Embed Size (px)
Citation preview

European Journal of Pharmaceutics and Biopharmaceutics 85 (2013) 42–52
Contents lists available at SciVerse ScienceDirect
European Journal of Pharmaceutics and Biopharmaceutics
journal homepage: www.elsevier .com/locate /e jpb
Research paper
Influence of methionine oxidation on the aggregation of recombinanthuman growth hormone
0939-6411/$ - see front matter � 2013 Elsevier B.V. All rights reserved.http://dx.doi.org/10.1016/j.ejpb.2013.03.015
Abbreviations: r-hGH, recombinant human growth hormone; UV–Vis, ultravio-let–visible; 1,8-ANS, 1-anilinonaphthalene-8-sulfonic acid; RP-HPLC, reverse phasehigh pressure liquid chromatography; SEC, size exclusion chromatography; RT,room temperature.⇑ Corresponding author. School of Pharmaceutical Sciences, University of Geneva,
University of Lausanne, CH-1211 Geneva 4, Switzerland. Tel.: +41 76 3910208; fax:+41 615440016.
E-mail address: [email protected] (T. Arvinte).
Filippo Mulinacci a, Emilie Poirier b, Martinus A.H. Capelle b, Robert Gurny a, Tudor Arvinte a,b,⇑a School of Pharmaceutical Sciences, University of Geneva, University of Lausanne, Geneva, Switzerlandb Therapeomic Inc., Bio Park Rosental, Mattenstrasse 22, Basel, Switzerland
a r t i c l e i n f o a b s t r a c t
Dedicated to Hans Peter Merkle on theoccasion of his 70th birthday
Keywords:AggregationMethionine oxidationHuman growth hormoneStabilityFreeze/thawing
Oxidation of methionine (Met) residues is one of the major chemical degradations of therapeutic pro-teins. This chemical degradation can occur at various stages during production and storage of a biother-apeutic drug. During the oxidation process, the side chain of methionine residue undergoes a chemicalmodification, with the thioether group substituted by a sulfoxide group. In previous papers, we showedthat oxidation of the two most accessible methionine residues of recombinant human growth hormone(r-hGH), Met14 and Met125, has no influence on the conformation of the protein [1]. However, the oxidizedr-hGH is less thermally stable than the native protein [2]. In the current work, the consequences of theoxidation of these two methionine residues on the aggregation of r-hGH were investigated. The aggrega-tion properties and kinetics of the native and oxidized r-hGH were measured in different buffers withboth spectroscopic and chromatographic methods. Stabilities of oxidized and non-oxidized r-hGH werestudied after storage at 37 �C and freeze/thawing cycles. Methionine oxidation influenced the aggregationproperties of r-hGH. In accelerated stability studies at 37 �C, oxidized hormone aggregated more and fas-ter than non-oxidized hormone. In freezing/thawing stability studies, it was found that oxidized r-hGHwas less stable than its non-oxidized counterpart. In case of hGH, we have shown that chemical degra-dations such as oxidation can affect its physical stability and can induce aggregation.
� 2013 Elsevier B.V. All rights reserved.
1. Introduction
Non-enzymatic degradations of proteins are in general subdi-vided into conformational changes (i.e. modifications of the tridi-mensional structure of the protein) and covalent modifications(i.e. chemical degradations of the primary structure of the protein)[3,4]. Examples of covalent modifications of proteins are oxidation,deamidation, disulfide bridges formation or scrambling, racemiza-tion, hydrolysis, glycation, and cross-linking [5,6]. These chemicaldegradations can involve different amino acid residues and canbe influenced by pH, temperature, and ionic strength [7–9]. Amongthe various chemical degradations, one of the most occurring is theoxidation of methionine (Met) residues. Oxidation produces amodification of the thioether group of the methionine side chain.In the first stage of the reaction, the thioether group is substituted
by a sulfoxide derivative; in the second step, the sulfoxide is fur-ther oxidized to sulfone. The sulfoxide derivative is commonly ob-served both in vivo and in vitro; on the contrary, the sulfonederivative product is rarely observed as it requires a much strongerchemical attack [10].
Oxidation of methionine residues has been reported to influ-ence the conformation and stability of proteins: (i) the secondarystructure of parathyroid hormone (PTH) and of its biological activefragment, 1–34 PTH, are modified as a consequence of the oxida-tion of the methionine residues [11]; (ii) oxidation of Met388 ofthrombomodulin reduces its anticoagulant cofactor activity of90% [12]; (iii) the structure of human IgG1 Fc is modified as a con-sequence of methionine oxidation and the melting temperature(Tm) is significantly reduced [13]; (iv) oxidation of the Met69 of re-combinant human leptin reduces to about 20% the bioactivity ofthe protein [14]; (v) oxidation of methionine residues in salmoncalcitonin produces morphologically different aggregates [15].However, there are a few examples where oxidation has no effecton the conformation, stability, and activity of proteins or wherethe effect of oxidation depends on the position of the methionineresidues involved: (i) oxidation of Met14 and Met125 of humangrowth hormone has minimal effect on the protein activity[16,17]; (ii) oxidation of Met314 and Met315 of antithrombin does

F. Mulinacci et al. / European Journal of Pharmaceutics and Biopharmaceutics 85 (2013) 42–52 43
not affect either the thrombin-inhibitor activity or the heparinbinding, while oxidation of Met17 and Met20 reduces the heparinaffinity [18].
Human growth hormone is a single-chain protein consisting of191 amino acid residues which contains three methionine residuesin position Met14, Met125, and Met170 [19]. The first two methio-nine residues, Met14 and Met125, are partially exposed on the sur-face of the protein and easily oxidized by chemical treatment withhydrogen peroxide (H2O2) [20]. On the contrary, the third residue,Met170, is partially buried and not easily oxidized [21,22]. In a pre-vious work of our group [1], the investigation of the conformationof an oxidized sample of recombinant human growth hormone (r-hGH) revealed that the oxidation of the methionine residues Met14
and Met125 does not induce conformation changes in the protein.Nevertheless, the thermal stability of the oxidized r-hGH is de-creased as a consequence of the oxidation [2]. In the current paper,the effect of the methionine residues oxidation on the aggregationof r-hGH is investigated. The physical stability of the oxidized r-hGH is directly compared with that of the native protein in differ-ent buffers. Kinetic of the aggregation, intensity of the aggregationin accelerated stability studies at 37 �C, and the effect of freeze/thawing are evaluated and discussed.
2. Materials and methods
2.1. Materials
Hydrogen peroxide (H2O2), L-Histidine, sodium hydroxide, 1-propanol, hydrochloric acid, and 7-diethylamino-3,4-benzophe-noxazine-2-one (Nile Red) were supplied by Sigma–Aldrich (Sig-ma–Aldrich Chemie GmbH, Buchs, Switzerland). Citric acidanhydrous, Tris(hydroxymethyl)aminomethane, and L-Methioninewere purchased from Fluka (Fluka GmbH, Buchs, Switzerland). So-dium phosphate mono- and di-basic and sodium chloride were ac-quired from Riedel-de Haën (Riedel-de Haën GmbH, Seelze,Germany). All chemicals and reagents were of analytical grade.Ultrapure water (Type I) was produced by a Millipore MilliQ Aca-demic system (Millipore AG, Zug, Switzerland). The recombinanthuman growth hormone was kindly provided by Merck-Serono(Merck-Serono SA, Italy). UV transparent 96-well Costar� Corning(Corning Inc., New York, NY, USA) microplates were supplied byVitaris (Vitaris SA, Baar, Switzerland). UV–Vis transparent GreinerVIEWseals™ (Greiner Bio-One GmbH, Frickenhausen, Germany)were purchased from Huber & Co. AG (Reinach, Switzerland).Quartz cuvettes for fluorescence spectroscopy were from Hellma(Hellma Schweiz AG, Zumikon, Switzerland). Slide-A-Lyzer� dialy-sis cassette (Pierce Biotechnology, Thermo Fisher Scientific Inc.,USA) were purchased from Perbio (Perbio Science SA, Lausanne,Switzerland). Disposable polystyrene cells were from VWR (VWRInternational, Nyon, Switzerland).
Table 1List of buffers used for the stability studies.
Buffers pH Concentration (mM) NaCl (mM)
Sodium citrate 3.0 20 –Sodium citrate 3.0 20 100Histidine/HCl 6.5 20 –Histidine/HCl 6.5 20 100Histidine/HCl 7.5 20Histidine/HCl 7.5 20 100Sodium phosphate 7.5 20 –Sodium phosphate 7.5 20 100
2.2. Preparation and chromatographic analysis of the oxidized growthhormone
The oxidation was performed as previously reported [1]. Theoxidized protein was characterized by reverse phase liquid chro-matography (RP-HPLC) using the method described in the Euro-pean Pharmacopoeia monograph for hGH [19]. The separationwas performed on a Waters Alliance HT 2790 HPLC system coupledto a Waters 2487 UV–Vis detector (Waters AG, Switzerland) and aBruker Esquire 3000+ mass spectrometer (Bruker Daltonics GmbH,Switzerland). The chromatographic peaks were identified by tryp-tic digestion and HPLC-MS as previously reported [1].
2.3. Intrinsic fluorescence spectroscopy
The intrinsic fluorescence of the protein was measured using aSPEX Fluoromax fluorescence spectrometer (Horiba Jobin YvonInc., US). Fluorescence spectra were recorded at 25 �C in a quartzcuvette with a 1 cm pathlength. Settings for the measurementswere the following: excitation wavelength at 280 nm, excitationslit of 0.5 mm, emission slit of 1.0 mm, and integration time of0.1 s. The emission of the tryptophan residue Trp86 was monitoredbetween 295 nm and 450 nm by using an optical filter (10% lighttransmission) to avoid saturation of the detector. The fluorescenceof both the native and oxidized protein was measured in differentbuffers, listed in Table 1, after preparation (t = 0) and after2 months storage at 37 �C. Solutions of oxidized and non-oxidizedr-hGH in different buffers were prepared by dilution from a stocksolution of r-hGH in water. The concentration of both oxidizedand native hGH was 4 mg/mL in the final formulations.
2.4. Aggregation kinetics at 37 �C measured by 90� light scattering
The kinetics of protein aggregation was monitored using the 90�static light scattering signal. The 90� light scattering of the sampleswas measured with a SPEX Fluoromax fluorescence spectrometer(Horiba Jobin Yvon Inc., US) operating in synchronous mode. In thistype of experiment, the light scattering signal at a chosen wave-length is monitored over a period of time; the resulting plot showsthe variations of the light scattering of the sample over time andprovides an indication of the aggregation kinetics. Disposable poly-styrene cells with a 1 cm pathlength were used; the cells werethermostated at 37 �C in a water bath, and the synchronous signalat 500 nm was recorded. The measurements were performed in thefollowing order: first, the light scattering of the buffer alone wasrecorded for 2 min to obtain a baseline signal; then, the proteinwas added to the buffer solution and mixed. The light scatteringsignal was measured continuously. Both excitation and emissionslits were 1 mm, the integration time was 1 s, and the signal wasrecorded every 10 s. The period of time for the analyses varied be-tween 1 h and 10 h based on the aggregation rate of the samples.All the measurements were carried out without stirring. Proteinconcentration was 8 mg/mL. For those solutions which becameturbid by eye during the experiment, an optical filter (10% lighttransmission) was used.
2.5. Size exclusion chromatography
The percentages of soluble dimers and higher molecular weightspecies of both proteins at different pH values were measured atthe beginning of the stability study (t = 0) and after 2 months ofstorage at 37 �C. For both oxidized and non-oxidized r-hGH, threereplicates per pH value were measured. Protein concentrationwas about 4.5 mg/mL in all samples. The separation was achievedon a Waters Alliance HT 2790 pump with a Waters BioSuite™ UHRSEC (4.6 � 300 mm, 125 Å, 4 lm) (Waters AG, Switzerland) column

44 F. Mulinacci et al. / European Journal of Pharmaceutics and Biopharmaceutics 85 (2013) 42–52
operating at room temperature with a flow rate of 0.3 mL/min. Themobile phase was the same as described in the European Pharma-copoeia monograph for hGH [19]: 63 mM phosphate buffer pH 7.0and 1-propanol (97:3 v/v). The elution was monitored at 280 nmwith a Waters 2487 UV–Vis detector (Waters AG, Switzerland),and the percentage of the different r-hGH species were given asarea-under-curve (AUC) at 280 nm.
2.6. Stability at 37 �C measured by Nile Red fluorescence spectroscopy
The intensity of Nile Red fluorescence in the different formula-tions containing oxidized and non-oxidized r-hGH was measuredwith a Tecan Safire2™ (Tecan Group Ltd., Männedorf, Switzerland)microplate reader. Prior to the start of the kinetic at 37 �C, Nile Redwas added to the protein samples to a final concentration of 1 lM.All the samples were incubated and measured in the Safire platereader at a temperature of 37 �C for 2 weeks. The dye was excitedat 550 nm, and its fluorescence emission was recorded at 620 nm;bandwidths of 7 nm were used for both the excitation and theemission. Five measurements per sample were acquired with anintegration time of 40 ls. The fluorescence emission spectra weremeasured using the bottom optics. Three replicates per samplewere measured. The concentration of both oxidized and non-oxi-dized r-hGH was 4 mg/mL.
2.7. Fluorescence microscopy with Nile Red staining
The presence of aggregates at the beginning of the stabilitystudy (t = 0) and after 2 months storage at 37 �C was investigatedby fluorescence microscopy with the use of a staining agent. Thedye used for the staining was Nile Red, at a concentration of1 lM. Analyses were performed as described by Demeule et al.[23]. The microscope employed for the observations was an AxioImager Z1 microscope (Zeiss, Germany) equipped with a mercurydischarge lamp for fluorescence microscopy. A Zeiss filter cubeno. 15 was used (EX BP 546/12, BS FT 580, EM LP 590). The pictureswere acquired with an AxioCam MRm camera (Zeiss, Germany),with a 10X objective and processed using the AxioVision v4.4(Zeiss, Germany).
Fig. 1. Structure of human growth hormone. The three methionine residues Met14,Met125, and Met170 are marked in yellow; the tryptophan residue Trp86 is marked inblue. Image is drawn from published coordinates (PDB ID 1A22) by using PyMOLsoftware (Delano Scientific, California, USA). (For interpretation of the references tocolor in this figure legend, the reader is referred to the web version of this article.)
2.8. Freezing/thawing stability
The role of the oxidation on the freeze/thaw induced aggrega-tion of r-hGH was investigated in different buffers. For the experi-ments, samples were frozen at �20 �C in a Whirlpool Easytronicfreezer (Bauknecht A.G., Lenzberg, Switzerland). Before each thaw-ing cycle, samples were stored at least 1 day at �20 �C. The effect ofthree successive freeze/thaw cycles was investigated by Nile Redfluorescence spectroscopy by using a Tecan Safire2™ (Tecan GroupLtd., Männedorf, Switzerland) microplate reader, Nile Red fluores-cence microscopy using an Axio Imager Z1 microscope (Zeiss, Ger-many) and 90� light scatter using a SPEX Fluoromax spectrometer(Horiba Jobin Yvon Inc, Stanmore, UK). The following settings wereused: (a) Nile Red fluorescence: excitation 550 nm and emissionbetween 570 and 750 nm; excitation and emission slits of 7 nmboth; 3 scans per sample with an integration time of 40 ls; mea-surements performed at RT with the bottom optics; (b) Nile Redmicroscopy: same settings used for the stability at 37 �C, previ-ously described; (c) 90� light scattering: acquisition range between450 and 750 nm, with excitation and emission slits of 1 mm both;protein concentration between 0.5 mg/mL and 8 mg/mL. All mea-surements were performed at room temperature. After thawing,samples were equilibrated at RT prior to analyses. Protein concen-tration was 4 mg/mL unless otherwise specified.
3. Results and discussions
3.1. Percentage of oxidation after treatment with hydrogen peroxide
As previously reported [1], the treatment with hydrogen perox-ide resulted in the oxidation of more than 90% of r-hGH. The oxida-tion by hydrogen peroxide involved only the methionine residuesin position Met14 and Met125; no oxidation of the methionine res-idue in position Met170 or of other amino acid residues was found.
3.2. Intrinsic fluorescence spectroscopy
Human growth hormone contains eight tyrosine residues andone tryptophan residue in position Trp86; this tryptophan residueis the major contributor to the intrinsic fluorescence of the protein.In the native conformation of hGH, the tryptophan residue is par-tially buried inside the protein and far from the methionine residueMet14 and Met125 (Fig. 1). The fluorescence emission of Trp86 wasused to investigate possible conformation changes and aggregationupon 2 months storage at 37 �C.
Intrinsic fluorescence spectroscopy is a powerful tool whichprovides conformational and structural information of proteins.The source of the intrinsic fluorescence of protein resides in thearomatic residues tryptophan, tyrosine, and phenylalanine. Eachof them has a typical fluorescence spectrum, with the position ofthe maximum that varies based on the residue and its electronicenvironment [24]. Due to their hydrophobic nature, these residuesare often buried or partially buried inside the protein core. How-ever, a conformation change can expose these residues to the sur-face of the protein, in closer contact with the aqueous polarenvironment. If this is the case, a shift of the kmax, the wavelengthat which the fluorescence intensity is maximal, toward lower fre-quencies (red shift) is often observed.
At the beginning of the stability study, the maximum of fluores-cence emission was at 337 nm for both native and oxidized
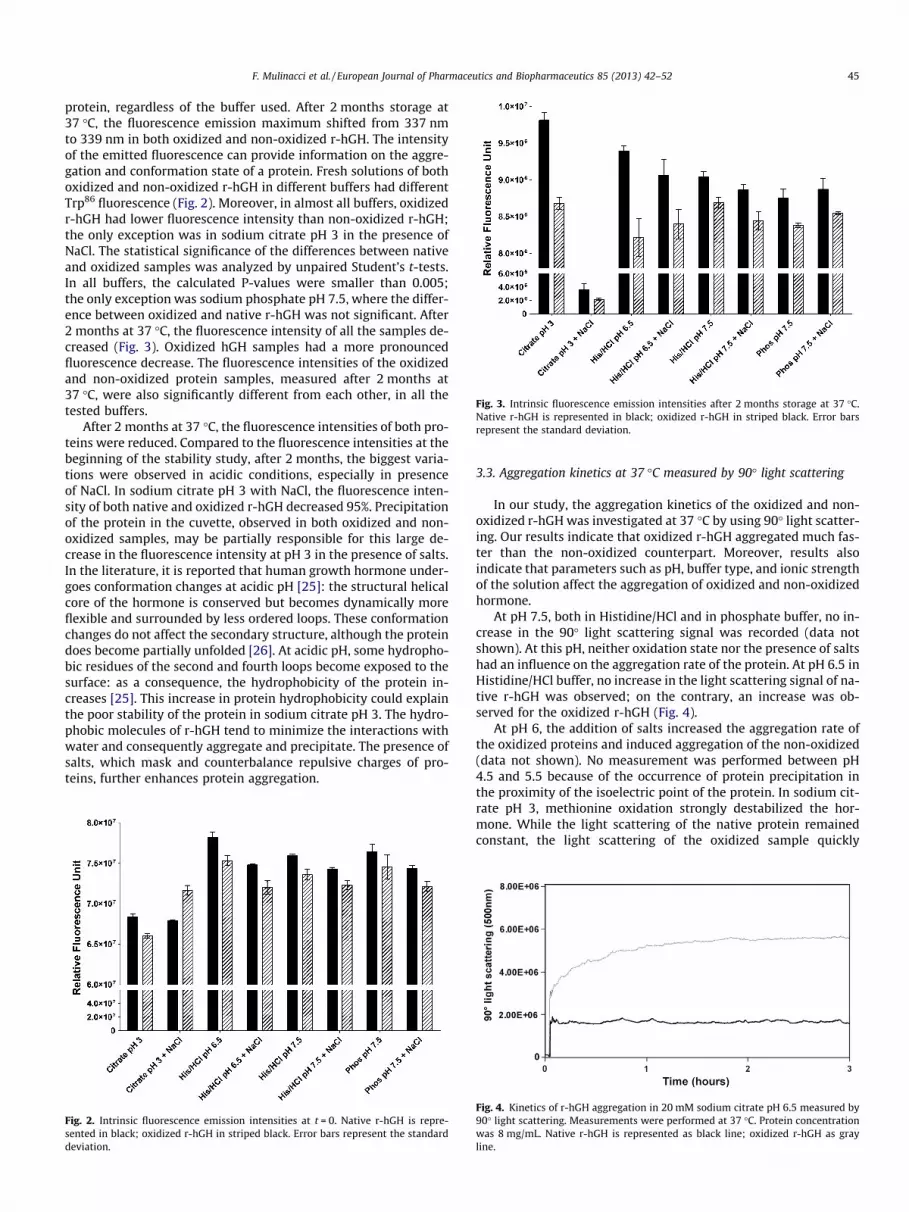
Fig. 3. Intrinsic fluorescence emission intensities after 2 months storage at 37 �C.Native r-hGH is represented in black; oxidized r-hGH in striped black. Error barsrepresent the standard deviation.
F. Mulinacci et al. / European Journal of Pharmaceutics and Biopharmaceutics 85 (2013) 42–52 45
protein, regardless of the buffer used. After 2 months storage at37 �C, the fluorescence emission maximum shifted from 337 nmto 339 nm in both oxidized and non-oxidized r-hGH. The intensityof the emitted fluorescence can provide information on the aggre-gation and conformation state of a protein. Fresh solutions of bothoxidized and non-oxidized r-hGH in different buffers had differentTrp86 fluorescence (Fig. 2). Moreover, in almost all buffers, oxidizedr-hGH had lower fluorescence intensity than non-oxidized r-hGH;the only exception was in sodium citrate pH 3 in the presence ofNaCl. The statistical significance of the differences between nativeand oxidized samples was analyzed by unpaired Student’s t-tests.In all buffers, the calculated P-values were smaller than 0.005;the only exception was sodium phosphate pH 7.5, where the differ-ence between oxidized and native r-hGH was not significant. After2 months at 37 �C, the fluorescence intensity of all the samples de-creased (Fig. 3). Oxidized hGH samples had a more pronouncedfluorescence decrease. The fluorescence intensities of the oxidizedand non-oxidized protein samples, measured after 2 months at37 �C, were also significantly different from each other, in all thetested buffers.
After 2 months at 37 �C, the fluorescence intensities of both pro-teins were reduced. Compared to the fluorescence intensities at thebeginning of the stability study, after 2 months, the biggest varia-tions were observed in acidic conditions, especially in presenceof NaCl. In sodium citrate pH 3 with NaCl, the fluorescence inten-sity of both native and oxidized r-hGH decreased 95%. Precipitationof the protein in the cuvette, observed in both oxidized and non-oxidized samples, may be partially responsible for this large de-crease in the fluorescence intensity at pH 3 in the presence of salts.In the literature, it is reported that human growth hormone under-goes conformation changes at acidic pH [25]: the structural helicalcore of the hormone is conserved but becomes dynamically moreflexible and surrounded by less ordered loops. These conformationchanges do not affect the secondary structure, although the proteindoes become partially unfolded [26]. At acidic pH, some hydropho-bic residues of the second and fourth loops become exposed to thesurface: as a consequence, the hydrophobicity of the protein in-creases [25]. This increase in protein hydrophobicity could explainthe poor stability of the protein in sodium citrate pH 3. The hydro-phobic molecules of r-hGH tend to minimize the interactions withwater and consequently aggregate and precipitate. The presence ofsalts, which mask and counterbalance repulsive charges of pro-teins, further enhances protein aggregation.
Fig. 2. Intrinsic fluorescence emission intensities at t = 0. Native r-hGH is repre-sented in black; oxidized r-hGH in striped black. Error bars represent the standarddeviation.
3.3. Aggregation kinetics at 37 �C measured by 90� light scattering
In our study, the aggregation kinetics of the oxidized and non-oxidized r-hGH was investigated at 37 �C by using 90� light scatter-ing. Our results indicate that oxidized r-hGH aggregated much fas-ter than the non-oxidized counterpart. Moreover, results alsoindicate that parameters such as pH, buffer type, and ionic strengthof the solution affect the aggregation of oxidized and non-oxidizedhormone.
At pH 7.5, both in Histidine/HCl and in phosphate buffer, no in-crease in the 90� light scattering signal was recorded (data notshown). At this pH, neither oxidation state nor the presence of saltshad an influence on the aggregation rate of the protein. At pH 6.5 inHistidine/HCl buffer, no increase in the light scattering signal of na-tive r-hGH was observed; on the contrary, an increase was ob-served for the oxidized r-hGH (Fig. 4).
At pH 6, the addition of salts increased the aggregation rate ofthe oxidized proteins and induced aggregation of the non-oxidized(data not shown). No measurement was performed between pH4.5 and 5.5 because of the occurrence of protein precipitation inthe proximity of the isoelectric point of the protein. In sodium cit-rate pH 3, methionine oxidation strongly destabilized the hor-mone. While the light scattering of the native protein remainedconstant, the light scattering of the oxidized sample quickly
Fig. 4. Kinetics of r-hGH aggregation in 20 mM sodium citrate pH 6.5 measured by90� light scattering. Measurements were performed at 37 �C. Protein concentrationwas 8 mg/mL. Native r-hGH is represented as black line; oxidized r-hGH as grayline.

Fig. 5. Kinetics of r-hGH aggregation in buffer pH 3 measured by 90� lightscattering. Measurements were performed at 37 �C. Protein concentration was8 mg/mL. Native r-hGH is represented as black line; oxidized r-hGH as gray line. (A)Sodium citrate 20 mM pH 3; (B) sodium citrate 20 mM pH 3 + 100 mM NaCl.
Fig. 6. Percentages of r-hGH monomer measured by size exclusion chromatogra-phy. (A) Results at t = 0; (B) results after 2 months storage at 37 �C. Native r-hGH isrepresented by black bars; oxidized r-hGH by white dotted bars. Three replicatesper sample were measured; error bars represent the standard deviation of theresults.
46 F. Mulinacci et al. / European Journal of Pharmaceutics and Biopharmaceutics 85 (2013) 42–52
increased (Fig. 5). At pH 6, the addition of salt enhanced the rateand intensity of aggregation of both oxidized and non-oxidized r-hGH. While in absence of NaCl, the light scattering of non-oxidizedr-hGH remained constant, in presence of NaCl an increase in thelight scattering of non-oxidized hormone was observed. The desta-bilizing effect of salt was even more pronounced on the oxidizedhGH, where light scattering of the sample increased more than10-folds (Fig. 5B).
Protein aggregation is an important phenomenon which hasconsequences both in vivo and in vitro. In vivo, protein aggregationis often associated with neurodegenerative diseases such as Alzhei-mer’s [27,28], Parkinson’s [29,30], or prion diseases [31]; in vitro, itinterferes with the production and storage of therapeutics.
An understanding of the mechanisms behind this phenomenoncan help to obtain more stable biotherapeutics. In solution, pro-teins adopt different conformations all in equilibrium with eachother; in some of these possible conformations, hydrophobic resi-dues of the proteins are more exposed to the surface. The aggrega-tion pathway of proteins is often described to proceed through anintermediate native-like state In where the protein have partiallylost of native conformations to expose some hydrophobic residuesto the surface [6,32]. This intermediate In can proceed back to thenative state N or further unfold to other intermediate states, asindicated in
N� In� In�1 . . . � I1�U! A ð1Þ
The final state is the unfolded state U. In this state, proteins tendto form aggregates [6] in order to minimize the interaction be-tween these hydrophobic residues and water. The reaction con-stants for the formation of the different native-like intermediatestates (In . . . I1), unfolded state (U), and aggregate (A) vary among
different proteins. When the rate constants for the intermediatestates are much smaller than for the unfolded or aggregated state,a lag-phase for the aggregation can be observed.
At pH 7.5, the oxidation of the two partially exposed methio-nine residue Met14 and Met125 did not have an impact on the kinet-ics of aggregation of r-hGH. This result can indicate that at neutralpH, both the native and oxidized r-hGH proceed through a similaraggregation pathway. In this pathway, the increase in polarity ofthe methionine side-chain of the oxidized r-hGH has no effect.Nevertheless, it cannot be excluded that the presence of lag-phasesin both oxidized and non-oxidized r-hGH samples prevented usfrom observing differences in the aggregation rate of the two sam-ples at this pH.
At lower pH values, the influence of the oxidized methionineresidues on r-hGH aggregation was more pronounced. The 90�light scattering signal of oxidized r-hGH increased at pH 6.5 andno lag-phase was observed; at the same pH value, the light scatter-ing signal of non-oxidized hormone remained constant. Enhance-ment of methionine-oxidation-induced r-hGH aggregation wasmaximal at pH 3, where a higher increase in the 90� light scatteringsignal of oxidized r-hGH was observed (Fig. 5A). In sodium citratepH 3, no increase in the light scattering signal of the non-oxidizedhormone was recorded after 3 h. Addition of NaCl increased thelight scattering signal of oxidized r-hGH by approximately 10-fold

Fig. 7. Total area of the SEC peaks of r-hGH. Protein elution was monitored at280 nm with a UV–Vis detector. (A) Results at t = 0; (B) results after 2 monthsstorage at 37 �C. Native r-hGH is represented by black bars; oxidized r-hGH bydotted bars. Each sample was measured in triplicate; error bars represent thestandard deviation of the results.
Fig. 8. Protein concentration in the various solutions of native and oxidized r-hGHafter 2 months at 37 �C. Concentrations were measured by UV spectroscopy. Nativer-hGH is represented by black bars; oxidized r-hGH by white bars. Each sample wasmeasured in triplicate; error bars represent the standard deviation of the results.
Fig. 9. Corrected percentages of monomer r-hGH in the various protein solutionsafter 2 months at 37 �C. Percentages are calculated by r-hGH monomer percentagesobtained by SEC analyses corrected for the area of the chromatographic peaks.Native r-hGH is represented by black bars; oxidized r-hGH by white dotted bars.Each sample was measured in triplicate; error bars represent the standard deviationof the results.
F. Mulinacci et al. / European Journal of Pharmaceutics and Biopharmaceutics 85 (2013) 42–52 47
(Fig. 5B). The presence of NaCl contributed to destabilize also thenon-oxidized hormone, as shown in Fig. 5B.
The destabilizing effect of acidic buffer on the human growthhormone has already been described [25,26]. Furthermore, ourprevious results indicated that the stability of the protein, analyzedby thermal methods, is strongly reduced in acidic buffer [2]. In ourexperiments, we found that the melting temperatures of the nativeand oxidized r-hGH at pH 3 were Tm = 69 �C and Tm = 49 �C, respec-tively, showing that the oxidized protein was less stable than thenon-oxidized [2]. Those findings are in agreement with the aggre-gation kinetics results. In the presence of NaCl, the aggregation rateof both proteins is increased. These findings are also in agreementwith the melting temperature values previously measured in pres-ence of NaCl: Tm = 56 �C for the native and Tm = 27 �C for the oxi-dized r-hGH [2].
3.4. Size exclusion chromatography
The content of soluble r-hGH aggregates after 2 months storageat 37 �C was addressed by SEC. At time t = 0, all the samples of bothnative and oxidized r-hGH had a monomer content between 96%and 99% (Fig. 6A). After 2 months, small variations in the percent-age of monomeric r-hGH were observed in all the samples; theonly exceptions were samples of both oxidized and native r-hGH
in sodium citrate pH 3 with NaCl. In this aqueous condition, bothprotein samples were almost completely aggregated: the averagemonomer contents for the native and oxidized r-hGH sampleswere 13% and 3%, respectively. At all other pH values, the averagemonomeric content was 95% (Fig. 6B). In our experiments, detec-tion was based on the measurement of absorbance at 280 nm; areaof the peaks was indicated as area under the curve at 280 nm. Atthe beginning of the stability study (t = 0), some difference in peakareas at 280 nm between the samples was observed (Fig. 7A); thesedifferences were due to the nature of the protein (i.e. oxidized ornon-oxidized) and to the pH value. For example, the areas of thehGH peaks in citrate pH 3 are less than half of the areas in phos-phate pH 7.5. These data show that in some buffers, aggregateswere already present at the beginning of the stability study sincenot all the injected protein was eluted from the column.
After 2 months at 37 �C, decreases in the peak areas of the var-ious solutions of oxidized and non-oxidized r-hGH were found(Fig. 7B). The biggest decreases were found in protein samples atpH 3 with NaCl, suggesting that formation of big and poorly solubleaggregates occurred. In the absence of NaCl at pH 3, the total peakarea of oxidized r-hGH was less than that of the native protein.

Fig. 10. Changes in time of Nile Red emission intensity in different solutions of native and oxidized r-hGH as function of time. In the left column, native r-hGH results arepresented; in the right column, oxidized r-hGH results. From the top to the bottom, Nile Red emission intensities in sodium citrate pH 3 (A and B); Histidine/HCl pH 6.5 (C andD); Histidine/HCl pH 7.5 (E and F); sodium phosphate pH 7.5 (G and H). Full line represent Nile Red emission in absence of NaCl; broken line in presence of 100 mM NaCl.Protein concentration was 4 mg/mL and temperature was 37 �C. Each sample was measured in triplicate; error bars represent the standard deviation of the results.
48 F. Mulinacci et al. / European Journal of Pharmaceutics and Biopharmaceutics 85 (2013) 42–52

Fig. 11. Human growth hormone aggregates formed in 20 mM phosphate buffer pH7.5 with 100 mM NaCl after 2 months at 37 �C. Aggregates are stained with 1 lMNile Red. On the left, native r-hGH; on the right, oxidized r-hGH.
Fig. 12. Oxidized r-hGH aggregates in 20 mM buffer pH 7.5 with 100 mM NaClstained with 1 lM Nile Red. On the left, protein in Histidine buffer; on the right,protein in phosphate buffer. Both samples were measured after 2 months at 37 �C.
F. Mulinacci et al. / European Journal of Pharmaceutics and Biopharmaceutics 85 (2013) 42–52 49
These results indicate that in acidic conditions, and in the absenceof salt, oxidized protein tends to aggregate more than native. Min-or differences in peaks areas between oxidized and non-oxidized r-hGH samples were also found at other pH values. As differenceswere observed in total peaks area of the various samples, proteinconcentration was checked by UV–Vis spectroscopy. For the mea-surements, a Nanodrop 1000 UV–Vis Spectrophotometer (Ther-mo-Scientific, Delaware, USA) was used. Protein concentrationwas calculated taking A0:1%
280nm ¼ 0:82 cm�1 [33]. No differences inconcentrations were measured, except for protein samples at pH3 in the presence of NaCl where reduced protein concentrationswere found (Fig. 8). These findings confirm the hypothesis of thepresence of poorly soluble aggregates that could not be eluted fromthe size exclusion column.
SEC analyses did not show significant differences in the aggre-gation of oxidized and native r-hGH at pH 6.5 and 7.5: all sampleshad a monomer percentage >95%. However, the outcome of a SECanalysis is a measure of the content of soluble monomers andaggregates; the presence of insoluble or poorly soluble aggregatescan lead to an underestimation of total aggregates percentages. Away to minimize this risk is by taking into account the total peaksareas. In our study, by considering the protein not eluted from thecolumn as aggregated r-hGH, it was possible to obtain a more pre-cise evaluation of the amount of aggregates present in all r-hGHsamples after 2 months at 37 �C (Fig. 9). The total peak area of afresh solution in water of r-hGH at 4.5 mg/mL was used as refer-ence area. In this way, it was possible, for example, to find thatafter 2 months at 37 �C, oxidized and non-oxidized r-hGH in so-dium phosphate pH 7.5 had a monomer content of, respectively,68% and 81% (Fig. 9) and not 98% and 96% as previously calculatedby SEC analyses (Fig. 6B).
3.5. Stability at 37 �C measured by Nile Red fluorescence spectroscopyand microscopy
Nile Red is a fluorescent probe known to bind to hydrophobicsurfaces [34,35]. It is widely used to analyze changes in proteinconformation and aggregation [23,35–37]. In this study, physicalstability of oxidized and non-oxidized r-hGH was investigated withNile Red upon 2 months storage at 37 �C; for the study, bufferslisted in Table 1 were used. In all samples, Nile Red fluorescenceintensity quickly increased until it reached equilibrium after 24 h(Fig. 10). No significant differences were observed between thesamples at pH 6.5 and pH 7.5: neither oxidation nor presence ofsalts had an effect on the intensity of Nile Red fluorescence of r-hGH. However, at pH 3, some differences in the intensity of NileRed emission were observed. At pH 3 with NaCl, Nile Red emissionincreased about 10-fold more than at any other pH values, both fornative and oxidized r-hGH. In the absence of NaCl, the increase wasmuch higher for the oxidized r-hGH. Data in acidic condition indi-cate, as reported before in this publication with other methods,that r-hGH is destabilized by acidic conditions, in particular afteroxidation. In all other pH solutions, results show that oxidationhas no strong effect on the Nile Red intensity emission.
The intensity of Nile Red fluorescence emission increases uponbinding of the dye to hydrophobic surfaces. When proteins aggre-gate, their native conformations are lost and hydrophobic residuesmay be exposed to the surface of the proteins. Nile Red binds tothese residues, and its fluorescence intensity increases and theemission is shifted. Similar Nile Red fluorescence intensities in oxi-dized and native r-hGH samples may suggest no effect of oxidationon the unfolding and aggregation of the protein. However, even ifthe intensity is similar, the morphology and the size of r-hGH

Fig. 13. Changes in Nile Red fluorescence emission intensities of different solutions of native and oxidized r-hGH as function of the number of freeze–thaw cycles. Eachsample was measured in triplicate; error bars represent the standard deviation of the results.
Fig. 14. Relative increase in Nile Red fluorescence emission of different solutions ofnative and oxidized r-hGH after one freeze–thaw cycle. Protein solutions werefrozen at �20 �C and thawed at RT. Native r-hGH is represented by black bars;oxidized are represented by black-squared bars. Each sample was measured intriplicate; error bars represent the standard deviation of the results.
50 F. Mulinacci et al. / European Journal of Pharmaceutics and Biopharmaceutics 85 (2013) 42–52
aggregates may be different upon oxidation. In the literature, fewexamples of the effect of oxidation on the size and shape of proteinaggregates are reported: salmon calcitonin upon oxidation formsaggregates with different morphologies [15]. To study the aggrega-tion morphology, the samples were investigated by fluorescencemicroscopy after Nile Red staining, as described by Demeuleet al. [23]. Fluorescence microscopy analyses revealed differencesin the number and the size of protein aggregates between variousr-hGH samples. All samples at pH 3, both oxidized and native, withand without NaCl, contained particles large in number and size.The high number of particles present in both samples at pH 3 madeit difficult to observe differences between the native and oxidizedprotein (data not shown). At higher pH values, both the numberand the size of the particles were reduced. At pH 6.5, oxidized r-hGH samples contained more particles than native r-hGH samples;particle size was similar in both samples (data not shown). Atphysiological pH, oxidized protein contained more particles thannative r-hGH, in particular in the presence of NaCl (Fig. 11).
Analyses at pH 7.5 also showed the effect of buffer type (phos-phate instead of Histidine/HCl) on the aggregation. For example, inpresence of NaCl, oxidized r-hGH contained less particles in Histi-dine/HCl buffer than in phosphate buffer (Fig. 12). As the Nile Redfluorescence emission was similar in the two samples (Fig. 10F andH, broken lines), Nile Red microscopy results suggest that oxidizedr-hGH in Histidine/HCl might contain smaller particles which aretoo small to be visualized.
3.6. Stability after freezing/thawing
Therapeutic proteins are routinely stored as frozen solutionsboth during intermediate stages of production of the bulk drugsubstance and, often, during the long term storage of the final drugproduct. However, freezing and thawing of these solutions can re-sult in denaturation and aggregation of the protein [38–40]. Pro-tein aggregation during freezing and thawing has been attributedto several factors, such as conformation changes and partialunfolding caused by low temperature [38,41], concentration of sol-utes upon freezing [42], pH changes due to buffer crystallization[43,44], and exposure of the protein to ice–liquid interface. Humangrowth hormone is known to be susceptible to freeze-inducedaggregation. Eckhardt et al. [45] reported that freezing hGH resultsin the formation of insoluble aggregates; they also reported that
pH and cooling rate have an effect on freeze-induced aggregationof the hormone. No significant increase in soluble aggregates wasreported after freezing. In our study, the effect of oxidation onthe freezing/thawing induced aggregation was investigated at dif-ferent pHs with three orthogonal methods: Nile Red fluorescencespectroscopy, Nile Red microscopy and 90� light scattering. Forboth native and oxidized r-hGH, three independent samples perpH condition were prepared and analyzed. The effect of repeatedfreezing/thawing cycles was visible in all native and oxidized sam-ples. After each freezing/thawing cycle, increases in Nile Red fluo-rescence intensities of all the samples were observed (Fig. 13).
During the stability studies at 37 �C, an effect of pH on theaggregation of the protein was observed. The same effect is presenton the freeze/thaw induced aggregation. As observed for the stabil-ity studies at 37 �C, acidic conditions are the most destabilizing forr-hGH, in particular after oxidation. The Nile Red fluorescence

Fig. 15. Aggregates of human growth hormone formed in 20 mM citrate buffer pH 3with 100 mM NaCl after 3 freeze–thaw cycles at �20 �C. Particles are stained with1 lM Nile Red.
F. Mulinacci et al. / European Journal of Pharmaceutics and Biopharmaceutics 85 (2013) 42–52 51
increase was greater for the oxidized r-hGH samples (Fig. 14). Sim-ilar results were obtained with 90� light scattering analyses (datanot shown). Staining of the protein samples with Nile Red allowedvisualization of insoluble r-hGH aggregates. Differences in shapeand number of particles were observed between the samples. Oxi-dized samples presented the larger number of particles. Further-more, the number of particles was greater in samples containingNaCl.
During freezing, solutes such as protein, buffers, salts, andexcipients are concentrated; the resulting increase in protein con-centration is one of the explanations for the freeze-induced aggre-gation of proteins. Consequently, it is possible to expect thatsamples with higher concentrations of r-hGH tend to aggregatemore after freezing. As our results indicated that low pH and thepresence of salts had a strong effect on the freeze-induced aggrega-tion of r-hGH, influence of protein concentration was studied in so-dium citrate pH 3 with NaCl. Results are shown in Fig. 15. Nosignificant differences in the number of particles were observedbetween non-oxidized hGH samples at different concentrations;on the contrary, samples containing higher concentrations of oxi-dized r-hGH showed a larger number of particles. The differencescan be explained by two mechanisms of aggregation for nativeand oxidized r-hGH during freezing and thawing.
4. Conclusions
Aggregation of therapeutic proteins is one of the main issuesduring the development of biopharmaceuticals. Aggregation canoccur in liquid, frozen, and lyophilized formulations. A completeunderstanding of the mechanisms of protein aggregation is still
missing, although numerous studies have identified some causesof this phenomenon [46–50].
In this study, we showed the influence of methionine oxidationon the aggregation properties of recombinant human growth hor-mone. Oxidation of Met14 and Met125 enhanced aggregation of r-hGH both in accelerated stability at 37 �C and in freeze/thawingstudies in different formulations.
The results presented here document the importance of investi-gating and minimizing chemical degradations early during bio-therapeutical drug development.
References
[1] F. Mulinacci, S.E.J. Bell, M.A.H. Capelle, R. Gurny, T. Arvinte, Oxidizedrecombinant human growth hormone that maintains conformationalintegrity, Journal of Pharmaceutical Sciences 100 (2011) 110–122.
[2] F. Mulinacci, M.A.H. Capelle, R. Gurny, A.F. Drake, T. Arvinte, Stability of humangrowth hormone: influence of methionine oxidation on thermal folding,Journal of Pharmaceutical Sciences 100 (2011) 451–463.
[3] P.A.C. Cloos, S. Christgau, Non-enzymatic covalent modifications of proteins:mechanisms, physiological consequences and clinical applications, MatrixBiology 21 (2002) 39–52.
[4] J.E. Visick, S. Clarke, Repair, refold, recycle – how bacteria can deal withspontaneous-damage and environmental-damage to proteins, MolecularMicrobiology 16 (1995) 835–845.
[5] C.M. Smales, D.S. Pepper, D.C. James, Protein modification during anti-viralheat-treatment bioprocessing of factor VIII concentrates, factor IXconcentrates, and model proteins in the presence of sucrose, Biotechnologyand Bioengineering 77 (2002) 37–48.
[6] J.L. Cleland, M.F. Powell, S.J. Shire, The development of stable proteinformulations – a close look at protein aggregation, deamidation, andoxidation, Critical Reviews in Therapeutic Drug Carrier Systems 10 (1993)307–377.
[7] M.L. Xie, D. Vandervelde, M. Morton, R.T. Borchardt, R.L. Schowen, PH-inducedchange in the rate-determining step for the hydrolysis of the Asp/Asn-derivedcyclic-imide intermediate in protein degradation, Journal of the AmericanChemical Society 118 (1996) 8955–8956.
[8] M.L. Xie, R.L. Schowen, Secondary structure and protein deamidation, Journalof Pharmaceutical Sciences 88 (1999) 8–13.
[9] R. Thirumangalathu, S. Krishnan, P. Bondarenko, M. Speed-Ricci, T.W.Randolph, J.F. Carpenter, D.N. Brems, Oxidation of methionine residues inrecombinant human interleukin-1 receptor antagonist: implications ofconformational stability on protein oxidation kinetics, Biochemistry 46(2007) 6213–6224.
[10] W. Vogt, Oxidation of methionyl residues in proteins – tools, targets, andreversal, Free Radical Biology and Medicine 18 (1995) 93–105.
[11] J.E. Zull, S.K. Smith, R. Wiltshire, Effect of methionine oxidation and deletion ofamino-terminal residues on the conformation of parathyroid-hormone –circular-dichroism studies, Journal of Biological Chemistry 265 (1990) 5671–5676.
[12] C.B. Glaser, J. Morser, J.H. Clarke, E. Blasko, K. Mclean, I. Kuhn, R.J. Chang, J.H.Lin, L. Vilander, W.H. Andrews, D.R. Light, Oxidation of a specific methionine inthrombomodulin by activated neutrophil products blocks cofactor activity – apotential rapid mechanism for modulation of coagulation, Journal of ClinicalInvestigation 90 (1992) 2565–2573.
[13] D. Liu, D. Ren, H. Huang, J. Dankberg, R. Rosenfeld, M.J. Cocco, L. Li, D.N. Brems,R.L. Remmele, Structure and stability changes of human IgG1 Fc as aconsequence of methionine oxidation, Biochemistry 47 (2008) 5088–5100.
[14] J.L. Liu, K.V. Lu, T. Eris, V. Katta, K.R. Westcott, L.O. Narhi, H.S. Lu, In vitromethionine oxidation of recombinant human leptin, Pharmaceutical Research15 (1998) 632–640.
[15] M.C. Gaudiano, M. Colone, C. Bombelli, P. Chistolini, L. Valvo, M. Diociaiuti,Early stages of salmon calcitonin aggregation: effect induced by ageing andoxidation processes in water and in the presence of model membranes,Biochimica et Biophysica Acta – Proteins and Proteomics 1750 (2005) 134–145.
[16] L.C. Teh, L.J. Murphy, N.L. Huq, A.S. Surus, H.G. Friesen, L. Lazarus, G.E.Chapman, Methionine oxidation in human growth-hormone and humanchorionic somatomammotropin – effects on receptor-binding and biological-activities, Journal of Biological Chemistry 262 (1987) 6472–6477.
[17] G.W. Becker, R.M. Riggin, Derivative of Human Growth Hormone, US Patent5068317, in: Eli Lilly and Company (Ed.), 208 ed., vol. 168, Indianapolis, USA,1991.
[18] S.M. Van Patten, E. Hanson, R. Bernasconi, K. Zhang, P. Manavalan, E.S. Cole,J.M. McPherson, T. Edmunds, Oxidation of methionine residues inantithrombin – effects on biological activity and heparin binding, Journal ofBiological Chemistry 274 (1999) 10268–10276.
[19] Somatropin, in: European Pharmacopoeia v6.4, 2008, pp. 2931–2937.[20] R.A. Houghten, C.B. Glaser, C.H. Li, Human somatotropin: reaction with
hydrogen peroxide, Archives of Biochemistry and Biophysics 178 (1977)350–355.

52 F. Mulinacci et al. / European Journal of Pharmaceutics and Biopharmaceutics 85 (2013) 42–52
[21] A.M. Devos, M. Ultsch, A.A. Kossiakoff, Human growth-hormone andextracellular domain of its receptor – crystal-structure of the complex,Science 255 (1992) 306–312.
[22] G. Teshima, E. Canovadavis, Separation of oxidized human growth-hormonevariants by reversed-phase high-performance liquid-chromatography – effectof mobile phase ph and organic modifier, Journal of Chromatography 625(1992) 207–215.
[23] B. Demeule, R. Gurny, T. Arvinte, Detection and characterization of proteinaggregates by fluorescence microscopy, International Journal of Pharmaceutics329 (2007) 37–45.
[24] J.R. Lakowicz, B.R. Masters, Principles of fluorescence spectroscopy (ThirdEdition), Journal of Biomedical Optics 13 (2008) 029901–029902.
[25] M.R. Kasimova, S.M. Kristensen, P.W.A. Howe, T. Christensen, F. Matthiesen, J.Petersen, H.H. Sorensen, J.J. Led, NMR studies of the backbone flexibility andstructure of human growth hormone: a comparison of high and low pHconformations, Journal of Molecular Biology 318 (2002) 679–695.
[26] M.R. Defelippis, M.A. Kilcomons, M.P. Lents, K.M. Youngman, H.A. Havel, Acidstabilization of human growth-hormone equilibrium folding intermediates,Biochimica et Biophysica Acta – Protein Structure and Molecular Enzymology1247 (1995) 35–45.
[27] L. Dumery, F. Bourdel, Y. Soussan, A. Fialkowsky, S. Viale, P. Nicolas, M.Reboud-Ravaux, Beta-amyloid protein aggregation: its implication in thephysiopathology of Alzheimer’s disease, Pathologie Biologie 49 (2001) 72–85.
[28] J. Avila, Tau phosphorylation and aggregation in Alzheimer’s diseasepathology, FEBS Letters 580 (2006) 2922–2927.
[29] M. Hashimoto, E. Rockenstein, L. Crews, E. Masliah, Role of protein aggregationin mitochondrial dysfunction and neurodegeneration in Alzheimer’s andParkinson’s diseases, Neuromolecular Medicine 4 (2003) 21–35.
[30] K.S.P. McNaught, C.W. Olanow, Protein aggregation in the pathogenesis offamilial and sporadic Parkinson’s disease, Neurobiology of Aging 27 (2006)530–545.
[31] S.B. Prusiner, Molecular-biology of Prion diseases, Science 252 (1991) 1515–1522.
[32] D. Shortle, H.S. Chan, K.A. Dill, Modeling the effects of mutations on thedenatured states of proteins, Protein Science 1 (1992) 201–215.
[33] S. Wicar, M.G. Mulkerrin, G. Bathory, L.H. Khundkar, B.L. Karger,Conformational-changes in the reversed-phase liquid-chromatography ofrecombinant human growth-hormone as a function of organic-solvent – themolten globule state, Analytical Chemistry 66 (1994) 3908–3915.
[34] D.L. Sackett, J. Wolff, Nile Red as a polarity-sensitive fluorescent-probe ofhydrophobic protein surfaces, Analytical Biochemistry 167 (1987) 228–234.
[35] A. Hawe, M. Sutter, W. Jiskoot, Extrinsic fluorescent dyes as tools for proteincharacterization, Pharmaceutical Research 25 (2008) 1487–1499.
[36] M.A.H. Capelle, R. Gurny, T. Arvinte, A high throughput protein formulationplatform: case study of salmon calcitonin, Pharmaceutical Research 26 (2008)118–128.
[37] M. Sutter, S. Oliveira, N.N. Sanders, B. Lucas, A. van Hoek, M.A. Hink, A.J.W.G.Visser, S.C. De Smedt, W.E. Hennink, W. Jiskoot, Sensitive spectroscopicdetection of large and denatured protein aggregates in solution by use of thefluorescent dye Nile red, Journal of Fluorescence 17 (2007) 181–192.
[38] B.S. Bhatnagar, R.H. Bogner, M.J. Pikal, Protein stability during freezing:separation of stresses and mechanisms of protein stabilization,Pharmaceutical Development and Technology 12 (2007) 505–523.
[39] C.F. Lopez, R.K. Darst, P.J. Rossky, Mechanistic elements of protein colddenaturation, Journal of Physical Chemistry B (2008).
[40] K.A. Pikal-Cleland, J.L. Cleland, T.J. Anchordoquy, J.F. Carpenter, Effect ofglycine on pH changes and protein stability during freeze-thawing inphosphate buffer systems, Journal of Pharmaceutical Sciences 91 (2002)1969–1979.
[41] P.L. Privalov, Cold denaturation of proteins, Critical Reviews in Biochemistryand Molecular Biology 25 (1990) 281–305.
[42] M.C. Heller, J.F. Carpenter, T.W. Randolph, Protein formulation andlyophilization cycle design: prevention of damage due to freeze-concentration induced phase separation, Biotechnology and Bioengineering63 (1999) 166–174.
[43] G. Gomez, M.J. Pikal, N. Rodriguez-Hornedo, Effect of initial buffer compositionon pH changes during far-from-equilibrium freezing of sodium phosphatebuffer solutions, Pharmaceutical Research 18 (2001) 90–97.
[44] L. Vandenberg, D. Rose, Effect of freezing on the ph and composition of sodiumand potassium phosphate solutions – the reciprocal system KH2PO4–Na2HPO4–H2O, Archives of Biochemistry and Biophysics 81 (1959) 319–329.
[45] B.M. Eckhardt, J.Q. Oeswein, T.A. Bewley, Effect of freezing on aggregation ofhuman growth-hormone, Pharmaceutical Research 8 (1991) 1360–1364.
[46] C. Frieden, Protein aggregation processes: in search of the mechanism, ProteinScience 16 (2007) 2334–2344.
[47] E.Y. Chi, S. Krishnan, T.W. Randolph, J.F. Carpenter, Physical stability ofproteins in aqueous solution: mechanism and driving forces in nonnativeprotein aggregation, Pharmaceutical Research 20 (2003) 1325–1336.
[48] S.Y. Fung, C. Keyes, J. Duhamel, P. Chen, Concentration effect on theaggregation of a self-assembling oligopeptide, Biophysical Journal 85 (2003)537–548.
[49] H.C. Mahler, W. Friess, U. Grauschopf, S. Kiese, Protein aggregation: pathways,induction factors and analysis, Journal of Pharmaceutical Sciences 98 (2009)2909–2934.
[50] W. Wang, Protein aggregation and its inhibition in biopharmaceutics,International Journal of Pharmaceutics 289 (2005) 1–30.





























