Embed Size (px)
Citation preview

.Molecular Immunaing~. Vol. 24, No. 12. pp. 1263-1211, 1987 Prmted m Great Britain.
0161-5@0/87 $3.00 +o.oo Pergmmn Journals Ltd
ROLE OF COMPLEMENT C9 AND CALCIUM IN THE GENERATION OF ARACHIDONIC ACID
AND ITS METABOLITES FROM RAT POLYMORPHONUCLEAR LEUKOCYTES*
DAVID K. IMAGAWA,?.$ SWANNE E. BARBOUR,? B. PAUL MoRGA~\~,?$ TIMOTHY M. WRIGHT,//
HYUN S. SHIN? and LOUISE E. RAMM~?~
tsubdepartment of Immunology, Department of Molecular Biology and Genetics and /IDepartment of Medicine, Johns Hopkins IJniversity School of Medicine. Baltimore, MD 21205, U.S.A.
Abstract-We have previously shown that antibody-sensitized mouse peritoneal macrophages release arachidonic acid (C20:4) and its oxygenated derivatives when treated with complement, and that the major part of the release depended on the terminal complement complexes (TCC). To further delineate the process(es) responsible for this release we have extended our studies to rat peritoneal poly- morphonuclear leukocytes (PMNs). Experiments were performed with antibody-sensitized rat PMNs labeled with [3H]C20:4 and carrying the TCC, C5b-7, C5b-8 or C5b-9. In contrast to the results of other studies, production of leukotriene B, (LTB,), the major radiolabeled derivative. was strictly dependent on the presence of C9. However, low levels of C20:4 and pros~glandins (PGs) were produced prior to the C5b-9 stage. Kinetic studies demonstrated that release of LTB, was rapid; the initial release occurred within 4-6 mm and a second rise in release coincided with cell death. Virtually all the LTB, produced was released as we found no evidence of retention of intracellular LTB, at either the C5b-8 or C5b-9 stages. In the absence of extracellular calcium, the release of LTB, was completely abolished and the release of C20:4 and PGs was drastically reduced. [‘H]C20:Clabeled PMNs carrying C5b-9 did release substantial amounts of radiolabeled material in the presence of EGTA; however, the majority of this lipid was in the form of intact phospholipid and triglyceride. These results indicate that release of C20:4 and its oxygenated derivatives from rat PMNs is (1) dependent on the partjcipation of C9 in’the preexisting CSb-8 compiex in the cell membrane, and (2) largely dependent on the presence of calcium.
INTRODUCTION
The release of arachidoni~ acid (C20:4) and its metabolites can be initiated by many types of stimuli. It is well documented that the complement cleavage products, C3b, C3a and CSa will induce phospholipid
*This work was supported in part by USPHS grants 2 ROl A102566 and 2 ROl AI19826 and grants PCM 8306963 and AM 01298 from the National Science Foundation.
$Supported by Medical Scientist Training Program GM 07309. Present address: UCLA Medical Center, Depart- ment of Surgery, Los Angeles, CA 90024, U.S.A.-
@‘resent address: Universitv of WakS College of Medicine, Department of Medical Biochemistry; Heath Park, Cardiff CF4 4XN, Cardiff, Wales, U.K.
:Please address correspondence to: L. E. Ramm, Depart- ment of Pathology, University of Maryland School of Medicine, Baltimore, MD 21201, U.S.A.
Abbreviations: C20: 4, arachidonic acid; TCC, terminal complement complexes; HPLC, high performance liquid chromatography; PGs, prostaglandins; 6-keto-PGF,,, 6-keto-prostaglandin F,,; TXB?, thromboxane B2; PMNs, polymorphonuclear leukocytes; LTB,. leuko- triene B,; CSD I-IS, C9D HS, human serum deficient in the eighth or ninth component of complement. respect- ively; PtdCho, phosphatidylcholine; PtdEtn, phospha- tidylethanolamine; TLC, thin layer chromatography; PL, phospholipid; TG, triglyceride; HETEs. hydroxy- eicosatetraenoic acids.
hydrolysis and C20:4 metabolite production from inflammatory cells bearing receptors for these frag- ments (Regal, 1982: Stimier el a!., 1982; Hartung ef
al., 1983a,b; Pison et al., 1983: Rutherford and Schenkein, 1983). Similarly, the inflammatory path- way can be initiated by the terminal complement complexes (Raisz et cd., 1974; Sandberg et al.. 1977: Polley et ai., 1981; Imagawa et al., 1983. 1986; goldsmith and McCormick, 1984; Hlnsch et al.. 1984, 1985; Seeger ei al., 1986: Shirazi et ul., 1987).
We have previously shown that C20:4 and pros- taglandins (PCs) are released from [‘H]C20:4- labeled, antibody (Ab)-sensitized mouse peritoneal macrophages treated with complement (Imagawa et
al., 1983). Treatment of these cells with C&deficient rabbit serum and various doses of C6 indicated that release of C20:4 and its metdbohtes is dependent on channel formation and not cell lysis (lmagawa rf nl., 1986). High-performance liquid chromatography (HPLC) analysis of the released radiolabeled material demonstrated that the major C20:4 derivative from macrophages was the prostacyclin breakdown prod- uct. &keto-prostaglandin F,,(6-keto-PGF,,). The release of &keto-PGF,, has also been demonstrated with vascular endothehal cells following treatment with heterologous Ab and complement (Goldsmith and McCormick, 1984).
1263

1264 DAVID K. IMAGAWA et al.
Hansch et al. (1984), using purified complement proteins in a reactive lysis system, showed the release of C20:4, PGE, and thromboxane B, (TXB2) from rat peritoneal macrophages and human monocytes following formation of C5b-9 and C5b-8, respect- ively. In human polymorphonuclear leukocytes (PMNs) (Seeger et al., 1986) and rat oligodendrocytes (Shirazi et al., 1987), the release of leukotriene B, (LTB,) has been shown to occur at the CSb-8 stage and addition of lytic doses of C9 did not amplify this release. Thus, it becomes abundantly clear that the release of lipid-derived inflammatory substances due to the terminal complement complexes (TCC) occurs in the absence of cell death, and appears to be the consequence of insertion of complement proteins and channel formation.
In order to better understand the process(es) by which the TCC causes the release of inflammatory mediators, we focused our studies on the release of C20:4, an early step required for the generation of cyclooxygenase and lipoxygenase products. In con- junction with this release, we simultaneously moni- tored the production of leukotrienes (LTs) and PGs. Our results indicate that release of C20:4 and its metabolites in rat PMNs is dependent on C9 and calcium.
MATERIALS AND METHODS
Reagents. [5,6.8,9,11,12,14,15-3H]C20:4, 91.2Ci/ mmol in ethanol (1Ci = 3.7 x IO” Bq) was purchased from New England Nuclear (Boston, MA). TXB,, 6-keto-PGF,,, PGE,, PGF,, and S(R, S)-HETE were generous gifts of Upjohn Diagnostics (Kalamazoo, MI). Leukotrienes B,, Cd, D, and E, (LTB,, LTC,, LTD, and LTE,) were generous gifts of Joshua Rokasch (Merck Frosst, Canada). I5-HETE was purchased from Seragen (Boston, MA). IZHETE was isolated from human platelets (McGuire et al., 1978) and 5-HETE from polymorphonuclear leuko- cytes (Borgeat and Samuelsson, 1979). Dipalmitoyl phosphatidylcholine was obtained from Serdary Re- search Labs (Port Huron, MI). Other phospholipid standards were purchased from Supelco (Bellefonte, PA). Melittin was purchased from Sigma (St. Louis, MO).
BufSers. The following buffers were used: RPM1 medium, RPM1 1640 (M.A. Bioproducts, Walkers- ville, MD) supplemented with 10% heat-inactivated fetal calf serum (Biofluids, Rockville, MD), 0.002 M glutamine (M.A. Bioproducts), 100 U/ml penicillin, and 100 pg/ml streptomycin (M.A. Bioproducts); RPM1 buffer, RPM1 1640 supplemented with 0.025 M HEPES (Sigma) and 0.05% gelatin, final pH: 7.4; Verona]-buffered saline (VBS), pH 7.4, ionic strength 0.15, prepared for use by diluting a stock solution 5-fold with water (Hammer et a/., 1977); DGVB, Verona]-buffered saline, pH 7.4, ionic strength 0.075, containing 0.139 M dextrose, 0.1% gelatin (Hammer et al., 1977); HBSS buffer, Hanks’
balanced salt solution without calcium and mag- nesium (Hazelton, Denver, PA) containing 0.025 hl HEPES and 0.05% gelatin.
Antibody. A polyclonal antisera pool against rat PMNs was obtained by immunizing rabbits with cell stroma mixed with complete Freunds adjuvant by intramuscular (i.m.) injection on a weekly basis for 4 weeks.
Purzjied complement proteins. Human C8 (I mg/ml, 2.2 x IO5 U/ml), human C9 (1 mg/ml, 1 .O x IO5 U/ml) and human Clq (1 mg/ml) were purchased from Cytotech (San Diego, CA). Human C9 (2.2 x 10’ U/ ml) was also prepared according to procedures pre- viously described (Hammer et al., 1981).
Depleted serum complement. Normal human serum (NHS) was obtained from healthy volunteers. Sera specifically depleted of C8 or C9 was produced by passage of NHS over an antibcdy-Sepharose column of appropriate specificity (Morgan et al., 1983: Dankert et al., 1985).
Titration of C8 and C9 in depleted serum. EAC I -3b were prepared by use of the C5 inhibitor K76 mono- carboxylic acid (kindly supplied by Otsuko Pharma-
ceutical, New York) according to Hong et al. (1981) with slight modification (Ramm et al., 1982~). The EACl-3b were converted to EACl-7 as de- scribed previously (Ramm et al., 1982a). The titer of C8 and C9 was determined by mixing 0.2 ml test samples with 0.1 ml of EACI-7 (1.5 x 10’ cells/ml) in DGVB buffer and then adding 0.2ml converting reagent. For the assay of residual C8 in C8D HS the converting reagent consisted of 50 CH,,, U/ml of guinea pig C9 (Cordis, Miami, FL). For the assay of C9 the converting reagent consisted of 50 CH,,, U;ml of guinea pig C8 (Cordis). After incubation for 60 min at 37°C the samples were centrifuged and the supernates analyzed for hemoglobin by reading ab- sorbance at 412 nm. C8D HS contained 0.3 U/ml of residual C8 (0.0008% of NHS), and C9D HS con- tained 120 U/ml of residual C9 (0.16% of NHS).
Dialysis of serum. Nonspecific activation of phos- pholipase A, and PG generation has been reported previously (Hsueh et al., 1983; Shier, 1980). Thus, all sera were dialyzed in Spectrapor 6 tubing, mol. wt cutoff 50,000 (Spectrum Medical Industries. L.os Angeles) against two changes in VBS at 4 C. This procedure substantially decreased nonspecitic activity.
Preparation and purification of rat polymorpho- nuclear leukocytes. Male rats (Sprague-Dawley) weighing 225-250 g were purchased from Charles River Labs (Wilmington, MA). Rat PMNs were prepared from a sodium caseinate-elicited peritoneal exudate as previously described (Newby, 1980). The purified leukocytes were washed once in RPM1 buffer, resuspended to the desired concn and pro- cessed as indicated below. Cells were greater than 98% PMNs by morphology.
Incorporation of [‘H]C20:4 into rut PMNs. Rat PMNs (3 x 106/ml) in RPM1 buffer were incubated

C9 and calcium requirement for C20:4 release from rat PMNs 1265
with 0.8 pCi/ml of [‘H]C20:4 for 1 hr at 37°C. Cells were washed twice in RPM1 buffer (containing 10% heat-inactivated fetal calf serum) to remove unin-
corporated label and resuspended in RPM1 buffer. Uptake of label was between 50 and 60%.
Preparation of Ab-sensitized rat PMNs. Rat PMNs (3 x 106/ml) in RPM1 buffer were incubated with an equal vol of anti-PMN Ab (final dilution l/1240) at 37°C for 30min. Cells were washed once, re- suspended in RPM1 buffer and processed as indicated below.
Preparation of rat PMN intermediates. Ab- sensitized rat PMNs (7.5 x lO’/ml) in RPM1 buffer were incubated with C8D HS (final dilution, l/10) or Ab-sensitized rat PMNs (1.5 x 106/ml) were incu- bated with C9D HS (final dilution, l/11). Clq (1 mg/ml) was added (10 PI/ml of undiluted sera) and the mixtures incubated for 20min at 34°C. Pre- liminary kinetic experiments indicated that these con-
ditions were optimal for intermediate formation. The resulting CSb-7 or C5b-8 intermediates were washed twice and then resuspended with the appropriate buffer (see below) and used immediately.
Release of C20:4 and oxygenated derivatives from
rat PMNs carrying C5b-7, CSb-8 or C5b-9. Ab- sensitized, [3H]C20:4-labeled rat PMNs carrying C5b-7 (3 x lob/ml) in HBSS buffer were aliquoted into each of a series of tubes. To one set of tubes, C8 (13 pg/ml, 2.9 x lo3 U/ml) was added. To a second set of tubes, C8 (13 pg/ml, 2.9 x IO-’ U/ml) and C9 (1 pg/ml, 1.0 x lO*U/ml) were added. The HBSS buffers contained 1 mM CaCI, or 1 mM EGTA with or without 2 mM CaCI,. The cells were incubated at 37°C and at suitable time intervals, aliquots were centrifuged in a Brinkmann Eppendorf microfuge for 1 min and 100 ~1 portions of the supernates were removed and assayed for lactate dehydrogenase (LDH) for cell lysis and tritium release. The 100% LDH activity was obtained by treating 1.5 x 10h cells with 100 ~1 melittin (5 mg/ml). Percent ‘H release was calculated as follows:
Total release (%)
cpm in aqueous phase
=cpm incorporated into cells x 100.
Extraction of 3H-labeled lipids for TLC analysis.
Supernatant fluids from complement-treated cells
were extracted according to the method of Bligh and Dyer (1959) with minor modification. Dipalmitoyl phosphatidylcholine (40 nmol/sample) was added as a carrier to optimize extraction efficiency. The aque- ous phase was washed 3 times with chloroform and the organic phases were combined and evaporated to dryness under a stream of nitrogen. The extraction efficiency for 3H-labeled lipids was 8&90%.
TLC analysis of [3H]C20:4 and metabolites. Lipid samples including the appropriate neutral and phos- pholipid reference standards were dissolved in chlo- roform and spotted on Anasil 0 TLC plates (Analabs,
North Haven, CT). After development in isopropyl ether/acetic acid, 96:4 (vol/vol), the plates were dried and sprayed with EN3HANCE (New England
Nuclear). After autoradiography at -70°C and ex- tensive drying to remove residual EN3HANCE, the lipid standards were detected by iodine vapor. Areas of the TLC plate corresponding to the radioactive spots on the autoradiogram, and identified by com- parison to the known standards, were scraped and counted.
HPLC analysis of C20:4 and oxygenated deriva-
tives. The supernatant fluids from complement- treated cells were extracted according to the pro- cedure of Unger et al. (1971) as modified by Scott et
al. (1982). Identification of C20:4 metabolites was performed on a Varian 5000 liquid chromatograph, equipped with variable U.V. detector, 250 mm x 4.6mm 5 h Cl8 column (Rainin Instruments, Woburn, MA), and a 2 cm Cl 8 precolumn (Supelco) using a method developed by Peters et al. (1983) as modified by Imagawa et al. (1986).
Release of LTB, from rat PMN intermediates.
PMN C5b-8 intermediates were suspended at 3.0 x lO”/ml in RPM1 buffer and mixed with equal vols of RPM1 buffer or RPM1 buffer containing purified C9. At suitable time intervals, 1 ml aliquots were removed and centrifuged in a Brinkmann Eppendorf microfuge for 1 min. One hundred micro- liters of supernate were removed from LDH deter- mination and the remainder assayed for LTB, by RIA. The cell pellets were extracted with 50 ~1 of 100% methanol at 4°C for 12 hr (Williams et al.,
1985) and analyzed for retained LTB,. Determination of LTB, release by RIA. Cells used
in these experiments were not labeled with [3H]C20 : 4. Following treatment with Ab and complement, the cells were centrifuged and 100 ~1 of the aqueous phase removed for LDH determination. The remain- der of the aqueous phase was immediately transferred to polypropylene test tubes containing 10 pg of indo- methacin (Sigma) to inhibit further catabolism of C20:4 metabolites and stored at -70°C. Mediator production was determined by RIA employing a commercially available kit (Amersham). The w-oxidation products of LTB, (Jubiz et al., 1982; Shak and Goldstein, 1984) are not cross-reactive with this RIA.
Cytotoxicity assay. Cell viability was assessed by release of LDH (Sigma Technical Bulletin No. 340-UV, May 1977).
RESULTS
The release of radiolabeled lipid from rat PMNs
carrying terminal complement complexes. In order to examine the role of terminal complement complexes in the release of C20:4 and its metabolites, Ab- sensitized rat PMNs, labeled with [3H]C20:4, were incubated with C8D HS for 20 min at 34°C and then washed to obtain cells carrying C5b-7 intermediates.
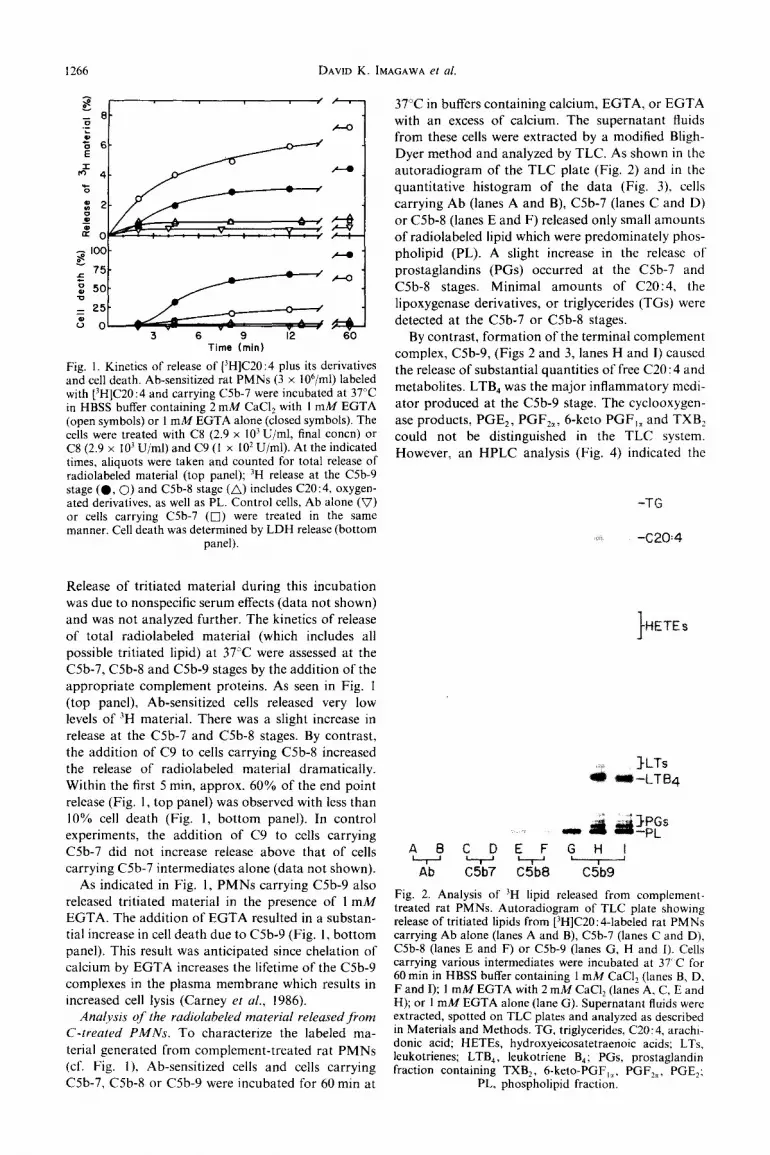
1266 DAVID K.
3 6 9 12 60 Time (min)
Fig. I. Kinetics of release of [‘H]C20:4 plus its derivatives and cell death. Ab-sensitized rat PMNs (3 x IOh/ml) labeled with [3H]C20:4 and carrying C5b-7 were incubated at 37°C in HBSS buffer containing 2 mM CaCI, with 1 mM EGTA (open symbols) or 1 mM EGTA alone (closed symbols). The cells were treated with C8 (2.9 x 10’ U/ml, final concn) or C8 (2.9 x IO3 U/ml) and C9 (1 x IO2 U/ml). At the indicated times, aliquots were taken and counted for total release of radiolabeled material (top panel); ‘H release at the C5b-9 stage (0, 0) and C5b-8 stage (a) includes C20:4, oxygen- ated derivatives, as well as PL. Control cells, Ab alone (0) or cells carrying C5b-7 (0) were treated in the same manner. Cell death was determined by LDH release (bottom
panel).
Release of tritiated material during this incubation
was due to nonspecific serum effects (data not shown) and was not analyzed further. The kinetics of release of total radiolabeled material (which includes all possible tritiated lipid) at 37°C were assessed at the C5b-7, CSb-S and C5b-9 stages by the addition of the appropriate complement proteins. As seen in Fig. 1 (top panel), Ab-sensitized cells released very low levels of ‘H material. There was a slight increase in release at the C5b-7 and C5b-8 stages. By contrast, the addition of C9 to cells carrying C5b-8 increased the release of radiolabeled material dramatically. Within the first 5 min, approx. 60% of the end point release (Fig. 1, top panel) was observed with less than 10% cell death (Fig. 1, bottom panel). In control experiments, the addition of C9 to cells carrying CSb-7 did not increase release above that of cells carrying C5b-7 intermediates alone (data not shown).
As indicated in Fig. 1, PMNs carrying C5b-9 also released tritiated material in the presence of I mM EGTA. The addition of EGTA resulted in a substan- tial increase in cell death due to C5b-9 (Fig. 1, bottom panel). This result was anticipated since chelation of calcium by EGTA increases the lifetime of the C5b-9 complexes in the plasma membrane which results in increased cell lysis (Carney ef al., 1986).
Analysis of’ the rudiolabeled material released jkom C-treated PMNs. To characterize the labeled ma-
terial generated from complement-treated rat PMNs (cf. Fig. I), Ab-sensitized cells and cells carrying C5b-7, C5b-8 or C5b-9 were incubated for 60 min at
I~~ACAWA et al
37°C in buffers containing calcium, EGTA, or EGTA with an excess of calcium. The supernatant fluids from these cells were extracted by a modified Bligh- Dyer method and analyzed by TLC. As shown in the autoradiogram of the TLC plate (Fig. 2) and in the quantitative histogram of the data (Fig. 3) cells carrying Ab (lanes A and B), C5b-7 (lanes C and D) or C5b-8 (lanes E and F) released only small amounts of radiolabeled lipid which were predominately phos- pholipid (PL). A slight increase in the release of prostaglandins (PGs) occurred at the C5b-7 and C5b-8 stages. Minimal amounts of C20:4, the lipoxygenase derivatives, or triglycerides (TGs) were detected at the C5b-7 or C5b-8 stages.
By contrast, formation of the terminal complement complex, C5b-9, (Figs 2 and 3, lanes H and I) caused the release of substantial quantities of free C20: 4 and metabolites. LTB, was the major inflammatory medi- ator produced at the C5b-9 stage. The cyclooxygen- ase products, PGE,, PGF,,, 6-keto PGF,, and TXBz could not be distinguished in the TLC system. However, an HPLC analysis (Fig. 4) indicated the
-TG
-c20:4
1 HETEs
>LTs *, rl-LT04
A-2 C-J EF G H ’ Al3 C5b7 C5b8 Gjb9
Fig. 2. Analysis of ‘H lipid released from complement- treated rat PMNs. Autoradiogram of TLC plate showing release of tritiated lipids from [3H]C20:4-labeled rat PMNs carrying Ab alone (lanes A and B), C5b-7 (lanes C and D), C5b-8 (lanes E and F) or C5b-9 (lanes G, H and I). Cells carrying various intermediates were incubated at 37’C for 60 min in HBSS buffer containing 1 mM CaCI, (lanes B, D. F and I); 1 mM EGTA with 2 mM CaCl, (lanes A, C, E and H); or 1 mM EGTA alone (lane G). Supernatant fluids were extracted, spotted on TLC plates and analyzed as described in Materials and Methods. TG, triglycerides, C20:4, arachi- donic acid; HETEs, hydroxyeicosatetraenoic acids; LTs, leukotrienes; LTB,, leukotriene B,; PCs, prostaglandin fraction containing TXB,, 6-keto-PGF,,, PGFzz. PGE,:
PL, phospholipid fraction.

C9 and calcium requirement for C20:4 release from rat PMNs
3~-c20:4 AND METABOLITES RELEASED BY COMPLEMENT
”
A B C D E F G H I
Fig. 3. Release of radiolabeled lipid from rat PMNs. Histogram of the data from the TLC plate of Fig. 2. Lanes A-I correspond to the respective lanes in Fig. 2. Cells carrying various terminal complexes were incubated in buffer containing I mM CaClz (lanes B, D, F and I); 1 mM EGTA with 2 mA4 CaCI, (lanes
A, C, E and H); or I mM EGTA alone (lane G).
presence of all four of these derivatives with 6-keto-PGF,, and TXB, the predominant cyclo- oxygenase derivatives generated.
The role of extracellular calcium in the release of
[‘H]C20:4 and its derivatives by C5b-9. The total release of radiolabeled lipids at the C5b-9 stage was reduced approx. 5&60% by 1 mM EGTA (cf. Fig. 1). As shown in Figs 2 and 3 (lane G), the lipids released in the presence of EGTA were primarily radiolabeled PLs and TGs. Interestingly, similar amounts of PL and TG were also released in the presence of calcium (Fig. 3, lanes H and I). On the other hand, the release
600
500
400
300
200
100
LTB4
0' I IO 20 30 40 50 60 70 80
Time (mid
Fig. 4. HPLC chromatogram of [‘HI-labeled products from rat PMNs treated with C9D HS plus C9. [‘H]C20:Clabeled Ab-sensitized rat PMNs (1.5 x 106/m1) were incubated with C9D HS (final concn l/11) for 20min at 34°C. Cells were washed and 2.6 x lo6 cells were treated with 22 U of purified human C9 for 90 min at 37°C. The cells were extracted and chromatographed as described in Materials and Methods. Cell death was 36.7%. The major radiolabeled products coeluted with 6-keto-PGF,, , TXB?, PGFlz, PGE, and LTB,
standards run on the same column.
1267
of C20: 4 and cyclooxygenase products by C5b-9 was reduced drastically, but not totally eliminated, in the
absence of extracellular calcium. It has been previously reported that 5-lipoxygenase
is a calcium-dependent enzyme (Jakschik et al., 1980). As expected, EGTA completely inhibited the release of LTB, by C5b-9 (Fig. 3, lane G). Addition of excess CaCl, (2mM) to the I mM EGTA buffer effectively restored release of LTB, and increased the release of C20:4 and PGs (lane H) from rat PMNs carrying C5b-9. Thus, release of LTB, by C5b-9 is strictly dependent upon the presence of calcium. However. these results indicate that the calcium requirement for the release of C20 : 4 and PGs may not be as stringent.
Production and release of LTB, from rat PMNs is
dependent on the presence of C9. As indicated in Figs 3 and 4, LTB, is the major arachidonic acid metabo- lite produced by C5b-9 stimulation of rat PMNs. Therefore, our studies on the mechanism of release of arachidonic acid and its derivatives focused on LTB,. To examine the C9 dependency of the generation ol LTB, from the endogenous membrane phosphohpid pool, kinetic experiments were performed with un- labeled rat PMNs carrying C5b-8 and various doses of purified C9. As shown in Fig. 5 (top panel), release of LTB, (assessed by RIA) was less than 0.5 ng/ml at the C5b-8 stage throughout the time course of the experiment. This very low level of release was also detected with cells treated with Ab alone or cells carrying C5b-7 (data not shown). By contrast, sub- stantial release of LTB, occurred at the C5b-9 stage and this release was dose-dependent on C9. Cells carrying C5b-9 displayed a biphasic kinetic release curve for LTB,; the first rise occurred within 46 minutes of the addition of C9 and a second phase in release was associated with cell lysis (Fig. 5, bottom panel). In confirmation of the data in Fig. 3, no LTB, was released by C5b-9 in the presence of I mM

1268 DAVID K. IMAGAWA et al
12 c9 u/ml
IO 220
6
6 110
4 55
2
Time (min)
Fig. 5. Kinetics of LTB, release and cell death from Ab-sensitized PMNs treated with C9D HS plus various doses of C9. Ab-sensitized rat PMNs (1.5 x 106/ml) were incubated with C9D HS (final concn l/11) and Clq for 20 min at 34°C. Cells were washed twice and resuspended to 1.5 x 106/ml in RPM1 buffer. The CSb-8 intermediates were treated with various doses of C9; 220 U/ml of C9 (O), 110 U/ml (A), 55 U/ml (O), or no C9 (V) at 37°C. LTB, release was determined by RIA (top), and cell death by LDH release (bottom). Endpoint lysis at 60 min was 23.4% for C5b-8 cells and 40.6, 52.1 and 62.3% for increasing concns of C9. Data shown are a representative experiment
of N =5.
EGTA over the time-course of this experiment (data not shown).
Williams et al. (1985) have reported intracellular retention of LTB, by PMNs stimulated with zymo-
san. In addition, other investigators have demon- strated LTB, production associated with C5b-8 for- mation (HHnsch et al., 1984; Seeger et a/., 1986; Shirazi et al., 1987). Thus, in order to ascertain whether PMNs were synthesizing LTB, without re- lease, cell pellets were extracted and analyzed by RIA. As seen in Fig. 6 (left panel) there was no appreciable production/retention of LTB, by C5b-8 treated cells. There was also no retention of the cyclooxygenase product, 6-keto-PGF,, at the C5b-8 stage as determined by RIA (data not shown). Similarly, extraction of C5b-9 cell pellets (Fig. 6, right panel) showed no accumulation of intracellular LTB,
DISCUSSION
Stimulation of nucleated cells by TCC can cause the release of C20:4 from membrane phospholipid and the conversion of this fatty acid to its oxygenated derivatives (Raisz et al., 1974; Sandberg et al., 1977; Polley et al., 1981; Imagawa et ul., 1983, 1986; Goldsmith and McCormick, 1984; Hlnsch et al.,
1984, 1985; Seeger et al., 1986; Shirazi et al., 1987). The present data (cf. Fig. 3) demonstrate that the release of C20:4, and the subsequent generation of cyclooxygenase and lipoxygenase derivatives from rat PMNs, are dependent on the complement protein C9 and calcium. Substantial quantities of labeled phos- pholipid are released as well. The release of phos- pholipid may be attributable to the exposure of lipid binding sites on the complement proteins upon their activation and subsequent release of complement protein-lipid complexes (Shin et al., 1977; HInsch et
al., 1984). The release of C20:4 from complement-treated
target cells may be explained by the activation of phospholipase A1 (Van den Bosch, 1980). Alter-
natively, C20: 4 release could follow the activation of
released 1 retained
40 52 56 60
Fig. 6. Left panel: retained vs released LTB, in rat PMNs at the C5b-8 stage. Rat PMNs (1.5 x 106/ml) were incubated with C9D HS (final concn l/l 1) for 20min at 34°C. Cells were washed twice and resuspended to 1.5 x 106/ml in RPM1 buffer and incubated at 37°C. At various times, 1 ml aliquots were centrifuged in a Brinkmann Eppendorf microfuge. The supernates were analyzed for LTB, by RIA and cell death by LDH. Cell pellets were extracted as described in Materials and Methods and resuspended in 100 gl of RPM1 buffer and assayed for LTB,. Endpoint cell death was 37%. Right panel: retained vs released LTB, in rat PMNs at the C5b-9 stage. Ab-sensitized rat PMNs (1.5 x 106/ml) carrying C5b-8 were suspended in RPM1 buffer containing 220 U/ml of C9 and incubated at 37’C. The supernates and cell pellets were analyzed as above. Endpoint cell death was 69%. Data shown are a representative experiment
of N =3.

C9 and calcium requirement for C20:4 release from rat PMNs 1269
phospholipase C (Cockcroft et al., 1984; Smith et al.,
1985) with the subsequent generation of free C20:4
by diacylglycerol lipase. Another possible mechanism is the inhibition of acyltransferase which would pre- vent the reincorporation of C20:4 into phospholipids (Hlnsch et al., 1985). Further investigation is neces- sary to confirm the pathway(s) involved in TCC- mediated C20 : 4 release.
We have observed low levels of C20:4 and PG release in the absence of extracellular calcium (Fig. 3). These data may reflect a role for intracellular calcium in the release of C20:4 and activation of the arachidonic acid cascade. On the other hand, there may be a calcium-independent process involved in the release of arachidonic acid, since it is possible that the
EGTA molecule traverses the complement channel (Ramm et al., 1982b) and chelates intracellular cal- cium as well. It must be emphasized, however, that the majority of C20: 4 and PG release is both C9 and calcium dependent. This is consistent with the lack of LTB, release in the presence of EGTA (Fig. 3) since the 5-lipoxygenase enzyme is strictly calcium- dependent (Jakschik et al., 1980). Seeger et al. (1986) also reported calcium-dependent LTB, release from human peripheral blood PMNs; however, the cal- cium dependence of C20: 4 release was not in- vestigated in their system.
Our finding that LTB, is the predominant C20:4 metabolite produced from complement attack on rat PMNs is in accord with the work of Seeger et al. (1986) in human PMNs and Shirazi et al. (1987) in rat oligodendrocytes. However, contrary to these reports of LTB, production at the C5b-8 stage, rat PMNs generate the vast majority of this metabolite upon the addition of C9 (cf. Fig. 6). One possible explanation for the difference between rat and human PMNs may be the presence of homologous restriction factor on the surface of PMNs. This membrane protein, present on erythrocytes (Schbnermark et al., 1986; Zalman et ul., 1986) and PMNs (Zalman et al., 1986), has an affinity for human CSjC9 and inhibits poly C9 formation in human species. This, however, would not explain the C5b-8 dependence on the release of LTB, by rat oligodendrocytes with heterologous strum (Shirazi et al., 1987). Thus, the likely expla- nation for the difference may be that the CSb-8 or C5b-9 dependent release of LTB, is cell-type specific.
The biphasic nature of the kinetic release curve for LTB, (Fig. 5) may be due to conversion of this metabolite to its w-oxidation products (Jubiz et al.,
1982; Shak and Goldstein, 1984) or other metabolites that do not cross-react in the RIA. The second rise in release of LTB, is most likely triggered by cell lysis.
The relationship of C9 and calcium in the C20:4 release process is unknown. Others have shown an early C9-induced calcium pulse (Hallet et al., 1981; Farzadegan et al., 1984). It is possible that the flux itself or the transitory increase in intracellular cal- cium concn may trigger the release through the activation of the phospholipases, as discussed above.
Alternatively, terminal complement complexes in the membranes of nucleated cells may affect the activity of the enzyme(s) involved in C20:4 release through the direct interaction of the complement proteins with the appropriate enzymes. The specific inter- action of C8 or C9 with the membrane-associated proteins of mol. wt 64,000 (Schonermark et al., 1986) or 38,000 (Zalman et al., 1986) respectively, and C5b-9 with fi-endorphin (Schweigerer et al., 1982) have been reported. Finally, the interaction of C5b-9 with the plasma membrane may produce alterations in membrane lipid architecture (Esser, 1982) and thereby affect enzyme activity. It is known that C5b-9 complexes disrupt membrane structure (Mayer et al.,
1981). In summary, we have demonstrated that rat PMNs
release the inflammatory mediator LTB, in response
to a terminal complement complex stimulus. This release is strictly dependent upon the presence of C9 and calcium. The data suggest that the requirement for calcium and C9 in the release of C20:4 and PGs may not be as stringent. These studies provide further evidence for a role of the membrane attack complex C5b-9 in the inflammatory process.
Acknowledgements--We express our thanks to Mr W. A. Paznekas for excellent technical assistance and to Mrs M. L. Glick for expert typing.
REFERENCES
Bligh E. G. and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911L917.
Borgeat P. and Samuelsson B. (1979) Arachidonic acid metabolism in polymorphonuclear leukocytes: effects of ionophore A23187. Proc. mtn. Acad. Sci. U.S.A. 76, 2148-2152.
Carney D. F., Hammer C. H. and Shin M. L. (1986) Elimination of terminal complement complexes in the plasma membrane of nucleated cells: influence of extra- cellular Ca’+ and association with cellular Ca’+. J. Immun. 317, 2633270.
Cockcroft S., Baldwin J. M. and Allan D. (1984) The Cal+-activated polyphosphoinositide phosphodiesterase of human and rabbit neutrophil membranes. Biochem. J. 221, 477482.
Dankert J. R., Shiver J. W. and Esser A. F. (1985) Ninth component of complement: self-aggregation and inter- action with lipids. Biochemistry 24, 27542762.
Esser A. F. (1982) Interactions between complement pro- teins and biological and model membranes. In Biological Membranes (Edited by Chapman D.), pp. 2777325. Academic Press, New York.
Farzadegan H., Johnson R. J. and Mayer M. M. (1984) Influx of 45Ca’+ into U937 cells after attack by sublytic doses of complement. Fedn. Proc. 43, 1763 (Abstr.).
Goldsmith J. C. and McCormick J. J. (1984) Immunologic injury to vascular endothelial cells: effects on release of prostacyclin. Blood 63, 984-989.
Hallett M. B., Luzio J. P. and Campbell A. K. (1981) Stimulation of Ca *+-dependent chemiluminescence in rat polymorphonuclear leucocytes by polystyrene beads and the non-lytic action of complement. Immunology 44, 569-576.
Hammer C. H., Shin M. L., Abramovitz A. S. and Mayer M. M. (1977) On the mechanism of cell membrane

1270 DAVID K. IMACAWA et al.
damage by complement: evidence on insertion of poly- peptide chains from C8 and C9 into the lipid bilayer of erythrocytes. .I. Immun. 119, l--8.
Hammer C. H., Wirtz G. H., Renfer L.. Gresham H. D. and Tack B. F. (1981) Large scale isolation of functionally active components of the human complement system. J. biol. Chem. 256, 39954006.
Hansch G. M., Gemsa D. and Resch K. (1985) In- duction of prostanoid synthesis in human platelets by the late complement components C5b-9 and channel forming antibiotic nystatin: inhibition of the reacyl- ation of liberated arachidonic acid. J. Immun. 135, 132&1324.
Hlnsch G. M., Seitz M., Martinotti G., Betz M., Rauterberg E. W. and Gemsa D. (1984) Macrophages release arachidonic acid, prostaglandin El, and throm- boxane in response to late complement components. J. Immun. 133, 2145-2150.
Hartung H. P., Bitter-Suermann D. and Hadding U. (1983~) Induction of thromboxane release from macro- phages by anaphylatoxic peptide C3a of complement and synthetic hexapeptide C3a 72-77. J. Immun. 130, 1345-I 349.
Hartung H. P., Hadding U., Bitter-Suermann D. and Gemsa D. (19836) Stimulation of prostaglandin E and thromboxane synthesis in macrophages by purified C3b. J. Immun. 130, 2861-2865.
Hong K., Kinoshita T. and Inoue K. (1981) Simple methods for preparing EAC 1,4b,2a,3b and EAC4b,3b with human or guinea pig complement components using an anti- complementary agent, K-76 monocarboxylic acid. J. Immun. 127, 1099114.
Hsueh W., Jordan R. L., Harrison H. H. and Cobb M. A. (1983) Serum and plasma stimulate prostaglandin pro- duction by alveolar macrophages. Pro.~taglundins 25, 793-808.
Imagawa D. K., Osifchin N. E., Paznekas W. A., Shin M. L. and Mayer M. M. (1983) Consequences of cell membrane attack by complement: release of arachidonate and formation of inflammatory derivatives. Proc. nutn. Acad. Sci. U.S.A. 80, 6647-6651.
Imagawa D. K., Osifchin N. E., Ramm L. E., Koga P. G., Hammer C. H., Shin H. S. and Mayer M. M. (1986) Release of arachidonic acid and formation of oxygenated derivatives after complement attack on macrophages: role of channel formation. J. Immun. 136, 4637-4643.
Jakschik B. A., Sun F. F., Lee L. and Steinhoff M. M. (1980) Calcium stimulation of a novel lipoxygenase. Biochem. biophys. Res. Commun. 95, 103~~110.
Jubiz W., Radmark O., Malmsten C., Hansson G., Lindgren J. A., Palmblad J., Udtn A. and Samuelsson B. (1982) A novel leukotriene produced by stimulation of leukocytes with formylmethionylleucylphenylalanine. J. biol. Chem. 257, 61066110.
Mayer M. M., Michaels D. W., Ramm L. E., Whitlow M. B., Willoughby J. B. and Shin M. L. (1981) Membrane damage by complement. Crit. Rec. Immun. 2, 1333166.
McGuire J. C., Kelly R. C., Gorman R. R. and Sun F. F. (1978) Preparation and spectral properties of 12-hydroxy- leicosatetraenoic acid (HETE). Prep. B&hem. 8, 147-153.
Morgan B. P., Daw R. A., Siddle K., Luzio J. P. and Campbell A. K. (1983) Immunoaffinity purification of human complement component C9 using monoclonal antibodies. J. Immun. Mefh. 64, 269-281.
Newby A. C. (1980) Role of adenosine deaminase, Ecto- (5’nucleotidase) and Ecto-(non-specific phosphatase) in cyanide-induced adenosine monophosphate catabolism in rat polymorphonuclear leukocytes. Biochem. J. 186, 907-918.
Peters S. P., Schulman E. S.. Liu M. C., Hayes E. C. and Lichtenstein L. M. (1983) Separation of major pros- taglandins, leukotrienes, and monoHETEs by high per-
formance liquid chromatography. J. Immun. Meth. 64, 335-343.
Pison U., Kunau W. H., Damerau B. and Konig W. (19831 Induction of leukotriene formation by the anaphylatoxin C3a and C5a. 10th Int. Complement Workshop. Immuno- biol. 164, 265 (Abstr.).
Polley M. J., Nachman R. L. and Weksler B. B. (1981) Human complement in the arachidonic acid trans- formation pathway in platelets. J. exp. Med. 153, 257-268.
Raisz L. G., Sandberg A. L., Goodson J. M., Simmons H. A. and Mergenhagen S. E. (1974) Complement- dependent stimulation of prostaglandin synthesis and bone resorption. Science 185, 7899791.
Ramm L. E., Whitlow M. B. and Mayer M. M. (1982~) Transmembrane channel formation by complement: func- tional analysis of the number of C5b6, C7, C8 and C9 molecules required for a single channel. Proc. norm. Acad. Sci. U.S.A. 79, 47514755.
Ramm L. E., Whitlow M. B. and Mayer M. M. (19826) Size of the transmembrane channels produced by complement proteins CS-8. J. Immun. 129, 114331146.
Regal J. F. (1982) CSa-induced aortic contraction: effect 01 an antihistamine and inhibitors of arachidonate metab- olism. J. Pharmac. exp. Ther. 220, 102Z107.
Rutherford B. and Schenkein H. A. (1983) C3 cleavage products stimulate release of prostaglandins by human mononuclear phagocytes in oitro. J. Immun. 874-877.
Sandberg A. L., Raisz L. G., Goodson J. M., Simmons H. A. and Mergenhagen S. E. (1977) Initiation of bone resorption by the classical and alternative C pathways and its mediation by prostaglandins. J. lmmun. 119, 1378.-1381.
Schonermark S., Rauterberg E. W., Shin M. L., Lake S.. Roelcke D. and Hansch G. M. (1986) Homologous species restriction in lysis of human erythrocytes: a membrane-derived protein with C8-binding capacity functions as an inhibitor. J. Immun. 136, 1772-1776.
Schweigerer L., Bhakdi S. and Teschemacher H. (19X2) Specific non-opiate binding sites for human /j-endorphin on the terminal complex of human complement. Nature, Lond. 296, 572-574.
Scott W. A., Pawlowski N. A.. Andreach M. and Cohn 2. A. (1982) Resting macrophages produce distinct metabolites from exogenous arachidonic acid. J. c’_vp. Med. 155, 535-547.
Seeger W., Suttorp N., Hellwig A. and Bhakdi S. (1986) Non-cytolytic terminal complement complexes may serve as calcium gates to elicit leukotriene B, generation m human polymorphonuclear leukocytes. J. Immun. 137, 12861293.
Shak S. and Goldstein 1. M. (1984) u-oxidation is the major pathway for the catabolism of leukotriene B, in human polymorphonuctear leukocytes. J. biol. Chrm. 259, 10181~10187.
Shier W. T. (1980) Serum stimulation of phospholipase AZ and prostaglandin release in 3T3 cells is associated with platelet-derived growth-promoting activity. Proc. natn. Acad. Sci. U.S.A. 77, 137-141.
Shin M. L., Paznekas W. A., Abramovitz A. S. and Mayer M. M. (1977) On the mechanism of membrane damage by C: exposure of hydrophobic sites on activated C proteins. J. Immun. 119, 135881364.
Shirazi Y., lmagawa D. K. and Shin M. L. (1987) Release of leukotriene B, from sublethally-injured oligodendro- cytes by terminal complement proteins, C5b-9. ./. Neurochem. 48, 271-278.
Smith C. D., Lane B. C., Kusaka I., Verghese M. W. and Snyderman R. (1985) Chemoattractant receptor-induced hydrolysis of phosphatidylinositol 4.5.bisphosphate in human polymorphonuclear leukocyte membranes. J. hiol. Chem. 260, 5875-5878.
Stimler N. P., Bach M. K., Bloor C. M. and Hugli T. E.

C9 and calcium requirement for C20:4 release from rat PMNs 1271
(1982) Release of leukotrienes from guinea pig lung stimulated by C5adrsAr8 anaphylatoxin. J. Immun. 128, 2247-2252.
Unger W. G.. Stamford 1. F. and Bennett A. (1971) Extraction of prostaglandins from human blood. Nature 233, 336 337.
Van den Bosch H. (1980) Intracellular phospholipase A. Biochim. hiophys. Acta 604, 191.-246.
Williams J. D., Lee T. H., Lewis R. A. and Austen F. (1985)
Intracellular retention of the 5lipoxygenase pathway product, leukotriene B,, by human neutrophils activated with unopsonized zymosan. J. Immun. 134, 2624-2630.
Zalman L. S., Wood L. M. and Miiller-Eberhard H. J. (1986) Isolation of a human erythrocyte membrane protein capable of inhibiting expression of homologous complement transmembrane channels. Proc. nafn. Acad. Sci. U.S.A. 83, 6975-6879.













![Comparison of PAF- and fMLP-induced [Ca2+]i transients in human polymorphonuclear leukocytes](https://img.dokumen.tips/doc/110x75/63607be75e6ba0ceb50ee373/comparison-of-paf-and-fmlp-induced-ca2i-transients-in-human-polymorphonuclear.jpg)















