Embed Size (px)
Citation preview

Pim-1 kinase antagonizes aspects of myocardialhypertrophy and compensation to pathologicalpressure overloadJohn A. Muraski*†, Kimberlee M. Fischer*†, Weitao Wu*, Christopher T. Cottage*, Pearl Quijada*, Matt Mason*,Shabana Din*, Natalie Gude*, Roberto Alvarez, Jr.*, Marcello Rota‡, Jan Kajstura‡, Zeping Wang§, Erik Schaefer§,Xiongen Chen¶, Scott MacDonnel¶, Nancy Magnuson�, Stephen R. Houser¶, Piero Anversa‡, and Mark A. Sussman*,**
*San Diego State Heart Institute, San Diego State University, 5500 Campanile Drive, San Diego, CA 92182; ‡Brigham and Women’s Hospital, Boston,MA 00311; §Invitrogen Corporation, Hopkinton, MA 01748; �School of Molecular Biosciences, Washington State University, Pullman, WA 99164;and ¶Cardiovascular Research Center, Temple University School of Medicine, 3420 North Broad Street, Philadelphia, PA 19140
Edited by Eric N. Olson, University of Texas Southwestern Medical Center, Dallas, TX, and approved July 7, 2008 (received for review September 25, 2007)
Pim-1 kinase exerts potent cardioprotective effects in the myocar-dium downstream of AKT, but the participation of Pim-1 in cardiachypertrophy requires investigation. Cardiac-specific expression ofPim-1 (Pim-WT) or the dominant-negative mutant of Pim-1 (Pim-DN) in transgenic mice together with adenoviral-mediated over-expression of these Pim-1 constructs was used to delineate the roleof Pim-1 in hypertrophy. Transgenic overexpression of Pim-1 pro-tects mice from pressure-overload-induced hypertrophy relative towild-type controls as evidenced by improved hemodynamic func-tion, decreased apoptosis, increases in antihypertrophic proteins,smaller myocyte size, and inhibition of hypertrophic signaling afterchallenge. Similarly, Pim-1 overexpression in neonatal rat cardio-myocyte cultures inhibits hypertrophy induced by endothelin-1. Onthe cellular level, hearts of Pim-WT mice show enhanced incorpo-ration of BrdU into myocytes and a hypercellular phenotypecompared to wild-type controls after hypertrophic challenge. Incomparison, transgenic overexpression of Pim-DN leads to dilatedcardiomyopathy characterized by increased apoptosis, fibrosis,and severely depressed cardiac function. Furthermore, overexpres-sion of Pim-DN leads to reduced contractility as evidenced byreduced Ca2� transient amplitude and decreased percentage of cellshortening in isolated myocytes. These data support a pivotal rolefor Pim-1 in modulation of hypertrophy by impacting responses onmolecular, cellular, and organ levels.
The serine/threonine kinase AKT serves as a nodal regulatorypoint for wide-ranging responses that mediate paracrine factor
production, apoptotic stimuli, mechanical stress, hormones, energyutilization, protein synthesis, proliferation, differentiation, motility,and gene transcription (to name a select few) (1–6). Influence ofAKT in the myocardial context upon hypertrophic responses is wellknown, either as an inducer (7, 8) or inhibitor (9, 10) dependingupon experimental design. Our group previously focused uponmyocardial nuclear accumulation of AKT as a mediator of cellsurvival (10), inhibitor of hypertrophic remodeling (9), facilitator ofstem cell proliferation (11), and upstream activator of Pim-1 kinase(12). The close interrelationship between AKT and Pim-1 activitieshas been delineated in the context of hematopoesis leading to agreater understanding of Pim-1 function (13, 14), but activities ofPim-1 in the myocardium remain obscure.
Pim-1 is a protooncogenic serine/threonine kinase and proviralintegration site for Moloney murine leukemia virus (15). Originaldescriptions in the hematopoetic system indicate the kinase po-tently increases cell survival and proliferation (reviewed in ref. 15).Recently, overexpression of Pim-1 was found to protect the myo-cardium following infarction injury and cardiomyocytes from apo-ptotic challenge by increasing cell-survival signaling (12). Geneticablation of Pim-1 results in larger infarct size and exacerbatedcardiac failure following pressure-overload aortic banding (12).These properties of Pim-1 were similar to AKT-mediated effects,but the impact of Pim-1 upon hypertrophic signaling and remod-
eling remained to be determined. Results show Pim-1 bluntshypertrophy, consistent with a role for Pim-1 expression down-stream of nuclear AKT activity.
ResultsCardiac-Specific Pim-1 Transgenesis. Wild-type human Pim-1 (Pim-WT) and a kinase-dead mutant that functions as a dominant-negative protein (Pim-DN) (16) were fused to GFP under controlof the cardiac-specific �-myosin heavy chain promoter [supportinginformation (SI) Fig. S1a]. PCR constructs show incorporation ofthe transgenes into the genome (Fig. S1b). Whole heart lysates fromtransgenic samples reveal a 64-kDa GFP-Pim-1 fusion protein thatis recognized by both Pim-1 and GFP antibodies (Fig. S1c). Bonafide inhibitory function of the Pim-DN construct was validatedusing the ability of Pim-1 to activate GATA-1 transcription (N.M.,unpublished data). Pim-WT phosphorylates the transcription fac-tor GATA-1 and induces GATA-1 luciferase reporter expression inC2C12 myoblasts, with increasing titration of Pim-DN inhibitingGATA-1 activity (Fig. S1d). On the basis of previous studies thatshowed Pim-1 phosphorylates p21 (17), in vitro kinase assaysconfirmed activity of Pim-WT construct using whole heart lysates,were prepared from GFP-Pim-1-WT, GFP-Pim-1-DN transgenicmice, and nontransgenic (NTG) mice. GFP-Pim-1 proteins [WT orkinase dead (KD)] were immunoprecipitated from whole heartlysates and incubated in the presence of [�-32P]ATP with GST-p21as substrates. Pim-WT overexpression phosphorylates p21 whilethis activity was abolished in the Pim-DN construct (Fig. S1e).
Pim-1 Inactivation Increases Cardiomyocyte Apoptosis and Fibrosis.Hearts created with genetic deletion of Pim-1 (Pim-1 KO) exhibitincreased apoptosis in myocytes relative to NTG controls but showno evidence of overt cardiomyopathic remodeling (12). In com-parison, Pim-DN overexpressing mice suffer from cardiomyopathycharacterized by progressive wall thinning beginning at 3–4 monthsof age (Fig. 1 A and B, *, P � 0.05, **, P � 0.01). Heart:body weightratio at 10 and 22 weeks after birth is also significantly increased(Fig. 1C, **, P � 0.01). Because Pim-DN overexpression inducescardiomyocyte apoptosis in vitro (12), assessment of apoptoticmyocytes in the myocardium of Pim-DN animals was performed by
Author contributions: J.A.M., K.M.F., and M.A.S. designed research; J.A.M., K.M.F., W.W.,C.T.C., P.Q., M.M., S.D., N.G., R.A., M.R., J.K., Z.W., and M.A.S. performed research; E.S., X.C.,S.M., N.M., S.R.H., and P.A contributed new reagents/analytic tools; and J.A.M., K.M.F., andM.A.S. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
†J.A.M. and K.M.F. contributed equally to this work.
**To whom correspondence should be addressed: E-mail: [email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0709135105/DCSupplemental.
© 2008 by The National Academy of Sciences of the USA
www.pnas.org�cgi�doi�10.1073�pnas.0709135105 PNAS � September 16, 2008 � vol. 105 � no. 37 � 13889–13894
CELL
BIO
LOG
Y

TUNEL staining. Pim-DN animals exhibit a twofold increase inapoptotic cardiomyocytes per mm2 relative to age-matched controls(1.2/mm2 and 2.4/mm2, respectively, Fig. 1D, **, P � 0.01) resultingin increased fibrosis and collagen deposits in the left ventricle (Fig.1E, blue). In addition, the amount of necrosis (Fig. S2a, **, P �0.01) was significantly increased at basal levels.
Pim-DN Hearts Exhibit Depressed Cardiac Function. Hearts ofPim-DN mice show progressive dilation from 17 weeks of age (Fig.2A, *, P � 0.05) with depression of fractional shortening andejection fraction (36.6 and 74.2%, respectively) by 27 weeks of age(Fig. 2 B and C, *, P � 0.05, **, P � 0.01) by echocardiographicanalyses. Morphometric analysis on both NTG and Pim-DN heartsconfirmed that Pim-DN hearts were significantly dilated (Table S1).In vivo hemodynamic assessments verified impaired hemodynamicswith diminished �dP/dt, increased left ventricular end diastolicpressure (LVEDP), and decreased left ventricular developed pres-sure (LVDP) (Fig. 2 D–F, **, P � 0.01). Mechanistically, Pim-DNmyocytes displayed reduced Ca2� transient amplitude with de-creased percentage of cell shortening with respect to NTG myo-cytes (Fig. 2G). Additionally, the time constant (�) of the Ca2�
transient decay was larger in Pim-DN myocytes (Fig. 2G). These
results indicate that depressed contractile function of Pim-DNmyocytes is mediated in part by a decline in Ca2� release from thesarcoplasmic reticulum together with a slower reuptake. Thusinactivation of Pim-1 by Pim-DN in the myocardium has a negativeeffect on cardiac function.
Overexpression of Pim-1 Inhibits Hypertrophy in Vitro. Induction ofPim-1 in the damaged myocardium is thought to be a protectivesurvival response (12) occurring in cardiomyocytes in the borderzone where Pim-1 colocalizes to cells expressing atrial natriureticpeptide (ANP, Fig. 3A). ANP is both antihypertrophic and cardio-protective (24), the coincidence of these proteins prompted assess-ment of the role that Pim-1 accumulation plays in mitigation ofhypertrophic signaling.
Pim-WT overexpression upon cardiomyocyte hypertrophy wasexamined using neonatal rat cardiomyocytes (NRCMs) infectedwith adenoviruses encoding EGFP-Pim-WT or EGFP proteinfollowed by stimulation with endothelin-1 (ET-1) for 24 h.Pim-WT overexpression inhibits ET-1-induced hypertrophy(Fig. 3B, *, P � 0.05, **, P � 0.01) as assessed by cell surface areameasurements relative to the increase in cell size seen in controland EGFP-infected myocytes treated with ET-1. Molecular
Fig. 1. Inactivation of Pim-1 in the myocardium increases apoptosis and fibrosis. (A and B) Echocardiographic measurement of posterior (A) and anterior (B)wall dimension (PWD and AWD, respectively) in NTG (n � 5) and Pim-DN (n � 7) animals at 2-week intervals (*, P � 0.05, **, P � 0.01). (C) Heart weight to bodyweight ratios in NTG and Pim-DN animals at 10 and 22 weeks of age (n � 6, **, P � 0.01). (D) Histogram representing counts of TUNEL positive myocytes permm2 in 17–22-week-old NTG and Pim-DN transgenics (n � 3, **, P � 0.01). (E) Masson trichrome staining of sections from paraffin embedded NTG and Pim-DNhearts at 17 weeks of age.
Fig. 2. Pim-DN hearts exhibit dilation and depressed function. (A–C) Echocardiographic assessment of end-diastolic diameter (EDD, A), percentage of fractionalshortening (FS, B), and ejection fraction (EF, C) at 27 weeks of age in NTG (n � 5) and Pim-DN (n � 7) hearts (*, P � 0.05, **, P � 0.01). (D–F ) In vivo hemodynamicmeasurements in27-week-oldNTGandPim-DNanimals (n�4).Positiveandnegativechange inpressure/change intime(�dP/dt,D), leftventriculardevelopedpressure(LVDP, E), and left ventricular end diastolic pressure (LVEDP, F ) are represented (*, P � 0.05, **, P � 0.01). (G) Calcium handling and contractile function of isolatedmyocytes from NTG and Pim-DN animals (*, P � 0.05).
13890 � www.pnas.org�cgi�doi�10.1073�pnas.0709135105 Muraski et al.
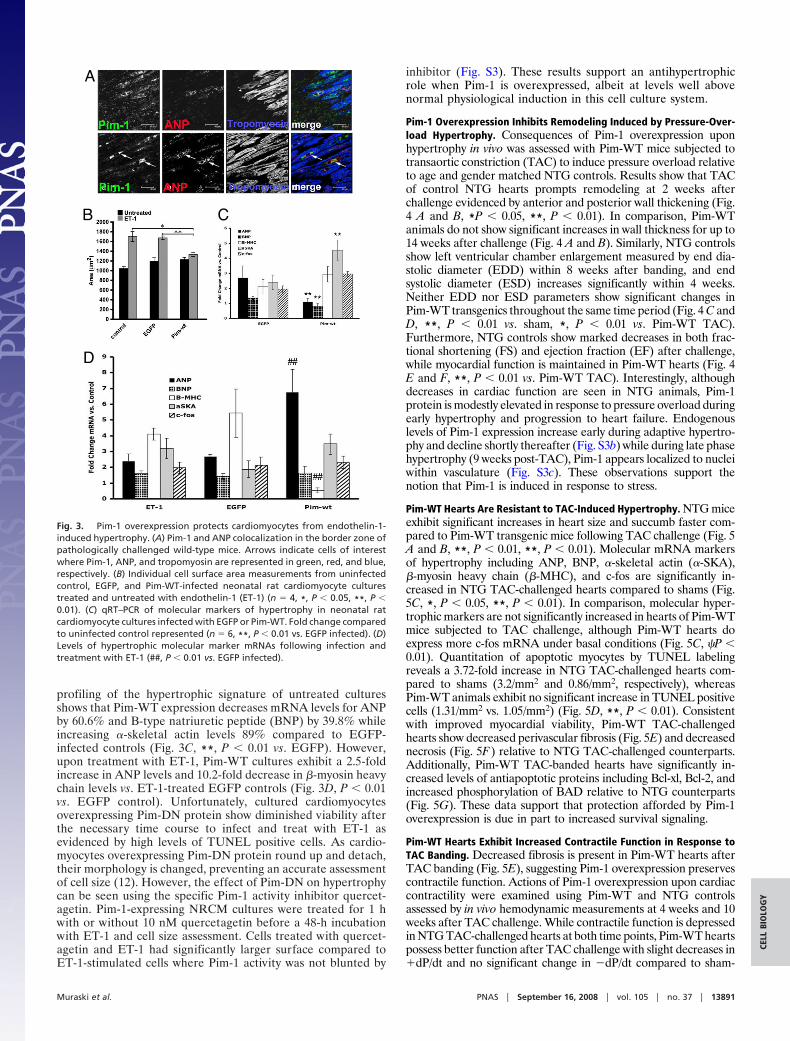
profiling of the hypertrophic signature of untreated culturesshows that Pim-WT expression decreases mRNA levels for ANPby 60.6% and B-type natriuretic peptide (BNP) by 39.8% whileincreasing �-skeletal actin levels 89% compared to EGFP-infected controls (Fig. 3C, **, P � 0.01 vs. EGFP). However,upon treatment with ET-1, Pim-WT cultures exhibit a 2.5-foldincrease in ANP levels and 10.2-fold decrease in �-myosin heavychain levels vs. ET-1-treated EGFP controls (Fig. 3D, P � 0.01vs. EGFP control). Unfortunately, cultured cardiomyocytesoverexpressing Pim-DN protein show diminished viability afterthe necessary time course to infect and treat with ET-1 asevidenced by high levels of TUNEL positive cells. As cardio-myocytes overexpressing Pim-DN protein round up and detach,their morphology is changed, preventing an accurate assessmentof cell size (12). However, the effect of Pim-DN on hypertrophycan be seen using the specific Pim-1 activity inhibitor quercet-agetin. Pim-1-expressing NRCM cultures were treated for 1 hwith or without 10 nM quercetagetin before a 48-h incubationwith ET-1 and cell size assessment. Cells treated with quercet-agetin and ET-1 had significantly larger surface compared toET-1-stimulated cells where Pim-1 activity was not blunted by
inhibitor (Fig. S3). These results support an antihypertrophicrole when Pim-1 is overexpressed, albeit at levels well abovenormal physiological induction in this cell culture system.
Pim-1 Overexpression Inhibits Remodeling Induced by Pressure-Over-load Hypertrophy. Consequences of Pim-1 overexpression uponhypertrophy in vivo was assessed with Pim-WT mice subjected totransaortic constriction (TAC) to induce pressure overload relativeto age and gender matched NTG controls. Results show that TACof control NTG hearts prompts remodeling at 2 weeks afterchallenge evidenced by anterior and posterior wall thickening (Fig.4 A and B, *P � 0.05, **, P � 0.01). In comparison, Pim-WTanimals do not show significant increases in wall thickness for up to14 weeks after challenge (Fig. 4 A and B). Similarly, NTG controlsshow left ventricular chamber enlargement measured by end dia-stolic diameter (EDD) within 8 weeks after banding, and endsystolic diameter (ESD) increases significantly within 4 weeks.Neither EDD nor ESD parameters show significant changes inPim-WT transgenics throughout the same time period (Fig. 4 C andD, **, P � 0.01 vs. sham, *, P � 0.01 vs. Pim-WT TAC).Furthermore, NTG controls show marked decreases in both frac-tional shortening (FS) and ejection fraction (EF) after challenge,while myocardial function is maintained in Pim-WT hearts (Fig. 4E and F, **, P � 0.01 vs. Pim-WT TAC). Interestingly, althoughdecreases in cardiac function are seen in NTG animals, Pim-1protein is modestly elevated in response to pressure overload duringearly hypertrophy and progression to heart failure. Endogenouslevels of Pim-1 expression increase early during adaptive hypertro-phy and decline shortly thereafter (Fig. S3b) while during late phasehypertrophy (9 weeks post-TAC), Pim-1 appears localized to nucleiwithin vasculature (Fig. S3c). These observations support thenotion that Pim-1 is induced in response to stress.
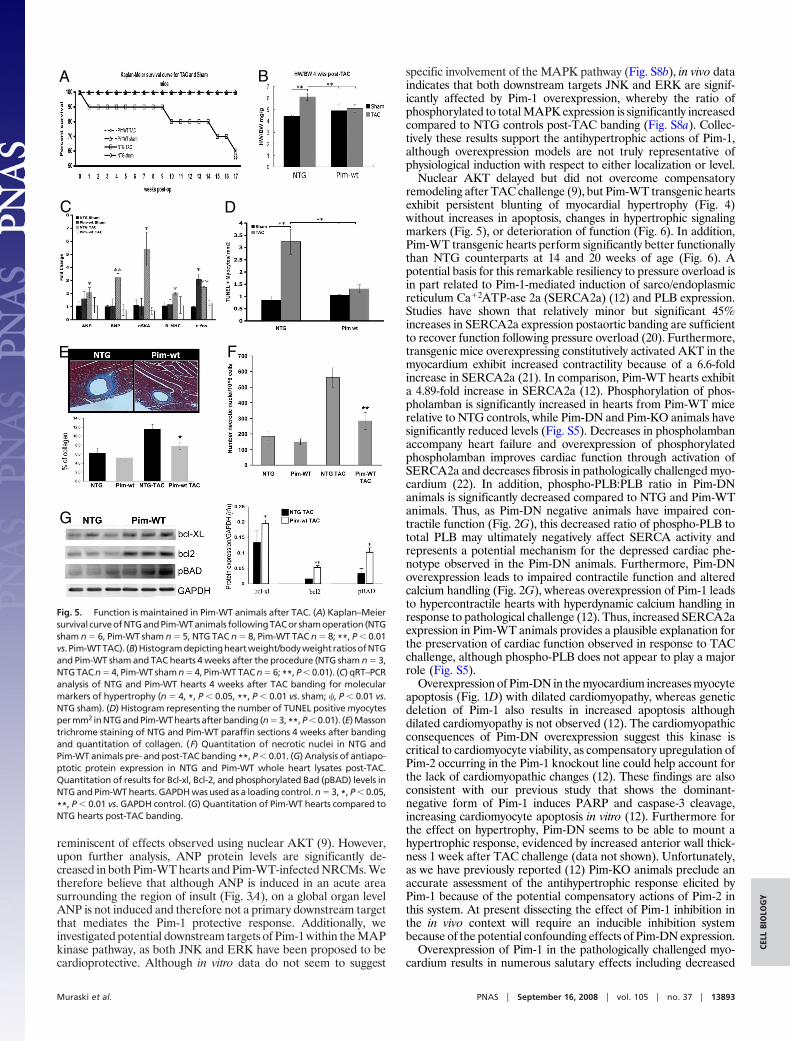
Pim-WT Hearts Are Resistant to TAC-Induced Hypertrophy. NTG miceexhibit significant increases in heart size and succumb faster com-pared to Pim-WT transgenic mice following TAC challenge (Fig. 5A and B, **, P � 0.01, **, P � 0.01). Molecular mRNA markersof hypertrophy including ANP, BNP, �-skeletal actin (�-SKA),�-myosin heavy chain (�-MHC), and c-fos are significantly in-creased in NTG TAC-challenged hearts compared to shams (Fig.5C, *, P � 0.05, **, P � 0.01). In comparison, molecular hyper-trophic markers are not significantly increased in hearts of Pim-WTmice subjected to TAC challenge, although Pim-WT hearts doexpress more c-fos mRNA under basal conditions (Fig. 5C, �P �0.01). Quantitation of apoptotic myocytes by TUNEL labelingreveals a 3.72-fold increase in NTG TAC-challenged hearts com-pared to shams (3.2/mm2 and 0.86/mm2, respectively), whereasPim-WT animals exhibit no significant increase in TUNEL positivecells (1.31/mm2 vs. 1.05/mm2) (Fig. 5D, **, P � 0.01). Consistentwith improved myocardial viability, Pim-WT TAC-challengedhearts show decreased perivascular fibrosis (Fig. 5E) and decreasednecrosis (Fig. 5F) relative to NTG TAC-challenged counterparts.Additionally, Pim-WT TAC-banded hearts have significantly in-creased levels of antiapoptotic proteins including Bcl-xl, Bcl-2, andincreased phosphorylation of BAD relative to NTG counterparts(Fig. 5G). These data support that protection afforded by Pim-1overexpression is due in part to increased survival signaling.
Pim-WT Hearts Exhibit Increased Contractile Function in Response toTAC Banding. Decreased fibrosis is present in Pim-WT hearts afterTAC banding (Fig. 5E), suggesting Pim-1 overexpression preservescontractile function. Actions of Pim-1 overexpression upon cardiaccontractility were examined using Pim-WT and NTG controlsassessed by in vivo hemodynamic measurements at 4 weeks and 10weeks after TAC challenge. While contractile function is depressedin NTG TAC-challenged hearts at both time points, Pim-WT heartspossess better function after TAC challenge with slight decreases in�dP/dt and no significant change in �dP/dt compared to sham-
A
B C
D
Fig. 3. Pim-1 overexpression protects cardiomyocytes from endothelin-1-induced hypertrophy. (A) Pim-1 and ANP colocalization in the border zone ofpathologically challenged wild-type mice. Arrows indicate cells of interestwhere Pim-1, ANP, and tropomyosin are represented in green, red, and blue,respectively. (B) Individual cell surface area measurements from uninfectedcontrol, EGFP, and Pim-WT-infected neonatal rat cardiomyocyte culturestreated and untreated with endothelin-1 (ET-1) (n � 4, *, P � 0.05, **, P �0.01). (C) qRT–PCR of molecular markers of hypertrophy in neonatal ratcardiomyocyte cultures infected with EGFP or Pim-WT. Fold change comparedto uninfected control represented (n � 6, **, P � 0.01 vs. EGFP infected). (D)Levels of hypertrophic molecular marker mRNAs following infection andtreatment with ET-1 (##, P � 0.01 vs. EGFP infected).
Muraski et al. PNAS � September 16, 2008 � vol. 105 � no. 37 � 13891
CELL
BIO
LOG
Y

operated NTG controls (Fig. 6A, *, P � 0.05 and **, P � 0.01 vs.sham, *, P � 0.05 and **, P � 0.01 vs. NTG TAC). Comparison of4-week and 10-week dP/dt assessments show significant decreasesin function for both NTG and Pim-WT TAC-challenged hearts,although performance of Pim-WT TAC-challenged hearts is rela-tively improved (Fig. 6A **, P � 0.01 vs. 4 weeks). Measurementsreveal increases in left ventricular developed pressure and enddiastolic pressure in NTG hearts 4 and 10 weeks after TAC, butPim-WT hearts show relative preservation of LVDP (Fig. 6B,Pim-WT 19.75% increase, **, P � 0.01) and no change in LVEDP(Fig. 6C **, P � 0.01, **, P � 0.01 vs. NTG TAC). Hemodynamicfunction reflected in �dP/dt and LVDP is improved in Pim-WThearts compared to NTG at 4- and 10-week time points (Fig. 6 Aand B, $P � 0.05, $$P � 0.01). The mechanistic basis for preser-vation of contractile function in Pim-WT hearts may rest with thecellular response in TAC-challenged animals. NTG and Pim-WTgroups injected with BrdU for 10 days were used to assess stimu-lation of DNA synthesis and potential cellular proliferation afterTAC challenge. Pim-WT hearts possess 67% more BrdU� myo-cytes relative to NTG controls after TAC challenge (Fig. S4a, *, P �0.05). The majority of BrdU� cells in Pim-WT hearts post-TAC arediploid (Fig. S4b) supporting the premise that increases in BrdU�cells stemmed from new myocyte formation and not enhancedDNA synthesis in preexisting cells. We now show that in additionto increased SERCA2a levels (12), Pim-WT hearts also showincreased levels of phosphorylated phospholamban (PLB) whilePim-DN animals show significant decreases in phospho-PLB com-pared to NTG control animals (Fig. S5). Interestingly, the ratio ofphospho-PLB over total PLB in Pim-WT animals is similar to theratio observed in NTG animals, while in Pim-DN animals this ratiois significantly decreased. These results suggest that in the face ofdecreased cardiac function, overexpression of Pim-1 allows theheart to maintain function through increased contractility throughelevation of SERCA2a (18) although phosphorylated PLB does notseem to play a major role in this protection.
Pim-1 Increases Myocardial Cellularity. The volume and cellularity ofmyocytes resulting from myocardial Pim-1 overexpression was assessedby quantitation of myocyte volume distribution. Results show Pim-WT
hearts possess an increased percentage of small myocytes (Fig. S6d)relative to NTG controls that is also reflected in decreased averagemyocyte size in these hearts (Fig. S6 a and b, **, P � 0.01), resulting ina hypercellular phenotype of �33% more myocytes in Pim-WT com-pared to NTG (Fig. S6c). Additionally, isolated Pim-DN myocytes were11% larger than NTG myocytes (Fig. S7) indicating an inverse effectwherein impaired Pim-1 activity prompts formation of larger myocytesin the transgenic heart.
DiscussionThe initial identification and characterization of myocardial Pim-1demonstrated this kinase is regulated by nuclear AKT accumula-tion and is cardioprotective in response to infarction challenge (12).Because nuclear Akt also possesses antihypertrophic properties (9),Pim-1 was circumstantially associated with possible inhibition ofhypertrophy. Inhibition of hypertrophy in vivo and in vitro indicatesPim-1 contributes to Akt-mediated blunting of hypertrophic re-modeling. Pim-1 is upregulated in localized regions close to acuteinjury or damage and is not increased throughout the myocardiumuntil initiation of transit to end-stage failure. Thus, Pim-1 likelyserves as a survival and protective response to blunt maladaptivehypertrophic remodeling in early phases of reactive signaling. Incomparison, Pim-1 elevation occurring in late-stage decompensa-tion probably represents a terminal effort to preserve function,although beneficial effects are overridden by the sequelae ofend-stage failure. The differential expression of endogenous Pim-1during transition from adaptive to maladaptive hypertrophy possi-bly represents a mechanism by which Pim-1 is partially protective.Targets of Pim-1 interaction contributing to protection are likely toinclude mediators of mitochondrial protection (12) and recentpublished findings regarding induction and stabilization of c-mycthrough Pim-1 (19), with ongoing studies exploring these mecha-nisms to determine their participation in the Pim-1 response.
Similar to our findings with nuclear AKT (9), Pim-1 overexpres-sion in cardiomyocytes inhibits ET-1-induced hypertrophy in vitro(Fig. 3) (Fig. S3a) and in vivo (Fig. 4). In addition, overexpressionof Pim-1 increases ANP transcript levels following endothelin-1administration while reducing �-MHC transcription and inhibitingincreases in other molecular markers of hypertrophy (Fig. 3D),
Fig. 4. Pim-WT transgenic animals are resistant to pressure-overload-induced hypertrophy. (A–F ) Line graphs representing weekly echocardiographicassessment of NTG and Pim-WT sham and TAC-banded hearts for anterior wall dimension (AWD, A), posterior wall dimension (PWD, B), end-diastolic dimension(EDD, C), end-systolic dimension (ESD, D), percentage of fractional shortening (FS, E), and ejection fraction (EF, F ) (NTG sham n � 6, Pim-WT sham n � 6, NTGTAC n � 9, Pim-WT TAC n � 9; *, P � 0.05, **, P � 0.01).
13892 � www.pnas.org�cgi�doi�10.1073�pnas.0709135105 Muraski et al.
reminiscent of effects observed using nuclear AKT (9). However,upon further analysis, ANP protein levels are significantly de-creased in both Pim-WT hearts and Pim-WT-infected NRCMs. Wetherefore believe that although ANP is induced in an acute areasurrounding the region of insult (Fig. 3A), on a global organ levelANP is not induced and therefore not a primary downstream targetthat mediates the Pim-1 protective response. Additionally, weinvestigated potential downstream targets of Pim-1 within the MAPkinase pathway, as both JNK and ERK have been proposed to becardioprotective. Although in vitro data do not seem to suggest
specific involvement of the MAPK pathway (Fig. S8b), in vivo dataindicates that both downstream targets JNK and ERK are signif-icantly affected by Pim-1 overexpression, whereby the ratio ofphosphorylated to total MAPK expression is significantly increasedcompared to NTG controls post-TAC banding (Fig. S8a). Collec-tively these results support the antihypertrophic actions of Pim-1,although overexpression models are not truly representative ofphysiological induction with respect to either localization or level.
Nuclear AKT delayed but did not overcome compensatoryremodeling after TAC challenge (9), but Pim-WT transgenic heartsexhibit persistent blunting of myocardial hypertrophy (Fig. 4)without increases in apoptosis, changes in hypertrophic signalingmarkers (Fig. 5), or deterioration of function (Fig. 6). In addition,Pim-WT transgenic hearts perform significantly better functionallythan NTG counterparts at 14 and 20 weeks of age (Fig. 6). Apotential basis for this remarkable resiliency to pressure overload isin part related to Pim-1-mediated induction of sarco/endoplasmicreticulum Ca�2ATP-ase 2a (SERCA2a) (12) and PLB expression.Studies have shown that relatively minor but significant 45%increases in SERCA2a expression postaortic banding are sufficientto recover function following pressure overload (20). Furthermore,transgenic mice overexpressing constitutively activated AKT in themyocardium exhibit increased contractility because of a 6.6-foldincrease in SERCA2a (21). In comparison, Pim-WT hearts exhibita 4.89-fold increase in SERCA2a (12). Phosphorylation of phos-pholamban is significantly increased in hearts from Pim-WT micerelative to NTG controls, while Pim-DN and Pim-KO animals havesignificantly reduced levels (Fig. S5). Decreases in phospholambanaccompany heart failure and overexpression of phosphorylatedphospholamban improves cardiac function through activation ofSERCA2a and decreases fibrosis in pathologically challenged myo-cardium (22). In addition, phospho-PLB:PLB ratio in Pim-DNanimals is significantly decreased compared to NTG and Pim-WTanimals. Thus, as Pim-DN negative animals have impaired con-tractile function (Fig. 2G), this decreased ratio of phospho-PLB tototal PLB may ultimately negatively affect SERCA activity andrepresents a potential mechanism for the depressed cardiac phe-notype observed in the Pim-DN animals. Furthermore, Pim-DNoverexpression leads to impaired contractile function and alteredcalcium handling (Fig. 2G), whereas overexpression of Pim-1 leadsto hypercontractile hearts with hyperdynamic calcium handling inresponse to pathological challenge (12). Thus, increased SERCA2aexpression in Pim-WT animals provides a plausible explanation forthe preservation of cardiac function observed in response to TACchallenge, although phospho-PLB does not appear to play a majorrole (Fig. S5).
Overexpression of Pim-DN in the myocardium increases myocyteapoptosis (Fig. 1D) with dilated cardiomyopathy, whereas geneticdeletion of Pim-1 also results in increased apoptosis althoughdilated cardiomyopathy is not observed (12). The cardiomyopathicconsequences of Pim-DN overexpression suggest this kinase iscritical to cardiomyocyte viability, as compensatory upregulation ofPim-2 occurring in the Pim-1 knockout line could help account forthe lack of cardiomyopathic changes (12). These findings are alsoconsistent with our previous study that shows the dominant-negative form of Pim-1 induces PARP and caspase-3 cleavage,increasing cardiomyocyte apoptosis in vitro (12). Furthermore forthe effect on hypertrophy, Pim-DN seems to be able to mount ahypertrophic response, evidenced by increased anterior wall thick-ness 1 week after TAC challenge (data not shown). Unfortunately,as we have previously reported (12) Pim-KO animals preclude anaccurate assessment of the antihypertrophic response elicited byPim-1 because of the potential compensatory actions of Pim-2 inthis system. At present dissecting the effect of Pim-1 inhibition inthe in vivo context will require an inducible inhibition systembecause of the potential confounding effects of Pim-DN expression.
Overexpression of Pim-1 in the pathologically challenged myo-cardium results in numerous salutary effects including decreased
A B
C D
E F
G
Fig. 5. Function is maintained in Pim-WT animals after TAC. (A) Kaplan–Meiersurvival curveofNTGandPim-WTanimals followingTACor shamoperation (NTGsham n � 6, Pim-WT sham n � 5, NTG TAC n � 8, Pim-WT TAC n � 8; **, P � 0.01vs. Pim-WTTAC). (B)Histogramdepictingheartweight/bodyweightratiosofNTGand Pim-WT sham and TAC hearts 4 weeks after the procedure (NTG sham n � 3,NTG TAC n � 4, Pim-WT sham n � 4, Pim-WT TAC n � 6; **, P � 0.01). (C) qRT–PCRanalysis of NTG and Pim-WT hearts 4 weeks after TAC banding for molecularmarkers of hypertrophy (n � 4, *, P � 0.05, **, P � 0.01 vs. sham; �, P � 0.01 vs.NTG sham). (D) Histogram representing the number of TUNEL positive myocytesper mm2 in NTG and Pim-WT hearts after banding (n � 3, **, P � 0.01). (E) Massontrichrome staining of NTG and Pim-WT paraffin sections 4 weeks after bandingand quantitation of collagen. (F) Quantitation of necrotic nuclei in NTG andPim-WT animals pre- and post-TAC banding **, P � 0.01. (G) Analysis of antiapo-ptotic protein expression in NTG and Pim-WT whole heart lysates post-TAC.Quantitation of results for Bcl-xl, Bcl-2, and phosphorylated Bad (pBAD) levels inNTG and Pim-WT hearts. GAPDH was used as a loading control. n � 3, *, P � 0.05,
**, P � 0.01 vs. GAPDH control. (G) Quantitation of Pim-WT hearts compared toNTG hearts post-TAC banding.
Muraski et al. PNAS � September 16, 2008 � vol. 105 � no. 37 � 13893
CELL
BIO
LOG
Y

apoptosis (Fig. 5D), increased expression of antiapoptotic proteins(Fig. 5G), and decreased fibrosis (Fig. 5E) and necrosis (Fig. S2).Pim-1 also increases the percentage of small myocytes and anoverall increase in the number of myocytes constituting the myo-cardium (Fig. S6c). Taken together, these data point to anotherpotential mechanism to account for increased capacity to withstandTAC challenge by virtue of increased cell numbers of small cells anddecreased cell death. These results were also previously postulatedfor the effect of nuclear AKT in the myocardium, which increasesthe resident cardiac progenitor pool, induces proproliferative cy-tokine expression, and increases the number of myocytes in theheart (11) (9). Future studies will more thoroughly elucidate theconnection between Pim-1 and cardiac stem cell pools to determinewhether Pim-1, like AKT, increases the resident pools of these cells.
On the basis of the collective results thus far, Pim-1 can beconsidered a potent cardioprotective and antihypertrophic mole-cule in the myocardium with effects mediated through increasedcell survival, decreased myocyte size, increased myocyte number,and increased Ca�2 reuptake. All of these properties have beenpreviously ascribed to AKT kinase (8–11, 21, 23), so with theconnection of Pim-1 as a downstream mediator of AKT activity itwill be important to determine the contribution of Pim-1 toactivities traditionally associated with AKT. By providing cardio-protective actions offered by AKT without undesired consequencesfor metabolic alterations and hypertrophic remodeling (8, 24),Pim-1 could become an important therapeutic agent in myocardialrepair and prevention of cardiac failure.
Materials and MethodsGeneration of Transgenic Animals and Animal Use. Pim-WT and Pim-DN cDNAfragments (16) were subcloned into the �-MHC plasmid for transgenesis. Priorpublications describe methods for TAC banding and echo (9), and HW:BW ratiodetermination and hemodynamics (12). Further details are provided in the onlinesupplement. All animal studies were approved by the Institutional Animal Useand Care Committee.
Confocal Microscopy, Immunoblotting, and Assays. GFP-Pim-1 proteins immu-noprecipitated from heart lysates were used in an in vitro kinase assay withGST-p21 as substrate. For luciferase assays, C2C12 cells transfected with indicatedplasmids and pGATA-Luc reporter construct were analyzed for GATA-dependentluciferase activity. Prior publications describe methods for immunofluorescencemicroscopy (25), immunoblotting (26), quantitative RT–PCR and TUNEL staining(9). Details are provided in SI Text.
In Vitro Cell Culture and Analyses. Neonatal rat cardiomyocyte cultures wereprepared as described previously (10). Adult myocyte isolation, volume calcula-tions, cell shortening, and Ca2� transient experiments were performed as previ-ously described (12, 22, 27). Additional details are provided in SI Text.
Statistical Analysis. Error bars are represented as plus and minus standard errorof the mean (SEM), except where indicated. Statistical analysis was performedusing Student’s t test and ANOVA with Tukey’s posthoc. P � 0.05 consideredsignificant.
ACKNOWLEDGMENTS. The authors thank members of the Sussman laboratoryfor helpful discussion and comments. Dr. Sussman is supported by NationalInstitutes of Health grants 5R01HL067245, 1R01HL091102, 1P01HL085577, and1P01AG023071 (to P.A.). J.M., K.F., and N.G. are Fellows of the Rees-StealyResearch Foundation and the San Diego State University Heart Institute. N.M. isfunded by National Institutes of Health Grant CA104470.
1. Neri LM, Borgatti P, Capitani S, Martelli AM (2002) The nuclear phosphoinositide 3-kinase/AKT pathway: A new second messenger system. Biochim Biophys Acta 1584:73–80.
2. Sugden PH (2003) Ras, Akt, and mechanotransduction in the cardiac myocyte. Circ Res93:1179–1192.
3. McGowan BS, Ciccimaro EF, Chan TO, Feldman AM (2003) The balance betweenpro-apoptotic and anti-apoptotic pathways in the failing myocardium. CardiovascToxicol 3:191–206.
4. Kumar D, Lou H, Singal PK (2002) Oxidative stress and apoptosis in heart dysfunction.Herz 27:662–668.
5. Hardt SE, Sadoshima J (2002) Glycogen synthase kinase-3beta: A novel regulator ofcardiac hypertrophy and development. Circ Res 90:1055–1063.
6. Sussman MA, Anversa P (2004) Myocardial aging and senescence: Where have the stemcells gone? Annu Rev Physiol 66:29–48.
7. Latronico MV, Costinean S, Lavitrano ML, Peschle C, Condorelli G (2004) Regulation of cellsize and contractile function by AKT in cardiomyocytes. Ann NY Acad Sci 1015:250–260.
8. Condorelli G, et al. (2002) Akt induces enhanced myocardial contractility and cell sizein vivo in transgenic mice. Proc Natl Acad Sci USA 99:12333–12338.
9. Tsujita Y, et al. (2006) Nuclear targeting of Akt antagonizes aspects of cardiomyocytehypertrophy. Proc Natl Acad Sci USA 103:11946–11951.
10. Shiraishi I, et al. (2004) Nuclear targeting of Akt enhances kinase activity and survivalof cardiomyocytes. Circ Res 94:884–891.
11. Gude N, et al. (2006) Akt promotes increased cardiomyocyte cycling and expansion ofthe cardiac progenitor cell population. Circ Res 99:381–388.
12. Muraski J, et al. (2007) Pim-1 regulates cardiomyocyte survival downstream of Akt. NatMed 13:1467–1475.
13. Hammerman PS, Fox CJ, Birnbaum MJ, Thompson CB (2005) Pim and Akt oncogenes areindependent regulators of hematopoietic cell growth and survival. Blood 105:4477–4483.
14. Fox CJ, et al. (2003) The serine/threonine kinase Pim-2 is a transcriptionally regulatedapoptotic inhibitor. Genes Dev 17:1841–1854.
15. Wang Z, et al. (2001) Pim-1: A serine/threonine kinase with a role in cell survival,proliferation, differentiation and tumorigenesis. J Vet Sci 2:167–179.
16. Bhattacharya N, et al. (2002) Pim-1 associates with protein complexes necessary formitosis. Chromosoma 111:80–95.
17. Zhang Y, Wang Z, Magnuson N (2007) Pim-1 kinase-dependent phosphorylation of p21Cip1/WAF1 regulates its stability and cellular localization in H1299 cells. Mol Cancer Res 5:909–922.
18. Prasad AM, et al. (2007) Phenylephrine hypertrophy, Ca2�-ATPase (SERCA2), and Ca2�signaling in neonatal rat cardiac myocytes. Am J Physiol Cell Physiol 292:C2269–2275.
19. Zhang Y, Wang Z, Li X, Magnuson N (2008) Pim kinase-dependent inhibition of c-Mycdegradation. Oncogene, 10.1038/onc.2008.123.
20. Suarez J, et al. (2004) Oxycycline inducible expression of SERCA2a improves calciumhandling and reverts cardiac dysfunction in pressure overload-induced cardiac hyper-trophy. Am J Physiol Heart Circ Physiol 287:H2164–2172.
21. Kim YK, et al. (2003) Mechanism of enhanced cardiac function in mice with hypertro-phy induced by overexpressed Akt. J Biol Chem 278:47622–47628.
22. Hoshijima M, et al. (2002) Chronic suppression of heart-failure progression by apseudophosphorylated mutant of phospholamban via in vivo cardiac rAAV genedelivery. Nat Med 8:864–871.
23. Rota M, et al. (2005) Nuclear targeting of Akt enhances ventricular function andmyocyte contractility. Circ Res 97:1332–1341.
24. Seimi SK, et al. (2004) Glycogen synthase kinase-3beta is involved in the process ofmyocardial hypertrophy stimulated by insulin-like growth factor-1. Circ J 68:247–253.
25. Fransioli J, et al. (2008) Evolution of the c-kit positive cell response to pathologicalchallenge in the myocardium. Stem Cells 26(5):1315–24.
26. Kato T, et al. (2005) ANP promotes cardiomyocyte survival by cGMP-dependent nuclearaccumulation of Zyxin and Akt. J Clin Invest 115:2716–2730.
27. Kajstura J, et al. (1995) The cellular basis of pacing-induced dilated cardiomyopathy.Myocyte cell loss and myocyte cellular reactive hypertrophy. Circulation 92:2306–2317.
Fig. 6. Pim-1 enhances cardiac function. (A–C) In vivohemodynamic assessment of NTG and Pim-WT hearts 4and 10 weeks (black and gray bars, respectively) aftersham or TAC operation (14 and 20 weeks of age, respec-tively). �dP/dt measurements (A), left ventricular devel-oped pressure (LVDP, B), left ventricular end diastolicpressure (LVEDP, C). For 4-week animals NTG sham n � 4,NTG TAC n � 3, Pim-WT sham n � 4, Pim-WT TAC n � 4.For 10-week animals NTG sham n � 5, NTG TAC n � 10,Pim-WT sham n � 14, Pim-WT TAC n � 7 (*, P � 0.05, **,P � 0.01 vs. sham, ##, P � 0.01 vs. 4-week TAC, $, P � 0.05,$$, P � 0.01 vs. NTG TAC, �, P � 0.05, ��, P � 0.01 vs. NTGsham).
13894 � www.pnas.org�cgi�doi�10.1073�pnas.0709135105 Muraski et al.





























