Embed Size (px)
Citation preview

N
NN
M
Copyright © 2003 Society of Porphyrins & Phthalocyanines Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
COBALT, IRON, MANGANESE, NICKEL AND ZINC PHTHALOCYANINES 509
Synthesis, spectral and electrochemical properties of a new family of pyrrole substituted cobalt, iron, manganese, nickel and zinc phthalocyanine complexes
Joe Obiraia, Nazaré Pereira Rodriguesb, Fethi Bediouib and Tebello Nyokong*a
a Department of Chemistry, Rhodes University, Grahamstown 6140, South Africab Laboratoire d’Electrochimie et Chimie Analytique, UMR CNRS-ENSCP n° 7575, Ecole Nationale Supérieure de Chimie de Paris, 11 rue Pierre et Marie Curie, 75231 Paris cedex 05, France
Received 12 May 2003Accepted 4 July 2003
ABSTRACT: A new family of pyrrole substituted metallophthalocyanine complexes, namely cobalt(II), iron(II), manganese(III), nickel(II) and zinc(II) tetrakis-4-(pyrrol-1-yl)phenoxy phthalocyanines (noted as M(TPhPyrPc), where M is the metallic cation) have been synthesized and fully characterized. In particular, the UV-visible spectra of the iron and nickel complexes showed extensive aggregation even at low concentrations. The cyclic voltammetry of the cobalt, iron and manganese complexes showed three to four redox couples assigned to metal and ring based processes. Spectroelectrochemistry of the manganese derivative confirmed that the synthesized complex is MnIII(TPhPyrPc-2) and that the reduction of MnII(TPhPyrPc-2) to be centred on the ring and rather than on the metal, forming the MnII(TPhPyrPc-4) species. Also, the electrochemical polymerization of these newly synthesized pyrrole-substituted phthalocyanines has been demonstrated in the case of the cobalt complex and the electrocatalytic activity of the obtained film has been tested towards the oxidation of L-cysteine. Copyright © 2003 Society of Porphyrins & Phthalocyanines.
KEYWORDS: cobalt, iron, manganese, nickel, phthalocyanine, pyrrole, voltammetry, spectroelectrochemistry, electropolymerization.
INTRODUCTIONPhthalocyanine derivatives, M(Pc), have attracted
attention due to their diverse electronic, optical and structural properties, which have resulted in their applications in fields such as photosensitization [1], nonlinear optics [2], catalysis [3], liquid crystals
[4] and sensing [5]. Thus, a large number of ring or axially substituted phthalocyanine complexes have been tailored and studied as homogeneous and heterogeneous catalysts [6-20].
Electrochemical polymerization of M(Pc) complexes onto electrodes is one of the most effective means of developing electroactive surfaces for electrocatalytic purposes. Pyrrole and its
derivatives are often employed for the formation of conducting polymer films [21] and negatively charged tetrasulfonated M(TSPc) complexes have been used to functionalize polypyrrole films by incorporating them into the oxidized polypyrrole films [22, 23]. But the major part of the work has been devoted to pyrrole-substituted metalloporphyrin complexes. Indeed, pyrrole-substituted porphyrins have been synthesized and used to form well structured multi-layer films through their direct electropolymerization onto electrodes [24-26]. In the various reported cases, the pyrrole groups were separated from the porphyrin ring via spacer chains. Recently there has been a report on the synthesis of a unique example of a pyrrole substituted phthalocyanine complex [27]. The electropolymerizable pyrrole group was separated from the phthalocyanine macrocycle by an insulating *Correspondence to: Tebello Nyokong, email:
[email protected], fax: +27 46-622-5109
Journal of Porphyrins and Phthalocyanines Published at http://www.u-bourgogne.fr/jpp/
J. Porphyrins Phthalocyanines 2003; 7: 508-520
N
NN
M
Copyright © 2003 Society of Porphyrins & Phthalocyanines Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
COBALT, IRON, MANGANESE, NICKEL AND ZINC PHTHALOCYANINES 509
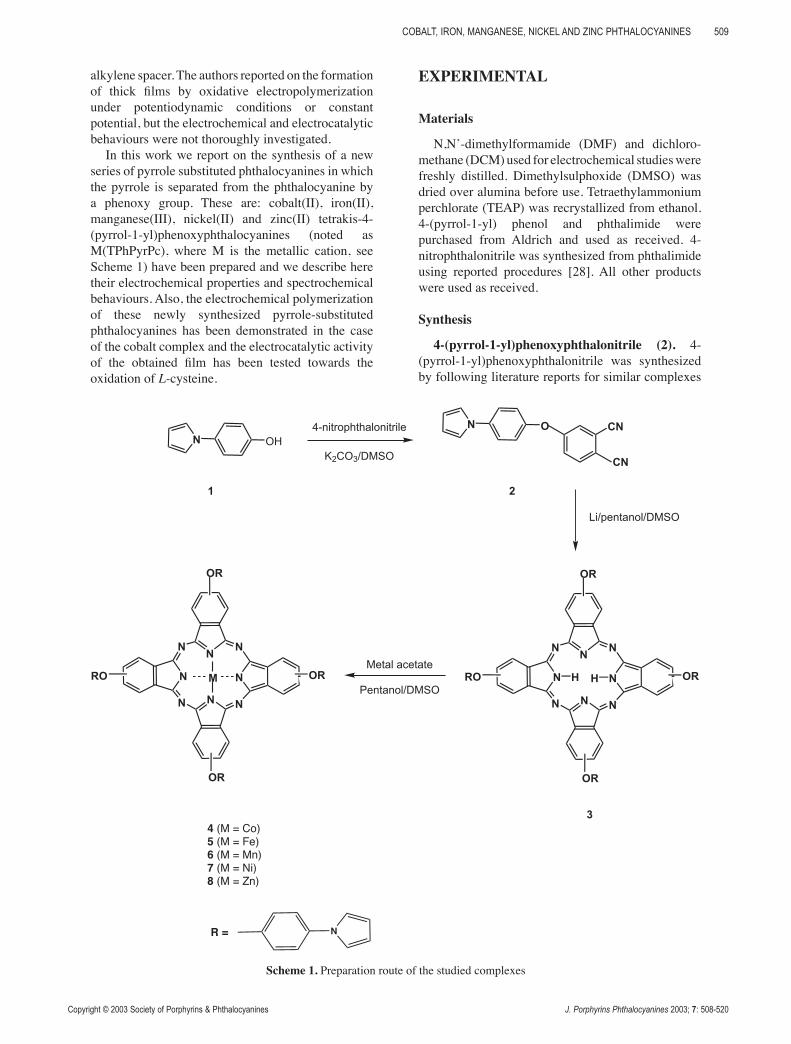
alkylene spacer. The authors reported on the formation of thick films by oxidative electropolymerization under potentiodynamic conditions or constant potential, but the electrochemical and electrocatalytic behaviours were not thoroughly investigated.
In this work we report on the synthesis of a new series of pyrrole substituted phthalocyanines in which the pyrrole is separated from the phthalocyanine by a phenoxy group. These are: cobalt(II), iron(II), manganese(III), nickel(II) and zinc(II) tetrakis-4-(pyrrol-1-yl)phenoxyphthalocyanines (noted as M(TPhPyrPc), where M is the metallic cation, see Scheme 1) have been prepared and we describe here their electrochemical properties and spectrochemical behaviours. Also, the electrochemical polymerization of these newly synthesized pyrrole-substituted phthalocyanines has been demonstrated in the case of the cobalt complex and the electrocatalytic activity of the obtained film has been tested towards the oxidation of L-cysteine.
EXPERIMENTAL
Materials
N,N’-dimethylformamide (DMF) and dichloro-methane (DCM) used for electrochemical studies were freshly distilled. Dimethylsulphoxide (DMSO) was dried over alumina before use. Tetraethylammonium perchlorate (TEAP) was recrystallized from ethanol. 4-(pyrrol-1-yl) phenol and phthalimide were purchased from Aldrich and used as received. 4-nitrophthalonitrile was synthesized from phthalimide using reported procedures [28]. All other products were used as received.
Synthesis
4-(pyrrol-1-yl)phenoxyphthalonitrile (2). 4-(pyrrol-1-yl)phenoxyphthalonitrile was synthesized by following literature reports for similar complexes
Scheme 1. Preparation route of the studied complexes
N
N OH4-nitrophthalonitrile
K2CO3/DMSO
N O CN
CN
N
N N
N
N
N
N
M OR
OR
OR
RO
NN
N N
NH
N
N
N
H OR
OR
OR
ROMetal acetate
Pentanol/DMSO
Li/pentanol/DMSO
1 2
34 (M = Co)5 (M = Fe)6 (M = Mn)7 (M = Ni)8 (M = Zn)
NR =

Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
J. OBIRAI ET AL.510
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
COBALT, IRON, MANGANESE, NICKEL AND ZINC PHTHALOCYANINES 511
[27]: 0.3 g (1.9 mmol) 4-(pyrrol-1-yl)phenol (1) and 0.27 g (1.56 mmol) 4-nitrophthalonitrile were added to 0.6 g dry K2CO3 in 15 ml dry DMSO. The mixture was stirred under nitrogen for 48 h at room temperature. The precipitated product was washed with cold water (four times) and finally with methanol (twice). The resulting solid was dried in the oven at 50 °C. IR (KBr): ν, cm-1 3442, 3106, 3063, 2229 (CN), 1598, 1516, 1486, 1244, 1205, 1076, 840, 727. 1H NMR (DMSO-d6): δ, ppm 8.10 (d, 1H); 7.85 (d, 1H); 7.50 (d, 2H); 7.44 (m, 1H); 7.38 (d, 2H); 7.30 (d, 2H); 6.29 (s, 2H).
Tetra-4-(pyrrol-1-yl)phenoxyphthalocyanine (3). A procedure similar to that reported [27] for the synthesis of tetrakis(3-pyrrol-1-yl)propoxy- phthalocyanine was adopted for the preparation of 3. A solution of 0.15 g (6 × 10-4 mol) of 2 in 5 ml dry pentanol/DMSO (4:1) was refluxed for 20 min in the presence of nitrogen. 4 mg of lithium was added to the refluxing solution. The solution immediately turned blue-green on addition of lithium metal. Refluxing was continued for another 1 h. The UV-visible spectrum of the refluxing solution was recorded at different time intervals to monitor formation of the characteristic Q-band of the phthalocyanine ring (Pc). Pentanol was removed by blowing out with nitrogen while the heating continued. The product was stirred for 10 min after adding water at pH 5. The unreacted phthalonitrile was removed by Soxhlet extraction with methanol. The purity of the Pc was checked using thin layer chromatography (TLC). TLC revealed two different spots (impurities) that were removed using diethylether and chloroform. The Pc was eluted with tetrahydrofuran (THF). Yield 50%. IR (KBr): ν, cm-1 3430, 3300, 2922, 2853, 1612, 1513, 1467, 1328, 1230, 1093, 1011, 923, 801, 725. 1H NMR (DMSO-d6): δ, ppm 9.10 (s, 8H, Pc); 8.60 (s, 4H, Pc); 7.5-7.9 (m, 16H, phenoxy); 7.40 (d, 8H, pyrrole); 6.30 (d, 8H, pyrrole). UV-vis (DMF): λmax, nm 344, 609, 637, 670 (ε = 8.85 × 104 M-1.cm-1), 700 (ε = 4.88 × 104 M-1.cm-1).
Cobalt(tetra-4-(pyrrol-1-yl)phenoxyphthalo-cyanine) (CoII(TPhPyrPc)). 0.027 g (0.023 mmol) of cobalt (II) acetate was added to a solution of 0.03 g (0.026 mmol) unmetallated phthalocyanine 3 in pentanol and stirred under reflux for 90 min. The pentanol was removed by bubbling nitrogen and the residue was treated with a 1:1 mixture of water-methanol to remove unreacted acetates. The product was washed 3 times with methanol. Column chromatography was employed to remove any remaining insoluble compounds. The product was first passed through an alumina column and eluted with THF, then through a second column and eluted with dichloromethane. The purity of the product was confirmed using TLC. Yield 21%. IR (KBr disk): ν, cm-1 2923, 2852, 1606, 1515, 1462, 1236, 1096, 835,
724. UV-vis (DMF): λmax, nm 328, 603, 665 (ε = 7.5 × 104 M-1.cm-1). MS (FAB): m/z 1200 [M +1]+.
Iron(tetra-4-(pyrrol-1-yl)phenoxyphthalocya-nine) (FeII(TPhPyrPc)). This compound was prepared using a similar procedure as for Co(TPhPyrPc) complex, using ferric acetate instead of cobalt acetate. The same amount (in moles) of reagents was used as for the cobalt complex. The complex was purified by column chromatography as described above for the cobalt complex and the purity confirmed with TLC. Yield 35%. IR (KBr disk): ν, cm-1 3436, 2955, 2922, 2859, 1610, 1510, 1472, 1233, 1077, 831, 726. UV-vis (DMF): λmax, nm 327, 457, 628 (ε = 2.4 × 104 M-1.cm-1), 660, 701.
Manganese(tetrakis-4-(pyrrol-1-yl)phenoxy)-phthalocyanine) (ClMnIII(TPhPyrPc)). This com-pound was prepared using a similar procedure as for complex Co(TPhPyrPc), using manganous chloride instead of cobalt acetate. The same amount (in moles) of reagents was used as for the cobalt complex. The complex was purified by column chromatography as described above for the cobalt complex and the purity confirmed with TLC. Yield 20%. IR (KBr disk): ν, cm-1 3437, 2944, 2859, 1606, 1514, 1468, 1234, 1071, 808, 720, 285 (MnIII-Cl). UV-vis (DMF): λmax, nm 380, 497, 649, 720 (ε = 6.36 × 104 M-1.cm-1).
Nickel(tetrakis-3-(pyrrol-1-yl)phenoxy)-phthalocyanine) (NiII(TPhPyrPc)). This com-pound was prepared using a similar procedure as for Co(TPhPyrPc) complex, using nickel sulphate instead of cobalt acetate. The same amount of reagents was used as for the cobalt complex. The complex was purified by column chromatography as described above for the cobalt complex and the purity confirmed with TLC. Yield 20%. IR (KBr disk): ν, cm-1 3423, 2958, 2923, 2845, 1609, 1514, 1461, 1236, 1096, 808, 720. UV-vis (DMF): λmax, nm 329, 621, 671 (ε = 2.84 × 104 M-1.cm-1)
Zinc(tetrakis-4-(pyrrol-1-yl)phenoxy)-phthalocyanine) (ZnII(TPhPyrPc)). This compound was prepared using a similar procedure as for Co(TPhPyrPc) complex, using zinc acetate instead of cobalt acetate. The same amount of reagents was used as for the cobalt complex. The complex was purified by column chromatography as described above for the cobalt complex and the purity confirmed with TLC. Yield 20%. IR (KBr disk): ν, cm-1 3433, 2924, 2845, 1609, 1511, 1478, 1232, 1090, 833, 728. UV-vis (DMF): λmax, nm 352, 609, 678 (ε = 1.1 × 105
M-1.cm-1).
Electrochemical methods
Electrochemical data were collected with either the BioAnalytical Systems (BAS) model 100 B/W electrochemical workstation or a Princeton Applied

Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
J. OBIRAI ET AL.510
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
COBALT, IRON, MANGANESE, NICKEL AND ZINC PHTHALOCYANINES 511
Research Inc. potentiostat/galvanostat model 263A. For cyclic voltammetry, platinum disk (2.0 mm diameter from BAS or Radiometer Analytical), glassy carbon disk (3 mm diameter from BAS) and vitreous carbon disk (3 mm diameter from Radiometer Analytical) were used as working electrodes. Platinum wire auxiliary and Ag wire pseudo-reference electrodes were employed for all electrochemical experiments in DMF and a Ag/AgCl pseudo reference was used in all other experiments. The ferrocinium/ferrocene (fc+/fc) redox couple was used as internal reference for metallophthalocyanine complexes in DMF and its potential was measured as being equal to ca. 0.74 ± 0.06 V versus the used pseudo reference silver wire in DMF. DMF and dichloromethane (DCM) containing 0.1 M tetraethyl ammonium perchlorate (TEAP) or tetrabutyl ammonium tetrafluoroborate (TBABF4), respectively, were employed as electrolytes. Nitrogen or argon gas was bubbled through the solutions prior to recording cyclic voltammograms, and the inert atmosphere was maintained throughout the cyclic voltammogram scans. Prior to cyclic voltammetry scans, the working electrodes were polished with various alumina pastes on a Buehler felt pad, followed by washing with deionised water and rinsing with methanol and DMF. Experiments were also conducted on ITO conductive glass electrodes (Solems, France) to obtain the transmission UV-visible spectra of electropolymerized films of different thickness.
Spectroelectrochemical data were recorded using an optically transparent thin-layer electrochemical (OTTLE) cell as previously reported [29]. The OTTLE cell was connected to a BAS CV 27 voltammograph.
UV-visible spectra were recorded on a Varian 500 UV-visible/NIR spectrophotometer. IR spectra (KBr pellets) were recorded on a Perkin-Elmer Spectrum 2000 FTIR spectrometer. 1H-nuclear magnetic resonance (NMR, 400 MHz) spectra were obtained in DMSO-d6 using the Bruker EMX 400 NMR spectrometer. Mass measurement was conducted with MASPEC II (FAB).
RESULTS AND DISCUSSION
Synthesis and UV-visible spectral characterization of the pyrrole substituted phthalocyanines
The 4-(pyrrol-1-yl)phenoxyphthalonitrile (2) was synthesized from 4-(pyrrol-1-yl)phenol and nitrophthalonitrile following the method employed by Trombach et al. [27] for the preparation of (pyrrol-1-ylalkoxy)phthalonitrile. The characteristic nitrile (C≡N) stretch at 2229 cm-1 of 2 disappears upon
formation of the phthalocyanine. The ether stretching frequencies are prominent for both the phthalonitrile (2) and the phthalocyanines in the range of 1230-1240 cm-1. The presence of axial chloride in (Cl)MnIII(TphPyrPc) was confirmed by the presence of a MnIII-Cl vibration at 285 cm-1. This value is very close to the value (285 cm-1) reported for MnIII-Cl in porphyrins [30].
The synthesis of the 4-(pyrrol-1-yl)phenoxy substituted metal phthalocyanine complexes was achieved using a method different from that previously reported [27], in that a metal free derivative was synthesized first, then metallated using the metal salt, instead of a direct reaction of the phthalonitrile with the metal salt. Also a solvent mixture of 1-pentanol/DMSO (4:1 v/v) was used instead of the single solvent (pentanol). Preparation of the complexes via the metal free derivative (3) resulted in high yield and purer compound. The cobalt complex Co(TPhPyrPc) was used as an example for mass spectral analysis, and the results obtained (m/z 1200 [M + 1]+) confirmed the formation of the pyrrole substituted derivatives. The Co(TPhPyrPc), Fe(TPhPyrPc), Mn(TPhPyrPc), Ni(TPhPyrPc) and Zn(TPhPyrPc) complexes were found to be soluble in common organic solvents such as DMF, DCM, tetrahydrofuran and dimethylsulfoxide. The complexes were further characterized by UV-vis, IR and NMR spectra.
1H NMR spectra of the free-base ligand 3 which was used in synthesizing all of the metallated complexes, gave characteristic resonances due to the peripheral protons of the phthalocyanines. These were observed as singlets at 9.1 ppm, integrating for a total of 8 protons. In addition, non-peripheral protons for the phthalocyanine ring were observed at 8.6 ppm and integrated for four protons. The protons for the pyrrol-phenoxy substituents integrated correctly.
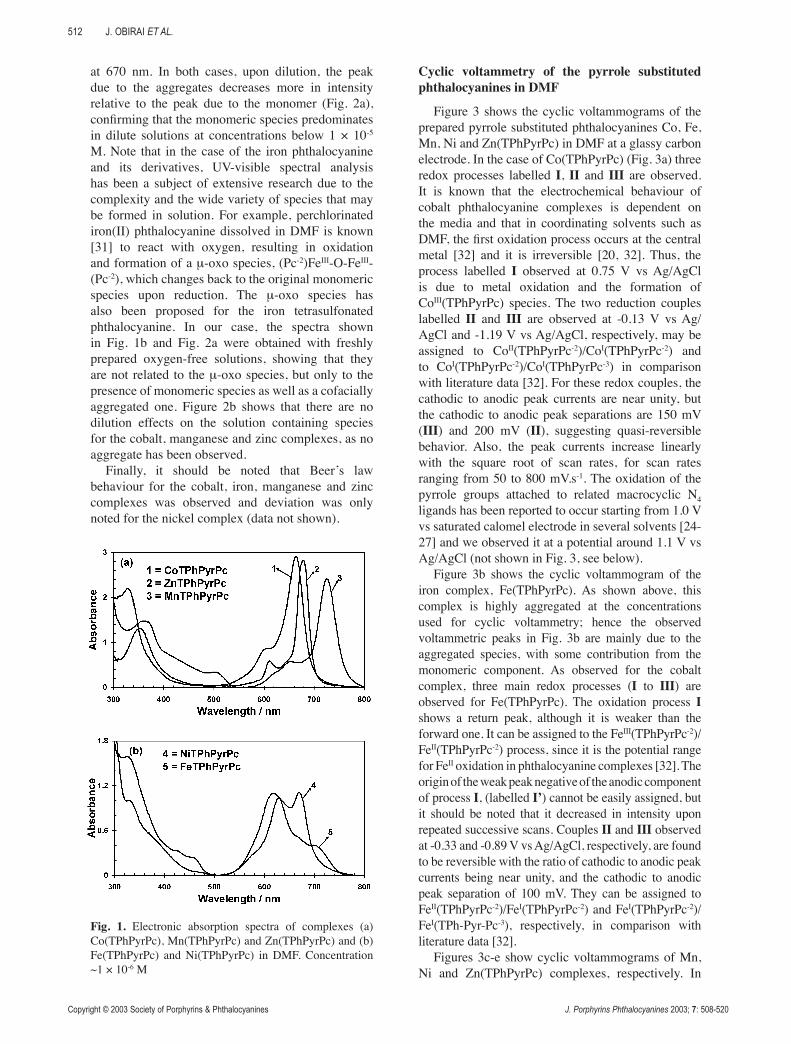
Figure 1 shows electronic absorption spectra of the 4-(pyrrol-1-yl)phenoxy substituted phthalocyanine complexes of cobalt, iron, manganese, nickel and zinc complexes in DMF. Note that the spectral features are indicative of MII(TPhPyrPc-2) species except in the case of the manganese complex where they are indicative of MnIII species. This is further discussed below in the spectroelectrochemical characterization section. The Q band of the cobalt, manganese and zinc complexes in DMF was typical of non-aggregated species, as it can be observed on Fig. 1a, at concentrations lower than 1 × 10-4 M. The iron and nickel complexes show aggregation as evidenced by the splitting of the Q band region observed in Fig. 1b. In the case of Fe(TPhPyrPc), the high energy band at 628 nm may be attributed to the aggregated species and the lower energy band at 665 nm to the monomeric species. Concerning Ni(TPhPyrPc) complex, the high energy peak due to the aggregated species is observed at 615 nm and the peak due to the monomeric species
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
J. OBIRAI ET AL.512
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
COBALT, IRON, MANGANESE, NICKEL AND ZINC PHTHALOCYANINES 513
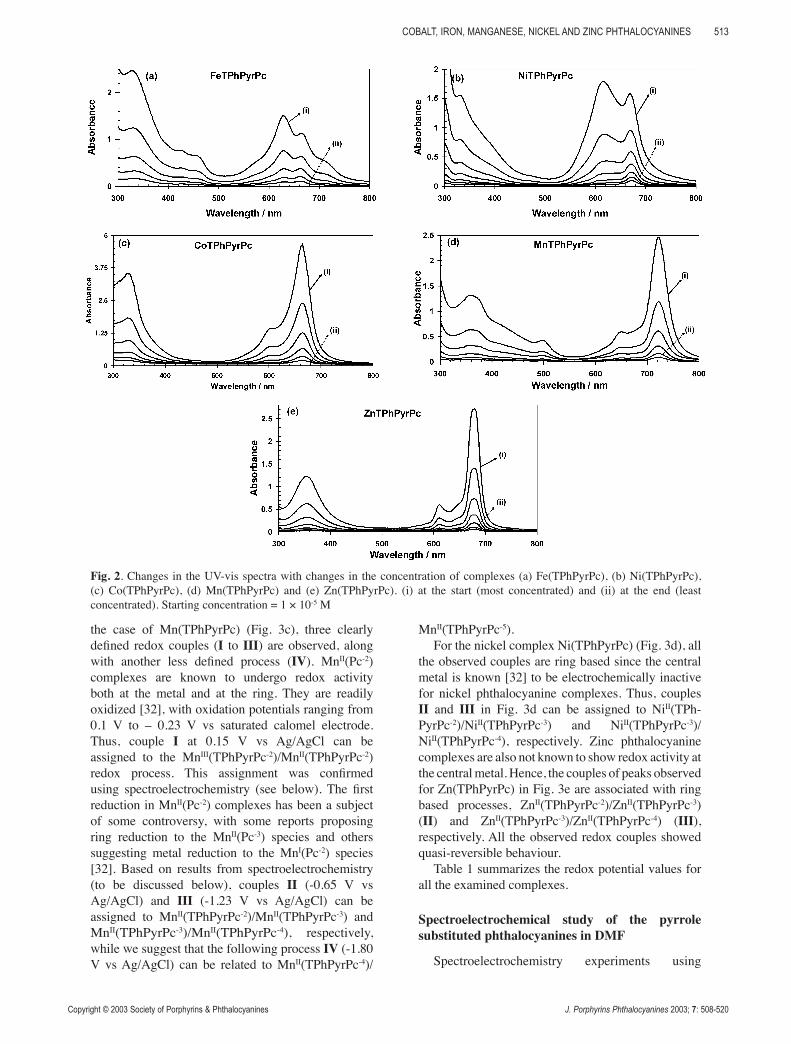
at 670 nm. In both cases, upon dilution, the peak due to the aggregates decreases more in intensity relative to the peak due to the monomer (Fig. 2a), confirming that the monomeric species predominates in dilute solutions at concentrations below 1 × 10-5 M. Note that in the case of the iron phthalocyanine and its derivatives, UV-visible spectral analysis has been a subject of extensive research due to the complexity and the wide variety of species that may be formed in solution. For example, perchlorinated iron(II) phthalocyanine dissolved in DMF is known
[31] to react with oxygen, resulting in oxidation and formation of a μ-oxo species, (Pc-2)FeIII-O-FeIII-(Pc-2), which changes back to the original monomeric species upon reduction. The μ-oxo species has also been proposed for the iron tetrasulfonated phthalocyanine. In our case, the spectra shown in Fig. 1b and Fig. 2a were obtained with freshly prepared oxygen-free solutions, showing that they are not related to the μ-oxo species, but only to the presence of monomeric species as well as a cofacially aggregated one. Figure 2b shows that there are no dilution effects on the solution containing species for the cobalt, manganese and zinc complexes, as no aggregate has been observed.
Finally, it should be noted that Beer’s law behaviour for the cobalt, iron, manganese and zinc complexes was observed and deviation was only noted for the nickel complex (data not shown).
Cyclic voltammetry of the pyrrole substituted phthalocyanines in DMF
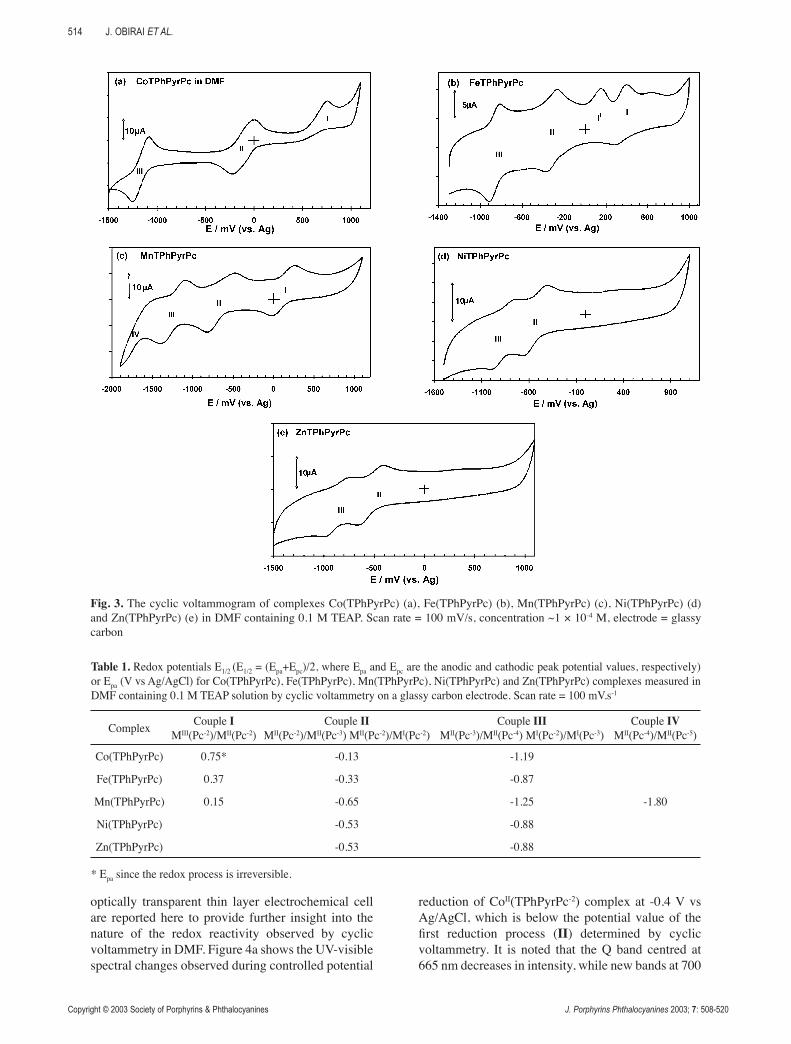
Figure 3 shows the cyclic voltammograms of the prepared pyrrole substituted phthalocyanines Co, Fe, Mn, Ni and Zn(TPhPyrPc) in DMF at a glassy carbon electrode. In the case of Co(TPhPyrPc) (Fig. 3a) three redox processes labelled I, II and III are observed. It is known that the electrochemical behaviour of cobalt phthalocyanine complexes is dependent on the media and that in coordinating solvents such as DMF, the first oxidation process occurs at the central metal [32] and it is irreversible [20, 32]. Thus, the process labelled I observed at 0.75 V vs Ag/AgCl is due to metal oxidation and the formation of CoIII(TPhPyrPc) species. The two reduction couples labelled II and III are observed at -0.13 V vs Ag/AgCl and -1.19 V vs Ag/AgCl, respectively, may be assigned to CoII(TPhPyrPc-2)/CoI(TPhPyrPc-2) and to CoI(TPhPyrPc-2)/CoI(TPhPyrPc-3) in comparison with literature data [32]. For these redox couples, the cathodic to anodic peak currents are near unity, but the cathodic to anodic peak separations are 150 mV (III) and 200 mV (II), suggesting quasi-reversible behavior. Also, the peak currents increase linearly with the square root of scan rates, for scan rates ranging from 50 to 800 mV.s-1. The oxidation of the pyrrole groups attached to related macrocyclic N4 ligands has been reported to occur starting from 1.0 V vs saturated calomel electrode in several solvents [24-27] and we observed it at a potential around 1.1 V vs Ag/AgCl (not shown in Fig. 3, see below).
Figure 3b shows the cyclic voltammogram of the iron complex, Fe(TPhPyrPc). As shown above, this complex is highly aggregated at the concentrations used for cyclic voltammetry; hence the observed voltammetric peaks in Fig. 3b are mainly due to the aggregated species, with some contribution from the monomeric component. As observed for the cobalt complex, three main redox processes (I to III) are observed for Fe(TPhPyrPc). The oxidation process I shows a return peak, although it is weaker than the forward one. It can be assigned to the FeIII(TPhPyrPc-2)/FeII(TPhPyrPc-2) process, since it is the potential range for FeII oxidation in phthalocyanine complexes [32]. The origin of the weak peak negative of the anodic component of process I, (labelled I’) cannot be easily assigned, but it should be noted that it decreased in intensity upon repeated successive scans. Couples II and III observed at -0.33 and -0.89 V vs Ag/AgCl, respectively, are found to be reversible with the ratio of cathodic to anodic peak currents being near unity, and the cathodic to anodic peak separation of 100 mV. They can be assigned to FeII(TPhPyrPc-2)/FeI(TPhPyrPc-2) and FeI(TPhPyrPc-2)/FeI(TPh-Pyr-Pc-3), respectively, in comparison with literature data [32].
Figures 3c-e show cyclic voltammograms of Mn, Ni and Zn(TPhPyrPc) complexes, respectively. In
Fig. 1. Electronic absorption spectra of complexes (a) Co(TPhPyrPc), Mn(TPhPyrPc) and Zn(TPhPyrPc) and (b) Fe(TPhPyrPc) and Ni(TPhPyrPc) in DMF. Concentration ~1 × 10-6 M
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
J. OBIRAI ET AL.512
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
COBALT, IRON, MANGANESE, NICKEL AND ZINC PHTHALOCYANINES 513
the case of Mn(TPhPyrPc) (Fig. 3c), three clearly defined redox couples (I to III) are observed, along with another less defined process (IV). MnII(Pc-2) complexes are known to undergo redox activity both at the metal and at the ring. They are readily oxidized [32], with oxidation potentials ranging from 0.1 V to – 0.23 V vs saturated calomel electrode. Thus, couple I at 0.15 V vs Ag/AgCl can be assigned to the MnIII(TPhPyrPc-2)/MnII(TPhPyrPc-2) redox process. This assignment was confirmed using spectroelectrochemistry (see below). The first reduction in MnII(Pc-2) complexes has been a subject of some controversy, with some reports proposing ring reduction to the MnII(Pc-3) species and others suggesting metal reduction to the MnI(Pc-2) species [32]. Based on results from spectroelectrochemistry (to be discussed below), couples II (-0.65 V vs Ag/AgCl) and III (-1.23 V vs Ag/AgCl) can be assigned to MnII(TPhPyrPc-2)/MnII(TPhPyrPc-3) and MnII(TPhPyrPc-3)/MnII(TPhPyrPc-4), respectively, while we suggest that the following process IV (-1.80 V vs Ag/AgCl) can be related to MnII(TPhPyrPc-4)/
MnII(TPhPyrPc-5). For the nickel complex Ni(TPhPyrPc) (Fig. 3d), all
the observed couples are ring based since the central metal is known [32] to be electrochemically inactive for nickel phthalocyanine complexes. Thus, couples II and III in Fig. 3d can be assigned to NiII(TPh-PyrPc-2)/NiII(TPhPyrPc-3) and NiII(TPhPyrPc-3)/NiII(TPhPyrPc-4), respectively. Zinc phthalocyanine complexes are also not known to show redox activity at the central metal. Hence, the couples of peaks observed for Zn(TPhPyrPc) in Fig. 3e are associated with ring based processes, ZnII(TPhPyrPc-2)/ZnII(TPhPyrPc-3) (II) and ZnII(TPhPyrPc-3)/ZnII(TPhPyrPc-4) (III), respectively. All the observed redox couples showed quasi-reversible behaviour.
Table 1 summarizes the redox potential values for all the examined complexes.
Spectroelectrochemical study of the pyrrole substituted phthalocyanines in DMF
Spectroelectrochemistry experiments using
Fig. 2. Changes in the UV-vis spectra with changes in the concentration of complexes (a) Fe(TPhPyrPc), (b) Ni(TPhPyrPc), (c) Co(TPhPyrPc), (d) Mn(TPhPyrPc) and (e) Zn(TPhPyrPc). (i) at the start (most concentrated) and (ii) at the end (least concentrated). Starting concentration = 1 × 10-5 M
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
J. OBIRAI ET AL.514
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
COBALT, IRON, MANGANESE, NICKEL AND ZINC PHTHALOCYANINES 515
optically transparent thin layer electrochemical cell are reported here to provide further insight into the nature of the redox reactivity observed by cyclic voltammetry in DMF. Figure 4a shows the UV-visible spectral changes observed during controlled potential
reduction of CoII(TPhPyrPc-2) complex at -0.4 V vs Ag/AgCl, which is below the potential value of the first reduction process (II) determined by cyclic voltammetry. It is noted that the Q band centred at 665 nm decreases in intensity, while new bands at 700
Fig. 3. The cyclic voltammogram of complexes Co(TPhPyrPc) (a), Fe(TPhPyrPc) (b), Mn(TPhPyrPc) (c), Ni(TPhPyrPc) (d) and Zn(TPhPyrPc) (e) in DMF containing 0.1 M TEAP. Scan rate = 100 mV/s, concentration ~1 × 10-4 M, electrode = glassy carbon
Table 1. Redox potentials E1/2 (E1/2 = (Epa+Epc)/2, where Epa and Epc are the anodic and cathodic peak potential values, respectively) or Epa (V vs Ag/AgCl) for Co(TPhPyrPc), Fe(TPhPyrPc), Mn(TPhPyrPc), Ni(TPhPyrPc) and Zn(TPhPyrPc) complexes measured in DMF containing 0.1 M TEAP solution by cyclic voltammetry on a glassy carbon electrode. Scan rate = 100 mV.s-1
Complex Couple IMIII(Pc-2)/MII(Pc-2)
Couple IIMII(Pc-2)/MII(Pc-3) MII(Pc-2)/MI(Pc-2)
Couple IIIMII(Pc-3)/MII(Pc-4) MI(Pc-2)/MI(Pc-3)
Couple IVMII(Pc-4)/MII(Pc-5)
Co(TPhPyrPc) 0.75* -0.13 -1.19
Fe(TPhPyrPc) 0.37 -0.33 -0.87
Mn(TPhPyrPc) 0.15 -0.65 -1.25 -1.80
Ni(TPhPyrPc) -0.53 -0.88
Zn(TPhPyrPc) -0.53 -0.88
* Epa since the redox process is irreversible.
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
J. OBIRAI ET AL.514
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
COBALT, IRON, MANGANESE, NICKEL AND ZINC PHTHALOCYANINES 515
nm and 475 nm appear. The band at 475 nm is typical
[33] of the formation of reduced CoI(TPhPyrPc-2) species, and it is then assigned to a charge transfer band. The Q band for CoI(Pc) species is generally weaker than that of the CoII and it is red shifted, hence the band at 700 nm is associated with the Q band for the CoI(TPhPyrPc-2) species. The envelope of bands near 450 nm is characteristic [34] of CoI species and comprises of several transitions. It can also be observed that the formation of the CoI(TPhPyrPc-2) species is accompanied by diffuse isosbestic points. The number of moles of electrons transferred was found to be near unity, confirming a one electron reduction process. The application of 0 V vs Ag/AgCl resulted in > 90% regeneration of the spectra of the starting species, hence showing that the reduction of CoII(TPhPyrPc-2) complex is reversible.
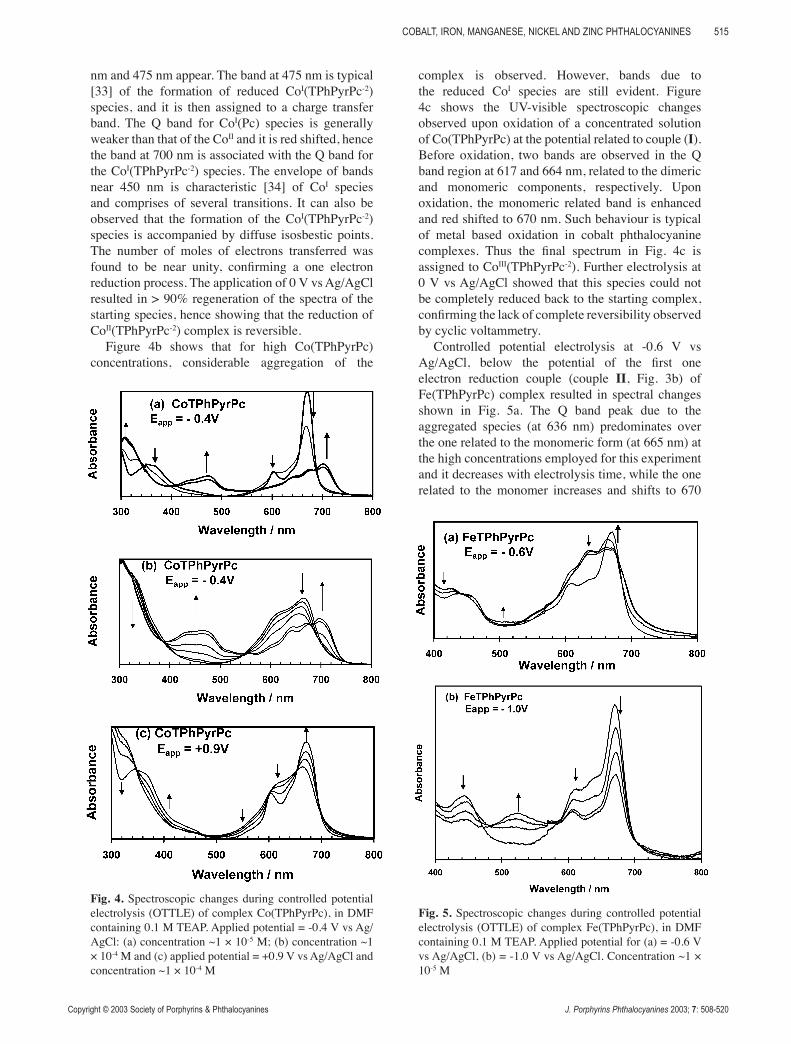
Figure 4b shows that for high Co(TPhPyrPc) concentrations, considerable aggregation of the
complex is observed. However, bands due to the reduced CoI species are still evident. Figure 4c shows the UV-visible spectroscopic changes observed upon oxidation of a concentrated solution of Co(TPhPyrPc) at the potential related to couple (I). Before oxidation, two bands are observed in the Q band region at 617 and 664 nm, related to the dimeric and monomeric components, respectively. Upon oxidation, the monomeric related band is enhanced and red shifted to 670 nm. Such behaviour is typical of metal based oxidation in cobalt phthalocyanine complexes. Thus the final spectrum in Fig. 4c is assigned to CoIII(TPhPyrPc-2). Further electrolysis at 0 V vs Ag/AgCl showed that this species could not be completely reduced back to the starting complex, confirming the lack of complete reversibility observed by cyclic voltammetry.
Controlled potential electrolysis at -0.6 V vs Ag/AgCl, below the potential of the first one electron reduction couple (couple II, Fig. 3b) of Fe(TPhPyrPc) complex resulted in spectral changes shown in Fig. 5a. The Q band peak due to the aggregated species (at 636 nm) predominates over the one related to the monomeric form (at 665 nm) at the high concentrations employed for this experiment and it decreases with electrolysis time, while the one related to the monomer increases and shifts to 670
Fig. 4. Spectroscopic changes during controlled potential electrolysis (OTTLE) of complex Co(TPhPyrPc), in DMF containing 0.1 M TEAP. Applied potential = -0.4 V vs Ag/AgCl: (a) concentration ~1 × 10-5 M; (b) concentration ~1 × 10-4 M and (c) applied potential = +0.9 V vs Ag/AgCl and concentration ~1 × 10-4 M
Fig. 5. Spectroscopic changes during controlled potential electrolysis (OTTLE) of complex Fe(TPhPyrPc), in DMF containing 0.1 M TEAP. Applied potential for (a) = -0.6 V vs Ag/AgCl, (b) = -1.0 V vs Ag/AgCl. Concentration ~1 × 10-5 M
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
J. OBIRAI ET AL.516
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
COBALT, IRON, MANGANESE, NICKEL AND ZINC PHTHALOCYANINES 517
nm. The observed spectral changes can be assigned to the formation of a monomeric species, as judged by the narrowing of the final spectrum compared to the starting one. Thus, it seems that the presence of the pyrrole substituents results in the formation of the reduced FeI(TPhPyrPc2-) species with a resolved Q band, contrary to literature reports for other Fe(Pc) complexes. Further reduction at -1.0 V vs Ag/AgCl, below the potential value of couple III, results in the spectral changes shown in Fig. 5b. A relatively strong peak appeared at 529 nm and a weaker one at around 600 nm. It is known that the first ring reduction in unsubstituted metallophthalocyanine complexes is accompanied by absorption bands between 550 and 650 nm [34]. Thus, the new formed species is most likely the FeI(TPhPyrPcPc3-) complex.
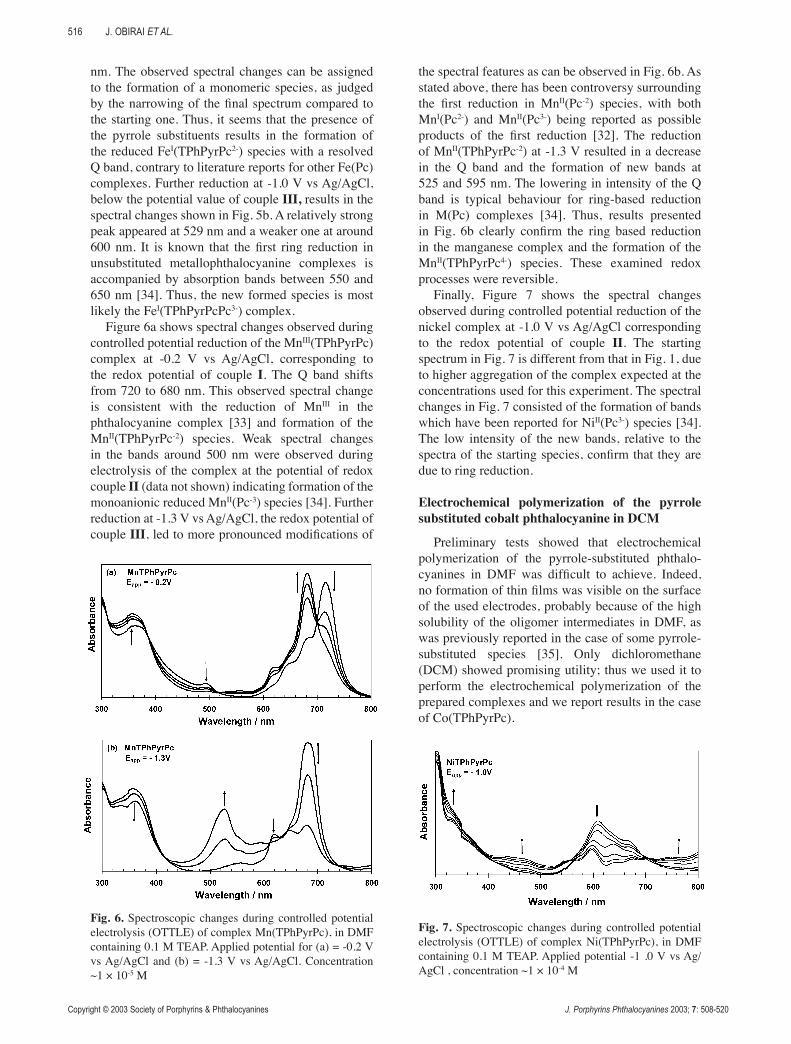
Figure 6a shows spectral changes observed during controlled potential reduction of the MnIII(TPhPyrPc) complex at -0.2 V vs Ag/AgCl, corresponding to the redox potential of couple I. The Q band shifts from 720 to 680 nm. This observed spectral change is consistent with the reduction of MnIII in the phthalocyanine complex [33] and formation of the MnII(TPhPyrPc-2) species. Weak spectral changes in the bands around 500 nm were observed during electrolysis of the complex at the potential of redox couple II (data not shown) indicating formation of the monoanionic reduced MnII(Pc-3) species [34]. Further reduction at -1.3 V vs Ag/AgCl, the redox potential of couple III, led to more pronounced modifications of
the spectral features as can be observed in Fig. 6b. As stated above, there has been controversy surrounding the first reduction in MnII(Pc-2) species, with both MnI(Pc2-) and MnII(Pc3-) being reported as possible products of the first reduction [32]. The reduction of MnII(TPhPyrPc-2) at -1.3 V resulted in a decrease in the Q band and the formation of new bands at 525 and 595 nm. The lowering in intensity of the Q band is typical behaviour for ring-based reduction in M(Pc) complexes [34]. Thus, results presented in Fig. 6b clearly confirm the ring based reduction in the manganese complex and the formation of the MnII(TPhPyrPc4-) species. These examined redox processes were reversible.
Finally, Figure 7 shows the spectral changes observed during controlled potential reduction of the nickel complex at -1.0 V vs Ag/AgCl corresponding to the redox potential of couple II. The starting spectrum in Fig. 7 is different from that in Fig. 1, due to higher aggregation of the complex expected at the concentrations used for this experiment. The spectral changes in Fig. 7 consisted of the formation of bands which have been reported for NiII(Pc3-) species [34]. The low intensity of the new bands, relative to the spectra of the starting species, confirm that they are due to ring reduction.
Electrochemical polymerization of the pyrrole substituted cobalt phthalocyanine in DCM
Preliminary tests showed that electrochemical polymerization of the pyrrole-substituted phthalo-cyanines in DMF was difficult to achieve. Indeed, no formation of thin films was visible on the surface of the used electrodes, probably because of the high solubility of the oligomer intermediates in DMF, as was previously reported in the case of some pyrrole-substituted species [35]. Only dichloromethane (DCM) showed promising utility; thus we used it to perform the electrochemical polymerization of the prepared complexes and we report results in the case of Co(TPhPyrPc).
Fig. 6. Spectroscopic changes during controlled potential electrolysis (OTTLE) of complex Mn(TPhPyrPc), in DMF containing 0.1 M TEAP. Applied potential for (a) = -0.2 V vs Ag/AgCl and (b) = -1.3 V vs Ag/AgCl. Concentration ~1 × 10-5 M
Fig. 7. Spectroscopic changes during controlled potential electrolysis (OTTLE) of complex Ni(TPhPyrPc), in DMF containing 0.1 M TEAP. Applied potential -1 .0 V vs Ag/AgCl , concentration ~1 × 10-4 M
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
J. OBIRAI ET AL.516
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
COBALT, IRON, MANGANESE, NICKEL AND ZINC PHTHALOCYANINES 517
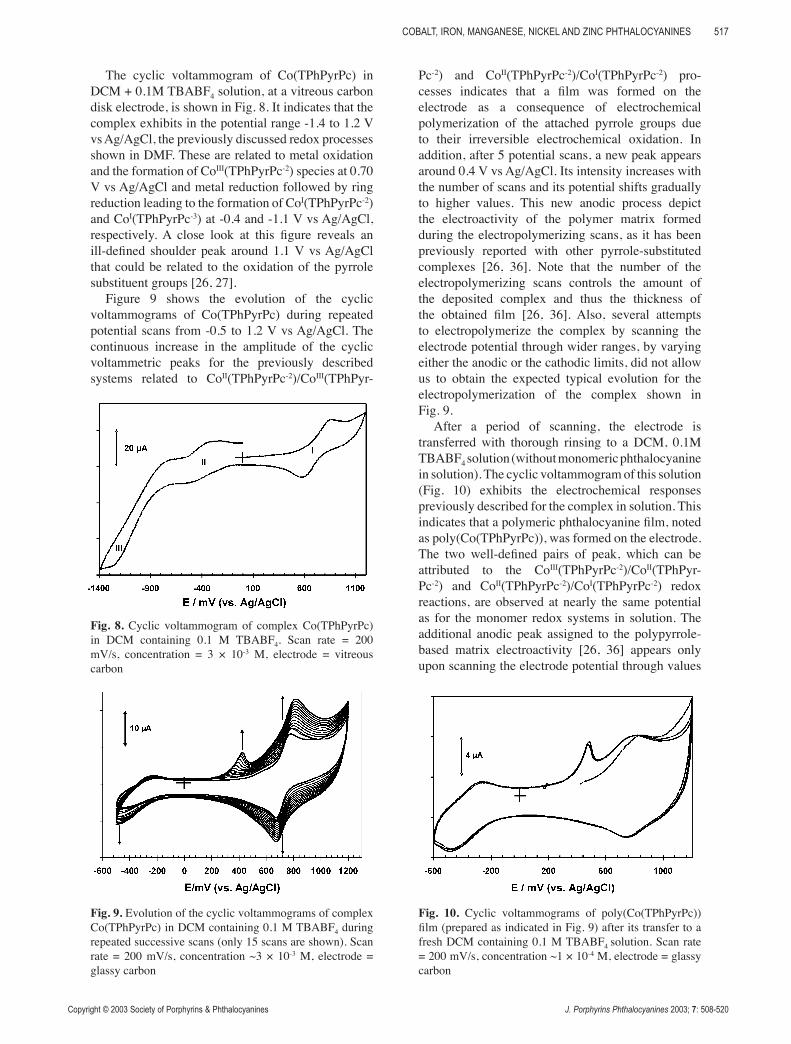
The cyclic voltammogram of Co(TPhPyrPc) in DCM + 0.1M TBABF4 solution, at a vitreous carbon disk electrode, is shown in Fig. 8. It indicates that the complex exhibits in the potential range -1.4 to 1.2 V vs Ag/AgCl, the previously discussed redox processes shown in DMF. These are related to metal oxidation and the formation of CoIII(TPhPyrPc-2) species at 0.70 V vs Ag/AgCl and metal reduction followed by ring reduction leading to the formation of CoI(TPhPyrPc-2) and CoI(TPhPyrPc-3) at -0.4 and -1.1 V vs Ag/AgCl, respectively. A close look at this figure reveals an ill-defined shoulder peak around 1.1 V vs Ag/AgCl that could be related to the oxidation of the pyrrole substituent groups [26, 27].
Figure 9 shows the evolution of the cyclic voltammograms of Co(TPhPyrPc) during repeated potential scans from -0.5 to 1.2 V vs Ag/AgCl. The continuous increase in the amplitude of the cyclic voltammetric peaks for the previously described systems related to CoII(TPhPyrPc-2)/CoIII(TPhPyr-
Pc-2) and CoII(TPhPyrPc-2)/CoI(TPhPyrPc-2) pro-cesses indicates that a film was formed on the electrode as a consequence of electrochemical polymerization of the attached pyrrole groups due to their irreversible electrochemical oxidation. In addition, after 5 potential scans, a new peak appears around 0.4 V vs Ag/AgCl. Its intensity increases with the number of scans and its potential shifts gradually to higher values. This new anodic process depict the electroactivity of the polymer matrix formed during the electropolymerizing scans, as it has been previously reported with other pyrrole-substituted complexes [26, 36]. Note that the number of the electropolymerizing scans controls the amount of the deposited complex and thus the thickness of the obtained film [26, 36]. Also, several attempts to electropolymerize the complex by scanning the electrode potential through wider ranges, by varying either the anodic or the cathodic limits, did not allow us to obtain the expected typical evolution for the electropolymerization of the complex shown in Fig. 9.
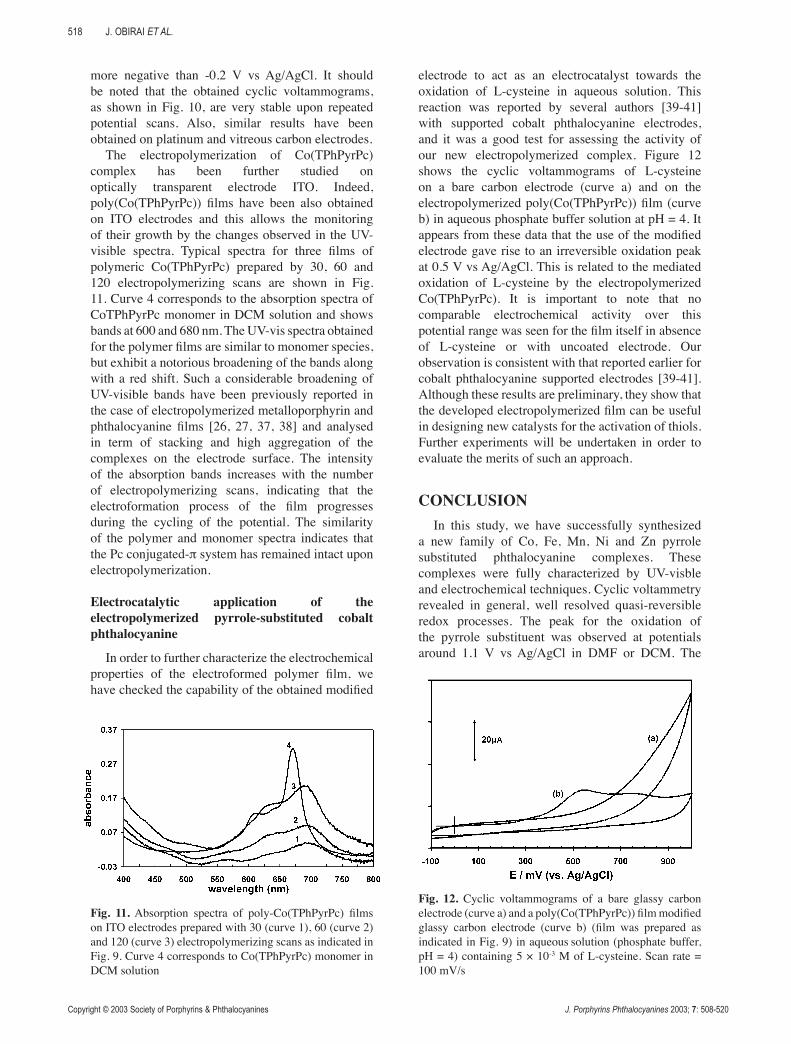
After a period of scanning, the electrode is transferred with thorough rinsing to a DCM, 0.1M TBABF4 solution (without monomeric phthalocyanine in solution). The cyclic voltammogram of this solution (Fig. 10) exhibits the electrochemical responses previously described for the complex in solution. This indicates that a polymeric phthalocyanine film, noted as poly(Co(TPhPyrPc)), was formed on the electrode. The two well-defined pairs of peak, which can be attributed to the CoIII(TPhPyrPc-2)/CoII(TPhPyr-Pc-2) and CoII(TPhPyrPc-2)/CoI(TPhPyrPc-2) redox reactions, are observed at nearly the same potential as for the monomer redox systems in solution. The additional anodic peak assigned to the polypyrrole-based matrix electroactivity [26, 36] appears only upon scanning the electrode potential through values
Fig. 9. Evolution of the cyclic voltammograms of complex Co(TPhPyrPc) in DCM containing 0.1 M TBABF4 during repeated successive scans (only 15 scans are shown). Scan rate = 200 mV/s, concentration ~3 × 10-3 M, electrode = glassy carbon
Fig. 8. Cyclic voltammogram of complex Co(TPhPyrPc) in DCM containing 0.1 M TBABF4. Scan rate = 200 mV/s, concentration = 3 × 10-3 M, electrode = vitreous carbon
Fig. 10. Cyclic voltammograms of poly(Co(TPhPyrPc)) film (prepared as indicated in Fig. 9) after its transfer to a fresh DCM containing 0.1 M TBABF4 solution. Scan rate = 200 mV/s, concentration ~1 × 10-4 M, electrode = glassy carbon
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
J. OBIRAI ET AL.518
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
COBALT, IRON, MANGANESE, NICKEL AND ZINC PHTHALOCYANINES 519
more negative than -0.2 V vs Ag/AgCl. It should be noted that the obtained cyclic voltammograms, as shown in Fig. 10, are very stable upon repeated potential scans. Also, similar results have been obtained on platinum and vitreous carbon electrodes.
The electropolymerization of Co(TPhPyrPc) complex has been further studied on optically transparent electrode ITO. Indeed, poly(Co(TPhPyrPc)) films have been also obtained on ITO electrodes and this allows the monitoring of their growth by the changes observed in the UV-visible spectra. Typical spectra for three films of polymeric Co(TPhPyrPc) prepared by 30, 60 and 120 electropolymerizing scans are shown in Fig. 11. Curve 4 corresponds to the absorption spectra of CoTPhPyrPc monomer in DCM solution and shows bands at 600 and 680 nm. The UV-vis spectra obtained for the polymer films are similar to monomer species, but exhibit a notorious broadening of the bands along with a red shift. Such a considerable broadening of UV-visible bands have been previously reported in the case of electropolymerized metalloporphyrin and phthalocyanine films [26, 27, 37, 38] and analysed in term of stacking and high aggregation of the complexes on the electrode surface. The intensity of the absorption bands increases with the number of electropolymerizing scans, indicating that the electroformation process of the film progresses during the cycling of the potential. The similarity of the polymer and monomer spectra indicates that the Pc conjugated-π system has remained intact upon electropolymerization.
Electrocatalytic application of the electropolymerized pyrrole-substituted cobalt phthalocyanine
In order to further characterize the electrochemical properties of the electroformed polymer film, we have checked the capability of the obtained modified
electrode to act as an electrocatalyst towards the oxidation of L-cysteine in aqueous solution. This reaction was reported by several authors [39-41] with supported cobalt phthalocyanine electrodes, and it was a good test for assessing the activity of our new electropolymerized complex. Figure 12 shows the cyclic voltammograms of L-cysteine on a bare carbon electrode (curve a) and on the electropolymerized poly(Co(TPhPyrPc)) film (curve b) in aqueous phosphate buffer solution at pH = 4. It appears from these data that the use of the modified electrode gave rise to an irreversible oxidation peak at 0.5 V vs Ag/AgCl. This is related to the mediated oxidation of L-cysteine by the electropolymerized Co(TPhPyrPc). It is important to note that no comparable electrochemical activity over this potential range was seen for the film itself in absence of L-cysteine or with uncoated electrode. Our observation is consistent with that reported earlier for cobalt phthalocyanine supported electrodes [39-41]. Although these results are preliminary, they show that the developed electropolymerized film can be useful in designing new catalysts for the activation of thiols. Further experiments will be undertaken in order to evaluate the merits of such an approach.
CONCLUSIONIn this study, we have successfully synthesized
a new family of Co, Fe, Mn, Ni and Zn pyrrole substituted phthalocyanine complexes. These complexes were fully characterized by UV-visble and electrochemical techniques. Cyclic voltammetry revealed in general, well resolved quasi-reversible redox processes. The peak for the oxidation of the pyrrole substituent was observed at potentials around 1.1 V vs Ag/AgCl in DMF or DCM. The
Fig. 11. Absorption spectra of poly-Co(TPhPyrPc) films on ITO electrodes prepared with 30 (curve 1), 60 (curve 2) and 120 (curve 3) electropolymerizing scans as indicated in Fig. 9. Curve 4 corresponds to Co(TPhPyrPc) monomer in DCM solution
Fig. 12. Cyclic voltammograms of a bare glassy carbon electrode (curve a) and a poly(Co(TPhPyrPc)) film modified glassy carbon electrode (curve b) (film was prepared as indicated in Fig. 9) in aqueous solution (phosphate buffer, pH = 4) containing 5 × 10-3 M of L-cysteine. Scan rate = 100 mV/s

Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
J. OBIRAI ET AL.518
Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
COBALT, IRON, MANGANESE, NICKEL AND ZINC PHTHALOCYANINES 519
cyclic voltammetry assignments for the observed electrochemical reactions were confirmed by spectroelectrochemistry and metal based redox processes were demonstrated for the cobalt, iron and manganese complexes. Also, the electrochemical polymerization of these newly synthesized pyrrole-substituted phthalocyanines has been demonstrated in the case of the cobalt complex and the electrocatalytic activity of the obtained film has been tested towards the oxidation of L-cysteine.
Acknowledgements
This work is supported by Rhodes University and by the Foundation of Research Development in South Africa. The authors are grateful to CNRS (France) and NRF (South Africa) for financial supports for travels and visits expenses (DRI grant n° 13273).
REFERENCES 1. Rosenthal I and Ben-Hur E. In Phthalocyanines:
Properties and Applications, Vol. 1, Leznoff CC, Lever ABP. (Eds.) Wiley-VCH: New York, 1989; Chapter 6.
2. Nalwa HS and Shirk JS. In Phthalocyanines: Properties and Applications, Vol. 4, Leznoff CC, Lever ABP. (Eds.) Wiley-VCH: New York, 1996; Chapter 4.
3. Hanabusa K and Shirai H. In Phthalocyanines: Properties and Applications, Vol. 2, Leznoff CC, Lever ABP. (Eds.) VCH: New York, 1993; Chapter 4.
4. Simon J and Bassoul P. In Phthalocyanines: Properties and applications, Vol. 2, Leznoff CC, Lever ABP. (Eds.) Wiley-VCH: New York, 1993; Chapter 5.
5. McKeown NB. Phthalocyanine Materials: Synthesis, Structure and Function; Cambridge University Press: London, 1998; p 211.
6. Zagal JH. Coord. Chem. Rev. 1992; 119: 89-136.
7. Zecevic S, Simic-Glavaski B, Yeager E, Lever ABP and Minor PC. J. Electroanal. Chem. 1985; 196: 339-358.
8. Janda P, Kobayashi N, Auburn PR, Lam H, Leznoff CC and Lever ABP. Can. J. Chem. 1989; 67: 1109-1119.
9. Maree S and Nyokong T. J. Electroanal. Chem. 2000; 492: 120-127.
10. Oni J and Nyokong T. Anal. Chim. Acta. 2001; 434: 9-21.
11. Vilakazi SL and Nyokong T. J. Electroanal. Chem. 2001; 512: 56-63.
12. Ozoemena K, Westbroek P and Nyokong T. Electrochem. Commun. 2001; 3: 529-534.
13. Huang X and Kok WT. Anal. Chim. Acta. 1993; 273: 245-253.
14. Halbert MK and Baldwin RP. Anal. Chem. 1985; 57: 591-595.
15. Napier A and Hart JP. Electroanalysis 1996; 8: 1006-1013.
16. Kobayashi N and Nevin WA. Appl. Organomet. Chem. 1996; 10: 579-590.
17. Chebotareva N and Nyokong T. J. Appl. Electrochem. 1997; 27: 975-981.
18. Thamae M and Nyokong T. J. Electroanal. Chem. 1999; 470: 126-135.
19. Limson J and Nyokong T. Electroanalysis 1998; 10: 988-993.
20. Limson J and Nyokong T. Electroanalysis 1997; 9: 255-260.
21. Linford RG, Ed. Electrochemical Science and Technology of Polymers; Elsevier Applied Science: London, 1987.
22. Elzing A, Van der Putten A, Visscher W and Barendrecht E. J. Electroanal. Chem. 1987; 233: 113-123.
23. Gulppi M, Bedioui F and Zagal JH. Electroanalysis 2001; 13: 1136-1139.
24. Diab N and Schuhmann W. Electrochim. Acta 2001; 47: 265-273.
25. Bedioui F, Devynck J and Bied-Charreton C. Acc. Chem. Res. 1995; 28: 30-36.
26. Cosnier S, Gondran C, Wessel R, Montforts FP and Wedel M. J. Electroanal. Chem. 2000; 488: 83-91.
27. Trombach N, Hild O, Schlettwein D and Wohrle D. J. Mater. Chem. 2002; 12: 879-885.
28. Huang XB, Liu YQ, Wang S, Zhou SQ and Zhu DB. Chem.--Eur. J. 2002; 8: 4179-4184.
29. Krejcik M, Danek M and Hartl F. J. Electroanal. Chem. 1991; 317: 179-187.
30. Cheng B, Cukiernik F, Fries PH, Marchon J-C and Scheidt WR. Inorg. Chem. 1995; 34: 4627-4639.
31. Golovin MN, Seymour P, Jayaraj K, Fu YS and Lever ABP. Inorg. Chem. 1990; 29: 1719-1727.
32. Lever ABP, Milaeva ER and Speier G. In Phthalocyanines: Properties and Applications, Vol. 3, Leznoff CC, Lever ABP. (Eds.) VCH: New York, 1993; Chapter 1.
33. Stillman MJ and Nyokong T. In Phthalocyanines: Properties and Applications, Vol. 1, Leznoff CC, Lever ABP. (Eds.) VCH: New York, 1989; Chapter 3.
34. Stillman MJ. Phthalocyanines: Properties and Applications; VCH: New York, 1993; Vol. 3; Chapter 5.
35. Diaz AF and Bargon J. In Handbook of Organic Conducting Polymers, Vol. 1, Staokheim TA. (Ed.) Marcel Dekker: New York, 1986; pp 87-

Copyright © 2003 Society of Porphyrins & Phthalocyanines J. Porphyrins Phthalocyanines 2003; 7: 508-520
J. OBIRAI ET AL.520
N
NN
M
Copyright © 2003 Society of Porphyrins & Phthalocyanines
88.36. Deronzier A and Moutet J-C. Coord. Chem.
Rev. 1996; 147: 339-371.37. Griveau S, Albin V, Pauporte T, Zagal JH and
Bedioui F. J. Mater. Chem. 2002; 12: 225-232.38. Griveau S, Pavez J, Zagal JH and Bedioui F. J.
Electroanal. Chem. 2001; 497: 75-83.
39. Qi X, Baldwin RP, Li H and Guarr TF. Electroanalysis 1991; 3: 119-124.
40. Zagal JH and Herrera P. Electrochim. Acta 1985; 30: 449-454.
41. Gulppi M, Griveau S, Pavez J, Zagal JH and Bedioui F. Electroanalysis 2003; 15: 779-785.



![Poly [(1-methylene-2-methylnaphthalene) -N-pyrrole] : a new fluorophore-containing electroactive conducting polymer 1](https://img.dokumen.tips/doc/110x75/63562a64922cbb7c550cfd62/poly-1-methylene-2-methylnaphthalene-n-pyrrole-a-new-fluorophore-containing.jpg)









![3,4-Dibromo-2,5-bis[(diethoxyphosphoryl)methyl]-1-phenylsulfonyl-1 H -pyrrole](https://img.dokumen.tips/doc/110x75/63488b07f4145ce0ba02c618/34-dibromo-25-bisdiethoxyphosphorylmethyl-1-phenylsulfonyl-1-h-pyrrole.jpg)















