formate confirmed that the peak was derived from
formate, showing that C2 of HEP is excised and oxidized to formate.
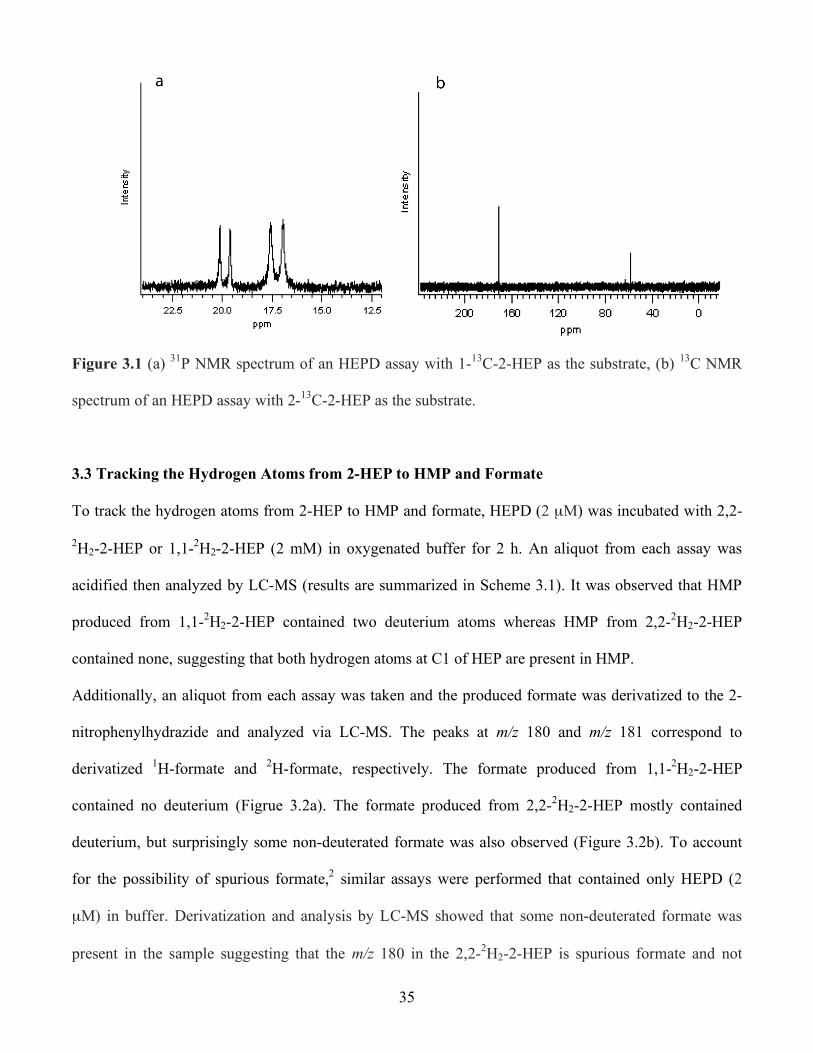
35
Figure 3.1 (a) 31P NMR spectrum of an HEPD assay with 1-13C-2-HEP as the substrate, (b) 13C NMR
spectrum of an HEPD assay with 2-13C-2-HEP as the substrate.
3.3 Tracking the Hydrogen Atoms from 2-HEP to HMP and Formate
To track the hydrogen atoms from 2-HEP to HMP and formate, HEPD (2 µM) was incubated with 2,2-
2H2-2-HEP or 1,1-2H2-2-HEP (2 mM) in oxygenated buffer for 2 h. An aliquot from each assay was
acidified then analyzed by LC-MS (results are summarized in Scheme 3.1). It was observed that HMP
produced from 1,1-2H2-2-HEP contained two deuterium atoms whereas HMP from 2,2-2H2-2-HEP
contained none, suggesting that both hydrogen atoms at C1 of HEP are present in HMP.
Additionally, an aliquot from each assay was taken and the produced formate was derivatized to the 2-
nitrophenylhydrazide and analyzed via LC-MS. The peaks at m/z 180 and m/z 181 correspond to
derivatized 1H-formate and 2H-formate, respectively. The formate produced from 1,1-2H2-2-HEP
contained no deuterium (Figrue 3.2a). The formate produced from 2,2-2H2-2-HEP mostly contained
deuterium, but surprisingly some non-deuterated formate was also observed (Figure 3.2b). To account
for the possibility of spurious formate,2 similar assays were performed that contained only HEPD (2
µM) in buffer. Derivatization and analysis by LC-MS showed that some non-deuterated formate was
present in the sample suggesting that the m/z 180 in the 2,2-2H2-2-HEP is spurious formate and not
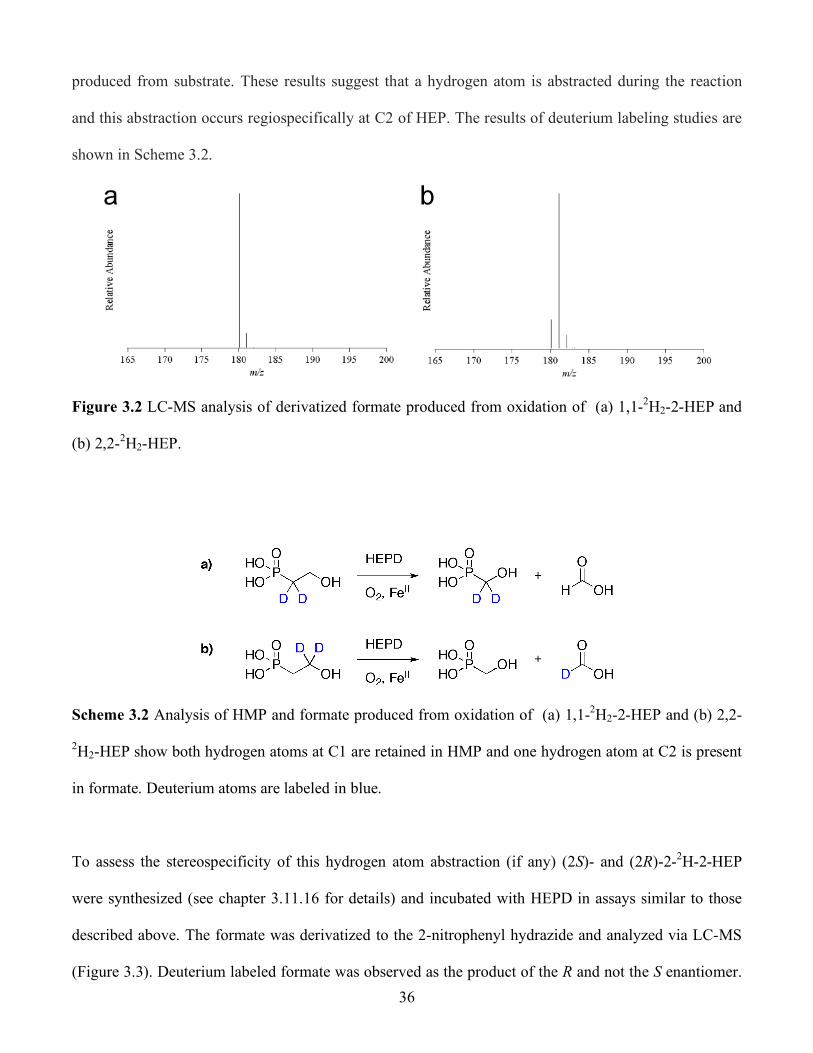
36
produced from substrate. These results suggest that a hydrogen atom is abstracted during the reaction
and this abstraction occurs regiospecifically at C2 of HEP. The results of deuterium labeling studies are
shown in Scheme 3.2.
Figure 3.2 LC-MS analysis of derivatized formate produced from oxidation of (a) 1,1-2H2-2-HEP and
(b) 2,2-2H2-HEP.
Scheme 3.2 Analysis of HMP and formate produced from oxidation of (a) 1,1-2H2-2-HEP and (b) 2,2-
2H2-HEP show both hydrogen atoms at C1 are retained in HMP and one hydrogen atom at C2 is present
in formate. Deuterium atoms are labeled in blue.
To assess the stereospecificity of this hydrogen atom abstraction (if any) (2S)- and (2R)-2-2H-2-HEP
were synthesized (see chapter 3.11.16 for details) and incubated with HEPD in assays similar to those
described above. The formate was derivatized to the 2-nitrophenyl hydrazide and analyzed via LC-MS
(Figure 3.3). Deuterium labeled formate was observed as the product of the R and not the S enantiomer.
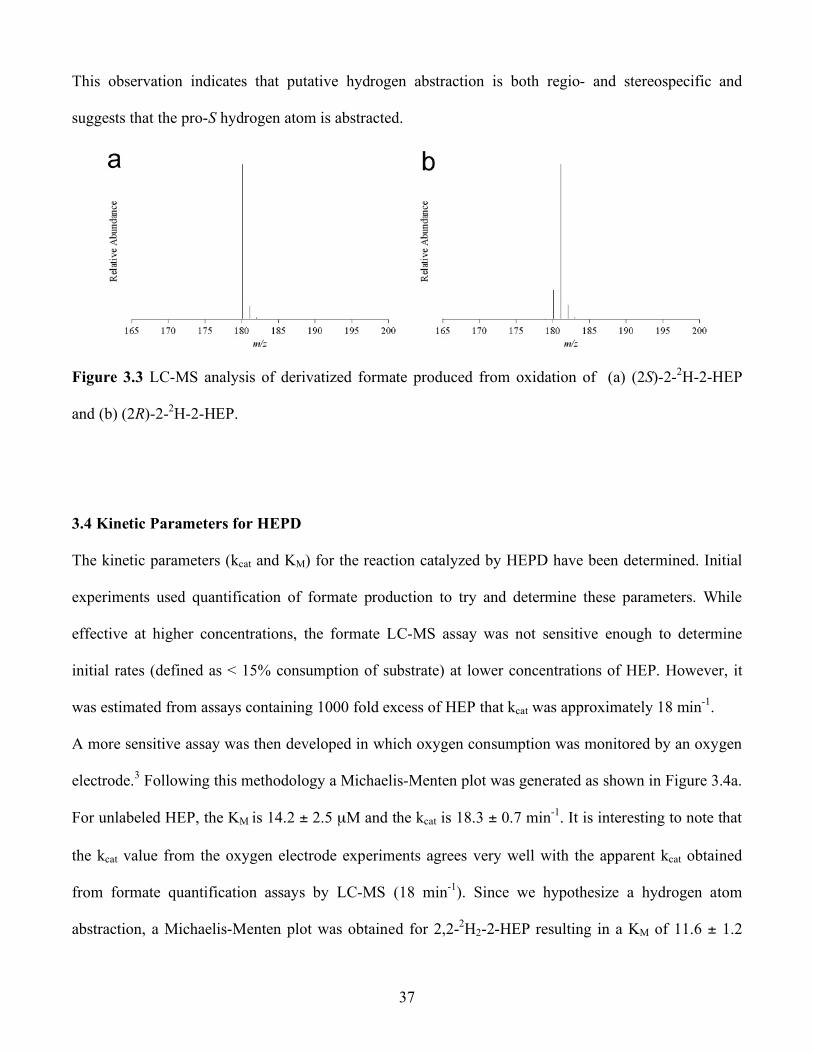
37
This observation indicates that putative hydrogen abstraction is both regio- and stereospecific and
suggests that the pro-S hydrogen atom is abstracted.
Figure 3.3 LC-MS analysis of derivatized formate produced from oxidation of (a) (2S)-2-2H-2-HEP
and (b) (2R)-2-2H-2-HEP.
3.4 Kinetic Parameters for HEPD
The kinetic parameters (kcat and KM) for the reaction catalyzed by HEPD have been determined. Initial
experiments used quantification of formate production to try and determine these parameters. While
effective at higher concentrations, the formate LC-MS assay was not sensitive enough to determine
initial rates (defined as < 15% consumption of substrate) at lower concentrations of HEP. However, it
was estimated from assays containing 1000 fold excess of HEP that kcat was approximately 18 min-1.
A more sensitive assay was then developed in which oxygen consumption was monitored by an oxygen
electrode.3 Following this methodology a Michaelis-Menten plot was generated as shown in Figure 3.4a.
For unlabeled HEP, the KM is 14.2 ± 2.5 µM and the kcat is 18.3 ± 0.7 min-1. It is interesting to note that
the kcat value from the oxygen electrode experiments agrees very well with the apparent kcat obtained
from formate quantification assays by LC-MS (18 min-1). Since we hypothesize a hydrogen atom
abstraction, a Michaelis-Menten plot was obtained for 2,2-2H2-2-HEP resulting in a KM of 11.6 ± 1.2
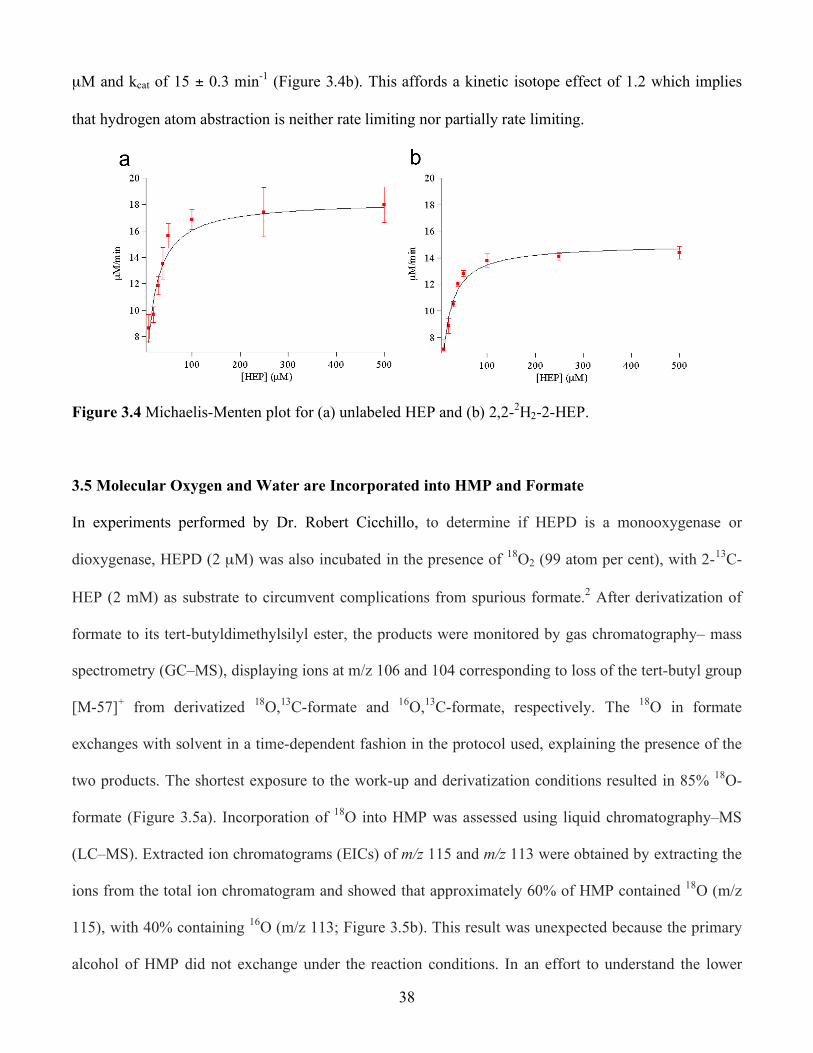
38
µM and kcat of 15 ± 0.3 min-1 (Figure 3.4b). This affords a kinetic isotope effect of 1.2 which implies
that hydrogen atom abstraction is neither rate limiting nor partially rate limiting.
Figure 3.4 Michaelis-Menten plot for (a) unlabeled HEP and (b) 2,2-2H2-2-HEP.
3.5 Molecular Oxygen and Water are Incorporated into HMP and Formate
In experiments performed by Dr. Robert Cicchillo, to determine if HEPD is a monooxygenase or
dioxygenase, HEPD (2 µM) was also incubated in the presence of 18O2 (99 atom per cent), with 2-13C-
HEP (2 mM) as substrate to circumvent complications from spurious formate.2 After derivatization of
formate to its tert-butyldimethylsilyl ester, the products were monitored by gas chromatography– mass
spectrometry (GC–MS), displaying ions at m/z 106 and 104 corresponding to loss of the tert-butyl group
[M-57]+ from derivatized 18O,13C-formate and 16O,13C-formate, respectively. The 18O in formate
exchanges with solvent in a time-dependent fashion in the protocol used, explaining the presence of the
two products. The shortest exposure to the work-up and derivatization conditions resulted in 85% 18O-
formate (Figure 3.5a). Incorporation of 18O into HMP was assessed using liquid chromatography–MS
(LC–MS). Extracted ion chromatograms (EICs) of m/z 115 and m/z 113 were obtained by extracting the
ions from the total ion chromatogram and showed that approximately 60% of HMP contained 18O (m/z
115), with 40% containing 16O (m/z 113; Figure 3.5b). This result was unexpected because the primary
alcohol of HMP did not exchange under the reaction conditions. In an effort to understand the lower
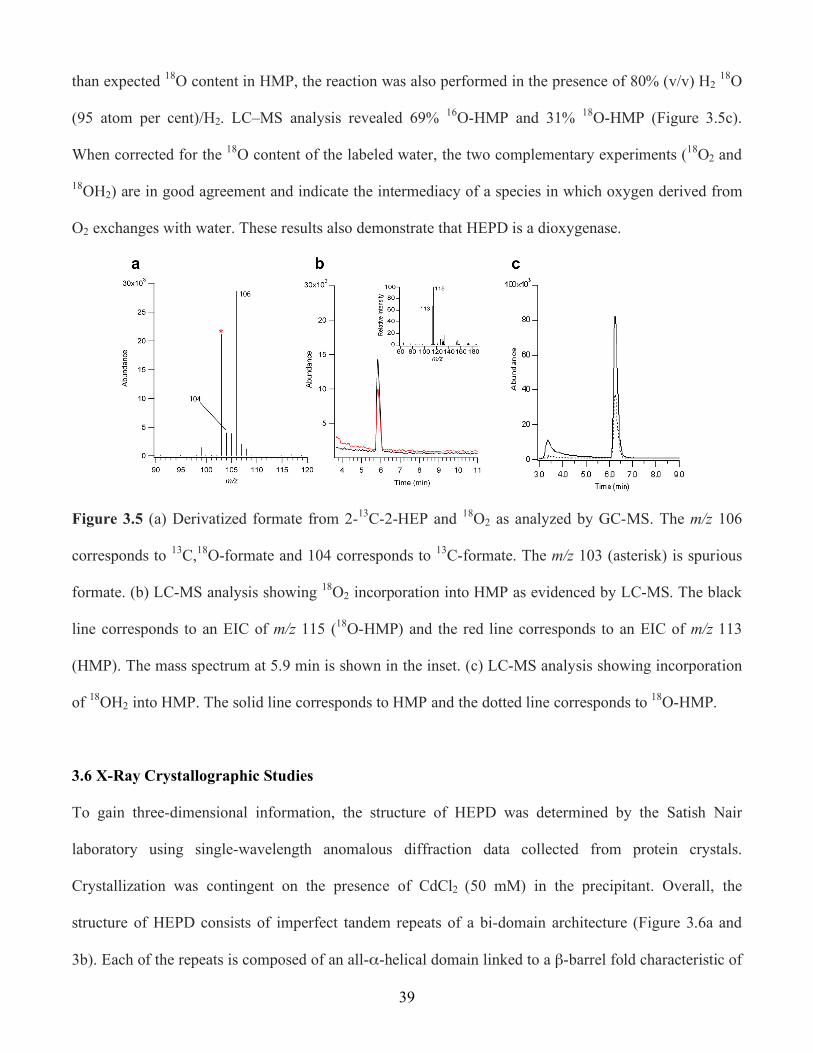
39
than expected 18O content in HMP, the reaction was also performed in the presence of 80% (v/v) H2 18O
(95 atom per cent)/H2. LC–MS analysis revealed 69% 16O-HMP and 31% 18O-HMP (Figure 3.5c).
When corrected for the 18O content of the labeled water, the two complementary experiments (18O2 and
18OH2) are in good agreement and indicate the intermediacy of a species in which oxygen derived from
O2 exchanges with water. These results also demonstrate that HEPD is a dioxygenase.
Figure 3.5 (a) Derivatized formate from 2-13C-2-HEP and 18O2 as analyzed by GC-MS. The m/z 106
corresponds to 13C,18O-formate and 104 corresponds to 13C-formate. The m/z 103 (asterisk) is spurious
formate. (b) LC-MS analysis showing 18O2 incorporation into HMP as evidenced by LC-MS. The black
line corresponds to an EIC of m/z 115 (18O-HMP) and the red line corresponds to an EIC of m/z 113
(HMP). The mass spectrum at 5.9 min is shown in the inset. (c) LC-MS analysis showing incorporation
of 18OH2 into HMP. The solid line corresponds to HMP and the dotted line corresponds to 18O-HMP.
3.6 X-Ray Crystallographic Studies
To gain three-dimensional information, the structure of HEPD was determined by the Satish Nair
laboratory using single-wavelength anomalous diffraction data collected from protein crystals.
Crystallization was contingent on the presence of CdCl2 (50 mM) in the precipitant. Overall, the
structure of HEPD consists of imperfect tandem repeats of a bi-domain architecture (Figure 3.6a and
3b). Each of the repeats is composed of an all-α-helical domain linked to a β-barrel fold characteristic of

40
the cupin superfamily.4 Despite the lack of appreciable sequence homology, each of the two repeats is
structurally homologous to the monomer of HppE,5 a nonheme iron-dependent enzyme that converts
(S)-2-hydroxypropylphosphonic acid to fosfomycin (see chapter 1 for further details). The β-barrel fold
of the first repeat contains all of the canonical active site elements of non-heme iron enzymes, including
a 2-His–1-carboxy facial triad.6 A Cd(II) metal ion is situated at the base of the active site and is
coordinated by His129, Glu176 and His182 (Figure 3.6c). The metal binding ligands are similar to that of
HppE, but the position of these ligands within the β-barrel are not conserved since Glu176 of HEPD is
situated on a different β-strand to the equivalent Glu142 of HppE.5 Furthermore, the spacing between the
first two metal ligands in HEPD (HX46E, where X represents any amino acid) is unique as these residues
are closely spaced (HX1–4E/D) in other facial triad enzymes.5 Attempts in the Nair laboratory to produce
crystals of Fe(II) HEPD have failed owing to the high concentrations of Cd(II) required for
crystallization. Additionally, crystals of Cd(II)–HEPD were grown in the presence of 2-HEP and
analysis of the structure has suggested bidentate coordination of substrate to Cd(II) (Figure 3.6d), which
is similar to that observed for binding of 2-HPP to Co(II)–HppE.5

41
Figure 3.6 a), b) Orthogonal views of Cd(II)–HEPD showing the tandem arrangement of the HppE fold
including cupin domains (blue and purple), α-helical domains (red and cyan), cadmium (white sphere)
and metal ligands (green). C) Stereoview of electron density maps using model phases (Fo – Fc). The
first map (contoured at 3σ in blue) was calculated by omitting metal-bound solvents (red sphere) before
one round of refinement. The second map, contoured at 5σ (purple mesh) and 14σ (yellow mesh), was
calculated by omitting the cadmium (gold sphere) before one round of refinement. d) Stereoview of
electron density maps (Fo – Fc) calculated using HEPD–HEP model phases. The map was calculated by
omitting non-protein metal-bound ligands before one round of refinement (contoured at 3σ in blue mesh
and 6σ in red). HEP carbons are shown in green. (Figure made by Prof. Satish Nair)

42
3.7 Proposed Mechanisms
Two mechanisms have been proposed to account for these data (Scheme 3.3). In the resting state,
ferrous iron is bound to HEPD by a 2-His/1-Carboxylate facial triad (His129, His182 and Glu176),1 which
is commonly found in many non-heme iron dependent enzymes.7, 8 The substrate is proposed to bind to
Fe(II) in a bidentate manner followed by reaction with O2, resulting in a Fe(III)–[O2]- intermediate.
This species is proposed to abstract the pro-S hydrogen atom from C2 of HEP to generate intermediate I,
akin to similar steps proposed for isopenicillin N synthase and myo-inositol oxygenase.9, 10 One
important difference between isopenicillin N synthase and HEPD is that the former contains a sulphur
ligand from the substrate that has been suggested on the basis of computational studies to stabilize the
Fe(III)–[O2] - intermediate and overcome the unfavourable energetics of O2 binding and activation.11
Although a thiolate ligand is lacking in HEPD, the observation that external electrons are not required
leaves no alternate pathways to convert the Fe(III)–[O2]- intermediate into a more reactive species
found in other non-heme iron oxygenases.12. 2-HEP does not have low-energy electrons that can be
provided by the substrate, as in the extradiol dioxygenases that use highly electron-rich catechol
substrates.6 HEPD also does not contain other nearby metals that are important for activity, in contrast to
the binuclear iron enzyme myo-inositol oxygenase.14 Hence, the only option for HEPD seems to be
hydrogen atom abstraction from C2 to produce the hydroperoxo complex II and a substrate radical.
Although the electronic consequences of bidentate substrate binding on molecular oxygen activation and
subsequent hydrogen atom abstraction are currently unknown, a recent biomimetic study by Prof.
Lawrence Que and coworkers showed synthetic mononuclear ferrous iron complexes were capable of
hydrogen atom abstraction from a bidentate α-hydroxy acid ligand.13 Further evidence has suggested a
Fe(III)–[O2]- intermediate is the hydrogen atom abstracting species.
From intermediate II, two different pathways can be envisioned to account for the labeling studies. The
substrate radical could attack the hydroperoxide generating a ferryl species (Fe(IV)=O) and a hemiacetal
(Scheme 3.3, intermediate II). The latter can undergo a retro-Claisen type-C-C bond scission with the

43
incipient negative charge on C1 attacking the electrophilic ferryl, potentially by means of a Horner
Wadsworth–Emmons-like intermediate stabilized by metal coordination (III). A ferryl would account
for the required intermediacy of a species in which oxygen derived from O2 can exchange with solvent
(Scheme 3.4).15-20 A second mechanism that can account for the observed data involves conversion of
intermediate II to a hydroperoxoacetal, which can undergo a Criegee-type rearrangement to provide the
formate ester of HMP (O-formylhydroxymethylphosphonate, OFHMP, IV). Hydrolysis of this ester is
expected to occur at the carbonyl carbon, but such a mechanism would not account for the incorporation
of oxygen derived from solvent into HMP. This observation can be explained if hydrolysis took place
via attack at C1 by solvent exchangeable hydroxide released in the Criegee rearrangement.
Scheme 3.3 Two proposed mechanisms for HEPD.

44
Scheme 3.4 Potential mechanism for exchange of a ferryl oxygen in HEPD with solvent.
3.8 Tracking the Hydroxyl Group from 2-HEP into HMP and Formate
These proposed mechanisms are restricted by the outcome of studies using either 18O2 or H218O, which
provided complementary evidence that the hydroxyl oxygen in the HMP product was derived in part
from molecular oxygen and in part from solvent. A third possibility that was not considered involves
partial incorporation of the hydroxyl oxygen from 2-HEP into HMP. A possible mechanism for such a
process is shown in Scheme 3.5.
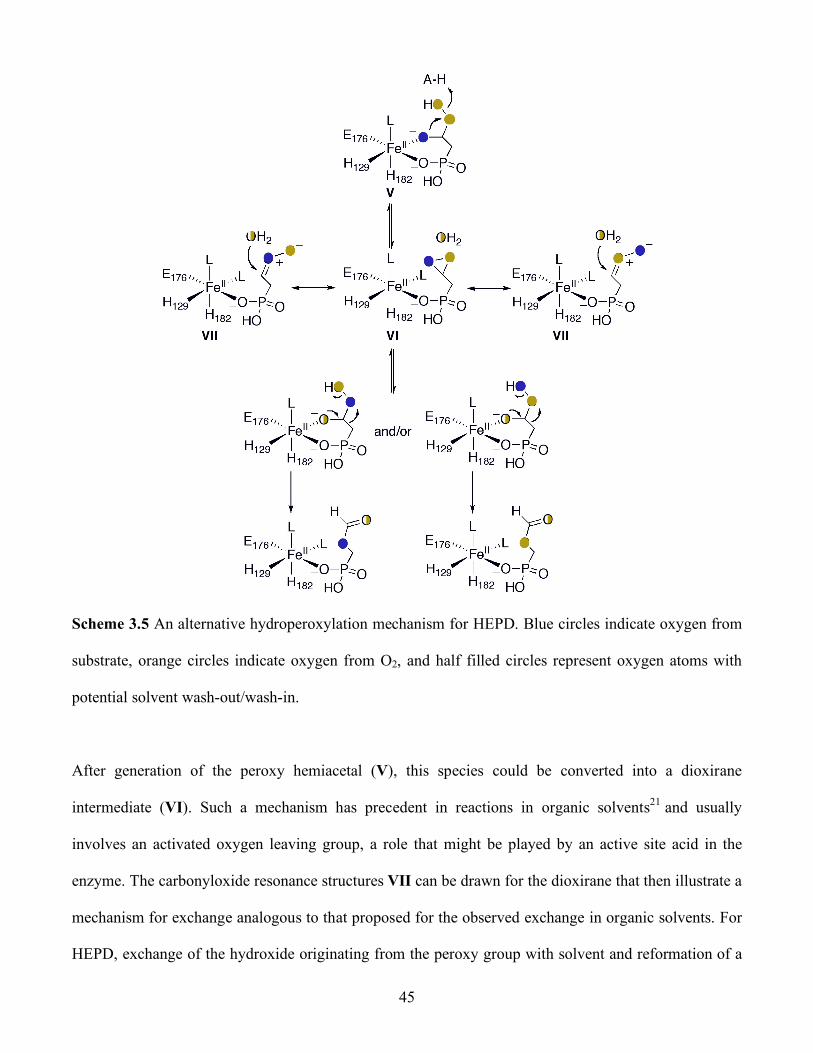
45
Scheme 3.5 An alternative hydroperoxylation mechanism for HEPD. Blue circles indicate oxygen from
substrate, orange circles indicate oxygen from O2, and half filled circles represent oxygen atoms with
potential solvent wash-out/wash-in.
After generation of the peroxy hemiacetal (V), this species could be converted into a dioxirane
intermediate (VI). Such a mechanism has precedent in reactions in organic solvents21 and usually
involves an activated oxygen leaving group, a role that might be played by an active site acid in the
enzyme. The carbonyloxide resonance structures VII can be drawn for the dioxirane that then illustrate a
mechanism for exchange analogous to that proposed for the observed exchange in organic solvents. For
HEPD, exchange of the hydroxide originating from the peroxy group with solvent and reformation of a
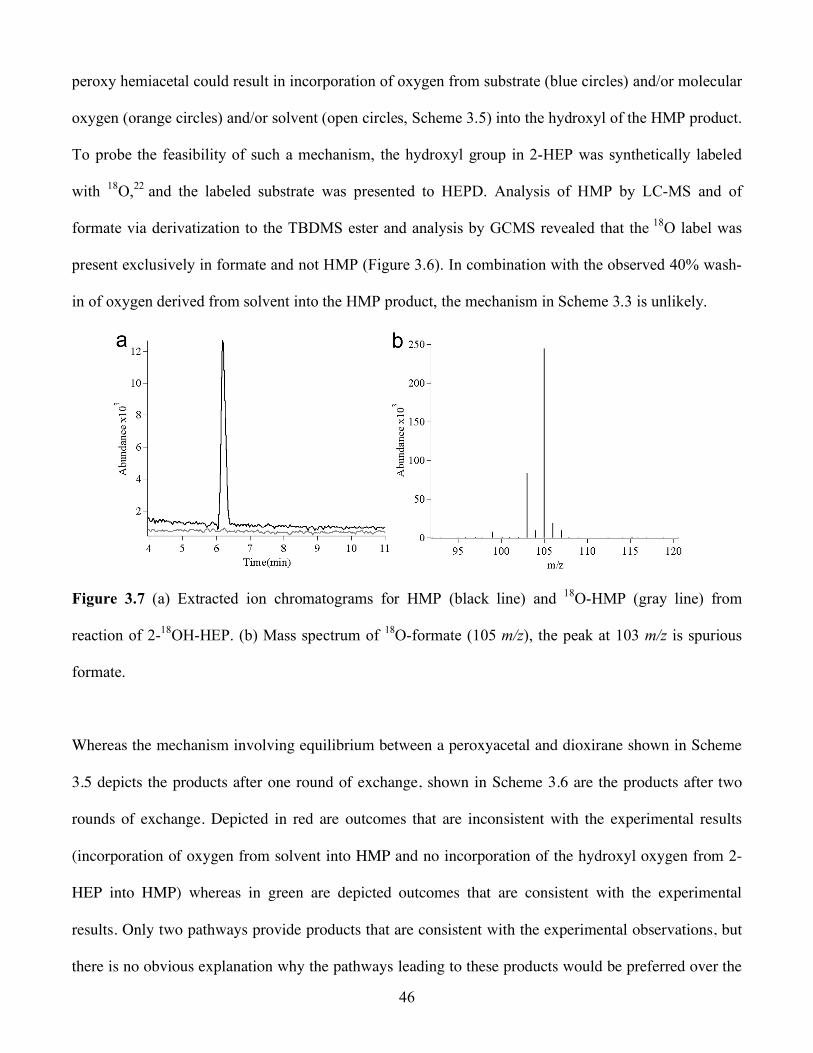
46
peroxy hemiacetal could result in incorporation of oxygen from substrate (blue circles) and/or molecular
oxygen (orange circles) and/or solvent (open circles, Scheme 3.5) into the hydroxyl of the HMP product.
To probe the feasibility of such a mechanism, the hydroxyl group in 2-HEP was synthetically labeled
with 18O,22 and the labeled substrate was presented to HEPD. Analysis of HMP by LC-MS and of
formate via derivatization to the TBDMS ester and analysis by GCMS revealed that the 18O label was
present exclusively in formate and not HMP (Figure 3.6). In combination with the observed 40% wash-
in of oxygen derived from solvent into the HMP product, the mechanism in Scheme 3.3 is unlikely.
Figure 3.7 (a) Extracted ion chromatograms for HMP (black line) and 18O-HMP (gray line) from
reaction of 2-18OH-HEP. (b) Mass spectrum of 18O-formate (105 m/z), the peak at 103 m/z is spurious
formate.
Whereas the mechanism involving equilibrium between a peroxyacetal and dioxirane shown in Scheme
3.5 depicts the products after one round of exchange, shown in Scheme 3.6 are the products after two
rounds of exchange. Depicted in red are outcomes that are inconsistent with the experimental results
(incorporation of oxygen from solvent into HMP and no incorporation of the hydroxyl oxygen from 2-
HEP into HMP) whereas in green are depicted outcomes that are consistent with the experimental
results. Only two pathways provide products that are consistent with the experimental observations, but
there is no obvious explanation why the pathways leading to these products would be preferred over the

47
other pathways. Whereas the asymmetric environment of the active site in principle could favor
exchange of one of the two dioxirane oxygens, both pathways that provide the observed products require
exchange of the alkoxide oxygen of the peroxyacetal in one round of exchange and then exchange of the
proximal peroxy oxygen in the other round of exchange. This requirement makes this mechanism highly
unlikely. In addition, both pathways result in exchange at both positions of formyl-HMP, adding other
constraints. These include complete lack of presence in HMP of the oxygen derived from the hydroxyl
group of 2-HEP but only exchange of 40% of the oxygen derived from molecular oxygen, as well as the
observation that none of the isolated formate product contains two oxygen atoms derived from solvent.
Collectively, the dioxirane mechanism does not agree with all experimental observations.
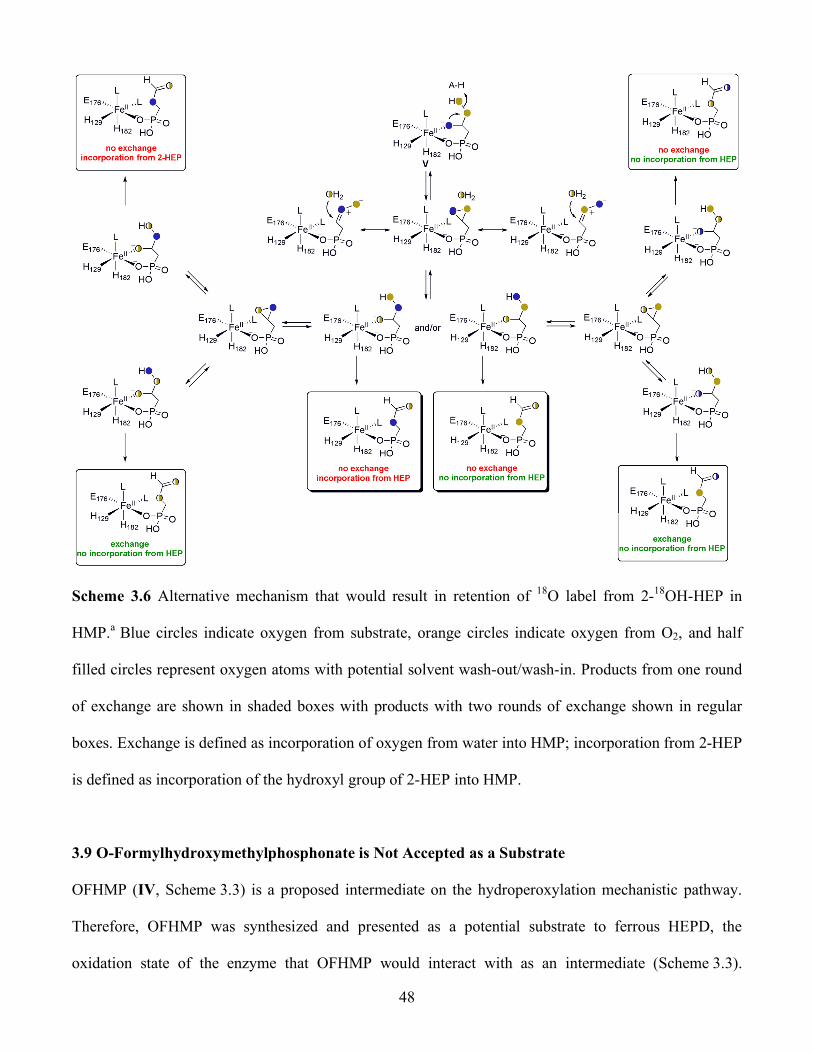
48
Scheme 3.6 Alternative mechanism that would result in retention of 18O label from 2-18OH-HEP in
HMP.a Blue circles indicate oxygen from substrate, orange circles indicate oxygen from O2, and half
filled circles represent oxygen atoms with potential solvent wash-out/wash-in. Products from one round
of exchange are shown in shaded boxes with products with two rounds of exchange shown in regular
boxes. Exchange is defined as incorporation of oxygen from water into HMP; incorporation from 2-HEP
is defined as incorporation of the hydroxyl group of 2-HEP into HMP.
3.9 O-Formylhydroxymethylphosphonate is Not Accepted as a Substrate
OFHMP (IV, Scheme 3.3) is a proposed intermediate on the hydroperoxylation mechanistic pathway.
Therefore, OFHMP was synthesized and presented as a potential substrate to ferrous HEPD, the
oxidation state of the enzyme that OFHMP would interact with as an intermediate (Scheme 3.3).

49
OFHMP is not particularly stable and was partially hydrolyzed to HMP and formate during workup of
the synthetic procedure. The sample presented to HEPD therefore contained both OFHMP and HMP in a
1:1 ratio. A control experiment showed that further hydrolysis is very slow under the HEPD assay
conditions without HEPD. Thus, a sample of OFHMP and HMP (1:1) was incubated with HEPD and the
reaction was monitored by 31P NMR spectroscopy revealing peaks corresponding to phosphate and
OFHMP, and a HMP peak that had decreased in area compared to OFHMP (Figure 3.8). The area of the
peaks corresponding to phosphate and OFHMP was 1.0:0.9 indicating that most of the HMP present in
the original mixture had been oxidized to phosphate, consistent with data that will be presented in
chapter 4.2. However, the experiment also showed that no enzyme catalyzed hydrolysis of OFHMP had
occurred.
Figure 3.8 31P NMR spectrum of (a) a standard of OFHMP and HMP in buffered solution, (b) the same
sample after incubation with HEPD for 2 h.
3.10 Stereochemistry of HMP from Stereospecifically Labeled 1-1H-2-HEP
The stereochemistry of HMP produced from 2-HEP stereoselectively labeled at C1 with deuterium is
mechanism dependent (Scheme 3.7). If HEPD proceeds through hydroperoxylation then overall
inversion of stereochemistry is predicted with retention during Baeyer-Villiger rearrangement23 and
inversion in SN2 hydrolysis. If HEPD proceeds through hydroxylation then overall retention or inversion

50
of configuration is predicted with attack of the Horner-Wadsworth-Emmons-like intermediate on the
Fe(IV)=O proceeding from the same or opposite face as the loss of formate, respectively. For inversion
of configuration, rotation around the P-C1 bond would occur prior to C-C bond scission.
Scheme 3.7 Proposed stereochemical outcomes for HEPD mechanisms.
To ascertain the stereochemistry of HMP produced from 2-HEP labeled at C1 with deuterium, (1R)- and
(1S)-2H-2-HEP were synthesized by Petra Malova in the Friedrich Hammerschmidt laboratory at the
University of Vienna. It will be shown in chapter 4.2 that HMP is oxidized to phosphate and formate by
HEPD and that 2-HEP is oxidized to phosphate and formate when incubated stoichiometrically with
HEPD, presumably via HMP (Scheme 3.8). Importantly, oxidation of HMP has been shown to occur via
stereospecific abstraction of the pro-R hydrogen atom.

51
Scheme 3.8 When incubated stoichiometrically, 2-HEP is oxidized by HEPD to HMP, which is then
converted to phosphate.
It is then possible to indirectly assess the stereochemistry of HMP produced by incubating (1R)- and
(1S)-2H-2-HEP with 1 equivalent of HEPD and comparing the ratio of produced 1H-formate and 2H-
formate (Table 3.1). Oxidation of 2-HEP resulted in production of only 1H-formate (entry 1). Oxidation
of 1,1-2H2-2-HEP (entry 2) resulted in production of approximately one equivalent of 1H-formate (from
oxidation to 2H2-HMP) and one equivalent of 2H-formate (from oxidation of 2H2-HMP). Surprisingly,
both enantiomers of 1-2H-2-HEP afforded approximately a 3:1 ratio of 1H-formate to 2H-formate
(entries 3 and 4). Since 1 equivalent of 1H-formate is produced from oxidation of 1-2H-2-HEP to HMP
only racemic 2H-HMP would afford an additional 0.5 equivalents of both 1H- and 2H-formate to afford a
1.5:0.5 (or 3:1) ratio.
Table 3.1 Ratio of 1H-:2H-formate from oxidation of various 2-HEP isotopologues to phosphate by
HEPD.
Entry Substrate 1H-formate:2H-formate
1 2-HEP 1:0
2 1,1-2H2-2-HEP 1.1:1
3 (1R)-1-2H-2-HEP 2.7:1
4 (1S)-1-2H-2-HEP 2.5:1

52
Production of racemic HMP is unexpected and is not predicted by either mechanism. To unambiguously
confirm this result, the stereochemistry of produced HMP was assessed directly by Petra Malova using
Mosher ester analysis. Either (1R)- and (1S)-2H-2-HEP were incubated in large scale with HEPD, the
produced 2H-HMP was derivatized to the dimethyl ester and then purified to afford milligram quantities
of dimethyl 2H-HMP. These samples were then shipped to Austria where they were converted to the (R)-
Mosher ester and the stereochemistry was assessed by 1H NMR spectroscopy (Scheme 3.9). Both (1R)-
and (1S)-2H-2-HEP afforded racemic 2H-HMP after HEPD oxidation, confirming the previous results.
The unexpected production of racemic HMP from stereoselectively labeled 1-2H-2-HEP is inconsistent
with the later steps of both of the proposed mechanisms.
Scheme 3.9 Both (1S) and (1R)-2H-2-HEP (reactions a and b, respectively) were converted to racemic
2H-HMP by HEPD as evidenced by conversion of the 2H-HMP product to dimethyl 2H-HMP and
Moshers ester analysis.
3.11 Revised Mechanisms
To account for racemic HMP produced from 2-HEP stereoselectively labeled at C1, an alternate
mechanism is proposed containing a single electron transfer step. This mechanism would proceed
through the originally proposed hydroxylation mechanism up until formation of the Horner-Wadsworth-
Emmons-like intermediate (Scheme 3.10, III). Transfer of one electron from the phosphonate anion to
the Fe(IV)=O would afford a ferric hydroxyl (Fe(III)-OH) intermediate and a phosphonate radical
(VIII). Oxygen rebound of the Fe(III)-OH with substrate affords HMP and Fe(II). Such mechanisms

53
involving single electron reduction of ferryl species have been proposed for heme dependent
cyctochrome c oxidases,24-26 peroxidases,27, 28 and horseradish peroxidase.29, 30 In addition,
differentiating whether HMP formation is from two electron attack on the ferryl (concerted) or through
electron transfer then oxygen rebound (step-wise) is reminiscent of the literature discussion of the
mechanisms proposed for P450 oxygenases.31, 32 Early studies of P450s showed that hydroxylation
proceeded with retention of configuration and suggested synchronous insertion of an oxygen atom from
the ferryl species.33, 34 However, further studies pioneered by Groves and coworkers35 showed that some
substrates undergo either inversion36 or loss of stereochemistry37 and have large kinetic isotope effects,38
which suggested a carbon radical intermediate and an asynchronous mechanism. This asynchronous
mechanism first involved hydrogen atom abstraction by the ferryl to afford Fe(III)-OH and a substrate
radical followed by radical rebound to afford the hydroxylated product.
An alternate mechanism to account for production of racemic HMP through hydroperoxylation is a
methylphosphonate intermediate. Studies on the stereochemistry of HMP are inconsistent with
formation of OFHMP followed by hydrolysis at C1 and are consistent with the obsevation that OFHMP
is not subject to enzymatic hydrolysis by HEPD. Though one explanation could be that hydrolysis of
OFHMP partitions between C1 and the carbonyl in a near 1:1 ratio, this would be inconsistent with
labeling studies showing near full incorporation of one 18O into formate and only ~60% incorporation of
18O into HMP from 18O2. To account for racemic HMP, then, instead of OFHMP formation, the Criegee
intermediate (V) could undergo retro-Claisen-like C-C bond cleavage to afford a Horner-Wadsworth-
Emmons-like intermediate and performic acid (IX). Recently, biomimetic studies have shown that
peracids can oxidize ferrous iron complexes to afford Fe(IV)=O species.39 It is then proposed that the
produced performic acid would oxidize the ferrous iron center to a ferryl which would afford formate
and methylphosphonate (intermediate X). The last two steps are reminiscent of α-ketoglutarate
dependent dioxygenases (typified by TauD40) and involve hydrogen atom abstraction to afford VIII then
oxygen rebound to afford HMP. It should be noted that a methylphosphonate intermediate can also be
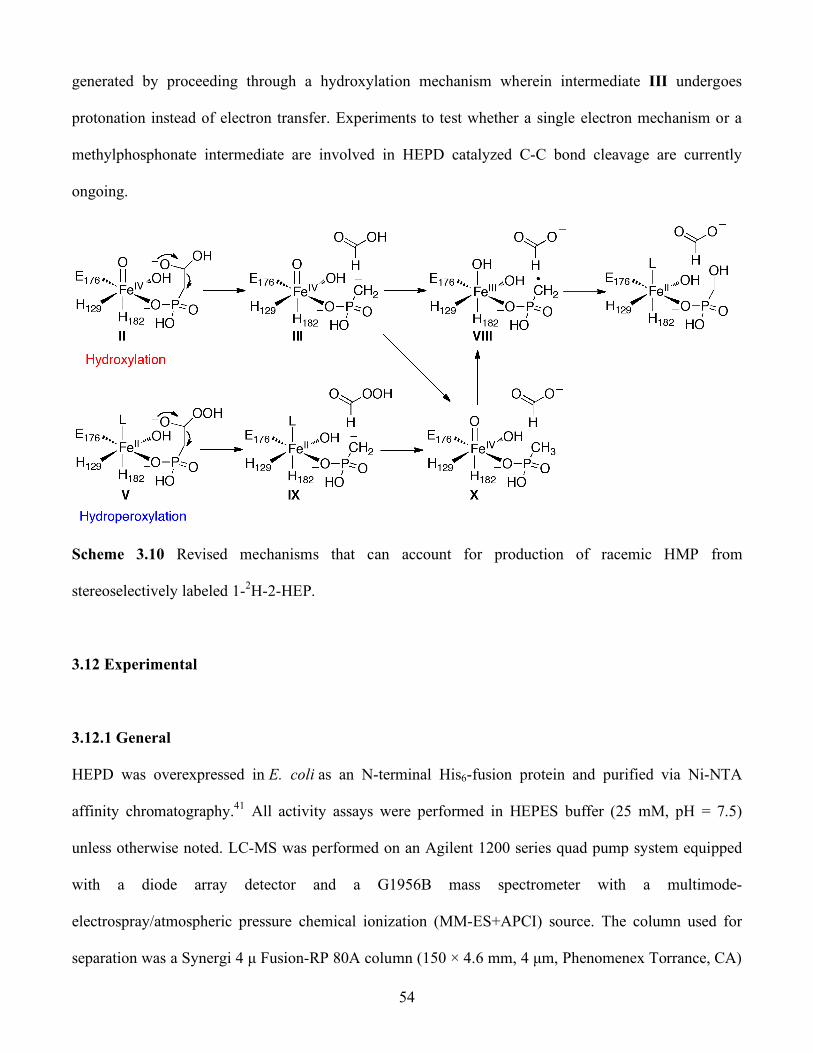
54
generated by proceeding through a hydroxylation mechanism wherein intermediate III undergoes
protonation instead of electron transfer. Experiments to test whether a single electron mechanism or a
methylphosphonate intermediate are involved in HEPD catalyzed C-C bond cleavage are currently
ongoing.
Scheme 3.10 Revised mechanisms that can account for production of racemic HMP from
stereoselectively labeled 1-2H-2-HEP.
3.12 Experimental
3.12.1 General
HEPD was overexpressed in E. coli as an N-terminal His6-fusion protein and purified via Ni-NTA
affinity chromatography.41 All activity assays were performed in HEPES buffer (25 mM, pH = 7.5)
unless otherwise noted. LC-MS was performed on an Agilent 1200 series quad pump system equipped
with a diode array detector and a G1956B mass spectrometer with a multimode-
electrospray/atmospheric pressure chemical ionization (MM-ES+APCI) source. The column used for
separation was a Synergi 4 µ Fusion-RP 80A column (150 × 4.6 mm, 4 µm, Phenomenex Torrance, CA)

55
using a flow rate of 0.5 mL/min. UV−visible absorption spectra were recorded on a Varian Cary 4000
UV−vis spectrophotometer. GC-MS was performed on an Agilent 6890N gas chromatograph equipped
with an electron impact (EI) ionization source.
NMR spectra were recorded on a Varian Unity 500 or Varian Unity Inova 600 spectrometer. Proton and
carbon chemical shifts are reported in δ values relative to an external standard of 0.1% tetramethylsilane
in CDCl3 (0.00 ppm). Phosphorus shifts are reported in δ values relative to an external standard of 85%
phosphoric acid (0.00 ppm).
Labeled 1-13C-2-HEP and 2-13C-2-HEP were synthesized by Gongyong Li as reported.1 Labeled (1R)-
and (1S)-2H-2-HEP were synthesized by Petra Malova in the Friedrich Hammerschmidt laboratory at the
University of Vienna as reported.42 Sodium 13C-formate (99 atom % 13C) and N-tert-butyldimethylsilyl-
N-methyltrifluoroacetamide (MTBSTFA) were purchased from Sigma-Aldrich. 2-Nitrophenylhydrazine
(NPH) was purchased from Acros Organics. N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide (EDC)
was purchased from Chem-Impex (Wood Dale, IL). Dichloromethane was distilled over CaH2.
Tetrahydrofuran was distilled over Na metal with benzophenone as a colorimetric indicator. All other
chemicals were purchased from Sigma Aldrich at the highest purity and used without further
purification.
3.12.2 Identification of Assay Products via LC-MS
Enzymatic assays were quenched by adding H2SO4 (30 mM) and the protein was pelleted via
microcentrifugation (1 min at 16.1g). The supernatant was analyzed via LC-MS (positive mode) using
aqueous 0.1% formic acid as an isocratic mobile phase and scanning a mass range of m/z 50−500.
3.12.3 Identification of Assay Products via 31P or 13C NMR Spectroscopy
HEPD was separated from the assay via centrifugal filtration (Millipore Micron YM-30, 30 kDa
nominal molecular weight limit). D2O (20% v/v) was added to the flow-through. The 31P NMR spectrum

56
was recorded on a Varian UI600 (frequency for phosphorus: 242 MHz). The 13C NMR spectrum was
recorded on a Varian U500 (frequency for carbon: 125 MHz).
3.12.4 Aqueous Derivatization and Quantification of Organic Acids
As detailed in Notebook IV, pages 236 and 266-268, following a modified literature procedure,43 to a
100 µL sample from standards or assays was added propionic acid as an internal standard (10 µL of a 1
mM stock solution in 1:1 pyridine:HCl), followed by EDC (10 µL of a 0.29 M stock solution), and
subsequently NPH (10 µL of a 0.12 M stock solution in 250 mM aqueous HCl). The orange solution was
heated to 60 °C in a water bath for 15 min and precipitated protein was pelleted via microcentrifugation
(1 min at 16.1g). The supernatant (20 µL) was analyzed via LC-MS using an isocratic mobile phase
(50:50 MeOH:H2O + 0.1% formic acid) for separation. Authentic standards of sodium 13C-formate,
sodium acetate, and propionic acid were also derivatized and analyzed by monitoring the absorbance at
400 nm and by MM-ES+APCI (negative mode) and found to have retention times of 5.81, 5.93, and
6.93 min, respectively. The observed m/z values for each derivatized compound corresponded to the
monodeprotonated hydrazide and were: 181 (13C-formate), 194 (acetate), and 208 (propionic acid).
Solutions of varying concentrations of each of these acids were made in HEPES buffer (25 mM, pH =
7.5), derivatized, and analyzed via LC-MS as described previously using single ion monitoring (SIM)
for the derivatized acid of interest. The area of the peak corresponding to derivatized acid was divided
by the area of the derivatized propionic acid peak to afford a response factor, which was plotted against
the acid concentration to afford a linear relationship. This equation was used to quantify the organic acid
concentration in the enzymatic assays.
3.12.5 Formate Derivatization and Analysis via GC-MS
As previously reported,1 formate production was monitored via GC-MS on an Agilent 6890N (Agilent
Technologies) gas chromatograph with helium carrier gas. Sample introduction was via split injection

57
onto a HP-5 (5% phenyl-methylpolylsiloxane) column (30 m, 0.32 mm inner diameter, 0.25 mm film
thickness). The injection temperature was 250 oC. The initial column temperature was 40 oC, and was
held for 5 min after injection before increasing to 230 oC at 15 oC/min. The temperature was held at 230
oC for the remainder of the 27 min program. The HEPD reaction was performed as described above.
Sulphuric acid was added to a final concentration of 0.12 M to quench the reaction. The acidified
solutions were subjected to three separate 0.5 mL diethyl ether extractions. The extracts were combined
and formate was derivatized using 0.5 mL MTBSTFA plus 0.1% TBDMSCl. Typical derivatizations
were performed for 20 min at 25 oC. The solution (1–2 mL) was then directly injected into the GC–MS.
Under the given GC conditions derivatized formate had a retention time of 7.4 min.
3.12.6 Assays using 1-13C-2-HEP and 2-13C-HEP
As previously reported,1 to determine the fate of the excised carbon atom HEPD (10 µM) in phosphate
buffer (50 mM, pH = 7.5) was incubated with 2-13C-2-HEP for 2 h in oxygen saturated buffer. Analysis
via 13C NMR spectroscopy showed a new peak at δ = 171 ppm, which was confirmed as 13C-formate by
spiking of a commercial standard of sodium 13C-formate. To verify the phosphorus containing product,
HEPD (10 µM) in HEPES buffer (50 mM, pH = 7.5) was incubated with 1-13C-2-HEP for 2 h in
oxygenated buffer. Analysis via 31P NMR spectroscopy showed two peaks: one at δ = 20 ppm (1-13C-2-
HEP) and a new peak at δ = 17.6 ppm (which is consistent with the chemical shift of HMP in pH = 7.5
buffer).
3.12.7 Assays using Deuterated 2-HEP
As previously reported,1 1,1-2H2-2-HEP was incubated with HEPD (10 µM) in oxygen saturated HEPES
buffer (50 mM, pH = 7.5) for 2 h. Analysis of the sample via LC-MS showed a new peak at 6 min with a
predominant m/z of 115 in the mass spectrum, which corresponded to [2H2-HMP+H+]+. Organic acid
analysis via LC-MS showed production of 1H-formate.

58
As reported in Notebook IV on page 227, 2,2-2H2-2-HEP was incubated with HEPD (10 µM) in
oxygenated HEPES buffer (50 mM, pH = 7.5) for 2 h. Analysis of the sample via LC-MS showed a new
peak at 6 min with a predominant m/z of 113 in the mass spectrum, which corresponded to [HMP+H+]+.
Organic acid analysis via LC-MS showed production of 2H-formate. To test the stereospecificity of
putative hydrogen atom abstraction at C2 of 2-HEP, (2R)- and (2S)-2H-2-HEP were incubated with
HEPD (10 µM) in oxygenated HEPES buffer (25 mM, pH = 7.5) for 2 h. Organic acid analysis showed
production from 1H-formate for (2S)-2H-2-HEP and 2H-formate from (2R)-2H-2-HEP.
3.12.8 Determining the Kinetic Parameters for HEPD
As described in Notebook V, pages 163-164, the activity of HEPD was monitored by monitoring the
substrate-dependent rate of oxygen consumption at 20 oC using an oxygen electrode (Oxytherm
Electrode Control Unit, Hansatech Instruments, Norfolk, England). Assays containing HEPES (25 mM,
pH = 7.5) and HEPD (2 mM) were allowed to equilibrate aerobically while stirring at 100 rpm before
initiation of reaction by addition of 2-HEP. Initial rates (defined as the rate before 10-15% substrate
consumption) were recorded and plotted against substrate concentration. These data were fit using the
Michaelis-Menton equation using Igor Pro software (v6.03, WaveMetrics Inc, Lake Oswego, OR) with a
nonlinear least-squares fit algorithm. The kinetic parameters for HEPD with unlabeled substrate are KM
= 14.2 ± 2.5 µM and kcat = 18.3 ± 0.7 min-1, and for 2,2-2H2-2-HEP are KM = 11.6 ± 1.2 µM and kcat =
15 ± 0.3 min-1 to afford a kinetic isotope effect of 1.2.
3.12.9 Assays Showing Incorporation of 18O2 and 18OH2 into HMP and Formate
As previously reported,1 assays using 18O2 were carried out as follows. All assay components were
prepared and mixed anaerobically. 2-13C-2-HEP was substituted for HEP so that formate derived from
substrate could be differentiated from spurious formate during GC–MS analysis. A glass vial containing
enzyme and substrate was fitted with a tight rubber septum and brought outside of the glove box. The

59
reaction was initiated by the introduction of 1 mL of 18O2 (99 atom per cent, Isotec) via a gastight
syringe. The solution was allowed to stir slowly at room temperature for 90 min at which time sulphuric
acid was added to a final concentration of 92 mM to quench the reaction. After centrifugation to remove
precipitated protein, HMP was directly analysed by LC–MS. Two EICs at m/z 115 and m/z 113 were
extracted from the TIC and integrated to afford the ratio of 18O-HMP and 16O-HMP, respectively.
Formate was extracted, derivatized and analyzed via GC–MS analysis. Two EICs at m/z 106 and m/z
104 were extracted from the TIC and integrated to afford the amounts of 18O13C-formate and 13C-
formate, respectively.
The potential for 18O incorporation from H2O into HMP was evaluated by running the reaction in 18OH2.
HEPD (10.5 µM, 150 µL volume) was added to an aerobic mixture of 18OH2 (832.6 ml, 95 atom per
cent) and HEP (dissolved in 500 mM HEPES, pH = 7.5, 3 mM, 17.4 µL volume) resulting in an 18OH2
dilution to 79% in the 1 mL reaction. Intermittent bubbling of O2 enabled the complete consumption of
HEP over 1 h. Produced HMP was analyzed via LC-MS. Two EICs at m/z 115 and m/z 113 were
extracted from the TIC and integrated to afford the ratio of 18O-HMP and 16O-HMP, respectively.
3.12.10 Tracking the Hydroxyl Group of 2-18O-HEP
As described in Notebook V on pages 160-161, HEPD (2 µM) and 2-18OH-HEP (2 mM) were added to
oxygen saturated HEPES buffer (25 mM, pH = 7.5), incubated at 25 °C for 2 h, and then HMP was
analyzed via LC-MS. The masses for protonated HMP and 18OH-HMP (115 and 117, respectively) were
extracted to afford a single peak in each EIC with a retention time of 5.4 min. Each monoisotopic
product peak was separately integrated to afford the isotopic composition. Additionally, formate was
analyzed via GC-MS as described in chapter 3.12.5 to determine the isotopic composition.

60
3.12.11 O-Formyl-HMP is not a Substrate
As described in Notebook V on pages 95-96, in a typical assay, O-formyl-2-hydroxymethylphosphonate
(OFHMP, 2 mM) was incubated with HEPD (10 µM) for 2 h and samples were analyzed via 31P NMR
spectroscopy. A control sample without HEPD was also analyzed in a similar fashion.
3.12.12 Determining the Stereochemistry of 2H-HMP from Stereospecifically Labeled 1-2H-2-HEP
by Oxidation of 2-HEP to Phosphate
As described in Notebook V on pages 166-167, HEPD (100 µM) was incubated with either 2-HEP, 2,2-
2H2-2-HEP, (2R)- or (2S)-2H-2-HEP (100 µM) in oxygenated HEPES buffer (25 mM, pH = 7.5) for 2 h.
Organic acid analysis was performed to obtain the ratio of 1H-formate and 2H-formate.
3.12.13 Enzymatic Milligram Production of HMP and Derivatization to Dimethyl HMP
As described in Notebook V on page 165, either (1R)- or (1S)-2H-2-HEP (1 mM, final) was dissolved in
ammonium acetate (AmAc) buffer (100 mM) that had been oxygenated for 1 h. Then HEPD (10 µM),
which had been buffer exchanged into AmAc (100 mM), was added and the solution is incubated at 25
oC for 2 h. The sample was then snap frozen and water removed by lyophilization. Water (10 mL) was
then added, snap frozen and the solvent again removed by lyophilization. Then H2SO4 (10 mL of a 100
mM solution) was added, the solution snap frozen and water removed by lyophilization. A solution of
10:1 Et2O:MeOH (6 mL) was then added followed by trimethylsilyldiazomethane (3.9 eq with respect to
HEP from a 2 M Et2O stock) resulting in evolution of gas. The yellow solution was agitated for 10 min
then filtered, and solvent was removed under reduced pressure. Purification via silica gel column
chromatography (10:1 Et2O:MeOH, isocratic, Rf = 0.4 (10:1 CHCl3:MeOH) afforded dimethyl HMP (2
mg) as a colorless oil.

61
3.12.14 Synthesis of 1,1-2H2-2-HEP
Labeled 1,1-2H2-2-HEP was synthesized as shown in Scheme 3.11.
Scheme 3.11 Synthesis of 1,1-2H2-2-HEP.
3.12.14.1 Diethyl 1,1-dideutero-2-hydroxyethylphosphonate
As reported in Notebook IV on page 112, under an N2 atmosphere, a three-necked round bottom flask,
equipped with a magnetic stir bar, was charged with borane-THF (1 M solution in THF, 7 mL, 7 mmol)
which was cooled to 0 °C on an ice–water bath. Ethyl (diethoxyphosphono)dideuteroacetate44 (1.53 g,
6.8 mmol) was added resulting in the evolution of gas. The solution was allowed to warm to room
temperature and vigorously stirred overnight. The solution then was cooled to 0 °C in an ice–water bath
and ethanol (1 mL) was added to quench excess borane. Purification via silica gel flash chromatography
(20:1 DCM:MeOH, Rf = 0.45) afforded diethyl 1,1-dideutero-2-hydroxyethylphosphonate (1 g, 5.9
mmol, 84%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 1.34 (t, J = 7.08 Hz, 6H, OCH2CH3), 2.80
(bs, 1H, OH), 3.48 (d, J = 19.8 Hz, PCD2CH2), 4.145 (m, 4 H, OCH2CH3); 13C NMR (100 MHz,
CDCl3) δ 16.47, 56.89 (d, J = 5.85 Hz), 62.05 (d, J = 6.56 Hz); 31P NMR (162 MHz, CDCl3) δ 31.6;
HRMS (ESI+) calcd (C61H132H2O4P+Na)+ 207.0731; found 207.0736.

62
3.12.14.2 1,1-Dideutero-2-hydroxyethylphosphonate
As reported in Notebook IV on page 113, under an N2 atmosphere, a round bottom flask equipped with a
magnetic stir bar was charged with diethyl 1,1-dideutero-2-hydroxyethylphosphonate (1 g, 5.4 mmol, 1
eq)) and was diluted with dry DCM (20 mL). Bromotrimethylsilane (2.86 mL, 21.6 mmol, 4 eq) was
added and the yellow solution was heated in an oil bath under reflux while stirring for 2 h. The reaction
was allowed to cool to room temperature followed by removal of the solvent under reduced pressure.
After addition of ethanol (20 mL), the solution was stirred for 30 min. The solvent was then removed via
rotary evaporation at 30 °C for 1 h. Ethanol (20 mL) and cyclohexylamine (616 µL, 5.4 mmol, 1 eq)
were added sequentially and the yellow solution was stirred for 30 min followed by removal of the
solvent via rotary evaporation. The white solid was recrystallized (hot ethanol:ethyl ether), filtered,
dissolved in water and passed through a column of DOWEX H+ (50WX8-200, column volume = 5 mL).
Elution with water (20 column volumes), which was subsequently removed overnight under reduced
pressure, afforded 1,1-dideutero-2-hydroxyethylphosphonate (502 mg, 3.9 mmol, 73%) as a yellow oil.
1H NMR (400 MHz, D2O) δ 3.62 (d, J = 12.3 Hz), 31P NMR (162 MHz, D2O) δ 27.6; HRMS (ESI+)
calcd (C21H62H2O4P)+ 129.0286; found 129.0288. The spectroscopic data is consistent with a
previously reported synthesis.42
3.12.15 Synthesis of 2,2-2H2-2-HEP
Labeled 2,2-2H2-2-HEP was synthesized as shown in Scheme 3.12.
Scheme 3.12 Synthesis of 2,2-2H2-2-HEP.

63
3.12.15.1 Synthesis of Diethyl 2,2-2H2-2-hydroxyethylphosphonate
As described in Notebook IV on page 111, under N2 a three-necked round bottom flask was charged
with borane-d3-THF complex (97.5 atom % D, 1 M in THF, 5 mmol, 3 reducing equivalents). To this
solution was added ethyl (diethoxyphosphono)acetate44 (1.1 g, 5 mmol, 1 eq) while stirring, which
resulted in evolution of gas. The clear solution was vigorously stirred at 25 oC overnight. Ethanol (1 mL)
was added to quench and stirred for 30 min. Purification via silica gel flash chromatography (20:1
DCM:MeOH) afforded the product (772 mg, 4.2 mmol, 84%) as a colorless oil. 1H NMR (400 MHz,
CDCl3) δ 1.33 (m, 6H, OCH2CH3), 2.03 (d, J = 17.4 Hz, 2H, P-CH2), 4.13 (m, 4H, OCH2CH3); 31P
NMR (162 MHz, CDCl3) δ 31.6. The spectroscopic data is consistent with a previously reported
synthesis.42
3.12.15.2 2,2-2H2-2-Hydroxyethylphosphonate
As described in Notebook IV on page 110, a round bottom flask equipped with a magnetic stir bar was
charged with diethyl 2,2-2H2-2-hydroxyethylphosphonate (316 mg, 1.7 mmol), which was dissolved in 6
M HCl (5 mL) then heated under reflux while stirring for 2 h. The yellow solution was allowed to cool
to 25 oC and solvent was removed under reduced pressure. The resulting residue was dissolved in H2O
(10 mL), stirred for 30 min and solvent was removed via lyopholization to afford 2,2-2H2-2-HEP (128
mg, 1.3 mmol, 76%) as a viscous oil. 1H NMR (400 MHz, D2O) δ 1.83 (d, J = 18.2 Hz, PCH2); 31P (400
MHz, D2O) δ 25.9; HRMS (ESI+) calcd (C21H6
2H2O4P)+ 129.0286; found 129.0287 The spectroscopic
data is consistent with a previously reported synthesis.42

64
3.12.16 Synthesis of (2R)- and (2S)-2H-2-HEP
Synthesis of (2R)- and (2S)-2H-2-HEP followed the method of Hammerschmidt and coworkers from
benzyloxydeuteroethanol forward and differs only in the syntheses of α-2H-benzyloxyacetaldehyde and
benzyloxydeuteroethanol, which are detailed below (Scheme 3.13).
Scheme 3.13 Synthesis of (2R)- and (2S)-2H-2-HEP.
3.12.16.1 Synthesis of 1,4-dibenzyloxy-2,3-dideutero-trans-2-butene
As described in Notebook IV on page 152, under N2, a two-necked round bottom flask equipped with a
magnetic stir bar and a reflux condenser was charged with lithium aluminum deuteride (96 atom % D, 1
M solution in THF, 50 mL, 50 mmol, 1.2 eq). A separate round bottom flask was charged under N2 with
1,4-dihydroxybut-2-yne (3.44 g, 40 mmol, 1 eq) which was dissolved in dry THF (100 mL) and slowly
transferred via double-ended needle to the solution of lithium aluminum deuteride resulting in evolution
of gas. The solution was then heated under reflux for 3 h while stirring before cooling to 0 oC on an
ice/water bath. Deuterium oxide (15 mL) was then slowly added to quench the reaction, which was then

65
allowed to warm to 25 oC. The white suspension was then passed over a column of Celite, which was
washed with Et2O (200 mL). The yellow solution was concentrated via rotary evaporation and then
passed over a silica plug, which was eluted with EtOAc (200 mL). This yellow solution was
concentrated to dryness under reduced pressure to afford the crude trans-2-butene, 1,4-diol.
Under N2 a two necked round bottom flask equipped with a magnetic stir bar and reflux condenser was
charged with sodium hydride (60% dispersion in mineral oil, 3.5 g, 88 mmol, 2.2 eq) which was
suspended in dry THF (80 mL). To this suspension was slowly added trans-2-butene, 1,4 diol while
stirring which resulted in evolution of gas. After 30 min, benzyl bromide (10.4 mL, 88 mmol, 2.2 eq)
was slowly added and the tan suspension was heated under reflux while stirring overnight. This
suspension was then allowed to cool to 25 oC, diluted with EtOAc (500 mL), and washed with H2O (400
mL). Both phases were saved and the aqueous phase was extracted with EtOAc (300 mL). Organic
phases were combined, washed with brine (200 mL), dried over Na2SO4, filtered, and concentrated to
dryness under reduced pressure. Purification via silica gel flash chromatography (100% hexanes then 1:1
hexanes:EtOAc (Rf = 0.25 Hexanes)) afforded 1,4-dibenzyloxy-2,3-dideutero-trans-2-butene (8.6 g, 32
mmol, 80%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 4.05 (s, 4H, CH2C=C), 4.53 (s, 4H, ArCH2),
7.34 (m, 10H, Ar); 13C NMR (400 MHz, CDCl3) δ 70.25, 72.45, 127.84, 127.96, 128.62, 138.44; HRMS
(EI+) calcd (C18H182H2O2)+ 270.15888; found 270.15870
3.12.16.2 Synthesis of α-2H-Benzyloxyacetaldehyde
As described in Notebook IV on page 153, under N2, a Schlenk flask equipped with a magnetic stir bar
was charged with 1,4-dibenzyloxy-2,3-dideutero-trans-2-butene (8.6 g, 32 mmol, 1 eq.) which was
dissolved with dry DCM (50 mL) and cooled to -78 oC on a dry ice/acetone bath. Ozone was then purged
through the yellow solution while stirring until the color changed to light green. Oxygen was then
purged through the solution until the color changed to yellow and the solution was placed in an ice/water
bath. Acetic acid (5 mL) was added followed by zinc powder (10.4 g, 160 mmol, 5 eq) was added

66
incrementally over 1 h while stirring vigorously for 1 h at 0 oC. Purification via short path distillation
(bp 79 oC, 0.1 Torr) afforded α-2H-benzyloxyacetaldehyde (6.76 g, 44.8 mmol, 71%) as a yellow oil. 1H
NMR (400 MHz, CDCl3) δ 4.18 (s, 2H, CH2CHO), 4.63 (s, 2H, ArCH2), 7.37 (m, 5H, Ar); 13C NMR
(100 MHz, CDCl3) δ 73.90, 75.36, 128.28, 128.46, 128.84, 137.01. The spectroscopic data is consistent
with a previously reported synthesis.42
3.12.16.3 Synthesis of (1S)-2-Benzyloxy-1-2H-ethanol
As described in Notebook IV on pages 154-155, following a published proceedure45 a three-necked flask
equipped with a magnetic stir bar and a reflux condenser under N2 was charged with 9-BBN (0.5 M in
THF, 24 mL, 12 mmol, 1.2 eq) followed by (+)-α-pinene (91+% ee/GLC, 2.3 mL, 14.4 mmol, 1.44 eq).
The colorless solution was heated on an oil bath under a gentle reflux while stirring for 2 h. The solution
was cooled to 25 oC and then α-2H-benzyloxyacetaldehyde (1.5 g, 10 mmol, 1 eq) was added and the
solution stirred for 10 min before heating under a gentle reflux for 1 h . Acetaldehyde (1 mL) was added
to quench the reaction and the yellow solution was allowed to cool to 25 oC, solvent was removed under
reduced pressure, and the resulting residue was dissolved in diethyl ether (20 mL) and cooled to 0 oC on
an ice/water bath. Ethanolamine (732 µL, 12 mmol, 1 eq) was added resulting in precipitation of a white
powder. This precipitate was filtered off using a Buckner funnel and the solid was washed with diethyl
ether (50 mL). The filtrate was washed with H2O (50 mL), brine (50 mL), dried over Na2SO4, filtered,
and concentrated via rotary evaporation. Purification via silica flash chromatography (3:1
hexanes:EtOAc then 2:1 hexanes:EtOAc then 1:1 hexanes:EtOAc, (Rf = 0.25, 1:1 hexanes:EtOAc)
afforded (1S)-2-benzyloxy-1-2H-ethanol (668 mg, 4.4 mmol, 44%) as a yellow oil. 1H NMR (400 MHz,
CDCl3) δ 2.18 (br. s, IH, OH), 3.61 (d, J = 4.2 Hz, 2H, OCH2),3.75 (tt, J = 4.2, J = 1.6 Hz, IH, CHD),
4.95 (s, 2H, PhCH2),7.35 (m, 5H, Ph), which is consistent with a previously reported synthesis.46

67
3.12.16.4 Synthesis of (1R)-2-Benzyloxy-2H-ethanol
As described in Notebook IV on page 191, (1R)-2-benzyloxy-2H-deuteroethanol was prepared in a
similar manner from (1S)-2-benzyloxy-1-2H-ethanol (1.6 g, 10.6 mmol, 1 eq), and (-)-alpine borane
generated from (-)-pinene (>87% ee, 2.4 mL, 1.5 eq) and 9-BBN (0.5 M in THF, 32 mL, 1.5 eq) to
afford the product as a colorless oil (1.4 g, 9.2 mmol, 86%). Spectroscopic characterization (1H NMR,
13C NMR, HMRS) was the same as (1S)-2-benzyloxy-1-2H-ethanol.
3.12.16.5 2-Benzyloxy-1-2H-ethanol
As described in Notebook IV on page 187, under N2 a three-necked flask equipped with a magnetic stir
bar and a reflux condenser was charged with α-2H-benzyloxyacetaldehyde (225 mg, 1.5 mmol, 1 eq),
which was dissolved in 1:1 DCM:EtOH (10 mL). To this solution was added, while stirring sodium
borodeuteride (228 mg) resulting in evolution of gas. After stirring for 30 min the white suspension was
cooled to 0 oC in an ice/water bath and 5% citric acid (5 mL) was added while vigorously stirring to
quench the reaction. This solution was then taken up in 5% citric acid (100 mL) and extracted with
DCM (3x150 mL). Organic layers were combined, dried over Na2SO4, filtered, and concentrated via
rotary evaporation. Purification via silica flash chromatography (1:1 hexanes:EtOAc (Rf = 0.25)
afforded 2-benzyloxy-1-2H-ethanol (214 mg, 1.4 mmol, 93$) as a yellow oil. Spectroscopic
characterization (1H NMR, 13C NMR, HMRS) was the same as (1S)-2-benzyloxy-1-2H-ethanol.
3.12.16.6 Assignment of stereochemistry and enantiomeric excess (ee) of (1R)- and (1S)-2-
Benzyloxy-2H-ethanol
A 5-mm NMR tube was charged with a solution of either racemic, (1S)- or (1R)-2-benzyloxy-2H-ethanol
(4.0 mg, 0.0180 mmol) in CDCl3 (0.5 mL). To the solution was added sequentially pyridine (0.2 mL,
0.196 mmol, 11 equiv) and (S)-α-mehoxy-α-trifluoromethyl-phenylacetyl chloride (8.4 µL, 0.045 mmol,
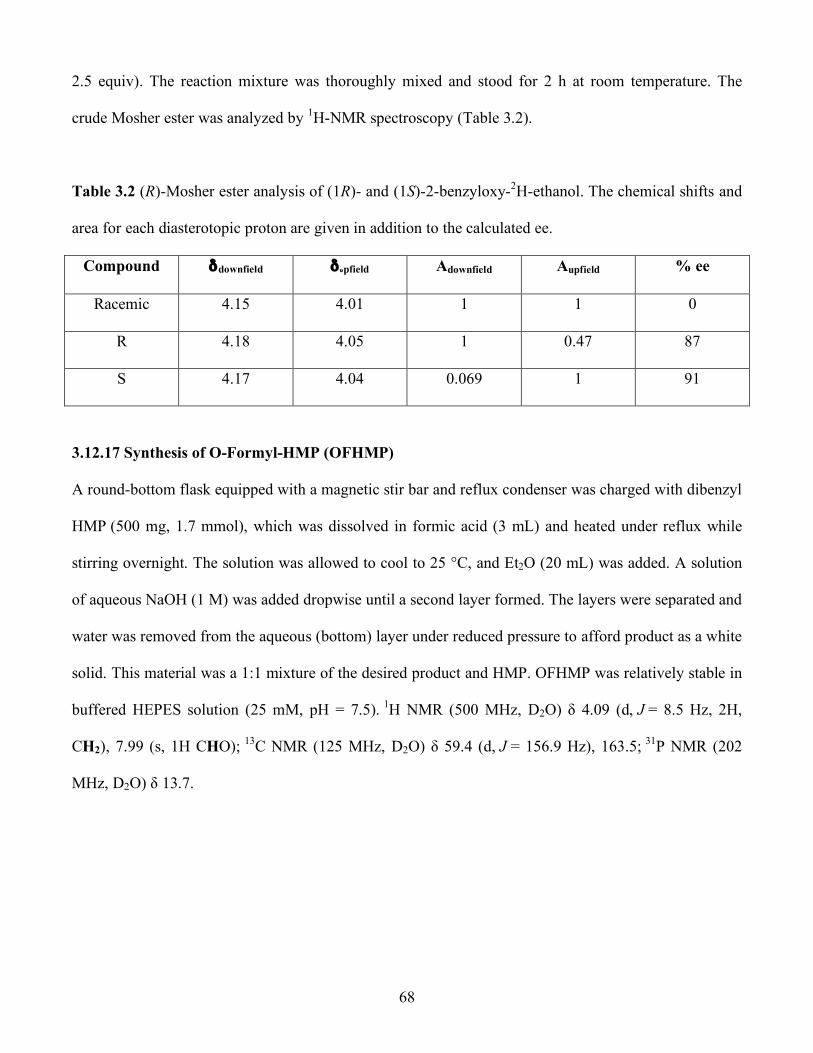
68
2.5 equiv). The reaction mixture was thoroughly mixed and stood for 2 h at room temperature. The
crude Mosher ester was analyzed by 1H-NMR spectroscopy (Table 3.2).
Table 3.2 (R)-Mosher ester analysis of (1R)- and (1S)-2-benzyloxy-2H-ethanol. The chemical shifts and
area for each diasterotopic proton are given in addition to the calculated ee.
Compound δdownfield δυpfield Adownfield Aupfield % ee
Racemic 4.15 4.01 1 1 0
R 4.18 4.05 1 0.47 87
S 4.17 4.04 0.069 1 91
3.12.17 Synthesis of O-Formyl-HMP (OFHMP)
A round-bottom flask equipped with a magnetic stir bar and reflux condenser was charged with dibenzyl
HMP (500 mg, 1.7 mmol), which was dissolved in formic acid (3 mL) and heated under reflux while
stirring overnight. The solution was allowed to cool to 25 °C, and Et2O (20 mL) was added. A solution
of aqueous NaOH (1 M) was added dropwise until a second layer formed. The layers were separated and
water was removed from the aqueous (bottom) layer under reduced pressure to afford product as a white
solid. This material was a 1:1 mixture of the desired product and HMP. OFHMP was relatively stable in
buffered HEPES solution (25 mM, pH = 7.5). 1H NMR (500 MHz, D2O) δ 4.09 (d, J = 8.5 Hz, 2H,
CH2), 7.99 (s, 1H CHO); 13C NMR (125 MHz, D2O) δ 59.4 (d, J = 156.9 Hz), 163.5; 31P NMR (202
MHz, D2O) δ 13.7.

69
3.13 References
1. Cicchillo, R. M.; Zhang, H.; Blodgett, J. A. V.; Whitteck, J. T.; Li, G.; Nair, S. K.; van der Donk, W. A.; Metcalf, W. W., Nature 2009, 459, (7248), 871-874. An unusual carbon-carbon bond cleavage reaction during phosphinothricin biosynthesis. 2. Shyadehi, A. Z.; Lamb, D. C.; Kelly, S. L.; Kelly, D. E.; Schunck, W.-H.; Wright, J. N.; Corina, D.; Akhtar, M., J. Biol. Chem. 1996, 271, (21), 12445-12450. The Mechanism of the Acyl-Carbon Bond Cleavage Reaction Catalyzed by Recombinant Sterol 14-Demethylase of Candida albicans (Other Names Are: Lanosterol 14-Demethylase, P-450, and CYP51). 3. Clark, L. C. J. R.; Wolf, R.; Granger, D.; Taylor, Z., J. Appl. Physiol. 1953, 6, (3), 189-193. Continuous Recording of Blood Oxygen Tensions by Polarography. 4. Dunwell, J. M.; Purvis, A.; Khuri, S., Phytochem. 2004, 65, (1), 7-17. Cupins: the most functionally diverse protein superfamily? 5. Higgins, L. J.; Yan, F.; Liu, P.; Liu, H.-w.; Drennan, C. L., Nature 2005, 437, (7060), 838-844. Structural insight into antibiotic fosfomycin biosynthesis by a mononuclear iron enzyme. 6. Costas, M.; Mehn, M. P.; Jensen, M. P.; Que, L., Chem. Rev. 2004, 104, (2), 939-986. Dioxygen activation at mononuclear nonheme iron active sites: Enzymes, models, and intermediates. 7. Ryle, M. J.; Hausinger, R. P., Curr. Opin. Chem. Biol. 2002, 6, (2), 193-201. Non-heme iron oxygenases. 8. Koehntop, K. D.; Emerson, J. P.; Que, L., J. Biol. Inorg. Chem. 2005, 10, (2), 87-93. The 2-His-1-carboxylate facial triad: a versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. 9. Xing, G.; Diao, Y.; Hoffart, L. M.; Barr, E. W.; Prabhu, K. S.; Arner, R. J.; Reddy, C. C.; Krebs, C.; Bollinger, J. M., Jr., Proc. Natl. Acad. Sci. U.S.A. 2006, 103, (16), 6130-6135. Evidence for C-H cleavage by an iron-superoxide complex in the glycol cleavage reaction catalyzed by myo-inositol oxygenase. 10. Burzlaff, N. I.; Rutledge, P. J.; Clifton, I. J.; Hensgens, C. M. H.; Pickford, M.; Adlington, R. M.; Roach, P. L.; Baldwin, J. E., Nature 1999, 401, (6754), 721-724. The reaction cycle of isopenicillin N synthase observed by X-ray diffraction. 11. Brown, C. D.; Neidig, M. L.; Neibergall, M. B.; Lipscomb, J. D.; Solomon, E. I., J. Am. Chem. Soc. 2007, 129, (23), 7427-7438. VTVH-MCD and DFT Studies of Thiolate Bonding to FeNO/FeO2 Complexes of Isopenicillin N Synthase: Substrate Determination of Oxidase versus Oxygenase Activity in Nonheme Fe Enzymes. 12. Kovaleva, E. G.; Lipscomb, J. D., Nat. Chem. Biol. 2008, 4, (3), 186-193. Versatility of biological non-heme Fe(II) centers in oxygen activation reactions.

70
13. Mukherjee, A.; Cranswick, M. A.; Chakrabarti, M.; Paine, T. K.; Fujisawa, K.; Munck, E.; Que, L., Inorg. Chem. 49, (8), 3618-3628. Oxygen Activation at Mononuclear Nonheme Iron Centers: A Superoxo Perspective. 14. Xing, G.; Barr, E. W.; Diao, Y.; Hoffart, L. M.; Prabhu, K. S.; Arner, R. J.; Reddy, C. C.; Krebs, C.; Bollinger, J. M., Jr., Biochemistry 2006, 45, (17), 5402-5412. Oxygen activation by a mixed-valent, diiron(II/III) cluster in the glycol cleavage reaction catalyzed by myo-inositol oxygenase. 15. Kikuchi, Y.; Suzuki, Y.; Tamiya, N., Biochem. J. 1983, 213, (2), 507-512. The source of oxygen in the reaction catalysed by collagen lysyl hydroxylase. 16. Baldwin, J. E.; Adlington, R. M.; Crouch, N. P.; Pereira, I. A. C., Tetrahedron 1993, 49, (34), 7499-7518. Incorporation of 18O-labelled water into oxygenated products produced by the enzyme deacetoxy/deacetylcephalosporin C synthase. 17. Sabourin, P. J.; Bieber, L. L., J. Biol. Chem. 1982, 257, (13), 7468-7471. The mechanism of alpha-ketoisocaproate oxygenase. Formation of beta-hydroxyisovalerate from alpha-ketoisocaproate. 18. Lindblad, B.; Lindstedt, G.; Lindstedt, S., J. Am. Chem. Soc. 1970, 92, (25), 7446-7449. Mechanism of enzymic formation of homogentisate from p-hydroxyphenylpyruvate. 19. Wackett, L. P.; Kwart, L. D.; Gibson, D. T., Biochemistry 1988, 27, (4), 1360-1367. Benzylic monooxygenation catalyzed by toluene dioxygenase from Pseudomonas putida. 20. Pestovsky, O.; Bakac, A., Inorg. Chem. 2005, 45, (2), 814-820. Aqueous Ferryl(IV) Ion: Kinetics of Oxygen Atom Transfer To Substrates and Oxo Exchange with Solvent Water. 21. John, O. E.; Ruth, H. P.; Ruggero, C.; Fulvio Di, F., Photochem. Photobiol. 1979, 30, (1), 63-70. On the Formation and Reactivity of Dioxirane Intermediates in the Reaction of Peroxoanions with Organic Substrates. 22. Hammerschmidt, F., Angew. Chem., Int. Ed. Engl. 1994, 33, 341-342. Incorporation of L-[Methyl-2H3]methionine and 2-[Hydroxy-18O]hydroxyethylphosphonic Acid into Fosfomycin in Streptomyces fradiae-An Unusual Methyl Transfer. 23. Gallagher, T. F.; Kritchevsky, T. H., J. Am. Chem. Soc. 1950, 72, (2), 882-885. Perbenzoic Acid Oxidation of 20-Ketosteroids and the Stereochemistry of C-171. 24. Wikstroem, M.; Morgan, J. E., J. Biol. Chem. 1992, 267, (15), 10266-10273. The dioxygen cycle. Spectral, kinetic, and thermodynamic characteristics of ferryl and peroxy intermediates observed by reversal of the cytochrome oxidase reaction. 25. Michel, H.; Behr, J.; Harrenga, A.; Kannt, A., Ann. Rev. Biophys. Biomol. Struct. 1998, 27, (27), 329-356. Cytochrome C Oxidase: Structure and Spectroscopy. 26. Wikstrom, M., Biochemistry 2000, 39, (13), 3515-3519. Proton Translocation by Cytochrome c Oxidase: A Rejoinder to Recent Criticism.

71
27. Coulson, A. F. W.; Erman, J. E.; Yonetani, T., J. Biol. Chem. 1971, 246, (4), 917-924. Studies on Cytochrome c Peroxidase. 28. Dunford, H. B.; Stillman, J. S., Coord. Chem. Rev. 1976, 19, (3), 187-251. On the function and mechanism of action of peroxidases. 29. Ator, M. A.; David, S. K.; Ortiz de Montellano, P. R., J. Biol. Chem. 1987, 262, (31), 14954-14960. Structure and catalytic mechanism of horseradish peroxidase. Regiospecific meso alkylation of the prosthetic heme group by alkylhydrazines. 30. Rodriguez-Lopez, J. N.; Lowe, D. J.; Hernandez-Ruiz, J.; Hiner, A. N. P.; Garcia-Canovas, F.; Thorneley, R. N. F., J. Am. Chem. Soc. 2001, 123, (48), 11838-11847. Mechanism of Reaction of Hydrogen Peroxide with Horseradish Peroxidase: Identification of Intermediates in the Catalytic Cycle. 31. Groves, J. T., Proc. Natl. Acad. Sci. U.S.A. 2003, 100, (7), 3569-3574. The bioinorganic chemistry of iron in oxygenases and supramolecular assemblies. 32. Ortiz de Montellano, P. R., Chem. Rev. 2009, 110, (2), 932-948. Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. 33. Bergstrom, S.; Lindstredt, S.; Samuelson, B.; Corey, E. J.; Gregoriou, G. A., J. Am. Chem. Soc. 1958, 80, (9), 2337-2338. The Stereochemistry of 7-Hydroxylation in the Biosynthesis of Cholic Acid from Cholesterol. 34. Corey, E. J.; Gregoriou, G. A.; Peterson, D. H., J. Am. Chem. Soc. 1958, 80, (9), 2338-2338. The Stereochemistry of 11-Hydroxylation of Steroids. 35. Groves, J. T.; McClusky, G. A.; White, R. E.; Coon, M. J., Biochem. Biophys. Res. Comm. 1978, 81, (1), 154-160. Aliphatic hydroxylation by highly purified liver microsomal cytochrome P-450. Evidence for a carbon radical intermediate. 36. Gelb, M. H.; Heimbrook, D. C.; Malkonen, P.; Sligar, S. G., Biochemistry 1982, 21, (2), 370-377. Stereochemistry and deuterium isotope effects in camphor hydroxylation by the cytochrome P450cam monooxygenase system. 37. McClanahan, R. H.; Huitric, A. C.; Pearson, P. G.; Desper, J. C.; Nelson, S. D., J. Am. Chem. Soc. 1988, 110, (6), 1979-1981. Evidence for a cytochrome P-450-catalyzed allylic rearrangement with double-bond topomerization. 38. Krauser, J. A.; Guengerich, F. P., J. Biol. Chem. 2005, 280, (20), 19496-19506. Cytochrome P450 3A4-catalyzed Testosterone 6-Hydroxylation Stereochemistry, Kinetic Deuterium Isotope Effects, and Rate-limiting Steps. 39. Lim, M. H.; Rohde, J.-U.; Stubna, A.; Bukowski, M. R.; Costas, M.; Ho, R. Y. N.; Munck, E.; Nam, W.; Que, L., Proc. Natl. Acad. Sci. U.S.A. 2003, 100, (7), 3665-3670. An Fe(IV)O complex of a tetradentate tripodal nonheme ligand.

72
40. Bollinger, J. M.; John, C. P.; Lee, M. H.; Eric, W. B.; Carsten, K., Eur. J. Inorg. Chem. 2005, 2005, (21), 4245-4254. Mechanism of Taurine: alpha-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. 41. Hengen, P. N., Trends Biochem. Sci. 1995, 20, (7), 285-286. Purification of His-Tag fusion proteins from Escherichia coli. 42. Hammerschmidt, F.; Kaehlig, H., J. Org. Chem. 1991, 56, (7), 2364-2370. Biosynthesis of natural products with a phosphorus-carbon bond. 7. Synthesis of [1,1-2H2]-, [2,2-2H2]-, (R)- and (S)-[1-2H1](2-hydroxyethyl)phosphonic acid and (R,S)-[1-2H1](1,2-dihydroxyethyl)phosphonic acid and incorporation studies into fosfomycin in Streptomyces fradiae. 43. Albert, D. B.; Martens, C. S., Marine Chem. 1997, 56, (1-2), 27-37. Determination of low-molecular-weight organic acid concentrations in seawater and pore-water samples via HPLC. 44. Goerger, M. M.; Hudson, B. S., J. Org. Chem. 1988, 53, (14), 3148-3153. Synthesis of all-trans-parinaric acid-d8 specifically deuterated at all vinyl positions. 45. Brown, H. C.; Chandrasekharan, J.; Ramachandran, P. V., J. Am. Chem. Soc. 1988, 110, (5), 1539-1546. Chiral synthesis via organoboranes. 14. Selective reductions. 41. Diisopinocampheylchloroborane, an exceptionally efficient chiral reducing agent. 46. Hammerschmidt, F., Liebigs Ann. Chem. 1988, 1988, (10), 955-960. Biosynthese von Naturstoffen mit einer P&bond;C-Bindung. III. Synthese der (R)- und (S)-(2-Amino[1-D-ethyl)phosphonsaure und deren Hydroxylierung zu (2-Amino-1-hydroxyethyl)phosphonsaure in Acanthamoeba castellanii (Neff).

73
CHAPTER 4: CHEMISTRY OF SUBSTRATE ANALOGUES WITH HEPD*
4.1 Background
The strategy of using substrate analogues to study enzymes has been well established.1 They have served
as reversible inhibitors of enzymes and have been the basis of many commercial therapeutics.2 Substrate
analogues have also been used to probe enzyme promiscuity and such non-native transformations have
served as the basis for directed divergent evolution of new catalysts.3
In the context of the current studies, substrate analogues were prepared and presented to HEPD in order
to gain further mechanistic insight. Analysis of the products produced from substrate analogues by
HEPD was envisioned to potentially provide support for one of the mechanisms discussed in chapter 3 if
one mechanism can best account for the observed transformations. For reference, the two proposed
mechanisms (hydroxylation and hydroperoxylation) for HEPD are shown in Scheme 4.1. More detailed
descriptions for each mechanism have been given in chapter 3.6.
* Parts of this chapter have been reproduced from 1) Whitteck, J.T., Cicchillo, R.M., van der Donk, W.A., J. Am. Chem. Soc. 2009, 131, 16225-16232 and 2) Cicchillo, R.M., Zhang, H., Blodgett, J.A.V., Whitteck, J.T., Li, G., Nair, S.K., van der Donk, W.A., Metaclf, W.W., Nature, 2009, 459, 871-874 with permission from the American Chemical Society and Nature Publishing Group, respectively.
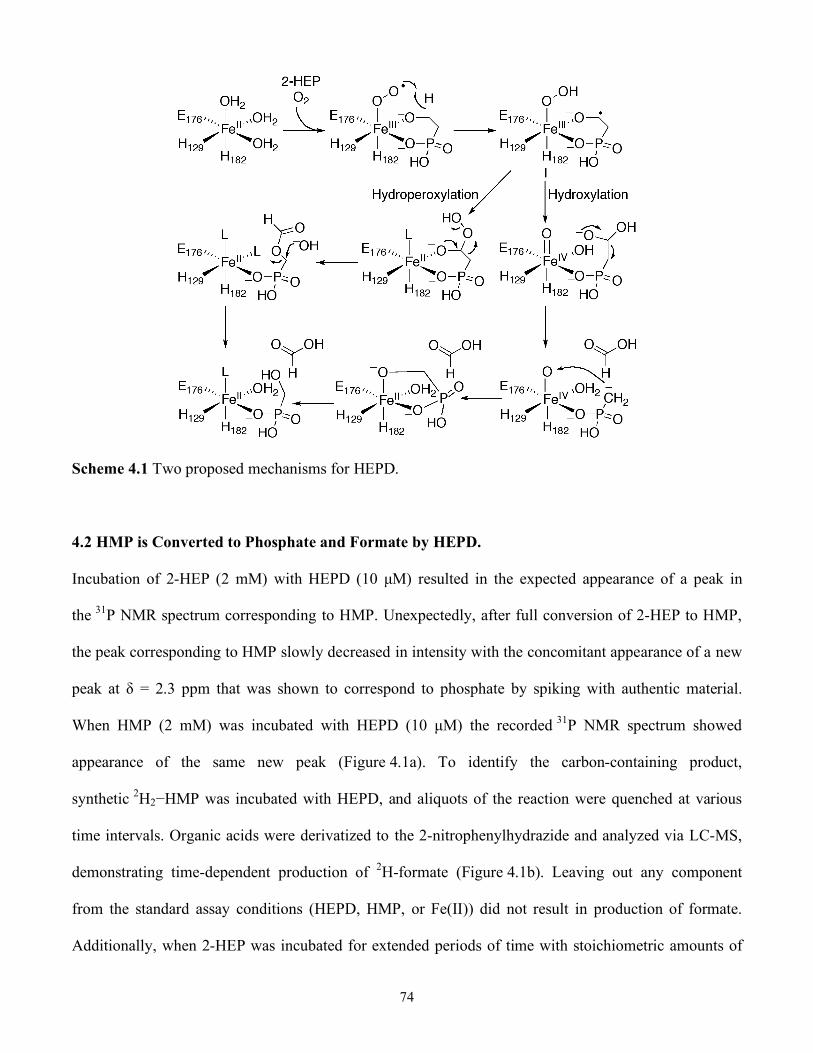
74
Scheme 4.1 Two proposed mechanisms for HEPD.
4.2 HMP is Converted to Phosphate and Formate by HEPD.
Incubation of 2-HEP (2 mM) with HEPD (10 µM) resulted in the expected appearance of a peak in
the 31P NMR spectrum corresponding to HMP. Unexpectedly, after full conversion of 2-HEP to HMP,
the peak corresponding to HMP slowly decreased in intensity with the concomitant appearance of a new
peak at δ = 2.3 ppm that was shown to correspond to phosphate by spiking with authentic material.
When HMP (2 mM) was incubated with HEPD (10 µM) the recorded 31P NMR spectrum showed
appearance of the same new peak (Figure 4.1a). To identify the carbon-containing product,
synthetic 2H2−HMP was incubated with HEPD, and aliquots of the reaction were quenched at various
time intervals. Organic acids were derivatized to the 2-nitrophenylhydrazide and analyzed via LC-MS,
demonstrating time-dependent production of 2H-formate (Figure 4.1b). Leaving out any component
from the standard assay conditions (HEPD, HMP, or Fe(II)) did not result in production of formate.
Additionally, when 2-HEP was incubated for extended periods of time with stoichiometric amounts of
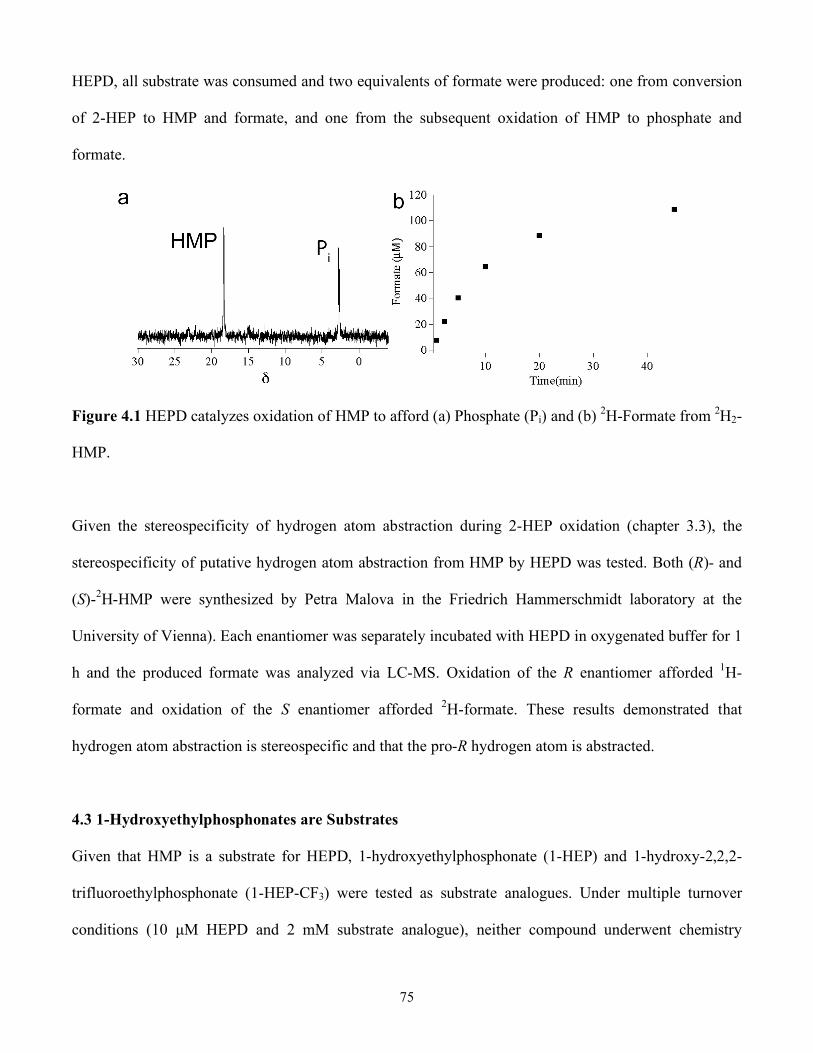
75
HEPD, all substrate was consumed and two equivalents of formate were produced: one from conversion
of 2-HEP to HMP and formate, and one from the subsequent oxidation of HMP to phosphate and
formate.
Figure 4.1 HEPD catalyzes oxidation of HMP to afford (a) Phosphate (Pi) and (b) 2H-Formate from 2H2-
HMP.
Given the stereospecificity of hydrogen atom abstraction during 2-HEP oxidation (chapter 3.3), the
stereospecificity of putative hydrogen atom abstraction from HMP by HEPD was tested. Both (R)- and
(S)-2H-HMP were synthesized by Petra Malova in the Friedrich Hammerschmidt laboratory at the
University of Vienna). Each enantiomer was separately incubated with HEPD in oxygenated buffer for 1
h and the produced formate was analyzed via LC-MS. Oxidation of the R enantiomer afforded 1H-
formate and oxidation of the S enantiomer afforded 2H-formate. These results demonstrated that
hydrogen atom abstraction is stereospecific and that the pro-R hydrogen atom is abstracted.
4.3 1-Hydroxyethylphosphonates are Substrates
Given that HMP is a substrate for HEPD, 1-hydroxyethylphosphonate (1-HEP) and 1-hydroxy-2,2,2-
trifluoroethylphosphonate (1-HEP-CF3) were tested as substrate analogues. Under multiple turnover
conditions (10 µM HEPD and 2 mM substrate analogue), neither compound underwent chemistry

76
discernible by 31P NMR spectroscopy. Each substrate analogue (1.0 mM) was then incubated with 0.5
equiv of HEPD and, after acidic workup, two peaks were observed in both experiments. One peak
corresponded to the starting material and the other peak at δ = 2.3 ppm corresponded to phosphate.
To determine the other products generated from 1-HEP, the substrate analogue (0.3 mM) was incubated
with HEPD (0.1 mM) for 2 h, followed by analysis of organic acids by LC-MS after derivatization as
described in the experimental section. A peak was observed in the chromatogram with a retention time
and mass consistent with derivatized acetate. The produced acetate was quantified (0.11 mM), closely
corresponding to the amount of HEPD in the assay. The phosphorus-containing product before acidic
workup was then determined by incubating the analogue with HEPD and analysis by 31P NMR
spectroscopy (Figure 4.2a). The spectrum showed three peaks: one for unreacted 1-HEP, a small peak
for phosphate and an additional peak at δ = −2.2 ppm. Sulphuric acid was then added to the assay, the
precipitated enzyme was removed by centrifugation, and another 31P NMR spectrum was recorded. The
spectrum showed a peak for 1-HEP and a peak for phosphate suggesting the third peak at δ = −2.2 ppm
was associated with an acid labile intermediate that can be converted to phosphate under acidic
conditions. This intermediate was then identified as acetyl phosphate (AP) by spiking with authentic
material. Because 1-HEP was only converted when incubated with high concentrations of HEPD, the
enzyme is likely inactivated during the conversion of 1-HEP to AP. Indeed, preincubation of HEPD (2
µM) with AP (100 µM) inhibited oxidation of its native substrate 2-HEP. Similarly, incubation of HEPD
(100 µM) with 1 equiv of AP decreased the rate of formate production by 67% compared to an assay
without AP.
The 31P NMR spectrum for the reaction of 1-HEP-CF3 with HEPD also showed a peak for the starting
material and a peak for phosphate (Figure 4.2b). To determine the identity of any nonphosphorus
containing products, 1-HEP-CF3 (0.3 mM) was incubated with HEPD (0.1 mM) and the 19F NMR
spectrum was recorded. Two peaks were observed, with one peak corresponding to unreacted 1-HEP-
CF3 and a second signal corresponding to trifluoroacetate, as demonstrated with an authentic sample.

77
Integration of the peaks in the spectrum showed a ratio of 1:2 for trifluoroacetate:1-HEP-CF3. The
amount of trifluoroacetate and phosphate formed was approximately equal to the amount of HEPD in
the reaction, suggesting that HEPD was inactivated after converting 1-HEP-CF3 to trifluoroacetate and
phosphate. However, no reduction of formate production from 2-HEP was observed after preincubation
of HEPD with trifluoroacetate by itself or with trifluoroacetate and phosphate.
Figure 4.2 31P NMR spectra after incubation of HEPD with (a) 1-HEP, (b) 1-HEP-CF3, Pi = inorganic
phosphate (AP = acetyl phosphate).
4.4 2-Hydroxypropylphosphonate is Oxidized by HEPD
Given the similarities between the structures of and substrates for HEPD and HppE, 2-
hydroxypropylphosphonate (2-HPP) was presented as a substrate for HEPD. When HEPD (0.1 mM) was
incubated with 2-HPP (0.2 mM) with and without reducing equivalents (0.15 mM FMN and 16 mM
NADH) the same result was observed. The protein was removed and the 31P NMR spectrum was
recorded and two peaks were observed: one corresponding to 2-HPP and a new peak at 10.8 ppm.
Analysis via LC-MS (MS-APCI positive mode) showed some 2-HPP remained and a new peak with an
m/z of 141 was produced. Two potential products, fosfomycin and 2-oxopropylphosphonate (OPP), have

78
identical 31P NMR chemical shifts and an m/z that is consistent with those observed during the reaction
of 2-HPP with HEPD.
To differentiate between these two products the protein free reaction mixture was analyzed via LC-
MS/MS (ESI negative mode) in Molecular Reaction Monitoring (MRM) mode. Under identical
conditions, OPP (m/z 139) fragments to an ion with m/z 139 to m/z 79, whereas fosfomycin (m/z 139)
fragments to provide ions with m/z 139 to m/z 63, m/z 79 and m/z 81. The observed fragmentation
pattern for the 2-HPP reaction was identical to OPP showing unambiguously that it is the product of
HEPD catalyzed oxidation of 2-HPP. Incubating 2-HPP with HEPD under multiple turnover conditions
only produced small amounts of OPP. Preincubating 2-HPP with HEPD before 2-HEP resulted in a
>90% loss of activity as determined by formate production. These results suggest that HEPD is being
inactivated during the conversion of 2-HPP to OPP.
Both the hydroxylation and hydroperoxylation mechanisms can account for the oxidation of 2-HPP and
the inactivation of HEPD, though the mechanisms of enzyme inactivation are different (Scheme 4.2).
During both mechanisms a C-O bond is broken instead of the C-C bond. This difference is likely due to
the introduction of a methyl group, which could disfavor the necessary antiperiplanar orbital geometry
that promotes C-C bond cleavage. In the hydroperoxylation mechanism, this change in orientation
results in formation of OPP and hydrogen peroxide, which is predicted to inactivate the enzyme via
oxidation of ferrous iron. Alternatively, in the hydroxylation mechanism OPP production and release
results in the metal being trapped in the inactive Fe(IV) state.
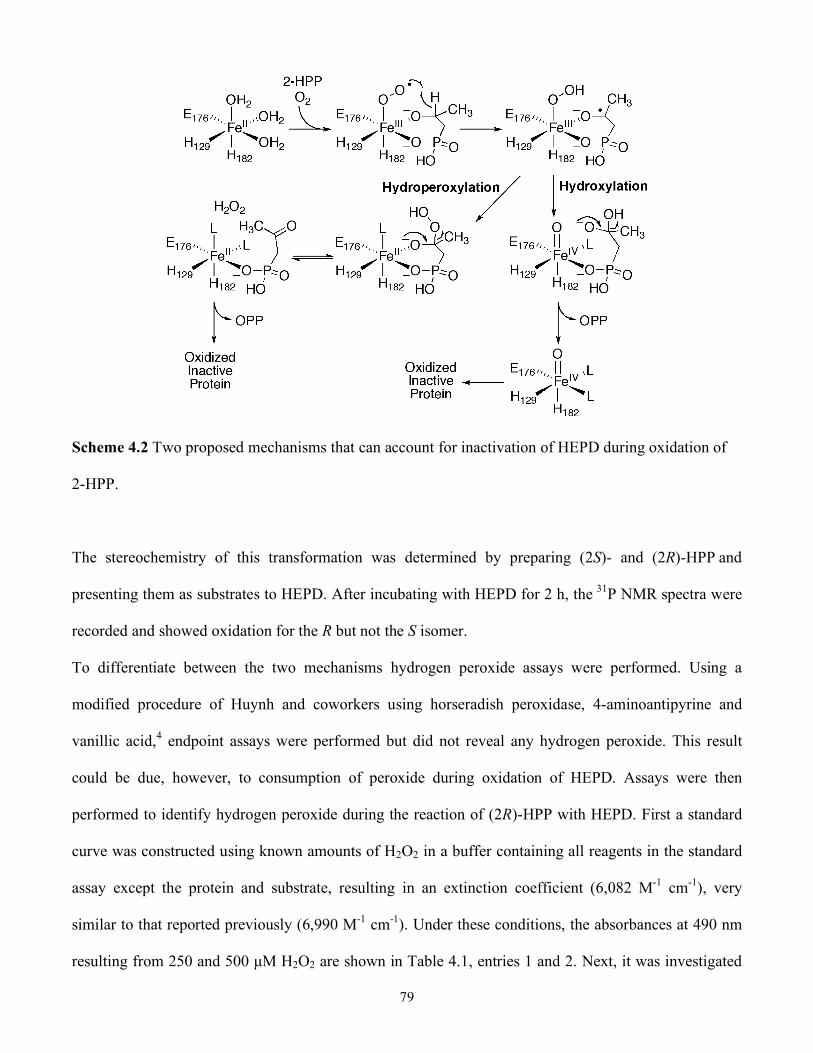
79
Scheme 4.2 Two proposed mechanisms that can account for inactivation of HEPD during oxidation of
2-HPP.
The stereochemistry of this transformation was determined by preparing (2S)- and (2R)-HPP and
presenting them as substrates to HEPD. After incubating with HEPD for 2 h, the 31P NMR spectra were
recorded and showed oxidation for the R but not the S isomer.
To differentiate between the two mechanisms hydrogen peroxide assays were performed. Using a
modified procedure of Huynh and coworkers using horseradish peroxidase, 4-aminoantipyrine and
vanillic acid,4 endpoint assays were performed but did not reveal any hydrogen peroxide. This result
could be due, however, to consumption of peroxide during oxidation of HEPD. Assays were then
performed to identify hydrogen peroxide during the reaction of (2R)-HPP with HEPD. First a standard
curve was constructed using known amounts of H2O2 in a buffer containing all reagents in the standard
assay except the protein and substrate, resulting in an extinction coefficient (6,082 M-1 cm-1), very
similar to that reported previously (6,990 M-1 cm-1). Under these conditions, the absorbances at 490 nm
resulting from 250 and 500 µM H2O2 are shown in Table 4.1, entries 1 and 2. Next, it was investigated

80
whether H2O2 would react with ferrous HEPD in the presence of the peroxide detection reagents. As
shown in entry 4, approximately half of the 500 µM H2O2 added to 500 µM ferrous HEPD is rapidly
consumed resulting in an absorbance at 490 nM corresponding to ~215 µM remaining H2O2; this initial
absorbance remains unchanged over the 2 h assay. Further addition of H2O2 results in the expected
increase in absorbance. Furthermore, HEPD does not show any activity after H2O2 treatment. These
observations therefore show that one equivalent of H2O2 oxidizes approximately two molecules of
ferrous HEPD.
With these control experiments established, the reactions of HEPD with substrates were investigated.
First, it was determined that addition of the peroxide assay reagents (horseradish peroxidase, 4-
aminoantipyrine, and vanillic acid) was compatible with HEPD activity. In these experiments
(performed stoichiometrically because HEPD did not appear to turnover multiple (2R)-HPP molecules)
2-HEP was converted to HMP by HEPD with the observed activity comparable to experiments in the
absence of the peroxide detection reagents. Similarly, these reagents did not affect the conversion of
(2R)-HPP to OPP by HEPD. Hydrogen peroxide was not detected in either the conversion of 2-HEP or
(2R)-HPP (Table 4.1, entries 5 and 6). If OPP had been formed stoichiometrically by the
hydroperoxylation mechanism, the conversion of 500 µM 2-HPP to OPP by 500 µM HEPD would have
resulted in 500 µM H2O2. Approximately half of the H2O2 formed would oxidize the ferrous HEPD
formed as shown in entry 4, but approximately 200-250 µM H2O2 should have remained and should
have been detected. Since this is not observed, a hydroperoxylation mechanism appears inconsistent
with the data. This argument assumes that all (2R)-HPP is converted to OPP.

81
Table 4.1 Hydrogen peroxide assays for the reaction 2-HEP and 2-HPP with HEPD.
Entry Reaction Conditions A490
1 250 µM H2O2 1.61
2 500 µM H2O2 3.04a
3 500 µM ferrous HEPD 0.18
4 500 µM ferrous HEPD + 500 µM H2O2 1.38
5 500 µM ferrous HEPD + 500 µM 2-HEP 0.18
6 500 µM ferrous HEPD + 500 µM (2R)-HPP 0.19
a) Value confirmed by absorbance measurements after defined dilution.
If the reaction does proceed via hydroperoxylation and H2O2 is oxidizing and inactivating HEPD, then it
may be possible to scavenge H2O2 and convert (2R)-HPP with multiple turnovers. To test this hypothesis
(2R)-HPP (1 mM) was incubated with HEPD (20 µM) and either catalase (2 mg/mL) or tris(2-
carboxyethyl)phosphine (TCEP, 3 mM) for 2 h. Analysis by both LC-MS and 31P NMR spectroscopy
showed that in both assays no additional OPP was produced. While this does not provide evidence for
H2O2 production it does not explicitly rule out hydroperoxylation or provide direct support for
hydroxylation.
The stoichiometry of (2R)-HPP consumed to OPP produced was then determined unambiguously by
incubation of HEPD with a 3-fold excess of (2R)-HPP and quantification of the OPP generated via LC-
MS. Reaction of 0.1 mM, 0.25 mM, or 0.5 mM HEPD with 0.3 mM, 0.75 mM, and 1.5 mM (2R)-HPP
produced OPP concentrations of 0.052 ± 0.011 mM, 0.122 ± 0.005 mM and 0.220 ± 0.016 mM,
respectively. These three experiments demonstrate stoichiometries of OPP produced to HEPD of 0.52:1,
0.49:1 and 0.44:1, respectively. In the hydroperoxylation mechanism, each conversion of (2R)-HPP to
OPP would produce one equivalent of H2O2. Since it has been previously shown that one equivalent of
H2O2 rapidly inactivates approximately two ferrous HEPD molecules, the observed stoichiometries at
first glance appear to agree very well with the hydroperoxylation mechanism (i.e., each turnover of

82
HEPD results in one OPP and 2 oxidized, inactive enzymes). As noted before, one caveat is that
complete conversion of (2R)-HPP to OPP is assumed in this model.
To probe if (2R)-HPP was cleanly converted to OPP, HEPD (0.5 mM) was incubated with (2R)-HPP
(1.5 mM). EDTA and dithionite were added to improve signal quality through sharpening of the peaks in
the 31P NMR spectrum (Figure 4.3) that were very broad at the high enzyme concentrations used. The
spectrum after 2 h showed peaks corresponding to (2R)-HPP, OPP, and phosphate in a 1:0.4:0.33 ratio;
this ratio did not change after the initial enzymatic conversion. Since all phosphorus-containing products
originated from (2R)-HPP, the concentrations of (2R)-HPP, OPP and phosphate are approximately 867,
346, and 286 µM, respectively. To account for the presence of phosphate, OPP was incubated under
identical conditions with stoichiometric amounts of HEPD and the 31P NMR spectrum was recorded, but
no conversion of OPP to phosphate was observed. Then (2R)-HPP (1.5 mM) was incubated with HEPD
(0.5 mM) for 2 h and the sample was analyzed for organic acids. Both acetate and formate (364 and 216
µM, respectively) were observed in significantly higher concentrations than in control reactions (62 and
16 µM of acetate and formate, respectively). Collectively, these results are consistent with partitioning
of (2R)-HPP to generate OPP, which is unreactive toward further enzymatic oxidation, in addition to
acetate and HMP (Scheme 4.3). Subsequent oxidation of HMP produces the observed formate and
phosphate products, as discussed above. It should be noted that small differences in ratios are possible
due to uncertainties in protein concentrations, but these potential variations are small enough that they
would not affect our interpretation of the data.

83
Figure 4.3 31P NMR spectrum after incubation of HEPD with (2R)-HPP, demonstrating formation of
OPP as well as inorganic phosphate (Pi).
Scheme 4.3 Reaction of (2R)-HPP partitions between formation of OPP and phosphate (presumably via
HMP).
4.5 Studies with Additional Substrate Analogues
Other analogues of HEP tested as potential substrates of HEPD are shown in Figure 4.4. The compounds
were incubated with HEPD under both catalytic and single turnover conditions and the assays were
analyzed via 31P NMR spectroscopy and LC-MS. Both methods showed that none of the tested
analogues gave rise to new products when compared to authentic standards.
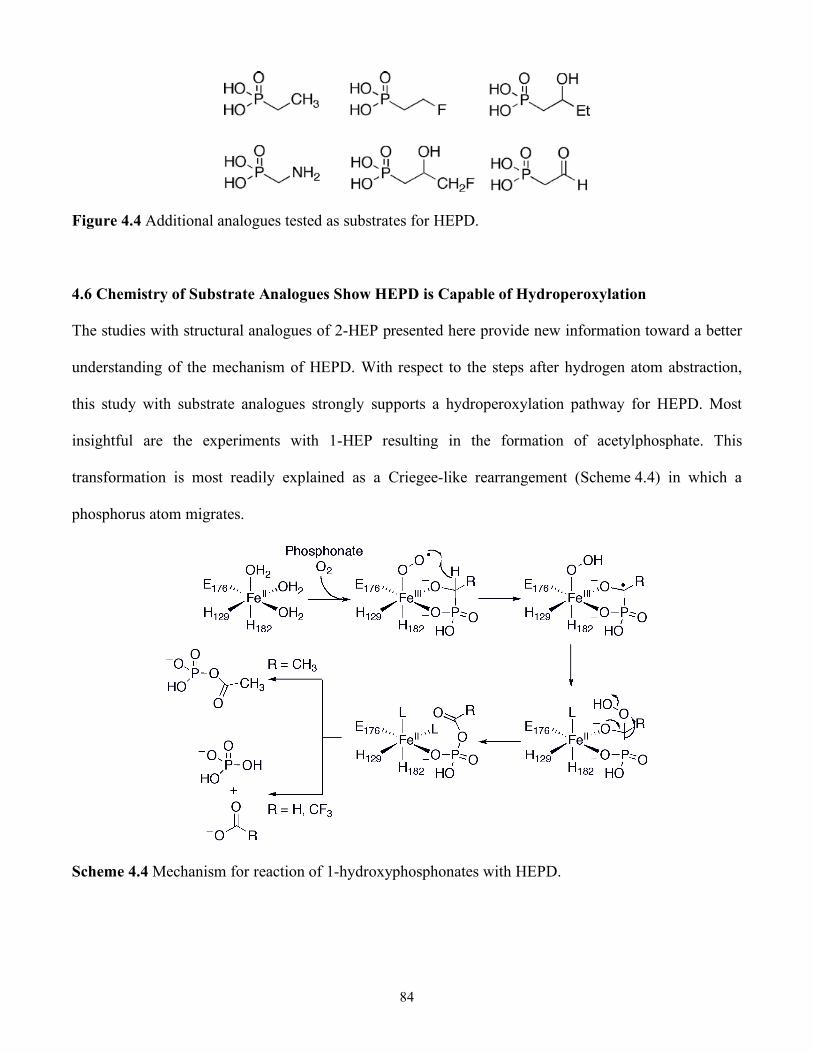
84
Figure 4.4 Additional analogues tested as substrates for HEPD.
4.6 Chemistry of Substrate Analogues Show HEPD is Capable of Hydroperoxylation
The studies with structural analogues of 2-HEP presented here provide new information toward a better
understanding of the mechanism of HEPD. With respect to the steps after hydrogen atom abstraction,
this study with substrate analogues strongly supports a hydroperoxylation pathway for HEPD. Most
insightful are the experiments with 1-HEP resulting in the formation of acetylphosphate. This
transformation is most readily explained as a Criegee-like rearrangement (Scheme 4.4) in which a
phosphorus atom migrates.
Scheme 4.4 Mechanism for reaction of 1-hydroxyphosphonates with HEPD.

85
Acetylphosphate is shown to be a potent inhibitor of HEPD explaining why only a single turnover
occurs, with the inhibition likely due to the resemblance of acetylphosphate to HMP-formyl ester. The
proposed phosphorus migration has precedent in Baeyer−Villiger oxidation of dialkyl acylphosphonates
in organic solvents that have been shown to proceed through a Criegee intermediate (Scheme 4.5).5 The
hydroperoxylation mechanism can also explain the observed conversion of HMP into phosphate and
formate with formylphosphate as a likely intermediate (Scheme 4.4). Unlike acetyl phosphate, which has
been shown to be stable at neutral pH for months,6 formylphosphate is unstable and readily hydrolyzes
nonenzymatically to phosphate and formate as observed by Benkovic and co-workers for the reaction
catalyzed by PurT GAR formyltransferase.7 HEPD is regenerated in its Fe(II) state in this model and,
unlike with 1-HEP, the enzyme is not inactivated due to the rapid hydrolysis of formylphosphate,
accounting for the catalytic turnover of HMP to phosphate and formate. Elucidating the details of the
mechanism by which the alkylhydroperoxides are formed from a ferric hydroperoxide and a carbon-
based radical requires further studies.
Scheme 4.5 Published example of formation of acylphosphates via Baeyer-Villiger oxidation of
acylphosphonates.5
The results with 1-HEP-CF3 can also be rationalized by the hydroperoxylation pathway. After initial
hydroperoxylation, Criegee-like rearrangement would result in trifluoroacetylphosphate, which is more
reactive toward hydrolysis than acetylphosphate and is not detected directly. Instead, the hydrolysis
products trifluoroacetate and phosphate were observed. Why HEPD is inactivated during this process
resulting in a single turnover is not clear as trifluoroacetate is not an inhibitor. It is possible that

86
trifluoroacetyl phosphate remains bound to the iron center after its formation in the active site and that
hydrolysis only occurs after protein denaturation associated with product analysis.
The results of the detailed investigation of the conversion of (2R)-HPP to OPP are also consistent with
initial hydroperoxylation followed by a Criegee rearrangement. Hydrogen peroxide production was not
observed, which appeared to be incompatible with the hydroperoxylation mechanism because
stoichiometric conversion of (2R)-HPP to OPP should produce 1 equivalent of H2O2. Half an equivalent
of H2O2 has been shown to inactivate approximately 1 equivalent of Fe(II)-HEPD,8 and thus 0.5
equivalents of H2O2 should remain upon oxidation of (2R)-HPP and should have been detected.
However, as mentioned caveat in this model is that all (2R)-HPP is converted to OPP. Analysis of the
products by 31P NMR spectroscopy after solving problems with peak broadening have shown that
conversion of (2R)-HPP to OPP is not quantitative and that (2R)-HPP oxidation partitions between
formation of OPP and HMP along with acetate (Scheme 4.6). The latter reaction is similar to the
physiological reaction, conversion of 2-HEP to HMP and formate. Presumably, an intermediate in this
process is O-acetyl-hydroxymethylphosphonate (I) that is hydrolyzed to acetate and HMP. HMP is then
converted to formate and phosphate as discussed above. The partitioning also explains why H2O2 was
not observed since only 0.6 equivalents of OPP (and hence H2O2) are formed with respect to HEPD.
This amount is sufficient to completely oxidize the enzyme, but very little H2O2 would remain after
HEPD oxidation. The partitioning between carbon migration and peroxide elimination suggests that
adding an additional methyl group to C2 results in less favorable orbital overlap for a Criegee
rearrangement such that peroxide elimination competes.
The observed conversion of (2R)-HPP to OPP is reminiscent of the conversion by HppE of (2R)-HPP,
the enantiomer of the physiological substrate, to OPP. Thus, when a hydrogen atom is available for
abstraction at C2, both HEPD and HppE carry out the same transformation with (2R)-HPP. On the other
hand, unlike HppE, HEPD cannot be coerced to abstract a hydrogen atom from C1 of (2S)-HPP. It
should be noted that other features are shared between HEPD and HppE. Three-dimensional analysis of

87
the proteins has revealed that they not only share similar tertiary structure (as noted in chapter 3.6) but
also very similar active site architecture.8, 9 A bidentate mode of substrate binding to the metal ion and
subsequent oxygen activation have been proposed as the initial steps in both mechanisms (see Scheme
1.5 for details).8,10 Additionally, an Fe(IV)=O intermediate has been proposed for both enzymes (see
chapter 1.4.1 and chapters 3.7 and 3.11). Given the similar substrates, enzyme structures and proposed
mechanisms, it is interesting that, at this time, HEPD has not been shown to catalyze epoxidation and
HppE has never been shown to catalyze C-C bond cleavage (though they do catalyze the same oxidation
of (2R)-HPP). Further studies will be required to provide insights into the factors that result in epoxide
formation by HppE and C-C bond scission by HEPD.
It should also be noted that the observed oxidation of (2R)-HPP and not (2S)-HPP by HEPD is
consistent with the active site geometry and bidentate binding mode of 2-HEP observed in the cocrystal
structure of Cd2+-HEPD with 2-HEP.8 As well, these observations are consistent with the
stereospecificity for abstraction of the pro-S hydrogen atom at C2 of 2-HEP as shown in Chapter 3.3.
Scheme 4.6 Mechanism for oxidation of (2R)-HPP to OPP as well as acetate, formate and phosphate.

88
In summary, the studies presented here with substrate analogues demonstrate that HEPD is capable of
hydroperoxylation. As such, the enzyme has similarities with flavin dependent Bayer-Villigerases,11-
14 except that the substrates for HEPD are at a lower oxidation level than the carbonyl-containing
substrates of these enzymes. The corollary of the hydroperoxylation mechanism for these substrates of
HEPD is that the O−O bond of molecular oxygen is not broken prior to substrate activation, as has been
proposed for the aforementioned non-heme iron enzymes MIOX and IPNS, for a diiron(II/III)
superoxide model complex,15 and for the copper-dependent enzymes dopamine β-monooxygenase,
peptidyl glycine α-hydroxylating monooxygenase, and galactose oxidase.16-19
These experiments may also lend support for a hydroperoxylation mechanism for oxidation of 2-HEP to
HMP. However, the experiments on the stereochemistry of HMP produced from stereoselectively
labeled 1-2H-2-HEP have ruled out the later steps of the hydroperoxlation mechanism shown in Scheme
4.1. Specifically, production of racemic HMP combined with 18O labeling studies rule out the hydrolysis
at C1 of OFHMP. An alternate mechanism has been proposed via hydroxylation and is shown in
Scheme 3.7. Additional experiments with substrate analogues may afford further insight into the natural
reaction.
4.7 Experimental
4.7.1 General
HEPD was overexpressed in E. coli as an N-terminal His6-fusion protein and purified via affinity
chromatography as previously described.8 All activity assays were performed in HEPES buffer (25 mM,
pH = 7.5) unless otherwise noted. LC-MS was performed on an Agilent 1200 series quad pump system
equipped with a diode array detector and a G1956B mass spectrometer with a multimode-
electrospray/atmospheric pressure chemical ionization (MM-ES+APCI) source. The column used for
separation was a Synergi 4 µ Fusion-RP 80A column (150 × 4.6 mm, 4 µm, Phenomenex Torrance, CA)

89
using a flow rate of 0.5 mL/min. UV−visible absorption spectra were recorded on a Varian Cary 4000
UV−vis spectrophotometer. GC-MS was performed on an Agilent 6890N gas chromatograph equipped
with an electron impact (EI) ionization source.
NMR spectra were recorded on a Varian Unity 500 or Varian Unity Inova 600 spectrometer. Proton and
carbon chemical shifts are reported in δ values relative to an external standard of 0.1% tetramethylsilane
in CDCl3 (0.00 ppm). Phosphorus shifts are reported in δ values relative to an external standard of 85%
phosphoric acid (0.00 ppm). Fluorine shifts are reported in δ values relative to an external standard of
CFCl3 (0.00 ppm). Fast atom bombardment (FAB) mass spectrometry for characterization of synthetic
compounds was performed by the University of Illinois Mass Spectrometry Center using a Waters 70-
SE-4F mass spectrometer.
The isotopologues (R)- and (S)-2H-HMP were provided by the Friedrich Hammerschmidt laboratory in
Vienna, Austria. The following substrate analogues were synthesized according to literature procedures:
2-HPP, (2R)- and (2S)-HPP,20 3-hydroxypropylphosphonate,21 2-hydroxybutylphosphonate,22 3-fluoro-
2-hydroxypropylphosphonate,23 2-fluoroethylphosphonate,24 phosphonoacetaldehyde,25 and 1-
hydroxyethylphosphonate.26 Ethylphosphonate and aminoethylphosphonate were purchased from
Sigma-Aldrich. 2-Nitrophenylhydrazine (NPH) was purchased from Acros Organics. N-(3-
Dimethylaminopropyl)-N′-ethylcarbodiimide (EDC) was purchased from Chem-Impex (Wood Dale, IL).
Dichloromethane was distilled over CaH2. All other chemicals were purchased from Sigma Aldrich at
the highest purity and used without further purification.
4.7.2 Synthesis of 1-Hydroxy-2,2,2-trifluoroethylphosphonate
As detailed in Notebook IV on pages 79-80, under a nitrogen atmosphere a round-bottom flask equipped
with a magnetic stir bar and reflux condenser was charged with commercially available diethyl 1-
hydroxy-2,2,2-trifluoroethylphosphonate (1 g, 4.2 mmol, 1 equiv), which was dissolved in dry DCM (10
mL). To the solution was added bromotrimethylsilane (2 mL, 15.1 mmol, 3.6 equiv) and the orange

90
solution was heated under reflux while stirring for 2 h. The solution was allowed to cool to 25 °C and
solvent was removed under reduced pressure. The resulting residue was taken up in 1:1 H2O:EtOH (10
mL) and solvent was removed under reduced pressure to afford product (663 mg, 3.7 mmol, 88%) as a
tan oil. 1H NMR (500 MHz, D2O) δ 4.09 (m); 13C NMR (125 MHz, D2O) δ 27.76 (d, J = 8.5), 66.01
(m); 31P NMR (202 MHz, D2O) δ 11.13 (bs); 19F NMR (215 MHz, D2O) δ 17.34 (at); HRMS (FAB+)
calcd for (C2H4F3O4P+H+) 180.9878 m/z found 180.9880 m/z.
4.7.3 Identification of Assay Products via LC-MS
Enzymatic assays were quenched by adding H2SO4 (30 mM) and the protein was pelleted via
microcentrifugation (1 min at 16.1g). The supernatant was analyzed via LC-MS (positive mode) using
aqueous 0.1% formic acid as an isocratic mobile phase and scanning masses of 50−500 m/z.
4.7.4 Identification of Assay Products via 31P NMR Spectroscopy
HEPD was separated from the assay via centrifugal filtration (Millipore Micron YM-30, 30 kDa
nominal molecular weight limit). D2O (20% v/v) was added to the flow-through and the 31P NMR
spectrum was recorded on a Varian UI600 (frequency for phosphorus: 242 MHz).
4.7.5 Aqueous Derivatization and Quantification of Organic Acids
As detailed in Notebook IV, pages 236 and 266-268, following a modified literature procedure,27 to a
100 µL sample was added propionic acid as an internal standard (10 µL of a 1 mM stock solution in 1:1
pyridine:HCl), followed by EDC (10 µL of a 0.29 M stock solution), and subsequently NPH (10 µL of a
0.12 M stock solution in 250 mM aqueous HCl). The orange solution was heated to 60 °C in a water
bath for 15 min and precipitated protein was pelleted via microcentrifugation (1 min at 16.1g). The
supernatant (20 µL) was analyzed via LC-MS using an isocratic mobile phase (50:50 MeOH:H2O +
0.1% formic acid) for separation. Authentic standards of sodium 13C-formate, sodium acetate, and

91
propionic acid were also derivatized and analyzed by monitoring the absorbance at 400 nm and by MM-
ES+APCI (negative mode) and found to have retention times of 5.81, 5.93, and 6.93 min, respectively.
The observed m/z values for each derivatized compound corresponded to the monodeprotonated
hydrazide and were: 181 (13C-formate), 194 (acetate), and 208 (propionic acid).
Solutions of varying concentrations of each of these acids were made in HEPES buffer (25 mM, pH =
7.5), derivatized, and analyzed via LC-MS as described in the previous paragraph using single ion
monitoring (SIM) for the derivatized acid of interest. The area of the peak corresponding to derivatized
acid was divided by the area of the derivatized propionic acid peak to afford a response factor, which
was plotted against the acid concentration to afford a linear relationship. This equation was used to
quantify the organic acid concentration in the enzymatic assays.
4.7.6 Analysis of HMP as a Substrate for HEPD
As detailed in Notebook V, pages 20-21, in a typical assay HEPD (10 µM) and HMP (2 mM) were
added to oxygen saturated buffer (25 mM HEPES, pH 7.5) and incubated at 25 °C for 2 h. The sample
was then analyzed via 31P NMR spectroscopy (proton coupled and decoupled). The carbon-containing
product was identified by running an assay similar to the one described above except using 2H2−HMP as
the substrate and subsequent derivatization and quantification of organic acids as described above.
As detailed in Notebook V, pages 168-169, in a typical assay HEPD (100 µM) and either (R)- or (S)-2H-
HMP (100 µM) were added to oxygenated buffer (25 mM HEPES, pH 7.5) and incubated at 25 °C for 2
h. Produced formate was analyzed via derivatization of organic acids as described above.
4.7.7 Oxidation of 2-HPP to OPP
As reported by Robert Cicchillo, the product of 2-HPP oxidation was identified by incubating 2-HPP (2
mM) with HEPD (500 µM) in oxygenated buffer for 2 h followed by analysis of the assay products via
31P NMR spectroscopy as described in chapter 4.7.3. A new peak was observed at 10.8 ppm

92
corresponding to either fosfomycin or OPP. LC-MS analysis identified a two peaks at 5.5 and 6 min
corresponding to 2-HPP and the oxidized product, respectively. To delineate between the two potential
products (OPP and fosfomycin), the sample was analyzed using an Agilent 1100 series quad pump
system equipped with an MSD Trap XBT Plus mass spectrometer with an ESI source run in negative
mode. Chromatography conditions were identical to those described in chapter 4.7.3. Fragmentation
patterns for authentic OPP and fosfomycin were obtained by direct infusion mass spectrometry (ESI
negative) and were m/z 139 to 63 for OPP and m/z 139 to 63, 79 and 81 for fosfomycin. A sample from
the assay was then analyzed via multiple reaction monitoring (MRM) mode for fragmentation of m/z
139 to m/z 63, 79 and 81. Only the m/z 139 to 79 fragmentation was observed in the sample from the
assay, which is consistent with OPP as the oxidized product.
As detailed in Notebook IV, page 287, to determine which enantiomer of HPP is oxidized, HEPD (500
µM) and either (2S)- or (2R)-HPP (1.5 mM) were incubated in oxygen saturated HEPES buffer (25 mM,
pH 7.5) for 2 h at 25 °C.
As described in Notebook V on pages 98-99, it was determined if HEPD could multiply turn over 2-HPP
by scavenging H2O2. In oxygen saturated buffer, HEPD (20 µM), scavenger (either catalase (2 mg/mL)
or TCEP (3 mM)) and 2-HPP (1 mM) were incubated at room temperature for 2 h. Analysis by 31P NMR
spectroscopy and LC-MS did not show additional production of OPP.
As described in Notebook V, pages 66-67, H2O2 concentration was determined spectrophotometrically
by modifying a procedure described by Huynh and coworkers.4 In this assay, horseradish peroxidase
uses H2O2 to oxidize 4-aminoantipyrine, the product of which condenses with vanillic acid to form a red
quinone imine with a broad absoption band at 490 nm, which was quantified by ultraviolet-visible
spectroscopy (Varian, Cary 4000 UV-Vis spectophotometer). The assay solution contained HEPES (25
mM, pH = 7.5), HEPD (500 µM), horseradish peroxidase (13 units/mL), 4-aminoantipyrine (1.7 mM),
vanillic acid (3.3 mM), and substrate (either 2-HEP or (2R)-HPP or H2O2, 500 µM). Substrate was added

93
last and the sample was incubated for 2 h. Then the absoption at 490 nm was recorded. Subsequently,
samples were analyzed via LC-MS.
Then the products were analyzed by 31P NMR spectroscopy and LC-MS. The masses for protonated 2-
HPP (141) and OPP (139) were extracted from the total ion chromatogram (TIC) to afford a single peak
in each extracted ion chromatogram (EIC) with retention times of 6 and 7 min, respectively, that were
identical to authentic standards of 2-HPP and OPP.
As described in Notebook V on pages 86-87, to determine the stoichiometry of the oxidation of (2R)-
HPP to OPP via LC-MS, first a linear relationship between OPP concentration and area of the peak in
the EIC was established by analyzing varying concentrations of synthetic OPP in buffer (50, 100, 250,
500, and 1000 µM). Similar assays as previously described were then carried out with varying
concentrations of HEPD (100, 250, and 500 µM) and (2R)-HPP (3-fold excess over HEPD), which were
analyzed via LC-MS.
As described in Notebook V on pages 127 and 129, to determine the stoichiometry of the oxidation of
(2R)-HPP to OPP by NMR spectroscopy, (2R)-HPP (1.5 mM) was incubated with HEPD (500 µM) for 2
h. EDTA (50 mM) followed by dithionite (10 mM) and D2O (20% v/v) were added and the 31P NMR
spectrum was recorded. The EDTA and dithionite were necessary to abrogate the line broadening effect
of a high concentration of paramagnetic Fe(III) in the sample. Additionally, (2R)-HPP (1.5 mM) was
incubated with HEPD (500 µM) for 2 h and the sample was analyzed for organic acids as described in
chapter 4.7.5.
4.7.8 Analysis of 1-Hydroxyethyl Phosphonates as Substrates for HEPD
As described in Notebook V, page 119, to identify the carbon-containing product of 1-
hydroxyethylphosphonate, HEPD (100 µM) was incubated in oxygenated buffer with 1-HEP (300 µM)
for 2 h at 25 °C. The organic acids were then derivatized and analyzed via LC-MS as described in
chapter 4.7.3.

94
As described in Notebook V, on pages 30-31, to identify the product of 1-HEP-CF3, HEPD (100 µM)
was incubated in oxygen saturated HEPES buffer with 1-HEP-CF3 (300 µM) for 2 h at 25 °C, followed
by addition of D2O (20% v/v) and recording of the 19F NMR spectrum. Standards of 1-HEP-CF3 and
sodium trifluoroacetate showed peaks at −72.5 ppm and −76.0 ppm, respectively.
As described in Notebook V on pages 127 and 129, to identify the phosphorus containing product, 1-
hydroxy ethylphosphonate or 2,2,2-trifluoro-1-hydroxyethylphosphonate (1.5 mM) were incubated with
HEPD (500 µM) for 2 h. EDTA (50 mM) followed by dithionite (10 mM) and D2O (20% v/v) were
added and the 31P NMR spectrum was recorded. Standards of 1-HEP and 1-HEP-CF3 under similar
conditions had chemical shifts of 19.2 ppm and 7.3 ppm, respectively. Phosphate, phosphite, and acetyl
phosphate under these conditions had chemical shifts of 2.3 ppm, 4 ppm, and −2.2 ppm respectively.
4.8 References
1. Kraut, D. A.; Carroll, K. S.; Herschlag, D., Ann. Rev. Biochem. 2003, 72, 517-571. Challenges in enzyme mechanism and energetics. 2. Smyth, T. P., Bioorg. Med. Chem. 2004, 12, (15), 4081-4088. Substrate variants versus transition state analogues as noncovalent reversible enzyme inhibitors. 3. Babtie, A.; Tokuriki, N.; Hollfelder, F., Curr. Opin. Chem. Biol. 2010, (Article in press), -. What makes an enzyme promiscuous? 4. Jameson, G. N. L.; Jin, W.; Krebs, C.; Perreira, A. S.; Tavares, P.; Liu, X.; Theil, E. C.; Huynh, B. H., Biochemistry 2002, 41, (45), 13435-13443. Stoichiometric Production of Hydrogen Peroxide and Parallel Formation of Ferric Multimers through Decay of the Diferric-Peroxo Complex, the First Detectable Intermediate in Ferritin Mineralization. 5. Gordon, N. J.; Evans, S. A., J. Org. Chem. 1993, 58, (17), 4516-4519. Acyl phosphates from acyl phosphonates - a novel baeyer villiger rearangement. 6. Lipmann, F.; Tuttle, L. C., J. Biol. Chem. 1944, 153, ((2)), 571-582. Acetyl phosphate: Chemistry, determination and synthesis. 7. Marolewski, A. E.; Mattia, K. M.; Warren, M. S.; Benkovic, S. J., Biochemistry 1997, 36, (22), 6709-6716. Formyl phosphate: A proposed intermediate in the reaction catalyzed by Escherichia coli PurT GAR transformylase.

95
8. Cicchillo, R. M.; Zhang, H.; Blodgett, J. A. V.; Whitteck, J. T.; Li, G.; Nair, S. K.; van der Donk, W. A.; Metcalf, W. W., Nature 2009, 459, (7248), 871-874. An unusual carbon-carbon bond cleavage reaction during phosphinothricin biosynthesis. 9. Higgins, L. J.; Yan, F.; Liu, P.; Liu, H.-w.; Drennan, C. L., Nature 2005, 437, (7060), 838-844. Structural insight into antibiotic fosfomycin biosynthesis by a mononuclear iron enzyme. 10. Yan, F.; Moon, S.-J.; Liu, P.; Zhao, Z.; Lipscomb, J. D.; Liu, A.; Liu, H.-w., Biochemistry 2007, 46, (44), 12628-12638. Determination of the Substrate Binding Mode to the Active Site Iron of (S)-2-Hydroxypropylphosphonic Acid Epoxidase Using 17O-Enriched Substrates and Substrate Analogues. 11. Ryerson, C. C.; Ballou, D. P.; Walsh, C., Biochemistry 1982, 21, (11), 2644-2655. Mechanistic studies on cyclohexanone oxygenase. 12. Nanne, M. K.; MariÎlle, J. H. M.; Jos, G. M. v. d. V.; Willem, J. H. v. B.; Marco, W. F.; Dick, B. J., Eur. J. Biochem. 2001, 268, (9), 2547-2557. 4-Hydroxyacetophenone monooxygenase from Pseudomonas fluorescens ACB. 13. Sheng, D.; Ballou, D. P.; Massey, V., Biochemistry 2001, 40, (37), 11156-11167. Mechanistic Studies of Cyclohexanone Monooxygenase: Chemical Properties of Intermediates Involved in Catalysis. 14. Malito, E.; Alfieri, A.; Fraaije, M. W.; Mattevi, A., Proc. Natl. Acad. Sci. U.S.A. 2004, 101, (36), 13157-13162. Crystal structure of a Baeyer-Villiger monooxygenase. 15. Shan, X.; Que, L., Proc. Natl. Acad. Sci. U.S.A. 2005, 102, (15), 5340-5345. Intermediates in the oxygenation of a nonheme diiron(II) complex, including the first evidence for a bound superoxo species. 16. Evans, J. P.; Blackburn, N. J.; Klinman, J. P., Biochemistry 2006, 45, (51), 15419-15429. The Catalytic Role of the Copper Ligand H172 of Peptidylglycine Hydroxylating Monooxygenase: A Kinetic Study of the H172A Mutant. 17. Chen, P.; Solomon, E. I., J. Am. Chem. Soc. 2004, 126, (15), 4991-5000. Oxygen Activation by the Noncoupled Binuclear Copper Site in Peptidylglycine-Hydroxylating Monooxygenase. Reaction Mechanism and Role of the Noncoupled Nature of the Active Site. 18. Chen, P.; Bell, J.; Eipper, B. A.; Solomon, E. I., Biochemistry 2004, 43, (19), 5735-5747. Oxygen Activation by the Noncoupled Binuclear Copper Site in Peptidylglycine-Hydroxylating Monooxygenase. Spectroscopic Definition of the Resting Sites and the Putative Cu(II)OOH Intermediate. 19. Humphreys, K. J.; Mirica, L. M.; Wang, Y.; Klinman, J. P., J. Am. Chem. Soc. 2009, 131, (13), 4657-4663. Galactose Oxidase as a Model for Reactivity at a Copper Superoxide Center. 20. Zhao, Z. B.; Liu, P. H.; Murakami, K.; Kuzuyama, T.; Seto, H.; Liu, H. W., Angew. Chem., Int. Ed. Engl. 2002, 41, (23), 4529-4530. Mechanistic studies of HPP epoxidase: Configuration of the substrate governs its enzymatic fate.

96
21. Capson, T. L.; Thompson, M. D.; Dixit, V. M.; Gaughan, R. G.; Poulter, C. D., J. Org. Chem. 1988, 53, (25), 5903-5908. Synthesis of ammonium analogs of carbocationic intermediates in the conversion of presqualene diphosphate to squalene. 22. Schweifer, A.; Hammerschmidt, F., Bioorg. Med. Chem. Lett. 2008, 18, (10), 3056-3059. On the conversion of structural analogues of (S)-2-hydroxypropylphosphonic acid to epoxides by the final enzyme of fosfomycin biosynthesis in S. fradiae. 23. Ridge, J. A.; Roberts, F.; Schaffer, M. H.; Stark, G. R., J. Biol. Chem. 1976, 251, (19), 5966-5975. Aspartate transcarbamylase of Escherichia coli. Heterogeneity of binding sites for carbamyl phosphate and fluorinated analogs of carbamyl phosphate. 24. Saunders, B. C.; Stacey, G. J., J. Chem. Soc. 1948, 1773-1779. 25. Isbell, A. F.; Englert, L. F.; Rosenberg, H., The Journal of Organic Chemistry 1969, 34, (3), 755-756. Phosphonoacetaldehyde. 26. Baraldi, P. G.; Guarneri, M.; Moroder, F.; Simoni, D.; Vicentini, C. B., Synthesis 1982, 1982, (01), 70-71. Synthesis of 3-Methyl-6-phenylamino-substituted-1H-pyrazolo[4,3-d]pyrimidine-7(6H)-ones. 27. Albert, D. B.; Martens, C. S., Marine Chemistry 1997, 56, (1-2), 27-37. Determination of low-molecular-weight organic acid concentrations in seawater and pore-water samples via HPLC.

97
APPENDIX A: CHEMISTRY OF 4-PHOSPHONOCARBOXYLIC ACIDS WITH HEPD
A.1 Glyphosate and 4-Phosphonobutyric Acid are Substrates for HEPD
Glyphosate (N-(phosphonomethyl)-glycine, 1) and 4-phosphonobutyric acid (PBA, 2) were
incubated in a 1:1 ratio with HEPD and the 31P NMR spectrum was recorded. In both assays the
31P resonance shifted upfield compared to substrate (from 12 ppm to 8 ppm for glyphosate and
from 28 ppm to 25 ppm for PBA). During analysis of the glyphosate reaction by LC-MS the
starting material was not observed. Instead a new peak was observed with m/z of 184, which
corresponds to starting material with loss of two hydrogen atoms and addition of one oxygen
atom. Two potential structures (3 and 4) can be drawn for the product of this reaction, which
differ only in the position of the ketone (Scheme A.1). During the same analysis of the PBA
reaction neither starting material nor oxidized product were observed. However, by analogy to
the glyphosate reaction, structures 5, 6 and 7 have been proposed for the products.
Scheme A.1 Potential products from glyphosate and PBA oxidation by HEPD under single
turnover conditions.
By analogy to the 31P NMR spectra of known phosphonates, structures 4 and 7 seem the most
likely products of glyphosate and PBA oxidation, respectively. Structures 3 and 5 are unlikely

98
since acylphosphonates tend to have a chemical shift in the range of ±3 ppm at neutral pH.1, 2
Structure 6 is unlikely since β-ketophosphonates tend to shift much further upfield than the
observed 25 ppm. For example, at pH = 7.5 the shifts of some known β-carbonylphosphonates
are: 2-oxopropylphosphonate (10.8 ppm), 2-oxobutylphosphonate (10 ppm),
phosphonoacetaldehyde (11 ppm) and phosphonoacetate (17 ppm). By process of elimination,
then, 4 and 7 are the most likely structures for the products.
While interesting, the observed oxidation of 4-phosphonocarbylic acids does not support either
of the two proposed mechanisms for HEPD, prima facie, as both mechanisms can account for the
observed products (Scheme A.2). The reaction proceeding via hydroxylation and formation of an
Fe(IV)=O intermediate is shown in Scheme A.2a. The Fe(IV)=O species could abstract a
hydrogen atom from the hydroxylated intermediate to afford a carbon centered radical and an
Fe(III)-OH species. The radical could then recombine with the iron species to afford an acetal
that would isomerize to afford the product. Alternatively, Scheme A.2b shows a
hydroperoxylated intermediate that could afford the observed product. Such hydroperoxylated or
peroxylated intermediates have been proposed for transition metal catalyzed oxidation of
benzylic3 and α-ester4 positions to ketones, though the mechanistic details remain unclear.
Interestingly, these mechanisms and chemistries are reminiscent of the reaction catalyzed by
myo-inositol oxygenase, which oxidizes an alcohol by two oxidation states to a carboxylic acid.5

99
Scheme A.2 Oxidation of 4-phosphonocarboxylic acids could proceed via (a) hydroxylation or
(b) hydroperoxylation.
This transformation is also of interest given the increased research into C-H functionalization
chemistry.6-9 Early research in the field of C-H functionalization identified so called “Gif”
chemistry, which utilizes ferric iron to catalyze selective oxidation of saturated secondary
hydrocarbons to ketones.10 More recently a designed system has been developed that utilizes
ferrous iron and can selectively hydroxylate tertiary C-H positions.11 Many of these studies are
either biomimetic or draw heavily from studies into the mechanisms of metalloenzymes for
design insights.12 Further study of the C-H oxidation catalyzed by HEPD may provide further
insight for use in solving synthetically important challenges.

100
A.2 Experimental
Glyphosate was purchased from Sigma-Aldrich and 4-phosphonobutyric acid was synthesized
according to a literature procedure.13 HEPD was overexpressed and purified according to the
procedure outlined in chapter 3. As detailed in Notebook V on page 47, in a typical assay, HEPD
(500 µM) and either glyphosate of PBA (500 µM) were incubated in oxygenated HEPES buffer
(25 mM, pH 7.5) for 2 h at 25 °C. Then the products were analyzed by 31P NMR spectroscopy
and LC-MS as detailed in chapter 4.7.4 and 4.7.3, respectively. The masses for protonated
glyphosate (m/z 170) and oxidized glyphosate (m/z 184) were extracted from the total ion
chromatogram (TIC) to afford a single peak in each extracted ion chromatogram (EIC) with
retention times of 6 and 5.5 min, respectively.
A.3 References
1. Katzhendler, J.; Ringel, I.; Karaman, R.; Zaher, H.; Breuer, E., J. Chem. Soc., Perkin Trans. 2 1997, 341-350. 2. Glabe, A. R.; Sturgeon, K. L.; Ghizzoni, S. B.; Musker, W. K.; Takahashi, J. N., J. Org. Chem. 1996, 61, (20), 7212-7216. Novel Functionalized Acylphosphonates as Phosphonoformate Analogs. 3. Velusamy, S.; Punniyamurthy, T., Tetrahedron Lett. 2003, 44, (50), 8955-8957. Copper(II)-catalyzed C---H oxidation of alkylbenzenes and cyclohexane with hydrogen peroxide. 4. Wentzel, B. B.; Donners, M. P. J.; Alsters, P. L.; Feiters, M. C.; Nolte, R. J. M., Tetrahedron 2000, 56, (39), 7797-7803. N-Hydroxyphthalimide/Cobalt(II) Catalyzed Low Temperature Benzylic Oxidation Using Molecular Oxygen. 5. Bollinger, J. M.; Diao, Y.; Matthews, M. L.; Xing, G.; Krebs, C., Dalton Trans. 2009, (6), 905-914. myo-Inositol oxygenase: a radical new pathway for O2 and C-H activation at a nonheme diiron cluster. 6. Diaz-Requejo, M. M.; Perez, P. J., Chem. Rev. 2008, 108, (8), 3379-3394. Coinage Metal Catalyzed C‚àíH Bond Functionalization of Hydrocarbons.

101
7. Lyons, T. W.; Sanford, M. S., Chem. Rev. 110, (2), 1147-1169. Palladium-Catalyzed Ligand-Directed C‚àíH Functionalization Reactions. 8. Gunay, A.; Theopold, K. H., Chem. Rev. 110, (2), 1060-1081. C-H Bond Activations by Metal Oxo Compounds. 9. Chen, M. S.; Prabagaran, N.; Labenz, N. A.; White, M. C., J. Am. Chem. Soc. 2005, 127, (19), 6970-6971. Serial Ligand Catalysis: A Highly Selective Allylic C‚àíH Oxidation. 10. Barton, D. H. R.; Doller, D., Acc. Chem. Res. 1992, 25, (11), 504-512. The selective functionalization of saturated hydrocarbons: Gif chemistry. 11. Chen, M. S.; White, M. C., Science 2007, 318, (5851), 783-787. A Predictably Selective Aliphatic C H Oxidation Reaction for Complex Molecule Synthesis. 12. Costas, M.; Mehn, M. P.; Jensen, M. P.; Que, L., Chem. Rev. 2004, 104, (2), 939-986. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. 13. Wasielewski, C. Ç.; Antczak, K., Synthesis 1981, 1981, (07), 540-541. Aminophosphonic Acids; XVI1. A New and Facile Synthesis of Phosphinothricine and 2-Amino-4-phosphonobutanoic Acid.

102
AUTHOR’S BIOGRAPHY
John T. Whitteck was born in Independence, MO (a suburb just outside Kansas City, MO) in
1982. He graduated from Truman High School in 2001 and then attended the University of
Missouri – Columbia. There he worked with Prof. Kent Gates on the synthesis and in vitro
evaluation of heterocyclic aromatic N-oxides as DNA damaging agents. Also during that time he
was able to visit the lab of Prof. Dr. Carsten Bolm at the RWTH Aachen for a summer then
attend the University of Stuttgart for a year. He graduated from the University of Missouri –
Columbia in 2005 with a B.S. in chemistry with departmental and general honors, summa cum
laude. He then attended the University of Illinois at Urbana-Champaign. There he studied under
Prof. Wilfred van der Donk studying phosphonate biosynthesis. He graduated in 2010 after
accepting a position at Dow AgroSciences in Indianapolis, IN.





























