Embed Size (px)
Citation preview

WFS1 mutations are frequent monogenic causesof juvenile-onset diabetes mellitus in Lebanon
Pierre A. Zalloua1,2,3, Sami T. Azar2,3, Marc Delepine4, Nadine J. Makhoul1, Herve Blanc5,
May Sanyoura1, Anne Lavergne5, Karmen Stankov6, Arnaud Lemainque4,
Patrick Baz2,7 and Cecile Julier5,6,�
1Lebanese American University, School of Medicine, Chouran, Beirut 1102 2801, Lebanon, 2Chronic Care Center,
Hazmieh, Lebanon, 3Division of Endocrinology, American University of Beirut, Bliss St, Beirut, Lebanon, 4Centre
National de Genotypage, Institut de Genomique, Commissariat a l’Energie Atomique, 2 rue Gaston Cremieux, CP
5721, 91057 Evry Cedex, France, 5Genetics of Infectious and Autoimmune Diseases, Institut Pasteur, 75015 Paris,
France, 6Genetics of Diabetes, Inserm U730, Centre National de Genotypage, 2 rue Gaston Cremieux, CP 5721,
91057 Evry Cedex, France and 7Department of Ophthalmology, Hotel Dieu Hospital, Beirut, Lebanon
Received August 11, 2008; Revised September 8, 2008; Accepted September 16, 2008
Most cases of juvenile-onset diabetes (JOD) are diagnosed as type 1 diabetes (T1D), for which genetic studiesconducted in outbred Caucasian populations support the concept of multifactorial inheritance. However, thisview may be partly challenged in particular population settings. In view of the suggestive evidence for a highprevalence of Wolfram syndrome (WFS) in Lebanon, the phenotypic variability associated with WFS1mutations, and the high consanguinity rate in Lebanon, we aimed to evaluate the contribution of WFS1mutations as monogenic determinants to JOD in Lebanon. We performed a family-based genetic study,with linkage analysis followed by systematic mutation screening of WFS1 exons in all JOD probands. Thestudy population consisted of an unbiased recruitment of all juvenile-onset insulin-dependent diabeticpatients from a specialized diabetes pediatric clinic in Beirut, Lebanon. Homozygous or compound heterozy-gous WFS1 mutations were found in 22 of the 399 JOD probands (5.5%), resulting in WFS (17 probands) or innon-syndromic non-autoimmune diabetes mellitus (DM, five probands). These accounted for 12.1% (21/174)of probands in consanguineous families, compared with 0.4% (1/225) in non-consanguineous families. Ofthe 38 patients identified with homozygous or compound heterozygous WFS1 mutations, 11 (29%) hadnon-syndromic DM, all of whom carried a particular WFS1 mutation, WFS1LIB, encoding a protein with anextended C-terminal domain. This mutation resulted in a delayed onset or absence of extrapancreaticfeatures. These results underscore the major impact of population-specific factors, such as population-specific mutations and founder effects, and family structure in the genetic determinism of JOD.
INTRODUCTION
Type 1 diabetes (T1D) is among the most common chronicchildhood diseases in Caucasian populations. Its etiology isthought to be principally autoimmune, resulting in the destruc-tion of insulin-producing pancreatic b cells, leading to com-plete dependence on insulin replacement therapy generallyat a young age. Epidemiologic and genetic studies supportthe hypothesis of multifactorial contributions, with genetic
and environmental risk factors (1). Genes located within themajor histocompatibility complex locus are major riskfactors of T1D, and genome-wide linkage and associationscans in outbred Caucasian populations consistently supportthe model of multiple additional genes with minor contri-bution to T1D risk (2,3). Hence, monogenic factors areunlikely to play a major role in T1D in these populations.However, the extent of monogenic contributions may varyconsiderably between populations, depending on ethnic or
�To whom correspondence should be addressed. Genetics of Diabetes, Inserm U730, Centre National de Genotypage, 2 rue Gaston Cremieux, CP 5721,91057 Evry Cedex, France. Tel: þ33 160878330; Fax: þ33 160878383; Email: [email protected]
# The Author 2008. Published by Oxford University Press. All rights reserved.For Permissions, please email: [email protected]
Human Molecular Genetics, 2008, Vol. 17, No. 24 4012–4021doi:10.1093/hmg/ddn304Advance Access published on September 20, 2008
at Chinese A
cademy of M
edical Sciences on M
ay 1, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

population-specific factors, including founder effects andthe presence of consanguinity. They may also vary betweenfamilies, depending on their structure (presence or not of mul-tiple affected siblings, and inter-parental consanguinity).
Several rare recessive syndromes in which insulin-dependent juvenile diabetes is associated with other clinicalmanifestations have been described, including Wolframsyndrome (WFS), Wolcott–Rallison syndrome and thiamine-responsive megaloblastic anemia (4). Although these syn-dromes are very rare in outbred Caucasian populations, theirincidence may be increased in some populations or contexts,due to founder effects and/or the high frequency of consangui-neous marriages. WFS (DIDMOAB, OMIM 222300) ischaracterized by juvenile-onset diabetes (JOD) mellitus,optic atrophy (OA), diabetes insipidus (DI) and sensorineuraldeafness, and is caused by recessive mutations in the Wolfra-min gene (WFS1) (5,6). It is generally considered to be veryrare, with an estimated prevalence of 1/770 000 in UK (7).However, based on previous reports, the prevalence of thissyndrome may be much higher in some countries, includingLebanon (8). In addition to WFS, WFS1 mutations andgenetic variants have been implicated in low-frequency non-syndromic hearing loss (dominant mutations) and psychiatricdiseases (9,10), and common WFS1 variants have recentlybeen shown to be associated with type 2 diabetes (T2D)(11,12). In view of the large phenotypic variability associatedwith WFS1 mutations with regard to diabetes and extrapan-creatic manifestations, the possibility of some geneticoverlap between typical WFS and non-syndromic diabetescould also be anticipated. In this study, we aimed to evaluatethe contribution of WFS1 mutations and variants in syndromicand non-syndromic JOD in Lebanese families.
RESULTS
Strong contribution of WFS1 locus to JOD in Lebanon inWFS and non-syndromic DM detected by linkage analysis
We studied 408 nuclear families from Lebanon, which hadbeen ascertained through a patient with insulin-dependentJOD, with a total of 455 JOD patients, including non-syndromic and syndromic cases. Twenty seven patients from17 of these families had clinical features characteristic ofWFS (Tables 1 and 2). The high frequency of WFS patientsamong Lebanese JOD probands (17/408, or 4.2%), based onour survey, confirms the high prevalence of this syndrome inLebanon that had been suggested in a previous study (8).
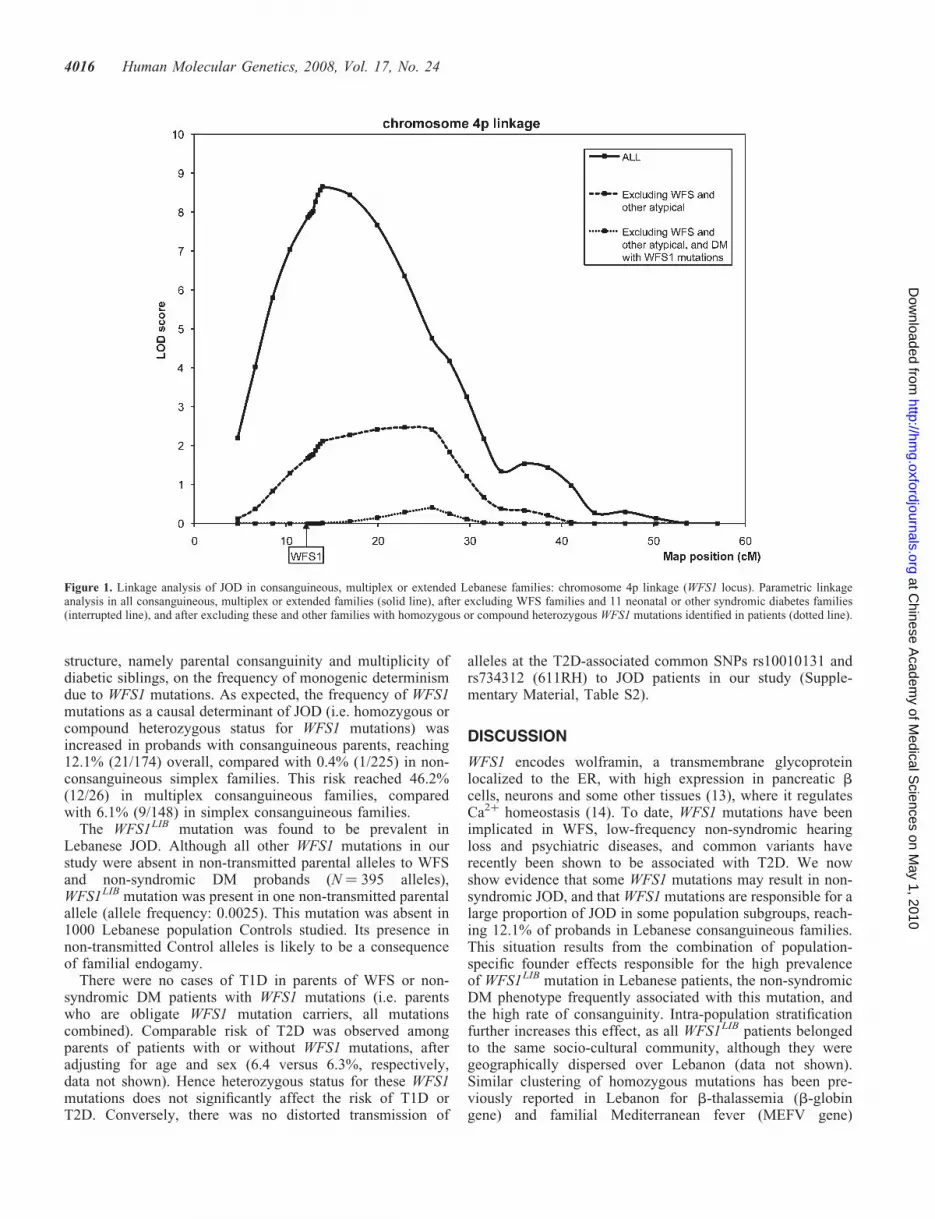
Linkage analysis performed under a highly penetrant rarerecessive model in all multiplex, consanguineous or extendedfamilies showed highly significant evidence of linkagenear the WFS1 locus on chromosome 4p (Fig. 1), with LOD(logarithm of odds) scores with heterogeneity of 8.66(estimated proportion of linked families: a ¼ 13.8%). Afterexcluding WFS families and the 11 neonatal or other syndro-mic diabetes families, there was evidence of residual linkageat WFS1 locus (LOD ¼ 2.47, a ¼ 7.1%) (Fig. 1), suggestingeither that some WFS patients may have been underdiagnosed,or that some WFS1 mutations may result in an incompletephenotype restricted to diabetes, or that frequent variants in
the WFS1 gene or another gene near WFS1 may contributeto diabetes susceptibility in Lebanon.
WFS1 mutations in WFS and non-syndromic DM patients
By sequencing WFS1 exons in WFS and non-syndromic DMprobands and their parents from multiplex, consanguineousor extended families, and in probands from simplex non-consanguineous families, we identified 173 WFS1 DNAvariants, 46 of which were predicted to affect the primarysequence of the protein, 38 of which were novel (Supple-mentary Material, Table S2).
In the WFS patients, we identified 10 variants compatiblewith the status of WFS mutations, nine of which were novel(Table 1). All the WFS patients carried a homozygousmutation (or a combination of two mutations in completelinkage disequilibrium) that was predicted to have deleteriousconsequences on the protein function, as it resulted in a trun-cated protein, deleted one amino acid or was predicted to bedamaging based on PolyPhen analysis. A double mutation,WFS1LIB, associating the 707VI non-synonymous changeand the F884fs951X frameshift on the same haplotype, washomozygous in 7/17 (41.2%) of the WFS probands, and10/27 of all WFS patients. F884fs951X frameshift affectsthe most C-terminal endoplasmic reticulum (ER) luminaldomain, replacing the last seven amino acids by 67 otheramino acids, while 707VI is predicted to have no majorfunctional consequence on protein function.
In non-syndromic DM patients, two variants were compatiblewith the status of mutations, based on our criteria (Methods):WFS1LIB and F646fs708X (Supplementary Material,Table S2). WFS1LIB was homozygous in four non-syndromicDM probands (10 non-syndromic DM patients) and compoundheterozygous with F646fs708X in one non-syndromic DMproband (Table 2). Re-analysis of linkage data after excludingthe corresponding families cancelled the residual linkagepeak near WFS1, indicating that WFS1 mutations in WFS andnon-syndromic DM patients accounted for the totality of theevidence of linkage detected at WFS1 locus (Fig. 1).
WFS1LIB mutation is associated with delayedor absent extrapancreatic manifestations
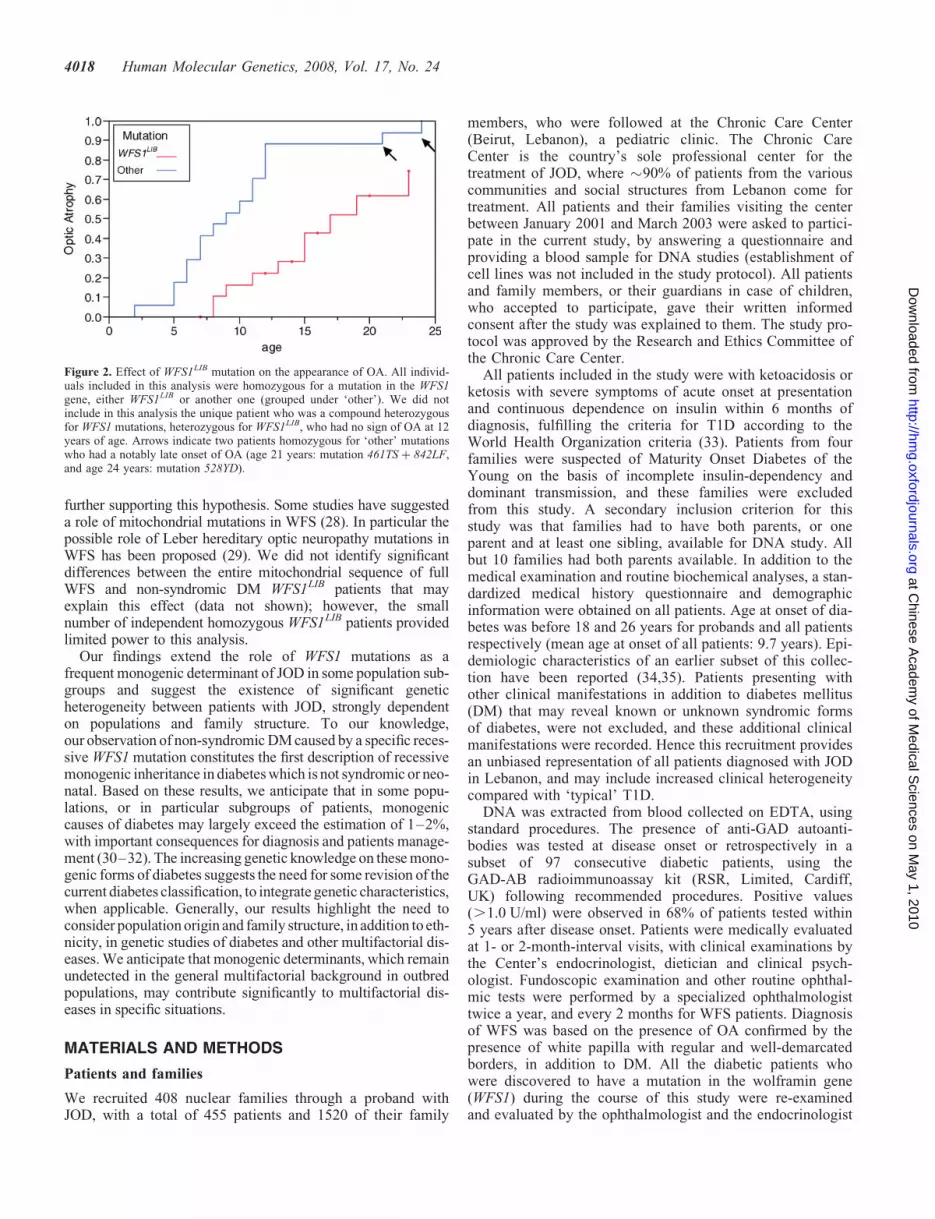
Of the 38 JOD patients from our study population diagnosedwith homozygous or compound heterozygous WFS1mutations, 11 (27%) had non-syndromic DM; 11/21 (52.4%)of the patients homozygous or compound heterozygous forWFS1LIB mutation had only DM, without any evidence ofOA, DI or deafness, compared with none (0/17) of the patientshomozygous for any other mutation (Table 2). Using logisticregression analysis, we showed that the absence of extrapan-creatic manifestations was dependant on the age of the patientsat last examination (P ¼ 0.036) and on the nature of themutation (P ¼ 0.0001) (Table 3A). In a Kaplan–Meier analy-sis, we showed that the appearance of OA was significantlydelayed in patients homozygous for the WFS1LIB mutationcompared with those homozygous for other WFS1 mutations(Fig. 2, P ¼ 0.0003). Patients homozygous for the WFS1LIB
mutation exhibited reduced expression of other extrapancrea-tic features, including hearing impairment, DI, cataract and
Human Molecular Genetics, 2008, Vol. 17, No. 24 4013
at Chinese A
cademy of M
edical Sciences on M
ay 1, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from
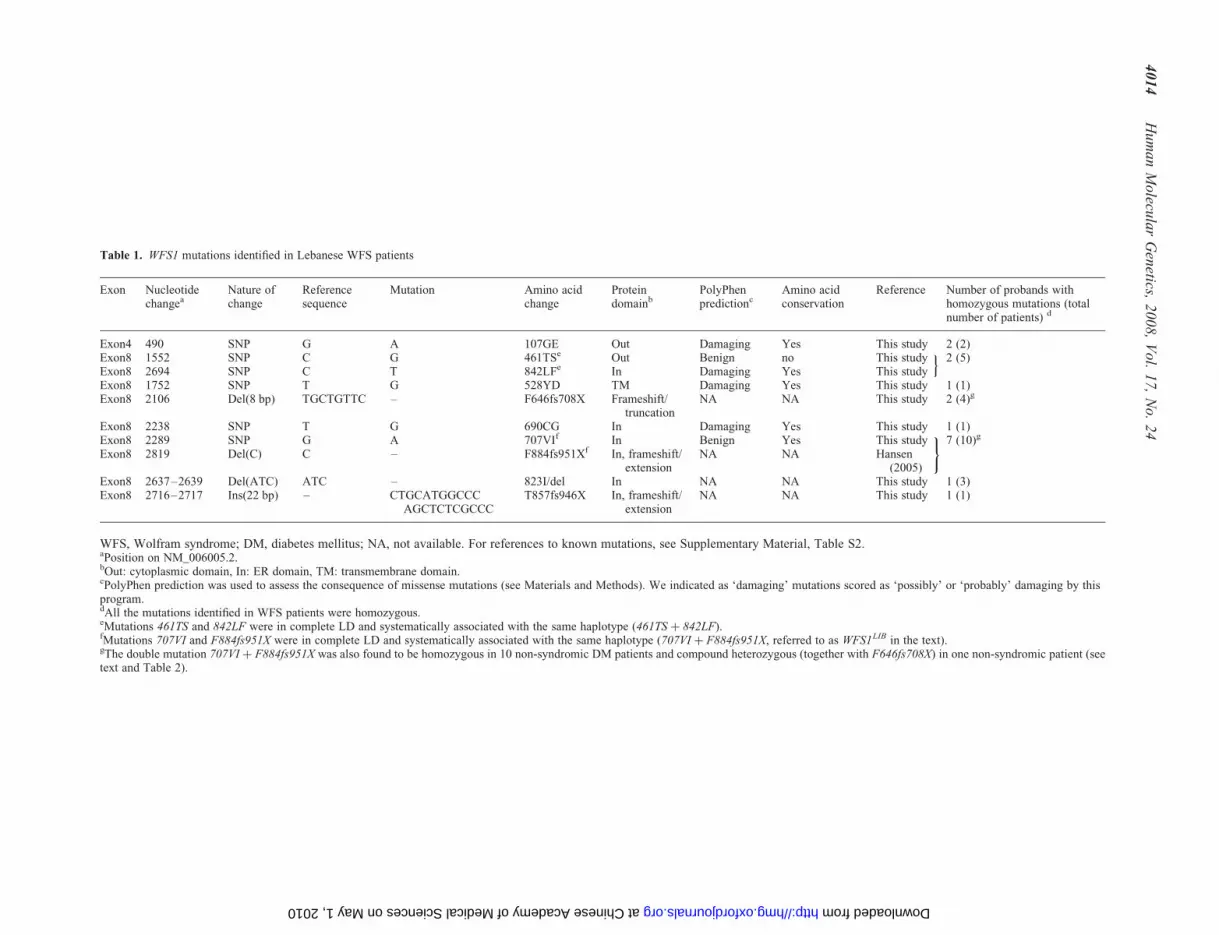
Table 1. WFS1 mutations identified in Lebanese WFS patients
Exon Nucleotidechangea
Nature ofchange
Referencesequence
Mutation Amino acidchange
Proteindomainb
PolyPhenpredictionc
Amino acidconservation
Reference Number of probands withhomozygous mutations (totalnumber of patients) d
Exon4 490 SNP G A 107GE Out Damaging Yes This study 2 (2)Exon8 1552 SNP C G 461TSe Out Benign no This study 2 (5)Exon8 2694 SNP C T 842LFe In Damaging Yes This study
oExon8 1752 SNP T G 528YD TM Damaging Yes This study 1 (1)Exon8 2106 Del(8 bp) TGCTGTTC – F646fs708X Frameshift/
truncationNA NA This study 2 (4)g
Exon8 2238 SNP T G 690CG In Damaging Yes This study 1 (1)Exon8 2289 SNP G A 707VIf In Benign Yes This study 7 (10)g
Exon8 2819 Del(C) C – F884fs951Xf In, frameshift/extension
NA NA Hansen(2005)
)
Exon8 2637–2639 Del(ATC) ATC – 823I/del In NA NA This study 1 (3)Exon8 2716–2717 Ins(22 bp) – CTGCATGGCCC
AGCTCTCGCCCT857fs946X In, frameshift/
extensionNA NA This study 1 (1)
WFS, Wolfram syndrome; DM, diabetes mellitus; NA, not available. For references to known mutations, see Supplementary Material, Table S2.aPosition on NM_006005.2.bOut: cytoplasmic domain, In: ER domain, TM: transmembrane domain.cPolyPhen prediction was used to assess the consequence of missense mutations (see Materials and Methods). We indicated as ‘damaging’ mutations scored as ‘possibly’ or ‘probably’ damaging by thisprogram.dAll the mutations identified in WFS patients were homozygous.eMutations 461TS and 842LF were in complete LD and systematically associated with the same haplotype (461TS þ 842LF).fMutations 707VI and F884fs951X were in complete LD and systematically associated with the same haplotype (707VI þ F884fs951X, referred to as WFS1LIB in the text).gThe double mutation 707VI þ F884fs951X was also found to be homozygous in 10 non-syndromic DM patients and compound heterozygous (together with F646fs708X) in one non-syndromic patient (seetext and Table 2).
40
14
Hu
ma
nM
olecu
lar
Gen
etics,
20
08
,V
ol.
17
,N
o.
24
at Chinese Academy of Medical Sciences on May 1, 2010 http://hmg.oxfordjournals.org Downloaded from

neuropsychiatric complications, with the possible exception ofepilepsy, showing the opposite trend (Table 3A); in contrast,ages at onset of diabetes were similar in both groups(WFS1LIB: 5.95, others: 5.24, P ¼ 0.32) (Table 3B). Withthe exception of epilepsy reported in one patient, non-syndromic DM patients with WFS1 mutations had no extra-pancreatic features, at age up to 23 years. Glutamic acid dec-arboxylase (GAD) autoantibody level was negative in allpatients with WFS1 mutations tested, seven WFS patientsand two non-syndromic DM patients, supporting that thedisease process is non-autoimmune in all cases.
Among WFS1LIB homozygous individuals, the risk ofhaving WFS (compared with non-syndromic DM) wasfamily dependent (P ¼ 0.002, after correcting for the age atlast examination). This observation could not be accountedfor by other WFS1 variants, since the entire WFS1 sequence
was identical in all the WFS1LIB haplotypes. This suggeststhe contribution of additional factors to the clinical outcomeof WFS1LIB patients, which may be variants located inflanking regions of the WFS1 gene. Alternatively, this couldbe explained by other genetic or environmental factors thatmay be shared within families; the role of a differential diag-nosis by family is unlikely, since all patients were examinedby the same ophthalmologist.
Impact of family structure on the risk ofWFS1-monogenic contribution
Based on our sequence screening survey, 5.5% (22/399) of allLebanese JOD probands (including all WFS and non-WFSsyndromic cases) were homozygous or compound heterozy-gous for WFS1 mutations. We tested the impact of family
Table 2. Clinical characteristics and nature of WFS1 mutations in Lebanses WFS and non-syndromic DM patients carrying WFS1 mutations
Family Patient Clinicaldiagnosis
Mutationa Age atonset ofdiabetes
Age at lastexamination
OA Age atonsetof OA
Deafness Age atonset ofdeafness
DI Age atonsetof DI
Cataract Epilepsy
LIB1 1 WFS 461TS þ 842LF 4 29 Yes 8 No – No – Yes NoLIB1 2 WFS 461TS þ 842LF 6 31 Yes 21 Yes 24 Yes 24 Yes NoLIB1 3 WFS 461TS þ 842LF 7 33 Yes 11 Yes 20 Yes 18 No NoLIB1 4 WFS 461TS þ 842LF 6 24 Yes 9 Yes – No – No NoLIB2 1 WFS 707VI þ F884fs951X 7 32 Yes 23 No – No – Yes YesLIB2 2 WFS 707VI þ F884fs951X 10 24 Yes 17 Yes 17 No – No NoLIB3 1 WFS F646fs708X 5 19 Yes 12 Yes 8 Yes 12 No NoLIB3 2 WFS F646fs708X 3 15 Yes 6 No – No No NoLIB4 1 WFS 107GE 4 23 Yes 5 Yes 1 Yes 2 Yes NoLIB5 1 WFS 707VI þ F884fs951X 7 29 Yes 19 No – Yes 24 No YesLIB5 2 WFS 707VI þ F884fs951X 4 21 Yes 13 No – No – No YesLIB6 1 WFS 823I/del 2 16 Yes 7 No – No – No NoLIB6 2 WFS 823I/del 1 13 Yes 2 No – No – Yes NoLIB6 3 WFS 823I/del 5 11 Yes 7 No – No – No NoLIB7 1 WFS F646fs708X 7 16 Yes 11 No – Yes 13 No NoLIB7 2 WFS F646fs708X 5 9 Yes 5 No – No – No NoLIB8 1 WFS 461TS þ 842LF 4 16 Yes 12 No – Yes 12 No NoLIB9 1 WFS 707VI þ F884fs951X 7 20 Yes 15 No – No – No NoLIB9 2 WFS 707VI þ F884fs951X 8 19 Yes 9 No – No – No NoLIB10 1 WFS 707VI þ F884fs951X 6 22 Yes 15 No – Yes 21 No NoLIB11 1 WFS 107GE 11 21 Yes 12 Yes 13 Yes 14 No YesLIB12 1 WFS 690CG 5 19 Yes 10 No – No – No NoLIB13 1 WFS 528YD 9 33 Yes 24 Yes 24 Yes 25 Yes NoLIB14 1 WFSb 707VI þ F884fs951X 3 12 Yes 11 No – No – No NoLIB14 2 DM 707VI þ F884fs951X 3 8 No – No – No – No NoLIB15 1 WFS 707VI þ F884fs951X 7 15 Yes 8 No – Yes 7 No YesLIB16 1 WFS T857fs946X 5 12 Yes 6 No – No – No NoLIB17 1 WFSb 707VI þ F884fs951X 6 13 Yes 8 No – No – No NoLIB18 1 DM 707VI þ F884fs951X 7 14 No – No – No – No NoLIB18 2 DM 707VI þ F884fs951X 5 12 No – No – No – No NoLIB18 3 DM 707VI þ F884fs951X 5 9 No – No – No – No NoLIB19 1 DM 707VI þ F884fs951X 3 23 No – No – No – No NoLIB19 2 DM 707VI þ F884fs951X 7 20 No – No – No – No NoLIB19 3 DM 707VI þ F884fs951X 5 16 No – No – No – No NoLIB20 1 DM 707VI þ F884fs951X 8 16 No – No – No – No NoLIB20 2 DM 707VI þ F884fs951X 4 7 No – No – No – No NoLIB21 1 DM 707VI þ F884fs951X 7 23 No – No – No – No YesLIB22 1 DM 707VI þ F884fs951X/
F646fs708X6 12 No – No – No – No No
WFS, Wolfram syndrome; DM, diabetes mellitus; OA, optic atrophy; DI, diabetes insipidus. For references to known mutations, see SupplementaryMaterial, Table S2.aWith the exception of LIB22-1 patient, all the mutations were found in the homozygous status.bThe initial diagnosis of these patients was DM, and was revised to WFS as OA developed during the course of this study.
Human Molecular Genetics, 2008, Vol. 17, No. 24 4015
at Chinese A
cademy of M
edical Sciences on M
ay 1, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from
structure, namely parental consanguinity and multiplicity ofdiabetic siblings, on the frequency of monogenic determinismdue to WFS1 mutations. As expected, the frequency of WFS1mutations as a causal determinant of JOD (i.e. homozygous orcompound heterozygous status for WFS1 mutations) wasincreased in probands with consanguineous parents, reaching12.1% (21/174) overall, compared with 0.4% (1/225) in non-consanguineous simplex families. This risk reached 46.2%(12/26) in multiplex consanguineous families, comparedwith 6.1% (9/148) in simplex consanguineous families.
The WFS1LIB mutation was found to be prevalent inLebanese JOD. Although all other WFS1 mutations in ourstudy were absent in non-transmitted parental alleles to WFSand non-syndromic DM probands (N ¼ 395 alleles),WFS1LIB mutation was present in one non-transmitted parentalallele (allele frequency: 0.0025). This mutation was absent in1000 Lebanese population Controls studied. Its presence innon-transmitted Control alleles is likely to be a consequenceof familial endogamy.
There were no cases of T1D in parents of WFS or non-syndromic DM patients with WFS1 mutations (i.e. parentswho are obligate WFS1 mutation carriers, all mutationscombined). Comparable risk of T2D was observed amongparents of patients with or without WFS1 mutations, afteradjusting for age and sex (6.4 versus 6.3%, respectively,data not shown). Hence heterozygous status for these WFS1mutations does not significantly affect the risk of T1D orT2D. Conversely, there was no distorted transmission of
alleles at the T2D-associated common SNPs rs10010131 andrs734312 (611RH) to JOD patients in our study (Supple-mentary Material, Table S2).
DISCUSSION
WFS1 encodes wolframin, a transmembrane glycoproteinlocalized to the ER, with high expression in pancreatic bcells, neurons and some other tissues (13), where it regulatesCa2þ homeostasis (14). To date, WFS1 mutations have beenimplicated in WFS, low-frequency non-syndromic hearingloss and psychiatric diseases, and common variants haverecently been shown to be associated with T2D. We nowshow evidence that some WFS1 mutations may result in non-syndromic JOD, and that WFS1 mutations are responsible for alarge proportion of JOD in some population subgroups, reach-ing 12.1% of probands in Lebanese consanguineous families.This situation results from the combination of population-specific founder effects responsible for the high prevalenceof WFS1LIB mutation in Lebanese patients, the non-syndromicDM phenotype frequently associated with this mutation, andthe high rate of consanguinity. Intra-population stratificationfurther increases this effect, as all WFS1LIB patients belongedto the same socio-cultural community, although they weregeographically dispersed over Lebanon (data not shown).Similar clustering of homozygous mutations has been pre-viously reported in Lebanon for b-thalassemia (b-globingene) and familial Mediterranean fever (MEFV gene)
Figure 1. Linkage analysis of JOD in consanguineous, multiplex or extended Lebanese families: chromosome 4p linkage (WFS1 locus). Parametric linkageanalysis in all consanguineous, multiplex or extended families (solid line), after excluding WFS families and 11 neonatal or other syndromic diabetes families(interrupted line), and after excluding these and other families with homozygous or compound heterozygous WFS1 mutations identified in patients (dotted line).
4016 Human Molecular Genetics, 2008, Vol. 17, No. 24
at Chinese A
cademy of M
edical Sciences on M
ay 1, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

(15,16), providing strong evidence for the combined impact ofconsanguinity and founder effect, resulting from the religiousand socio-cultural ethnic endogamy in the Lebanese popu-lation. Hence, the impact of WFS1 mutations on JOD isstrongly dependent on population-specific factors, rangingfrom almost none in outbred Caucasians patients (7), consist-ent with the absence of genome-wide significant linkage andSNP association observed at WFS1 locus in outbred Caucasianpopulations (16,17), to very high in Lebanese consanguineousfamilies, and especially those with multiple affected siblings.WFS1 mutations may also have a strong impact in other popu-lations or patients’ subgroups with high frequency of autoanti-body negative T1D. Interestingly, association of commonpopulation-specific WFS1 variants with T1D has been reportedin Japan (18), although the role of monogenic determinantshas not been demonstrated in this population.
Although we were able to show that WFS1LIB mutation isassociated with significant delay in the appearance of OA,compared with other WFS1 mutations, a longer follow-up ofthese patients, or a specific study of adult patient populations,would be needed in order to determine if a subset of the non-syndromic WFS1LIB patients may be exempted from extrapan-creatic manifestations during their lifetime. Remarkably, someof these patients were still with non-syndromic DM, with nosign of OA and full visual acuity, up to 23 years of age.
A mild WFS phenotype has been previously described intwo siblings homozygous for F884fs951X, included in theWFS1LIB mutation (707VI mutation was newly identified inour study and was not described in these patients), who hadDM and OA, but no deafness, DI or neurologic abnormalities(19). A compound heterozygous F884fs951X mutation hasalso been reported in another WFS patient who had no deaf-ness or DI (20). WFS patients carrying a homozygous 4 bp
deletion with similar protein consequences (F883fs950X)also had no DI or deafness (21,22). All these patients hadOA and had been diagnosed as having WFS. Two hypothesesmay explain the selective mild WFS/non-syndromic DM fea-tures of WFS1LIB mutation: (i) b cells may be more sensitivethan other cells to the pathway affected in WFS; b cells arehighly secreting cells, which makes them particularly sensitiveto ER stress (23), a proposed mechanism by which WFS1mutations may lead to WFS (24,25). This may result in anobligate diabetes phenotype, but no or delayed extrapancreaticfeatures in case of ‘mild’ mutations; (ii) the C-terminal regionof wolframin affected in the WFS1LIB mutation may be criticalfor b-cell-specific function. Recently, this region has beenshown to interact with Naþ/Kþ ATPase b1 subunit, whichis critical for b cell function and may involve mechanismsthat are partly independent of ER stress pathways (26).Evidence for genotype/phenotype correlation on the clinicalvariability of WFS was shown in a recent meta-analysis, with adose–effect association of the number of inactivating mutationson disease severity (20). However, the effect of specific mutationson this variability could not be detected in the previous study,owing to the large variety of mutations. In addition, theWFS1LIB mutation, which would have been considered as aninactivating mutation in this study (frameshift) is actually associ-ated with a milder disease phenotype. Additional molecularstudies in patients and Controls cell lines will be needed to deter-mine the underlying mutation-dependant mechanisms resultingin variable clinical expression.
The full WFS or non-syndromic DM expression of WFS1LIB
showed familial clustering, suggesting the contribution ofadditional modifier factors, most likely genetic; Wfs1-deficientmice appear to have a ‘milder’ disease than human WFS (27),as extrapancreatic features have not been described to date,
Table 3. Effect of the nature of the mutation on clinical manifestations of WFS
(A) Extrapancreatic clinical manifestations in patients homozygous for WFS1 mutations
Trait Number positive for the trait (%) P-valuea
WFS1LIB (N ¼ 20) Other (N ¼ 17)
Optic atrophy 10 (50%) 17 (100%) 0.0001Hearing impairment 1 (5%) 7 (41.2%) 0.008Diabetes insipidus 3 (15%) 8 (47.1%) 0.06Cataract 1 (5%) 5 (29.4%) 0.08Epilepsy 5 (25%) 1 (5.9%) 0.03Neuropsychiatric complicationsb 3 (15%) 9 (52.9%) 0.01
(B) Age at onset of diabetes
Phenotype (number) Homozygous for WFS1LIB mutation;numbers, mean (95% CI)
Homozygous for ‘other’ mutations;numbers, mean (95% CI)
P-valuec
WFS (N ¼ 27) N ¼ 10, 6.50 (5.03–7.97) N ¼ 17, 5.24 (4.11–6.36) 0.17DM (N ¼ 10) N ¼ 10, 5.40 (4.13–6.67) N ¼ 0All (N ¼ 37) N ¼ 20, 5.95 (4.97–6.93) N ¼ 17, 5.24 (4.18–6.29) 0.32
Notes: All individuals were homozygous for WFS1 mutations, either WFS1LIB, or all others grouped (other). We excluded one compound heterozygousWFS1LIB/other from this analysis.CI, confidence interval.aLogistic regression test (likelihood ratio test), taking into account the age at last examination.bNeuropsychiatric complications were: memory loss, disorientation to time, place or person, delusions or hallucinations, or confusion.cANOVA test comparing patients homozygous for WFS1LIB with those homozygous for other mutations.
Human Molecular Genetics, 2008, Vol. 17, No. 24 4017
at Chinese A
cademy of M
edical Sciences on M
ay 1, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from
further supporting this hypothesis. Some studies have suggesteda role of mitochondrial mutations in WFS (28). In particular thepossible role of Leber hereditary optic neuropathy mutations inWFS has been proposed (29). We did not identify significantdifferences between the entire mitochondrial sequence of fullWFS and non-syndromic DM WFS1LIB patients that mayexplain this effect (data not shown); however, the smallnumber of independent homozygous WFS1LIB patients providedlimited power to this analysis.
Our findings extend the role of WFS1 mutations as afrequent monogenic determinant of JOD in some population sub-groups and suggest the existence of significant geneticheterogeneity between patients with JOD, strongly dependenton populations and family structure. To our knowledge,our observation of non-syndromic DM caused by a specific reces-sive WFS1 mutation constitutes the first description of recessivemonogenic inheritance in diabetes which is not syndromic or neo-natal. Based on these results, we anticipate that in some popu-lations, or in particular subgroups of patients, monogeniccauses of diabetes may largely exceed the estimation of 1–2%,with important consequences for diagnosis and patients manage-ment (30–32). The increasing genetic knowledge on these mono-genic forms of diabetes suggests the need for some revision of thecurrent diabetes classification, to integrate genetic characteristics,when applicable. Generally, our results highlight the need toconsider population origin and family structure, in addition to eth-nicity, in genetic studies of diabetes and other multifactorial dis-eases. We anticipate that monogenic determinants, which remainundetected in the general multifactorial background in outbredpopulations, may contribute significantly to multifactorial dis-eases in specific situations.
MATERIALS AND METHODS
Patients and families
We recruited 408 nuclear families through a proband withJOD, with a total of 455 patients and 1520 of their family
members, who were followed at the Chronic Care Center(Beirut, Lebanon), a pediatric clinic. The Chronic CareCenter is the country’s sole professional center for thetreatment of JOD, where �90% of patients from the variouscommunities and social structures from Lebanon come fortreatment. All patients and their families visiting the centerbetween January 2001 and March 2003 were asked to partici-pate in the current study, by answering a questionnaire andproviding a blood sample for DNA studies (establishment ofcell lines was not included in the study protocol). All patientsand family members, or their guardians in case of children,who accepted to participate, gave their written informedconsent after the study was explained to them. The study pro-tocol was approved by the Research and Ethics Committee ofthe Chronic Care Center.
All patients included in the study were with ketoacidosis orketosis with severe symptoms of acute onset at presentationand continuous dependence on insulin within 6 months ofdiagnosis, fulfilling the criteria for T1D according to theWorld Health Organization criteria (33). Patients from fourfamilies were suspected of Maturity Onset Diabetes of theYoung on the basis of incomplete insulin-dependency anddominant transmission, and these families were excludedfrom this study. A secondary inclusion criterion for thisstudy was that families had to have both parents, or oneparent and at least one sibling, available for DNA study. Allbut 10 families had both parents available. In addition to themedical examination and routine biochemical analyses, a stan-dardized medical history questionnaire and demographicinformation were obtained on all patients. Age at onset of dia-betes was before 18 and 26 years for probands and all patientsrespectively (mean age at onset of all patients: 9.7 years). Epi-demiologic characteristics of an earlier subset of this collec-tion have been reported (34,35). Patients presenting withother clinical manifestations in addition to diabetes mellitus(DM) that may reveal known or unknown syndromic formsof diabetes, were not excluded, and these additional clinicalmanifestations were recorded. Hence this recruitment providesan unbiased representation of all patients diagnosed with JODin Lebanon, and may include increased clinical heterogeneitycompared with ‘typical’ T1D.
DNA was extracted from blood collected on EDTA, usingstandard procedures. The presence of anti-GAD autoanti-bodies was tested at disease onset or retrospectively in asubset of 97 consecutive diabetic patients, using theGAD-AB radioimmunoassay kit (RSR, Limited, Cardiff,UK) following recommended procedures. Positive values(.1.0 U/ml) were observed in 68% of patients tested within5 years after disease onset. Patients were medically evaluatedat 1- or 2-month-interval visits, with clinical examinations bythe Center’s endocrinologist, dietician and clinical psych-ologist. Fundoscopic examination and other routine ophthal-mic tests were performed by a specialized ophthalmologisttwice a year, and every 2 months for WFS patients. Diagnosisof WFS was based on the presence of OA confirmed by thepresence of white papilla with regular and well-demarcatedborders, in addition to DM. All the diabetic patients whowere discovered to have a mutation in the wolframin gene(WFS1) during the course of this study were re-examinedand evaluated by the ophthalmologist and the endocrinologist
Figure 2. Effect of WFS1LIB mutation on the appearance of OA. All individ-uals included in this analysis were homozygous for a mutation in the WFS1gene, either WFS1LIB or another one (grouped under ‘other’). We did notinclude in this analysis the unique patient who was a compound heterozygousfor WFS1 mutations, heterozygous for WFS1LIB, who had no sign of OA at 12years of age. Arrows indicate two patients homozygous for ‘other’ mutationswho had a notably late onset of OA (age 21 years: mutation 461TS þ 842LF,and age 24 years: mutation 528YD).
4018 Human Molecular Genetics, 2008, Vol. 17, No. 24
at Chinese A
cademy of M
edical Sciences on M
ay 1, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

for any clinical symptoms of WFS every 2 months followinggenetic diagnosis, and throughout the duration of the study.
Information on consanguinity between probands’ parents wasobtained through a questionnaire, and statistically re-evaluatedbased on genome-wide microsatellite genotyping done on allmultiplex, consanguineous or extended families, and on 40additional probands from non-consanguineous families andtheir parents. Information on consanguinity obtained byquestionnaire and inferred from microsatellite data werehighly concordant. In particular, no consanguinity was detectedin the 40 non-consanguineous probands in this analysis. Fre-quencies of first and all degree consanguineous marriagesbetween probands’ parents were 25.7 and 43.4% respectively(general population estimates: 14 and 25%, respectively, 36).
Families were classified as consanguineous, multiplex(multiple affected siblings), extended (multiple relatednuclear families), or simplex (single affected child) non-consanguineous. Of a total of 408 nuclear families, 220belonged to consanguineous, multiplex or extended families,and 192 were consanguineous or multiplex. Several non-typicalT1D patients were identified based on clinical diagnosis or/andsubsequent genetic studies and re-examination of patients. Theseincluded 17 families with WFS, and 11 other families wherepatients had neonatal diabetes or were affected by additionalextrapancreatic clinical manifestations, suggesting recessivemonogenic inheritance. A role of WFS1 gene has been excludedin these 11 families, based on linkage analysis, WFS1 mutationscreening and/or direct gene identification of the causativedefect. These 11 families were included in our study in orderto achieve an exhaustive prevalence survey of genetic causesof JOD in Lebanon.
One thousand unrelated consenting healthy adults (.40years) representative of the socio-cultural diversity of theLebanese population were used as population Controls.These individuals are part of an ongoing larger populationstudy in Lebanon (37).
Microsatellite genotyping in WFS1 gene region
We combined microsatellite genotyping data from a micro-satellite genome scan performed in all consanguineous,multiplex or extended families, excluding those initiallydiagnosed as having WFS, and additional genotyping ofthree microsatellite markers located near WFS1 gene,D4S431, D4S2366 and D4S2935, was performed in all thesefamilies, including those with WFS. The microsatellitegenome scan used .400 microsatellite markers (LinkageMapping Set 2, Applied Biosystems, with modifications).
Mutation and polymorphisms screening in WFS1 gene
Screening of WFS1 mutations and polymorphisms was per-formed by DNA sequencing in all WFS probands (N ¼ 17)and their parents (N ¼ 32), in all non-syndromic diabetic pro-bands except nine due to technical failure (N ¼ 371) and allavailable parents from multiplex, consanguineous or extendedfamilies (N ¼ 363). We amplified all WFS1 exons, includingintron/exon boundaries, from genomic DNA by PCR usingprimers shown in Supplementary Material, Table S1A.Sequence interpretation was performed using the Genalys soft-
ware (38). After identification of a prevalent WFS1 Lebanesemutation (WFS1LIB), we genotyped it in duplicate in all studyindividuals, using a TaqMan assay (Applied Biosystems), withprimers described in Supplementary Material, Table S1B.There was 100% concordance between sequencing and geno-typing results. We also screened 1000 unrelated LebaneseControls for this mutation using the same TaqMan assay.
Defining the mutation status of variants, in WFSand non-syndromic DM patients
The mutation status of variants was assessed based on WFS1sequence data generated on patients (WFS: N ¼ 17 and non-syndromic DM: N ¼ 371) and their parents (N ¼ 32 and 363respectively). WFS1 variants were considered to be compatiblewith WFS mutations if they satisfied simultaneously the follow-ing four criteria: (i) being homozygous or compound heterozy-gous with other mutations in WFS patients; (ii) never beinghomozygous or compound heterozygous with other mutationsin non-diabetic individuals (parents); (iii) being absent (orpresent at a very low allele frequency, in the case of moreprevalent mutations) in parental alleles that were not transmittedto diabetic children (Control alleles); (iv) predicted to affect thecoding sequence of the protein (coding variant or alteration ofsplice site). We used the same strategy to assess WFS1 mutationstatus for non-syndromic DM (test population: non-syndromicDM probands). In addition to the Control alleles from thestudy population (non-transmitted parental alleles), a largeControl population (N ¼ 1000) was used to assess the frequencyof a more prevalent WFS1 mutation in the Lebanese population.
Prediction of the functional consequencesof WFS1 missense variants
Missense variants that were compatible with WFS or non-syndromic DM mutations, based on the criteria mentionedearlier, were evaluated for their predicted impact on proteinfunction using the PolyPhen computational program (39).We also assessed the species-conservation of amino acidsby sequence alignment. Scores of ‘possibly damaging’ or‘probably damaging’ given by this program for a conservedamino acid were estimated to be likely deleterious to proteinfunction.
Linkage and association studies, and statistical analyses
We revised the family structures and corrected genotypingerrors resulting in Mendelian inconsistencies, using the PED-CHECK program (40). Family structures and consanguinityestimates were determined using the IBSCHECK program(S. Heath, unpublished). Linkage analyses were performedunder a highly penetrant recessive model (penetrance: 0.95,disease allele frequency: 0.001, no phenocopy), allowing forheterogeneity, using MERLIN (41). Complex or extendedpedigrees were simplified when necessary to enable compu-tation with this program, while maintaining the necessary con-sanguinity information. Hardy–Weinberg equilibrium testing,and family-based association and transmission disequilibriumtest studies were performed using the PLINK package (42).Additional statistical analyses, including logistic regression
Human Molecular Genetics, 2008, Vol. 17, No. 24 4019
at Chinese A
cademy of M
edical Sciences on M
ay 1, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

analyses and Kaplan–Meier survival analysis were done usingSTATVIEW or JMP7 statistical packages.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
FUNDING
This study was supported by institutional support from Insermand the Pasteur Institute of Paris, and a joined grant from theJuvenile Diabetes Research Foundation, the Fondation pour laRecherche Medicale and Inserm to C.J. We thank the HospicesCivils de Lyon for their support.
ACKNOWLEDGEMENTS
We thank the patients and their families for agreeing to partici-pate in this study. We thank Valerie Senee and Anne Bolandfor their help with DNA samples management, and FumihikoMatsuda and Victor Renault for their help in database inte-gration. We thank the Chronic Care Centre, Beirut, Lebanon,for their collaboration and support. We are very grateful toDouglas Cavener, Bernhard Boehm and Marc Nicolino fordiscussions and critical reading of the manuscript.
Conflict of Interest statement. None declared.
REFERENCES
1. Rich, S.S. (1990) Mapping genes in diabetes. Genetic epidemiologicalperspective. Diabetes, 39, 1315–1319.
2. Concannon, P., Erlich, H.A., Julier, C., Morahan, G., Nerup, J., Pociot, F.,Todd, J.A. and Rich, S.S. (2005) Type 1 diabetes: evidence forsusceptibility loci from four genome-wide linkage scans in 1,435multiplex families. Diabetes, 54, 2995–3001.
3. Todd, J.A., Walker, N.M., Cooper, J.D., Smyth, D.J., Downes, K.,Plagnol, V., Bailey, R., Nejentsev, S., Field, S.F., Payne, F. et al. (2007)Robust associations of four new chromosome regions from genome-wideanalyses of type 1 diabetes. Nat. Genet., 39, 857–864.
4. Barrett, T.G. (2007) Differential diagnosis of type 1 diabetes: whichgenetic syndromes need to be considered? Pediatr. Diabetes, 8, 15–23.
5. Inoue, H., Tanizawa, Y., Wasson, J., Behn, P., Kalidas, K.,Bernal-Mizrachi, E., Mueckler, M., Marshall, H., Donis-Keller, H.,Crock, P. et al. (1998) A gene encoding a transmembrane protein ismutated in patients with diabetes mellitus and optic atrophy (Wolframsyndrome). Nat. Genet., 20, 143–148.
6. Strom, T.M., Hortnagel, K., Hofmann, S., Gekeler, F., Scharfe, C., Rabl, W.,Gerbitz, K.D. and Meitinger, T. (1998) Diabetes insipidus, diabetes mellitus,optic atrophy and deafness (DIDMOAD) caused by mutations in a novelgene (wolframin) coding for a predicted transmembrane protein. Hum. Mol.Genet., 7, 2021–2028.
7. Barrett, T.G., Bundey, S.E. and Macleod, A.F. (1995) Neurodegenerationand diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome.Lancet, 346, 1458–1463.
8. Medlej, R., Wasson, J., Baz, P., Azar, S., Salti, I., Loiselet, J., Permutt, A.and Halaby, G. (2004) Diabetes mellitus and optic atrophy: a study ofWolfram syndrome in the Lebanese population. J. Clin. Endocrinol.Metab., 89, 1656–1661.
9. Khanim, F., Kirk, J., Latif, F. and Barrett, T.G. (2001) WFS1/Wolframinmutations, Wolfram syndrome, and associated diseases. Hum. Mutat., 17,357–367.
10. Cryns, K., Sivakumaran, T.A., Van den Ouweland, J.M., Pennings, R.J.,Cremers, C.W., Flothmann, K., Young, T.L., Smith, R.J., Lesperance,M.M. and Van Camp, G. (2003) Mutational spectrum of the gene in
Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus,and psychiatric disease. Hum. Mutat., 22, 275–287.
11. Sandhu, M.S., Weedon, M.N., Fawcett, K.A., Wasson, J., Debenham,S.L., Daly, A., Lango, H., Frayling, T.M., Neumann, R.J., Sherva, R. et al.
(2007) Common variants in confer risk of type 2 diabetes. Nat. Genet., 39,951–953.
12. Franks, P.W., Rolandsson, O., Debenham, S.L., Fawcett, K.A., Payne, F.,Dina, C., Froguel, P., Mohlke, K.L., Willer, C., Olsson, T. et al. (2008)Replication of the association between variants in WFS1 and risk of type 2diabetes in European populations. Diabetologia, 51, 458–463.
13. Hofmann, S., Philbrook, C., Gerbitz, K.D. and Bauer, M.F. (2003)Wolfram syndrome: structural and functional analyses of mutant andwild-type wolframin, the WFS1 gene product. Hum. Mol. Genet., 12,2003–2012.
14. Osman, A.A., Saito, M., Makepeace, C., Permutt, M.A., Schlesinger, P.and Mueckler, M. (2003) Wolframin expression induces novel ion channelactivity in endoplasmic reticulum membranes and increases intracellularcalcium. J. Biol. Chem., 278, 52755–52762.
15. Makhoul, N.J., Wells, R.S., Kaspar, H., Shbaklo, H., Taher, A., Chakar, N.and Zalloua, P.A. (2005) Genetic heterogeneity of Beta thalassemia inLebanon reflects historic and recent population migration. Ann. Hum.Genet., 69, 55–66.
16. Jalkh, N., Genin, E., Chouery, E., Delague, V., Medlej-Hashim, M.,Idrac, C.A., Megarbane, A. and Serre, J.L. (2008) Familial Mediterraneanfever in Lebanon: founder effects for different MEFV mutations. Ann.Hum. Genet., 72, 41–47.
17. Qu, H.Q., Grant, S.F., Bradfield, J.P., Kim, C., Frackelton, E.,Hakonarson, H. and Polychronakos, C. (2008) Association analysis of thetype 2 diabetes loci in type 1 diabetes. Diabetes, 57, 1983–1986.
18. Awata, T., Inoue, K., Kurihara, S., Ohkubo, T., Inoue, I., Abe, T., Takino,H., Kanazawa, Y. and Katayama, S. (2000) Missense variations of thegene responsible for Wolfram syndrome (WFS1/wolframin) in Japanese:possible contribution of the Arg456His mutation to type 1 diabetes as anonautoimmune genetic basis. Biochem. Biophys. Res. Commun., 268,612–616.
19. Hansen, L., Eiberg, H., Barrett, T., Bek, T., Kjaersgaard, P., Tranebjaerg,L. and Rosenberg, T. (2005) Mutation analysis of the WFS1 gene in sevenDanish Wolfram syndrome families; four new mutations identified.Eur. J. Hum. Genet., 13, 1275–1284.
20. Cano, A., Rouzier, C., Monnot, S., Chabrol, B., Conrath, J., Lecomte, P.,Delobel, B., Boileau, P., Valero, R., Procaccio, V. et al. (2007)Identification of novel mutations in WFS1 and genotype-phenotypecorrelation in Wolfram syndrome. Am. J. Med. Genet. A, 143A,1605–1612.
21. Hardy, C., Khanim, F., Torres, R., Scott-Brown, M., Seller, A., Poulton,J., Collier, D., Kirk, J., Polymeropoulos, M., Latif, F. et al. (1999) Clinicaland molecular genetic analysis of 19 Wolfram syndrome kindredsdemonstrating a wide spectrum of mutations in WFS1. Am. J. Hum.Genet., 65, 1279–1290.
22. Sam, W., Qin, H., Crawford, B., Yue, D. and Yu, S. (2001) Homozygosityfor a 4-bp deletion in a patient with Wolfram syndrome suggestingpossible phenotype and genotype correlation. Clin. Genet., 59, 136–138.
23. Shi, Y., Taylor, S.I., Tan, S.L. and Sonenberg, N. (2003) When translationmeets metabolism: multiple links to diabetes. Endocr. Rev., 24, 91–101.
24. Yamada, T., Ishihara, H., Tamura, A., Takahashi, R., Yamaguchi, S.,Takei, D., Tokita, A., Satake, C., Tashiro, F., Katagiri, H. et al. (2006)WFS1-deficiency increases endoplasmic reticulum stress, impairs cellcycle progression and triggers the apoptotic pathway specifically inpancreatic beta-cells. Hum. Mol. Genet., 15, 1600–1609.
25. Fonseca, S.G., Fukuma, M., Lipson, K.L., Nguyen, L.X., Allen, J.R., Oka,Y. and Urano, F. (2005) WFS1 is a novel component of the unfoldedprotein response and maintains homeostasis of the endoplasmic reticulumin pancreatic beta-cells. J. Biol. Chem., 280, 39609–39615.
26. Zatyka, M., Ricketts, C., da Silva Xavier, G., Minton, J., Fenton, S.,Hofmann-Thiel, S., Rutter, G.A. and Barrett, T.G. (2008)Sodium-potassium ATPase 1 subunit is a molecular partner of Wolframin,an endoplasmic reticulum protein involved in ER stress. Hum. Mol.Genet., 17, 190–200.
27. Ishihara, H., Takeda, S., Tamura, A., Takahashi, R., Yamaguchi, S.,Takei, D., Yamada, T., Inoue, H., Soga, H., Katagiri, H. et al. (2004)Disruption of the WFS1 gene in mice causes progressive beta-cell loss andimpaired stimulus-secretion coupling in insulin secretion. Hum. Mol.
Genet., 13, 1159–1170.
4020 Human Molecular Genetics, 2008, Vol. 17, No. 24
at Chinese A
cademy of M
edical Sciences on M
ay 1, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from

28. Bu, X. and Rotter, J.I. (1993) Wolfram syndrome: amitochondrial-mediated disorder? Lancet, 342, 598–600.
29. Hofmann, S., Bezold, R., Jaksch, M., Obermaier-Kusser, B., Mertens, S.,Kaufhold, P., Rabl, W., Hecker, W. and Gerbitz, K.D. (1997) Wolfram(DIDMOAD) syndrome and Leber hereditary optic neuropathy (LHON)are associated with distinct mitochondrial DNA haplotypes. Genomics,39, 8–18.
30. Barrett, T.G. (2007) Differential diagnosis of type 1 diabetes: which geneticsyndromes need to be considered? Pediatr. Diabetes, 8 (Suppl. 6), 15–23.
31. Murphy, R., Ellard, S. and Hattersley, A.T. (2008) Clinical implicationsof a molecular genetic classification of monogenic beta-cell diabetes.Nat. Clin. Pract. Endocrinol. Metab., 4, 200–213.
32. Slingerland, A.S. (2006) Monogenic diabetes in children and youngadults: challenges for researcher, clinician and patient. Rev. Endocr.Metab. Disord., 7, 171–185.
33. Alberti, K.G. and Zimmet, P.Z. (1998) Definition, diagnosis andclassification of diabetes mellitus and its complications. Part 1: diagnosisand classification of diabetes mellitus provisional report of a WHOconsultation. Diabet. Med., 15, 539–553.
34. Zalloua, P.A., Shbaklo, H., Halaby, G., Terwedow, H., Xu, X. andAzar, S.T. (2002) Type-2 diabetes family history delays the onset oftype-1 diabetes. J. Clin. Endocrinol. Metab., 87, 3192–3196.
35. Zalloua, P.A., Terwedow, H., Shbaklo, H., Halaby, G., Xu, X. andAzar, S.T. (2003) Host and environmental factors defining the
epidemiology of type 1 diabetes mellitus in a group of Lebanese childrenand young adults. J. Pediatr. Endocrinol. Metab., 16, 759–769.
36. Khlat, M. (1988) Consanguineous marriage and reproduction in Beirut,Lebanon. Am. J. Hum. Genet., 43, 188–196.
37. Abchee, A., Puzantian, H., Azar, S.T., Shbaklo, H., Nasrallah, A.,
Sawaya, F.J., Alam, S. and Zalloua, P.A. (2006) Predictors of coronary arterydisease in the Lebanese population. Thromb. Res., 117, 631–637.
38. Takahashi, M., Matsuda, F., Margetic, N. and Lathrop, M. (2003)Automated identification of single nucleotide polymorphisms from
sequencing data. J. Bioinform. Comput. Biol., 1, 253–265.
39. Sunyaev, S., Ramensky, V., Koch, I., Lathe, W. III, Kondrashov, A.S. andBork, P. (2001) Prediction of deleterious human alleles. Hum. Mol.
Genet., 10, 591–597.
40. O’Connell, J.R. and Weeks, D.E. (1998) PedCheck: a program foridentification of genotype incompatibilities in linkage analysis.Am. J. Hum. Genet., 63, 259–266.
41. Abecasis, G.R., Cherny, S.S., Cookson, W.O. and Cardon, L.R. (2002)Merlin – rapid analysis of dense genetic maps using sparse gene flowtrees. Nat. Genet., 30, 97–101.
42. Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M.A.,Bender, D., Maller, J., Sklar, P., de Bakker, P.I., Daly, M.J. et al. (2007)PLINK: a tool set for whole-genome association and population-basedlinkage analyses. Am. J. Hum. Genet., 81, 559–575.
Human Molecular Genetics, 2008, Vol. 17, No. 24 4021
at Chinese A
cademy of M
edical Sciences on M
ay 1, 2010 http://hm
g.oxfordjournals.orgD
ownloaded from





























