Embed Size (px)
Citation preview

Proton�/metal exchange processes in synthetic and naturalpolyelectrolyte solution systems
Julio C. Benegas a, Rodolfo D. Porasso a, Marc A.G.T. van den Hoop b,*a Department of Physics �/ IMASL, National University of San Luis, 5700 San Luis, Argentina
b National Institute of Public Health and the Environment, P.O. Box 1, 3720 BA Bilthoven, The Netherlands
Received 11 November 2002; accepted 12 June 2003
Abstract
Proton�/metal exchange processes that take place in polyelectrolyte solutions have been studied using previously
reported potentiometric titration data for polyacrylic acid (PAA) and humic acid (HA) systems obtained at different
degrees of polymer deprotonation, for various metals and at different added metal concentrations. It is shown that the
extent of the exchange process, quantified by the parameter nexch, strongly depends on the way it has been determined,
i.e. at constant polyelectrolyte characteristics or at constant pH, being significantly larger under constant pH
conditions. In addition, it is found that the exchange process depends upon polyelectrolyte structural charge density, the
degree of ionization, the type of metal and the total metal concentration in solution. For the experiments reported here,
in general the larger exchange coefficients are found for PAA at the lowest reported degree of ionization (a�/0.2) and
low pH values, while the lower values correspond to HA at the highest studied degree of ionization (a�/0.6) and high
pH values. The ability of the heavy metal ions to induce H� release increase following the order Ca:/Cd:/NiB/Pb5/
Cu. The present analysis shows that counterion condensation theory of linear polyelectrolytes appropriately describes
the experimental data, provided chemical bonding interactions are not too strong to disrupt the general polyelectrolytic
behavior.
# 2003 Elsevier B.V. All rights reserved.
Keywords: Proton�/metal exchange processes; Natural polyelectrolyte solution; Polyelectrolyte characteristics
1. Introduction
Knowledge of the interaction processes of
(heavy and proton) metal ions with macromole-
cular ligands is of importance for the understand-
ing of their physicochemical behavior in
environmental and biological systems. In natural
aquatic systems the speciation of metals, i.e. their
distribution over different physicochemical forms,
is highly dependent on the charge characteristics of
the complexing macromolecules. In general,
macromolecules of natural origin consist of var-
ious different functional groups, like carboxylic
and phenolic ones [1�/4], which can be charged due
to dissociation. Ionization characteristics of these
* Corresponding author. Tel.: �/31-30-274-4013; fax: �/31-
30-274-4411.
E-mail address: [email protected] (M.A.G.T. van
den Hoop).
Colloids and Surfaces A: Physicochem. Eng. Aspects 224 (2003) 107�/117
www.elsevier.com/locate/colsurfa
0927-7757/03/$ - see front matter # 2003 Elsevier B.V. All rights reserved.
doi:10.1016/S0927-7757(03)00327-3

macromolecules have been studied quite exten-sively and are mainly controlled by the free
protons in the system.
For weak polyacids, like polyacrylic acid (PAA)
and humic acids (HAs), these charge character-
istics can be controlled or adjusted by acid/base
titrations of the polyacids in solution. Although
the experimental approach is rather simple, the
modeling and interpretation of the data can berather involved, especially for the case of hetero-
geneous polyelectrolytes. In this case, the analysis
of the concentration and distribution of ionizable
polymeric sites, as well as the concentration of all
species of small ions in solution and the interaction
processes between all charged species with the
functional group(s) of the polyelectrolyte should
be taken into account. Metal ion interactions withthe complexing ligands should therefore have to
compete with and/or modify the proton binding by
the (different) functional groups.
In the literature, two different experimental
approaches are found to study H�/Me2� ex-
change processes in polyelectrolyte systems: ana-
lysis at constant degree of ionization a or at
constant pH [5�/7]. In both cases, the startingpoint is the reference solution containing a par-
tially ionized polyelectrolyte with no (heavy) metal
added. Titrations at constant a are obtained by
just adding, in successive steps, a solution contain-
ing the metal ion. The amount of released H� is
given in these experiments by the change in pH. In
the experiments at constant pH, after adding the
solution containing the metal ion, hydroxidetitrant is added to return the solution to the
original pH. In this case, the amount of released
H� is given by the amount of hydroxide added.
Tipping et al. [6] observed a decrease in the
experimentally obtained H�/Cu2� molar ex-
change ratio of Sable HA with increasing pH
values at constant ionic strength from 1.38 down
to 1.12 in the pH range 4�/5. Kinniburgh et al. [7]have analyzed metal purified peat HA titration
curves at constant pH for Ca, Cd, Pb, Cu and Al,
and found that proton�/metal molar exchange
ratios varied strongly with the metal, the pH of
the solution and the free metal ion concentration.
Different models have been presented in the
literature to deal with these processes [2,7�/13]. In
the present case, the interactions and their con-sequences in polyelectrolytic solutions are ana-
lyzed within the framework of a recent extension
of counterion condensation (CC) theory [8,9] of
linear polyelectrolytes that includes, besides purely
polyelectrolytic interactions [12], chemical binding
of counterions to the polyelectrolyte [13]. In terms
of polyelectrolyte/counterion association of inter-
est here, polyelectrolytic interactions lead to delo-calized trapping of counterions in the immediate
vicinity of the polymer (CC), while the latter
results in chemical association to specific polymer
binding sites. The model has been shown to be able
to predict very well the outcome of various
experiments under different solution conditions,
for a number a heavy metal/polyelectrolyte sys-
tems, including metals like Cd, Cu, Zn, Pb and Niin solution with monoprotic polyelectrolytes like
PAA and polymethacrylic acid and multifunc-
tional polyelectrolytes like HA [14�/17].
In the present paper, experimentally obtained
potentiometric titration data of the high charged
synthetic polyelectrolyte PAA and the low charged
natural polyelectrolyte HA is analyzed with re-
spect to the H�/Me2� molar exchange process. Anumber of different experimental data are ana-
lyzed including different polyelectrolytic charge
densities and different metals at various concen-
trations using the theoretical determination of
both the changes in pKa due to ionization of the
polyelectrolyte involved and the resulting metal
speciation in solution. In the modeling section, the
theoretical procedure for determining the proton/(heavy) metal exchange that occurs upon binding
of the metal ions to a partially ionized polyelec-
trolyte will be described in some detail. In addi-
tion, it will be shown that appropriate description
of the applied procedure for obtaining the H�/
Me2� molar exchange coefficient is of great
importance for the interpretation of the data.
2. Modeling
In the literature, the interactions between the
different counterion species and the polyelectrolyte
in solution have been considered from various
theoretical frameworks, including CC theory of
J.C. Benegas et al. / Colloids and Surfaces A: Physicochem. Eng. Aspects 224 (2003) 107�/117108

linear polyelectrolytes [8,9], solution of the
Poissson�/Boltzmann equation [10,11] and differ-
ent types of parametric models that include some
of relevant interactions [2,7]. These models have
varying degrees of convenience and shortcomings
when dealing with the different experimental
approaches used in these very complex polyelec-
trolytic systems (see e.g. [18]). Our group has been
involved in developing extensions of the CC
theory, with the aim of including important
aspects of polyelectrolytes in natural systems
such as counterion competition [12], chemical
binding [13,14] and heterogeneity of functional
groups [15]. This extended model presents the
following convenient characteristics:
i) polyelectrolytic interactions, which include the
interactions between all ionic species and theeffect of the solvent, are taken into account
from first principles;
ii) different thermodynamic states for the small
ions (free in solution, restrained to a ‘con-
densation’ volume in the immediate neighbor-
hood of the polyelectrolyte and chemically
bound to particular binding sites) that are
crucial for counterion speciation are built intoor are a direct consequence of the model;
iii) entropic effects are explicitly included;
iv) the possibility of dealing with linear polyelec-
trolytes of two or more functional groups is
considered in the model;
v) the minimization of the total (excess) free
energy of the solution system determines,
among other properties, the population dis-tribution of small ions in the different thermo-
dynamic states, i.e. their speciation;
vi) the proper derivation of the total (excess) free
energy of the system allows for the straightfor-
ward determination of the functional form of
the thermodynamic variable of interest.
Within this general framework, the present
paper deals with the central problem of counterion
exchange processes, which are driven by the inter-
play of all relevant interactions. When a salt
containing a (heavy) metal is added to a solution
containing a weak polyelectrolyte, some of the
resulting metal ions are usually bound to binding
sites on the polyelectrolyte. Consequently, protonsare released from the polymer resulting in a H�/
Me2� ion exchange process that may strongly
modify the polyelectrolytic characteristics of the
solution. In the present modeling, it is assumed
that the binding sites are the ionizable groups on
the polymer. Competition with H� ions for these
binding sites and/or the shift in the pK0 upon
heavy metal binding is expected to drive the H�/Me2� exchange process of the polyelectrolyte
neighboring undissociated functional groups.
What is determined, both experimentally and
theoretically, is the change in pH that occurs
when the divalent metal salt is added to the
reference solution. Theoretically, pH is obtained
from the theoretically calculated pKa value by the
equation,
pH�pKa� log
�1 � a
a
�(1)
where pKa is calculated according to Eq. (A8) or
Eq. (A10) and Eq. (A9), respectively, in Appendix
A, and a is the degree of ionization of thepolyelectrolyte.
3. Data analysis
In the present paper, we focus on the following
experimental conditions: (i) the characteristic
properties of the polyelectrolyte itself are kept
constant by applying the experimental procedure
at constant degree of ionization of the polyacid,and (ii) the solution composition is kept constant
with respect to one of the counterions under
investigation (protons). Under the latter condi-
tions, changes of the properties of the studied
polyelectrolyte may be induced, affecting as well
the distribution of monovalent and divalent coun-
terions over free and bound states. To illustrate the
different approaches, in Fig. 1 (calculated) poten-tiometric titration curves are plotted for a weak
heterogeneous polyacid in the absence (upper solid
line) and in the presence (lower dotted line) of a
given concentration of divalent metal ions. These
potentiometric titration curves show the typical
monotonic increase over almost the whole range of
J.C. Benegas et al. / Colloids and Surfaces A: Physicochem. Eng. Aspects 224 (2003) 107�/117 109
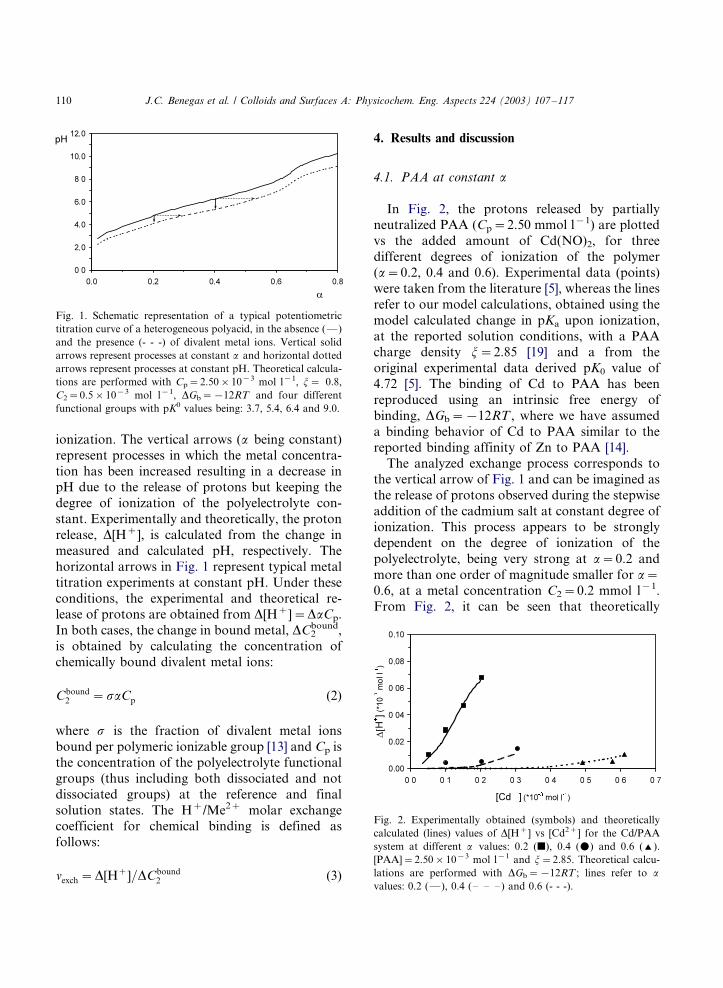
ionization. The vertical arrows (a being constant)
represent processes in which the metal concentra-
tion has been increased resulting in a decrease inpH due to the release of protons but keeping the
degree of ionization of the polyelectrolyte con-
stant. Experimentally and theoretically, the proton
release, D[H�], is calculated from the change in
measured and calculated pH, respectively. The
horizontal arrows in Fig. 1 represent typical metal
titration experiments at constant pH. Under these
conditions, the experimental and theoretical re-lease of protons are obtained from D[H�]�/DaCp.
In both cases, the change in bound metal, DC2bound,
is obtained by calculating the concentration of
chemically bound divalent metal ions:
Cbound2 �saCp (2)
where s is the fraction of divalent metal ions
bound per polymeric ionizable group [13] and Cp is
the concentration of the polyelectrolyte functional
groups (thus including both dissociated and not
dissociated groups) at the reference and final
solution states. The H�/Me2� molar exchangecoefficient for chemical binding is defined as
follows:
nexch�D[H�]=DCbound2 (3)
4. Results and discussion
4.1. PAA at constant a
In Fig. 2, the protons released by partiallyneutralized PAA (Cp�/2.50 mmol l�1) are plotted
vs the added amount of Cd(NO)2, for three
different degrees of ionization of the polymer
(a�/0.2, 0.4 and 0.6). Experimental data (points)
were taken from the literature [5], whereas the lines
refer to our model calculations, obtained using the
model calculated change in pKa upon ionization,
at the reported solution conditions, with a PAAcharge density j�/2.85 [19] and a from the
original experimental data derived pK0 value of
4.72 [5]. The binding of Cd to PAA has been
reproduced using an intrinsic free energy of
binding, DGb�/�/12RT , where we have assumed
a binding behavior of Cd to PAA similar to the
reported binding affinity of Zn to PAA [14].
The analyzed exchange process corresponds tothe vertical arrow of Fig. 1 and can be imagined as
the release of protons observed during the stepwise
addition of the cadmium salt at constant degree of
ionization. This process appears to be strongly
dependent on the degree of ionization of the
polyelectrolyte, being very strong at a�/0.2 and
more than one order of magnitude smaller for a�/
0.6, at a metal concentration C2�/0.2 mmol l�1.From Fig. 2, it can be seen that theoretically
Fig. 1. Schematic representation of a typical potentiometric
titration curve of a heterogeneous polyacid, in the absence (*/)
and the presence (- - -) of divalent metal ions. Vertical solid
arrows represent processes at constant a and horizontal dotted
arrows represent processes at constant pH. Theoretical calcula-
tions are performed with Cp�/2.50�/10�3 mol l�1, j�/ 0.8,
C2�/0.5�/10�3 mol l�1, DGb�/�/12RT and four different
functional groups with pK0 values being: 3.7, 5.4, 6.4 and 9.0.
Fig. 2. Experimentally obtained (symbols) and theoretically
calculated (lines) values of D[H�] vs [Cd2�] for the Cd/PAA
system at different a values: 0.2 (j), 0.4 (m) and 0.6 (').
[PAA]�/2.50�/10�3 mol l�1 and j�/2.85. Theoretical calcu-
lations are performed with DGb�/�/12RT ; lines refer to a
values: 0.2 (*/), 0.4 (�/ �/ �/) and 0.6 (- - -).
J.C. Benegas et al. / Colloids and Surfaces A: Physicochem. Eng. Aspects 224 (2003) 107�/117110
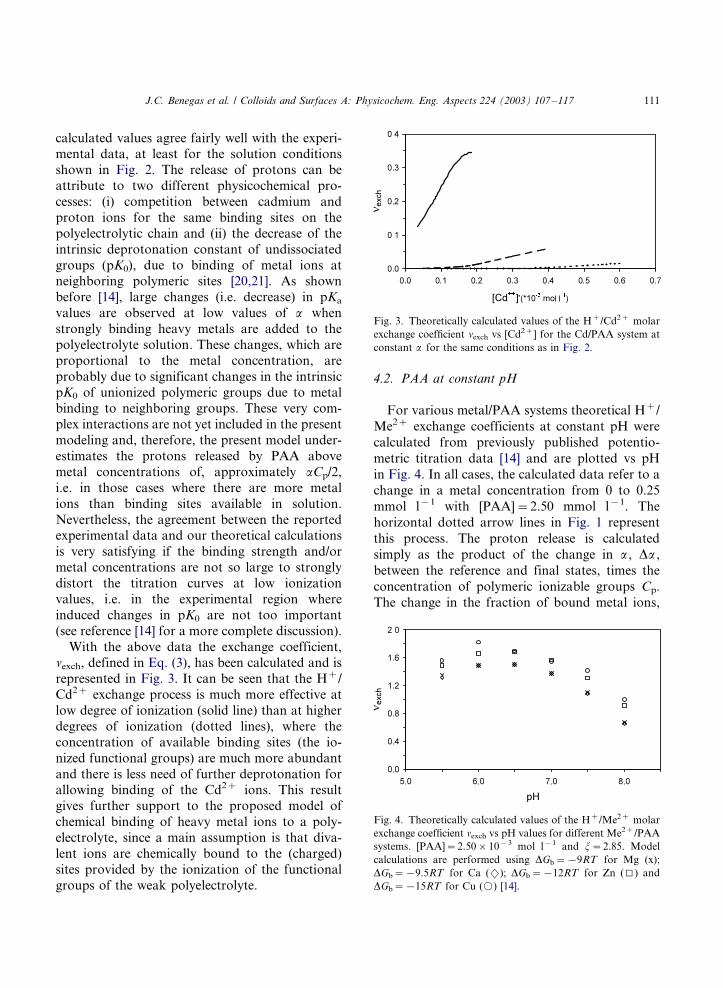
calculated values agree fairly well with the experi-mental data, at least for the solution conditions
shown in Fig. 2. The release of protons can be
attribute to two different physicochemical pro-
cesses: (i) competition between cadmium and
proton ions for the same binding sites on the
polyelectrolytic chain and (ii) the decrease of the
intrinsic deprotonation constant of undissociated
groups (pK0), due to binding of metal ions atneighboring polymeric sites [20,21]. As shown
before [14], large changes (i.e. decrease) in pKa
values are observed at low values of a when
strongly binding heavy metals are added to the
polyelectrolyte solution. These changes, which are
proportional to the metal concentration, are
probably due to significant changes in the intrinsic
pK0 of unionized polymeric groups due to metalbinding to neighboring groups. These very com-
plex interactions are not yet included in the present
modeling and, therefore, the present model under-
estimates the protons released by PAA above
metal concentrations of, approximately aCp/2,
i.e. in those cases where there are more metal
ions than binding sites available in solution.
Nevertheless, the agreement between the reportedexperimental data and our theoretical calculations
is very satisfying if the binding strength and/or
metal concentrations are not so large to strongly
distort the titration curves at low ionization
values, i.e. in the experimental region where
induced changes in pK0 are not too important
(see reference [14] for a more complete discussion).
With the above data the exchange coefficient,nexch, defined in Eq. (3), has been calculated and is
represented in Fig. 3. It can be seen that the H�/
Cd2� exchange process is much more effective at
low degree of ionization (solid line) than at higher
degrees of ionization (dotted lines), where the
concentration of available binding sites (the io-
nized functional groups) are much more abundant
and there is less need of further deprotonation forallowing binding of the Cd2� ions. This result
gives further support to the proposed model of
chemical binding of heavy metal ions to a poly-
electrolyte, since a main assumption is that diva-
lent ions are chemically bound to the (charged)
sites provided by the ionization of the functional
groups of the weak polyelectrolyte.
4.2. PAA at constant pH
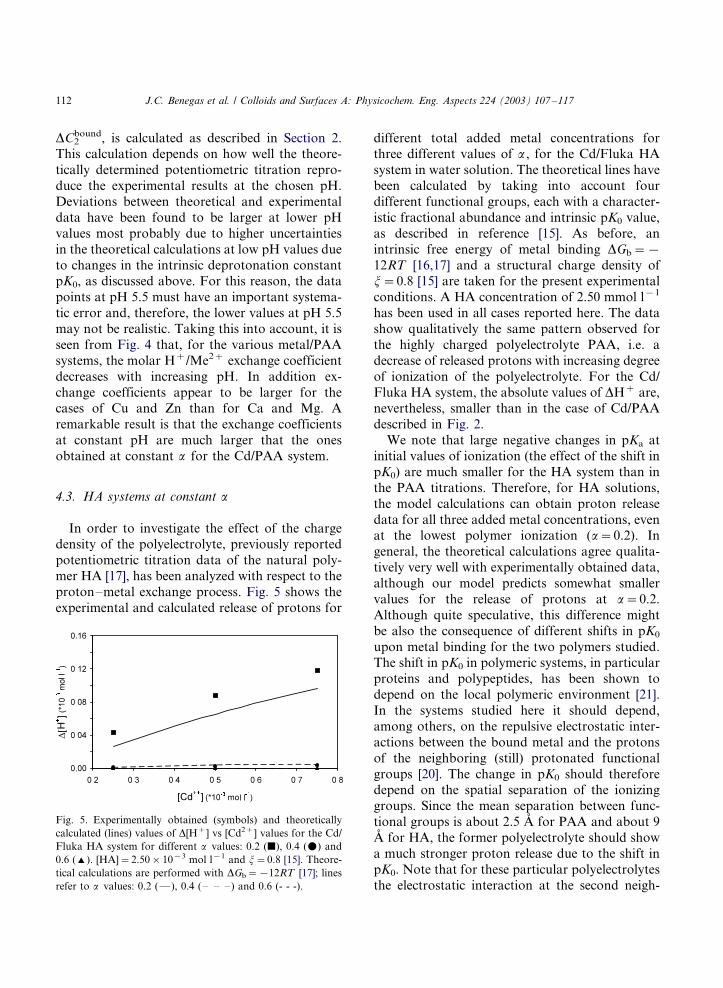
For various metal/PAA systems theoretical H�/
Me2� exchange coefficients at constant pH were
calculated from previously published potentio-
metric titration data [14] and are plotted vs pH
in Fig. 4. In all cases, the calculated data refer to a
change in a metal concentration from 0 to 0.25
mmol l�1 with [PAA]�/2.50 mmol l�1. The
horizontal dotted arrow lines in Fig. 1 represent
this process. The proton release is calculated
simply as the product of the change in a , Da ,
between the reference and final states, times the
concentration of polymeric ionizable groups Cp.
The change in the fraction of bound metal ions,
Fig. 3. Theoretically calculated values of the H�/Cd2� molar
exchange coefficient nexch vs [Cd2�] for the Cd/PAA system at
constant a for the same conditions as in Fig. 2.
Fig. 4. Theoretically calculated values of the H�/Me2� molar
exchange coefficient nexch vs pH values for different Me2�/PAA
systems. [PAA]�/2.50�/10�3 mol l�1 and j�/2.85. Model
calculations are performed using DGb�/�/9RT for Mg (x);
DGb�/�/9.5RT for Ca (2); DGb�/�/12RT for Zn (I) and
DGb�/�/15RT for Cu (k) [14].
J.C. Benegas et al. / Colloids and Surfaces A: Physicochem. Eng. Aspects 224 (2003) 107�/117 111
DC2bound, is calculated as described in Section 2.
This calculation depends on how well the theore-
tically determined potentiometric titration repro-
duce the experimental results at the chosen pH.
Deviations between theoretical and experimental
data have been found to be larger at lower pH
values most probably due to higher uncertainties
in the theoretical calculations at low pH values due
to changes in the intrinsic deprotonation constantpK0, as discussed above. For this reason, the data
points at pH 5.5 must have an important systema-
tic error and, therefore, the lower values at pH 5.5
may not be realistic. Taking this into account, it is
seen from Fig. 4 that, for the various metal/PAA
systems, the molar H�/Me2� exchange coefficient
decreases with increasing pH. In addition ex-
change coefficients appear to be larger for thecases of Cu and Zn than for Ca and Mg. A
remarkable result is that the exchange coefficients
at constant pH are much larger that the ones
obtained at constant a for the Cd/PAA system.
4.3. HA systems at constant a
In order to investigate the effect of the chargedensity of the polyelectrolyte, previously reported
potentiometric titration data of the natural poly-
mer HA [17], has been analyzed with respect to the
proton�/metal exchange process. Fig. 5 shows the
experimental and calculated release of protons for
different total added metal concentrations forthree different values of a , for the Cd/Fluka HA
system in water solution. The theoretical lines have
been calculated by taking into account four
different functional groups, each with a character-
istic fractional abundance and intrinsic pK0 value,
as described in reference [15]. As before, an
intrinsic free energy of metal binding DGb�/�/
12RT [16,17] and a structural charge density ofj�/0.8 [15] are taken for the present experimental
conditions. A HA concentration of 2.50 mmol l�1
has been used in all cases reported here. The data
show qualitatively the same pattern observed for
the highly charged polyelectrolyte PAA, i.e. a
decrease of released protons with increasing degree
of ionization of the polyelectrolyte. For the Cd/
Fluka HA system, the absolute values of DH� are,nevertheless, smaller than in the case of Cd/PAA
described in Fig. 2.
We note that large negative changes in pKa at
initial values of ionization (the effect of the shift in
pK0) are much smaller for the HA system than in
the PAA titrations. Therefore, for HA solutions,
the model calculations can obtain proton release
data for all three added metal concentrations, evenat the lowest polymer ionization (a�/0.2). In
general, the theoretical calculations agree qualita-
tively very well with experimentally obtained data,
although our model predicts somewhat smaller
values for the release of protons at a�/0.2.
Although quite speculative, this difference might
be also the consequence of different shifts in pK0
upon metal binding for the two polymers studied.The shift in pK0 in polymeric systems, in particular
proteins and polypeptides, has been shown to
depend on the local polymeric environment [21].
In the systems studied here it should depend,
among others, on the repulsive electrostatic inter-
actions between the bound metal and the protons
of the neighboring (still) protonated functional
groups [20]. The change in pK0 should thereforedepend on the spatial separation of the ionizing
groups. Since the mean separation between func-
tional groups is about 2.5 A for PAA and about 9
A for HA, the former polyelectrolyte should show
a much stronger proton release due to the shift in
pK0. Note that for these particular polyelectrolytes
the electrostatic interaction at the second neigh-
Fig. 5. Experimentally obtained (symbols) and theoretically
calculated (lines) values of D[H�] vs [Cd2�] values for the Cd/
Fluka HA system for different a values: 0.2 (j), 0.4 (m) and
0.6 ('). [HA]�/2.50�/10�3 mol l�1 and j�/0.8 [15]. Theore-
tical calculations are performed with DGb�/�/12RT [17]; lines
refer to a values: 0.2 (*/), 0.4 (�/ �/ �/) and 0.6 (- - -).
J.C. Benegas et al. / Colloids and Surfaces A: Physicochem. Eng. Aspects 224 (2003) 107�/117112

boring PAA site is stronger than at the firstneighbor in HA.
In Table 1, both experimentally obtained [17]
and theoretically calculated proton release data as
well as theoretically calculated H�/Me2� molar
exchange coefficients are summarized for different
metal/Fluka HA systems at different degrees of
ionization, different metal concentrations but for
the same polymer concentration of Fig. 5. Calcu-lations have been made using the intrinsic pK0 of
the different functional groups and intrinsic free
energies of binding, DGb, as reported in Ref. [17].
The following pattern is observed:
i) Higher levels of released protons are found for
all metal/Fluka HA systems at the lowest
degree of ionization;
ii) The release of protons appears to increase in
the order Ca:/Cd:/NiB/Pb5/Cu;
iii) The H�/Me2� exchange coefficients cover awide range of values. Very small values are
found for highly ionized HA (high a ), while
more significant values are obtained for the
HA at low degree of ionization (a�/0.2).
Exchange coefficients increase with the con-
centration of added heavy metal for a�/0.2
and 0.4, while for a�/0.6 the exchange coeffi-
cients appear to be more or less constant overthe covered metal range. The highest values of
nexch(�/1.5) are obtained for Cu and Pb ions
at a�/0.2 and a metal concentration of 0.75
mmol l�1. Note that in all these cases, there is
not complete charge compensation, i.e. the
(effective) net polyelectrolyte charge is lower
after adding metal ion to the solution.
iv) Theoretically calculated data are in goodagreement with the experimentally obtained
ones, in particular for the lower binding
strength and lower concentrations of heavy
metal ions, i.e. in the region where the shift in
pK0 upon metal ion binding is less important.
4.4. HA at constant pH
For various metal/HA systems theoretical H�/
Me2� exchange coefficients at constant pH were
calculated by considering points of equal pH from
previously published potentiometric titration data
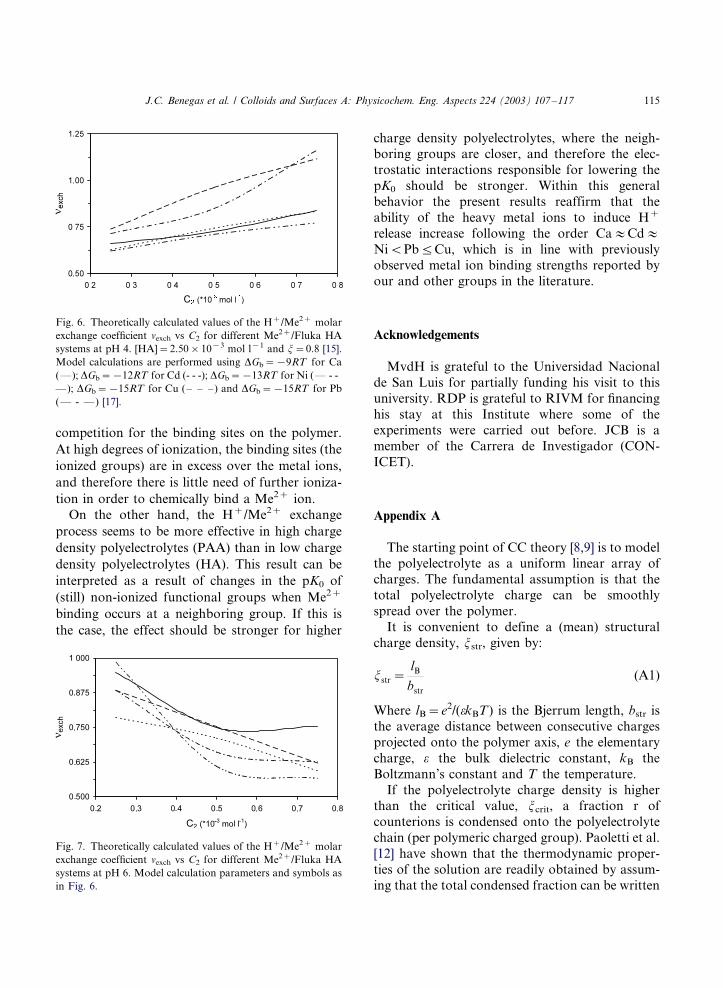
[17] and are plotted vs the total metal concentra-tion at pH 4 and 6 in Fig. 6 and Fig. 7,
respectively. The horizontal dotted arrow lines in
Fig. 1 represent this process. For the case of pH 4
there is a tendency for the exchange coefficient to
increase with the concentration of the added metal
ion. In the concentration range of our experi-
ments, the exchange process is stronger for Cu and
Pb than for Cd, Ni and Ca. In general under theseconditions, the calculated exchange coefficients
appear to be larger than the ones obtained at
constant a , similar to our observations for the
different metal/PAA systems. At pH 6, exchange
coefficients appear to be smaller than at pH 4 and
there is a tendency for the exchange coefficient to
decrease with the concentration of the added metal
ion. Kinniburgh et al. [7] found a similar patternfor various metal/PPHA systems in this metal
concentration range at both pH 6 and 8 in 0.1
mol l�1 KNO3.
In general, it can be seen that the exchange
process is stronger at low pH values, i.e. in the
titration region where there are more metal ions in
solution than binding sites available, therefore the
competition of H� and Me2� ions for these sitesis stronger. This result is completely in line with
the analysis at constant degree of ionization.
5. Conclusions
The H�/Me2� exchange process that occurs
when a simple salt containing heavy metal ions is
added to a polyelectrolyte solution has beenmodeled and described successfully with the ex-
tensions of the CC theory of linear polyelectrolytes
previously proposed by our group. Two important
polymers have been considered: a high charge
density, monoprotic synthetic polyelectrolyte
(PAA) and an environmentally important low
charge density, multifunctional polyelectrolyte
(HA). In both cases, it is shown that the exchangeprocess is much stronger at low ionization degrees
of the polyelectrolyte (in the Me2� ion concentra-
tion region studied here). It is furthermore found
that ion exchange coefficients increase for higher
concentrations of the Me2� added. This result can
be interpreted as a manifestation of the H�/Me2�
J.C. Benegas et al. / Colloids and Surfaces A: Physicochem. Eng. Aspects 224 (2003) 107�/117 113
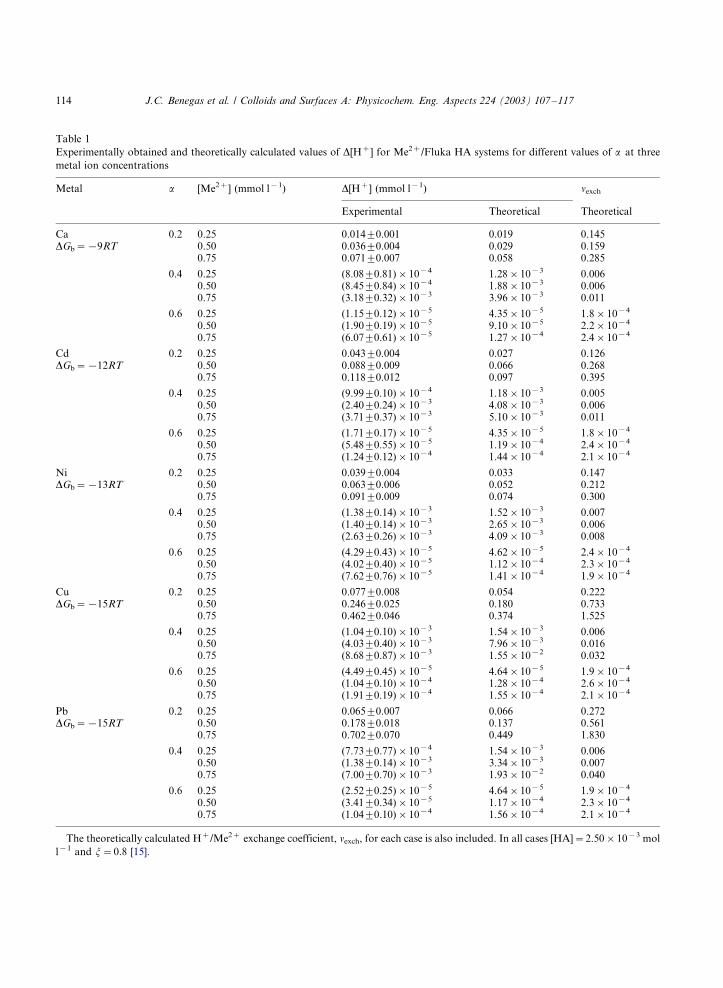
Table 1
Experimentally obtained and theoretically calculated values of D[H�] for Me2�/Fluka HA systems for different values of a at three
metal ion concentrations
Metal a [Me2�] (mmol l�1) D[H�] (mmol l�1) nexch
Experimental Theoretical Theoretical
Ca 0.2 0.25 0.0149/0.001 0.019 0.145DGb�/�/9RT 0.50 0.0369/0.004 0.029 0.159
0.75 0.0719/0.007 0.058 0.285
0.4 0.25 (8.089/0.81)�/10�4 1.28�/10�3 0.0060.50 (8.459/0.84)�/10�4 1.88�/10�3 0.0060.75 (3.189/0.32)�/10�3 3.96�/10�3 0.011
0.6 0.25 (1.159/0.12)�/10�5 4.35�/10�5 1.8�/10�4
0.50 (1.909/0.19)�/10�5 9.10�/10�5 2.2�/10�4
0.75 (6.079/0.61)�/10�5 1.27�/10�4 2.4�/10�4
Cd 0.2 0.25 0.0439/0.004 0.027 0.126DGb�/�/12RT 0.50 0.0889/0.009 0.066 0.268
0.75 0.1189/0.012 0.097 0.395
0.4 0.25 (9.999/0.10)�/10�4 1.18�/10�3 0.0050.50 (2.409/0.24)�/10�3 4.08�/10�3 0.0060.75 (3.719/0.37)�/10�3 5.10�/10�3 0.011
0.6 0.25 (1.719/0.17)�/10�5 4.35�/10�5 1.8�/10�4
0.50 (5.489/0.55)�/10�5 1.19�/10�4 2.4�/10�4
0.75 (1.249/0.12)�/10�4 1.44�/10�4 2.1�/10�4
Ni 0.2 0.25 0.0399/0.004 0.033 0.147DGb�/�/13RT 0.50 0.0639/0.006 0.052 0.212
0.75 0.0919/0.009 0.074 0.300
0.4 0.25 (1.389/0.14)�/10�3 1.52�/10�3 0.0070.50 (1.409/0.14)�/10�3 2.65�/10�3 0.0060.75 (2.639/0.26)�/10�3 4.09�/10�3 0.008
0.6 0.25 (4.299/0.43)�/10�5 4.62�/10�5 2.4�/10�4
0.50 (4.029/0.40)�/10�5 1.12�/10�4 2.3�/10�4
0.75 (7.629/0.76)�/10�5 1.41�/10�4 1.9�/10�4
Cu 0.2 0.25 0.0779/0.008 0.054 0.222DGb�/�/15RT 0.50 0.2469/0.025 0.180 0.733
0.75 0.4629/0.046 0.374 1.525
0.4 0.25 (1.049/0.10)�/10�3 1.54�/10�3 0.0060.50 (4.039/0.40)�/10�3 7.96�/10�3 0.0160.75 (8.689/0.87)�/10�3 1.55�/10�2 0.032
0.6 0.25 (4.499/0.45)�/10�5 4.64�/10�5 1.9�/10�4
0.50 (1.049/0.10)�/10�4 1.28�/10�4 2.6�/10�4
0.75 (1.919/0.19)�/10�4 1.55�/10�4 2.1�/10�4
Pb 0.2 0.25 0.0659/0.007 0.066 0.272DGb�/�/15RT 0.50 0.1789/0.018 0.137 0.561
0.75 0.7029/0.070 0.449 1.830
0.4 0.25 (7.739/0.77)�/10�4 1.54�/10�3 0.0060.50 (1.389/0.14)�/10�3 3.34�/10�3 0.0070.75 (7.009/0.70)�/10�3 1.93�/10�2 0.040
0.6 0.25 (2.529/0.25)�/10�5 4.64�/10�5 1.9�/10�4
0.50 (3.419/0.34)�/10�5 1.17�/10�4 2.3�/10�4
0.75 (1.049/0.10)�/10�4 1.56�/10�4 2.1�/10�4
The theoretically calculated H�/Me2� exchange coefficient, nexch, for each case is also included. In all cases [HA]�/2.50�/10�3 mol
l�1 and j�/0.8 [15].
J.C. Benegas et al. / Colloids and Surfaces A: Physicochem. Eng. Aspects 224 (2003) 107�/117114
competition for the binding sites on the polymer.
At high degrees of ionization, the binding sites (the
ionized groups) are in excess over the metal ions,
and therefore there is little need of further ioniza-
tion in order to chemically bind a Me2� ion.
On the other hand, the H�/Me2� exchange
process seems to be more effective in high charge
density polyelectrolytes (PAA) than in low charge
density polyelectrolytes (HA). This result can be
interpreted as a result of changes in the pK0 of
(still) non-ionized functional groups when Me2�
binding occurs at a neighboring group. If this is
the case, the effect should be stronger for higher
charge density polyelectrolytes, where the neigh-boring groups are closer, and therefore the elec-
trostatic interactions responsible for lowering the
pK0 should be stronger. Within this general
behavior the present results reaffirm that the
ability of the heavy metal ions to induce H�
release increase following the order Ca:/Cd:/
NiB/Pb5/Cu, which is in line with previously
observed metal ion binding strengths reported byour and other groups in the literature.
Acknowledgements
MvdH is grateful to the Universidad Nacional
de San Luis for partially funding his visit to this
university. RDP is grateful to RIVM for financing
his stay at this Institute where some of theexperiments were carried out before. JCB is a
member of the Carrera de Investigador (CON-
ICET).
Appendix A
The starting point of CC theory [8,9] is to model
the polyelectrolyte as a uniform linear array ofcharges. The fundamental assumption is that the
total polyelectrolyte charge can be smoothly
spread over the polymer.
It is convenient to define a (mean) structural
charge density, jstr, given by:
jstr�lB
bstr
(A1)
Where lB�/e2/(okBT ) is the Bjerrum length, bstr is
the average distance between consecutive charges
projected onto the polymer axis, e the elementary
charge, o the bulk dielectric constant, kB the
Boltzmann’s constant and T the temperature.
If the polyelectrolyte charge density is higherthan the critical value, jcrit, a fraction r of
counterions is condensed onto the polyelectrolyte
chain (per polymeric charged group). Paoletti et al.
[12] have shown that the thermodynamic proper-
ties of the solution are readily obtained by assum-
ing that the total condensed fraction can be written
Fig. 6. Theoretically calculated values of the H�/Me2� molar
exchange coefficient nexch vs C2 for different Me2�/Fluka HA
systems at pH 4. [HA]�/2.50�/10�3 mol l�1 and j�/0.8 [15].
Model calculations are performed using DGb�/�/9RT for Ca
(*/); DGb�/�/12RT for Cd (- - -); DGb�/�/13RT for Ni (*/ - -
*/); DGb�/�/15RT for Cu (�/ �/ �/) and DGb�/�/15RT for Pb
(*/ - */) [17].
Fig. 7. Theoretically calculated values of the H�/Me2� molar
exchange coefficient nexch vs C2 for different Me2�/Fluka HA
systems at pH 6. Model calculation parameters and symbols as
in Fig. 6.
J.C. Benegas et al. / Colloids and Surfaces A: Physicochem. Eng. Aspects 224 (2003) 107�/117 115

in the following form:
r�ri�rj �r(xi�xj) (xi�xj �1) (A2)
where ri and rj stand for the fractions of con-
densed counterions of valence zi and zj , respec-
tively. According to this model, in the
condensation regime, both species of counterions
are found in the condensation volume.
Chemical binding of counterions of valence zj tothe polyelectrolyte is considered by reducing the
average charge density effectively by an amount
equivalent to a fraction s per polymeric charge
[13]:
js�jstr(1�zjs) (A3)
where s�/ [Me]b/Cp, and [Me]b is the concentra-
tion of bound metal ions. The effective chargedensity thus becomes the central characteristic of
the polyelectrolytic solution.
Following the procedure described in Refs.
[12,13], one can write analytical expressions for
the polyelectrolytic (Gpol), entropic (Gentr) and
binding (Gb) contributions to the total (excess)
free energy of the system:
Gtot�Gpol�Gentr�Gb�Gion�Gb (A4)
The contribution of each counterion species to
the total condensed fraction (xi and xj), is
calculated [12] by minimizing the total reduced
free energy of the system:
@gtot
@r�0 (A5)
@gtot
@xi
�0 (A6)
where gtot�/Gtot/RT and R is the gas constant. In
the framework of CC theory, the limiting behavior
at infinite dilution of Eq. (A5) determines the
value of the total fraction of condensed counter-
ions, r , which, for the particular case of a mixtureof monovalent and divalent counterions, it is given
by:
r�1
2 � x
�1�
1
jstr(2 � x)(1 � 2s)
�(A7)
where we have set xi �/x1�/x and xj �/x2�/1�/x .
Notice that Eq. (A7) properly reproduces the
functional form of the condensed fraction givenin Ref. [12] for the case in which no bonding
occurs (s�/0).
For the present study the thermodynamic func-
tion of interest is the apparent dissociation con-
stant, pKa, given in general by:
pKa�pK0�DpKa (A8)
where pK0 is the intrinsic pKa, characteristic of the
(isolated) ionizing group making up the polymer,and DpKa is the change in pKa due to the
ionization of the polyelectrolyte. Once the excess
free energy function, Gion, is known, we can
readily calculate:
DpKa�1
np2:303RT
@Gion
@a
�G(a; j; Cp; C1; C2; T ; o; s0) (A9)
where np is the number of polymeric charge units,
C1 and C2 stand for the analytical concentrations
of monovalent and divalent counterions, respec-
tively, and s0 is the maximum (mean) fraction of
chemically bounded counterions per polymericcharge unit, i.e. the mean stoichiometry of the
binding process.
For a heterogeneous weak polyelectrolyte like
HA, constituted by N functional groups of frac-
tional abundance (Xi ) and intrinsic pK , (pK0i ), the
overall intrinsic pK0 is a function of the degree of
ionization. Porasso et al. [14] have shown that:
pK0(a)�pK0i � log
�bi
(1 � bi)
(1 � a)
a
�(A10)
where bi stands for the ionization degree of the ith
functional group.
References
[1] M.J. Gomez, M. Iris, D. Loreto, Fresenius J. Anal. Chem.
339 (1991) 664�/668.
[2] E. Tipping, M.A. Hurley, Geochim. Cosmochim. Acta 56
(1992) 3627�/3641.
[3] S.J. Marshall, S.D. Young, K. Gregson, Eur. J. Soil Sci. 46
(1995) 471�/480.
[4] J.C. Masini, G. Abate, E.C. Lima, L.C. Hahn, M.S.
Nakamura, J. Lichtig, H.R. Nagatomy, Anal. Chim.
Acta 364 (1998) 223�/233.
J.C. Benegas et al. / Colloids and Surfaces A: Physicochem. Eng. Aspects 224 (2003) 107�/117116

[5] R.M.J. Cleven, Ph.D. Thesis, Wageningen University, The
Netherlands, 1984.
[6] E. Tipping, A. Fitch, F.J. Stevenson, Eur. J. Soil Sci. 46
(1995) 95�/101.
[7] D.G. Kinniburgh, W.H. van Riemsdijk, L.K. Koopal, M.
Borkovec, M.F. Benedetti, M.J. Avena, Colloids Surf. A:
Physiochem. Eng. Aspects 151 (1999) 147.
[8] G.S. Manning, J. Chem. Phys. 51 (1969) 924.
[9] G.S. Manning, Q. Rev. Biophys. 11 (1978) 179.
[10] Ch.F. Anderson, M.Th. Record, Jr., Annu. Rev. Phys.
Chem. 33 (1982) 191.
[11] Ch.F. Anderson, M.Th. Record, Jr., Annu. Rev. Biophys.
Chem. 19 (1990) 423.
[12] S. Paoletti, J.C. Benegas, A. Cesaro, G. Manzini, F.
Fogolari, V. Crescenzi, Biophys. Chem. 41 (1991) 73.
[13] R.D. Porasso, J.C. Benegas, M.A.G.T. Van den Hoop,
S. Paoletti, Phys. Chem. Chem. Phys. 3 (2001)
1057.
[14] R.D. Porasso, J.C. Benegas, M.A.G.T. van den Hoop, J.
Phys. Chem. B 103 (1999) 2361.
[15] R.D. Porasso, J.C. Benegas, M.A.G.T. Van den Hoop, S.
Paoletti, Biophys. Chem. 86 (2000) 59.
[16] M.A.G.T. van den Hoop, R.D. Porasso, J.C. Benegas,
Colloids Surf. A: Physicochem. Eng. Aspects 203 (2002)
105.
[17] R.D. Porasso, J.C. Benegas, S. Paoletti, M.A.G.T. van den
Hoop, Environ. Sci. Technol. 36 (2002) 3815�/3821.
[18] D.G. Kinniburgh, K.J. Milne, M.F. Benedetti, J.P. Pin-
heiro, J. Filius, L.K. Koopal, W.H. van Riemsdijk,
Environ. Sci. Technol. 30 (1996) 1687�/1698.
[19] M. Mandel, Encyclopedia of Polymer Science and En-
gineering, vol. 11, second ed., Wiley, New York, 1988, p.
739.
[20] D. Bashford, M. Karplus, Biochemistry 29 (1990) 10219.
[21] D.R. Ripoll, Y.N. Vorobjeb, A. Liwo, J.A. Vila, H.A.
Sheraga, J. Mol. Biol. 264 (1996) 770.
J.C. Benegas et al. / Colloids and Surfaces A: Physicochem. Eng. Aspects 224 (2003) 107�/117 117





























