Embed Size (px)
Citation preview

The FASEB Journal • FJ Express Full-Length Article
Transgenic expression of �-APP in fast-twitch skeletalmuscle leads to calcium dyshomeostasisand IBM-like pathology
Charbel E-H. Moussa,* Qinghao Fu,* Pravir Kumar,* Alexander Shtifman,*,‡
Jose R. Lopez,‡ Paul D. Allen,‡ Frank LaFerla,† David Weinberg,* Jordi Magrane,*Tamar Aprahamian,§ Kenneth Walsh,§ Kenneth M. Rosen,* and Henry W. Querfurth*,1
*Department of Neurology, Caritas St. Elizabeth’s Medical Center, Tufts University School ofMedicine, Boston, Massachusetts, USA; †Department of Neurobiology and Behaviour, University ofCalifornia, Irvine, California, USA; §Whitaker Cardiovascular Institute, Boston University School ofMedicine, Boston, Massachusetts, USA; and ‡Department of Anaesthesia, Brigham and Women’sHospital, Harvard Medical School, Boston, Massachusetts, USA
ABSTRACT Intracellular deposition of the �-amyloid(A�) peptide is an increasingly recognized pathologicalhallmark associated with neurodegeneration and mus-cle wasting in Alzheimer’s disease (AD) and inclusionbody myositis (IBM), respectively. Previous reportshave implicated dysregulation of �-amyloid precursorprotein (�APP) expression in IBM. Accumulation offull-length �APP, its various proteolytic derivativesincluding A�, and phospho-tau into vacuolated inclu-sions is an early pathogenic event. We previously re-ported on a statistical tendency favoring fast twitchfiber involvement in IBM, reminiscent of the tissuespecific patterns of misfolded protein deposition seenin neurodegenerative diseases. To test this principle,we generated an animal model in which human wild-type (WT) �APP expression was limited to postnataltype II skeletal muscle. Hemizygous transgenic miceharboring increased levels of holo�APP751 and A� inskeletal muscle fibers became significantly weaker withage compared with nontransgenic littermates and ex-hibited typical myopathic features. A subpopulation ofdissociated muscle fibers from transgenic mice exhib-ited a 2-fold increase in resting calcium and membranedepolarization compared with nontransgenic litter-mates. Taken together, these data indicate that overex-pression of human �APP in fast twitch skeletal muscleof transgenic mice is sufficient for the development ofsome features characteristic of IBM, including abnor-mal tau histochemistry. The increase in resting calciumand depolarization are novel findings, suggesting both amechanism for the weakness and an avenue for thera-peutic intervention in IBM.—Moussa, C. E-H., Fu, Q.,Kumar, P., Shtifman, A., Lopez, J. R., Allen, P. D.,LaFerla, F., Weinberg, D., Magrane, J., Aprahamian, T.,Walsh, K., Rosen, K. M., Querfurth, H. W. Transgenicexpression of �-APP in fast-twitch skeletal muscle leadsto calcium dyshomeostasis and IBM-like pathology.FASEB J. 20, E1570–E1578 (2006)
Key Words: �-amyloid � inclusion body myositis � skeletal muscle
Human inclusion body myositis (IBM) is the mostcommon muscle disorder affecting the elderly. Despiteintense efforts to understand IBM, its etiology remainsunknown. Sporadic IBM (sIBM) is an inflammatorycondition, but clinical and pathological features alsosupport a primary degenerative cause (1). IBM sharesseveral pathological hallmarks with Alzheimer’s disease(AD). Deposits of nonmutant AD-associated proteins�-amyloid precursor protein (�APP), �-amyloid (A�),hyperphosphorylated neurofilaments and Tau, ubiq-uitin, and various shared chaperones and kinases arethought to play pathological roles in the cognitivedecline (2) and muscular failure (3) that define spo-radic AD and IBM, respectively. An interesting distinc-tion between neurodegeneration in AD and IBM in-volves the accumulation of fibrillar A� in extracellularbrain parenchyma and intracellular myoplasm, respec-tively (4).
Misfolded mutant gene products appear to attackspecific brain regions in the various inherited neurode-generative diseases. For instance, mutant �-synucleincauses degeneration of the substantia nigra in PD (5,6), specific sets of striatal neurons are affected inHuntington’s disease by expansion of polyglutaminerepeat within the Huntingtin protein (7, 8), and motorneurons are sensitive to mutant superoxide dismutase(SOD) in amytrophic lateral sclerosis (9–13). In Alzhei-mer’s disease, early degeneration preferentially occursin the entorhinal cortex in association with increasingamyloid load (14–17). In IBM, evidence points to anexcess of �APP transcripts (18) and protein (19).Induction of �APP overexpression by myotubes inculture can recreate some of the hallmarks of IBM (20,21). Intracellular inclusions bearing AD-associated pro-
1 Correspondence: Department of Neurology, Caritas St.Elizabeth’s Medical Center of Boston, Tufts University Schoolof Medicine, 736 Cambridge St., CBR419, Boston. MA, USA.E-mail: [email protected]
doi: 10.1096/fj.06-5763fje
E1570 0892-6638/06/0020-1570 © FASEB

teins are relatively sparse, occurring in scattered, non-necrotic angulated myofibers (22). These and otherdata suggest that clinical muscle weakness arises from amore widespread metabolic defect (23). In previouswork, we suggested that a more generalized mismetabo-lism of calcium could be one such defect (24). Exces-sive calcium could result in muscle weakness and arisefrom an exaggerated release or leak involving ryano-dine receptors (24), reduced sarcoplasmic, or endo-plasmic reticulum calcium ATPase (SERCA) reuptakeor influx from A�-forming pores (25).
In two previous reports, transgenic mice that overex-press the C-terminal 100 amino acid (C100) fragmentof �APP displayed some of the pathology associatedwith IBM (26, 27). More recently we have shown inhumans and in a transgenic mouse line that expresses anon-IBM related mutant �-APP under the control of ageneral muscle creatine kinase promoter that type IIfibers are to a modest degree more vulnerable topathological changes (28).
To study the hypothesis that �APP gene expression,when confined to a specific muscle fiber type, canreproduce both the disease phenotype and defect incalcium homeostasis in vivo, we generated a transgenicmouse in which �APP production is restricted to fast-twitch fibers through control by a myosin light chain(MLC) 1/3 promoter/enhancer (29). These mice de-velop myopathological and clinical features resemblingthose associated with IBM, including colocalization ofimmunoreactivities to �APP and A�-sequence-contain-ing peptides, and ultrastructural and histopathologicalchanges that indicate myodegeneration and skeletalmuscle weakness. In addition, an alteration in musclemembrane potential and dysregulation of calcium ho-meostasis was discovered.
MATERIALS AND METHODS
Generation of transgenic mice
The complete �APP 751 open reading frame (in Bluescript,gift of C. Abraham) was cloned into pMEX-M2-myc3 expres-sion vector downstream of the 800 bp 5� flanking sequence ofthe myosin light chain (MLC) 1/3 promoter (SacI restrictionsite) and upstream of the Simian virus 40 polyadenylationsequence and the MLC enhancer flanking the 3� end (gift ofDrs C. Neville and N. Rosenthal, Massachusetts GeneralHospital). The entire expression cassette containing thepromoter, the cDNA, and the enhancer can be isolated as asingle fragment using one of the infrequent Srf I cuttingenzymes engineered on either side of the cassette (Fig. 1A).
Pronuclear stage zygotes were harvested from B6/C3F1mice (Charles River Laboratories, Cambridge. MA, USA).The linearized expression cassette was injected into the malepronucleus and between 25 and 30 embryos each wereimplanted per pseudopregnant CD1 foster mother. Afterdelivery of litters, distal tail samples were collected from eachoffspring and the DNA was extracted in Hot Shot reagent (2.5mM NaOH, 0.2 mM disodium EDTA) and neutralized in 40mM Tris-HCl. Transgenic mice were identified using a poly-merase chain reaction (PCR) -based assay. To avoid theamplification of endogenous mouse DNA, we designed the
sense primer from the rat MLC 1/3 promoter (5�-GCG TGTGTC AAG GTT CTA TTA GGC-3�) and antisense strandprimer (5�-ACA TCC GCC GTA AAA GAA TG-3�) from theKPI domain of human �APP 751. Both PCR-derived cDNAsgenerated from transgenic and control mice genomic DNAsand total genomic DNA digested with EcoR1, were electro-phoresed and transferred to a charged nylon membrane.Southern blots were hybridized with a probe generated usingthe 1.2 kb PCR product derived from the MLC-APP751 cDNAconstruct (Fig. 1A) as template and random priming with�-32P dATP (Perkin Elmer Life Sciences, Boston, MA, USA).
Immunocytochemical and histological analysis ofskeletal muscle
Immunohistochemistry was performed on 10 micron-thickmuscle sections. �APP 751 was probed with 22C11 (1:800)mouse monoclonal antibody (mAb) (Chemicon Interna-tional; Temecula, CA, USA). A� was probed with 6E10(1:600) mouse mAb (Signet; Dedham, MA, USA). Specificanti-A�1–42 antibodies included a rabbit polyclonal (1:60)(Chemicon International) and a mouse monoclonal (21F12;1:60) (gift of Dr. Dennis Selkoe, Brigham and Women’sHospital. Boston) to probe for A�1–42 immunoreactivity.Total tau was probed with tau-5 (1:500) mouse mAb (Bio-source International, Inc. Camarillo, CA, USA). Immunola-beled specimens were immunoperoxidase stained using aVECTASTAIN avidin-biotin complex (ABC) system (VectorLaboratories, Inc.; Burlingame, CA, USA) and counterstainedwith hematoxylin. Further histological staining was per-formed, including routine hematoxylin and eosin (H&E),modified Gomori trichrome, ATPase at pH 4.6, and thioflavinS. Serially sectioned muscles were stained with (1:40) myosinheavy chain (fast fiber specific) antibody (Ab) (Vector Labo-ratories) and counterstained with hematoxylin. mAb againstmouse LCA (CD45, PharMingen, Inc.; San Diego, CA, USA)was used (1:100) as a marker for cellular inflammation.
Immunoblot analysis
Tissues were snap-frozen in liquid nitrogen, powdered usinga tissue grind pestle (Kontes; Vineland, NJ, USA), homoge-nized 1:5 (w:v) in homogenization buffer (150 mM NaCl, 15mM EDTA, 10 mM Tris�Cl, pH 7.4 and protease cocktailinhibitor; Roche Diagnostics; Mannheim, Germany), andcentrifuged at 20,000 g for 20 min at 4°C. To probe for �APP,the supernatant was dried by speed vacuum and the pellet wasresuspended in 1:5 (w:v) Laemmli sample buffer (2.5% SDS)and analyzed by Western blot using mouse monoclonal 22C11Ab. To probe for A�, 400 mg of skeletal muscle tissue wassnap-frozen in liquid nitrogen, mechanically homogenized,and suspended in 70% formic acid for 2–3 h on ice. Formicacid was evaporated using speed vacuum and the tissuehomogenate was washed (3�) in 0.1 M Tris/HCl (pH 7.4), 2mM EDTA, and 0.5% SDS in the presence of protease cocktailinhibitor. A� was immunoprecipitated in 300 �l 1 � STENbuffer (50 mM Tris (pH 7.6), 150 mM NaCl, 2 mM EDTA,0.2% Nonidet P-40, 0.2% BSA, 20 mM PMSF and proteaseinhibitor cocktail) using (1:100) rabbit polyclonal R1282 Ab(gift of Dr. Dennis Selkoe, Brigham and Women’s Hospital;Boston, MA, USA) and analyzed by Western blot using 6E10mouse mAb.
Maximal Force Generation Strength Test
Isometric grip strength was evaluated using a Shimpo digitalforce gauge (Shimpo Instruments, FGV-1; Itasca, Japan).Prior to testing, the gauge was zeroed and set horizontally in
E1571APP EXPRESSION IN FAST TWITCH MUSCLE FIBERS AND IBM

recording mode to measure maximal strength in gram force(g-force). The animal was trained to grasp with either fore- orhind-paw a T-bar or triangular grasping ring, respectively.Using one hand, the animal is grasped about three-fourths ofthe way up toward the base of the tail and steadily pulled (�1in/s) away from the ring/T-bar in line with the transduceraxis until the grip is broken and maximal force is digitallyrecorded. Typically 15 recordings were taken for each animalwith an intertrial interval long enough to record the data andzero the gauge meter for next trial. All statistical analyses wereperformed using GraphPad Prism version 4 (GraphPad Soft-ware, Inc.; San Diego, CA, USA).
In vivo electromyography (EMG) recordings
Electrically grounded animals were gently restrained while aconcentric needle (30G, DCF25, Medtronic, Inc., MN, USA)was inserted into the hamstring to measure individualspontaneous motor unit action potentials (MUAP) usingMedtronic’s keypoint™. MUAP quantification was per-formed through decomposition analysis (30). Each MUAPwas reviewed blinded to the animal transgene status and thecursors reset manually. Duplicates were detected and re-moved. Criteria for MUAP selection were rise time: �0.8 ms,amplitude: �50 �V.
Membrane potential and resting [Ca2�]i recording
Flexor digitorum brevis (FDB) muscles were dissected outand fibers were enzymatically dissociated. Dissociated fiberswere plated onto plastic dishes for microelectrode recordingand myoplasmic resting [Ca2�]i and the plasma membranepotentials were recorded simultaneously using double-bar-reled Ca2�-selective microelectrodes as described previously(24).
Electron microscopy
Muscle tissue was fixed in (1:4, v:v) 4% paraformaldehyde-picric acid solution and 25% glutaraldehyde overnight, thenwashed 3 � in 0.1M cacodylate buffer and osmicated in 1%osmium tetroxide/1.5% potassium ferrocyanide for 3 h,followed by another 3 � wash in distilled water. Samples werenext treated with 1% uranyl acetate in maleate buffer for 1 h,washed 3 � in maleate buffer (pH 5.2), then exposed to agraded cold ethanol series up to 100% and ending with apropylene oxide treatment. Samples were embedded in pureplastic and incubated at 60°C for 1–2 days. Blocks weresectioned on a Leica ultracut microtome at 95 nm, picked uponto 100 nm formvar-coated copper grids, and analyzed usinga Philips Technai Spirit transmission electron microscope.
Figure 1. Genomic screening and expression ofthe �APP 751 transgene in mouse skeletalmuscle. A) Schematic of the MLC 1/3-�APPenhancer construct injected into the male pro-nucleus of each zygote to generate MLC-�APPtransgenic mice. B) PCR products of total DNAfrom distal tail samples (top panel, ethidiumstain 1% agarose gel) and Southern hybridiza-tion signal (bottom panel). Transgene-bearinganimals are represented in lanes 3, 6, 7, 10, and11 corresponding to animals nos. 15, 10, 24, 27,and 28. Negative control is mouse liver DNAfrom an unrelated animal and positive controlis plasmid MLC-�APP751 DNA. C) Southernblot analysis of genomic DNA in 2 transgenicand 2 nontransgenic mice showing transmis-sion of the expected transgene at �2.1 kb. A 1.4kb EcoR1 digested endogenous mouse APPgenomic DNA signal is shown. D) Immunoblotanalysis of �APP from skeletal muscle of 2.5-year-old mice, fractionated alongside controlextracts of C2C12 myotubes and K275 stablytransfected HEK cell line expressing �APP on8% PAGE gel. E) Immunoprecipitated A� fromskeletal muscle of 3 transgenic mice vs. 2 non-transgenic littermates (4–12% Bis-Tris gel).Synthetic A�42 peptide as a control. F) Immu-noblot analysis of �APP from skeletal muscle of4 transgenic and 4 nontransgenic littermates,and G) immunoprecipitated A� from skeletalmuscle of 4 transgenic and 4 nontransgeniclittermates, ages as shown. H) Comparative im-munoblot analysis of �APP expression fromnonskeletal muscle tissues from both transgenicand nontransgenic littermates.
E1572 Vol. 20 October 2006 MOUSSA ET AL.The FASEB Journal

Creatine kinase and muscle area measurements
To measure creatine kinase levels, blood was collected fromthe tail artery and immediately analyzed by an enzymatic ratemethod using the SYNCHRON LX20 system according to themanufacturer’s protocol (Beckman Coulter, Inc., Fullerton,CA, USA). Whole muscle area was measured by manuallytracing the perimeter of mid belly of cross sections using Spotprogram (Version 3.4, Diagnostic Instruments, Inc., SterlingHeights, MI, USA).
RESULTS
Identification of transgenic mice
Transgene integration was analyzed in total DNA ex-tracted from distal tail samples both by PCR andSouthern blot hybridization (Fig. 1B, C). Transgenicfounders were backcrossed to B6/C3F1 mice (JacksonLaboratories; Bar Harbor, ME, USA). One femalefounder gave rise to a line consisting of at least 5transgenic males nos. 27 and 28 (DOB 6/1/2002), nos.10 and 15 (DOB 7/20/03) and no. 19 (DOB 7/25/03).One male founder gave rise to at least one transgenicfemale no. 24 (DOB 6/12/2002). Transgenic 10, 15,and 19 were backcrossed with WT B6/C3F1. Trans-genic mice and control littermates were aged for up to2.5 years.
Transgene-derived expression of �APP inMLC-�APP mice
Steady-state levels of transgene-derived human �APPwere determined by Western blot analysis on totalprotein extracted from skeletal muscle of transgenicmice. Human �APP was detected in skeletal muscleextracts from transgenic mice (nos. 24, 27, and 28)aged 2.5 years (Fig. 1D). We detected 4- to 5-fold more�APP in skeletal muscle homogenates of transgenicmice compared with nontransgenic littermates. The�-amyloid peptide derived from �- and �-secretasemediated �APP proteolysis was also detected in totalprotein extracted from skeletal muscle of transgenicmice (nos. 24, 27, and 28) (Fig. 1E) compared withnontransgenic littermates. Age-dependent analysis oftransgene expression showed a steady presence of�APP in transgenic mice as early as 3 months of agecompared with age-matched nontransgenic littermates(Fig. 1F). �APP levels remain constant throughoutdevelopment. Monomeric A�, on the other hand, accu-mulated in an age-dependent manner, first detected at 3months of age. Oligomeric A� was also increasinglyevident in an age-dependent manner (Fig. 1G). No no-ticeable difference in �APP expression in the brain, liver,kidney, and heart (Fig. 1H) was detected between trans-genic and nontransgenic littermates.
IBM-like histopathology in MLC-�APP mice
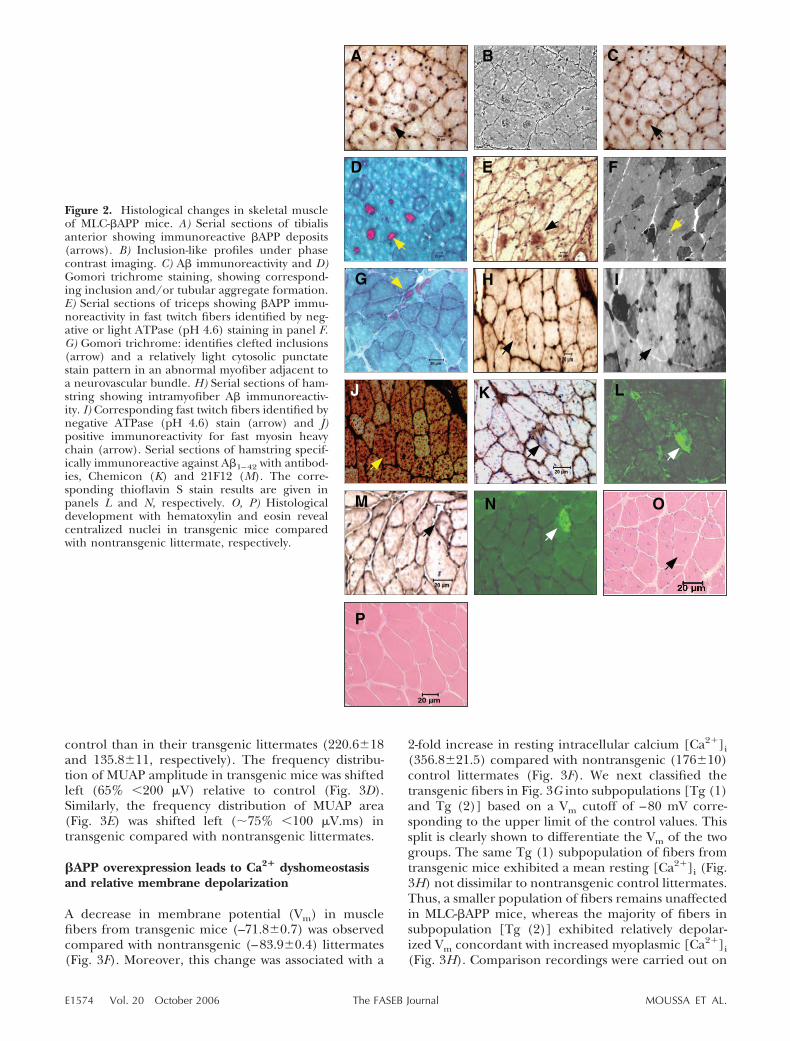
Serially sectioned tibialis anterior dissected from trans-genic mouse no. 10 showed intracellular deposits of
immunoreactive �APP (Fig. 2A). The same depositswere immunoreactive for antibodies vs. A� (Fig 2C).The proteinaceous aggregates were shown to corre-spond with large intramyofiber vacuoles easily identi-fied on Gomori trichrome-developed sections (Fig.2D). We confirmed �APP immunoreactivity in seriallysectioned triceps from transgenic mouse no. 27, where�APP deposits (Fig. 2E) were exclusively localized tofast twitch fibers (Fig. 2F). Subsarcolemmal inclusionswere confirmed by Gomori trichrome staining in across section of tibialis anterior of transgenic animal 27(Fig. 2G). The paler cytosolic stain suggests relativemitochondrial paucity, such as characteristic of type IIfibers, which are expected to bear the transgene. Wenext probed for immunoreactive A� deposits (Fig. 2H)in serially sectioned hamstring from animal no. 15 andconfirmed that A� immunoreactivity was localized tofast-twitch muscle fibers. Thus, A�-bearing myofiberswere ATPase 4.6 negative (Fig. 2I) and were fast myosinheavy chain positive (Fig. 2J). We probed for A�1–42-specific immunoreactivity using either of two antibod-ies, followed by thioflavin S staining, in cross sections ofhamstring from transgenic animals 27 (Fig. 2K) and 28(Fig. 2M). A�1–42-positive fibers were also thioflavin Spositive (Fig 2L, N), suggesting the presence of beta-sheet structures. �APP and A� immunoreactivity werenot widely distributed throughout tissue, but tended toaffect clusters of fast-twitch muscle fibers within discretefascicles. H&E staining of hamstring sections fromtransgenic mouse no. 27 reveals centralized nuclei (Fig.2O) compared with the normal peripheral location ofnuclei in nontransgenic control littermates (Fig. 2P).
MLC-�APP mice exhibit electromyographicabnormalities and muscle weakness.
Transgenic mice did not display any obvious behavioralor movement disorders, and their body weight and lifespan were not diminished compared with nontrans-genic littermates (data not shown). Transgenic micewith increased levels of �APP in skeletal muscle weresignificantly weaker (84.91.3 g-force) as early as 6months of age (Fig. 3A) in an isometric forelimbstrength test compared with nontransgenic controllittermates (124.93.5 g-force). Transgene harboringanimals remained weak and developed palpable muscleatrophy and waddling gait changes by 2.5 years of age(77.14.2 g-force) compared with nontransgenic con-trol littermates (133.94.8 g-force). Hind limb isomet-ric grip strength (Fig. 3B) was also decreased by 6 months(77.46 g-force) and up to 2.5 years of age (98.54.5g-force) in transgenic compared with nontransgenic con-trol littermates (140.34.8 and 159.97.9 g-force, re-spectively). EMG recordings (Fig. 3) revealed a greaterproportion of smaller MUAP in the transgene-affectedmice. These were typically small in amplitude andduration (Fig. 3C, middle) and, in some instances,polyphasic (Fig. 3C, bottom). Both the mean amplitude(309.423) and mean area (179.614) of MUAP (Fig.3D and E, respectively) were significantly larger in
E1573APP EXPRESSION IN FAST TWITCH MUSCLE FIBERS AND IBM
control than in their transgenic littermates (220.618and 135.811, respectively). The frequency distribu-tion of MUAP amplitude in transgenic mice was shiftedleft (65% 200 �V) relative to control (Fig. 3D).Similarly, the frequency distribution of MUAP area(Fig. 3E) was shifted left (�75% 100 �V.ms) intransgenic compared with nontransgenic littermates.
�APP overexpression leads to Ca2� dyshomeostasisand relative membrane depolarization
A decrease in membrane potential (Vm) in musclefibers from transgenic mice (–71.80.7) was observedcompared with nontransgenic (–83.90.4) littermates(Fig. 3F). Moreover, this change was associated with a
2-fold increase in resting intracellular calcium [Ca2�]i(356.821.5) compared with nontransgenic (17610)control littermates (Fig. 3F). We next classified thetransgenic fibers in Fig. 3G into subpopulations [Tg (1)and Tg (2)] based on a Vm cutoff of –80 mV corre-sponding to the upper limit of the control values. Thissplit is clearly shown to differentiate the Vm of the twogroups. The same Tg (1) subpopulation of fibers fromtransgenic mice exhibited a mean resting [Ca2�]i (Fig.3H) not dissimilar to nontransgenic control littermates.Thus, a smaller population of fibers remains unaffectedin MLC-�APP mice, whereas the majority of fibers insubpopulation [Tg (2)] exhibited relatively depolar-ized Vm concordant with increased myoplasmic [Ca2�]i(Fig. 3H). Comparison recordings were carried out on
Figure 2. Histological changes in skeletal muscleof MLC-�APP mice. A) Serial sections of tibialisanterior showing immunoreactive �APP deposits(arrows). B) Inclusion-like profiles under phasecontrast imaging. C) A� immunoreactivity and D)Gomori trichrome staining, showing correspond-ing inclusion and/or tubular aggregate formation.E) Serial sections of triceps showing �APP immu-noreactivity in fast twitch fibers identified by neg-ative or light ATPase (pH 4.6) staining in panel F.G) Gomori trichrome: identifies clefted inclusions(arrow) and a relatively light cytosolic punctatestain pattern in an abnormal myofiber adjacent toa neurovascular bundle. H) Serial sections of ham-string showing intramyofiber A� immunoreactiv-ity. I) Corresponding fast twitch fibers identified bynegative ATPase (pH 4.6) stain (arrow) and J)positive immunoreactivity for fast myosin heavychain (arrow). Serial sections of hamstring specif-ically immunoreactive against A�1–42 with antibod-ies, Chemicon (K) and 21F12 (M). The corre-sponding thioflavin S stain results are given inpanels L and N, respectively. O, P) Histologicaldevelopment with hematoxylin and eosin revealcentralized nuclei in transgenic mice comparedwith nontransgenic littermate, respectively.
E1574 Vol. 20 October 2006 MOUSSA ET AL.The FASEB Journal

previously reported muscle creatine kinase (MCK)-�APP mice, which express a non-IBM pathogenicSwedish double mutation of �APP and without speci-ficity for muscle fiber type (31). These mice exhibited acomparable increase in resting [Ca2�]i in all fiberstested (Supplemental Fig. 1). Thus, the distribution ofcalcium abnormalities into two fiber populations [Tg(1) and (2)] observed in our MLC-�APP mice isconsistent with the expected chimeric gene expression,type II transgene-bearing fibers having the abnormallyraised [Ca2�]i.
Ultrastructural changes in the muscle of the MLC-�APP mice
Electron microscope analysis of cross sections of ham-string from animal 29 (13 months old) revealed adistortion in Z line clarity, a substantial decrease inmitochondrion number and disrupted junctional triads(Fig 4A) compared with nontransgenic littermates (Fig.
4B). A higher magnification view (Fig. 4A, insert)showed glycogen granule dispersion around abnormalmitochondria. Further examination of cross sections ofhamstring from a less affected 6-month-old transgenicanimal showed the presence of what may either repre-sent inclusions and/or grossly dilated cisternae (aster-isk), as well as tubular aggregates (arrow, Fig. 4C).Centralized or invading nuclei with early membranechanges were also frequent (Fig. 4D). A magnified viewof a representative vacuolated inclusion, lacking incontinuous membrane boundary, from a 13-monthtransgenic animal revealed the presence of strangelyformed multivesicular body (MVB) -like structures andmembranous whorls (Fig. 4E). The content consisted ofamorphous material and short 6–10 nm filaments inbundles (Fig. 4F, insert).
Significant hamstring muscle atrophy was noted intransgenic (13.51 mm2) compared with nontrans-genic (20.14.6) littermates (Fig. 4G). Serum creatinekinase (CK) measurement revealed a 4-fold increase in
Figure 3. Clinical and physiological changes intransgenic mice. A, B) Histograms of isometriclimb strength demonstrating weakness in fore-limb and hindlimb of transgenic mice, respec-tively (n�13 Tg and 10 non-Tg). C) Waveformsindicating typical changes in MUAP in skeletalmuscle of transgenic mice (middle and lowertraces) compared with nontransgenic littermates(top trace). D, E) Frequency histogram of MUAPamplitude and area, respectively. Inserts showmean MUAP amplitude (D) and area (E) (n�3Tg and 3 non-Tg). F) Changes in membranepotential and resting [Ca2�]i (black bar, Tg;clear bar, non-Tg). G) Plot of individual fiber Vm.Using a non-Tg upper cutoff of –80 mV (Vm),two subpopulations of Tg (1 and 2) according toVm are created. H) Graph showing changes inresting [Ca2�]i and the corresponding distribu-tion of transgene-bearing fibers into two popu-lations according to the cutoff established inpanel G (n�3 Tg and 3 non-Tg animals, all Tgare from the same line). *Significantly differentfrom control (P0.05), independent t test,mean se.
E1575APP EXPRESSION IN FAST TWITCH MUSCLE FIBERS AND IBM

the serum CK levels in the transgenic (1176.25379U/dl) compared with nontransgenic (217.25116) lit-termates (Fig. 4H). This is consistent with 2- to 5-foldincreases noted in sIBM patients. Inflammatory cellinfiltrates were searched for using anti-CD45 recogniz-ing mouse leukocyte common antigen, but were notfound in our transgenic samples (data not shown).
DISCUSSION
The MLC1/3-�APP transgenic mouse exhibited manyof the clinical and myopathological features character-
istic of the human IBM condition. Among thesechanges, �APP and A� deposition, inclusion formation,centralized nuclei, increased creatine kinase levels, andskeletal muscle weakness with atrophy featured promi-nently. This is the first demonstration that WTholo�APP expression in skeletal muscle tissue is suffi-cient to produce these disease markers in vivo and, asshown in Fig. 1E, G, to lead to the age-dependentaccumulation of A� monomer and oligomers. Theexpression of �APP, then gradual accumulation of A�into inclusions in an age-dependent manner, also re-semble sIBM, a disorder affecting aged adults, and wasorchestrated by the MLC 1/3-promoter/enhancer,which is expressed postnatally (29). These results areconsistent with culture-based models of �APP misme-tabolism in muscle (20, 21). Furthermore, the observedincrease in resting calcium and relative membranedepolarization in muscle fibers of MLC-�APP trans-genic mice represents a mechanism relating �APPmismetabolism to altered calcium homeostasis andclinical weakness. Abnormalities in calcium metabolismare significant factors in other chronic myopathic con-ditions, central core disease, and muscular dystrophy(32–36). We were able to confirm these physiologicalfindings in the less restricted MCK-�APPSwed transgenicmouse of Sugarman et al. (31).
WT �APP expression was selectively targeted to post-natal fast-twitch skeletal muscle by choosing the MLC1/3 promoter/enhancer. In previous models, the mu-tant �APP and mutant C99 fragment transgene prod-ucts were contrived to accelerate A� genesis (25, 26,31). Accordingly, they are removed to a degree fromthe central �APP-sIBM hypothesis (3, 4). Moreover,their pattern of expression was not directed to fibertype or postnatal development as the MLC-�APP mousewas.
The reported ultrastructural findings are informativewith respect to IBM pathology. Features such as mem-brane whorls, glycogen granules, incompletely vacuo-lated deposits containing amorphous material and ar-rays of small filaments, and mitochondrial cytopathyare all reported in IBM (37). Except for the filaments,many of these are seen in other neuromuscular condi-tions. Additional profiles such as the proliferation anddilatation of sarcoplasmic reticulum networks (e.g.,tubular aggregates) are also a nonspecific finding seenin a number of human neuromuscular diseases andclinically normal inbred male mice (38). These and/orthe inclusion material may account for the uptake ofthe modified Gomori stain. The abnormal multivesicu-lar structures may be more unique to this model, alsonoting that mutlivesicular bodies or endosomes areassociated with intraneuronal �-amyloid in AD (17).
Several features of human sIBM, however, were notrepresented in this mouse model, including the lack ofendomysial inflammatory infiltrates, severely atrophicangulated myofibers, and dense aggregates of phos-phorylated neurofilaments. However, we detected re-gional increases of ‘thread-like’ immunoreactivity totau-5 Ab in transgenic compared with nontransgenic
Figure 4. Ultrastructure and muscle damage. A) Electronmicrograph of a cross section of hamstring from 13-month-old transgenic animal showing decreased mitochondrialnumber, disrupted triads, and glycogen dispersion. Glycogengranule accumulation around an abnormal mitochondrion(insert). B) Control littermate. C) An electron micrograph ofa cross section of hamstring from 6-month-old transgenicshowing a possible inclusion or dilated cisternae (asterisk)abutting a collection of tubular aggregates (arrow). D) Acentralized nucleus. E) An intersarcomeric inclusion (aster-isk) shown in direct contact with a sarcomere lacking any rimor membrane (double arrows) in a 13-month-old transgenic.The affected area contains a complex multivesicular body-likestructure (arrowhead) and membranous whorls (arrow). M,mitochondrion. F) Higher power view of “inclusion” sub-stance containing arrays of short filaments (arrows). G) Plotof muscle area (n�3) depicting atrophy. H) Elevation ofsystemic creatine kinase levels (n�4). Mean sd, Mann-Whitney, P 0.05.
E1576 Vol. 20 October 2006 MOUSSA ET AL.The FASEB Journal

littermates (Supplemental Fig. 2). These changes in tauwere accompanied by the appearance of immunoreac-tivity to the MC1 epitope, an AD conformation-specifictau Ab (39), in transgenic compared with nontrans-genic littermates. Moreover, an increase in the signal ofa high MW tau isoform was noted in the insolublefraction (Supplemental Fig. 2E). One explanation forthese findings is that endogenous tau is partitioned intoa less soluble and more aggregate-prone fraction in thepresence of the transgene products. More experimen-tal work is needed to fully characterize changes in tauphosphorylation in the MLC-�APP mouse. The reasonfor these variations relative to human IBM is likely dueto factors intrinsic to the murine host, as most singleand double transgenic models for AD also have similarshortcomings in brain neurofibrillary tangle formation(40, 41). It is noteworthy to mention that some tauisoforms that characterize muscle tau are not microtu-bule associated and lack certain phospho-epitopes im-portant to AD pathology (e.g., AT8) (42).
The MLC-�APP mouse model specifying type II fastfiber involvement supports the notion that tissue sub-type vulnerability can guide the degenerative pheno-type according to the intersection of specific metaboliccharacteristics with toxic properties particular to thetransgene. These results suggest that, to a degree,vulnerability of fast twitch fibers to A� inclusion forma-tion may contribute to disease in humans (28, 43). Wedo not, however, prove or claim with this model thathuman IBM is primarily a disorder of type II fibers,since type I fibers are also affected (28, 44). Onepossibility for future investigation is that A� is moreprone to aggregation under conditions favoring anaer-obic acidic environments (45), such as fast twitchmyofibers engaged in glycolytic metabolism (46, 47).
The authors thank Dr. Wendy Robinson for assistance withelectromyography, Dr. Dennis Selkoe for antibodies R1282and 21F12, and Dr. Peter Davies for the MC1 Ab. Thanks toMaria Ericsson and Louise Trakimas of the Harvard MedicalSchool electron microscopy facility. Our thanks also to JamesHappel for assistance with the creatine kinase assays. Thiswork was supported by grants from the Myositis Associationand NIH 41373 to H.W.Q.
REFERENCES
1. Tawil, R., and Griggs, R. (2002) Inclusion body myositis. Curr.Opin. Rheumatol. 14, 653–657
2. Selkoe, D. J. (2001) Alzheimer’s disease: gene, proteins andtherapy. Physiol. Rev. 81, 751–766
3. Askanas, V., and Engel, W. (2002) Inclusion body myositis andMyopathies: different etiologies, possibly similar pathogenicmechanisms. Curr. Opin. Neurol. 15, 525–531
4. Askanas, V., and Engel. W. K., and Alvarez, R. B. (1992) Lightand electron microscopic localization of �-amyloid protein inmuscle biopsies of patients with inclusion body myositis. Am. J.Physiol. 141, 31–36
5. Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E.,Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer,R., et al. (1997) Mutation in the alpha-synuclein gene identifiedin families with Parkinson’s disease. Science 276, 2045–2047
6. Kruger, R., Khun, W., Muller, T., Woitalla, D., Graeber, M.,Kosel, S., Przuntek, H., Epplen, J. T., Schols, L., and Reiss, O.
(1998) Ala30Pro mutation in the gene encoding �-synuclein inParkinson’s disease. Nat. Genet. 18, 106–108
7. Becher, M. W., Kotzuk, J. A., Sharp, A. H., Davies, S. W., Bates,G. P., Price, D. L., and Ross, C. A. (1997) Superoxide dismutasemutations in Huntington disease. Neurobiol. Dis. 4, 1–11
8. DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P.,Vonsattel, J. P., and Aronin, N. (1997) Aggregation of Hunting-tin in neuronal intranuclear inclusions and dystrophic neuritesin Brain. Science 277, 1990–1993
9. Kunst, C. B., Mezey, E., Brownstein, M. J., and Patterson. D.(1997). Mutations in SOD1 associated with amyotrophic lateralsclerosis cause novel protein interactions. Nat. Genet. 15, 91–94
10. Johnston. J. A., Dalton, M. J., Gurney, M. E., and Kopito, R. R.(2000) Formation of high molecular weight complexes ofmutant Cu,Zn-superoxide dismutase in a mouse model forfamilial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci.U. S. A. 97, 12571–12576
11. Shinder, G. A., Lacourse, M. C., Minotti, S., and Durham, H. D.(2001) Mutant Cu/Zn-superoxide dismutase proteins have al-tered solubility and interact with heat shock/stress proteins inmodels of amyotrophic lateral sclerosis. J. Biol. Chem. 276,12791–12796
12. Lindberg, M. J., Tibell, L., and Oliveberg, M. (2002) Folding ofhuman superoxide dismutase: disulfide reduction preventsdimerization and produces marginally stable monomers. Proc.Natl. Acad. Sci. U. S. A. 99, 16607–16612
13. Tiwari, A., and Hayward, L. J. (2003) Familial amyotrophiclateral sclerosis mutants of copper/zinc superoxide dismutaseare susceptible to disulfide reduction. J. Biol. Chem. 278, 5984–5992
14. Mufson, E. J., Chen, E-Y., Cochran, E. J., Beckett, L. A., Bennett,D. A., and Kordower, J. H. (1999) Entorhinal cortex �-amyloidload in individuals with mild cognitive impairment. Exp. Neurol.158, 469–490
15. Braak, H., and Braak, E. (1991) Neuropathological staggeringof Alzheimer-related changes. Acta Neuropathol. (Berlin) 82, 239–259
16. Arriagada, P. V., Growdon, J. H., Hedley-Whyte, E. T., andHyman, B. T. (1992) Neurofibrillary tangles but not senileplaques parallel duration and severity of Alzheimer’s disease.Neurology 42, 631–639
17. Gouras, G. K., Almeida, C. G., and Takahashi, R. H. (2005)Intraneuronal Abeta accumulation and origin of plaques inAlzheimer’s disease. Neurobiol. Aging 26, 1235–1244
18. Sarkozi, E., Askanas, V., Johnston, S. A., Engel, W. K., andAlvarez, R. B. (1993) Beta-amyloid precursor protein mRNA isincreased in inclusion-body myositis. NeuroReport 4, 815–818
19. McFerrin, J., Engel, W. K., and Askanas, V. (1998) Impairedinnervation of cultured human muscle overexpressing betaAPPexperimentally and genetically, relevance to inclusion bodymyopathies. NeuroReport 9, 3201–3205
20. Askanas, V., McFerrin, J., Baque, S., Alvarez, R. B., Sarkozi, E.,and Engel. W. K. (1996). Transfer of beta-amyloid precursorprotein gene using adenovirus vector causes mitochondrialabnormalities in cultured normal human muscle. Proc. Natl,Acad. Sci. U. S. A. 93, 1314–1319
21. Askanas, V., McFerrin, J., Alvarez, R. B., Baque, S., and Engel,W. K. (1997) Beta APP gene transfer into cultured humanmuscle induces inclusion-body myositis aspects. NeuroReport 8,2155–2158
22. Askanas, V., and Engel, W. K. (1998). In Inclusion Body Myopa-thies, (Askanas, V., Serratrice, G., and Engel, W., eds) pp. 3–78,Cambridge University Press
23. Askanas, V., and Engel, W. K. (1998) Sporadic inclusion bodymyositis and hereditary inclusion-body myopathies: diseases ofoxidative stress and aging? Arch. Neurol. 55, 915–920
24. Christensen, R. A., Shtifman, A., Allen, P. D., Lopez, J. R., andQuerfurth, H. W. (2004) Calcium dyshomeostasis in beta-amyloid and tau-bearing skeletal myotubes. J. Biol. Chem. 51,53524–53532
25. Arispe, N., Pollard, H., and Rojas, E. (1994) Beta-AmyloidCa2�-channel hypothesis for neuronal death in Alzheimer dis-ease. Mol. Cell. Biochem. 140, 119–125
26. Fukuchi, K-I., Pham, D., Hart, M., Li, L., and Lindsey. R. (1998).Amyloid-� deposition in skeletal muscle of transgenic mice.Am. J. Pathol 153, 1687–1693
E1577APP EXPRESSION IN FAST TWITCH MUSCLE FIBERS AND IBM

27. Jin, L-W., Hearm, M. G., Ogburn, C. E., Dang, N., Nochlin, D.,Ladgies, W. C., and Martin, G. M. (1998) Transgenic mouseover-expressing the C-99 fragment of �APP with an �-secretasesite mutation develop a myopathy similar to human inclusionbody myositis. Am. J. Pathol. 153, 1679–1686
28. Sugarman, C. M., Kitazawa, M., Baker, M., Caiozzo, V, J.,Querfurth, W. H., and LaFerla, M. F. (2006). Pathogenicaccumulation of APP in fast twitch muscle of IBM patients anda transgenic model. Neurobiol. Aging 27, 423–32.
29. Neville, G., Gonzales, D., Houghton, L., McGrew, M. J., andRosenthal, N. (1996) Modular elements of the MLC 1f/3f locusconfer fiber-specific transcription regulation in transgenic mice.Dev. Genet. 19, 157–162
30. Stalberg, E., Falck, B., Sonoo, M., Stalberg, S., and Astrom, M.(1995) Multi-MUP EMG analysis—a two year experience in dailyclinical work. Electroencephalogr. Clin. Neurophysiol. 97, 145–154
31. Sugarman, M. C., Yamasaki, T. R., Oddo, S., Echegoyen, J. C.,Murphy, M. P., Golde, T. E., Jannatipour, M., Leissring, M. A.,and LaFerla, F. M. (2002) Inclusion body myositis-like pheno-type induced by transgenic overexpression of �APP in skeletalmuscle Proc. Natl. Acad. Sci. U. S. A. 99, 6334–6339
32. Imbert, N., Cognard, C., Duport, G., Guillou, C., and Raymond,G. (1995) Abnormal calcium homeostasis in Duchenne muscu-lar dystrophy myotubes contracting in vitro. Cell Calcium 18,177–186
33. Berchtold, M. W., Brinkmeier, H., and Muntener, M. (2000)Calcium ion in skeletal muscle: its crucial role for musclefunction, plasticity and disease. Physiol. Rev. 80, 1215–1330
34. MacLennan, D. H. (2000) Ca2� signaling and muscle disease.Eur. J. Biochem. 267, 5291–5297
35. Dirksen, R. T., and Avila, G. (2002) Altered ryanodine receptorfunction in central core disease: leaky or uncoupled Ca2�
release channels? Trends. Cardiovascul. Med. 12, 189–19736. Rizzuto, R., and Pozzan, T. (2003) When calcium goes wrong:
genetic alterations of a ubiquitous signaling route. Nat. Genet.34, 135–141
37. Carpenter, S. (1996) Inclusion body myositis, a review. J. Neuro-pathol. Exp. Neurol. 55, 1105–1114
38. Rosenberg, N. L., Neville, H. E., and Ringel, S. P. (1985) Tubularaggregates. Their association with neuromuscular diseases, includ-ing the syndrome of myalgias/cramps. Arch. Neurol. 42, 973–976
39. Weaver, C. L., Espinoza, M., Kress, Y., and Davies. P. (2000).Conformational change as one of the earliest alterations of tauin Alzheimer’s disease. Neurobiol Aging 21, 719–727
40. Bornemann, K. D., and Staufenbiel, M. (2000) Transgenicmouse models of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 908,260–266
41. Emilien. G., Maloteaux, J-M., Beyreuther, K., and Masters, L. C.(2000) Alzheimer disease: mouse models pave the way fortherapeutic opportunities. Arch. Neurol. 57, 176–181
42. Gu, Y., Oyama, F., and Ihara, Y. (1996) Tau is widely expressedin rat tissues. J. Neurochem. 67, 1235–44
43. Arnardottir, S., Borg, K., and Answed, T. (2004) Sporadicinclusion body myositis: morphology, regeneration, and cy-toskeletal structure of muscle fibres. J. Neurol. Neurosurg. Psychi-atry 75, 917–920
44. Serratrice, G. (1998). Evolving concepts of inclusion bodymyositis. In Inclusion Body Myopathies (Askanas, V., and Engel,W. K., eds) pp. 81–104, Cambridge University Press
45. Wood, J. S., Maleeff, B., Hart, T., and Wetzel, R. (1996) Physical,morphological and functional differences between pH 5.8 and7.4 aggregates of the Alzheimer’s amyloid peptide A�. J. Mol.Biol. 256, 870–877
46. Tesch, P., Sjodin, B., and Karlsson, J. (1978) Relationshipbetween lactate accumulation, LDH activity, LDH isozyme andfiber type distribution in human skeletal muscle. Acta Physiol.Scand. 103, 40–46
47. Pette, D., and Spamer, C. (1986) Metabolic properties of musclefibers. Federation Proc. 45, 2910–2914
Received for publication January 20, 2006.Accepted for publication May 8, 2006.
E1578 Vol. 20 October 2006 MOUSSA ET AL.The FASEB Journal

The FASEB Journal • FJ Express Summary
Transgenic expression of �-APP in fast-twitch skeletalmuscle leads to calcium dyshomeostasis andIBM-like pathology
Charbel E-H. Moussa,* Qinghao Fu,* Pravir Kumar,* Alexander Shtifman,*,‡
Jose R Lopez,‡ Paul D Allen,‡ Frank LaFerla,† David Weinberg,* Jordi Magrane,*Tamar Aprahamian,§ Kenneth Walsh,§ Kenneth M. Rosen,* and Henry W. Querfurth*,1
*Department of Neurology, Caritas St. Elizabeth’s Medical Center, Tufts University School ofMedicine, †Department of Neurobiology and Behaviour, University of California, Irvine, California,USA; §Whitaker Cardiovascular Institute, Boston University School of Medicine, and ‡Department ofAnaesthesia, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA
To read the full text of this article, go to http://www.fasebj.org/cgi/doi/10.1096/fj.06-5763fje
1. SPECIFIC AIMS
The intracellular deposition of the �-amyloid (A�) peptidein skeletal muscle fibers is a recognized pathological featureof human inclusion body myositis (IBM). An abnormalup-regulation of �APP gene expression is one possiblecontributing factor. We found that overexpression of A� isdetrimental to the ionic homeostasis and viability of myo-tubes in culture. Since deposits of misfolded proteins areselectively toxic to subpopulations of cells in the brain,presumably based on metabolic vulnerabilities, we reasonedthat �APP gene expression confined to muscle fiber typecan reproduce some aspects of the disease phenotype in anage-dependent manner. We generated a transgenic mousein which wild-type (WT) �APP751 production is restricted tofast-twitch fibers through control by a myosin light chain(MLC) 1/3 promoter/enhancer. Hemizygous transgenicmice (MLC-�APP) were examined for deposits of �APP,A�42, and inclusions in type II skeletal muscle fibers, as wellas electrophysiological abnormalities and calcium deregula-tion in a corresponding subpopulation of myofibers. Corre-lation to ultrastructural changes was made. The functionalsignificance of these findings to clinical weakness with ad-vancing age in IBM is discussed.
2. PRINCIPAL FINDINGS
2.1. IBM-like histopathology in MLC-�APP mice
Transgene integration was analyzed in total DNA ex-tracted from distal tail samples by polymerase chainreaction (PCR) and genomic Southern blot hybridiza-tions. Most results are reported from a single hemizygousline. Steady-state levels of transgene-derived human �APPwere determined by Western blot on total protein ex-tracted from skeletal muscle of transgenic mice comparedwith aged-matched control littermates. Serially sectionedtibialis anterior dissected from transgenic mouse no. 10showed intracellular deposits of immunoreactive �APP(Fig. 1A). The same deposits were immunoreactive forantibodies vs. A� (Fig 1C). The proteinaceous aggregates
were shown to correspond with large intramyofiber vacu-oles or tubular aggregates easily identified on Gomoritrichrome-developed sections (Fig. 1D). We confirmed�APP immunoreactivity in serially sectioned triceps fromtransgenic mouse no. 27, where �APP deposits (Fig. 1E)were exclusively localized to fast twitch fibers (Fig. 1F).Subsarcolemmal inclusions were confirmed by Gomoritrichrome staining in a cross section of tibialis anterior oftransgenic animal 27 (Fig. 1G). The paler cytosolic stainsuggests relative mitochondrial paucity, such as character-ized type II fibers that are expected to bear the transgene.We next probed for immunoreactive A� deposits (Fig.1H) in serially sectioned hamstring from animal no. 15and confirmed that A� immunoreactivity was localized tofast-twitch muscle fibers. Thus, A�-bearing myofibers wereATPase 4.6 negative (Fig. 1I) and were fast myosin heavychain positive (Fig. 1J ). We probed for A�1–42 immuno-reactivity using either of two end-specific antibodies, fol-lowed by thioflavin S staining, in cross sections of ham-string from transgenic animals 27 (Fig. 1K) and 28 (Fig.1M). Several A�1–42-positive fibers were also thioflavin Spositive (Fig 1L, N), suggesting the presence of beta-sheetstructure. �APP and A� immunoreactivity were not widelydistributed throughout the tissue but tended to affectclusters of fast-twitch muscle fibers within discrete fasci-cles. H and E staining of hamstring sections from trans-genic mouse no. 27 revealed centralized nuclei (Fig. 1O)compared with the normal peripheral location of nucleiin nontransgenic control littermates (Fig. 1P).
2.2. MLC-�APP mice exhibit electromyographicabnormalities and muscle weakness
Transgenic mice did not display any obvious behavioral ormovement disorders, and their body weight and life span
1 Correspondence: Department of Neurology, Caritas St.Elizabeth’s Medical Center of Boston, Tufts University Schoolof Medicine, 736 Cambridge St., CBR419, Boston. MA 02135,USA. E-mail: [email protected]
doi: 10.1096/fj.06-5763fje
21650892-6638/06/0020-2165 © FASEB

were not diminished compared with nontransgenic litter-mates (data not shown). Transgenic mice with increasedlevels of �APP in skeletal muscle were significantly weaker(84.9�1.3 g-force) as early as 6 months of age (Fig. 2A) inan isometric forelimb strength test compared with non-transgenic control littermates (124.9�3.5 g-force). Trans-gene harboring animals remained weak and developedpalpable muscle atrophy and waddling gait changes by 2.5years of age (77.1�4.2 g-force) compared with nontrans-genic control littermates (133.9�4.8 g-force). Hind limbisometric grip strength (Fig. 2B) was also decreased by 6
Figure 2. Clinical and physiological changes in transgenicmice. A, B) Histograms of isometric limb strength demonstrat-ing weakness in forelimb and hind limb of transgenic mice,respectively (n�13 Tg and 10 non-Tg). C) Waveforms indi-cating typical changes in MUAP in skeletal muscle of trans-genic mice (middle and lower traces) compared with non-transgenic littermates (top trace). D, E) Frequency histogramof MUAP amplitude and area, respectively. Inserts show meanMUAP amplitude (D) and area (E) (n�3 Tg and 3 non-Tg).F) Changes in membrane potential and resting [Ca2�]i (blackbar, Tg; open bar, non-Tg). G) Plot of individual fiber Vm.Using a non-Tg upper cutoff of –80 mV (Vm), two subpopu-lations of Tg (1 and 2) according to Vm are created. H) Graphshowing changes in resting [Ca2�]i and the correspondingdistribution of transgene-bearing fibers into two populationsaccording to the cutoff established in panel G (n�3 Tg and 3non-Tg animals, all Tg are from the same line). *Significantlydifferent from control (P�0.05), independent t test, mean �se. See full text for quantification of muscle injury markers.
Figure 1. Histological changes in skeletal muscle of MLC-�APP mice. A) Serial sections of tibialis anterior showingimmunoreactive �APP deposits (arrows). B) Inclusion-likeprofiles under phase contrast imaging. C) A� immunoreactiv-ity and D) Gomori trichrome staining showing correspondinginclusion and/or tubular aggregate formation. E) Serial sec-tions of triceps showing �APP immunoreactivity in fast twitchfibers are identified in panel F by negative or light ATPase(pH 4.6) staining. G) Gomori trichrome histochemistry iden-tifies clefted inclusions (arrow) and a relatively light cytosolicreaction in an abnormal myofiber adjacent to a neurovascularbundle. H) Serial sections of hamstring showing intramyofi-ber A� immunoreactivity. I) Corresponding fast twitch fibersidentified by negative ATPase (pH 4.6) stain (arrow) and byJ) positive immunoreactivity for fast myosin heavy chain(arrow). K–N) Serial sections of hamstring demonstratingimmunoreactive A�1–42 deposits using specific antibodies,Chemicon (K) and 21F12 (M). The corresponding thioflavinS reactions are given in panels L and N, respectively. O, P)Histological development with hematoxylin and eosin revealcentralized nuclei in transgenic mice compared with non-transgenic littermate, respectively. See full text for ultrastruc-tural studies.
2166 Vol. 20 October 2006 MOUSSA ET AL.The FASEB Journal

months (77.4�6 g-force) and up to 2.5 years of age(98.5�4.5 g-force) in transgenic compared with nontrans-genic control littermates (140.3�4.8 and 159.9�7.9 g-force, respectively). Muscle atrophy and markers of injuryand inflammation were documented (see full text). EMGrecordings (Fig. 2) revealed a greater proportion ofsmaller MUAP in the transgene-affected mice. These weretypically small in amplitude and duration (Fig. 2C, mid-dle) and in some instances, polyphasic (Fig. 2C, bottom).Both the mean amplitude (309.4�23) and mean area(179.6�14) of MUAP (Fig. 2D, E, respectively) weresignificantly larger in control mice than their transgeniclittermates (220.6�18 and 135.8�11, respectively). Thefrequency distribution of MUAP amplitude in transgenicmice was shifted left (65% �200 �V) relative to control(Fig. 2D). Similarly, the frequency distribution of MUAParea (Fig. 2E) was shifted left (�75% �100 �V.ms) intransgenic compared with nontransgenic littermates.
2.3. �APP overexpression leads to Ca2�
dyshomeostasis and relative membrane depolarization
A decrease in membrane potential (Vm) in muscle fibersfrom transgenic mice (–71.8�0.7) was observed com-pared with nontransgenic (– 83.9�0.4) littermates(Fig. 2F). Moreover, this change was associated with a2-fold increase in resting intracellular calcium [Ca2�]i(356.8�21.5) compared with nontransgenic (176.0�10.0) control littermates (Fig. 2F). We next classifiedthe transgenic fibers in Fig 2G into subpopulations [Tg(1) and Tg (2)], based on a Vm cutoff of –80 mV,corresponding to the upper limit of the control values.This split is clearly shown to differentiate the Vm of thetwo groups. The same subpopulation of Tg (1) fibersfrom transgenic mice exhibited a mean resting [Ca2�]i(Fig. 2H) not dissimilar to nontransgenic control litter-mates. Thus, a smaller population of fibers remainunaffected in MLC-�APP mice, whereas the majority of
fibers in subpopulation Tg (2), all having relativelydepolarized Vm, displayed an increase in mean myo-plasmic [Ca2�]i (Fig. 2H). Comparison recordings werecarried out on previously reported muscle creatinekinase (MCK)-�APP mice, which express a non-IBMpathogenic Swedish double mutation of �APP withoutspecificity for muscle fiber type involvement. Thesemice exhibited a comparable increase in resting[Ca2�]i in all fibers tested (Supplemental Fig. 1 of fulltext). Thus, the distribution of resting calcium valuesinto two fiber populations [Tg (1) and (2)] observed inour MLC-�APP mice is consistent with the expectedchimeric gene expression; type II transgene-bearingfibers having an abnormally raised [Ca2�]i.
Transgenic mice were examined for ultrastructuralchanges (see full text). The findings include mitochon-drial cytopathy, tubular aggregates, vacuolar inclusionsbearing amorphous and filamentous material, mem-brane whorls, and multivesicular body-like structures.
3. CONCLUSIONS AND SIGNIFICANCE
The MLC-�APP transgenic mouse exhibited many of theclinical and myopathological features characteristic of thehuman IBM condition. Directed overexpression of hu-man WT �APP751 into type II skeletal muscle leads to A�accumulation into vacuolar inclusions affecting only fast-twitch skeletal muscle. Intramyofiber accumulation of�-amyloid is associated with membrane depolarizationand an increase in the level of resting calcium in skeletalmuscle (Fig. 2). This is the first demonstration that WTholo�APP expression in skeletal muscle tissue is not onlysufficient to produce these disease markers in vivo, butalso when restricted to fast twitch myofibers. The gradualaccumulation of A�42 over time into aggregates alsoresembles sIBM, a disorder affecting aged adults. Ourresults are consistent with culture-based models of �APPand A� mismetabolism in muscle. Furthermore, the ob-served increase in resting calcium and relative membranedepolarization in muscle fibers of MLC-�APP transgenicmice provides a mechanism relating �APP mismetabolismto abnormal excitation-contraction coupling and clinicalweakness (Fig. 3). Myocellular degeneration, muscle in-jury, and atrophy also contribute.
WT �APP expression was selectively targeted to post-natal fast-twitch skeletal muscle by choosing the develop-mentally sensitive MLC 1/3 promoter/enhancer. In pre-vious models, the mutant �APP and mutant C99 fragmenttransgene products were controlled by a general �-actin/cytomegalovirus (CMV) promoter and were contrived toaccelerate A� genesis. Accordingly, they are removed to adegree from the central WT �APP-IBM hypothesis. Sev-eral features of IBM, however, were not represented inthis mouse model, including a lack of endomysial inflam-matory infiltrates, small angulated myofibers and denseaggregates of phosphorylated neurofilaments. How-ever, changes in tau expression and conformation wereobserved in our model (see full text). Further studiesare planned to identify the molecular cause of calciumderangement and correct the phenotypic changes bymanipulations of calcium handling, amyloid load, andsurvival signal pathways.
Figure 3. A schematic of the proposed events that lead toclinical weakness in the MLC1/3-�APP mice. Overexpressionof human WT �APP with age results in A� accumulation intoinclusions in type II fast twitch skeletal muscle. Intramyofiberaccumulation of �-amyloid into inclusions is associated withloss of membrane depolarization, an increase in the level ofresting calcium, and degeneration of skeletal muscle.
2167APP EXPRESSION IN FAST TWITCH MUSCLE FIBERS AND IBM





























