Embed Size (px)
Citation preview

Biochimica et Biophysica Acta 1812 (2011) 919–928
Contents lists available at ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r.com/ locate /bbad is
Review
Role of nuclear receptor corepressor RIP140 in metabolic syndrome☆
Meritxell Rosell 1, Marius C. Jones 1, Malcolm G. Parker ⁎Institute of Reproductive and Developmental Biology, Imperial College London, Faculty of Medicine, Hammersmith Campus 158 Du Cane Road, W12 0NN, UK
Abbreviations: AR, androgen receptor; ATF, activatCRE-binding protein; CIDEA, cell death-inducing DNA feffector A; CPT, 1β carnitine palmitoyltransferase 1β; CRC-terminal binding protein; Dnmt, DNA methyltransfelongus; ERR, estrogen related receptor; FA, fatty acid; FAforkhead-box C2; GLUT4, glucose transporter 4; GR, ghistone deacetylase; HFD, high-fat diet; IL, interleukin;MEF, myocyte-specific enhancer binding factor; NCoR,NF-κB, nuclear factor κB; NR, nuclear receptor; NRF, nupolycystic ovary syndrome; PEPCK, phosphoenolpyruvacoactivator; PPAR, peroxixome proliferator-activatedregulatory domain-containing 16; RAR, retinoic acid reprotein 4; ROS, reactive oxygen species; RXR, retinoidmediator of retinoic acid and thyroid hormone reccoactivator; SREBP1c, sterol regulatory element bindingide; TLR, Toll-like receptor; TNFα, tumour necrosis fareceptor; UCP1, uncoupling protein 1; VLDL, very low d☆ This article is part of a Special Issue entitled: Tranhealth to disease.⁎ Corresponding author. Fax: +44 0 20 7594 2184.
E-mail address: [email protected] (M.G. Park1 M.R and M.J. contributed equally to this publication
0925-4439/$ – see front matter © 2010 Elsevier B.V. Aldoi:10.1016/j.bbadis.2010.12.016
a b s t r a c t
a r t i c l e i n f oArticle history:Received 29 October 2010Received in revised form 15 December 2010Accepted 17 December 2010Available online 28 December 2010
Keywords:Metabolic syndromeRIP140White adipose tissueInflammation
Obesity and its associated complications, which can lead to the development of metabolic syndrome, are aworldwidemajor public health concern especially indevelopedcountrieswhere they have a veryhigh prevalence.RIP140 is a nuclear coregulator with a pivotal role in controlling lipid and glucose metabolism. Geneticallymanipulatedmicedevoid of RIP140 are leanwith increasedoxygenconsumption and are resistant to high-fat diet-inducedobesity andhepatic steatosiswith improved insulin sensitivity.Moreover,white adipocyteswith targeteddisruption of RIP140 express genes characteristic of brown fat including CIDEA and UCP1 while skeletal musclesshow a shift in fibre type composition enriched in more oxidative fibres. Thus, RIP140 is a potential therapeutictarget inmetabolic disorders. In this articlewewill review the role of RIP140 in tissues relevant to the appearanceand progression of the metabolic syndrome and discuss how the manipulation of RIP140 levels or activity mightrepresent a therapeutic approach to combat obesity and associated metabolic disorders. This article is part of aSpecial Issue entitled: Translating nuclear receptors from health to disease.
ing transcription factors; CBP,ragmentation factor alpha-likeEB, CRE-binding protein; CtBP,rase; EDL, extensor digitorumS, fatty acid synthase; FOXC2,lucocorticoid receptor; HDAC,JNK, Janus N-terminal kinase;nuclear receptor corepressor;clear respiratory factor; PCOS,te carboxykinase; PGC, PPARγreceptor; PRDM16, positiveceptor; RBP4, retinol bindingX receptor; SMRT, silencing
eptor; SRC, steroid receptorprotein 1c; TAG, triacylglycer-ctor α; TR, thyroid hormoneensity lipoproteinslating nuclear receptors from
er)..
l rights reserved.
© 2010 Elsevier B.V. All rights reserved.
1. Introduction
Obesity has reached worldwide epidemic proportions, especiallyin Western countries, often associated with metabolic dysfunctionssuch as insulin resistance, dyslipidaemia, cardiovascular disease andeven some cancers [1–3]. The clustering of these metabolic dysfunc-tions is recognized as the metabolic syndrome and the componentdisorders are essentially caused by a positive energy imbalance,where nutrient uptake is greater than energy expenditure. Strongepidemiological links between obesity, type II diabetes and cardio-
vascular diseases have suggested a common molecular pathogenesis.A limit on adipose tissue expandability has been proposed to provide aunifying causal link to the development of metabolic syndrome [4–6].Challenging the body with nutrient excess for a prolonged period oftime results in lipid deposition in adipose tissue. Adipose tissueexpands in order to accommodate the excess of energy withadipocytes undergoing hypertrophy (increase in size) and hyperplasia(increase in number) to fulfil the increased demand for lipid storage.Hypertrophic adipocytes have an altered profile of adipokinesecretion: a decrease in hormones/cytokines that promote insulinsensitivity, including adiponectin, and an increase in those thatpromote insulin resistance, such as resisitin and retinol bindingprotein (RBP) 4, and inflammation, such as interleukin (IL) 6 andtumour necrosis factor (TNF) α [7–9]. These inflammatory moleculesrecruit and activate macrophages present in the tissue to trigger low-grade inflammation in adipose tissue. Furthermore, activated macro-phages block preadipocyte recruitment and limit hyperplasic expan-sion of the adipose tissue. Once the storage capacity of the adiposetissue is exceeded, lipids start to accumulate in non-adipose tissues,leading to lipotoxicity [10,11]. Steatosis of the liver, muscle, pancreas,heart, and kidney can lead to organ failure or exacerbate whole-bodyinsulin resistance. In addition, increased levels of circulating free fattyacids (FAs) and development of type II diabetes are major risk factorsfor development of atherosclerosis and cardiovascular complications[12].
Several nuclear receptors (NRs), including peroxisome prolifera-tor-activated receptors (PPARs), thyroid hormone receptors (TRs) andestrogen-related receptors (ERRs), have emerged as importantmetabolic regulators [13]. These receptors control the transcriptionof genes involved in metabolic pathways underpinning the

920 M. Rosell et al. / Biochimica et Biophysica Acta 1812 (2011) 919–928
pathophysiology of the metabolic syndrome. The ability of NRs toregulate gene transcription depends on the recruitment of coactiva-tors and corepressors that remodel chromatin in the vicinity of thepromoters. Both the expression and activity of cofactors are subject toregulation by a number of signalling pathways in a tissue-specificmanner. The activity of many receptors is suppressed in the absence ofligand passively, in complexes with heat shock proteins in thecytoplasm, as in the case of androgen receptor (AR) and glucocorti-coid receptor (GR) [14,15]. Some NRs, including TR, retinoid Xreceptor (RXR) and PPARs, can be actively repressed by recruitment ofcorepressors, such as nuclear receptor corepressor (NCoR) andsilencing mediator of retinoic acid and thyroid hormone receptor(SMRT) [16,17]. Ligand binding results in conformational changeswhich lead to the dissociation of repressive complexes and therecruitment of coactivators, such as p300/CBP, p160/steroid receptorcoactivator (SRC) and PPARγ coactivator (PGC) families, to activatetranscription [18,19]. Ligand binding may not only result in theactivation of subsets of genes but also the repression of certain genes.Receptor interacting protein (RIP) 140 was found to function as acorepressor for a number of ligand bound nuclear receptors with arole in metabolism (TR2, PPARs, LXRα, GR, retinoic acid receptor(RAR)/RXR) [20–25]. RIP140 is itself highly expressed in metabolictissue like adipose tissue, skeletal muscle and liver [26]. Genome-wideexpression arrays studies have highlighted its role as a global regulatorof energy balance, repressing multiple metabolic pathways to reduceenergy expenditure. On the other hand, recent studies have shown thatRIP140 also functions as a coactivator for NR [22,27] or other types oftranscription factors [28,29]. The molecular basis for these opposingfunctions is poorly understood and this review will focus on thebiological roles of RIP140 in metabolism and inflammation andconsiders how targeting its function may provide an approach totreatment of metabolic syndrome.
AS160
genes involved in:mitochondrial uncouplingFA β-oxidationoxidative phosphorylationTCA cycleglycolysis
energyconsumption
Glut4
RIP140
RIP140
ERR
PPARγ
nucleuscytoplasm
ADIPOCYTE
PKCε14-3-3/PRMT1
exportin1
?
HFD
Fig. 1. Actions of RIP140 in adipocytes. Nuclear RIP140 is recruited by nuclear receptorsto repress sets of genes that promote energy consumption. Glut4 gene is alsotransrepressed and the action of Glut4 protein is inhibited by cytoplasmic RIP140,contributing to insulin resistance. Cytoplasmic translocation of RIP140 is stimulated byPKCε-mediated phosphorylation, followed by 14-3-3-dependent recruitment ofPRMT1, arginine methylation and export through exportin1. This sequence of post-translational modifications is promoted under a high-fat diet. PKCε, protein kinase Cε;PRTM1, protein arginine methyl transferase 1; HFD, high-fat diet.
2. Role of RIP140 in adipose tissue
Both white (WAT) and brown (BAT) adipose tissue share manycommon features such as their ability to take up glucose, synthesizeand store triacylglycerides (TAG), and mobilize those TAG insituations of energy demand. However, they play largely opposingroles in mammalian physiology.WAT serves as the prime reservoir forstoring excess caloric energy in the form of TAG. It is also an importantsource of endocrine hormones involved in metabolic regulation suchas leptin, adiponectin, and resistin [7,30]. In contrast, BAT is the majorsite of adaptive thermogenesis capable of generating heat from fattyacid oxidation to maintain body temperature, particularly in responseto cold exposure [31]. Thus, it is a net consumer of caloric energy. BATis also a site of diet-induced thermogenesis [31]. The function ofbrown adipocytes is critically dependent on the expression andactivity of uncoupling protein (UCP) 1, a mitochondrial protein almostexclusively found in BAT and therefore considered as a key molecularmarker [31]. That WAT and BAT play an important role in themaintenance of energy homeostasis is reflected in the observationthat small alterations in their function can have a large impact onwhole-body metabolism [31]. Moreover, the ability of white andbrown adipocytes in each depot to reversibly switch into one anotherhas been reported [30,32,33], but the extent to which this occurs andthe precise mechanisms involved are not fully understood (reviewedin [34]). The search for regulators that could mediate conversion ofwhite adipocytes (energy storing) into brown adipocytes (energyconsuming) has led to the identification of PGC1α, forkhead-box(FOX) C2 and positive regulatory domain-containing (PRDM) 16 astranscriptional regulators that have been found to promote a brownfat genetic program, while retinoblastoma protein and RIP140 havebeen described to favour awhite adipose phenotype (reviewed in refs.[35,36]).
RIP140 is most highly expressed in WAT where it regulates theexpression of many genes involved in catabolic pathways (energyconsuming), especially those involved in lipid and glucose metabo-lism. Mice devoid of the coregulator express higher levels of genesthat regulate fatty acid oxidation and proteins involved in energydissipation, highlighting its role as a corepressor and its rolecontrolling the balance between energy consumption and energyexpenditure [26]. On the contrary, genes involved in FA synthesis andgluconeogenesis seem to be downregulated in the absence of RIP140.As a consequence, RIP140 null mice are lean, with a 70% reduction ofbody fat and a 20% reduction in body weight compared to wild-typemice and they are resistant to high-fat diet (HFD) induced obesity andhepatic steatosis so that the mice maintain their insulin sensitivity[26,37]. It is particularly noteworthy that in the RIP140 KO animalsWAT expresses genes characteristic of BAT including UCP1, fatty acidtransporter carnitine palmitoyltransferase (CPT-1β) and the lipiddroplet protein cell death-inducing DNA fragmentation factor alpha-like effector A (CIDEA). Targeted disruption in mice of several genesdirectly involved in energy metabolism and fat accumulation (LXRα,ERRα and retinoblastoma protein) also leads to a lean phenotypewitha marked increase in UCP1 expression in adipocytes, particularly inwhite fat depots (see ref. [38] for a review). RIP140 expression is verylow in preadipocytes and induced upon differentiation [37,39,40].Studies in different adipocyte cell models (3T3-L1, MEFs (mouseembryonic fibroblasts), immortalised white adipocytes) have shownthat RIP140 is not required for adipocyte differentiation. Analysis ofgene expression profiles from the RIP140 null cell lines show that,RIP140 modulates the expression of similar genes involved in fattyacid and carbohydrate metabolism found in WAT, indicating that itfunctions as a cell autonomous corepressor. Most of the upregulatedgenes in the RIP140-null adipocytes are genes involved in catabolicpathways including fatty acid oxidation, oxidative phosphorylation,glycolysis and the tricarboxilic acid cycle (Fig. 1). Again, some genesnormally restricted to expression in BAT, including UCP1, CIDEA andCPT1-β, are elevated in RIP140-null adipocytes [37,39]. Many of thegenes regulated by RIP140 are also targets for the coactivator PGC1α

921M. Rosell et al. / Biochimica et Biophysica Acta 1812 (2011) 919–928
that was originally identified in BAT as factor that is elevated inresponse to cold. PGC1α is a key regulator of mitochondrial biogenesisand oxidative metabolism in tissues with high oxidative capacity suchas skeletal muscle, liver and brown fat by acting as a coactivator for anumber of different transcription factors involved in metaboliccontrol [41]. Since RIP140 also can bind to and repress many ofthese transcription factors, including PPARα, PPARγ and ERRα [21,39]it appears that the PGC1α and RIP140 coregulators may play mutuallyopposing roles in controlling sets of metabolic genes. We have foundthat both coregulators can be recruited simultaneously to the CIDEApromoter and it's noteworthy that they are able to physically interact.These observations suggest that RIP140 may be acting as atranscriptional brake that could be overcomed under conditionswhen the expression or activity of the coactivator is increased [42].
Recent studies by Li Na Wei and her colleagues indicate thatRIP140 may not only be a transcriptional coregulator but may alsofunction in the cell cytoplasm. They have found that cytoplasmicRIP140 inhibits glucose metabolism by reducing insulin-stimulatedglucose transporter 4 (GLUT4) trafficking and glucose uptake [43](Fig. 1). Importantly, the same study shows that high-fat feedingresults in cytoplasmic localization of RIP140 in epididymal whiteadipocytes, highlighting the biological relevance of a function forRIP140 in the cytoplasm. The cytoplasmic role of RIP140 is in additionto the direct regulation of GLUT4 mRNA expression by RIP140 inmouse and human adipocytes [37,40]). These findings provide thebasis for a novel mechanism by which RIP140 might impair glucoseutilization and promote insulin resistance. The observations alsosuggest that irrespective of RIP140 expression levels it may also beimportant to establish whether there are changes in compartmental-ization of the protein.
Very few studies have been carried out in humans. A decrease inRIP140 mRNA in biopsies of visceral WAT depots from obese patientshas been reported with a strong correlation between bodymass indexand RIP140mRNA levels [44]. It is conceivable that decreased levels ofRIP140 serve as a compensatory mechanism to favour energyexpenditure to reduce fat accumulation. In another study nodifference was found in RIP140 expression between obese and leanwomen with polycystic ovary syndrome (PCOS), or between obesePCOS and lean controls [45]. Finally, a recent study shows that RIP140is decreased in subcutaneous adipose tissue of obese women andincreased by weight loss. In the same study, in primary culture ofhuman adipocytes, RIP140 expression increased during adipocytedifferentiation and its knockdown increased basal glucose transportandmRNA levels of GLUT4 and UCP1, a similar behaviour to that of themouse ortholog [46]. Overall, high levels of human RIP140 in WAT oflean subjects may minimise energy utilization from depleted fatstores. At first sight, the overexpression of RIP140 in tissues fromobese individuals would be predicted from the mouse studies, whereits absence promotes reduced TAG accumulation; but on the otherhand, the subcellular localisation of RIP140was not examined in thesestudies and we have yet to elucidate signalling pathways that maycontrol of the activity of RIP140 by post-translational modifications.
RIP140 is highly expressed in BAT albeit to a lesser extent than inWAT. Interest in the study of BAT physiology has been renewed byrecent demonstration of considerable amounts of active tissue inmany adult humans [47–50]. In adult knock-out mice, the size andappearance of BAT is similar to the wild-type animals [26]. At themolecular level, UCP1 expression, together with the expression ofnuclear receptors PPARα, PPARγ, and fatty acid transporter aP2 issimilar in both knock-out and wild-type animals. These findingssuggested that BAT might not be a major site for RIP140 function or atleast its lack of expression would not seem to have a big impact underbasal conditions. Nevertheless, some recent experiments seem topoint out a role for RIP140 in BAT. It has been shown that adultRIP140-null mice exhibit a reduced body core temperature andreduced mRNA expression of coregulator PGC1α in BAT, although
response to an adrenergic activator does not seem impaired in theseanimals [51]. This is consistent with in vitro observations [21].However, newborn and young RIP140-null mice exhibit a significantreduction in BAT mass and PGC1α mRNA expression, which might beassociated with poor thermogenesis and that this in turn mightaccount for the poor rate of postnatal survival [51]. More recently, in acell line model of brown adipocytes, RIP140 was found to be recruitedto and repress the CIDEA promoter through binding to both ERRα andnuclear respiratory factor (NRF) 1 [42]. Moreover, RIP140 has alsobeen described to target and repress UCP1 enhancer in brownadipocytes trough LXR binding [23]. These findings suggest a rolefor RIP140 in BAT physiology, which might not be as evident as inWAT, but which could have important implication regarding energyexpenditure. Further studies need to be carried out in both adults andnewborn mice in order to establish the exact role of RIP140 in thistissue.
The amount of BAT and the expression of UCP1 correlate withincreased basal metabolic rate and protection from obesity in bothmice and humans. Genetic ablation of BAT induces obesity in mice[52]. Likewise, genetic deletion of UCP1 in mice causes weight gainwhen mice are kept at thermoneutrality [53]. On the other hand,ectopic expression of UCP1 in WAT results in resistance to obesity[54]. Mice with an increase in the amount of active BAT gain lessweight and are more insulin-sensitive. Indeed, BAT mass and activityis greatly decreased in obese patients, particularly those with morbidobesity [50,55]. Therefore, the increased expression of genes in WATfrom RIP140 null mice that are usually restricted to BAT seems tocompromise the function of this tissue as the site of energy storage.Total energy consumption is increased significantly in RIP140-nullmice, presumably as a consequence of energy dissipation in WATresulting from the expression of UCP1 and increased mitochondrialactivity [26]. BAT, on the other hand, utilises energy not only lipidoxidation but also glucose metabolism. Given the high metaboliccapacity of BAT, any reduction in the amount or activity of BAT couldperturb the ability of the body to dispose of glucose. Accordingly, themaintenance of a high glucose-utilizing activity by BAT could preventthe development of glucose intolerance and insulin resistance. AsRIP140 has been found to control glucose metabolism in whiteadipocytes it will be of high interest to study its role in glucosemetabolism in BAT. Glucosemetabolism in BAT is under insulin and β-adrenergic control. β-Adrenergic responses were found to beunaltered, at least in primary cultures of brown adipocytes fromRIP140 null mice [21]. It will be interesting to determine whetherinsulin signalling is affected when RIP140 levels are modified andwhether high levels of RIP140 inhibit oxidative metabolism andexpression of genes involved in thermogenesis, which could have avast impact on the total energy expenditure in an individual.
3. Role of RIP140 in inflammation
Lipotoxicity and inflammation are intimately involved in theevolution of a number of metabolic diseases. Lipid overload ofmacrophages in vascular endothelium leads to phenotypic transfor-mation into foam cells, secretion of pro-inflammatory cytokines, andapoptosis, contributing to atherosclerosis [56]. In steatotic liver,activation of kupffer cells, the residentmacrophages, can drive hepaticinflammation and transition to steatohepatitis [57]. Accumulation oflipoproteins and lipid derivatives in the kidney induces macrophageinfiltration and activation to initiate renal inflammation in chronickidney disease [58]. Obesity in mice and humans is associated with alow-grade subclinical inflammation of adipose tissue [59–62]. Non-obese adipose tissue contains a quiescent population of localmacrophages with little pro-inflammatory activity to support adipo-cyte function and maintain insulin sensitivity [63]. When hypertro-phic, adipocytes secrete higher amounts of chemo-attractants topromote macrophage infiltration [9]. Additional macrophages may
922 M. Rosell et al. / Biochimica et Biophysica Acta 1812 (2011) 919–928
also be recruited simply to promote disposal of necrotic cells, whichare more abundant in obese adipose tissue [30,64]. Free FA spill-overfrom enlarged adipocytes is able to activate Toll-like receptor (TLR)signalling and downstream Janus N-terminal kinase (JNK) and nuclearfactor κB (NF-κB) pathways in the resident macrophages to elicit aswitch towards a classically activated, pro-inflammatory phenotype[65,66]. The subsequent production of pro-inflammatory cytokines,including IL6 and TNFα, will interfere with insulin signalling leadingto type 2 diabetes and further propagate the state of chronicinflammation [67,68]. The importance of inflammation in metabolicdiseases has been reinforced by studies showing that limitingmacrophage activation [67,69–71] and production of pro-inflamma-tory signals [72] is of value in reducing insulin resistance.
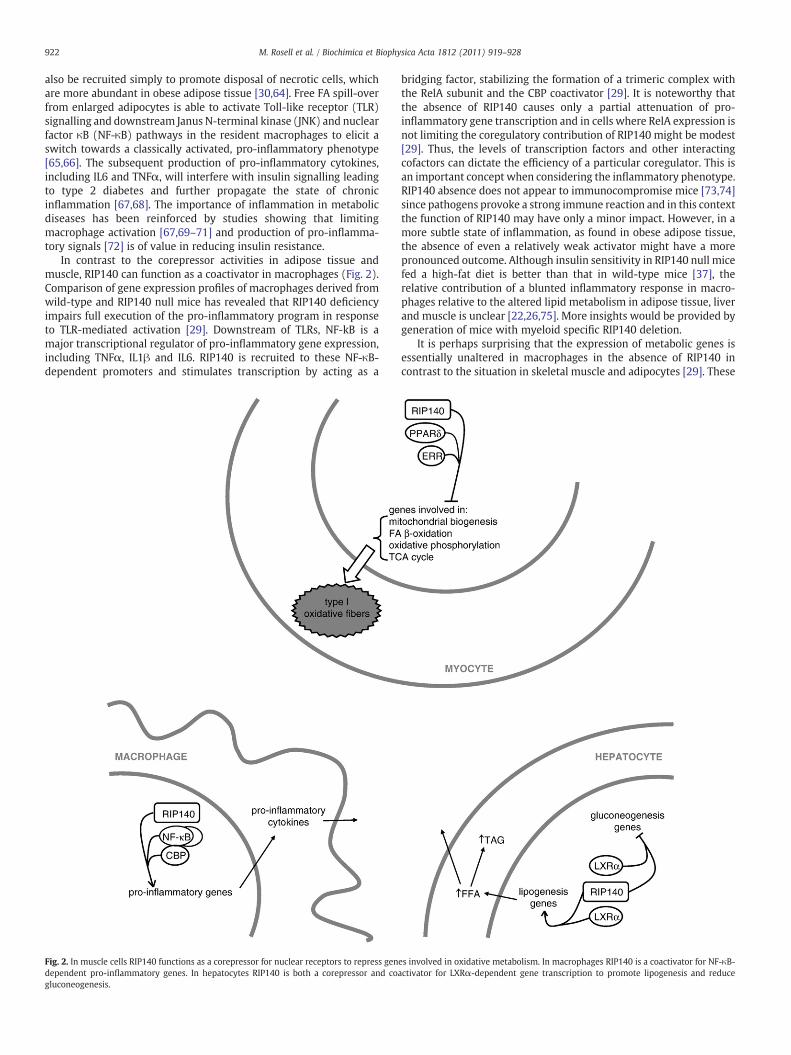
In contrast to the corepressor activities in adipose tissue andmuscle, RIP140 can function as a coactivator in macrophages (Fig. 2).Comparison of gene expression profiles of macrophages derived fromwild-type and RIP140 null mice has revealed that RIP140 deficiencyimpairs full execution of the pro-inflammatory program in responseto TLR-mediated activation [29]. Downstream of TLRs, NF-kB is amajor transcriptional regulator of pro-inflammatory gene expression,including TNFα, IL1β and IL6. RIP140 is recruited to these NF-κB-dependent promoters and stimulates transcription by acting as a
Fig. 2. In muscle cells RIP140 functions as a corepressor for nuclear receptors to repress gendependent pro-inflammatory genes. In hepatocytes RIP140 is both a corepressor and coagluconeogenesis.
bridging factor, stabilizing the formation of a trimeric complex withthe RelA subunit and the CBP coactivator [29]. It is noteworthy thatthe absence of RIP140 causes only a partial attenuation of pro-inflammatory gene transcription and in cells where RelA expression isnot limiting the coregulatory contribution of RIP140 might be modest[29]. Thus, the levels of transcription factors and other interactingcofactors can dictate the efficiency of a particular coregulator. This isan important concept when considering the inflammatory phenotype.RIP140 absence does not appear to immunocompromise mice [73,74]since pathogens provoke a strong immune reaction and in this contextthe function of RIP140 may have only a minor impact. However, in amore subtle state of inflammation, as found in obese adipose tissue,the absence of even a relatively weak activator might have a morepronounced outcome. Although insulin sensitivity in RIP140 null micefed a high-fat diet is better than that in wild-type mice [37], therelative contribution of a blunted inflammatory response in macro-phages relative to the altered lipid metabolism in adipose tissue, liverand muscle is unclear [22,26,75]. More insights would be provided bygeneration of mice with myeloid specific RIP140 deletion.
It is perhaps surprising that the expression of metabolic genes isessentially unaltered in macrophages in the absence of RIP140 incontrast to the situation in skeletal muscle and adipocytes [29]. These
es involved in oxidative metabolism. In macrophages RIP140 is a coactivator for NF-κB-ctivator for LXRα-dependent gene transcription to promote lipogenesis and reduce

923M. Rosell et al. / Biochimica et Biophysica Acta 1812 (2011) 919–928
genes are regulated by PPARδ [75], PPARα and PPARγ [21] and LXRα[22], which play important roles in the control of lipid homeostasisand inflammation in response to endogenous fatty acid or sterolligands. These NRs have been documented to mediate repressiveeffects on inflammatory genes in an NCoR- and SMRT-dependentmanner [76–78]. It remains to be investigated if in macrophages,PPARs and LXRs can also form repression complexes with RIP140 onsome genes, which would provide an interesting setting, where thecoregulator has negative as well as positive functions.
Communications between adipocytes and macrophages are im-portant in triggering inflammation in adipose tissue and it isincreasingly recognized that both cell types contribute to thedevelopment of obesity-induced insulin resistance. Although theinfiltrated macrophages are the main source of TNFα, adipocytes areresponsible for a sizable fraction of the IL6 concentration in thecirculation of obese patients [79,80]. The importance of an inflamma-tory response within adipocytes themselves is clear from a study withmice with adipocyte-specific JNK1 deficiency, which have improvedinsulin sensitivity in response to high-fat diet and normal levels ofserum IL6 [67]. Interestingly, microarray interrogation of geneexpression profiles shows that depletion of RIP140 in adipocytescauses genes encoding proteins involved in catabolic pathways forcarbohydrates and fatty acids to be upregulated, but a subset of pro-inflammatory genes, including IL6, are downregulated (D. Morgen-stein, unpublished observations;[39]). Therefore the activity of RIP140as a coactivator of pro-inflammatory gene expression is unlikely to berestricted to macrophages and may also be found in adipocytes andpossibly other cell types. Thismerits further investigation, for example,in vascular endothelial cells, where inflammation is the basis foratherosclerosis, or in the muscle, where macrophage infiltration andactivation can give rise to muscle insulin resistance [81,82], andwherecopious amounts of IL6 are produced during exercise [83].
4. Role of RIP140 in muscle
4.1. Skeletal muscle
In both mouse and human, skeletal muscle contains distinct typesof fibres, characterized by differential expression of specific myosinheavy chains. This difference in fibre composition accounts fordifferent types of contractile function. Type I fibres (slow twitchfibres) are rich in mitochondria and show high levels of oxidativephosphorylation relaying mainly on fatty acid oxidation. These are thefibers resistant to fatigue and predominant in red muscles, such as thesoleous. Type II (fast twitch fibers) contain a low number ofmitochondria and relay more on anaerobic respiration via glycolysisor glycogenolysis that enable them to undergo rapid and shortcontractile bursts. In comparison to type I fibers, type II are moreprone to fatigue. These types of fibers are predominant in muscles likethe gastrocnemius and extensor digitorum longus (EDL) [84]. There isalso an intermediate type of fibers, type IIX, that have fast twitchcharacteristics, but depend on oxidative metabolism that is similar totype I fibres [85]. Muscle fiber type composition can be modulated inresponse to several factors such as exercise, aging, hormonal changesor disease [86,87]. An increase in exercise has been shown to induce afibre composition switch in the amount of type I fibres inmuscles withpredominantly Type II fibers such as the gastrocnemius. The switch infiber type is reflected by changes in the expression of the myosinisoforms and other fiber type markers, as well as an increasedexpression of genes involved in mitochondrial myogenesis, fatty acidoxidation and oxidative phosphorylation. At a molecular level, anumber of transcriptional regulators that are able to remodel skeletalmuscle have been identified but it is still uncertain how the pathwaysare regulated. Nuclear receptors including PPARs, ERRα and othertranscription factors such as CRE-binding protein (CREB), myocyte-specific enhancer binding factors (MEFs), activating transcription
factors (ATFs) and NRFs have been found to activate expression ofmetabolic genes in skeletalmusclewhile the PGC1αplays a pivotal roleas a coactivator for the aforementioned transcription factors [88,89].
Skeletal muscles play an important role inwhole-bodymetabolismas they account for the 70% of the total insulin-stimulated glucoseuptake. Alterations in its function or fibre composition can have a bigimpact on the whole-body metabolism. For example, in insulin-resistant states, such as obesity and type II diabetes, insulin-stimulated glucose uptake is markedly impaired [90], while increasingthe number of type I fibres enhances insulin-mediated glucose uptakeand protects against diabetes and other metabolic diseases [91].Moreover, in obese patients, skeletal muscles have reduced metabolicoxidative capacities with morphologic changes that reveal decreasedtype I slow twitch fibers [92].
In skeletal muscle, RIP140 is expressed in a fiber type specificmanner. RIP140 mRNA levels seem to be higher in glycolytic muscles,like gastrocnemius and EDL, rather than in oxidative muscles, likesoleus and diaphragm [75]. In the absence of RIP140, EDL andgastrocnemius muscles exhibit a marked increase in mitochondrialactivity accompanied by a corresponding shift in myofibers favouringthe more oxidative type. Muscles appear redder in colour and expressincreased levels of type I fibres markers. These changes areaccompanied by an increase in mitochondrial number and oxidativemetabolism (most notably fatty acid oxidation) and are coordinatedfollowing upregulation of genes involved in these processes [75].PPARδ and ERRα are direct targets for RIP140 action at least for someof the genes. Conversely, in transgenic animals, elevated RIP140expression resulted in a decrease in both mitochondrial activity andthe number of oxidative myofibers [75]. In contrast, muscles thatconsist of predominantly type I fibres (usually expressing low RIP140levels) show an unaltered phenotype in the RIP140 null mice. This isconsistent with the function of RIP140 as a corepressor and suggests arole for RIP140 as an inhibitor of oxidative metabolism in skeletalmuscle [75] (Fig. 2). Considering the mass of skeletal muscle it canhave an important impact on thewhole-body energetic metabolism. Itis conceivable that alterations in RIP140 expression may contribute tocertain metabolic diseases such as obesity, insulin resistance and typeII diabetes but this will require further investigation.
4.2. Cardiac muscle
Recent evidence suggests that impaired myocardial mitochondrialbiogenesis, fatty acid metabolism, and antioxidant defence mecha-nisms lead to diminished cardiac substrate flexibility, decreasedcardiac energetic efficiency, and diastolic dysfunction. These mito-chondrial abnormalities can predispose to a metabolic cardiomyop-athy and have been observed in insulin-resistant animal models andpersons that are at higher risk of developing type 2 diabetes mellitus,hypertension, and cardiovascular disease [93].
RIP140 mRNA expression is high in cardiac muscle [26,94]. Studiesin transgenic mice overexpressing RIP140 show that these mice arecharacterized by rapid onset of cardiac hypertrophy and ventricularfibrosis which results in an increase in themortality rate from 4weeksof age [26,94]. Interestingly, females are less sensitive to thedeleterious effects of exogenous RIP140 expression compared tomales, suggesting that estrogens might play a protective role againstcardiac hypertrophy [95]. In these mice, overexpression of RIP140leads to reduced expression of genes involved in fatty acid transport/oxidation and mitochondrial activity. This is accompanied by adecrease in mitochondrial number and activity, as demonstrated bydecreased state III and state IV membrane potential and oxygenconsumption, associated with abnormal morphology [94]. Thus,decreased energy production associated to increased fibrosis mightbe responsible for impaired cardiac function and decreased survivalobserved in the RIP140 transgenic mice. This study is consistent withdata for skeletal muscle and myotubes obtained from RIP140-null

Rip140
ADIPOCYTE MYOCYTE
HEPATOCYTEMACROPHAGE
pro-energy storageinsulin resistance
pro-type II glycolytic fibre type
pro-inflammatory pro-lipogenicanti-gluconeogenic
Fig. 3. Actions of RIP140 in different metabolic tissue.
924 M. Rosell et al. / Biochimica et Biophysica Acta 1812 (2011) 919–928
mice where absence of RIP140 leads to increased oxidative metab-olism [75]. Moreover, it highlights the importance of RIP140 inpostnatal cardiac function and the possible implication in adultmitochondrial dysfunction leading to myopathies and cardiacmalfunction.
5. Role of RIP140 in the liver
The hepatic manifestation of the Metabolic Syndrome is non-alcoholic fatty liver disease (NAFLD) and it is associated withinsulin resistance although the cause and consequence relationshipis debated [96,97]. NAFLD is diagnosed as the histological presenceof lipid in more than 5% of hepatocytes, which is mostly in theform of TAG derived from the circulating pool of non-esterifiedfatty acids (59%), diet (15%) or de novo synthesis (26%) [98].Hepatic lipogenesis is regulated by LXRα [99,100] and two studieshave identified roles for RIP140 in this process. Our lab has foundthat in response to high-fat diet or agonist treatment, RIP140 isable to act as a coactivator for LXR-dependent induction of sterolregulatory element binding protein (SREBP) 1c and fatty acidsynthase (FAS), which promote TAG synthesis [22]. Interestingly,RIP140 is also required for LXR-dependent repression of phospho-enolpyruvate carboxykinase (PEPCK) gene to limit gluconeogenesisin hepatocytes. Therefore, RIP140 and LXR can partner to formdistinct activator or repressor complexes to either promote lipidgeneration or reduce glucose production, respectively [22] (Fig. 2).In contrast, a second study has shown that RIP140 contributed tohepatic steatosis during starvation and cancer cachexia byrepressing SREBP1c [73]. In addition, RIP140 knockdown in theliver of starved mice altered hepatic FA oxidation, very low densitylipoprotein (VLDL) production, FA uptake, serum TAG andperipheral LPL activity, to have a wider effect on local andsystemic lipid homeostasis [73]. The reason for the contradictingresults is not clear but differences in the experimental modelsused may have a contribution. We have utilized high-fat diet-induced liver steatosis with whole-animal RIP140 knockouts, asopposed to starvation or cachexia-triggered steatosis accompaniedby shRNA-mediated liver-specific RIP140 knockdown. Nevertheless,both studies have found that RIP140 absence is beneficial bylowering hepatic TAG content in the given experimental context.Further experiments, perhaps making use of hepatocyte-specificRIP140-null animals, are required to understand the precisefunctions of the cofactor in this tissue.
6. Targeting RIP140 for treatment of metabolic syndrome
Lipotoxic events are the underlying cause of cellular damage in themetabolic syndrome and therefore tackling the oversupply of lipid toaffected organs is important. Reducing caloric intake and increasingexercise to favour a negative energy balance should lead to clearanceof ectopic lipid deposition and improve an array of metabolicparameters [101,102]. Nevertheless, although lifestyle changes arebeneficial, obesity remains difficult to combat. A pharmacologicalapproach is to increase the lipid storage capacity of subcutaneousadipose depots with a view to prevent overspill and accumulation innon-adipose organs. This is exploited by the thiazolidinedione (TDZ)class of drugs, which, by activating PPARγ, promote preadipocytedifferentiation in addition to its insulin sensitizing effects [103]. Thus,the improved whole-body insulin sensitivity and reduced hepatic andvisceral lipid content is attributed at least in part to the hyperplasicincrease in subcutaneous adipose tissue seen in patients or animalmodels treated with TDZ [104–106]. In this case however, animprovement in metabolic well-being comes with increased obesityas a side-effect.
Excess fatty acids reaching non-adipose tissue can be activated tofatty acyl-CoA (FACoA) that undergo a series of esterification reaction
to form lysophosphatidic acid (LPA), phosphatidic acid (PA),diacylglycerol (DAG) and finally TAG. Alternatively, FACoA can bechannelled down the sphingosine synthesis pathway, where cera-mide is an intermediate, or it can undergo β-oxidation to feed themitochondrial electron transport chain [107]. All three fates have thepotential to present the cell with toxic challenges. LPA, PA, DAG andceramide can activate pro-inflammatory kinases to contribute toinsulin resistance [108–111]. Indeed, ceramide levels are elevated inskeletalmuscle of obese humans [112,113] and can further antagonizeinsulin signalling by inhibition of Akt [114]. Increasing the ability tooxidize FA, especially in the mitochondria of skeletal muscle, wouldreduce the accumulation of such intermediates and therefore protectfrom insulin resistance. It is important to note that several lines ofevidence have questioned the role ofmitochondrial function in insulinsensitivity and the cause and consequence relationship between thetwo remains highly debated [115]. Nevertheless, mitochondrialoverload with lipid supply would increase reactive oxygen species(ROS) production, which has been shown to have a causal role ininsulin resistance [116]. An important facet of treatment of metabolicdisorders is limiting the damaging effects of the three possible fates offatty acids, and RIP140 is involved in regulation of all three.
The pathways underlying the metabolic syndrome are interlinkedin a complex network and an effective approach to treatment mayneed to target multiple points. For example, TDZ treatment, whichactivates PPARγ to reduce inflammation and promote adipogenesisand enhances adiponectin secretion to increase lipid oxidation[117,118], is often prescribed in conjunction with metformin toreduce gluconeogenesis in the liver, improve glucose uptake inperipheral tissue and overall insulin sensitivity. The evidenceaccumulated form studying RIP140 suggests that targeting thefunction of this cofactor for treatment of metabolic disorders wouldbe of particular value since it would also have multiple beneficialconsequences (Fig. 3). As detailed above, RIP140 null mice have ahypermetabolic phenotype, largely as a result of upregulation of genesinvolved in FA β-oxidation, oxidative phosphorylation, TCA cycle andmitochondrial biogenesis in adipose tissue and muscle [37,75]. Thiscauses WAT to function in a manner akin to BAT to expend stored fatfor thermogenesis, while in skeletal muscle the proportion of type Ioxidative fibres, where TAGs are mainly used as fuel, is increased.Consequently, lipids are removed from the circulation, and bothvisceral and subcutaneous fat depots are reduced [26]. The upregula-tion of UCP1 inWAT [119] may also limit the amount of ROS producedby the mitochondria [120] to prevent inhibition of insulin signalling.Absence of RIP140 may have an additional protective effect on insulinsignalling by preventing downregulation and inhibition of GLUT4[43]. Impairing RIP140 function would protect against hepaticsteatosis during a lipid challenge by promoting gluconeogenesis andreducing lipogenesis. Here, downregulation of SREBP1c and FASrestricts de novo FA synthesis [22], therefore reducing both TAG andsphingolipid biosynthetic pathways and the production of harmfulintermediates. Furthermore, absence of RIP140 in macrophageswould decrease the inflammation associated with obesity and insulinresistance [29]. The net results are improved lipid partitioning

925M. Rosell et al. / Biochimica et Biophysica Acta 1812 (2011) 919–928
(decrease in circulating and ectopic load), decrease in adipose tissuesize to allow it to regain normal lipid homeostatic function, andconsequently, improved insulin sensitivity. The phenotype of RIP140null mice is proof of principle that targeting RIP140might constitute asuccessful approach for the treatment of metabolic syndrome.Although caution must be exercised since RIP140 function is essentialfor female fertility [28,121,122], this might not necessarily be apriority since metabolic disorders prevail at an age past sexualreproduction. Drugs seldom approach 100% efficiency, however, evena 50% inhibition of RIP140 activity would be beneficial, sinceheterozygous mice challenged with a high-fat diet also displayimproved metabolic parameters at a level intermediate betweenwild-type and null animals [74,75].
What pharmacological approaches could be used to modulate thefunction of RIP140? Three possibilities are outlined: (a) regulation ofRIP140 expression, (b) regulation of RIP140 activity and (c) regulationof NR-dependent recruitment. In the first instance, several NR ligandshave been shown to control RIP140 expression. In breast cancer cellsestrogens [123], retinoic acid [124] and androgens [125] upregulateRIP140 mRNA levels thus providing potential feedback loops to limitnuclear receptor action. Estrogen antagonists may therefore be ofvalue in preventing an induction in RIP140 expression [125], whileprogestins have been found to modestly downregulate RIP140 mRNA[126]. However, the use of such compounds would not offer efficientaction in metabolic tissues that are largely non-responsive to steroidsex hormones. More interestingly, ERRs, which have been implicatedin control of energy metabolism, can activate RIP140 gene expressionby directly binding to an ERRE and indirectly through SP1 bindingsites in the promoter [127]. ERRα-null mice have reduced bodyweight and peripheral fat deposits [128], reminiscent of the RIP140-null phenotype, yet the lack of a known ligand leavesmodulation of itsactivity a difficult proposition. Recently miRNA have become veryattractive tools in manipulating gene expression. The 5′ UTR of theRIP140 mRNA has been found to be targeted by microRNA (miRNA)mir-346. Rather than silencing RIP140 expression, mir-346 has beenfound to function in an atypical manner, increasing the protein levelsand therefore the repressive activity of the cofactor, but a miRNAantagomir can be used to dampen RIP140 activity [129]. Furthermore,RIP140 mRNA contains a large 3′ UTR of ~3.5 kb that remains to beinvestigated for target sites of additional miRNAs.
In its role as a transcriptional regulator, RIP140 lacks any intrinsicenzymatic activities but is thought to function as a scaffold protein,providing a platform for the recruitment of enzymes such as C-terminal binding protein (CtBP) [130], histone deacetylases (HDACs)[131], DNAmethyltransferese (Dnmt) [119], CBP [29], that change theaccessibility of the promoter to the transcription machinery. Post-translational modifications (PTMs) lie at the basis of regulating theactivity of non-enzymatic proteins. By favouring or disrupting specificinteractions, PTMs can alter not only the recruitment of transcrip-tional complex partners but may also have wide-ranging conse-quences on the behaviour of proteins, including changes in subcellularlocalization and half-life. RIP140 is subject to a diverse array of PTMs.Multiple site of phosphorylation [132], acetylation [133], arginine[134] and lysine methylation [135] have been identified in RIP140 bymass spectrometry-based studies. Vitamin B6 [136] and SUMOconjugation [137] have also been described and may also modulateRIP140 function. Several PTMsmay engage in an interplay tomodulateRIP140 activity and affect cellular function. For example, PKCε-stimulated phosphorylation promotes PRTM1-directed argininemethylation to reduce RIP140 gene repressive activity by preventinginteraction with HDAC3 and facilitating its export to the cytoplasm[134,138]. Conversely, MAPK-mediated RIP140 phosphorylation mayenhance repressive activity [139,140]. Post-translational modifica-tions are often investigated by ablating or mimicking them with theuse of point mutations of modification sites. A caveat to suchexperiments is that a single amino acid can be subject to several
modifications. For example, interpretation of results of a lysinemutantis complicated by the potential of the amino acid to undergoacetylation, methylation, SUMOylation or ubiquitination. However, ifwell validated, mechanistic knowledge of PTMs in RIP140 might beexploited for therapeutic purposes by targeting upstream signallingpathways that control its activity.
A third method of regulating RIP140 function involves takingadvantage of its recruitment by NR. Partial agonists or antagonistsmay selectively prevent interaction with RIP140 while not interferingwith other activities of the receptor. Since the metabolic repressoraction of RIP140 is supported by NR-dependent recruitment, thediscovery of such compounds would prove valuable for limiting thedampening effect on catabolism in adipose tissue and skeletal muscle.Notably, MRL24 has been recently identified as a PPARγ minimalagonist that inhibits Cdk5-mediated receptor phosphorylation toprevent obesity-associated gene dysregulation in fat cells [141]. Sincechromatin occupancy of PPARγ was not altered, it is likely thatdifferential recruitment of coregulators is responsible for the observedeffect. Such a finding should encourage screening of ligands for PPARγand other nuclear receptors for selective interaction with RIP140.
7. Conclusions
Overall this article summarizes the regulatory effects of RIP140 intissues with an important role in the development and progression ofthe metabolic syndrome including white and brown adipose tissue,inflammatory cells, skeletal and cardiac muscles and liver. We haveoutlined signalling pathways that are subject to regulation by RIP140and contribute to the development of the metabolic syndrome andspeculated about the possibility of targeting RIP140 to ameliorateassociated metabolic disorders.
In our opinion, therapeutic strategies targeting RIP140 have to beconsidered with caution, considering the broad spectrum of actions ofRIP140 as well as its recently described role as a coactivator. Given thatRIP140 is likely to be a difficult metabolic target to finely tune it wouldbe necessary to screen for compounds that modify the recruitment ofRIP140 to specific nuclear receptors at a subset of genes, oralternatively, compounds that canalter its binding toother componentsof the transcriptional machinery, such as histone modifying enzymes.
Acknowledgments
This work was supported by the Welcome Trust number 079200/Z/06/Z, the BBSRC number 504327/and Genesis Trust.
References
[1] C. Vernochet, S.B. Peres, S.R. Farmer, Mechanisms of obesity and relatedpathologies: transcriptional control of adipose tissue development, FEBS J. 276(2009) 5729–5737.
[2] M.A. Cornier, D. Dabelea, T.L. Hernandez, R.C. Lindstrom, A.J. Steig, N.R. Stob, R.E.Van Pelt, H. Wang, R.H. Eckel, The metabolic syndrome, Endocr. Rev. 29 (2008)777–822.
[3] G.A. Bray, T. Bellanger, Epidemiology, trends, and morbidities of obesity and themetabolic syndrome, Endocr. 29 (2006) 109–117.
[4] S. Virtue, A. Vidal-Puig, Adipose tissue expandability, lipotoxicity and themetabolic syndrome—an allostatic perspective, Biochim Biophys Acta 1801,338–349.
[5] S. Virtue, A. Vidal-Puig, It's not how fat you are, it's what you do with it thatcounts, PLoS Biol. 6 (2008) e237.
[6] J.K. Sethi, A.J. Vidal-Puig, Targeting fat to prevent diabetes, Cell Metab. 5 (2007)323–325.
[7] R.S. Ahima, J.S. Flier, Adipose tissue as an endocrine organ, Trends Endocrinol.Metab. 11 (2000) 327–332.
[8] S. Galic, J.S. Oakhill, G.R. Steinberg, Adipose tissue as an endocrine organ, Mol CellEndocrinol 316, 129–139.
[9] T. Skurk, C. Alberti-Huber, C. Herder, H. Hauner, Relationship between adipocytesize and adipokine expression and secretion, J. Clin. Endocrinol. Metab. 92(2007) 1023–1033.
[10] R.H. Unger, G.O. Clark, P.E. Scherer, L. Orci, Lipid homeostasis, lipotoxicity and themetabolic syndrome, Biochim. Biophys. Acta (1801) 209–214.

926 M. Rosell et al. / Biochimica et Biophysica Acta 1812 (2011) 919–928
[11] A. Vidal-Puig, R.H. Unger, Special issue on lipotoxicity, Biochim. Biophys. Acta(1801) 207–208.
[12] J.P. Wilding, The importance of free fatty acids in the development of type 2diabetes, Diabet. Med. 24 (2007) 934–945.
[13] B. Desvergne, L. Michalik, W. Wahli, Transcriptional regulation of metabolism,Physiol. Rev. 86 (2006) 465–514.
[14] J. Edwards, J.M. Bartlett, The androgen receptor and signal-transductionpathways in hormone-refractory prostate cancer. Part 2: Androgen-receptorcofactors and bypass pathways, BJU Int. 95 (2005) 1327–1335.
[15] J. Yang, D.B. DeFranco, Assessment of glucocorticoid receptor-heat shock protein90 interactions in vivo during nucleocytoplasmic trafficking, Mol. Endocrinol. 10(1996) 3–13.
[16] J.D. Chen, R.M. Evans, A transcriptional co-repressor that interacts with nuclearhormone receptors, Nature 377 (1995) 454–457.
[17] P. Dowell, J.E. Ishmael, D. Avram, V.J. Peterson, D.J. Nevrivy, M. Leid, Identificationof nuclear receptor corepressor as a peroxisome proliferator-activated receptoralpha interacting protein, J. Biol. Chem. 274 (1999) 15901–15907.
[18] A.J. Bannister, T. Kouzarides, The CBP co-activator is a histone acetyltransferase,Nature 384 (1996) 641–643.
[19] T.E. Spencer, G. Jenster, M.M. Burcin, C.D. Allis, J. Zhou, C.A. Mizzen, N.J. McKenna,S.A. Onate, S.Y. Tsai, M.J. Tsai, B.W. O'Malley, Steroid receptor coactivator-1 is ahistone acetyltransferase, Nature 389 (1997) 194–198.
[20] C.H. Lee, C. Chinpaisal, L.N. Wei, Cloning and characterization of mouse RIP140, acorepressor for nuclear orphan receptor TR2, Mol. Cell. Biol. 18 (1998)6745–6755.
[21] D. Debevec, M. Christian, D. Morganstein, A. Seth, B. Herzog, M. Parker, R. White,Receptor interacting protein 140 regulates expression of uncoupling protein 1 inadipocytes through specific peroxisome proliferator activated receptor isoformsand estrogen-related receptor alpha, Mol. Endocrinol. 21 (2007) 1581–1592.
[22] B. Herzog, M. Hallberg, A. Seth, A. Woods, R. White, M.G. Parker, The nuclearreceptor cofactor, receptor-interacting protein 140, is required for the regulationof hepatic lipid and glucose metabolism by liver X receptor, Mol. Endocrinol. 21(2007) 2687–2697.
[23] H. Wang, Y. Zhang, E. Yehuda-Shnaidman, A.V. Medvedev, N. Kumar, K.W.Daniel, J. Robidoux, M.P. Czech, D.J. Mangelsdorf, S. Collins, Liver X receptor alphais a transcriptional repressor of the uncoupling protein 1 gene and the brown fatphenotype, Mol. Cell. Biol. 28 (2008) 2187–2200.
[24] S.H. Windahl, E. Treuter, J. Ford, J. Zilliacus, J.A. Gustafsson, I.J. McEwan, Thenuclear-receptor interacting protein (RIP) 140 binds to the human glucocorti-coid receptor and modulates hormone-dependent transactivation, J. SteroidBiochem. Mol. Biol. 71 (1999) 93–102.
[25] C.H. Lee, L.N. Wei, Characterization of receptor-interacting protein 140 inretinoid receptor activities, J. Biol. Chem. 274 (1999) 31320–31326.
[26] G. Leonardsson, J.H. Steel, M. Christian, V. Pocock, S. Milligan, J. Bell, P.W. So, G.Medina-Gomez, A. Vidal-Puig, R. White, M.G. Parker, Nuclear receptorcorepressor RIP140 regulates fat accumulation, Proc. Natl Acad. Sci. USA 101(2004) 8437–8442.
[27] D.C. Harnish, M.J. Evans, M.S. Scicchitano, R.A. Bhat, S.K. Karathanasis, Estrogenregulation of the apolipoprotein AI gene promoter through transcriptioncofactor sharing, J. Biol. Chem. 273 (1998) 9270–9278.
[28] J. Nautiyal, J.H. Steel, M.M. Rosell, E. Nikolopoulou, K. Lee, F.J. Demayo, R.White, J.S. Richards, M.G. Parker, The nuclear receptor cofactor receptor-interacting protein 140 is a positive regulator of amphiregulin expression andcumulus cell-oocyte complex expansion in the mouse ovary, Endocrinology151, 2923–2932.
[29] I. Zschiedrich, U. Hardeland, A. Krones-Herzig, M. Berriel Diaz, A. Vegiopoulos, J.Muggenburg, D. Sombroek, T.G. Hofmann, R. Zawatzky, X. Yu, N. Gretz, M.Christian, R. White, M.G. Parker, S. Herzig, Coactivator function of RIP140 forNFkappaB/RelA-dependent cytokine gene expression, Blood 112 (2008)264–276.
[30] S. Cinti, The adipose organ, Prostaglandins Leukot. Essent. Fatty Acids 73 (2005)9–15.
[31] B. Cannon, J. Nedergaard, Brown adipose tissue: function and physiologicalsignificance, Physiol. Rev. 84 (2004) 277–359.
[32] J. Himms-Hagen, A. Melnyk, M.C. Zingaretti, E. Ceresi, G. Barbatelli, S. Cinti,Multilocular fat cells in WAT of CL-316243-treated rats derive directly fromwhite adipocytes, Am. J. Physiol. Cell Physiol. 279 (2000) C670–C681.
[33] J.G. Granneman, P. Li, Z. Zhu, Y. Lu, Metabolic and cellular plasticity in whiteadipose tissue I: effects of beta3-adrenergic receptor activation, Am. J. Physiol.Endocrinol. Metab. 289 (2005) E608–E616.
[34] S. Gesta, Y.H. Tseng, C.R. Kahn, Developmental origin of fat: tracking obesity to itssource, Cell 131 (2007) 242–256.
[35] S. Kajimura, P. Seale, B.M. Spiegelman, Transcriptional control of brown fatdevelopment, Cell. Metab. 11, 257–262.
[36] P. Seale, S. Kajimura, B.M. Spiegelman, Transcriptional control of brownadipocyte development and physiological function–of mice and men, GenesDev. 23 (2009) 788–797.
[37] A.M. Powelka, A. Seth, J.V. Virbasius, E. Kiskinis, S.M. Nicoloro, A. Guilherme, X.Tang, J. Straubhaar, A.D. Cherniack, M.G. Parker, M.P. Czech, Suppression ofoxidative metabolism and mitochondrial biogenesis by the transcriptionalcorepressor RIP140 in mouse adipocytes, J. Clin. Invest. 116 (2006) 125–136.
[38] J.B. Hansen, K. Kristiansen, Regulatory circuits controlling white versus brownadipocyte differentiation, Biochem. J. 398 (2006) 153–168.
[39] M. Christian, E. Kiskinis, D. Debevec, G. Leonardsson, R. White, M.G. Parker,RIP140-targeted repression of gene expression in adipocytes, Mol. Cell. Biol. 25(2005) 9383–9391.
[40] D.L. Morganstein, M. Christian, J.J. Turner, M.G. Parker, R. White, Conditionallyimmortalized white preadipocytes: a novel adipocyte model, J. Lipid Res. 49(2008) 679–685.
[41] P. Puigserver, Z. Wu, C.W. Park, R. Graves, M. Wright, B.M. Spiegelman, A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis, Cell92 (1998) 829–839.
[42] M. Hallberg, D.L. Morganstein, E. Kiskinis, K. Shah, A. Kralli, S.M. Dilworth, R.White, M.G. Parker, M. Christian, A functional interaction between RIP140 andPGC-1alpha regulates the expression of the lipid droplet protein CIDEA, Mol. Cell.Biol. 28 (2008) 6785–6795.
[43] P.C. Ho, Y.W. Lin, Y.C. Tsui, P. Gupta, L.N. Wei, A negative regulatory pathway ofGLUT4 trafficking in adipocyte: new function of RIP140 in the cytoplasm viaAS160, Cell Metab. 10 (2009) 516–523.
[44] V. Catalan, J. Gomez-Ambrosi, A. Lizanzu, A. Rodriguez, C. Silva, F. Rotellar, M.J.Gil, J.A. Cienfuegos, J. Salvador, G. Fruhbeck, RIP140 gene and protein expressionlevels are downregulated in visceral adipose tissue in human morbid obesity,Obes. Surg. 19 (2009) 771–776.
[45] K.M. Seow, Y.L. Tsai, C.C. Juan, L.W. Huang, J.L. Hwang, L.T. Ho, Omental fatreceptor interacting protein 140 mRNA expression in women with polycysticovary syndrome, Gynecol. Obstet. Invest. 69, 51–56.
[46] N. Mejhert, J. Laurencikiene, A.T. Pettersson, M. Kaaman, B.M. Stenson, M. Ryden,I. Dahlman, Role of Receptor-Interacting Protein 140 in human fat cells, BMCEndocr. Disord. 10, 1.
[47] A.M. Cypess, S. Lehman, G. Williams, I. Tal, D. Rodman, A.B. Goldfine, F.C. Kuo, E.L.Palmer, Y.H. Tseng, A. Doria, G.M. Kolodny, C.R. Kahn, Identification andimportance of brown adipose tissue in adult humans, N. Engl. J. Med. 360(2009) 1509–1517.
[48] M. Saito, Y. Okamatsu-Ogura, M. Matsushita, K. Watanabe, T. Yoneshiro, J. Nio-Kobayashi, T. Iwanaga, M. Miyagawa, T. Kameya, K. Nakada, Y. Kawai, M.Tsujisaki, High incidence of metabolically active brown adipose tissue in healthyadult humans: effects of cold exposure and adiposity, Diabetes 58 (2009)1526–1531.
[49] K.A. Virtanen, M.E. Lidell, J. Orava, M. Heglind, R. Westergren, T. Niemi, M.Taittonen, J. Laine, N.J. Savisto, S. Enerback, P. Nuutila, Functional brown adiposetissue in healthy adults, N. Engl. J. Med. 360 (2009) 1518–1525.
[50] W.D. van Marken Lichtenbelt, J.W. Vanhommerig, N.M. Smulders, J.M. Dros-saerts, G.J. Kemerink, N.D. Bouvy, P. Schrauwen, G.J. Teule, Cold-activated brownadipose tissue in healthy men, N. Engl. J. Med. 360 (2009) 1500–1508.
[51] M. Parker, G. Leonardsson, R. White, J. Steel, S. Milligan, Identification of RIP140as a nuclear receptor cofactor with a role in female reproduction, FEBS Lett. 546(2003) 149–153.
[52] B.B. Lowell, S.S. V, A. Hamann, J.A. Lawitts, J. Himms-Hagen, B.B. Boyer, L.P. Kozak,J.S. Flier, Development of obesity in transgenic mice after genetic ablation ofbrown adipose tissue, Nature 366 (1993) 740–742.
[53] H.M. Feldmann, V. Golozoubova, B. Cannon, J. Nedergaard, UCP1 ablation inducesobesity and abolishes diet-induced thermogenesis in mice exempt from thermalstress by living at thermoneutrality, Cell Metab. 9 (2009) 203–209.
[54] J. Kopecky, G. Clarke, S. Enerback, B. Spiegelman, L.P. Kozak, Expression of themitochondrial uncoupling protein gene from the aP2 gene promoter preventsgenetic obesity, J. Clin. Invest. 96 (1995) 2914–2923.
[55] H. Esterbauer, H. Oberkofler, Y.M. Liu, D. Breban, E. Hell, F. Krempler, W. Patsch,Uncoupling protein-1 mRNA expression in obese human subjects: the role ofsequence variations at the uncoupling protein-1 gene locus, J. Lipid Res. 39(1998) 834–844.
[56] N. Wang, I. Tabas, R. Winchester, S. Ravalli, L.E. Rabbani, A. Tall, Interleukin8 is induced by cholesterol loading of macrophages and expressed bymacrophage foam cells in human atheroma, J. Biol. Chem. 271 (1996)8837–8842.
[57] J.M. Olefsky, C.K. Glass, Macrophages, inflammation, and insulin resistance,Annu. Rev. Physiol. 72, 219–246.
[58] I.M. Wahba, R.H. Mak, Obesity and obesity-initiated metabolic syndrome:mechanistic links to chronic kidney disease, Clin. J. Am. Soc. Nephrol. 2 (2007)550–562.
[59] G.S. Hotamisligil, N.S. Shargill, B.M. Spiegelman, Adipose expression of tumornecrosis factor-alpha: direct role in obesity-linked insulin resistance, Science259 (1993) 87–91.
[60] P.A. Kern, M. Saghizadeh, J.M. Ong, R.J. Bosch, R. Deem, R.B. Simsolo, Theexpression of tumor necrosis factor in human adipose tissue. Regulation byobesity, weight loss, and relationship to lipoprotein lipase, J. Clin. Invest. 95(1995) 2111–2119.
[61] S.P. Weisberg, D. McCann, M. Desai, M. Rosenbaum, R.L. Leibel, A.W. Ferrante Jr.,Obesity is associated with macrophage accumulation in adipose tissue, J. Clin.Invest. 112 (2003) 1796–1808.
[62] K.E. Wellen, G.S. Hotamisligil, Inflammation, stress, and diabetes, J. Clin. Invest.115 (2005) 1111–1119.
[63] C.N. Lumeng, J.L. Bodzin, A.R. Saltiel, Obesity induces a phenotypic switch inadipose tissue macrophage polarization, J. Clin. Invest. 117 (2007) 175–184.
[64] I. Murano, G. Barbatelli, V. Parisani, C. Latini, G. Muzzonigro, M. Castellucci, S.Cinti, Dead adipocytes, detected as crown-like structures, are prevalent invisceral fat depots of genetically obese mice, J. Lipid Res. 49 (2008) 1562–1568.
[65] H. Shi, M.V. Kokoeva, K. Inouye, I. Tzameli, H. Yin, J.S. Flier, TLR4 links innateimmunity and fatty acid-induced insulin resistance, J. Clin. Invest. 116 (2006)3015–3025.
[66] M.T. Nguyen, S. Favelyukis, A.K. Nguyen, D. Reichart, P.A. Scott, A. Jenn, R. Liu-Bryan, C.K. Glass, J.G. Neels, J.M. Olefsky, A subpopulation of macrophagesinfiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-

927M. Rosell et al. / Biochimica et Biophysica Acta 1812 (2011) 919–928
like receptors 2 and 4 and JNK-dependent pathways, J. Biol. Chem. 282 (2007)35279–35292.
[67] G. Solinas, C. Vilcu, J.G. Neels, G.K. Bandyopadhyay, J.L. Luo, W. Naugler, S.Grivennikov, A. Wynshaw-Boris, M. Scadeng, J.M. Olefsky, M. Karin, JNK1 inhematopoietically derived cells contributes to diet-induced inflammation andinsulin resistance without affecting obesity, Cell Metab. 6 (2007) 386–397.
[68] G. Sabio, M. Das, A. Mora, Z. Zhang, J.Y. Jun, H.J. Ko, T. Barrett, J.K. Kim, R.J. Davis, Astress signaling pathway in adipose tissue regulates hepatic insulin resistance,Science 322 (2008) 1539–1543.
[69] A.L. Hevener, J.M. Olefsky, D. Reichart, M.T. Nguyen, G. Bandyopadyhay, H.Y.Leung, M.J. Watt, C. Benner, M.A. Febbraio, A.K. Nguyen, B. Folian, S.Subramaniam, F.J. Gonzalez, C.K. Glass, M. Ricote, Macrophage PPAR gamma isrequired for normal skeletal muscle and hepatic insulin sensitivity and fullantidiabetic effects of thiazolidinediones, J. Clin. Invest. 117 (2007) 1658–1669.
[70] J.I. Odegaard, R.R. Ricardo-Gonzalez, M.H. Goforth, C.R. Morel, V. Subramanian, L.Mukundan, A. Red Eagle, D. Vats, F. Brombacher, A.W. Ferrante, A. Chawla,Macrophage-specific PPARgamma controls alternative activation and improvesinsulin resistance, Nature 447 (2007) 1116–1120.
[71] M.C. Arkan, A.L. Hevener, F.R. Greten, S. Maeda, Z.W. Li, J.M. Long, A. Wynshaw-Boris, G. Poli, J. Olefsky, M. Karin, IKK-beta links inflammation to obesity-inducedinsulin resistance, Nat. Med. 11 (2005) 191–198.
[72] K.T. Uysal, S.M. Wiesbrock, M.W. Marino, G.S. Hotamisligil, Protection fromobesity-induced insulin resistance in mice lacking TNF-alpha function, Nature389 (1997) 610–614.
[73] M. Berriel Diaz, A. Krones-Herzig, D. Metzger, A. Ziegler, A. Vegiopoulos, M.Klingenspor, K. Muller-Decker, S. Herzig, Nuclear receptor cofactor receptorinteracting protein 140 controls hepatic triglyceride metabolism during wastingin mice, Hepatology 48 (2008) 782–791.
[74] R. White, G. Leonardsson, I. Rosewell, M. Ann Jacobs, S. Milligan, M. Parker, Thenuclear receptor co-repressor nrip1 (RIP140) is essential for female fertility, Nat.Med. 6 (2000) 1368–1374.
[75] A. Seth, J.H. Steel, D. Nichol, V. Pocock,M.K. Kumaran, A. Fritah,M.Mobberley, T.A.Ryder, A. Rowlerson, J. Scott,M. Poutanen, R.White,M. Parker, The transcriptionalcorepressor RIP140 regulates oxidative metabolism in skeletal muscle, CellMetab. 6 (2007) 236–245.
[76] G. Pascual, A.L. Fong, S. Ogawa, A. Gamliel, A.C. Li, V. Perissi, D.W. Rose, T.M.Willson, M.G. Rosenfeld, C.K. Glass, A SUMOylation-dependent pathwaymediates transrepression of inflammatory response genes by PPAR-gamma,Nature 437 (2005) 759–763.
[77] S. Ghisletti, W. Huang, K. Jepsen, C. Benner, G. Hardiman, M.G. Rosenfeld, C.K.Glass, Cooperative NCoR/SMRT interactions establish a corepressor-basedstrategy for integration of inflammatory and anti-inflammatory signalingpathways, Genes Dev. 23 (2009) 681–693.
[78] S. Ghisletti, W. Huang, S. Ogawa, G. Pascual, M.E. Lin, T.M. Willson, M.G.Rosenfeld, C.K. Glass, Parallel SUMOylation-dependent pathways mediate gene-and signal-specific transrepression by LXRs and PPARgamma, Mol. Cell 25(2007) 57–70.
[79] J.N. Fain, A.K. Madan, M.L. Hiler, P. Cheema, S.W. Bahouth, Comparison of therelease of adipokines by adipose tissue, adipose tissue matrix, and adipocytesfrom visceral and subcutaneous abdominal adipose tissues of obese humans,Endocrinology 145 (2004) 2273–2282.
[80] S.K. Fried, D.A. Bunkin, A.S. Greenberg, Omental and subcutaneous adiposetissues of obese subjects release interleukin-6: depot difference and regulationby glucocorticoid, J. Clin. Endocrinol. Metab. 83 (1998) 847–850.
[81] D.M. Muoio, T.R. Koves, Lipid-induced metabolic dysfunction in skeletal muscle,Novartis Found. Symp. 286 (2007) 24–388 discussion 38–46, 162–163, 196–203.
[82] M.W. Hulver, G.L. Dohm, The molecular mechanism linking muscle fataccumulation to insulin resistance, Proc. Nutr. Soc. 63 (2004) 375–380.
[83] B.K. Pedersen, M.A. Febbraio, Muscle as an endocrine organ: focus on muscle-derived interleukin-6, Physiol. Rev. 88 (2008) 1379–1406.
[84] R. Bassel-Duby, E.N. Olson, Signaling pathways in skeletal muscle remodeling,Annu. Rev. Biochem. 75 (2006) 19–37.
[85] L. Larsson, L. Edstrom, B. Lindegren, L. Gorza, S. Schiaffino, MHC composition andenzyme-histochemical and physiological properties of a novel fast-twitch motorunit type, Am. J. Physiol. 261 (1991) C93–C101.
[86] D.R. Thomas, Loss of skeletal muscle mass in aging: examining the relationship ofstarvation, sarcopenia and cachexia, Clin. Nutr. 26 (2007) 389–399.
[87] H. Wu, T. Gallardo, E.N. Olson, R.S. Williams, R.V. Shohet, Transcriptional analysisof mouse skeletal myofiber diversity and adaptation to endurance exercise, J.Muscle Res. Cell Motil. 24 (2003) 587–592.
[88] C.R. Benton, D.C. Wright, A. Bonen, PGC-1alpha-mediated regulation of geneexpression and metabolism: implications for nutrition and exercise prescrip-tions, Appl. Physiol. Nutr. Metab. 33 (2008) 843–862.
[89] C. Handschin, Regulation of skeletal muscle cell plasticity by the peroxisomeproliferator-activated receptor gamma coactivator 1alpha, J Recept SignalTransduct Res.
[90] M.A. Abdul-Ghani, R.A. DeFronzo, Pathogenesis of insulin resistance in skeletalmuscle, J. Biomed. Biotechnol. (2010) 476279.
[91] J.W. Ryder, R. Bassel-Duby, E.N. Olson, J.R. Zierath, Skeletal muscle reprogram-ming by activation of calcineurin improves insulin action on metabolicpathways, J. Biol. Chem. 278 (2003) 44298–44304.
[92] C.J. Tanner, H.A. Barakat, G.L. Dohm, W.J. Pories, K.G. MacDonald, P.R. Cunning-ham, M.S. Swanson, J.A. Houmard, Muscle fiber type is associated with obesityand weight loss, Am. J. Physiol. Endocrinol. Metab. 282 (2002) E1191–E1196.
[93] J. Ren, L. Pulakat, A. Whaley-Connell, J.R. Sowers, Mitochondrial biogenesis in themetabolic syndrome and cardiovascular disease, J. Mol. Med.
[94] A. Fritah, J.H. Steel, D. Nichol, N. Parker, S. Williams, A. Price, L. Strauss, T.A. Ryder,M.A. Mobberley, M. Poutanen, M. Parker, R. White, Elevated expression of themetabolic regulator receptor-interacting protein 140 results in cardiac hyper-trophy and impaired cardiac function, Cardiovasc. Res. 86, 443–451.
[95] O. Vuolteenaho, H. Ruskoaho, Gender matters: estrogen protects from cardiachypertrophy, Trends Endocrinol. Metab. 14 (2003) 52–54.
[96] Q. Liu, S. Bengmark, S. Qu, The role of hepatic fat accumulation in pathogenesis ofnon-alcoholic fatty liver disease (NAFLD), Lipids Health Dis. 9, 42.
[97] E. Vanni, E. Bugianesi, A. Kotronen, S. De Minicis, H. Yki-Jarvinen, G. Svegliati-Baroni, From the metabolic syndrome to NAFLD or vice versa?, Dig. Liver Dis. 42,320–330.
[98] K.L. Donnelly, C.I. Smith, S.J. Schwarzenberg, J. Jessurun, M.D. Boldt, E.J. Parks,Sources of fatty acids stored in liver and secreted via lipoproteins in patientswith nonalcoholic fatty liver disease, J. Clin. Invest. 115 (2005) 1343–1351.
[99] J.J. Repa, G. Liang, J. Ou, Y. Bashmakov, J.M. Lobaccaro, I. Shimomura, B. Shan, M.S.Brown, J.L. Goldstein, D.J. Mangelsdorf, Regulation of mouse sterol regulatoryelement-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalphaand LXRbeta, Genes Dev, 14 (2000) 2819–2830.
[100] E.G. Lund, L.B. Peterson, A.D. Adams, M.H. Lam, C.A. Burton, J. Chin, Q. Guo, S.Huang,M. Latham, J.C. Lopez, J.G.Menke,D.P.Milot, L.J.Mitnaul, S.E. Rex-Rabe, R.L.Rosa, J.Y. Tian, S.D. Wright, C.P. Sparrow, Different roles of liver X receptor alphaand beta in lipid metabolism: effects of an alpha-selective and a dual agonist inmice deficient in each subtype, Biochem. Pharmacol. 71 (2006) 453–463.
[101] W.C. Knowler, E. Barrett-Connor, S.E. Fowler, R.F. Hamman, J.M. Lachin, E.A.Walker, D.M. Nathan, Reduction in the incidence of type 2 diabetes with lifestyleintervention or metformin, N. Engl. J. Med. 346 (2002) 393–403.
[102] C.R. Bruce, A.B. Thrush, V.A. Mertz, V. Bezaire, A. Chabowski, G.J. Heigenhauser, D.J.Dyck, Endurance training in obese humans improves glucose tolerance andmitochondrial fatty acid oxidation and alters muscle lipid content, Am. J. Physiol.Endocrinol. Metab. 291 (2006) E99–E107.
[103] P. Tontonoz, B.M. Spiegelman, Fat and beyond: the diverse biology ofPPARgamma, Annu. Rev. Biochem. 77 (2008) 289–312.
[104] R. Belfort, S.A. Harrison, K. Brown, C. Darland, J. Finch, J. Hardies, B. Balas, A.Gastaldelli, F. Tio, J. Pulcini, R. Berria, J.Z. Ma, S. Dwivedi, R. Havranek, C. Fincke, R.DeFronzo, G.A. Bannayan, S. Schenker, K. Cusi, A placebo-controlled trial ofpioglitazone in subjects with nonalcoholic steatohepatitis, N. Engl. J. Med. 355(2006) 2297–2307.
[105] A. Hammarstedt, V.R. Sopasakis, S. Gogg, P.A. Jansson, U. Smith, Improved insulinsensitivity and adipose tissue dysregulation after short-term treatment withpioglitazone in non-diabetic, insulin-resistant subjects, Diabetologia 48 (2005)96–104.
[106] C.J. de Souza, M. Eckhardt, K. Gagen, M. Dong, W. Chen, D. Laurent, B.F. Burkey,Effects of pioglitazone on adipose tissue remodeling within the setting of obesityand insulin resistance, Diabetes 50 (2001) 1863–1871.
[107] M. Prentki, S.R. Madiraju, Glycerolipid metabolism and signaling in health anddisease, Endocr. Rev. 29 (2008) 647–676.
[108] P. Sathyanarayana, M.K. Barthwal, C.N. Kundu, M.E. Lane, A. Bergmann, G.Tzivion, A. Rana, Activation of the Drosophila MLK by ceramide reveals TNF-alpha and ceramide as agonists of mammalian MLK3, Mol. Cell 10 (2002)1527–1533.
[109] X. Wang, S.P. Devaiah, W. Zhang, R. Welti, Signaling functions of phosphatidicacid, Prog. Lipid Res. 45 (2006) 250–278.
[110] G. Jean-Baptiste, Z. Yang, C. Khoury, M.T. Greenwood, Lysophosphatidic acidmediates pleiotropic responses in skeletal muscle cells, Biochem. Biophys. Res.Commun. 335 (2005) 1155–1162.
[111] S.R. Sampson, D.R. Cooper, Specific protein kinase C isoforms as transducers andmodulators of insulin signaling, Mol. Genet. Metab. 89 (2006) 32–47.
[112] J.M. Adams 2nd, T. Pratipanawatr, R. Berria, E. Wang, R.A. DeFronzo, M.C.Sullards, L.J. Mandarino, Ceramide content is increased in skeletal muscle fromobese insulin-resistant humans, Diabetes 53 (2004) 25–31.
[113] P.M. Coen, J.J. Dube, F. Amati, M. Stefanovic-Racic, R.E. Ferrell, F.G. Toledo, B.H.Goodpaster, Insulin resistance is associated with higher intramyocellulartriglycerides in type I but not type II myocytes concomitant with higherceramide content, Diabetes 59, 80–88.
[114] S. Stratford, K.L. Hoehn, F. Liu, S.A. Summers, Regulation of insulin action byceramide: dual mechanisms linking ceramide accumulation to the inhibition ofAkt/protein kinase B, J. Biol. Chem. 279 (2004) 36608–36615.
[115] D.M. Muoio, Intramuscular triacylglycerol and insulin resistance: guilty ascharged or wrongly accused, Biochim. Biophys. Acta (1801) 281–288.
[116] N. Houstis, E.D. Rosen, E.S. Lander, Reactive oxygen species have a causal role inmultiple forms of insulin resistance, Nature 440 (2006) 944–948.
[117] A.R. Nawrocki, M.W. Rajala, E. Tomas, U.B. Pajvani, A.K. Saha, M.E. Trumbauer, Z.Pang, A.S. Chen, N.B. Ruderman, H. Chen, L. Rossetti, P.E. Scherer, Mice lackingadiponectin show decreased hepatic insulin sensitivity and reduced respon-siveness to peroxisome proliferator-activated receptor gamma agonists, J. Biol.Chem. 281 (2006) 2654–2660.
[118] T. Yamauchi, T. Kadowaki, The molecular mechanisms by which PPAR gamma/RXR inhibitors improve insulin resistance, Nippon Rinsho 59 (2001) 2245–2254.
[119] E. Kiskinis, M. Hallberg, M. Christian, M. Olofsson, S.M. Dilworth, R. White, M.G.Parker, RIP140 directs histone and DNA methylation to silence Ucp1 expressionin white adipocytes, EMBO J. 26 (2007) 4831–4840.
[120] A. Dlaskova, K.J. Clarke, R.K. Porter, The role of UCP 1 in production of reactiveoxygen species by mitochondria isolated from brown adipose tissue, Biochim.Biophys. Acta (1797) 1470–1476.
[121] G. Leonardsson, M.A. Jacobs, R. White, R. Jeffery, R. Poulsom, S. Milligan, M.Parker, Embryo transfer experiments and ovarian transplantation identify the

928 M. Rosell et al. / Biochimica et Biophysica Acta 1812 (2011) 919–928
ovary as the only site in which nuclear receptor interacting protein 1/RIP140action is crucial for female fertility, Endocrinology 143 (2002) 700–707.
[122] J.M. Tullet, V. Pocock, J.H. Steel, R. White, S. Milligan, M.G. Parker, Multiplesignaling defects in the absence of RIP140 impair both cumulus expansion andfollicle rupture, Endocrinology 146 (2005) 4127–4137.
[123] P. Augereau, E. Badia, M. Fuentes, F. Rabenoelina, M. Corniou, D. Derocq, P.Balaguer, V. Cavailles, Transcriptional regulation of the human NRIP1/RIP140gene by estrogen is modulated by dioxin signalling, Mol. Pharmacol. 69 (2006)1338–1346.
[124] J.S. Kerley, S.L. Olsen, S.J. Freemantle, M.J. Spinella, Transcriptional activation ofthe nuclear receptor corepressor RIP140 by retinoic acid: a potential negative-feedback regulatory mechanism, Biochem. Biophys. Res. Commun. 285 (2001)969–975.
[125] S. Carascossa, J. Gobinet, V. Georget, A. Lucas, E. Badia, A. Castet, R. White, J.C.Nicolas, V. Cavailles, S. Jalaguier, Receptor-interacting protein 140 is a repressorof the androgen receptor activity, Mol. Endocrinol. 20 (2006) 1506–1518.
[126] J.D. Graham, M.L. Yager, H.D. Hill, K. Byth, G.M. O'Neill, C.L. Clarke, Alteredprogesterone receptor isoform expression remodels progestin responsiveness ofbreast cancer cells, Mol. Endocrinol. 19 (2005) 2713–2735.
[127] D. Nichol, M. Christian, J.H. Steel, R. White, M.G. Parker, RIP140 expression isstimulated by estrogen-related receptor alpha during adipogenesis, J. Biol. Chem.281 (2006) 32140–32147.
[128] J. Luo, R. Sladek, J. Carrier, J.A. Bader, D. Richard, V. Giguere, Reduced fat mass inmice lacking orphan nuclear receptor estrogen-related receptor alpha, Mol. Cell.Biol. 23 (2003) 7947–7956.
[129] N.P. Tsai, Y.L. Lin, L.N. Wei, MicroRNA mir-346 targets the 5′-untranslated regionof receptor-interacting protein 140 (RIP140) mRNA and up-regulates its proteinexpression, Biochem. J. 424 (2009) 411–418.
[130] N. Vo, C. Fjeld, R.H. Goodman, Acetylation of nuclear hormone receptor-interacting protein RIP140 regulates binding of the transcriptional corepressorCtBP, Mol. Cell. Biol. 21 (2001) 6181–6188.
[131] L.N. Wei, X. Hu, D. Chandra, E. Seto, M. Farooqui, Receptor-interacting protein
140 directly recruits histone deacetylases for gene silencing, J. Biol. Chem. 275(2000) 40782–40787.
[132] M.D. Huq, S.A. Khan, S.W. Park, L.N. Wei, Mapping of phosphorylation sites ofnuclear corepressor receptor interacting protein 140 by liquid chromatography-tandem mass spectroscopy, Proteomics 5 (2005) 2157–2166.
[133] M.D. Huq, L.N. Wei, Post-translational modification of nuclear co-repressorreceptor-interacting protein 140 by acetylation, Mol. Cell. Proteomics 4 (2005)975–983.
[134] M.D. Mostaqul Huq, P. Gupta, N.P. Tsai, R. White, M.G. Parker, L.N. Wei,Suppression of receptor interacting protein 140 repressive activity by proteinarginine methylation, EMBO J. 25 (2006) 5094–5104.
[135] M.D. Huq, S.G. Ha, H. Barcelona, L.N. Wei, Lysine methylation of nuclear co-repressor receptor interacting protein 140, J. Proteome Res. 8 (2009) 1156–1167.
[136] M.D. Huq, N.P. Tsai, Y.P. Lin, L. Higgins, L.N. Wei, Vitamin B6 conjugation tonuclear corepressor RIP140 and its role in gene regulation, Nat. Chem. Biol. 3(2007) 161–165.
[137] M.M. Rytinki, J.J. Palvimo, SUMOylation modulates the transcription repressorfunction of RIP140, J. Biol. Chem. 283 (2008) 11586–11595.
[138] P. Gupta, P.C. Ho, M.M. Huq, S.G. Ha, S.W. Park, A.A. Khan, N.P. Tsai, L.N. Wei,Retinoic acid-stimulated sequential phosphorylation, PML recruitment, andSUMOylation of nuclear receptor TR2 to suppress Oct4 expression, Proc. NatlAcad. Sci. USA 105 (2008) 11424–11429.
[139] P. Gupta, M.D. Huq, S.A. Khan, N.P. Tsai, L.N. Wei, Regulation of co-repressiveactivity of and HDAC recruitment to RIP140 by site-specific phosphorylation,Mol. Cell. Proteomics 4 (2005) 1776–1784.
[140] P.C. Ho, P. Gupta, Y.C. Tsui, S.G. Ha, M. Huq, L.N. Wei, Modulation of lysineacetylation-stimulated repressive activity by Erk2-mediated phosphorylation ofRIP140 in adipocyte differentiation, Cell. Signal. 20 (2008) 1911–1919.
[141] J.H. Choi, A.S. Banks, J.L. Estall, S. Kajimura, P. Bostrom, D. Laznik, J.L. Ruas, M.J.Chalmers, T.M. Kamenecka, M. Bluher, P.R. Griffin, B.M. Spiegelman, Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5,Nature 466 451–456.





























