Embed Size (px)
Citation preview

Pyrosequencing the salivary transcriptome of Haemadipsa interrupta(Annelida: Clitellata: Haemadipsidae): anticoagulant diversity and insight
into the evolution of anticoagulation capabilities in leeches
Sebastian Kvist,1,a Mercer R. Brugler,2,3 Thary G. Goh,4
Gonzalo Giribet,1 and Mark E. Siddall2,3
1 Museum of Comparative Zoology, Department of Organismic and Evolutionary Biology,
Harvard University, Cambridge, Massachusetts 02138, USA2 Division of Invertebrate Zoology, American Museum of Natural History, New York, New York 10024, USA
3 Sackler Institute for Comparative Genomics, American Museum of Natural History,
New York, New York 10024, USA4 Institute of Biological Sciences, Science Faculty, University of Malaya, Kuala Lumpur 50603, Malaysia
Abstract. Freshwater, marine, and terrestrial leeches that depend on a diet of fresh bloodhave evolved salivary peptide components that inhibit normal thrombus formation by prey.Although bloodfeeding in leeches has long been of interest to biologists and medical practi-tioners alike, only a few studies have comprehensively examined the anticoagulant reper-toires of hematophagous leeches, and these have largely been confined to representatives ofGlossiphoniidae, Hirudinidae, and Macrobdellidae. Here, we present 454 pyrosequencingdata from the salivary transcriptome of the hematophagous terrestrial leech Haemadipsainterrupta (Haemadipsidae). Assembled transcripts were annotated using both similarityscores (BLAST) and gene ontology (Blast2GO). Subsequently, transcripts were examinedwithin alignments containing well-characterized anticoagulants and other select bioactive(i.e., affecting the living cells of the prey) salivary proteins and phylogenies were recon-structed for each protein data set to verify orthology predictions. In total, transcripts signi-ficantly matching 20 salivary proteins of interest were found in the transcriptome,representing several different antagonistic pathways. After reviewing gene ontologies, align-ments, and phylogenetic trees, sequences for 15 out of the 20 hits were deemed correctlyannotated. Additionally, we recovered matches against several proteins that have previouslybeen linked to anticoagulation (e.g., cathepsin and disintegrins), but the specific function ofwhich in leeches needs further investigation. Finally, in light of these data as well as thosepreviously published, we discuss our current understanding of the distribution and evolutionof anticoagulants in leeches.
Additional key words: transcriptomics, Hirudinida, bioactive proteins, hematophagy
Thought to be a cure for a variety of ailmentssuch as fever, hysteria, obesity, insomnia, ulcers,and an imbalance of the four humors (blood,phlegm, choler, and melancholy), hematophagousleeches have been used in human phlebotomy formillennia (Sawyer 1981; Min et al. 2010; Elliott &Kutschera 2011). Concurrent with our growingunderstanding of the structure and function of leechanticoagulation factors that are responsible for per-sistent blood loss in patients, leeches have become
more authoritatively used in modern medicine(Markwardt 1985; Adams 1988; Wells et al. 1993;Whitaker et al. 2004; Stange et al. 2012). Medicinalleeches are most frequently used as relievers ofvenous congestion to salvage microvascular freeflaps or following digit replantation surgery. Theirusefulness in this regard stems from their intensebloodfeeding behavior in combination with potentanticoagulation factors that they secrete into thewound (Salzet 2001). Although the European medic-inal leech, Hirudo verbana CARENA 1820, has becomethe centerpiece for clinical hirudinology (Trontelj &Utevsky 2005; Siddall et al. 2007; Shain 2009), its
aAuthor for correspondence.E-mail: [email protected]
Invertebrate Biology x(x): 1–25.© 2013, The American Microscopical Society, Inc.DOI: 10.1111/ivb.12039

anticoagulant repertoire has only recently beeninvestigated in a comprehensive manner (Kvist et al.2013); that study suggests that bioactive (i.e., affect-ing the living cells of the prey) salivary proteins withat least four different antagonistic pathways (factorXa inhibitors, elastase inhibitors, plasmin inhibitors,and Kazal-type serine proteinase inhibitors) areexpressed and secreted by the leech. In addition toH. verbana, salivary transcriptomes of other repre-sentatives of Hirudinidae and Macrobdellidae haverecently been screened for anticoagulation factors(Min et al. 2010; Siddall et al. 2011; Kvist et al.2013), revealing an astonishing array of anticoagu-lants even within a single individual. However, sev-eral other leech families, terrestrial and aquatic, arehematophagous but largely or entirely unstudied interms of the expressed anticoagulation factors. Spe-cifically targeting the anticoagulant repertoires ofterrestrial leeches will increase our understanding ofthe evolution of hirudinid bloodfeeding in general,and specific anticoagulation factors in particular. Inthis regard, the family Haemadipsidae is of specialinterest because of its biology and natural history.
Haemadipsidae includes terrestrial leeches, mostof which are obligate bloodfeeders. The clade can bebroadly divided into two main groups, two-jawed(duognathous) and three-jawed (trignathous) species.Recent phylogenetic reconstructions show thatduognathous species represent a monophyleticgroup, nesting within trignathous species, renderingthe latter paraphyletic (Borda & Siddall 2011).There are ~70 described haemadipsid species, whichis likely an underestimate of their true diversity(Keegan et al. 1968; Richardson 1975; Sawyer 1986;Nesemann & Sharma 1996, 2001; Borda & Siddall2011). Roughly 24 of these are representatives of thetrignathous genus Haemadipsa, whereas the remain-ing species have been placed in 30 (Nybelin 1943;Richardson 1975, 1978) or 15 different genera (Saw-yer 1986; see Borda & Siddall 2011 for full discus-sion). Interestingly, the family shows an unusualbiogeographic distribution, with the trignathousleeches found only in East and Southeast Asia andon the Indian subcontinent, and duognathousleeches found on Sahul, Wallacea, Melanesia, Ocea-nia, Juan Fern�andez, Madagascar, and the Sey-chelles. Haemadipsids are not known from Africa orSouth America; most other leech families have aglobal distribution. Haemadipsids are well known tofield biologists of the Indo-Pacific tropics as smallyet intensely aggressive leeches that infiltrate pants,boots, and shirts. In fact, no other group of annelidshas inspired such passionate accounts by travelersor naturalists. They are typically found in “dank
tropical jungles, misty ravines and showery, forestedmountain-sides [where] they are among the mostdominant and self-assertive elements” (Moore 1927).Because of their terrestrial preference, haemadipsidleeches have different dietary options than freshwa-ter and marine leeches, and this provides a uniqueopportunity to compare how their mode of blood-feeding, as it pertains to anticoagulants, has evolvedin general, and if it evolved in parallel with theirchoice of diet. That is, because of differing physiol-ogy and anatomy of their prey, it is likely thathaemadipsid leeches possess chemical components intheir pharmacological cocktails that differ fromthose of their aquatic counterparts.
Understanding the phylogenetic position of terres-trial hematophagous leeches is particularly impor-tant when investigating the evolution ofbloodfeeding in Hirudinida. Using morphology, twonuclear ribosomal markers 18S rRNA and 28SrRNA, and mitochondrial 12S rRNA and cyto-chrome c oxidase subunit I (COI), Borda & Siddall(2004) showed that Cylicobdellidae (also terrestrialleeches) is sister to the remaining hirudiniformleeches. Within the latter clade, Haemadipsidae issister to the remaining families (Semiscolecidae,Xerobdellidae, Macrobdellidae and Hirudinidae). Ina later study with increased taxon sampling, Phillips& Siddall (2009) found a slightly shifted topologywith Haemadipsidae grouping sister to Xerobdelli-dae and Hirudinidae, while Semiscolecidae (nowincluding Macrobdellidae) grouped as sister to thatclade. As such, it appears as though terrestrialleeches occur toward the base of the evolutionarytree of jaw-bearing (arhynchobdellid) leeches. Inves-tigations into the anticoagulant repertoires of hae-madipsid leeches should be useful in elucidatingboth the presence of anticoagulants in early Hiru-diniformes and potential specializations of this rep-ertoire in response to the disparate prey diversity.
Methods
Specimen collection and cDNA synthesis
Leech specimens were collected from exposed skinduring field studies in the tropical rainforest at theUlu Gombak Field Study Centre located northeastof Kuala Lumpur, Malaysia (03°19′29.15″ N,101°45′08.27″E), and were immediately preserved inRNAlater� (Qiagen, Valencia, CA, USA). Speci-mens were identified as Haemadipsa interrupta(MOORE 1935) using a stereomicroscope whilesubmerged in RNAlater; identifications were laterverified by molecular sequences. Prior to RNA
Invertebrate Biologyvol. x, no. x, xxx 2013
2 Kvist, Brugler, Goh, Giribet, & Siddall

extraction, leeches were washed in 0.5% bleach for1 min and subsequently rinsed in deionized waterfor 1 min to minimize contamination with surfacebacteria. Using sterilized dissecting tools, salivarytissue masses (glandular tissue) from three leecheswere removed aseptically and subsequently rinsed in0.5% bleach for 1 min followed by rinsing in deion-ized water for 1 min. Total RNA then was isolatedusing a hybrid protocol based both on that of Haleet al. (2009) and that of the RNeasy Plus Mini Kit(Qiagen, Valencia, CA, USA). A 1.5 mL microcen-trifuge tube containing leech salivary gland tissuewas submerged in liquid nitrogen, followed immedi-ately by homogenization of the frozen tissue using asterile pestle. To maintain the integrity of the RNA,500 lL of TRIzol� (Life Technologies, Gaithers-burg, MD, USA) was added as soon as the samplebegan to thaw; homogenization continued after theaddition of TRIzol. An additional 500 lL of TRIzolwas then added to the 1.5 mL tube (1 mL in total),and tissue was further macerated using a sterile syr-inge. Immediately following maceration, the tubewas vortexed and maintained at room temperature(RT) for 5 min. Thereafter, we added 200 lL ofchloroform, inverted the tube for 20 s, and let it sitat RT for 3 min. The tube then was centrifuged at12,800 g for 15 min at 4°C. The top colorless layer(containing RNA) was transferred to a new 1.5 mLtube; at this point, we transitioned from the proto-col by Hale et al. (2009) to step 3 of the RNeasy�
Plus Mini Kit (which begins by adding 600 lL of70% ethanol to the tube, and subsequently transfer-ring the sample to an RNeasy spin column) and fol-lowed the manufacturer’s protocols, including bothoptional steps (i.e., an extra spin to dry the mem-brane and repeat elution in RNase-free water [40 lLtotal]). The resulting concentration of total RNAwas determined using an Agilent 2100 BioAnalyzer(Agilent Technologies, Palo Alto, CA, USA), and inparticular, the Agilent RNA 6000 Nano Kit. First-strand cDNA synthesis was carried out in a 5 lLreaction consisting of 1.0 lg total RNA, 1 lL of10 lmol L�1 SMART IV oligo (5′-AAG CAGTGG TAT CAA CGC AGA GTG GCC ATTACG GCC GGG-3′), 1 lL of 10 lmol L�1 MODI-FIED CDS III (i.e., the 3′ cDNA synthesis primer;5′-TAG AGG CCG AGG CGG CCG ACA TGTTTT GTT TTT TTT TCT TTT TTT TTT VN-3′),and brought to final volume with RNase-free water.Contents were mixed, spun briefly, and incubated at72°C for 2 min. Following incubation, the tube wasimmediately placed on ice for 2 min, spun briefly,and the following reagents were added to the tube:2 lL of 5X First-Strand Buffer (Clontech Laborato-
ries, Palo Alto, CA, USA), 1 lL of 20 mmol L�1
DTT, 1 lL of 10 mmol L�1 dNTP mix, and 1 lLof SuperScript II Reverse Transcriptase (Invitrogen,Carlsbad, CA, USA) (10 lL total volume). Contentswere mixed, spun briefly, and incubated at 42°C for1.5 h. Thereafter, the tube was immediately placedon ice to terminate first-strand synthesis. The fol-lowing reagents were combined to initiate second-strand cDNA synthesis: 2 lL of first-strand cDNA,18.5 lL RNase-free water, 2.5 lL 10X Advantage 2PCR Buffer, 0.5 lL of 10 mmol L�1 dNTP mix,0.5 lL of 10 lmol L�1 5′ PCR Primer (5′-AAGCAG TGG TAT CAA CGC AGA GT-3′), 0.5 lLof 10 lmol L�1 CDS III/3′ PCR Primer, and 0.5 lL50X Advantage 2 Polymerase Mix (25 lL totalvolume). Contents were gently mixed, spun briefly,and placed in a preheated (95°C) thermocyler. Thefollowing thermal cycling program was used: 95°Cfor 1 min, 95°C for 15 s, 68°C for 6 min, repeatsteps 2–3 for 21 cycles. Approximately 3 lL of sec-ond-strand cDNA was visualized on a 1.1% agarosegel. Second-strand cDNA then underwent SfiI diges-tion by transferring 22 lL of second-strand cDNAto a 0.2 mL PCR tube, adding 2.79 lL 10X SfiIBuffer, 2.79 lL SfiI enzyme, and 0.28 lL of10 mg mL�1 (=100X) BSA, and incubating at 50°Cfor 2 h. Lastly, the sample was run through a Qia-gen QIAquick PCR Purification Kit column (andeluted in Buffer EB), quantified on a NanoDrop1000 Spectrophotometer (Thermo Fisher Scientific,Wilmington, DE, USA), and visualized using anAgilent 2100 BioAnalyzer High Sensitivity DNAKit.
Library construction, emPCR, and pyrosequencing
The cDNA Rapid Library (RL) was preparedusing a Roche 454 GS RL Prep Kit by followingmanufacturer’s protocols for individual samplecleanup as outlined in the Roche 454 RL Prepara-tion Method Manual (Roche Applied Sciences,Indianapolis, IN, USA). Using a QuantiFluorTM-ST Fluorometer (Promega, Madison, WI, USA) incombination with the RL Quantitation Calculator,we calculated a sample concentration of 4.219108 molecules lL�1 (R2=0.99356), which was dilutedto a working stock of 19107 molecules lL�1. Emul-sion-based clonal amplification (PCR) was carriedout using the GS Junior Titanium emPCR (Lib-L)Kit and following manufacturer’s protocols asoutlined in the emPCR Amplification Method Man-ual (Lib-L). This manual was also used for beadrecovery, DNA library bead enrichment, andsequence primer annealing. Enriched beads were
Invertebrate Biologyvol. x, no. x, xxx 2013
Anticoagulant diversity in Haemadipsa interrupta 3

prepared for sequencing on a GS Junior TitaniumPicoTitrePlate Device using the GS Junior TitaniumSequencing Kit and following manufacturer’s proto-cols as outlined in the Sequencing Method Manual.Massively parallel single-end pyrosequencing wasconducted on a 454 GS Junior at the Sackler Insti-tute for Comparative Genomics, American Museumof Natural History, New York, NY, USA.
Filtering, trimming, and assembly
Post-sequencing processing involved a multi-tieredapproach to assure the quality of downstreamsequence data, and was initiated using the five stan-dard 454 quality filters on the GS Junior (Dot,Mixed, Signal Intensity, Primer and TrimBack Val-ley). Thereafter, sff_extract (http://bioinf.comav.upv.es/sff_extract/index.html) was used to create .fasta,.fasta.qual, .fastq, and .xml files. In addition, sff_extract trimmed key/adaptor sequences and removedlow quality reads (i.e., any base listed in lower case).After visualizing the results of sff_extract usingFastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), two binaries, FASTX_trimmer(-f 20 –l 500 –Q 33) and FASTQ_quality_trimmer(-v –t 25) (both part of the FASTX toolkit [http://hannonlab.cshl.edu/fastx_toolkit/]), were used tofurther trim low quality regions; only bases betweenpositions 20–500 and those with a Phred qualityscore ≥25 were retained in the data set (–Q 33 indi-cates that the Phred quality values are standard 454/Sanger scores [0–93] encoded as ASCI characters33–126). After utilizing FASTX_trimmer andFASTQ_quality_trimmer, FastQC was again used tovisualize and verify the overall quality of the reads.
As of July 2013, there were no available tran-scriptomes for species closely related to H. inter-rupta. Thus, a de novo assembly of the filtered readswas conducted using MIRA v. 3.4.0 (Chevreux et al.1999); MIRA was called as follows: mira –project=MyFile –job=denovo,est,accurate,454 454_SETTINGS–CL:qc=no:cpat=no >&log_assembly.txt (where qc=quality clip; cpat=poly-A clipping).
Open reading frames and BLAST queries
To minimize the risk of introns embedded withinthe data, the program getorf, which is part of theEMBOSS package (Rice et al. 2000), was used toextract open reading frames (ORFs) greater than200 nucleotides in length within the contigs. Getorfemployed the standard genetic code, and only ORFsbeginning with a methionine (Met) and ending in astop codon were extracted.
Uninterrupted contigs then were annotated utiliz-ing a series of BLAST searches against various data-bases (all BLAST searches were carried out usingBLAST 2.2.22). First, all contigs were queriedagainst three databases constructed from GenBanknon-redundant protein sequences (nr), nucleotidesequences (nt), and expressed sequence tags (EST)using blastall (-p BLASTx against nr and BLASTnagainst nt and EST). Results of these searches thenwere used as authoritative cross-controls (reciprocalbest hit approach: Ge et al. 2005; Fang et al. 2010)for BLASTx searches against a locally compileddata set of amino acids from both well-characterizedanticoagulants (predominantly from leeches) andother select salivary proteins from previous leechtranscriptome studies (Table 1). That is, the contigsthat best matched the sequences in the local data setwere also BLASTed against GenBank nr, nt, andEST to confirm that they did not match other pro-teins at a better e-value. Positive reciprocal hitsinvolved significant matches of the contigs tosequences annotated as the same bioactive proteinas its best match in the locally compiled data set. Bycontrast, negative reciprocal hits occurred when thecontigs matched a different bioactive protein thanthe best hit against the local data set. In addition,the raw FilterPass reads were both queried againstGenBank nt (via BLASTn) and against the assem-bled contigs using BLASTn to get a better under-standing of the distribution and coverage of thereads and the expression levels of the proteins.
Amino acid sequences for each of the bestmatches (those that scored the lowest e-values)against the known salivary proteins then werealigned in a pairwise fashion with those of therespective archetypal sequence using MAFFT(Katoh et al. 2005) on the European BioinformaticsInstitute website (http://www.ebi.ac.uk/Tools/msa/mafft/) applying default settings. The term arche-typal here refers either to the corresponding well-characterized anticoagulant (but not always thatfrom the first investigation of the respective proteinin leeches) or other bioactive proteins that have pre-viously been considered of interest based on theirmatches against salivary peptides (see, e.g., Minet al. 2010). Our rationale behind pairwise align-ments, as opposed to multiple sequence alignments,is an in-depth comparison to the known and, inmost cases, functional anticoagulant sequence.Whereas multiple sequence alignments could be usedto understand mutational rates across clades orselection pressures acting on the proteins, here wemerely attempt to annotate sequences based on theirsimilarity and composition as compared to a single,
Invertebrate Biologyvol. x, no. x, xxx 2013
4 Kvist, Brugler, Goh, Giribet, & Siddall

Table 1. Anticoagulants included in the locally compiled data set used for the BLASTx comparisons.
Organism Bioactiveprotein
Antagonistic pathway Genbankaccessionnumber
Reference
Hirudo medicinalis Hirudin Thrombin inhibitor Q07558 Scacheri et al. (1993)Poecilobdella viridis Hirudin Thrombin inhibitor P84590 Vankhede et al. (2005)Hirudo medicinalis Hirudin II Thrombin inhibitor P28504 Scharf et al. (1989)Hirudinaria manillensis Hirullin Thrombin inhibitor P26631 Tulinsky & Qiu (1993)Haemadipsa sylvestris Unidentified
thrombininhibitor
Thrombin inhibitor CAA79672 Strube et al. (1993)
Hirudo medicinalis Bdellin Trypsin, plasmininhibitor
P09865 Fink et al. (1986)
Hirudo medicinalis Destabillase I Destabilized fibrinde-polymerase
AAA96144 Zavalova et al. (1996)
Hirudo medicinalis Destabillase II Destabilized fibrinde-polymerase
AAA96143 Zavalova et al. (1996)
Theromyzon tessulatum Cystatin Cystein proteinaseinhibitor
AAN28679 Lefebvre et al. (2004)
Hirudo medicinalis Eglin c Leukocyte elastase,cathepsin G inhibitor
0905140A Knecht et al. (1983)
Haementeria officinalis LAPP Platelet aggregationinhibitor
Q01747 Keller et al. (1992)
Macrobdella decora Decorsin Glycoprotein IIb/IIIainhibitor
P17350 Seymour et al. (1990)
Theromyzon tessulatum Therostasin Factor Xa inhibitor Q9NBW4 Chopin et al. (2000)Haementeria ghilianii Ghilanten Factor Xa inhibitor AAB21233 Brankamp et al. (1991)Haementeria ghilianii Ghilanten Factor Xa inhibitor P16242 Blankenship et al. (1990)Haementeria officinalis Antistasin Factor Xa inhibitor AAA29193 Han et al. (1989)Haementeria officinalis Antistasin Factor Xa inhibitor P15358 Han et al. (1989)Hirudo nipponia Guamerin Factor Xa (elastase)
inhibitorAAD09442 Jung et al. (1995)
Hirudo medicinalis Hirustasin Thrombin, trypsin,chymotrypsin,cathepsin G, kallikreininhibitor
P80302 S€ollner et al. (1994)
Haementeria officinalis Saratin Platelet aggregationinhibitor
2K13-X Gronwald et al. (2008)
Hirudinaria manillensis Manillase Hyaluronic acid-specifichyaluronidase
N/A US Patent:2006_US_7.049.124_B1
Macrobdella decora Ficolin Complementary cascadestimulator, immunoresponse
N/A Min et al. (2010);cluster 686
Hirudo medicinalis Leech derivedtryptaseinhibitor
Tryptase, trypsin, chymotrypsininhibitor
AAB33769 Sommerhoff et al. (1994)
Placobdella ornata Ornatin Glycoprotein IIb/IIIa inhibitor AAB28218 Mazur et al. (1993)Haemadipsa sylvestris Haemadin Thrombin inhibitor AAA15044 Strube et al. (1993)Hirudinaria manillensis Bufrudin Thrombin inhibitor P81492 Scacheri et al. (1993)Theromyzon tessulatum Theromin Thrombin inhibitor P82354 Salzet et al. (2000)Haementeria ghilianii Haementin Fibrinogenolytic protease Q7M3P9 Swadesh et al. (1990)Hirudinaria manillensis Gelin Elastase, cathepsin G,
chymotrypsin, subtilisininhibitor
AAB27871 Electricwala et al. (1992)
Hirudo nipponia Piguamerin Factor Xa, trypsin,kallikrein inhibitor
P81499 Kim & Kang (1998)
(continued)
Invertebrate Biologyvol. x, no. x, xxx 2013
Anticoagulant diversity in Haemadipsa interrupta 5

established sequence. The alignments then were visu-alized using Jalview ver. 2 (Waterhouse et al. 2009),and prediction of secretory signal peptides wasperformed on the SignalP 4.1 (Petersen et al. 2011)server at http://www.cbs.dtu.dk/services/SignalP/.Following alignment, in cases where the sequencesof the archetypal bioactive proteins were substan-tially longer than the contigs, these were truncatedat the 5′ end, 3′ end, or both. Moreover, in somecases, the contigs were truncated in the 3′-end. Thiswas done to investigate the conservation and diver-gences in only the sequence regions that were repre-sented by orthologous regions in both thearchetypal protein and the contigs.
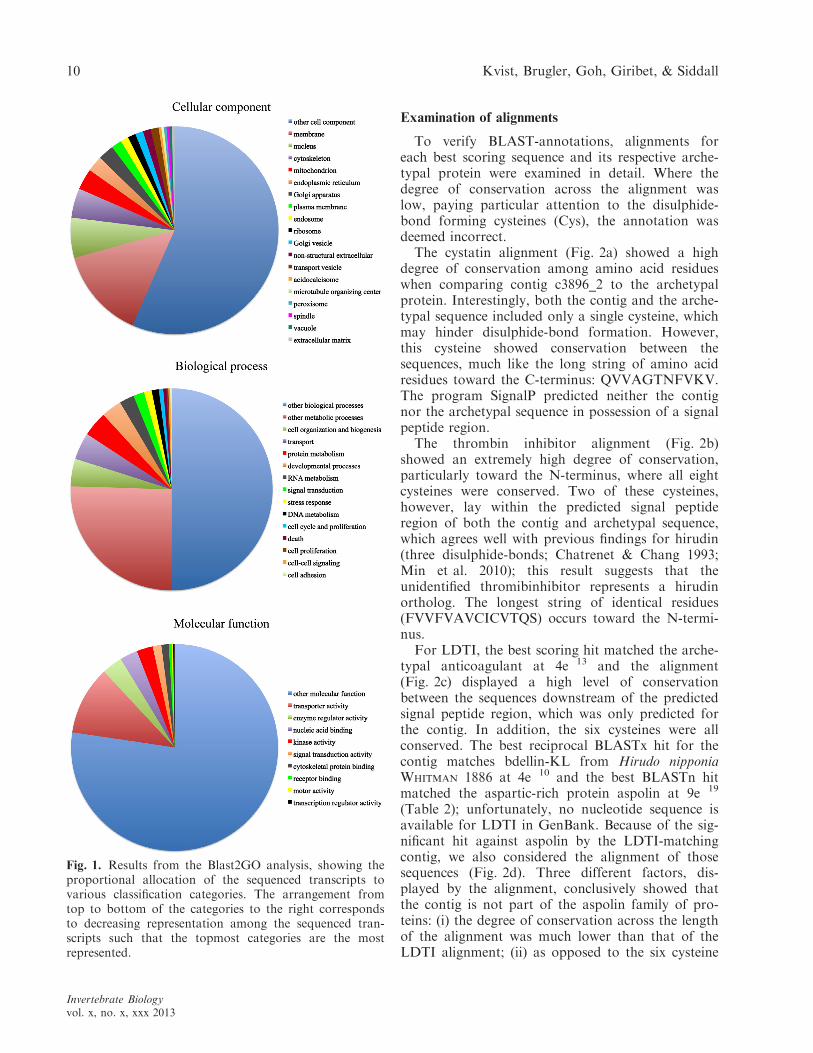
Blast2GO (Conesa et al. 2005) was used to inferfunctional annotations using gene ontology. Theassembled transcripts were jointly submitted toBlast2GO to clarify the proportional allocation ofthe partial transcriptome to various classificationcategories. The resulting GO terms were subse-quently interpreted using the CateGOrizer (Hu et al.2008) platform at http://www.animalgenome.org/bioinfo/tools/countgo/, and pie charts illustratingthe proportions were created in Microsoft Excel.
Phylogenetic analyses
Putative orthology and evolutionary relationshipswithin each cluster of select bioactive salivary pro-teins were also examined by creating phylogenetichypotheses of aligned amino acid sequences. In addi-tion to the sequences of the best scoring contig andthe archetypal bioactive protein, the final matricesincluded amino acid sequences employed by Kvistet al. (2013) in a similar study on the evolution ofbloodfeeding in three species of medicinal leeches.Amino acid sequences were aligned using MAFFTemploying the G-INS-i strategy with a gap-openingcost of 3.0 and default settings for all other parame-
ters. Phylogenetic analyses were conducted in TNT(Goloboff et al. 2008) under the parsimony criterion,where a traditional search was performed for eachdata set employing 100 initial addition sequencesand TBR branch swapping. All characters were un-weighted and non-additive and gaps were treated asmissing data. Because of the lack of appropriate out-groups, all trees were left unrooted.
For the leech derived tryptase inhibitor (LDTI)data set, which was not represented in the study byKvist et al. (2013), a potentially orthologous proteinsequence for aspolin (accession number AB189965;see below) was included and the following acces-sions were downloaded from GenBank and addedto the matrix: P80424, 1LDT_L, 1LDT_T,2KMR_A, 2KMQ_A, 2KMO_A, 2KMP_A. Thebest scoring contig matching bufrudin (also not rep-resented in the study by Kvist et al. 2013), as wellas the archetypal sequence (accession numberP81492), was added to the hirudin data set. Unfor-tunately, too few sequences are available on Gen-Bank for leech-derived cystatin and theromin toconstitute workable data sets for phylogenetic infer-ences (only a single sequence of each). Therefore,phylogenies were not estimated for these data sets.Similarly, phylogenies were not estimated for theHis-rich proteins (as their affiliation in terms of pro-tein family and their function remains unknown) orthe Kazal-type serine-protease inhibitors.
Results
Raw reads: BLAST, trimming, and assembly
Pyrosequencing of the cDNA library resulted in200,316 Raw wells, 190,511 KeyPass wells, and141,765 (74.4%) FilterPass reads (the latter of whichpassed all five quality filters). Average read lengthwas 427.5�109.8 base pairs with a total of
Table 1. (continued)
Organism Bioactiveprotein
Antagonistic pathway Genbankaccessionnumber
Reference
Macrobdella decora C-type lectin Potential fibrin inhibitor N/A Min et al. (2010); cluster 38Macrobdella decora Kazal-type serpin Serin protease inhibitor N/A Min et al. (2010); cluster 26Macrobdella decora His ASP rich Unknown N/A Min et al. (2010); cluster 18Macrobdella decora His rich Unknown N/A Min et al. (2010); cluster 7Macrobdella decora KGD disintegrin Potential inhibitor of
platelet aggregationN/A Min et al. (2010); cluster 45
Macrobdella decora Leukocyteelastaseinhibitor
Elastase inhibitor N/A Min et al. (2010); cluster 41
Invertebrate Biologyvol. x, no. x, xxx 2013
6 Kvist, Brugler, Goh, Giribet, & Siddall

60,598,465 bases sequenced. MIRA assembled125,474 reads (16,291 reads did not assemble) into14,873 contigs with a N50 of 478 bases for the totalpool of contigs, and an N50 of 753 bases for largecontigs (>500 nucleotides). Getorf counted a total of22,005 ORFs longer than 200 nucleotides. The num-ber of ORFs exceeds that of the contigs becausegetorf explores all possible ORFs for each contig,often outputting two or more possible ORFs. AllORFs were used in subsequent BLAST searches;those with the best match to protein and nucleotidedatabases were considered to be in the correct read-ing frame. Raw reads (i.e., non-truncated data; seeabove) from the GS Junior as well as the MIRAassembly are deposited in the GenBank SequenceRead Archive (submission number SRX246952).
BLAST and Blast2GO annotations
Complete lists of all results from the BLASTxand BLASTn searches using the contigs as queriesare presented in Tables S1 and S2, respectively. Onthe one hand, the BLASTn searches using the rawreads as queries indicated that fully 16,062 readsfound no match in the nt database, 2,100 matchedleech nuclear 18S rDNA at 1e�10 or below, 746matched leech nuclear 28S rDNA at 1e�10 or below,and 2,843 matched leech mtDNA transcripts includ-ing 12S rDNA, 16S rDNA, ATP6, Cox1, Cox2,Cox3, CytB, ND1, ND4, and ND6. On the otherhand, only a very small number of the assembledcontigs scored significantly against sequences anno-tated as mitochondrial, suggesting that MIRA effi-ciently trimmed these data from the data set duringassembly. Our initial morphological identification ofthe specimens was confirmed by several contigBLASTn and BLASTx hits matching sequencesderived from Haemadipsa interrupta (e.g., c232matching 18S rDNA from H. interrupta at 1e�127).
Through BLASTx searches against the locallycompiled data set, unique ORFs matching the fol-lowing proteins at an e-value of 1e�5 or below werefound in the H. interrupta transcriptome (Table 2):cystatin, manillase, an unidentified thrombin inhibi-tor, leukocyte elastase inhibitor, lectoxin-like c-typelectin, antistasin, ficolin, guamerin, piguamerin,bdellin, Kazal-type serine protease inhibitors, ghil-anten, HIS-rich proteins, therostasin, leech-derivedtryptase inhibitor (LDTI), bufrudin, eglin c, saratin,theromin, and leech antiplatelet protein (LAPP).After reciprocally BLASTing sequences against Gen-Bank nr, nt, and EST databases, nine of the best 20bioactive protein-matching ORFs show identicalannotations to those of the best reciprocal BLAST(x
and/or n) hit; for six of the ORFs, the best recipro-cal BLAST(x and/or n) hits were against unannotat-ed sequences; for four of the ORFs, the bestreciprocal BLAST(x and/or n) hits were againstsequences annotated for the same family of proteinsas the best BLASTx hit against the local data set;and for only one ORF did the best reciprocalBLASTx and BLASTn hit match a sequence anno-tated as a different anticoagulant or other bioactiveprotein but at a much higher e-value (Table 2).Using the nr database, no significant reciprocalBLASTx hit was found for contig c3900_1, whichinitially matched a highly expressed HIS-rich proteinderived from the salivary glands of the North Amer-ican medicinal leech Macrobdella decora (SAY 1824)(see Table 2). In addition, the BLASTn searchesagainst GenBank EST offered no further informa-tion concerning annotations as all contigs, save forc5389_2, matched unannotated sequences; also notethat most of the hits were against hirudinid taxa.Several e-values resulting from the reciprocalBLASTx/n searches were insignificant, in that theywere several orders of magnitude higher thanthe best match found against the locally compileddatabase of bioactive proteins, regardless of theannotation. In addition to the matches against well-characterized anticoagulants and other bioactive sal-ivary proteins, numerous ORFs returned significanthits against proteins with putative involvement inanticoagulation activity, such as cathepsin, disinte-grins, galactose-binding lectin, and leech carboxy-peptidase inhibitors. When mapping the raw readsback to the putatively anticoagulation-related con-tigs, more than half of the reads that found a signifi-cant match mapped against disintegrins (185 rawreads), antistasins (167 raw reads), and c-type/galac-tose-binding lectin (144 raw reads), and this mayserve as a coarse, initial indication of expression lev-els within the transcriptome. The number of rawreads mapped against each of the bioactive proteinsis listed in Table 2.
Blast2GO and CateGOrizer mapped the contigsto 75 of the MGI_GO_slim1 ancestor terms by sin-gle count; 17 contigs found no match in the GOcategory database. Other biological process, othermolecular function, and other cellular componentwere the most frequently encountered hits for eachof the three categories (biological process, molecularfunction, and cellular component, respectively), indi-cating that the gene ontology for several of our pro-teins remains largely unknown (Fig. 1). Thestructures and functions of these BLAST and Blas-t2GO annotated, putative anticoagulation-relatedproteins, are discussed further below.
Invertebrate Biologyvol. x, no. x, xxx 2013
Anticoagulant diversity in Haemadipsa interrupta 7
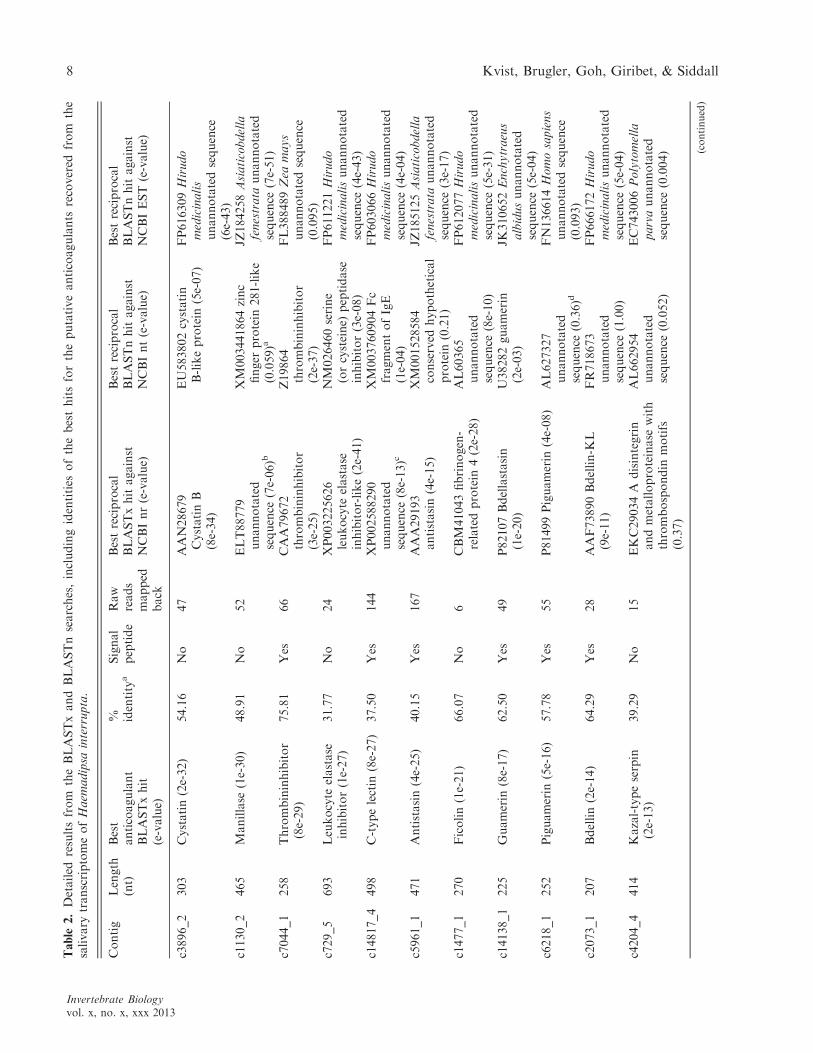
Table
2.Detailed
resultsfrom
theBLASTxandBLASTnsearches,includingidentities
ofthebesthitsfortheputativeanticoagulants
recovered
from
the
salivary
transcriptomeofHaem
adipsa
interrupta.
Contig
Length
(nt)
Best
anticoagulant
BLASTxhit
(e-value)
% identity
aSignal
peptide
Raw
reads
mapped
back
Bestreciprocal
BLASTxhitagainst
NCBInr(e-value)
Bestreciprocal
BLASTnhitagainst
NCBInt(e-value)
Bestreciprocal
BLASTnhitagainst
NCBIEST
(e-value)
c3896_2
303
Cystatin(2e-32)
54.16
No
47
AAN28679
CystatinB
(8e-34)
EU583802cystatin
B-likeprotein
(5e-07)
FP616309Hirudo
medicinalis
unannotatedsequence
(6e-43)
c1130_2
465
Manillase
(1e-30)
48.91
No
52
ELT88779
unannotated
sequence
(7e-06)b
XM003441864zinc
finger
protein
281-like
(0.059)a
JZ184258Asiaticobdella
fenestrata
unannotated
sequence
(7e-51)
c7044_1
258
Thrombininhibitor
(8e-29)
75.81
Yes
66
CAA79672
thrombininhibitor
(3e-25)
Z19864
thrombininhibitor
(2e-37)
FL388489Zea
mays
unannotatedsequence
(0.095)
c729_5
693
Leukocyte
elastase
inhibitor(1e-27)
31.77
No
24
XP003225626
leukocyte
elastase
inhibitor-like(2e-41)
NM026460serine
(orcysteine)
peptidase
inhibitor(3e-08)
FP611221Hirudo
medicinalisunannotated
sequence
(4e-43)
c14817_4
498
C-typelectin
(8e-27)
37.50
Yes
144
XP002588290
unannotated
sequence
(8e-13)c
XM003760904Fc
fragmentofIgE
(1e-04)
FP603066Hirudo
medicinalisunannotated
sequence
(4e-04)
c5961_1
471
Antistasin(4e-25)
40.15
Yes
167
AAA29193
antistasin(4e-15)
XM001528584
conserved
hypothetical
protein
(0.21)
JZ185125Asiaticobdella
fenestrata
unannotated
sequence
(3e-17)
c1477_1
270
Ficolin(1e-21)
66.07
No
6CBM41043fibrinogen-
relatedprotein
4(2e-28)
AL60365
unannotated
sequence
(8e-10)
FP612077Hirudo
medicinalisunannotated
sequence
(5e-31)
c14138_1
225
Guamerin
(8e-17)
62.50
Yes
49
P82107Bdellastasin
(1e-20)
U38282guamerin
(2e-03)
JK310652Enchytraeus
albidusunannotated
sequence
(5e-04)
c6218_1
252
Piguamerin
(5e-16)
57.78
Yes
55
P81499Piguamerin
(4e-08)
AL627327
unannotated
sequence
(0.36)d
FN136614Homosapiens
unannotatedsequence
(0.093)
c2073_1
207
Bdellin(2e-14)
64.29
Yes
28
AAF73890Bdellin-K
L(9e-11)
FR718673
unannotated
sequence
(1.00)
FP666172Hirudo
medicinalisunannotated
sequence
(5e-04)
c4204_4
414
Kazal-typeserpin
(2e-13)
39.29
No
15
EKC29034A
disintegrin
andmetalloproteinase
with
thrombospondin
motifs
(0.37)
AL662954
unannotated
sequence
(0.052)
EC743006Polytomella
parvaunannotated
sequence
(0.004) (continued)
Invertebrate Biologyvol. x, no. x, xxx 2013
8 Kvist, Brugler, Goh, Giribet, & Siddall
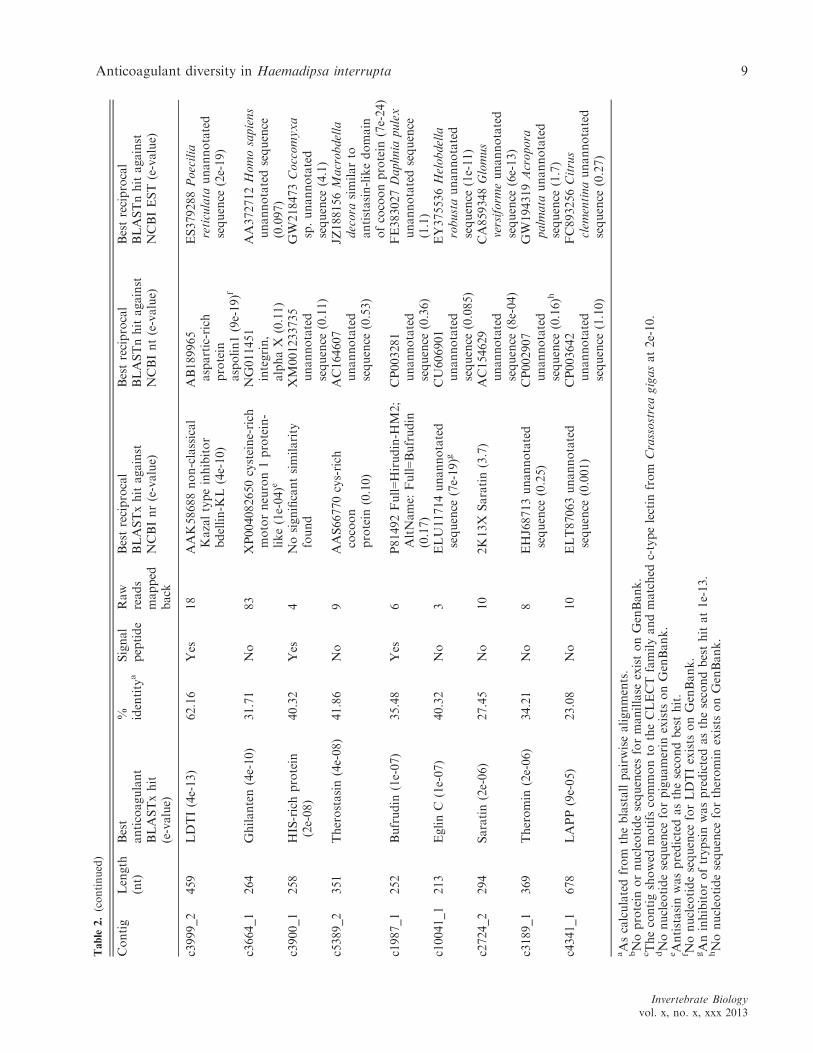
Table
2.(continued)
Contig
Length
(nt)
Best
anticoagulant
BLASTxhit
(e-value)
% identity
aSignal
peptide
Raw
reads
mapped
back
Bestreciprocal
BLASTxhitagainst
NCBInr(e-value)
Bestreciprocal
BLASTnhitagainst
NCBInt(e-value)
Bestreciprocal
BLASTnhitagainst
NCBIEST(e-value)
c3999_2
459
LDTI(4e-13)
62.16
Yes
18
AAK58688non-classical
Kazaltypeinhibitor
bdellin-K
L(4e-10)
AB189965
aspartic-rich
protein
aspolin1(9e-19)f
ES379288Poecilia
reticulata
unannotated
sequence
(2e-19)
c3664_1
264
Ghilanten(4e-10)
31.71
No
83
XP004082650cysteine-rich
motorneuron1protein-
like(1e-04)e
NG011451
integrin,
alphaX
(0.11)
AA372712Homosapiens
unannotatedsequence
(0.097)
c3900_1
258
HIS-richprotein
(2e-08)
40.32
Yes
4Nosignificantsimilarity
found
XM001233735
unannotated
sequence
(0.11)
GW218473Coccomyxa
sp.unannotated
sequence
(4.1)
c5389_2
351
Therostasin(4e-08)
41.86
No
9AAS66770cys-rich
cocoon
protein
(0.10)
AC164607
unannotated
sequence
(0.53)
JZ188156Macrobdella
decora
similarto
antistasin-likedomain
ofcocoonprotein
(7e-24)
c1987_1
252
Bufrudin
(1e-07)
35.48
Yes
6P81492Full=Hirudin-H
M2;
AltName:
Full=Bufrudin
(0.17)
CP003281
unannotated
sequence
(0.36)
FE383027Daphnia
pulex
unannotatedsequence
(1.1)
c10041_1
213
EglinC
(1e-07)
40.32
No
3ELU11714unannotated
sequence
(7e-19)g
CU606901
unannotated
sequence
(0.085)
EY375536Helobdella
robustaunannotated
sequence
(1e-11)
c2724_2
294
Saratin(2e-06)
27.45
No
10
2K13X
Saratin(3.7)
AC154629
unannotated
sequence
(8e-04)
CA859348Glomus
versiform
eunannotated
sequence
(6e-13)
c3189_1
369
Theromin
(2e-06)
34.21
No
8EHJ68713unannotated
sequence
(0.25)
CP002907
unannotated
sequence
(0.16)h
GW194319Acropora
palm
ata
unannotated
sequence
(1.7)
c4341_1
678
LAPP(9e-05)
23.08
No
10
ELT87063unannotated
sequence
(0.001)
CP003642
unannotated
sequence
(1.10)
FC893256Citrus
clem
entinaunannotated
sequence
(0.27)
aAscalculatedfrom
theblastallpairwisealignments.
bNoprotein
ornucleotidesequencesformanillase
existonGenBank.
cThecontigshowed
motifs
commonto
theCLECTfamilyandmatched
c-typelectin
from
Crassostreagigasat2e-10.
dNonucleotidesequence
forpiguamerin
exists
onGenBank.
eAntistasinwaspredictedasthesecondbesthit.
f Nonucleotidesequence
forLDTIexists
onGenBank.
gAninhibitoroftrypsinwaspredictedasthesecondbesthitat1e-13.
hNonucleotidesequence
fortheromin
exists
onGenBank.
Invertebrate Biologyvol. x, no. x, xxx 2013
Anticoagulant diversity in Haemadipsa interrupta 9
Examination of alignments
To verify BLAST-annotations, alignments foreach best scoring sequence and its respective arche-typal protein were examined in detail. Where thedegree of conservation across the alignment waslow, paying particular attention to the disulphide-bond forming cysteines (Cys), the annotation wasdeemed incorrect.
The cystatin alignment (Fig. 2a) showed a highdegree of conservation among amino acid residueswhen comparing contig c3896_2 to the archetypalprotein. Interestingly, both the contig and the arche-typal sequence included only a single cysteine, whichmay hinder disulphide-bond formation. However,this cysteine showed conservation between thesequences, much like the long string of amino acidresidues toward the C-terminus: QVVAGTNFVKV.The program SignalP predicted neither the contignor the archetypal sequence in possession of a signalpeptide region.
The thrombin inhibitor alignment (Fig. 2b)showed an extremely high degree of conservation,particularly toward the N-terminus, where all eightcysteines were conserved. Two of these cysteines,however, lay within the predicted signal peptideregion of both the contig and archetypal sequence,which agrees well with previous findings for hirudin(three disulphide-bonds; Chatrenet & Chang 1993;Min et al. 2010); this result suggests that theunidentified thromibinhibitor represents a hirudinortholog. The longest string of identical residues(FVVFVAVCICVTQS) occurs toward the N-termi-nus.
For LDTI, the best scoring hit matched the arche-typal anticoagulant at 4e�13 and the alignment(Fig. 2c) displayed a high level of conservationbetween the sequences downstream of the predictedsignal peptide region, which was only predicted forthe contig. In addition, the six cysteines were allconserved. The best reciprocal BLASTx hit for thecontig matches bdellin-KL from Hirudo nipponiaWHITMAN 1886 at 4e�10 and the best BLASTn hitmatched the aspartic-rich protein aspolin at 9e�19
(Table 2); unfortunately, no nucleotide sequence isavailable for LDTI in GenBank. Because of the sig-nificant hit against aspolin by the LDTI-matchingcontig, we also considered the alignment of thosesequences (Fig. 2d). Three different factors, dis-played by the alignment, conclusively showed thatthe contig is not part of the aspolin family of pro-teins: (i) the degree of conservation across the lengthof the alignment was much lower than that of theLDTI alignment; (ii) as opposed to the six cysteine
Fig. 1. Results from the Blast2GO analysis, showing theproportional allocation of the sequenced transcripts tovarious classification categories. The arrangement fromtop to bottom of the categories to the right correspondsto decreasing representation among the sequenced tran-scripts such that the topmost categories are the mostrepresented.
Invertebrate Biologyvol. x, no. x, xxx 2013
10 Kvist, Brugler, Goh, Giribet, & Siddall
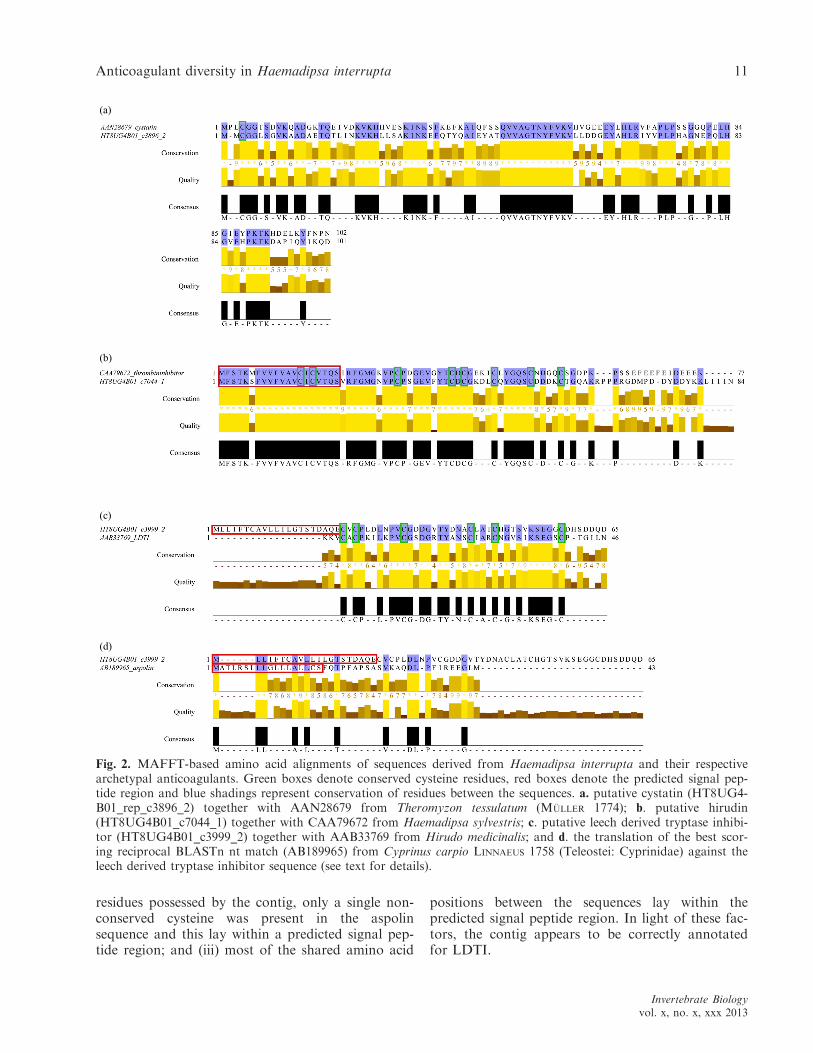
residues possessed by the contig, only a single non-conserved cysteine was present in the aspolinsequence and this lay within a predicted signal pep-tide region; and (iii) most of the shared amino acid
positions between the sequences lay within thepredicted signal peptide region. In light of these fac-tors, the contig appears to be correctly annotatedfor LDTI.
(a)
(b)
(c)
(d)
Fig. 2. MAFFT-based amino acid alignments of sequences derived from Haemadipsa interrupta and their respectivearchetypal anticoagulants. Green boxes denote conserved cysteine residues, red boxes denote the predicted signal pep-tide region and blue shadings represent conservation of residues between the sequences. a. putative cystatin (HT8UG4-B01_rep_c3896_2) together with AAN28679 from Theromyzon tessulatum (M €ULLER 1774); b. putative hirudin(HT8UG4B01_c7044_1) together with CAA79672 from Haemadipsa sylvestris; c. putative leech derived tryptase inhibi-tor (HT8UG4B01_c3999_2) together with AAB33769 from Hirudo medicinalis; and d. the translation of the best scor-ing reciprocal BLASTn nt match (AB189965) from Cyprinus carpio LINNAEUS 1758 (Teleostei: Cyprinidae) against theleech derived tryptase inhibitor sequence (see text for details).
Invertebrate Biologyvol. x, no. x, xxx 2013
Anticoagulant diversity in Haemadipsa interrupta 11

The eglin c alignment (Fig. S1) displayed a highdegree of conservation across the molecule. Incontrast to findings that eglin c is devoid of cyste-ines (Min et al. 2010; Kvist et al. 2013), the bestscoring eglin c contig herein showed a single cys-teine at position 43. No signal peptides were pre-dicted.
For c-type lectin, the alignment (Fig. S2) betweenthe contig and the archetypal protein showed arather high degree of conservation with eight con-served cysteines shared between the sequences; threeadditional cysteines were present in the archetypalsequence derived from the salivary EST library ofM. decora. Both the contig and archetypal sequencecontained a predicted signal peptide region at theN-terminus.
The antiplatelet alignment (Fig. S3) contained thebest hits for saratin and LAPP as well as botharchetypal anticoagulants. The alignment showed ahigh degree of conservation primarily in the middomain of the molecule, between codon positions85–169, whereas the conservation decreased towardthe termini. Closer examination of the alignmentrevealed less conservation between the best scoringputative saratin contig and the archetypal anticoag-ulants when compared to the best scoring putativeLAPP contig. This was especially true in terms ofcysteine conservation; all six known cysteines ofsaratin/LAPP (Gronwald et al. 2008; Kvist et al.2011a) were conserved within the putative LAPPcontig, whereas only one was present in the puta-tive saratin contig, suggesting that the annotationof the latter is incorrect. A signal peptide regionwas only predicted for the archetypal LAPPsequence.
The best scoring putative manillase contig alignedwell with the archetypal sequence and showed ahigh degree of conservation, in particular towardthe C-terminus, where the longest string of identicalresidues is SKMRLLFDLNAE (Fig. S4). The arche-typal sequence did not include cysteines; however, asingle cysteine was present in the contig (position61). No signal peptide regions were predicted foreither sequence.
For His-rich protein, the archetypal sequencewas highly expressed in the salivary transcriptomeof M. decora (Min et al. 2010), but its function isstill unknown. Although the archetypal sequenceincluded two mid domain insertions when comparedto the contig (codon positions 31–37 and 44–52), thedegree of conservation across shared amino acidpositions was high (Fig. S5). The single cysteine wasconserved and predicted signal peptide regions werepresent in both sequences.
The degree of amino acid conservation was highin the first domain of the ficolin alignment (Fig. S6)and two cysteine residues shared identical positionsbetween the sequences; an additional cysteine waspresent in the archetypal sequence. In the seconddomain, the contig included a large insertion, whichwas lacking in the archetypal sequence. Judgingfrom the levels of shared similarity, however, thecontig appears to be correctly annotated for ficolin.No signal peptide regions were predicted.
The bdellin alignment (Fig. S7) also displayedhigh degree of conservation between the sequencesacross their length. This included conservation of sixcysteines that are thought to be involved in forma-tion of three disulphide bonds (Min et al. 2010;Kvist et al. 2013). In accordance with Min et al.(2010) and Kvist et al. (2013), the present study alsofound two proline (Pro) residues (positions 19 and24), confirming that this unexpected finding in paststudies was indeed correct (bdellin was long thoughtto be devoid of proline residues [Fritz et al. 1971]).The contig included a signal peptide region, suggest-ing that it is secreted.
In the alignment of the Kazal-type serine proteaseinhibitors (Fig. S8), the degree of conservation washigh at the 3′-end and lower in the 5′-end. Thearchetypal anticoagulant, represented by an ESTfrom M. decora, possessed 12 cysteines, but the con-tig only possessed six. Whereas the six cysteinesshowed conservation between the sequences, theputative annotation based on the BLAST search isstill questionable.
The anti-factor Xa alignment (Fig. S9) includedboth matching contigs and archetypal sequencesfor the following anticoagulants: antistasin, guam-erin, piguamerin, ghilanten, and therostasin. Thenumber of fully, or nearly fully, conserved cyste-ines across the alignment (n=20) agreed well withprevious findings in leech antistasin (n=22; Kvistet al. 2013). Only 10 of these occurred outside ofany predicted signal peptide region; except forcontig c5389_2, which matched therostasin at4e�8, the cysteines were fully conserved in allsequences. After closer examination, H. interruptaappears to possess sequences with significant simi-larity to antistasin, guamerin, piguamerin and ghil-anten, whereas the therostasin match was toodisparate to confidently infer annotation. Indeed,the best reciprocal BLASTn hit (7e�24) for contigc5389_2 against GenBank EST was a sequencefrom M. decora annotated for a protein similar toan antistasin-like domain of cysteine-rich cocoonproteins. Some cocoon proteins are paralogous toantistasin family proteins in leeches (Mason et al.
Invertebrate Biologyvol. x, no. x, xxx 2013
12 Kvist, Brugler, Goh, Giribet, & Siddall

2004), and this is probably the reason that contigc5389_2 also matched therostasin, but at a muchhigher e-value.
Conservation in the bufrudin alignment (Fig.S10) was largely centered on the six conserved cy-steines that occurred outside of the predicted signalpeptide region (predicted for both the contig andarchetypal sequence). Bufrudin, a thrombin inhibi-tor, is similar in length and cysteine distribution tohirudin, and the best scoring hit for bufrudin alsomatched hirudin but at a higher e-value (3e�6 vs.1e�7).
In contrast to the well-conserved bioactive pro-tein sequences noted above, some alignments werenot as conserved. For example, the theromin align-ment (Fig. S11) contained two large insertions inthe contig when compared with the archetypal anti-coagulant; even in regions of shared amino acids,conservation was remarkably low. Whereas thearchetypal anticoagulant contained 16 cysteineresidues, the contig only included one, which wasnot shared with the anticoagulant. No signal pep-tides were predicted. Although the e-value score(2e�6) for contig c3189_1 suggests a close affinityto theromin, the alignment clearly indicates thatthe contig is not related to the theromin family ofproteins.
The same holds true for the leukocyte elastaseinhibitor alignment (Fig. S12), which also con-tradicted the low e-value score (1e�27) obtainedfrom a BLASTx analysis against our local data set.Except for the second domain of the molecule, con-servation across the alignment was minimal. Asignal peptide was predicted for the archetypalsequence but no cyteines (n=1 in the anticoagulant,n=5 in the contig) were shared by the sequences.Thus, details of the alignment suggest that thesesequences are most likely not orthologous and thatthe contig does not belong to the leukocyte elastaseinhibitor family.
In conclusion, sequences from the H. interruptatranscriptome matched each of cystatin, hirudin,LDTI, eglin c, c-type lectin, LAPP, manillase, His-rich protein, ficolin, bdellin, Kazal-type serine prote-ase inhibitor, antistasin, guamerin, piguamerin, andbufrudin with low (well scoring) e-values, identicalor near identical best reciprocal BLASTx and/orBLASTn hits, and high conservation across thealignments. These three factors suggest that annota-tions for the contigs are correct. In addition, regionsindicative of signal peptides were predicted forcontigs that matched hirudin, LDTI, c-type lectin,His-rich protein, bdellin, antistasin, guamerin, pigu-amerin, and bufrudin, supporting the hypothesis
that these proteins are actively secreted and usedduring bloodfeeding by the leech.
Other highly represented transcripts
The connectin titin, originally isolated from thehemichordate Saccoglossus kowalevskii (AGASSIZ
1873), was not only the most frequent hit amongthe assembled contigs, but was also present in threedifferent forms among the ten most frequent hits(best hit=4e�38; see Table S1). Whereas the remain-ing seven hits were largely unannotated sequences(hypothetical proteins) from various organisms, twomatched sequences annotated as collagen alpha 2(IV) at 1e�17. In fact, among the 50 most frequenthits matching an annotated sequence, titin, variouscollagen forms, filamin (3e�96), twitchin-like proteins(3e�22) and connectin (4e�20) dominate; this preva-lence is only interrupted by isolated hits matchingtumor differentially expressed protein (9e�17), calpo-nin (2e�91), protein LET-2 (2e�28), glycine-richsecreted cement protein (2e�34), zinc finger protein(1e�50), and granulins (3e�28) (Table S1).
A surprisingly high number of hits were recoveredthat matched sequences derived from other blood-feeding organisms, such as the yellow fever mos-quito (Aedes aegypti [LINNAEUS 1758]), southernhouse mosquito (Culex quinquefasciatus [SAY 1823]),and the malaria-bearing mosquito Anopheles gam-biae GILES 1902. The vast majority of these matcheswere against unannotated sequences, but some wereof potential interest to anticoagulation activities,such as calpain (Cong et al. 1989) at an e-value of3e�26 and vitellogenic carboxypeptidase (Ribeiro2003) at 9e�16.
In addition to these select matches against eukary-otic taxa, several matches were found againstsequences derived from bacteria, predominatelyalphaproteobacteria (Agrobacterium spp., Rhizobiumspp.; see Tables S1, S2). The three highest scoringbacterial BLASTx hits were against sequences anno-tated as aconitate hydratase from Agrobacteriumtumefaciens CONN 1942 (e-value of 0), proteins fromthe porin subfamily from Rhizobium leguminosarum(FRANK 1879) (1e�155), and outer membrane proteinOmpA from an Agrobacterium spp. (1e�154). Inter-estingly, several significant hits were found againstbacteria-derived protein catalysts, specifically thoseregulating the biosynthesis of riboflavin (Richteret al. 1997; Ludwig et al. 1987). These includebifunctional riboflavin deaminase-reductase (at4e�35), riboflavin synthase subunit beta (at 4e�38),and 6,7-dimethyl-8-ribityllumazine synthase (at1e�56), all from A. tumefaciens.
Invertebrate Biologyvol. x, no. x, xxx 2013
Anticoagulant diversity in Haemadipsa interrupta 13

Phylogenies
As mentioned above, phylogenetic analyses werelimited to those bioactive protein families for whichthree or more amino acid sequences with probableorthology were available, which excluded cystatin,theromin, His-rich proteins, and Kazal-type serineprotease inhibitors. The terminology used todescribe the unrooted trees shown here follows thatof Wilkinson et al. (2007), such that a “clan” is theequivalent to a “monophyletic group” in a rootedtree and “adjacent group” is equivalent to “sister-group” in a rooted tree. Summary statistics for thetrees are presented in Table 3. By and large, theclans in the trees do not conform to species clusters,i.e., there is little species fidelity among the selectproteins.
In the hirudin tree (Fig. 3), contig c7044_1 fromH. interrupta forms a clan with three archetypalsequences; one annotated as haemadin and two asthrombin inhibitors, and all from H. sylvestris BLAN-
CHARD 1894. Moreover, contig c1987_1, whichscored well against both hirudin and bufrudin in theBLASTx and BLASTn searches, places as the adja-cent group to a larger set of sequences includingthose mentioned above as well as a putative hirudinortholog derived from a M. decora EST library. Thecontrast between the position of this sequence andthe archetypal bufrudin sequence indicates that bothH. interrupta sequences are hirudin (or possiblyhaemadin) orthologs.
The LDTI tree (Fig. 4) included an aspolinsequence due to its high scoring reciprocal BLASTnhit against contig c3999_2 from H. interrupta. How-ever, the long branches in the tree, both leading tothe aspolin sequence and a putative LDTI sequencefrom Hirudo medicinalis LINNAEUS 1758, evince thenon-homology between these sequences and theremainder of the tree. The H. interrupta contigforms a clan with an unresolved clan of archetypalsequences derived from Hi. medicinalis, verifying theBLAST-based orthology between these sequences.
The eglin c putative ortholog from H. interruptareceived a rather long branch length in the tree (Fig.S13). However, branch lengths are generally short inthe tree and the branch leading to contig c10041_1only corresponds to about 25 amino acid substitu-tions. Judging from the tree alone, it remainsunclear whether or not the H. interrupta-derivedcontig shows orthology with the archetypalsequence, but it is contained within a larger clanconsisting of the archetypal sequence and putativeorthologs from Hi. medicinalis and Aliolimnatis fene-strata (MOORE 1939).
For c-type lectin, the tree (Fig. S14) shows two lar-ger clans, one including the archetypal sequence, andthe other including the H. interrupta-derived contig(c14817_4). The contig places as the adjacent groupto a c-type lectin putative ortholog from Hi. medici-nalis and, together, these place as the adjacent groupof two sequences derived from Hi. medicinalis and A.fenestrata, respectively. Although the branch leadingto contig c14817_4 is proportionally longer thanmost other branches, neither its length nor its place-ment in the tree does necessarily indicate paralogy.
The antiplatelet tree (Fig. S15), including putativesaratin and LAPP orthologs as well as the arche-typal sequences, shows a high amount of structurein terms of both taxonomic fidelity and separationof the different proteins. The two main clans in thetree represent the saratin orthologs and the LAPPorthologs, respectively. The two contigs from H. in-terrupta (c2724_2 and c4341_1) form a clan adjacentto the archetypal sequences of LAPP derived fromglossiphoniid leeches. This solidifies the results fromthe examination of the alignments, in that no saratinorthologs are present in the H. interrupta data set.Instead, both contigs seem to represent LAPP puta-tive orthologs.
Unfortunately, the manillase tree (Fig. S16) offerslittle help in assigning orthology as the tree is com-pletely unresolved. What this might suggest, how-ever, is that the included sequences are all part ofthe same radiation, with inconsiderable subsequentdifferentiation. To this end, the putative manillase
Table 3. Summary statistics for the aligned amino aciddata sets for each bioactive protein family and resultingphylogenetic trees.
Bioactiveprotein
No.alignedsites
No. ofmostparsimonioustrees
Length CI RI
Hirudin 136 2 206 0.971 0.937LDTI 225 2 101 0.990 0.857Eglin c 124 2 177 0.977 0.826C-typelectin
193 1 459 0.950 0.763
Saratin/LAPP
241 6 899 0.811 0.833
Manillase 434 19 469 0.989 0.884Ficolin 208 1 304 0.964 0.621Bdellin 79 1 211 0.919 0.874Antistasin 222 28 748 0.848 0.797Elastaseinhibitor
331 3 492 0.980 0.412
CI, consistency index; RI, retention index.
Invertebrate Biologyvol. x, no. x, xxx 2013
14 Kvist, Brugler, Goh, Giribet, & Siddall

ortholog derived from H. interrupta seems correctlyannotated.
Three main clans are presented in the ficolin tree(Fig. S17), the most interesting of which includesboth the archetypal sequence from Hi. medicinalisand the H. interrupta contig (c1477_1). Thebranches within this clan are also somewhat shorterthan the remaining branches, which suggests thatthe H. interrupta contig indeed represents a ficolinortholog.
There is little taxonomic structure in the bdellintree (Fig. S18), with sequences from the various taxaplacing in several different clans. Together with asequence from M. decora, the H. interrupta contig(c2073_1) places as the adjacent group to a clusterof Hirudo-derived sequences, including the arche-typal sequence. Judging from this placement, thecontig seems to represent a true bdellin ortholog.
The tree from the factor Xa inhibitor data set(Fig. S19) is almost completely unresolved, offering
no substantial information on the putative orthol-ogy determinations of the H. interrupta contigs(c5389_2, 5961_1, 6218_1, c14138_1 and c3664_1).Much like the manillase data set, it seems as thoughthere is either too low phylogenetic signal amongthe polymorphic sites or that there are too few poly-morphic sites in the data set. Regardless of this, itseems as though there is no evidence for paralogyor non-homology of each of the putative factor Xainhibitors derived from the H. interrupta transcrip-tome (therostasin, antistasin, guamerin, piguamerin,and ghilanten).
The tree generated from the leukocyte elastaseinhibitor data set (Fig. S20) is completely unre-solved, which is somewhat surprising as the align-ment of the H. interrupta-derived contig (c729_5)and the archetypal sequence from Hi. medicinalissuggested a non-affiliation between the sequences.Thus, it remains unclear whether or not contigc729_5 is a true leukocyte elastase ortholog.
Fig. 3. Unrooted strict consensus of two equally parsimonious trees recovered from analysis of the hirudin data set(see also Table 3). When appropriate, GenBank accession numbers follow taxon names. Branch lengths are drawn pro-portional to change.
Invertebrate Biologyvol. x, no. x, xxx 2013
Anticoagulant diversity in Haemadipsa interrupta 15

Discussion
Pyrosequencing of the salivary gland transcrip-tome of the terrestrial leech Haemadipsa interruptagenerated sequences that show strong resemblanceto a large diversity of proteins related to anticoagu-lation. Transcripts significantly matched 20 differentknown bioactive proteins, many of which employdifferent antagonistic pathways; 13 of these hadidentical or near identical best reciprocal BLASThits when targeted against GenBank, six matchedunannotated sequences best, and only one matcheda different annotation when reciprocally BLASTed.For several of the contigs, the best reciprocalBLAST hits matched at insignificant e-values, typi-cally orders of magnitude higher than the best hit
against the local data set. This can be attributed toat least two factors. First, some proteins in thelocally compiled database lack a nucleotide, EST,and/or protein counterpart in GenBank (thisimpelled our performing BLAST searches against avariety of databases). Second, as the size of theBLAST target database increases, the probability ofrecovering a match by chance alone increases,thereby increasing the e-values for each hit (Altschulet al. 1997); the same is true when the nucleotidelength of the query is low, and this may also lead toinflated e-values.
Subsequent to BLAST-based analyses, detailedexamination of alignments further supported ourhypothesis that cystatin, hirudin, LDTI, eglin c, c-type lectin, LAPP, manillase, His-rich protein, fico-lin, bdellin, Kazal-type serine protease inhibitors,antistasin, guamerin, piguamerin, and bufrudin arepossessed by H. interrupta, and that roughly half ofthese include a signal peptide region (Table 2). Theapparent lack of secretory signal peptides for someof the sequences can be set in context when review-ing previous work on leech salivary proteins. Ourimposed restriction on getorf to return only ORFsstarting with an initiation codon (Met) meant that itis possible that some of the three domains of thesignal peptide region (Von Heijne 1990) have notbeen recovered, such that SignalP does not predict asignal peptide. Indeed, both Min et al. (2010) andKvist et al. (2013) found several predicted secretorysignal peptides in sequences without an initial methi-onine. To this end, the non-sampled full length ofthe proteins investigated in the present study mightstill be endowed with a transient extension beyondthe methionine that would allow for the mature pro-tein to be translocated across the cell membrane.
The final security tier for our orthology inferenceswas to examine the phylogenetic trees derived fromthe different protein data sets. Although some ofthe trees offered little information due to their lowresolution, others verified the BLAST-based orthol-ogy. In particular, the trees generated from the hiru-din, LDTI, antiplatelet, and ficolin data sets solidifyour orthology determinations. It can be concludedfrom these analyses that our alignment-basedhypotheses of orthology for almost all of the bioac-tive proteins were correct, as they were also con-firmed by the phylogenetic analyses, except for thesequence best matching bufrudin, which is likely ahirudin ortholog.
Below we discuss the significance of matches toknown bioactive proteins and frame them in anevolutionary context by comparing the relativeplacement of their antagonistic pathways in a recent,
Fig. 4. Unrooted strict consensus of two equally parsimo-nious trees recovered from analysis of the LDTI data set(see also Table 3). When appropriate, GenBank accessionnumbers follow taxon names. Branch lengths are drawnproportional to change.
Invertebrate Biologyvol. x, no. x, xxx 2013
16 Kvist, Brugler, Goh, Giribet, & Siddall

comprehensive leech phylogeny (Min et al. 2010;Siddall et al. 2011).
The evolution of bloodfeeding in leeches
Although the evolution of hematophagy in leecheshas been studied extensively, hypotheses related tothis subject have been somewhat contradictorydepending on the type and amount of data includedin the study. Sawyer (1986) first suggested that themost recent common ancestor of extant leeches didnot employ hematophagy and that rhyncobdellid(proboscis-bearing) leeches and hirudiniformsevolved their bloodfeeding behavior independently.Siddall & Burreson (1995) reconstructed one of theearliest molecular phylogenies for leeches (based onmitochondrial COI) and found that their results didnot directly conflict with that of Sawyer (1986); theauthors noted that bloodfeeding seems to haveevolved independently at least twice in leeches—once along the branch leading to Rhynchobdellida,and another time along the branch leading to Hiru-diniformes (Siddall & Burreson 1995, 1996). Inde-pendent origins of this life-history strategy in leechesare further supported by differences in the anatomi-cal features through which bloodfeeding is carriedout. Rhynchobdellid leeches possess an eversibleproboscis that pierces the skin of the prey, whereasarhynchobdellid leeches are armed with three jawsand create an incision wound using a sawing motion(Siddall & Burreson 1996). Regardless of this funda-mental discrepancy, more contemporary and data-rich studies have converged on the opinion thatbloodfeeding is a plesiotypic strategy among leeches.For example, using nuclear 18S rRNA and mito-chondrial 12S rRNA, Trontelj et al. (1999) recov-ered the families Piscicolidae and Glossiphoniidae asevolving on two subsequent branches. The most par-simonious character optimization of bloodfeedingon that tree involved a hematophagous most recentcommon ancestor of leeches and three subsequentlosses in predaceous and liquidosomatophagous(those that feed on haemolymphal fluids) glossi-phoniids, erpobdelliforms and haemopid hirudini-forms. Two independent lines of evidence supportthe hypothesis that the ancestral leech was blood-feeding. First, Kvist et al. (2011a) showed that leechantiplatelet proteins, which inhibit von Willebrandfactor-mediated activation of subendothelial plate-lets, are present in the genome of the non-blood-feeding glossiphoniid leech Helobdella robustaSHANKLAND ET AL. 1992. As Glossiphoniidae is com-monly recovered at the base of the leech phylogeny(Siddall & Burreson 1998; Apakupakul et al. 1999;
Light & Siddall 1999; Siddall et al. 2001; but seeTrontelj et al. 1999), it is likely that He. robustapossesses anticoagulants simply by virtue of theirbeing passed down from a common ancestor. Sec-ond, Hirabayashi et al. (1998), Joo et al. (2009), andLee et al. (2010) each isolated and characterizedanticoagulation-related proteins from lumbricid cli-tellates (galactose-binding lectin from Lumbricusterrestris LINNAEUS 1758 and factor Xa inhibiting ei-senstasin from Eisenia andrei BOUCH�E 1972). Becauseleeches are derived oligochaetous annelids (Roussetet al. 2007; Zrzav�y et al. 2009; Struck et al. 2011;Kvist & Siddall 2013), it can be hypothesized thatthe archetypal leech possessed some “oligochaete”-inherited anticoagulants similar in composition tothose of modern leeches. The last point, as it relatesto the evolution of hematophagy in leeches, haslargely been neglected in the literature, yet may havea fundamental impact on our understanding of it.
Recent advancements in the generation of dataconcerning the presence of anticoagulants in variousleeches (Min et al. 2010; Kvist et al. 2011a, 2013;Siddall et al. 2011) have increased our understand-ing of the evolution of bloodfeeding, inasmuch asanticoagulants are directly related to bloodfeedingcapacity. These data also lend themselves well to atimely discussion on the presence and diversity ofanticoagulants across leech phylogeny. Figure 5illustrates our current understanding of leech phy-logeny (Min et al. 2010; Siddall et al. 2011), as wellas our contemporary knowledge of the distributionof the antagonistic pathways used by several bio-active proteins across the diversity of leeches. Thefigure bears witness to a growing interest in salivarytranscriptomics in the larger and perhaps more char-ismatic leech families; Glossiphoniidae, Macrobdelli-dae, Haemadispidae, and Hirudinidae have beenheavily studied when compared to other bloodfeed-ing families such as Piscicolidae, Praobdellidae, andXerobdellidae. Whereas the absence of bioactivesalivary proteins related to bloodfeeding in Ameri-cobdellidae, Gastrostomobdellidae, Salifidae, Haemo-pidae, Semiscolecidae, and to a large extent,Erpobdellidae may be reflective of their predatorylifestyle, their absence in the bloodfeeding Piscicoli-dae, Praobdellidae, and Xerobdellidae may simplyreflect a lack of data for these groups. One of themore general insights provided by the tree is thatmost of the pathways used by bioactive proteinspossessed by representatives of arhynchobdellidfamilies (all families except Glossiphoniidae andPiscicolidae) are also present in Glossiphoniidae, thelatter of which represents the earliest diverging line-age of leeches. Therefore, it seems likely that these
Invertebrate Biologyvol. x, no. x, xxx 2013
Anticoagulant diversity in Haemadipsa interrupta 17
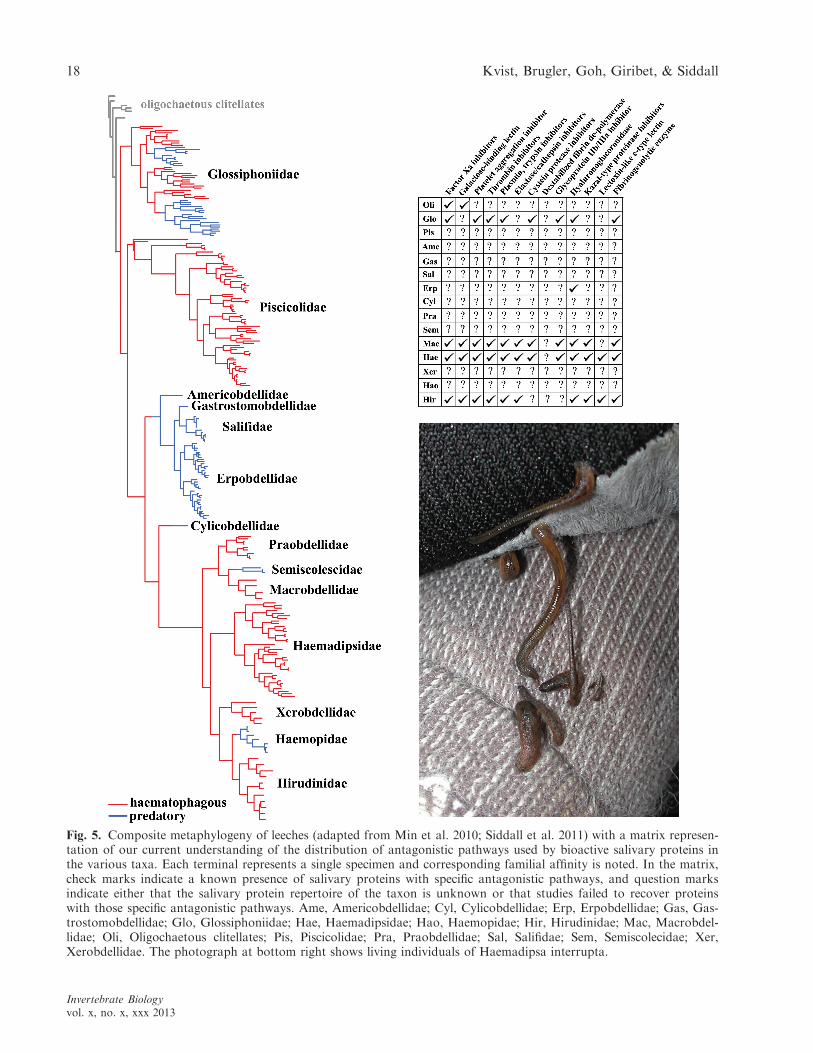
Fig. 5. Composite metaphylogeny of leeches (adapted from Min et al. 2010; Siddall et al. 2011) with a matrix represen-tation of our current understanding of the distribution of antagonistic pathways used by bioactive salivary proteins inthe various taxa. Each terminal represents a single specimen and corresponding familial affinity is noted. In the matrix,check marks indicate a known presence of salivary proteins with specific antagonistic pathways, and question marksindicate either that the salivary protein repertoire of the taxon is unknown or that studies failed to recover proteinswith those specific antagonistic pathways. Ame, Americobdellidae; Cyl, Cylicobdellidae; Erp, Erpobdellidae; Gas, Gas-trostomobdellidae; Glo, Glossiphoniidae; Hae, Haemadipsidae; Hao, Haemopidae; Hir, Hirudinidae; Mac, Macrobdel-lidae; Oli, Oligochaetous clitellates; Pis, Piscicolidae; Pra, Praobdellidae; Sal, Salifidae; Sem, Semiscolecidae; Xer,Xerobdellidae. The photograph at bottom right shows living individuals of Haemadipsa interrupta.
Invertebrate Biologyvol. x, no. x, xxx 2013
18 Kvist, Brugler, Goh, Giribet, & Siddall

proteins are present in arhynchobdellid taxa simplyby virtue of their presence in a common glossipho-niid or piscicolid ancestor, or both (Fig. 5). Insofaras some of these pathways refer to those used byknown and well-characterized anticoagulants, thisfurther supports the hypothesis of a bloodfeedingancestral leech.
Based on our current knowledge of bioactive pro-tein pathway distribution, it appears that someglycoprotein IIb/IIIa inhibitors may not be presentin arhyncobdellid taxa outside of Macrobdellidae.For example, prior studies have failed to obtain dec-orsin-like sequences from representatives of Hirudi-nidae (Kvist et al. 2013), and the present studyshows that the same holds true for a haemadipsidspecies. However, ornatin, a paralog of decorsin,has been isolated from at least one glossiphoniidleech, Placobdella ornata (VERRILL 1872) (Mazuret al. 1993), which could suggest its subsequent lossin Hirudinidae and Haemadipsidae and further evo-lution in Macrobdellidae.
Anticoagulant-associated proteins and bacterial hits
A number of ORFs significantly matchedsequences that were annotated as bacterial; the pre-dominant match was to alphaproteobacteria (Agro-bacterium spp., Rhizobium spp.; see Tables S1, S2).While this may be the result of external contamina-tion during dissection or RNA extraction, great carewas taken to avoid such contamination, and wehypothesize that these sequences originated from aninternal infection. Several families of leeches hostendosymbiotic alpha- and gammaproteobacteria(Kikuchi & Fukatsu 2002; Siddall et al. 2004, 2011;Perkins et al. 2005; Graf et al. 2006; Kvist et al.2011b). It has even been hypothesized that bacteriasupply leeches with nutrients and essential vitaminsthat are deficient in their diet. Vertebrate blood isnotoriously low in vitamin B and, in the presentstudy, we found several high-scoring hits against pro-teins that play a direct role in the biosynthesis ofriboflavin (vitamin B2). These findings provide yetanother line of evidence in support of a symbioticrelationship between leeches and bacteria that likelyis beneficial to the host (Kvist et al. 2011b). Priorstudies have indicated that leeches of the familyGlossiphoniidae enjoy symbiotic relations with myce-tome-associated gamma- and alphaproteobacteria,whereas hirudiniforms are most often associatedstrictly with gammaproteobacterial symbionts (Grafet al. 2006; Siddall et al. 2011). Fluorescent in situhybridization studies, using class-specific probes,would reveal whether or not the bacterial contigs
found here stem from an external contamination orinternal infection; such studies are a necessity beforeany robust conclusions can be drawn about the asso-ciation between haemadipsid leeches and alphaprote-obacterial endosymbionts.
As mentioned above, several other bioactive sali-vary proteins of interest were recovered in the H. in-terrupta salivary transcriptome. The majority ofBLAST annotations were to a variety of differentforms of anticoagulant-related proteins and all ofthem scored significant e-values. The specific struc-ture and function of these proteins remain unclear,but it seems likely that they are related to blood-feeding and anticoagulation. The most significanthits are briefly discussed below in the context oftheir potential contribution to anticoagulation.
At a range of e-values (the lowest scoring at1e�16), a total of 98 transcripts matched sequencesthat were annotated as various forms of cathepsin.Cathepsin is thought to play a role in inhibiting thecoagulation cascade by cleaving and thus inactivat-ing several coagulation factors, including fibrinogen.The function of cathepsin is similar to that of hiru-din, in that it cleaves, and thereby potentially modu-lates, the function of the thrombin receptor.
Disintegrins are small yet potent proteins thatinhibit platelet aggregation and integrin-dependentcell adhesion (McLane et al. 2004). Various disinte-grins have been isolated from snake venom wherethey also act as metalloproteinases (Kini & Evans1992; Jia et al. 1996). A number of contigs (n=24)from the H. interrupta salivary transcriptomematched various forms of disintegrins, the best hitscoring an e-value of 2e�22.
In addition, several transcripts matched sequencesannotated as a 29 kDa galactose-binding lectin (at1e�57 and above). As with disintegrins, galactose-binding lectins are frequently recovered from snakevenoms where they are involved in anticoagulationand antiplatelet aggregation (Xu et al. 1999). Inter-estingly, the best scoring transcript matched a galac-tose-binding lectin isolated from the lumbricidearthworm, L. terrestris, supporting the aforemen-tioned hypothesis that ancestors of the archetypalleech did indeed possess anticoagulants.
In response to parasitic infections, host mast cellsrelease a granule-associated enzyme to augmentwound healing and defend against pathogens (Rever-ter et al. 1998; Prussin & Metcalfe 2003). Leech car-boxypeptidase inhibitors (LCIs) are thought to blocka host’s defenses by inhibiting proteases of mast cells(Reverter et al. 1998). A sequence annotated as LCIwas retrieved as the best match for several transcriptsfrom H. interrupta, the best hit scoring at 6e�18.
Invertebrate Biologyvol. x, no. x, xxx 2013
Anticoagulant diversity in Haemadipsa interrupta 19

Anticoagulant repertoire versus diet
In contrast to the dietary preferences of manyfreshwater leeches (poikilotherm blood), terrestrialleeches are typically less fastidious in their preychoice. Considering their aptitude for locating suit-able sources of blood in tropical jungles, one cansurmise that these leeches must be able to feed onanything that crosses their path: mammals, reptiles,and amphibians alike (e.g., Fogden & Proctor 1985;Schnell et al. 2012). A strategy such as this wouldnecessitate a wide array of anticoagulants to coun-teract the wide variety of coagulation factors presentin different prey. Specimens of H. interrupta arenotoriously aggressive and feed on a variety of prey(Seagrave & Salsman 1946; Stammers 1950; Bhatia1975; Sawyer 1986; Kutschera et al. 2007). Prior tothis study, it was unknown whether the diversity ofanticoagulants in the repertoire of a single leech wascorrelated with dietary choices. The results pre-sented herein provide a first line of evidence insupport of a positive relationship between the diver-sity of anticoagulants and variety of prey items, asH. interrupta possesses one of the most inclusiveanticoagulant repertoires recovered in a single spe-cies of leech. This may suggest that the broad anti-coagulant repertoire of H. interrupta has evolved inresponse to the dietary strategy of the species, andthat other terrestrial leeches, particularly those withfewer dietary restrictions, will probably contain asimilarly broad repertoire. This is further supportedby the fact that H. interrupta secretes a wide varietyof proteins that may have anticoagulatory proper-ties, even beyond the obvious anticoagulants (seeabove). Min et al. (2010) and Kvist et al. (2013) didnot recover all of these proteins from EST librariesof the aquatic Hirudo verbana, Macrobdella decora,and Aliolimnatis fenestrata. However, differentialgene expression during various life-stages of theleeches may alter the potential of finding anticoagu-lants; therefore, this result should be viewed withappropriate caution. That is, it is possible that eachof the aforementioned leech species does produce awider array of anticoagulants than found in previ-ous studies but that they were not expressed at thetime of RNA extraction. In terms of bioactive pro-tein pathways, the repertoire of H. interrupta doesnot possess unique or significantly broader compo-nents when compared to aquatic leeches.
Conclusions
Through molecular characterization and phyloge-netic inference, we demonstrate that the terrestrial,
Southeast Asian haemadipsid leech H. interruptapossesses a diverse set of salivary proteins putativelyinvolved in anticoagulation. Furthermore, severaladditional bioactive proteins were found in the tran-scriptome, most of which are lacking completeinformation regarding structure and function inleeches. This suggests that leeches may contain moreanticoagulation factors than previously predicted.Forthcoming salivary transcriptomic studies shouldfocus on increasing taxon sampling in an effort tomap the presence of anticoagulants across the taxo-nomic diversity of leeches and thus gain a betterunderstanding of the evolution of salivary proteinsin leeches. Whereas H. interrupta possesses a diverseanticoagulant repertoire, the antagonistic pathwaysof putative anticoagulants do not fundamentally dif-fer from those of their aquatic counterparts. Asputative anticoagulants have also been found in oli-gochaetous clitellates, a complete picture of anti-coagulant evolution cannot be obtained withoutspecifically investigating and integrating data fromthese key taxa.
Acknowledgments. This research was funded by gener-ous support from the Wenner-Gren Foundations, theSixten Gemz�eus Foundation, Helge Ax:son Johnson’sFoundation, and the Olle Engkvist Byggm€astare Foun-dation to S.K. Fieldwork was funded by the Museumof Zoology, University of Malaya Grant 2011. Wethank the Willi Hennig Society for making TNT freelyavailable. Editor Michael Hart and two anonymousreviewers provided important comments that greatlyimproved the manuscript. The authors declare no con-flict of interest.
References
Adams SL 1988. The medicinal leech. A page from theannelids of internal medicine. Ann. Intern. Med. 109:399–405.
Altschul SF, Madden TL, Sch€affer AA, Zhang J, ZhangZ, Miller W, & Lipman DJ 1997. Gapped BLASTand PSI-BLAST: a new generation of protein data-base search programs. Nucleic Acids Res. 25: 3389–3402.
Apakupakul K, Siddall ME, & Burreson EM 1999.Higher level relationships of leeches (Annelida: Clitella-ta: Euhirudinea) based on morphology and genesequences. Mol. Phylogenet. Evol. 12: 350–359.
Bhatia ML 1975. Land leeches, their adaptation, andresponses to external stimuli. Zool. Pol. 25: 31–53.
Blankenship DT, Brankamp RG, Manley GD, & CardinAD 1990. Amino acid sequence of ghilanten: antico-agulant-antimetastatic principle of the South Americanleech, Haementeria ghilianii. Biochem. Biophys. Res.Commun. 166: 1384–1389.
Invertebrate Biologyvol. x, no. x, xxx 2013
20 Kvist, Brugler, Goh, Giribet, & Siddall

Borda E & Siddall ME 2004. Arhynchobdellida(Annelida: Oligochaeta: Hirudinida): phylogenetic rela-tionships and evolution. Mol. Phylogenet. Evol. 30:213–225.
———— 2011. Insights into the evolutionary history ofIndo-Pacific bloodfeeding terrestrial leeches (Hirudin-ida: Arhynchobdellida: Haemadipsidae). Invertebr.Syst. 24: 456–472.
Brankamp RG, Manley GG, Blankenship DT, BowlinTL, & Cardin AD 1991. Studies in the anticoagulant,antimetastatic and heparin-binding properties of ghilan-ten-related inhibitors. Blood Coagul. Fibrinolysis 2:161–166.
Chatrenet B & Chang J-Y 1993. The disulphide foldingpathway of hirudin elucidated by stop/go folding exper-iments. J. Biol. Chem. 268: 20988–20996.
Chevreux B, Wetter T, & Suhai S 1999. Genome sequenceassembly using trace signals and additional sequenceinformation. Sci. Biol. Proc. Ger. Conf. Bioinform. 99:45–56.
Chopin V, Salzet M, Baert J, Vandenbulcke F, SautierePE, Kerckaert JP, & Malecha J 2000. Therostasin, anovel clotting factor Xa inhibitor from the rhynchob-dellid leech, Theromyzon tessulatum. J. Biol. Chem.275: 32701–32707.
Conesa A, G€otz S, Garc�ıa-G�omez JM, Terol J, Tal�on M,& Robles M 2005. Blast2GO: a universal tool forannotation, visualization and analysis in functionalgenomics research. Bioinformatics 21: 3674–3676.
Cong J, Goll DE, Peterson AM, & Kapprell HP 1989.The role of autolysis in activity of the Ca2+-dependentproteinases (mu-calpain and m-calpain). J. Biol. Chem.264: 10096–10103.
Electricwala A, von Sicard NA, Sawyer RT, & AtkinsonT 1992. Biochemical characterization of a pancreaticelastase inhibitor from the leech Hirudinaria manillensis.J. Enzym. Inhib. 6: 293–302.
Elliott JM & Kutschera U 2011. Medicinal Leeches: his-torical use, ecology, genetics and conservation. Freshw.Rev. 4: 21–41.
Fang G, Bhardwaj N, Robilotto R, & Gerstein MB 2010.Getting started in gene orthology and functional analy-sis. PLoS Comput. Biol. 6: e1000703.
Fink E, Rehm H, Gippner C, Bode W, Eulitz M, Mach-leidt W, & Fritz H 1986. The primary structure ofbdellin B-3 from the leech Hirudo medicinalis. BdellinB-3 is a compact proteinase inhibitor of a ‘non-classi-cal’ Kazal type. It is present in the leech in a highmolecular mass form. Biol. Chem. Hoppe-Seyler 367:1235–1242.
Fogden SLC & Proctor J 1985. Notes on the feeding ofland leeches (Haemadipsa zeylanica Moore and H. pictaMoore) in Gunung Mulu National Park, Sarawak. Bio-tropica 17: 172–174.
Fritz H, Gebhardt M, Meister R, & Fink E 1971. Tryp-sin-plasmin inhibitors from leeches – isolation, aminoacid composition, inhibitory characteristics. In: Pro-ceedings of the International Research Conference on
Proteinase Inhibitors. Fritz H & Tschesche H, eds., pp.271–280. Walter de Gruyter, Berlin, Germany.
Ge F, Wang L-S, & Kim J 2005. The cobweb of liferevealed by genome-scale estimates of horizontal genetransfer. PLoS Biol. 3: 1709–1718.
Goloboff PA, Farris JS, & Nixon KC 2008. TNT, a freeprogram for phylogenetic analysis. Cladistics 24: 774–786.
Graf J, Kikuchi Y, & Rio RVM 2006. Leeches and theirmicrobiota: naturally simple symbiosis models. TrendsMicrobiol. 14: 365–371.
Gronwald W, Bomke J, Maurer T, Domogalla B, HuberF, Schumann F, Kremer W, Fink F, Rysiok T, FrechM, & Kalbitzer HR 2008. Structure of the leech proteinsaratin and characterization o fits binding to collagen.J. Mol. Biol. 381: 913–927.
Hale MC, McCormick CR, Jackson JR, & DeWoody JA2009. Next-generation pyrosequencing of gonad tran-scriptomes in the polyploid lake sturgeon (Acipenserfulvescens): the relative merits of normalization andrarefaction in gene discovery. BMC Genomics 10: 203.
Han JH, Law SW, Keller PM, Kniskern PJ, SilberklangM, Tung JS, Gasic TB, Gasic GJ, Friedman PA, &Ellis RW 1989. Cloning and expression of cDNAencoding antistasin, a leech-derived protein havinganti-coagulant and anti-metastatic properties. Gene 75:47–57.
Hirabayashi J, Dutta SK, & Kasai K-I 1998. Novelgalactose-binding proteins in Annelida. Characteriza-tion of 29-kDa tandem repeat-type lectins from theearthworm Lumbricus terrestris. J. Biol. Chem. 273:14450–14460.
Hu Z-L, Bao J, & Reecy JM 2008. CateGOrizer: a web-based program to batch analyze gene ontology classifi-cation categories. Online J. Bioinform. 9: 108–112.
Jia LG, Shimokawa K, Bjarnason JB, & Fox JW 1996.Snake venom metalloproteinases: structure, functionand relationship to the ADAMs family of proteins.Toxicon 34: 1269–1276.
Joo SS, Won TJ, Kim JS, Yoo YM, Tak ES, Park SY,Hwang KW, Park SC, & Lee DI 2009. Inhibition ofcoagulation activation and inflammation by a novelfactor Xa inhibitor synthesized from the earthwormEisenia andrei. Biol. Pharm. Bull. 32: 253–258.
Jung HI, Kim SI, Ha KS, Joe CO, & Kang KW 1995.Isolation and characterization of guamerin, a newhuman leukocyte elastase inhibitor from Hirudo nippo-nia. J. Biol. Chem. 270: 13879–13884.
Katoh K, Kuma K-I, Toh H, & Miyata T 2005. MAFFTversion 5: improvement in accuracy of multiplesequence alignment. Nucleic Acids Res. 33: 511–518.
Keegan HL, Toshioka S, & Suzuki H 1968. Blood suck-ing Asian leeches of families Hirudinidae and Haemad-ipsidae. Biomedical Reports of The 406 MedicalLaboratory No. 16. U.S. Army Medical Command,Japan.
Keller PM, Schultz LD, Condra C, Karczewski J, & Con-nolly TM 1992. An inhibitor of collagen-stimulatedplatelet activation from the salivary glands of the Hae-
Invertebrate Biologyvol. x, no. x, xxx 2013
Anticoagulant diversity in Haemadipsa interrupta 21

menteria officinalis leech. II. Cloning of the cDNA andexpression. J. Biol. Chem. 267: 6899–6904.
Kikuchi Y & Fukatsu T 2002. Endosymbiotic bacteria inthe esophageal organ of glossiphoniid leeches. Appl.Environ. Microbiol. 68: 4637–4641.
Kim DR & Kang KW 1998. Amino acid sequence ofpiguamerin, an antistasin-type protease inhibitor fromthe blood sucking leech Hirudo nipponia. Eur. J. Bio-chem. 254: 692–697.
Kini RM & Evans HJ 1992. Structural domains invenom proteins: evidence that metalloproteinasesand nonenzymatic platelet aggregation inhibitors(disintegrins) from snake venoms are derived by pro-teolysis from a common precursor. Toxicon 30: 265–293.
Knecht R, Seemuller U, Liersch M, Fritz H, Braun DG,& Chang JY 1983. Sequence determination of eglin Cusing combined microtechniques of amino acid analy-sis, peptide isolation, and automatic Edman degrada-tion. Anal. Biochem. 130: 65–71.
Kutschera U, Pfeiffer I, & Ebermann E 2007. The Euro-pean land leech: biology and DNA-based taxonomy ofa rare species that is threatened by climate warming.Naturwissenschaften 94: 967–974.
Kvist S & Siddall ME 2013. Phylogenomics of Annelidarevisited: a cladistics approach using genome-wideexpressed sequence tag data mining and examining theeffects of missing data. Cladistics 29: 435–448.
Kvist S, Sarkar IN, & Siddall ME 2011a. Genome-widesearch for leech antiplatelet proteins in the non-blood-feeding leech Helobdella robusta (Rhyncobdellida:Glossiphoniidae) reveals evidence of secreted antico-agulants. Invertebr. Biol. 130: 344–350.
Kvist S, Narechania A, Oceguera-Figueroa A, Fuks B, &Siddall ME 2011b. Phylogenomics of Reichenowia par-asitica, an alphaproteobacterial endosymbiont of thefreshwater leech Placobdella parasitica. PLoS ONE 6:e28192.
Kvist S, Min G-S, & Siddall ME 2013. Diversity andselective pressures of anticoagulants in three medicinalleeches (Hirudinida: Hirudinidae, Macrobdellidae).Ecol. Evol. 3: 918–933.
Lee MS, Tak ES, Park SK, Cho SJ, Hahn Y, Joo SS, LeeDI, Ahn CH, & Park SC 2010. Eisenstasin, new anti-stasin family inhibitor from the earthworm. Biologia65: 284–288.
Lefebvre C, Cocquerelle C, Vandenbulcke F, Hot D,Huot L, Lemoine Y, & Salzet M 2004. Transcriptomicanalysis in the leech Theromyzon tessulatum: involve-ment of cystatin B in innate immunity. Biochem. J.380: 617–625.
Light JE & Siddall ME 1999. Phylogeny of the leech familyGlossiphoniidae based on mitochondrial gene sequencesand morphological data. J. Parasitol. 85: 815–823.
Ludwig HC, Lottspeich F, Henschen A, Ladenstein R, &Bacher A 1987. Heavy riboflavin synthase of Bacillussubtilis. Primary structure of the beta subunit. J. Biol.Chem. 262: 1016–1021.
Markwardt F 1985. Pharmacology of hirudin: one hun-dred years after the first report of the anticoagulantagent in medicinal leeches. Biomed. Biochim. Acta 44:1007–1013.
Mason TA, McIlroy PJ, & Shain DH 2004. A cysteine-rich protein in the Theromyzon (Annelida: Hirudinea)cocoon membrane. FEBS Lett. 561: 167–172.
Mazur P, Dennis MS, Seymour JL, & Lazarus RA 1993.Expression, purification, and characterization of recom-binant ornatin E, a potent glycoprotein IIb-IIIa anta-gonist. Protein Expression Purif. 4: 282–289.
McLane MA, Sanchez EE, Wong A, Paquette-Straub C,& Perez JC 2004. Disintegrins. Curr. Drug TargetsCardiovasc. Haematol. Disord. 4: 327–355.
Min G-S, Sarkar IN, & Siddall ME 2010. Salivary tran-scriptome of the North American medicinal leech,Macrobdella decora. J. Parasitol. 96: 1211–1221.
Moore JP 1927. The segmentation (metamerism andannulation) of the Hirudinea; Arhynchobdellae. In:The Fauna of British India Hirudinea. Harding WA &Moore JP, eds., pp. 97–302. Taylor & Francis, Lon-don.
Nesemann H & Sharma S 1996. Contribution to theknowledge of the leeches of Nepal (Annelida: Hirudi-nea). Acta Zool. Acad. Sci. Hung. 42: 231–249.
———— 2001. Leeches of the suborder Hirudiniformes(Hirudinea: Haemopidae, Hirudinidae, Haemadipsidae)from the Ganga watershed (Nepal, India: Bihar). Ann.Naturhist. Mus. Wien (B Bot. Zool.) 103B: 77–88.
Nybelin O 1943. Nesophilaemon n. g. fur Philaemonskottsbergi L. Johansson aus den Juan FernandezInseln. Zool. Anz. 142: 249–250.
Perkins SL, Budinoff RB, & Siddall ME 2005. NewGammaproteobacteria associated with blood-feedingleeches and a broad phylogenetic analysis of leech en-dosymbionts. Appl. Environ. Microbiol. 71: 5219–5224.
Petersen TN, Brunak S, von Heijne G, & Nielsen H 2011.SignalP 4.0: discriminating signal peptides from trans-membrane regions. Nat. Methods 8: 785–786.
Phillips AJ & Siddall ME 2009. Poly-paraphyly of Hirudi-nidae: many lineages of medicinal leeches. BMC Evol.Biol. 9: 246.
Prussin C & Metcalfe DD 2003. IgE, mast cells, baso-phils, and eosinophils. J. Allergy Clin. Immunol. 111(Suppl. 2): S486–S494.
Reverter D, Vendrell J, Canals F, Horstmann J, Avil�esFX, Fritz H, & Sommerhoff CP 1998. A carboxypepti-dase inhibitor from the medical leech Hirudo medicinal-is isolation, sequence analysis, cDNA cloning,recombinant expression, and characterization. J. Biol.Chem. 273: 32927–32933.
Ribeiro JM 2003. A catalogue of Anopheles gambiae tran-scripts significantly more or less expressed following ablood meal. Insect Biochem. Mol. Biol. 33: 865–882.
Rice P, Longden I, & Bleasby A 2000. EMBOSS: theEuropean Molecular Biology Open Software Suite.Trends Genet. 16: 276–277.
Invertebrate Biologyvol. x, no. x, xxx 2013
22 Kvist, Brugler, Goh, Giribet, & Siddall

Richardson LR 1975. A contribution to the general zool-ogy of the land-leeches (Hirudinea: Haemadipsoideasuperfam. nov.). Acta Zool. Acad. Sci. Hung. 21: 119–152.
———— 1978. On the zoological nature of land leeches inthe Seychelles Islands, and a consequential revision ofthe status of land leeches in Madagascar (Hirudinea:Haemadipsoidea). Rev. Zool. Afr. 92: 837–866.
Richter G, Fischer M, Krieger C, Eberhardt S, L€uttgenH, Gerstenschl€ager I, & Bacher A 1997. Biosynthesis ofriboflavin: characterization of the bifunctional deami-nase-reductase of Escherichia coli and Bacillus subtilis.J. Bacteriol. 179: 2022–2028.
Rousset V, Pleijel F, Rouse GW, Ers�eus C, & Siddall ME2007. A molecular phylogeny of annelids. Cladistics 23:41–63.
Salzet M 2001. Anticoagulants and inhibitors of plateletaggregation derived from leeches. FEBS Lett. 429: 187–192.
Salzet M, Chopin V, Baert J, Matias I, & Malecha J2000. Theromin, a novel leech thrombin inhibitor. J.Biol. Chem. 275: 30774–30780.
Sawyer RT 1981. Why we need to save the medicinalleech. Oryx 16: 165–168.
———— 1986. Leech Biology and Behavior. ClarendonPress, Oxford, UK. 1065 pp.
Scacheri E, Nitti G, Valsasina B, Orsini G, Visco C, Fer-rera M, Sawyer RT, & Sarmientos P 1993. Novel hiru-din variants from the leech Hirudinaria manillensis.Amino acid sequence, cDNA cloning and genomicorganization. Eur. J. Biochem. 214: 295–304.
Scharf M, Engels J, & Tripier D 1989. Primary structuresof new “iso-hirudins”. FEBS Lett. 255: 105–110.
Schnell IB, Thomsen PF, Wilkinson N, Rasmussen M,Jensen LRD, Willerslev E, Berteksen MF, & GilbertMTP 2012. Screening mammal biodiversity using DNAfrom leeches. Curr. Biol. 22: R262–R263.
Seagrave GS & Salsman LV 1946. Burma surgeonreturns. Am. J. Nurs. 46: 724.
Seymour JL, Henzel WJ, Nevins B, Stults JT, & LazarusRA 1990. Decorins. A potent gycoprotein IIb-IIIaantagonist and platelet aggregation inhibitor from theleech Macrobdella decora. J. Biol. Chem. 265: 10143–10147.
Shain DH 2009. Annelids in Modern Biology. JohnWiley, Sons, New York, NY.
Siddall ME & Burreson EM 1995. Phylogeny of the Euhi-rudinea: independent evolution of blood feeding byleeches? Can. J. Zool. 73: 1048–1064.
———— 1996. Leeches (Oligochaeta?: Euhirudinea), theirphylogeny and the evolution of life history strategies.Hydrobiologia 334: 277–285.
———— 1998. Phylogeny of leeches (Hirudinea) based onmitochondrial cytochrome c oxidase subunit I. Mol.Phylogenet. Evol. 9: 156–162.
Siddall ME, Apakupakul K, Burreson EM, Coates KA,Ers�eus C, Gelder SR, K€allersj€o M, & Trapido-Rosen-thal H 2001. Validating Livanow: molecular data agreethat leeches, branchiobdellidans, and Acanthobdella
peledina form a monophyletic group of oligochaetes.Mol. Phylogenet. Evol. 21: 346–351.
Siddall ME, Perkins SL, & Desser SS 2004. Leech myce-tome endosymbionts are a new lineage of alphaproteo-bacteria related to the Rhizobiaceae. Mol. Phylogenet.Evol. 30: 178–186.
Siddall ME, Trontelj P, Utevsky SY, Nkamany M, &Macdonald KS III 2007. Diverse molecular data dem-onstrate that commercially available medicinal leechesare not Hirudo medicinalis. Proc. R. Soc. Lond. B 274:1481–1487.
Siddall ME, Min G-S, Fontanella FM, Phillips AJ, &Watson SC 2011. Bacterial symbiont and salivary pep-tide evolution in the context of leech phylogeny. Parasi-tology 138: 1815–1827.
S€ollner C, Mentele R, Eckerskorn C, Fritz H, & Sommer-hoff CP 1994. Isolation and characterization of hirusta-sin, an antistasin-type serine-protease inhibitor fromthe medicinal leech Hirudo medicinalis. Eur. J. Bio-chem. 219: 937–943.
Sommerhoff CP, S€ollner C, Mentele R, Piechottka GP,Auerswald EA, & Fritz H 1994. A Kazal-type inhibitorof human mast cell tryptase: isolation from the medicalleech Hirudo medicinalis, characterization, and sequenceanalysis. Biol. Chem. Hoppe-Seyler 375: 685–694.
Stammers FMG 1950. Observations on the behavior ofland-leeches (genus Haemadipsa). Parasitology 40: 237–246.
Stange R, Moser C, Hopfenmueller W, Mansmann U,Buehring M, & Uehleke B 2012. Randomised con-trolled trial with medical leeches for osteoarthritis ofthe knee. Complement Ther. Med. 20: 1–7.
Strube KH, Kroger B, Bialojan S, Otte M, & Dodt J1993. Isolation, sequence analysis, and cloning ofhaemadin. An anticoagulant peptide from the Indianleech. J. Biol. Chem. 268: 8590–8595.
Struck TH, Paul C, Hill N, Hartmann S, H€osel C, KubeM, Lieb B, Meyer A, Tiedemann R, Purschke G, &Bleidorn C 2011. Phylogenomic analyses unravel anne-lid evolution. Nature 471: 95–98.
Swadesh JK, Huang IY, & Budzynski AZ 1990. Purifica-tion and characterization of hementin, a fibrinogenoly-tic protease from the leech Haementeria ghilianii. J.Chromatogr. 502: 359–369.
Trontelj P & Utevsky SY 2005. Celebrity with a neglectedtaxonomy: molecular systematics of the medicinal leech(genus Hirudo). Mol. Phylogenet. Evol. 34: 616–624.
Trontelj P, Sket B, & Steinbr€uck G 1999. Molecular phy-logeny of leeches: congruence of nuclear and mitochon-drial rDNA data sets and the origin of bloodsucking.J. Zool. Syst. Evol. Res. 37: 141–147.
Tulinsky A & Qiu X 1993. Active site and exosite bindingof alpha-thrombin. Blood Coagul. Fibrinolysis 4: 305–312.
Vankhede GN, Gomase VS, Deshmukh SV, & ChikhaleNJ 2005. Prediction of antigenic epitope of hirudinfrom Poecilobdella viridis. J. Comp. Toxicol. Physiol. 2:110–118.
Invertebrate Biologyvol. x, no. x, xxx 2013
Anticoagulant diversity in Haemadipsa interrupta 23

Von Heijne G 1990. The signal peptide. J. Membr. Biol.115: 195–201.
Waterhouse AM, Procter JB, Martin DMA, Clamp M, &Barton GJ 2009. Jalview version 2 – a multiplesequence alignment editor and analysis workbench.Bioinformatics 25: 1189–1191.
Wells MD, Manktelow RT, Boyd JB, & Bowen V 1993.The medical leech: an old treatment revisited. Micro-surgery 14: 183–186.
Whitaker IS, Izadi D, Oliver DW, Monteath GM, & But-ler PE 2004. Hirudo Medicinalis and the plastic sur-geon. Br. J. Plast. Surg. 57: 348–353.
Wilkinson M, McInerney JO, Hirt RP, Foster PG, &Embley TM 2007. Of clades and clans: terms for phylo-genetic relationships in unrooted trees. Trends Ecol.Evol. 22: 114–115.
Xu Q, Wu X-F, Xia Q-C, & Wang K-Y 1999. Cloning ofa galactose-binding lectin from the venom of Tri-meresurus stejnegeri. Biochem. J. 341: 733–737.
Zavalova L, Lukyanov S, Baskova I, Snezhkov E, Ako-pov S, Berezhnoy S, Bogdanova E, Basova E, & Sverd-lov ED 1996. Genes from the medicinal leech (Hirudomedicinalis) coding for unusual enzymes that specifi-cally cleave endo-epsilon (gamma-Glu)-Lys isopeptidebonds and help dissolve blood clots. Mol. Gen. Genet.253: 20–25.
Zrzav�y J, �R�ıha P, Pi�alek L, & Janou�skovec J 2009. Phylog-eny of Annelida (Lophotrochozoa): total-evidence analy-sis of morphology and six genes. BMC Evol. Biol. 9: 189.
Supporting information
Additional Supporting information may be found in theonline version of this article:
Fig. S1. MAFFT-based amino acid alignment of putativeeglin-c from Haemadipsa interrupta (HT8UG4B01_c10041_1) against the known sequence of the bioactiveprotein (0905140A) from an unknown hirudinid. Blueshadings represent conservation of residues between thesequences.Fig. S2. MAFFT-based amino acid alignment of putativelectoxin-like c-type lectin from Haemadipsa interrupta(HT8UG4B01_c14817_4) against a sequence of the puta-tive bioactive protein (Macrobdella2B10strict38) fromMacrobdella decora. Red boxes denote the predicted sig-nal peptide region, green boxes denote conserved cyste-ines, and blue shadings represent conservation of residuesbetween the sequences.Fig. S3. MAFFT-based amino acid alignment of putativesaratin and LAPP from Haemadipsa interrupta (HT8UG-4B01_c2724_2 and HT8UG4B01_rep_c4341_1, respec-tively) against the known sequences of the anticoagulants(2K13_X and Q01747) from Haementeria officinalis DE
FILIPPI 1849. Red boxes denote the predicted signal pep-tide region, green boxes denote conserved cysteines, andblue shadings represent conservation of residues betweenthe sequences.
Fig. S4. MAFFT-based amino acid alignment of putativemanillase from Haemadipsa interrupta (HT8UG4B01_c1130_2) against the known sequence of the bioactiveprotein (Patent no. 2006 US 7.049.124 B1) from Hirudina-ria manillensis (LESSON 1842). Blue shadings representconservation of residues between the sequences.Fig. S5. MAFFT-based amino acid alignment of putativeHis-rich protein from Haemadipsa interrupta (HT8UG4-B01_rep_c3900_1) against a sequence of the putative bio-active protein (Macrobdella14F06strict7) fromMacrobdella decora. Red boxes denote the predicted signalpeptide region, green boxes denote conserved cysteines,and blue shadings represent conservation of residuesbetween the sequences.Fig. S6. MAFFT-based amino acid alignment of putativeficolin from Haemadipsa interrupta (HT8UG4B01_c1477_1) against a sequence of the putative bioactive pro-tein (Macrobdella10A03strict686) from Macrobdelladecora. Green boxes denote conserved cysteines and blueshadings represent conservation of residues between thesequences.Fig. S7. MAFFT-based amino acid alignment of putativebdellin from Haemadipsa interrupta (HT8UG4B01_c2073_1) against the known sequence of the anticoagulant(P09865) from Hirudo medicinalis. Red boxes denote thepredicted signal peptide region, green boxes denoteconserved cysteines, and blue shadings representconservation of residues between the sequences.Fig. S8. MAFFT-based amino acid alignment of putativeKazal-type serine proteinase inhibitor from Haemadipsainterrupta (HT8UG4B01_rep_c4204_4) against a sequenceof the putative bioactive protein (Macrobdella14D11strict26) from Macrobdella decora. Green boxesdenote conserved cysteines and blue shadings representconservation of residues between the sequences.Fig. S9. MAFFT-based amino acid alignment of putativefactor Xa inhibitors (HT8UG4B01_c14138_1 guamerin,HT8UG4B01_rep_c6218_1 piguamerin, HT8UG4B01_rep_c5961_1 antistasin, HT8UG4B01_c3664_1 ghilantenand HT8UG4B01_rep_c5389_2 therostasin) from Hae-madipsa interrupta against the known sequences of theanticoagulants (AAD09442 guamerin, P81499 piguamerin,AAA29193 antistasin, P16242 ghilanten, and Q9NBW4therostasin) from Hirudo nipponia (guamerin and pigua-merin), Haementeria officinalis, Haementeria ghilianii DE
FILIPPI 1849, and Theromyzon tessulatum, respectively.Red boxes denote the predicted signal peptide region,green boxes denote conserved cysteines, and intensity ofblue shadings corresponds to BLOSUM62 conservation ofresidues.Fig. S10. MAFFT-based amino acid alignment of putativebufrudin from Haemadipsa interrupta (HT8UG4B01_c1987_1) against the known sequence of the anticoagulant(P81492) from Poecilobdella manillensis (LESSON 1842). Redboxes denote the predicted signal peptide region, greenboxes denote conserved cysteines, and blue shadings repre-sent conservation of residues between the sequences.
Invertebrate Biologyvol. x, no. x, xxx 2013
24 Kvist, Brugler, Goh, Giribet, & Siddall

Fig. S11. MAFFT-based amino acid alignment of putativetheromin from Haemadipsa interrupta (HT8UG4B01_c3189_1) against the known sequence of the anticoagulant(P82354) from Theromyzon tessulatum. Blue shadings rep-resent conservation of residues between the sequences.Fig. S12. MAFFT-based amino acid alignment of putativeleukocyte elastase inhibitor from Haemadipsa interrupta(HT8UG4B01_c729_5) against a sequence of the putativebioactive protein (MacrobdellaElastaseInhib) from Mac-robdella decora. Red boxes denote the predicted signal pep-tide region, green boxes denote conserved cysteines, andblue shadings represent conservation of residues betweenthe sequences.Fig. S13. Unrooted strict consensus of two equally parsi-monious trees recovered from the analysis of the eglin cdata set (see also Table 3). When appropriate, GenBankaccession numbers follow taxon names. Branch lengths areproportional to change.Fig. S14. Unrooted single most parsimonious tree recov-ered from the analysis of the c-type lectin data set (see alsoTable 3). When appropriate, GenBank accession numbersfollow taxon names. Branch lengths are proportional tochange.Fig. S15. Unrooted strict consensus of six equally parsimo-nious trees recovered from the analysis of the antiplatelet(saratin and LAPP) data set (see also Table 3). Whenappropriate, GenBank accession numbers follow taxonnames. Branch lengths are proportional to change.
Fig. S16. Unrooted strict consensus of 19 equally parsimo-nious trees recovered from the analysis of the manillasedata set (see also Table 3). When appropriate, GenBankaccession numbers follow taxon names. Branch lengths areproportional to change.Fig. S17. Unrooted single most parsimonious tree recov-ered from the analysis of the ficolin data set (see alsoTable 3). Branch lengths are proportional to change.Fig. S18. Unrooted single most parsimonious tree recov-ered from the analysis of the bdellin data set (see alsoTable 3). When appropriate, GenBank accession numbersfollow taxon names. Branch lengths are proportional tochange.Fig. S19. Unrooted strict consensus of 28 equally parsi-monious trees recovered from the analysis of the factorXa inhibitor data set (antistasin, therostasin, guamerin,piguamerin, ghilanten; see also Table 3). When appropri-ate, GenBank accession numbers follow taxon names.Branch lengths are proportional to change.Fig. S20. Unrooted strict consensus of three equally par-simonious trees recovered from the analysis of the leuko-cyte elastase inhibitor data set (see also Table 3). Whenappropriate, GenBank accession numbers follow taxonnames. Branch lengths are proportional to change.Table S1. Results from the BLASTx searches againstNCBI, using the contigs as queries.Table S2. Results from the BLASTn searches againstNCBI, using the contigs as queries.
Invertebrate Biologyvol. x, no. x, xxx 2013
Anticoagulant diversity in Haemadipsa interrupta 25





























