Embed Size (px)
Citation preview

Synthesis and field-effect properties of a,v -disubstituted sexithiophenesbearing polar groups
Antonio Dell’Aquila,ac Piero Mastrorilli,a Cosimo Francesco Nobile,*a Giuseppe Romanazzi,b
Gian Paolo Suranna,a Luisa Torsi,*c Maria Cristina Tanese,c Domenico Acierno,d Eugenio Amendolae andPiero Moralesf
Received 2nd November 2005, Accepted 5th January 2006
First published as an Advance Article on the web 23rd January 2006
DOI: 10.1039/b515583e
The synthesis of sexithiophenes bearing amide or ester groups in the a,v-terminal positions is
described, along with their characterization in the solid state. The influence of the functional
group on mobilities and on/off ratios of the organic FET devices was investigated. The oligomer
bearing the ester functional group separated from the sexithiophene core by an ethylene spacer
showed a hole field-effect mobility as high as 0.012 cm2 V21 s21, which is among the highest
reported so far for organic FETs using sexithiophenes modified with polar groups.
Introduction
An ever increasing effort is being successfully directed towards
the synthesis of organic materials based on p-conjugated
oligomers, as they hold easily controllable properties that
render them suitable as conducting or semiconducting active
layers in devices such as photovoltaic cells,1 light emitting
diodes2 and organic field effect transistors (OFET).3,4 The first
reports on OFET date back to the mid-eighties,5 but the idea
that these devices could reach mobilities and on/off ratios
comparable to those of amorphous silicon was consolidated
only after the charge transport properties of a-sexithiophene
were discovered.6 Since then on, continuously increasing con-
sideration has been given to OFET based on both unsub-
stituted and substituted thiophene oligomers.7 In particular,
after the seminal work by Garnier et al.,8 several reports have
dealt with the synthesis and characterization of well-defined
a,v-disubstituted sexithiophenes aiming at obtaining more
soluble and processable derivatives, while preserving or even
improving their electronic properties: for instance, field-effect
mobilities as high as 1.0 cm2 V21 s21 have been achieved using
a,v-dihexyl-sexithiophene.9
An intriguing and fast developing application of organic
thin-film transistors is their use as chemical sensors.10,11 In this
area, the role of functional groups attached to the polymer/
oligomer chain is very important, as it allows modulation of
the device selectivity towards chemically homologous
species.12 On the other hand, the key role of the side chains
in promoting the organic active layer adhesion/orientation on
the gate dielectric surface or in guiding the oligomer stacking
is well known.8 In this respect the presence of suitable side
groups might critically influence the molecular packing by
means, for instance, of hydrogen-type interactions. Despite
the scientific and technological interest towards oligothio-
phenes, to date only a few studies have dealt with the use
of thiophene oligomers bearing polar side groups in p-type
OFET devices , reporting hole mobilities not higher than
1024–1023 cm2 V21 s21.13 Comparable mobilities have been
recently reported also for OFET comprising a carbonyl sub-
stituted quaterthiophene as channel material. This figure of
merit became as high as 1022 cm2 V21 s21 when the substrate
was held at 70 uC during the organic thin-film evaporation.14
In this paper we report on our studies on the synthesis of
new a,v-disubstituted sexithiophenes, bearing polar functional
groups as amides or esters. The influence of these moieties on
the field effect properties of sexithiophenes was also addressed.
Experimental
Materials
The reactants were purchased by Aldrich, Acros or Fluka and
used without further purification. All manipulations were
carried out under inert nitrogen atmosphere using Schlenk
techniques unless otherwise specified and the solvents
used were carefully dried and freshly distilled according to
common laboratory practice. Flash chromatography was
performed using Merck Kieselgel 60 (230–400 mesh) silica
gel or Macherey-Nagel polygoprep 60-50 C18 silica gel
reversed phase.
Methods
1H NMR and 13C{1H} NMR spectra were recorded on a
Bruker Avance 400 and are reported in ppm using tetra-
methylsilane as standard. FT-IR spectra were recorded in KBr
aDepartment of Water Engineering and Chemistry (DIAC), Polytechnicof Bari, via Orabona 4, I-70125, Bari, Italy. E-mail: [email protected];Fax: +39 080 5963611; Tel: +39 080 5963608bDepartment of Civil and Environmental Engineering (DICA),Polytechnic of Bari, via Orabona 4, I-70125, Bari, ItalycDepartment of Chemistry, T.I.R.E.S. Centre of Excellence, Universityof Bari, via Orabona-4, I-70126, Bari, Italy.E-mail: [email protected]; Fax: +39 080 5442026;Tel: +39 080 5442092dDepartment of Materials and Production Engineering (DIMP),University of Naples Federico II, p.le Tecchio 80, I-80125, Naples, ItalyeInstitute of Composite and Biomedical Materials (IMCB), Italy’sNational Research Council, p.le Tecchio 80, I-80125, Naples, ItalyfENEA, Unita Materiali e Nuove Tecnologie, Centro Ricerche dellaCasaccia, 00060 S. Maria di Galeria, Roma, Italy
PAPER www.rsc.org/materials | Journal of Materials Chemistry
This journal is � The Royal Society of Chemistry 2006 J. Mater. Chem., 2006, 16, 1183–1191 | 1183
Publ
ishe
d on
23
Janu
ary
2006
. Dow
nloa
ded
by U
nive
rsita
Deg
li St
udi d
i Nap
oli F
eder
ico
II o
n 27
/07/
2013
16:
24:0
6.
View Article Online / Journal Homepage / Table of Contents for this issue

on a Bruker Vector 22 spectrometer. UV-Vis spectra were
measured with a Kontron Uvikon 942. GC-MS data (EI,
70 eV) were acquired on a HP 6890 instrument equipped with a
HP-5MS capillary column (crosslinked 5% Ph Me siloxane)
30.0 m 6 250 mm 6 0.25 mm coupled with a HP 5973 mass
spectrometer. LC-MS analyses were performed on selected
compounds by direct injection on an Agilent HPLC system
equipped with DAD, autosampler and MS system (Agilent
1100 LC-MS SL series) using an atmospheric pressure
chemical ionization interface (APCI). APCI conditions:
positive ion mode, flow rate 0.5 mL min21, nitrogen as
nebulizing and drying gas, nebulizer pressure 60 psi, vaporizer
temperature 350 uC, corona current 4.0 mA, drying-gas flow
5 L min21, drying gas temperature 350 uC, capillary voltage
4000 V. Mass spectrometry data were acquired in the scan
mode (mass range m/z = 200–3000). C, H, N elemental
analyses were carried out with a Eurovector CHNS-O
elemental analyser.
Thermogravimetric analyses (TGA) were carried out under
a nitrogen flow with a TA Instruments 2950 thermobalance at a
scanning rate of 10 uC min21; differential scanning calorimetry
(DSC) analyses were carried out with a TA Instruments 2920
thermosystem at a scanning rate of 10 uC min21. Polarised
optical microscopy (POM) investigations were performed
using a Reichert-Jung Polyvar II polarizing microscope
equipped with a Linkam TH600 heating stage.
An atomic force microscope (AFM, Quesant Nomad) was
used to study the surface morphology of the sublimed 6T films.
Surface images were collected in intermittent contact mode,
with typically 20 N m21 silicon cantilever probes. Care was
taken to minimize the cantilever oscillation amplitude, in order
to avoid surface damage. This was checked by subsequent
imaging of the same area by first smaller and then larger scans.
Device fabrication
Field effect transistors were fabricated in a bottom gate top
contact configuration. A cross-sectional schematic of the
device configuration is reported in Fig. 1.
The devices comprised a heavily n-doped silicon substrate
(resistivity: 0.02–1 V cm21) coated by a 300 nm thick SiO2
thermal oxide layer (Ci = 10 nF cm2). A gold ohmic contact
created directly on the silicon substrate functions as the gate
contact (G). The organic 6T layers were deposited by vacuum
sublimation at room temperature (1026 Torr) onto the SiO2
layer until a 200–300 nm film was formed. The film thickness
was measured by a KLA Tencor T10 profilometer. No
patterning of the active layer was performed in order to
confine it to the channel region. This is a very convenient and
easy thin-film transistor fabrication procedure as no litho-
graphic pattering is required. It has, however, the disadvantage
that a small leakage current can add to the source–drain
current (Ids) flowing in the channel region. The gold source (S)
and drain (D) contacts were defined by thermal evaporation
on the organic layer through a shadow mask. The resulting
channel length is L = 200 mm and the width is W = 4 mm. The
current–voltage (Ids–Vds) characteristics were evaluated with
an Agilent 4155B semiconductor parameter analyzer operating
the device in a common source configuration and in the
accumulation mode and the potentials (both Vds and Vg) were
ranged between 0 V and 2100 V. Cyclic voltammetry
measurements were carried out under inert nitrogen atmo-
sphere with an Autolab potentiostat PGSTAT 10 using a
three-electrode cell (a glassy carbon as working electrode, an
Ag wire as reference electrode, and a platinum counter
electrode). The rate scan was 200 mV s21 and the supporting
electrolyte was a n-Bu4NClO4 (0.1 M) solution in CHCl3.
Potentials are referenced to the Fc/Fc+ couple.
Synthesis
Some of the precursors were synthesized following literature
procedures, namely: 59-bromo-2,29-bithiophene-5-carbonyl
chloride (1),15 5,59-bis-trimethylstannyl-2,29-bithiophene (8),16
5-bromo-2,29-bithiophene (9),17 decylmethylamine,18 and
Pd0(PPh3)4.19 The syntheses of the other compounds were
carried out as reported below.
N-Decyl-59-bromo-2,29-bithiophene-5-carboxamide (2). A
solution of 1 (720 mg, 2.34 mmol) in methylene chloride
(180 cm3) was added dropwise over 1 h to an ice-cooled
solution of decylamine (369 mg, 2.34 mmol) in CH2Cl2 (20 cm3)
containing triethylamine (710 mg, 7.02 mmol). After the
addition, the reaction was allowed to reach room temperature
and kept under stirring overnight. The reaction mixture was
subsequently washed with 100 cm3 of 5%w HCl, 100 cm3 of
saturated NaHCO3 solution, and 100 cm3 of brine. The
organic layer was dried over anhydrous Na2SO4, and the
solvent removed in vacuo. The crude was purified by flash
chromatography on silica gel using methylene chloride as
eluent yielding 2 (802 mg, 80%) as a white solid. Mp 135.7–
136.2 uC; nmax(KBr pellet)/cm21 3331, 3096, 3094, 2956, 2930,
2916, 2850, 1628, 1545, 1534, 1510, 1478, 1470, 1454, 1416,
1319, 1293, 1268, 809, 793 and 742; dH(400 MHz; CDCl3;
Me4Si) 0.87 (t, J = 7.0 Hz, 3H, CH3), 1.21–1.38 (m, 14H), 1.60
(pseudoquintet, J = 7.0 Hz, 2H, -CH2CH2CH2NHCO), 3.42
(m, 2H, -CH2NHCO), 5.90 (br t, J = 5.7 Hz, 1H, -NH-), 6.98–
7.00 (m, 2H), 7.04 (d, J = 4.0 Hz, 1H) and 7.34 (d, J = 4.0 Hz,
1 H); dC(100.6 MHz; CDCl3; Me4Si) 14.12, 22.67, 26.95, 29.30,
29.53, 29.68, 31.87, 40.12, 112.43, 123.93, 124.95, 128.26,
130.85, 137.66, 137.86, 140.42 and 161.32; m/z (EI, 70 eV) 429
(M+, 29), 289 (M+ 2 CH2LCH(CH2)7CH3, 41), 273 (M+ 2
NH(CH2)9CH3, 100) and 245 (M+ 2 NH(CH2)9CH3 2 CO, 8).
N-Decyl-N-methyl-59-bromo-2,29-bithiophene-5-carboxamide
(3). The synthesis of 3 was carried out by the same procedure
outlined for 2 using 697 mg (2.26 mmol) of 1, 388 mg
(2.26 mmol) of decylmethylamine, and triethylamine (686 mg,
6.78 mmol) respectively. The product 3 was obtained as a white
solid (850 mg, 85%). Mp 53.0–54.0 uC; nmax(KBr pellet)/cm21Fig. 1 Bottom gate top contact OFET device structure.
1184 | J. Mater. Chem., 2006, 16, 1183–1191 This journal is � The Royal Society of Chemistry 2006
Publ
ishe
d on
23
Janu
ary
2006
. Dow
nloa
ded
by U
nive
rsita
Deg
li St
udi d
i Nap
oli F
eder
ico
II o
n 27
/07/
2013
16:
24:0
6.
View Article Online

3074, 2952, 2914, 2850, 1589, 1515, 1495, 1459, 1428, 1404,
1315, 1260, 1199, 1109, 1082, 1048, 971, 864, 785, 735 and 462;
dH(400 MHz; CDCl3; Me4Si) 0.88 (t, J = 7.0 Hz, 3H, CH3),
1.23–1.33 (m, 14H), 1.65 (pseudoquintet, J = 7.0 Hz, 2H,
-CH2CH2CH2N-), 3.16 (br s, 3H, N-CH3), 3.52 (m, 2H,
-CH2N-), 6.96–6.99(m, 2H), 7.02 (d, J = 4.0 Hz, 1H) and 7.22
(br s, 1H); dC(100.6 MHz; CDCl3; Me4Si) 14.10, 22.67, 26.69,
29.28, 29.32, 29.52, 31.87, 112.10, 123.29, 124.76, 129.48,
130.85, 137.90, 138.16, 139.45 and 163.50; m/z (EI, 70 eV)
443 (M+, 15), 303 (M+2 CH2LCH(CH2)7CH3, 14), 273 (M+ 2
CH3N(CH2)9CH3, 100) and 245 (M+ 2 CH3N(CH2)9CH3 2
CMO, 10).
N-Butyl-59-bromo-2,29-bithiophene-5-carboxamide (4). The
synthesis of 4 was carried out by the same procedure outlined
for 2 using 922 mg (3.00 mmol) of 1, 227 mg (3.10 mmol) of
n-butylamine, and triethylamine (910 mg, 9.00 mmol) respec-
tively. The product 4 was obtained as a white solid (692 mg,
67%). Mp 113.2–115.8 uC; nmax(KBr pellet)/cm21 3332, 3095,
3078, 2955, 2929, 2901, 2870 , 1629, 1536, 1506, 1455, 1302,
1256, 809, 794, 742 and 595; dH(400 MHz; CDCl3; Me4Si) 0.95
(t, J = 7.4 Hz, 3H, CH3), 1.40 (m, 2H, CH2CH2CH3), 1.59 (m,
2H, CH3CH2CH2CH2N-), 3.43 (td, J1 = 7.1 Hz, J2 = 5.8 Hz,
2H, CH3CH2CH2CH2NH-), 5.96 (br s, 1H, -NH-), 6.97–6.99
(m, 2H), 7.03 (d, J = 4.0 Hz, 1H) and 7.35 (d, J = 4.0 Hz, 1H);
dC(100.6 MHz; CDCl3; Me4Si) 13.77, 20.12, 31.75, 39.72,
112.41, 123.91, 124.93, 128.27, 130.8, 137.65, 137.85, 140.42
and 161.36; m/z (EI, 70 eV) 345 (M+, 38), 289 (M+ 2
CH2LCHCH2CH3, 42), 273 (M+ 2 NH(CH2)3CH3, 100) and
245 (M+ 2 NH(CH2)3CH3 2 CMO, 10).
N-Butyl-N-methyl-59-bromo-2,29-bithiophene-5-carboxamide
(5). The synthesis of 5 was carried out by the same procedure
reported for 2 using 671 mg (2.18 mmol) of 1, 200 mg
(2.29 mmol) of butylmethylamine, and triethylamine (662 mg,
6.54 mmol) respectively. The product 5 was obtained as a white
solid (494 mg, 63%). Mp 48.1–49.3 uC; nmax(KBr pellet)/cm21
3102, 3074, 2949, 2925, 2869, 2857, 1597, 1513, 1486, 1452,
1424, 1401, 795 and 731; dH(400 MHz; CDCl3; Me4Si) 0.95 (t,
J = 7.4 Hz, 3H, CH3), 1.35 (m, 2H, CH2CH2CH3), 1.64
(pseudoquintet, J = 7 Hz, 2H, CH3CH2CH2CH2N-), 3.3 (br s,
3H, N-CH3), 3.53 (t, J = 7.6 Hz, 2H, CH3CH2CH2CH2N-),
6.97–6.99 (m, 2H), 7.02 (d, J = 4.0 Hz, 1H) and 7.22 (br d, J =
3.8 Hz, 1H); dC(100.6 MHz; CDCl3; Me4Si) 13.83, 19.94,
22.68, 29.68, 37.35, 49.20, 112.11, 123.31, 124.79, 129.56,
130.81, 137.17, 137.88, 139.49 and 163.56; m/z (EI, 70 eV)
359 (M+, 20), 303 (M+ 2 CH2LCHCH2CH3, 9), 273 (M+ 2
CH3N(CH2)3CH3, 100) and 245 (M+ 2 CH3N(CH2)3CH3 2
CMO, 12).
Butyl-59-bromo-2,29-bithiophene-5-carboxylate (6). A toluene
solution (17 cm3) containing butan-1-ol (246 mg, 3.32 mmol), 1
(1.022 g, 3.32 mmol), and pyridine (295 mg, 3.73 mmol) was
refluxed under stirring overnight. After cooling to room
temperature, the solution was poured into water and extracted
with diethyl ether (3 6 25 cm3). The collected organic layers
were dried over anhydrous Na2SO4, and the solvent removed
in vacuo. The crude residue was then purified by flash chro-
matography on silica gel using methylene chloride as eluent
yielding 6 (1.126 g, 98%) as a pale yellow solid. Mp 26–28 uC;
nmax(KBr pellet)/cm21 3101, 3069, 2956, 2938, 2874, 1705,
1514, 1446, 1419, 1295, 1281, 1251, 1097, 783 and 748;
dH(400 MHz; CDCl3; Me4Si) 0.98 (t, J = 7.4 Hz, 3H, CH3),
1.46 (m, 2H, CH2CH2CH3), 1.73 (m, 2H, CH3CH2CH2CH2O-),
4.3 (t, J = 6.7 Hz, 2H, CH3CH2CH2CH2O-), 6.99–7.02 (m,
2H), 7.06 (d, J = 3.7 Hz, 1H) and 7.67 (d, J = 3.9 Hz, 1H);
dC(100.6 MHz; CDCl3; Me4Si) 13.73, 19.18, 30.71, 65.16,
112.85, 124.05, 125.21, 130.91, 132.25, 133.98, 137.80, 142.74
and 162.06; m/z (EI, 70 eV) 346 (M+, 32), 290 (M+ 2
CH2LCHCH2CH3, 100), 273 (M+ 2 O(CH2)3CH3, 35) and
245 (M+ 2 O(CH2)3CH3 2 CMO, 14).
Decyl-59-bromo-2,29-bithiophene-5-carboxylate (7). The
synthesis of 7 was carried out by the same procedure reported
for 6 using 1.012 g (3.29 mmol) of 1, 522 mg (3.29 mmol) of
decan-1-ol, and pyridine (300 mg, 3.79 mmol) respectively.
Product 7 was obtained as a pale yellow solid (1.174 g, 83%).
Mp 47–48.5 uC; nmax(KBr pellet)/cm21 3093, 3076, 2960, 2927,
2853, 1695, 1510, 1454, 1420, 1286, 1106, 787 and 744;
dH(400 MHz; CDCl3; Me4Si) 0.88 (t, J = 7.0 Hz, 3H, CH3),
1.27–1.45 (m, 14H), 1.74 (pseudoquintet, J = 6.7 Hz, 2H,
CH3(CH2)7CH2CH2O-), 4.28 (t, J = 6.4 Hz, 2H, CH3(CH2)7-
CH2CH2O-), 6.99–7.03 (m, 2H), 7.07 (d, J = 4.0 Hz, 1H) and
7.67 (d, J = 4.0 Hz, 1H); dC(100.6 MHz; CDCl3; Me4Si) 14.11,
22.67, 25.93, 28.65, 29.23, 29.29, 29.50, 29.52, 31.88, 65.43,
112.85, 124.05, 125.20, 130.91, 132.27, 133.98, 137.81, 142.73
and 162.01; m/z (EI, 70 eV) 430 (M+, 27), 290 (M+ 2
CH2LCH(CH2)7CH3, 100), 273 (M+ 2 O(CH2)9CH3, 34), 245
(M+ 2 O(CH2)9CH3 2 CMO, 10).
N,N9-Bis-decyl-2,29;59,20;50,2-;5-,29-;59-,20--sexithiophene-
5,50--dicarboxamide (6Ta). To a 25 cm3 three-necked round-
bottomed flask were added 754 mg (1.76 mmol) of 2, 431 mg
(0.88 mmol) of 5,59-bis-trimethylstannyl-2,29-bithiophene (8),
and dry dimethylacetamide (DMA, 7.0 cm3). The solution was
purged from residual traces of oxygen by three freeze–pump–
thaw cycles and subsequently 20 mg (1.7 6 1022 mmol) of
Pd(PPh3)4 were added. The mixture was then heated at 120 uCand kept under stirring overnight. After cooling to room
temperature, the product formed as a red precipitate was
collected and washed on a Soxhlet using hot methanol,
acetone, chloroform and hexane to afford 6Ta as a red powder
(455 mg, 60%). Further purification was achieved by vacuum
sublimation. nmax(KBr pellet)/cm21 3360, 3073, 3061, 2955,
2918, 2872, 2850, 1627, 1556, 1537, 1252, 1496, 1467, 1439,
1322, 1266, 1074, 841, 818, 793 and 741; Found: C, 64.12; H,
6.30; N, 3.14%. Calc. for C46H56N2O2S6: C, 64.14; H, 6.55; N,
3.25%.
N,N9-Bis-decyl-N,N9-bis-methyl-2,29:59,20:50,2-:5-,29-:59-,
20--sexithiophene-5,50--dicarboxamide (6Tb). To a 25 cm3
three-necked round-bottomed flask was added 800 mg
(1.81 mmol) of 3, 443 mg (0.90 mmol) of 5,59-bis-trimethyl-
stannyl-2,29-bithiophene (8), and dry DMA (7.5 cm3). The
solution was purged from residual traces of oxygen by
three freeze–pump–thaw cycles and subsequently 21 mg
(1.8 6 1022 mmol) of Pd(PPh3)4 were added. The mixture
was then heated at 120 uC and kept under stirring overnight.
This journal is � The Royal Society of Chemistry 2006 J. Mater. Chem., 2006, 16, 1183–1191 | 1185
Publ
ishe
d on
23
Janu
ary
2006
. Dow
nloa
ded
by U
nive
rsita
Deg
li St
udi d
i Nap
oli F
eder
ico
II o
n 27
/07/
2013
16:
24:0
6.
View Article Online

After cooling to room temperature, the solid was collected
and washed several times on a Soxhlet with hot methanol,
acetone, and hexane to afford 6Tb as a red powder (576 mg,
72%). Further purification was achieved by vacuum sublima-
tion. nmax(KBr pellet)/cm21 3075, 3057, 2957, 2919, 2873,
2850, 1600, 1529, 1502, 1485, 1454, 1437, 1404, 1322,
1306, 1080, 1069, 840, 791 and 735; lmax(CHCl3)/nm 443;
m/z(APCI) 889.3 ([M + H]+ requires 889.31); Found: C, 64.59;
H, 6.76; N, 3.14%. Calc. for C48H60N2O2S6: C, 64.82; H, 6.80;
N, 3.15%.
N,N9-Bis-butyl-2,29:59,20:50,2-:5-,29-:59-,20--sexithiophene-
5,50--dicarboxamide (6Tc). The synthesis of 6Tc was carried
out by the same procedure reported for 6Ta using 650 mg
(1.89 mmol) of 4, 462 mg (0.94 mmol) of 8, 22 mg (1.9 61022 mmol) of Pd(PPh3)4, and 8.0 cm3 of dry DMA. The
title product was obtained as a red powder (423 mg, 65%).
Further purification was achieved by vacuum sublimation.
nmax(KBr pellet)/cm21 3357, 3071, 3059, 2957, 2931, 2871,
1628 , 1538, 1495, 1438, 1295, 841, 792 and 742; Found: C,
58.84; H, 4.56; N, 3.97%. Calcd for C34H32N2O2S6: C, 58.92;
H, 4.65; N, 4.04%.
N,N9-Bis-butyl-N,N9-bis-methyl-2,29:59,20:50,2-:5-,29-:59-,
20--sexithiophene-5,50--dicarboxamide (6Td). The synthesis of
6Td was carried out by the same procedure reported for 6Tb
using 452 mg (1.26 mmol) of 5, 310 mg (0.63 mmol) of 8, 15 mg
(1.3 6 1022 mmol) of Pd(PPh3)4, and 5.0 cm3 of dry DMA.
The title product was obtained as a red powder (363 mg, 80%).
Further purification was achieved by vacuum sublimation.
nmax(KBr pellet)/cm21 3071, 3058, 2955, 2928, 2871, 2859,
1599, 1528, 1501, 1485, 1454, 1403, 1078, 841, 791 and 734;
m/z(APCI) 721.1 ([M + H]+ requires 721.12); Found: C, 59.87;
H, 4.98; N, 3.87%. Calc. for C36H36N2O2S6: C, 59.96; H, 5.03;
N, 3.88; O, 4.44%.
Bis-butyl 2,29:59,20:50,2-:5-,29-:59-,20--sexithiophene-5,50--
dicarboxylate (6Te). The synthesis of 6Te was carried out by
the same procedure reported for 6Tb using 1.020 g (2.95 mmol)
of 6, 726 mg (1.47 mmol) of 8, 34 mg (2.9 6 1022 mmol) of
Pd(PPh3)4, and 12.0 cm3 of dry DMA. The title product was
obtained as a red powder (766 mg, 75%). Further purification
was achieved by vacuum sublimation. nmax(KBr pellet)/cm21
3081, 3063, 2960, 2930, 2900, 2870, 1691, 1527, 1454, 1437,
1286, 1093, 835, 787 and 744; lmax(CHCl3)/nm 451; m/z
(APCI) 695.1 ([M + H]+ requires 695.05); Found: C, 58.45; H,
4.32%. Calc. for C34H30O4S6: C, 58.76; H, 4.35%.
Bis-decyl 2,29:59,20:50,2-:5-,29-:59-,20--sexithiophene-5,50--
dicarboxylate (6Tf). The synthesis of 6Tf was carried out by
the same procedure reported for 6Tb using 974 mg (2.27 mmol)
of 7, 558 mg (1.13 mmol) of 8, 27 mg (2.3 6 1022 mmol) of
Pd(PPh3)4, and 10.0 cm3 of dry DMA. The title product was
obtained as a red powder (878 mg, 90%). Further purification
was achieved by vacuum sublimation. nmax(KBr pellet)/cm21
3071, 3035, 2951, 2917, 2844, 1712, 1532, 1458, 1441, 1321,
1278, 1248, 1097, 839, 796 and 749; lmax(CHCl3)/nm 451;
m/z(APCI) 863.3 ([M + H]+ requires 863.24); Found: C, 63.95;
H, 6.25%. Calc. for C46H54O4S6: C, 64.00; H, 6.30%.
Ethyl (2E)-3-(2,29-bithien-5-yl)acrylate (10). To a solution of
5-bromo-2,29-bithiophene 9 (2.000 g, 8.16 mmol) in DMF
(12 cm3), ethyl acrylate (1.144 g, 11.42 mmol), triethylamine
(2.32 g, 22.84 mmol) and Pd(PPh3)4 (94.3 mg, 8.2 61022 mmol) were added respectively. The mixture was heated
at 120 uC and kept under stirring overnight. After cooling to
room temperature, the solution was poured into water and
extracted with diethyl ether. The organic layers were collected,
washed with water and dried over anhydrous Na2SO4.
Evaporation of the solvent and flash chromatography on
silica gel using petroleum ether–methylene chloride (60 : 40) as
eluent afforded pure 10 as a bright orange solid (1.51 g, 70%).
Mp 60.0–61.0 uC; nmax(KBr pellet)/cm21 3108, 3076, 3048,
3023, 2970, 2922, 2901, 2863, 1763, 1752, 1707, 1617, 1547,
1476, 1464, 1453, 1441, 1281, 1274, 1263, 1209, 1164, 1112,
1099, 1044, 1032, 915, 873, 800 and 695; dH(400 MHz; CDCl3;
Me4Si) 1.32 (t, J = 7.2 Hz, 3H, CH3), 4.25 (q, J = 7.2 Hz, 2H,
OCH2CH3), 6.18 (d, J = 15.7 Hz, 1H, LCH), 7.03 (dd, J1 =
5.1 Hz, J2 = 3.7 Hz, 1H), 7.11 (d, J = 3.8 Hz, 1H), 7.15 (d,
J = 3.8 Hz, 1H), 7.22 (dd, J1 = 3.7 Hz, J2 = 1.1 Hz), 7.26 (dd ,
J1 = 5.1 Hz, J2 = 1.1 Hz, 1H) and 7.71 (d, J = 15.7 Hz, 1H,
LCH); dC(100.6 MHz; CDCl3; Me4Si) 14.32, 60.49, 116.61,
124.30, 124.74, 125.56, 128.06, 132.05, 136.69, 136.81, 138.16,
140.25 and 166.83; m/z(EI, 70 eV) 264 (M+, 100), 236 (15), 219
(M+ 2 OCH2CH3, 58) and 192 (74).
Ethyl 3-(2,29-bithien-5-yl)propanoate (11). To a solution of
10 (661 mg, 2.5 mmol) in MeOH (50 cm3) was added 266 mg of
supported Pd (10 wt% on activated charcoal) and hydro-
genated in a stainless-steel autoclave at 25 bar of pressure; the
substrate conversion was monitored by GC-MS. After reaction
completion, the solution was filtered through a short plug of
Celite and the solvent evaporated off. The residue was purified
by flash chromatography on silica gel using petroleum ether–
methylene chloride (50 : 50) as eluent affording 11 as a
yellowish oil (653 mg, 98%). nmax(KBr pellet)/cm21 3107, 3069,
2980, 2960, 2928, 2871, 2855, 1732, 1638; 1519, 1465, 1444,
1427, 1374, 1351, 1298, 1256, 1176, 1122, 1095, 1081, 1037,
941, 910, 887, 872, 838, 800 and 695; dH(400 MHz; CDCl3;
Me4Si) 1.26 (t, J = 7.2 Hz, 3H, CH3), 2.68 (t, J = 7.5 Hz, 2H,
CH2CH2), 3.13 (t, J = 7.5 Hz, 2H, CH2CH2), 4.16 (q, J =
7.2 Hz, 2H, OCH2CH3), 6.72 (br d, J = 3.7 Hz, 1H), 6.97–6.99
(m, 2H), 7.09 (dd, J1 = 3.7 Hz, J2 = 1.1 Hz, 1H) and 7.17 (dd,
J1 = 5.1 Hz, J2 = 1.1 Hz, 1H); dC(400 MHz; CDCl3; Me4Si)
14.21, 25.37, 35.94, 60.63, 123.26, 123.45, 125.42, 127.69,
135.54, 137.60, 142.45 and 172.28; m/z(EI, 70 eV) 266 (M+, 98),
237 (M+ 2 CH2CH3, 20), 192 (81) and 179 (100).
Ethyl 3-(59-bromo-2,29-bithien-5-yl)propanoate (12). A solu-
tion of NBS (392 mg, 2,2 mmol) in CH2Cl2 (30 cm3) was added
dropwise over a period of 2 h to a solution of 11 (586 mg,
2.2 mmol) in CH2Cl2 (25 cm3) kept at 0 uC and in the dark.
After the addition the solution was allowed to reach room
temperature and was held overnight. The mixture was then
poured onto ice and extracted several times with CH2Cl2. The
organic phases were collected, washed with water, and dried
over Na2SO4. After solvent evaporation, the crude residue was
purified by chromatography on reverse phase (RP-18) silica
using methanol as eluent affording pure 12 as a pale yellow
1186 | J. Mater. Chem., 2006, 16, 1183–1191 This journal is � The Royal Society of Chemistry 2006
Publ
ishe
d on
23
Janu
ary
2006
. Dow
nloa
ded
by U
nive
rsita
Deg
li St
udi d
i Nap
oli F
eder
ico
II o
n 27
/07/
2013
16:
24:0
6.
View Article Online

solid (722 mg, 95%). Mp 49–50 uC; nmax(KBr pellet)/cm21
3072, 3056, 2980, 2960, 2928, 2871, 2855, 1725, 1520, 1463,
1446, 1428, 1396, 1380, 1345, 1314, 1277, 1199, 1059, 1037,
969, 873, 803 and 789; dH(400 MHz; CDCl3; Me4Si) 1.25 (t, J =
7.2 Hz, 3H, CH3), 2.67 (t, J = 7.5, 2H, CH2CH2), 3.11 (t, J =
7.5, 2H, CH2CH2), 4.15 (q, J = 7.2 Hz, 2H, OCH2CH3), 6.75
(d, J = 3.7 Hz, 1H), 6.83 (d, J = 3.8 Hz, 1H), 6.90 (d, J =
3.7 Hz, 1H) and 6.93 (d, J = 3.8 Hz, 1H); dC(100.6 MHz;
CDCl3; Me4Si) 14.19, 25.34, 35.86, 60.65, 110.45, 123.31,
123.75, 125.50, 130.49, 134.50, 139.08, 143.03 and 172.18;
m/z(EI, 70 eV) 344 (M+, 58), 315 (7), 270 (30) and 257 (100).
Bis-ethyl 3-(2,29:59,20:50,2-:5-,29-:59-,20--sexithien-5,50--
diyl)dipropanoate (6Tg). The synthesis of 6Tg was carried out
by the same procedure reported for 6Tb using 656 mg
(1.90 mmol) of 12, 467 mg (0.95 mmol) of 8, 22 mg (1.9 61022 mmol) of Pd(PPh3)4, and 8 cm3 of dry DMA. The
product 6Tg was obtained as a red powder (462 mg, 70%).
Further purification was achieved by vacuum sublimation.
nmax(KBr pellet)/cm21 3060, 2979, 2908, 2851, 1735, 1505,
1441, 1380, 1.352, 1.321, 1298, 1.179, 1097, 1070, 1034, 858,
838 and 793; lmax(CHCl3)/nm 439; m/z(APCI) 695.1 ([M + H]+
requires 695.05); Found: C, 58.43; H, 4.38%. Calc. for
C34H30O4S6: C, 58.76; H, 4.35%.
Results and discussion
Synthesis
The structure of the novel sexithiophenes synthesized in this
study are reported in Fig. 2.
The sexithiophene 6Ta is functionalized in the a,v-positions
with two amide groups bearing decyl alkyl chains. In order to
investigate the possible effect of hydrogen bonding between
amide groups of different sexithiophene functionalities on the
properties of the materials, compound 6Tb, bearing a methyl
group instead of the amide hydrogen, was synthesized. The
next target was to synthesise the oligomers 6Tc and 6Td
bearing a shorter (butyl) alkyl chain. The synthesis of 6Te
and 6Tf was carried out in order to compare the effect of the
ester group with respect to the amide group. Eventually, the
effect of a spacer between the ester functional group and
the sexithiophene core was investigated with the synthesis
of 6Tg.
The synthetic approach to the preparation of the 6Ta-g
sexithiophenes comprised a Stille coupling (vide infra) between
5,59-bis-trimethylstannyl-2,29-bithiophene (8) and suitable
bithiophene derivatives. The synthetic approach followed
for the 2-bromo-bithiophene precursors 2–7 is reported in
Scheme 1.
The bithiophene derivatives 2–7 were obtained in yields
ranging from 63 to 98% by reacting 59-bromo-2,29-bithio-
phene-5-carbonyl chloride (1)15 with the corresponding amines
or alcohols under Schotten–Baumann conditions. The synth-
esis of the 2-bromo-bithiophene derivative 12 required a
different approach, depicted in Scheme 2.
5-Bromo-2,29-bithiophene (9)17 was submitted to a Heck
coupling with ethyl acrylate affording ester 10 in 70% yield.
Hydrogenation of 10 was carried out in the presence of
supported palladium under a constant hydrogen pressure
(25 bar) yielding 11 in 98% yield. Bromination of 11 with
stoichiometric NBS afforded 12 in 95% yield. The structures of
1–12 were confirmed by IR, 1H and 13C NMR spectroscopy as
well as GC-MS.
The target sexithiophenes were obtained by means of a Stille
coupling, due to its versatility and its compatibility towards
amide and ester functional groups.20 The synthesis of the
functionalized sexithiophenes 6Ta-g is sketched in Scheme 3
along with the product yields (60–90%).
Fig. 2 Structures of the synthesised a,v-sexithiophenes.
Scheme 1 (i) Conditions for 2–5: reaction with 1.0 eq. of the
corresponding amines in the presence of triethylamine in CH2Cl2 at
rt. (ii) For 6 and 7: reaction with 1.0 eq. of the corresponding alcohols
in the presence of pyridine in toluene at reflux.
Scheme 2 (i) 1% mol/mol Pd(PPh3)4 with respect to the bromide 9 in
the presence of triethylamine in DMF at 120 uC; (ii) P(H2) = 25 bar,
10% mol/mol Pd on active charcoal in MeOH at rt; (iii) NBS (1 eq) in
CH2Cl2.
This journal is � The Royal Society of Chemistry 2006 J. Mater. Chem., 2006, 16, 1183–1191 | 1187
Publ
ishe
d on
23
Janu
ary
2006
. Dow
nloa
ded
by U
nive
rsita
Deg
li St
udi d
i Nap
oli F
eder
ico
II o
n 27
/07/
2013
16:
24:0
6.
View Article Online

The products precipitated from the reaction medium as
orange or red powders and were purified from soluble
byproducts by subsequent Soxhlet washings with methanol,
chloroform, acetone and n-hexane.
Compounds 6Tb, 6Td, 6Te, 6Tf and 6Tg were characterised
by APCI-mass spectrometry that confirmed the achievement
of the desired products. Elemental analyses of all oligomers
were in accordance with the proposed structures and
confirmed the high purity of all sexithiophenes. As expected,
all synthesised sexithiophenes are poorly soluble in common
organic solvents except hot DMSO, chlorobenzene and
o-dichlorobenzene. However, the solubility of 6Tb, 6Te, 6Tf,
and 6Tg in chloroform was good enough to measure their
UV-Vis spectra. The lmax are 443 (6Tb), 451 (6Te), 451 (6Tf)
and 439 (6Tg) nm. These absorptions are red-shifted with
respect to the lmax of unsubstituted sexithiophene (lit.,21
432 nm) because of the increase of conjugation length brought
about by the carbonyl groups directly attached to the
sexithiophene core in the case of 6Tb, 6Te, 6Tf. In the case
of 6Tg the inductive effect of the substituent may account for
the observed slight red shift.
Thermal behaviour
The thermal properties of 6Ta-g are summarized in Table 1.
The stability of all sexithiophenes was investigated by TGA,
that revealed high decomposition temperatures (at 5% weight
loss) between 333 and 399 uC. It can be noted, comparing the
Tdec of 6Ta,6Tb and those of 6Tc,6Td, that the substitution of
a hydrogen atom with a methyl group increased the thermal
stability of the sexithiophenes. On the other hand the
introduction of the ester groups lowered the decomposition
temperature. The thermal behaviour of all obtained sexithio-
phenes was also investigated by means of polarizing optical
microscopy (POM) and differential scanning calorimetry
(DSC) methods, and the main results are also summarized in
Table 1.
6Tb is the only compound that presented an evident fusion
point at 295.9 uC without passing through mesophases. For all
the other compounds, isotropization was concomitant to
partial decomposition. POM observation permitted the charac-
terisation of several observed thermal events as transitions to
unidentified smectic phases (SmX). In other cases, in particular
for 6Tg, the thermal events could not be further characterised
because the POM texture did not change in the course of
the transition. This behaviour can be interpreted either by
invoking a solid–solid transition or with the fusion of small
portions of crystalline sample deriving from the synthesis.
The difference in thermal behaviour between 6Ta and 6Tb is
noteworthy. The possibility of hydrogen bonding in solid 6Ta
seems to be responsible for the more complex thermal
behaviour probably related to a more structured organization.
Comparing the behaviour of 6Ta to that of 6Tc it can be
inferred that the shorter alkyl chain prevents the occurrence of
mesophases in the latter. Similar behaviour was observed also
for the ester substituted compounds, namely 6Te and 6Tf: both
sexithiophenes showed multiple transitions to smectic phases,
before isotropization occurs concomitantly to decomposition.
Increasing the ester chain length led to a lowering of the
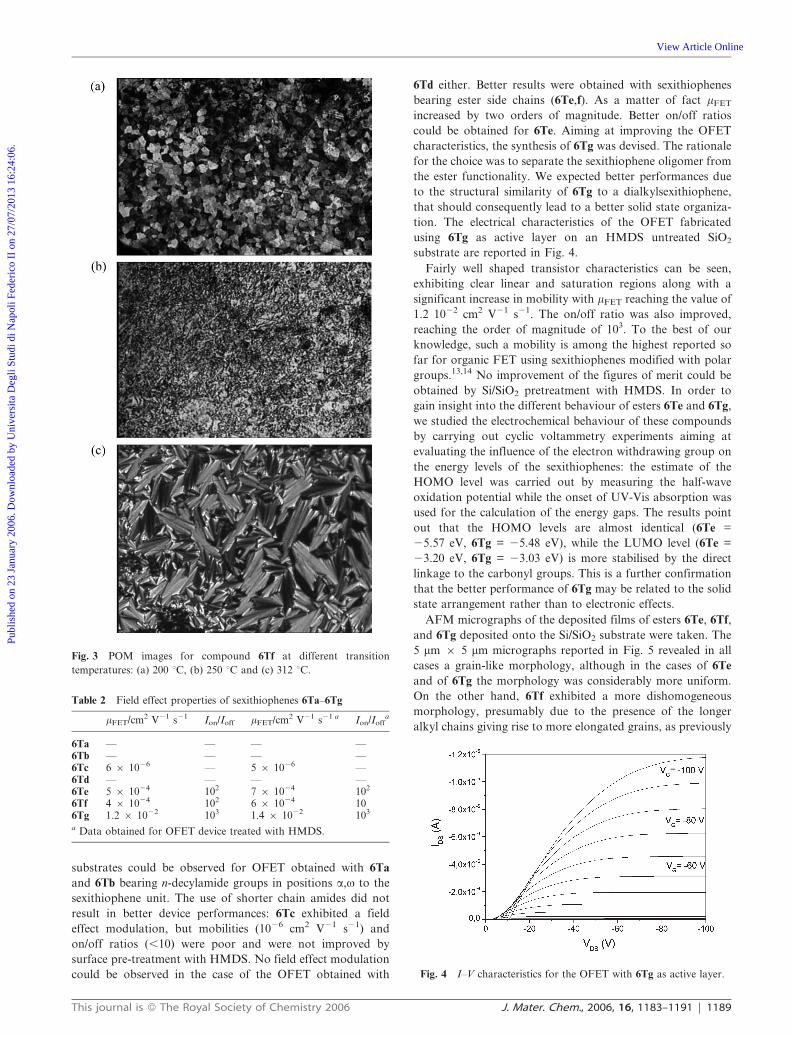
transition temperatures. In Fig. 3 is displayed the change in
optical texture of 6Tf passing from the three observed SmX
phases.
The observation of such smectic mesophases indicates a
degree of order of the materials that reflects an ordered solid
state.22
Field-effect properties and surface morphology
The OFET devices have been fabricated as reported in the
Experimental section. The main figures of merit, namely the
on/off ratio and the field effect mobility (mFET), for the devices
are reported in Table 2. The transistor characteristics were
measured by polarizing the devices up to 2100 V. Negative
source–drain and gate voltage biases were applied as p-type
behavior was expected. All sexithiophene films were evapo-
rated on two sets of substrates, one of which was pre-treated
with hexamethyldisilazane (HMDS), aiming at improving the
adhesion of the organic layer to the SiO2 dielectric surface.4
No field-effect modulation on either treated or untreated
Scheme 3 (i) 2% mol/mol Pd(PPh3)4 with respect to the organo-
distannane (8) in dimethylacetamide at 120 uC.
Table 1 Thermal data for sexithiophenes 6Ta-6Tg
Compound Transition temperatures (uC) and enthalpies of transitions (in parentheses, J g21) TGAa/uC
6Ta 203.5b (6.1) 285.0b (3.2) 340.9c (68.9) 371.7d (dec.) 368.16Tb 295.9d (109.3) 398.76Tc 367.0d (dec.) 372.56Td 311.2c (111.2) 329.0d 384.36Te 210.5c (18.6) 267.9c (32.5) 343.4c (dec.) 341.26Tf 177.0c (26.9) 239.5c (27.2) 300.0c (10.8) 344.56Tg 159.2b (7.1) 178.9b (10.3) 332.9a At 5% weight loss. b The transitions observed cannot be evidenced in the POM, they may be solid–solid transitions or the fusion of a smallportion of the crystalline phases obtained in the course of the synthesis of the material. c Transition to a SmX mesophase. d Transition toisotropic phase.
1188 | J. Mater. Chem., 2006, 16, 1183–1191 This journal is � The Royal Society of Chemistry 2006
Publ
ishe
d on
23
Janu
ary
2006
. Dow
nloa
ded
by U
nive
rsita
Deg
li St
udi d
i Nap
oli F
eder
ico
II o
n 27
/07/
2013
16:
24:0
6.
View Article Online
substrates could be observed for OFET obtained with 6Ta
and 6Tb bearing n-decylamide groups in positions a,v to the
sexithiophene unit. The use of shorter chain amides did not
result in better device performances: 6Tc exhibited a field
effect modulation, but mobilities (1026 cm2 V21 s21) and
on/off ratios (,10) were poor and were not improved by
surface pre-treatment with HMDS. No field effect modulation
could be observed in the case of the OFET obtained with
6Td either. Better results were obtained with sexithiophenes
bearing ester side chains (6Te,f). As a matter of fact mFET
increased by two orders of magnitude. Better on/off ratios
could be obtained for 6Te. Aiming at improving the OFET
characteristics, the synthesis of 6Tg was devised. The rationale
for the choice was to separate the sexithiophene oligomer from
the ester functionality. We expected better performances due
to the structural similarity of 6Tg to a dialkylsexithiophene,
that should consequently lead to a better solid state organiza-
tion. The electrical characteristics of the OFET fabricated
using 6Tg as active layer on an HMDS untreated SiO2
substrate are reported in Fig. 4.
Fairly well shaped transistor characteristics can be seen,
exhibiting clear linear and saturation regions along with a
significant increase in mobility with mFET reaching the value of
1.2 1022 cm2 V21 s21. The on/off ratio was also improved,
reaching the order of magnitude of 103. To the best of our
knowledge, such a mobility is among the highest reported so
far for organic FET using sexithiophenes modified with polar
groups.13,14 No improvement of the figures of merit could be
obtained by Si/SiO2 pretreatment with HMDS. In order to
gain insight into the different behaviour of esters 6Te and 6Tg,
we studied the electrochemical behaviour of these compounds
by carrying out cyclic voltammetry experiments aiming at
evaluating the influence of the electron withdrawing group on
the energy levels of the sexithiophenes: the estimate of the
HOMO level was carried out by measuring the half-wave
oxidation potential while the onset of UV-Vis absorption was
used for the calculation of the energy gaps. The results point
out that the HOMO levels are almost identical (6Te =
25.57 eV, 6Tg = 25.48 eV), while the LUMO level (6Te =
23.20 eV, 6Tg = 23.03 eV) is more stabilised by the direct
linkage to the carbonyl groups. This is a further confirmation
that the better performance of 6Tg may be related to the solid
state arrangement rather than to electronic effects.
AFM micrographs of the deposited films of esters 6Te, 6Tf,
and 6Tg deposited onto the Si/SiO2 substrate were taken. The
5 mm 6 5 mm micrographs reported in Fig. 5 revealed in all
cases a grain-like morphology, although in the cases of 6Te
and of 6Tg the morphology was considerably more uniform.
On the other hand, 6Tf exhibited a more dishomogeneous
morphology, presumably due to the presence of the longer
alkyl chains giving rise to more elongated grains, as previously
Fig. 3 POM images for compound 6Tf at different transition
temperatures: (a) 200 uC, (b) 250 uC and (c) 312 uC.
Table 2 Field effect properties of sexithiophenes 6Ta–6Tg
mFET/cm2 V21 s21 Ion/Ioff mFET/cm2 V21 s21 a Ion/Ioffa
6Ta — — — —6Tb — — — —6Tc 6 6 1026 — 5 6 1026 —6Td — — — —6Te 5 6 1024 102 7 6 1024 102
6Tf 4 6 1024 102 6 6 1024 106Tg 1.2 6 1022 103 1.4 6 1022 103
a Data obtained for OFET device treated with HMDS.
Fig. 4 I–V characteristics for the OFET with 6Tg as active layer.
This journal is � The Royal Society of Chemistry 2006 J. Mater. Chem., 2006, 16, 1183–1191 | 1189
Publ
ishe
d on
23
Janu
ary
2006
. Dow
nloa
ded
by U
nive
rsita
Deg
li St
udi d
i Nap
oli F
eder
ico
II o
n 27
/07/
2013
16:
24:0
6.
View Article Online

reported.11 The root mean square (RMS) deviations of the
measured topography are 0.87 nm for 6Te, 1.7 nm for 6Tg and
42 nm for 6Tf. AFM observations were also carried out on the
other 6T films, that showed a microstructured morphology as
well, the RMS values being 14.96 (6Ta), 2.266 (6Tb), 1.681
(6Tc), 0.6 nm (6Td). No morphological differences were
observed for thin films deposited on treated or untreated
Si/SiO2.
Conclusions
Seven new sexithiophenes bearing amide or ester function-
alities in the a,v-positions were synthesised and characterised,
and their properties as OFET active layers were investigated.
The amide substituted materials resulted in poor charge
transport features. Use of sexithiophenes functionalised with
ester moieties permitted the construction of OFET with
moderate hole mobilities. Isolation of the ester functionalities
from the sexithiophene unit by an ethylene spacer led to a
device with good modulation, which showed hole mobilities of
1.2 6 1022 cm2 V21 s21 and on/off ratios of 103. The latter
sexithiophene holds interesting properties for future use as an
active layer in chemically selective OFET sensors.
Acknowledgements
The FIRB project Micropolys (Italian MIUR) is gratefully
acknowledged for funding. We also thank Dr Giuseppe
Ciccarella for the APCI-mass spectrometric measurements,
Dr Francesco Marinelli for laboratory assistance and Dr
Roberto Grisorio for helpful discussions. We are indebted to
Professor Pynalisa Cosma (University of Bari) for carrying out
the cyclic voltammetry experiments.
References
1 J. Roncali, Chem. Soc. Rev., 2005, 34, 483; K. M. Coakley andM. D. McGehee, Chem. Mater., 2004, 16, 4533; S. E. Shaheen,C. J. Brabec, N. S. Sariciftci, F. Padinger, T. Fromherz andJ. C. Hummelen, Appl. Phys. Lett., 2001, 78, 841; M. Granstroem,K. Petritsch, A. C. Arias, A. Lux, M. R. Andersson andR. H. Friend, Nature, 1998, 395, 257.
2 P. F. H. Schwab, J. R. Smith and J. Michl, Chem. Rev., 2005, 105,1197; Y. Shirota, J. Mater. Chem., 2005, 15, 75; J. L. Bredas,D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104,4971; L. S. Hung and C. H. Chen, Mater. Sci. Eng. R., 2002, 39,143.
3 Y. Sun, Y. Liu and D. Zhu, J. Mater. Chem., 2005, 15, 53;G. Horowitz, J. Mater. Res., 2004, 19, 1946; H. E. Katz, Chem.Mater., 2004, 16, 4748; H. Sirringhaus, Adv. Mater., 2005, 17,2411; K. Mullen and G. Wegner, Electronic Materials: TheOligomer Approach, Wiley-VCH, Weinheim, Germany, 1998.
4 C. D. Dimitrakopoulos and P. R. L. Malenfant, Adv. Mater., 2002,14, 99.
5 A. Tsumura, H. Koezuka and K. Ando, Appl. Phys. Lett., 1986,49, 1210; M. Madru, G. Guillaud, M. Al Saduon, M. Maitrot,C. Clarisse, C. Le Contellec, J. J. Andre and J. Simon, Chem. Phys.Lett., 1987, 142, 103; A. Assadi, M. Svensson, M. Willander andO. Inganas, Appl. Phys. Lett., 1988, 53, 195.
6 G. Horowitz, D. Fichou, X. Peng, Z. Xu and F. Garnier, SolidState Commun., 1989, 72, 381.
7 D. Fichou, Handbook of Oligo and Polythiophenes, Wiley-VCH,Weinheim, Germany, 1999; H. E. Katz, Z. Bao and S. L. Gilat,Acc. Chem. Res., 2001, 34, 359.
8 F. Garnier, A. Yassar, R. Hajlaoui, G. Horowitz, F. Deloffre,B. Servet, S. Ries and P. Alnot, J. Am. Chem. Soc., 1993, 115,8716.
9 M. Halik, U. T. Klauk, G. Schmid, S. Ponomarenko,S. Kirchmeyer and W. Weber, Adv. Mater., 2003, 15, 917.
10 G. Guillaud, J. Simon and J. P. Germain, Coord. Chem. Rev., 1998,178–180, 1433–1484; L. Torsi, A. Dodabalapur, L. Sabbatini andP. G. Zambonin, Sens. Actuators, B, 2000, 67, 312; B. Crone,A. Dodabalapur, A. Gelperin, L. Torsi, H. E. Katz, A. J. Lovingerand Z. Bao, Appl. Phys. Lett., 2001, 78, 2229; M. Bouvet,G. Guillaud, A. Leroy, A. Maillard, S. Spirkovitch andF. G. Tournilhac, Sens. Actuators, B, 2001, 79, 63; C. Bartic,B. Palan, A. Campitelli and G. Borghs, Sens. Actuators, B, 2002,83, 115; Z. T. Zhu, J. T. Mason, R. Dieckman and G. G. Malliaras,Appl. Phys. Lett., 2002, 81, 4643; C. Bartic, A. Campitelli andS. Borghs, Appl. Phys. Lett., 2003, 82, 475.
11 L. Torsi, A. J. Lovinger, B. Crone, T. Someya, A. Dodabalapur,H. E. Katz and A. Gelperin, J. Phys. Chem. B, 2002, 106, 12563.
Fig. 5 AFM images of thin films of 6Te (a), 6Tf (b) and 6Tg (c) as
evaporated on the Si/SiO2 substrate.
1190 | J. Mater. Chem., 2006, 16, 1183–1191 This journal is � The Royal Society of Chemistry 2006
Publ
ishe
d on
23
Janu
ary
2006
. Dow
nloa
ded
by U
nive
rsita
Deg
li St
udi d
i Nap
oli F
eder
ico
II o
n 27
/07/
2013
16:
24:0
6.
View Article Online

12 L. Torsi, M. C. Tanese, N. Cioffi, M. C. Gallazzi, L. Sabbatini,P. G. Zambonin, G. Raos, S. V. Meille and M. M. Giangregorio,J. Phys. Chem. B, 2003, 107, 7589.
13 A. Afzali, T. L. Breen and C. R. Kagan, Chem. Mater., 2002, 14,1742; H. E. Katz, J. G. Laquindanum and A. Lovinger, Chem.Mater., 1998, 10, 633; H. E. Katz, A. Dodabalapur, L. Torsi andD. Elder, Chem. Mater., 1995, 7, 2238; L. Antolini, M. Borsari,F. Goldoni, D. Iarossi, A. Mucci and L. Schenetti, J. Chem. Soc.,Perkin Trans. 1, 1999, 3207; B. H. Huisman, J. J. P. Valeton,W. Nijssen, J. Lub and W. ten Hoeve, Adv. Mater., 2003, 15, 2002;A. R. Murphy, J. M. J. Frechet, P. Chang, J. Lee andV. Subramanian, J. Am. Chem. Soc., 2004, 126, 1596.
14 M. H. Yoon, S. Di Benedetto, A. Facchetti and T. J. Marks, J. Am.Chem. Soc., 2005, 127, 1348.
15 A. F. M. Kilbinger, A. P. H. J. Schenning, F. Goldoni, W. J. Feastand E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 1820.
16 S. Kotani, K. Shiina and K. Sonoghashira, J. Organomet. Chem.,1992, 429, 403.
17 P. Baurle, F. Wurthner, G. Gots and F. Effenberger, Synthesis,1993, 1099.
18 D. Enjalbert, C. Bassilana, V. Krier, S. Szonyi and A. Cambon,J. Fluorine Chem., 1999, 93, 145.
19 D. R. Coulson, Inorg. Synth., 1972, 13, 121.20 J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508; V. Farina,
V. Krishnamurthy and W. J. Scott, The Stille Reaction, Wiley-Interscience, New York, 2000.
21 C. Van Pham, A. Burkhardt, R. Shabana, D. D. Cunningham Jr.,H. B. Mark and H. Zimmer, Phosphorus, Sulfur Silicon Relat.Elem., 1989, 46, 153.
22 D. Byron, A. Matharu, R. Wilson and G. Wright, Mol. Cryst. Liq.Cryst., 1995, 265, 61; R. Azumi, G. Gotz and P. Baurle, Synth.Met., 1999, 101, 5544; T. Yamada, R. Azumi, H. Tachibana,H. Sakai, M. Abe, P. Baurle and M. Matsumoto, Chem. Lett.,2001, 10, 1022; S. Ponomarenko and S. Kirchmeyer, J. Mater.Chem., 2003, 13, 197; M. Funahashi and J.-I. Hanna, Adv. Mater.,2005, 17, 594.
This journal is � The Royal Society of Chemistry 2006 J. Mater. Chem., 2006, 16, 1183–1191 | 1191
Publ
ishe
d on
23
Janu
ary
2006
. Dow
nloa
ded
by U
nive
rsita
Deg
li St
udi d
i Nap
oli F
eder
ico
II o
n 27
/07/
2013
16:
24:0
6.
View Article Online





























