Embed Size (px)
Citation preview

Neurotoxicimpactofprotein
fragmentationandaggregationin
tauopathymousemodels
Inauguraldissertationzur
ErlangungderWürdeeinesDoktorsderPhilosophie
vorgelegtder
Philosophisch-NaturwissenschaftlichenFakultät
derUniversitätBasel
von
FrederikSprenger
AusSalzkotten,Deutschland
Basel,2017
OriginaldokumentgespeichertaufdemDokumentenserverderUniversitätBasel
edoc.unibas.ch

II
GenehmigtvonderPhilosophisch-NaturwissenschaftlichenFakultät
aufAntragvon
Fakultätsverantwortlicher: Prof.Dr.MarkusRüegg
Dissertationsleiter: PDDr.Dr.DavidT.Winkler
Koreferent: Prof.Dr.BernhardBettler
Basel,den18.04.2017
____________________________________
UnterschriftdesFakultätsverantwortlichen
Prof.Dr.MartinSpiess
(Dekan)

Preface
III
Preface
The following dissertation was written by the author. The “Introduction” is based on an
extendedversionofareviewmanuscriptinpreparation(Sprenger,Winkler,2017expected).
The“Results”sectionconsistsoftwopublishedmanuscriptsandadditionalpreliminarydata.
Intheco-first-authorshippublication(Ozcelik,Sprengeretal.,2016)theauthorsignificantly
contributedtoexperiments,analysis,andwritingprocess.Inthesecondco-authorpublication
(Skachokova,Sprengeretal.,2015)theauthorcontributedtosomeanalysisandfinalwriting.
Theadditionaldatasectionistheresultofownwork.


Acknowledgements
V
Acknowledgements
Mymostheartfeltthanksgotomyboss,DavidWinkler,forhisvaluablementoringandhis
substantialsupport.
I would like to expressmy deep gratitude toMarkus Tolnay and Stephan Frank for their
supportofmywork.
Iamgratefultoourcollaborators,inparticularMichelGoedertandGrahamFraser,fortheir
expertise.
Iparticularlywishtothankcolleaguesandtechnicians,fortheirusefuldiscussions,advices,
andpracticalsupportthroughoutmywork.Myspecialthanksareextendedtothestaffatthe
ZLFanimalfacilityfortheirsupport.
Finally,Iwishtothankmyfamily,mywifeLimaandmysonSam.Thankyouforeverything
thatmakethispossible.


Abstract
VII
Abstract
The microtubule-associated protein tau and its pathological modification constitute the
central pathology of various human neurodegenerative diseases, including Alzheimer’s
disease (AD), collectively termed ‘tauopathies’. Abundant hyperphosphorylation and
aggregation of tau is a disease-defining hallmark, yet the underlying pathogenic and
pathophysiological processes have remained only partly understood. In addition, protein
fragmentation is a frequently observed phenomenon in the course of various
neurodegenerative diseases; however, the contribution of tau fragmentation to the
pathogenesisoftauopathiesisstillamatterofdebate.
Inournovel induciblemousemodel,co-expressionoftruncatedandfull-lengthhumantau
provokes axonal transport failure, mitochondrial mislocalization, disruption of the Golgi
apparatusanddysregulationofsynapticproteinsassociatedwithextensivenervecelllossand
asevereneurologicalphenotypeasearlyas3weeksofage.Ofnote,thiswasparalleledonly
by the formation of soluble oligomeric tau species, and no insoluble filamentous tau
aggregates;therewith,identifyingoligomerictauspeciesastoxickeyplayersintaupathology.
Despitecontinuousfull-lengthtauexpression,micerecoveredfromtheneurotoxicinsultonce
truncated tau expressionwas halted. The inductionof drastic but reversible neurotoxicity
highlights the neurotoxic potential of tau fragments as pathogenic mediators in
neurodegenerativedisorders.
Thepresentworkimplicatesthecomplexityofproteinfragmentationandoligomerizationand
theirneurotoxicimpactinthecontextoftauopathiesandaimsforabetterunderstandingof
thecellularmechanismsunderlyingtautoxicity.

VIII

Index
IX
Index
Preface III
Acknowledgements V
Abstract VII
Index IX
1 Introduction 13
1.1 Chapter1:Tauproteinandneurodegeneration............................................................13
1.1.1 Neurodegeneration.........................................................................................................131.1.2 Proteinfragmentationandneurodegeneration..............................................................141.1.3 Caspasesandcalpainsandneurodegeneration..............................................................151.1.4 Neurodegenerativedisease-associatedproteins............................................................171.1.5 Tauprotein......................................................................................................................18
1.1.5.1 Cellularlocalizationanddomainorganizationoftau..........................................................181.1.5.2 Tauisoforms........................................................................................................................191.1.5.3 Taufunction.........................................................................................................................201.1.5.4 Post-translationalmodificationsoftau................................................................................21
1.1.5.4.1 Tauphosphorylation.......................................................................................................221.1.5.4.2 Tautruncation.................................................................................................................23
1.1.6 Tauopathies.....................................................................................................................251.1.7 Prion-likeseedinginneurodegenerativeproteinopathies..............................................26
1.1.7.1 CSFAβ..................................................................................................................................26
1.2 Chapter2:Proteinfragmentationinneurodegenerativedisorders................................28
1.2.1 Neurodegenerativedisorders..........................................................................................281.2.2 ProteinfragmentationinAD:Aβ.....................................................................................30
1.2.2.1 Alzheimer’sdisease(AD)......................................................................................................301.2.2.2 Aβ.........................................................................................................................................31
1.2.3 ProteinfragmentationinfamilialCAA:ABriandADan....................................................331.2.3.1 Cerebralamyloidangiopathies:familialBritishandDanishdementia................................331.2.3.2 ABriandADan......................................................................................................................34
1.2.4 ProteinfragmentationinPD:α-synuclein.......................................................................351.2.4.1 Parkinson'sdisease(PD)......................................................................................................351.2.4.2 α-synuclein...........................................................................................................................36
1.2.5 ProteinfragmentationinTRD:Htt,ataxin,andatrophin................................................371.2.5.1 Huntington’sdisease(HD)...................................................................................................371.2.5.2 htt........................................................................................................................................371.2.5.3 Otherpolyglutaminediseases.............................................................................................38
1.2.6 ProteinfragmentationinPriondiseases:PrPCandPrPSc.................................................391.2.6.1 Priondiseases......................................................................................................................391.2.6.2 Prionprotein........................................................................................................................39

Index
X
1.2.7 ProteinfragmentationinFTLDandALS:TDP-43.............................................................411.2.7.1 TDP-43proteinopathies.......................................................................................................411.2.7.2 TDP-43..................................................................................................................................42
2 Aimsofthework 45
3 Results 47
3.1 PublicationNo.1:
Co-expressionoftruncatedandfull-lengthtauinducessevereneurotoxicity................47
3.2 Preliminarydata:
Protectiveeffectofearlytauburdenonlateneurotoxicdistresslevel–Mechanisms underlyingtauopathyandconsequencesforfuturetherapies.......................................67
3.2.1 DelayedmotorphenotypeinagedP301SxTAU62on-offmiceafterrecoveryofsevereneurotoxicity....................................................................................................................68
3.2.2 ReducedtaupathologyinagedP301SxTAU62on-offmiceafterrecoveryofsevereneurotoxicity....................................................................................................................70
3.2.3 ReducedtauproteinlevelsinagedP301SxTAU62on-offmiceafterrecoveryofsevereneurotoxicity....................................................................................................................71
3.2.4 Supplementalmaterial....................................................................................................73
3.3 PublicationNo.2:
Amyloid-betainthecerebrospinalfluidofAPPtransgenicmicedoesnotshowprion-like properties.....................................................................................................................75
4 Discussion 84
5 MaterialsandMethods 97
5.1 Animals.........................................................................................................................97
5.1.1 Housingoftransgenicmice.............................................................................................975.1.2 TAU62mice.....................................................................................................................975.1.3 P301Smice......................................................................................................................985.1.4 ALZ17mice......................................................................................................................985.1.5 ALZ31mice......................................................................................................................995.1.6 P301SxTAU62mice..........................................................................................................995.1.7 ALZ17xTAU62mice..........................................................................................................995.1.8 ALZ31xTAU62mice..........................................................................................................995.1.9 P301SxALZ31mice...........................................................................................................995.1.10 ALZ17xALZ31mice.....................................................................................................1005.1.11 APP23mice................................................................................................................100
5.2 DNAisolationandgenotyping.....................................................................................100
5.3 Histologyandimmunohistochemistry.........................................................................102
5.3.1 Tissuepreparationandprocessing:Brain,SpinalcordandSciaticnerve......................1025.3.2 HematoxylinandEosinStaining....................................................................................103

Index
XI
5.3.3 Gallyassilverstaining.....................................................................................................1045.3.4 HolmesSilverNitrate-LuxolFastBluestaining..............................................................1055.3.5 MassonTrichromestaining(Sciaticnerve)....................................................................1065.3.6 Musclespreparation......................................................................................................1065.3.7 Myosin-ATPase(Adenosintriphosphatase)staining(pH4.2).........................................1075.3.8 Semithinsections(Sciaticnerve)...................................................................................1085.3.9 Para-Phenylendiamine(Sciaticnerve)...........................................................................1085.3.10 Electronmicroscopy..................................................................................................108
5.4 Sarkosylextraction.....................................................................................................109
5.5 WesternBlot...............................................................................................................111
5.6 Antibodies..................................................................................................................112
5.7 Behavioralassessment................................................................................................113
5.7.1 Grid-test.........................................................................................................................1135.7.2 Rotarodtest...................................................................................................................1135.7.3 Objectrecognitiontest..................................................................................................113
5.8 Statistics.....................................................................................................................114
6 References 115
7 Abbreviations 135


Introduction
13
1 Introduction
1.1 Chapter1
Tauproteinandneurodegeneration
1.1.1 Neurodegeneration
In human beings, neurodegenerative diseases are commonly characterized by progressive
dysfunctionanddeceaseofneuronsassociatedwithpathologicaldepositsofalteredproteins
inthebrainaswellasinperipheralorgans.Inpatientswithneurologicaldisorders,theclinical
manifestationscausedbymalfunctionofindividualgeneexpressionproductscorrelatewith
theaffectedbrain regions, linkingaparticulardisease-type to itspredominantphenotype.
Unique pathological conformers or misfolded proteins with modified native physiological
properties are integral parts and the core concept of diverse human ’proteinopathies’;
nevertheless, the understanding of the cellular and molecular bases underlying the
pathogenesisofneurodegenerativediseasesgraduallywidenedovertheyears,yetbeingfar
fromfullydisclosed.
The pathological conformation and subsequent aggregation of proteins is not solely
responsible for neuronal degeneration, rather a complex network of molecular events
ultimatelyleadingtoprogressiveneuronaldysfunctionanddeath.Forinstance,theubiquitine

Introduction
14
proteasome system (UPS) and autophagy-lysosomal pathways, asmajormechanisms for
degradation of numerous proteins, have been found to be relevant for the genesis and
progressionofseveralneurodegenerativediseases(Kelleretal.,2000;Nixon,2007;reviewed
inOddo,2008;Pickfordetal.,2008).Indeed,variousaggregatedproteinsaswellasinduction
of proteasome inhibitors have been shown to interfere with the highly regulated cell
physiologyby impairingUPSfunction(Benceetal.,2001;Davidetal.,2002;Gregorietal.,
1995;Kecketal.,2003;Leeetal.,2010;Snyderetal.,2003;Tsengetal.,2007).Incontrastto
their ability of non-aggregated and soluble unfolded protein degradation, oligomeric or
aggregated species are rather inaccessible to the catalytic core of the UPS; an efficient
autophagy-lysosomalmachinery isneededtodegradeaggregation-proneproteins, thereby
preventingneuropathologicalprocesses(Angladeetal.,1997;Bolandetal.,2008;Qinetal.,
2003). In addition, stimulationof autophagyhasbeen shown to reduce the generationof
pathologicalproteininclusionsinnervecells(Ozceliketal.,2013;Schaefferetal.,2012;Wang
etal.,2009b).
Aside further neurodegenerative disease-causing processes such as glutamate-induced
exocytoxic insults (Marchetti et al., 2004) or neuroinflammatory processes (Harry et al.,
2000), another event detrimental to neuronal homeostasis is mitochondrial injury by
neurodegeneration-associatedproteins;mitochondrialdysfunction,i.e.intermsofimpaired
mitochondrial trafficking insideneurons (Ruietal.,2010;Ruietal.,2006)oralterations in
mitochondrialdynamics (Wangetal.,2009a), iscrucially linkedtooxidativeandnitrosativ
stress (Cho et al., 2009; Hirai et al., 2001; Lustbader et al., 2004), contributing to
neuropathologicalprocesses.
1.1.2 Proteinfragmentationandneurodegeneration
Protein fragmentation is a frequently observed phenomenon in the course of various
neurodegenerativeprocesses.Indeed,small,aggregationpronecleavedproteinsareintegral
partsofaplethoraofdisordersincludingAlzheimer’sdiseases(AD);familialBritishandDanish
dementia(FBD,FDD);Parkinson’sdiseases(PD);TDP-43relateddisorders;andinothertriplet
expansiondisorderssuchasHuntingtondisease(HD)andspinocerebellar-ataxias(SCAs).The
initiationoffragmentationremainsmostlyelusive;mutationsrepresentthecauseofaltered

Introduction
15
cleavage in some hereditary variants of neurodegenerative diseases such as presenilin
mutationsinADortheframeshiftmutationsinFBD;bycontrast,inmostsporadicformsthe
causeforincreasedoraberrantfragmentationissimplynotknown.
However,thecontributionofproteinfragmentationtothepathogenesisofproteinopathiesis
not always evident and the neurotoxic potential of cleavage products is still a matter of
debate.Cleavageoccursatmultiplesitesofsinglelargeproteinswithresearchersconsiderthe
question of a causal relationship between protein fragmentation and disease, orwhether
fragmentation just being an epiphenomenon. Protein aggregates in neurodegenerative
disordersmayeitherconsistof(I)fragmentsalonederivedfromlargerprecursorproteins:as
in case of amyloid-β (Aβ) in AD or amyloid-Bri (ABri) in FBD (Vidal et al., 1999); or they
comprise(II)full-lengthandfragmentedproteinsinparallel:asincaseofα-synuclein(α-Syn)
inPD(Duftyetal.,2007;Liuetal.,2005)orinTDP-43relateddisorders(Neumannetal.,2006;
Zhangetal.,2009b).
Neurodegeneration-associatedproteinscanbesubstratetoaplethoraofproteolyticenzymes
includingmembersoftheα-,β-,and!-secretasefamiliesaswellascysteineproteases.Aside
thrombin (Arai et al., 2005), cathepsins (Kenessey et al., 1997) and puromycin-sensitive
aminopeptidase(PSA)(Senguptaetal.,2006),membersofthecaspaseandcalpainfamilyare
themost prominent enzymes involved in tauprotein cleavage and therefore of particular
interestinthepresentwork.
1.1.3 Caspasesandcalpainsandneurodegeneration
Caspasesareintracellularcysteine-aspartatic-specificproteasesthatcleavetheirsubstrates
at specific sites. First expressed as latent zymogens, these pro-caspases get post-
translationallyactivatedthateithercanleadtotheinactivationofthesubstrateortoatoxic
gainoffunctionintheformofactiveproteinfragmentsintheproteolyticprocess.Asideother
non-apoptoticandpro-inflammatorymembers,apoptoticcaspasescanbesubdividedinto(I)
initiatorcaspases(caspases-2,8,9and10)that,inresponsetoastimulus,directthesignalto
(II) executioner caspases (caspases-3, 6, and 7) (Pop and Salvesen, 2009). These caspase
candidates are associated with programmed apoptotic cell death in various
neurodegenerative disorders including AD, HD, and PD. Also, calpains are intracellular

Introduction
16
calcium-activated (papain-like) neutral proteases and have been implicated in the
pathogenesisofneurodegenerativediseasessuchasADorPD;althoughtheircalcium-induced
apoptoticroleislesscharacterizedcomparedtothatofapoptoticcaspases.
Multipleneurodegeneration-relatedproteinsaresubstratetocaspase-andcalpain-mediated
cleavage. Indeed, caspases-3 (Metcalfe et al., 2012), caspase-6, and calpains are themain
enzymesinvolvedintauproteinfragmentation,withtheAsp421beingthemostprominent
caspasecleavagesite(forreviewseeAvila,2010;Fasuloetal.,2005;Guillozet-Bongaartset
al., 2005; Guo et al., 2004) (Figure 1.1). Furthermore, site-directed mutagenesis at two
caspase-7cleavagesitesofneurotoxicataxin-7proteininSpinocerebellarataxiatype7(SCA7)
polyglutamine(polyQ)disorderresults inanon-cleavableformofpolyQ-expandedataxin-7
displayingattenuatedneuronaldeath,aggregateformation,andtranscriptionalinterference
(Youngetal.,2007).Ofnote,caspase-3andcaspase-6havebeenimplicatedincytoskeletal
disintegrationthroughactinandtubulincleavageultimatelyleadingtoaxonaldegeneration
invitroandinvivo(Sokolowskietal.,2014).Aside,calpainactivationhasbeendemonstrated
to play a crucial role in Aβ-triggered pathological cascade in AD (Higuchi et al., 2012).
Moreover,N-terminalcalpaincleavedataxin-3fragmenthasbeenshowntoprovokealtered
behaviouralandmotorphenotypeassociatedwithpathologicalproteininclusionsandnerve
celldeathinvivo(Hubeneretal.,2011).
Figure1.1Proteolyticprocessingoftau.Caspaseandcalpainsarethemainproteasesinvolvedintauproteincleavage. Truncationof tau at distinct proteolytic cleavage sites can either lead to preservationof neuronalstructureand/orfunction,and/orexacerbationoftautoxicity.(Chesseretal.,2013)

Introduction
17
1.1.4 Neurodegenerativedisease-associatedproteins
Awidevarietyofcellularandmoleculareventscanbeascribedtotheindividualpathological
profile of a vast number of neurodegenerative diseases. However, pathologically altered
proteins including their characteristic structure and morphology remain to be the most
prominententity.Proteinsthatundergofragmentationinparalleltopathologicalaggregation
are not only found in neurodegenerative proteopathies, but also in systemic amyloidosis.
Proteinopathieswithassociatedproteinfragmentationinclude:
(I) Themicrotubule-associatedproteintauthatisencodedbyagene(MAPT)located
onchromosome17(Weingartenetal.,1975);
(II) The amyloid-beta peptide (Aβ) that is encoded by a gene (APP) located in
chromosome21(Alzheimer,1906,1907;Kangetal.,1987;Tanzietal.,1987);
(III) Theamyloid-Bri(ABri)andamyloid-Dan(ADan)peptidesthatarebothencoded
byagene(BRI)locatedonchromosome13(Vidaletal.,1999;Vidaletal.,2000);
(IV) Theneuronalproteinalpha-synuclein (α-Syn) that isencodedbyagene (SNCA)
locatedonchromosome4(Spillantinietal.,1997);
(V) Proteinsencodedbygeneslinkedtocytosine-adenine-guanine(CAG)trinucleotide
repeatsincludinghuntingtin(Htt),ataxins(1,2,3,6,7,and17),andatrophin-1
(Fanetal.,2014);
(VI) Prionprotein(PrP)thatisencodedbyagene(PRNP)locatedonchromosome20
(AguzziandO'Connor,2010);
(VII) Transactiveresponse(TAR)DNA-bindingprotein43(TDP-43)thatisencodedbya
gene(TARDBP)locatedonchromosome1(Ouetal.,1995);
(VIII) AndothersincludingproteinsthatbelongtotheFETfamilyincluding(F)usedin
sarcomaprotein(FUS),(E)wing’ssarcomaprotein(EWS),(T)ATA-bindingprotein-
associatedfactor15(TAF15)(Kwiatkowskietal.,2009;Lawetal.,2006);charged
multivescularbodyprotein2B(CHMP2B)(Ghazi-Noorietal.,2012);glycoprotein
reelin(D'Arcangeloetal.,1997);globularproteintransthyretin(TTR)(Conceicao
etal.,2016);actinbindingproteingelsolin(Chenetal.,2001;Solomonetal.,2012);
hormone islet amyloid peptide (IAPP) (Akter et al., 2016); and human serum
amyloidA(SAA)(Egashiraetal.,2011).

Introduction
18
Themostnotableneurodegeneration-associatedproteinswillbeaddressedinthefollowing
paragraphs;however,thetauproteinanditsroleintauopathiesandtheneuropathological
relevanceofproteinfragmentationwillbeofparticularinterest.
1.1.5 Tauprotein
Tau belongs to the natively unfoldedmicrotubule-associated protein family (MAP) and is
abundantinthecentralandperipheralnervoussystem.Inthe1970s,amicrotubulebinding
activityoftauproteinhasfirstbeenshownbyWeingartenetal.,whoisolatedaheatstable
proteinmostabundantly found topromotemicrotubuleassemblyand stability in cell-free
conditions (Weingarten et al., 1975). Microtubules are protein polymers and a major
component of the cytoskeleton with an essential role in regulated motor-driven axonal
transport. Later, evidence frombrainsofpatientswithAD suggested that tau is actual an
integralpartofthepathologyinneurodegenerativedisorders(Goedertetal.,1988;Grundke-
Iqbaletal.,1986;Kondoetal.,1988;Kosiketal.,1986;Wischiketal.,1988a).Effortsfora
better understanding of the physiological role and identity of tau protein have been
intensifiedsince.
1.1.5.1 Cellularlocalizationanddomainorganizationoftau
Thetauprotein,synthesizedandproducedinallneurons, ispredominantlyfoundinaxons
(Binder et al., 1985). However, it also has been shown to be located in the dendritic
compartmentalbeit in lowerconcentrationaswellasunderpathologicalconditions in the
somatodendriticdomain(Ittneretal.,2010).Structurally,tauisanaturallyunfoldedprotein
thatcontainsfourmajorregions.Oncetauisboundtomultipletubulindimers,theN-terminal
acidicprojectionregionprotrudesoutwardfromthesurfaceofthemicrotubuleandinthis
way, being able to serve as a spacer between the individual components within the
microtubulenetwork(Chenetal.,1992;Frappieretal.,1994).Furthermore,itwasfoundthat
thisregionappearstointeractwithmembrane-bindingproteinssuchasannexinA2(AnxA2)
and thus retain the tauproteinat thedistal tipofneurites (Brandtetal., 1995;Gauthier-

Introduction
19
Kemperetal.,2011;Weissmannetal.,2009).Theproline-richregion(PRR)harboursmany
phosphorylationsitesandcontributestothemicrotubule-bindingaffinityoftau(Augustinack
et al., 2002; Biernat et al., 1992; Brandt and Lee, 1993;Goodeet al., 1997).Moreover, it
enablesinteractionwithotherproteinssuchastheSH3domaincontainingtyrosinekinaseFyn
(Leeetal.,1998).TheC-terminalregionoftaucontainsamicrotubulebindingdomain(MTB)
composedof18-aminoacid(aa)tandemrepeatsseparatedbysequencesof13-or14-aathat
encouragetautobindtothemicrotubulesandashorttailsequence,whichisinvolvedinthe
regulationofmicrotubulepolymerization(BrandtandLee,1993;Leeetal.,1988;Leeetal.,
1989).
1.1.5.2 Tauisoforms
Thetauproteinisencodedbyasinglegenethatcontainsatotalof16exons(Andreadisetal.,
1992).Six isoforms of tau are expressed in the adult humanbrain (Goedert et al., 1989).
ProducedbycomplexalternativemRNAsplicingoftheMAPTgenelocatedonchromosome
17q21.31,eachisoformdiffersinitsspecificrepresentationassuchinthepresenceorabsence
of both, amino-terminal inserts (0N, 1N, 2N) and carboxy-terminal microtubule-binding
repeatdomains(3R,4R).Thus,splicingoftheneuron-specifictautranscriptattwo29-or58-
aainsertsandone31-aarepeatdomainencodedbyexons2,3and10,respectively,resultsin
asetofproteinswiththerangefrom352-aainlengthfor3R0N,381for3R1Nand410for
3R2Nto383-aainlengthfor4R0N,412for4R1Nand441for4R2N(Figure1.2).

Introduction
20
Figure1.2SchematicrepresentationofthehumanTAUgene,TAUmRNA,andthesixtauproteinisoforms.Sixisoformsaregeneratedthroughalternativesplicingintheadulthumanbrain.(AdaptedfromBueeetal.,2000)
1.1.5.3 Taufunction
Tau,asanativelyunfoldedprotein,lacksawell-definedsecondaryortertiarystructurethat
allowstheproteintointeractwithavarietyofpartnersintheenvironmentofacell(Schweers
et al., 1994). The main physiological function of tau is stabilization of the microtubule
network bypromoting thepolymerizationof tubulinand thus themaintenanceofnormal
axonaltransport(Bohmetal.,1990;BrandtandLee,1993;Clevelandetal.,1977;Shahaniand
Brandt, 2002;Weingarten et al., 1975). As amain cytoskeleton component,microtubules
contributetomorphogenesis,divisionandintracellulartraffickinginthecell(Mitchisonand
Kirschner,1984).

Introduction
21
Atanygivenmoment,about80%oftauisindirectinteractionwithmicrotubules(Weissmann
etal.,2009).Taubindingtomicrotubulesisregulatedbyadelicateequilibriumofkinasesand
phosphatasesandboth,over-stabilizationbytauordetachedtaufromthemicrotubulecan
impaircellviability(Bramblettetal.,1993;Pandaetal.,2003;ThiesandMandelkow,2007;
Wangetal.,2007).
Infact,neuronalfunctioniscriticallydependentonanintactmicrotubulenetwork.Specific
cellular compartments suchas thepre-andpost-synaptic cell structureshavehighenergy
requirementsandaccumulatedwasteproductstomanage;therefore,cellularmotorsofthe
kinesinanddyneinsuperfamilyutilizeenergyderivedfromATPhydrolysistotransportcargo-
filledvesiclesoverlongdistancesonmicrotubuletracksintheaxon(DeVosetal.,2008).In
this way, mitochondria (Hollenbeck and Saxton, 2005), lysosomes (Harada et al., 1998),
peroxisomes(Walietal.,2016)andvariousotherorganellescanbelocalizedtodistinctareas
intheneuronalrealminordertoaccomplishtheirveryownfunction.Thetightbindingoftau
alters the intracellular traffic as well as ensures the dynamic instability of microtubules
(Mitchison and Kirschner, 1984; Trinczek et al., 1995). The latter is distinguished by their
capability to switch between slow growth and rapid shrinking duringmicrotubule growth
(Binder et al., 1985; Mitchison and Kirschner, 1984). Aside from its predominant
neurophysiological activity – binding to microtubule – numerous functions have been
attributedtotau.Amongthem,modulationofbiochemicalcascades,suchthattaucanactas
a protein scaffold (Brandt et al., 1995; Ittner et al., 2010; Reynolds et al., 2008) or direct
enzymeinhibitor(Perezetal.,2009).Inparallel,tauappearstointeractwithnucleicacidsas
wellasmitochondria(Jancsiketal.,1989;Kampersetal.,1996;Loomisetal.,1990;Sultanet
al.,2011);suggestingaroleasamultifunctionalcommunicationinstrumentwithinthecell.
1.1.5.4 Post-translationalmodificationsoftau
Inphysiologicalconditionsanduntypicallyformostcytosolicproteins,tauresistsacompact
foldedstructure;theentiretaumoleculeisconsideredtobeintrinsicallydisorderedandits
function is tightly regulated by a host of post-translational modification such as
phosphorylation(Grundke-Iqbaletal.,1986),acetylation(Cohenetal.,2011),glycosylation
(Gongetal.,2005),glycation(Ledesmaetal.,1994),sumoylation(DorvalandFraser,2006),

Introduction
22
ubiquitination(Morietal.,1987),nitration(Reynoldsetal.,2006),andtruncation(Gamblinet
al., 2003; Wischik et al., 1988b). The contribution of various cellular mechanisms to tau
pathogenesisarenotknownandithasyetremainedunclear,whichmodificationiscrucialfor
thedevelopmentoftauopathies.
Theindividualpost-translationalmodificationsoftaucanonlybeoutlinedinthepresentwork;
however, tauphosphorylation (I) and tau truncation (II)will be discussed in detail in the
followingparagraph.
1.1.5.4.1 Tauphosphorylation
Tauphosphorylationoccurs inbothpathologicalandphysiologicalconditions. Itwasfound
that isolated tau molecules from healthy human brains contained roughly two moles of
phosphate permole of tau,whereas tau proteins associatedwith paired helical filaments
(PHFs)ofpatientswithADcontainedsixtoeightmolesofphosphatepermoleoftau(Ksiezak-
Redingetal.,1992).
Giventheloosedisorderedcharacteroftau,manyknownpotentialphosphorylationsitesare
sensitive to numerous protein kinases and phosphatases; indeed, these phosphorylatable
domainsconsistof80serineandthreonineresiduesandfivetyrosineresiduesarekeyplayers
intheregulationofthemicrotubulebindingactivityoftau(Figure1.3).Themostprominent
candidatekinasesfortauphosphorylationincludeproline-directedkinasesglycogensynthase
kinase3(GSK3),cyclin-dependentproteinkinase(cdk5),thep38mitogen-activatedprotein
kinase(MAPK);orc-JunN-terminalkinases(JNK) families,aswellasotherstressactivated
kinases,suchascdc2;andnon-proline-directedkinasessuchasproteinkinaseA(PKA),protein
kinaseC(PKC),calmodulin(CaM)kinaseII,microtubule-affinityregulatingkinase(MARK),and
caseinkinaseII(CKII)(Correasetal.,1992).
Asidefromkinases,variousphosphatases(PP)havebeenidentifiedtodephosphorylatetau
protein(Sergeantetal.,2005).IthasbeenshownthatPP1,PP2AandPP2B,predominantly
dephosphorylate tau in vitro (Wangetal., 1995;Yamamotoetal., 1988);moreover,PP2A
expressionandactivitywasfoundtobereducedinthebrainsofADpatients,suggestingthat
dephosphorylation defects play a vital role in the pathological cascade in tau mediated
neurodegenerativedisorders(Gongetal.,1993).

Introduction
23
Indeed,abnormaltauphosphorylationisconsideredasanearlyeventinthepathologyoftau
(Bramblettetal.,1993).Asmentionedabove,theadulthumanbraintauharboursmorethan
80 potential phosphorylation sites and a disequilibrium in candidate protein kinase and
phosphatase activity results in tau hyperphosphorylation with subsequently increased
amountoftaudetachedfrommicrotubule.
Figure1.3Dysregulationofaxonaltransport.Destabilizationofmicrotubulesbydecreasedkinaseactivityresultsinhyperphoshorylatedandaggregatedtau.Subsequentdisintegrationofthemicrotubuletracksleadstokinesin-mediatedanterogradeanddynein-mediatedretrogradeaxonaltransportdysfunction.(AdaptedfromDeVosetal.,2008)
1.1.5.4.2 Tautruncation
Phosphorylationistypicallyregardedasoneofthemostrelevantmodificationresponsiblefor
changesoftauprotein.Alternatively,proteolyticprocessingoftaubyavarietyofendogenous

Introduction
24
proteaseshasbeenpostulatedtobe involved in thepathologicalcascade in taumediated
neurodegenerativedisorders(Gamblinetal.,2003;Rissmanetal.,2004).
Tauproteinexhibitaphysiologicalrandomcoilstructure;however,itisabletoassembleinto
orderedfilamentousaggregatesbytheformationofβ-sheetstructuralelements(vonBergen
et al., 2005). Given that the aggregation of tau correlateswith its propensity for β-sheet
structure,analteredshapeoftauproteinbyproteolyticfragmentationpotentiallyaffectits
aggregation capacity. The architecture of paired helical and straight filaments of AD is
predominantly defined by all six full-length isoforms of tau (Goedert et al., 1992);
nevertheless,studiesontauaggregationhadrevealedthattruncatedformsoftauare,infact,
presentinthecoreofPHFs(Gamblinetal.,2003;Menaetal.,1996;Rissmanetal.,2004).
Likewise,fragmentedtauhasbeenreportedtofacilitateandpromotefasterpolymerization
intofibrils,inthisway,highlightagreateraggregationpropensitycomparedtofull-lengthtau
(Abrahaetal.,2000).
InAlzheimer’sdisease, tau truncationwas found tobeanearlyeventandappearsbefore
neurofibrillary tangle (NFT) formation albeit after hyperphophorylation of tau (Guillozet-
Bongaartsetal.,2005;Mondragon-Rodriguezetal.,2008;Rohnetal.,2002;Saitoetal.,2010).
Various types of tau fragmentation have been reported in cells and brain tissue; cysteine
proteases suchascaspasesandcalpainshavebeen implicated inproteolysisof tauduring
apoptosis (Canuet al., 1998). In vitro and in vivoexperimentspredictedmultipleputative
cleavage sites of tau at both, its C-terminal aswell as N-terminal domain by a variety of
caspases(Delobeletal.,2008;Gamblinetal.,2003;Guillozet-Bongaartsetal.,2005;Horowitz
etal.,2004).However,notallproteolyticcleavagesitescanbeassignedtoadistinctprotease:
C-terminaltruncatedtauatglutamicacid391(E391)islinkedtoclinicaldementiayetcatalyzed
byanunknownproteolyticenzyme(Basurto-Islasetal.,2008;Novaketal.,1993).
However,presentlythemoststudied,caspasescleavetaupreferentiallyataspartaticacid421
(D421);both,caspase3andcaspase6havebeenfoundtobeinvolvedinthiscleavageatthe
C-terminal domain of tau (Guo et al., 2004; Rissman et al., 2004; Zhang et al., 2009a).
Moreover, accumulation of caspase-cleaved C-terminal fragments have been reported to
correlatewiththeprogressioninADandinvivomousemodelsoftauopathy(Basurto-Islaset
al.,2008;Centeetal.,2006;deCalignonetal.,2010;Delobeletal.,2008;Guillozet-Bongaarts
etal.,2005).

Introduction
25
1.1.6 Tauopathies
The term “Tauopathy” summarizes a heterogeneous group of disorders with
hyperphosphorylated,insoluble,filamentoustauproteininclusionsinneuronsandglialcells
(SpillantiniandGoedert,2013)(Figure1.4).Thesehumanneurodegenerativediseasesarein
most cases sporadic, and clinically characterized by dementia, often associated with
movement impairment. Most frequent tauopathies include AD (see paragraph 1.2.2.1),
Progressive supranuclearpalsy (PSP) (Steeleetal., 1964),Corticobasaldegeneration (CBD)
(Rebeizetal.,1968),Pick’sdisease(PiD)(Constantinidisetal.,1974),Argyrophilicgraindisease
(AgD) (Braak and Braak, 1989), and Frontotemporal dementia and parkinsonism linked to
chromosome17(FTDP-17T)(Wilhelmsenetal.,1994).
Figure1.4Differenttypesoftauimmunoreactivity intauopathies.Hyperphosphorylatedtau,AD(A);gallyaspositiveglobosetangle,AD(B);gallyaspositivetuftedastrocyte,PSP(C);hyperphosphorylatedtauinastrocyticplaques, CBD (D); hyperphosphorylated tau from pick bodies, PiD (E); hyperphosphorylated tau, AgD (F).(AdaptedfromNeumannetal.,2009)

Introduction
26
1.1.7 Prion-likeseedinginneurodegenerativeproteinopathies
Prion-like seedingor transmission isageneralmechanismobserved inneurodegeneration
proteinopathiesincludingCreutzfeldt-Jakobdisease(CjD),kuru,andscrapie.Inthisprocess
involvedaremisfoldedandaggregatedproteinsthatpotentiallyactasinfectiousagentsby
structurallycorruptingotherproteins,andthustriggertheirpathogenicaggregation(Jucker
andWalker,2013).Variousneurodegeneration-associatedproteinsincludingAβ,tau,α-Syn,
andTDP-43 are increasinglyemergingas considerable candidateswithpotential prion-like
properties(Figure1.5).IncaseofpotentialAβseeding,thisisreflectedinstudiesonmouse
models, where brain tissues from AD patients of APP transgenic mice can indeed seed
amyloidosisinvivo,indicatingaprion-likebehaviorofpathologicAβ(Kaneetal.,2000;Meyer-
Luehmannetal.,2006).Moreover, inoculationofbrainhomogenatesderivedfromhuman
tauopathypatientsandNFTsbearingP301Stautransgenicmiceintowild-typetauexpressing
ALZ17hostmicedemonstratedaprion-likespreadingpotentialof insolubletauaggregates
(Clavagueraetal.,2013;Clavagueraetal.,2009).
1.1.7.1 CSFAβ
PreviouslyithasbeenshownthatsmallandsolubleAβspeciesfoundinthebrainofAPP23
transgenicmiceexhibitprion-likebehaviour(Langeretal.,2011).GiventhatAβisalsopresent
in the cerebrospinal fluid (CSF), CSF biomarkers constitute a valuable tool for early AD
diagnosis(Cummings,2004;McKhannetal.,2011);currentCSFanalysis,though,suffersfrom
measurementsvariabilities(Scheltensetal.,2016).Studiesonpost-mortemCSFhavebeen
demonstratedthatAβ42levelsinverselycorrelatewithcorticalplaquedeposition(Tapiolaet
al.,2009).Further,inparalleltotheprogressionofsenileplaques,Aβ40andAβ42levelsinthe
CSFappeartodecreasewithage(Maiaetal.,2013).However,thepotencyofhumanCSFfor
amyloidaggregationinductioninmousemodelsofdementiaremainselusive.

Introduction
27
Figure 1.5 Spreading of neurodegeneration-associated proteins. Progressive prion-like propagation andspreadingofAβ(A),tau(B),α-Syn(C),andTDP-43(D)basedonbrainautopsystudiesofhumandiseasepatients.(AdaptedfromJuckerandWalker,2013)

Introduction
28
1.2 Chapter2:
Proteinfragmentationinneurodegenerativedisorders
FrederikSprenger,DavidT.Winkler
Reviewinpreparation
1.2.1 Neurodegenerativedisorders
The majority of neurodegenerative disorders is associated with pathological protein
aggregation.Theterm“proteinopathies”hasthereforebeenestablishedforthesediseases.
Themechanismsunderlyingproteinaggregation inproteinopathieshas inmanycasesonly
been partially understood. Different factors may eventually lead to pathological protein
aggregation.Firstofall,an increasedaggregationpropensityofagivenproteincan induce
pathological folding and aggregation. Proteins can be rendered aggregation prone by
patholocigal mutations, e.g. in the case of hereditary forms of proteinopathies, or by
truncation.Insporadicdiseases,factorsi.e.decreasedproteinclearance,e.g.byautophagy
disruption, are furthermore suspected to be underlying the aggregation process. These
processes involve protein cleavage and may thereby not only be beneficial by removing
misfoldingproteins,butalso indirectly contribute to render theaggregationpropensityby
cuttingproteinsintosmallpeptideswithseed-likecharacter.Inmostcases,theaggregating
proteins are rather small, often derived of a precursor protein. All this points towards a
unifying characteristic of protein cleavage as a key factor leading to aggregation prone
fragments. Such fragments often possess an inherently higher aggregation propensity
comparedtotheirprimaryfull-lengthproteinofwhichtheyarederived.
Proteincleavageisawidelyobservedprocessinneurodegenerativedisorders.Basedonthe
specificneurodegeneration-associatedproteins and theirmodifications leading todisease-
typicalhallmarksattheneuropathologicallevel,variousneurodegenerativediseasescanbe
linkedtooneanotherandassignedtothefollowingterms:

Introduction
29
(I) Tauopathy; including Alzheimer’s disease (AD), frontotemporal dementia and
parkinsonism linked to chromosome 17 (FTDP-17T), progressive supranuclear
palsy(PSP),cortocobasaldegeneration(CBD),andargyrophilicgraindisease(AGD);
Pickdisiease(PiD),andglobularglialtauopathy(GGT)(Fasuloetal.,2005;Ferreira
andBigio,2011;Guoetal.,2004;Parketal.,2007;Zhangetal.,2009a)(seealso
paragraph1.1.6.);
(II) Heriditary amyloidoses/Cerebral amyloid angiopathy (CAA); including AD and
familialCAArelatedtoAβvariantssuchasfamilialBritish(FBD)andDanish(FDD)
dementias;gelsolin;andtransthyterin(Chenetal.,2001;DeStrooperetal.,1999;
Ihseetal.,2013;Vidaletal.,1999;Vidaletal.,2000);
(III) α-Synucleinopathy;includingParkinsondisease(PD),dementiawithLewybodies
(DLB),andmultiplesystematrophy(MSA)(Duftyetal.,2007;Kessleretal.,2003;
Mishizen-Eberzetal.,2005);
(IV) Trinucleotiderepeatexpansiondisorder(TRD);includingHuntingtindisease(HD),
andspinocerebellar-ataxia-1,2,3,6,7,17(SCA3andSCA7)(Grahametal.,2006;
Hubeneretal.,2011;Mookerjeeetal.,2009;Wellingtonetal.,2002);
(V) Priondisease; includingCreutzfeldt-Jakobdisease(CjD)(Altmeppenetal.,2012;
Notarietal.,2008);
(VI) TDP-43proteinopathy;TDP-43relateddisorders including frontotemporal lobar
degeneration(FTDP-TDP)(Araietal.,2010;Igazetal.,2009;Nonakaetal.,2009);
(VII) FUS/FETproteinopathy; includingbasophilicinclusionbodydisease(BIBD)(Kent
etal.,2014;RademakersandRovelet-Lecrux,2009).
Aggregatesofaberrantmodifiedproteinsmayeitherconsistofmainlyfragmentedprecursor
protein remnants, or contain co-aggregated full-length proteins and cleaved fragments in
parallel. Cleavage derived species of themost notable entities and their potential role in
neurodegeneration will be addressed separately in the following paragraphs; a detailed
discussion, though, of other individual protein modifications is beyond the scope of the
presentwork.

Introduction
30
1.2.2 ProteinfragmentationinAD:Aβ
1.2.2.1 Alzheimer’sdisease(AD)
In2015,theworldwideprevalenceofdementiawasestimatedtobe46.8million(Princeet
al.,2015).Thisnumberisforecasttodoubleevery20years,reachingover130millionaffected
people in 2050.Over90%of allAD casesoccur sporadically; foremost amongothers, age
constitutesthemainriskfactorfordementia(Blennowetal.,2006);inaddition,thefrequency
oftheapolipoproteinE4(ApoE4)allele,amajorgeneticriskfactor,onchromosome19inlate-
onsetAD(LOAD)patientshasbeenshowntobesignificantly increased(Strittmatteretal.,
1993).Incontrast,about5to10%arefamilialcases(FAD)andresultsinaggressiveearly-onset
progression of AD (EOAD) (Tanzi, 1999).Mutations in genes coding for amyloid precursor
protein (APP; located on chromosome21; presenilin 1 (PS1; located on chromosome14),
presenilin2(PS2;locatedonchromosome1)havebeenreportedinFAD(Goateetal.,1991;
Sherringtonetal.,1995).
Alzheimer’s disease is a most probably heterogeneous neurodegenerative disorder. It is
clinically characterizedby progressive cognitive decline, typically delineatedby short-term
andlong-termmemoryimpairment,aswellaslossofsocialabilitiesincludingsymptomslike
confusion,irritability,aggressionandlanguagebreakdown(Tabertetal.,2005;Waldemaret
al.,2007);inparallel,patientsareaffectedbymusculardeteriorationandmotordisabilities
(Scarmeasetal.,2004).Furthermore,AD isneuropathologicallydefinedbyextracellularβ-
amyloid (Aβ; see paragraph 1.2.2.2) plaques, vascular Aβ deposits (cerebral amyloid
angiopathy;CAA;seeparagraph1.2.3.)andintracellularaggregatesoftauprotein(NFTs;see
paragraph1.1.6)(Alzheimer,1906,1907)(Figure1.6).
Massive neuronal and dendritic loss is the primary cause of cortical atrophy during the
progressionofAD.Atrophicchangesresultsincorticalthinningandenlargementofthelateral
cerebral ventricles. Reduced neuronal numbers in a variety of brain regions such as the
temporal, parietal and enthorhinal cortex (Gomez-Isla et al., 1997), theCA1 regionof the
hippocampus (West et al., 1994) and amygdala (Vereecken et al., 1994) have been
documented;however,thecauseofneurondeathisdisputable.
Infact,thepathogenicmechanismsarepoorlyunderstood,withsomestudiessuggestthat

Introduction
31
intraneuronaland/oroligomericAβ-peptidesactaskeymediatorsofneurotoxicityandcell
death(BayerandWirths,2010;LarsonandLesne,2012).However,pathologicaltauprotein
increasinglygainedattentioninthepathogenesisoftauopathiesincludingAD.Otherstudies
provide evidence of an intimate coherence between neuron loss and the appearance of
neurofibrillarytangles(Crasetal.,1995;Gomez-Islaetal.,1997);indeed,incontrasttothe
degreeofAβpathologyinformofsenileplaques,theextentoftaupathologyhasbeenshown
tocorrelatebestwiththeclinicalstateofAD(Arriagadaetal.,1992;Giannakopoulosetal.,
2007;Landauetal.,2016;Ossenkoppeleetal.,2016).
Figure 1.6 The pathological hallmarks of AD. Senile Aβ plaque (A), cerebrovascular amyloid (B), andneurofibrillarytangles(C).(AandBadaptedfromCastellanietal.,2010)
1.2.2.2 Aβ
Amyloid-beta(Aβ)isgeneratedfromtheamyloidprecursorprotein(APP)(Kangetal.,1987;
Tanzi et al., 1987), which is a single transmembrane glycoprotein consisting of an
exocytoplasmic domain and a short cytoplasmic tail. This large precursor protein is
sequentially processed releasing distinct secreted derivatives into vesicle lumens and the
extracellularspace.AssummarizedinFigure1.7,APPisproteolyticprocessedvia(I)thenon-
amyloidogenicpathway;or(II)theamyloidogenicpathway(Selkoe,2001b);ofnote,thelatter
iscrucialforAβliberationandthusofneuropathologicalrelevance.

Introduction
32
(I) Thenon-amyloidogenicpathway
This processing prevents an accumulation of Aβ peptides by demolishing the complete
amyloid sequence. APP is sequentially cleaved by α-and !-secretases to release a smaller
fragment(p3),thathasnoneuropathologicalrecognizedrole.Theα-sitecleavageofAPPby
adamalysin protease (ADAM) cuts 12-aa at the N-terminal single transmembrane domain
(Roberts et al., 1994); consequently, a C-terminally truncated formof soluble ectodomain
fragment (α-sAPP) is released from the membrane. The remaining 83-residue C-terminal
fragment(C83;CTFα)inthemembraneisfurtherprocessedby!-secretaseswhichthenleads
tothereleaseoftheshortextracellularp3fragment(Selkoe,2001b)andtheAPPintracellular
domain(AICD)(Zhangetal.,2011).
(II) Theamyloidogenicpathway
Aβ protein is liberated by sequential proteolytic processing of APP involving β-and !-
secretases. Initially,proteolytic cleavageoccurs16-aa residues to theN-terminalof theα-
cleavagesitebytheβ-siteAPPcleavingenzyme1(BACE1)(Hussainetal.,1999;Sinhaetal.,
1999;Vassaretal.,1999),generatingasolubleaminoterminalAPPderivate(β-sAPP)anda
membrane-associated 99-residue C-terminal (C99, CTFβ). Further, !-secretase activity at
different intramembranousCTFβsitesgeneratesAβspeciesofvarying lengthbetween35-
and43-aa.ForemostamongthemarepeptidesofAβ40orAβ42aainlengths,thatare,together
with theAICD,most abundantly produced (Citron et al., 1995;Haass et al., 1994; Selkoe,
2001a;Tang,2009).The!-secretaseisahighlyhydrophobiccatalyticenzymeandcomposed
ofdistinctcomponents:presenilinproteins(PS1orPS2),nicastrin,anteriorpharynxdefective
1 (APH1)andpresenilinenhancer2 (PEN-2).Asideofbeing involved inAPPprocessing,!-
secretase cleaves further substrates, such as Notch, cadherins, CD44 and neuregulin (De
StrooperandAnnaert,2010).

Introduction
33
Figure1.7SchematicrepresentationofAPPprocessing. Innon-amyloidogenicprocessing,APPissequentiallycleavedbyα-secretaseand!-secretases to releasethep3 fragment. Inamyloidogenicprocessing,sequentialcleavagebyβ-and!-secretasesreleasestheAβpeptide.(ThathiahandDeStrooper,2011)
1.2.3 ProteinfragmentationinfamilialCAA:ABriandADan
1.2.3.1 Cerebralamyloidangiopathies:familialBritishandDanishdementia
Cerebralamyloidangiopathy(CAA)summarizesentitieswithdepositionofamyloidwithinthe
wallsofbloodvesseloftheCNS(Ghisoetal.,2001;Mandybur,1986);itoccursassporadic
and/orfamilialdiseaseformswithvariousproteinsinvolvedincludingAPPinAD,amyloid-Bri
precursor protein (ABriPP) in familial British dementia (FBD), and amyloid-Dan precursor
protein(ADanPP)infamilialDanishdementia(FDD)(Rensinketal.,2003;Reveszetal.,1999;
Reveszetal.,2002).

Introduction
34
FBDandFDDare late-onsetdiseasesand clinically characterizedbyprogressivedementia,
spastictetraparesis,andcerebellarataxia(Meadetal.,2000;Stromgrenetal.,1970);with
neuropathologicalhallmarkssimilartothoseinADincludingsevereCAA,neuroinflammation,
and neurofibrillary tangle formation (Coomaraswamy et al., 2010; Revesz et al., 2002;
RostagnoandGhiso,2008).
1.2.3.2 ABriandADan
MutationintheintegralmembraneproteinBRI2leadstoaccumulationofhighlysolubleABri
andADanpeptides.BRI2isatype-IIsingle-spanningtrans-membraneprecursorproteinof266-
aainlength.Amissensemutationordecamerduplicationmutationproducesaframe-shiftin
theBRIgenesequencearegeneratingtwolarger,277-residueprecursorproteinsABriPPand
ADanPP,respectively(Vidaletal.,1999;Vidaletal.,2000).
TheimmatureBRI2precursorproteiniscleavedinamulti-stepproteolyticprocessresultingin
distinct intermediate- and end-products: (I) several proteases such as furin and other
subtilisin/kexin-likeproproteinconvertases(PPCs) leadstothesecretionofa23-residueC-
terminalpeptide(Bri2-23)(Kimetal.,1999).(II)theremainingmembraneboundmatureBRI2
proteinisfurtherprocessedbyα-secretasesADAM10releasingtheBRICHOSdomaintothe
extracellularspace(Martinetal.,2008).(III)finally,anN-terminalfragment(NTF)undergoes
proteolyticcleavagebysignalpeptidepeptidase-like2(SPPL2)resultinginasmallextracellular
BRI2-C-terminalpeptideandinparalleltoanintracellulardomain(ICD)(Martinetal.,2008).
Notably, instead of Bri2-23, proteolytic processing of ABriPP or ADanPP by PPCs leads to
liberationofhighlysoluble34-residueC-terminalpeptidesABriandADan,respectively(Figure
1.8).
Unliketheshorterwild-typepeptideBri,cleavedABrishowedanincreasedpropensityinthe
formationoftoxicoligomersthroughinter-moleculedisulfidebonds invitro(Cantlonetal.,
2015; El-Agnaf et al., 2001).Overexpressionof different amyloid peptides in the retinaof
DrosophilademonstratedthatADanappearstobemoretoxiccomparedtoe.g.Aβ42peptides
(Marcoraetal.,2014).Thetwoamyloidogenicpeptides,ABriandADan,whichdonotoccurin
nature intheabsenceof thediseasecausingchanges intherespectiveprecursorproteins,
demonstrate that the de novo creation of short peptides is sufficient to induce amyloid

Introduction
35
formation and associated neurodegeneration. ABri and ADan therefore serve as principal
models for anupstream roleofprotein cleavage inneurodegeneration. In addition to the
inductionofamyloiddeposits,thesenovelpeptidesalsoinducedownstreamtaupathologyin
FBDandFDD(DelCampoetal.,2015).Inmurinemodels,co-expressionofsolubleADanand
P301Stauexacerbatestautoxicityassociatedsynapticdysfunction,andresultsinincreased
solublehyperphosphorylatedandinsolubleaggregatedtauspeciesaswellastautruncation
atAsp421(Coomaraswamyetal.,2010;Garringeretal.,2013).
Figure1.8SchematicrepresentationofBRI2processing.(Cantlonetal.,2015)
1.2.4 ProteinfragmentationinPD:α-synuclein
1.2.4.1 Parkinson’sdisease(PD)
Parkinson’s disease (PD) is characterized by tremors and locomotion abnormalities and
constitutes the most frequent movement disorder among α-synucleinopathies including
dementiawith Lewy body (DLB) andMultiple SystemAtrophy (MSA). PD is pathologically
defined by the presence of aberrantα-synuclein (α-Syn) in intracellular deposits of Lewy
bodies (LB) and Lewy neurites (LN) and by neuronal degeneration in the substantia nigra
(Forno,1996;reviewedinTofarisandSpillantini,2005).

Introduction
36
1.2.4.2 α-synuclein
Proteolyticprocessingofα-synucleinisthoughttobeconsiderablyrelevantfortheformation
offibrillogenicproteininclusionsandneurotoxicity(Duftyetal.,2007)(Figure1.9); indeed,
truncatedα-synucleinspecieshavebeenfound inbrainsPDandDLBpatients (Babaetal.,
1998;Liuetal.,2005).Infact,C-terminallytruncatedα-Synhasbeenfoundtoincreasethe
aggregationpropensityoffull-lengthα-Syn,resultinginenhancedneurotoxicityinvivoandin
vitro(Giassonetal.,2002;Kandaetal.,2000;Lietal.,2005).Italsohasbeendemonstrated
that C-terminal truncated α-Syn aggregates more rapidly compared to its full-length
counterpart;moreover,truncatedα-Synspeciesexhibitseedingpotentialoffull-lengthα-Syn
aggregation in vitro (Murray et al., 2003). Of note, manganese (Mn), a cofactor for
homeostaticandtrophicenzymesintheCNS,hasbeenshowntoinducecleavageofα-Syn
proteininvitroandprovoketoxicα-Synoligomers,ultimatelyleadingtoneuronalinjury(Xu
etal.,2015).Aside,matrixmetalloproteinases(MMPs)generatingaggregation-enhancingα-
SynfragmentsinvitroarebelievedtobealsorelevantforPDpathogenesisinvivo(Levinetal.,
2009).Further,N-terminaltruncationofα-Synproteinpreventsβ-sheetandfibrilformation
(Kessler et al., 2003). Also, in transgenic mice overexpressing a calpain-specific inhibitor,
reducedproteolyticcleavageofα-Synproteinleadtoadecreaseofα-Syn-positiveaggregates
andastrogliosis;indicatingacrucialroleoftruncatedα-Synspecies,butalsoofcalpainsinthe
pathogenesisofPD(Diepenbroeketal.,2014).
Figure1.9Schematic representationofα-Synproteinprocessing. (Adapted fromEmanueleandChieregatti,2015)

Introduction
37
1.2.5 ProteinfragmentationinTRD:Htt,ataxin,andatrophin
1.2.5.1 Huntington’sdisease(HD)
Huntington’s disease (HD) is a late-onset, autosomal dominant, and progressive
neurodegenerativedisordercausedbyaCAGtrinucleotiderepeatsexpansioninthecoding
region of the huntingtin (htt) gene,which encodes an polyglutamine (polyQ) stretch that
altersthefunctionofthehttprotein;indeed,themutanthttpossiblyaffectscellularprocesses
includingmitochondrialdysfunctionandvesicletransportfailure(Browne,2008).Inaddition,
polyQ induced conformational changemakes thehtt proteinmoreaggregation-proneand
causeinclusionsinthecytoplasm,dendritesandaxonterminalsofneurons(Fanetal.,2014).
Interestingly,ithasbeenshownthatwild-typehuntingtincandiminishtheneurotoxicityof
mutanthuntingtin(Leavittetal.,2001;Leavittetal.,2006;Zhangetal.,2003)andalsoactas
anautophagyscaffold(Ruietal.,2015).
The clinical picture of HD includes psychiatric features and dementia; progressive chorea
developmentandothermovementabnormalities.Inparallel,severedegenerationoccursin
thestriatalmedium-sizedspinyneurons,andsubsequentlyinthedeeplayersofthecortex
(NovakandTabrizi,2011;VonsattelandDiFiglia,1998).
1.2.5.2 htt
Thecorrelationbetweenhuntingtinlengthandneurotoxicityispoorlyunderstood;however,
httfragmentshavebeenfoundinthebrainsofHDpatients,aswellasintransgenicmouse
modelsofHD(Kimetal.,2001;Wellingtonetal.,2002).Indeed,proteolyticcleavageofmutant
httbycaspases,calpainsandotherproteasessuchasMMPs(Milleretal.,2010)releasean
aggregation-pronepolyQ tract containingN-terminal fragment, andultimately leading to
intracellularinclusionsandneuronaldysfunction(Wellingtonetal.,2000)(Figure1.10);aswell
as to impairedmitochondrial trafficking,precedingthe formationofaggregates (Orretal.,
2008). Further,mousemodels transgenic for truncated formsofhtthave showna rapidly
progressiveandlethalphenotype(Mangiarinietal.,1996;Schillingetal.,1999);incontrast,
miceexpressingfull-lengthmutantconstructsexhibitonlyamildpathologicalphenotype(Van
Raamsdonketal.,2005).

Introduction
38
1.2.5.3 Otherpolyglutaminediseases
Other trinuleotide repeat expansion disorders, including various forms of Spinocerebellar
ataxia (SCA types1,2,3,6,7, and17)andDentatorubral-pallidoluysianatrophy (DRPLA)
sharecommonpropertiesofHD,albeitbeing less frequent;mutations inpolyQexpansion
altersthenormalfunctionofataxinproteins(MargolisandRoss,2001;Palhanetal.,2005)
and the transcription co-regulator atrophin-1 (Sato et al., 2009), respectively, leading to
intranuclearaggregationincerebellarPurkinjecellsandcorticalneurons(Rolfsetal.,2003).
Among the other types, an important neuropathological role of ataxin cleavage has been
showninaDrosophilamodelofSCA3andforSCA7 invitroand invivo.Specifically,polyQ-
containing fragments derived from caspase-dependent cleavage of ataxin-3 have been
demonstratedtoinduceneurotoxicity,thuscontributetoSCA3diseaseprogression(Junget
al.,2009)Inaddition,ithasbeenshownthattoxicpolyQ-containingfragmentsgeneratedby
caspase-7 mediated N-terminal cleavage of ataxin-7 are subject to regulated clearance
mechanisms; however, distinct post-translational modifications such as
acetylation/deacetylationappear tomodulate fragmentstability/clearance,and thusbeing
crucialformediatingpolyQ-fragmentinducedtoxicity.(Mookerjeeetal.,2009).
Figure1.10Schematicrepresentationofhttproteinprocessing.(AdaptedfromRossandTabrizi,2011)

Introduction
39
1.2.6 ProteinfragmentationinPriondiseases:PrPCandPrPSc
1.2.6.1 Priondiseases
Priondiseasesareagroupofneurodegenerativedisordersthatmayoccursporadically,but
also manifest familial and, infectious forms. In human beings, prion diseases such as
Creutzfeldt-Jakobdisease(CjD)andkuruareclinicallycharacterizedbyprogressivedementia
andmotordysfunction(Prusiner,1991).
Prions are infectious, pathogenic proteins that, in contrast to viruses, are encoded by a
chromosomal gene (PRNP) and are devoid of nucleic acid; they are crucially linked to the
conversion of the physiological cellular prion protein (PrPC), a membrane-associated
extracellularglycoproteinanchoredbyglycosylphosphatidylinositol(GPI),intotheabnormal,
self-propagating‘scrapie’prionisoform(PrPSc)(Prusineretal.,1998).Thepost-translational
modification froma structural α-helical sheet to insoluble β-sheet structures results in an
increased aggregation propensity of PrPSc; ultimately, polymerizing into amyloidogenic
depositsandencouragingprionpropagation(DeArmondetal.,1985).
1.2.6.2 Prionprotein
Biologically active fragments derived from proteolytic processing are thought to have
implicationsinthecourseofpriondiseases.PrPCissubjecttodiverseproteolyticeventsunder
physiologicalconditions:(I)α-cleavagewithintheneurotoxicdomainproducessolubleN1-
andamembraneboundC1-fragment;(II)β-cleavagegivesrisetoaN2-andC2-fragment;and
(III) shedding close to the plasma membrane releases almost full-length PrPC into the
extracellularspace(reviewedinAltmeppenetal.,2012)(Figure1.11).
GiventhattheneurotoxicdomainofPrPCisessentialfortheabnormalconversiontothePrPSc
isoform,α-cleavageandtheresultinginactivationofthisstructuralpartisassumedtobea
protectivemechanismastoprionpropagation(Lewisetal.,2009;Turnbaughetal.,2012).In
addition,α-cleavedC1-fragmenthasbeen linked toapoptotic caspase-3activation in vitro
(Sunyachetal.,2007).Ofnote,thecleavagederivedN1-fragmenthasbeendemonstratedto
beneuroprotectiveinvitroandinvivo(Guillot-Sestieretal.,2009).

Introduction
40
The role of PrPC shedding by a desintegrin and metalloproteinase 10 (ADAM10) in
neurodegenerativediseasesremainunclear.However,thereisevidencethatsheddingofnot
onlyPrPC,butalsoofalreadymisfoldedPrPScleadstoaccumulationofanchorlessPrPSc;thus,
encouragingneurotoxicspreading inthebrain(Chesebroetal.,2005;Rogersetal.,1993).
Notably,sheddedPrPScinthecerebrospinalfluid(CSF)isalsothoughttoenhancepathological
transmission(Tagliavinietal.,1992).
N-terminallytruncatedPrPspecieshasbeenfoundtoco-localizedwithfull-lengthformsin
infectiousPrPScaggregatesinmousebrains(Panetal.,2005).Indeed,varioustruncatedforms
offull-lengthPrPSchasbeenshowntobeinvolvedinpriondiseases:themostcommonPrPSc
fragment PrP27-30 (Parchi et al., 1996); PrP7-8 truncated forms in Gerstmann-Sträussler-
Scheinker(GSS)diseases(Parchietal.,1998);PrP16-17truncatedspeciesinscrapie-infected
animals(Caugheyetal.,1998);orPrP-CTF12/13insporadicCjD(Zouetal.,2003).Aside,itis
thoughtthatnon-fibrillarPrPoligomershaveimplicationsintheprocessofpriondiseases.
However,N-terminaltruncatedPrPleadtonon-specificaggregates,butfailedtoformtoxic
oligomericspecies invitroand invivo; indicatingthatthestructuralN-terminusplaysakey
roleintheinfectiousandneurotoxicprocess(Trevittetal.,2014).Moreover,transgenicmice
expressing a truncated form of mutant PrP lacking a short N-terminal segment of 9-aa
displayedaneurotoxicphenotype(Westergardetal.,2011).

Introduction
41
Figure1.11SchematicrepresentationofPrPprocessing.(AdaptedfromAltmeppenetal.,2012)
1.2.7 ProteinfragmentationinFTLDandALS:TDP-43
1.2.7.1 TDP-43proteinopathies
TDP-43proteinopathies including sporadic and familial frontotemporal lobardegeneration
(FTLD)(Araietal.,2006)andamyotrophiclateralsclerosis(ALS)(Neumannetal.,2006)area
groupof neurodegenerativedisorder causedbypathological neuritic inclusions containing
transactiveresponse(TAR)DNA-bindingprotein43.AsideAD,FTLDisthemostcommonform
ofprogressivedementiainhumanbeingsundertheageof65years(Nearyetal.,1998).ALS,
however, is the most common cause of motor neuron degeneration accompanied by
progressivemusclewasting.Moreover,FTLDpatientsmayalsodevelopALS;andviceversa
(Murphyetal.,2007).Aside,mutationintheprogranulingene(PGRN)arethoughttocause
familialformofFTLD(Ghidonietal.,2008).

Introduction
42
1.2.7.2 TDP-43
Physiologically,TDP-43isthoughttoplayaroleintheregulationofvariouscellularprocesses
including alternate splicing, transcription, apoptosis, microRNA biogenesis, and mRNA
transportandstability(reviewedinBurattiandBaralle,2008;Ouetal.,1995).HumanTDP-43
is a 414-aa nuclear protein, ubiquitously expressed and highly conserved; structurally, it
harbors two RNA-recognition motifs (RRM1; RRM2) and a protein-protein interaction
mediating C-terminal glycine-rich region (Wang et al., 2004). Furthermore, the TDP-43
moleculeexhibitthreepotentialcaspase-3cleavagesites(Zhangetal.,2007).
Both,hyperphosphorylatedandubiquitinatedfull-lengthandN-terminallytruncatedTDP-43
are themajor component ofneuritic inclusions in brain tissue of FTLD and ALS patients.
Caspase 3/7-mediated proteolytic cleavage at Asp89 and Asp220 of wild-type TDP-43
generatesa35-kDa(CTF35)anda25-kDaC-terminal fragment (CTF25) respectively (Figure
1.12); moreover, different cleavage sites at Arg208, Asp219, and Asp247 of CTF25 were
further identified(Igazetal.,2009;Neumannetal.,2006;Nonakaetal.,2009).Moreover,
eachindividualTDP-43CTFhasbeenshowntopossessdistinctmolecularpropertiesincluding
intracellulardistributionandphosphorylationstatus,contributingtothepathogenicdiversity
ofTDP-43proteinopathies(Furukawaetal.,2011).
Sofar,accumulationoftruncatedspeciesmightmediatetoxicity,orissimplyaconsequence
inthecourseofneurodegeneration;inanycase,thepathologicalroleofTDP-43fragments
remains elusive. However, several studies implicate a neurotoxic relevance of TDP-43
fragmentation. Indeed, expressionofcaspase-cleavedCTF25 fragment inhumancell lines
lead tocelldeathaccompaniedby the formationof toxic insolubleaggregates;aside, this
fragmentdidnotinteractwithfull-lengthnuclearTDP-43oraffectitsfunction,thus,indicating
atoxicgain-of-function(Zhangetal.,2009b).Furthermore,thisfragmentshowedanincreased
hyperphosphorylation propensity at sites not required for neurotoxic inclusion formation
comparedtofull-lengthTDP-43.Incontrast,anotherstudydemonstratedthatTDP-43CTFs
provokeboth,first,theformationofaberrantphosphorylatedandubiquitinatedaggregates,
and second, the incorporation of newly synthesized endogenous full-lengths species into
thesecytoplasmicinclusions(Nonakaetal.,2009).Interestingly,giventhatregulationofexon
splicingisadefinedfunctionofTDP-43,aberrantsplicinghasbeendemonstratedincultured

Introduction
43
cellsexpressingTDP-43CTFscleavedatArg208; thus, strengthenedthepathogenic roleof
TDP-43fragmentsinFTLDandALS(Igazetal.,2009).
However,thereisalsoevidenceoffragmentation-mediatedclearanceoffull-lengthTDP-43:
the activation of endoplasmatic reticulum (ER) membrane-bound caspase-4 cleavage
diminishesERstresscausedbyabundantaccumulationofTDP-43;subsequentactivationof
the downstream caspase-3/7 pathway ultimately results in reduced full-length TDP-43
cytotoxicityandnecroticcelldeath(Lietal.,2015).
Figure1.12SchematicrepresentationofTDP-43domainprocessing.(AdaptedfromChiang2016)

Introduction
44
Wehaveoutlinedaboveseveralexamplesofproteinopathies,whereproteincleaveageresults
in aggregation prone fragments, pointing towards a general pathogenic role of protein
fragmentation in neurodegenerative diseases. This can’t be said without mentioning the
ongoingdebateonthetoxicityofaggregatesinproteinopathies.
Thereexisttwoopposingviewsonamyloidlesionsinmostneurodegenerativediseases:high
molecular,fibrillaryaggregatescaneitherbeconsideredasinertintracellulargarbageor,as
specifically toxic entities. In most proteinopathies, the current evidence points towards
considerableneurotoxicityofearly,non-fibrillaryoligomericaggregates(Bergeretal.,2007;
Gersonetal.,2014;Pattersonetal.,2011;Usenovicetal.,2015).Thisconceptofoligomer
toxicity in neurodegenerative disorders is compatible with a primary pathogenic role of
proteinfragmentsthatinducetheaggregationofproteinsintooligomericspecies.Wehave
recentlyreportedsuchatoxicityinducingeffectofataufragmentintautransgenicmouse
models(Ozceliketal.,2016),providingexperimentalsupportofthishypothesis.

Aimsofwork
45
2 Aimsofthework
Attributabletotheincreaseinlifeexpectancy,theprevalenceoftauopathiesincludingADis
continually growingwith underlying pathologicalmechanisms partly unknown. Given that
currenttherapeutictreatmentsarelimitedtosymptomaticapproaches,globalsocietyisfaced
withenormoussocioeconomicchallenges.
Yet,neurodegenerativediseaseresearchhighlightedvariousaspectsofthepathogenesisof
tauopathiessuchasproteinfragmentationandspreading.
Protein fragmentation is increasinglyemergingasan integralevent in thepathogenesisof
neurodegenerativediseases.Numerousinvitroandinvivostudieshaveaddressedwhether
cleavageofdisease-associatedproteinsissufficienttoexacerbateanongoingdiseaseprocess.
Indeed, the relevance of protein fragmentation has been highlighted in the course of a
plethora of proteinopathies with an emphasis on the neurotoxic potential of protein
fragments (Altmeppen et al., 2012; Furukawa et al., 2011; Masters and Selkoe, 2012;
Mookerjeeetal.,2009;Vidaletal.,1999;Wellingtonetal.,2000).Asfortauprotein,truncated
formshavebeenshowntofacilitatetauaggregation;andthus,appeartopossessanincreased
neurotoxicpotentialcomparedtofull-lengthtauspecies(Abrahaetal.,2000).Intauopathies,
thedynamicsofvariousneurotoxictauspeciesisstillamatterofdebate.Todate, it isnot
knownwhichtauspeciesisthemosttoxicandhowthetoxicityismediated(Goedert,2015).
However, hyperphosphorylated and aggregated tau species are considered as the
fundamentalneurodegenerativecomponentsintauopathies(SpillantiniandGoedert,2013).

Aimsofwork
46
Directlydeterminingthepathologicalfunctionofdistincttauspeciesandcellularmechanisms
underlyingtautoxicity,invivo,remainschallenging.Numeroustautransgenicmousemodels
havebeencreatedtoexplainchangesintheregulationoftaumetabolism(Duyckaertsetal.,
2008). Accordingly, we generated multiple transgenic mouse lines in order to study the
relevanceoftaufragmentationfortheneurodegenerativeprocessintauopathies.Thesemice
eitherexpressatruncatedtausequence(3RΔtau151–421)aloneorincombinationwithwild-
type(0N3Ror2N4R)ormutant(0N4R)full-lengthtauisoforms.Ofnote,animalmodelsthat
expresswild-type full-length formsof taugenerallystaydevoidof insoluble tau inclusions,
whereasmodelsthatoverexpressmutatedfull-lengthtauexhibitNFTs(Allenetal.,2002;Gotz
etal.,1995).
Aside tauprotein, theaggregationofAβspeciesgeneratedbyproteolyticcleavageofAPP
constitutesakeypathologicalhallmarkofAD.Lately,aprion-likeroleofmousebrainderived
Aβ species has been demonstrated (Langer et al., 2011); thus indicating that Aβ species
presentintheCSFmayexhibitsimilarseed-likepropertiesinvivo.
Within this framework, we sought, to longitudinally analyze the effects of a human tau
fragmentinamurinemodelandspecificallyitspotentialinteractionwithhumanfull-length
tau,andinparallel,toanalyzepotentialseed-potentCSFderivedAβspeciesinvivo.

Results
47
3 Results
3.1 PublicationNo.1
Co-expression of truncated and full-length tau inducessevereneurotoxicity
Ozcelik S*,Sprenger F*, Skachokova Z, FraserG, Abramowski D, Clavaguera F,ProbstA,FrankS,MüllerM,StaufenbielM,GoedertM,TolnayM,WinklerDT.
*Bothauthorscontributedequallytothework
MolPsychiatry.2016

Results
48

Results
49

Results
50

Results
51

Results
52

Results
53

Results
54

Results
55

Results
56

Results
57
SupplementalInformation
SupplementaryInformationaccompaniesthepaperontheMolecularPsychiatrywebsite
(http://www.nature.com/mp)
SupplementaryVideos:
VideoS1
A3-week-oldP301SxTAU62ondoubletransgenicmouse isshown.Notetheparalysisof the
hindlimbs,whilethemouseisstillcapableofmovingbyusingtheforelimbs.
VideoS2
Videoofa5-month-oldhomozygousP301Smouse.HomozygousP301Smicedevelopovert
motor impairmentsstartingatagesof3-4months.Thismouse iswalkingslowly,however,
thereisnoseverehindlimbpalsyoccurringinP301Smiceuptoagesof5months.
VideoS3
ThesameP301SxTAU62on-offmouseseeninVideoS1isshown,butnow38daysafterDtau
expressionhasbeenstoppedbywithdrawalofdoxycycline.Thehindlimbpalsyissignificantly
improvedandthemouseisagainwalkingalmostnormally.
VideoS4
A3-week-oldALZ17xTAU62ondoubletransgenicmouse isshown.Notetheparalysisof the
hindlimbs,whilethemouseisstillcapableofmovingbyusingtheforelimbs.
VideoS5
ArecoveredALZ17xTAU62on-offmouse isshownonemonthafterDtauexpressionhasbeen
stoppedbywithdrawalofdoxycycline.
VideoS6
ParalyzedALZ31xTAU62onmouseagedthreeweeks.

Results
58
VideoS7
12-month-oldP301SxALZ31mouse.Notethenormalgridclimbingcapabilityofthisdouble
transgenicmouse.
VideoS8
4-month-oldALZ17SxALZ31mouse.Notethenormalgridclimbingcapabilityofthisdouble
transgenicmouse.

Results
59
SupplementaryTableandFigures:
TableS1
(a-c) Overview on the transgenic and co-transgenicmouse lines used for the present studies. Tau isoformsexpressedareshown.Darkgreyboxesindicatethetaurepeatdomains(3Ror4Risoforms),andlightgreyboxesN-terminalinserts(e.g.ALZ17�2N4R).The tau cDNA constructs are either driven by a standard Thy1.2minigene (Thy1.2) or by amodified Thy1.2minigene that contains a tetracycline controlled transcriptional silencer element (Thy1.2-tTS) in case of theTAU62mouse. (a) shows the tau isoformsexpressed in single-transgenic lines. (b) shows the tau formsof3mouselinesco-expressingfull-lengthtauwithDtau.(c)depicts2mouselinesco-expressingdifferentfull-lengthtauisoforms.

Results
60
FigureS1
TAU62micedevelopaslowlyprogressivemotorphenotype.Motorfitnesswasassessedonaverticalmeshgrid.TimespentonthegridprogressivelydeclinedinTAU62mice,whileB6controlswereunimpairedat12monthsandshowedamilddeclineat18monthsofage.

Results
61
FigureS2
(a)ExtensivehyperphosphorylationoftauwasseeninthebrainstemofoldhomozygousP301Smicebystainingwithantibodiestargetinglatephospho-epitopes.MultipletautanglesandgranularaggregatesweredetectableinthesemicebyGallyassilverstain.Thescalebarinacorrespondsto50µm.(b)Westernblottingundernon-reducingconditionsrevealedhigh-moleculartauspeciesinparalyzedP301SxTAU62onmice(lanes6&7);similartauspecieswereseeninagedtanglebearinghomozygousP301Smice(lane5).Whentauexpressionwashalted,nomorehigh-moleculartauformsweredetectableinP301SxTAU62on-offmice(lane3;Westernblotperformedwithanti-tauantibodyHT7).(c)StainingwiththeRD3antibodytargetingAsp421showsthepresenceofDtauinthe highmolecular weight tau species. (d) Sarkosyl-extraction detects only soluble tau species in paralyzedP301SxTAU62on mice (“sol”: sarkosyl-soluble tau; “insol”: sarkosyl-insoluble fraction). (e-g)Dtau was widelyexpressed in the spinal cord of P301SxTAU62on mice (e) and phosphorylated at the AT8 epitope (f). Uponcessation of Dtau expression,Dtau- and AT8-positive tau was no longer detectable (g). The scale bar in ecorrespondsto100µmine-g.

Results
62
FigureS3
(a-m)P301SxTAU62miceexhibitsignsofGolgidisruption,proteinmissortingandmitochondrialclustering.ThesesignsarereversibleuponcessationofDtauexpression.ImmunohistochemistryusingantibodiesagainstMG160(a-f), synaptophysin (g-i), VAMP2 (k-m), and cytochromeCoxidase (COX) (n-p), in thehippocampusof non-transgenicmice(B6),3-week-oldparalyzedmice(P301SxTAU62on)andrecoveredmice6weeksaftercessationofDtauexpression(P301SxTAU62on-off).Thescalebarinacorrespondsto19µmind-f,63µmina-candg-i,and400µm in k-m. The scale bar in n corresponds to 30µm for n-p. Arrows in (e) indicate fragmented Golgistructures.

Results
63
FigureS4
(a-c)YoungTAU62miceexhibitnormalsciaticnerves(Masson’strichromstain(a);para-Phenylenediamine(b);immunohistochemistry using 2f11 antibody (c). The scale bar in c corresponds to 30 µm in a-c. (d-g) M.gastrocnemiusstainedforATPase(pH4.2).Darktype1fibres(1)andlighttype2fibres(2).Thescalebaringcorresponds to 50µm (ford-g). P301S: heterozygousmice transgenic for humanmutant P301S tau, aged 3weeks;TAU62:heterozygousmiceexpressing3Rtau151-421,aged3weeks;P301SxTAU62
on:paralyzedmice,aged3weeks;P301SxTAU62on-off:recoveredmice,6weeksaftercessationoftheexpressionofDtau.

Results
64
FigureS5
(a-i)Co-expressionof3Rwild-typetauandDtau(ALZ31xTAU62mice)causesparalysisandneuropathy,whicharenotreverseduponcessationofDtauexpression.(a)Paralyzed(aged3weeks)andnon-recovered(3weeksaftercessationofDtauexpression)ALZ31xTAU62mice (seealsovideoS6). (b)Absenceof recoveryofmotorfunctionasassessedbyagrid-testofALZ31xTAU62micefollowingtheremovalofdoxycyclinebetween14and16days(blueline;trianglesindicatethetimesofeuthanasia,n=6).MotorfunctionofheterozygousALZ31mice(green line,n=7). (c)WesternblotwithHT7ofbrainstemtissue fromnon-transgenicmice (B6),TAU62mice,ALZ31miceandALZ31xTAU62mice.Actinstainingwasusedastheloadingcontrol.(d-i)HistologicalanalysisofparalyzedALZ31xTAU62miceaged3weeks,usingAT8(d),AT100(e),NF200(f),Holmes-Luxol(HL)(g),Masson’strichrome(h),andHematoxylin-eosin(HE)staining(i).Thearrowin(g)pointstoaspheroid.Thescalebarindcorrespondsto200µmindande;100µminf;33µming;50µminh,i.

Results
65
FigureS6
(a,b)Robustexpressionofthetwofull-lengthtauisoformsinP301SxALZ31(a)andALZ17xALZ31(b)co-transgenicmice.Forcomparison,expressionofmiceco-transgenicforDtauwithfull-lengthtau,aswellastherespectivesingletransgenicmiceisshown.WesternblotsrununderreducingconditionsusingHT7antibody.

Results
66
SupplementalExperimentalProcedures Antibodiesusedforimmunohistochemistry(IHC)andWesternblotting(WB)(speciesismouse,unlessindicatedotherwise): Antibody Target Dilution SourceHT7 humantau
aa159-163WB1:4000IHC1:800
Pierce,Rockford,IL#MN1000
BR134 humantau WB1:1000 (Goedertetal.,1989)
Tau-C3 TaucleavedatresidueAsp421
WB1:1000IHC1:1000
SantaCruzBiotechnology,Inc,Dallas,TX#sc-32240
AT8 TaupSer202/Thr205
WB1:1000IHC1:800
Pierce,Rockford,IL#MN1020
AT100 TaupThr212/Ser214
WB1:1000IHC1:500
Pierce,Rockford,IL#MN1060
PHF-1 TaupSer396/404
WB1:2000IHC1:1000
PeterDavies,AlbertEinsteinCollegeofMedecine,Bronx,NY
MC1 Tauaa5-15,312-322
IHC1:100 PeterDavies,AlbertEinsteinCollegeofMedecine,Bronx,NY
2F11 neurofilament(NF)NF-L,NF-H(70kD)
IHC1:800 Dako,Glostrup,DK#M0762
NF200 neurofilament(200kD) IHC1:100 (Probstetal.,2000)GFAP glialfibrillaryacidic
proteinIHC1:500 ThermoFisherScientificInc.,Kalamazoo,
MI#MS-1407-R7
Synaptophysin synaptophysin IHC1:1000 MilliporeCorporation,Billerica,MA#MAB5258
MG160(rabbit) Golgiapparatus IHC1:1000 NicholasGonatas,PathologyandLaboratoryMedicine,UniversityofPennsylvania,PA
VAMP2/Synaptobrevin2(rabbit)
transportvesicles IHC1:1000 Synapticsystem,Goettingen,Germany#104202
GAPDH(6C5) GAPDH WB1:1000 SantaCruzBiotechnology,SantaCruz,CA,#32233
ß-actin actin WB1:5000 Sigma-Aldrich,SaintLouis,MO#A5316Coxsubunit1a mitochondrial
stainingIHC1:200 Abcamplc,Cambridge,UK
#ab14705

Results
67
3.2 Preliminarydata
Protective effect of early tau burden on late neurotoxicdistress level – Mechanisms underlying tauopathy andconsequencesforfuturetherapies

Results
68
Protectiveeffectofearlytauburdenonlateneurotoxicdistresslevel–Mechanisms
underlyingdelayedtauopathyandconsequencesforfuturetherapies
Preliminaryresults
Wehaverecentlyprovidedevidencethatassemblyoffilamentoustauaggregatesisnotsolely
responsibleforthecharacteristictautoxicityinneurodegenerativedisorders.Wecouldshow
thatboth,co-expressionoffull-lengthP301Smutant,aswellasfull-lengthwildtypetauwith
Δtau151-421resultsinahighinvivoneurotoxicityassociatedwiththeformationofsolublehigh
molecular weight tau oligomers, extensive nerve cell dysfunction and severemotor palsy
albeitinabsenceofinsolubletauaggregatesortangles(Ozceliketal.,2016).
GiventhatourP301SxTAU62on-offmicewereexposedtodrasticneurotoxicstressduringearly
postnatallife,weaskedwhetherthiseventhasanypossibleimpactontaupathologylaterin
life,evenaftertheexpressionoftruncatedtauishalted.Initially,wehypothesizedthatthe
observedprevalentoligomerictauspeciesmightactasaformoftoxicseedformoredrastic
tau pathology in later stages, ultimately manifesting in earlier tau tangle formation.
Interestingly, compared to their heterozygous P301S tau transgenic littermates that have
neverbeenparalyzed,recoveredP301SxTAU62on-offmicethat,incontrast,experiencedearly
neurotoxicity tau burden however exhibit a better motor phenotype associated with
considerablylesstauproteinlevelsandanoverallattenuatedtaupathology.
3.2.1 DelayedmotorphenotypeinagedP301SxTAU62on-offmiceafterrecovery
ofsevereneurotoxicity
TomonitorthebehaviouralphenotypeinP301SxTAU62on-offmiceaftercessationofΔtau151-421
butcontinuousfull-lengthP301Stauexpression,weperformedabatteryofmotorfunction
testsatagesof12to16months.Giventhatthepathologicalhindlimbpostureisnormalized
inrecoveredP301SxTAU62on-offmice(Ozceliketal,2016,Figure3.1a),wechosetorepeatthe
tailsuspensiontest:14-month-oldheterozygousP301Stransgeniclittermatesshowedalready
first signsof hind limb clasping that aggravated at 16monthsof age,while thehind limb
spreadingofP301SxTAU62on-offmicestayedalmostnormalupto16monthsofage(Figure3.1

Results
69
bandc).Indeed,comparedtotheirheterozygousP301Slittermates,P301SxTAU62on-offmice
maintainasignificantlybettermotorstrength,coordinationandbalanceat14monthsofage
thatisstillpronounced,butnon-significantattheageof16months(Figure3.1dande).Of
note,bothtransgeniclinesshowedcomparableperformancesonthegridandrotarodatthe
age of 12months, aswell as no differences in sex-specific bodyweightwere found at 16
monthsofage(Figure3.1d,eandf).
months months 16 months
0
10
20
30
40
50
male female
Bodyweight
(g)
0
30
60
90
120
150
180
161412
***
***
***
Tim
e on
Grid
(sec
)
0
10
20
30
40
***
*** ***
Tim
e on
Rot
arod
(sec
)
161412
d e f
P301S heterozygous P301SxTAU62on-off P301SxTAU62on-offP301S heterozygous
aged 14 months
P301SxTAU62onparalyzed,
P301SxTAU62on-offrecovered,
aged 16 monthsaged 6 weeksaged 3 weeks
a b c
P301S heterozygous
P301SxTAU62on-off
B6
Figure3.1.FormerlyparalyzedP301SxTAU62on-offmicedevelopasignificantdelayedmotorphenotype. Incontrast to their heterozygous P301S transgenic littermates, P301SxTAU62on-off mice exhibit a better gridreflex,motorstrengthandmotorcoordinationatagesof12,14,and16months.(a-c)Pathologicalhindlimbspreadingwasassessedbyatailsuspensiontest.(a)ParalyzedP301SxTAU62onmiceatageof3weeksexhibitpathological hind limb posture. 3 weeks after cessation of Δtau151-421 expression, P301SxTAU62
on-off micerecoverfromtheirpalsy,albeitcontinuativeexpressionofP301Smutanttau(fordetailsseeOzceliketal2016).(bandc)LessvisibleuponP301SxTAU62on-offmice,pathologicalhindlimbspreadingmanifestedpredominantlyinheterozygousP301Stransgenic littermatesat14to16monthsofage. (d-f)Basedonthegrid(d,P301Sheterozygous:12months,n=15;14months,n=21;16months,n=21;P301SxTAU62on-off:12months,n=5;14months,n=11;16months,n=13;non-transgenicB6:12months,n=5;14months,n=13;16months,n=12)androtarod(e,P301Sheterozygous:12months,n=15;14months,n=25;16months,n=21;P301SxTAU62on-off:12months,n=5;14months,n=14;16months,n=13;non-transgenicB6:12months,n=5;14months,n=13;16months,n=12)performancetest,P301SxTAU62on-offmiceexhibitsignificantlybettermotorfitnessatagesof14 to 16 months, compared to heterozygous P301S transgenic littermates. (f) Sex-sorted body weightmonitoringat16monthsofage(P301Sheterozygous:male,n=6;female,n=17;P301SxTAU62on-off:male,n=5;female,n=6).Datarepresentsthemeanands.d.ofindicatedanimalspergroup.***P<0.001.

Results
70
3.2.2 ReducedtaupathologyinagedP301SxTAU62on-offmiceafterrecoveryof
severeneurotoxicity
InordertoelucidatewhethertheconsiderablydelayedmotorphenotypeinP301SxTAU62on-
offmicewasassociatedwithavaryingdegreeofseverityoftaupathology,wescreenedbrain
sections of both, 16-months-old P301SxTAU62on-off mice and their heterozygous P301S
littermates,forthepresenceofcertaintauspecies.Indeed,wecouldshowthatneurofibrillary
tanglesparalleledthepreviouslyobservedimpairedmotorphenotypeinheterozygousP301S
littermates;specifically,GallyasBraakSilverstainingshowedmassivetautangleformationin
the brainstem of heterozygous P301S mutant littermates, whereas formerly paralyzed
P301SxTAU62on-offmiceremainedalmostdevoidoffibrillarytauinclusions(Figure3.2a,band
e).
Furthermore, in linewith these histopathological findings, anti-tau antibody AT8 revealed
attenuated tau hyperphosphorylation in P301SxTAU62on-off mice when compared to their
Figure3.2.FormerlyparalyzedP301SxTAU62on-offmicerevealsignificantlesstaupathology.ComparedtotheirheterozygousP301Slittermates,P301SxTAU62on-offmicethatunderwentsevere,earlytaustressremainalmostdevoid of tau tangle pathology in the brainstem and exhibit attenuated tau hyperphosphorylation. (a - d)Histological analysis of 16-month-old P301SxTAU62on-off mice (b and d) and heterozygous P301S transgeniclittermates(aandc)usingGallyas–Braaksilver(aandb)andanti-tauantibodyAT8(candd).Thescalebarin(d)correspondsto500μmin(aandb)and200μmin(candd).QuantificationofGallyaspositivetautanglesinthebrainstemofP301Sheterozygous(n=10)andP301SxTAU62on-off(n=10)mice.***P<0.001.

Results
71
heterozygousP301Slittermates(Figure3.2candd).Though, tauproteinismostexpressedin
thebrainstemareaunderthecontrolofneuron-specificThy1.2-promoterelement, wealso
foundsignificantlesstaupathologyinthecervicalspinalcord(SupplementalFigureS3.1c-d)
andtheforebrainarea(SupplementalFigureS3.1d-f)ofP301SxTAU62on-off;asopposedtothis,
nosignificantdifferenceinfilamentoustauformationcouldbedetectedinthehippocampal
area(SupplementalFigureS3.1g-i).
Further,onlyheterozygousP301Slittermatesshowedhyperphosphorylationoflateepitopes
inthebrainstem,visualizedbyanti-tauAT100antibody,consistentwiththepresenceoftau
filaments (SupplementalFigureS3.2aandc);ofnote,Δtau151-421wasnotexpressed inthe
brainstemofeitherthetransgenicmouselines(SupplementalFigureS3.2bandd).
3.2.3 ReducedtauproteinlevelsinagedP301SxTAU62on-offmiceafterrecovery
ofsevereneurotoxicity
Giventheobtaineddatasofaravailableweconsideredthatchangesintauproteinexpression
levels correspond to the present histopathological phenotypes in formerly paralyzed
P301SxTAU62on-offmiceandtheirheterozygousP301Smutantlittermates.ThroughWestern
Blot analysis,we confirmed, asexpected, a significant reduction inboth the total tauand
sarkosyl-soluble tauexpression level inP301SxTAU62on-offmice (Figure3.3a-d);notably, in
heterozygous P301S littermates, the marked histopathological tau tangle pathology is
reflected in a significant increase of the sarkosyl-insoluble tau fraction expression levels
(Figure3.3aandb,stars).

Results
72
0.0
0.5
1.0
1.5
2.0
2.5
3.0 *
Total tau
HT7
/GAP
DH
0.0
0.2
0.4
0.6
0.8***
Insoluble tau fraction
HT7
/GAP
DH
aP301S heterozygous P301SxTAU62on-off
aged 16 months
P301Shomo
B6
HT7
GAPDH
HT7
GAPDH
c
0.0
0.5
1.0
1.5
2.0 *
Soluble tau
HT7
/GAP
DH
P301S heterozygousP301SxTAU62on-off
totaltau
solubletau
*
*
b
d
Figure3.3.FormerlyparalyzedP301SxTAU62on-offmicerevealsignificantlesstotal, insolubleandsolubletauproteinlevels.(a-d)Westernblottingwithhuman-specificanti-tauantibodyHT7ofbrainstemtissuefromP301Shomozygous(lane1),non-transgenicB6(lane2),P301Sheterozygous(lanes3-7)andP301SxTAU62on-off(lanes8-12)mice (a and c); GAPDH staining was used for protein normalization (b andd). At the age of 16months,significantly increased total tau protein levels were detected in heterozygous P301S transgenic littermates;associatedwithelevatedinsolubletaufractionlevels(aandb,stars).(candd)Sarkosyl-extractiondetectsonlysolubletauspeciesthatwerefoundtobesignificantlyattenuatedinP301SxTAU62on-offmice.Datarepresentsthemeanands.d.of5animalspergroup.*P<0.05and***P<0.001.

Results
73
3.2.4 Supplementalmaterial
P301S
heter
ozyg
ous
P301S
xTAU62
on-o
ff0.00000
0.00005
0.00010
0.00015
0.00020
0.00025 ***
Cervical Spinal Cord
Gal
lyas
pos
itive
par
ticle
/pix
el
P301S
heter
ozyg
ous
P301S
xTAU62
on-o
ff0.00000
0.00002
0.00004
0.00006
0.00008 **
Forebrain
Gal
lyas
pos
itive
par
ticle
/pix
el
P301S
heter
ozyg
ous
P301S
xTAU62
on-of
f0.000000
0.000001
0.000002
0.000003
0.000004 ns
Hippocampus
Gal
lyas
pos
itive
par
ticle
/pix
el
P301S heterozygous P301SxTAU62on-off
aged 16 months
a b
g
c
fd e
h iGallyas
FigureS3.1.FormerlyparalyzedP301SxTAU62on-offmicerevealsignificantlesstaupathologyinthecervicalspinalcordandforebrainarea.(a-i)Histologicalanalysisof16-month-oldP301SxTAU62on-offmice(b,e,andh)andheterozygousP301Stransgeniclittermates(a,dandc)usingGallyas–Braaksilverstainofthecervicalspinalcord(a-c),forebrain(d-f)andhippocampalarea(g-i).Dottedsquaresindicateareaofmagnificationinthelower-right corner (a,b,d ande). The scalebar in (h) corresponds to500μm in (a,b,d,e, gand h). (c, f and i)QuantificationofGallyaspositivetautanglesinthecervicalspinalcord(c),forebrain(f)andhippocampus(i)ofP301Sheterozygous(n=10)andP301SxTAU62on-off(n=10)mice.ns=non-significant,**P<0.01and***P<0.001.

Results
74
FigureS3.2.(aandb) Immunohistochemistrywithanti-tauantibodiestargeting latephospho-epitopesandDtau.AftercessationofDtauexpression,extensivehyperphosphorylationofAT100-positivetauwasonlyseeninthebrainstemofheterozygousP301Slittermates(a),butnotinP301SxTAU62on-offmice(b).Inthebrainstemofbothtransgeniclines,noC3-positiveDtaucouldbedetectedattheageof16months(aandb).Thescalebarin(d)correspondsto100μmin(aandd).

Results
75
3.3 PublicationNo.2
Amyloid-beta in the cerebrospinal fluid of APP transgenicmicedoesnotshowprion-likeproperties
Skachokova Z, Sprenger F, Breu K, Abramowski D, Clavaguera F, Hench J,StaufenbielM,TolnayM,WinklerDT.
CurrAlzheimerRes.2015

Results
76

Results
77

Results
78
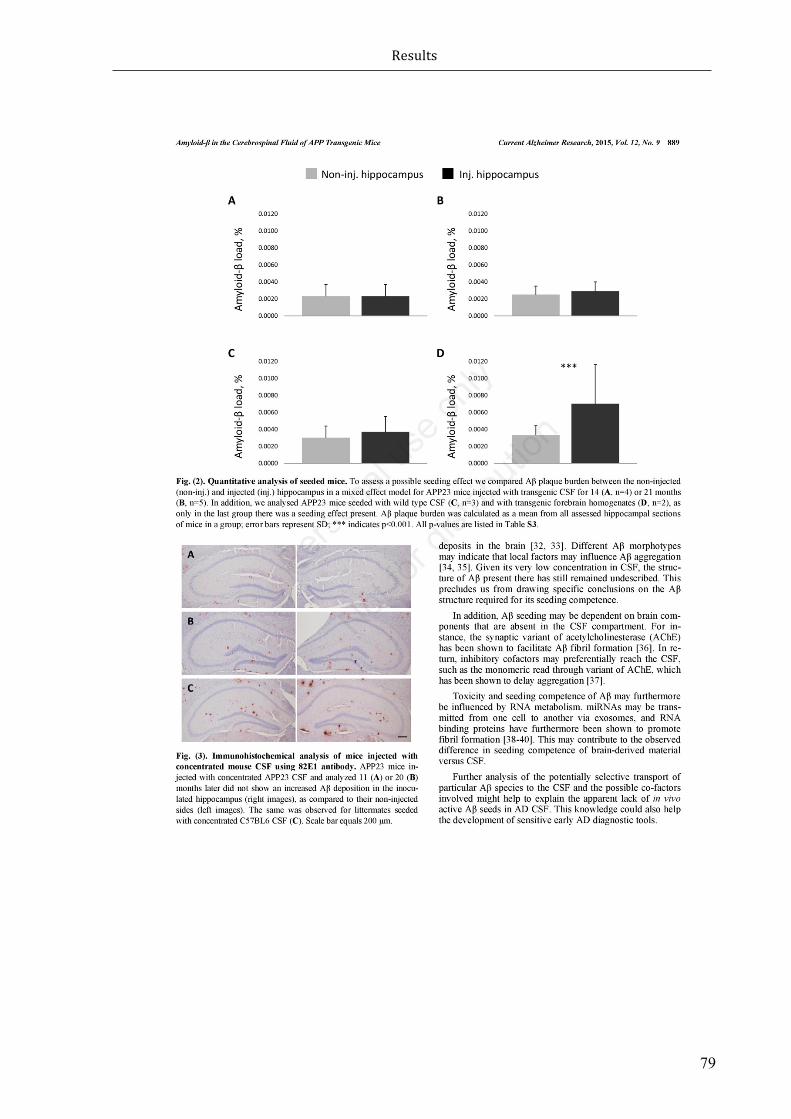
Results
79

Results
80

Results
81

Results
82
SupplementaryInformation1.1. ELISA
Abeta40andsAPPlevelsweremeasuredusingHuman6E10andAPPalpha/sAPPbetaKitsrespectively,bothfromMesoScaleDiscovery,accordingtothemanufacturer’sinstructions.1.2. Mice
Genotype Seed Seeddonorage,mo
Seedingtime,mo
App23 tgCSF 18 14App23 tgCSF 24 14App23 tgCSF 24 14App23 tgCSF 24 14App23 tgCSF 18 21App23 tgCSF 18 21App23 tgCSF 24 21App23 tgCSF 24 21App23 tgCSF 24 21App23 wtCSF 3 21App23 wtCSF 24 21App23 wtCSF 24 21App23 FB 24 20App23 FB 24 20C57BL6 tgCSF 24 21C57BL6 tgCSF 18 21C57BL6 tgCSF 18 21
App23conc. tgCSF
24 11
App23conc. wtCSF
24 11
App23conc. tgCSF
24 20
Table S1. Table of all mice used for quantitative analysis in the study. Abbreviations used: tg=transgenic,wt=wildtype,FB=forebrainhomogenate,conc.=concentrated.

Results
83
Supplementaryresults1.1. ELISA
Sample Aβ40,pg/μlCSF sAPPα,pg/μlCSF sAPPβ,pg/μlCSFAPP23CSF 4.2 92 121ConcentratedAPP23CSF 92.6 2594 3659
TableS2.ELISAtableofresults.
1.2. QuantificationofAβpathologycomparinginjectedvsnon-injectedhippocampus
Genotype Treatmentgroup
Seedingtime,mo N
Lowerratio GMR Upper
ratio Pvalue
APP23 tgCSF 14 4 0.77 0.94 1.15 0.56
APP23 tgCSF 21 5 0.79 0.91 1.05 0.19
APP23 wtCSF 21 3 0.65 0.81 1.01 0.06
APP23 FB 20 2 0.41 0.54 0.70 0.00***
C57BL6 tgCSF 21 3 0.00 0.00 0.00 NA
APP23conc. tgCSF
11 1 0.59 0.88 1.30 0.52
APP23conc. wtCSF
11 1 0.72 1.10 1.67 0.66
APP23conc. tgCSF
20 1 0.61 0.92 1.40 0.70
TableS3.Geometricmeanratios(GMR)ofamyloid-βratioscomparingnon-injectedvsinjectedhippocampus.N indicates the number of mice used; tg=transgenic, wt=wild type, FB=forebrain homogenate,conc.=concentrated;***indicatesp<0.001.

Discussion
84
4 Discussion
Alzheimer’s disease represents the most prevalent form of a major subclass of various
neurodegenerativediseases:thetauopathies.InSwitzerlandalone,over100’000peopleare
estimatedtosufferfromdementiaincludingAD;andthisnumberisexpectedtotripleby2050
(Blankmanet.al, 2012). Todate, the lackof therapeutic treatments limitspharmacological
therapies to symptomatic interventions and given that millions of people have been
diagnosed worldwide, AD constitutes a crucial source of social and economic problems
(Scheltensetal.,2016).
ThediseasespectruminpatientswithneurodegenerativedisordersincludingADextendsfrom
genetic alterations to protein modifications and altered inner life of a cell in affected
anatomical regions through to clinical manifestations. Years of research has led to the
discovery of a plethora of proteins being crucial in the course of neurodegenerative
syndromes;buteversinceresearchersarepuzzledbypiecesofthebigpicture.Thecleavage
of larger, neurodegeneration-associated proteins into smaller, potentially neurotoxic
fragmentshasbeensubjectofnumerousstudies; indeed,academicresearchsuggeststhat
fragmentationandothersubstantialeventsleadingtoneuronaldysfunctionatthesametime
haveledmanypeopletosufferfromvarietyofneurodegenerativediseasessuchasADand
other tauopathies, PD, HD, or CjD. However, the relevance of protein fragmentation in
neurodegenerationisstillunderdebate.

Discussion
85
Inhuman tauopathies, thedistinctmolecular and cellularmechanisms contributing to the
pathogenesisandprogressionoftauopathiesremainstillopaque.Precisely, the initiatorof
neurotoxiceventsandhowneurotoxicity ismediated isnotknown indetail.Aside theAβ
peptide and its pathological role in AD, the closer correlation between tau and disease
progressionarouseattentionandpromotedtau-focusedresearch.Thus,thedevelopmentof
transgenic mouse models allowed to investigate core aspects of the pathogenesis of
tauopathies by modeling particular disease steps in vivo; ultimately aiming for a better
understanding of the neuropathological mechanisms behind the complex nature of
neurodegenerativeprocesses.
Thepursuitofneurotoxictauspecies
The neuropathological identity of AD falls into two broad categories: extracellular senile
plaquescomposedof theAPPcleavageproductAβand filamentous tau inclusionsderived
fromaccumulationofmodifiedtauspecies.
Tauplaysacentralroleinmultipleneurodegenerativedisorders;however,itscontributionto
theonsetandprogressionofADisstillamatterofdebate.ButastaugenemutationinFTDP-
17T(Huttonetal.,1998;Poorkajetal.,1998;Spillantinietal.,1998)pavedthewayforinvivo
expression ofmutant human tau protein, transgenicmousemodels were generated that
exhibit distinct aspects of tau pathology such as age-related filamentous tau deposits,
neuronaldegenerationandaxonaltransportdysfunction(Gotzetal.,2004).
The formation of tau aggregates is depending on the conformational switch to β-sheet
structure;indeed,pointmutationsbutalsovariouspost-translationalmodificationsincluding
phosphorylation,acetylation,glycosylation,ubiquitination,andtruncationstructurallyalter
tauandthusfavouritsaccumulation.Whileaberrantaggregationandhyperphosphorylation
oftauareproposedtobethepivotalneurotoxicelementsinhumantauopathies,themost
toxicspeciesandhowtoxicityismediatedisofongoingdebate(BraakandDelTredici,2011;
ColemanandYao,2003;Goedert,2015;Terryetal.,1991).
Notably,proteolyticcleavageoftauhasbeenshowntobeanearlyeventinthepathological
cascadewithconsiderableimpactontautoxicity;indeed,fragmentationoftauoccursbefore

Discussion
86
toxictangleformationandfacilitateandpromotetauaggregationcomparedtofull-lengthtau
species (Abraha et al., 2000; Berger et al., 2007; Cowan et al., 2010). Various proteases
includingcaspase3,caspase6andcalpainshavebeenidentifiedtocleavefull-lengthtauinto
fragments which accumulation correlates with the progression of AD; notably, D421
constitutesthemostprominentcaspasecleavagesite inall formsof tauopathies (Basurto-
Islasetal.,2008;Fasuloetal.,2005;Guillozet-Bongaartsetal.,2005;Guoetal.,2004).
Withinthisframework,wehaveestablishedaninduciblemouseline(TAU62)overexpressing
a short, human 3R tau151–421 fragment (Δtau) that develop a mildly progressive motor
phenotypewithataxiaatabout18monthsofage.Interestingly,TAU62micedidnotdevelop
filamentous tau inclusions but exhibited neuron specific pretangle pathology in form of
abnormallyphosphorylatedtau;thisisinlinewithfindingsinothertaufragmentexpressing
mousemodels (McMillanetal.,2011),butcontradicts thepredominantneuropathological
phenotypedefinedbycorticalinsolubletaufilamentsinratmodelsoverexpressingtruncated
tau(Filipciketal.,2012).However,theobservedmildneurotoxicitywascomparabletothat
seen inmice expressing full-length wild-type forms of tau (Probst et al., 2000, 2N4R tau
transgenicALZ17mice;0N3RtautransgenicALZ31).
Inthecourseofthepresentwork,Ihavefocusedonthedrastic,butreversibleneurotoxicity
inourownnovelmousemodel co-expressing truncatedand full-length tau (Ozceliket al.,
2016). In linewith this,we set out to complete the neuropathological characterization of
several transgenic mouse models that recapitulate different aspects of AD to study the
neurodegenerativerelevanceoftaufragmentationandoligomerizationinvivo.
Truncatedtauinducessevereneurotoxicityinpresenceoffull-lengthformsoftau
InAD,PHFsarecomposedofnotonlyfull-lengthtauspeciesbutalsotruncatedformsoftau
(Gamblinetal.,2003;Goedertetal.,1992;Menaetal.,1996;Rissmanetal.,2004;Wischiket
al.,1988b).Wehaveapproachedthisneuropathologicalconditionwithco-expressionofΔtau
anddifferentfull-lengthformsoftauinvivo.

Discussion
87
First,we generateddouble transgenic P301SxTAU62onmice co-expressingΔtau andP301S
mutant 0N4R tau. Aside, full-length tau expressing human mutant P301S heterozygous
transgenicmicedevelopinsolubletaufilamentsandparalysisaround12monthsofage(Allen
etal., 2002). Strikingly,ourP301SxTAU62on exhibitedextensivenerve cell dysfunctionand
severemotorpalsyalreadybytheageof3weeks;consistently,inspinalcordneurons,axonal
transportdisruptionwasassociatedwithaccumulationofneurofilamentandaxonalspheroids
formation;furthermore,peripheralnervesshowedsignsofWalleriandegenerationwithfiber
lossandmyelindebrisfollowedbydrasticneurogenicmuscleatrophy.Itisinterestingtonote
thatdespitemassivefunctionalandstructuraldefects,onlyoligomerictauandnoformation
ofinsolubletauaggregatesortanglescouldhavebeendetected;infact,thepresenceoftoxic
tauoligomersaloneisastronghintthattheneurotoxiccascadeleadingtoneurodegeneration
mightnotdependonfilamentoustauinclusions.
Remarkably,thepronounceddrasticneurotoxicityinP301SxTAU62onmicewasphenotypically
andpathohistologicallyreversible:despitecontinuousexpressionoffull-lengthP301Stau,we
could confirm the reversibility of Δtau induced neuronal dysfunction in severely affected
P301SxTAU62on mice. When only Δtau expression was halted, former paralyzed
P301SxTAU62on-off mice rapidly regained full motor control and re-established nerve fiber
function and restored muscle fiber atrophy was observed. Moreover, the functional and
structuralrecoverywasparalleledbythedisappearanceofoligomerictauspecies.
Second,giventheartificialnatureofP301Smutanttau,weaimedforan invivoanalysisof
Δtau inpresenceof full-lengthwild-typetau. Indeed,wecould reproducethepronounced
reversible neurotoxicity in double transgenic ALZ17xTAU62onmice co-expressing Δtau and
full-lengthwild-type2N4Rtau.
Third,weanalyzedTAU62xALZ31onmiceexpressing3RΔtauandtheshortestformofwild-
type human 0N3R tau; however, these mice exhibited the most pronounced neurotoxic
phenotypethatwasnotreversiblewhenΔtauexpressionwasceased.Apossibleexplanation
isthatfunctionalandstructuralrecoverywouldrequirethepresenceofmixed3R/4Rratioof
tauoligomericspecies.
Of note, co-expression of only full-length tau species was not sufficient to induce severe
neurotoxicity: both, P301SxALZ31 and ALZ17xALZ31 double transgenic mice only exhibit
pretangletaupathologyanddidnotdisplayparalysis.

Discussion
88
Overall,thesefindingshighlighttwoessentialaspectsoftauopathies:taufragmentationand
oligomerization. In accordance with previous findings, we could highlight a neurotoxic
potentialoftaufragmentsaspathogenicmediatorsintauopathies.Indeed,recentworkhas
shownthattruncationoftaunotonlyfacilitatestauassemblyintofilaments(Abrahaetal.,
2000;Mocanuetal.,2008;Sydowetal.,2011),butalsoexacerbatesthetoxicityoffull-length
tau(Fasuloetal.,2005).Inparallel,weidentifiedoligomericformsoftauasakeyneurotoxic
species contributing to the neurodegenerative process in tauopathies; in fact, tau toxicity
precedestheformationof insolubleaggregates inourmousemodelandthus fibrillarytau
formation appears to be not required for tau toxicity, similar to findings in other
proteinopathies (Eisenberg and Jucker, 2012). Indeed, this confirms the recognized
importanceofsolubleoligomerictauspeciesforthepathogenesisoftauopathies(Lasagna-
Reevesetal.,2010;Spires-Jonesetal.,2011;Spires-Jonesetal.,2009).
Targetingtruncatedtauspecies
Theneurotoxicpotential of protein fragments in the courseof variousneurodegenerative
disorders remains to be established in detail. However, neurodegeneration-associated
proteins are substrate to various proteolytic enzymes that cleave designated proteins at
individuallydifferentsites(Figure4.1).Theneurotoxiceffectofproteinfragmentshasbeen
showni.e.fortheintramebranouslocatedAPP(Selkoe,2001a),thetransmembranouslocated
BRI2 (RostagnoandGhiso, 2008;Vidal et al., 1999;Vidal et al., 2000), intracellular and/or
extracellularcleavedα-Syn(Duftyetal.,2007;Kimetal.,2012;Mishizen-Eberzetal.,2005),
intracellularcleavedhtt(Grahametal.,2006;Waldron-Robyetal.,2012;Wellingtonetal.,
2002) and ataxins (Guyenet et al., 2015; Hubener et al., 2013; Mookerjee et al., 2009),
membraneanchoredPrP(Altmeppenetal.,2012;Trevittetal.,2014),andnuclearTDP-43
(Furukawa et al., 2011; Nonaka et al., 2009). Across the different neurodegenerative
disorders,majorenzymesinvolvedintheproteolyticprocessesarecaspasesandcalpains;in
particular, tau protein is cleaved by caspases-3/6 (Fasulo et al., 2005; Guo et al., 2004;
Metcalfeetal.,2012)andcalpains(FerreiraandBigio,2011;Higuchietal.,2012;Parketal.,
2007).

Discussion
89
Inlightofthis,inhibitionofcaspasesandcalpainscleavageemergedasapotentialtargetto
prevent disease pathology andmight be a promising therapeutic strategy considering the
observed neurotoxicity in ourmousemodel. Indeed, calpain-specific inhibitors have been
showntoattenuateAD-likepathologyin3xTgADmice(Medeirosetal.,2012);furtherreduce
ataxin cleavage (Haacke et al., 2007) and attenuate α-Syn induced synaptic impairments
(Diepenbroeketal.,2014).Inaddition,caspaseinhibitionhasalsobeenshowntomitigateα-
Synpathology(Bassiletal.,2016).
Nevertheless,thereisalsoevidencethatcleavageofneurodegeneration-associatedprotein
speciesmayconstitutearegularcellularprocess(Lietal.,2005).Forinstance,N-terminally
truncatedα-SynspeciesarepresentincaseswithorwithoutLewypathologyandcorrelating
withthetotalamountofα-Syn(Muntaneetal.,2012).
Figure4.1Overviewof the cellular locationofneurodegeneration-associatedproteins and their individualcleavagesites.

Discussion
90
Targetingtoxicoligomerictauspecies
The spectrumof neurotoxic potentials of tau species is still under debate.Oligomeric tau
formationoccursearlyintheprogressionofADpathology(Pattersonetal.,2011).InAD,late
insolubletauaggregatesarebelievedtocontributetonervecelldeathandcloselycorrelate
withcognitivedecline(Arriagadaetal.,1992;Gomez-Islaetal.,1997).Incontrast,thetoxicity
of oligomeric tau has been shown to correlate betterwith neuronal dysfunction than the
extentoffilamentoustau(Bergeretal.,2007).
Ourdataprovideevidencethatsolubleoligomerictauspeciesplayakeyroleintheneurotoxic
cascadeof taupathology leading toneuronal loss (Blairetal.,2013;Brundenetal.,2008;
Gerson and Kayed, 2013; Gerson et al., 2014; Usenovic et al., 2015). Indeed, progressive
neuronal degeneration in absence of tau filament formation was reported not only in
tauopathymousemodels (Oddo et al., 2003; Spires et al., 2006; Sydow andMandelkow,
2010), but also in tau transgenic drosophilamodels (Cowanet al., 2010;Wittmann et al.,
2001).Aside,oligomericproteinspecieshavebeenimplicatedinthepathogenesisofvarious
neurodegenerativediseases: inbrainsofADpatients, oligomericAβpeptides appeared to
participateintheneurotoxiccascadeevenbeforetheonsetofsymptoms(Lesneetal.,2013);
moreover, toxic extracellular α-Syn oligomers have been shown to favour prion-like
propagationinPDinvitro(Danzeretal.,2011);further,doperminergiclossinthesubstantia
nigracausedbyα-Synoligomerformationratherthanamyloidfibrilsinvivohighlightedthe
toxicityofsolubleoligomers(Winneretal.,2011);notably,oligomericAβandα-Synspecies
werepostulatedtobecompetenttoprovokeneurotoxictauoligomerization(Lasagna-Reeves
etal.,2010);inaddition,toxicoligomersformsofPrPhavebeensuggestedtomediatethe
infectiousprocessinpriondiseases(Silveiraetal.,2005).
Oligomeric tau species are considerably interesting as a potential immune target. The
observedsevereneurotoxicityassociatedwithabundanttauoligomerformationsuggestsour
mouse model of special relevance for this type of pharmacological treatment. Indeed, a
growingbodyofevidencesupportsthestrategyofdiminishingtaupathologywiththerapeutic
antibodies.Studiesonactiveimmunizationusingphospho-taupeptideshasbeenshownto

Discussion
91
mitigate tau aggregation in P301L transgenicmice (Asuni et al., 2007).Moreover, passive
immunization approaches with anti-tau directed monoclonal antibodies were shown to
reduce taupathology,whenadministratedprior to thediseaseonset (Boutajangoutetal.,
2011;Chaietal.,2011).However,therapidonsetoftautoxicityinourmousemodelfavours
it greatly for immunotherapeutic approaches and investigating clearance mechanisms of
pathologicaltauproteininvivo.
Targetingtheaxonaltransportsystemandoligomerictauinducedneurotoxicity
Herewereportthat fragmentedtauexacerbatesthetoxicityof full-lengthtau invivo.Our
mousemodelexhibitpronounceddrastic,butremarkably,reversibletoxiceffectofoligomeric
tau species associated with extensive neuronal dysfunction and a severe neurological
phenotype. In P301SxTAU62on mice, apparent mitochondrial dislocation and clumping,
disruption of the Golgi network and missorting of synaptic proteins strongly suggests a
widespreaddisruptionofthecellulartransportsystem(Kopeikinaetal.,2011;Liazoghlietal.,
2005). Interestingly, comparable perinuclear mitochondrial clumping associated with the
presence of only soluble tau specieswas reflected in studies on rTg4510 transgenicmice
(Kopeikinaetal.,2011).FragmentationoftheGolgiapparatus(GA)disturbsthecentralsorting
machineryforallnewlysynthesizedproteinsdestinedforfastaxonaltransportandisbelieved
to be an early event in various neurodegenerative disorders (Gonatas et al., 2006;
Hammerschlagetal.,1982;Liazoghlietal.,2005).InAD,giventhattheGAwasfoundtobe
fragmenteduponcdk5dysregulation,thisprocessappearstodirectlyprecedecelldeath(Sun
etal.,2008).Ofnote,overexpressionofα-SynhasbeenshowntocauseGolgifragmentation
andblockER-Golgitraffickinginvitroandinvivo(Cooperetal.,2006;Winslowetal.,2010).
However,somaticaccumulationofsynaptophysininthehippocampusofP301SxTAU62onmice
and degradation of VAMP2, a binding partner of synaptophysin, indicate congested Golgi
pathways and synaptic transmission disturbances (Nakashiba et al., 2008; Pennuto et al.,
2003).
Inaccordancewithourfindings,damagetotheaxonaltransportsystemhavebeendescribed
inmultipleneurodegenerativedisordersincludingAD(Kopeikinaetal.,2011),ALS(DeVoset

Discussion
92
al.,2007),PD(Sahaetal.,2004),andHD(Changetal.,2006).Further,inAD,Aβwasreported
tointerferewithmitochondrialtransportinculturedhippocampalneurons(Ruietal.,2006).
Intauopathies,over-expressionoftheshortesttauisoformhasbeenshowntoimpairaxonal
transportinvivo;inaddition,thiswasparalleledwithinsolubletauinclusions(Ishiharaetal.,
1999).Incontrast,solubletauspeciesweresufficienttoinducedisruptionofthemicrotubule
cytoskeletonindrosophila(Cowanetal.,2010).
Inphysiologicalconditions,tauistightlyassociatedwithmicrotubulesandcriticallyinvolved
in the dynamically assembly and stability of this cytoskeleton component; and thus,
instrumental for viability of the intracellular transport system (Kosik, 1993). However,
modificationsoftauarelikelyresponsibleforthedysregulationofthemicrotubulenetwork.
Therearetwopossibleexplanationsforthedisruptionofpolarizedmicrotubuletracks:one
source of interference might constitute abnormal high levels of tau and its concomitant
excessive binding to microtubules results in the detachment of kinesin; thus, ultimately
preventsanterogradetransportofindividualcargoes(BaasandQiang,2005;Dixitetal.,2008;
Dubeyetal.,2008). Inthiscase,hyperphosphorylationandsubsequentdissociationoftau,
typicallyresponsibleforthedestabilizationofmicrotubulesinthefirstplace,mightbeeven
of benefit for themicrotubule network operability.Of note, co-expression of various full-
lengthsformsoftauinourP301SxALZ31andALZ17xALZ31revealedcomparablelevelsoftau
hyperphosphorylation, without a pronounced motor phenotype; thus, tau
hyperphosphorylationappearnot to correlatedirectlywithnerve cell death inourmouse
modelsandstrengthenthepossibilitythatabnormalphosphorylationmightnotbeaprimary
pathoglogicalevent.
Microtubulenetworkdisruptionisamajorpathogeniceventintheevolutionoftauopathies.
Our findings inP301SxTAU62onmice suggest that, initiatedbyΔtauoverexpression, highly
toxicoligomerictauspeciesinterferewithmicrotubuleassemblyandstabilization,ultimately
contributetoaxonaltransportdisruption;however,theunderlyingmechanismsremaintobe
establishedindetail.Apossibleapproachtodissecttheprocessofaxonaltransportdisruption
wouldbetoassessvariouspotentmicrotubulestabilizingcompoundssuchasepothiloneD

Discussion
93
andpaclitaxelthathavebeenshowntopartlyrescuetauinducedaxonaltransportdysfunction
(Ballatoreetal.,2012;Bartenetal.,2012;Brundenetal.,2012;DasandMiller,2012).
Anotherapproachwouldbetotargettauoligomericformationdirectlythroughaggregation
inhibitors including methylene blue (MB) (Violet et al., 2015; Wischik et al., 1996) and
curcumin(Maetal.,2013).However,whileMBhasbeenshowntoblocktauproteinassembly
throughtherepeatdomainandbeingcompetenttoreverseearlytauaggregation,however,
itfailedtoremoverobusttanglepathologyintherTG4510mousemodeloftauopathy(Spires-
Jonesetal.,2014).Inaddition,areductionofAβlevelsandimprovedcognitivedeficitsupon
MBtreatmentwasreported(Medinaetal.,2011).
Protectiveeffectsofearlytauburdenonlateneurotoxicdistresslevel
ThisworkoriginatesfromourmostrecentpublishedobservationthatoverexpressionofΔtau
inpresenceoffull-lengthtaucausesdrastic,butreversibleneurotoxicityinvivo(Ozceliketal.,
2016).Basedonour results,we setout to investigate the long-termconsequencesof the
extensiveneurotoxicstressduringearlypostnatallifeinformerlyparalyzedP301SxTAU62on-
offmice.Remarkably,wefoundthatthesemiceexhibitalesspronouncedmotorphenotype
at14 to16monthsofagecomparedto theirheterozygousP301S littermates thatdidnot
experiencecompellingneurotoxicityearlyinlife.Thisisparalleledbysignificantreducedtau
pathology in aged P301SxTAU62on-off mice: the histopathological phenotype of formerly
paralyzedmiceshowedmitigatedtauhyperphosphorylationandalmostnosignsofinsoluble
tau inclusions. In linewith these findings, significant less total, insoluble, and soluble tau
proteinlevelscouldbedetected.
OurobservationsadvocatealatepreventiveeffectofearlyΔtauinducedneurotoxicityand
raisesthepossibilityofdiversesources,aloneor incombination,beingresponsibleforthe
underlying delayed tau pathology in P301SxTAU62on-off mice. As aforementioned, various
active immunizationattemptsusingrecombinanttaupeptideshavebeenshowntoreduce
tau pathology in various tau transgenicmousemodels (Asuni et al., 2007; Bi et al., 2011;
Boutajangoutetal.,2010).Apossiblescenarioisthattheexcessivetoxictauoligomerization
inyoungP301SxTAU62on-offmicewouldtriggeranautoimmunereactionintheformofarising
anti-tauantibodies.Alternatively,otherclearancemachineriesmaybeinvolvedinremoving

Discussion
94
oligomericorhyperphosphorylatedtaufromthecell.Forinstance,cytosolicproteasessuch
as puromycin sensitive aminopeptidase (PSA) were observed to attenuate tau levels and
motordeficitsinvariousmodelsoftauopathyincludingDrosophila(Karstenetal.,2006)and
mice(Kudoetal.,2011).Consideringthis,PSAmightregulatetaudegradationintwoways:
directlyviaproteolyticdegradationoftauasreportedinotherinvitrostudies(Senguptaetal.,
2006),orindirectlybyinducingautophagy(Menziesetal.,2010);specifically,theinhibitionof
PSAhasledtoincreasedlevelsoftoxicpolyQ-containinghttandataxin-3fragmentsaswellas
mutantα-Synassociatedwithadecreaseinautophagosomeformation.Indeed,theprotective
effects of early tau stress in P301SxTAU62on-offmice could point towards the induction of
autophagicpathways ingeneral.The removalofaggregation-proneproteins throughbasal
autophagyhasbeenshowntobeofimportanceforcellsurvival;indeed,theconsensusinthe
field is that impairmentof theautophagyprocessandsubsequent lossofacytoprotective
functionofautophagyleadstoneurodegeneration(Haraetal.,2006;Komatsuetal.,2006).
ProteinclearancesystemsandautophagydysfunctioninADhavebeensubjecttonumerous
studies(Nixon,2013;Nixonetal.,2005;Pirasetal.,2016);specifically,thereductionofBeclin1
expressionhasbeenshowntoincreaseAβpathologyanddisruptintheautophagyfluxinvivo
(Pickfordetal.,2008).InPD,α-Synoverexpressioncausesmislocalizationoftheautophagy
protein Atg9, which controls the formation of phagophore in the process of autophagy,
throughRab1ainhibition;however,Rab1aoverexpressionhasbeenshowntoreverseα-Syn
inducedautophagyimpairment(Winslowetal.,2010).Inaddition,stimulationofinsulinand
insulin-likegrowth factor1 signaling cascades results in increasedautophagic clearanceof
toxic huntingtin aggregates (Yamamoto et al., 2006). Furthermore, mTORC1-independent
activationofautophagybytrehaloseand inhibitionof themTORC1pathwaybyrapamycin
havenbeenshowntopromotetheclearanceofmutanthuntingtinandα-Syninvitro(Sarkar
etal.,2007).Inlinewiththesefindings,ourgroupcouldpreviouslyshowthattreatmentwith
theseautophagy-inducingagentsresultsindelayedprogressionoftaupathologyandsignsof
a restored autophagic flux associated with reduced accumulation of autophagy related
proteinsp62andLC3inP301Smutantmice(Ozceliketal.,2013;Schaefferetal.,2012).Of
note, microtubule-associated protein 1 light chain 3 protein (LC3) is required in the
autophagosomeformationprocess(MizushimaandKomatsu,2011);andp62isanubiquitin-
LC3-binding protein that links various substrates including protein aggregates to the

Discussion
95
autophagosome-lysosome degradation machinery (Kim et al., 2008). However, we lack a
completeunderstandingoftheautophagymachineryinourmousemodelsatthistimepoint;
thus,itwouldbeinterestingtoinvestigateapotentialautophagic,rectifyingresponsetoΔtau
inducedsevereneurotoxicityassociatedwiththeformationoftoxictauoligomers.Forthis,
experimentswithautophagyinducingagentsincludingrapamycinwouldyieldnewinsightinto
the degradation process of toxic tau species and the potential late preventive,
neuroprotectiveeffectofearlytaustress inagedP301SxTAU62on-offmice. Inparticular,the
assessmentofautophagicmarkersincludingLC3,p62,andBeclin1wouldallowtoanalyzeand
defineaclearerpictureoftheautophagyfluxphenomenon.
MouseCSFderivedAβspeciesdonotinduceprion-likeamyloidogenicpropagation
inAPP23transgenichostmice
ThisworkextendsrecentpublishedfindingsreportedonbrainandCSFderivedAβseeding
(Fritschi et al., 2014). Here we set out to inoculate forebrain homogenate or CSF in the
hippocampusofAPP23transgenicmice,andanalysetheseedingeffectofAβintheCSFand
brain.Consistentwiththispreviousreport,wefoundthatAβspeciesinbrainhomogenates
harbours seed-like potential, whereas CSF derived Aβ species lack prion-like propagation
activity(Skachokovaetal.,2015).Indeed,APP23transgenicmiceseededwithbrainderived
Aβfor20monthsexhibitedandincreasedplaque-loadinthehippocampus;incontrast,inthe
sameexperimentalset-up,CSFderivedAβdidnotresultinachangeinplaque-load.Apossible
explanationmightconstitutetheabsenceorstructuraldifferenceofseed-potentAβspecies
in the CSF. Whereas small and soluble Aβ species derived from APP23 mice have been
demonstratedtobehighlyseed-potent(Langeretal.,2011),theseAβspeciesappearnotto
bepresentintheCSF;possiblyduetodownregulatedtransporttotheCSFcompartment,or
upregulated degradation within the CSF compartment. Further, N-terminal truncated Aβ
specieshavebeenfoundlargelyinthebrain,butlessintheCSFofADpatients(Langeretal.,
2011).However,ifitsstructuraldiversityaffectstheseedingpotentialofAβremainselusive.
Inaddition,analysisoftheselectiveAβtransportandpotentialco-factorsinvolvedmightbe
neededtoexplainthelackofseed-potentAβspeciesinADCSF.

Discussion
96
Conclusionandperspectives
Ourdatashowthattruncatedtau,whenco-expressedwithfull-lengthformsoftau,resultsin
drastic,butreversibleearlytaustress.Wefurtherhave identifiedsolubletauoligomersas
toxickeyspecies,ratherthaninsolubletauaggregates,intheneuropathologicalcascadeof
tauopathies. This information is in line with the neurotoxic potential of protein cleavage
products and oligomeric species observed in diverse neurodegenerative disorders and
strengthenstheideathattruncatedtauindeedcontributestothepathogenesisoftauopathies
(HangerandWray,2010;RossandPoirier,2004).
In fact, ourmousemodel offers a remarkable setting to asses therapeutic strategies and
elucidatefurtherthepathomechanismsleadingtotauopathy.Forinstance,modulationoftau
fragmentationandassociated formationofoligomeric tauspeciesmaypotentiallyprevent
neurodegenerativeprocesses;ultimately,beingofcrucialrelevanceforthedevelopmentof
noveltherapeuticapproaches.
WefurthershowthatCSFderivedAβspeciesisnotcompetentenoughtoinduceprion-like
propagation. Instead, an interesting issue emerged from our work is that abundant toxic
oligomerictauspeciespresentinourmousemodelmightactuallyexhibitprion-likepotential.
Thishypothesisraisesthepossibilitytobetterunderstandprion-likeprocessesintauopathies
andneedstobecorroboratedbyfurtherexperiments.
In conclusion, ourwork underlines the role of fragmented tau and soluble oligomeric tau
aggregationintheneurodegenerativeprocessoftauopathies.

MaterialsandMethods
97
5 MaterialsandMethods
5.1 Animals
Alltransgenicmicewereheterozygousforthetransgenesofinterest,unlessspecifically
mentionedotherwise.ThenumberofmiceusedwasminimizedaccordingtotheSwiss
regulation on Animal Experimentation. All animal experiments were conducted in
accordancewiththeSwissguidelinesforanimalcareandapprovedbythe local legal
authorities.
5.1.1 Housingoftransgenicmice
Femalemiceweregroup-housedwithamaximumoffiveindividualspercage.Malemice
werehousedindividuallyastoavoidaggressive interactions.Freeaccesstofoodand
waterwasprovidedandanimalswerekeptona12hour/12hourinvertedlightcycle.
5.1.2 TAU62mice
Inducible and neuron-specific 3R tau151–421 (Δtau) expressing TAU62 mice were
generated by co-injection of two Thy 1.2 minigene-based constructs into C57BL/6J
oocytes.TheThy1.2tTSconstructwasobtainedbyreplacingexon2ofthemurineThy

MaterialsandMethods
98
1.2promoterbyatetracyclinecontrolledtranscriptionalsilencerelement(tTS).TheThy
1.2-TRE-Δtau construct contained a tetracycline responsive element (TRE) �800bp
upstreamofthehumanwild-typeΔtaucDNAencodingaminoacids151to421ofa3-
repeatdomainspanninghumanwild-typetaufragment(0N3Rtau151-421).Atotalofsix
positive transgenic founder TAU62 mice (C57BL/6J-TgN(tTS-Thy1-Δtau151-421) were
identifiedandtheinducibleexpressionofhumanΔtauwasassessedbywesternblotand
immunohistochemistry. The Lines 62-2 and 62-48 expressed comparable and robust
Δtau(‘on’)andstoppedexpressionfollowingtheremovalofdoxycycline(‘on-off’).Most
experimentswereperformedusing theTAU62/48 line,abbreviatedTAU62.TAU62/2
micewereusedtoruleoutaninsertionsiteeffect.
5.1.3 P301Smice
The production of P301S mutant 0N4R tau transgenic mice (C57BL/6J-TgN (Thy1
hTauP301S))hasbeenpreviouslydescribed(Allenetal.,2002).Miceweregeneratedby
subcloningP301SmutatedcDNAencodingtheshortesthumanfour-repeattauisoform
(383aminoacids isoformsofhuman tau) into amurineThy1.2 genomicexpression
vectorusingXhoIrestrictionsite.Transgenicmiceweregeneratedbymicroinjectioninto
pronucleiof(C57BL/6JxCBA/ca)F1generation.PCRanalysiswasperformedtoidentify
the founders, which then were interbred with C57BL/6J mice. Homozygous and
heterozygousP301Smicewereselectedaccordingtospecificexperiment.
5.1.4 ALZ17mice
Theproductionoffull-lengthwild-type2N4RtautransgenicALZ17mice(C57BL/6J-TgN
(Thy1hTau)17)hasbeenpreviouslydescribed(Probstetal.,2000).Miceweregenerated
bysubcloningcDNAencodingthelongesthumantauisoformintotheXhoIrestriction
siteof themurineThy1.2minigene.Aftermicroinjection intopronucleiofB6D2F1x
B6D2F1embryos,thefounderswereanalyzedbyPCRandtheninterbredwithC57BL/6J
mice.

MaterialsandMethods
99
5.1.5 ALZ31mice
ForthegenerationofALZ31wild-typehuman0N3Rtautransgenicmice(C57BL/6J-TgN
(Thy1hTau)31), 0N3R human tau complementary DNA was cloned into the neuron-
specificThy1.2promoterelementandinjectedintoC57BL/6Joocytes.
5.1.6 P301SxTAU62mice
P301SxTAU62doubletransgenicmiceco-expressatau151-421fragment(Δtau)withfull-
lengthmutantP301S(0N4R)tauas longas500mgkg−1doxycyclinewasprovidedad
libitum.
5.1.7 ALZ17xTAU62mice
ALZ17xTAU62doubletransgenicmiceco-expressatau151-421fragment(Δtau)withfull-
lengthwildtype(2N4R)tauaslongas500mgkg−1doxycyclinewasprovidedadlibitum.
5.1.8 ALZ31xTAU62mice
ALZ31xTAU62doubletransgenicmiceco-expressatau151-421fragment(Δtau)withfull-
lengthwildtype(0N3R)tauaslongas500mgkg−1doxycyclinewasprovidedadlibitum.
5.1.9 P301SxALZ31mice
P301SxALZ31doubletransgenicmicehavebeenobtainedbycrossingfull-lengthmutant
P301S(0N4R)tauwithfull-lengthwildtype(0N3R)tauexpressingmice.

MaterialsandMethods
100
5.1.10 ALZ17xALZ31mice
ALZ17xALZ31 double transgenic mice have been obtained by crossing full-length
wildtype(2N4R)tauwithfull-lengthwildtype(0N3R)tauexpressingmice.
5.1.11 APP23mice
APP23miceoverexpressthehumanfull-lengthAPPgene(751-aaisoform)harbouring
theSwedishdoublemutationatpositions670/671(KM->NL),undercontroloftheThy
1.2promoter.TheAPP751cDNAwasinsertedintotheblunt-endedXhoIrestrictionsite
of themurine Thy 1.2minigene that wasmicroinjected into C57BL/6J oocytes. The
founderswereanalyzedbyPCRandtheninterbredwithC57BL/6Jmice.
5.2 DNAisolationandgenotyping
GenomicDNAisolatedfromtoebiopsieswasusedtoidentifyandanalyzethetransgene
inheritance of the respective mouse line. Biopsies were incubated in lysis buffer
containing0.1mg/mlProteinaseK (Macherey-Nagel,Germany) incubatedovernight
(O/Nat55°Cand600rpminEppendorfThermomixercomfortshaker.Thesolutionwas
centrifuged(13000rpm,5min)and750μlofthesupernatantwasmixedwith750μl
isopropanolbyinvertingthetubes.Thesolutionwascentrifuged(13000rpm,10min),
thesupernatantwasdiscardedandthepelletwaswashedwith200μlof75%ethanol
(EtOH).After thesolutionwascentrifugedagain (13000rpm,10min), theremaining
DNApelletwasdriedonEppendorfThermomixercomfortshaker(55°C,5-10min)and
dissolved in 250μl dH2O (50°C, 1h).DNA sampleswere kept at 4°C until analysis of
transgeneinheritancebyPCR.
Reagent Concentration SupplierTris 100mM(pH8.0) Biomol#08003EDTA 5mM FlukaBioChemika#03690NaCl 200mM Merck#1.06404.10000SDS 0.20% BioRad#1610301
Table5.1:Lysisbuffercomposition

MaterialsandMethods
101
PCR conditionswere determined independently for each transgenes of interest and
eachindividualPCRprogramwasrunonaPTC100machine(MJResearch,Canada).PCR
productswererunon1.5%agarosegelsat150Vfor90min.
Reagent VolumeForwardprimer(10pmol/μl) 2μlReverseprimer(10pmol/μl) 2μlTopTaqMastermix(Qiagen,Germany) 12.5μlSterileH2O 6.5μlDNA 2μl
Table5.2:PCRmixture
Temperature Time Cycles95°C 2min 195°C 1min
3060°C 1min72°C 2min72°C 10min 14°C ∞
Table5.3:TAUF151PCRprogram
Temperature Time Cycles95°C 4min 195°C 1min
3060°C 1min72°C 3min72°C 10min 14°C ∞
Table5.4:P301SPCRprogram
Temperature Time Cycles95°C 10min 195°C 20s
3054°C 15s72°C 1min72°C 10min 14°C ∞
Table5.5:P301SxALZ31PCRprogram

MaterialsandMethods
102
Temperature Time Cycles94°C 2min 194°C 45s 160°C 1min
3572°C 45s94°C 45s72°C 5min 14°C ∞
Table5.6:APP23PCRprogram
Primersusedforgenotypingweredesignedindividuallyfortransgeneofinterest.PCRprogram Forwardprimer ReverseprimerTAUF151 GTGGATCTCAAGCCCTCAAG GGCGACTTGGGTGGAGTAP301S GGTTTTTGCTGGAATCCTGG GGAGTTCGAAGTGATGGAAGALZ31xP301S CCTCTCCCGTCCTCGCCTCTG
TCGAAGACAGACCACGGGGCGGAGATC
APP23 CCGATGGGTAGTGAAGCAATGGTT
GAATTCCGACATGACTCAGG
Table5.7:Designedprimersequences(5’-3’)
5.3 Histologyandimmunohistochemistry
Micewereanesthetizedwithamixtureof100mgkg−1ketamine(Ketalar®,Pfizer)and
10mg kg−1 xylazine (Rompun® 2%, Bayer) intraperitoneally and after deep sleeping,
micewereinjectedby100mgkg−1sodiumpentobarbital(Pentothal®0.5g,Ospedalia
AG)andtranscardiallyperfusedwith0.01Mcoldphosphate-bufferedsaline(PBS).
5.3.1 Tissue preparation and processing: Brain, Spinal cord and Sciatic
nerve
After perfusion, the spinal cord, sciatic nerve and the brain were quickly removed,
immersionfixedin4%paraformaldehydeO/N,andembeddedinparaffin.Brainswere
cuteithersagittally(4–20μmtransverseserialsections)orincoronalplaneanteriorto
the hippocampus (4–20 μm transverse serial sections starting at -2 Bregma to -3
Bregma,asdefinedbytheMouseBrainAtlasbyG.PaxinosandK.Franklin)byasliding

MaterialsandMethods
103
microtome(LeicaSM2000R,Leica,Germany).Paraffinsectionsweretransferredona
floatingbath(60°C),mountedonhistologicalslides(LeicaIPS,Leica,Germany)anddried
O/Nat37°Cbeforefurtherusage.ParaffinsectionsweredeparaffinisedinXylol(20min;
fromBiosystems,Switzerland),rehydratedin100%EtOH(3min),96%EtOH(2x3min)
and70%EtOH(2x3min),followedbywashinginPBS.Inordertomaskantigenicsites,
antigenretrievalwasperformedinCitricacidbufferpH6.0(ProTaps®)for30minat90
°C.Afterblockingin2.5%normalhorseserum(VectorLaboratories)for30minatroom
temperature(RT),sectionswereincubatedwithprimaryantibodydilutedinPBSO/Nat
4°C.Thenextday,thesectionswerewashed inTris/PBSsolution(3x5min)andthen
incubated with ImmPRESTM Reagent peroxidase for anti-mouse antibody (Vector
Laboratories, USA) for 1h at RT. After the incubation, sections were washed with
Tris/PBS (3x5 min) and developed by using chromogen ImmPACTTM NovaREDTM
Peroxidasesubstratekit(VectorLaboratories,USA).Thedevelopingtimewascontrolled
underamicroscopeandsectionswashedwithH2Otostopthereaction.Additionally,
slideswerethencounterstainedwithhematoxyline(J.T.Baker).Finally,sectionswere
rehydratedin70%EtOH(1min),96%and100%EtOH(each2x1min)andkeptinxylol,
beforeusingPertex®mountingmedium(Biosystems,Switzerland).Picturesofsection
were takenusing anOlympusDP73 (Olympus,USA)microscope. The content of the
originalimageswasnotneglectedbychangesofprocessedimages.
5.3.2 HematoxylinandEosinStaining
Afterdeparaffinisingandrehydration,sectionswererinsedincoldtapwaterandstained
inhematoxylinfor5-8min.Afterwashingincoldtapwateranddecolorizingshortlyin
alcohol-HCl,theslideswerewashedagainincoldtapwaterandkeptinwarmwateruntil
bluecolourappears.Then,sectionswereimmersedin1%erythrosineBsolution(RAL
diagnosis)for2-3min,washedagainshortlyincoldtapwater,followedbydehydration
inEtOH(70%,96%and100%)andmountingaspreviouslydescribed.

MaterialsandMethods
104
5.3.3 Gallyassilverstaining
Sectionsweresilver-impregnatedbyapreviouslydescribedmethod(Braaketal.,1988;
Gallyas,1971).Afterdeparaffinizationandrehydrationthesectionswereincubatedin
3%PeriodicAcid(Sigma-Aldrich,USA)for30minatRT.SlidesthenwerewashedindH2O
(2 min) and incubated in 1% Alkaline Silver solution (1M sodium hydroxide; 0.6M
potassiumiodide;1%silvernitratesolution;allfromMerck,Germany),for10minatRT.
After incubation in ABC solution and simultaneousmonitoring of the reaction time,
sectionswere treatedwith0.5%AceticAcid (Merck,Germany) toblock the reaction
during30mininRT.TreatedslideswerewashedindH2Oandincubatedin5%Sodium
thiosulfate(Merck,Germany)for5minatRT.Then,slideswerewashedincoldtapwater
andnucleistained(hematoxylineosinstaining),followedbydehydrationinEtOH(70%,
96%and100%)andmountingaspreviouslydescribed.
Solution Composition Concentration Supplier
A SodiumCarbonateAnhydride 0.5M Merck,Germany
BAmmoniumnitrate 24mM Merck,GermanySilverNitrate 0.01M Merck,GermanyTungstosilicicAcidHydrate 3.5mM Sigma-Aldrich,USA
C
Ammoniumnitrate 24mM Merck,GermanySilverNitrate 0.01M Merck,GermanyTungstosilicicAcidHydrate 3.5mM Sigma-Aldrich,USAFormalinSolution 0,26% Merck,Germany
Table5.8:ABCsolution

MaterialsandMethods
105
5.3.4 HolmesSilverNitrate-LuxolFastBluestaining
Reagents Concentration Supplier
Impregnationsolution
Boricacid 1,24% Merck,GermanyDinatriumtetraborat(Borax)
1,90% Merck,Germany
Silvernitrate 1% Merck,GermanyPyridinsolution 10% J.T.Baker
Reductionsolution
Sodiumsulfite 0.8M Merck,GermanyHydrochinon 90mM Merck,GermanyGoldchloride 0,25% Sigma-Aldrich,USAAcidoxalic 2% Merck,GermanySodiumthiosulfate 5% Merck,Germany
LuxolFastBluesolution
Luxolfastblue 24mM Medite
Ethanol 0.01M PharmacyUSBAceticacid 3.5mM Merck,Germany
Table5.9:HolmesSilverNitrate-LuxolFastBluestainingsolutions
Sectionsweredeparaffinisedandrehydratedaspreviouslydescribedandplacedin20%
Silvernitratesolution(Merck,Germany),for90mininthedarkatRT.Slidesthenwere
washed indH2O (3x)and incubated in impregnation solutionat37°CO/N.Nextday,
sections were placed onto filter paper to remove superfluous fluid and transferred
directlyinreductionsolutionfor10min.Thesectionswerethenwashedintapwater(5
min)andplacedindH2O.After,thesectionsweremovedin0.25%Goldchloridesolution
(5min)andthenrinsed indH2O(10min).Washedslideswere incubated in2%Acid
oxalic(10min)andthereactionwerestoppedbyrinsingthesectionsindH2O.Slides
werethenplacedin5%sodiumthiosulfatesolutionfor5minandrinsedintapwater
beforeplacingtheslidesbrieflyin70%(1x)and96%EtOH(2x).Next,thesectionswere
incubatedfor2hoursat60°CinLuxolfastbluesolutionandbrieflywashedin96%EtOH
(2x) to remove excess stain, then placed in running cold tap water for 5 min and
transferred into dH2O. Slides then were placed in 0.1% Lithium carbonate for few
secondsanddistainedin70%EtOHbeforetobewashedindH2O.Finally,theslideswere
immersed in Cresyl violet solution (10min; at RT) andwashed in 96% EtOH (2x) to
removeexcessstainandthenin100%EtOH(2x)beforetoplaceinxylolandapplyPertex
mountingmedium.

MaterialsandMethods
106
5.3.5 MassonTrichromestaining(Sciaticnerve)
Reagents Concentration Supplier
Weigert'sIronHematoxylinsolutionI
SolutionI Hematoxyline 33mM Merck,Germany
Ethanol 96% PharmacyUSB
Ferricchloridesolution
SolutionII Iron(III)chloride
32mM Merck,Germany
Acid-HCl 25% Merck,Germany
AcidfuchsinPonceau
SolutionI Fuchsinacid 17mM Merck,Germany
Aceticacid 1% Merck,Germany
SolutionII Ponceau 23mM Chroma,Germany
Aceticacid 1% Merck,Germany
Anilinebluesolution
Anilineblue 34mM Chroma,Germany
Aceticacid 2,5% Merck,Germany
Table5.10:MassonTrichromestainingsolutions
ThesectionsweredeparaffinisedandincubatedinWeigert’sIronHematoxylinsolution
(1min)andwashed inrunningcoldwaterfollowedbywarmwater(5-10min).Next,
slideswereplacedinAcidfuchsinPonceausolution(5min),followedbywashingintap
water.Thesectionswerethenincubatedin1%AcidPhosphomolibdic(Merck,Germany)
for5minandAnilinebluesolutionwasaddeddirectlyontheslidesfor3min(without
discardingthe1%AcidPhosphomolibdic)undershaking.Then,thesectionswererinsed
three-fivetimes indH2Oandtransferred in1%aceticacid (5sec).Finally, therinsed
slidesweredehydratedinabsoluteEtOH(2x),theninxylol,andcoverslipped.
5.3.6 Musclespreparation
After perfusion, gastrocnemius (GC), soleus (Sol), tibialis anterior (TA) and extensor
digitorum longus (EDL) were removed and snap frozen in liquid nitrogen cooled
isopentane.MuscleswereembeddedonacorkdisconO.C.TTMcompound(Tissue-Tek®

MaterialsandMethods
107
SakuraTM,USA)andstoredat-80°C.Coronalcryosections(10-12μm)werecutwitha
cryostat(HydraxC,HistocomAG,Switzerland)maintainedat-20°C.
5.3.7 Myosin-ATPase(Adenosintriphosphatase)staining(pH4.2)
Reagents Concentration SupplierVeronalacetatebuffer(stocksolution)
Sodiumacetate
0.1M Merck,Germany
Sodiumbarbiturate(Veronal)
0.1M Sigma-Aldrich,USA
ATPpH4.2solution
VeronalacetatebufferpH4.2
0.1M Sigma-Aldrich,USA
HCl 0.1M VeronalCabuffer(stocksolution)
Sodiumbarbiturate(Veronal)
0.1M Sigma-Aldrich,USA
CaCl2
0.03M
Merck,Germany
VeronalCaATPpH9.4solution
Adenosin-5-triphosphatedisodium(ATP)
4.5mM
Fluka#02060
Cobaltchloride
2%
Merck,Germany
HCl 0.1M
NaOH 1M Merck,Germany
Ammoniumsulfidesolution
21%
Sigma-Aldrich,USA
Table5.11:Myosin-ATPasestainingsolutions
FrozensectionsofO.C.TembeddedsampleswereequilibratedO/Nat-20°C,andthen
placedintothecryostatforminimum20minutes.Thesamplesweremountedonthe
cryostatwithO.C.Tand10μmsectionswerecutandcollectedonwarmslidesatRT.
Beforetoproceedthestaining,thesectionsmustbedryat least1hatRT.Theslides
weretransferredinATPpH4.2solution(10min;RT)andplacedshortlyforwashingin
Veronal-CapH9.4buffer.Next,thesectionswereincubatedinVeronal-Ca-ATPpH9.4
solution(45minatRT).Theslideswerethenwashedintapwater(2x)andmovedin

MaterialsandMethods
108
dH2Obeforetransferringin2%CobaltChloridefor5min.Afterwashingintapwater
(3x),thesectionswereplacedinAmmoniumsulfidesolution(14sec)andrinsedbefore
to be rehydrated in ascending EtOH (1x in 70%, 2x in 96% and 2x in 100%) and
transferredinxylolandapplyPertexmountingmedium.
5.3.8 Semithinsections(Sciaticnerve)
Sciaticnervesweredissectedandfixedforatleast2hin2.5%ofGlutaraldehydeatRT,
followed bywashing samples in 10mM PBSO/N. The tissueswere reduced in 1%
Osmium tetroxide (Oxkem Limited 10x1g) for 2h at RT. After a dehydration step in
histologicalgradeEtOH(70%,80%,90%and2x100%)for20mineachand2xinAcetone
for30min,thetissuewasincubated,first,inAcetone-Durcupan(1:1)solutionfor60min
and,second,inAcetone-Durcupane(1:3)O/Nat4°C.Then,thetissuesweremountedin
Durcupanresin,containing150mlofDurcupanA(Fluka),150mlofDurcupanB(Fluka),
3.1mlofDurcupanC(Fluka),4mlofDurcupanD(Fluka),andcookedat60°Cfor2-3
days,beforeprocessedforlightmicroscopy.Semithinsectionswerecut(1.5μm)using
aglasstripthatwasequippedwithaReichert-Jungapparatus.
5.3.9 Para-Phenylendiamine(Sciaticnerve)
Ultrathincryosectionsontheslidesweredriedonawarmplatebeforeincubatedin1%
p-Phenylenediamine(Sigma-Aldrich,USA)solutionfor2-3h.Theslidesthenwererinsed
6-10timesindH2OanddriedatRT.
5.3.10 Electronmicroscopy
Micewereanesthetizedaspreviouslydesrcibed.AftertranscardiallyperfusionwithPBS
for2-4min,animalswereperfusedfurtherwithafixativesolutioncomposedofwith2%
paraformaldehyde,2%glutaraldehydeand10mMPBS(pH7.4)for1h.Brainsandspinal
cordswereremovedandpostfixedfor1h,followedbyrinsingofthetissuesin10mM
PBS.

MaterialsandMethods
109
Thetissueswerereducedin1%Osmiumtetroxideand1.5%PotassiumFerrocyanide
for40min;after,thetissuesweretransferredin1%Osmiumtetroxidfor40min.The
tissues were dehydrated as previously described and embedded in Epon. During
dehydration, the sectionswere treatedwith 1% uranyl acetate in 70% EtOH for 1h.
Ultrathin sections from selected areaswere cutwith amicrotome (Ultracut E; Leica
MicrosystemsGmbH,Wetzlar,Germany),collectedonsingle-shotgrids,stainedin6%
uranylacetatefor1h.SectionswereexaminedandphotographedwithaMorgagniFEI
80kVelectronmicroscope(FEICompany,Eindhoven,TheNetherlands).
5.4 Sarkosylextraction
Reagent Concentration SupplierTris 25mM(pH7.4) BiomolNaCl 150mM Merck,GermanyEDTA 1mM FlukaBioChemikaEGTA 1mM Sigma-Aldrich,USASodiumpyrophosphate 5mM Sigma-Aldrich,USAPhosSTOP®, Phosphatase inhibitor cocktailtablet
1tablet Roche,Switzerland
Sodiumfluoride 30mM Merck,GermanyComplete Mini, Protease inhibitor cocktailtablets
1tablet Roche,Switzerland
Phenylmethylsulfonylfluoride(PMSF) 1mM Sigma-Aldrich,USALeupeptine 10μg/ml Sigma-Aldrich,USAAprotinine 10μg/ml Sigma-Aldrich,USAPepstatine 10μg/ml Sigma-Aldrich,USA
Table5.12:Extractionbuffer

MaterialsandMethods
110
Reagent Concentration Supplier
Tris 10mM(pH7.4) Biomol
NaCl 800mM Merck,Germany
Sucrose 10% FlukaBioChemika
EGTA 1mM Sigma-Aldrich,USA
Phenylmethylsulfonylfluoride(PMSF) 1mM Sigma-Aldrich,USA
Leupeptine 10μg/ml Sigma-Aldrich,USA
Aprotinine 10μg/ml Sigma-Aldrich,USA
Pepstatine 10μg/ml Sigma-Aldrich,USA
Table5.13:A68buffer
Phosphataseandproteaseinhibitorcocktailtabletswerefreshlyaddedtoeachbuffer.
Sarkosyl extraction was performed as described previously (Delobel et al., 2008).
FollowingPBSperfusion,onehalfofthemousebrainwasdissectedintoforebrainand
brainstemandfrozeninliquidnitrogenorondryice.Braintissuewashomogenizedin
coldExtractionbuffer1:3(w/v)byusingUltraturraxT8(IKA labortechnik)andbriefly
sonicated(BandelinSONOPULS,90
%power,10%cycle,10secpulses).Thesampleswerecentrifugedat4000g(5000rpm)
for15minand10%ofthesupernatant(=totaltau)wascollectedandaliquoted.Then,
samples were further centrifuged at 80 000g (28 000 rpm) for 15 min by using
ultracentrifuge (Beckman Coulter, OptimaTM L-70K Ultracentrifuge) by using SW55Ti
rotor(BeckmanCoulter).Thesupernatant(=solubletau)wascollectedandaliquoted.
TheremainingpelletswerehomogenizedinA68buffer1:3(w/v)andcentrifugedat4
000g(5000rpm)for20minand1%ofsarkosyl(N-laurylsarcosine,Sigma-Aldrich,USA)
added for 1h30 at 37 °C in thermoshaker (Eppendorf Thermomixer comfort shaker)
undershaking(maxrpm).Thesampleswerefurthercentrifugedat80000g(28000rpm)
for30minandthepelletswasresuspendedin150μlg−1of50mMTris-HCl,pH7.4and
aliquoted(=sarkosylinsolubletau).

MaterialsandMethods
111
5.5 WesternBlot
Reagent Concentration SupplierTris 20mM(pH7.5) BiomolNaCl 137mM Merck,GermanyComplete Mini, Protease inhibitor cocktailtablets
1tablet Roche,Switzerland
Table5.14:TBS-Completebuffer
Reagent Reducedsample(μl)
Non-reducedsample(μl)
Sample x xNuPAGE®LDSSampleBuffer(4X) 5 xNuPAGE®ReducingAgent(10X) 2 —DeionizedWater x xTotalVolume 20 20
Table5.15:Reducedandnon-reducedsamplepreparation
FollowingPBSperfusion,onehalfofthemousebrainwasdissectedintoforebrainand
brainstemandfrozeninliquidnitrogenorondryice.Thebraintissuewashomogenized
in1:10volumeofTBS-CompletebufferbyusingUltraturraxT8(IKAlabortechnik)and
briefly sonicated (Bandelin SONOPULS, 90% power, 10% cycle, 10 sec pulses). The
sampleswerespundownat4000g(5000rpm)for30minandthesupernatantwas
collectedandaliquoted.Westernblotswereperformedeitherunderreducingornon-
reducingconditions(seeTable5.15).Followingappropriatepreparation,sampleswere
heatedfor5minat95°Candshortlyspindownandloadedontoa7%NuPAGE®Tris-
acetate gel. Gels were run first at 100V for 30min, then subsequently at 120V for
additional60min.AftertheremovalofgelsfromthecassetteandactivationofPVDF
membrane (AmershamBiosciences) for first, 30 sec inmethanol and then, 5min in
transferbuffer,samplesweretransferredonthePVDFmembraneat30Vfor2hbyusing
theXCellIITMBlotModule.Next,Unspecificbindingepitopeswereblockedwith5%non-
fat milk in PBS-T (0.01M PBS pH 7.4; 0.05 % Tween-20) for 1h at RT, followed by
incubationwithprimaryantibodyO/Nat4°Conashaker.AfterwashingwithPBS-T(3x5
min)atRT,themembranewasincubatedwithhorseradishperoxidase(HRP)-conjugated
anti-mouseor-rabbitsecondaryantibodyfor1hatRT.Then,themembranewaswashed
again inPBS-T (3x5min) atRT anddetectedbyelectrochemiluminescence (ECL) (GE
Healthcare, USA). Western blot was performed with the NuPAGE® System from
Invitrogen.

MaterialsandMethods
112
5.6 Antibodies
Antibody Target Dilution SourceHT7 humantau
aa159-163WB1:4000IHC1:800
Pierce,Rockford,IL#MN1000
BR134 humantau WB1:1000 (Goedertetal.,1989)
Tau-C3 TaucleavedatresidueAsp421
WB1:1000IHC1:1000
SantaCruzBiotechnology,Inc,Dallas,TX#sc-32240
AT8 TaupSer202/Thr205
WB1:1000IHC1:800
Pierce,Rockford,IL#MN1020
AT100 TaupThr212/Ser214
WB1:1000IHC1:500
Pierce,Rockford,IL#MN1060
PHF-1 TaupSer396/404
WB1:2000IHC1:1000
PeterDavies,AlbertEinsteinCollegeofMedecine,Bronx,NY
MC1 Tauaa5-15,312-322
IHC1:100 PeterDavies,AlbertEinsteinCollegeofMedecine,Bronx,NY
2F11 neurofilament(NF)NF-L,NF-H(70kD)
IHC1:800 Dako,Glostrup,DK#M0762
NF200 neurofilament(200kD) IHC1:100 (Probstetal.,2000)
GFAP glialfibrillaryacidicprotein
IHC1:500 ThermoFisherScientificInc.,Kalamazoo,MI#MS-1407-R7
Synaptophysin synaptophysin IHC1:1000 MilliporeCorporation,Billerica,MA#MAB5258
MG160(rabbit) Golgiapparatus IHC1:1000 NicholasGonatas,PathologyandLaboratoryMedicine,UniversityofPennsylvania,PA
VAMP2/Synaptobrevin2(rabbit)
transportvesicles IHC1:1000 Synapticsystem,Goettingen,Germany#104202
GAPDH(6C5) GAPDH WB1:1000 SantaCruzBiotechnology,SantaCruz,CA,#32233
ß-actin actin WB1:5000 Sigma-Aldrich,SaintLouis,MO#A5316
Coxsubunit1a mitochondrialstaining
IHC1:200 Abcamplc,Cambridge,UK#ab14705
RD3,clone8E6/C11 Humantau,recognize3R,residue209-224
WB1:4000;IHC1:3000
MilliporeCorporation,Billerica,MA#05-803
RD4,clone1E1/A6 Humantauandmouse,recognize4R,aa275-291
WB1:4000;IHC1:100
MilliporeCorporation,Billerica,MA#05-804
T49 Specificforrodenttau
WB1:10000
VirginiaLee,CNDR,UniversityofPennsylvaniaSchoolofMedicine,Philadelphia,PA
Table 5.16: Antibodies used for immunohistochemistry (IHC) and Western blotting (WB). Species ismouse,unlessindicatedotherwise)

MaterialsandMethods
113
5.7 Behavioralassessment
5.7.1 Grid-test
Grid reflex andmotor strengthofmicewas assessedby the grid test.Animalswere
placedonaverticalmeshgridandthe latencyto falloff fromthegridwasrecorded
duringamaximumtimeof180sec.Eachmousewastested3timeswithatleasta5min
restintervalinbetweentrails.Anaveragescoreperdaywasmade.
5.7.2 Rotarodtest
ThemotorcoordinationandbalanceofmicewereassessedusingthePanlabHarvard
Rotarod.TheRotarodstartsataspeedof4rpmandacceleratesconsistentlywith1rpm
every3sec.Thetestingphaseconsistedof4consecutivedays.Witheachanimal,an
acclimation period consisting of 3 sessions with a rest interval of at least 5 min in
betweentrailswasperformed.Subsequently,miceweretestedfor3consecutivedays
with3trialseachdayandarestintervalof5minminimumandthemeanlatencytofall
wasdocumented.
5.7.3 Objectrecognitiontest
Shorttermmemorywasassessedbytheobjectrecognitiontest.Animalswereplacedin
asquaredopenfieldbox(48×48×40cm)underdimlightconditions.Micewereallowed
tofreelyexploretheboxduringahabituationphaseover3consecutivedaysfor15min,
untilnosignsofstresswerepresent.Duringatrainingphaseover2days,twoidentical
objectswereintroducedatdiagonalcornersofthefieldfortrainingsessionsof10min
duration.Thetrainingwashaltedwhenthemicehadcloselyexploredtheobjectsfor20
sec,foramaximumtrainingdurationof10min(Legeretal.,2013)).Inthetestphase,
theanimals’short-termmemorywastestedbyreplacingoneofthefamiliarobjectswith

MaterialsandMethods
114
anovelone,andthetimespentexploringeachobjectduringaperiodof6minwasvideo
recorded.VideoscoringVLCvideoplayer,VideoLAN,France)wasdonebyaresearcher
blindtothegenotype,andasexplorationcriterianosesniffing/touchingoftheobjectat
2cmorlessdistance(Legeretal.,2013))wereused.Forbothtrainingandtestphases,
10cmhighobjectscomposedofthesamematerialwereused,andthepositionofthe
novelandfamiliarobjectswererandomizedacrossgroups.
5.8 Statistics
All statistical analysiswasperformedusingone-wayanalysisof variance followedby
Bonferroni’s multiple comparison test and Student’s t-tests with GraphPad Prism
SoftwareVersion5.0a(GraphPadSoftware,LaJolla,CA,USA).P-valuesareestablished
and outlined as follows: *P<0.05, **P<0.01 and ***P<0.001. Themean and s.d. are
indicated.

References
115
6 References
Abraha,A.,Ghoshal,N.,Gamblin,T.C.,Cryns,V.,Berry,R.W.,Kuret,J.,andBinder,L.I.(2000).C-terminalinhibitionoftauassemblyinvitroandinAlzheimer'sdisease.JCellSci113Pt21,3737-3745.
Aguzzi, A., and O'Connor, T. (2010). Protein aggregation diseases: pathogenicity and therapeuticperspectives.NatRevDrugDiscov9,237-248.
Akter,R.,Cao,P.,Noor,H.,Ridgway,Z.,Tu,L.H.,Wang,H.,Wong,A.G.,Zhang,X.,Abedini,A.,Schmidt,A.M.,etal.(2016).IsletAmyloidPolypeptide:Structure,Function,andPathophysiology.JDiabetesRes2016,2798269.
Allen,B., Ingram,E.,Takao,M.,Smith,M.J., Jakes,R.,Virdee,K.,Yoshida,H.,Holzer,M.,Craxton,M.,Emson,P.C., etal. (2002).Abundant tau filamentsandnonapoptoticneurodegeneration in transgenicmiceexpressinghumanP301Stauprotein.JNeurosci22,9340-9351.
Altmeppen, H.C., Puig, B., Dohler, F., Thurm, D.K., Falker, C., Krasemann, S., and Glatzel, M. (2012).Proteolyticprocessingoftheprionproteininhealthanddisease.AmJNeurodegenerDis1,15-31.
Alzheimer, A. (1906). Über einen eigenartigen schweren Erkrankungsprozess der Hirnrinde.NeurologischesCentralblatt25,1134.
Alzheimer,A.(1907).ÜbereineeigenartigeErkrankungderHirnrinde.AllgZPsychiatPsych-GerichtlMed64,146-148.
Andreadis,A.,Brown,W.M.,andKosik,K.S.(1992).Structureandnovelexonsofthehumantaugene.Biochemistry31,10626-10633.
Anglade,P.,Vyas,S.,Javoy-Agid,F.,Herrero,M.T.,Michel,P.P.,Marquez,J.,Mouatt-Prigent,A.,Ruberg,M., Hirsch, E.C., and Agid, Y. (1997). Apoptosis and autophagy in nigral neurons of patients withParkinson'sdisease.HistolHistopathol12,25-31.
Arai,T.,Guo,J.P.,andMcGeer,P.L.(2005).Proteolysisofnon-phosphorylatedandphosphorylatedtaubythrombin.JBiolChem280,5145-5153.
Arai,T.,Hasegawa,M.,Akiyama,H.,Ikeda,K.,Nonaka,T.,Mori,H.,Mann,D.,Tsuchiya,K.,Yoshida,M.,Hashizume, Y., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions infrontotemporallobardegenerationandamyotrophiclateralsclerosis.BiochemBiophysResCommun351,602-611.
Arai,T.,Hasegawa,M.,Nonoka,T.,Kametani,F.,Yamashita,M.,Hosokawa,M.,Niizato,K.,Tsuchiya,K.,Kobayashi, Z., Ikeda, K., et al. (2010). Phosphorylated and cleaved TDP-43 in ALS, FTLD and otherneurodegenerativedisordersandincellularmodelsofTDP-43proteinopathy.Neuropathology30,170-181.

References
116
Arriagada,P.V.,Growdon,J.H.,Hedley-Whyte,E.T.,andHyman,B.T.(1992).NeurofibrillarytanglesbutnotsenileplaquesparalleldurationandseverityofAlzheimer'sdisease.Neurology42,631-639.
Asuni,A.A.,Boutajangout,A.,Quartermain,D.,andSigurdsson,E.M.(2007). Immunotherapytargetingpathologicaltauconformersinatanglemousemodelreducesbrainpathologywithassociatedfunctionalimprovements.JNeurosci27,9115-9129.
Augustinack,J.C.,Schneider,A.,Mandelkow,E.M.,andHyman,B.T.(2002).SpecifictauphosphorylationsitescorrelatewithseverityofneuronalcytopathologyinAlzheimer'sdisease.ActaNeuropathol103,26-35.
Avila,J.(2010).Alzheimerdisease:caspasesfirst.NatRevNeurol6,587-588.
Baas,P.W.,andQiang,L.(2005).Neuronalmicrotubules:whentheMAPistheroadblock.TrendsCellBiol15,183-187.
Baba,M.,Nakajo,S.,Tu,P.H.,Tomita,T.,Nakaya,K.,Lee,V.M.,Trojanowski,J.Q.,andIwatsubo,T.(1998).Aggregationofalpha-synucleininLewybodiesofsporadicParkinson'sdiseaseanddementiawithLewybodies.AmJPathol152,879-884.
Ballatore, C., Brunden, K.R., Huryn, D.M., Trojanowski, J.Q., Lee, V.M., and Smith, A.B., 3rd (2012).Microtubule stabilizing agents as potential treatment for Alzheimer's disease and relatedneurodegenerativetauopathies.JMedChem55,8979-8996.
Barten,D.M.,Fanara,P.,Andorfer,C.,Hoque,N.,Wong,P.Y.,Husted,K.H.,Cadelina,G.W.,Decarr,L.B.,Yang,L.,Liu,V.,etal.(2012).Hyperdynamicmicrotubules,cognitivedeficits,andpathologyareimprovedintautransgenicmicewithlowdosesofthemicrotubule-stabilizingagentBMS-241027.JNeurosci32,7137-7145.
Bassil,F.,Fernagut,P.O.,Bezard,E.,Pruvost,A.,Leste-Lasserre,T.,Hoang,Q.Q.,Ringe,D.,Petsko,G.A.,and Meissner, W.G. (2016). Reducing C-terminal truncation mitigates synucleinopathy andneurodegenerationinatransgenicmodelofmultiplesystematrophy.ProcNatlAcadSciUSA113,9593-9598.
Basurto-Islas,G., Luna-Munoz, J.,Guillozet-Bongaarts,A.L.,Binder, L.I.,Mena,R., andGarcia-Sierra, F.(2008).AccumulationofAsparticAcid421-andGlutamicAcid391-CleavedTauinNeurofibrillaryTanglesCorrelatesWithProgressioninAlzheimerDisease.JNeuropatholExpNeurol67,470-483.
Bayer,T.A.,andWirths,O.(2010).Intracellularaccumulationofamyloid-Beta-apredictorforsynapticdysfunctionandneuronlossinAlzheimer'sdisease.FrontAgingNeurosci2,8.
Bence,N.F.,Sampat,R.M.,andKopito,R.R.(2001).Impairmentoftheubiquitin-proteasomesystembyproteinaggregation.Science292,1552-1555.
Berger, Z., Roder,H.,Hanna,A., Carlson,A., Rangachari, V., Yue,M.,Wszolek, Z., Ashe, K., Knight, J.,Dickson,D., et al. (2007). Accumulationof pathological tau species andmemory loss in a conditionalmodeloftauopathy.JNeurosci27,3650-3662.
Bi, M., Ittner, A., Ke, Y.D., Gotz, J., and Ittner, L.M. (2011). Tau-targeted immunization impedesprogressionofneurofibrillaryhistopathologyinagedP301Ltautransgenicmice.PLoSOne6,e26860.
Biernat, J., Mandelkow, E.M., Schroter, C., Lichtenberg-Kraag, B., Steiner, B., Berling, B., Meyer, H.,Mercken,M.,Vandermeeren,A.,Goedert,M.,etal.(1992).TheswitchoftauproteintoanAlzheimer-likestate includes the phosphorylation of two serine-prolinemotifs upstreamof themicrotubule bindingregion.EMBOJ11,1593-1597.
Binder, L.I., Frankfurter,A., andRebhun, L.I. (1985). Thedistributionof tau in themammaliancentralnervoussystem.JCellBiol101,1371-1378.
Blair,L.J.,Nordhues,B.A.,Hill,S.E.,Scaglione,K.M.,O'Leary,J.C.,3rd,Fontaine,S.N.,Breydo,L.,Zhang,B.,Li, P., Wang, L., et al. (2013). Accelerated neurodegeneration through chaperone-mediatedoligomerizationoftau.JClinInvest123,4158-4169.
Blennow,K.,deLeon,M.J.,andZetterberg,H.(2006).Alzheimer'sdisease.Lancet368,387-403.

References
117
Bohm,K.J.,Vater,W.,Steinmetzer,P.,Kusnetsov,S.A.,Rodionov,V.I.,Gelfand,V.I.,andUnger,E.(1990).EffectofMAP1,MAP2,andtau-proteinsonstructuralparametersoftubulinassemblies.ActaHistochemSuppl39,357-364.
Boland,B.,Kumar,A.,Lee,S.,Platt,F.M.,Wegiel,J.,Yu,W.H.,andNixon,R.A.(2008).Autophagyinductionandautophagosomeclearanceinneurons:relationshiptoautophagicpathologyinAlzheimer'sdisease.JNeurosci28,6926-6937.
Boutajangout,A.,Ingadottir,J.,Davies,P.,andSigurdsson,E.M.(2011).Passiveimmunizationtargetingpathologicalphospho-tauproteininamousemodelreducesfunctionaldeclineandclearstauaggregatesfromthebrain.JNeurochem118,658-667.
Boutajangout,A.,Quartermain,D.,andSigurdsson,E.M.(2010).Immunotherapytargetingpathologicaltaupreventscognitivedeclineinanewtanglemousemodel.JNeurosci30,16559-16566.
Braak, H., and Braak, E. (1989). Cortical and subcortical argyrophilic grains characterize a diseaseassociatedwithadultonsetdementia.NeuropatholApplNeurobiol15,13-26.
Braak,H.,Braak,E.,Ohm,T.,andBohl,J.(1988).SilverimpregnationofAlzheimer'sneurofibrillarychangescounterstainedforbasophilicmaterialandlipofuscinpigment.StainTechnol63,197-200.
Braak,H.,andDelTredici,K.(2011).ThepathologicalprocessunderlyingAlzheimer'sdiseaseinindividualsunderthirty.ActaNeuropathol121,171-181.
Bramblett,G.T.,Goedert,M.,Jakes,R.,Merrick,S.E.,Trojanowski,J.Q.,andLee,V.M.(1993).Abnormaltau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes toreducedmicrotubulebinding.Neuron10,1089-1099.
Brandt, R., and Lee, G. (1993). Functional organization of microtubule-associated protein tau.Identificationofregionswhichaffectmicrotubulegrowth,nucleation,andbundleformationinvitro.JBiolChem268,3414-3419.
Brandt,R.,Leger,J.,andLee,G.(1995).Interactionoftauwiththeneuralplasmamembranemediatedbytau'samino-terminalprojectiondomain.JCellBiol131,1327-1340.
Browne, S.E. (2008). Mitochondria and Huntington's disease pathogenesis: insight from genetic andchemicalmodels.AnnNYAcadSci1147,358-382.
Brunden,K.R.,Ballatore,C., Lee,V.M., Smith,A.B., 3rd, andTrojanowski, J.Q. (2012).Brain-penetrantmicrotubule-stabilizingcompoundsaspotentialtherapeuticagentsfortauopathies.BiochemSocTrans40,661-666.
Brunden,K.R.,Trojanowski,J.Q.,andLee,V.M.(2008).Evidencethatnon-fibrillartaucausespathologylinkedtoneurodegenerationandbehavioralimpairments.JAlzheimersDis14,393-399.
Buee, L., Bussiere, T., Buee-Scherrer, V., Delacourte, A., and Hof, P.R. (2000). Tau protein isoforms,phosphorylationandroleinneurodegenerativedisorders.BrainResBrainResRev33,95-130.
Buratti,E.,andBaralle,F.E.(2008).MultiplerolesofTDP-43ingeneexpression,splicingregulation,andhumandisease.FrontBiosci13,867-878.
Cantlon,A.,Frigerio,C.S.,andWalsh,D.M.(2015).LessonsfromaRareFamilialDementia:AmyloidandBeyond.JParkinsonsDisAlzheimersDis2.
Canu,N.,Dus,L.,Barbato,C.,Ciotti,M.T.,Brancolini,C.,Rinaldi,A.M.,Novak,M.,Cattaneo,A.,Bradbury,A., and Calissano, P. (1998). Tau cleavage and dephosphorylation in cerebellar granule neuronsundergoingapoptosis.JNeurosci18,7061-7074.
Castellani,R.J.,Rolston,R.K.,andSmith,M.A.(2010).Alzheimerdisease.DisMon56,484-546.
Caughey, B., Raymond, G.J., and Bessen, R.A. (1998). Strain-dependent differences in beta-sheetconformationsofabnormalprionprotein.JBiolChem273,32230-32235.
Cente,M.,Filipcik,P.,Pevalova,M.,andNovak,M.(2006).Expressionofatruncatedtauproteininducesoxidativestressinarodentmodeloftauopathy.EurJNeurosci24,1085-1090.

References
118
Chai,X.,Wu,S.,Murray,T.K.,Kinley,R.,Cella,C.V.,Sims,H.,Buckner,N.,Hanmer,J.,Davies,P.,O'Neill,M.J.,etal.(2011).Passiveimmunizationwithanti-Tauantibodiesintwotransgenicmodels:reductionofTaupathologyanddelayofdiseaseprogression.JBiolChem286,34457-34467.
Chang,D.T.,Rintoul,G.L.,Pandipati,S.,andReynolds, I.J. (2006).Mutanthuntingtinaggregates impairmitochondrialmovementandtraffickingincorticalneurons.NeurobiolDis22,388-400.
Chen,C.D.,Huff,M.E.,Matteson,J.,Page,L.,Phillips,R.,Kelly,J.W.,andBalch,W.E.(2001).FurininitiatesgelsolinfamilialamyloidosisintheGolgithroughadefectinCa(2+)stabilization.EMBOJ20,6277-6287.
Chen,J.,Kanai,Y.,Cowan,N.J.,andHirokawa,N.(1992).ProjectiondomainsofMAP2andtaudeterminespacingsbetweenmicrotubulesindendritesandaxons.Nature360,674-677.
Chesebro,B.,Trifilo,M.,Race,R.,Meade-White,K.,Teng,C.,LaCasse,R.,Raymond,L.,Favara,C.,Baron,G.,Priola,S.,etal.(2005).Anchorlessprionproteinresultsininfectiousamyloiddiseasewithoutclinicalscrapie.Science308,1435-1439.
Chesser,A.S.,Pritchard,S.M.,andJohnson,G.V.(2013).TauclearancemechanismsandtheirpossibleroleinthepathogenesisofAlzheimerdisease.FrontNeurol4,122.
Cho,D.H.,Nakamura,T.,Fang,J.,Cieplak,P.,Godzik,A.,Gu,Z.,andLipton,S.A.(2009).S-nitrosylationofDrp1mediatesbeta-amyloid-relatedmitochondrialfissionandneuronalinjury.Science324,102-105.
Citron,M.,Teplow,D.B.,andSelkoe,D.J.(1995).Generationofamyloidbetaproteinfromitsprecursorissequencespecific.Neuron14,661-670.
Clavaguera, F., Akatsu, H., Fraser, G., Crowther, R.A., Frank, S., Hench, J., Probst, A., Winkler, D.T.,Reichwald, J., Staufenbiel,M., et al. (2013). Brain homogenates from human tauopathies induce tauinclusionsinmousebrain.ProcNatlAcadSciUSA110,9535-9540.
Clavaguera,F.,Bolmont,T.,Crowther,R.A.,Abramowski,D.,Frank,S.,Probst,A.,Fraser,G.,Stalder,A.K.,Beibel,M.,Staufenbiel,M.,etal.(2009).Transmissionandspreadingoftauopathyintransgenicmousebrain.NatCellBiol11,909-913.
Cleveland,D.W.,Hwo,S.Y.,andKirschner,M.W.(1977).Physicalandchemicalpropertiesofpurifiedtaufactorandtheroleoftauinmicrotubuleassembly.JMolBiol116,227-247.
Cohen,T.J.,Guo,J.L.,Hurtado,D.E.,Kwong,L.K.,Mills,I.P.,Trojanowski,J.Q.,andLee,V.M.(2011).Theacetylationoftauinhibitsitsfunctionandpromotespathologicaltauaggregation.NatCommun2,252.
Coleman,P.D.,andYao,P.J.(2003).SynapticslaughterinAlzheimer'sdisease.NeurobiolAging24,1023-1027.
Conceicao,I.,Gonzalez-Duarte,A.,Obici,L.,Schmidt,H.H.,Simoneau,D.,Ong,M.L.,andAmass,L.(2016)."Red-flag"symptomclustersintransthyretinfamilialamyloidpolyneuropathy.JPeripherNervSyst21,5-9.
Constantinidis,J.,Richard,J.,andTissot,R.(1974).Pick'sdisease.Histologicalandclinicalcorrelations.EurNeurol11,208-217.
Coomaraswamy,J.,Kilger,E.,Wolfing,H.,Schafer,C.,Kaeser,S.A.,Wegenast-Braun,B.M.,Hefendehl,J.K.,Wolburg,H.,Mazzella,M.,Ghiso,J.,etal.(2010).ModelingfamilialDanishdementiainmicesupportstheconceptoftheamyloidhypothesisofAlzheimer'sdisease.ProcNatlAcadSciUSA107,7969-7974.
Cooper,A.A.,Gitler,A.D.,Cashikar,A.,Haynes,C.M.,Hill,K.J.,Bhullar,B.,Liu,K.,Xu,K.,Strathearn,K.E.,Liu,F.,etal.(2006).Alpha-synucleinblocksER-GolgitrafficandRab1rescuesneuronlossinParkinson'smodels.Science313,324-328.
Correas, I.,Diaz-Nido, J.,andAvila, J. (1992).Microtubule-associatedprotein tau isphosphorylatedbyproteinkinaseConitstubulinbindingdomain.JBiolChem267,15721-15728.
Cowan,C.M.,Bossing,T.,Page,A.,Shepherd,D.,andMudher,A.(2010).Solublehyper-phosphorylatedtaucausesmicrotubulebreakdownandfunctionallycompromisesnormaltauinvivo.ActaNeuropathol120,593-604.

References
119
Cras, P., Smith, M.A., Richey, P.L., Siedlak, S.L., Mulvihill, P., and Perry, G. (1995). Extracellularneurofibrillary tangles reflect neuronal loss and provide further evidence of extensive protein cross-linkinginAlzheimerdisease.ActaNeuropathol89,291-295.
Cummings,J.L.(2004).Alzheimer'sdisease.NEnglJMed351,56-67.
D'Arcangelo,G.,Nakajima,K.,Miyata,T.,Ogawa,M.,Mikoshiba,K., andCurran,T. (1997).Reelin is asecretedglycoproteinrecognizedbytheCR-50monoclonalantibody.JNeurosci17,23-31.
Danzer,K.M.,Ruf,W.P.,Putcha,P.,Joyner,D.,Hashimoto,T.,Glabe,C.,Hyman,B.T.,andMcLean,P.J.(2011).Heat-shockprotein70modulatestoxicextracellularalpha-synucleinoligomersandrescuestrans-synaptictoxicity.FASEBJ25,326-336.
Das, V., and Miller, J.H. (2012). Microtubule stabilization by peloruside A and paclitaxel rescuesdegeneratingneuronsfromokadaicacid-inducedtauphosphorylation.EurJNeurosci35,1705-1717.
David,D.C., Layfield, R., Serpell, L.,Narain, Y.,Goedert,M., and Spillantini,M.G. (2002). Proteasomaldegradationoftauprotein.JNeurochem83,176-185.
de Calignon, A., Fox, L.M., Pitstick, R., Carlson, G.A., Bacskai, B.J., Spires-Jones, T.L., andHyman, B.T.(2010).Caspaseactivationprecedesandleadstotangles.Nature464,1201-1204.
DeStrooper,B.,andAnnaert,W.(2010).Novelresearchhorizonsforpresenilinsandgamma-secretasesincellbiologyanddisease.AnnuRevCellDevBiol26,235-260.
DeStrooper,B.,Annaert,W.,Cupers,P.,Saftig,P.,Craessaerts,K.,Mumm,J.S.,Schroeter,E.H.,Schrijvers,V., Wolfe, M.S., Ray, W.J., et al. (1999). A presenilin-1-dependent gamma-secretase-like proteasemediatesreleaseofNotchintracellulardomain[seecomments].Nature398,518-522.
DeVos,K.J.,Chapman,A.L.,Tennant,M.E.,Manser,C.,Tudor,E.L.,Lau,K.F.,Brownlees,J.,Ackerley,S.,Shaw,P.J.,McLoughlin,D.M.,etal. (2007).Familialamyotrophic lateralsclerosis-linkedSOD1mutantsperturbfastaxonaltransporttoreduceaxonalmitochondriacontent.HumMolGenet16,2720-2728.
De Vos, K.J., Grierson, A.J., Ackerley, S., and Miller, C.C. (2008). Role of axonal transport inneurodegenerativediseases.AnnuRevNeurosci31,151-173.
DeArmond,S.J.,McKinley,M.P.,Barry,R.A.,Braunfeld,M.B.,McColloch,J.R.,andPrusiner,S.B.(1985).Identificationofprionamyloidfilamentsinscrapie-infectedbrain.Cell41,221-235.
Del Campo,M., Oliveira, C.R., Scheper,W., Zwart, R., Korth, C., Muller-Schiffmann, A., Kostallas, G.,Biverstal,H.,Presto,J.,Johansson,J.,etal.(2015).BRI2ectodomainaffectsAbeta42fibrillationandtautruncationinhumanneuroblastomacells.CellMolLifeSci72,1599-1611.
Delobel,P.,Lavenir,I.,Fraser,G.,Ingram,E.,Holzer,M.,Ghetti,B.,Spillantini,M.G.,Crowther,R.A.,andGoedert, M. (2008). Analysis of tau phosphorylation and truncation in a mouse model of humantauopathy.AmJPathol172,123-131.
Diepenbroek,M.,Casadei,N.,Esmer,H.,Saido,T.C.,Takano,J.,Kahle,P.J.,Nixon,R.A.,Rao,M.V.,Melki,R.,Pieri,L.,etal.(2014).Overexpressionofthecalpain-specificinhibitorcalpastatinreduceshumanalpha-Synucleinprocessing,aggregationandsynapticimpairmentin[A30P]alphaSyntransgenicmice.HumMolGenet23,3975-3989.
Dixit,R.,Ross,J.L.,Goldman,Y.E.,andHolzbaur,E.L.(2008).Differentialregulationofdyneinandkinesinmotorproteinsbytau.Science319,1086-1089.
Dorval,V.,andFraser,P.E.(2006).Smallubiquitin-likemodifier(SUMO)modificationofnativelyunfoldedproteinstauandalpha-synuclein.JBiolChem281,9919-9924.
Dubey,M.,Chaudhury,P.,Kabiru,H.,andShea,T.B.(2008).Tauinhibitsanterogradeaxonaltransportandperturbsstabilityingrowingaxonalneuritesinpartbydisplacingkinesincargo:neurofilamentsattenuatetau-mediatedneuriteinstability.CellMotilCytoskeleton65,89-99.
Dufty,B.M.,Warner,L.R.,Hou,S.T.,Jiang,S.X.,Gomez-Isla,T.,Leenhouts,K.M.,Oxford,J.T.,Feany,M.B.,Masliah,E.,andRohn,T.T.(2007).Calpain-cleavageofalpha-synuclein:connectingproteolyticprocessingtodisease-linkedaggregation.AmJPathol170,1725-1738.

References
120
Duyckaerts, C., Potier, M.C., and Delatour, B. (2008). Alzheimer disease models and humanneuropathology:similaritiesanddifferences.ActaNeuropathol115,5-38.
Egashira, M., Takase, H., Yamamoto, I., Tanaka, M., and Saito, H. (2011). Identification of regionsresponsibleforheparin-inducedamyloidogenesisofhumanserumamyloidAusingitsfragmentpeptides.ArchBiochemBiophys511,101-106.
Eisenberg,D.,andJucker,M.(2012).Theamyloidstateofproteins inhumandiseases.Cell148,1188-1203.
El-Agnaf,O.M.,Nagala, S.,Patel,B.P., andAusten,B.M. (2001).Non-fibrillaroligomeric speciesof theamyloidABripeptide,implicatedinfamilialBritishdementia,aremorepotentatinducingapoptoticcelldeaththanprotofibrilsormaturefibrils.JMolBiol310,157-168.
Emanuele,M.,andChieregatti,E.(2015).Mechanismsofalpha-synucleinactiononneurotransmission:cell-autonomousandnon-cellautonomousrole.Biomolecules5,865-892.
Fan,H.C.,Ho,L.I.,Chi,C.S.,Chen,S.J.,Peng,G.S.,Chan,T.M.,Lin,S.Z.,andHarn,H.J.(2014).Polyglutamine(PolyQ)diseases:geneticstotreatments.CellTransplant23,441-458.
Fasulo,L.,Ugolini,G.,andCattaneo,A.(2005).Apoptoticeffectofcaspase-3cleavedtauinhippocampalneuronsanditspotentiationbytauFTDP-mutationN279K.JAlzheimersDis7,3-13.
Ferreira, A., and Bigio, E.H. (2011). Calpain-mediated tau cleavage: a mechanism leading toneurodegenerationsharedbymultipletauopathies.MolMed17,676-685.
Filipcik,P.,Zilka,N.,Bugos,O.,Kucerak,J.,Koson,P.,Novak,P.,andNovak,M.(2012).Firsttransgenicratmodeldevelopingprogressivecorticalneurofibrillarytangles.NeurobiolAging33,1448-1456.
Forno,L.S.(1996).NeuropathologyofParkinson'sdisease.JNeuropatholExpNeurol55,259-272.
Frappier, T.F., Georgieff, I.S., Brown, K., and Shelanski, M.L. (1994). tau Regulation of microtubule-microtubulespacingandbundling.JNeurochem63,2288-2294.
Fritschi, S.K., Langer, F.,Kaeser, S.A.,Maia, L.F., Portelius, E., Pinotsi,D.,Kaminski,C.F.,Winkler,D.T.,Maetzler,W.,Keyvani,K.,etal. (2014).Highlypotentsolubleamyloid-betaseeds inhumanAlzheimerbrainbutnotcerebrospinalfluid.Brain137,2909-2915.
Furukawa,Y.,Kaneko,K.,andNukina,N. (2011).MolecularpropertiesofTARDNAbindingprotein-43fragmentsaredependentuponitscleavagesite.BiochimBiophysActa1812,1577-1583.
Gallyas,F.(1971).SilverstainingofAlzheimer'sneurofibrillarychangesbymeansofphysicaldevelopment.ActaMorpholAcadSciHung19,1-8.
Gamblin,T.C.,Chen,F.,Zambrano,A.,Abraha,A.,Lagalwar,S.,Guillozet,A.L.,Lu,M.,Fu,Y.,Garcia-Sierra,F., LaPointe, N., et al. (2003). Caspase cleavage of tau: linking amyloid and neurofibrillary tangles inAlzheimer'sdisease.ProcNatlAcadSciUSA100,10032-10037.
Garringer, H.J., Murrell, J., Sammeta, N., Gnezda, A., Ghetti, B., and Vidal, R. (2013). Increased tauphosphorylationandtautruncation,anddecreasedsynaptophysinlevelsinmutantBRI2/tautransgenicmice.PLoSOne8,e56426.
Gauthier-Kemper,A.,Weissmann,C.,Golovyashkina,N.,Sebo-Lemke,Z.,Drewes,G.,Gerke,V.,Heinisch,J.J.,andBrandt,R.(2011).ThefrontotemporaldementiamutationR406Wblockstau'sinteractionwiththemembraneinanannexinA2-dependentmanner.JCellBiol192,647-661.
Gerson,J.E.,andKayed,R.(2013).Formationandpropagationoftauoligomericseeds.FrontNeurol4,93.
Gerson, J.E., Sengupta, U., Lasagna-Reeves, C.A., Guerrero-Munoz, M.J., Troncoso, J., and Kayed, R.(2014). Characterization of tau oligomeric seeds in progressive supranuclear palsy. Acta NeuropatholCommun2,73.
Ghazi-Noori,S.,Froud,K.E.,Mizielinska,S.,Powell,C.,Smidak,M.,FernandezdeMarco,M.,O'Malley,C.,Farmer,M.,Parkinson,N.,Fisher,E.M.,etal.(2012).ProgressiveneuronalinclusionformationandaxonaldegenerationinCHMP2Bmutanttransgenicmice.Brain135,819-832.

References
121
Ghidoni,R.,Benussi,L.,Glionna,M.,Franzoni,M.,andBinetti,G.(2008).Lowplasmaprogranulinlevelspredictprogranulinmutationsinfrontotemporallobardegeneration.Neurology71,1235-1239.
Ghiso,J.,Holton,J.,Miravalle,L.,Calero,M.,Lashley,T.,Vidal,R.,Houlden,H.,Wood,N.,Neubert,T.,Rostagno,A.,etal.(2001).SystemicamyloiddepositsinfamilialBritishdementia.JBiolChem13,13.
Giannakopoulos,P.,vonGunten,A.,Kovari,E.,Gold,G.,Herrmann,F.R.,Hof,P.R.,andBouras,C.(2007).Stereologicalanalysisofneuropilthreadsinthehippocampalformation:relationshipswithAlzheimer'sdiseaseneuronalpathologyandcognition.NeuropatholApplNeurobiol33,334-343.
Giasson,B.I.,Duda,J.E.,Quinn,S.M.,Zhang,B.,Trojanowski,J.Q.,andLee,V.M.(2002).Neuronalalpha-synucleinopathywithseveremovementdisorderinmiceexpressingA53Thumanalpha-synuclein.Neuron34,521-533.
Goate,A., Chartier-Harlin,M.C.,Mullan,M., Brown, J., Crawford, F., Fidani, L.,Giuffra, L.,Haynes,A.,Irving,N.,James,L.,etal.(1991).SegregationofamissensemutationintheamyloidprecursorproteingenewithfamilialAlzheimer'sdisease.Nature349,704-706.
Goedert,M.(2015).NEURODEGENERATION.Alzheimer'sandParkinson'sdiseases:TheprionconceptinrelationtoassembledAbeta,tau,andalpha-synuclein.Science349,1255555.
Goedert,M.,Spillantini,M.G.,Cairns,N.J.,andCrowther,R.A.(1992).TauproteinsofAlzheimerpairedhelicalfilaments:abnormalphosphorylationofallsixbrainisoforms.Neuron8,159-168.
Goedert,M.,Spillantini,M.G.,Jakes,R.,Rutherford,D.,andCrowther,R.A.(1989).Multipleisoformsofhuman microtubule-associated protein tau: sequences and localization in neurofibrillary tangles ofAlzheimer'sdisease.Neuron3,519-526.
Goedert,M.,Wischik,C.M.,Crowther,R.A.,Walker,J.E.,andKlug,A.(1988).CloningandsequencingofthecDNAencodingacoreproteinofthepairedhelicalfilamentofAlzheimerdisease:identificationasthemicrotubule-associatedproteintau.ProcNatlAcadSciUSA85,4051-4055.
Gomez-Isla,T.,Hollister,R.,West,H.,Mui,S.,Growdon,J.H.,Petersen,R.C.,Parisi,J.E.,andHyman,B.T.(1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. AnnNeurol41,17-24.
Gonatas, N.K., Stieber, A., and Gonatas, J.O. (2006). Fragmentation of the Golgi apparatus inneurodegenerativediseasesandcelldeath.JNeurolSci246,21-30.
Gong,C.X.,Liu,F.,Grundke-Iqbal,I.,andIqbal,K.(2005).Post-translationalmodificationsoftauproteininAlzheimer'sdisease.JNeuralTransm112,813-838.
Gong,C.X.,Singh,T.J.,Grundke-Iqbal, I.,andIqbal,K.(1993).PhosphoproteinphosphataseactivitiesinAlzheimerdiseasebrain.JNeurochem61,921-927.
Goode, B.L., Denis, P.E., Panda, D., Radeke, M.J., Miller, H.P., Wilson, L., and Feinstein, S.C. (1997).Functionalinteractionsbetweentheproline-richandrepeatregionsoftauenhancemicrotubulebindingandassembly.MolBiolCell8,353-365.
Gotz, J., Probst, A., Spillantini, M.G., Schafer, T., Jakes, R., Burki, K., and Goedert, M. (1995).Somatodendriticlocalizationandhyperphosphorylationoftauproteinintransgenicmiceexpressingthelongesthumanbraintauisoform.EmboJ14,1304-1313.
Gotz,J.,Streffer,J.R.,David,D.,Schild,A.,Hoerndli,F.,Pennanen,L.,Kurosinski,P.,andChen,F.(2004).Transgenic animalmodels of Alzheimer's disease and related disorders: histopathology, behavior andtherapy.MolPsychiatry9,664-683.
Graham,R.K.,Deng,Y., Slow,E.J.,Haigh,B.,Bissada,N., Lu,G.,Pearson, J., Shehadeh, J.,Bertram, L.,Murphy, Z., et al. (2006). Cleavage at the caspase-6 site is required for neuronal dysfunction anddegenerationduetomutanthuntingtin.Cell125,1179-1191.
Gregori,L.,Fuchs,C.,Figueiredo-Pereira,M.E.,VanNostrand,W.E.,andGoldgaber,D.(1995).Amyloidbeta-proteininhibitsubiquitin-dependentproteindegradationinvitro.JBiolChem270,19702-19708.
Grundke-Iqbal,I.,Iqbal,K.,Tung,Y.C.,Quinlan,M.,Wisniewski,H.M.,andBinder,L.I.(1986).Abnormalphosphorylationofthemicrotubule-associatedproteintau(tau)inAlzheimercytoskeletalpathology.ProcNatlAcadSciUSA83,4913-4917.

References
122
Guillot-Sestier,M.V., Sunyach,C.,Druon,C., Scarzello, S., andChecler, F. (2009).Thealpha-secretase-derivedN-terminalproductofcellularprion,N1,displaysneuroprotectivefunctioninvitroandinvivo.JBiolChem284,35973-35986.
Guillozet-Bongaarts,A.L.,Garcia-Sierra,F.,Reynolds,M.R.,Horowitz,P.M.,Fu,Y.,Wang,T.,Cahill,M.E.,Bigio,E.H.,Berry,R.W.,andBinder,L.I.(2005).TautruncationduringneurofibrillarytangleevolutioninAlzheimer'sdisease.NeurobiolAging26,1015-1022.
Guo,H.,Albrecht,S.,Bourdeau,M.,Petzke,T.,Bergeron,C.,andLeBlanc,A.C.(2004).Activecaspase-6andcaspase-6-cleavedtauinneuropilthreads,neuriticplaques,andneurofibrillarytanglesofAlzheimer'sdisease.AmJPathol165,523-531.
Guyenet,S.J.,Mookerjee,S.S.,Lin,A.,Custer,S.K.,Chen,S.F.,Sopher,B.L.,LaSpada,A.R.,andEllerby,L.M. (2015). Proteolytic cleavage of ataxin-7 promotes SCA7 retinal degeneration and neurologicaldysfunction.HumMolGenet24,3908-3917.
Haacke,A.,Hartl,F.U.,andBreuer,P.(2007).Calpaininhibitionissufficienttosuppressaggregationofpolyglutamine-expandedataxin-3.JBiolChem282,18851-18856.
Haass,C.,Hung,A.Y.,Selkoe,D.J.,andTeplow,D.B.(1994).MutationsassociatedwithalocusforfamilialAlzheimer'sdiseaseresultinalternativeprocessingofamyloidbeta-proteinprecursor.JBiolChem269,17741-17748.
Hammerschlag,R.,Stone,G.C.,Bolen,F.A.,Lindsey,J.D.,andEllisman,M.H.(1982).EvidencethatallnewlysynthesizedproteinsdestinedforfastaxonaltransportpassthroughtheGolgiapparatus.JCellBiol93,568-575.
Hanger, D.P., and Wray, S. (2010). Tau cleavage and tau aggregation in neurodegenerative disease.BiochemSocTrans38,1016-1020.
Hara,T.,Nakamura,K.,Matsui,M.,Yamamoto,A.,Nakahara,Y., Suzuki-Migishima,R.,Yokoyama,M.,Mishima, K., Saito, I., Okano, H., et al. (2006). Suppression of basal autophagy in neural cells causesneurodegenerativediseaseinmice.Nature441,885-889.
Harada,A.,Takei,Y.,Kanai,Y.,Tanaka,Y.,Nonaka,S.,andHirokawa,N. (1998).Golgivesiculationandlysosomedispersionincellslackingcytoplasmicdynein.JCellBiol141,51-59.
Harry,G.J.,Lefebvred'Hellencourt,C.,Bruccoleri,A.,andSchmechel,D.(2000).Age-dependentcytokineresponses:trimethyltinhippocampalinjuryinwild-type,APOEknockout,andAPOE4mice.BrainBehavImmun14,288-304.
Higuchi,M., Iwata,N.,Matsuba,Y.,Takano,J.,Suemoto,T.,Maeda,J.,Ji,B.,Ono,M.,Staufenbiel,M.,Suhara, T., et al. (2012). Mechanistic involvement of the calpain-calpastatin system in Alzheimerneuropathology.FASEBJ26,1204-1217.
Hirai,K.,Aliev,G.,Nunomura,A.,Fujioka,H.,Russell,R.L.,Atwood,C.S.,Johnson,A.B.,Kress,Y.,Vinters,H.V.,Tabaton,M.,etal.(2001).MitochondrialabnormalitiesinAlzheimer'sdisease.JNeurosci21,3017-3023.
Hollenbeck,P.J.,andSaxton,W.M. (2005).Theaxonal transportofmitochondria. JCellSci118,5411-5419.
Horowitz,P.M.,Patterson,K.R.,Guillozet-Bongaarts,A.L.,Reynolds,M.R.,Carroll,C.A.,Weintraub,S.T.,Bennett,D.A.,Cryns,V.L.,Berry,R.W.,andBinder,L.I. (2004).EarlyN-terminalchangesandcaspase-6cleavageoftauinAlzheimer'sdisease.JNeurosci24,7895-7902.
Hubener, J., Vauti, F., Funke, C., Wolburg, H., Ye, Y., Schmidt, T., Wolburg-Buchholz, K., Schmitt, I.,Gardyan,A.,Driessen,S.,etal.(2011).N-terminalataxin-3causesneurologicalsymptomswithinclusions,endoplasmicreticulumstressandribosomaldislocation.Brain134,1925-1942.
Hubener,J.,Weber,J.J.,Richter,C.,Honold,L.,Weiss,A.,Murad,F.,Breuer,P.,Wullner,U.,Bellstedt,P.,Paquet-Durand,F., etal. (2013).Calpain-mediatedataxin-3cleavage in themolecularpathogenesisofspinocerebellarataxiatype3(SCA3).HumMolGenet22,508-518.

References
123
Hussain, I., Powell, D., Howlett, D.R., Tew, D.G.,Meek, T.D., Chapman, C., Gloger, I.S.,Murphy, K.E.,Southan, C.D., Ryan, D.M., et al. (1999). Identification of a novel aspartic protease (Asp 2) as beta-secretase.MolCellNeurosci14,419-427.
Hutton,M.,Lendon,C.L.,Rizzu,P.,Baker,M.,Froelich,S.,Houlden,H.,Pickering-Brown,S.,Chakraverty,S.,Isaacs,A.,Grover,A.,etal.(1998).Associationofmissenseand5'-splice-sitemutationsintauwiththeinheriteddementiaFTDP-17.Nature393,702-705.
Igaz,L.M.,Kwong,L.K.,Chen-Plotkin,A.,Winton,M.J.,Unger,T.L.,Xu,Y.,Neumann,M.,Trojanowski,J.Q.,and Lee, V.M. (2009). Expression of TDP-43 C-terminal Fragments in Vitro Recapitulates PathologicalFeaturesofTDP-43Proteinopathies.JBiolChem284,8516-8524.
Ihse,E.,Rapezzi,C.,Merlini,G.,Benson,M.D.,Ando,Y.,Suhr,O.B.,Ikeda,S.,Lavatelli,F.,Obici,L.,Quarta,C.C.,etal.(2013).AmyloidfibrilscontainingfragmentedATTRmaybethestandardfibrilcompositioninATTRamyloidosis.Amyloid20,142-150.
Ishihara,T.,Hong,M.,Zhang,B.,Nakagawa,Y.,Lee,M.K.,Trojanowski,J.Q.,andLee,V.M.(1999).Age-dependentemergenceandprogressionofa tauopathy in transgenicmiceoverexpressing theshortesthumantauisoform.Neuron24,751-762.
Ittner,L.M.,Ke,Y.D.,Delerue,F.,Bi,M.,Gladbach,A.,vanEersel,J.,Wolfing,H.,Chieng,B.C.,Christie,M.J.,Napier, I.A.,etal. (2010).Dendritic functionof taumediatesamyloid-betatoxicity inAlzheimer'sdiseasemousemodels.Cell142,387-397.
Jancsik,V.,Filliol,D.,Felter,S.,andRendon,A.(1989).Bindingofmicrotubule-associatedproteins(MAPs)to rat brainmitochondria: a comparative study of the binding ofMAP2, itsmicrotubule-binding andprojectiondomains,andtauproteins.CellMotilCytoskeleton14,372-381.
Jucker, M., and Walker, L.C. (2013). Self-propagation of pathogenic protein aggregates inneurodegenerativediseases.Nature501,45-51.
Jung, J., Xu, K., Lessing, D., and Bonini, N.M. (2009). Preventing Ataxin-3 protein cleavage mitigatesdegenerationinaDrosophilamodelofSCA3.HumMolGenet18,4843-4852.
Kampers, T., Friedhoff, P., Biernat, J., Mandelkow, E.M., and Mandelkow, E. (1996). RNA stimulatesaggregationofmicrotubule-associatedproteintauintoAlzheimer-likepairedhelicalfilaments.FEBSLett399,344-349.
Kanda, S., Bishop, J.F., Eglitis,M.A., Yang, Y., andMouradian,M.M. (2000). Enhanced vulnerability tooxidativestressbyalpha-synucleinmutationsandC-terminaltruncation.Neuroscience97,279-284.
Kane,M.D.,Lipinski,W.J.,Callahan,M.J.,Bian,F.,Durham,R.A.,Schwarz,R.D.,Roher,A.E.,andWalker,L.C.(2000).Evidenceforseedingofbeta-amyloidbyintracerebralinfusionofAlzheimerbrainextractsinbeta-amyloidprecursorprotein-transgenicmice.JNeurosci20,3606-3611.
Kang, J., Lemaire, H.G., Unterbeck, A., Salbaum, J.M., Masters, C.L., Grzeschik, K.H., Multhaup, G.,Beyreuther, K., and Muller-Hill, B. (1987). The precursor of Alzheimer's disease amyloid A4 proteinresemblesacell-surfacereceptor.Nature325,733-736.
Karsten, S.L., Sang, T.K.,Gehman, L.T., Chatterjee, S., Liu, J., Lawless,G.M., Sengupta, S., Berry, R.W.,Pomakian,J.,Oh,H.S.,etal.(2006).Agenomicscreenformodifiersoftauopathyidentifiespuromycin-sensitiveaminopeptidaseasaninhibitoroftau-inducedneurodegeneration.Neuron51,549-560.
Keck,S.,Nitsch,R.,Grune,T.,andUllrich,O.(2003).Proteasomeinhibitionbypairedhelicalfilament-tauinbrainsofpatientswithAlzheimer'sdisease.JNeurochem85,115-122.
Keller, J.N., Hanni, K.B., and Markesbery, W.R. (2000). Impaired proteasome function in Alzheimer'sdisease.JNeurochem75,436-439.
Kenessey, A., Nacharaju, P., Ko, L.W., and Yen, S.H. (1997). Degradation of tau by lysosomal enzymecathepsinD:implicationforAlzheimerneurofibrillarydegeneration.JNeurochem69,2026-2038.
Kent, L.,Vizard,T.N., Smith,B.N.,Topp,S.D.,Vance,C.,Gkazi,A.,Miller, J., Shaw,C.E., andTalbot,K.(2014). Autosomal dominant inheritance of rapidly progressive amyotrophic lateral sclerosis due to atruncationmutationinthefusedinsarcoma(FUS)gene.AmyotrophLateralSclerFrontotemporalDegener15,557-562.

References
124
Kessler,J.C.,Rochet,J.C.,andLansbury,P.T.,Jr.(2003).TheN-terminalrepeatdomainofalpha-synucleininhibitsbeta-sheetandamyloidfibrilformation.Biochemistry42,672-678.
Kim, K.S., Choi, Y.R., Park, J.Y., Lee, J.H., Kim, D.K., Lee, S.J., Paik, S.R., Jou, I., and Park, S.M. (2012).Proteolyticcleavageofextracellularalpha-synucleinbyplasmin:implicationsforParkinsondisease.JBiolChem287,24862-24872.
Kim, P.K., Hailey, D.W.,Mullen, R.T., and Lippincott-Schwartz, J. (2008). Ubiquitin signals autophagicdegradationofcytosolicproteinsandperoxisomes.ProcNatlAcadSciUSA105,20567-20574.
Kim,S.H.,Wang,R.,Gordon,D.J.,Bass,J.,Steiner,D.F.,Lynn,D.G.,Thinakaran,G.,Meredith,S.C.,andSisodia,S.S.(1999).FurinmediatesenhancedproductionoffibrillogenicABripeptidesinfamilialBritishdementia.NatNeurosci2,984-988.
Kim,Y.J.,Yi,Y.,Sapp,E.,Wang,Y.,Cuiffo,B.,Kegel,K.B.,Qin,Z.H.,Aronin,N.,andDiFiglia,M. (2001).Caspase3-cleavedN-terminalfragmentsofwild-typeandmutanthuntingtinarepresentinnormalandHuntington'sdiseasebrains,associatewithmembranes,andundergocalpain-dependentproteolysis.ProcNatlAcadSciUSA98,12784-12789.
Komatsu,M.,Waguri, S., Chiba, T.,Murata, S., Iwata, J., Tanida, I.,Ueno, T., Koike,M.,Uchiyama, Y.,Kominami,E.,etal.(2006).Lossofautophagyinthecentralnervoussystemcausesneurodegenerationinmice.Nature441,880-884.
Kondo,J.,Honda,T.,Mori,H.,Hamada,Y.,Miura,R.,Ogawara,M.,andIhara,Y.(1988).Thecarboxylthirdoftauistightlyboundtopairedhelicalfilaments.Neuron1,827-834.
Kopeikina,K.J.,Carlson,G.A.,Pitstick,R.,Ludvigson,A.E.,Peters,A.,Luebke,J.I.,Koffie,R.M.,Frosch,M.P.,Hyman,B.T.,andSpires-Jones,T.L.(2011).TauaccumulationcausesmitochondrialdistributiondeficitsinneuronsinamousemodeloftauopathyandinhumanAlzheimer'sdiseasebrain.AmJPathol179,2071-2082.
Kosik,K.S.(1993).Themolecularandcellularbiologyoftau.BrainPathol3,39-43.
Kosik, K.S., Joachim, C.L., and Selkoe,D.J. (1986).Microtubule-associated protein tau (tau) is amajorantigeniccomponentofpairedhelicalfilamentsinAlzheimerdisease.ProcNatlAcadSciUSA83,4044-4048.
Ksiezak-Reding,H.,Liu,W.K.,andYen,S.H.(1992).Phosphateanalysisanddephosphorylationofmodifiedtauassociatedwithpairedhelicalfilaments.BrainRes597,209-219.
Kudo,L.C.,Parfenova,L.,Ren,G.,Vi,N.,Hui,M.,Ma,Z.,Lau,K.,Gray,M.,Bardag-Gorce,F.,Wiedau-Pazos,M., et al. (2011). Puromycin-sensitive aminopeptidase (PSA/NPEPPS) impedes development ofneuropathologyinhPSA/TAU(P301L)double-transgenicmice.HumMolGenet20,1820-1833.
Kwiatkowski, T.J., Jr., Bosco, D.A., Leclerc, A.L., Tamrazian, E., Vanderburg, C.R., Russ, C., Davis, A.,Gilchrist,J.,Kasarskis,E.J.,Munsat,T.,etal.(2009).MutationsintheFUS/TLSgeneonchromosome16causefamilialamyotrophiclateralsclerosis.Science323,1205-1208.
Landau,S.M.,Horng,A.,Fero,A.,Jagust,W.J.,andAlzheimer'sDiseaseNeuroimaging,I.(2016).AmyloidnegativityinpatientswithclinicallydiagnosedAlzheimerdiseaseandMCI.Neurology.
Langer,F.,Eisele,Y.S.,Fritschi,S.K.,Staufenbiel,M.,Walker,L.C.,andJucker,M.(2011).SolubleAbetaseedsarepotentinducersofcerebralbeta-amyloiddeposition.JNeurosci31,14488-14495.
Larson,M.E.,andLesne,S.E.(2012).SolubleAbetaoligomerproductionandtoxicity.JNeurochem120Suppl1,125-139.
Lasagna-Reeves,C.A.,Castillo-Carranza,D.L.,Guerrero-Muoz,M.J.,Jackson,G.R.,andKayed,R.(2010).Preparationandcharacterizationofneurotoxictauoligomers.Biochemistry49,10039-10041.
Law,W.J.,Cann,K.L.,andHicks,G.G.(2006).TLS,EWSandTAF15:amodelfortranscriptionalintegrationofgeneexpression.BriefFunctGenomicProteomic5,8-14.
Leavitt,B.R.,Guttman,J.A.,Hodgson,J.G.,Kimel,G.H.,Singaraja,R.,Vogl,A.W.,andHayden,M.R.(2001).Wild-typehuntingtinreducesthecellulartoxicityofmutanthuntingtininvivo.AmJHumGenet68,313-324.

References
125
Leavitt,B.R.,vanRaamsdonk,J.M.,Shehadeh,J.,Fernandes,H.,Murphy,Z.,Graham,R.K.,Wellington,C.L.,Raymond,L.A.,andHayden,M.R.(2006).Wild-typehuntingtinprotectsneuronsfromexcitotoxicity.JNeurochem96,1121-1129.
Ledesma,M.D.,Bonay,P.,Colaco,C.,andAvila,J.(1994).Analysisofmicrotubule-associatedproteintauglycationinpairedhelicalfilaments.JBiolChem269,21614-21619.
Lee,B.H.,Lee,M.J.,Park,S.,Oh,D.C.,Elsasser,S.,Chen,P.C.,Gartner,C.,Dimova,N.,Hanna,J.,Gygi,S.P.,etal.(2010).Enhancementofproteasomeactivitybyasmall-moleculeinhibitorofUSP14.Nature467,179-184.
Lee,G.,Cowan,N.,andKirschner,M.(1988).Theprimarystructureandheterogeneityoftauproteinfrommousebrain.Science239,285-288.
Lee,G.,Neve,R.L.,andKosik,K.S.(1989).Themicrotubulebindingdomainoftauprotein.Neuron2,1615-1624.
Lee,G.,Newman,S.T.,Gard,D.L.,Band,H.,andPanchamoorthy,G.(1998).Tauinteractswithsrc-familynon-receptortyrosinekinases.JCellSci111(Pt21),3167-3177.
Leger,M., Quiedeville, A., Bouet, V., Haelewyn, B., Boulouard,M., Schumann-Bard, P., and Freret, T.(2013).Objectrecognitiontestinmice.NatProtoc8,2531-2537.
Lesne,S.E.,Sherman,M.A.,Grant,M.,Kuskowski,M.,Schneider,J.A.,Bennett,D.A.,andAshe,K.H.(2013).Brainamyloid-betaoligomersinageingandAlzheimer'sdisease.Brain136,1383-1398.
Levin,J.,Giese,A.,Boetzel,K.,Israel,L.,Hogen,T.,Nubling,G.,Kretzschmar,H.,andLorenzl,S.(2009).Increasedalpha-synucleinaggregationfollowinglimitedcleavagebycertainmatrixmetalloproteinases.ExpNeurol215,201-208.
Lewis,V.,Hill,A.F.,Haigh,C.L.,Klug,G.M.,Masters,C.L.,Lawson,V.A.,andCollins,S.J.(2009).IncreasedproportionsofC1truncatedprionproteinprotectagainstcellularM1000prioninfection.JNeuropatholExpNeurol68,1125-1135.
Li,Q.,Yokoshi,M.,Okada,H.,andKawahara,Y.(2015).ThecleavagepatternofTDP-43determinesitsrateofclearanceandcytotoxicity.NatCommun6,6183.
Li,W.,West,N.,Colla,E.,Pletnikova,O.,Troncoso,J.C.,Marsh,L.,Dawson,T.M.,Jakala,P.,Hartmann,T.,Price, D.L., et al. (2005). Aggregation promoting C-terminal truncation of alpha-synuclein is a normalcellularprocessandisenhancedbythefamilialParkinson'sdisease-linkedmutations.ProcNatlAcadSciUSA102,2162-2167.
Liazoghli,D.,Perreault,S.,Micheva,K.D.,Desjardins,M.,andLeclerc,N. (2005).Fragmentationof theGolgiapparatusinducedbytheoverexpressionofwild-typeandmutanthumantauformsinneurons.AmJPathol166,1499-1514.
Liu,C.W.,Giasson,B.I.,Lewis,K.A.,Lee,V.M.,Demartino,G.N.,andThomas,P.J.(2005).Aprecipitatingrolefortruncatedalpha-synucleinandtheproteasomeinalpha-synucleinaggregation:implicationsforpathogenesisofParkinsondisease.JBiolChem280,22670-22678.
Loomis,P.A.,Howard,T.H.,Castleberry,R.P.,andBinder,L.I.(1990).Identificationofnucleartauisoformsinhumanneuroblastomacells.ProcNatlAcadSciUSA87,8422-8426.
Lustbader, J.W., Cirilli,M., Lin, C., Xu,H.W., Takuma,K.,Wang,N., Caspersen,C., Chen, X., Pollak, S.,Chaney,M., et al. (2004). ABAD directly links Abeta tomitochondrial toxicity in Alzheimer's disease.Science304,448-452.
Ma,Q.L.,Zuo,X.,Yang,F.,Ubeda,O.J.,Gant,D.J.,Alaverdyan,M.,Teng,E.,Hu,S.,Chen,P.P.,Maiti,P.,etal. (2013). Curcumin suppresses soluble tau dimers and correctsmolecular chaperone, synaptic, andbehavioraldeficitsinagedhumantautransgenicmice.JBiolChem288,4056-4065.
Maia,L.F.,Kaeser,S.A.,Reichwald, J.,Hruscha,M.,Martus,P.,Staufenbiel,M.,and Jucker,M. (2013).ChangesinAmyloid-betaandTauintheCerebrospinalFluidofTransgenicMiceOverexpressingAmyloidPrecursorProtein.SciTranslMed5,194re192.
Mandybur, T.I. (1986). Cerebral amyloid angiopathy: the vascular pathology and complications. JNeuropatholExpNeurol45,79-90.

References
126
Mangiarini,L.,Sathasivam,K.,Seller,M.,Cozens,B.,Harper,A.,Hetherington,C.,Lawton,M.,Trottier,Y.,Lehrach,H.,Davies,S.W.,etal.(1996).Exon1oftheHDgenewithanexpandedCAGrepeatissufficienttocauseaprogressiveneurologicalphenotypeintransgenicmice.Cell87,493-506.
Marchetti,L.,Klein,M.,Schlett,K.,Pfizenmaier,K.,andEisel,U.L.(2004).Tumornecrosisfactor(TNF)-mediatedneuroprotectionagainstglutamate-inducedexcitotoxicityisenhancedbyN-methyl-D-aspartatereceptoractivation.EssentialroleofaTNFreceptor2-mediatedphosphatidylinositol3-kinase-dependentNF-kappaBpathway.JBiolChem279,32869-32881.
Marcora,M.S.,Fernandez-Gamba,A.C.,Avendano,L.A.,Rotondaro,C.,Podhajcer,O.L.,Vidal,R.,Morelli,L., Ceriani, M.F., and Castano, E.M. (2014). Amyloid peptides ABri and ADan show differentialneurotoxicityintransgenicDrosophilamodelsoffamilialBritishandDanishdementia.MolNeurodegener9,5.
Margolis, R.L., and Ross, C.A. (2001). Expansion explosion: new clues to the pathogenesis of repeatexpansionneurodegenerativediseases.TrendsMolMed7,479-482.
Martin,L.,Fluhrer,R.,Reiss,K.,Kremmer,E.,Saftig,P.,andHaass,C.(2008).RegulatedintramembraneproteolysisofBri2(Itm2b)byADAM10andSPPL2a/SPPL2b.JBiolChem283,1644-1652.
Masters, C.L., and Selkoe, D.J. (2012). Biochemistry of amyloid beta-protein and amyloid deposits inAlzheimerdisease.ColdSpringHarbPerspectMed2,a006262.
McKhann, G.M., Knopman, D.S., Chertkow, H., Hyman, B.T., Jack, C.R., Jr., Kawas, C.H., Klunk, W.E.,Koroshetz,W.J.,Manly, J.J.,Mayeux, R., et al. (2011). The diagnosis of dementia due to Alzheimer'sdisease:recommendationsfromtheNationalInstituteonAging-Alzheimer'sAssociationworkgroupsondiagnosticguidelinesforAlzheimer'sdisease.AlzheimersDement7,263-269.
McMillan, P.J., Kraemer, B.C., Robinson, L., Leverenz, J.B., Raskind, M., and Schellenberg, G. (2011).TruncationoftauatE391promotesearlypathologicchangesintransgenicmice.JNeuropatholExpNeurol70,1006-1019.
Mead,S.,James-Galton,M.,Revesz,T.,Doshi,R.B.,Harwood,G.,Pan,E.L.,Ghiso,J.,Frangione,B.,andPlant,G.(2000).FamilialBritishdementiawithamyloidangiopathy:earlyclinical,neuropsychologicalandimagingfindings.Brain123,975-991.
Medeiros,R.,Kitazawa,M.,Chabrier,M.A.,Cheng,D.,Baglietto-Vargas,D.,Kling,A.,Moeller,A.,Green,K.N.,andLaFerla,F.M. (2012).Calpain inhibitorA-705253mitigatesAlzheimer'sdisease-likepathologyandcognitivedeclineinaged3xTgADmice.AmJPathol181,616-625.
Medina,D.X.,Caccamo,A.,andOddo,S.(2011).Methylenebluereducesabetalevelsandrescuesearlycognitivedeficitbyincreasingproteasomeactivity.BrainPathol21,140-149.
Mena,R.,Edwards,P.C.,Harrington,C.R.,Mukaetova-Ladinska,E.B.,andWischik,C.M.(1996).StagingthepathologicalassemblyoftruncatedtauproteinintopairedhelicalfilamentsinAlzheimer'sdisease.ActaNeuropathol91,633-641.
Menzies,F.M.,Hourez,R.,Imarisio,S.,Raspe,M.,Sadiq,O.,Chandraratna,D.,O'Kane,C.,Rock,K.L.,Reits,E.,Goldberg,A.L.,etal.(2010).Puromycin-sensitiveaminopeptidaseprotectsagainstaggregation-proneproteinsviaautophagy.HumMolGenet19,4573-4586.
Metcalfe, M.J., Huang, Q., and Figueiredo-Pereira, M.E. (2012). Coordination between proteasomeimpairmentandcaspaseactivationleadingtoTAUpathology:neuroprotectionbycAMP.CellDeathDis3,e326.
Meyer-Luehmann, M., Coomaraswamy, J., Bolmont, T., Kaeser, S., Schaefer, C., Kilger, E.,Neuenschwander,A.,Abramowski,D.,Frey,P.,Jaton,A.L.,etal.(2006).Exogenousinductionofcerebralbeta-amyloidogenesisisgovernedbyagentandhost.Science313,1781-1784.
Miller,J.P.,Holcomb,J.,Al-Ramahi,I.,deHaro,M.,Gafni,J.,Zhang,N.,Kim,E.,Sanhueza,M.,Torcassi,C.,Kwak,S.,etal.(2010).MatrixmetalloproteinasesaremodifiersofhuntingtinproteolysisandtoxicityinHuntington'sdisease.Neuron67,199-212.
Mishizen-Eberz,A.J.,Norris,E.H.,Giasson,B.I.,Hodara,R.,Ischiropoulos,H.,Lee,V.M.,Trojanowski,J.Q.,andLynch,D.R.(2005).Cleavageofalpha-synucleinbycalpain:potentialroleindegradationoffibrillizedandnitratedspeciesofalpha-synuclein.Biochemistry44,7818-7829.

References
127
Mitchison,T.,andKirschner,M.(1984).Dynamicinstabilityofmicrotubulegrowth.Nature312,237-242.
Mizushima,N.,andKomatsu,M.(2011).Autophagy:renovationofcellsandtissues.Cell147,728-741.
Mocanu,M.M.,Nissen,A.,Eckermann,K.,Khlistunova,I.,Biernat,J.,Drexler,D.,Petrova,O.,Schonig,K.,Bujard,H.,Mandelkow,E., etal. (2008). Thepotential forbeta-structure in the repeatdomainof tauproteindeterminesaggregation,synapticdecay,neuronalloss,andcoassemblywithendogenousTauininduciblemousemodelsoftauopathy.JNeurosci28,737-748.
Mondragon-Rodriguez,S.,Basurto-Islas,G.,Santa-Maria,I.,Mena,R.,Binder,L.I.,Avila,J.,Smith,M.A.,Perry, G., and Garcia-Sierra, F. (2008). Cleavage and conformational changes of tau protein followphosphorylationduringAlzheimer'sdisease.IntJExpPathol89,81-90.
Mookerjee,S.,Papanikolaou,T.,Guyenet,S.J.,Sampath,V.,Lin,A.,Vitelli,C.,DeGiacomo,F.,Sopher,B.L.,Chen,S.F.,LaSpada,A.R.,etal.(2009).Posttranslationalmodificationofataxin-7atlysine257preventsautophagy-mediated turnover of an N-terminal caspase-7 cleavage fragment. J Neurosci 29, 15134-15144.
Mori,H.,Kondo,J.,andIhara,Y.(1987).UbiquitinisacomponentofpairedhelicalfilamentsinAlzheimer'sdisease.Science235,1641-1644.
Muntane,G.,Ferrer,I.,andMartinez-Vicente,M.(2012).alpha-synucleinphosphorylationandtruncationarenormaleventsintheadulthumanbrain.Neuroscience200,106-119.
Murphy, J.M., Henry, R.G., Langmore, S., Kramer, J.H., Miller, B.L., and Lomen-Hoerth, C. (2007).Continuumoffrontallobeimpairmentinamyotrophiclateralsclerosis.ArchNeurol64,530-534.
Murray, I.V.,Giasson,B.I.,Quinn,S.M.,Koppaka,V.,Axelsen,P.H., Ischiropoulos,H.,Trojanowski, J.Q.,andLee,V.M.(2003).Roleofalpha-synucleincarboxy-terminusonfibrilformationinvitro.Biochemistry42,8530-8540.
Nakashiba, T., Young, J.Z.,McHugh, T.J., Buhl, D.L., and Tonegawa, S. (2008). Transgenic inhibition ofsynaptictransmissionrevealsroleofCA3outputinhippocampallearning.Science319,1260-1264.
Neary,D.,Snowden,J.S.,Gustafson,L.,Passant,U.,Stuss,D.,Black,S.,Freedman,M.,Kertesz,A.,Robert,P.H., Albert,M., et al. (1998). Frontotemporal lobar degeneration: a consensus on clinical diagnosticcriteria.Neurology51,1546-1554.
Neumann,M.,Sampathu,D.M.,Kwong,L.K.,Truax,A.C.,Micsenyi,M.C.,Chou,T.T.,Bruce,J.,Schuck,T.,Grossman,M.,Clark,C.M.,etal.(2006).UbiquitinatedTDP-43infrontotemporallobardegenerationandamyotrophiclateralsclerosis.Science314,130-133.
Nixon,R.A.(2007).Autophagy,amyloidogenesisandAlzheimerdisease.JCellSci120,4081-4091.
Nixon,R.A.(2013).Theroleofautophagyinneurodegenerativedisease.NatMed19,983-997.
Nixon,R.A.,Wegiel,J.,Kumar,A.,Yu,W.H.,Peterhoff,C.,Cataldo,A.,andCuervo,A.M.(2005).Extensiveinvolvementofautophagy inAlzheimerdisease:an immuno-electronmicroscopystudy.JNeuropatholExpNeurol64,113-122.
Nonaka, T., Kametani, F., Arai, T., Akiyama,H., andHasegawa,M. (2009). Truncation and pathogenicmutationsfacilitatetheformationofintracellularaggregatesofTDP-43.HumMolGenet18,3353-3364.
Notari,S.,Strammiello,R.,Capellari,S.,Giese,A.,Cescatti,M.,Grassi,J.,Ghetti,B.,Langeveld,J.P.,Zou,W.Q., Gambetti, P., et al. (2008). Characterization of truncated forms of abnormal prion protein inCreutzfeldt-Jakobdisease.JBiolChem283,30557-30565.
Novak, M., Kabat, J., and Wischik, C.M. (1993). Molecular characterization of the minimal proteaseresistanttauunitoftheAlzheimer'sdiseasepairedhelicalfilament.EMBOJ12,365-370.
Novak,M.J.,andTabrizi,S.J. (2011).Huntington'sdisease:clinicalpresentationandtreatment. IntRevNeurobiol98,297-323.
Oddo,S.(2008).Theubiquitin-proteasomesysteminAlzheimer'sdisease.JCellMolMed12,363-373.
Oddo,S.,Caccamo,A.,Shepherd,J.D.,Murphy,M.P.,Golde,T.E.,Kayed,R.,Metherate,R.,Mattson,M.P.,Akbari, Y., and LaFerla, F.M. (2003). Triple-transgenicmodel of Alzheimer's diseasewith plaques andtangles:intracellularAbetaandsynapticdysfunction.Neuron39,409-421.

References
128
Orr,A.L.,Li,S.,Wang,C.E.,Li,H.,Wang,J.,Rong,J.,Xu,X.,Mastroberardino,P.G.,Greenamyre,J.T.,andLi, X.J. (2008). N-terminalmutant huntingtin associateswithmitochondria and impairsmitochondrialtrafficking.JNeurosci28,2783-2792.
Ossenkoppele,R.,Schonhaut,D.R.,Scholl,M.,Lockhart,S.N.,Ayakta,N.,Baker,S.L.,O'Neil,J.P.,Janabi,M.,Lazaris,A.,Cantwell,A.,etal.(2016).TauPETpatternsmirrorclinicalandneuroanatomicalvariabilityinAlzheimer'sdisease.Brain.
Ou,S.H.,Wu,F.,Harrich,D.,Garcia-Martinez,L.F.,andGaynor,R.B.(1995).Cloningandcharacterizationofanovelcellularprotein,TDP-43,thatbindstohumanimmunodeficiencyvirustype1TARDNAsequencemotifs.JVirol69,3584-3596.
Ozcelik,S.,Fraser,G.,Castets,P.,Schaeffer,V.,Skachokova,Z.,Breu,K.,Clavaguera,F.,Sinnreich,M.,Kappos,L.,Goedert,M.,etal.(2013).RapamycinAttenuatestheProgressionofTauPathologyinP301STauTransgenicMice.PLoSOne8,e62459.
Ozcelik,S.,Sprenger,F.,Skachokova,Z.,Fraser,G.,Abramowski,D.,Clavaguera,F.,Probst,A.,Frank,S.,Muller,M.,Staufenbiel,M.,etal.(2016).Co-expressionoftruncatedandfull-lengthtauinducessevereneurotoxicity.MolPsychiatry.
Palhan,V.B.,Chen,S.,Peng,G.H.,Tjernberg,A.,Gamper,A.M.,Fan,Y.,Chait,B.T.,LaSpada,A.R.,andRoeder,R.G.(2005).Polyglutamine-expandedataxin-7inhibitsSTAGAhistoneacetyltransferaseactivitytoproduceretinaldegeneration.ProcNatlAcadSciUSA102,8472-8477.
Pan, T.,Wong, P., Chang, B., Li, C., Li, R., Kang, S.C.,Wisniewski, T., and Sy,M.S. (2005). Biochemicalfingerprints of prion infection: accumulations of aberrant full-length and N-terminally truncated PrPspeciesarecommonfeaturesinmousepriondisease.JVirol79,934-943.
Panda, D., Samuel, J.C., Massie, M., Feinstein, S.C., and Wilson, L. (2003). Differential regulation ofmicrotubuledynamicsby three-and four-repeat tau: implications for theonsetofneurodegenerativedisease.ProcNatlAcadSciUSA100,9548-9553.
Parchi,P.,Castellani,R.,Capellari,S.,Ghetti,B.,Young,K.,Chen,S.G.,Farlow,M.,Dickson,D.W.,Sima,A.A., Trojanowski, J.Q., etal. (1996).Molecularbasisofphenotypic variability in sporadicCreutzfeldt-Jakobdisease.AnnNeurol39,767-778.
Parchi,P.,Chen,S.G.,Brown,P.,Zou,W.,Capellari,S.,Budka,H.,Hainfellner,J.,Reyes,P.F.,Golden,G.T.,Hauw,J.J.,etal.(1998).DifferentpatternsoftruncatedprionproteinfragmentscorrelatewithdistinctphenotypesinP102LGerstmann-Straussler-Scheinkerdisease.ProcNatlAcadSciUSA95,8322-8327.
Park, S.Y., Tournell, C., Sinjoanu, R.C., and Ferreira, A. (2007). Caspase-3- and calpain-mediated taucleavagearedifferentiallypreventedbyestrogenandtestosteroneinbeta-amyloid-treatedhippocampalneurons.Neuroscience144,119-127.
Patterson,K.R.,Remmers,C.,Fu,Y.,Brooker,S.,Kanaan,N.M.,Vana,L.,Ward,S.,Reyes,J.F.,Philibert,K.,Glucksman,M.J., et al. (2011). Characterizationofprefibrillar Tauoligomers in vitro and inAlzheimerdisease.JBiolChem286,23063-23076.
Pennuto,M.,Bonanomi,D.,Benfenati,F.,andValtorta,F.(2003).SynaptophysinIcontrolsthetargetingofVAMP2/synaptobrevinIItosynapticvesicles.MolBiolCell14,4909-4919.
Perez,M.,Santa-Maria,I.,GomezdeBarreda,E.,Zhu,X.,Cuadros,R.,Cabrero,J.R.,Sanchez-Madrid,F.,Dawson,H.N., Vitek,M.P., Perry,G., et al. (2009). Tau--an inhibitor of deacetylaseHDAC6 function. JNeurochem109,1756-1766.
Pickford, F., Masliah, E., Britschgi, M., Lucin, K., Narasimhan, R., Jaeger, P.A., Small, S., Spencer, B.,Rockenstein,E.,Levine,B.,etal.(2008).Theautophagy-relatedproteinbeclin1showsreducedexpressioninearlyAlzheimerdiseaseandregulatesamyloidbetaaccumulationinmice.JClinInvest118,2190-2199.
Piras,A.,Collin,L.,Gruninger,F.,Graff,C.,andRonnback,A.(2016).Autophagicandlysosomaldefectsinhuman tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease,corticobasaldegenerationandprogressivesupranuclearpalsy.ActaNeuropatholCommun4,22.
Poorkaj,P.,Bird,T.D.,Wijsman,E.,Nemens,E.,Garruto,R.M.,Anderson,L.,Andreadis,A.,Wiederholt,W.C., Raskind, M., and Schellenberg, G.D. (1998). Tau is a candidate gene for chromosome 17frontotemporaldementia.AnnNeurol43,815-825.

References
129
Pop,C.,andSalvesen,G.S. (2009).Humancaspases:activation,specificity,andregulation.JBiolChem284,21777-21781.
Probst,A.,Gotz,J.,Wiederhold,K.H.,Tolnay,M.,Mistl,C.,Jaton,A.L.,Hong,M.,Ishihara,T.,Lee,V.M.,Trojanowski,J.Q.,etal.(2000).Axonopathyandamyotrophyinmicetransgenicforhumanfour-repeattauprotein.ActaNeuropathol99,469-481.
Prusiner,S.B.(1991).Molecularbiologyofpriondiseases.Science252,1515-1522.
Prusiner,S.B.,Scott,M.R.,DeArmond,S.J.,andCohen,F.E.(1998).Prionproteinbiology.Cell93,337-348.
Qin,Z.H.,Wang,Y.,Kegel,K.B.,Kazantsev,A.,Apostol,B.L.,Thompson,L.M.,Yoder,J.,Aronin,N.,andDiFiglia,M.(2003).Autophagyregulatestheprocessingofaminoterminalhuntingtinfragments.HumMolGenet12,3231-3244.
Rademakers,R.,andRovelet-Lecrux,A.(2009).Recentinsightsintothemoleculargeneticsofdementia.TrendsNeurosci32,451-461.
Rebeiz, J.J., Kolodny, E.H., and Richardson, E.P., Jr. (1968). Corticodentatonigral degeneration withneuronalachromasia.ArchNeurol18,20-33.
Rensink,A.A.,deWaal,R.M.,Kremer,B.,andVerbeek,M.M.(2003).Pathogenesisofcerebralamyloidangiopathy.BrainResBrainResRev43,207-223.
Revesz, T., Holton, J.L., Doshi, B., Anderton, B.H., Scaravilli, F., and Plant, G.T. (1999). Cytoskeletalpathology in familial cerebral amyloid angiopathy (British type) with non-neuritic amyloid plaqueformation.ActaNeuropathol(Berl)97,170-176.
Revesz,T.,Holton,J.L.,Lashley,T.,Plant,G.,Rostagno,A.,Ghiso,J.,andFrangione,B.(2002).Sporadicandfamilialcerebralamyloidangiopathies.BrainPathol12,343-357.
Reynolds,C.H.,Garwood,C.J.,Wray,S.,Price,C.,Kellie,S.,Perera,T.,Zvelebil,M.,Yang,A.,Sheppard,P.W.,Varndell,I.M.,etal.(2008).PhosphorylationregulatestauinteractionswithSrchomology3domainsofphosphatidylinositol3-kinase,phospholipaseCgamma1,Grb2,andSrcfamilykinases.JBiolChem283,18177-18186.
Reynolds,M.R.,Reyes,J.F.,Fu,Y.,Bigio,E.H.,Guillozet-Bongaarts,A.L.,Berry,R.W.,andBinder,L.I.(2006).Taunitrationoccursattyrosine29inthefibrillarlesionsofAlzheimer'sdiseaseandothertauopathies.JNeurosci26,10636-10645.
Rissman,R.A.,Poon,W.W.,Blurton-Jones,M.,Oddo,S.,Torp,R.,Vitek,M.P.,LaFerla,F.M.,Rohn,T.T.,andCotman,C.W.(2004).Caspase-cleavageoftauisanearlyeventinAlzheimerdiseasetanglepathology.JClinInvest114,121-130.
Roberts,S.B.,Ripellino,J.A.,Ingalls,K.M.,Robakis,N.K.,andFelsenstein,K.M.(1994).Non-amyloidogeniccleavageofthebeta-amyloidprecursorproteinbyanintegralmembranemetalloendopeptidase.JBiolChem269,3111-3116.
Rogers,M.,Yehiely,F.,Scott,M.,andPrusiner,S.B.(1993).Conversionoftruncatedandelongatedprionproteinsintothescrapieisoforminculturedcells.ProcNatlAcadSciUSA90,3182-3186.
Rohn,T.T.,Rissman,R.A.,Davis,M.C.,Kim,Y.E.,Cotman,C.W.,andHead,E.(2002).Caspase-9activationandcaspasecleavageoftauintheAlzheimer'sdiseasebrain.NeurobiolDis11,341-354.
Rolfs,A.,Koeppen,A.H.,Bauer, I.,Bauer,P.,Buhlmann,S., Topka,H., Schols, L., andRiess,O. (2003).Clinicalfeaturesandneuropathologyofautosomaldominantspinocerebellarataxia(SCA17).AnnNeurol54,367-375.
Ross, C.A., andPoirier,M.A. (2004). Protein aggregationandneurodegenerativedisease.NatMed 10Suppl,S10-17.
Ross, C.A., and Tabrizi, S.J. (2011). Huntington's disease: from molecular pathogenesis to clinicaltreatment.LancetNeurol10,83-98.
Rostagno,A.,andGhiso,J.(2008).Preamyloidlesionsandcerebrovasculardepositsinthemechanismofdementia:lessonsfromnon-beta-amyloidcerebralamyloidosis.NeurodegenerDis5,173-175.

References
130
Rui, Y.,Gu, J., Yu,K.,Hartzell,H.C., andZheng, J.Q. (2010). InhibitionofAMPA receptor traffickingathippocampalsynapsesbybeta-amyloidoligomers:themitochondrialcontribution.MolBrain3,10.
Rui,Y.,Tiwari,P.,Xie,Z.,andZheng,J.Q.(2006).Acuteimpairmentofmitochondrialtraffickingbybeta-amyloidpeptidesinhippocampalneurons.JNeurosci26,10480-10487.
Rui,Y.N.,Xu,Z.,Patel,B.,Chen,Z.,Chen,D.,Tito,A.,David,G.,Sun,Y.,Stimming,E.F.,Bellen,H.J.,etal.(2015).Huntingtinfunctionsasascaffoldforselectivemacroautophagy.NatCellBiol17,262-275.
Saha,A.R.,Hill,J.,Utton,M.A.,Asuni,A.A.,Ackerley,S.,Grierson,A.J.,Miller,C.C.,Davies,A.M.,Buchman,V.L.,Anderton,B.H.,etal.(2004).Parkinson'sdiseasealpha-synucleinmutationsexhibitdefectiveaxonaltransportinculturedneurons.JCellSci117,1017-1024.
Saito,M.,Chakraborty,G.,Mao,R.F.,Paik,S.M.,Vadasz,C.,andSaito,M.(2010).Tauphosphorylationandcleavageinethanol-inducedneurodegenerationinthedevelopingmousebrain.NeurochemRes35,651-659.
Sarkar,S.,Davies,J.E.,Huang,Z.,Tunnacliffe,A.,andRubinsztein,D.C.(2007).Trehalose,anovelmTOR-independentautophagyenhancer,acceleratestheclearanceofmutanthuntingtinandalpha-synuclein.JBiolChem282,5641-5652.
Sato,T.,Miura,M.,Yamada,M.,Yoshida,T.,Wood,J.D.,Yazawa,I.,Masuda,M.,Suzuki,T.,Shin,R.M.,Yau,H.J.,etal.(2009).SevereneurologicalphenotypesofQ129DRPLAtransgenicmiceserendipitouslycreatedbyenmasseexpansionofCAGrepeatsinQ76DRPLAmice.HumMolGenet18,723-736.
Scarmeas, N., Hadjigeorgiou, G.M., Papadimitriou, A., Dubois, B., Sarazin, M., Brandt, J., Albert, M.,Marder, K., Bell, K., Honig, L.S., et al. (2004). Motor signs during the course of Alzheimer disease.Neurology63,975-982.
Schaeffer,V., Lavenir, I.,Ozcelik, S., Tolnay,M.,Winkler,D.T., andGoedert,M. (2012). Stimulationofautophagyreducesneurodegenerationinamousemodelofhumantauopathy.Brain135,2169-2177.
Scheltens,P.,Blennow,K.,Breteler,M.M.,deStrooper,B.,Frisoni,G.B.,Salloway,S.,andVanderFlier,W.M.(2016).Alzheimer'sdisease.Lancet.
Schilling,G., Becher,M.W., Sharp,A.H., Jinnah,H.A.,Duan, K., Kotzuk, J.A., Slunt,H.H., Ratovitski, T.,Cooper,J.K.,Jenkins,N.A.,etal.(1999).IntranuclearinclusionsandneuriticaggregatesintransgenicmiceexpressingamutantN-terminalfragmentofhuntingtin.HumMolGenet8,397-407.
Schweers,O.,Schonbrunn-Hanebeck,E.,Marx,A.,andMandelkow,E.(1994).StructuralstudiesoftauproteinandAlzheimerpairedhelical filaments shownoevidence forbeta-structure. JBiolChem 269,24290-24297.
Selkoe,D.J.(2001a).Alzheimer'sdisease:genes,proteins,andtherapy.PhysiolRev81,741-766.
Selkoe,D.J.(2001b).Presenilin,Notch,andthegenesisandtreatmentofAlzheimer'sdisease.ProcNatlAcadSciUSA98,11039-11041.
Sengupta,S.,Horowitz,P.M.,Karsten,S.L.,Jackson,G.R.,Geschwind,D.H.,Fu,Y.,Berry,R.W.,andBinder,L.I.(2006).Degradationoftauproteinbypuromycin-sensitiveaminopeptidaseinvitro.Biochemistry45,15111-15119.
Sergeant,N.,Delacourte,A.,andBuee,L.(2005).Tauproteinasadifferentialbiomarkeroftauopathies.BiochimBiophysActa1739,179-197.
Shahani,N.,andBrandt,R.(2002).Functionsandmalfunctionsofthetauproteins.CellMolLifeSci59,1668-1680.
Sherrington,R.,Rogaev,E.I.,Liang,Y.,Rogaeva,E.A.,Levesque,G.,Ikeda,M.,Chi,H.,Lin,C.,Li,G.,Holman,K.,etal.(1995).Cloningofagenebearingmissensemutationsinearly-onsetfamilialAlzheimer'sdisease.Nature375,754-760.
Silveira,J.R.,Raymond,G.J.,Hughson,A.G.,Race,R.E.,Sim,V.L.,Hayes,S.F.,andCaughey,B.(2005).Themostinfectiousprionproteinparticles.Nature437,257-261.

References
131
Sinha,S.,Anderson,J.P.,Barbour,R.,Basi,G.S.,Caccavello,R.,Davis,D.,Doan,M.,Dovey,H.F.,Frigon,N.,Hong,J.,etal.(1999).Purificationandcloningofamyloidprecursorproteinbeta-secretasefromhumanbrain[seecomments].Nature402,537-540.
Skachokova,Z.,Sprenger,F.,Breu,K.,Abramowski,D.,Clavaguera,F.,Hench,J.,Staufenbiel,M.,Tolnay,M.,andWinkler,D.T.(2015).Amyloid-betaintheCerebrospinalFluidofAPPTransgenicMiceDoesnotShowPrion-likeProperties.CurrAlzheimerRes12,886-891.
Snyder, H.,Mensah, K., Theisler, C., Lee, J.,Matouschek, A., andWolozin, B. (2003). Aggregated andmonomericalpha-synucleinbindtotheS6'proteasomalproteinandinhibitproteasomalfunction.JBiolChem278,11753-11759.
Sokolowski,J.D.,Gamage,K.K.,Heffron,D.S.,Leblanc,A.C.,Deppmann,C.D.,andMandell,J.W.(2014).Caspase-mediated cleavage of actin and tubulin is a common feature and sensitivemarker of axonaldegenerationinneuraldevelopmentandinjury.ActaNeuropatholCommun2,16.
Solomon,J.P.,Page,L.J.,Balch,W.E.,andKelly,J.W.(2012).Gelsolinamyloidosis:genetics,biochemistry,pathologyandpossiblestrategiesfortherapeuticintervention.CritRevBiochemMolBiol47,282-296.
Spillantini,M.G.,andGoedert,M.(2013).Taupathologyandneurodegeneration.LancetNeurol12,609-622.
Spillantini,M.G.,Murrell,J.R.,Goedert,M.,Farlow,M.R.,Klug,A.,andGhetti,B.(1998).Mutationinthetaugeneinfamilialmultiplesystemtauopathywithpreseniledementia.ProcNatlAcadSciUSA95,7737-7741.
Spillantini,M.G.,Schmidt,M.L.,Lee,V.M.,Trojanowski, J.Q., Jakes,R.,andGoedert,M. (1997).Alpha-synucleininLewybodies.Nature388,839-840.
Spires,T.L.,Orne,J.D.,SantaCruz,K.,Pitstick,R.,Carlson,G.A.,Ashe,K.H.,andHyman,B.T.(2006).Region-specificdissociationofneuronallossandneurofibrillarypathologyinamousemodeloftauopathy.AmJPathol168,1598-1607.
Spires-Jones,T.L.,Friedman,T.,Pitstick,R.,Polydoro,M.,Roe,A.,Carlson,G.A.,andHyman,B.T.(2014).MethylenebluedoesnotreverseexistingneurofibrillarytanglepathologyintherTg4510mousemodeloftauopathy.NeurosciLett562,63-68.
Spires-Jones,T.L.,Kopeikina,K.J.,Koffie,R.M.,deCalignon,A.,andHyman,B.T. (2011).Aretanglesastoxicastheylook?JMolNeurosci45,438-444.
Spires-Jones, T.L., Stoothoff, W.H., de Calignon, A., Jones, P.B., and Hyman, B.T. (2009). Taupathophysiologyinneurodegeneration:atangledissue.TrendsNeurosci32,150-159.
Steele,J.C.,Richardson,J.C.,andOlszewski,J.(1964).ProgressiveSupranuclearPalsy.AHeterogeneousDegeneration Involving the Brain Stem, Basal Ganglia and Cerebellum with Vertical Gaze andPseudobulbarPalsy,NuchalDystoniaandDementia.ArchNeurol10,333-359.
Strittmatter,W.J.,Saunders,A.M.,Schmechel,D.,Pericak-Vance,M.,Enghild,J.,Salvesen,G.S.,andRoses,A.D. (1993).ApolipoproteinE:high-aviditybinding tobeta-amyloidand increased frequencyof type4alleleinlate-onsetfamilialAlzheimerdisease.ProcNatlAcadSciUSA90,1977-1981.
Stromgren, E., Dalby, A., Dalby, M.A., and Ranheim, B. (1970). Cataract, deafness, cerebellar ataxia,psychosisanddementia--anewsyndrome.ActaNeurolScand46,Suppl43:261+.
Sultan,A.,Nesslany,F.,Violet,M.,Begard,S.,Loyens,A.,Talahari,S.,Mansuroglu,Z.,Marzin,D.,Sergeant,N.,Humez,S.,etal.(2011).Nucleartau,akeyplayerinneuronalDNAprotection.JBiolChem286,4566-4575.
Sun,K.H.,dePablo,Y.,Vincent,F.,Johnson,E.O.,Chavers,A.K.,andShah,K.(2008).NovelgenetictoolsrevealCdk5'smajorroleinGolgifragmentationinAlzheimer'sdisease.MolBiolCell19,3052-3069.
Sunyach,C.,Cisse,M.A.,daCosta,C.A.,Vincent,B.,andChecler,F.(2007).TheC-terminalproductsofcellularprionproteinprocessing,C1andC2,exertdistinct influenceonp53-dependentstaurosporine-inducedcaspase-3activation.JBiolChem282,1956-1963.
Sydow,A.,andMandelkow,E.M.(2010).'Prion-like'propagationofmouseandhumantauaggregatesinaninduciblemousemodeloftauopathy.NeurodegenerDis7,28-31.

References
132
Sydow,A.,VanderJeugd,A.,Zheng,F.,Ahmed,T.,Balschun,D.,Petrova,O.,Drexler,D.,Zhou,L.,Rune,G.,Mandelkow,E., etal. (2011). Tau-induceddefects in synapticplasticity, learning, andmemoryarereversibleintransgenicmiceafterswitchingoffthetoxicTaumutant.JNeurosci31,2511-2525.
Tabert,M.H.,Liu,X.,Doty,R.L.,Serby,M.,Zamora,D.,Pelton,G.H.,Marder,K.,Albers,M.W.,Stern,Y.,andDevanand,D.P.(2005).A10-itemsmellidentificationscalerelatedtoriskforAlzheimer'sdisease.AnnNeurol58,155-160.
Tagliavini,F.,Prelli,F.,Porro,M.,Salmona,M.,Bugiani,O.,andFrangione,B.(1992).Asolubleformofprion protein in human cerebrospinal fluid: implications for prion-related encephalopathies. BiochemBiophysResCommun184,1398-1404.
Tang,B.L.(2009).NeuronalproteintraffickingassociatedwithAlzheimerdisease:fromAPPandBACE1toglutamatereceptors.CellAdhMigr3,118-128.
Tanzi,R.E. (1999).Ageneticdichotomymodel forthe inheritanceofAlzheimer'sdiseaseandcommonage-relateddisorders.JClinInvest104,1175-1179.
Tanzi,R.E.,Gusella,J.F.,Watkins,P.C.,Bruns,G.A.,StGeorge-Hyslop,P.,VanKeuren,M.L.,Patterson,D.,Pagan,S.,Kurnit,D.M.,andNeve,R.L.(1987).Amyloidbetaproteingene:cDNA,mRNAdistribution,andgeneticlinkageneartheAlzheimerlocus.Science235,880-884.
Tapiola,T.,Alafuzoff,I.,Herukka,S.K.,Parkkinen,L.,Hartikainen,P.,Soininen,H.,andPirttila,T.(2009).Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologicchangesinthebrain.ArchNeurol66,382-389.
Terry,R.D.,Masliah,E., Salmon,D.P.,Butters,N.,DeTeresa,R.,Hill,R.,Hansen, L.A., andKatzman,R.(1991).PhysicalbasisofcognitivealterationsinAlzheimer'sdisease:synapselossisthemajorcorrelateofcognitiveimpairment.AnnNeurol30,572-580.
Thathiah,A., andDeStrooper,B. (2011). The roleofGprotein-coupled receptors in thepathologyofAlzheimer'sdisease.NatRevNeurosci12,73-87.
Thies,E.,andMandelkow,E.M.(2007).MissortingoftauinneuronscausesdegenerationofsynapsesthatcanberescuedbythekinaseMARK2/Par-1.JNeurosci27,2896-2907.
Tofaris,G.K.,andSpillantini,M.G.(2005).Alpha-synucleindysfunctioninLewybodydiseases.MovDisord20Suppl12,S37-44.
Trevitt,C.R.,Hosszu,L.L.,Batchelor,M.,Panico,S.,Terry,C.,Nicoll,A.J.,Risse,E.,Taylor,W.A.,Sandberg,M.K.,Al-Doujaily,H.,etal.(2014).N-terminaldomainofprionproteindirectsitsoligomericassociation.JBiolChem289,25497-25508.
Trinczek, B., Biernat, J., Baumann, K., Mandelkow, E.M., andMandelkow, E. (1995). Domains of tauprotein,differentialphosphorylation,anddynamicinstabilityofmicrotubules.MolBiolCell6,1887-1902.
Tseng, B.P., Green, K.N., Chan, J.L., Blurton-Jones, M., and Laferla, F.M. (2007). Abeta inhibits theproteasomeandenhancesamyloidandtauaccumulation.NeurobiolAging.
Turnbaugh, J.A., Unterberger, U., Saa, P., Massignan, T., Fluharty, B.R., Bowman, F.P., Miller, M.B.,Supattapone,S.,Biasini,E.,andHarris,D.A.(2012).TheN-terminal,polybasicregionofPrP(C)dictatestheefficiencyofprionpropagationbybindingtoPrP(Sc).JNeurosci32,8817-8830.
Usenovic,M.,Niroomand,S.,Drolet,R.E.,Yao,L.,Gaspar,R.C.,Hatcher,N.G.,Schachter,J.,Renger,J.J.,and Parmentier-Batteur, S. (2015). Internalized Tau Oligomers Cause Neurodegeneration by InducingAccumulation of Pathogenic Tau in Human Neurons Derived from Induced Pluripotent Stem Cells. JNeurosci35,14234-14250.
Van Raamsdonk, J.M., Pearson, J., Slow, E.J., Hossain, S.M., Leavitt, B.R., and Hayden, M.R. (2005).CognitivedysfunctionprecedesneuropathologyandmotorabnormalitiesintheYAC128mousemodelofHuntington'sdisease.JNeurosci25,4169-4180.
Vassar, R., Bennett, B.D., Babu-Khan, S., Kahn, S., Mendiaz, E.A., Denis, P., Teplow, D.B., Ross, S.,Amarante,P.,Loeloff,R.,etal.(1999).Beta-secretasecleavageofAlzheimer'samyloidprecursorproteinbythetransmembraneasparticproteaseBACE.Science286,735-741.

References
133
Vereecken,T.H.,Vogels,O.J.,andNieuwenhuys,R.(1994).NeuronlossandshrinkageintheamygdalainAlzheimer'sdisease.NeurobiolAging15,45-54.
Vidal,R.,Frangione,B.,Rostagno,A.,Mead,S.,Revesz,T.,Plant,G.,andGhiso,J.(1999).Astop-codonmutationintheBRIgeneassociatedwithfamilialBritishdementia.Nature399,776-781.
Vidal,R.,Revesz,T.,Rostagno,A.,Kim,E.,Holton,J.L.,Bek,T.,Bojsen-Moller,M.,Braendgaard,H.,Plant,G.,Ghiso,J.,etal.(2000).Adecamerduplicationinthe3'regionoftheBRIgeneoriginatesanamyloidpeptidethatisassociatedwithdementiainaDanishkindred.ProcNatlAcadSciUSA97,4920-4925.
Violet,M.,Chauderlier,A.,Delattre,L.,Tardivel,M.,Chouala,M.S.,Sultan,A.,Marciniak,E.,Humez,S.,Binder,L.,Kayed,R.,etal.(2015).PrefibrillarTauoligomersalterthenucleicacidprotectivefunctionofTauinhippocampalneuronsinvivo.NeurobiolDis82,540-551.
vonBergen,M.,Barghorn,S.,Biernat,J.,Mandelkow,E.M.,andMandelkow,E.(2005).Tauaggregationisdrivenbyatransitionfromrandomcoiltobetasheetstructure.BiochimBiophysActa1739,158-166.
Vonsattel,J.P.,andDiFiglia,M.(1998).Huntingtondisease.JNeuropatholExpNeurol57,369-384.
Waldemar, G., Dubois, B., Emre,M., Georges, J.,McKeith, I.G., Rossor,M., Scheltens, P., Tariska, P.,Winblad,B.,andEfns(2007).RecommendationsforthediagnosisandmanagementofAlzheimer'sdiseaseandotherdisordersassociatedwithdementia:EFNSguideline.EurJNeurol14,e1-26.
Waldron-Roby,E.,Ratovitski,T.,Wang,X.,Jiang,M.,Watkin,E.,Arbez,N.,Graham,R.K.,Hayden,M.R.,Hou,Z.,Mori,S.,etal. (2012).Transgenicmousemodelexpressingthecaspase6 fragmentofmutanthuntingtin.JNeurosci32,183-193.
Wali,G.,Sutharsan,R.,Fan,Y.,Stewart,R.,TelloVelasquez,J.,Sue,C.M.,Crane,D.I.,andMackay-Sim,A.(2016).Mechanismof impairedmicrotubule-dependentperoxisome trafficking andoxidative stress inSPAST-mutatedcellsfrompatientswithHereditarySpasticParaplegia.SciRep6,27004.
Wang,H.Y.,Wang,I.F.,Bose,J.,andShen,C.K.(2004).StructuraldiversityandfunctionalimplicationsoftheeukaryoticTDPgenefamily.Genomics83,130-139.
Wang,J.Z.,Gong,C.X.,Zaidi,T.,Grundke-Iqbal,I.,andIqbal,K.(1995).DephosphorylationofAlzheimerpairedhelicalfilamentsbyproteinphosphatase-2Aand-2B.JBiolChem270,4854-4860.
Wang, J.Z.,Grundke-Iqbal, I., and Iqbal,K. (2007).Kinasesandphosphatasesand tau sites involved inAlzheimerneurofibrillarydegeneration.EurJNeurosci25,59-68.
Wang, X., Su, B., Lee, H.G., Li, X., Perry, G., Smith, M.A., and Zhu, X. (2009a). Impaired balance ofmitochondrialfissionandfusioninAlzheimer'sdisease.JNeurosci29,9090-9103.
Wang,Y.,Martinez-Vicente,M.,Kruger,U.,Kaushik,S.,Wong,E.,Mandelkow,E.M.,Cuervo,A.M.,andMandelkow, E. (2009b). Tau fragmentation, aggregation and clearance: the dual role of lysosomalprocessing.HumMolGenet18,4153-4170.
Weingarten,M.D.,Lockwood,A.H.,Hwo,S.Y.,andKirschner,M.W.(1975).Aproteinfactoressentialformicrotubuleassembly.ProcNatlAcadSciUSA72,1858-1862.
Weissmann,C.,Reyher,H.J.,Gauthier,A.,Steinhoff,H.J.,Junge,W.,andBrandt,R.(2009).Microtubulebindingandtrappingatthetipofneuritesregulatetaumotioninlivingneurons.Traffic10,1655-1668.
Wellington, C.L., Ellerby, L.M., Gutekunst, C.A., Rogers, D.,Warby, S., Graham, R.K., Loubser, O., vanRaamsdonk, J.,Singaraja,R.,Yang,Y.Z.,etal. (2002).CaspasecleavageofmutanthuntingtinprecedesneurodegenerationinHuntington'sdisease.JNeurosci22,7862-7872.
Wellington,C.L.,Singaraja,R.,Ellerby,L.,Savill,J.,Roy,S.,Leavitt,B.,Cattaneo,E.,Hackam,A.,Sharp,A.,Thornberry,N., et al. (2000). Inhibiting caspase cleavageofhuntingtin reduces toxicity andaggregateformationinneuronalandnonneuronalcells.JBiolChem275,19831-19838.
West, M.J., Coleman, P.D., Flood, D.G., and Troncoso, J.C. (1994). Differences in the pattern ofhippocampalneuronallossinnormalageingandAlzheimer'sdisease.Lancet344,769-772.
Westergard,L.,Turnbaugh,J.A.,andHarris,D.A.(2011).AnaturallyoccurringC-terminalfragmentoftheprionprotein (PrP)delaysdiseaseandactsasadominant-negative inhibitorofPrPSc formation. JBiolChem286,44234-44242.

References
134
Wilhelmsen,K.C.,Lynch,T.,Pavlou,E.,Higgins,M.,andNygaard,T.G.(1994).Localizationofdisinhibition-dementia-parkinsonism-amyotrophycomplexto17q21-22.AmJHumGenet55,1159-1165.
Winner, B., Jappelli, R.,Maji, S.K., Desplats, P.A., Boyer, L., Aigner, S., Hetzer, C., Loher, T., Vilar,M.,Campioni,S.,etal.(2011).Invivodemonstrationthatalpha-synucleinoligomersaretoxic.ProcNatlAcadSciUSA108,4194-4199.
Winslow,A.R.,Chen,C.W.,Corrochano,S.,Acevedo-Arozena,A.,Gordon,D.E.,Peden,A.A.,Lichtenberg,M.,Menzies, F.M.,Ravikumar,B., Imarisio, S., etal. (2010). alpha-Synuclein impairsmacroautophagy:implicationsforParkinson'sdisease.JCellBiol190,1023-1037.
Wischik, C.M., Edwards, P.C., Lai, R.Y., Roth, M., and Harrington, C.R. (1996). Selective inhibition ofAlzheimerdisease-liketauaggregationbyphenothiazines.ProcNatlAcadSciUSA93,11213-11218.
Wischik,C.M.,Novak,M.,Edwards,P.C.,Klug,A.,Tichelaar,W.,andCrowther,R.A.(1988a).StructuralcharacterizationofthecoreofthepairedhelicalfilamentofAlzheimerdisease.ProcNatlAcadSciUSA85,4884-4888.
Wischik,C.M.,Novak,M.,Thogersen,H.C.,Edwards,P.C.,Runswick,M.J.,Jakes,R.,Walker,J.E.,Milstein,C.,Roth,M.,andKlug,A.(1988b).IsolationofafragmentoftauderivedfromthecoreofthepairedhelicalfilamentofAlzheimerdisease.ProcNatlAcadSciUSA85,4506-4510.
Wittmann,C.W.,Wszolek,M.F.,Shulman,J.M.,Salvaterra,P.M.,Lewis,J.,Hutton,M.,andFeany,M.B.(2001).Tauopathy inDrosophila:neurodegenerationwithoutneurofibrillarytangles.Science293,711-714.
Xu,B.,Liu,W.,Deng,Y.,Yang,T.Y.,Feng,S.,andXu,Z.F.(2015).Inhibitionofcalpainpreventsmanganese-inducedcellinjuryandalpha-synucleinoligomerizationinorganotypicbrainslicecultures.PLoSOne10,e0119205.
Yamamoto,A.,Cremona,M.L.,andRothman,J.E. (2006).Autophagy-mediatedclearanceofhuntingtinaggregatestriggeredbytheinsulin-signalingpathway.JCellBiol172,719-731.
Yamamoto,H.,Saitoh,Y.,Fukunaga,K.,Nishimura,H.,andMiyamoto,E.(1988).Dephosphorylationofmicrotubuleproteinsbybrainproteinphosphatases1and2A,anditseffectonmicrotubuleassembly.JNeurochem50,1614-1623.
Young,J.E.,Gouw,L.,Propp,S.,Sopher,B.L.,Taylor,J.,Lin,A.,Hermel,E.,Logvinova,A.,Chen,S.F.,Chen,S., et al. (2007). Proteolytic cleavage of ataxin-7 by caspase-7 modulates cellular toxicity andtranscriptionaldysregulation.JBiolChem282,30150-30160.
Zhang,Q.,Zhang,X.,andSun,A.(2009a).TruncatedtauatD421isassociatedwithneurodegenerationandtangleformationinthebrainofAlzheimertransgenicmodels.ActaNeuropathol117,687-697.
Zhang,Y.,Li,M.,Drozda,M.,Chen,M.,Ren,S.,MejiaSanchez,R.O.,Leavitt,B.R.,Cattaneo,E.,Ferrante,R.J.,Hayden,M.R.,etal.(2003).Depletionofwild-typehuntingtininmousemodelsofneurologicdiseases.JNeurochem87,101-106.
Zhang,Y.J.,Xu,Y.F.,Cook,C.,Gendron,T.F.,Roettges,P.,Link,C.D.,Lin,W.L.,Tong,J.,Castanedes-Casey,M.,Ash,P.,etal.(2009b).AberrantcleavageofTDP-43enhancesaggregationandcellulartoxicity.ProcNatlAcadSciUSA106,7607-7612.
Zhang,Y.J.,Xu,Y.F.,Dickey,C.A.,Buratti,E.,Baralle,F.,Bailey,R.,Pickering-Brown,S.,Dickson,D.,andPetrucelli,L.(2007).Progranulinmediatescaspase-dependentcleavageofTARDNAbindingprotein-43.JNeurosci27,10530-10534.
Zhang,Y.W.,Thompson,R.,Zhang,H.,andXu,H.(2011).APPprocessinginAlzheimer'sdisease.MolBrain4,3.
Zou,W.Q.,Capellari,S.,Parchi,P.,Sy,M.S.,Gambetti,P.,andChen,S.G.(2003).IdentificationofnovelproteinaseK-resistantC-terminalfragmentsofPrPinCreutzfeldt-Jakobdisease.JBiolChem278,40429-40436.
Princeetal.2015:http://www.worldalzreport2015.org/downloads/world-alzheimer-report-2015.pdf

Abbreviations
135
7 Abbreviations
α-SynaaAβABriABriPPADAMADAM10ADanADanPPADAgDAICDALSAnxA2APH1ApoE4APPAspATPBRI2C-terminalCAACAGCaMCBDcdk;cdcCHMP2BCjDCKCNSCSFCTF
α-synucleinaminoacidsamyloidbetaamyloid-Briamyloid-Briprecursoradamalysinproteaseadesintegrinandmetalloproteinase10amyloid-Danamyloid-DanprecursorAlzheimer’sdiseaseargyrophilicgraindiseaseAPPintracellulardomainamyotrophiclateralsclerosisannexinA2anteriorpharynxdefective1apolipoproteinE4amyloidprecursorproteinasparticacidadenosinetriphosphateBRICHOSdomaincontaining2Bcarboxyterminalcerebralamyloidangiopathycytosine-adenine-guaninetrinucleotiderepeatscalmodulincorticobasaldegenerationcyclin-dependentkinasechargedmultivescularbodyprotein2BCreutzfeldt-JakobdiseasecaseinkinasecentralnervoussystemcerebrospinalfluidC-terminalfragment

Abbreviations
136
DLBDNADRPLAE391ECLEOADERgGAGGTGPIGSK3GSSFADFBDFDDFTDFTLDFTDP-17TGSK3hHDhttIAPPIHCJNKkDaLBLNLOADMAPKMAPTMARKmRNAMSAMTBµmmmmMMMPMnN-terminalNTFnmnmolNFTPBSPCR
dementiawithLewybodiesdeoxyribonucleicacidDentatorubral-pallidoluysianatrophyC-terminaltruncatedtauatGlutamicAcid391enhancedchemiluminescenceearly-onsetADendoplasmaticreticulumgramgolgiapparatusglobularglialtauopathyglycosylphosphatidylinositolglycogensynthasekinase3Gerstmann-Sträussler-ScheinkerdiseasesfamilialADfamilialBritishdementiafamilialDanishdementiafrontotemporaldementiafrontotemporallobardegenerationFTDandparkinsonismlinkedtochromosome17glycogensynthasekinase3hourHuntingtondiseasehuntingtinhormoneisletamyloidpeptideimmunohistochemistryc-JunN-terminalkinasekilodaltonsLewybodiesLewyneuriteslate-onsetADmicrotubuleassociatedproteinkinasemicrotubuleassociatedproteintaumicrotubule-affinityregulatingkinasemessengerribunucleicacidmultiplesystematrophymicrotubulebindingdomainmicrometer(s)millimeter(s)millimolarmatrixmetalloproteinasemanganeseaminoterminalaminoterminalfragmentnanometer(s)nanomolarneurofibrillarytanglephosphatebufferedsalinepolymerasechainreaction

Abbreviations
137
PCsPDPEN-2PGRNPHFPiDPKPolyQPPPRNPPrPCPrPScPRRPS1/2PSAPSPRRMsAPPSAASCASH3SPPL2TARDBPTDP-43TRDTTRUPStTsVAMP2
proproteinconvertasesParkinson’sdiseasepresenilinenhancer2progranulinpairedhelicalfilamentsPick’sdiseaseproteinkinasePolyglutaminediseasephosphatasePRioNProteincellularprionprotein scrapieprionproteinpronline-richregionpresenilin1and2puromycin-sensitiveaminopeptidaseprogressivesupranuclearpalsyRNA-recognitionmotifssolubleectodomainofAPPserumamyloidAspinocerebellar-ataxiaSCRHomology-3signalpeptidepeptidase-like2transactiveresponseDNAbindingproteintransactiveresponseDNAbindingprotein43kDATrinucleotiderepeatexpansiondisorderGlobularproteintransthyretinubiquitineproteasomesystemtetracyclinecontrolledtranscriptionalsilencerelementvesicleassociatedmembraneprotein
![[O-alkyl-O-methylchlorformiminophenylphosphonates--effective inhibitors of neurotoxic esterases in chicken brains]](https://img.dokumen.tips/doc/110x75/634bf5e1526bce8bbe0bf579/o-alkyl-o-methylchlorformiminophenylphosphonates-effective-inhibitors-of-neurotoxic.jpg)




























