Embed Size (px)
Citation preview

Today’s work, tomorrow’s possibility.
www.pvri.infoan official publication.
ISSN 2057-5354Volume 3 - Issue 2, October 2016 - March 2017
PVRI Chronicle

1
The Journal
PVRI Chronicle (ISSN 2057-5353) is a non-peer reviewed journal published on behalf of the Pulmonary Vascular Research Institute. The journal publishes articles, reviews and commentaries on the subject of pulmonary vascular diseases and actions within the PVRI. The journal is published bi-annually online and is available in print upon request.
Information for Authors
There are no page charges for submission to the journal. All manuscripts are solicited by the Editorial Boards, but submissions may also be made to the PVRI Team.
Subscription Information
Copies are provided to members of the PVRI free of charge. Members must notify our administrative office of a change of address in order to continue receiving the journal. The PVRI Chronicle is published and distributed by the Pulmonary Vascular Research Institute. Requested print copies are sent to subscribers directly from the publisher’s address. It is illegal to acquire copies from any other source. If a copy is recieved for personal use as a member of the PVRI, it cannot be resold or given away for commercial or library use.
Advertising Policies
PVRI Chronicle accepts display and classified advertsing. Frequency discounts and special positions are available. Enquiries about advertising should be sent to the administrative office. The PVRI Chronicle reserves the right to reject any advertisment considered unsuitable according to the set policies of the journal. The appearance of advertising or product information within the journal does not constitute an endorsement or approval of the journal and/or its publisher of the quality or value of said product or of claims made by its manufacturer.
Copyright
The entire contents of the PVRI Chronicle are protected under international copyrights. PVRI Chronicle, however, grants all users a free, irrevocable, worldwide, perpetual right of access to and license to copy, use, distribute, perform and display the work publicly and to make and distribute works in any digital medium for any reasonable, non-commercial purpose, subject to proper attribution of authorship and ownership of the rights. The journal also grants the right to make small numbers of printed copies for their personal, non-commercial use.
Permissions
To request permission for the reproduction of articles or information from this journal, please contact the administrative office (see contact).
Disclaimer
The information and opinions presented in the Chronicle reflect the views of the authors and not of the PVRI. Publication does not constitute endorsement by the Chronicle. Neither the PVRI Chronicle nor its publishers or anyone else involved in the preparation of the material contained in the Chronicle represents or warrants that the information herein is in every respect accurate or complete, and they are not responsible for any errors or ommissions or for the results obtained from such material. Readers are encouraged to confirm the information contained herein with other sources.
Contact Us
Pulmonary Vascular Research Institute 33 St George’s PlaceCanterburyKentCT1 1UTUnited Kingdom [email protected]
General Information
PVRI Chronicle

2
Editorial Board
Chief Editor
Eileen BauerDept of Surgery
PittsburghPA 15228 USA
Co-Editor in Chief
Zahara AliUniversity of Kent
CanterburyKent UK
Senior Editor
Ghazwan ButrousProfessor of Cardio-Pulmonary Sciences
University of KentCanterbury, Kent, United Kingdom
Executive Editor & Designer
Aaron ShefrasMarketing Manager
Pulmonary Vascular Research Institute33 St George’s Place
Canterbury Kent, United Kingdom
Cover Art credit
Rachel Wortman
Editorial Board
Rebecca Vanderpool, USAStylianos Orfanos, Greece
Zeenat Safdar, USAMamotbo Matshela, South AfricaMichiel de Raaf, The Netherlands
Eileen Bauer, USAEwa Kolosionek, Sweden
Djuro Kosanovic, Germany
Zahara Ali, UKMichael Seimetz, Germany
Mariella Velez-Martinez, USA
PVRI Chronicle

3
Table of contents
1. Editorial
Accepting The Torch Eileen Bauer
2. Art Club
Infogrpahic adapted from Oldham: Unexplained exertional dyspnea caused by low ventricular filling pressures: results from clinical invasive cardiopulmonary exercise testing Rebecca Vanderpool
3. Clinical Corner
Interview: Stephen Chan is interviewed by Imad Al Ghouleh Imad al Ghouleh
4. Journal Club
A colloquim on HIV and pulmonary diseases Jose Luis Sandoval Gutierrez, Sharilyn Almodovar
Interactive Discussion: COPD and aging Michael Seimetz
Interactive Discussion: Targeting PPARγ in IPF Srikanth Karnati
Interactive Discussion: Diagnosis and markers for the assessment of COPD; can a consensus be reached? Srikanth Karnati
5. Learner’s Corner
Did you know... “Stress on Lungs: a search through 8-Iso Prostaglandin-F2α” Zahara Ali
Did you know... Therapy of Pulmonary Hypertension – What’s New Natascha Somner
Cover Art credit
Rachel Wortman

4
Table of contents
Review... Targeting the cause, affecting the course – PhD defense Michiel Alexander de Raaf
6. PVRI News and Activities
Rome Walking Tour Michiel Alexander de Raaf
CYCS Waldeck Castle Retreat Michiel Alexander de Raaf
Obituary of Almaz Aldashev Martin Wilkins

5
Accepting The Torch - it needs many to keep the flame burning
As the Olympic Games in Rio are coming to an end, I will stay with the theme and happily accept the torch Sachindra Joshi passed on to me. I will try to fill his shoes as new Editor in Chief for the PVRI Chronicle - though they seem a little large.
Who am I?
Well, I am still struggling with the same question myself, but here the easy answer: I live in the US in Pittsburgh Pennsylvania and try to uncover the mysteries underlying pulmonary hypertension from my perspective – focused on the innate immune system. While it may seem obvious to assume that I belong to the Department of Pulmonary Medicine (or at least cardiology), I am actually in the Department of Surgery, which became well known for their work in regards to understanding the innate immune system long before I ever learned to hold a pipette. I carry the official title of Assistant Professor and am lucky enough to have my own laboratory.On a personal level I am a YOUNG 45 year old woman – and I guess thus making me still eligible to belong to the Committee of Young Clinicians and Scientists. I was born and raised in Germany and came to the US as young adult. I have three children, all teenagers, and just recently got divorced. And yes, as family, we are dealing with our own fair share of teenage related problems.
This current issue has all of the sections you have come to know previously and hopefully enjoy. Without listing all of the articles here I would like to thank all of the contributors who took time from their busy schedules to write and share their work here with us. I also like to make one point: If you look in detail at the contributors of this issues (and previous ones), you will notice that a large percentage of our articles come from the German group in Giessen. I cannot thank them all enough! However, where would we be if our friends in Giessen decided to stop writing articles for the chronicle? The PVRI is a global group creating connections between lung scientists and clinicians throughout the world. I urge you all to consider writing for us! While lung disease may be the same everywhere, every country has different approaches and experiences dealing with it and it would be lovely to have more international members share their work or comments with us. Some of my favorite talks at the meeting in Rome in January were the ones that presented on their approaches of treating patients with lung disease in countries struggling with obtaining all the “right and recommended” medicines. I was very impressed by their creativity and innovation. Wouldn’t it be great if we had ten articles from ten different countries? I will leave you with this quote as we struggle along to try and understand the underlying mechanisms of pulmonary vascular disease and trying to find a way to cure patients:
“Mistakes are, after all, the foundations of truth, and if a man does not know what a thing is, it is at least an increase in knowledge if he knows what it is not.” - Carl Jung
Editorial
Accepting the torch
Dr. Eileen Bauer
Dept of SurgeryPittsburgh
PA 15228 USA

6
Infographic Oldham
Rebecca Vanderpool
University of Pittsburgh Medical CenterPittsburgh
United States
ART CLUB

7
Interview with Stephen Chan
Dr. Imad al Ghouleh
Assistant Professor of MedicineDivision of Cardiology
CLINICAL CORNER
Interview with Stephen Chan MD-PhD Clinician Scientist and Director of the University of Pittsburgh Center for Pulmonary Vascular Biology and Medicine
I recently had the pleasure of interviewing Dr. Stephen Chan for the PVRI Chronicle. Dr. Chan is an MD-PhD Clinician Scientist and Director of the University of Pittsburgh Center for Pul-monary Vascular Biology and Medicine. He is a practising cardiologist and a well funded, prolif-ic scientist with numerous high impact publica-tions. He also is a warm, welcoming and down-to-earth person who was happy to meet with me and answer all of my questions. The following conversation ensued:
Q: Tell me a little about your background and your career path leading to your current posi-tion.
A: I am a physician scientist and was originally interested in biomedical research that would di-rectly impact the quality and service of patients in need. I had this desire since I was a young adult back in college and it is what drove me to pursue medical school and join the MD-PhD program at UCSF. I initially trained as a molec-ular biologist in virology and infectious disease. Though the training was very good, I ultimately decided to switch fields, when I became exposed to clinical cardiology and vascular disease in general. I believed I could make the most impact in this area. After graduating with both degrees, MD-PhD, I joined the internal medicine resi-dency program in Boston at BWH followed by a fellowship in cardiology at Massachusetts Gen-
eral Hospital. It was during my fellowship that I became much more acquainted with the disease of pulmonary hypertension (PH). PH struck me from two perspectives: 1) our understanding of what this disease is, how it occurs and 2) how it is being treated. At the same time, I witnessed the devastating effects of this disease in our pa-tients. This drove me, at least clinically, to delve deeper into exploration of the disease. Now, as a basic research scientist, PH became very appeal-ing to me because I realized that we knew very little about the disease. It had been historically neglected and is also often considered an or-phan disease. It was very exciting to me to feel I could make an impact in an area where there was still unmet need. That’s how I began to train as a physician scientist in pulmonary hyperten-sion. I became a postdoctoral fellow with Joseph Loscalzo, who is the Chairman of Medicine in BWH. Dr. Loscalzo has been interested in the study of PH for decades. I also trained clinically at BWH with Aaron Waxman in the Center for Pulmonary Vascular Medicine. This was truly the start of my independent career both as a physician scientist as well as a clinician. I recent-ly moved to the University of Pittsburgh where I now direct the Center for Pulmonary Vascular Biology and Medicine with the goal to combine both clinical and research operations to opti-mize patient care.
Q: Looking at the PH field right now, both from the perspective of a clinician and a physician scientist, would you consider that we have made a lot of headway in the past years and where do you see the field going in the next few years?
A: Over the past five to ten years it definite-ly seems that there has been an influx of new information, particularly from the molecular aspects of understanding this disease. I believe even from the clinical perspective we have now a better perspective of our patients, how they develop the disease and how it progresses. We seem to be on the upslope and I believe that we

8
Interview with Stephen Chan
CLINICAL CORNER
are hitting an inflection point of gaining great traction into establishing new paradigms for this disease, both from a clinical and research per-spective. It has been helpful not only in terms of the interests from our trainees who embraced the tasks of pushing the field to the next level, but also by increasing community awareness. The industry and pharma are also more aware and willing to invest into PH research, as is the federal government via the NIH. We are now beginning to see the payoff of this focus, at least in small form and still have a long way to go. Certainly other fields such as cancer and ather-osclerosis for instance, are light-years ahead of us in terms of disease understanding and how to best approach the complexities of patient care and pathogenesis. I am convinced we are on the right track but we still have a long way to go.
Q: What do you think is the next big thing - or should be - in terms of PH research?
A: It depends on where you look. There are still a number of holes in our understanding of how we treat PH. I will highlight a few of the ones our program is interested in. The first is about early diagnosis and our understanding of ear-ly pathogenesis of PH. For years we have been fixated on trying to understand the end stages of this chronic disease. This has given us a limited understanding of where the disease is coming from. But we now know that probably what is going to drive the next era of therapeutics and management of PH is trying to understand how to identify those patients who are at risk or those who are just developing this disease. We would love to prevent PH rather than having to treat something that has developed for many years. We may want to understand the molecular processes initiating PH. We may want to try to design new diagnostics in order to understand the beginning stages of the disease. Investigating
exercise physiology, for example, is one particu-lar notion that may be addressed this way. Also, we may want to pursue novel imaging modalities of looking at early stage disease in the context of the pathways that we have yet to discover. I be-lieve that early diagnostics will be an important area to focus on.Secondly, it would be interesting to develop personalized medicine for PH. We are certain-ly behind the times compared to other areas in medicine, such as cancer or atherosclerosis, where “big data” are already in play in trying to allow for individualized management and pa-tient care. I’m not just focused on genomics, but also other types of high throughput analyses, be it at the molecular level, such as metabolomix or expression data, or in the clinical realm where clinical analytics can be utilized to understand big population type data from electronic health patient. This is going to be a very important concept. I believe the integration of computa-tional methods, bioinformatics, high throughput molecular understanding is going to allow us to drive that particular phase forward. Third, we would be happy to usher in the next generation of therapeutics, be it small molecule inhibitors or otherwise, that would allow us to treat PH in a better fashion. This will also entail understanding the disease at a much finer level in order to target pathways other than the big three that we already have drugs for (prostacyc-lin, endothelin, and nitric oxide).
Q: Can you tell us a little about the PH program here at the University of Pittsburgh?
A: I arrived about 7 months ago as the Di-rector for the Center for Pulmonary Vascular Biology and Medicine. It was an added bonus for me was that this program already had such phenomenal basic scientists and clinicians in place. This is an exciting time for us here at the

9
One Event.
Interview with Stephen Chan
CLINICAL CORNER
University of Pittsburgh because we feel that we have a critical mass of scientists and clinicians that will allow us to integrate those disciplines to optimize patient care. We focus on innovation and research and we pride ourselves on research opportunities at the basic, translational and clin-ical levels. We believe we have the manpower as well as the commitment from the institution in order to pursue those types of endeavors. Sev-eral of our investigators are looking at many dif-ferent pathways that we think are important in PH. At the molecular level, we are thinking not only of vasomotor tone, proliferation and sur-vival mechanisms (all important in pulmonary vasculature and right ventricle), but also of other pathways that have not been previously studied. This includes metabolism, vascular stiffness, other phenotypes that we think are also playing an important role in this disease. We also have a number of translational compo-nents that we are very proud of in terms of mak-ing sure that our basic science is really translated to the clinical realm not only in the therapeutic arm but also diagnostically. Accomplishing this doesn’t happen just by accident and oftentimes in the field of PH it really relies upon the indi-vidual investigator to establish those types of collaborations. Because we have a critical mass of investigators who will be here long term, we have instituted a number of structural modal-ities to allow for that type of collaboration to occur. One of those, certainly, is the idea that we are very intent on building a very unique data-base of centralized information not only from our electronic health records system but also in combination with high throughput molecular data directly from bio-banked patient samples. This includes blood, plasma, tissue and cells. We believe that that will be a very prominent resource for what we think is going to be im-portant in personalized medicine in pulmonary hypertension. We also have a very strong un-
derstanding that in order for our home-grown science to be translated to patient care we need to be on the forefront of that effort, rather than simply making the discovery and leaving it up to others who may or may not want to take it on. In that sense we’ve also put in a lot of infrastruc-ture and resources in order to ensure that we have the ability to act as our own facility of de-veloping drugs from the ground up, i.e. from the pre-clinical modeling, pharmacodynamics and pharmacokinetics all the way to first-in-human studies. We believe we have an opportunity to really take it from soup to nuts. So that’s another very big push not only for our center but also for the institution in general to ensure that drug discovery remains a very high priority. We also would like to ensure that the integration of multiple different disciplines continues to be prioritized here. We pride ourselves on hav-ing a very integrated program at least between pulmonary and cardiology clinicians for the care of pulmonary hypertension. We also part-ner with a number of other disciplines such as rheumatology, sickle cell disease, interstitial lung disease, transplant, surgery and so on. That’s a very big component of what we pride ourselves on and we want to train the next generation of investigators, both clinicians and researchers, in order to allow for them to have that type of training. Right now, we have both pulmonary and cardiology fellows training in PH in a num-ber of different regards. We want to make sure that’s a real dedicated program. That will take a little bit of institutional and/or philanthropic money to do so but we are intent on making sure that we prioritize the training and mentor-ship of our physician scientists going forward. Finally, one of the things that we also want to ensure is that we have a very meaningful rela-tionship with our community of patients and advocacy groups in order to make sure that we offer the most holistic care to our patients. We

10
Interview with Stephen Chan
CLINICAL CORNER
have a very large program of physician and patient outreach locally, nationally and inter-nationally. We are interested in determining whether new innovative techniques such as tele-medicine services may also be something that we could offer not only in the region but also to patients perhaps in resource-poor environ-ments throughout the world. We think that our expertise may actually have an impact there as well. So those are some examples of where our program is going in addition to the individual efforts of our investigators to really advance the care of these patients.
Q: Being part of the PVRI you have probably seen the great contribution they make to the field. Where do you see the PVRI going in the future and what is an area that you would think this institute can have an instrumental role in pushing the field forward?
A: The PVRI is a great group and institution be-cause I believe, at least from my work with them, two things are very apparent. One of which it is a very international program and I think that one of the key areas from what I’ve heard from the leadership of the PVRI is that they want to maintain true international collaboration and not just be dominated by the same groups in the developed regions such as the US, Europe and some of the countries in Asia but certain-ly everywhere else including resource poor environments. That is a huge unmet need for this disease in terms of making the appropriate diagnosis and treatment of patients. Again, as I mentioned, we would love to be able to partner with someone like the PVRI in order to institute a program of telemedicine of sorts in order for us to really allow for communication between experts in the field as well as patients through-out the world. That will allow for a much more connected group and I think it would optimize
the care of those patients that really deserve better treatment then what they are getting right now. I think that is one very laudable goal of the PVRI that speaks to why we would love to partner with them. Secondly I noticed that the PVRI is very invest-ed in research and innovation and that it is very apparent when going to their meetings. Their research component is a very big priority and we appreciate that here in our program because innovation is probably our top priority in terms of research and development. We would love to work in concert and conjunction with the PVRI as it goes forward. I think that the PVRI also has a great opportunity to really invest more re-sources for research in general. I think that that program is still getting off the ground in terms of it but I think that it really puts itself in a good position to do so and we’d love to partner in that realm as well.
Q: For the young junior faculty and post-docs, given the current environment of difficult fund-ing and limited resources not just here in the US but worldwide, what advice do you have for them and what do you think they should focus on the most in securing a transition to the next step and having a successful future as research-ers and physician scientists?
A: It certainly it is a very difficult time for everybody in terms of getting funding. Often-times it is difficult for a person, especially at a young stage, to be optimistic if they hear all of the stories of difficulties in funding and how hard a road it really is. On the flip side, however, I actually feel quite a bit of optimism being in this field at this stage of the game. I think that there are two reasons for that. One of which, is that it is a phenomenal time to be a scientist and a research investigator in pulmonary hy-pertension. The technologies have gotten so

11
Interview with Stephen Chan
CLINICAL CORNER
much better than they had been ten or fifteen years ago. There are so many more opportuni-ties for a young investigator to really sink their teeth into and truly make an impact. Around fifteen years ago, there had been a very limit-ed amount of research, so you’re going to have to start from scratch. At this stage, I feel there is so much technology and advancement, not just in the PH field, but also in other disciplines that we can learn from and leverage in order to make important discoveries in this field. I think there is great excitement and optimism to be had in terms of making profound discoveries at this stage in the next five to ten years. I like the direction in that regard. In terms of rais-ing money I think that ironically, even though financial investment in research and research endeavors in general throughout the world has decreased, the PH field is actually on an upswing. If you were to look at the amount of money that is being invested and is available to the PH researcher and the young investigator included, I think that it is actually more than it was fifteen years ago. Fifteen years ago, there was less awareness and there was less interest from industry and federal partners to understand this disease better. Now I think we have critical mass and an inflection point of interest and persona in order to make this happen. I think that it is actually a really good time for a young investi-gator to be in this field. Even though we don’t get as much money on an absolute level as say cancer or other types of cardiovascular disease, I still feel the ratio of really good investigators to the amount of money that is being spent is very favorable for the person who is doing very strong science and making very strong discover-ies. If you are to compare it to some of the other fields where there is a lot more saturation in terms of the competition, I feel perhaps even the competition is less in this regard and that, if you are well trained and you have really good ide-
as, you will have a very viable outlet for raising money for yourself in this purpose. Now with that being said, I don’t think that it is easy to do by any means. I think that one of the things that I would certainly advise any young investiga-tor at any stage of their career is to ensure that they have a very strong, devoted and committed mentor who knows how the system works and understands how to train young investigators in order to usher them through this particular time of their career. This is a very fragile time in any person’s career when they are trying to make the step from a trainee to an independent investiga-tor. You do not want to have too many missteps or mistakes in that line. I think having men-torship and having a person who you really can trust is paramount to making sure you have that success. That success is very available to you if you put in the time, effort and strategize well.

12
A Colloquim on HIV and Pulmonary Diseases
A Colloquim on HIV and Pulmonary Diseases
Summary
Infection with human immunodeficiency virus (HIV) is now a chronic disease, thanks to the unquestioned success of antiretroviral therapies. Nevertheless, patients, clinicians and researchers are still facing challenges thirty years after the discovery of the virus. HIV has cleverly tricked both the host immune system and antiretroviral therapy (ART). As a first instance, the many HIV subtypes and recombinant forms have different susceptibilities to antiretroviral drugs, which may represent an issue in countries where ART is just being made available. Second, even under ART-indiced viral suppression, HIV still promotes inflammation, deregulates bystander cell biology, and induces oxidative stress in the host. Third, the preference of HIV for CXCR4 as co-receptor may also have noxious outcomes including potential malignancies. Furthermore, HIV still replicates cryptically in anatomical reservoirs like the lung and impairs bronchoalveolar cell immune responses, rendering the lung susceptible to co-morbidities. Hence, it is becoming evident that HIV-infected individuals are significantly more susceptible to long-term
HIV-associated complications, particularly now that HIV-infected individuals on ART live as long as the uninfected population. With this review, we will focus on chronic obstructive pulmonary disease (COPD), pulmonary arterial hypertension and lung cancer to hopefully braid concepts, give good starting points, and food for thought to pulmonologists, HIV specialists, cardiologists, and the new generations of scientists to jumpstart new efforts towards HIV-associated pulmonary diseases as common goal.
Introduction
Our respiratory tract is exposed to a myriad of everything from gases, dusts, pollens, oxidants, gastric contents, live pathogenic and non-pathogenic bacteria, fungi, and viruses. Together, these represent relentless challenges to the respiratory immune system, which relies in physical aerodynamic and immune barriers to maintain the lungs in good health; this would ensure an undisturbed gas exchange process, which is the ultimate physiologic goal of the lung. In addition, systemic diseases like infection with human immunodeficiency virus (HIV) may affect the lung. This colloquial review will focus on the impact of HIV in pulmonary immunology and the new challenges in both developed and developing countries, focusing on the non-infectious complications of HIV disease.
Human Immunodeficiency Virus: A Constant Challenge
HIV causes AIDS: that’s not new; it has been the topic of intense research efforts all over the world for over 30 years now. HIV produces billions of virions per day and has a rapid turnover with new generations every 2.6 days [1]. Due to its extensive genetic variability, the main group of HIV type 1 is subtyped into nine genetic variants (A, B, C, D, F, G, H, J, and K), all of which recombine, which in turn introduce differences in mutation rates and fitness. Understanding HIV interactions with the host is
Dr. JOSE LUIS SANDOVAL GUTIERREZ
Instituto Nacional de Enfermedades Respiratorias Ismael Coslo Villegas
Mexico City, Mexico
Corresponding AuthorSharylin Almodovar, PhD
University of Colorado Anschutz Medical CampusPulmonary Sciences and Cricial Care Medicine
Aurora, Colorado, USA
Journal club

13
A Colloquim on HIV and Pulmonary Diseases
essential to learn about the viral strategies to induce pathogenicity and to identify potential additional therapeutic targets. Let’s start a discussion of key concepts on HIV entry, persistence and pathogenesis.
HIV receptors. HIV enters the cells via interactions with CD4 receptor in the host cell and C-C chemokine receptor-5 (CCR5) and C-X-C chemokine receptor-4 (CXCR4). The CCR5 is a receptor for RANTES/CCL5, MIP-α/CCL3, and MIP-β/CCL4 in primary macrophages [2]. The CCR5 receptor is expressed in microglia, T lymphocytes, macrophages and dendritic cells (DC). On the other hand, CXCR4 is a 7-transmembrane G protein-coupled receptor used by HIV as co-receptor for preferential entry to T cells lines [3]. Its natural ligand is stromal derived factor-1 (SDF-1/CXCL12) [4] Conventionally, HIV virions that use CCR5 as portal of entry are designated as “R5”, while virions using CXCR4 are referred to as “X4”. The HIV preference for CCR5 co-receptor switches to a preference for CXCR4 over the course of HIV infection; this co-receptor switch predicts progression to AIDS in ~50% of HIV+ individuals [5].
HIV-mediated evasion of immune surveillance. HIV hides in cells by downregulation of key host receptors to evade immune surveillance [6]. For example, HIV-Nef is a key player in HIV pathogenesis by enhancing infectivity and downregulating critical molecules such as major histocompatibility complex-1 (MHC-1) and CD4 receptors [7]. It is known that Nef downregulates the CD4 receptor by targeting it to the endocytic degradation pathway in clathrin-coated vesicles. CD4 downregulation stimulates viral replication in primary T cells [8]. HIV Nef also downregulates MHC-1 by sequestering it in the trans-Golgi and hence, it prevents the recycling of this receptor from the Golgi to the membrane. Nef has highly conserved protein-protein interaction domains essential for these functions [9]. In addition, the Nef signature motifs used to downregulate MHC-1 are also used to downregulate CXCR4 and CCR5
which decreases the chances of HIV superinfection [10, 11] and the SOS call to the immune system.
HIV Hiding Places: Reservoirs. HIV-infected individuals who are compliant to antiretroviral therapy (ART) show an apparent clearance of the virus in the peripheral blood shortly after initiating therapy. Nevertheless, viral particles can be detected after interruption of antiretrovirals [12-14] and there is genetic HIV evolution over time in patients with undetectable viremia, suggesting a continuous low level replication of HIV even below the limits of clinical detection. This notion has been supported by the finding that in the presence of suppressive ART, the integrated HIV (AKA. archival, proviral HIV) and extrachromosomal HIV (episomal, surrogate for recent infection) belong to different viral populations [15].
Where does the virus hide? Resting T lymphocytes (memory cells) or long-lived myeloid cells (macrophages and DC) remain transcriptionally silent for long periods of time while having integrated copies of the HIV genome, especially in the presence of antiretroviral therapy [16-19]. The activation of these cells resumes the production of infectious particles and hence, the story repeats all over again by infection of new cells ⇔ reseeding of the reservoir ⇔ return to resting state and perpetuation of the persistence of HIV. At the organ/system level, anatomic compartments that may serve as reservoirs of HIV include the central nervous system [20, 21], the genitourinary tract [22, 23], and the gut-associated lymphoid tissue [24, 25].
The pulmonary microenvironment can also embrace high levels of HIV replication [26] [27] [28]. In the alveoli, lymphocytes are more susceptible to HIV infection than macrophages. HIV infects 1 in 100 of CD4+ alveolar lymphocytes [29] and 1 in 1,000 alveolar macrophages (AM) [30]. The alveolar space harbors small and large sub-populations of macrophages, which differ not only in morphology
A Colloquim on HIV and Pulmonary Diseases
Journal club

14
A Colloquim on HIV and Pulmonary Diseases
but also in cell surface markers [31, 32]. HIV preferentially infects the small macrophages, which exhibit more of highly active inflammatory phenotypes [33].
May the lungs act as anatomical reservoirs for HIV? Studies in the 90’s showed significantly complete phylogenetic separation of the HIV lineages in the lung, blood, brain and testis [34] [35]. A decade later, studies that compared HIV env sequences spanning the second constant and the fifth variable region (C2-V5) from matched blood and lung samples (either lung sputa or BALc) found lung-specific evolution in up to 56% of HIV-infected individuals [36]. The existence of HIV reservoirs demonstrates that ART does not eliminate from the host. Although evidence points to lung-specific viral evolution, the lung is not as anatomically enclosed as the brain and hence, viruses circulate freely - aided by the active blood flow through the cardiopulmonary system. In light of this, it is clear that HIV may contribute to immune disturbances leading to pulmonary complications.
HIV as an intrapulmonary pathogen: While some studies suggest that alveolar macrophages are resilient to HIV infection and remain competent to respond to Streptococcus pneumoniae [37, 38] and Cryptococcus neoformans [39], others suggest that HIV alters the pulmonary cell biology to the point that compared to HIV-uninfected counterparts, HIV-infected individuals have lower secretion of IFN-γ and tumor necrosis factor-alpha (TNF-α) in the lung, significantly increased RANTES and lysozyme in the BALf [40], marked cellular activation and accumulation of inflammatory mediators in the alveolar space, including increased HIV-specific CD8+ T cells. All these get complicated by smoking, which certainly complicates the immunologic landscape in the lungs [41], [42].The presence of HIV in the lungs also impacts bystander pulmonary resident cells like endothelial
cells. Although pulmonary endothelial cells are resistant to HIV infection [43], EC remain susceptible to apoptosis when exposed to HIV proteins [43-46] which suggests that while EC may not represent a cellular source of HIV in the lung, they certainly remain susceptible to the cytopathic effects of HIV that may eventually lead to HIV-associated pulmonary complications including chronic obstructive pulmonary disease (COPD), lung cancer, pulmonary arterial hypertension (PAH), fibrosis and infections [47, 48]. For instance, COPD is characterized by limited expiratory airflow, affecting individuals from 45 to 52 years of age [49], is the third leading cause of mortality in the world[50], and a significant risk factor for hospitalizations in the HIV-infected population [51-53] regardless of smoking status and antiretroviral therapy [51, 54], and is associated with inflammatory markers [48] [55, 56], increased oxidative stress [57] Of note, contrasting studies reported that there is not a significant risk for COPD (odds ratio (OR)= 1.61) or lung cancer (OR= 2.65) in HIV-infected individuals, particularly in the era post-antiretrovirals [58]. However, it is very likely that the reported findings were masked by the presence of unrecognized COPD because that study relied on self-reported pulmonary diagnoses, which were not clinically confirmed. Together, this suggests that the presence of infectious pathogens like HIV, coupled with abnormal inflammatory responses and oxidative stress all contribute mechanistically to HIV-COPD. Emphysema is a form of COPD characterized by apoptosis of epithelial and alveolar cells, with various degrees of inflammation. While cigarette smoking is a major cause of COPD/emphysema [59], HIV is another risk factor for COPD, regardless of smoking status. Histologically, HIV is mostly found in the emphysematous regions of the lung, while very rare HIV+ cells are present in normal lung areas [60], suggesting a direct role of HIV and/or HIV proteins in emphysema. One of the mechanistic insights offered for the lung endothelial cell apoptosis is the upregulation of the inflammatory cytokine
Journal club

15
A Colloquim on HIV and Pulmonary Diseases
A Colloquim on HIV and Pulmonary Diseases
endothelial monocyte activating polypeptide II (EMAP II) [61] and that such upregulation is induced by gp120 signaling through the CXCR4 receptor and activation of p38 MAPK [45]. Relevant to chronic bronchitis, CXCR4 and HIV-X4 viruses are implicated in the overproduction of mucus and mucous cell metaplasia in human bronchial epithelial cells in vitro, via the CXCR4/α7-nicotinic acetylcholine receptor/ γ-aminobutyric acid (GABA)-A receptor axis [62]. These results further support a potential role of HIV in higher incidence of COPD. 2) Pulmonary arterial hypertension is a rare disease in the general population, affecting 1-2 persons per million individuals but is significantly more frequent in the HIV-infected population, regardless of gender, age, socio-demographic characteristics, duration of HIV diagnosis, and interventions with ART. Listed in the Group 1 of clinical classification of the 5th World Symposium of Pulmonary Hypertension [63] HIV-associated PAH is characterized by increased inflammatory cytokines, atypical pulmonary vascular remodeling featured quasi-malignant phenotype of pulmonary endothelial cells [64, 65], and highly glycolytic pulmonary artery vascular cells [66, 67]. In addition, a signature of PAH is the presence of cells that obliterate the lumina of pulmonary arteries (plexiform lesions) [68]; therefore, the mean pulmonary artery pressures increase (mPAP >25 mmHg), ending fatally due to right heart failure. PAH can be screened by echocardiography, which measures PASP and diagnosed by right heart catheterization (RHC), which measures mPAP; final diagnosis is made based on mPAP > 25 mm Hg, pulmonary arteriolar wedge pressure <15 mmHg and pulmonary vascular resistance > 3 Wood units. Clinically, HIV-PAH presents as any idiopathic PAH. Symptoms are often nonspecific and insidious, so they are attributed to other complications of HIV or HIV itself. The time of presentation to the diagnosis is often long, from 6 to 2 years[69], or many times it is just overlooked. The prevalence of PAH in HIV-infected population
is usually reported to be 1 in 200 (0.5%) individuals. Nonetheless, the awareness of HIV-PAH has increased in medical communities worldwide, as evidenced by the coordination of taskforces aimed to screen patients who are asymptomatic for pulmonary arterial hypertension. PAH has been recently reported to affect 0.2-12.7% of HIV-infected individuals in several countries, based on either echocardiographic PASP or RHC. Based on these results, and the fact that PAH screening and diagnostic tools are not part of the routine clinical care to HIV-infected individuals, there are two questions on the table: should all patients with PAH should undergo HIV testing [70]? or should all patients with HIV undergo screening for PAH? A study comparing pressures measured by Doppler-based echocardiography vs right heart catheterization showed that 19.7% of the Doppler-based measurements were inaccurate, missing the PAH phenotype in 1/3 patients [71] Despite this, the reality is that many patients may just decline the RHC procedure, are ineligible or it may just not be available, especially in low-resource settings. Hence, the best scenario in many instances is to retrieve echocardiography data and use PASP 30-35 mm Hg as a cutoff for echocardiographic abnormalities associated with PAH, with the caveat that some of these data may still be underestimated but at least, accounted for. The increased prevalence of HIV-PAH (whether accurate or undestimated) has been documented by several studies worldwide but, do we have new ideas about new mechanisms and targets for therapy? Do we really know what in HIV increases the chances of PAH? There is no definitive proof that HIV causes PAH and no evidence that HIV infects lung endothelial cells [57]. However, viral proteins and their interactions with molecular partners in the infected cells may damage endothelial cells, induce inflammation and deregulate apoptosis and proliferation of vascular endothelial cells in the lung, resulting in pulmonary vascular remodeling featured in PAH patients [44, 45, 72-74]. Primate
Journal club

16

17
J ourn al C lu bA Colloquim on HIV and Pulmonary Diseases
models recapitulate intimal and medial hyperplasia along with elevated pulmonary pressures associated with human PAH, as reported after infection of the animals with chimeric SHIVenv virions [75]. Additional HIV proteins like Nef co-localizes with EC in PAH-like plexiform lesions and promotes severe dysfunction of the Golgi tethers at the sub-cellular level [76-78]. Additional studies found Nef signature sequences associated with the PAH phenotype in humans [79]. Together, these studies suggest that HIV proteins play key roles in the pathogenesis of HIV-PAH. The combination of HIV –and/or its proteins- and recreational drugs like cocaine exacerbates pulmonary arteriopathies. For example, HIV Tat and cocaine disrupts tight junction proteins, increases the expression of platelet-derived growth factor and increases the proliferation of pulmonary smooth muscle cells particularly when Tat and cocaine are combined[80]. Moreover, macaques exposed to the simian immunodeficiency virus (simian homologue of HIV) and morphine exhibit significant pulmonary vascular remodeling and oxidative-stress mediated apoptosis of endothelial cells followed by proliferation of apoptosis-resistant cells [81]. 3) HIV-associated malignancies that define AIDS (e.g. Kaposi’s sarcoma, non-Hodgkin lymphoma, and cervical cancer) have decreased in the post-ART era; nonetheless, the incidence of non-AIDS –defining cancers (NADC) have tripled: lung cancer is now the leading cause of NADC. Up to 52% of deaths in the post-ART era have been ascribed to NADC, including liver, gastric, colorectal and lung malignancies, which occurred in patients with fairly well-controlled HIV disease [82-84]. The most common type of lung cancer in the HIV-infected population is the adenocarcinoma [85], although non–small cell lung cancer (NSCLC) was found in 88% of cases with HIV-associated lung cancer [86]. Compared to the HIV-uninfected population, patients with HIV present with a younger age (mean 50 years) at the time of diagnosis with
lung cancer [87]. In addition, most of the affected patients are smokers and unfortunately, present with symptoms of advanced cancer [88]. HIV itself is an independent risk factor for lung cancer, regardless of smoking, COPD and bacterial pneumonia [85]. One of the mechanisms proposed for HIV-associated lung cancer is immunosuppression [83, 84, 89] [90]. Separate studies found a 2.2 relative risk of lung cancer in HIV-infected immunosuppressed patients (with CD4 counts <200 cells/mL), compared to uninfected [83]. Contrasting data from several groups show that lung cancer in HIV-infected individuals is not associated with CD4 counts [91-93], despite the inverse relationship with HIV viral load [94]. Additional mechanistic views into lung malignancies associated with HIV infection are provided by respiratory infections and genomic instability. The HIV-infected population is particularly prone to bacterial pneumonia, in addition to mycobacterial Pneumocystis, and viral respiratory infections. Pulmonary infections, in turn, may increase the risk of lung malignancies in the HIV-infected population [91, 95, 96]. In addition, genomic instability, reflected by microsatellite alterations, has been hypothesized to increase the risk of lung cancer in HIV. Microsatellite alterations, but not the loss of heterozygosity, were significantly increased (6-fold higher) in HV-associated lung carcinomas [97]. What produces this HIV-related genomic instability? A previously unrecognized interaction between HIV and endogenous retrotransposable elements has been uncovered with the finding that HIV infection results in accumulation of Type 1 long-interspersed nuclear elements (L1s) DNA in primary CD4+ lymphocytes [98]. Inflammation is also an ingredient in the recipe for HIV-associated cancers. A study that evaluated activated inflammatory pathways (IL-6 and C-reactive protein) and coagulation pathways (D-dimer) in HIV-infected patients found that individuals with higher levels of IL-6 had significantly higher risk for cancer [99].
Journal club

18
A Colloquim on HIV and Pulmonary Diseases
Journal club
Food For Thought: THE CARDS ON THE TABLE
Despite the potential underestimations due calculations based on self-reported disorders, the use of screening tools for diagnosis or even undocumented patient cohorts, the higher susceptibility of HIV+ patients to serious lung complications including COPD, PAH and lung
cancers is evident. Inflammation, oxidative stress, de-regulated apoptosis and proliferation, and malignant phenotypes are common denominators in the quest for mechanistic hints. Many of the specifics regarding the direct and indirect role of the virus in these diseases remain undetermined.

19
Journal club
A Colloquim on HIV and Pulmonary Diseases
We have won many battles against HIV/ADS aided by antiretrovirals but not the war. We are still learning about the HIV molecular tricks and facing challenges thirty years after its discovery. This review presents key concepts of HIV persistence and focused on how the lung resents HIV, echoing to HIV-associated pulmonary complications like COPD, pulmonary arterial hypertension and lung malignancies. We still need systematic epidemiological surveillance to document HIV-associated pulmonary complications globally; therefore, we insist that the crosstalk between pulmonologists, cardiologists, and HIV specialists is essential to document the true prevalence of these diseases, which otherwise would go unnoticed and untreated before the patient’s quality of life is seriously deteriorated. Antiretroviral drug toxicity, resistance and drug-drug interactions are issues affecting both the developed and the developing world, which certainly require extensive research, pharmacological formulations, and implementation of revised therapeutic strategies. New research enterprises are certainly warranted at the basic science level. For instance, the role of HIV and HIV-proteins in PAH, particularly at the sub-cellular level and the impact in cellular cross-talk (e.g. EC, macrophages, T cells, SMC) remain as opportunities to carve deeper niches to eventually identify novel therapeutic targets. In addition, it is necessary learn more about the modulation of cellular sources of viruses within reservoirs in order to strategize for a functional eradication of HIV reservoirs. Finally, the role of chronic inflammation in HIV-associated pulmonary diseases is certainly a unifying, hypothesis-generating topic that can take us to the next level.
AUTHOR DISCLOSURE STATEMENTNo competing financial interests exist.
References
1. Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 1996,271:1582-1586.2. Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996,381:661-666.3. Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996,272:872-877.4. Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, et al. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 1996,382:829-833.5. Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change in coreceptor use correlates with disease progression in HIV-1--infected individuals. The Journal of Experimental Medicine 1997,185:621-628.6. Guha D, Ayyavoo V. Innate Immune Evasion Strategies by Human Immunodeficiency Virus Type 1. ISRN AIDS 2013,2013:954806.7. Miller MD, Warmerdam MT, Gaston I, Greene WC, Feinberg MB. The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. Journal of Experimental Medicine 1994,179:101-113.8. Lundquist CA, Tobiume M, Zhou J, Unutmaz D, Aiken C. Nef-mediated downregulation of CD4 enhances human immunodeficiency virus type 1 replication in primary T lymphocytes. Journal of Virology 2002,76:4625-4633.9. Geyer M, Fackler OT, Peterlin BM. Structure--function relationships in HIV-1 Nef. EMBO Rep. 2001,2:580-585.10. Venzke S, Michel N, Allespach I, Fackler OT, Keppler OT. Expression of Nef downregulates CXCR4 the major coreceptor of human

20
immunodeficiency virus, from the surfaces of target cells and thereby enhances resistance to superinfection. Journal of Virology 2006,80:11141-11152.11. Chandrasekaran P, Moore V, Buckley M, Spurrier J, Kehrl JH, Venkatesan S. HIV-1 Nef down-modulates C-C and C-X-C chemokine receptors via ubiquitin and ubiquitin-independent mechanism. PloS one 2014,9:e86998.12. Zugna D, Geskus RB, De Stavola B, Rosinska M, Bartmeyer B, Boufassa F, et al. Time to virological failure, treatment change and interruption for individuals treated within 12 months of HIV seroconversion and in chronic infection. Antiviral therapy 2012,17:1039-1048.13. Costiniuk CT, Kovacs C, Routy JP, Singer J, Gurunathan S, Sekaly RP, et al. Short communication: human immunodeficiency virus rebound in blood and seminal plasma following discontinuation of antiretroviral therapy. Aids Research and Human Retroviruses 2013,29:266-269.14. Goujard C, Emilie D, Roussillon C, Godot V, Rouzioux C, Venet A, et al. Continuous versus intermittent treatment strategies during primary HIV-1 infection: the randomized ANRS INTERPRIM Trial. AIDS 2012,26:1895-1905.15. Buzon MJ, Codoner FM, Frost SD, Pou C, Puertas MC, Massanella M, et al. Deep molecular characterization of HIV-1 dynamics under suppressive HAART. PLoS pathogens 2011,7:e1002314.16. Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997,387:183-188.17. Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, Siliciano RF. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat.Med 1995,1:1284-1290.18. Demoustier A, Gubler B, Lambotte O, de Goer MG, Wallon C, Goujard C, et al. In patients on prolonged HAART, a significant pool of HIV infected CD4 T cells are HIV-specific. AIDS 2002,16:1749-
1754.19. Gunthard HF, Frost SD, Leigh-Brown AJ, Ignacio CC, Kee K, Perelson AS, et al. Evolution of envelope sequences of human immunodeficiency virus type 1 in cellular reservoirs in the setting of potent antiviral therapy. Journal of Virology 1999,73:9404-9412.20. Clements JE, Gama L, Graham DR, Mankowski JL, Zink MC. A simian immunodeficiency virus macaque model of highly active antiretroviral treatment: viral latency in the periphery and the central nervous system. Current opinion in HIV and AIDS 2011,6:37-42.21. Kramer-Hammerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Research 2005,111:194-213.22. Nunnari G, Leto D, Sullivan J, Xu Y, Mehlman KE, Kulkosky J, et al. Seminal reservoirs during an HIV type 1 eradication trial. Aids Research and Human Retroviruses 2005,21:768-775.23. Craigo JK, Patterson BK, Paranjpe S, Kulka K, Ding M, Mellors J, et al. Persistent HIV type 1 infection in semen and blood compartments in patients after long-term potent antiretroviral therapy. Aids Research and Human Retroviruses 2004,20:1196-1209.24. Chun TW, Nickle DC, Justement JS, Meyers JH, Roby G, Hallahan CW, et al. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. The Journal of infectious diseases 2008,197:714-720.25. Poles MA, Boscardin WJ, Elliott J, Taing P, Fuerst MM, McGowan I, et al. Lack of decay of HIV-1 in gut-associated lymphoid tissue reservoirs in maximally suppressed individuals. Journal of acquired immune deficiency syndromes 2006,43:65-68.26. Clarke JR, Gates AJ, Coker RJ, Douglass JA, Williamson JD, Mitchell DM. HIV-1 proviral DNA copy number in peripheral blood leucocytes and bronchoalveolar lavage cells of AIDS patients.
A Colloquim on HIV and Pulmonary Diseases
Journal club

21
A Colloquim on HIV and Pulmonary Diseases
the major coreceptor of human immunodeficiency virus, from the surfaces of target cells and thereby enhances resistance to superinfection. Journal of Virology 2006,80:11141-11152.11. Chandrasekaran P, Moore V, Buckley M, Spurrier J, Kehrl JH, Venkatesan S. HIV-1 Nef down-modulates C-C and C-X-C chemokine receptors via ubiquitin and ubiquitin-independent mechanism. PloS one 2014,9:e86998.12. Zugna D, Geskus RB, De Stavola B, Rosinska M, Bartmeyer B, Boufassa F, et al. Time to virological failure, treatment change and interruption for individuals treated within 12 months of HIV seroconversion and in chronic infection. Antiviral therapy 2012,17:1039-1048.13. Costiniuk CT, Kovacs C, Routy JP, Singer J, Gurunathan S, Sekaly RP, et al. Short communication: human immunodeficiency virus rebound in blood and seminal plasma following discontinuation of antiretroviral therapy. Aids Research and Human Retroviruses 2013,29:266-269.14. Goujard C, Emilie D, Roussillon C, Godot V, Rouzioux C, Venet A, et al. Continuous versus intermittent treatment strategies during primary HIV-1 infection: the randomized ANRS INTERPRIM Trial. AIDS 2012,26:1895-1905.15. Buzon MJ, Codoner FM, Frost SD, Pou C, Puertas MC, Massanella M, et al. Deep molecular characterization of HIV-1 dynamics under suppressive HAART. PLoS pathogens 2011,7:e1002314.16. Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997,387:183-188.17. Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, Siliciano RF. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat.Med 1995,1:1284-1290.18. Demoustier A, Gubler B, Lambotte O, de Goer MG, Wallon C, Goujard C, et al. In patients on prolonged HAART, a significant pool of HIV infected CD4 T cells are HIV-specific. AIDS 2002,16:1749-
1754.19. Gunthard HF, Frost SD, Leigh-Brown AJ, Ignacio CC, Kee K, Perelson AS, et al. Evolution of envelope sequences of human immunodeficiency virus type 1 in cellular reservoirs in the setting of potent antiviral therapy. Journal of Virology 1999,73:9404-9412.20. Clements JE, Gama L, Graham DR, Mankowski JL, Zink MC. A simian immunodeficiency virus macaque model of highly active antiretroviral treatment: viral latency in the periphery and the central nervous system. Current opinion in HIV and AIDS 2011,6:37-42.21. Kramer-Hammerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Research 2005,111:194-213.22. Nunnari G, Leto D, Sullivan J, Xu Y, Mehlman KE, Kulkosky J, et al. Seminal reservoirs during an HIV type 1 eradication trial. Aids Research and Human Retroviruses 2005,21:768-775.23. Craigo JK, Patterson BK, Paranjpe S, Kulka K, Ding M, Mellors J, et al. Persistent HIV type 1 infection in semen and blood compartments in patients after long-term potent antiretroviral therapy. Aids Research and Human Retroviruses 2004,20:1196-1209.24. Chun TW, Nickle DC, Justement JS, Meyers JH, Roby G, Hallahan CW, et al. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. The Journal of infectious diseases 2008,197:714-720.25. Poles MA, Boscardin WJ, Elliott J, Taing P, Fuerst MM, McGowan I, et al. Lack of decay of HIV-1 in gut-associated lymphoid tissue reservoirs in maximally suppressed individuals. Journal of acquired immune deficiency syndromes 2006,43:65-68.26. Clarke JR, Gates AJ, Coker RJ, Douglass JA, Williamson JD, Mitchell DM. HIV-1 proviral DNA copy number in peripheral blood leucocytes and bronchoalveolar lavage cells of AIDS patients.
A Colloquim on HIV and Pulmonary Diseases
Journal club

22
Clinical and experimental immunology 1994,96:182-186.27. Brenchley JM, Knox KS, Asher AI, Price DA, Kohli LM, Gostick E, et al. High frequencies of polyfunctional HIV-specific T cells are associated with preservation of mucosal CD4 T cells in bronchoalveolar lavage. Mucosal immunology 2008,1:49-58.28. Horiike M, Iwami S, Kodama M, Sato A, Watanabe Y, Yasui M, et al. Lymph nodes harbor viral reservoirs that cause rebound of plasma viremia in SIV-infected macaques upon cessation of combined antiretroviral therapy. Virology 2012,423:107-118.29. Jeffrey AA, Israel-Biet D, Andrieu JM, Even P, Venet A. HIV isolation from pulmonary cells derived from bronchoalveolar lavage. Clinical and experimental immunology 1991,84:488-492.30. Clarke JR, Israel-Biet D. Interactions between opportunistic micro-organisms and HIV in the lung. Thorax 1996,51:875-877.31. Frankenberger M, Menzel M, Betz R, Kassner G, Weber N, Kohlhaufl M, et al. Characterization of a population of small macrophages in induced sputum of patients with chronic obstructive pulmonary disease and healthy volunteers. Clinical and experimental immunology 2004,138:507-516.32. Wright AK, Rao S, Range S, Eder C, Hofer TP, Frankenberger M, et al. Pivotal Advance: Expansion of small sputum macrophages in CF: failure to express MARCO and mannose receptors. Journal of Leukocyte Biology 2009,86:479-489.33. Jambo KC, Banda DH, Kankwatira AM, Sukumar N, Allain TJ, Heyderman RS, et al. Small alveolar macrophages are infected preferentially by HIV and exhibit impaired phagocytic function. Mucosal immunology 2014.34. Nakata K, Weiden M, Harkin T, Ho D, Rom WN. Low copy number and limited variability of proviral DNA in alveolar macrophages from HIV-1-infected patients: evidence for genetic differences in HIV-1 between lung and blood macrophage populations. Mol.Med. 1995,1:744-757.
35. van’t Wout AB, Ran LJ, Kuiken CL, Kootstra NA, Pals ST, Schuitemaker H. Analysis of the temporal relationship between human immunodeficiency virus type 1 quasispecies in sequential blood samples and various organs obtained at autopsy. The Journal of Virology 1998,72:488-496.36. Heath L, Fox A, McClure J, Diem K, van ‘t Wout AB, Zhao H, et al. Evidence for limited genetic compartmentalization of HIV-1 between lung and blood. PLoS.ONE. 2009,4:e6949.37. Gordon SB, Jagoe RT, Jarman ER, North JC, Pridmore A, Musaya J, et al. The alveolar microenvironment of patients infected with human immunodeficiency virus does not modify alveolar macrophage interactions with Streptococcus pneumoniae. Clinical and vaccine immunology : CVI 2013,20:882-891.38. Gordon SB, Molyneux ME, Boeree MJ, Kanyanda S, Chaponda M, Squire SB, et al. Opsonic phagocytosis of Streptococcus pneumoniae by alveolar macrophages is not impaired in human immunodeficiency virus-infected Malawian adults. The Journal of infectious diseases 2001,184:1345-1349.39. Cameron ML, Granger DL, Matthews TJ, Weinberg JB. Human immunodeficiency virus (HIV)-infected human blood monocytes and peritoneal macrophages have reduced anticryptococcal activity whereas HIV-infected alveolar macrophages retain normal activity. The Journal of infectious diseases 1994,170:60-67.40. Gordon SB, Janoff EN, Sloper D, Zhang Q, Read RC, Zijlstra EE, et al. HIV-1 infection is associated with altered innate pulmonary immunity. The Journal of infectious diseases 2005,192:1412-1416.41. Reardon CC, Kim SJ, Wagner RP, Koziel H, Kornfeld H. Phagocytosis and growth inhibition of Cryptococcus neoformans by human alveolar macrophages: effects of HIV-1 infection. AIDS 1996,10:613-618.42. Helleberg M, Afzal S, Kronborg G, Larsen CS,
A Colloquim on HIV and Pulmonary Diseases
Journal club

23
A Colloquim on HIV and Pulmonary Diseases
Pedersen G, Pedersen C, et al. Mortality attributable to smoking among HIV-1-infected individuals: a nationwide, population-based cohort study. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 2013,56:727-734.43. Kanmogne GD, Kennedy RC, Grammas P. Analysis of human lung endothelial cells for susceptibility to HIV type 1 infection, coreceptor expression, and cytotoxicity of gp120 protein. Aids Research and Human Retroviruses 2001,17:45-53.44. Wang T, Green LA, Gupta SK, Kim C, Wang L, Almodovar S, et al. Transfer of Intracellular HIV Nef to Endothelium Causes Endothelial Dysfunction. PloS one 2014,9:e91063.45. Green LA, Yi R, Petrusca D, Wang T, Elghouche A, Gupta SK, et al. HIV envelope protein gp120-induced apoptosis in lung microvascular endothelial cells by concerted upregulation of EMAP II and its receptor, CXCR3. American journal of physiology. Lung cellular and molecular physiology 2014,306:L372-382.46. Park IW, Ullrich CK, Schoenberger E, Ganju RK, Groopman JE. HIV-1 Tat induces microvascular endothelial apoptosis through caspase activation. Journal of Immunology 2001,167:2766-2771.47. Crothers K, Huang L, Goulet JL, Goetz MB, Brown ST, Rodriguez-Barradas MC, et al. HIV infection and risk for incident pulmonary diseases in the combination antiretroviral therapy era. American Journal of Respiratory and Critical Care Medicine 2011,183:388-395.48. Morris A, Gingo MR, George MP, Lucht L, Kessinger C, Singh V, et al. Cardiopulmonary function in individuals with HIV infection in the antiretroviral therapy era. AIDS 2012,26:731-740.49. Gingo MR, Morris A, Crothers K. Human immunodeficiency virus-associated obstructive lung diseases. Clin Chest Med 2013,34:273-282.50. Organization WH. The top 10 causes of death. 2014.51. Madeddu G, Fois AG, Calia GM, Babudieri S, Soddu V, Becciu F, et al. Chronic obstructive pulmonary disease: an emerging comorbidity in
HIV-infected patients in the HAART era? Infection 2013,41:347-353.52. Drummond MB, Kirk GD, Astemborski J, McCormack MC, Marshall MM, Mehta SH, et al. Prevalence and risk factors for unrecognized obstructive lung disease among urban drug users. International journal of chronic obstructive pulmonary disease 2011,6:89-95.53. Akgun KM, Gordon K, Pisani M, Fried T, McGinnis KA, Tate JP, et al. Risk factors for hospitalization and medical intensive care unit (MICU) admission among HIV-infected Veterans. Journal of acquired immune deficiency syndromes 2013,62:52-59.54. Cui Q, Carruthers S, McIvor A, Smaill F, Thabane L, Smieja M. Effect of smoking on lung function, respiratory symptoms and respiratory diseases amongst HIV-positive subjects: a cross-sectional study. AIDS research and therapy 2010,7:6.55. Guillon JM, Autran B, Denis M, Fouret P, Plata F, Mayaud CM, et al. Human immunodeficiency virus-related lymphocytic alveolitis. Chest 1988,94:1264-1270.56. Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. The European respiratory journal 2003,22:672-688.57. Lassiter C, Fan X, Joshi PC, Jacob BA, Sutliff RL, Jones DP, et al. HIV-1 transgene expression in rats causes oxidant stress and alveolar epithelial barrier dysfunction. AIDS research and therapy 2009,6:1.58. Gingo MR, Balasubramani GK, Kingsley L, Rinaldo CR, Jr., Alden CB, Detels R, et al. The impact of HAART on the respiratory complications of HIV infection: longitudinal trends in the MACS and WIHS cohorts. PloS one 2013,8:e58812.59. Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet 2012,379:1341-1351.60. Yearsley MM, Diaz PT, Knoell D, Nuovo GJ. Correlation of HIV-1 detection and histology in AIDS-associated emphysema. Diagnostic molecular pathology : the American journal of surgical
A Colloquim on HIV and Pulmonary Diseases
Journal club

24
pathology, part B 2005,14:48-52.61. Clauss M, Voswinckel R, Rajashekhar G, Sigua NL, Fehrenbach H, Rush NI, et al. Lung endothelial monocyte-activating protein 2 is a mediator of cigarette smoke-induced emphysema in mice. The Journal of clinical investigation 2011,121:2470-2479.62. Gundavarapu S, Mishra NC, Singh SP, Langley RJ, Saeed AI, Feghali-Bostwick CA, et al. HIV gp120 Induces Mucus Formation in Human Bronchial Epithelial Cells through CXCR4/alpha7-Nicotinic Acetylcholine Receptors. PloS one 2013,8:e77160.63. Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013,62:D34-41.64. Rai PR, Cool CD, King JA, Stevens T, Burns N, Winn RA, et al. The cancer paradigm of severe pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine 2008,178:558-564.65. Lee SD, Shroyer KR, Markham NE, Cool CD, Voelkel NF, Tuder RM. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. The Journal of clinical investigation 1998,101:927-934.66. Tuder RM, Davis LA, Graham BB. Targeting energetic metabolism: a new frontier in the pathogenesis and treatment of pulmonary hypertension. American Journal of Respiratory and Critical Care Medicine 2012,185:260-266.67. Goncharov DA, Kudryashova TV, Ziai H, Ihida-Stansbury K, Delisser H, Krymskaya VP, et al. mTORC2 Coordinates Pulmonary Artery Smooth Muscle Cell Metabolism, Proliferation and Survival in Pulmonary Arterial Hypertension. Circulation 2013.68. Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. The American journal of pathology 1994,144:275-285.69. Barnett CF, Hsue PY. Human
immunodeficiency virus-associated pulmonary arterial hypertension. Clin Chest Med 2013,34:283-292.70. Vazquez AA, Aduen JF, Aduen P, Heckman MG, Burger CD. Should all patients with pulmonary hypertension undergo HIV serologic testing? Southern medical journal 2011,104:589-592.71. Selby VN, Scherzer R, Barnett CF, MacGregor JS, Morelli J, Donovan C, et al. Doppler echocardiography does not accurately estimate pulmonary artery systolic pressure in HIV-infected patients. AIDS 2012,26:1967-1969.72. Kanmogne GD, Primeaux C, Grammas P. Induction of apoptosis and endothelin-1 secretion in primary human lung endothelial cells by HIV-1 gp120 proteins. Biochem.Biophys.Res.Commun. 2005,333:1107-1115.73. Liu K, Chi DS, Li C, Hall HK, Milhorn DM, Krishnaswamy G. HIV-1 Tat protein-induced VCAM-1 expression in human pulmonary artery endothelial cells and its signaling. American journal of physiology. Lung cellular and molecular physiology 2005,289:L252-260.74. Mermis J, Gu H, Xue B, Li F, Tawfik O, Buch S, et al. Hypoxia-inducible factor-1 alpha/platelet derived growth factor axis in HIV-associated pulmonary vascular remodeling. Respiratory research 2011,12:103.75. George MP, Champion HC, Simon M, Guyach S, Tarantelli R, Kling HM, et al. Physiologic changes in a nonhuman primate model of HIV-associated pulmonary arterial hypertension. American journal of respiratory cell and molecular biology 2013,48:374-381.76. Marecki JC, Cool CD, Parr JE, Beckey VE, Luciw PA, Tarantal AF, et al. HIV-1 Nef is associated with complex pulmonary vascular lesions in SHIV-nef-infected macaques. Am.J.Respir.Crit Care Med. 2006,174:437-445.77. Marecki J, Cool C, Voelkel N, Luciw P, Flores S. Evidence for vascular remodeling in the lungs of macaques infected with simian immunodeficiency
A Colloquim on HIV and Pulmonary Diseases
Journal club

25
A Colloquim on HIV and Pulmonary Diseases
J ourn al C lu b
virus/HIV NEF recombinant virus. Chest 2005,128:621S-622S.78. Sehgal PB, Mukhopadhyay S, Patel K, Xu F, Almodovar S, Tuder RM, et al. Golgi dysfunction is a common feature in idiopathic human pulmonary hypertension and vascular lesions in SHIV-nef-infected macaques. Am.J Physiol Lung Cell Mol.Physiol 2009.79. Almodovar S, Knight R, Allshouse AA, Roemer S, Lozupone C, McDonald D, et al. Human Immunodeficiency Virus nef signature sequences are associated with pulmonary hypertension. Aids Research and Human Retroviruses 2012,28:607-618.80. Dhillon NK, Li F, Xue B, Tawfik O, Morgello S, Buch S, et al. Effect of cocaine on human immunodeficiency virus-mediated pulmonary endothelial and smooth muscle dysfunction. American journal of respiratory cell and molecular biology 2011,45:40-52.81. Spikes L, Dalvi P, Tawfik O, Gu H, Voelkel NF, Cheney P, et al. Enhanced pulmonary arteriopathy in simian immunodeficiency virus-infected macaques exposed to morphine. American Journal of Respiratory and Critical Care Medicine 2012,185:1235-1243.82. Stewart A, Chan Carusone S, To K, Schaefer-McDaniel N, Halman M, Grimes R. Causes of Death in HIV Patients and the Evolution of an AIDS Hospice: 1988-2008. AIDS research and treatment 2012,2012:390406.83. Silverberg MJ, Chao C, Leyden WA, Xu L, Horberg MA, Klein D, et al. HIV infection, immunodeficiency, viral replication, and the risk of cancer. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 2011,20:2551-2559.84. Reekie J, Kosa C, Engsig F, Monforte A, Wiercinska-Drapalo A, Domingo P, et al. Relationship between current level of immunodeficiency and non-acquired immunodeficiency syndrome-defining malignancies. Cancer 2010,116:5306-5315.
85. Sigel K, Wisnivesky J, Gordon K, Dubrow R, Justice A, Brown ST, et al. HIV as an independent risk factor for incident lung cancer. AIDS 2012,26:1017-1025.86. Hakimian R, Fang H, Thomas L, Edelman MJ. Lung cancer in HIV-infected patients in the era of highly active antiretroviral therapy. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2007,2:268-272.87. Demopoulos BP, Vamvakas E, Ehrlich JE, Demopoulos R. Non-acquired immunodeficiency syndrome-defining malignancies in patients infected with human immunodeficiency virus. Archives of pathology & laboratory medicine 2003,127:589-592.88. Shcherba M, Shuter J, Haigentz M, Jr. Current questions in HIV-associated lung cancer. Current opinion in oncology 2013,25:511-517.89. Guiguet M, Boue F, Cadranel J, Lang JM, Rosenthal E, Costagliola D. Effect of immunodeficiency, HIV viral load, and antiretroviral therapy on the risk of individual malignancies (FHDH-ANRS CO4): a prospective cohort study. The lancet oncology 2009,10:1152-1159.90. Shiels MS, Cole SR, Kirk GD, Poole C. A meta-analysis of the incidence of non-AIDS cancers in HIV-infected individuals. Journal of acquired immune deficiency syndromes 2009,52:611-622.91. Kirk GD, Merlo C, O’ Driscoll P, Mehta SH, Galai N, Vlahov D, et al. HIV infection is associated with an increased risk for lung cancer, independent of smoking. Clin.Infect.Dis. 2007,45:103-110.92. Shiels MS, Cole SR, Mehta SH, Kirk GD. Lung cancer incidence and mortality among HIV-infected and HIV-uninfected injection drug users. Journal of acquired immune deficiency syndromes 2010,55:510-515.93. Chaturvedi AK, Pfeiffer RM, Chang L, Goedert JJ, Biggar RJ, Engels EA. Elevated risk of lung cancer among people with AIDS. AIDS 2007,21:207-213.94. Engels EA, Biggar RJ, Hall HI, Cross H, Crutchfield A, Finch JL, et al. Cancer risk in people
A Colloquim on HIV and Pulmonary Diseases
Journal club

26
virus/HIV NEF recombinant virus. Chest 2005,128:621S-622S.78. Sehgal PB, Mukhopadhyay S, Patel K, Xu F, Almodovar S, Tuder RM, et al. Golgi dysfunction is a common feature in idiopathic human pulmonary hypertension and vascular lesions in SHIV-nef-infected macaques. Am.J Physiol Lung Cell Mol.Physiol 2009.79. Almodovar S, Knight R, Allshouse AA, Roemer S, Lozupone C, McDonald D, et al. Human Immunodeficiency Virus nef signature sequences are associated with pulmonary hypertension. Aids Research and Human Retroviruses 2012,28:607-618.80. Dhillon NK, Li F, Xue B, Tawfik O, Morgello S, Buch S, et al. Effect of cocaine on human immunodeficiency virus-mediated pulmonary endothelial and smooth muscle dysfunction. American journal of respiratory cell and molecular biology 2011,45:40-52.81. Spikes L, Dalvi P, Tawfik O, Gu H, Voelkel NF, Cheney P, et al. Enhanced pulmonary arteriopathy in simian immunodeficiency virus-infected macaques exposed to morphine. American Journal of Respiratory and Critical Care Medicine 2012,185:1235-1243.82. Stewart A, Chan Carusone S, To K, Schaefer-McDaniel N, Halman M, Grimes R. Causes of Death in HIV Patients and the Evolution of an AIDS Hospice: 1988-2008. AIDS research and treatment 2012,2012:390406.83. Silverberg MJ, Chao C, Leyden WA, Xu L, Horberg MA, Klein D, et al. HIV infection, immunodeficiency, viral replication, and the risk of cancer. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 2011,20:2551-2559.84. Reekie J, Kosa C, Engsig F, Monforte A, Wiercinska-Drapalo A, Domingo P, et al. Relationship between current level of immunodeficiency and non-acquired immunodeficiency syndrome-defining malignancies. Cancer 2010,116:5306-5315.
85. Sigel K, Wisnivesky J, Gordon K, Dubrow R, Justice A, Brown ST, et al. HIV as an independent risk factor for incident lung cancer. AIDS 2012,26:1017-1025.86. Hakimian R, Fang H, Thomas L, Edelman MJ. Lung cancer in HIV-infected patients in the era of highly active antiretroviral therapy. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2007,2:268-272.87. Demopoulos BP, Vamvakas E, Ehrlich JE, Demopoulos R. Non-acquired immunodeficiency syndrome-defining malignancies in patients infected with human immunodeficiency virus. Archives of pathology & laboratory medicine 2003,127:589-592.88. Shcherba M, Shuter J, Haigentz M, Jr. Current questions in HIV-associated lung cancer. Current opinion in oncology 2013,25:511-517.89. Guiguet M, Boue F, Cadranel J, Lang JM, Rosenthal E, Costagliola D. Effect of immunodeficiency, HIV viral load, and antiretroviral therapy on the risk of individual malignancies (FHDH-ANRS CO4): a prospective cohort study. The lancet oncology 2009,10:1152-1159.90. Shiels MS, Cole SR, Kirk GD, Poole C. A meta-analysis of the incidence of non-AIDS cancers in HIV-infected individuals. Journal of acquired immune deficiency syndromes 2009,52:611-622.91. Kirk GD, Merlo C, O’ Driscoll P, Mehta SH, Galai N, Vlahov D, et al. HIV infection is associated with an increased risk for lung cancer, independent of smoking. Clin.Infect.Dis. 2007,45:103-110.92. Shiels MS, Cole SR, Mehta SH, Kirk GD. Lung cancer incidence and mortality among HIV-infected and HIV-uninfected injection drug users. Journal of acquired immune deficiency syndromes 2010,55:510-515.93. Chaturvedi AK, Pfeiffer RM, Chang L, Goedert JJ, Biggar RJ, Engels EA. Elevated risk of lung cancer among people with AIDS. AIDS 2007,21:207-213.94. Engels EA, Biggar RJ, Hall HI, Cross H, Crutchfield A, Finch JL, et al. Cancer risk in people
A Colloquim on HIV and Pulmonary Diseases
Journal club

27
A Colloquim on HIV and Pulmonary Diseases
infected with human immunodeficiency virus in the United States. International journal of cancer. Journal international du cancer 2008,123:187-194.95. Engels EA. Inflammation in the development of lung cancer: epidemiological evidence. Expert review of anticancer therapy 2008,8:605-615.96. Shebl FM, Engels EA, Goedert JJ, Chaturvedi AK. Pulmonary infections and risk of lung cancer among persons with AIDS. Journal of acquired immune deficiency syndromes 2010,55:375-379.97. Wistuba, II, Behrens C, Milchgrub S, Virmani AK, Jagirdar J, Thomas B, et al. Comparison of molecular changes in lung cancers in HIV-positive and HIV-indeterminate subjects. JAMA : the journal of the American Medical Association 1998,279:1554-1559.98. Jones RB, Song H, Xu Y, Garrison KE, Buzdin AA, Anwar N, et al. LINE-1 retrotransposable element DNA accumulates in HIV-1-infected cells. Journal of Virology 2013,87:13307-13320.99. Borges AH, Silverberg MJ, Wentworth D, Grulich AE, Fatkenheuer G, Mitsuyasu R, et al. Predicting risk of cancer during HIV infection: the role of inflammatory and coagulation biomarkers. AIDS 2013,27:1433-1441.
J ourn al C lu bA Colloquim on HIV and Pulmonary Diseases
Journal club

28
COPD/emphysema: cause or consequence of aging/senescence?
COPD/emphysema cause or consequence of aging/senescence?
Prelude
Chronic obstructive pulmonary disease (COPD) is described as treatable and preventable, but also incurable disease, manifested as persistent airflow limitation with alveolar wall destruction resulting in emphysema which is not fully reversible (1). It is proven to involve inflammation, for
instance induced by cigarette smoke, including oxidant-antioxidant and protease-antiprotease imbalances (2, 3). COPD is an increasing global health problem, which is predicted to become the third most common cause of death by 2030. On the one hand, tobacco smoke and air pollution in industrial countries, on the other hand, in house cooking with wood in developing countries are the major causes for COPD. However, in the time of increasing life expectancy, age may contribute to the development of COPD as well (Fig. 1). This is supported by observations that incidence of COPD positively correlates with advanced age (1, 4). Aging is without doubt an important factor in many diseases (5,6), but there is also an intensive discussion about characterization of premature aging (7) and senescence, cellular aging and its involvement in diseases (8). This article aims to discuss if aging and senescence are critical events for the development of COPD/emphysema. Based on our previous article “Pulmonary arterial hypertension and aging: Is there a connection?” (Vol. 3, Issue 2) describing an involvement of aging in the context of pulmonary hypertension (PH), we now discuss its role in the context of COPD, a disease often associated with PH.
Main article
Aging is a natural and inevitable process represented as progressive loss of physiological integrity, leading to impaired function and lower adaptive capacity to stress (5, 9). Even though aging is one of the most obvious processes, clearly visible at all levels from the cell to organism, it still remains poorly understood. Despite intensive research, there is no unique strong, well-accepted hypothesis providing comprehensive explanation for the mechanism of aging (5, 6), in particular in the context of diseases. However, there are hypotheses which survived constant scientific
Corresponding AuthorDr. rer. nat. Michael Seimetz
Justus-Liebig-University GiessenExcellence Cluster Cardio-Pulmonary System (ECCPS)
Universities of Giessen and Marburg Lung Center (UGMLC) Member of the German Center for Lung Research (DZL)
Aulweg 130D-35392 Giessen, Germany
Tel.: +49-(0)641/99-46803 (office)Fax: +49-(0)641/99-46839
E-mail: [email protected]
Stefan Hadzic (1)Srikanth Karnati (2)Djuro Kosanovic (1)Michael Seimetz (1)
(1)Excellence Cluster Cardio-Pulmonary System (ECCPS)
Universities of Giessen and Marburg Lung Center (UGMLC)Member of the German Center for Lung Research (DZL)
GiessenGermany
(2)Institute for Anatomy and Cell Biology II
Division of Medical Cell BiologyJustus Liebig University
Aulweg 123D-35385 Giessen
Germany
Journal club

29
COPD/emphysema: cause or consequence of aging/senescence?
COPD/emphysema: cause or consequence of aging/senescence?
challenging. Somatic mutation theory is one of them, which suggests mutations to be accumulated in somatic cells resulting in senescent phenotype (10). Mitochondrial theory suggests similar mechanism, but in mitochondrial DNA causing leaky electron transport chain and increased ROS release (11). Replicative senescence is supported by telomere loss theory, placing telomere shortening as hallmark of senescence (12). Indeed, stress and in particular oxidative stress (shown to be increased in COPD patients) are proven to cause telomere shortening (13). Altered protein and accumulated waste theories have also been postulated associated with decline in activities of proteasomes (14, 15) and chaperones (16), and cellular waste accumulation (17). Kirkwood´s network theory (disposable soma theory) indicates a connection of the mentioned hypotheses. He believes that they occur simultaneously, causing, or at least supporting, each other (5, 18). Aging is not linked to the chronological age in all cases. For that reason the term premature aging and senescence are used (5, 8). Major efforts are made in distinguishing senescence and premature aging from the actual, chronological, aging of an organism (7). This gives a solid ground to continue discovering triggers (e.g. ROS, cigarette smoke) (4, 9, 19) and markers (e.g. telomerase activity, cell cycle arrest) (12, 16) of senescence. The lung, like any other organ, ages with progressive functional impairment and reduced capacity to respond to environmental stresses and injury (20, 21). Lung function declines with increasing age and that decline is even greater in smoking individuals (3, 22). Physiologic aging of the lung is associated with dilatation of alveoli with an enlargement of airspaces, a decrease in gas exchange surface area and elasticity (24). This age-dependent loss of elastin fibers is similar to the loss of skin elasticity and wrinkling of the skin that occurs in elderly (25). Age-related changes in lung, also called ‘senile emphysema’, do not include alveolar wall destruction or bronchial
inflammation, the usual pathological changes seen in COPD patients (22). Also COPD in non-smokers is not just due to accelerated aging, but aging could contribute to the development of COPD in both, smokers and nonsmokers (22, 26). Many studies show that telomeres are shorter in COPD patients, but it is unknown whether telomere shortening is a cause or a consequence of COPD (13). Telomere shortening occurs in normal aging and in smokers and patients with COPD. It provides a biological marker for aging and has a critical role in cellular senescence (7, 13, 19). Studies suggest that telomere shortening could make individuals more susceptible to the development of emphysema and lowers the threshold for damage induced by cigarettes (13). Aging cells accumulate damaged proteins due to decreased autophagy (degradation and removal of damaged protein and organelles in lysosomes), and exposure to cigarette smoke can lead to additional cellular damage pushing cells towards senescence (17).
As mentioned above, the most known hallmarks of senescence include telomere attrition (11), cumulative DNA damage, impaired DNA repair (5, 10), protein damage (14, 16, 29) and accumulation of waste products (17). It is believed that tthese hallmarks lead to terminal cell cycle arrest as an evolutionary preserved defense mechanism preventing malignant transformation (27, 28). In lung alveoli, cells replicate to replace damaged cells but eventually cannot keep up with replacement because of replicative senescence. The consequences of the lack of alveolar cell replacement, which is the case in senescent lung, can be an increase in alveoli size and a decrease of the surface area (13). In aged tissue, this repair system may become inefficient due to the impaired regenerative capacity of stem cells, leading to the accumulation of senescent cells and lowering threshold towards damage which cannot be repaired (6, 26).
Journal club

30
COPD/emphysema: cause or consequence of aging/senescence?
COPD pathology and aging are both complex processes involving many mechanisms on cellular level as well as on the level of whole organism. Despite the obvious connection, it still remains unclear which players are really important and in which conditions. Our brief discussion is aimed to stimulate thinking about a core riddle behind the COPD pathology and senescence/aging.
The question for interactive discussionBased on the above described facts, ideas and suggestions, additionally we would like to raise few questions: Is there a strong connection between COPD/emphysema and aging? Is COPD/emphysema a cause or consequence of aging? Senescence and aging are shown to severely impair the regenerative potential of cells and tissues. Thus, does COPD/emphysema develop due to accumulated stress or inadequate repair? Is this disease age-related, or just the lung cannot recover anymore that efficiently? Should the focus of ongoing research be directed to the mechanisms of insult or repair? These questions are directed to all the scientists, clinicians and others interested in this topic across the world to try to answer and expose their own views, perspectives and visions in the next volume/issue of the PVRI Chronicle.
References:
1. Rabe KF, Hurd S, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2007; 176:532-55.2. Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 2003; 22:672-88.
3. Fletcher C, Peto R. The natural history of chronic airflow obstruction. BMJ 1977; 1:1645–1648.4. Karrasch S, Holz O, Jorres RA. Aging and induced senescence as factors in the pathogenesis of lung emphysema. Respiratory Medicine 2008; 102, 1215-1230.5. Kirkwood, T.B. Understanding the odd science of aging. Cell 2005; 120, 437–447. 6. Lopez-Otin C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell 2013; 153:1194–2177. Johnson TE. Recent results: biomarkers of aging. Exp Gerontol 2006; 41:1243-6.8. Vidak S and Foisner R. Molecular insights into the premature aging disease progeria. Histochem Cell Biol 2016; 145:401–417.9. Bratic A, Larsson NG. The role of mitochondria in aging. J. Clin. Investig. 2013; 123, 951–957.10. Promislow DE. DNA repair and the evolution of longevity: a critical analysis. J. Theor. Biol. 1994; 170, 291–300.11. Wallace DC. Mitochondrial diseases in man and mouse. Science 1999; 283, 1482–1488.12. Kim S, Kaminker P, and Campisi J. Telomeres, aging and cancer: in search of a happy ending. Oncogene 2002; 21, 503–511. 13. Amiri HM, Cooper JP, Nugent K. The role of telomere shortening in the development and progression of COPD. The Southwest Respiratory and Critical Care Chronicles 2013.14. Carrard G, Bulteau AL, Petropoulos I, and Friguet B. Impairment of proteasome structure and function in aging. Int. J. Biochem. Cell Biol. 2002; 34, 1461–1474. 15. Friguet B. Oxidized protein degradation and repair in ageing and oxidative stress. FEBS Lett 2006; 580:2910-6.16. Soti C and Csermely P. Aging and molecular chaperones. Exp. Gerontol. 2003; 38, 1037–1040.
Journal club
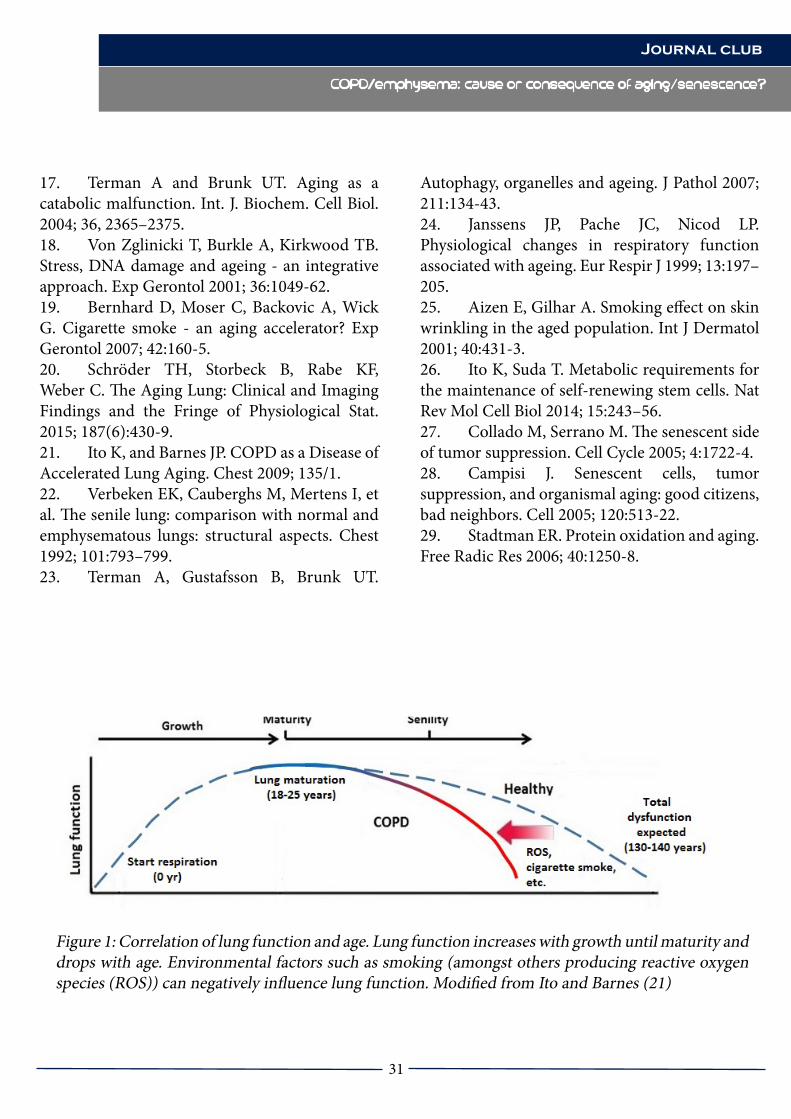
31
COPD/emphysema: cause or consequence of aging/senescence?
COPD/emphysema: cause or consequence of aging/senescence?
17. Terman A and Brunk UT. Aging as a catabolic malfunction. Int. J. Biochem. Cell Biol. 2004; 36, 2365–2375.18. Von Zglinicki T, Burkle A, Kirkwood TB. Stress, DNA damage and ageing - an integrative approach. Exp Gerontol 2001; 36:1049-62.19. Bernhard D, Moser C, Backovic A, Wick G. Cigarette smoke - an aging accelerator? Exp Gerontol 2007; 42:160-5.20. Schröder TH, Storbeck B, Rabe KF, Weber C. The Aging Lung: Clinical and Imaging Findings and the Fringe of Physiological Stat. 2015; 187(6):430-9.21. Ito K, and Barnes JP. COPD as a Disease of Accelerated Lung Aging. Chest 2009; 135/1.22. Verbeken EK, Cauberghs M, Mertens I, et al. The senile lung: comparison with normal and emphysematous lungs: structural aspects. Chest 1992; 101:793–799.23. Terman A, Gustafsson B, Brunk UT.
Autophagy, organelles and ageing. J Pathol 2007; 211:134-43.24. Janssens JP, Pache JC, Nicod LP. Physiological changes in respiratory function associated with ageing. Eur Respir J 1999; 13:197–205.25. Aizen E, Gilhar A. Smoking effect on skin wrinkling in the aged population. Int J Dermatol 2001; 40:431-3.26. Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol 2014; 15:243–56.27. Collado M, Serrano M. The senescent side of tumor suppression. Cell Cycle 2005; 4:1722-4.28. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005; 120:513-22.29. Stadtman ER. Protein oxidation and aging. Free Radic Res 2006; 40:1250-8.
Journal club
Figure 1: Correlation of lung function and age. Lung function increases with growth until maturity and drops with age. Environmental factors such as smoking (amongst others producing reactive oxygen species (ROS)) can negatively influence lung function. Modified from Ito and Barnes (21)

32
Targeting PPAR in Lung fibroblasts: Prospects of therapeutic treatment for IPF
Targeting PPAR in Lung fibroblasts: Prospects of therapeutic treatment for IPF Prelude
Idiopathic pulmonary fibrosis (IPF) is a devastating disease and the most common form of idiopathic interstitial pneumonias1. Formerly, IPF was described as a chronic inflammatory disease resulting from a response to an unknown stimulus which later progresses to lung injury and fibrosis [2, 3] Median survival rate of patients diagnosed with IPF is reported to be from 2.5 to 3.5 years [1, 4] and most available treatment compounds have not proved effective [5] including the antioxidant N‐acetylcysteine which is only known to slow the decline in lung functions though has no impact on mortality 6. Another approach to therapeutically target IPF was by using agonists of the peroxisome proliferator-activated receptor γ (PPARγ), as they were reported to possess anti-inflammatory and antifibrotic potentials [7, 8, 9, 10]. However, first trials of PPARγ agonists in pulmonary fibrosis did not yield conclusive results, possibly due to vastly diverse experimental approaches used to assess the efficacy of the treatment. Thus, the role of PPARγ in the pathomechanisms of IPF remains elusive. This interactive discussion therefore summarizes the therapeutic role of PPARγ in IPF model systems and proposes new experimental strategies for treatment of IPF with PPARγ agonists. Our aim is also to challenge all the researchers interested in this field to express their opinions on the potential of using PPARγ-associated treatments for IPF patients.IPF is a progressive disease that can result in respiratory failure11. IPF is characterized by scarring of lung tissues due to uncontrolled deposition of extracellular matrix proteins (ECM) and further disruption of the lung architecture and normal function [12]. Clinical staging of IPF has been well delineated to enforce management practices and clinical
Corresponding AuthorDr. Srikanth Karnati, PhD
Institute for Anatomy and Cell Biology II, Division of Medical Cell Biology,
Justus Liebig University, Aulweg 123, D-35385
GiessenGermany
Eistine Boateng (1)Natalia El-Merhie (1) Omelyan Trompak (2)
Srinu Tumpara(1)Michael Seimetz(3)
Adrian PilatzEveline Baumgart-Vogt (1)
Srikanth Karnati (1)
(1)Institute for Anatomy and Cell Biology II
Division of Medical Cell BiologyJustus Liebig UniversityAulweg 123, D-35385
Giessen, Germany
(2)Institute of Neuropathology
University of GiessenGermany
(3)Excellence Cluster Cardio-Pulmonary System (ECCPS)
Universities of Giessen and Marburg Lung Center (UGMLC) Member of the German Center for Lung Research (DZL)
Giessen, Germany
(4)Department of Urology
Pediatric Urology and Andrology, Justus Liebig University Giessen
Giessen, Germany
Journal club Targeting PPAR in Lung fibroblasts: Prospects of therapeutic treatment for IPF

33
Targeting PPAR in Lung fibroblasts: Prospects of therapeutic treatment for IPF
Targeting PPAR in Lung fibroblasts: Prospects of therapeutic treatment for IPF
Journal club
trials [26]. Clinical development of IPF starts with a slow progression over time followed by acute exacerbation of symptoms, loss of lung function, and early death [11, 27, 28]. Devastating effects of IPF and high mortality rates therefore demand biological studies to experimentally model the disease, find the underlying molecular mechanisms, and target the pathways involved in IPF progression as means of potential treatment. Despite the clear clinical staging of the disease, pathogenesis of IPF has not yet been fully elucidated. Current hypothesis suggests that an initial lung injury disrupts the alveolar-capillary basement membrane leading to a deterioration of re-epithelialisation and re-endothelialisation and a further cytokine-mediated fibroblast proliferation [13]. One of the major cytokines involved is the transforming growth factor β 1 (TGFβ1) [14]. TGFβ1 is a ubiquitously expressed cytokine that functions in numerous biological pathways and was shown to be both important for normal physiology, and implicated in playing a role in diverse diseases [15, 16]. Particularly, this cytokine is known to mediate the recruitment and activation of fibroblasts during injury17, stimulate regeneration of connective tissue, and inhibit its degradation18. Additionally, TGFβ1 stimulation leads to transdifferention of fibroblast into aggressive myofibroblast which are responsible for the production and secretion of components of the ECM [19, 20, 21, 22]. Consequently, interfering with the pathways that lead to myofibroblast expansion have been proposed as potential means of treatment for IPF patients [23] since regimens targeting inflammation have been ineffective [24, 25]. Antifibrotic potential of PPARγ agonists in IPF In previous studies PPARs were shown to control multiple physiological activities [29] and disease conditions. PPARs are part of a superfamily of nuclear receptors activated by lipid-derived substrates [30] and regulate gene expression
by binding to specific nucleotide sequences called PPAR response elements (PPREs) within promoters31 together with their heterodimeric partners, retinoid X receptors (RXRs) [32]. Three isotypes of PPARs characterized in lower vertebrates and mammals are PPARα, PPARβ, and PPARγ [33], of which the latter is subdivided into γ1, γ234, γ335, and γ436 isoforms. PPARs are ubiquitously and specifically expressed in various tissues and cell types [37]. Recently natural and synthetic activators of the PPARγ have been reported to elicit antifibrotic properties and inhibit myofibroblast differentiation and collagen secretion in cultured human lung fibroblasts [10, 38, 39, 40]. Burgess et al. (2005) suggested that endogenous and synthetic activators of PPARγ can control TGFβ1-mediated profibrotic effects, reverse primary human pulmonary myofibroblast differentiation and collagen production10. In this study, primary human fibroblasts were treated with different PPARγ agonists concomitant with recombinant human TGFβ1. The therapeutic effect of PPARγ ligands on myofibroblast after exogenous addition of TGFβ1 to fibroblasts in culture would possibly model the pathogenic mechanism in patients though this may not be the focus of the authors. Furthermore, in this study, PPARγ antagonist could not reverse the inhibition of differentiation of fibroblasts into myofibroblasts by PPARγ agonists, indicating towards an existence of a PPARγ-independent mechanism of agonist activity. Another study by Milam and colleagues et al (2008) demonstrated that treatment of lung fibroblasts in culture with PPARγ agonist and in vivo administration of the agonists to mice in the bleomycin-induced lung fibrosis model inhibited TGFβ1-induced myofibroblast differentiation and collagen secretion, showing the potential of PPARγ agonist as novel therapeutic agents for the treatment of fibrotic lung diseases

34
Targeting PPAR in Lung fibroblasts: Prospects of therapeutic treatment for IPF
[38]. Noteworthy is the fact that in this study, mechanism of the inhibition of the profibrotic phenotype caused by TGFβ1 is addressed. In the models used PPARγ agonists were administered in a pre-treatment setting: before TGFβ1 stimulation in vitro, and before induction of lung injury in vivo. Despite the important findings of the described study, a drawback in regards to the in vivo model used has to be mentioned: it is known that inflammation caused by lung injury may lead to many disease outcomes and not IPF alone41. In addition, pre-treatment with the PPARγ ligand before administration of bleomycin resulted in the interference with the inflammatory process before a possible lung remodeling could occur. Thus, the experimental approach could only confirm the anti-inflammatory potential of PPARγ agonists, which was already reported earlier29, 42. In attempts to assess the efficacy of agonist treatment in an established IPF model, troglitazone was given to another group of mice on day 11 following bleomycin instillation. However, authors provided no evidence of the extent of tissue remodeling and fibrosis at the day of the beginning of treatment. In all, in this study the authors investigated only the preventive role of PPARγ agonists and not the proposed therapeutic effects on an established disease. Nevertheless, the experimental models described highlight new ways of investigating the therapeutic potential of PPARγ ligands in vitro and in vivo.Ferguson et al. (2009) also reported that PPARγ ligands and 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid (CDDO) can block critical TGFβ1-mediated profibrogenic activities through pathways independent of PPARγ after co-treating human lung fibroblasts with TGFβ1 and PPARγ ligands39. The study did not investigate the exogenously secreted collagens and fibronectin in the ECM. Moreover, it is well known that IPF progression is characterized by aberrant cellular release of collagens, proteoglycans,
tenascin, laminin, and fibronectin into the ECM43. The study also proposed a possible role of PPARγ agonists and CDDO in the inhibition of myofibroblast differentiation in IPF, though it would be interesting to know the regulation of the fibrosis markers by PPAR agonists after TGFβ1 stimulation. A recent study by Lin et al. (2010) defined the role of rosiglitazone, a PPARγ agonist, in suppressing myofibroblast transdifferentiation40. The study results indicate towards a potential preventive role of the PPARγ agonist in pulmonary fibrosis, as observed while co-treating human fetal lung fibroblasts with TGFβ1 and rosiglitazone. Rosiglitazone downregulated αSMA, however other IPF markers, specifically expression of ECM components such as collagens, were not investigated.
Questions for the interactive discussion
Translation of in vitro and in vivo studies to clinical applications may be very challenging from the viewpoint of predicting the efficacy of the developed experimental approaches in alleviating clinical symptoms of real patients. Though much progress has been made towards understanding of the pathogenesis of IPF with regards to PPARγ activity, studies are often limited by focusing on the inhibition of fibroblast to myofibroblast conversion using PPARγ agonists (figure 1A). Cells accumulated at the fibroblastic foci of IPF patients present a mixed population of fibroblasts and myofibroblasts44. We therefore query the molecular effects of PPARγ agonists on the already existing myofibroblasts or TGF-β1 stimulated fibroblasts. Additionally, we propose that experimental models should be developed to investigate the role of PPARγ ligands on ECM deposition and reversal of the progressive fibrotic phenotype after TGFβ1 stimulation in vitro (figure
Journal club Targeting PPAR in Lung fibroblasts: Prospects of therapeutic treatment for IPF
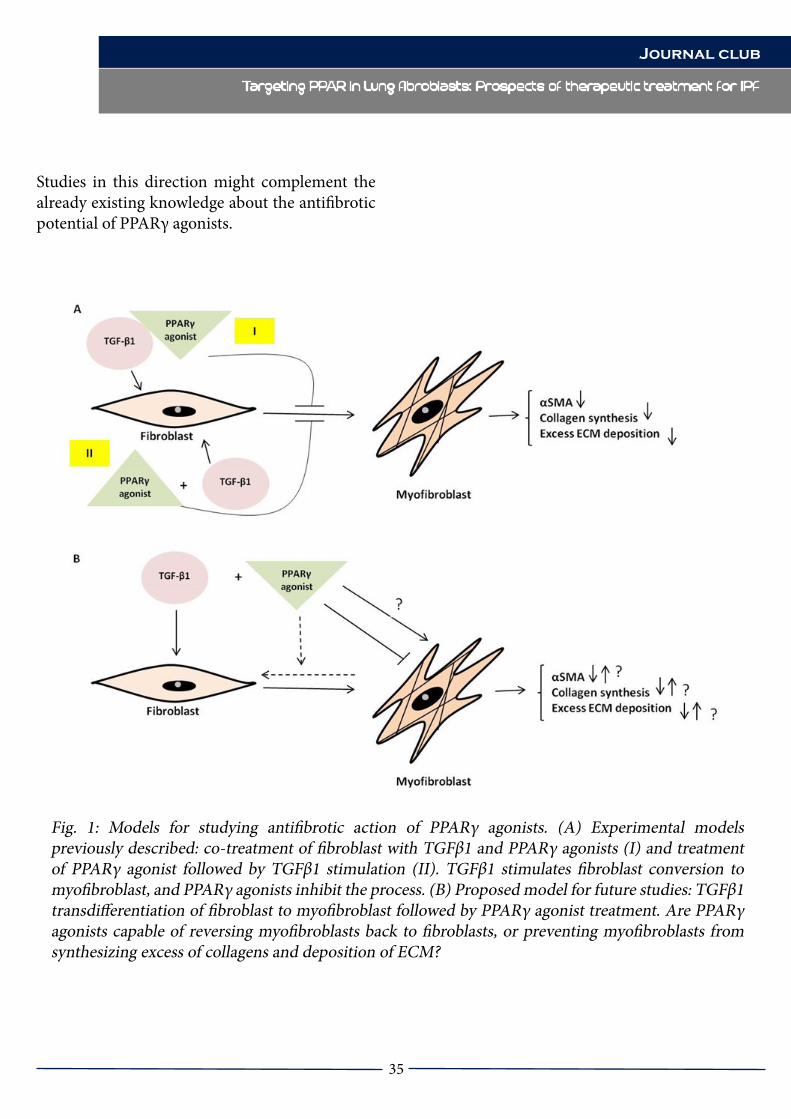
35
Targeting PPAR in Lung fibroblasts: Prospects of therapeutic treatment for IPF
Studies in this direction might complement the already existing knowledge about the antifibrotic potential of PPARγ agonists.
Fig. 1: Models for studying antifibrotic action of PPARγ agonists. (A) Experimental models previously described: co-treatment of fibroblast with TGFβ1 and PPARγ agonists (I) and treatment of PPARγ agonist followed by TGFβ1 stimulation (II). TGFβ1 stimulates fibroblast conversion to myofibroblast, and PPARγ agonists inhibit the process. (B) Proposed model for future studies: TGFβ1 transdifferentiation of fibroblast to myofibroblast followed by PPARγ agonist treatment. Are PPARγ agonists capable of reversing myofibroblasts back to fibroblasts, or preventing myofibroblasts from synthesizing excess of collagens and deposition of ECM?
Journal club

36
References1. American Thoracic Society (2000). Idiopathic pulmonary fibrosis: diagnosis and treatment. International Consensus Statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med.161:646–64.2. Hamman L, Rich A R (1994). Acute diffuse interstitial fibrosis of the lungs. Bull John Hopkins Hosp. 74:177–212.3. Noble P W, Homer R J (2005). Back to the future: historical perspective on the pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol. 33:113–20.4. American Thoracic Society/European Respiratory Society (2002). International multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 165:277–304.5. Bourke S J (2006). Interstitial lung disease: progress and problems. Postgrad Med J. 82(970):494-9.6. Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen H M, MacNee W, Thomeer M, Wallaert B, Laurent F, Nicholson A G, Verbeken E K, Verschakelen J, Flower C D, Capron F, Petruzzelli S, De Vuyst P, van den Bosch J M, Rodriguez-Becerra E, Corvasce G, Lankhorst I, Sardina M, Montanari M; IFIGENIA Study Group (2005). High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 353(21):2229-42. 7. Chinetti G, Fruchart J C, Staels B (2000). Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm Res. 49: 497–505.8. Chinetti G, Fruchart J C, Staels B (2003). Peroxisome proliferator-activated receptors and inflammation: from basic science to clinical applications. Int J Obes Relat Metab Disord. Suppl 3: S41–45.
9. Delerive P, Fruchart J C, Staels B (2001). Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol. 169: 453–459.10. Burgess H A, Daugherty L E, Thatcher T H, Lakatos H F, Ray D M, Redonnet M, Phipps R P, Sime P J (2005). PPAR gamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 288: L1146-L1153.11. Ley B, Collard H R, King T E Jr (2011). Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 183(4):431-40.12. Todd N W, Luzina I G, Atamas S P (2012). Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair. 5(1):11. 13. Strieter R M, Mehrad B (2009). New mechanisms of pulmonary fibrosis. Chest. 136(5):1364-70.14. Sheppard D (2006). Transforming growth factor beta: a central modulator of pulmonary and airway inflammation and fibrosis. Proc Am Thorac Soc. 3(5):413-7.15. Lawrence D A (1996). Transforming growth factor-beta: a general review. Eur Cytokine Network. 7:363–4.16. Border W A, Noble N A (1994). Transforming growth factor-beta‚ In tissue fibrosis. N Engl J Med. 331:1286–92.17. Sureshbabu A, Tonner E, Allan G J, Flint D J (2011). Relative Roles of TGF-β and IGFBP-5 in Idiopathic Pulmonary Fibrosis. Pulm Med. 2011:517687.18. Gharaee-Kermani M, Hu B, Phan S H, Gyetko M R (2009). Recent advances in molecular targets and treatment of Idiopathic Pulmonary Fibrosis: focus on TGFβ signaling and the myofibroblast. Curr Med Chem. 16(11):1400-17.19. Phan S H (2002). The myofibroblast in pulmonary fibrosis. Chest 122: 286S– 289S.
Journal club
Targeting PPAR in Lung fibroblasts: Prospects of therapeutic treatment for IPF
Targeting PPAR in Lung fibroblasts: Prospects of therapeutic treatment for IPF

37
20. Vaughan M B, Howard E W, Tomasek J J (2000). Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res. 257: 180–189. 21. Leask A, Abraham D J (2004). TGF-beta signaling and the fibrotic response. FASEB J. 18: 816–827.22. Moore M W, Herzog E L (2013). Regulation and Relevance of Myofibroblast Responses in Idiopathic Pulmonary Fibrosis. Current pathobiology reports. 1(3):199-208.23. Bagnato G, Harari S (2015). Cellular interactions in the pathogenesis of interstitial lung diseases. Eur Respir Rev. 24: 102–114.24. Richeldi L, Davies H R, Ferrara G, Franco F (2003). Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev. 3:CD002880.25. Davies H R, Richeldi L, Walters E H (2003). Immunomodulatory agents for idiopathic pulmonary fibrosis. Cochrane Data-base Syst Rev. 3:CD003134.26. Kolb M, Collard H R (2014). Staging of idiopathic pulmonary fibrosis: past, present and future. Eur Respir Rev. 23: 220–224.27. Fernández Pérez E R, Daniels C E, Schroeder D R, St Sauver J, Hartman T E, Bartholmai B J, Yi E S, Ryu J H (2010). Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest. 137: 129–137.28. Collard H R, Moore B B, Flaherty K R, Brown K K, Kaner R J, King T E Jr, Lasky J A, Loyd J E, Noth I, Olman M A, Raghu G, Roman J, Ryu J H, Zisman D A, Hunninghake G W, Colby T V, Egan J J, Hansell D M, Johkoh T, Kaminski N, Kim D S, Kondoh Y, Lynch D A, Müller-Quernheim J, Myers J L, Nicholson A G, Selman M, Toews G B, Wells A U, Martinez F J; Idiopathic Pulmonary Fibrosis Clinical Research Network Investigators (2007). Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit
Care Med. 176: 636–643.29. Lakatos H F, Thatcher T H, Kottmann R M, Garcia T M, Phipps R P, Sime P J (2007). The Role of PPARs in Lung Fibrosis. PPAR Research. 2007:71323. 30. Tyagi S, Gupta P, Saini A S, Kaushal C, Sharma S (2011). The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J Adv Pharm Technol Res. 2(4): 236–240.31. Berger J, Moller D E. (2002). The mechanisms of action of PPARs. Annu Rev Med. 53:409-35.32. Krey G, Keller H, Mahfoudi A, Medin J, Ozato K, Dreyer C, Wahli W J (1993). Xenopus peroxisome proliferator activated receptors: genomic organization, response element recognition, heterodimer formation with retinoid X receptor and activation by fatty acids. Steroid Biochem Mol Biol. 47(1-6):65-73.33. Wahli W (2002). Peroxisome proliferator-activated receptors (PPARs): from metabolic control to epidermal wound healing. Swiss Med Wkly. 132(7-8):83-91.34. Fajas L, Auboeuf D, Raspe E, Schoonjans C, Lefebvre A M, Saladin R, Najib Laville J M, Fruchart J C, Deeb S, Vidal-Puig A, Flier J, Briggs M R, Staels B, Vidal H, Auwerx J (1997). Organization, promoter analysis, and expression of the human PPARg gene. J. Biol. Chem. 272, 18779–18789.35. Fajas L, Fruchart J C, Auwerx J (1998). PPARgamma3 mRNA: A distinct PPAR gamma mRNA subtype transcribed from an independent promoter. FEBS Lett. 438, 55–60.36. Sundvold H, Lien S (2001). Identification of a novel peroxisome proliferator-activated receptor (PPAR) gamma promoter in man and transactivation by the nuclear receptor RORalpha1. Biochem Biophys Res Commun. 287(2):383-90.37. Evans R M, Barish G D, Wang Y X (2004).
Journal club
Targeting PPAR in Lung fibroblasts: Prospects of therapeutic treatment for IPF

38
PPARs and the complex journey to obesity. Nat Med. 10:355–61.38. Milam J E, Keshamouni V G, Phan S H, Hu B, Gangireddy S R, Hogaboam C M, Standiford T J, Thannickal V J, Reddy R C (2008). PPAR-gamma agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 294:L891-901.39. Ferguson H E, Kulkarni A, Lehmann G M, Garcia-Bates T M, Thatcher T H, Huxlin K R, Phipps R P, Sime P J (2009). Electrophilic peroxisome proliferator-activated receptor-gamma ligands have potent antifibrotic effects in human lung fibroblasts. Am J Respir Cell Mol Biol. 41:722-730. 40. Lin Q, Fang L P, Zhou W W, Liu X M (2010). Rosiglitazone inhibits migration, proliferation, and phenotypic differentiation in cultured human lung fibroblasts. Exp Lung Res. 36:120-128.41. Wallace W A, Fitch P M, Simpson A J, Howie S E (2007). Inflammation-associated remodelling and fibrosis in the lung – a process and an end point. Int J Exp Pathol. 88(2):103-110.42. Rizzo G, Fiorucci S (2006). PPARs and other nuclear receptors in inflammation. Curr Opin Pharmacol. 6:421–427.43. Clarke D L, Carruthers A M, Mustelin T, Murray L A (2013). Matrix regulation of idiopathic pulmonary fibrosis: the role of enzymes. Fibrogenesis and Tissue Repair. 26:6(1):20.44. Scotton C J, Chambers R C (2007). Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 132:1311–1321.
Journal club
Targeting PPAR in Lung fibroblasts: Prospects of therapeutic treatment for IPF

39

40
- current opinions about hot topics and state-of-the-art
Journal club
Diagnosis and markers for the assessment of COPD; can a consensus be reached?
Prelude
Chronic obstructive pulmonary disease (COPD) is a progressive disorder which is characterized by airflow limitation that is not fully reversible [1]. It affects central airways causing chronic bronchitis, peripheral airways leading to small airway disease, and the lung parenchyma giving rise to emphysema [2]. Further, COPD has already become the third leading cause of death worldwide [3]. The major therapeutic goal is prevention of further exacerbations, yet the currently available treatment options are not considered to be very effective. Hence, implementing the onset biomarkers for the early diagnosis of the disease could improve the clinical assessment of COPD. Here we summarize the currently available markers and methods for COPD evaluation and diagnosis.Generally, the assessment of COPD is based on physiological, symptomatic and nowadays – on newly emerging biological markers. Physiological markers are not straightforward to identify and validate, further requiring a standardized approach. These markers include:
Spirometry – this is used to measure the volume of forcibly exhaled air in 1 second (FEV1)/ forced vital capacity FVC [4], which is a marker for determination of staging, diagnosis, and treatment of the disease [5]. A fixed ratio of (FEV1/FVC)<70% defines airflow limitation. COPD patients lose lung function over a period of time, thus, a decline in FEV1 is used as an indicator of disease progression [6]. However, spirometry has its limitations. Since lung volumes are also affected during the normal process of aging, this method tends to be biased towards over diagnosis of the disease in some elderly patients [4]. Moreover, COPD is a complex disease which is accompanied by numerous pathological complications that are often ignored due to the poor understanding of
Corresponding AuthorDr. Srikanth Karnati, PhD
Institute for Anatomy and Cell Biology II, Division of Medical Cell Biology,
Justus Liebig University, Aulweg 123, D-35385
GiessenGermany
Eistine Boateng (1)Natalia El-Merhie (1) Omelyan Trompak (2)
Srinu Tumpara(1)Michael Seimetz(3)
Adrian PilatzEveline Baumgart-Vogt (1)
Srikanth Karnati (1)
(1)Institute for Anatomy and Cell Biology II
Division of Medical Cell BiologyJustus Liebig UniversityAulweg 123, D-35385
Giessen, Germany
(2)Institute of Neuropathology
University of GiessenGermany
(3)Excellence Cluster Cardio-Pulmonary System (ECCPS)
Universities of Giessen and Marburg Lung Center (UGMLC) Member of the German Center for Lung Research (DZL)
Giessen, Germany
(4)Department of Urology
Pediatric Urology and Andrology, Justus Liebig University Giessen
Giessen, Germany

41
- current opinions about hot topics and state-of-the-art
- current opinions about hot topics and state-of-the-art
Journal club
the relationship between spirometry and the symptoms [6].
Lung volume or lung hyperinflation characterizes an increase in the gas volume in the lungs in comparison to the predicted value [7]. It is another marker for the evaluation of COPD and determination of disease severity. Absolute lung volume is evaluated by measuring the increase in total lung capacity (TLC), functional residual capacity (FRC), residual volume (RV) and the decrease in inspiratory capacity (IC) [8]. Conventionally, lung hyperinflation is said to exist when the values of TLC, FRC and RV exceed 120–130% of the predicted volume [7]. These values are considered to be clinically relevant, however, cut-offs remain invalidated [7]. Moreover, high variability among COPD patients is a major problem in reproducing the cut-off values, and remains to be addressed in future studies [7].
Gas exchange (DL,CO) relies on the diffusing capacity of the lungs and is measured via the transfer of carbon monoxide from the airspace to the pulmonary capillary blood [9]. This method is used in the diagnosis of patients with emphysema [10]. A major draw-back in assessing DL,CO is the high price of the necessary equipment making it less available in the primary care units. Moreover, it requires specific training for the technical staff and its reproducibility depends on the expertise of the staff [9]. Furthermore, Salzman and colleagues argue that DL,CO cannot be definitive for either confirming or excluding the diagnosis of emphysema because a reduction in DL,CO accompanied by normal lung mechanics values (FEV1, FVC, FEV1/FVC, and lung volumes) might be suggestive of an emphysema, whereas normal DL,CO does not rule it out [11].
Exercise capacity or a 6-Minute Walk Test (6MWT) is the estimation of the distance covered
during a 6-min walk where oxygen saturation, cardiac frequency, and respiratory intensity are recorded [12]. There are many sources of variability for this test, such as patients’ age, sex, weight, height, etc., making this test highly unreliable in diagnosing COPD [8, 13].
The body mass index (BMI) and the BODE index. The BMI is calculated as weigh/height squared in kg/m2 in the journal the superscript is written wrongly. Body mass in this case is assumed to be comprised of fat mass and fat free mass (FFM). After accounting for BMI, FFM, airflow limitation (expressed by the FEV1), Dyspnea (expressed with the modified MRC scale), and exercise capacity (expressed with the 6-minute walking distance) an integral value is calculated – the so called “BODE index”, which provides information for COPD staging. In addition, the BODE index can serve as a prognostic factor of COPD where the decrease in BMI and FFM are associated with the impairment of the muscle function, health status, exercise capacity, and decreased survival of COPD patients [14, 15].
Imaging includes computed tomography (CT), functional positron emission tomography (PET) and magnetic resonance imaging (MRI). CT reflects the morphologic changes in the lung parenchyma, pulmonary vasculature, central and peripheral airways [16]. In addition, CT is the best way to assess the severity of emphysema [17]. A disadvantage of this technique is associated with radiation risk, limiting the multiple longitudinal scans in clinical trials. In contrast, MRI does not involve radiation and can be used as a substitute in the clinics [18]. However, MRI is more time-consuming and expensive [19]. Finally, functional imaging through (MRI) and (PET) using hyperpolarized helium and xenon can be done [20, 21]. However, further standardization is necessary to achieve optimal results.

42
Exacerbations are short periods of worsening of the patients’ symptoms and characterized by as sputum hyper-production, increased cough, and dyspnoea [22]. Exacerbations represent the disease progression and clinical instability, and are associated with an increased risk of mortality [23]. Exacerbations are recorded by the patients by using a questionnaire or a diary card where they provide the information on the frequency, time of onset, severity, and duration of the periods of worsening symptoms. However, lack of standardized criteria makes the evaluation and comparison of clinical studies difficult [4], resulting in a poor correlation between manifestation of exacerbations and pathological changes occurring in the lung [19].
Other markers used for the assessment of COPD belong to the symptomatic group. These markers are assessed by medical doctors by recording frequency and nature of coughing, colouring of sputum, shortness of breath, and wheeze [6]. However, there is no agreed form for quantifying symptomatic data, response options and the equivalence between different questionnaires [6]. The symptomatic measures of COPD comprise the following:
Baseline/Transition Dyspnea Index (BDI/TDI), Borg-Scale, and MRC (Medical Research Council) scale are the most frequently used scales for the identification of dyspnoea in clinical practice. The BDI/TDI is based on an interview conducted by the staff which converts the patients’ experience of dyspnoea into numerical parameters [8]. Such studies are limited, biased, and lack standardization when translating patient’s answers into parameters [24]. The Borg-scale (or CR-10), is a 10 point category scale that incorporates the description of the severity corresponding numbers [25]. CR-10 requires a detailed instruction for the patients on how to
use this scaling system [26]. The MRC scale is a five-point scale that describes breathlessness and the severity of dyspnoea [27]. This method is also limited by bias [28].
St. George’s Respiratory Questionnaire (SGRQ) covers three domains: frequency and severity of symptoms, activities that cause or are limited by breathlessness, and the impact these disturbances have on patients’ life, which is reflected in the end as an integral score. However, these scores are influenced by patient’s age, sex, education, and comorbidities [29].
Moreover, pulmonary biomarkers can also be used to assess the COPD disease status. These group of markers comprises an estimate of the presence and involvement of inflammatory cells (macrophages, neutrophils, and lymphocytes), levels of cytokines (IL-6, IL-8, TNF-α) myeloperoxidases (MPO), neutrophil elastases (NE), expired nitric oxide (NO), and carbon monoxide (CO), which can be sampled from either sputum, blood, bronchoalveolar lavage (BAL), exhaled breath, or bronchial biopsies [12]. Bronchial biopsies provide the information on the structural changes in the epithelium, muscles, and glands where structural components can be dissected and studied separately [30]. The interaction between inflammatory and resident cells can be investigated by immunostaining [12]. However, there are several drawbacks to bronchial biopsies. First, this procedure is invasive and unsafe to patients with a severe disease stage [31]. Second, the airway wall examined during biopsy might not reflect the pathological alterations in the lung parenchyma and peripheral airways.
Bronchoalveolar lavage (BAL) allows the study of chemical and cellular components in the epithelial lining fluid of the peripheral airways [32]. Unfortunately, the low sample numbers
- current opinions about hot topics and state-of-the-art
Journal club

43
obtained from BAL are not sufficient for statistical analysis.
Sputum analysis provides the information of the mediators in the central airways [33]. However, this analysis is not representing the environment in the distal airways that might be important for the diagnosis of COPD. Moreover, sputum contains thick mucus that has to be removed before processing the sample, and dithiothreitol (DTT) used to solubilize the sputum alters the proteins making them unrecognizable by the antibodies [34, 35].
Exhaled gas analysis is a non-invasive method for monitoring COPD airways inflammation [12]. It measures exhaled nitric oxide (eNO) and exhaled carbon monoxide (CO) in breath. Unfortunately, reproducibility and sensitivity issues are not yet been clarified [12].
Blood samples; there are several studies suggesting that CRP, TNF-α, IL-6 and IL-8 play a role in COPD and in its exacerbations [36, 37]. However, blood sampling requires more randomized studies with larger sample sizes in order to confirm the sensitivity and specificity of this method [38].
Summary and questions for interactive discussion
We summarized the available COPD assessment methods and their limitations. Currently, there exists no consensus concerning the best criteria to be used for the assessment of COPD despite the variety of markers and methods available for diagnosing this disease. However, reproducibility and effectiveness of the methods were shown to be limited. Moreover, by the time COPD patients seek medical help, they already manifest significant (and often severe) symptoms of the
disease. Therefore, we would like to forward the following questions to the scientific community worldwide: how reliable are the biological markers for COPD? What are the best and most specific markers to be used for assessment of COPD? What are the markers that will allow the early diagnosis of COPD? We would like to invite all those interested in this field to reply and express their views on this topic in the next volume of PVRI Chronicle.
Current available methods and markers of COPD assessmentAbbreviations: IL interleukin, MPO myeloperoxidase, NE neutrophil elastase, TNFα tumor necrosis factor alpha, CRP C-reactive protein, BAL bronchoalveolar lavage, NO nitric oxide, CO carbon monoxide, SGRQ St. George’s Respiratory Questionnaire, MRC medical research council, CT computed tomography, MRI magnetic resonance imaging, DL,CO diffusion capacity of the lung for carbon monoxide, PET functional positron emission tomography.
- current opinions about hot topics and state-of-the-art
- current opinions about hot topics and state-of-the-art
Journal club
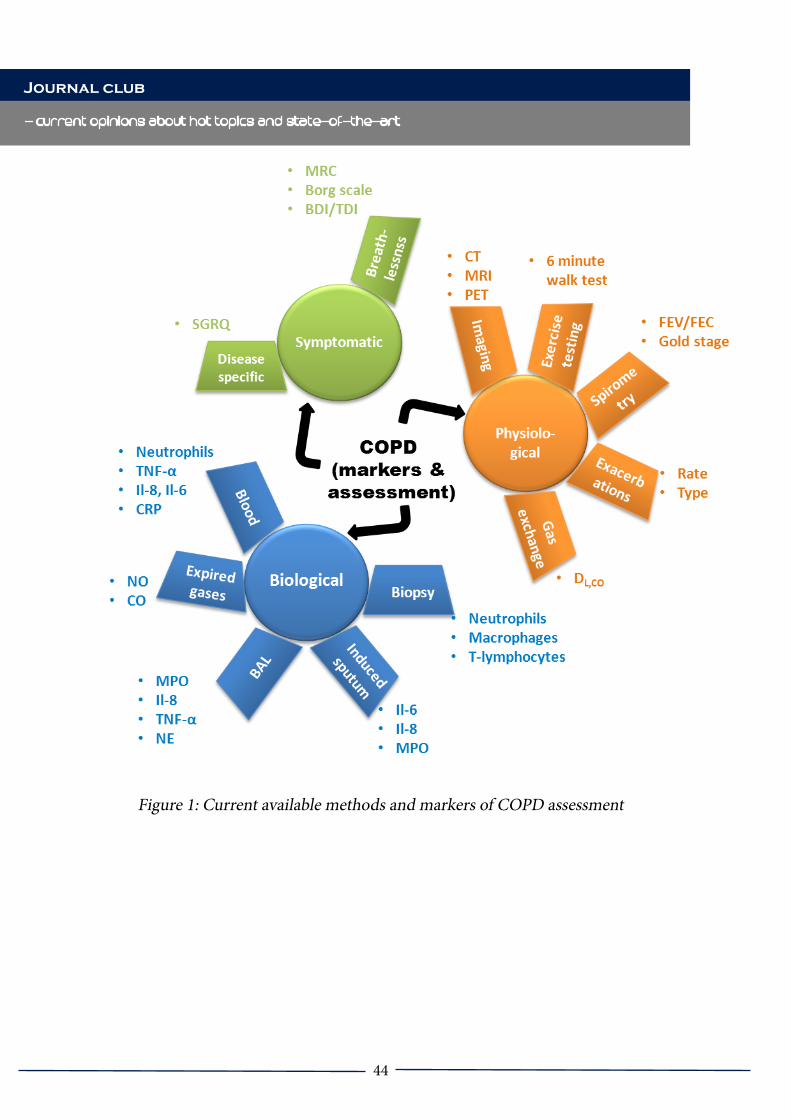
44
- current opinions about hot topics and state-of-the-art
Journal club
Figure 1: Current available methods and markers of COPD assessment

45
- current opinions about hot topics and state-of-the-art
- current opinions about hot topics and state-of-the-art
Journal club
Figure 1: Current available methods and markers of COPD assessment
References
1. Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS: Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med 2001, 163(5):1256-1276.2. Chronic obstructive pulmonary disease. National clinical guideline on management of chronic obstructive pulmonary disease in adults in primary and secondary care. Thorax 2004, 59 Suppl 1:1-232.3. WHO WHO: The top 10 causes of death Fact sheet N°310 Deaths across the globe: an overview. 2013.4. Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, Fukuchi Y, Jenkins C, Rodriguez-Roisin R, van Weel C et al: Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2007, 176(6):532-555.5. Hankinson JL, Crapo RO, Jensen RL: Spirometric reference values for the 6-s FVC maneuver. Chest 2003, 124(5):1805-1811.6. Jones PW, Agusti AG: Outcomes and markers in the assessment of chronic obstructive pulmonary disease. Eur Respir J 2006, 27(4):822-832.7. O’Donnell DE, Laveneziana P: The clinical importance of dynamic lung hyperinflation in COPD. COPD 2006, 3(4):219-232.8. Glaab T, Vogelmeier C, Buhl R: Outcome measures in chronic obstructive pulmonary disease (COPD): strengths and limitations. Respir Res, 11:79.9. American Thoracic Society. Single-breath carbon monoxide diffusing capacity (transfer factor). Recommendations for a standard technique--1995 update. Am J Respir Crit Care
Med 1995, 152(6 Pt 1):2185-2198.10. Sherrill DL, Enright PL, Kaltenborn WT, Lebowitz MD: Predictors of longitudinal change in diffusing capacity over 8 years. Am J Respir Crit Care Med 1999, 160(6):1883-1887.11. Salzman SH: Which pulmonary function tests best differentiate between COPD phenotypes? Respir Care, 57(1):50-57; discussion 58-60.12. Cazzola M, MacNee W, Martinez FJ, Rabe KF, Franciosi LG, Barnes PJ, Brusasco V, Burge PS, Calverley PM, Celli BR et al: Outcomes for COPD pharmacological trials: from lung function to biomarkers. Eur Respir J 2008, 31(2):416-469.13. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med 2002, 166(1):111-117.14. Landbo C, Prescott E, Lange P, Vestbo J, Almdal TP: Prognostic value of nutritional status in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999, 160(6):1856-1861.15. Schols AM, Broekhuizen R, Weling-Scheepers CA, Wouters EF: Body composition and mortality in chronic obstructive pulmonary disease. Am J Clin Nutr 2005, 82(1):53-59.16. Sverzellati N, Molinari F, Pirronti T, Bonomo L, Spagnolo P, Zompatori M: New insights on COPD imaging via CT and MRI. Int J Chron Obstruct Pulmon Dis 2007, 2(3):301-312.17. Newell JD, Jr., Hogg JC, Snider GL: Report of a workshop: quantitative computed tomography scanning in longitudinal studies of emphysema. Eur Respir J 2004, 23(5):769-775.18. de Lange EE, Altes TA, Patrie JT, Battiston JJ, Juersivich AP, Mugler JP, 3rd, Platts-Mills TA: Changes in regional airflow obstruction over time in the lungs of patients with asthma: evaluation with 3He MR imaging. Radiology 2009, 250(2):567-575.19. Barker BL, Brightling CE: Phenotyping the heterogeneity of chronic obstructive pulmonary disease. Clin Sci (Lond), 124(6):371-387.

46
20. Jones HA, Marino PS, Shakur BH, Morrell NW: In vivo assessment of lung inflammatory cell activity in patients with COPD and asthma. Eur Respir J 2003, 21(4):567-573.21. Suga K, Tsukuda T, Awaya H, Matsunaga N, Sugi K, Esato K: Interactions of regional respiratory mechanics and pulmonary ventilatory impairment in pulmonary emphysema: assessment with dynamic MRI and xenon-133 single-photon emission CT. Chest 2000, 117(6):1646-1655.22. Schmier JK, Halpern MT, Higashi MK, Bakst A: The quality of life impact of acute exacerbations of chronic bronchitis (AECB): a literature review. Qual Life Res 2005, 14(2):329-347.23. Anzueto A, Sethi S, Martinez FJ: Exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc 2007, 4(7):554-564.24. Campbell ML: Dyspnea prevalence, trajectories, and measurement in critical care and at life’s end. Curr Opin Support Palliat Care, 6(2):168-171.25. Borg GA: Psychophysical bases of perceived exertion. Med Sci Sports Exerc 1982, 14(5):377-381.26. Mador MJ, Rodis A, Magalang UJ: Reproducibility of Borg scale measurements of dyspnea during exercise in patients with COPD. Chest 1995, 107(6):1590-1597.27. Fletcher CM, Elmes PC, Fairbairn AS, Wood CH: The significance of respiratory symptoms and the diagnosis of chronic bronchitis in a working population. Br Med J 1959, 2(5147):257-266.28. Rennard S, Decramer M, Calverley PM, Pride NB, Soriano JB, Vermeire PA, Vestbo J: Impact of COPD in North America and Europe in 2000: subjects’ perspective of Confronting COPD International Survey. Eur Respir J 2002, 20(4):799-805.29. Ferrer M, Villasante C, Alonso J, Sobradillo
V, Gabriel R, Vilagut G, Masa JF, Viejo JL, Jimenez-Ruiz CA, Miravitlles M: Interpretation of quality of life scores from the St George’s Respiratory Questionnaire. Eur Respir J 2002, 19(3):405-413.30. Fuke S, Betsuyaku T, Nasuhara Y, Morikawa T, Katoh H, Nishimura M: Chemokines in bronchiolar epithelium in the development of chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2004, 31(4):405-412.31. Hattotuwa K, Gamble EA, O’Shaughnessy T, Jeffery PK, Barnes NC: Safety of bronchoscopy, biopsy, and BAL in research patients with COPD. Chest 2002, 122(6):1909-1912.32. Koutsokera A, Kostikas K, Nicod LP, Fitting JW: Pulmonary biomarkers in COPD exacerbations: a systematic review. Respir Res, 14:111.33. Alexis NE, Hu SC, Zeman K, Alter T, Bennett WD: Induced sputum derives from the central airways: confirmation using a radiolabeled aerosol bolus delivery technique. Am J Respir Crit Care Med 2001, 164(10 Pt 1):1964-1970.34. Djukanovic R: Induced sputum--a tool with great potential but not without problems. J Allergy Clin Immunol 2000, 105(6 Pt 1):1071-1073.35. Kelly MM, Keatings V, Leigh R, Peterson C, Shute J, Venge P, Djukanovic R: Analysis of fluid-phase mediators. Eur Respir J Suppl 2002, 37:24s-39s.36. Gan WQ, Man SF, Senthilselvan A, Sin DD: Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax 2004, 59(7):574-580.37. Agusti AG: Systemic effects of chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005, 2(4):367-370; discussion 371-362.38. Wouters EF: Local and systemic inflammation in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005, 2(1):26-33.
- current opinions about hot topics and state-of-the-art
Journal club

47
- current opinions about hot topics and state-of-the-art

48
Stress on Lungs: a search through 8-Iso Prostaglandin-F2
cardiovascular
High Altitude Pulmonary Edema (HAPE) is a form of altitude illness that develops in travellers on rapid ascent to or physical exertion at altitudes (> 2500 m; 8200 ft) [1,2]. The altitude, speed and mode of ascent and, above all, individual susceptibility are the most important determinants for the occurrence of HAPE. It commonly strikes the second night at a new altitude and rarely occurs after more than 4 days at a given altitude, owing to adaptive cellular and biochemical changes in pulmonary vessels [1,2]. Briefly, HAPE is characterized by hypoxia-induced pulmonary hypertension (PH), oxygen sensing redox switches and intravascular over perfusion [1-4]. Prolonged exposure to HA increases oxidative damage, which could be the consequence of increased reactive oxygen species (ROS), decreased antioxidants and free radical-mediated reduction in pulmonary NO bioavailability [4,5]. Ladakhi highlanders, living for several thousand years at such a great height have adapted mechanisms different from lowlanders for combating oxidative stress, which is the most severe repercussion of HA [5,6]. Hence, the relevant pathways and its constituents that maintain redox balance become an important focus of attention. In this regard 8-iso prostaglandin F2α (8-isoPGF2α) is believed to be one of the most accurate markers to estimate oxidative stress, providing an important
tool to explore the role of oxidative stress in the pathogenesis of human disease such as HAPE [3,5] .
8-Iso Prostaglandin-F2αIsoprostanes are prostaglandin(PG)-like substances that are produced in vivo independently of cyclooxygenase (COX) enzymes, mainly by free radical-induced peroxidation of arachidonic acid [7]. The formation of PG-like compounds during auto-oxidation of polyunsaturated fatty acids was first reported in the mid-1970s, but isoprostanes were not discovered to be formed in vivo in humans until 1990 [8]. F2-isoprostanes are a group of 64 compounds isomeric in structure to cyclooxygenase-derived PGF2 and one of the most important isoforms is 8-isoPGF2α [7,8]. They are released in response to cellular activation; circulate in plasma and are excreted in the urine. Morrow et al. first described the non-enzymatic production of a series of prostaglandin-like compounds during peroxidation of membrane phospholipids by free radicals and reactive oxygen species. Further, isoprostanes, appear to act through tyrosine kinase, Rho, and Rho kinase (ROCK), leading to decreased activity of myosin light chain phosphatase (MLCP) [8]. The measurement of 8-iso-PGF2α in tissues and/or biological fluids provides a valuable approach to the quantification of oxidative stress as well as a biochemical basis for assessing therapeutic intervention, as there is tremendous increase in reactive oxygen species at hypobaric hypoxia thereby predisposing the healthy sojourners towards HAPE [3]. Increased formation of 8-isoPGF2α has been detected in human cardiovascular diseases; it associates with enhanced plasma levels of noradrenaline and angiotensin II contributing to elevated vasoconstrictor effects and induces mitogenesis in vascular smooth muscle cells [3,5]. Furthermore,
Corresponding AuthorZahra Ali
University of Kent, School of Pharmacy, Chatham, ME4 4TB, UK
Stress on Lungs: A Search Through 8-ISO Prostaglandin-F2A
Learner’s Corner

49
8-Isoprostaglandin concentration was found to be elevated in asthma, COPD [9], cystic fibrosis (CF) [10] and pulmonary sarcoidosis [11]. A considerable body of literature exists documenting an increased production of indicators of oxidative stress in breath, blood, urine and tissue of laboratory rats in response to hypoxia [3, 12-13].In conclusion, increased 8-iso-PGF2α level has been associated with oxidative stress and pulmonary stress that, in turn, contributes to endothelial dysfunction, which is vital in the development of HAPE. Thus, this molecule holds immense potential for providing a major improvement in the field of high altitude.
References
1. Bärtsch P, Mairbäurl H, Maggiorini M, Swenson ER. Physiological aspects of high-altitude pulmonary edema. J Appl Physiol (1985). 2005;98(3):1101-10.2. Swenson ER, Maggiorini M, Mongovin S, Gibbs JS, Greve I, Mairbäurl H, Bärtsch P. Pathogenesis of high-altitude pulmonary edema: inflammation is not an etiologic factor. JAMA 2002;287:2228-35.3. Mishra A, Ali Z, Vibhuti A, Kumar R, Alam P, Ram R, Thinlas T, Mohammad G, Pasha MA. CYBA and GSTP1 variants associate with oxidative stress under hypobaric hypoxia as observed in high-altitude pulmonary oedema. Clin Sci (Lond). 2012;122(6):299-309.4. Bailey DM, Dehnert C, Luks AM, Menold E, Castell C, Schendler G, Faoro V, Gutowski M, Evans KA, Taudorf S, James PE, McEneny J, Young IS, Swenson ER, Mairbäurl H, Bärtsch P, Berger MM.5. Ali Z, Waseem M, Kumar R, Pandey P, Mohammad G, Qadar Pasha MA. Unveiling the interactions among BMPR-2, ALK-1 and 5-HTT genes in the pathophysiology of HAPE. Gene.
2016 22;588(2):163-72.6. Pasha MA, Newman JH. High-altitude disorders: pulmonary hypertension: pulmonary vascular disease: the global perspective. Chest. 2010;137(6 Suppl):13S-19S.7. Roberts LJ, Morrow JD. Measurement of F(2)-isoprostanes as an index of oxidative stress in vivo. Free Radic Biol Med. 2000 15;28(4):505-13.8. Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ 2nd. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci USA. 1990;87(23):9383-7.9. Biernacki WA, Kharitonov SA, Barnes PJ. Increased leukotriene B4 and 8-isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax. 2003;58(4):294-8.10. Montuschi P, Kharitonov SA, Ciabattoni G, Corradi M, van Rensen L, Geddes DM, Hodson ME, Barnes PJ. Exhaled 8-isoprostane as a new non-invasive biomarker of oxidative stress in cystic fibrosis. Thorax. 2000;55(3):205-9.11. Psathakis K, Papatheodorou G, Plataki M, Panagou P, Loukides S, Siafakas NM, Bouros D. 8-Isoprostane, a marker of oxidative stress, is increased in the expired breath condensate of patients with pulmonary sarcoidosis. Chest. 2004;125(3):1005-11.12. Askew EW. Work at high altitude and oxidative stress: antioxidant nutrients. Toxicology. 2002 15;180(2):107-19.13. Jefferson JA, Simoni J, Escudero E, Hurtado ME, Swenson ER, Wesson DE, Schreiner GF, Schoene RB, Johnson RJ, Hurtado A. Increased oxidative stress following acute and chronic high altitude exposure. High Alt Med Biol. 2004;5(1):61-9.
Stress on Lungs: A Search Through 8-ISO Prostaglandin-F2A
Learner’s Corner

50
Therapy of Pulmonary Hypertension - What’s New?
Learner’s Corner
Therapy of Pulmonary Hyperten-sion – What’s New?
Therapeutic options for pulmonary hyperten-sion (PH) have increased over the past years and include pharmacological approaches as well as surgical procedures. Pharmacological therapies approved for pulmonary arterial hypertension (PAH, group 2 according Nice classification, Table 1) include drugs addressing the signaling pathways of nitric oxide (NO), prostacyclin and endothelin (Figure 1), well known to be phosphodiesterase-5- (PDE5) inhibitors (sildenafil, tadalafil), prostacyclins/prostacyclin analoga (iloprost, epoprostenol, treprostinil) or endothelin receptor antagonists (ERAs: bosentan, ambrisentan). In the last years the spectrum of drugs was extended by a stimulator of the soluble guanylate cyclase (sGC) “riociguat” (Adempas, FDA approval: 2013), the prostacyclin receptor agonist “selexi-pag” (FDA approval: 2015) and the ERA “mac-icentan” (Opsumit, FDA approval: 2013). Most importantly, for the first time pharmacological therapy for another form of PH besides PAH became available, as riociguat was approved for the treatment of chronic thromboembol-ic PH (CTEPH). For continuous intravenous treatment with treprostinil the application of an implantable pump has been developed. This article provides an overview about currently approved new therapies for PH.
New pharmacological therapies in PH
Macitentan for treatment of PAHMacitentan (Opsumit, 10mg, once per day, oral) was approved by FDA in 2013 and acts as dual ERA with better tissue penetration and less interaction with other drugs [3]. In contrast to other ERAs liver toxicity seems to be negligible, as the respective clinical trial showed liver toxicity on the level of placebo. The placebo controlled study “SERAPHIN” (Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Im-prove Clinical Outcome) enrolled 742 patients with a mean follow up of 3.5 years. For the first time a composite endpoint of morbidity and mortality (e.g. lung transplantation, initiation of treatment with i.v. or subcutaneous pros-tanoids or worsening of PAH) was chosen for an approval-relevant clinical phase III trial. The trial showed that application of 3 or 10 mg Macitentan reduced the relative risk to reach the combined primary endpoint by 30 or 45%, respectively, compared to the placebo group. The combined endpoint was mainly reduced by decreasing PAH related hospitalizations and worsening of PAH, but not mortality. More-over, Macitentan was the first ERA, which showed positive effects in combination with PDE5 inhibitors. Main side effect at a dose of 10 mg was a decreased a hemoglobin value [10]. In contrast to bosentan, Macitentan nei-ther interacts with Warfarin [11] nor with oral contraceptiva [7].
Riociguat for treatment of PAHRiociguat (Adempas, standard dose 2.5 mg, three times per day, oral) was approved for the therapy of PAH in 2013 and acts as sGC stimulator. Thus, Riociguat addresses sim-
Natascha somner md, phd
New York Excellence Cluster Cardiopulmonary System, University of Giessen and Marburg Lung Center (UGMLC),
member of the German Center for Lung Research (DZL). Justus-Liebig-University, 35392 Giessen, Germany Medical
College

51
Therapy of Pulmonary Hypertension - What’s New?
Learner’s Corner
monophosphate (cGMP) signaling pathway. In contrast to sildenafil, which increases the concentration of cGMP by inhibition of its deg-radation, Riociguat enhances the sensitivity of the sGC for NO and stimulates production of cGMP independently of NO [14]. In the study conducted for approval of Riociguat for PAH (“PATENT-1” Pulmonary Arterial Hyperten-sion Soluble Guanylate Cyclase–Stimulator Tri-al 1) Riociguat improved the primary endpoint of 6-minute-walk-distance as well as secondary endpoints such as exercise capacity, pulmonary vascular resistance and time to clinical wors-ening. Adverse effects included particularly systemic arterial hypotony [5]. Therefore, in clinical practice Riociguat is up-titrated during a time period of approximately 6 weeks accord-ing to a titration scheme supervised by mon-itoring of blood pressure (starting dose 1 mg, three times per day) until reaching the stand-ard dose, if tolerated. Combination of Riociguat and sildenafil is contraindicated due to arterial hypotony [2].
Riociguat for treatment of CTEPHCTEPH is the only form of PH for which a potential curative approach is available, which is the removal of thromboembolic material by the surgical procedure of pulmonary endarter-ectomy (PEA). PEA is performed during cir-culatory arrest in deep hypothermia [9]. Fur-thermore, life-long sufficient anticoagulation is considered standard basic therapy.Since approval of Riociguat in April 2014, the first pharmacological therapy for CTEPH was approved. However, Riociguat is only approved for technical inoperable CTEPH, or if the bene-fit-risk ratio limits PEA, as well as in persistent or recurrent PH after PEA. First line therapy for CTEPH is still PEA. Thus every patient should be evaluated for PEA after diagnosis of CTEPH, before considering pharmacological therapy. Moreover, pharmacological therapy
with Riociguat for the purpose of bridging the time until PEA in conditions of severe CTEPH has not yet been evaluated in clinical studies. In the randomized controlled CHEST trial (Chronic Thromboembolic Pulmonary Hy-pertension Soluble Guanylate Cyclase–Stim-ulator Trial) application of Riociguat resulted in improved exercise capacity determined by the six minute walk distance, and decreased pulmonary vascular resistance [4]. However, time-to-clinical worsening (defined as com-bined end point of e.g. mortality, transplan-tation) was unchanged. In a prior study in CTEPH, Bosentan improved hemodynamics, but not exercise capacity, allowing this drug to only be used as off-label medication [8].
Selexipag for treatment of PAHSelexipag is an oral, selective prostacyclin receptor agonist with long half-life (6,2-13,5 h) and was approved by FDA at the end of 2015. In the clinical phase III trial GRIPHON PGI2 (Receptor agonist In Pulmonary arterial Hypertension) Selexipag -as mono- or combi-nation-therapy with ERAs and/or PDE5-inhib-itors- reduced the mixed endpoint of morbidity and mortality [12].
New application forms in PAH
Treprostinil: implantable pumpIntravenous application of prostacyclins can be cumbersome due to the persistent indwell-ing i.v. line which predisposes for infections and dislocations. A more preferable way of application was achieved by development of a fully implantable pump system with a titanium reservoir which can be re-filled from outside every 2-4 weeks percutaneously by an injection through a membrane with a special needle by trained personnel. The pump is implanted in the subcutaneous tissue of the abdominal wall and delivers the treprostinil in the superior
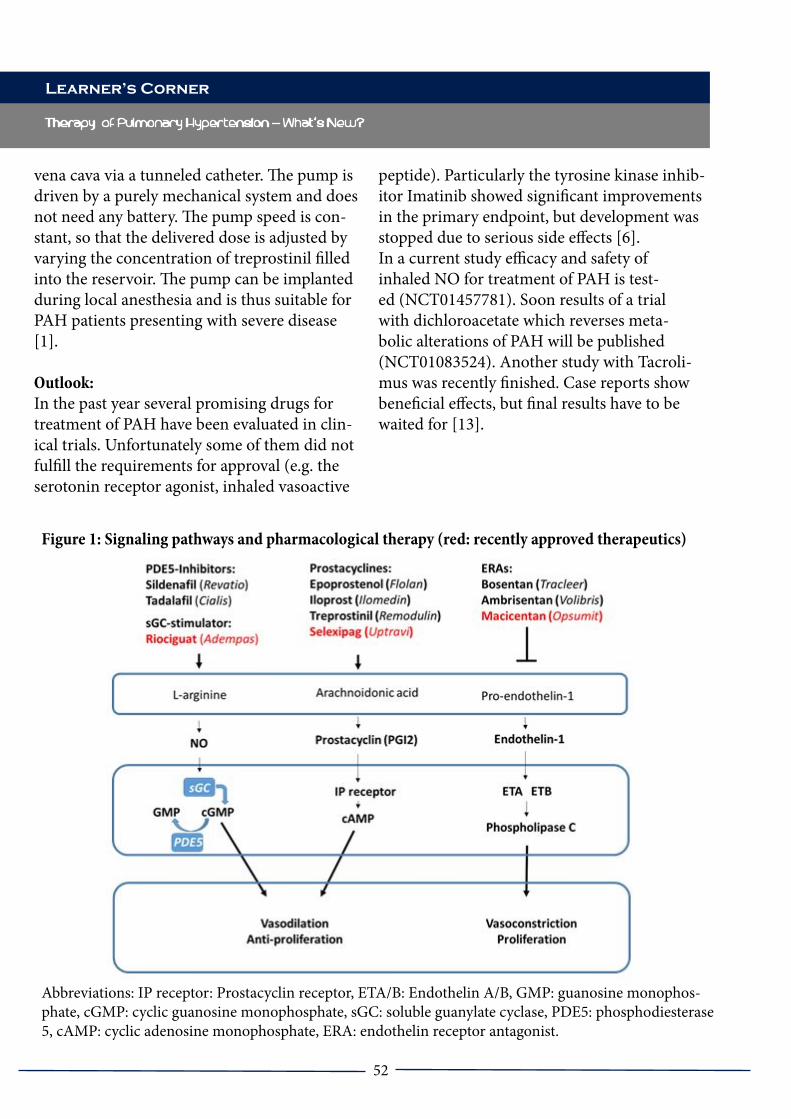
52
vena cava via a tunneled catheter. The pump is driven by a purely mechanical system and does not need any battery. The pump speed is con-stant, so that the delivered dose is adjusted by varying the concentration of treprostinil filled into the reservoir. The pump can be implanted during local anesthesia and is thus suitable for PAH patients presenting with severe disease [1].
Outlook:In the past year several promising drugs for treatment of PAH have been evaluated in clin-ical trials. Unfortunately some of them did not fulfill the requirements for approval (e.g. the serotonin receptor agonist, inhaled vasoactive
peptide). Particularly the tyrosine kinase inhib-itor Imatinib showed significant improvements in the primary endpoint, but development was stopped due to serious side effects [6]. In a current study efficacy and safety of inhaled NO for treatment of PAH is test-ed (NCT01457781). Soon results of a trial with dichloroacetate which reverses meta-bolic alterations of PAH will be published (NCT01083524). Another study with Tacroli-mus was recently finished. Case reports show beneficial effects, but final results have to be waited for [13].
Therapy of Pulmonary Hypertension - What’s New?
Learner’s Corner
Figure 1: Signaling pathways and pharmacological therapy (red: recently approved therapeutics)
Abbreviations: IP receptor: Prostacyclin receptor, ETA/B: Endothelin A/B, GMP: guanosine monophos-phate, cGMP: cyclic guanosine monophosphate, sGC: soluble guanylate cyclase, PDE5: phosphodiesterase 5, cAMP: cyclic adenosine monophosphate, ERA: endothelin receptor antagonist.

53
Therapy of Pulmonary Hypertension - What’s New?
Learner’s Corner
References:
1. Ewert R, Halank M, Bruch L et al. (2012) A case series of patients with severe pulmonary hypertension receiving an implantable pump for intravenous prostanoid therapy. American journal of respiratory and critical care medicine 186:1196-11982. Galie N, Muller K, Scalise AV et al. (2015) PATENT PLUS: a blinded, randomised and exten-sion study of riociguat plus sildenafil in pulmonary arterial hypertension. The European respiratory journal 45:1314-13223. Gatfield J, Mueller Grandjean C, Sasse T et al. (2012) Slow receptor dissociation kinetics differ-entiate macitentan from other endothelin receptor antagonists in pulmonary arterial smooth muscle cells. PloS one 7:e476624. Ghofrani HA, D'armini AM, Grimminger F et al. (2013) Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. The New England journal of medicine 369:319-3295. Ghofrani HA, Galie N, Grimminger F et al. (2013) Riociguat for the treatment of pulmonary arterial hypertension. The New England journal of medicine 369:330-3406. Hoeper MM, Barst RJ, Bourge RC et al. (2013) Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 127:1128-11387. Hurst N, Pellek M, Dingemanse J et al. (2015) Lack of pharmacokinetic interactions be-tween macitentan and a combined oral contracep-tive in healthy female subjects. Journal of clinical pharmacology8. Jais X, D'armini AM, Jansa P et al. (2008) Bosentan for treatment of inoperable chronic thromboembolic pulmonary hypertension: BEN-EFiT (Bosentan Effects in iNopErable Forms of chronIc Thromboembolic pulmonary hyperten-sion), a randomized, placebo-controlled trial. Journal of the American College of Cardiology 52:2127-21349. Jenkins D, Mayer E, Screaton N et al. (2012)
State-of-the-art chronic thromboembolic pulmo-nary hypertension diagnosis and management. European respiratory review : an official journal of the European Respiratory Society 21:32-3910. Pulido T, Adzerikho I, Channick RN et al. (2013) Macitentan and morbidity and mortality in pulmonary arterial hypertension. The New Eng-land journal of medicine 369:809-81811. Sidharta PN, Dietrich H, Dingemanse J (2014) Investigation of the effect of macitentan on the pharmacokinetics and pharmacodynamics of warfarin in healthy male subjects. Clinical drug investigation 34:545-55212. Sitbon O, Channick R, Chin KM et al. (2015) Selexipag for the Treatment of Pulmonary Arterial Hypertension. The New England journal of medicine 373:2522-253313. Spiekerkoetter E, Sung YK, Sudheendra D et al. (2015) Low-Dose FK506 (Tacrolimus) in End-Stage Pulmonary Arterial Hypertension. American journal of respiratory and critical care medicine 192:254-25714. Stasch JP, Evgenov OV (2013) Soluble guanylate cyclase stimulators in pulmonary hyper-tension. Handbook of experimental pharmacology 218:279-313
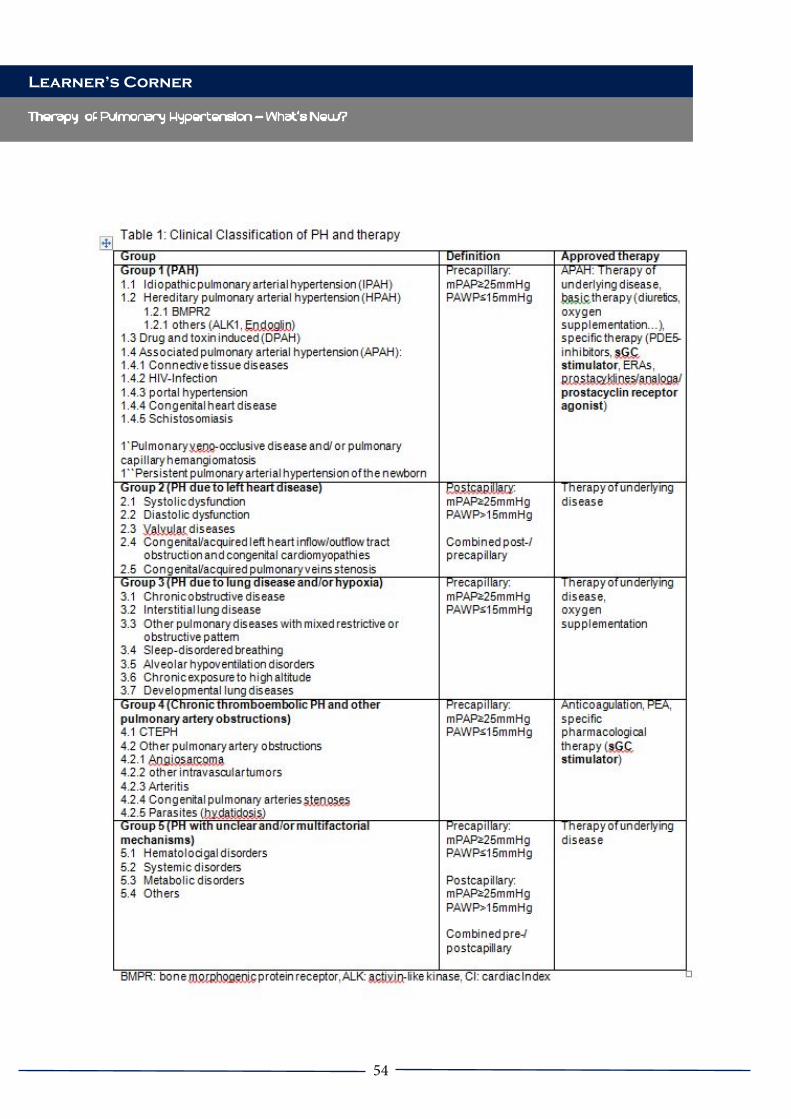
54
Therapy of Pulmonary Hypertension - What’s New?
Learner’s Corner

55
Therapy of Pulmonary Hypertension - What’s New?
Learner’s Corner
References:
1. Ewert R, Halank M, Bruch L et al. (2012) A case series of patients with severe pulmonary hypertension receiving an implant-able pump for intravenous prostanoid therapy. American journal of respiratory and critical care medicine 186:1196-11982. Galie N, Muller K, Scalise AV et al. (2015) PATENT PLUS: a blinded, randomised and extension study of riociguat plus sildenafil in pulmonary arterial hypertension. The Euro-pean respiratory journal 45:1314-13223. Gatfield J, Mueller Grandjean C, Sasse T et al. (2012) Slow receptor dissociation kinetics differentiate macitentan from other endothe-lin receptor antagonists in pulmonary arterial smooth muscle cells. PloS one 7:e476624. Ghofrani HA, D'armini AM, Grim-minger F et al. (2013) Riociguat for the treat-ment of chronic thromboembolic pulmonary hypertension. The New England journal of medicine 369:319-3295. Ghofrani HA, Galie N, Grimminger F et al. (2013) Riociguat for the treatment of pul-monary arterial hypertension. The New Eng-land journal of medicine 369:330-3406. Hoeper MM, Barst RJ, Bourge RC et al. (2013) Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 127:1128-11387. Hurst N, Pellek M, Dingemanse J et al. (2015) Lack of pharmacokinetic interactions between macitentan and a combined oral con-traceptive in healthy female subjects. Journal of clinical pharmacology8. Jais X, D'armini AM, Jansa P et al. (2008) Bosentan for treatment of inoperable chronic thromboembolic pulmonary hyperten-sion: BENEFiT (Bosentan Effects in iNopErable Forms of chronIc Thromboembolic pulmonary hypertension), a randomized, placebo-con-
trolled trial. Journal of the American College of Cardiology 52:2127-21349. Jenkins D, Mayer E, Screaton N et al. (2012) State-of-the-art chronic thromboem-bolic pulmonary hypertension diagnosis and management. European respiratory review : an official journal of the European Respiratory Society 21:32-3910. Pulido T, Adzerikho I, Channick RN et al. (2013) Macitentan and morbidity and mor-tality in pulmonary arterial hypertension. The New England journal of medicine 369:809-81811. Sidharta PN, Dietrich H, Dingemanse J (2014) Investigation of the effect of macitentan on the pharmacokinetics and pharmacody-namics of warfarin in healthy male subjects. Clinical drug investigation 34:545-55212. Sitbon O, Channick R, Chin KM et al. (2015) Selexipag for the Treatment of Pulmo-nary Arterial Hypertension. The New England journal of medicine 373:2522-253313. Spiekerkoetter E, Sung YK, Sudheendra D et al. (2015) Low-Dose FK506 (Tacrolimus) in End-Stage Pulmonary Arterial Hyperten-sion. American journal of respiratory and critical care medicine 192:254-25714. Stasch JP, Evgenov OV (2013) Soluble guanylate cyclase stimulators in pulmonary hypertension. Handbook of experimental phar-macology 218:279-313

56
PhD Defense: Targeting the cause, affecting the course
Targeting the cause, affecting the course
Pulmonary Arterial Hypertension (PAH) is a progressive disease, characterized by pulmonary vascular remodeling and right ventricular remodeling in hypertrophy, but eventually dilatation and right heart failure [1]. Today, there are several drug classes registered to hamper the disease progression [2]. However, no curative medication for PAH is available. Due to dedicated research, several factors playing an important role in the pathobiology of PAH are identified [3]. Many of these factors also play an important role for the compensatory cardiac adaptation, which poses a treatment paradox – what is beneficial to the lungs, might be harmful for the heart - when it comes to medication targeting these factors.From the PH center at the VU University medical center, Michiel Alexander de Raaf defended his PhD entitled: “targeting the cause, affecting the course”. The thesis discussed the treatment paradox using experimental models resembling important aspects of PAH.Important conclusions which could be made from the PhD program were that the different experimental models, like (chronic) hypoxia, monocrotaline and Sugen Hypoxia for example, supply different answers to the treatment paradox [4]. This is due to the fact that every animal model resembles only a ‘set’ of aspects seen in the human disease. With better understanding of endothelial dysfunction, cellular signaling
and ‘quasi-malignant phenotypes’ of the disease, the interpretation of what we understand about PAH is still evolving. The scientific progress that has been made also translates to the evolution of animal models; there is a continuous need to characterize and improve animal models by including the representation of aspects which has become more appreciated to have an important role in PAH [5]. This also means, that using older animal models such as monocrotaline or chronic hypoxia to study the efficacy of medication targeting endothelial proliferation – an aspect not represented in these animal models -, may lead to unpredictable translation to the clinic [4, 6, 7].The best solution to solve the treatment paradox pre-clinically, making the translation gap smaller, is to use multiple animal models; pulmonary vascular remodeling models representing the drug target for ‘targeting the cause’, and right ventricular pressure overload models to understand if the drug is not worsening cardiac function by ‘affecting the course’. To answer the latter, Pulmonary Artery Banding could be used. With Pulmonary Artery Banding, right ventricular adaptation under exposure of the medication can be measured and evaluated. A good example of the understanding gained from using multiple animal models is the evaluation of histone deacetylases (HDACs) for PAH. In animal models not representing the hyperproliferative endothelium, several HDACs showed a beneficial treatment effect. The thesis shows that HDAC activity in these experimental models is already low in the lungs and elevated in the right heart. Testing these HDAC’s in the Pulmonary Artery Banding model resulted in cardiac worsening and within the Sugen Hypoxia model, no treatment effect was visible [6, 8]. This would conclude that using multiple animal models, predictability of the contemplated beneficial treatment potential
Corresponding AuthorMICHIEL ALEXANDER DE RAAF
VU Medical CenterDe Boelelaan 1117, 1081 HV Amsterdam, Netherlands
Learner’s Corner

57
PhD Defense: Targeting the cause, affecting the course
can be made. Bridges were made to the field of the developmental biology to understand the treatment paradox in PAH [9]. During the prenatal phase, the increased pulmonary vascular resistance is needed for the development of the right heart. Medication which lowers the pulmonary vascular resistance results in cardiovascular malformations. Indeed, with the reactivation of fetal aspects in PAH in both pulmonary vascular remodeling as cardiac adaptation, is shows many parallels with cardiopulmonary development.
References
1. Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013; 62: D34-41.2. Humbert M, Ghofrani H-A. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax 2016; 71: 73–83.3. Voelkel NF, Gomez-Arroyo J, Abbate A, Bogaard HJ, Nicolls MR. Pathobiology of pulmonary arterial hypertension and right ventricular failure. Eur. Respir. J. 2012; 40: 1555–1565.4. De Raaf MA, Voelkel NF, Bogaard HJ. Advances in understanding of pulmonary arterial hypertension and the evolution of Experimental pulmonary hypertension models. PVRI Chron. Vol 2: Issue 2.5. de Raaf MA, Schalij I, Gomez-Arroyo J, Rol N, Happé C, de Man FS, Vonk-Noordegraaf A, Westerhof N, Voelkel NF, Bogaard HJ. SuHx rat model: partly reversible pulmonary hypertension and progressive intima obstruction. Eur. Respir. J. 2014; 44: 160–168.6. De Raaf MA, Hussaini AA, Gomez-Arroyo
J, Kraskaukas D, Farkas D, Happé C, Voelkel NF, Bogaard HJ. Histone deacetylase inhibition with trichostatin A does not reverse severe angioproliferative pulmonary hypertension in rats (2013 Grover Conference series). Pulm. Circ. 2014; 4: 237–243.7. De Raaf MA, Kroeze Y, Middelman A, de Man FS, de Jong H, Vonk Noordegraaf A, de Korte C, Voelkel NF, Homberg J, Bogaard HJ. Serotonin transporter is not required for the development of severe pulmonary hypertension in the Sugen hypoxia rat model. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015; : ajplung.00127.2015.8. Bogaard HJ, Mizuno S, Hussaini AAA, Toldo S, Abbate A, Kraskauskas D, Kasper M, Natarajan R, Voelkel NF. Suppression of Histone Deacetylases Worsens Right Ventricular Dysfunction after Pulmonary Artery Banding in Rats. Am. J. Respir. Crit. Care Med. 2011; 183: 1402–1410.9. de Raaf MA, Beekhuijzen M, Guignabert C, Vonk Noordegraaf A, Bogaard HJ. Endothelin-1 receptor antagonists in fetal development and pulmonary arterial hypertension. Reprod. Toxicol. Elmsford N 2015; .
Learner’s Corner

58
Rome Tour
Rome Tour
This walking tour through Rome was made as social event at the PVRI Annual Meeting in Rome 2016. The tour was guided by PVRI member and medical biologist Michiel Alexander de Raaf, who lived in Rome for one year during an internship. Romans consider their squares as living rooms, full with memories of nice and romantic stories of the city. Historically, not all stories can be exactly true as they are told nowadays. However, with a city full of history, the Romans don’t care or even know about this and these stories became treasures of the Roman ambiance and culture. The stories are often not known by tourists and provide a nice glimpse in the history of Rome.
Piazza MatteiThe tour started at “Piazza Mattei”, the square called after the Mattei family who had several palaces close to this square. The Duke of the Mattei family was desperately in love with a young woman. The father of the young woman did not approve the proposed marriage, as the Duke was ‘famous’ about his gambling addiction and lost all his money. On a market day, the father visited the Duke to tell him the news that he is not willing to marry his daughter to someone without money. Of course, the Duke was desperately in love and told the father to return the next day. The next day, there was no market and the father saw for
the first time the fountain of the turtles (“Fontane Felle Tartarughe”). The Duke lied and told him that he ordered to build the fountain in one night to convince the father about his richness. The father was convinced and the couple married. As the father did not want other people to see the beauty of the fountain as he did for the first time, the window facing the fountain is immured.
Area Sacrale Largo Torre ArgentinaAt “Area Sacrale Largo Torre Argentina” we saw the four oldest temples of the Roman Empire. They were probably already built by the Etruskians, who lived in this region before recorded Roman history. It is said that the temples were forgotten during the Roman reign, to whose Gods the temples were built for. To overcome the problem of dishonoring the gods , the complete area was called ‘sacred’. During the reign of Julius Gaius Caesar, this area was the playground of world history. As Julius Caesar was renovating the curia (senate house) at the “Forum Romanum”, the Senate of Rome used the area “sacrale” for their daily meetings. It was here, in the temple on the right side, where Caesar was murdered. Due to the age of these temples, we know that this area is also one of the oldest building sites of Rome. The area of the Roman forum and the Colosseum were still swamps in that time. It was known that a monster was living in that swampy area which could ‘grasp’ people by its bad breath; dying a horrible death. Therefore, the area was said to have ‘sick air’, which is in Latin ‘male aria’ and the base for the disease malaria.
Piazza NavonaThe “Piazza Navona” square is built on the foundations of the hippodrome built by Dominitianus. Horse races were very popular in ancient Rome. There were four teams (blue,
Corresponding AuthorMICHIEL ALEKSANDER DE RAAF
VU Medical CenterDe Boelelaan 1117, 1081 HV Amsterdam, Netherlands
PVRI NEWS AND ACTIVITIES

59
Rome Tour
white, red, green) and racing with chariots they needed to make 7 turns around the “spina”, which is a structure in the middle of the hippodrome. In the centre of the “spina”, an Egyptian obelisk was placed and later moved to “Piazza del Popolo”. The obelisk to be found nowadays at “Piazza Navona” is a replica made by the students of the architect, Bernini (they were not familiar with the Egyptian hieroglyphs, and placed them upside down). Bernini was not invited to the competition to make this fountain in the center, but he submitted his proposal and was chosen to construct it. The other architect, Borromini, was very angry, as he wanted the assignment. To ease the situation, he was ordered to build the church next to the fountain. The fountain consists of four men who represent the four ‘biggest’ rivers at that time; Donau, Ganges, Nile and Rio de la Plata. As the beginning of the Nile was not known at that time, the head of the ‘river god’ is covered by a cloth. The river god of “Rio de la Plata” is also special as the face is not of a human adult. One story tells that this face is a monkey as the ‘New World’ was not Christian yet. Another story tells the face is a baby because the ‘New World’ was new and immature. Borromini found a mathematic mistake in the drawings of Bernini and the fountain would collapse by the weight of the obelisk. Bernini went through his calculations several times but was not able to find his mistake. The only way to find the error and solve it, was to rely on one of his students. The daughter of Borromini was desperately in love with one of the students of Bernini. This student was sent to declare his love to Borromini’s daughter who let him in the house. At night he copied the calculations and the next day they were given to his master Bernini. Bernini was able to fix the mathematical error and the fountain was saved from collapsing. To annoy Borromini, who was still not aware that Bernini knew about the mistake and fixed it, he ‘saved’ the fountain from collapsing by stabilizing the
obelisk with silk wires. As “Piazza Navona” is one of lowest areas of the city, there is the possibility to make an artificial lake of 60 cm deep, which was used in summertime during the 18th-19th century.
San Luigi dei FrancesiWe passed this church and in the back and on the left, we were able to see original Caravaggio paintings. Please note that the paintings depict stories from the Bible, while the ‘setting’ is not from Ancient Rome era, but Medieval, so ‘contemporary’ in the time of Caravaggio himself. This is probably done, as in many outside Italy, to narrow the ‘translational’ gap for the Medieval people to understand better the stories told in the Bible.
Piazza della Rotonda – PantheonThe Pantheon is built on the fields of Mars. This field was sacred in Ancient Rome and it was permitted to build buildings here. The fields were used for elections; Roman people walked to the area dedicated to a specific consul to vote for him. Also the two Roman legions in order to keep ambitious Roman generals with their legions out of the governmental center of the Empire, had their camps here. However, Rome was growing and held at its maximum approximately 1.3 million inhabitants. So, the field of Mars needed to be used for residential buildings. To keep all gods happy with this need, the first building built was a temple for all the gods; the Pantheon. And there were a lot of Gods to please, partly due to the following aspect. To stabilize the power of Rome and to destabilize Roman generals to become too powerful (the divide and rule idea by Caesar), Roman legions needed to move every 6 months from their garrison to another. This was good for stabilizing the political and military power but by this strategy the legionnaires were exposed to a lot of new cultures and religions (keep in mind
PVRI NEWS AND ACTIVITIES

60
Pulmonary hypertension in Constrictive Pericarditis or Systemic Lupus Erythematosus patients?
PVRI NEWS AND ACTIVITIES
1 out of 10 Romans were in the army). Many of these religions were spread through the empire. For example, the Persian Mithras became very popular among soldiers and altars were found throughout the Empire (England, Spain, Rome, etc.). With the Pantheon, the Romans kept the ‘promise’ that all gods were represented in the city of Rome.
Sant’Ignazio di Loyola in Campo MarzioThis is one of the most beautiful churches in Rome and not so famous nor touristic. Every catholic province (with a cardinal) was allowed to build a church to represent themselves in Rome. However, most popes did not want that the church became more beautiful than their own churches as the Saint Peter in the Vatican. Therefore, the pope stopped the budget of the construction. Major problem was that without the money, the dome on top of the church could not be completed. Giovanni Tristano, the architect, ‘constructed’ the dome by painting the dome (which interior is the observatory of Galilei). When opening the church, the pope walked to the ‘yellow’ stone and was astonished by seeing the dome, not realizing that it was fake, and walked -very angry - out. Also the other breathtaking fresco which makes the church optical much higher is painted by Andrea Pozzo.
Fontana di TreviThe Trevi fountain is probably the most well known tourist hotspot in Rome. It is said that it is called “Trevi” as this means ‘three’ and the square is the connection point of three main streets. Another story tells of the Trevi Fountain getting its water from a river with 3 different springs. When Ancient Rome was flowering, the city had numerous aqueducts feeding the city with fresh water. After the ‘Sacco di Roma’, the Gauls destroyed all aqueducts except one. This same one still functions today and gives its water to the
Trevi Fountain. From there, all other fountains in the city are fed by the Trevi Fountain. Therefore, the water from the Trevi Fountain is the freshest water of the city. Drinking from the water together with your partner assures an eternal relationship. Two anecdotes were told at the Trevi Fountain. The first was about a little girl who was living next to the fountain when it was built. As she was sick, she was afraid that she was not able to see the final result. Indeed, she died before the construction was finished and therefore the architect, Nicola Salvi (but the fountain was drawn by Bernini 50 years earlier), made a little statue of her head and placed it on the façade of the church next to the fountain, as she could now ‘see’ the Trevi fountain for centuries.
Another very nice story is revolves around the construction of the fountain. At the time, there was a barbershop on the right side of the square. Every day, the barber came to talk with Nicola Salvi. He was full of ideas -all of which had one goal. He wanted to make his barbershop more appealing; when you were looking to the Trevi fountain, your eyes were directed to his barbershop. The architect Salvi was increasingly frustrated by these recommendations, and as a result, on the very last day of the construction, he placed a big rock on the right side of the square, including a big vase on top, so that when you were looking at the Trevi fountain, you could not see anything of the barbershop.

61

62
CYCS Waldeck Castle Retreat
PVRI NEWS AND ACTIVITIES
MICHIEL ALEXANDER DE RAAF
VU Medical CenterDe Boelelaan 1117, 1081 HV Amsterdam, Netherlands
When Djuro Kosanovic sent me an e-mail say-ing that he had rented a castle for the weekend and asking me to join him, I had mixed feelings of surprise and excitement. Would this finally the moment to push our scientific collaborative friendship further? Perhaps I had misjudged the situation a bit, I thought. Especially, when I no-ticed that I was not the only one being invited. And then I understood. It was the PVRI retreat for the Committee of Young Clinicians and Scientists. It was work. Science is a tough game.
The first -and therefore historic - PVRI CYCS Retreat was located at the castle of Waldeck in Germany. Waldeck castle is known to have had a substantial influence on world history. In 1858 for example, Emma of Waldeck-Pyrmont was born there. She married the Dutch king William the Third who needed to stabilize his legacy after his unsuccessful previous marriage with his niece Sophia. Emma gave birth to Queen Wilhelmina and after the death of the king, she was regent in the period when Wilhelmina was still a child. As we feel the influence of the historic Waldeck castle around us, we hope that this can be reflected in our efforts to support and evolve the PVRI.
The program started on Friday afternoon by wel-coming everybody. We had a lovely dinner and the CYCS board discussed the agenda. Later that evening, we had a wonderful tour of the castle, seeing the dungeons, prisons and the well. The well was dug by two prisoners who were released when they found water. After 27 years, and 140
meters of well, they lost their eyesight but finally found water. Upon release, one of the prisoners was so excited that he had a heart attack and un-fortunately died!
On Saturday, we had a CYCS plenary council meeting in the morning, addressing all ongoing topics of our committee. Some of these topics were discussed immediately and others were postponed to Sunday. In the afternoon we had a splendid so-cial teambuilding event by leaving the castle using a cable track and went sailing on the lake. In the evening, after dinner, we started our scientific ses-sion which was full of interactive discussions. In total we had 14 presentations (from the 11 attend-ees) and covered a broad spectrum of today’s cut-ting-edge science in the field of PVD. On Sunday, the day started with the second half of the CYCS plenary council meeting, where we discussed the remaining topics. My Waldeck Retreat finished a bit earlier; as my wife is also a scientist and was on her way to a congress herself, I headed back home to continue my duties as father. The others remained and continued in the same spirit and ambiance as the days before, having the second part of the scientific sessions closed by a dinner at the castle.
Aside from the scientific success, we had also oth-er successes. Regarding the PVRI CYCS tasks, the timing of this retreat was superb, as we were able to discuss our ongoing projects and start prepara-tions for the Annual Meeting in Miami. We had two ‘plenary council meetings’ (and one board meeting) to discuss and evaluate all ongoing pro-jects. We have never had so many topics discussed and never had so many ideas to explore and im-prove the council. I sincerely believe that we took a vast number of steps forward and that the things discussed at Waldeck will echo in a more effective CYCS for a long time.
Regarding friendship, this retreat had a marvelous atmosphere; giving ample possibility to start and

63
CYCS Waldeck Castle Retreat
PVRI NEWS AND ACTIVITIES
strengthen friendships. This is already reflect-ing in the increased dedication and activity we experience now among our members. All those who took part have been very enthusiastic about the retreat and look forward to the possibility of making such an event in the future an even bigger success.
All attendees of the PVRI CYCS Retreat at Wal-deck, would like to acknowledge Mariola Bednorz for the wonderful organization of this historic meeting. This retreat showed the strength of our CYCS, and personally I feel very fortunate to have experienced this.

64
Obituary of Almaz Aldashev
PVRI NEWS AND ACTIVITIES
Obituary of Professor Almaz A Aldashev (1953-2016)
Vice-President of National Academy of Sciences of Kyrgyz RepublicDirector of the Institute of Molecular Biology and MedicineBishkek, Kyrgyz Republic
We are saddened to announce the death of Al-maz Aldashev, a leading scientist in the world of pulmonary vascular research.
Almaz was born and much of spent his life in Kyrgyzstan where he devoted his academic life to pulmonary disease. He is best known to us for his work in pulmonary hypertension but he had many other interests, both with-in and outside of science. While Kyrgyzstan was part of the Soviet Union his travels were confined to Russia and Central Asia, but with the independence of Kyrgyzstan in 1991 he was able to reach out more freely to western
scientists. He developed a network of collabo-rators in Europe and America and linked them to physicians and scientists within the newly independent Central Asian states. A man of integrity and politically astute, it was natural that he should rise to high office in his coun-try’s leading science community.He was steeped in the history of Central Asia and a warm and engaging guide to Kyrgyzstan, now the Kyrgyz Republic. He was proud of his country and was happy and at home travel-ling the highlands, meeting local villagers and sharing stories and songs. His loss will be felt at home and abroad.
Prof martin WilkinsMD FRCP FBPhS FMedSci
Head of Department of Medicine, Imperial College London

The platform for global collaboration...
... that enables us to challenge what’s possible.
www.pvri.info
The Pulmonary Vascular Research Institute is a registered charity in the United Kingdom (Charity No: 1127115). A private limited company. Registered company in the United Kingdom (5780068). Registered address in the UK:
33 St George's Place, Canterbury, Kent, CT1 1UT
The Pulmonary Vascular Research Institute is registered in the United States of America as a not-for-profit organisation (501c3) in the State of Illinois (IN26: 0420959).
Registered address in the USA: 3621 Grove Street, Skokie, Illinois 60076-1901
An official publication.





























