Embed Size (px)
Citation preview

MAPK-dependent actin cytoskeletal reorganizationunderlies BK channel activation by insulin
Dervla O’Malley and Jenni HarveyNeurosciences Institute, Division of Pathology & Neuroscience, Ninewells Hospital & Medical School, University of Dundee, DundeeDD1 9SY, UK
Keywords: hippocampus, hyperexcitability, insulin signalling, rat
Abstract
Numerous brain regions are enriched with insulin and insulin receptors, and several lines of evidence indicate that insulin is animportant modulator of neuronal function. Indeed, recent studies have demonstrated that insulin inhibits hippocampal epileptiform-likeactivity, in part by activating large-conductance Ca2+-activated potassium (BK) channels. Moreover, the mitogen-activated proteinkinase (MAPK) signalling cascade has been found to couple insulin to BK channel activation. However, the cellular eventsdownstream of MAPK that underlie this action of insulin are unknown. Here we demonstrate that in hippocampal neurons, BK channelactivation by insulin is blocked by actin filament stabilization, suggesting that this process is dependent on the actin cytoskeleton.Stabilizing actin filaments also markedly attenuated the ability of insulin to inhibit the aberrant hippocampal synaptic activity evokedfollowing Mg2+ removal. Insulin also promoted rapid reorganization of fluorescently labelled polymerized actin filaments; an actionthat was prevented by inhibitors of MAPK activation. Moreover, in parallel studies, insulin increased the level of phospho-MAPKimmunostaining in hippocampal neurons. These data are consistent with BK channel activation by insulin involving MAPK-dependentalterations in actin dynamics. This process may have important implications for the role of insulin in regulating hippocampalexcitability.
Introduction
It is well established that insulin regulates energy metabolism via itsactions on peripheral glucose homeostasis. Insulin also enters theCNS, where it acts on hypothalamic neurons to regulate energybalance (Schwartz et al., 2000). However, insulin and insulin receptorsare expressed in many brain regions, including the hippocampus,cerebellum, cortex and olfactory bulb (Folli et al., 1994). Duringdevelopment and in adult tissue, insulin receptors have a distinct andhighly regionalized expression pattern (Heidenreich et al., 1988).However, there is still a lack of knowledge about the precise role ofinsulin in neuronal function.
Several lines of evidence implicate insulin in hippocampalassociative learning and memory processes (Wickelgren, 1998).Indeed, patients with diabetes with insulin deficiency or resistanceoften display cognitive impairments (Gispen & Biessels, 2000). Inrodent models of diabetes, deficits have been observed in spatialmemory tasks (Gispen & Biessels, 2000). Furthermore, alterations inhippocampal insulin receptor levels have been found followingspatial memory tasks (Zhao & Alkon, 2001). Insulin also modulatesthe surface expression of N-methyl-d-aspartate (NMDA), AMPA andc-aminobutyric acid (GABA)A receptors (Wan et al., 1997; Skeber-dis et al., 2001; Zhou et al., 2001). Another cellular target for insulinare large-conductance Ca2+-activated K+ (BK) channels, as recom-binant and native BK channel activity is enhanced by insulin(O’Malley & Harvey, 2004). One functional consequence of this isattenuation of hippocampal hyperexcitability, as insulin inhibits
epileptiform-like activity in a hippocampal culture model (O’Malleyet al., 2003).The insulin receptor is a receptor tyrosine kinase, which once
stimulated promotes phosphorylation of insulin receptor substrate (IRS)proteins that recruit SH2-domain kinases such as phosphoinositide3-kinase (PI 3-kinase; Cantrell, 2001). PI 3-kinase promotes phos-phorylation of phosphoinositides on the 3-position resulting predom-inately in PtdIns(3,4,5)P3, which is utilized by various secondmessengers including protein kinase B, p38 mitogen-activated proteinkinase and stress-activated protein kinase 2 (Cantrell, 2001). Insulin canalso stimulate the Ras-Raf-mitogen-activated protein kinase (MAPK)cascade. In hippocampal neurons, the MAPK pathway plays a key rolein insulin modulation of learning and memory processes (Zhao et al.,1999). BK channel activation and inhibition of hippocampal epilepti-form-like activity by insulin also involves a MAPK-dependent process(O’Malley et al., 2003; O’Malley & Harvey, 2004).The activity of numerous ion channels, including BK (Huang et al.,
2002; Piao et al., 2003; O’Malley et al., 2005), ATP-sensitive K+ (KATP;Terzic & Kurachi, 1996; Harvey et al., 2000) and NMDA channels(Rosenmund &Westbrook, 1993), are modulated by alterations in actindynamics (Janmey, 1998). Moreover, activation of KATP channels byinsulin and leptin is dependent on actin filament dynamics (Harveyet al., 2000;Mirshamsi et al., 2004). Reorganization of actin filaments isalso pivotal for BK channel activation by leptin (O’Malley et al., 2005).However, it is not clear whether insulin activation of BK channels andinhibition of hippocampal epileptiform-like activity is dependent on theactin cytoskeleton. Here we show that MAPK-dependent reorganiza-tion of actin filaments underlies BK channel activation by insulin.Moreover, the ability of insulin to inhibit hippocampal epileptiform-likeactivity is also driven by alterations in actin dynamics.
Correspondence: Dr J. Harvey, as above.E-mail: [email protected]
Received 9 August 2006, accepted 7 December 2006
European Journal of Neuroscience, Vol. 25, pp. 673–682, 2007 doi:10.1111/j.1460-9568.2007.05347.x
ª The Authors (2007). Journal Compilation ª Federation of European Neuroscience Societies and Blackwell Publishing Ltd

Materials and methods
Cell culture
Primary cultures of hippocampal neurons were prepared as describedpreviously (Shanley et al., 2002b). In brief, 1–3-day-old rat pups werekilled by cervical dislocation and the hippocampi removed, prior towashing in standard HEPES-buffered saline (HBS) consisting of (inmm): NaCl, 135; KCl, 5; CaCl2, 1; MgCl2, 1; HEPES, 10; d-glucose,25; pH 7.4. The hippocampi were then treated with a mixture ofprotease type XIVand type X (both at 0.5 mg ⁄ L; Sigma) for 25 min atroom temperature. Dissociated cells were plated onto sterile culturedishes (Falcon 3001), pretreated with poly-l-lysine (20 lg ⁄ L for1–2 h). Cultures were maintained in serum replacement medium(SR2; Sigma) in a humidified atmosphere of 5% CO2 and 95% O2 at37 �C for up to 3 weeks. Experiments were performed on neuronsbetween 4 and 14 days in culture.
Electrophysiology
Experiments were performed using cell-attached recordings to examinesingle-channel responses, as described previously (O’Malley &Harvey,2004). Currents and voltages were measured using an Axopatch 200Bamplifier. Single-channel data were analysed for current amplitude (I)and average channel activity (NfÆPo) where Nf is the number offunctional channels and Po is the open state probability. In most single-channel recordings, one–two channels were observed under control(no drugs) conditions. Single-channel data were recorded onto digitalaudiotapes and replayed for illustration on a Gould chart recorder.Recording electrodes were made from borosilicate glass capillaries andhad resistances of 7–12 MW when filled with an electrolyte solutionconsisting of (in mm): KCl, 140; CaCl2, 1; MgCl2, 1; HEPES, 10;pH 7.2. For cell-attached recordings, the bath solution was normal HBS(as described above). All solution changes were achieved by superfus-ing the bath with a gravity feed system (stop flow system) at a rate of10 mL ⁄ min, which allowed complete bath exchange within 2 min. Allonset times were corrected for exchange beginning after a dead time of20–30 s. All experiments were performed at room temperature(22–25 �C). The mean channel activity (NfÆPo) was calculated from a2-min segment or a minimum of 200 transitions.
Calcium imaging techniques
A digital epifluorescence imaging system (12 bit; Perkin-Elmer,Emeryville, CA, USA) mounted on an Olympus BX50W1 microscope(· 40 objective) was used to measure somatic changes in intracellular[Ca2+]i. Cells were incubated with the Ca2+-sensitive dye, fura-2 AM(6 lm; 40–60 min; room temperature). Dye loading and subsequentexperiments were performed in HBS consisting of (in mm): NaCl,135; HEPES, 10; KCl, 5; CaCl2, 1; glycine, 0.01; d-glucose, 25;pH 7.4, with or without added MgCl2 (1 mm), at room temperature(22–25 �C). Ratiometric images (340 ⁄ 380 nm excitation; 510 emis-sion) were collected at 2-s intervals and data are expressed as changesin fluorescence ratio.
Cytoskeletal imaging and analysis
Initially cultured neurons were incubated with leptin (50 nm), HBS (forcontrol conditions) or the cytoskeletal disrupters, cytochalasin D(10 lm) or latrunculin B (10 lm) prior to washing the neurons rapidlyin HBS. Cells were then fixed in 4% paraformaldehyde (12–15 min)and permeabilized with Triton X-100 (0.1%) for 5 min, then treated
with 10% blocking milk for 15 min. For staining of polymerized actinfilaments, cells were incubated with Alexa 488-conjugated phalloidin(Alexa-phalloidin, Molecular Probes; 26.4 nm in HBS) for 30 min andthen rinsed in HBS. Labelling was visualized using a confocal imagingsystem (Zeiss LSM 510). Alexa-phalloidin was excited using a 488 nmline, whereas fluorescence emission was detected at 519 nm. In controlexperiments, Alexa-phalloidin labelling was completely abolished bypretreatment with phalloidin (10 lm; Sigma Aldrich, St Louis, MO,USA). The intensity of Alexa-phalloidin staining was determined usingLasersharp software (Carl Zeiss). Analysis lines (50 lm) were drawnalong randomly selected regions corresponding to the somatic plasmamembrane and processes, and the mean fluorescence intensity wascalculated. Data were obtained from at least three cells selected atrandom for each condition. Within a given experimental series allconditions for capturing images, including illumination intensity andphotomultiplier gains, were constant. In order to allow for quantifica-tion of experimental data obtained from separate days, the data werenormalized relative to the mean (somatic or process) fluorescenceintensity measured in the control neurons for each day.
Immunocytochemistry
For phospho-MAPK labelling, neurons were fixed with 4% parafor-maldehyde (12–15 min), washed with HBS, permeabilized with 0.1%Triton X-100 (5 min) and blocked with 10% blocking milk (15 min).Neurons were incubated with the primary antibody (polyclonal rabbitanti-phospho p42 ⁄ p44 MAPK antibody; Cell Signalling, USA) at adilution of 1 : 250 for 1 h at room temperature. Following washingwith HBS, neurons were then incubated with 5.6 lg ⁄ mL of an Alexa488-conjugated goat anti-rabbit secondary antibody (MolecularProbes) for 30 min at room temperature. Analysis lines (50 lm) weredrawn along randomly selected regions corresponding to the somaticplasma membrane and processes, and the mean fluorescence intensitywas calculated. Cytosolic labelling was analysed by comparing themean intensity of defined regions within neuronal somata (excludingthe nucleus). Immunostaining was visualized using a laser scanningconfocal microscope (LSM 510: Carl Zeiss MicroImaging) andanalysed using Zeiss Lasersharp software.
Drugs
Human recombinant insulin (Sigma Aldrich) prepared in 0.01–0.02%bovine serum albumin as a carrier was used in all experiments.LY294002, wortmannin, latrunculin B, jasplakinolide, U0126 (Calbi-ochem, La Jolla, CA, USA), PD98059 (Tocris Cookson, UK) andcytochalasin D (Sigma Aldrich) were all obtained commercially.
Analyses
All data are expressed as means ± SEM, and statistical analyses wereperformed using paired or Student’s t-test (two-tailed; 95% confidenceinterval) for comparison of means, or one-way anova (analysis ofvariance) for comparisons between multiple groups (unless otherwisestated). P < 0.05 was considered significant.
Results
Stabilization of actin filaments prevents insulin activationof BK channels
It is well known that the actin cytoskeleton anchors ion channels inthe cell membrane, and modulating actin dynamics is one way of
674 D. O’Malley and J. Harvey
ª The Authors (2007). Journal Compilation ª Federation of European Neuroscience Societies and Blackwell Publishing LtdEuropean Journal of Neuroscience, 25, 673–682

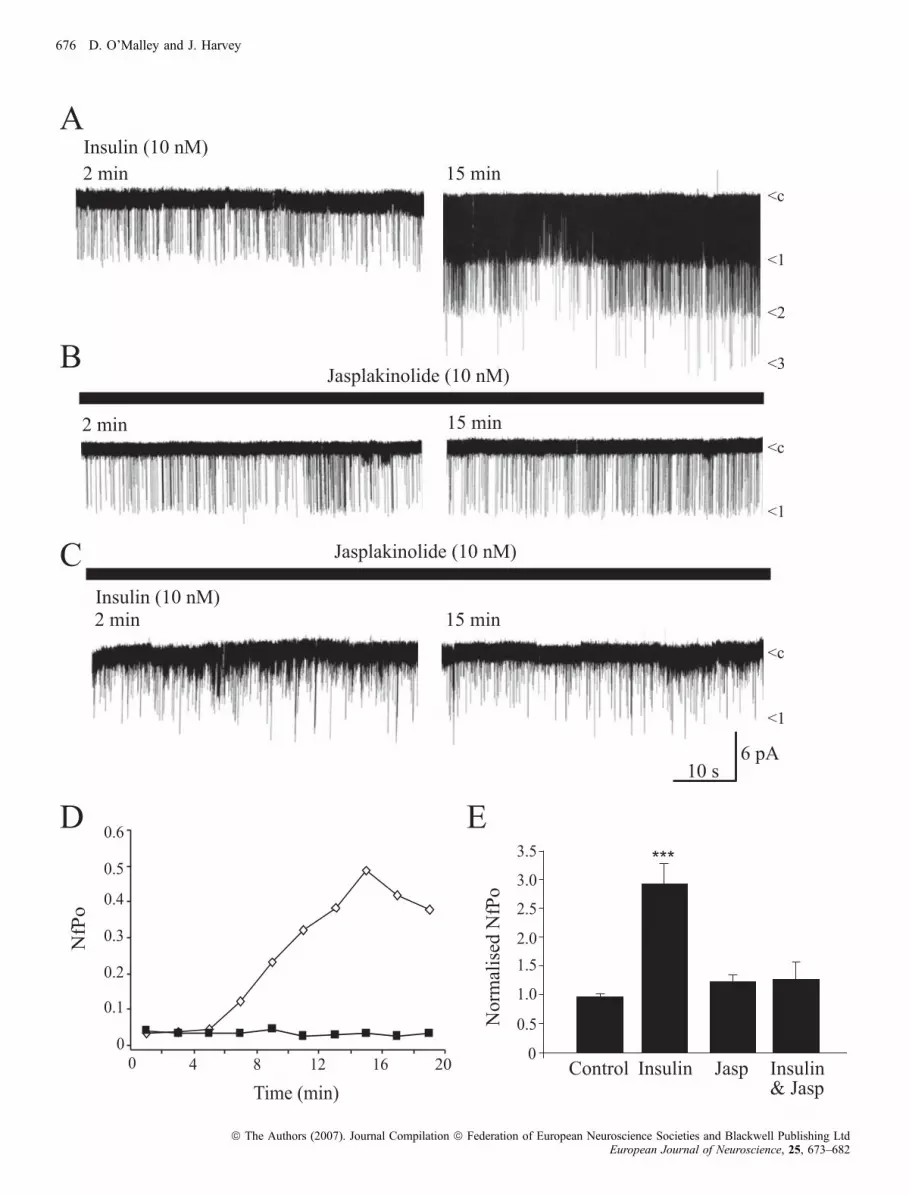
altering the activity of numerous ion channels, including neuronalBK channels (Huang et al., 2002; O’Malley et al., 2005). Thus, inorder to examine whether the actin cytoskeleton is involved in BKchannel activation by insulin, the effects of jasplakinolide, amembrane-permeable agent that stabilizes actin filaments in theirpolymerized form (Bubb et al., 1994), were examined. Duringcontrol cell-attached recordings (in the absence of jasplakinolide),application of insulin (10–50 nm; via the bath) induced a slowlydeveloping increase in BK channel activity such that the meanchannel activity (NfÆPo) increased from 0.05 ± 0.01 (at 2–4 min) to0.19 ± 0.04 (15 min after addition of insulin; n ¼ 5; P < 0.05;Fig. 1A and D). These data correlate well with our previous findings(O’Malley & Harvey, 2004). Bath application of jasplakinolide(10 nm) for 30 min had no effect on BK channels per se, such thatthe NfÆPo values obtained at 2–4 min (0.06 ± 0.01) were notsignificantly different to those obtained 15–17 min later(0.05 ± 0.02; n ¼ 6; P > 0.05; Fig. 1B). However, the ability ofinsulin to activate BK channels was prevented by jasplakinolide suchthat in neurons exposed to jasplakinolide (10 nm) for 30 min, theNfÆPo values obtained after application of insulin (10 nm) for15–17 min (0.04 ± 0.004; n ¼ 7) did not differ significantly fromthose obtained in its absence (0.04 ± 0.01; n ¼ 7; P > 0.05;Fig. 1C–E). These data are consistent with BK channel activationby insulin involving disruption of actin filaments.
Insulin inhibition of epileptiform-like activity involves actindisruption
We have shown previously that one functional consequence of BKchannel activation by insulin is the inhibition of epileptiform-likeactivity induced in hippocampal neurons by removal of extracellularMg2+ (O’Malley et al., 2003). Thus, we also examined the role ofthe actin cytoskeleton in this action of insulin using standard digitalepifluorescence imaging techniques. In agreement with our previousstudies, perfusion of cultured hippocampal neurons with Mg2+-freemedium caused an increase in somatic Ca2+ levels, and subsequentgeneration of spontaneous Ca2+ oscillations. Application of insulin(1–10 nm) depressed the global increase in Ca2+ induced followingMg2+ removal (by 51.7 ± 3.0%; n ¼ 275; P < 0.001; Fig. 2A andC), and this action was readily reversible on washout. Stabilizationof actin filaments with jasplakinolide (10 nm; 30 min) had no effecton the enhanced levels of Ca2+, or the spontaneous Ca2+ oscillationsper se; however, prior exposure to jasplakinolide attenuated theactions of insulin. Thus, in the presence of jasplakinolide, insulindepressed the Ca2+ levels by only 7.1 ± 2.1% (n ¼ 84; P < 0.001;Fig. 2A and C), suggesting that depolymerization of actin filamentsunderlies insulin-induced depression of this aberrant activity. Inparallel studies the effect of actin depolymerization on the sponta-neous Ca2+ oscillations was assessed using cytochalasin D. In amanner similar to insulin, application of cytochalasin D (50 lm)reversibly depressed the enhanced levels of Ca2+ (by 64 ± 6.2%;n ¼ 63; P < 0.01; Fig. 2B) induced following Mg2+ removal.Moreover, this action of cytochalasin D was significantly attenuatedafter exposure to jasplakinolide, such that cytochalasin D depressedthe Ca2+ levels by 4.5 ± 2.5% in the presence of jasplakinolide(n ¼ 58, P < 0.01; Fig. 2B and C). Application of insulin (10 nm),following prior exposure to cytochalasin D, resulted in 61 ± 7.9%depression in the enhanced Ca2+ levels (n ¼ 38; Fig. 2C). Thus,these data indicate that disruption of actin filaments with cytocha-lasin D inhibits the enhanced Ca2+ levels in a Mg2+-free model ofepileptiform-like activity.
Insulin promotes disruption of actin filaments in hippocampalneurons
Our data suggest that the ability of insulin to activate BK channels andto inhibit aberrant synaptic activity in the hippocampus involvesrearrangement of the actin cytoskeleton. In order to demonstratedirectly that insulin can promote actin disruption in hippocampalneurons, a fluorescently labelled marker of actin filaments, Alexa 488-conjugated phalloidin (Alexa-phalloidin) was utilized. In controlconditions (no insulin), Alexa-phalloidin staining was associated withthe plasma membrane and with actin-rich structures such as filopodia,spines and synapses. Application of insulin (50 nm; 30 min) caused asignificant decrease in Alexa-phalloidin staining in somata (reduced to68 ± 11% of control; n ¼ 9; P < 0.05) and in processes (74 ± 14% ofcontrol; n ¼ 9; P < 0.05; not illustrated), suggesting that insulinpromotes depolymerization of actin filaments. Exposure of hippo-campal neurons to lower concentrations of insulin (10 nm: 30 min)also caused actin depolymerization as the intensity of Alexa-phalloidinstaining was markedly reduced in somata (to 80 ± 13% of control;n ¼ 12; P < 0.05) and processes (to 76 ± 12% of control; n ¼ 12;P < 0.05; Fig. 3A and B), respectively. The effects of insulin onAlexa-phalloidin staining were attributable to depolymerization ofactin filaments, as the effects of insulin were markedly attenuated inthe presence of jasplakinolide, an inhibitor of actin disassembly. Thus,after exposure to jasplakinolide (10 nm) insulin reduced Alexa-phalloidin staining by 4.2 ± 1.7% (n ¼ 15; P < 0.05) and 3.1 ± 0.9%(n ¼ 19; P < 0.05) in the somata and processes, respectively. Thesedata are in agreement with other studies (Zhou et al., 2001) andindicate that insulin promotes depolymerization of the actincytoskeleton.
Disruption of actin filaments by insulin involves activation of aMAPK-dependent process
It is well documented that PI 3-kinase is a key component of insulinreceptor-driven signalling (Cantrell, 2001), and activation of thisenzyme has been linked to insulin-dependent alterations in actindynamics (Tobe et al., 2003). Thus, we compared the effects of twostructurally unrelated inhibitors of PI 3-kinase, namely LY294002 andwortmannin. Incubation of hippocampal neurons with eitherLY294002 (10 lm; 60 min; n ¼ 9) or wortmannin (50 nm; 60 min;n ¼ 12) had no effect on the intensity of Alexa-phalloidin staining atthe cell membrane or in neuronal processes. Moreover, incubationwith either agent did not attenuate the ability of insulin to promoteactin depolymerization. Thus, in the presence of LY294002, insulin(50 nm) decreased the intensity of Alexa-phalloidin staining to71 ± 6.0% of control (cell membrane; n ¼ 9; P < 0.05) and75 ± 7.2% of control (processes; n ¼ 9; P < 0.05; Fig. 3D). Similarly,insulin reduced the intensity of Alexa-phalloidin staining to 80 ± 10%at the cell membrane (n ¼ 9; P < 0.05) and 83 ± 7.0% in processes(n ¼ 9; P < 0.05) in wortmannin-treated neurons (Fig. 3C and D).The effects of PI 3-kinase inhibition on the ability of insulin to activateBK channels were also examined using cell-attached recordings. Inaccordance with our previous findings (O’Malley & Harvey, 2004),exposure to the PI 3-kinase inhibitor, wortmannin, failed to attenuateBK channel activation by insulin. Thus, in the presence of 50 nm
wortmannin, bath application of insulin (10 nm) resulted in anincrease in the mean channel activity from 0.06 ± 0.02 (at 2–4 min) to0.26 ± 0.05 (15–17 min after insulin addition; n ¼ 3; P < 0.05;Fig. 3E). Thus, together these data indicate that a PI 3-kinase-drivenprocess is unlikely to contribute to depolymerization of actin filamentsand the activation of BK channels by insulin.
MAPK-dependent actin cytoskeletal reorganization 675
ª The Authors (2007). Journal Compilation ª Federation of European Neuroscience Societies and Blackwell Publishing LtdEuropean Journal of Neuroscience, 25, 673–682
676 D. O’Malley and J. Harvey
ª The Authors (2007). Journal Compilation ª Federation of European Neuroscience Societies and Blackwell Publishing LtdEuropean Journal of Neuroscience, 25, 673–682

We have shown previously that the ability of insulin to activate BKchannels (O’Malley & Harvey, 2004) and to inhibit Mg2+-free-inducedepileptiform-like activity (O’Malley et al., 2003) involves a MAPK-dependent process. In agreement with these studies, prior inhibition ofMAPK activation with PD98059 (10 lm) completely inhibited theability of insulin to activate BK channels during cell-attachedrecordings, such that the NfÆPo values obtained after 2–4 min and15–17 min exposure to insulin (10 nm; via the bath) were 0.05 ± 0.02and 0.04 ± 0.01, respectively (n ¼ 3; P > 0.05; Fig. 4A). Asreorganization of actin filaments is also required for both actions ofinsulin, we examined if MAPK was involved in the disruption of actinfilaments induced by insulin. To explore this possibility, the effects oftwo distinct inhibitors of MAPK activation were examined. Incubationwith PD98059 (10 lm) for 60 min had no effect on Alexa-phalloidinstaining per se, such that the mean intensity of labelling at the cellmembrane and processes were 99 ± 13% (n ¼ 9; P > 0.05) and99 ± 22% (n ¼ 9; P > 0.05) of control, respectively. Similarly,U0126 (1 lm; 60 min) failed to influence the intensity of Alexa-phalloidin staining at the cell membrane (98 ± 10% of control; n ¼ 8;P > 0.05) or processes (99 ± 9.0% of control; n ¼ 8; P > 0.05).However, prior incubation with either PD98059 or U0126 blocked theeffects of insulin (50 nm) on actin filaments. Thus, in PD98059-treated neurons (Fig. 4A and B), the intensity of Alexa-phalloidinstaining after insulin (50 nm; 30 min) was 105 ± 13% of control (cellmembrane; n ¼ 9; P > 0.05) and 108 ± 9% of control (processes;n ¼ 9; P > 0.05), whereas in neurons exposed to U0126 the valueswere 104 ± 12% (cell membrane; n ¼ 8; P > 0.05) and 107 ± 11%(processes; n ¼ 8; P > 0.05; Fig. 4B), respectively. Thus, these dataare consistent with a MAPK-dependent process underlying disruptionof actin filaments by insulin.
Insulin increases phospho-MAPK immunostaining inhippocampal neurons.
Our previous studies (O’Malley et al., 2003; O’Malley & Harvey,2004) have shown that activation of a MAPK-driven pathway isessential for BK channel activation and inhibition of hippocampalexcitability by insulin. The present data also indicate that insulin-induced reorganization of actin filaments underlies both theseprocesses, and that activation of MAPK drives the changes in actindynamics by insulin. As MAPK is implicated in these processes, weexamined whether we could visualize a change in the levels ofphospho-MAPK in response to insulin. To quantify the levelsof phospho-MAPK, hippocampal neurons were incubated with ananti-phospho p42 ⁄ 44 MAPK antibody, and the intensity of stainingquantified using a combination of immunocytochemical techniquesand confocal microscopy. In control conditions, phospho-MAPKlabelling was distributed diffusely throughout the neuron, withlabelling detected at somatic regions and in neuronal processes.Intense phospho-MAPK was also observed in the nucleus, where itmay be involved in protein phosphorylation and gene transcriptionalchanges. Exposure of neurons to insulin (50 nm; 30 min) resulted in a
marked increase in the intensity of phospho-MAPK immunofluores-cence both in the cell body and in processes relative to control,although the distribution of phospho-MAPK was unaffected by insulin(Fig. 4C and D). Thus, the intensity of staining increased to121 ± 7.0% of control (n ¼ 16; P < 0.05) at the cell membrane,and 135 ± 12% of control (n ¼ 16; P < 0.05) in neuronal processes.Thus, these data demonstrate that insulin increases the levels ofphosphorylated MAPK in hippocampal neurons, which furthersupports the important role that MAPK plays in the actions of insulin.
Discussion
Several lines of evidence indicate that the hormone insulin plays aprominent role in numerous CNS functions. Indeed, insulin isimplicated in hippocampal learning and memory processes (Wickel-gren, 1998; Zhao & Alkon, 2001) and neurodegenerative diseasessuch as Alzheimer’s disease (Plum et al., 2005). Insulin can alsomodulate glutamatergic (Liu et al., 1995; Skeberdis et al., 2001; Zhouet al., 2001) and GABAergic (Wan et al., 1997; Wang et al., 2003)synaptic transmission in the hippocampus. Our previous studies havedemonstrated that another cellular target for insulin in hippocampalneurons is BK channels (O’Malley & Harvey, 2004). Moreover,activation of BK and ATP-sensitive K+ (KATP) channels by insulinresults in inhibition of aberrant synaptic induced in hippocampalneurons by Mg2+ removal (O’Malley et al., 2003). In this study wedemonstrate a role for MAPK-dependent reorganization of the actincytoskeleton in the activation of BK channels by insulin andsubsequent inhibition of Mg2+-free-induced hippocampal excitability.This insulin receptor-driven process may be a novel mechanism forregulating hippocampal excitability.It is well documented that ion channel proteins are anchored in the
cell membrane via association with the actin cytoskeleton, and thatchanges in actin dynamics can influence the activity and localizationof ion channels. Indeed, the activity of a number of different potassiumconductances, including BK, KATP and Kv4.2 channels, is influencedby dynamic changes in the actin cytoskeleton. We have shownrecently that depolymerization of actin filaments by the hormoneleptin results in BK channel activation in hippocampal culturesprepared from neonatal animals (O’Malley et al., 2005). Similarly inthis study, insulin receptor-driven actin depolymerization stimulatesBK channels in neonatal cultures. In contrast, disruption of actinfilaments attenuates BK channel activity in neurons dissociated fromadult rats (Huang et al., 2002). This suggests that developmentaldifferences may exist in the modulatory actions of the actincytoskeleton on these channels, which in turn suggests that theanchoring properties of the actin cytoskeleton with respect to BKchannels are distinct in the adult and developing brain. Alternativelythese differences may simply reflect differences in the recordingconfigurations used in these studies, as Huang et al. (2002) utilizedexcised inside-out single-channel recordings, whereas in this and ourprevious studies (O’Malley et al., 2005) channel activity wasmonitored using the cell-attached configuration.
Fig. 1. Actin filament stabilization prevents insulin-induced activation of BK channels. (A–C) Representative traces (taken at 2 and 15 min after the start ofrecording) of cell-attached recordings from hippocampal neurons with + 20 mVapplied to the recording pipette. Note that under these recording conditions, channelopenings are denoted as downward deflections (inward currents) and ‘c’ indicates the closed state of the channel. (A) Application of insulin (10 nm) via the bathcaused an increase in BK channel activity that was maintained in the presence of insulin. (B) Stabilization of actin filaments with jasplakinolide (10 nm) had noeffect on channel activity per se. However, actin filament stabilization with jasplakinolide prevented the insulin-induced increase in BK channel activity.(D) The relationship between NfÆPo (mean channel activity) and time for traces depicted in (A and C). Bath application of insulin resulted in the activation of BKchannels (open diamond), whereas prior exposure to jasplakinolide (filled squares) prevented BK channel activation by insulin. (E) Histogram of the pooled dataillustrating the normalized effects of insulin, jasplakinolide, and a combination of jasplakinolide and insulin on BK channel activity. In this figure *** representsP < 0.001.
MAPK-dependent actin cytoskeletal reorganization 677
ª The Authors (2007). Journal Compilation ª Federation of European Neuroscience Societies and Blackwell Publishing LtdEuropean Journal of Neuroscience, 25, 673–682
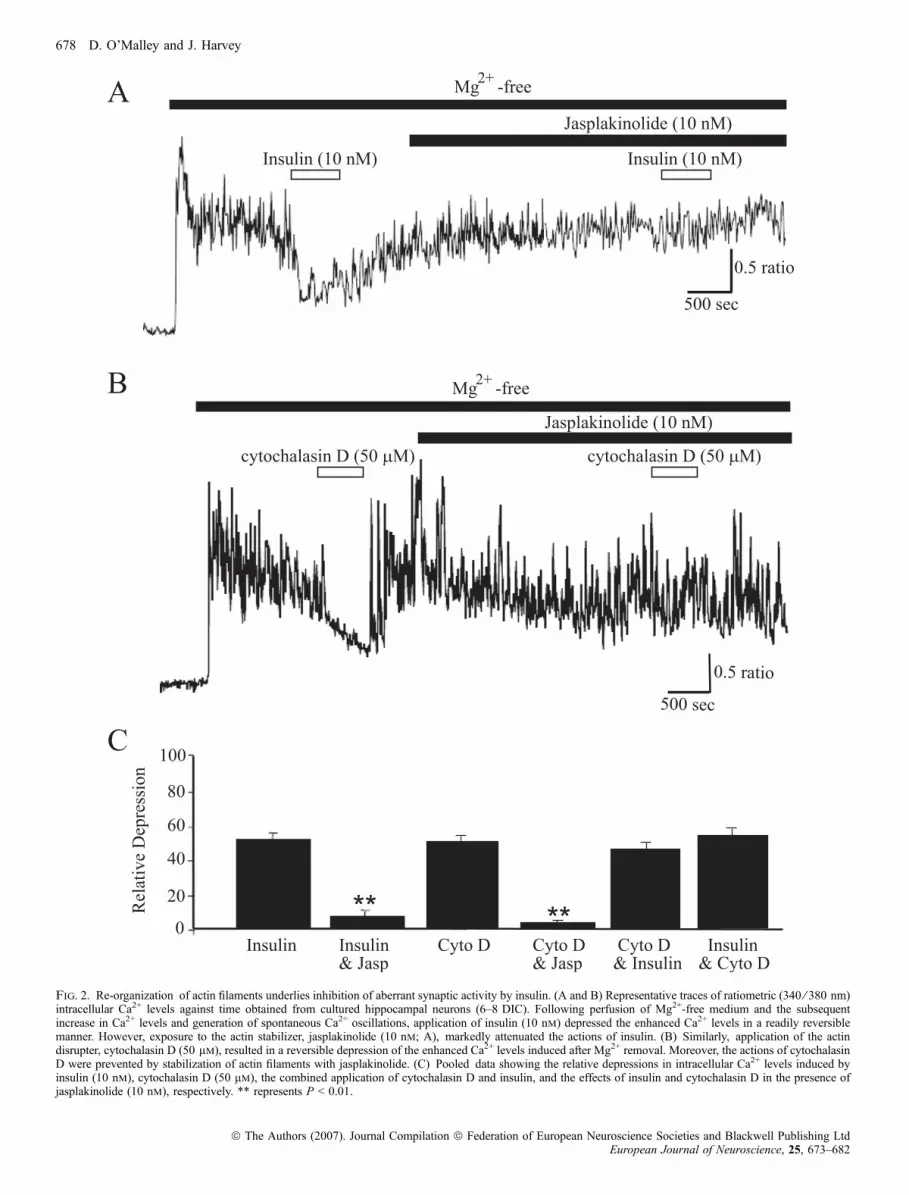
Fig. 2. Re-organization of actin filaments underlies inhibition of aberrant synaptic activity by insulin. (A and B) Representative traces of ratiometric (340 ⁄ 380 nm)intracellular Ca2+ levels against time obtained from cultured hippocampal neurons (6–8 DIC). Following perfusion of Mg2+-free medium and the subsequentincrease in Ca2+ levels and generation of spontaneous Ca2+ oscillations, application of insulin (10 nm) depressed the enhanced Ca2+ levels in a readily reversiblemanner. However, exposure to the actin stabilizer, jasplakinolide (10 nm; A), markedly attenuated the actions of insulin. (B) Similarly, application of the actindisrupter, cytochalasin D (50 lm), resulted in a reversible depression of the enhanced Ca2+ levels induced after Mg2+ removal. Moreover, the actions of cytochalasinD were prevented by stabilization of actin filaments with jasplakinolide. (C) Pooled data showing the relative depressions in intracellular Ca2+ levels induced byinsulin (10 nm), cytochalasin D (50 lm), the combined application of cytochalasin D and insulin, and the effects of insulin and cytochalasin D in the presence ofjasplakinolide (10 nm), respectively. ** represents P < 0.01.
678 D. O’Malley and J. Harvey
ª The Authors (2007). Journal Compilation ª Federation of European Neuroscience Societies and Blackwell Publishing LtdEuropean Journal of Neuroscience, 25, 673–682
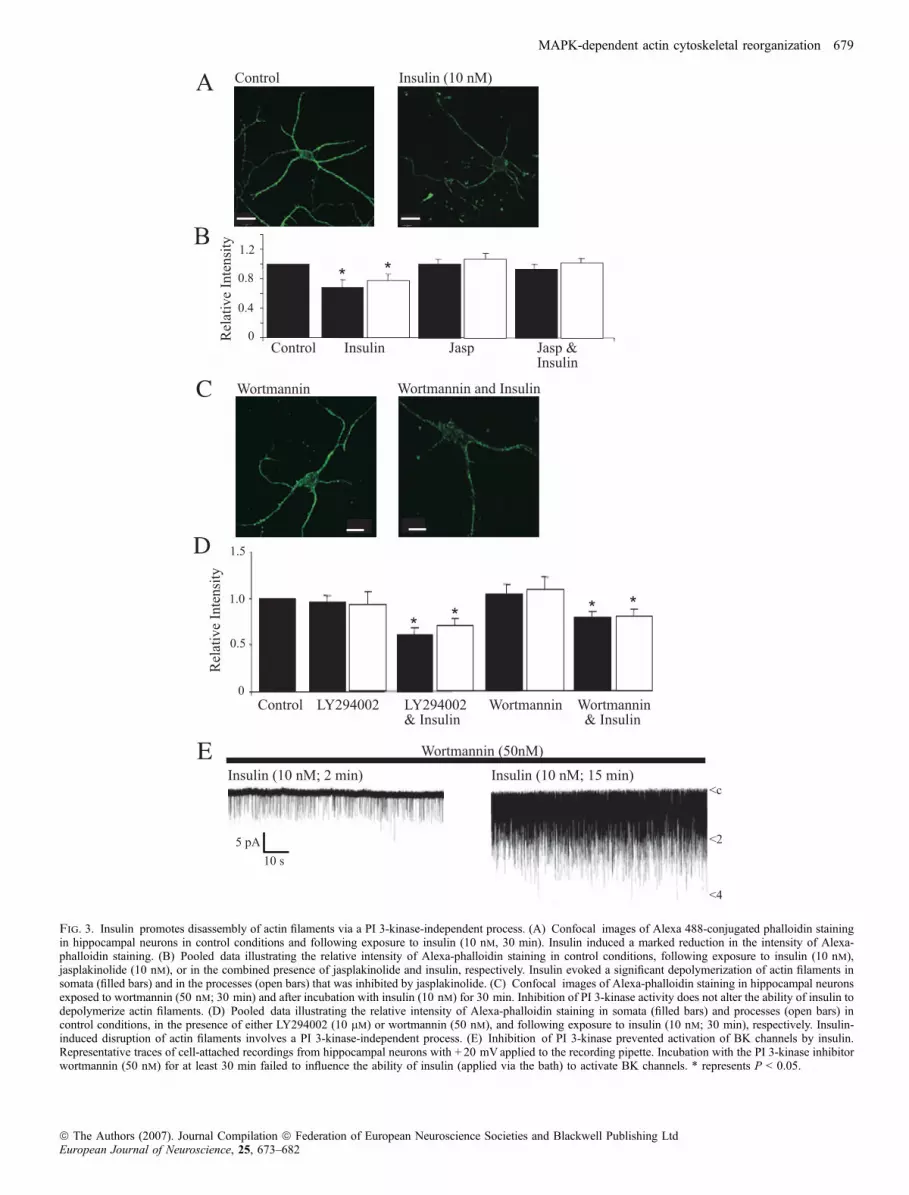
Fig. 3. Insulin promotes disassembly of actin filaments via a PI 3-kinase-independent process. (A) Confocal images of Alexa 488-conjugated phalloidin stainingin hippocampal neurons in control conditions and following exposure to insulin (10 nm, 30 min). Insulin induced a marked reduction in the intensity of Alexa-phalloidin staining. (B) Pooled data illustrating the relative intensity of Alexa-phalloidin staining in control conditions, following exposure to insulin (10 nm),jasplakinolide (10 nm), or in the combined presence of jasplakinolide and insulin, respectively. Insulin evoked a significant depolymerization of actin filaments insomata (filled bars) and in the processes (open bars) that was inhibited by jasplakinolide. (C) Confocal images of Alexa-phalloidin staining in hippocampal neuronsexposed to wortmannin (50 nm; 30 min) and after incubation with insulin (10 nm) for 30 min. Inhibition of PI 3-kinase activity does not alter the ability of insulin todepolymerize actin filaments. (D) Pooled data illustrating the relative intensity of Alexa-phalloidin staining in somata (filled bars) and processes (open bars) incontrol conditions, in the presence of either LY294002 (10 lm) or wortmannin (50 nm), and following exposure to insulin (10 nm; 30 min), respectively. Insulin-induced disruption of actin filaments involves a PI 3-kinase-independent process. (E) Inhibition of PI 3-kinase prevented activation of BK channels by insulin.Representative traces of cell-attached recordings from hippocampal neurons with + 20 mVapplied to the recording pipette. Incubation with the PI 3-kinase inhibitorwortmannin (50 nm) for at least 30 min failed to influence the ability of insulin (applied via the bath) to activate BK channels. * represents P < 0.05.
MAPK-dependent actin cytoskeletal reorganization 679
ª The Authors (2007). Journal Compilation ª Federation of European Neuroscience Societies and Blackwell Publishing LtdEuropean Journal of Neuroscience, 25, 673–682
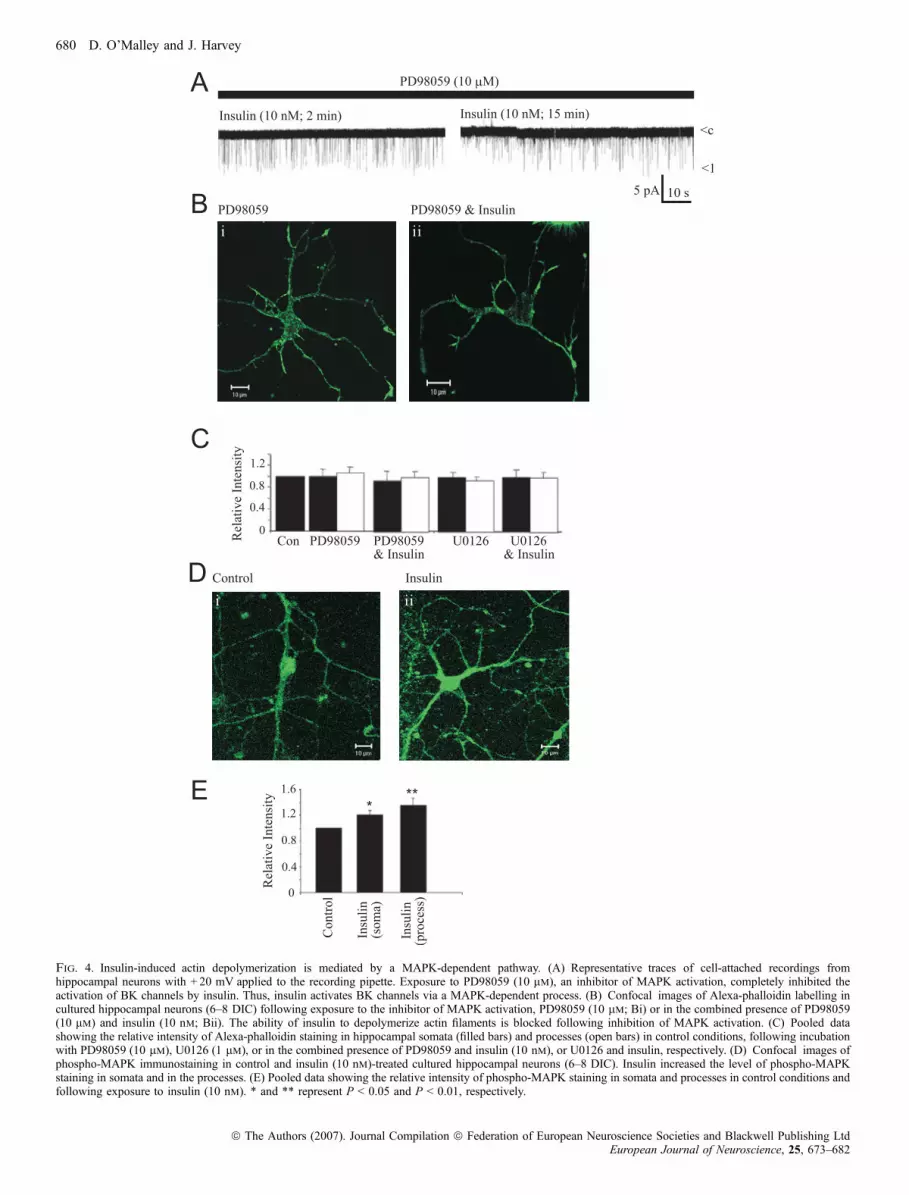
Fig. 4. Insulin-induced actin depolymerization is mediated by a MAPK-dependent pathway. (A) Representative traces of cell-attached recordings fromhippocampal neurons with + 20 mV applied to the recording pipette. Exposure to PD98059 (10 lm), an inhibitor of MAPK activation, completely inhibited theactivation of BK channels by insulin. Thus, insulin activates BK channels via a MAPK-dependent process. (B) Confocal images of Alexa-phalloidin labelling incultured hippocampal neurons (6–8 DIC) following exposure to the inhibitor of MAPK activation, PD98059 (10 lm; Bi) or in the combined presence of PD98059(10 lm) and insulin (10 nm; Bii). The ability of insulin to depolymerize actin filaments is blocked following inhibition of MAPK activation. (C) Pooled datashowing the relative intensity of Alexa-phalloidin staining in hippocampal somata (filled bars) and processes (open bars) in control conditions, following incubationwith PD98059 (10 lm), U0126 (1 lm), or in the combined presence of PD98059 and insulin (10 nm), or U0126 and insulin, respectively. (D) Confocal images ofphospho-MAPK immunostaining in control and insulin (10 nm)-treated cultured hippocampal neurons (6–8 DIC). Insulin increased the level of phospho-MAPKstaining in somata and in the processes. (E) Pooled data showing the relative intensity of phospho-MAPK staining in somata and processes in control conditions andfollowing exposure to insulin (10 nm). * and ** represent P < 0.05 and P < 0.01, respectively.
680 D. O’Malley and J. Harvey
ª The Authors (2007). Journal Compilation ª Federation of European Neuroscience Societies and Blackwell Publishing LtdEuropean Journal of Neuroscience, 25, 673–682

It is well documented that the actin cytoskeleton plays a pivotal roleas an intermediate signalling molecule for cytokine and growth factorreceptors. Indeed, our recent studies indicate that leptin activates BKchannels in the hippocampus, via PI 3-kinase-driven actin filamentdepolymerization (O’Malley et al., 2005). In this study, dynamicchanges in the actin cytoskeleton also underlie insulin-inducedactivation of hippocampal BK channels. However, in contrast to theactions of leptin, activation of a MAPK-, but not PI 3-kinase-,dependent process is critical for the insulin-driven changes in actindynamics, as specific inhibitors of MAPK activation blocked thisaction of insulin. In support of a role for MAPK in this process,treatment with insulin increased the levels of phosphorylated MAPKimmunolabelling in hippocampal neurons. Moreover, in agreementwith our previous studies (O’Malley et al., 2003; O’Malley & Harvey,2004), stimulation of BK channel activity by insulin is mediated bythe MAPK signalling cascade as the effects of insulin were markedlyattenuated by PD98059, but not wortmannin. The role of MAPK-dependent signalling in insulin-induced alterations in actin dynamicsin hippocampal neurons shows many parallels to the depolymerizationof actin filaments induced in neutrophils by cytokines such as tumournecrosis factor-alpha, as this also involves a MAPK-dependent process(Kutsuna et al., 2004). However, it is not clear at this stage howactivation of the MAPK signalling cascade by insulin influences actindynamics in hippocampal neurons. It is possible that MEKK-1 mayassociate with and alter the activity of the actin cross-linking proteina-actinin, thereby influencing actin dynamics (Christerson et al.,1999). Disruption of the actin cytoskeleton could also result frominactivation of the Rho ⁄ ROCK-LIM kinase pathway by MEK(Pawlak & Helfman, 2002). Alternatively, insulin activation of theRas-Raf-MAPK signalling pathway may promote actin cytoskeletalremodelling by dephosphorylation of cofilin and subsequent actinfilament depolymerization (Nebl et al., 2004; Pritchard et al., 2004).
Evidence is emerging that insulin and leptin receptor-drivensignalling cascades are interconnected at a number of levels inperipheral tissues. For instance, leptin attenuates the ability of insulinto phosphorylate IRS-1 in human hepatic cells (Cohen et al., 1996),whereas in insulinoma cells insulin occludes activation of KATP
channels by leptin (Harvey & Ashford, 1998). Moreover, in glucose-responsive hypothalamic neurons leptin and insulin stimulate KATP
channel activation via the PI 3-kinase signalling pathway (Spanswicket al., 1997; 2000), indicating that there is cross-talk between thesehormones in this brain region. In addition, there is evidence that PI3-kinase-driven actin filament reorganization underlies activation ofhypothalamic KATP channels by both leptin and insulin (Mirshamsiet al., 2004). In contrast, in the hippocampal formation, divergentsignalling pathways couple insulin and leptin to the activation of BKchannels (Shanley et al., 2002a; O’Malley & Harvey, 2004; O’Malleyet al., 2005) and subsequent inhibition of epileptiform-like activity(Shanley et al., 2002a; O’Malley et al., 2003), as PI 3-kinase andMAPK underlie the effects of leptin and insulin on these events,respectively. However, it is interesting to note that although distinctsignalling cascades mediate the effects of these hormones onhippocampal neurons, there is some degree of convergence asultimately reorganization of the actin cytoskeleton plays a key rolein both BK channel activation and inhibition of epileptiform-likeactivity by insulin and leptin (O’Malley et al., 2005).
It is well documented that in hippocampal neurons, BK channels arehighly expressed at somatodendritic regions and presynaptic terminals,and the role of these channels in regulating neuronal excitability isinfluenced by their cellular distribution (Hu et al., 2001). Moreover,the distribution of hippocampal BK channels appears to correlate wellwith regions that express high levels of actin such as somata, dendrites
and synapses. This suggests not only that BK channels closelyassociate with the actin cytoskeleton, but also that in agreement withthe present study dynamic alterations in the actin cytoskeletoninfluence the properties and function of hippocampal BK channels.In further support of a key role for the actin cytoskeleton in regulatingBK channel function, a recent study has shown functional linkage ofBK channels with the actin-binding protein, cortactin (Tian et al.,2006). Indeed, disruption of the cortactin–BK channel interactionprevents the regulation of BK channels by actin, suggesting thatcortactin directly links BK channels to the actin cytoskeleton and thatthis interaction is important for channel regulation. However, it islikely that the linkage of BK channels to the actin cytoskeleton viaassociation with cortactin is only a feature of postsynaptically locatedBK channels, as cortactin is predominantly expressed on cell bodiesand apical dendrites (Racz & Weinberg, 2004). Thus, this raises theinteresting possibility that distinct cellular mechanisms regulate theproperties and function of presynaptic BK channels.
Physiological significance
There is mounting evidence that the hormone insulin has widespreadactions in the CNS. A number of studies have highlighted a role forinsulin in the cellular events underlying learning and memory asinsulin-resistant rodents display deficits in hippocampal synapticplasticity and in spatial memory tasks (Gispen & Biessels, 2000). Inhumans, defects in insulin-dependent signalling cascades as well asresistance to insulin have been linked to the development ofneurodegenerative diseases such as Alzheimer’s disease (Plum et al.,2005). Our previous studies have shown that insulin also markedlyinfluences the excitability of hippocampal neurons as this hormone hasthe ability to stimulate the activation of BK channels (O’Malley et al.,2003; O’Malley & Harvey, 2004). In hippocampal neurons,postsynaptically located BK channels play an important role inregulating action potential firing rates and burst firing patterns(Lancaster & Nicoll, 1987). Moreover, as unregulated excitability inthe hippocampus is one underlying cause of temporal lobe epilepsy,activation of BK channels by insulin may be one potential way ofameliorating this hyperexcitable state. Indeed, activation of BKchannels contributes to insulin-induced inhibition of epileptiform-likeactivity evoked in hippocampal neurons (O’Malley et al., 2003). Thus,functionally, insulin receptor-driven BK channel activation may beimportant in diseases such as epilepsy. Thus, a greater understandingof neuronal insulin receptor-driven signalling pathways and thepotential CNS targets for insulin may provide novel therapeutic targetsfor treating diseases associated with deficiencies or resistance toinsulin.
Abbreviations
BK, large-conductance Ca2+-activated potassium; GABA, c-aminobutyric acid;HBS, HEPES-buffered saline; IRS, insulin receptor substrate; MAPK, mitogen-activated protein kinase; NMDA, N-methyl-d-aspartate; PI 3-kinase, phos-phoinositide 3-kinase.
References
Bubb, M.R., Senderowicz, A.M., Sausville, E.A., Duncan, K.L. & Korn, E.D.(1994) Jasplakinolide, a cytotoxic natural product, induces actin polymer-ization and competitively inhibits the binding of phalloidin to F-actin.J. Biol. Chem., 269, 14869–14871.
Cantrell, D.A. (2001) Phosphoinositide 3-kinase signalling pathways. J. CellSci., 114, 1439–1445.
MAPK-dependent actin cytoskeletal reorganization 681
ª The Authors (2007). Journal Compilation ª Federation of European Neuroscience Societies and Blackwell Publishing LtdEuropean Journal of Neuroscience, 25, 673–682

Christerson, L.B., Vanderbilt, C.A. & Cobb, M.H. (1999) MEKK1 interactswith alpha-actinin and localizes to stress fibers and focal adhesions. Cell.Motil. Cytoskeleton, 43, 186–198.
Cohen, B., Novick, D. & Rubinstein, M. (1996) Modulation of insulin activitiesby leptin. Science, 274, 1185–1188.
Folli, F., Bonfanti, L., Renard, E., Kahn, C.R. & Merighi, A. (1994) Insulinreceptor substrate-1 (IRS-1) distribution in the rat central nervous system.J. Neurosci., 14, 6412–6422.
Gispen, W.H. & Biessels, G.J. (2000) Cognition and synaptic plasticity indiabetes mellitus. Trends Neurosci., 23, 542–549.
Harvey, J. & Ashford, M.L. (1998) Insulin occludes leptin activation of ATP-sensitive K+ channels in rat CRI-G1 insulin-secreting cells. J. Physiol., 511,695–706.
Harvey, J., Hardy, S.C., Irving, A.J. & Ashford, M.L. (2000) Leptin activationof ATP-sensitive K+ (KATP) channels in rat CRI-G1 insulinoma cellsinvolves disruption of the actin cytoskeleton. J. Physiol., 527, 95–107.
Heidenreich, K.A., De Vellis, G & Gilmore, P.R. (1988) Functional propertiesof the subtype of insulin receptor found on neurons. J. Neurochem., 51, 878–887.
Hu, H., Shao, L.R., Chavoshy, S., Gu, N., Trieb, M., Behrens, R., Laake, P.,Pongs, O., Knaus, H.G., Ottersen, O.P. & Storm, J.F. (2001) PresynapticCa2+-activated K+ channels in glutamatergic hippocampal terminals andtheir role in spike repolarization and regulation of transmitter release.J. Neurosci., 21, 9585–9597.
Huang, H., Rao, Y., Sun, P. & Gong, L.W. (2002) Involvement of actincytoskeleton in modulation of Ca2+-activated K+ channels from rathippocampal CA1 pyramidal neurons. Neurosci. Lett., 332, 141–145.
Janmey, P.A. (1998) The cytoskeleton and cell signaling: component localiza-tion and mechanical coupling. Physiol. Rev., 78, 763–781.
Kutsuna, H., Suzuki, K., Kamata, N., Kato, T., Hato, F., Mizuno, K.,Kobayashi, H., Ishii, M. & Kitagawa, S. (2004) Actin reorganization andmorphological changes in human neutrophils stimulated by TNF, GM-CSF,and G-CSF: the role of MAP kinases. Am. J. Physiol. Cell. Physiol., 286,C55–C64.
Lancaster, B. & Nicoll, R.A. (1987) Properties of two calcium-activatedhyperpolarizations in rat hippocampal neurones. J. Physiol., 389, 187–203.
Liu, L., Brown, J.C. 3rd, Webster, W.W., Morrisett, R.A. & Monaghan, D.T.(1995) Insulin potentiates N-methyl-D-aspartate receptor activity in Xenopusoocytes and rat hippocampus. Neurosci. Lett., 192, 5–8.
Mirshamsi, S., Laidlaw, H.A., Ning, K., Anderson, E., Burgess, L.A., Gray, A.,Sutherland, C. & Ashford, M.L. (2004) Leptin and insulin stimulation ofsignalling pathways in arcuate nucleus neurones: PI3K dependent actinreorganization and KATP channel activation. BMC Neurosci., 5, 54.
Nebl, G., Fischer, S., Penzel, R. & Samstag, Y. (2004) Dephosphorylation ofcofilin is regulated through Ras and requires the combined activities of theRas-effectors MEK and PI3K. Cell Signal., 16, 235–243.
O’Malley, D. & Harvey, J. (2004) Insulin activates native and recombinantlarge conductance Ca2+-activated potassium channels via a mitogen-activa-ted protein kinase-dependent process. Mol. Pharmacol., 65, 1352–1363.
O’Malley, D., Irving, A.J. & Harvey, J. (2005) Leptin-induced dynamicalterations in the actin cytoskeleton mediate the activation and synapticclustering of BK channels. FASEB J., 19, 1917–1919.
O’Malley, D., Shanley, L.J. & Harvey, J. (2003) Insulin inhibits rathippocampal neurones via activation of ATP-sensitive K+ and largeconductance Ca2+-activated K+ channels. Neuropharmacology, 44, 855–863.
Pawlak, G. & Helfman, D.M. (2002) MEK mediates v-Src-induced disruptionof the actin cytoskeleton via inactivation of the Rho-ROCK-LIM kinasepathway. J. Biol. Chem., 277, 26927–26933.
Piao, L., Ho, W.K. & Earm, Y.E. (2003) Actin filaments regulate the stretchsensitivity of large-conductance, Ca2+-activated K+ channels in coronaryartery smooth muscle cells. Pflugers Arch., 446, 523–528.
Plum, L., Schubert, M. & Bruning, J.C. (2005) The role of insulin receptorsignaling in the brain. Trends Endocrinol. Metab., 16, 59–65.
Pritchard, C.A., Hayes, L., Wojnowski, L., Zimmer, A., Marais, R.M. &Norman, J.C. (2004) B-Raf acts via the ROCKII ⁄ LIMK ⁄ cofilin pathway tomaintain actin stress fibers in fibroblasts. Mol. Cell Biol., 24, 5937–5952.
Racz, B. & Weinberg, R.J. (2004) The subcellular organization of cortactin inhippocampus. J. Neurosci., 24, 10310–10317.
Rosenmund, C. & Westbrook, G.L. (1993) Calcium-induced actin depolymer-ization reduces NMDA channel activity. Neuron, 10, 805–814.
Schwartz, M.W., Woods, S.C., Porte, D. Jr, Seeley, R.J. & Baskin, D.G. (2000)Central nervous system control of food intake. Nature, 404, 661–671.
Shanley, L.J., Irving, A.J., Rae, M.G., Ashford, M.L. & Harvey, J. (2002a)Leptin inhibits rat hippocampal neurons via activation of large conductancecalcium-activated K+ channels. Nat. Neurosci., 5, 299–300.
Shanley, L.J., O’Malley, D., Irving, A.J., Ashford, M.L. & Harvey, J. (2002b)Leptin inhibits epileptiform-like activity in rat hippocampal neurons via PI3-kinase-driven activation of BK channels. J. Physiol., 545, 933–944.
Skeberdis, V.A., Lan, J., Zheng, X., Zukin, R.S. & Bennett, M.V. (2001) Insulinpromotes rapid delivery of N-methyl-D-aspartate receptors to the cell surfaceby exocytosis. Proc. Natl Acad. Sci. USA, 98, 3561–3566.
Spanswick, D., Smith, M.A., Groppi, V.E., Logan, S.D. & Ashford, M.L.(1997) Leptin inhibits hypothalamic neurons by activation of ATP-sensitivepotassium channels. Nature, 390, 521–525.
Spanswick, D., Smith, M.A., Mirshamsi, S., Routh, V.H. & Ashford, M.L.(2000) Insulin activates ATP-sensitive K+ channels in hypothalamic neuronsof lean, but not obese rats. Nat. Neurosci., 3, 757–758.
Terzic, A. & Kurachi, Y. (1996) Actin microfilament disrupters enhance KATP
channel opening in patches from guinea-pig cardiomyocytes. J. Physiol.,492, 395–404.
Tian, L., Chen, L., McClafferty, H., Sailer, C.A., Ruth, P., Knaus, H.G. &Shipston, M.J. (2006) A noncanonical SH3 domain binding motif links BKchannels to the actin cytoskeleton via the SH3 adapter cortactin. FASEB J.October 25 [Epub ahead of print].
Tobe, K., Asai, S., Matuoka, K., Yamamoto, T., Chida, K., Kaburagi, Y.,Akanuma, Y., Kuroki, T., Takenawa, T., Kimura, S., Nagai, R. &Kadowaki, T.(2003) Cytoskeletal reorganization induced by insulin: involvement ofGrb2 ⁄ Ash, Ras and phosphatidylinositol 3-kinase signalling. Genes Cells,8, 29–40.
Wan, Q., Xiong, Z.G., Man, H.Y., Ackerley, C.A., Braunton, J., Lu, W.Y.,Becker, L.E., MacDonald, J.F. & Wang, Y.T. (1997) Recruitment offunctional GABAA receptors to postsynaptic domains by insulin. Nature,388, 686–690.
Wang, Q., Liu, L., Pei, L., Ju, W., Ahmadian, G., Lu, J., Wang, Y., Liu, F. &Wang, Y.T. (2003) Control of synaptic strength, a novel function of Akt.Neuron, 38, 915–928.
Wickelgren, I. (1998) Tracking insulin to the mind. Science, 280, 517–519.Zhao, W.Q. & Alkon, D.L. (2001) Role of insulin and insulin receptor in
learning and memory. Mol. Cell. Endocrinol., 177, 125–134.Zhao, W., Chen, H., Xu, H., Moore, E., Meiri, N., Quon, M.J. & Alkon, D.L.
(1999) Brain insulin receptors and spatial memory: correlated changes ingene expression, tyrosine phosphorylation, and signaling molecules in thehippocampus of water maze trained rats. J. Biol. Chem., 274, 34893–34902.
Zhou, Q., Xiao, M. & Nicoll, R.A. (2001) Contribution of cytoskeleton to theinternalization of AMPA receptors. Proc. Natl Acad. Sci. USA, 98, 1261–1266.
682 D. O’Malley and J. Harvey
ª The Authors (2007). Journal Compilation ª Federation of European Neuroscience Societies and Blackwell Publishing LtdEuropean Journal of Neuroscience, 25, 673–682





























