Embed Size (px)
Citation preview

Pc
EEa
Hb
a
ARRAA
KBAPRDD
1
gpdPbfTst
I1
S
0d
Mutation Research 686 (2010) 57–67
Contents lists available at ScienceDirect
Mutation Research/Fundamental and MolecularMechanisms of Mutagenesis
journa l homepage: www.e lsev ier .com/ locate /molmutCommuni ty address : www.e lsev ier .com/ locate /mutres
arp1 activation in mouse embryonic fibroblasts promotes Pol �-dependentellular hypersensitivity to alkylation damage
lena Jelezcovaa, Ram N. Trivedia, Xiao-hong Wanga, Jiang-bo Tanga,b, Ashley R. Browna,va M. Goellnera, Sandy Schamusa, Jamie L. Fornsaglioa,1, Robert W. Sobola,b,∗
Department of Pharmacology & Chemical Biology, University of Pittsburgh School of Medicine & University of Pittsburgh Cancer Institute,illman Cancer Center, Pittsburgh, PA 15213, United StatesDepartment of Human Genetics, University of Pittsburgh Graduate School of Public Health, Pittsburgh, PA 15261, United States
r t i c l e i n f o
rticle history:eceived 11 November 2009eceived in revised form 10 January 2010ccepted 14 January 2010vailable online 22 January 2010
eywords:ase excision repairlkylating agentsarp1NAi
a b s t r a c t
Alkylating agents induce cell death in wild-type (WT) mouse embryonic fibroblasts (MEFs) by multiplemechanisms, including apoptosis, autophagy and necrosis. DNA polymerase � (Pol �) knockout (KO)MEFs are hypersensitive to the cytotoxic effect of alkylating agents, as compared to WT MEFs. To testthe hypothesis that Parp1 is preferentially activated by methyl methanesulfonate (MMS) exposure ofPol � KO MEFs, we have examined the relationship between Pol � expression, Parp1 activation and cellsurvival following MMS exposure in a series of WT and Pol � deficient MEF cell lines. Consistent withour hypothesis, we observed elevated Parp1 activation in Pol ˇ KO MEFs as compared to matched WTMEFs. Both the MMS-induced activation of Parp1 and the MMS-induced cytotoxicity of Pol ˇ KO MEFsare attenuated by pre-treatment with the Parp1/Parp2 inhibitor PJ34. Further, elevated Parp1 activationis observed following knockdown (KD) of endogenous Pol �, as compared to WT cells. Pol � KD MEFs are
NA glycosylaseNA polymerase beta
hypersensitive to MMS and both the MMS-induced hypersensitivity and Parp1 activation is prevented bypre-treatment with PJ34. In addition, the MMS-induced cellular sensitivity of Pol ˇ KO MEFs is reversedwhen Parp1 is also deleted (Pol ˇ/Parp1 double KO MEFs) and we observe no MMS sensitivity differentialbetween Pol ˇ/Parp1 double KO MEFs and those that express recombinant mouse Pol �. These studiessuggest that Parp1 may function as a sensor of BER to initiate cell death when BER is aborted or fails.Parp1 may therefore function in BER as a tumor suppressor by initiating cell death and preventing the
h chr
accumulation of cells wit. Introduction
DNA polymerase � (Pol �) plays a critical role in the repair ofenomic base damage and is considered the predominant DNAolymerase in the base excision repair (BER) pathway [1]. Theevelopment of mouse embryonic fibroblasts (MEFs) deficient inol � clearly defined a requirement for Pol � in the repair ofase damage mediated by the BER pathway [2]. Pol � is a bi-
unctional, two-domain, single-polypeptide 39 kDa enzyme [3].he polymerase activity responsible for gap-filling DNA synthe-is in BER resides in the C-terminal 31 kDa domain whereashe N-terminal 8 kDa domain encodes a 5′deoxyribose phosphate∗ Corresponding author at: Hillman Cancer Center, University of Pittsburgh Cancernstitute, Research Pavilion, Suite 2.6a, 5117 Centre Avenue, Pittsburgh, PA 15213-863, United States. Tel.: +1 412 623 7764; fax: +1 412 623 7761.
E-mail addresses: [email protected], [email protected] (R.W. Sobol).1 Current address: Division of Natural Health Sciences, Seton Hill University, One
eton Hill Drive, Greensburg, PA 15601, United States.
027-5107/$ – see front matter © 2010 Elsevier B.V. All rights reserved.oi:10.1016/j.mrfmmm.2010.01.016
omosomal damage due to a BER defect.© 2010 Elsevier B.V. All rights reserved.
(5′dRP) lyase activity that removes the sugar-phosphate lesion(5′dRP) [3]. The 5′dRP lesion is the product of AP endonuclease 1(Ape1)-mediated hydrolysis of the abasic site generated follow-ing DNA glycosylase action [3]. The polymerase function of Pol� is responsible for gap-filling DNA synthesis in both the short-patch and long-patch sub-pathways of BER [4] yet alternate DNApolymerases have been demonstrated to complement the long-patch BER related DNA synthesis activity of Pol � in its absencewhen evaluated in cell extracts in vitro [5]. It is possible that thepolymerase activity of Pol � is also complemented in vivo, as itis the 5′dRP lyase function of Pol � [6] that is essential and suf-ficient for alkylating agent resistance in MEFs [7] and in humanbreast cancer cells [8]. In the absence of Pol �, cells are unableto efficiently repair the highly toxic 5′dRP moiety and thereforeare hypersensitive to the cytotoxic effect of different alkylating
agents such as methylmethane sulfonate (MMS), N-methyl-N-nitrosourea and N-methyl-N′-nitro-N-nitrosoguanidine (MNNG)[2], the thymidine analog 5-hydroxymethyl-2′-deoxyuridine [9]as well as the therapeutic alkylating agent temozolomide[8–10].
5 on Res
tettseamitet(ptutt
dgpdr[tsdtrs([ois5b
erbbSpiCsaiPPtwtlsBPcibo
t
8 E. Jelezcova et al. / Mutati
The 5′dRP lyase enzymatic activity, an essential BER gap-ailoring function, was first identified in mammalian cells as annzymatic activity due to a 47 kDa enzyme [10] but was later showno be encoded by Pol � [6]. It is somewhat surprising howeverhat the 5′dRP lyase function of Pol � is not complemented in vivoince there are several mammalian proteins that have 5′dRP lyasenzymatic activity. In addition to Pol �, DNA polymerase � (Pol �)nd DNA polymerase � (Pol �), as well as the mitochondrial poly-erase Pol � have 5′dRP lyase activity and can function (in vitro)
n the removal of the 5′dRP BER intermediate [11–16]. Further,he endonuclease VIII-like DNA glycosylases NEIL1 and NEIL2 bothxcise 5′dRP lesions with similar efficiency to Pol � [17]. However,he severe 5′dRP-mediated cell death observed in Pol ˇ knockoutKO) MEFs [7] and Pol � deficient human cells [8] indicates thatotential complementing activities provide little, if any, repair ofhe 5′dRP lesion generated during BER. This would suggest that then-repaired 5′dRP lesion is not freely and readily available by addi-ional cellular proteins outside of the BER complex that forms athe lesion site [1].
The molecular mechanism responsible for 5′dRP-mediated celleath is unknown. However, left un-repaired, 5′dRP lesions trig-er mouse fibroblast and human breast cancer cell death via a53-independent mechanism that does not involve apoptosis andoes not induce the formation of autophagosomes [8,18]. Initialeports suggested a possible apoptotic mechanism of cell death19] concomitant with an elevated level of chromosome aberra-ions following alkylating agent exposure [19]. It was subsequentlyuggested that the observed apoptosis was the result of DNAouble-strand breaks (DSBs) that form secondary to failed BER (inhe absence of Pol �) following replication [20]. The formation ofeplication-dependent DSBs in Pol ˇ KO MEFs following MMS expo-ure is consistent with the increase in sister chromatid exchangeSCE) events observed in Pol ˇ KO MEFs following MMS treatment18]. However, the significantly long time-frame for the appearancef DSBs is not consistent with the relatively rapid (2 hrs) alterationsn the cell cycle observed in Pol ˇ KO MEFs following MMS expo-ure [18]. We have therefore hypothesized that the un-repaired′dRP lesion functions as a signal for cell death, possibly via proteininding and activation.
The DNA binding and signaling molecules Parp1 and Parp2 haveach been implicated in BER [1], although the precise enzymaticole of either Parp isoform in BER is un-defined. Robust BER cane recapitulated in vitro in the absence of Parp1 [21] and neitheracteria (Escherichia coli) nor yeast (Saccharomyces cerevisiae orchizosaccharomyces pombe) expresses a Parp homolog yet each isroficient in BER [22]. Interestingly, BER has been reported as both
mpaired [23] and proficient [24] in the absence of Parp1 [23,24].urrent models suggest that Parp1 and possibly Parp2 function ascaffold proteins in BER, facilitating the formation of BER complexest the site of DNA damage via multiple protein–protein interactions,ncluding interactions with Pol �, Xrcc1 and DNA LigIII� [1,25].arp1 was observed to bind to the lesion site prior to Xrcc1 orol � [26,27]. Further, Parp1 was found in a complex of BER pro-eins cross-linked to a BER intermediate [28] and more recently itas observed that Parp1 and Ape1 from MEF extracts compete for
he same 5′dRP BER intermediate [29]. It has been postulated thatocal, strand-break induced activation of Parp1 and the resultantynthesis of poly(ADP-ribose) (PAR) facilitates recruitment of theER proteins Xrcc1 and Pol � to stimulate DNA repair [30]. In short,arp1 (and Parp2) are involved in BER complex formation [1], Parp1an recognize and bind to BER intermediates [29], Parp1 activation
s observed upon BER initiation [30] and in un-related studies, it haseen shown that Parp1 hyperactivation is recognized as an inducerf necrotic cell death [31].We have hypothesized that Parp1 is the signaling protein thatriggers cell death in MEFs when 5′dRP lesions are not repaired.
earch 686 (2010) 57–67
In this study, we used genetically defined Pol ˇ KO and knock-down (KD) mouse cell lines to define a role for Parp1 in the cellularresponse to alkylation damage in MEFs when BER fails or is aborted.These studies suggest that Parp1 (and likely Parp2) may functionnot only in BER protein recruitment but may also act as a sensor ofcompleted BER and therefore Parp1 may be considered a BER sensorprotein. As a BER sensor or checkpoint protein, Parp1 may there-fore function as a tumor suppressor by preventing chromosomaldamage that could accumulate due to a BER defect.
2. Materials and methods
2.1. Chemicals and reagents
Cell culture media and supplies where from InVitrogen-Gibco (Carlsbad, CA).Fetal bovine serum was from Atlanta Biologicals (Lawrenceville, GA). Methylmethanesulfonate (MMS), DTT, Phosphocreatine di(tris) salt, Creatine Phospho-kinase and NAD+ were purchased from Sigma-Aldrich (St. Louis, MO). ATP andParp Inhibitor VIII (PJ34) were obtained from GE Healthcare (Piscatway, NJ) andCalbiochem (San Diego, CA), respectively. The hairpin DNA substrate with a 3′-biotinylated end was synthesized by Integrated DNA Technologies (IDT; Coralville,IA). We used the following primary antibodies: DNA polymerase beta Ab-2; clone 61(NeoMarker, Fremont, CA); Ape1 Ab (EMD Biosciences, Inc., San Diego, CA); Xrcc1 Ab(Bethyl Labs, Montgomery, TX); Parp1 mAb (Novus Biologicals, Littleton, CO); PCNAmAb (Santa Cruz Biotechnology, Santa Cruz, CA); PAR Ab (BD Biosciences, San Jose,CA); anti-human MPG (Mab; clone 506-3D)[8]. Secondary antibodies GAM-HRP andGAR-HRP conjugates and signal generation substrates were from Bio-Rad (Hercules,CA) and Pierce (Rockford, IL), respectively. All electrophoresis reagents were fromBio-Rad (Hercules, CA).
2.2. Cell lines and culture conditions
Transformed WT MEF cells (16tsA, clone 1B5; 36.3; 53TAg and 92TAg), Pol ˇKO MEF cells (38�4; 50TAg and 88TAg), Pol ˇ/Pol � double KO MEF cells (19tsA,clone 2B2) and Pol ˇ/Parp1 double KO MEF cells have been described previously[2]. Transformed WT MEFs HCC384 were derived from c57Bl/6 day 14 embryos, asdescribed previously [2]. The cell lines 16tsA, clone 1B5; 19tsA, clone 2B2; 88TAgand 92TAg are available from the ATCC. 92TAg cells engineered to express MPG havebeen reported previously [32]. The Pol ˇ/Parp1 double KO MEF cells were kindlyprovided by Dr. J. Menissier-de Murcia (CNRS). MEFs were cultured at 37 ◦C in ahumidified incubator with 10% CO2 in DMEM supplemented with 10% fetal bovineserum, penicillin (50 units/mL), streptomycin (50 �g/mL) and Glutamax (4 mmol/L).The Pol ˇ/Parp1 double KO MEF cells were then immortalized with SV40 T-antigen,as we have previously described [32].
2.3. Cloning and expression of mouse Pol ˇ
Total RNA was isolated from 5 × 106 92TAg cells using Qiagen RNAeasy reagents.Total cDNA was prepared using a first-strand cDNA synthesis kit (InVitrogen) asdescribed by the manufacturer, using oligo-dT as the primer. The mouse Pol �cDNA was then amplified by PCR using primers mBetaF (caccatgagcaaacgcaag-gcgccg) and mBetaR (tcattcacttctatccttggg). EGFP cDNA was also amplified by PCRfrom pIRES2-EGFP (Clontech) using primers FEGFP (caccatggtgagcaagggcgagga) andREGFP (ttacttgtacagctcgtccatgccgagag). Each cDNA was then cloned via a Topoiso-merase cloning procedure into the pENTR-D cloning plasmid (InVitrogen), as perthe manufacturers protocol. The cloned mouse Pol � cDNA and EGFP cDNA wereboth sequence verified to be identical to that reported earlier (NM 011130.1 andwww.addgene.com; respectively). The mouse Pol � open reading frame (ORF) andthe EGFP ORF were each transferred from pENTR-mPol � or pENTR-EGFP; respec-tively, to a Gateway modified pIRES-Puro plasmid via LR recombination, as per themanufacturer (InVitrogen).
Pol ˇ KO (88TAg) and Pol ˇ/Parp1 double KO MEF cells were modified to expresstransgenic mouse Pol � or EGFP as follows: briefly, 1.5 × 105 cells were seededinto 60 mm dishes and incubated for 24–30 h at 10% CO2 at 37 ◦C. The mouse Pol� expression plasmid (pIRES-Puro-mBeta) or the EGFP expression plasmid (pIRES-Puro-EGFP) was transfected using FuGene 6 Transfection Reagent (Roche DiagnosticCorp, Indianapolis, IN) according to the manufacturer’s instructions. Stable cell lineswere selected in puromycin (7 �g/ml) for 2 weeks, individual clones (stably express-ing mouse Pol � or EGFP) were amplified and 30 �g of nuclear extract was analyzedby immunoblotting for the expression of mouse Pol � protein as described below.
2.4. Lentivirus expressing mouse Pol ˇ specific shRNA
We have previously described a highly effective retroviral shRNA vector spe-cific for mouse Pol � [32]. Using the same target sequence (gatcagtactactgtggtg),we cloned the corresponding hairpin sequence into the lentiviral vector pLL3.7[33]. Lentiviral particles were generated by transfection of four plasmids (the con-trol plasmid pLL3.7 or the mouse Pol �-specific shRNA plasmid pLLOXmPol �,

on Research 686 (2010) 57–67 59
pFmps(a2ccbaa
2
dp01paK
rawtwe(Ppww
2
2w2ripLfbg
2
tdPpPtt2MzRr
3
3a
Pcrac
Fig. 1. WT and Pol � KO MEFs used in this study. Expression of the DNA repair
E. Jelezcova et al. / Mutati
lus pMD2.g (VSVG), pRSV-REV and pMDLg/pRRE) into 293-FT cells [33,34] usinguGene 6 transfection reagent (Roche Diagnostic Corp., Indianapolis, IN). Cultureedia from transfected cells was collected 48 h after transfection to isolate the viral
articles, passed through 0.45 �m filters, used immediately or stored at −80◦C iningle-use aliquots. Transduction of 92TAg and HCC384 cells with control lentivirusGFP expression only) and mouse Pol � specific shRNA lentivirus was completeds follows: briefly, 6.0 × 104 cells were seeded into 6-well plates and incubated for4–30 h at 10% CO2 at 37 ◦C. Cells were transduced for 18 h with virus at 32 ◦C andultured for 72 h at 37 ◦C and then seeded in 96-well plates at limiting dilution (0.3ells/well) for the isolation of single-cell clones. Individual clones (control and sta-le KD of mouse Pol � protein) were amplified and 30 �g of nuclear extract wasnalyzed by immunoblotting for the expression of endogenous mouse Pol � proteins described below.
.5. Cell extract preparation and immunoblot assays
Preparation of whole cell extract from 92TAg cells expressing MPG was asescribed [35]. Briefly, cells were washed three times with ice cold PBS and theellet was suspended in buffer containing 20 mM Hepes-KCl, pH 7.9, 100 mM KCl,.2 mM EDTA, 10% glycerol and 1 mM DTT. Cells were disrupted by sonication for0 s (four times) with 30-s incubations on ice in between each burst. The cell sus-ension was centrifuged for 20 min at 16400 RPM (4 ◦C) to obtain whole cell extractnd dialyzed overnight against re-suspension buffer (25 mM Hepes pH 7.9, 100 mMCl, 12 mM MgCl2, 1 mM EDTA, 5% Glycerol, 2 mM DTT).
Nuclear extracts were prepared using the NucBuster nuclear protein extractioneagent (Novagen). Protein concentration was determined using Bio-Rad proteinssay reagents according to the manufacturer’s instructions. Nuclear protein (30 �g)as separated by electrophoresis in a 4–12% SDS-polyacrylamide gel and electro-
ransferred to a 0.45 �m nitrocellulose membrane (Trans-Blot, Bio-Rad). Antigensere detected using standard protocols. Primary antibodies: anti-Pol � (NeoMark-
rs, #MS-1402-P0), 1000×; anti-Ape1 (Novus, #NB-100-116), 3000×; anti–Xrcc1Bethyl Labs, #A300-065A) 1000×; anti-Parp (Novus, #NB-100-112) 3000×; anti-CNA (Santa Cruz, #sc-56) 1000×; anti-MPG [8] 1000× and the horseradisheroxidase (HRP)-conjugated secondary antibody (GAM-HRP or GAR-HRP; Bio-Rad)ere diluted 10,000× in TBST/5% milk. Each membrane was stripped and re-probedith anti-PCNA antibodies to correct for differences in protein loading.
.6. PAR assay
Cells (1.5 × 106) where seeded in 100 mm dishes 24 h before treatment. After4 h, media was removed and replaced with fresh media or media supplementedith Parp Inhibitor VIII (PJ34; Calbiochem, #528150) at a final concentration of�M. After 30 min, cells were lysed immediately (0 time point) or media was
eplaced with MMS at a final concentration of 1.25 mM (diluted in media) andncubated for 15 or 30 min, as indicated in the figure legends. Extracts were pre-ared by washing the cells with PBS and preparing cell extract with 400 �l of 2×aemmli Buffer. 20 �l of the cell extract was analyzed by immunoblot with a 1000-old dilution of an anti-PAR primary antibody (BD Pharmingen, #551813) followedy a 5000-fold dilution of the horseradish peroxidase (HRP)-conjugated secondaryoat anti-rabbit Ab (Pierce; cat#1858415).
.7. Cell cytotoxicity assays
MMS-induced cytotoxicity was determined by growth inhibition assays, essen-ially as we have described previously [32]. Briefly, cells were seeded in 96-wellishes at 1200 cells per well in two sets. After 24 h one set was pre-treated with thearp inhibitor PJ34 at a final concentration of 2 �M. After 30 min of media or PJ34re-treatment, cells were treated with a range of concentrations of MMS for 1 h (+/−J34, 2 �M). Drug-containing medium was replaced with fresh media or media con-aining PJ34 (2 �M) and the plates were incubated at 37◦C for 48 h at which pointhe total cell number was determined by a modified 3-(4,5-dimethylthiazol-2-yl)-,5-diphenyltetrazolium bromide (MTT) assay (MTS; Promega, Madison, WI) [36].etabolically active cells were quantified by the bioreduction of the MTS tetra-
olium compound by recording absorbance at 490 nm using a microplate reader.esults were calculated from the average of four separate experiments and areeported as the % of treated cells relative to the cells in control wells (% Control).
. Results
.1. Increased Parp1 activation following MMS exposure in thebsence of Pol ˇ
These studies were designed to address our hypothesis that
arp1 is the signaling protein that triggers cell death in MEFs whenells are deficient in Pol � and the BER intermediate 5′dRP is notepaired. We utilized a series of SV40 T-Antigen transformed WTnd Pol ˇ KO MEF cell lines. As depicted in Fig. 1, each of theell lines have similar expression levels of the BER proteins Parp1,proteins Pol �, Ape1, Xrcc1, Parp1 and PCNA in MEFs as determined by immunoblotanalysis of nuclear proteins isolated from the following MEF cells: 92TAg (lane 1),88TAg (lane 2), 53TAg (lane 3), 50TAg (lane 4), 36.3 (lane 5), 38�4 (lane 6), 16tsa(lane 7) and 19tsA (lane 8). .
Xrcc1 and Ape1. Our human MPG antibody [8] does not cross-reactwith mouse Mpg/Aag and repeated attempts to validate antibodiesspecific for mouse Mpg were unsuccessful. However, as we havereported [32], Mpg glycosylase activity of both MEF WT and MEFPol ˇ knockout (KO) cell lines is low but uniform and suggests thatMEFs generally have a low, yet sufficient level of expression of Mpgto initiate repair of the MMS-induced lesions, in-line with a previ-ous report from one of us [18]. Further, these studies confirm thatthe WT cells all express similar levels of Pol � protein (lanes 1, 3,5 and 7) and as expected, no Pol � protein is detected in the Pol ˇKO cell lines (lanes 2, 4, 6 and 8). All of the cell lines express Pol �except for the Pol ˇ/Pol � double KO cell line 19tsA, as we and othershave reported [37,38].
Parp1 is part of the BER machinery [37] and can be found ina complex with other BER proteins, as was shown previously bytwo different experimental methodologies [26,28]. Using a simi-lar approach, we confirmed that Parp1, as well as the BER proteinsApe1, Pol �, MPG and Xrcc1 can bind to a BER substrate by incu-bating a hairpin oligonucleotide containing a single MPG substrate(Etheno-2′-deoxyAdenosine) with whole cell extracts preparedfrom MEFs expressing human MPG [32]. Proteins bound to thesubstrate were cross-linked by incubation with formaldehyde (seeSupplementary data and methods, Appendix A) and purified fromunbound proteins by affinity purification via the biotin moietylinked to the 3′ end of the DNA hairpin oligonucleotide, essentiallyas described [26]. Purified proteins were released from the DNA byheating to reverse the formaldehyde-induced cross-links, therebyseparating the protein complexes. The purified BER proteins werethen analyzed by immunoblot, as shown in Fig. S1. This is simi-lar to that reported by Parsons and Dianov [26] and importantly,
demonstrates that Parp1 binds to a BER substrate, as do the otherBER proteins MPG, Ape1, Xrcc1 and Pol �.Parp1 binding to a BER substrate supports the current BERmodel in which Parp1 is recruited to the lesion site to facilitateBER complex formation, including the recruitment of Xrcc1/DNA

60 E. Jelezcova et al. / Mutation Research 686 (2010) 57–67
Fig. 2. Preferential Parp1 activation in Pol ˇ KO cells following MMS exposure. Parp1 activation was determined in MEF cell extract by immunoblot analysis, measuring thes anel),( �4 (PoP own
lt(tWaatibp
ynthesis of PAR in the absence (left panel) or in the presence of PJ34 (2 �M, right pPol ˇ KO); Panel A], [53TAg (WT) and 50TAg (Pol ˇ KO); Panel B], [36.3 (WT) and 38arp1 and PCNA expression was determined by immunoblot. PCNA expression is sh
igase III and Pol � to complete repair [1,26,40]. Once bound tohe lesion site, Parp1 undergoes rapid, low-level auto-modificationADP-ribosylation). This self-imposed post-translational modifica-ion helps facilitate recruitment of Xrcc1 to complete repair [38].
e therefore reasoned that inability to complete repair due to thebsence of Pol � would perpetuate the DNA strand-break induced
ctivation of Parp1, preferentially in the Pol � KO cells. Activa-ion of Parp1 in cells can be monitored by the presence of PARn whole cell lysates, probing for PAR synthesis and accumulationy immunoblot analysis. In support of this hypothesis, DNA repairroficient cells (WT) show little or no evidence of Parp1 activa-after exposure with MMS (1.25 mM) for 0, 15 or 30 min in [92TAg (WT) and 88TAgl ˇ KO); Panel C] and [16tsa (WT) and 19tsA (Pol ˇ/Pol � double KO); Panel D] MEFs.
as a loading control.
tion following challenge to 1.25 mM MMS (Fig. 2A–D, left panel).Note however that over-exposure of these immunoblots revealsa very low level of Parp1 activation in WT cells (Fig. 4C and datanot shown). However, cells deficient in Pol � expression show arapid and robust activation of Parp1 within 15 min after exposureof the cells to the same low dose of MMS (1.25 mM). Further, an
even greater level of Parp1 activation is observed 30 min after MMStreatment (Fig. 2A–D, left panel). In 3 of the 4 Pol ˇ KO cell lines, allof the observed Parp1 activation can be blocked by a 30 min pre-treatment with the Parp1/Parp2 inhibitor PJ34 (2 �M) (Fig. 2A–C,right panel). In the fourth Pol ˇ KO cell line (19tsA), a majority of
on Res
t(df
3r
cMmaaotiHrpKptcvc4le[cetˇMwacwfsicPwcc�To(ttcts
3a
mecfa
E. Jelezcova et al. / Mutati
he Parp1 activation can be blocked by pre-incubation with PJ34Fig. 2D, right panel). These data show very clearly that failed BERue to loss of Pol � leads to rapid and robust Parp1 activationollowing DNA damage induction mediated by MMS exposure.
.2. Inhibition of Parp1 in Pol ˇ null MEFs provides partialesistance to MMS
The hallmark phenotype of Pol ˇ KO MEFs is an increase inytotoxicity following exposure to alkylating agents such as MMS,NNG and temozolomide [2]. We therefore evaluated the fouratched WT and Pol ˇ KO cell pairs for sensitivity to the alkylating
gent MMS, as this has been the most commonly used alkylatinggent for the characterization of Pol ˇ KO cells [2]. Based on ourbservation that MMS induces an elevated level of Parp1 activa-ion in Pol ˇ KO MEFs (as compared to WT MEFs) and that Parp1nhibition can protect MEFs, cortical neurons, PC12, SH-SY5Y andeLa cells from DNA damage-induced cell death [31,39–42], we
easoned that a potent Parp inhibitor may provide some level ofhenotypic rescue, leading to an increase in the survival of Pol ˇO cells exposed to MMS. Parp1 and Parp2 were inhibited by eitherre-treating cells (30 min) with PJ34 before addition of MMS orreating cells simultaneously with PJ34 and MMS. PJ34 was used at aoncentration (2 �M) sufficient to prevent MMS-induced Parp acti-ation in the Pol ˇ KO cells (Fig. 2). After an hour of MMS exposure,ells were re-fed normal growth media (+/− PJ34, 2 �M), grown for8 h and surviving cells were measured by an MTS assay (Fig. 3). In-
ine with reports that Parp inhibition can potentiate the cell killingffect of many DNA damaging agents, including alkylating agents38,43–45], blocking Parp activation in the WT MEFs increased theell killing effect of MMS at most doses (Fig. 3). This potentiationffect was observed for both the pre-treatment and simultaneousreatment regimen. However, the MMS-induced cytotoxicity of Pol
KO cells was not increased by Parp inhibition. Conversely theMS-induced cytotoxicity of Pol ˇ KO cells was reduced when Parpas inhibited. Two of the Pol ˇ KO MEF cell lines (88TAg; Fig. 3A
nd E and 50TAg; Fig. 3B and F) showed a marked improvement inell survival when Parp was inhibited and the cells were challengedith MMS, as evidenced by as much as a 10-fold increase in survival
or the +PJ34 treated Pol ˇ KO cells at the highest doses of MMS. Inome cases, the survival curve of the Pol ˇ KO cells +PJ34 was sim-lar to the WT cells treated with PJ34 (Fig. 3A and F). One Pol ˇ KOell line (38�4) showed an intermediate or variable response to thearp inhibitor. Whereas simultaneous treatment of the 38�4 cellsith PJ34 and MMS restored survival equal to that of the WT (36.3)
ells (MMS + PJ34, Fig. 3G), pre-treatment with PJ34 provided res-ue at only the highest doses of MMS (Fig. 3C). Finally, the Pol ˇ/Poldouble KO cell line was only mildly rescued by Parp inhibition.his was not entirely unexpected, as PJ34 treatment was found tonly prevent some (a majority) of the MMS-induced Parp activationFig. 2D, right panel). The involvement of Parp1 in the hypersensi-ive phenotype of Pol ˇ KO MEFs is consistent with earlier reportshat un-repaired 5′dRP lesions trigger MEF [7,32] and human breastancer [8] cell death via a mechanism that does not involve apop-osis and does not induce the formation of autophagosomes [8],uggesting a necrotic form of cell death [31].
.3. Pol ˇ down-regulation in WT MEFs leads to elevated Parpctivation and Parp-dependent cell death following MMS exposure
The general trend observed in Pol ˇ KO cells is that MMS treat-
ent leads to hyperactivation of Parp1 that contributes to thelevated level of cell death in Pol ˇ KO cells as compared to WTells (Figs. 2 and 3). As with many studies using MEF cells isolatedrom gene-targeted KO mice, direct comparison is made betweenPol ˇ KO cell line and a WT cell line, each derived from embryos
earch 686 (2010) 57–67 61
of the same litter to minimize genetic variation (i.e., an isogeniccell line pair). However, this may not guarantee a true isogenic sys-tem. For example, the WT cell line 16tsA is WT for Pol � while thelittermate-derived Pol ˇ KO cell line 19tsA is null for Pol � [37,38].To avoid any suggestion of genetic variation that may complicateinterpretation of the role of Parp1 in the hypersensitive phenotypeof Pol ˇ KO MEFs, we developed a lentiviral system for expres-sion of mouse Pol � specific shRNA, based on the target sequencefor our retroviral shRNA system reported earlier [32]. We trans-duced two different WT MEF cells: 92TAg and HCC384. Stable celllines expressing the mouse Pol � specific shRNA are devoid ofdetectable Pol � expression, as determined by immunoblot anal-ysis (Fig. 4A and B). Expression of the BER proteins Ape1, Xrcc1 andParp1 are similar when comparing the 92TAg and HCC384 cells andthe corresponding lentiviral-transduced cells that show a Pol � KDphenotype (Fig. 4A and B). Similar to the results discussed above forthe Pol ˇ KO cells, the Pol � KD cells have a highly elevated level ofParp1 activation following MMS exposure (Fig. 4C and D) whereasthe 92TAg and HCC384 WT cells show little or no Parp1 activa-tion following MMS treatment. The Pol � KD cells are functionallysimilar to the gene-targeted KO cells. Both the 92TAg/Pol�KD andHCC384/Pol�KD cells are hypersensitive to MMS as compared tothe corresponding WT cell line (Fig. 4E and F). This is consistent withour previous results and others that strong knockdown of Pol � inMEFs leads to hypersensitivity to alkylating agents [32,46]. Mostimportantly, this isogenic system clearly demonstrates the role ofParp1 activation in the hypersensitivity of Pol � deficient MEFs. Forboth the 92TAg/Pol�KD and HCC384/Pol�KD cells, Parp1 inhibitionby pre-treatment with the Parp1/Parp2 inhibitor PJ34 significantlyenhances survival following MMS exposure (Fig. 4E and F). Inter-estingly, there is some slight variation in response even whencomparing the 92TAg cells (Fig. 4E) and the HCC384 cells (Fig. 4F),supporting the significance of developing a truly isogenic systemusing shRNA-mediated gene expression knockdown. For example,the 92TAg cells were slightly sensitized to MMS when Parp1 wasinhibited whereas the viability of the HCC384 cells was not alteredby Parp1 inhibition (Fig. 4E and F). In both cases however, Parp1inhibition provides a significant survival advantage for the cor-responding Pol � KD cells (92TAg/Pol�KD and HCC384/Pol�KD),supporting our hypothesis that Parp1 activation is involved in thehypersensitivity of Pol � deficient MEFs to alkylating agents.
3.4. Complementation of Pol ˇ KO MEFs and Pol ˇ/Parp1 doubleKO MEFs with mouse Pol ˇ—impact of Parp1 expression on MMSsensitivity in the absence of Pol ˇ
The use of small molecule Parp inhibitors such as PJ34 to inhibitParp1 and Parp2 activation provides significant support for ourhypothesis that Parp1 activation promotes the hypersensitivity ofPol � deficient MEFs to alkylating agents. However, there are 18Parp-family members that are involved in many biological pro-cesses [47]. Although only Parp1 and Parp2 have been implicatedin BER and in response to DNA damage [37], potential off-targeteffects of inhibitors such as PJ34 (and others) is always a distinctpossibility. Therefore, we reasoned that if Parp1 was the primaryParp-family member that regulates the cellular response to failedrepair of the BER intermediate 5′dRP, then we should take advan-tage of the previously developed Pol ˇ/Parp1 double KO MEF cells[51]. To directly compare the cellular response of the Pol ˇ KO andPol ˇ/Parp1 double KO MEFs to those that express Pol �, we clonedthe mouse Pol � cDNA and developed stable cell lines that express
either GFP (as a control) or mouse Pol �. An immunoblot analy-sis of the Pol ˇ KO and Pol ˇ/Parp1 double KO cells as well as thecorresponding GFP or mouse Pol � expressing stable cell lines areshown (Fig. 5A and B). Pol � is only expressed in the transfected,stable cell lines (shown in lanes 4 and 5) and Parp1 is only expressed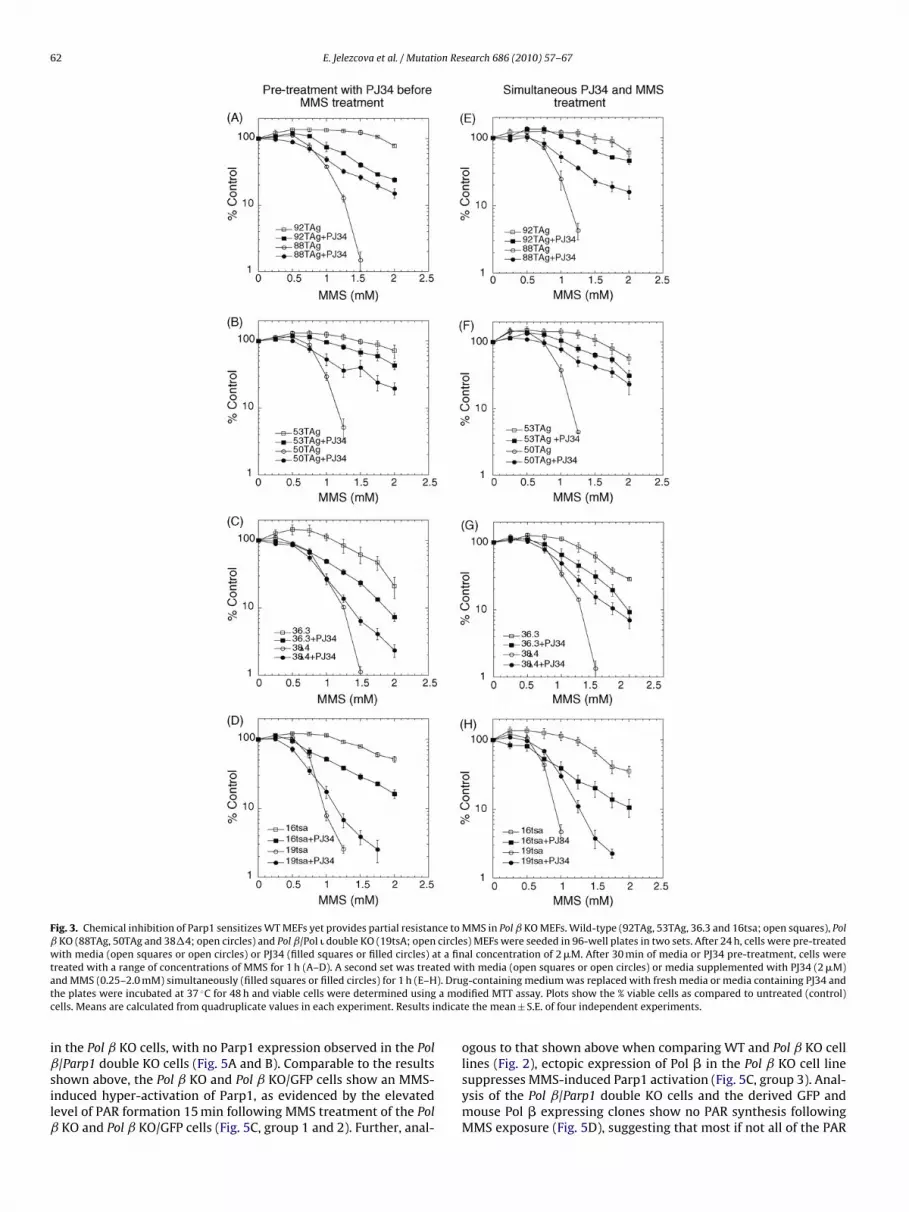
62 E. Jelezcova et al. / Mutation Research 686 (2010) 57–67
Fig. 3. Chemical inhibition of Parp1 sensitizes WT MEFs yet provides partial resistance to MMS in Pol ˇ KO MEFs. Wild-type (92TAg, 53TAg, 36.3 and 16tsa; open squares), Polˇ KO (88TAg, 50TAg and 38�4; open circles) and Pol ˇ/Pol � double KO (19tsA; open circles) MEFs were seeded in 96-well plates in two sets. After 24 h, cells were pre-treatedwith media (open squares or open circles) or PJ34 (filled squares or filled circles) at a final concentration of 2 �M. After 30 min of media or PJ34 pre-treatment, cells weret ted wa ). Drut a moc ndicat
iˇsilˇ
reated with a range of concentrations of MMS for 1 h (A–D). A second set was treand MMS (0.25–2.0 mM) simultaneously (filled squares or filled circles) for 1 h (E–Hhe plates were incubated at 37 ◦C for 48 h and viable cells were determined usingells. Means are calculated from quadruplicate values in each experiment. Results i
n the Pol ˇ KO cells, with no Parp1 expression observed in the Pol
/Parp1 double KO cells (Fig. 5A and B). Comparable to the resultshown above, the Pol ˇ KO and Pol ˇ KO/GFP cells show an MMS-nduced hyper-activation of Parp1, as evidenced by the elevatedevel of PAR formation 15 min following MMS treatment of the PolKO and Pol ˇ KO/GFP cells (Fig. 5C, group 1 and 2). Further, anal-
ith media (open squares or open circles) or media supplemented with PJ34 (2 �M)g-containing medium was replaced with fresh media or media containing PJ34 anddified MTT assay. Plots show the % viable cells as compared to untreated (control)e the mean ± S.E. of four independent experiments.
ogous to that shown above when comparing WT and Pol ˇ KO cell
lines (Fig. 2), ectopic expression of Pol � in the Pol ˇ KO cell linesuppresses MMS-induced Parp1 activation (Fig. 5C, group 3). Anal-ysis of the Pol ˇ/Parp1 double KO cells and the derived GFP andmouse Pol � expressing clones show no PAR synthesis followingMMS exposure (Fig. 5D), suggesting that most if not all of the PAR
E. Jelezcova et al. / Mutation Research 686 (2010) 57–67 63
Fig. 4. Mouse Pol � knockdown causes robust Parp1 activation and Parp1 dependent cell death following MMS exposure in MEFs. (A and B) BER protein expression asdetermined by immunoblot analysis of nuclear proteins isolated from the 92TAg cells or 92TAg cells transduced with a mouse Pol � shRNA lentiviral vector (A) or theHCC384 cells or HCC384 cells transduced with a mouse Pol � shRNA lentiviral vector (B). (A) Proteins isolated from the shRNA expressing clone (92TAg/Pol�-KD(3); lane2) as compared to proteins isolated from control cells (92TAg; lane 1) are shown in (A) and from the shRNA expressing clone (HCC384/Pol�-KD(1); lane 2) as compared toproteins isolated from control cells (HCC384; lane 1) are shown in (B). Pol �, Ape1, Xrcc1 and Parp1 expression was determined by immunoblot. PCNA expression is shownas a loading control. (C and D) Parp1 activation was determined in cell extracts by immunoblot analysis, measuring the synthesis of PAR after exposure with MMS (1.25 mM)for 0, 15 or 30 min in (C) 92TAg cells [WT] or 92TAg cells transduced with a mouse Pol � shRNA lentiviral vector, clone 3 [Pol � KD] or (D) HCC384 cells [WT] or HCC384 cellstransduced with a mouse Pol � shRNA lentiviral vector, clone 1 [Pol � KD]. PCNA expression is shown as a loading control. (E and F) MMS-induced cytotoxicity following PJ34p cellsc e Pol2 ares ow ed in F
sm
aoMoccm98bsOMe
re-treatment was evaluated by culturing (E) 92TAg cells (open squares) or 92TAgircles) or (F) HCC384 cells (open squares) or HCC384 cells transduced with a mous4 h prior to exposure with media (open squares or open circles) or PJ34 (filled squith MMS and viable cells were determined using a modified MTT assay as describ
ynthesized following MMS exposure of Pol ˇ KO cells (Fig. 5C) isediated by Parp1.We next extended our functional comparison of the Pol ˇ KO
nd Pol ˇ/Parp1 double KO cells, as well as, the corresponding GFPr mouse Pol � expressing stable cell lines by exposing each toMS (1.5 mM) and measuring survival, as described in the Meth-
ds section. Both the 88TAg cells and the derived GFP-expressingells (88TAg/GFP) were highly sensitive to MMS whereas the 88TAgells expressing recombinant mouse Pol � (88TAg/mPol �) showedarked resistance, similar to other genetically WT cells such as
2TAg (Fig. 5E). Similar to the WT cells described above (Fig. 4E), the8TAg/mPol � cells were slightly sensitized to MMS by Parp1 inhi-
ition (Fig. 5E). However, Parp1 inhibition provided a significanturvival advantage for the 88TAg and GFP-expressing cells (Fig. 5E).n the contrary, there is little or no difference in survival followingMS exposure when comparing GFP-expressing and mouse Pol �xpressing Pol ˇ/Parp1 double KO cells (Pol ˇ/Parp1 KO/GFP vs. Pol
transduced with a mouse Pol � shRNA lentiviral vector (92TAg/Pol�-KD(3); open� shRNA lentiviral vector (HCC384/Pol�-KD(1); open circles), in 96-well plates forr filled circles; 2 �M) for 30 min. After PJ34 pre-treatment, cells were then treatedig. 3.
ˇ/Parp1 KO/mPol �) (Fig. 5F). In support of the overall hypothesisput forth herein, the MMS-induced hypersensitive phenotype ofPol ˇ KO cells (as compared to Pol � expressing cells) requires or isexacerbated by Parp1 activation (Fig. 5).
4. Discussion
Alkylators such as MMS induce a spectrum of DNA base lesions[48] as well as single-strand and double-strand DNA breaks. Fail-ure to repair these lesions or DNA breaks triggers cell death viaapoptosis, autophagy and necrosis. The role of Pol � in repair ofMMS-induced lesions is restricted to base lesions removed by the
methylation-specific DNA glycosylase MPG (AAG) [18]. Once repairis initiated, Pol � is essential for repair gap tailoring to remove the5′dRP BER intermediate. Although it is clear that the un-repaired5′dRP BER intermediate is the cytotoxic lesion that renders Pol ˇKO MEFs hypersensitive to alkylating agents [7], the specific cell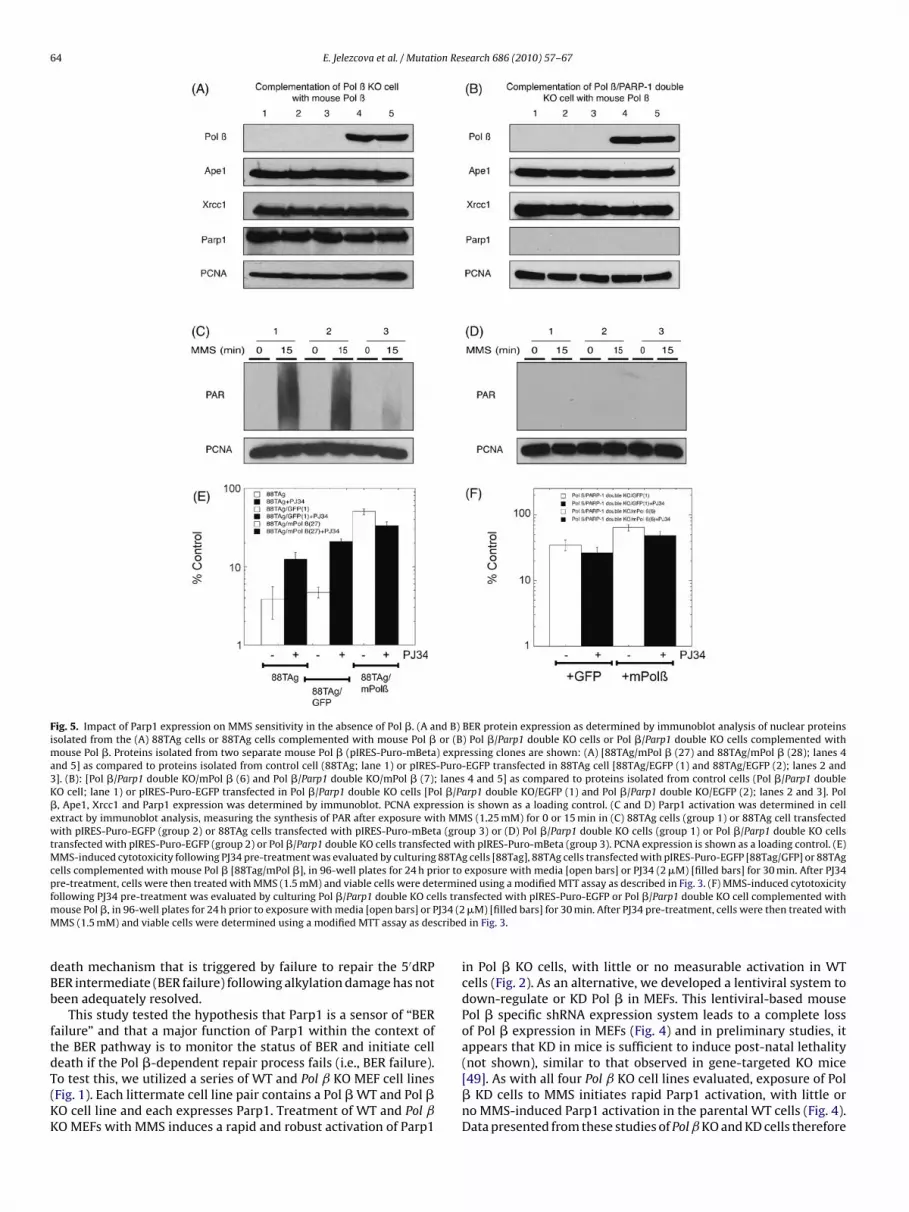
64 E. Jelezcova et al. / Mutation Research 686 (2010) 57–67
Fig. 5. Impact of Parp1 expression on MMS sensitivity in the absence of Pol �. (A and B) BER protein expression as determined by immunoblot analysis of nuclear proteinsisolated from the (A) 88TAg cells or 88TAg cells complemented with mouse Pol � or (B) Pol �/Parp1 double KO cells or Pol �/Parp1 double KO cells complemented withmouse Pol �. Proteins isolated from two separate mouse Pol � (pIRES-Puro-mBeta) expressing clones are shown: (A) [88TAg/mPol � (27) and 88TAg/mPol � (28); lanes 4and 5] as compared to proteins isolated from control cell (88TAg; lane 1) or pIRES-Puro-EGFP transfected in 88TAg cell [88TAg/EGFP (1) and 88TAg/EGFP (2); lanes 2 and3]. (B): [Pol �/Parp1 double KO/mPol � (6) and Pol �/Parp1 double KO/mPol � (7); lanes 4 and 5] as compared to proteins isolated from control cells (Pol �/Parp1 doubleKO cell; lane 1) or pIRES-Puro-EGFP transfected in Pol �/Parp1 double KO cells [Pol �/Parp1 double KO/EGFP (1) and Pol �/Parp1 double KO/EGFP (2); lanes 2 and 3]. Pol�, Ape1, Xrcc1 and Parp1 expression was determined by immunoblot. PCNA expression is shown as a loading control. (C and D) Parp1 activation was determined in cellextract by immunoblot analysis, measuring the synthesis of PAR after exposure with MMS (1.25 mM) for 0 or 15 min in (C) 88TAg cells (group 1) or 88TAg cell transfectedwith pIRES-Puro-EGFP (group 2) or 88TAg cells transfected with pIRES-Puro-mBeta (group 3) or (D) Pol �/Parp1 double KO cells (group 1) or Pol �/Parp1 double KO cellstransfected with pIRES-Puro-EGFP (group 2) or Pol �/Parp1 double KO cells transfected with pIRES-Puro-mBeta (group 3). PCNA expression is shown as a loading control. (E)MMS-induced cytotoxicity following PJ34 pre-treatment was evaluated by culturing 88TAg cells [88Tag], 88TAg cells transfected with pIRES-Puro-EGFP [88Tag/GFP] or 88TAgcells complemented with mouse Pol � [88Tag/mPol �], in 96-well plates for 24 h prior to exposure with media [open bars] or PJ34 (2 �M) [filled bars] for 30 min. After PJ34pre-treatment, cells were then treated with MMS (1.5 mM) and viable cells were determined using a modified MTT assay as described in Fig. 3. (F) MMS-induced cytotoxicityf lls tram J34 (2M cribed
dBb
ftdT(KK
ollowing PJ34 pre-treatment was evaluated by culturing Pol �/Parp1 double KO ceouse Pol �, in 96-well plates for 24 h prior to exposure with media [open bars] or PMS (1.5 mM) and viable cells were determined using a modified MTT assay as des
eath mechanism that is triggered by failure to repair the 5′dRPER intermediate (BER failure) following alkylation damage has noteen adequately resolved.
This study tested the hypothesis that Parp1 is a sensor of “BERailure” and that a major function of Parp1 within the context ofhe BER pathway is to monitor the status of BER and initiate cell
eath if the Pol �-dependent repair process fails (i.e., BER failure).o test this, we utilized a series of WT and Pol ˇ KO MEF cell linesFig. 1). Each littermate cell line pair contains a Pol � WT and Pol �O cell line and each expresses Parp1. Treatment of WT and Pol ˇO MEFs with MMS induces a rapid and robust activation of Parp1nsfected with pIRES-Puro-EGFP or Pol �/Parp1 double KO cell complemented with�M) [filled bars] for 30 min. After PJ34 pre-treatment, cells were then treated within Fig. 3.
in Pol � KO cells, with little or no measurable activation in WTcells (Fig. 2). As an alternative, we developed a lentiviral system todown-regulate or KD Pol � in MEFs. This lentiviral-based mousePol � specific shRNA expression system leads to a complete lossof Pol � expression in MEFs (Fig. 4) and in preliminary studies, itappears that KD in mice is sufficient to induce post-natal lethality
(not shown), similar to that observed in gene-targeted KO mice[49]. As with all four Pol ˇ KO cell lines evaluated, exposure of Pol� KD cells to MMS initiates rapid Parp1 activation, with little orno MMS-induced Parp1 activation in the parental WT cells (Fig. 4).Data presented from these studies of Pol ˇ KO and KD cells therefore
on Res
sf
dpdot[PimtciaddnutPephfpp�oceoslta
siPvtiipammptpi(tftKpawa
mcd
E. Jelezcova et al. / Mutati
upports our hypothesis that Parp1 provides a cellular signal ofailed BER via PAR synthesis.
Alkylation damage can induce cell death via a mismatch repairependent process [50,51], activation of caspases [52] or cal-ains [53], induction of autophagy [54] or the generation of DNAouble-strand breaks and H2AX recruitment [55]. In a multitudef studies of alkylation-induced cell death in MEFs, Parp1 activa-ion and the onset of necrosis is the primary means of cell death45,47,52,56–58]. Importantly, the specific molecular activator ofarp1 due to alkylation damage has not been clearly defined. Parp1s activated by breaks to ssDNA and dsDNA [47,58], by Erk1/2-
ediated phosphorylation [59] or by a direct interaction with ERK2,he latter independent of DNA damage [60]. In DNA repair proficientells, the expectation is that alkylation-induced Parp1 activations via DNA breaks [47,58], but it is not clear if activation is medi-ted by failed BER or via replication-induced specific ssDNA breaks,sDNA breaks or a spectrum of both. It is also possible that celleath arises from mitotic catastrophe [61]. To address the con-ection between Pol � status, Parp1 activation and cell death, wetilized Parp1/Parp2 inhibitors [31,39,62] to prevent Parp1 activa-ion resulting from BER failure. Since we found that a deficiency inol � caused DNA damage-induced Parp1 activation, we hypoth-sized that blocking Parp1 activation may provide some level ofhenotypic rescue in the Pol ˇ KO and KD cells. In support of thisypothesis, we find that the alkylation-sensitive phenotype of all
our Pol ˇ KO cells used in this study was rescued, either com-letely or partially, by Parp1 inhibition (Fig. 3). An even strongerhenotypic rescue is observed in two independently derived PolKD MEF cell lines (Fig. 4). The phenotypic (cell death) rescue
bserved herein is similar to that observed in neurons [63,64],ardiac myocytes [65–69] and ischemia-reperfusion stroke mod-ls [63,64] whereby cell death is Parp1 dependent and inhibitionf Parp1 activation prevents stress-induced cell death. Additionaltudies will be required to determine if this resistant phenotype isong-lived or if the resulting un-repaired BER intermediates leado the formation of DNA double-strand breaks and the onset ofpoptosis after several rounds of replication [70].
Complicating the interpretation of the DNA damage-inducedurvival curves of the Pol ˇ KO and Pol � KD cell lines (Figs. 3 and 4)s the well-documented potentiation of DNA damaging agents byarp1 inhibition [43]. It is hypothesized that blocking Parp1 acti-ation prevents Parp1-regulated DNA repair processes, increasinghe cell killing and anti-tumor effect of many DNA damaging agents,ncluding alkylating agents, cisplatin, radiation and topoisomerase-nhibitors [71,72]. Many of these earlier studies utilized DNA repairroficient MEFs and exposure to extremely high doses of DNA dam-ging agent. It is likely that Parp1 inhibition shifts the cell deathechanism from necrosis to apoptosis [45]. In the context of BER-ediated repair of alkylated bases, blocking Parp1 should therefore
revent BER complex recruitment [73,74], leading to accumula-ion of cytotoxic replication-induced DSBs [75]. In-line with theotentiating effect of Parp1 inhibition, we do observe an increase
n MMS-induced cell killing in WT cells when Parp1 is inhibitedFigs. 3 and 4). However, Parp1 inhibition provides a survival advan-age to Pol ˇ KO cells, equal to that of the Parp1 inhibited WT cellsor most doses of MMS (Figs. 3 and 4). This led us to speculate thathe alkylation damage mediated hypersensitive phenotype of Pol ˇO (and KD) cells requires Parp1 activation. Although we observe ahenotypic rescue by Parp1 inhibition, we recognize that the ‘dam-ge’ from a combination of alkylation treatment and BER failureill undoubtedly lead to an accumulation of DSBs and enhanced
poptotic cell death [43,45].To directly test the role of Parp1 in the alkylation damage
ediated hypersensitive phenotype of Pol � deficient cells, weompared the alkylation response of Pol ˇ KO cells and Pol ˇ/Parp1ouble KO cells. In support of our hypothesis that Parp1 activation is
earch 686 (2010) 57–67 65
the result of BER failure (i.e., Pol � deficiency), ectopic expression ofmouse Pol � in Pol � KO MEFs restored MMS resistance and attenu-ated MMS-induced Parp1 activation (Fig. 5). The Pol ˇ/Parp1 doubleKO cells did not show any MMS-induced PAR synthesis, implicatingParp1 as the major DNA damage response Parp-family member, asexpected [47]. In support of our hypothesis that Parp1 is requiredfor the Pol � hypersensitive phenotype, there was no MMS-inducedcytotoxic difference when comparing Pol ˇ/Parp1 double KO cellsexpressing GFP and those that express mouse Pol � (Fig. 5).
In summary, we propose that Pol ˇ KO MEFs are hypersensitiveto alkylating agents due to hyper-activation of Parp1. Once repairis initiated and the DNA backbone is hydrolyzed, Parp1 is engagedto recruit the necessary proteins required for the completion ofrepair [1], including Xrcc1 and Pol � [73,74]. However, we showthat BER failure (due to Pol � deficiency) leads to a rapid, elevatedlevel of Parp1 activation, implicating Parp1-mediated necrosis asthe cause of the Pol � hypersensitive phenotype [8,10]. This workshows one way by which defective BER can lead to cell death (viaParp1 activation), but there may be other ways as well. These stud-ies are in-line with our recent report that Pol � deficiency in humancells gives rise to cellular sensitivity to the chemotherapeutic alky-lating agent temozolomide by a mechanism that does not involveapoptosis and does not lead to the formation of autophagosomes[8]. Together with the Parp1 activation described here, it is ourexpectation that cell death due to failed BER is mediated by Parp1activation. Continuing studies will explore how Parp1 and relatedPAR- and NAD+-metabolic enzymes impact BER and in turn, affectDNA damage or chemotherapeutic response in BER-proficient ascompared to BER-deficient or BER-inhibited cells.
5. Conflict of interest
The authors state that there is no conflict of interest.
Acknowledgements
This research was supported by a Research Scholar grant(RSG-05-246-01-GMC) from the American Cancer Society, grantsfrom the Susan G. Komen Breast Cancer Foundation (Grant #BCTR0403276), NIH (1 R01 AG24364-01; P20 CA103730, 1 P20CA132385-01 and 1P50 CA 097190 01A1), the Brain Tumor Society,the UPMC Health System Competitive Medical Research Fund andthe University of Pittsburgh Cancer Institute to RWS. This project isalso funded, in part, under a grant with the Pennsylvania Depart-ment of Health. The Department of Health specifically disclaimsresponsibility for any analyses, interpretations or conclusions. Wewould like to thank Dr. Ben Van Houten (University of Pittsburgh)and Dr. Christi A. Walter (University of Texas Health Science CenterSan Antonio) for critically reading his manuscript.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.mrfmmm.2010.01.016.
References
[1] K.H. Almeida, R.W. Sobol, A unified view of base excision repair: lesion-dependent protein complexes regulated by post-translational modification,DNA Repair 6 (2007) 695–711.
[2] R.W. Sobol, J.K. Horton, R. Kuhn, H. Gu, R.K. Singhal, R. Prasad, K. Rajewsky,S.H. Wilson, Requirement of mammalian DNA polymerase-� in base-excision
repair, Nature 379 (1996) 183–186.[3] W.A. Beard, S.H. Wilson, Structure and mechanism of DNA polymerase Beta,Chem. Rev. 106 (2006) 361–382.
[4] J.K. Horton, R. Prasad, E. Hou, S.H. Wilson, Protection against methylation-induced cytotoxicity by DNA polymerase �-dependent long patch base excisionrepair, J. Biol. Chem. 275 (2000) 2211–2218.

6 on Res
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
6 E. Jelezcova et al. / Mutati
[5] P. Fortini, B. Pascucci, E. Parlanti, R.W. Sobol, S.H. Wilson, E. Dogliotti, DifferentDNA polymerases are involved in the short- and long-patch base excision repairin mammalian cells, Biochemistry 37 (1998) 3575–3580.
[6] Y. Matsumoto, K. Kim, Excision of deoxyribose phosphate residues by DNApolymerase � during DNA repair, Science 269 (1995) 699–702.
[7] R.W. Sobol, R. Prasad, A. Evenski, A. Baker, X.P. Yang, J.K. Horton, S.H. Wilson,The lyase activity of the DNA repair protein �-polymerase protects from DNA-damage-induced cytotoxicity, Nature 405 (2000) 807–810.
[8] R.N. Trivedi, X.H. Wang, E. Jelezcova, E.M. Goellner, J. Tang, R.W. Sobol, Humanmethyl purine DNA glycosylase and DNA polymerase � expression collectivelypredict sensitivity to temozolomide, Mol. Pharmacol. 74 (2008) 505–516.
[9] J.K. Horton, D.F. Joyce-Gray, B.F. Pachkowski, J.A. Swenberg, S.H. Wilson,Hypersensitivity of DNA polymerase beta null mouse fibroblasts reflects accu-mulation of cytotoxic repair intermediates from site-specific alkyl DNA lesions,DNA Repair (Amst.) 2 (2003) 27–48.
10] A. Price, T. Lindahl, Enzymatic release of 5′-terminal deoxyribose phos-phate residues from damaged DNA in human cells, Biochemistry 30 (1991)8631–8637.
11] K. Bebenek, A. Tissier, E.G. Frank, J.P. McDonald, R. Prasad, S.H. Wilson, R.Woodgate, T.A. Kunkel, 5′-Deoxyribose phosphate lyase activity of human DNApolymerase iota in vitro, Science 291 (2001) 2156–2159.
12] R. Prasad, K. Bebenek, E. Hou, D.D. Shock, W.A. Beard, R. Woodgate, T.A. Kunkel,S.H. Wilson, Localization of the deoxyribose phosphate lyase active site inhuman DNA polymerase iota by controlled proteolysis, J. Biol. Chem. 278 (2003)29649–29654.
13] E.K. Braithwaite, R. Prasad, D.D. Shock, E.W. Hou, W.A. Beard, S.H. Wilson, DNApolymerase lambda mediates a back-up base excision repair activity in extractsof mouse embryonic fibroblasts, J. Biol. Chem. 280 (2005) 18469–18475.
14] E.K. Braithwaite, P.S. Kedar, L. Lan, Y.Y. Polosina, K. Asagoshi, V.P. Poltoratsky,J.K. Horton, H. Miller, G.W. Teebor, A. Yasui, S.H. Wilson, DNA poly-merase lambda protects mouse fibroblasts against oxidative DNA damageand is recruited to sites of DNA damage/repair, J. Biol. Chem. 280 (2005)31641–31647.
15] M. Garcia-Diaz, K. Bebenek, G. Gao, L.C. Pedersen, R.E. London, T.A. Kunkel,Structure–function studies of DNA polymerase lambda, DNA Repair (Amst.) 4(2005) 1358–1367.
16] M. Garcia-Diaz, K. Bebenek, T.A. Kunkel, L. Blanco, Identification of an intrinsic5′-deoxyribose-5-phosphate lyase activity in human DNA polymerase lambda:a possible role in base excision repair, J. Biol. Chem. 276 (2001) 34659–34663.
17] I.R. Grin, S.N. Khodyreva, G.A. Nevinsky, D.O. Zharkov, Deoxyribophosphatelyase activity of mammalian endonuclease VIII-like proteins, FEBS Lett. 580(2006) 4916–4922.
18] R.W. Sobol, M. Kartalou, K.H. Almeida, D.F. Joyce, B.P. Engelward, J.K. Horton,R. Prasad, L.D. Samson, S.H. Wilson, Base excision repair intermediates inducep53-independent cytotoxic and genotoxic responses, J. Biol. Chem. 278 (2003)39951–39959.
19] K. Ochs, R.W. Sobol, S.H. Wilson, B. Kaina, Cells deficient in DNA polymerase� are hypersensitive to alkylating agent-induced apoptosis and chromosomalbreakage, Cancer Res. 59 (1999) 1544–1551.
20] K. Ochs, J. Lips, S. Profittlich, B. Kaina, Deficiency in DNA polymerase � provokesreplication-dependent apoptosis via DNA breakage, Bcl-2 decline and caspase-3/9 activation, Cancer Res. 62 (2002) 1524–1530.
21] D.K. Srivastava, B.J. Berg, R. Prasad, J.T. Molina, W.A. Beard, A.E. Tomkinson,S.H. Wilson, Mammalian abasic site base excision repair. Identification ofthe reaction sequence and rate-determining steps, J. Biol. Chem. 273 (1998)21203–21209.
22] E.C. Friedberg, G.C. Walker, W. Siede, R.D. Wood, R.A. Schultz, T. Ellen-berger, DNA Repair and Mutagenesis, 2nd ed., ASM Press, Washington, DC,2006.
23] F. Dantzer, G. de La Rubia, J. Menissier-De Murcia, Z. Hostomsky, G. de Mur-cia, V. Schreiber, Base excision repair is impaired in mammalian cells lackingpoly(ADP-ribose) polymerase-1, Biochemistry 39 (2000) 7559–7569.
24] M.D. Vodenicharov, F.R. Sallmann, M.S. Satoh, G.G. Poirier, Base excision repairis efficient in cells lacking poly(ADP-ribose) polymerase 1, Nucleic Acids Res.28 (2000) 3887–3896.
25] K.H. Almeida, R.W. Sobol, Increased specificity and efficiency of base excisionrepair through complex formation, in: W. Siede, P.W. Doetsch, Y.W. Kow (Eds.),DNA Damage Recognition, Marcel Dekker Inc., New York, 2005, pp. 33–64.
26] J.L. Parsons, G.L. Dianov, Monitoring base excision repair proteins on damagedDNA using human cell extracts, Biochem. Soc. Trans. 32 (2004) 962–963.
27] J.L. Parsons, I.I. Dianova, S.L. Allinson, G.L. Dianov, Poly(ADP-ribose)polymerase-1 protects excessive DNA strand breaks from deterioration duringrepair in human cell extracts, FEBS J. 272 (2005) 2012–2021.
28] O.I. Lavrik, R. Prasad, R.W. Sobol, J.K. Horton, E.J. Ackerman, S.H. Wilson, Pho-toaffinity labeling of mouse fibroblast enzymes by a base excision repairintermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNArepair, J. Biol. Chem. 276 (2001) 25541–25548.
29] C. Cistulli, O.I. Lavrik, R. Prasad, E. Hou, S.H. Wilson, AP endonuclease andpoly(ADP-ribose) polymerase-1 interact with the same base excision repairintermediate, DNA Repair (Amst.) 3 (2004) 581–591.
30] F. Dantzer, J.C. Ame, V. Schreiber, J. Nakamura, J. Menissier-de Murcia, G. deMurcia, Poly(ADP-ribose) polymerase-1 activation during DNA damage andrepair, Methods Enzymol. 409 (2006) 493–510.
31] W.X. Zong, D. Ditsworth, D.E. Bauer, Z.Q. Wang, C.B. Thompson, Alkylating DNAdamage stimulates a regulated form of necrotic cell death, Genes Dev. 18 (2004)1272–1282.
[
[
[
earch 686 (2010) 57–67
32] R.N. Trivedi, K.H. Almeida, J.L. Fornsaglio, S. Schamus, R.W. Sobol, The role ofbase excision repair in the sensitivity and resistance to temozolomide mediatedcell death, Cancer Res. 65 (2005) 6394–6400.
33] D.A. Rubinson, C.P. Dillon, A.V. Kwiatkowski, C. Sievers, L. Yang, J. Kopinja, M.Zhang, M.T. McManus, F.B. Gertler, M.L. Scott, L. Van Parijs, A lentivirus-basedsystem to functionally silence genes in primary mammalian cells, stem cellsand transgenic mice by RNA interference, Nat. Genet. 33 (2003) 401–406.
34] E.M. Poeschla, F. Wong-Staal, D.J. Looney, Efficient transduction of nondividinghuman cells by feline immunodeficiency virus lentiviral vectors, Nat. Med. 4(1998) 354–357.
35] N. Kataoka, G. Dreyfuss, A simple whole cell lysate system for in vitro splicingreveals a stepwise assembly of the exon–exon junction complex, J. Biol. Chem.279 (2004) 7009–7013.
36] M.V. Berridge, A.S. Tan, Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellularlocalization, substrate dependence, and involvement of mitochondrial electrontransport in MTT reduction, Arch. Biochem. Biophys. 303 (1993) 474–482.
37] R.D. Wood, M. Mitchell, J. Sgouros, T. Lindahl, Human DNA repair genes, Science291 (2001) 1284–1289.
38] B.C. Woodhouse, G.L. Dianov, Poly ADP-ribose polymerase-1: an internationalmolecule of mystery, DNA Repair (Amst.) 7 (2008) 1077–1086.
39] S.W. Yu, H. Wang, M.F. Poitras, C. Coombs, W.J. Bowers, H.J. Federoff, G.G.Poirier, T.M. Dawson, V.L. Dawson, Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor, Science 297 (2002)259–263.
40] C. Ethier, Y. Labelle, G.G. Poirier, PARP-1-induced cell death through inhibitionof the MEK/ERK pathway in MNNG-treated HeLa cells, Apoptosis 12 (2007)2037–2049.
41] F. Paquet-Durand, J. Silva, T. Talukdar, L.E. Johnson, S. Azadi, T. van Veen, M.Ueffing, S.M. Hauck, P.A. Ekstrom, Excessive activation of poly(ADP-ribose)polymerase contributes to inherited photoreceptor degeneration in the retinaldegeneration 1 mouse, J. Neurosci. 27 (2007) 10311–10319.
42] A. Iwashita, S. Yamazaki, K. Mihara, K. Hattori, H. Yamamoto, J. Ishida, N.Matsuoka, S. Mutoh, Neuroprotective effects of a novel poly(ADP-ribose)polymerase-1 inhibitor, 2-[3-[4-(4-chlorophenyl)-1-piperazinyl] propyl]-4(3H)-quinazolinone (FR255595), in an in vitro model of cell death and inmouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’sdisease, J. Pharmacol. Exp. Ther. 309 (2004) 1067–1078.
43] L. Tentori, G. Graziani, Chemopotentiation by PARP inhibitors in cancer therapy,Pharmacol. Res. 52 (2005) 25–33.
44] J.K. Horton, D.F. Stefanick, J.M. Naron, P.S. Kedar, S.H. Wilson, Poly(ADP-ribose) polymerase activity prevents signaling pathways for cell cycle arrestfollowing DNA methylating agent exposure, J. Biol. Chem. 280 (2005)15773–15785.
45] J.K. Horton, D.F. Stefanick, S.H. Wilson, Involvement of poly(ADP-ribose) poly-merase activity in regulating Chk1-dependent apoptotic cell death, DNA Repair(Amst.) 4 (2005) 1111–1120.
46] Y.Y. Polosina, T.A. Rosenquist, A.P. Grollman, H. Miller, ‘Knock down’ of DNApolymerase � by RNA interference: recapitulation of null phenotype, DNARepair (Amst.) 3 (2004) 1469–1474.
47] P.O. Hassa, S.S. Haenni, M. Elser, M.O. Hottiger, Nuclear ADP-ribosylation reac-tions in mammalian cells: where are we today and where are we going?Microbiol. Mol. Biol. Rev. 70 (2006) 789–829.
48] D.T. Beranek, Distribution of methyl and ethyl adducts following alkylationwith monofunctional alkylating agents, Mutat. Res. 231 (1990) 11–30.
49] H. Gu, J.D. Marth, P.C. Orban, H. Mossmann, K. Rajewsky, Deletion of a DNApolymerase � gene segment in T cells using cell type-specific gene targeting,Science 265 (1994) 103–106.
50] I. Casorelli, E. Pelosi, M. Biffoni, A.M. Cerio, C. Peschle, U. Testa, M. Bignami,Methylation damage response in hematopoietic progenitor cells, DNA Repair(Amst.) 6 (2007) 1170–1178.
51] B. Kaina, M. Christmann, S. Naumann, W.P. Roos, MGMT: key node in the bat-tle against genotoxicity, carcinogenicity and apoptosis induced by alkylatingagents, DNA Repair (Amst.) 6 (2007) 1079–1099.
52] O. Cohausz, C. Blenn, M. Malanga, F.R. Althaus, The roles of poly(ADP-ribose)-metabolizing enzymes in alkylation-induced cell death, Cell. Mol. Life Sci. 65(2008) 644–655.
53] R.S. Moubarak, V.J. Yuste, C. Artus, A. Bouharrour, P.A. Greer, J. Menissier-deMurcia, S.A. Susin, Sequential activation of poly(ADP-ribose) polymerase 1, cal-pains, and Bax is essential in apoptosis-inducing factor-mediated programmednecrosis, Mol. Cell. Biol. 27 (2007) 4844–4862.
54] T. Kanzawa, I.M. Germano, T. Komata, H. Ito, Y. Kondo, S. Kondo, Role ofautophagy in temozolomide-induced cytotoxicity for malignant glioma cells,Cell Death Differ. 11 (2004) 448–457.
55] J.A. Meador, M. Zhao, Y. Su, G. Narayan, C.R. Geard, A.S. Balajee, Histone H2AX isa critical factor for cellular protection against DNA alkylating agents, Oncogene27 (2008) 5662–5671.
56] B. Affar el, R.G. Shah, A.K. Dallaire, V. Castonguay, G.M. Shah, Role of poly(ADP-ribose) polymerase in rapid intracellular acidification induced by alkylatingDNA damage, Proc. Natl. Acad. Sci. U.S.A. 99 (2002) 245–250.
57] H.C. Ha, S.H. Snyder, Poly(ADP-ribose) polymerase is a mediator of necrotic celldeath by ATP depletion, Proc. Natl. Acad. Sci. U.S.A. 96 (1999) 13978–13982.
58] V. Schreiber, F. Dantzer, J.C. Ame, G. de Murcia, Poly(ADP-ribose): novel func-tions for an old molecule, Nat. Rev. Mol. Cell. Biol. 7 (2006) 517–528.
59] T.M. Kauppinen, W.Y. Chan, S.W. Suh, A.K. Wiggins, E.J. Huang, R.A. Swan-son, Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1

on Res
[
[
[
[
[
[
[
[
[
[
[
[
[
[
E. Jelezcova et al. / Mutati
by extracellular signal-regulated kinases 1/2, Proc. Natl. Acad. Sci. U.S.A. 103(2006) 7136–7141.
60] M. Cohen-Armon, L. Visochek, D. Rozensal, A. Kalal, I. Geistrikh, R. Klein, S.Bendetz-Nezer, Z. Yao, R. Seger, DNA-independent PARP-1 activation by phos-phorylated ERK2 increases Elk1 activity: a link to histone acetylation, Mol. Cell25 (2007) 297–308.
61] A.L. Russo, H.C. Kwon, W.E. Burgan, D. Carter, K. Beam, X. Weizheng, J. Zhang,B.S. Slusher, A. Chakravarti, P.J. Tofilon, K. Camphausen, In vitro and in vivoradiosensitization of glioblastoma cells by the poly (ADP-ribose) polymeraseinhibitor E7016, Clin. Cancer Res. 15 (2009) 607–612.
62] S.A. Andrabi, N.S. Kim, S.W. Yu, H. Wang, D.W. Koh, M. Sasaki, J.A. Klaus, T.Otsuka, Z. Zhang, R.C. Koehler, P.D. Hurn, G.G. Poirier, V.L. Dawson, T.M. Dawson,Poly(ADP-ribose) (PAR) polymer is a death signal, Proc. Natl. Acad. Sci. U.S.A.103 (2006) 18308–18313.
63] M.J. Eliasson, K. Sampei, A.S. Mandir, P.D. Hurn, R.J. Traystman, J. Bao, A. Pieper,Z.Q. Wang, T.M. Dawson, S.H. Snyder, V.L. Dawson, Poly(ADP-ribose) poly-merase gene disruption renders mice resistant to cerebral ischemia, Nat. Med.3 (1997) 1089–1095.
64] M. Endres, Z.Q. Wang, S. Namura, C. Waeber, M.A. Moskowitz, Ischemic braininjury is mediated by the activation of poly(ADP-ribose)polymerase, J. Cereb.Blood Flow Metab. 17 (1997) 1143–1151.
65] C. Fiorillo, V. Ponziani, L. Giannini, C. Cecchi, A. Celli, N. Nassi, L. Lanzi-lao, R. Caporale, P. Nassi, Protective effects of the PARP-1 inhibitor PJ34in hypoxic-reoxygenated cardiomyoblasts, Cell. Mol. Life Sci. 63 (2006)3061–3071.
66] K.S. Oh, S. Lee, K.Y. Yi, H.W. Seo, H.N. Koo, B.H. Lee, A novel and orally
active poly (ADP-ribose) polymerase-1 inhibitor, 2-[methoxycarbonyl(4-methoxyphenyl)methyl sulfanyl]-1H-benzimidazole-4-carboxylic acid amide(KR-33889), attenuates injury in in vitro model of cell death and in vivo modelof cardiac ischemia, J. Pharmacol. Exp. Ther. (2008).67] J.B. Pillai, M. Gupta, S.B. Rajamohan, R. Lang, J. Raman, M.P. Gupta,Poly(ADP-ribose) polymerase-1-deficient mice are protected from angiotensin
[
[
earch 686 (2010) 57–67 67
II-induced cardiac hypertrophy, Am. J. Physiol. Heart Circ. Physiol. 291 (2006)H1545–1553.
68] J.B. Pillai, A. Isbatan, S. Imai, M.P. Gupta, Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+depletion and reduced Sir2alpha deacetylase activity, J. Biol. Chem. 280 (2005)43121–43130.
69] K.T. Yang, W.L. Chang, P.C. Yang, C.L. Chien, M.S. Lai, M.J. Su, M.L. Wu, Acti-vation of the transient receptor potential M2 channel and poly(ADP-ribose)polymerase is involved in oxidative stress-induced cardiomyocyte death, CellDeath Differ. 13 (2006) 1815–1826.
70] X. Liu, Y. Shi, R. Guan, C. Donawho, Y. Luo, J. Palma, G.D. Zhu, E.F. Johnson, L.E.Rodriguez, N. Ghoreishi-Haack, K. Jarvis, V.P. Hradil, M. Colon-Lopez, B.F. Cox,V. Klinghofer, T. Penning, S.H. Rosenberg, D. Frost, V.L. Giranda, Y. Luo, Poten-tiation of temozolomide cytotoxicity by poly(ADP)ribose polymerase inhibitorABT-888 requires a conversion of single-stranded DNA damages to double-stranded DNA breaks, Mol. Cancer Res. 6 (2008) 1621–1629.
71] J.F. Haince, M. Rouleau, M.J. Hendzel, J.Y. Masson, G.G. Poirier, Targetingpoly(ADP-ribosyl)ation: a promising approach in cancer therapy, Trends Mol.Med. 11 (2005) 456–463.
72] S.J. Miknyoczki, S. Jones-Bolin, S. Pritchard, K. Hunter, H. Zhao, W. Wan, M.Ator, R. Bihovsky, R. Hudkins, S. Chatterjee, A. Klein-Szanto, C. Dionne, B. Rug-geri, Chemopotentiation of temozolomide, irinotecan, and cisplatin activity byCEP-6800, a poly(ADP-ribose) polymerase inhibitor, Mol. Cancer Ther. 2 (2003)371–382.
73] S.F. El-Khamisy, M. Masutani, H. Suzuki, K.W. Caldecott, A requirement forPARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative
DNA damage, Nucl. Acids Res. 31 (2003) 5526–5533.74] O. Mortusewicz, J.C. Ame, V. Schreiber, H. Leonhardt, Feedback-regulatedpoly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA dam-age in living cells, Nucl. Acids Res. 35 (2007) 7665–7675.
75] T. Helleday, E. Petermann, C. Lundin, B. Hodgson, R.A. Sharma, DNA repairpathways as targets for cancer therapy, Nat. Rev. Cancer 8 (2008) 193–204.





























