Embed Size (px)
Citation preview

SHINE Manual of Procedures i Version 6 – December 19, 2016
STROKE HYPERGLYCEMIA INSULIN NETWORK EFFORT
Manual of Procedures December 19, 2016
STUDY GROUP CHAIR
Karen C. Johnston, MD, MSc
CLINICAL PRINCIPAL INVESTIGATORS Kevin Barrett, MD, MSc; Askiel Bruno, MD, MS; Christiana E. Hall, MD, MS; Karen C. Johnston,
MD, MSc
Co-INVESTIGATORS Rattan Juneja, MD; Mercedes Falciglia, MD
Mark Conaway, PhD
The NETT Statistical and Data Management Center PRINCIPAL INVESTIGATORS
Valerie Durkalski, PhD; Yuko Palesch, PhD
The NETT Clinical Coordinating Center PRINCIPAL INVESTIGATORS
William Barsan, MD; William Meurer, MD, MS
GLUCOSE MONITORING TEAM Medical Decision Network, LLC
Sei Kim
SPONSOR NIH - NINDS (U01 NS069498)
NIH-NINDS (U01-NS056975)-NETT CCC NIH-NINDS U01-NS059041-NETT SDMC

SHINE Manual of Procedures 1 Version 6– December 19, 2016
Table of Contents December 19, 2016 ....................................................................................... i
1. Introduction ......................................................................................................................... 7
1.1 Study Overview ............................................................................................................ 7
1.2 Study Objectives .......................................................................................................... 7
2. Study Process Flowchart .................................................................................................... 8
3. Screening............................................................................................................................ 9
3.1 Subject Identification .................................................................................................... 9
3.2 Screening Overview ..................................................................................................... 9
3.3 Eligibility Criteria .........................................................................................................10
3.3.1 Inclusion Criteria ..................................................................................................10
3.3.2 Exclusion Criteria .................................................................................................10
3.4 Justification and Explanation of Eligibility Criteria ........................................................11
3.5 Screen Failure Log......................................................................................................14
3.6 Prohibited Therapy......................................................................................................15
4. Enrollment Procedures .......................................................................................................15
4.1 Obtaining Informed Consent .......................................................................................15
4.2 Sample Informed Consent ..........................................................................................16
4.3 Enrollment ..................................................................................................................16
4.3.1 Randomization by WebDCU™ .............................................................................16
4.3.2 Emergency Randomization Procedures ...............................................................17
4.4 Successful Enrollment ................................................................................................17
5. Study Treatment Procedures .............................................................................................17
5.1 Treatment Procedures ................................................................................................17
5.1.1 Intervention and control group protocols ..............................................................17
5.1.2 Intervention Group ...............................................................................................18
5.1.3 Control Group Protocol ........................................................................................51
5.1.4 Considerations for Study Coordinators during the Treatment Period ....................74
5.1.5 Concomitant or Ancillary Therapy ........................................................................74
6. Event Visits ........................................................................................................................75
6.1 Study Visit Schedule ...................................................................................................75
6.2 Assessments ..............................................................................................................75
6.2.1 Blinding of follow up visits ....................................................................................75
6.2.2 mRS29-31 ...............................................................................................................75

SHINE Manual of Procedures 2 Version 6– December 19, 2016
6.2.3 BI32 ......................................................................................................................77
6.2.4 NIHSS33-35 ............................................................................................................77
6.2.5 SSQOL36 ..............................................................................................................78
6.2.6 SSQOL Administration .........................................................................................78
6.2.7 Neuro Checks and Neurological Worsening .........................................................78
6.2.8 Lacunar Stroke Etiology .......................................................................................79
7. Data Collection and Management ......................................................................................80
7.1 SHINE CRF Collection Schedule ................................................................................80
7.2 Overview .....................................................................................................................80
7.3 Data Acquisition and Central Study Database .............................................................80
7.4 Case Report Forms and Worksheets ..........................................................................81
7.4.1 Overview of Forms and Requirements .................................................................81
7.5 Guidelines for Completion of Specific CRFs (See Appendix 8) ...................................83
8. Adverse Events ..................................................................................................................83
8.1 Definitions ...................................................................................................................83
8.1.1 Adverse Event .....................................................................................................83
8.1.2 Serious Adverse Event ........................................................................................83
8.2 Anticipated Adverse Events ........................................................................................84
8.2.1 Clinically Important Adverse Events .....................................................................84
8.2.2 Assessing Relatedness of Hypoglycemic Events After End of Treatment Period .85
8.3 Adverse Event Exceptions ..........................................................................................86
8.4 Algorithm to Determine Relatedness ...........................................................................86
8.5 Adverse Event Collection and Reporting .....................................................................87
8.5.1 Study Site AE Screening ......................................................................................87
8.5.2 Adverse Event Reporting .....................................................................................87
8.5.3 SAE Narrative and Templates ..............................................................................88
9. Protocol Adherence ...........................................................................................................89
9.1 Protocol Deviations .....................................................................................................89
9.2 Corrective Action/Preventative Action Plans ...............................................................89
9.3 Protocol Adherence Monitoring Procedures ................................................................89
10. Management of Hypoglycemia .......................................................................................90
10.1 Hypoglycemia Protocols..............................................................................................90
10.1.1 Initiating hypoglycemia protocol (Glucose concentration < 80 mg/dL) ..................90

SHINE Manual of Procedures 3 Version 6– December 19, 2016
10.1.2 Extra steps for hypoglycemia < 70 mg/dL ............................................................90
10.1.3 Severe Hypoglycemia (Glucose concentration < 40 mg/dL) .................................91
10.1.4 Determining Symptomatic Hypoglycemia .............................................................91
10.1.5 Additional steps for ≥ 3 episodes of hypoglycemia (Glucose concentration < 70 mg/dL) in 24 hour period ....................................................................................................91
10.1.6 Additional steps for severe hyperglycemia (Glucose concentration ≥ 500 mg/dL) 92
11. Safety Monitoring ...........................................................................................................92
11.1 Safety Reporting .........................................................................................................92
11.2 Safety Review Process ...............................................................................................92
11.3 Blinding .......................................................................................................................93
11.4 Emergency Unblinding ................................................................................................93
12. Pharmacy Manual ..........................................................................................................93
12.1 Study Design ..............................................................................................................93
12.2 Study Drug Descriptions .............................................................................................94
12.3 Method for Assigning Patients to the Treatment Group ...............................................94
12.4 Drug Labeling .............................................................................................................95
13. Source Documentation and Monitoring ...........................................................................95
13.1 Source Documentation ...............................................................................................95
13.2 Site Monitoring ............................................................................................................95
13.3 Queries/Data Clarification Requests (DCRs)...............................................................96
13.4 Regulatory Documentation and E-Binder ....................................................................97
13.5 Change in Study Personnel ........................................................................................97
13.5.1 Change in Site PI .................................................................................................97
13.5.2 Change in Other Study Personnel ........................................................................97
13.5.3 Notifying SDMC about Changes in Study Personnel ............................................97
14. Reports...........................................................................................................................97
14.1 Monthly Report ...........................................................................................................97
14.2 Enrollment Reports .....................................................................................................97
14.3 Newsletter ...................................................................................................................97
14.4 DSMB Reports ............................................................................................................98
14.5 Recruitment and Retention Reports ............................................................................98
15. Site Recruitment, Training and Initiation .........................................................................98
15.1 Site Start-Up and Initiation ..........................................................................................98

SHINE Manual of Procedures 4 Version 6– December 19, 2016
15.2 Training for Study Personnel .......................................................................................99
15.3 Training for Site Clinical Personnel (Non-Study Team Members) .............................. 100
15.4 Training Investigator Meeting .................................................................................... 100
15.5 Site Re-Training (Due to Lack of Subject Enrollments in 6 Months) .......................... 101
16. Patient Recruitment and Retention ............................................................................... 102
16.1 Recruitment .............................................................................................................. 102
16.2 Retention .................................................................................................................. 102
16.2.1 Retention and Study Participation ...................................................................... 102
16.2.2 Lost to Follow Up ............................................................................................... 102
16.3 Use of People Locating Service (OmniTrace) ........................................................... 103
16.4 Discontinuation of Study Participation ....................................................................... 103
16.4.1 Discontinuation of Study Participation ................................................................ 104
16.4.2 Procedures for Withdrawal of Informed Consent ................................................ 104
16.5 Subject Payments to Clinical Sites ............................................................................ 105
16.5.1 Per Subject Payments ....................................................................................... 106
17. Close-Out and Termination Stages ............................................................................... 106
17.1 Site Closeout ............................................................................................................ 106
17.2 Retention of Study Records ...................................................................................... 106
18. Study Policies and Research Conduct .......................................................................... 107
18.1 Protection of Human Subjects ................................................................................... 107
18.2 The HIPAA Privacy Rule ........................................................................................... 107
18.3 Protocol Amendments ............................................................................................... 108
18.4 Ancillary Studies ....................................................................................................... 108
18.5 Publications and Presentations Policies and Procedures .......................................... 109
18.6 Data Sharing Policy and Procedures ........................................................................ 109
19. Study Organization and Contacts ................................................................................. 110
19.1 Administrative Structure ............................................................................................ 110
19.1.1 SHINE Organizational Chart .............................................................................. 110
19.1.2 SHINE Committee Responsibilities and Charges ............................................... 110
19.1.3 Medical Decision Network (GlucoStabilizer®) .................................................... 113
19.2 SHINE Trial Contacts ................................................................................................ 114
19.3 Quick Reference Table ............................................................................................. 118
20. References ................................................................................................................... 119

SHINE Manual of Procedures 5 Version 6– December 19, 2016
21. Appendices .................................................................................................................. 123

SHINE Manual of Procedures 6 Version 6– December 19, 2016
ABBREVIATIONS Abbreviation Description ADA American Diabetes Association AE Adverse Event AHA American Heart Association ALIAS High-Dose Albumin Therapy for Neuroprotection in Acute
Ischemic Stroke ASAP Acute Stroke Accurate Prediction ATLANTIS Alteplase Thrombolysis for Acute Noninterventional Therapy in
Ischemic Stroke BG baseline glucose BI Barthel Index CCC Clinical Coordinating Center CI confidence interval CRF case report form D/C discharge DSMB Data and Safety Monitoring Board DVT deep venous thrombosis EC Executive Committee ED Emergency Department FDA Food and Drug Administration GIST-UK Glucose Insulin in Stroke Trial – United Kingdom GRASP Glucose Regulation in Acute Stroke Patients IA intra-arterial ICH intracranial hemorrhage IND Investigational New Drug IRB Institutional Review Board IQSR Internal Quality and Safety Reviewer IV intravenous LAR Legally Authorized Representative MOP Manual of Procedures NETT Neurological Emergency Treatment Trials NIH National Institutes of Health NIHSS NIH Stroke Scale NINDS National Institute of Neurological Disorders and Stroke mRS modified Rankin Scale PI Principal Investigator PO by mouth q every RX treatment SAP Statistical Analysis Plan SAE Serious Adverse Event SDMC Statistical Data Management Center SHINE Stroke Hyperglycemia Insulin Network Effort SQ subcutaneous SSQOL Stroke Specific Quality Of Life THIS Treatment of Hyperglycemia in Ischemic Stroke TOAST Trial of ORG 10172 in Acute Stroke Treatment tPA Tissue Plasminogen Activator

SHINE Manual of Procedures 7 Version 6– December 19, 2016
1. Introduction 1.1 Study Overview There is an increasing need for improved treatments for stroke patients as stroke is the most common cause of serious long term adult disability and the third most common cause of death in the United States.1 Hyperglycemia is seen in approximately 40% of acute ischemic stroke patients2,3 and has been associated with worse clinical outcomes.4,5 Intravenous (IV) insulin therapy with tight glucose control has been found to improve clinical outcomes in some non-stroke acute illness trials.6,7 Current stroke guidelines emphasize the need for definitive clinical trials to determine best practice for managing hyperglycemia in acute stroke patients.8 A clear determination of the risk and benefit of glucose control with IV insulin would have a dramatic impact on acute ischemic stroke patient therapy.
This Phase III multicenter, randomized, controlled trial will determine the efficacy of and provide further safety data on glycemic control in stroke patients. The hyperglycemic acute ischemic stroke patients that meet all eligibility criteria will receive up to 72 hours of hyperglycemia control with IV insulin therapy or control therapy with subcutaneous (SQ) insulin. Treatment will be started within 12 hours of symptom onset and is recommended, but not required, to begin within 3 hours of arrival to the emergency department (ED). The primary efficacy outcome to be assessed at 90 days will be the severity adjusted difference in favorable outcome between the groups. Favorable outcome will be defined by a previously described baseline severity adjusted dichotomized modified Rankin scale (mRS).9-11 Outcome success will depend on the severity of the initial stroke (per NIH Stroke Scale Score (NIHSS)). The primary safety outcome will be the severe hypoglycemic event rate. Secondary outcomes will assess additional neurological and functional status using stroke severity, functional and quality scales12-14 as well as glucose control success and adherence to the protocol dosing recommendations of the computerized decision support tool. This trial launches a highly collaborative model for stroke research providing a foundation for maximally generalizable results based on performance at academic, community, urban, rural, large and small hospitals throughout North America to produce a highly representative national population sample. A validated computer decision support tool will guide delivery of IV insulin therapy. A baseline severity-adjusted dichotomized outcome analysis (responder analysis)9 will adjust for variability of individual patient characteristics to allow detection of the true clinically relevant treatment effect. In this setting an absolute 7% treatment effect is recognized as a threshold at or above which a profound effect on a large stroke population would be realized.
1.2 Study Objectives Specific Aim 1 To determine the efficacy of tight glucose control to a target range of 80-130 mg/dL with IV insulin infusion in hyperglycemic acute ischemic stroke patients within 12 hours of symptom onset as measured by mRS at 90 days after stroke.
o Hypothesis 1: Tight glucose control (target 80-130 mg/dL) with IV insulin infusion therapy using a validated computerized decision support tool will increase the severity adjusted 90 day favorable outcome on the mRS by an absolute 7% or more, as compared to the control group.
SHINE Manual of Procedures 8 Version 6– December 19, 2016
Specific Aim 2 To determine the safety of tight glucose control with IV insulin infusion in hyperglycemic acute ischemic stroke patients treated for up to 72 hrs.
o Hypothesis 1: Tight glucose control with IV insulin infusion therapy using a decision support tool is safe as determined by a severe hypoglycemia (< 40 mg/dL) rate that does not exceed that of the control group by more than 4%.
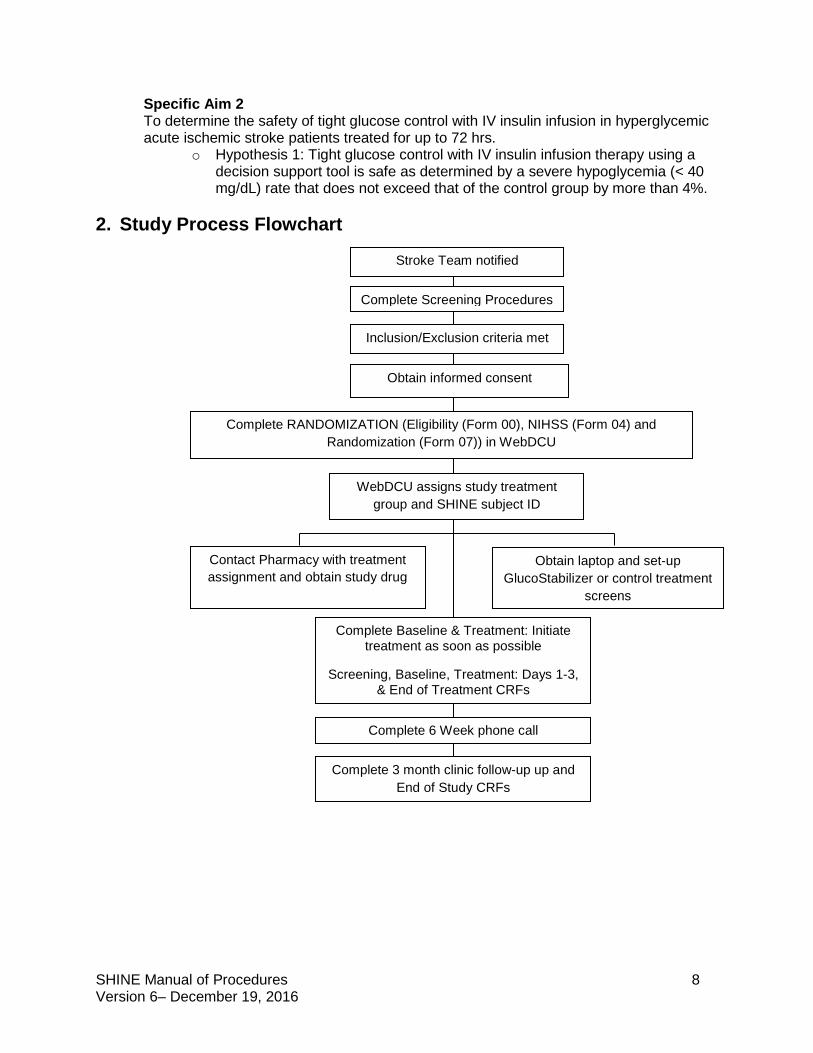
2. Study Process Flowchart
Complete Screening Procedures
Inclusion/Exclusion criteria met
Obtain informed consent
Complete RANDOMIZATION (Eligibility (Form 00), NIHSS (Form 04) and Randomization (Form 07)) in WebDCU
WebDCU assigns study treatment group and SHINE subject ID
Contact Pharmacy with treatment assignment and obtain study drug
Complete Baseline & Treatment: Initiate treatment as soon as possible
Screening, Baseline, Treatment: Days 1-3, & End of Treatment CRFs
Complete 6 Week phone call
Complete 3 month clinic follow-up up and End of Study CRFs
Stroke Team notified
Obtain laptop and set-up GlucoStabilizer or control treatment
screens

SHINE Manual of Procedures 9 Version 6– December 19, 2016
3. Screening 3.1 Subject Identification Each site will be responsible for identifying and recruiting participants into the study. It is known that screening methods vary across sites. It is, however, important to have multiple recruitment strategies to reach all potential patients (e.g., overnight, weekends, and holidays). The site PI, coordinator, and other support staff will agree to a screening plan before being eligible to enroll patients. Once potential participants are identified, the site will collect information about them to make a determination of their eligibility for the study. The determination of eligibility will be made using the data collected from patient history, assessments and examinations performed as part of the potential participant’s routine care. Potential participants who are eligible for and are interested in the study will be asked to sign an informed consent with subsequent enrollment into the study. The site will track potential participants from the time they are identified until they are enrolled or not enrolled. Each site will document and report a summary of recruitment and enrollment progress. The methods used for recruitment of subjects in the study will be devoid of any procedures that may be construed as coercive. The recruitment process will not involve any restrictions on sociodemographic factors including gender or racial and ethnic characteristics of the patient population. However, the composition of the study population will depend on patient sources available to the enrolling sites.
Patients will be recruited by members of the research teams at enrolling sites. Stroke teams and Emergency Medicine teams involved in the immediate evaluation and management of acute stroke patients will be trained to recognize potentially eligible candidates and to rapidly refer them for formal screening by appropriate study team personnel. When a patient is found eligible, they or their Legally Authorized Representative (LAR) as appropriate to the situation will be approached for discussion of the trial and informed consent. Recruitment and enrollment will usually occur at the acute portal of entry to the enrolling site. Most often this will be in the Emergency Department, but this could also occur in a hospital inpatient unit directly receiving an acute stroke patient in transfer from another facility as long as arrival is within the specified enrollment window (12 hours from symptom onset and is recommended within 3 hours from arrival to the hospital). Specifically randomization must be within 12 hours of onset and treatment must be started as soon as possible after enrollment AND is recommended to begin within 3 hours of arrival to the designated enrolling center.
3.2 Screening Overview Screening is defined as any procedure done solely for the purpose of determining a potential subject’s eligibility or to enter a subject into a research study. Federal regulations and institutional policy must be followed when screening subjects to determine potential eligibility. Potential subjects may be identified by neurologists, local emergency department, or clinic staff in conjunction with study personnel. The patient with “suspected stroke” could include both Acute Ischemic Stroke (AIS) and Intracerebral Hemorrhage (ICH) as the etiologies have a similar clinical presentation. When a potential candidate is identified, the site PI and/or the study coordinator (or their designee as clearly delineated by a call schedule system) should be contacted to begin the screening process. Patients should be screened and enrolled as quickly as possible

SHINE Manual of Procedures 10 Version 6– December 19, 2016
after presentation to the acute portal of entry. All acute ischemic stroke patients arriving within 12 hours of onset with glucose identified >110 must be referred to the study team for formal screening and must be captured in the Screen Failure Log.
In “FDA Information Sheets” dated 01 October 1995, the following specifications are made: Procedures that are to be performed as part of the practice of medicine and which would be done whether or not study entry was contemplated, such as for diagnosis or treatment of a disease or medical condition, may be performed and the results subsequently used for determining study eligibility without first obtaining consent.
For the SHINE study there is no activity required in the screening process that could not reasonably be considered routine care for acute stroke patients. Therefore, unless required by local IRB, patients would be approached for consent only after the clinical screening process had established eligibility.
3.3 Eligibility Criteria 3.3.1 Inclusion Criteria
(1) Age 18 years or older
(2) Clinical diagnosis of ischemic stroke defined as acute neurological deficit occurring in one or more cerebral vascular territories. Neuroimaging must be done to exclude intracranial hemorrhage (ICH).
(3) Protocol treatment must begin within 12 hours after stroke symptom onset and is recommended, but not required, to begin within 3 hours after hospital arrival. If time of symptom onset is unclear or patient is awakening with stroke symptoms, the time of onset will be the time the patient was last known to be normal.
(4) Known history of type 2 diabetes mellitus and glucose >110 mg/dL OR admission blood glucose ≥150 mg/dL in those without known diabetes mellitus
(5) Baseline NIHSS score of at least 3 but no more than 22
(6) Pre-stroke modified Rankin Scale score = 0 for patients with an NIHSS score of 3-7. Pre-stroke modified Rankin Scale score = 0 or 1 for patients with an NIHSS score of 8-22.
(7) Able to provide a valid informed consent to be in the study (self or their authorized legally accepted representative).
3.3.2 Exclusion Criteria
(1) Known history of type 1 diabetes mellitus
(2) Substantial pre-existing neurological or psychiatric illness that would confound the neurological assessment or other outcome assessment
(3) Having received experimental therapy for the enrollment stroke. IV tPA (up to 4.5 hrs) or IA tPA are allowed as are IA therapies including use of FDA cleared devices. Non FDA cleared devices are considered experimental and are excluded.

SHINE Manual of Procedures 11 Version 6– December 19, 2016
(4) Known to be pregnant or breast-feeding at the time of study entry
(5) Other serious conditions that make the patient unlikely to survive 90 days
(6) Inability to follow the protocol or return for the 90 day follow up
(7) Renal dialysis, including hemo- or peritoneal dialysis
3.4 Justification and Explanation of Eligibility Criteria Inclusion Criteria Age 18 years or older Only ≥ 18 year old ischemic stroke subjects will be included since ischemic strokes in the pediatric population are substantially different from adult strokes. The population under age 18 is excluded to avoid confounding the results. Diagnosis of ischemic stroke Ischemic stroke is defined as acute neurological deficit occurring in one or more cerebral vascular territories. Neuroimaging must be done to exclude intracranial hemorrhage (ICH). Primary ICH is excluded, but primary ischemic stroke with hemorrhagic conversion is not excluded. If the diagnosis was ischemic stroke at the time of randomization but the diagnosis changes during the 72 hour treatment period, follow instructions in Section 5.1.2 (12) -stopping study treatment when there is a change in diagnosis of ischemic stroke/stroke mimic. TIA is not considered a stroke mimic, and the study protocol should be followed. Protocol treatment recommended within 3 hours of hospital arrival & required within 12 hours of stroke symptom onset The 12-hour eligibility requirement was chosen as this is almost universally before the development of maximum edema in acute ischemic stroke patients, but is a wide enough time window to be inclusive of most patients allowing generalizable results and is supported by the preliminary data. Treatment is recommended but not required to begin within 3 hours of arrival to the Emergency Department to assure the avoidance of treatment delays in hopes of maximizing treatment effect as suggested by much of the animal and human data in acute ischemic stroke. This will also allow the patients to be treated with standard IV tPA as per published eligibility criteria and then enrolled in the trial. The start time of protocol treatment is defined as the time of randomization. To be eligible, randomization is recommended to occur within 3 hours of hospital arrival and must be within 12 hours of symptom onset. Glucose >110 mg/dL in patients with type 2 diabetes or ≥150 mg/dL in patients without diabetes An enrollment blood glucose >110 mg/dL in patients with type 2 diabetes or ≥150 mg/dL in patients without diabetes is based on our preliminary data and other data suggesting this group is most likely to benefit. As demonstrated by GIST-UK15, THIS16, GRASP17, Walters18 and Kriesel19 hyperglycemia frequently resolves spontaneously in most patients without diabetes. Thus, patients without diabetes mellitus or admission

SHINE Manual of Procedures 12 Version 6– December 19, 2016
hyperglycemia will be excluded. Both THIS16 and GRASP17 trials and an observational study20 demonstrated that most patients enrolled with hyperglycemia ≥150 mg/dL remained hyperglycemic during hospitalization unless they received intravenous insulin. The vast majority of patients with admission glucose ≥150 mg/dL have undiagnosed diabetes or impaired glucose metabolism (insulin resistance) as has been reported,21-23 thus making them good subjects for this trial. The admission blood glucose is the first finger stick point of care (POC) glucose measurement at the enrolling hospital. The most recent POC glucose at the time of randomization will be used to confirm eligibility. It is not necessary for study purposes to re-check glucose prior to randomization. Only finger stick POC glucose measurements (not serum laboratory glucose measurements) are used for randomization. Glucose measurements from outside hospitals or EMS cannot be used to determine eligibility. A diagnosis of type 2 diabetes will be based on the medical history provided and the medical record. Patients with current or past treatment with an oral agent with or without insulin therapy and/or a previous diagnosis of type 2 diabetes mellitus will be defined as having type 2 diabetes. Reports of ‘borderline diabetes mellitus’ will require clarification as to presence or absence of true diabetes mellitus. Patients determined to have a history of diagnosed type 2 diabetes must have a glucose level greater than 110 mg/dL to be eligible. Patients with no known history of type 2 diabetes must have a glucose level of greater than or equal to 150 mg/dL to be eligible. If there is documentation in the medical record of a HbA1c of > 6.5%, the patient will be assumed to have a diagnosis of diabetes mellitus. Baseline NIHSS score of at least 3 but no more than 22 Previous data suggest that patient with an NIHSS score of < 3 have overwhelmingly good recovery and those with an NIHSS score of > 22 have overwhelmingly poor recovery. The intervention of glucose control was not felt to be likely to alter these extreme outcomes. The NIHSS score used to determine eligibility must be completed by a certified investigator. The NIHSS score that is closest to the time of randomization should be used and must have been done within 30 minutes before the time of randomization. Patients who do not have a complete NIHSS are not eligible (e.g. untestable item per NIHSS scoring criteria). Based on the NIHSS scoring instructions, untestable is only considered for the following: Question 5: Motor Arm (UN: Amputation or joint fusion) Question 6: Motor Leg (UN: Amputation or joint fusion) Question 7: Limb Ataxia (UN: Amputation or joint fusion) Question 10: Dysarthria (UN: Intubation or other physical barrier) This only includes patients who are intubated or who have other physical barriers to assess the NIHSS during the entire screening period. If the NIHSS cannot be scored for any of these reasons within 30 minutes prior to randomization, the patient is not eligible. Urgent or emergent intubation during the screening period will be exclusionary if the NIHSS cannot be assessed with 30 minutes of randomization or if medications or neurologic worsening are confounding neurological assessment.

SHINE Manual of Procedures 13 Version 6– December 19, 2016
Prestroke mRS of 0 or 1 Patients with an NIHSS of 3-7 who do not have pre-stroke mRS score of 0 will be excluded as they may be unable to reach the success criteria defined by the stratified dichotomy outcome. Patients with an NIHSS of 8-22 will be eligible if they have a pre-stroke mRS score of 0 or 1. Note that only patients with a prior stroke can have a mRS score of 1. Use SHINE instructions for scoring the pre-stroke modified Rankin Scale found in Section 6. Able to provide a valid informed consent Informed consent is to be obtained from the patient or patient’s Legally Authorized Representative (LAR). Eligibility of a person to serve as a subject’s LAR is determined in accordance with local law at the study site. Consent from non-English speaking patients must be obtained according to site IRB procedures.
Exclusion Criteria Patients with type 1 diabetes Patients with type 1 diabetes mellitus are excluded for safety reasons. Usual care for type 1 diabetes patients during acute illness usually includes intravenous insulin infusion accompanied by dextrose otherwise these patients would be at risk of diabetic ketoacidosis. Since withholding standard care is unacceptable, these patients are excluded. The number of subjects excluded based on this criterion is likely to be very small given that only 2% of all patients with diabetes mellitus are classified as type 1. During the stress of hospitalization, type 1 diabetics and pregnant women routinely require IV insulin. Type 2 diabetics who are insulin dependent may be enrolled in the study. It will be per the discretion of the enrolling investigator to determine if the potential subject is one that could be safely randomized to either treatment group. Patients with type 1 diabetes will be identified based on medical history which may include patient/family report, medical records or conversations with treating medical personnel a HbA1c of > 6.5% is diagnostic of diabetes mellitus in the SHINE trial, and with no evidence of type 1 diabetes, these patients will be assumed to be eligible. If there is any uncertainty regarding type 1 or type 2, those having been started on insulin treatment without first being treated with any oral agent will be defined as having type 1 diabetes. Neurological or psychiatric illness likely to confound the final outcome assessment Patients with a neurological or psychiatric illness likely to confound the final outcome assessment will be excluded since their baseline deficits and outcomes cannot be accurately obtained. Any patient deemed by the enrolling physician to have any condition that confounds the enrollment neurological exam will be excluded. This includes diagnoses other than neurological or psychiatric illnesses (i.e. medication effect during the entire screening period). Experimental therapies Patients receiving experimental stroke therapies will be excluded due to uncertain effects of such therapies on outcomes. Experimental stroke therapies include any therapies that are being studied in an interventional research program.

SHINE Manual of Procedures 14 Version 6– December 19, 2016
Patients who are enrolled in SHINE must not be enrolled in another experimental trial during the entire period of enrollment (Baseline to End of Study). If the other study has an intervention/treatment arm and a control arm, even if the patient would be in the control arm, enrollment during the time that the patient is in SHINE is not permitted. Participation in observational trials of standard care would not be cause for exclusion but would require approval prior to enrollment both by the overall SHINE leadership team and the PI of the other trial.. Standard care IV tPA or IA tPA according to the AHA/ASA guidelines will be allowed.24 The pilot trials demonstrated safety in the population treated with IV tPA. The increased risk of symptomatic hemorrhagic transformation of infarcts observed in IV tPA treated stroke patients with hyperglycemia25, 26, 27 may be reduced with glucose control. Intra-arterial (IA) treatments that are standard care, including the use of FDA cleared devices, will be allowed. FDA cleared devices must be employed according to their Instructions for Use. Non FDA cleared devices or other experimental interventions will not be allowed. No clear data are available on the risk/benefit ratio of these interventions and they could confound the results. Pregnant or breastfeeding Pregnant women will be excluded since the standard care for this population often includes IV insulin treatment for hyperglycemia. Other serious conditions that make the patient unlikely to survive or unable to return at 90 days Patients with conditions other than the enrolling stroke who are unlikely to survive for 90 days will be excluded. Inability to follow the protocol or return for the 90 day follow up Patients known to be unable or unwilling to follow the protocol will be excluded. Patients unlikely to return at 90 days will be excluded since the primary efficacy outcome is measured at that time. Renal dialysis Renal dialysis patients will be excluded including those requiring hemodialysis and peritoneal dialysis due to inability to accurately follow glucose levels and variability in insulin requirements that would put patients at risk. Only patients requiring dialysis will be excluded.
3.5 Screen Failure Log To maintain compliance with recruitment procedures, a Screen Failure Log will be completed for all NON-RANDOMIZED patients who were screened for the SHINE study. All patients diagnosed with ischemic stroke who present within 12 hours of onset with glucose >110 mg/dL will be considered potentially eligible. All potentially eligible subjects that were not randomized into the SHINE trial will be recorded on the Screen Failure Log. These data will be entered into WebDCU™ monthly, by the 10th day of the following month. The coordinating center and the recruitment/executive committee will use the data on the screening forms to support the site in screening and recruitment in the trial. Monthly screening data are submitted to the Clinical Coordinating Center on a continuous basis. Information in the SHINE Screen Failure Log will be reviewed by the Recruitment and Executive teams and will be considered in site screening and

SHINE Manual of Procedures 15 Version 6– December 19, 2016
recruitment evaluation. See Appendix 8 for the Completion of Specific CRF Guidelines for instructions on completing the Screen Failure Log.
The Screen Failure Log will be maintained to document all patients considered for enrollment but not randomized to provide basic information on this population. This log will allow a better understanding of the population considered, the population enrolled and the reasons for ineligibility. See Appendix 2 for Screen Failure Log and decision tree tool.
3.6 Prohibited Therapy No other diabetes treatment medications (i.e., oral agents, IV or subcutaneous insulin) besides the assigned protocol treatment will be allowed during the 72-hour treatment period because such medications would confound the study. The use of non FDA cleared devices for IA therapy is not allowed. Patients taking PO diets must eat protocol-specified diets as defined in Section 5.
4. Enrollment Procedures Enrollment will be overseen by an enrolling investigator as designated on the site Delegation of Authority Log.
4.1 Obtaining Informed Consent Consent will be obtained by either the Principal Investigator or by a designated member of the study team prior to performing any procedures solely for the purpose of research. In every case, alternative available treatments will be explained and it will be made clear that the patient is under no obligation to participate in any research project being offered. In obtaining and documenting informed consent, each investigator will comply with the applicable regulatory requirements and adhere to GCP and to the ethical principles that have their origin in the Declaration of Helsinki. The consent must be the IRB-approved version corresponding to the version of the protocol approved when the screening was initiated. The name of the study team member obtaining consent should be clearly documented, and this person should sign the informed consent document and provide the date of their signature and time as required per local site procedures.
Informed consent is to be obtained from the patient or patient’s LAR. Eligibility of a person to serve as a subject’s LAR is determined in accordance with local law at the study site. The consent document should include the subject’s name, LAR’s name and relation to the subject (if a LAR provides consent), as well as the date the consent was signed. This information should be completed by the subject or the LAR. The study team should not fill in the date the consent was signed for the subject or LAR.
Additional informed consent procedures may be required for ancillary studies.
HIPAA consent must be similarly obtained and documented in keeping with local institutional and IRB regulation for form format (i.e. contained in body of ICF or a separate document.)
Sites must follow local IRB procedures when consenting non-English speaking patients. The informed consent process should be clearly documented. This documentation may occur in the patient’s medical record or research record. Templates for documenting the informed consent process are located on the SHINE website (www.SHINETrial.org) in the Study Toolbox. The patient or LAR must receive a copy of the signed consent form.

SHINE Manual of Procedures 16 Version 6– December 19, 2016
Signed ICF copies are typically filed in the clinical chart and are required to be stored in the secure study file and in accordance with local site regulations.
4.2 Sample Informed Consent A sample informed consent document is located in Appendix 3.
4.3 Enrollment Once eligibility criteria have been met and the informed consent procedure is complete the patient can then be randomized by accessing the trial web site (https://webdcu.musc.edu/). A patient who has completed the informed consent procedure and has been randomized is considered enrolled and must be followed until the end of the study per intention to treat (ITT) principles. Enrollment in appropriate ancillary studies may also need to be considered.
4.3.1 Randomization by WebDCU™ The SDMC developed web-based randomization module will be used by all authorized Hub/Spoke and SHINE Ancillary sites to randomize eligible patients. The WebDCU™ subject randomization module automatically generates unique subject IDs without storing any personal identifying information.
Randomization is available 24/7 at https://webdcu.musc.edu/ . To randomize a patient, study site personnel will log onto the WebDCU™ SHINE™ web-based system using a unique username and confidential password. The user then confirms eligibility (based on the predetermined inclusion and exclusion criteria) by data entering and submitting the Eligibility (Form 00), NIHSS (Form 04) and Randomization (Form 07) forms into WebDCU™, with all eligibility criteria met.
Required information for the Eligibility form includes date of birth, date/time of symptom onset, date/time of arrival at enrolling hospital, inclusion and exclusion criteria. The admission blood glucose is the first finger stick POC glucose measurement. The most recent finger stick POC glucose at the time of randomization will be used to confirm eligibility. It is not necessary for study purposes to re-check glucose prior to randomization. Only finger stick POC glucose measurements (not serum laboratory glucose measurements) are used for randomization and all insulin dosing calculations.
The full NIHSS and the date/time of assessment must be entered on the NIHSS form. The third form that is required to randomize a subject is the Randomization form. This form includes fields to indicate whether IV tPA was initiated or ordered(y/n) and name of enrolling investigator, which should be an investigator who is listed on the delegation of authority log. The date/time of randomization will automatically be generated by WebDCUTM. If all eligibility criteria are not met, randomization will be blocked.
After the Eligibility and Randomization form is submitted, a subject ID and treatment assignment will automatically be created in the WebDCU™ system. This is reported directly to the enrolling investigator via the web based screen.
In addition, an automatic e-mail notification of enrollment is sent to the appropriate parties (e.g., the SHINE leadership team). If, under rare

SHINE Manual of Procedures 17 Version 6– December 19, 2016
circumstances the web system is not available, use the emergency randomization hotline to obtain a randomization treatment assignment and subject ID (866-450-2016).
4.3.2 Emergency Randomization Procedures In the event that WebDCU™ cannot be accessed (either by direct computer access, or if during normal business hours, by contacting DCU personnel), emergency randomization may occur.
Should a site have randomization questions during business hours, please contact Kavita Patel at [email protected] (843-876-1167) or Catherine Dillon at [email protected] (843-876-1942) to request a treatment assignment and subject ID. If these parties are unavailable, please call DCU’s emergency randomization hotline: 866-450-2016.
If a site has emergency randomization questions after hours or is unable to reach the DCU, the site should call the emergency randomization hotline (866-450-2016) to request the treatment assignment and subject ID.
4.4 Successful Enrollment All NETT hubs and affiliate hubs (independent sites) were surveyed using a formal data-driven instrument or evaluated by a data driven formula to establish their individual annual recruitment estimates. Annual recruitment estimates are reviewed, discussed and amended during the readiness call. As recruitment trends have been established across all sites, adjustments have been made to the annual enrollment targets based upon site stroke volume, diabetes penetrance by geographic location, study team coverage and competing stroke trials. Sites are expected to meet and/or exceed their previous quarter’s enrollment. For sites enrolling less than their established estimates, a collaborative process of developing new strategies for recruitment in a close working partnership with the recruitment team will take place. Appendix 9 includes details of the Site Recruitment Performance and Milestone Plan and Recruitment Milestones.
5. Study Treatment Procedures 5.1 Treatment Procedures The study treatment time begins at the time of randomization. Treatment should be initiated as quickly as possible following randomization.
5.1.1 Intervention and control group protocols
Intervention Group Study treatment: continuous IV insulin + subcutaneous meal insulin or
saline injections Target blood glucose: 80-130 mg/dL

SHINE Manual of Procedures 18 Version 6– December 19, 2016
Control Group Study treatment: IV saline + subcutaneous sliding scale insulin injections
Target blood glucose: <180 mg/dL
The SHINE trial is a single-blinded study. Study patients will not know whether they are in the intervention or control group but will know that they will get both an IV solution and subcutaneous shots to manage high blood sugar. Both the treating and study teams will know the randomization. Care should be taken to maintain the blind. The study team is available to answer questions that study patients and their families or the treating teams have about the randomization and blinding.
5.1.2 Intervention Group Study treatment: IV insulin + subcutaneous meal insulin (or saline injections for patients who are not eating) Target blood glucose: 80-130 mg/dL Once randomized to the intervention group (IV insulin + subcutaneous meal insulin or saline injections with a target blood glucose of 80-130 mg/dL), the GlucoStabilizer® (decision support tool) program will be activated on the study laptop. The determination of IV and subcutaneous meal insulin dosing and instructions will be indicated by the decision support tool. The required timing of blood glucose checks will be displayed by the decision support tool. Patients will continue on study treatment for 72 hours. Discontinuation prior to 72 hour is allowed when discharge is clinically indicated and requires that all study protocol treatments stop at least 6 hours in advance of scheduled discharge.
(1) IV insulin preparation (a) Drug preparation
(i) Upon randomization, the SHINE site study team will contact the pharmacy to communicate the randomization allocation. Study treatment must begin as soon as possible following randomization.
(ii) Orders for study intervention group protocol should be entered by study investigator per site procedures.
(iii) The pharmacy will provide the study IV insulin per site procedures. (iv) The concentration of the IV insulin infusion will be 1:1 (e.g. 100 Units of
human regular insulin in 100 ml of normal saline (0.9% NaCl)) unless a site-specific exception is approved. See Section 12-Pharmacy Manual.
(b) Study patient preparation
(i) The study team/clinical nurse will gather supplies for the IV insulin infusion protocol (IV catheter, IV tubing, infusion pump, syringes, etc.).
(ii) The SHINE study will encourage that 1 vial of Dextrose 50% in Water (D50) be stored to allow immediate availability at the bedside. If hypoglycemia occurs, patients in the intervention group will receive an individualized dose of D50 as indicated by the GlucoStabilizer® tool. For this reason, syringes with graduated marks for volume (mL) must be used.
(iii) An IV for study medication only will be placed. All other IV medications should be administered separately from the IV for study insulin infusion.

SHINE Manual of Procedures 19 Version 6– December 19, 2016
(iv) Upon receipt of the IV insulin bag from the pharmacy, the study team/clinical nurse will hang it with an infusion pump. The IV infusion pump should be programmed to maintain the blind (i.e., the display on the infusion pump should read SHINE Study Drug rather than insulin).
(v) The IV insulin bag will be labeled with a BLUE sticker that says SHINE study drug. The label MUST only be applied by the pharmacist who is preparing the IV infusion.
(vi) The background for the GlucoStabilizer® screens will be BLUE for the intervention group. Verify that these match to assure that the correct study drug has been provided.
(vii) The initial IV infusion rate and subsequent rate adjustments will be based on POC finger stick glucose measurements and calculated by the GlucoStabilizer® decision support tool.
(viii) Implementation of the recommended dose is by the patient’s clinical or research nurse.
(2) Blood glucose monitoring
(a) The SHINE protocol recommends the use of capillary blood for the POC glucose tests. Finger stick POC glucose is required for randomization and should be the primary source for most patients, but other sources of blood may be used based on site standard practice.
(b) The Accu-Chek System is the SHINE study preferred system. Other blood glucose monitoring systems can be used per site procedures.
(c) The decision support tool requires POC glucose checks at least every 1-2 hours. The testing frequency is determined by the GlucoStabilizer® tool.
(d) If the blood glucose drops < 80 mg/dL, the GlucoStabilizer® tool will initiate the hypoglycemia prevention and management protocol. GlucoStabilizer® will recommend stopping all insulin, administering an individualized corrective dose of D50 and more frequent blood glucose monitoring (every 15 minutes).
(e) Glucose must be checked within +/-15 minutes of the scheduled time for a regular check and within +/-5 minutes of the scheduled time when the glucose is < 80 mg/dL..
(f) Only the results of the point of POC are glucose levels should be recorded in GlucoStabilizer®. Lab glucose levels should NOT be entered!

SHINE Manual of Procedures 20 Version 6– December 19, 2016
(3) GlucoStabilizer® and IV insulin initiation (a) Overview
SHINE study laptops will be supplied to all participating sites and must be used to access the GlucoStabilizer® decision support tool. The icon for the SHINE Trial Portal will be located on the desktop of the laptop. Study laptops are to remain powered on and open throughout the treatment period. The screens for the intervention group will allow the clinical nurses/study team to see the time countdown to the next glucose check and to enter the current glucose levels and see the consequent GlucoStabilizer® IV insulin rate recommendations. The time between glucose checks will be determined by the GlucoStabilizer® tool.
(b) Administrative set up instructions 1. Retrieve the study laptop and ensure internet connectivity per site
procedures. 2. The initial setup for each subject will be done by the site study team. 3. The login for the computer is SHINE user profile, and the password is
shine. 4. The GlucoStabilizer® login will be User ID: shine, Password: shine
for all personnel and all sites. Contact the site study team with questions about the login.
5. The SHINE study ID is unique identification number for each randomized patient. This will be provided by the study team after the patient has been randomized in WebDCUTM.
(c) Initiating GlucoStabilizer® (Starting a New Drip)
(i)Select SHINE icon on the desktop of the study laptop and the SHINE Trial Portal will open.

SHINE Manual of Procedures 21 Version 6– December 19, 2016
(ii)Check the hard copy of the randomization to confirm that the patient is in the intervention group. Click on the blue box Click Here for Intervention Group.
(iii) Enter User ID and Password (shine/shine). Click on Log In button.
(iv) If this is the first time that GlucoStabilizer® is used for this study patient, select Start New Drip button.

SHINE Manual of Procedures 22 Version 6– December 19, 2016
(v) Enter the SHINE Subject ID. Select Next.
(vi) Re-enter the SHINE Subject ID. Select Done.
(vii) The next screen will display Patient Information and Drip Settings. Because this is a study patient, NO IDENTIFIABLE PATIENT INFORMATION SHOULD BE ENTERED. (viii) DO NOT CHANGE ANY OF THE PREPOPULATED ITEMS ON THIS SCREEN. THESE ARE First Name: SHINE, Last name: Patient, DOB: 01/01/1900; Sex: Female). Drip settings will also be populated (Multiplier .02,
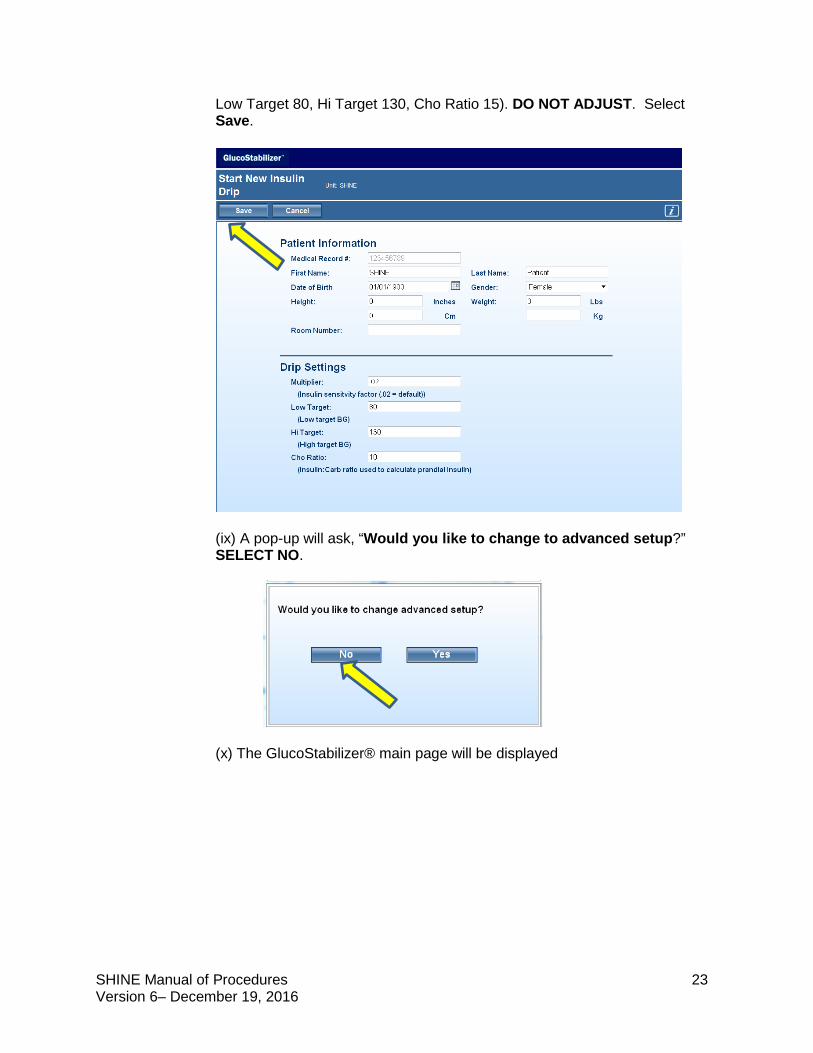
SHINE Manual of Procedures 23 Version 6– December 19, 2016
Low Target 80, Hi Target 130, Cho Ratio 15). DO NOT ADJUST. Select Save.
(ix) A pop-up will ask, “Would you like to change to advanced setup?” SELECT NO.
(x) The GlucoStabilizer® main page will be displayed

SHINE Manual of Procedures 24 Version 6– December 19, 2016
(xi) Remember that the study patient is blinded to his or her treatment group. Care should be taken to maintain the blind. Note that GlucoStabilizer® will display dosing recommendations for IV insulin. Use the lock program option if the study laptop will remain in the patient room so that this will only be visible to the treating and study teams and not the study patient and his or her family/friends.
(xii) Once the IV insulin infusion is ready to be started, check the POC glucose immediately prior to starting infusion. This value will be entered into the GlucoStabilizer® tool.
(d) IV insulin initiation
(i) A new finger stick glucose measurement will be checked just prior to initiation of therapy.
(ii) On the GlucoStabilizer® main page, select Enter Glucose.
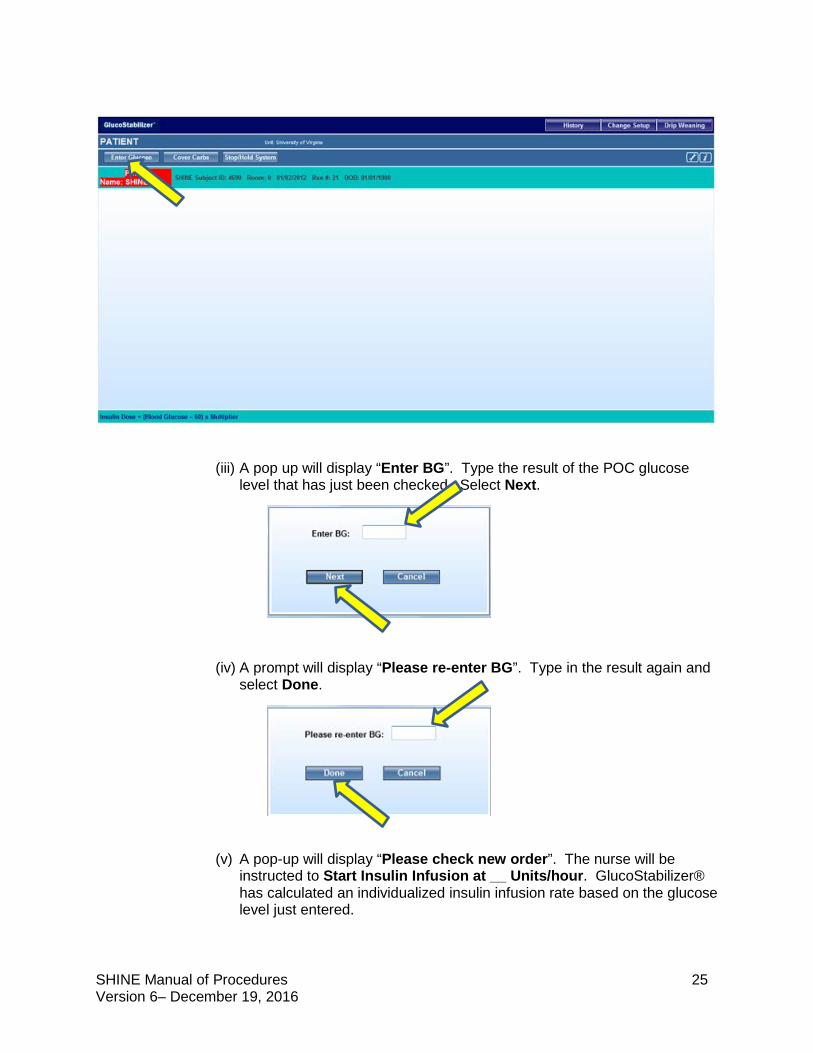
SHINE Manual of Procedures 25 Version 6– December 19, 2016
(iii) A pop up will display “Enter BG”. Type the result of the POC glucose level that has just been checked. Select Next.
(iv) A prompt will display “Please re-enter BG”. Type in the result again and select Done.
(v) A pop-up will display “Please check new order”. The nurse will be
instructed to Start Insulin Infusion at __ Units/hour. GlucoStabilizer® has calculated an individualized insulin infusion rate based on the glucose level just entered.
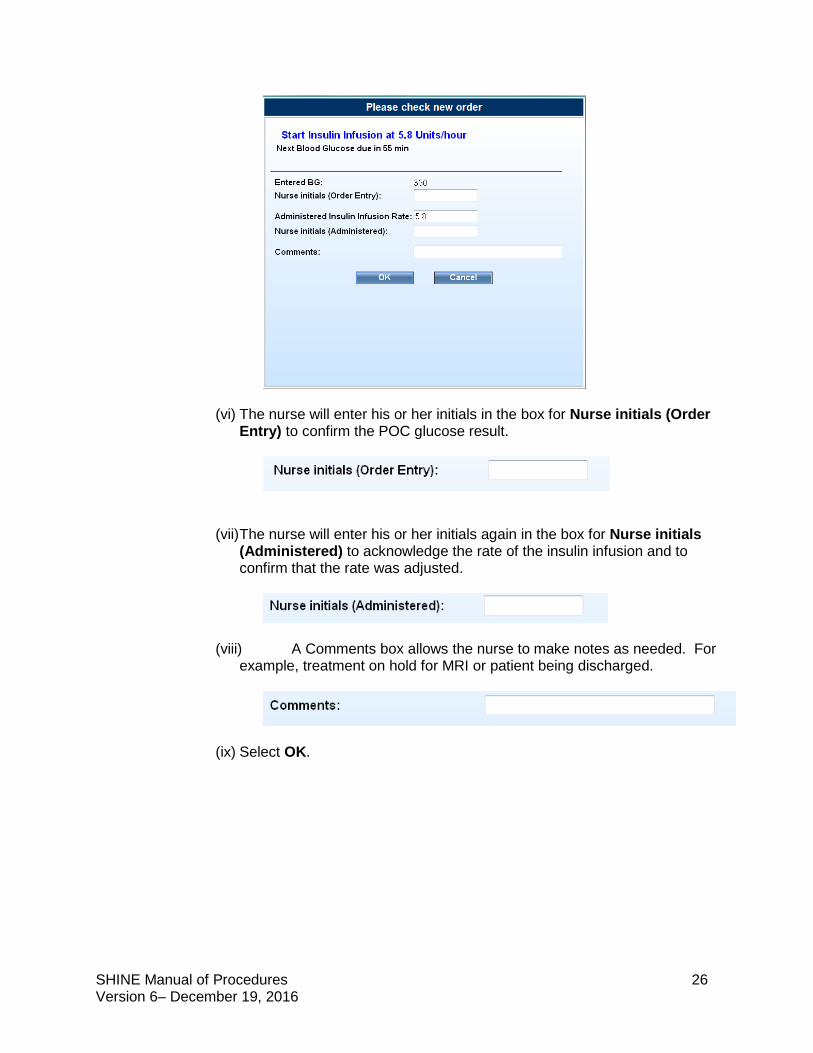
SHINE Manual of Procedures 26 Version 6– December 19, 2016
(vi) The nurse will enter his or her initials in the box for Nurse initials (Order Entry) to confirm the POC glucose result.
(vii) The nurse will enter his or her initials again in the box for Nurse initials (Administered) to acknowledge the rate of the insulin infusion and to confirm that the rate was adjusted.
(viii) A Comments box allows the nurse to make notes as needed. For example, treatment on hold for MRI or patient being discharged.
(ix) Select OK.
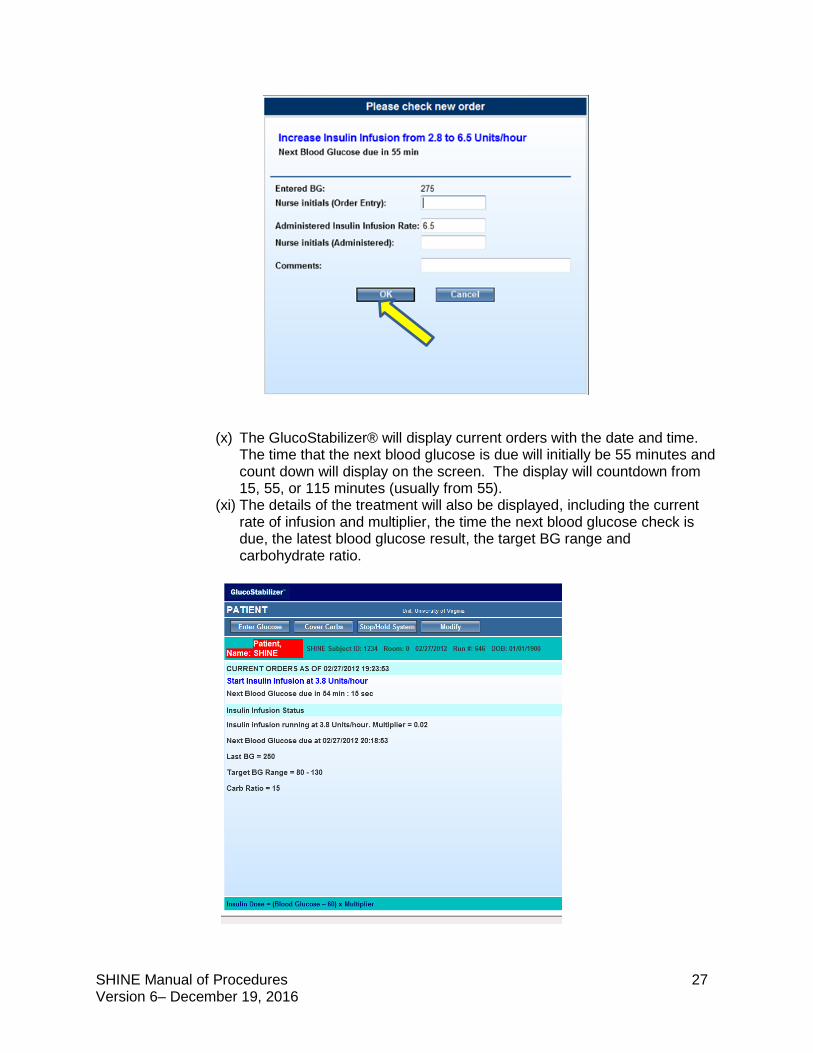
SHINE Manual of Procedures 27 Version 6– December 19, 2016
(x) The GlucoStabilizer® will display current orders with the date and time. The time that the next blood glucose is due will initially be 55 minutes and count down will display on the screen. The display will countdown from 15, 55, or 115 minutes (usually from 55).
(xi) The details of the treatment will also be displayed, including the current rate of infusion and multiplier, the time the next blood glucose check is due, the latest blood glucose result, the target BG range and carbohydrate ratio.
SHINE Manual of Procedures 28 Version 6– December 19, 2016
(xii) The second blood glucose check will be due in 60 minutes, but the after
will sound after 55 minutes.
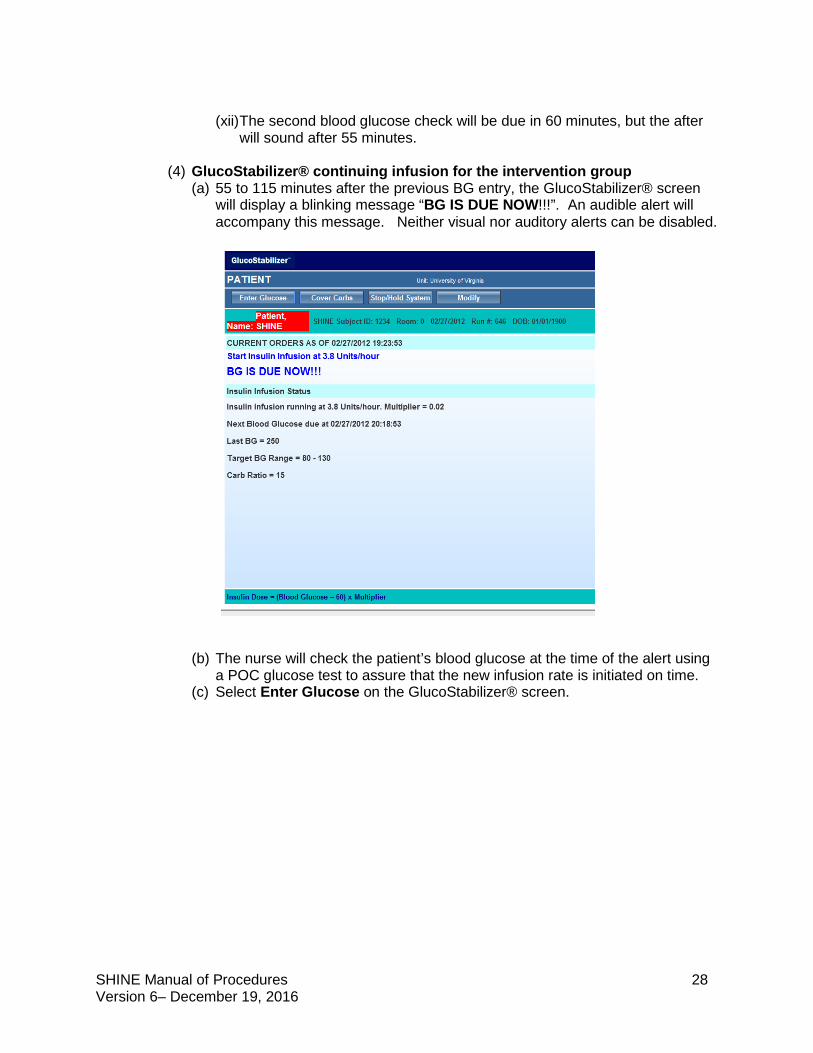
(4) GlucoStabilizer® continuing infusion for the intervention group (a) 55 to 115 minutes after the previous BG entry, the GlucoStabilizer® screen
will display a blinking message “BG IS DUE NOW!!!”. An audible alert will accompany this message. Neither visual nor auditory alerts can be disabled.
(b) The nurse will check the patient’s blood glucose at the time of the alert using
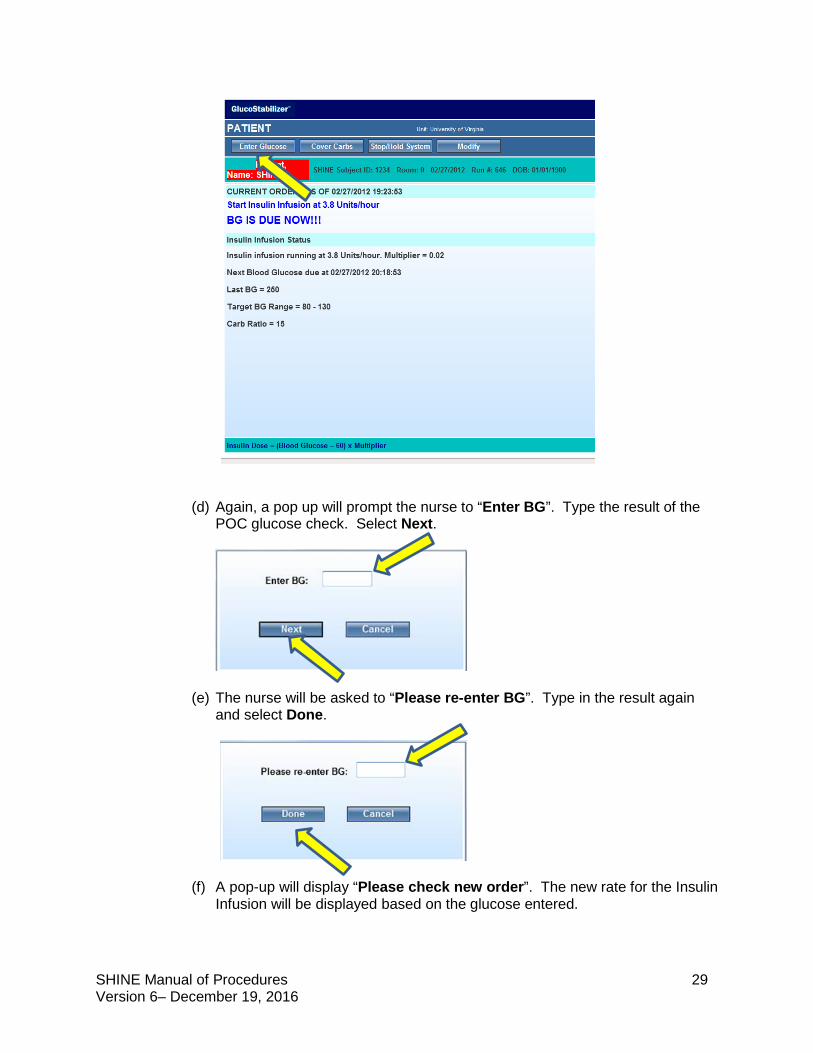
a POC glucose test to assure that the new infusion rate is initiated on time. (c) Select Enter Glucose on the GlucoStabilizer® screen.
SHINE Manual of Procedures 29 Version 6– December 19, 2016
(d) Again, a pop up will prompt the nurse to “Enter BG”. Type the result of the
POC glucose check. Select Next.
(e) The nurse will be asked to “Please re-enter BG”. Type in the result again and select Done.
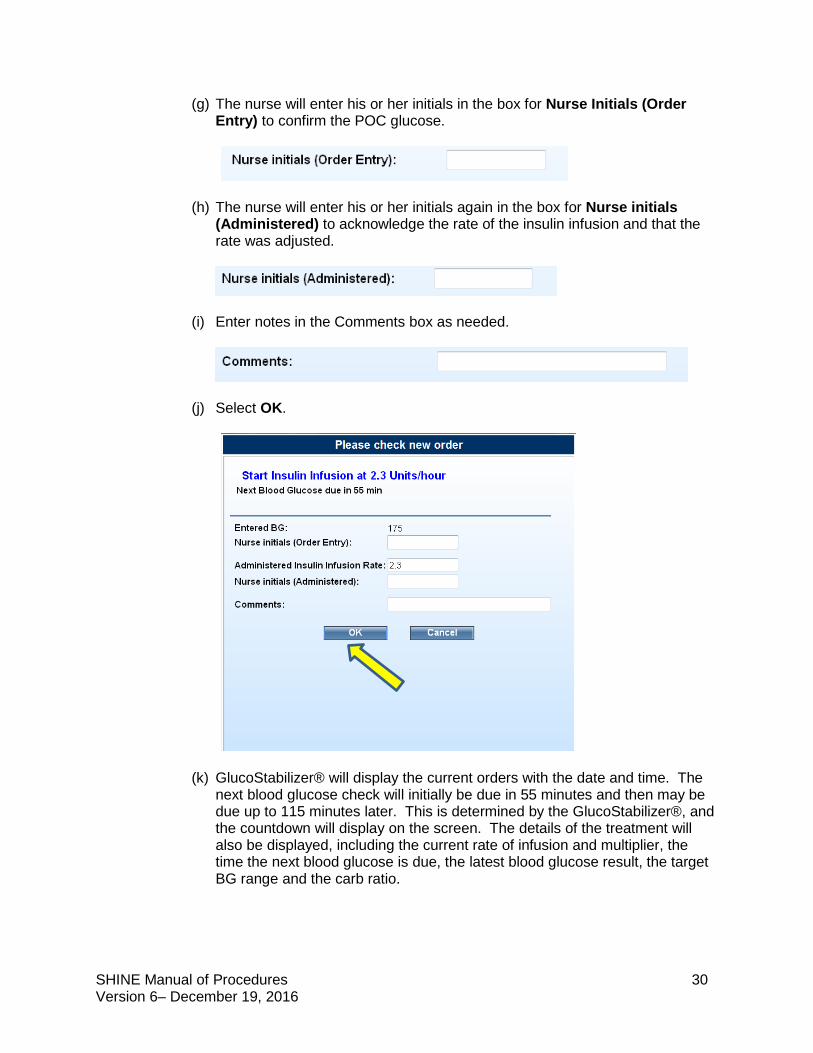
(f) A pop-up will display “Please check new order”. The new rate for the Insulin Infusion will be displayed based on the glucose entered.
SHINE Manual of Procedures 30 Version 6– December 19, 2016
(g) The nurse will enter his or her initials in the box for Nurse Initials (Order Entry) to confirm the POC glucose.
(h) The nurse will enter his or her initials again in the box for Nurse initials (Administered) to acknowledge the rate of the insulin infusion and that the rate was adjusted.
(i) Enter notes in the Comments box as needed.
(j) Select OK.
(k) GlucoStabilizer® will display the current orders with the date and time. The next blood glucose check will initially be due in 55 minutes and then may be due up to 115 minutes later. This is determined by the GlucoStabilizer®, and the countdown will display on the screen. The details of the treatment will also be displayed, including the current rate of infusion and multiplier, the time the next blood glucose is due, the latest blood glucose result, the target BG range and the carb ratio.

SHINE Manual of Procedures 31 Version 6– December 19, 2016
(l) 55 to 115 minutes after the previous BG entry, the GlucoStabilizer® screen
will display blinking message “BG IS DUE NOW!!!”. An audible alert will accompany this message.
(m) At the time of the alert, the nurse will check the patient’s blood glucose using a POC test to assure that the new infusion rate is initiated on time.
(n) For each glucose check and insulin infusion rate adjustment (if indicated) throughout the protocol, these steps will be repeated.
(o) If the glucose drops below 80 mg/dL at any time, GlucoStabilizer® will direct the nurse to follow specific hypoglycemia prevention and management instructions.
The clinical nurse or study team may choose to decline the GlucoStabilizer® infusion recommendation. To do so, document the actual infusion rate in the Administered Infusion Rate field and enter a comment to explain. A pop up box will display “The administered insulin infusion rate is different from the ordered rate. Would you like to continue?” Click OK. The SHINE Study Hotline should be notified when rate recommendations are declined by calling 800-915-7320 (ext.1).
FAQ Q: We have a patient in the intervention arm and just did a glucose check. GlucoStabilizer is recommending that the insulin infusion rate is decreased to 0.3 u/hr. The pump doesn’t go any lower than 0.5 u/hr.
A: In any case when the GlucoStabilizer recommendation is lower than the lowest infusion rate of the pump, decline the recommendation. Enter 0 for the infusion rate when confirming the blood glucose and administration rate. Turn off the drip and document that the infusion was stopped in the medical record. The GlucoStabilizer will continue to count down to next check, and the visible alert will appear and the audible alert will sound when the glucose check is due. Check the glucose, enter and re-enter the value, and if the recommended rate is one that your infusion pump can accommodate, restart the infusion at that rate. If the recommended rate continues to be lower than the lowest rate for your infusion pump, repeat the steps above to decline the recommendation and check again per GlucoStabilizer.

SHINE Manual of Procedures 32 Version 6– December 19, 2016
(5) Hypoglycemia prevention and management (a) The hypoglycemia prevention and management protocol will be initiated for
any patient whose glucose concentration drops below 80 mg/dL. (b) Definitions
(i) Blood glucose < 80 mg/dL initiate hypoglycemia prevention and management protocol
(ii) Hypoglycemia: defined as a blood glucose < 70 mg/dL (iii) Severe hypoglycemia: defined as a blood glucose < 40 mg/dL
(c) Hypoglycemia protocol for intervention group
(i) If the blood glucose drops to < 80 mg/dL the GlucoStabilizer® will automatically instruct the nurse to follow special hypoglycemia prevention instructions (stop all insulin treatments, give an individualized IV dose of 50% glucose (D50).
(ii) Stop the insulin infusion and hold all subcutaneous insulin/saline injections when the glucose is < 80 mg/dL! Meal insulin should not be given when glucose is < 80 mg/dL at any point during the meal.
(iii) To record the glucose measurement and receive D50 dose recommendation, select Enter Glucose.
(iv) A pop up will prompt the nurse to “Enter BG”. Type the result of the
POC glucose check. Select Next.
(v) Follow instructions to Please re-enter BG and select Done.
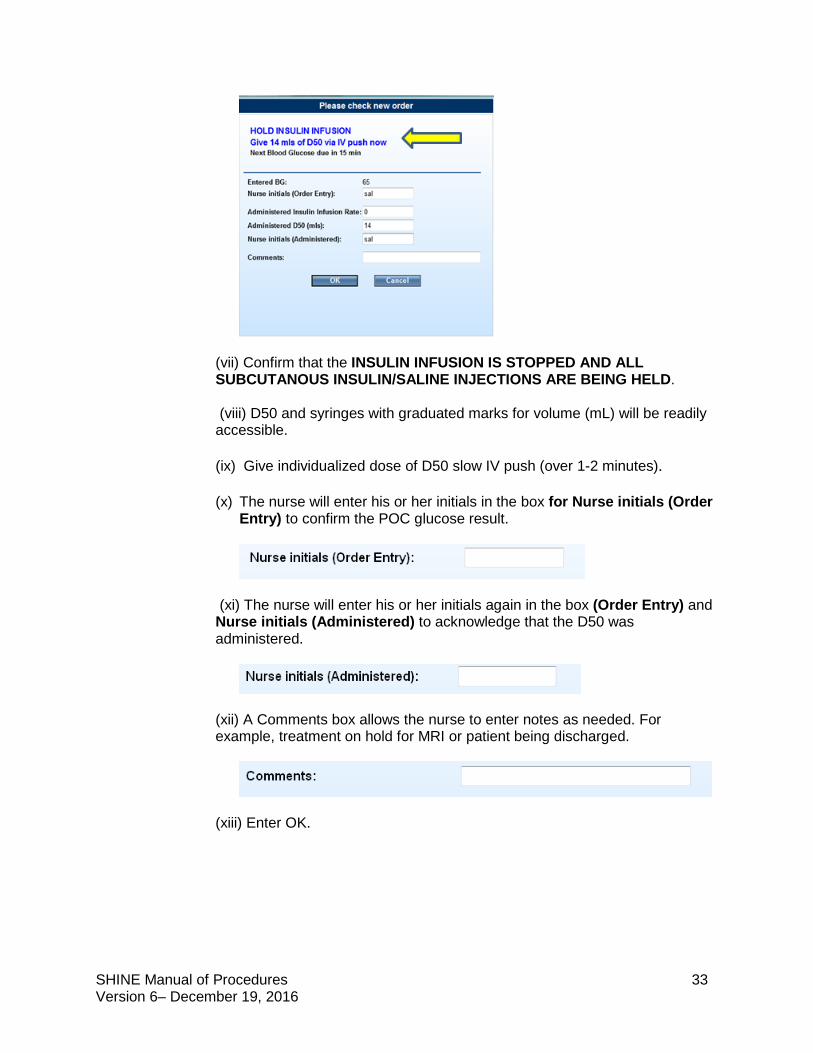
(vi) A pop-up will display “Please check new order”. When the blood glucose is <80 mg/dL, a message will display HOLD INSULIN INFUSION. The nurse will be instructed to “Give an individualized dose of D50 via IV push now”. This dose will be based on the glucose level just entered.
SHINE Manual of Procedures 33 Version 6– December 19, 2016
(vii) Confirm that the INSULIN INFUSION IS STOPPED AND ALL SUBCUTANOUS INSULIN/SALINE INJECTIONS ARE BEING HELD. (viii) D50 and syringes with graduated marks for volume (mL) will be readily accessible. (ix) Give individualized dose of D50 slow IV push (over 1-2 minutes).
(x) The nurse will enter his or her initials in the box for Nurse initials (Order
Entry) to confirm the POC glucose result.
(xi) The nurse will enter his or her initials again in the box (Order Entry) and Nurse initials (Administered) to acknowledge that the D50 was administered.
(xii) A Comments box allows the nurse to enter notes as needed. For example, treatment on hold for MRI or patient being discharged.
(xiii) Enter OK.
SHINE Manual of Procedures 34 Version 6– December 19, 2016
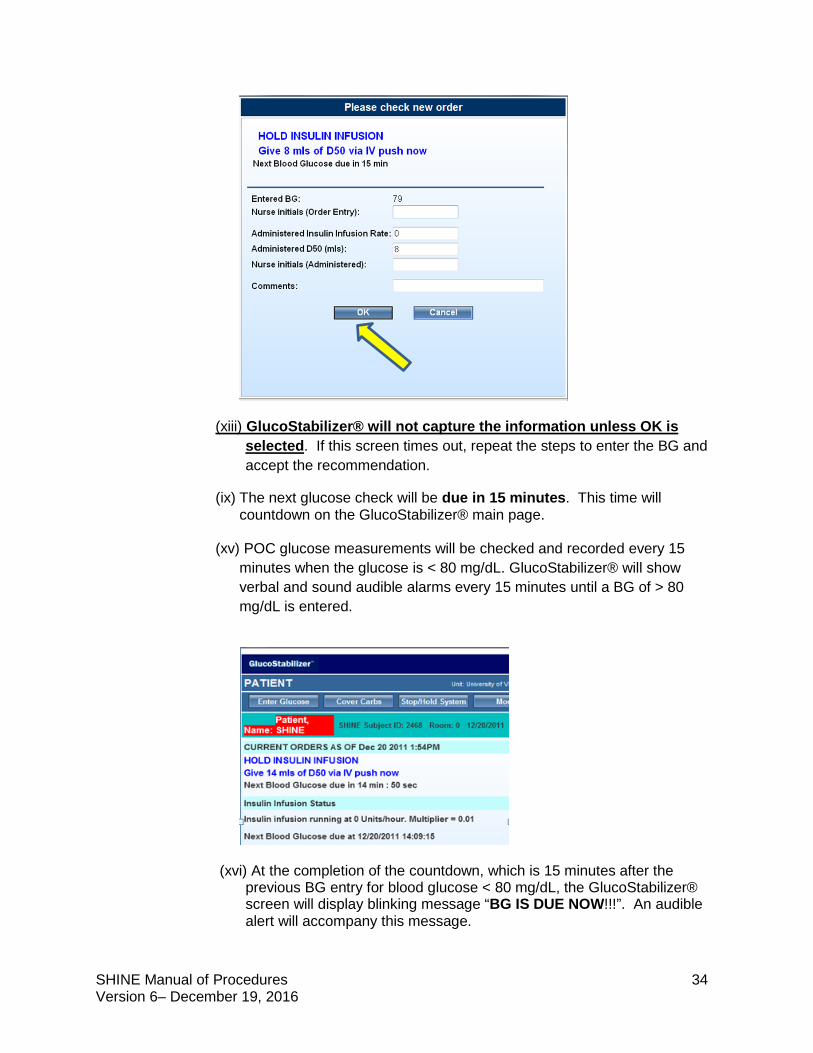
(xiii) GlucoStabilizer® will not capture the information unless OK is selected. If this screen times out, repeat the steps to enter the BG and accept the recommendation.
(ix) The next glucose check will be due in 15 minutes. This time will countdown on the GlucoStabilizer® main page.
(xv) POC glucose measurements will be checked and recorded every 15
minutes when the glucose is < 80 mg/dL. GlucoStabilizer® will show verbal and sound audible alarms every 15 minutes until a BG of > 80 mg/dL is entered.
(xvi) At the completion of the countdown, which is 15 minutes after the previous BG entry for blood glucose < 80 mg/dL, the GlucoStabilizer® screen will display blinking message “BG IS DUE NOW!!!”. An audible alert will accompany this message.

SHINE Manual of Procedures 35 Version 6– December 19, 2016
(xvii) The nurse must check the POC glucose at the time of the alert. These
steps will be repeated for every glucose measurement that is < 80 mg/dL.
(xviii) If the glucose drops below 70 mg/dL, additional steps are required and described below in Section 6 below (Additional steps for blood glucose < 70 mg/dL).
(xix) For any study patient in the intervention group who experiences 3 or more episodes of hypoglycemia (glucose concentration of < 70 mg/dL in a 24 hour period, the SHINE Safety Hotline must be notified (800-915-7320 ext 2).
(6) Additional steps for blood glucose < 70 mg/dL If the blood glucose drops < 70 mg/dL, the following additional actions are required:
(i) Continue to use the hypoglycemia protocol above (e.g. hold insulin, give D50, repeat glucose checks every 15 minutes).
(ii) Draw a STAT laboratory serum glucose measurement but do not delay treatment with D50 by waiting for this blood draw result. Only the results from POC glucose checks should be used for study treatment dosing. The study coordinator will document the result of the serum glucose measurement.
(iii) Screen the patient for hypoglycemia symptoms using Hypoglycemia Symptomatic Questionnaire. 1. The worksheet and instructions will be available on paper and on the
desktop of the laptops. 2. The Hypoglycemia Symptomatic Questionnaire must be repeated
every 15 minutes when glucose is < 70 mg/dL. 3. Once the glucose is ≥ 70 mg/dL, no additional assessments with the
Hypoglycemia Symptomatic Questionnaire are required 4. Once the glucose is ≥ 80 mg/dL, the timing of glucose checks and
insulin infusion rate will again be determined by the GlucoStabilizer®.
(iv) Screen the patient for neurological worsening. 1. An assessment of neurological change is required per site standard
care (“neuro check”) every 15 minutes when glucose is < 70 mg/dL even if there are no new neurological findings

SHINE Manual of Procedures 36 Version 6– December 19, 2016
2. The SHINE study definition of neurological worsening is any clinical change that is associated with a ≥ 4 point increase from baseline on the NIHSS score. (Baseline is considered to be the most recent daily NIHSS.)
3. Any patient with neurological worsening will be assessed for hypoglycemia and for relatedness of the hypoglycemia to neurological worsening if present.
4. If the patient has not returned to neurological baseline within 24 hours, a NIHSS assessment is required within 24 (+/-4) hours from onset of hypoglycemic event (< 70 mg/dL). If the patient has returned to neurological baseline at any point in less than 24 hours, the NIHSS is not required at 24 hours.
5. If neurological worsening (persists for ≥ 24 hours) is associated with a glucose concentration < 55 mg/dL, an SAE form is required.
(v) If the glucose falls below 40 mg/dL at any time, an SAE form and a call to the SHINE safety hotline are required.
(7) Meal insulin for the intervention group (a) General concepts-SHINE protocol-recommended diet
1. For patients who are cleared for a PO diet, a 60 grams/meal carbohydrate diet will be ordered for breakfast, lunch and dinner as part of standard care. Meals will be encouraged, but not required, to be delivered at approximately 06:00, 12:00 and 18:00.
2. Patients receiving tube feeds may receive bolus or continuous tube feeds per standard care. Bolus tube feeds will also be encouraged to be given at around 06:00, 12:00 and 18:00.
3. Dietary teams should be involved in assuring that the 60 grams/meal carbohydrate diets are maintained throughout the study protocol.
4. About twenty minutes after the initiation of a meal estimate meal consumption. If the patient is not finished eating, an estimate of the likely total consumption is required.
5. Estimate the proportion of the meal consumed by the patient. This estimate is based on the entire meal irrespective of its subparts or food groups.
i. Full or near full consumption 60 grams carbohydrates
FAQ Q: Our subject in the intervention group had a worrisome hypoglycemia of 49 mg/dL, which corrected with D50. Is there anything that we can do to help avoid additional episodes of hypoglycemia? A: Perhaps yes. Sometimes a reason can be identified, such as errors in insulin dosing (too much), taking non-study antidiabetic medications by mistake, sneaking in high sugar snacks that can mislead the Glucostabilizer program into recommending too much insulin, or a change in medical status that can affect insulin sensitivity (e.g. infection). Consider whether any protocol deviations occurred as they can be corrected. Call the SHINE hotline for instructions on hypoglycemia related to protocol deviations (might be best to start a new drip). Also, some patients on the Glucostabilizer protocol occasionally do have recurrent hypoglycemia episodes (<70 mg/dL) for no apparent reason. Remember to call the SHINE hotline for 3 episodes of hypoglycemia (<70 mg/dL) within any 24 hour period, or for a single measurement of <40 mg/dL.

SHINE Manual of Procedures 37 Version 6– December 19, 2016
ii. No or nearly no consumption 0 grams carbohydrates (no entry should be made in GlucoStabilizer ® and no meal insulin should be administered)
iii. Partial consumption 30 grams carbohydrates 6. Note that only 30 grams or 60 grams can be entered into
GlucoStabilizer®. No other values should be entered or GlucoStabilizer® will recommend an incorrect meal insulin dose.
7. If no or nearly no consumption is estimated, no meal insulin (carb coverage) is required. Because no meal insulin is required, no entry in GlucoStabilizer® should be made. No injection is given.
8. Patients should not consume additional food not included on the meal tray from the hospital kitchen aside from protocol-approved snacks.
9. Family, friends and visitors should be instructed not to consume food from the patient’s trays unless approved by the nurses, after patients finish eating.
(b) Protocol-approved snacks SHINE patients may also consume up to 2 low carbohydrate snacks (< 5 grams carbohydrates per serving) from the list below between meals (up to 6 low carbohydrate snacks daily). The study protocol diet does not limit the consumption of sugar free foods and drinks listed in the unlimited category below. Patients should only consume food included on the meal tray from the hospital kitchen or the protocol approved snacks during the 72 hour treatment period. For patients in the intervention group, there is NO estimate of consumption for snacks, NO entry in GlucoStabilizer® and NO insulin coverage. Snacks must be documented in the medical record. The study protocol should be followed for meal consumption estimates and meal insulin dosing only for breakfast, lunch and dinner. Low carbohydrate snack options (up to 2 between meals) 5 celery sticks + Tablespoon peanut butter 5 baby carrots 5 cherry tomatoes + 1 Tablespoon ranch 1 hard-boiled egg ½ cup raw broccoli + 1 Tablespoon ranch 1 cup cucumber slices + 1 Tablespoon ranch dressing
FAQ Q: What do we do if the glucose check is due at the same time we're getting ready to enter the carb coverage in GlucoStabilizer?
A: When you make an entry in the ‘Cover Carbs’ function in GlucoStabilizer, the timing of the next check is automatically pushed out to 1 hour later (even if the clock has been counting down and a glucose check is almost due). If a glucose check is almost due and you enter the carb consumption, you will not be prompted to check the glucose for another hour. If you have a patient who is on the q2 hour schedule for checks, this means that it could be nearly 3 hours between glucose checks. Because we want to avoid this, clinical nurses are encouraged to use the +/-15 minute window for glucose checks to ensure that a check is done at the time that it is due. If the clock is counting down and the meal has arrived, make every effort to hold the meal until the glucose check is due (+/-15 minutes). Once you have entered the glucose and received the insulin infusion rate recommendation, the patient can begin the meal. This will help to avoid a gap in glucose checks.

SHINE Manual of Procedures 38 Version 6– December 19, 2016
¼ cup of fresh blueberries 1 cup of salad greens, 1/2 cup of diced cucumber, and with vinegar and oil 2 saltine crackers 1 piece of string cheese stick ½ cup of egg salad, tuna salad or chicken salad 3 oz of deli ham, chicken or turkey slices 1 serving of cubed or sliced cheese (1 oz) ½ cup cottage cheese ½ cup tofu 1 slice deli ham, chicken or turkey + 1 slice cheese Unlimited Bouillon and broth Club soda, unsweetened Coffee, unsweetened or artificial sweeteners only Diet soft drinks Flavoring extracts Horseradish Mineral water Mustard Pickles Soy sauce Spices Sugar-free drink mixes Sugar-free gum Sugar-free Jell-O Tabasco or hot sauce Unsweetened lemon or lime juice Unsweetened tea Vinegar
(c) Subcutaneous meal insulin dosing using GlucoStabilizer® 1. The GlucoStabilizer® will indicate the meal insulin dose for patients who
are eating based on the quantity of carbohydrates in a meal and the specified insulin to carbohydrate ratio (1:15). This means that one unit of insulin will cover 15 grams of carbohydrates consumed.
2. Only rapid acting analog insulin (Humalog, Novolog, and Apidra are all acceptable rapid acting analog insulins) will be used to cover meal carbohydrates. Human regular insulin (Humulin R or Novolin R) MUST NOT be used for subcutaneous injections in the intervention group. (See Section 12- Pharmacy Manual)
3. Follow study site protocol for obtaining rapid acting analog insulin and syringes for subcutaneous injections. a. Nurses will prepare and inject the rapid acting analog insulin
according to the recommendation of the GlucoStabilizer®. b. The meal insulin will be given after assessment of consumption. c. The GlucoStabilizer® will determine the meal insulin dose. If the
patient has consumed part or all of the meal, select the option to Cover Carbs on the GlucoStabilizer® main page.

SHINE Manual of Procedures 39 Version 6– December 19, 2016
d. A pop up will display “Enter Grams Carbohydrate Eaten” e. Based on the estimated portion of the meal eaten, enter the following
in GlucoStabilizer®: f. Partial consumptionEnter 30. Full or near full consumption Enter
60, No or nearly no consumption No entry should be made, g. Select Next.
h. A pop up will display “Re-enter Grams Carbs Eaten”. Re-enter and select Done.

SHINE Manual of Procedures 40 Version 6– December 19, 2016
i. A pop-up will display “Please check new order”. This will include instructions to maintain insulin infusion and to give an individualized dose of subcutaneous meal insulin.
j. The nurse will enter his or her initials in the box for Nurse initials (Order Entry) and Nurse initials (Administered) to confirm the entered carbohydrates, to acknowledge the maintenance of the insulin infusion rate (this will NOT change) and the administration of the subcutaneous meal insulin. Select OK.
k. Note that the nurse must select OK within 2 minutes or this pop-up box will time out and the meal consumption and rapid acting insulin dose will not be captured, and the time of the next glucose check will not update as needed. The nurse must confirm that the order is accepted by clicking OK.
n. Upon acceptance of the recommendation and clicking OK, the GlucoStabilizer® will display the current orders for the insulin infusion rate and if applicable the dose of the subcutaneous insulin injection.
o. The time that the next blood glucose is due will continue to count down on the screen. The details of the treatment status will also be displayed, including the current rate of infusion and multiplier, the time the next blood glucose is due, the last blood glucose result, the target BG range and carb ratio. p. The nurse will give the subcutaneous injection of the rapid acting
insulin. xii. 60 grams carbohydrates consumed = 4 units of SQ rapid
acting analog (meal) insulin will be recommended xiii. 30 grams carbohydrates consumed =2 units of SQ rapid acting
analog (meal) insulin will be recommended q. Continue the steps described in GlucoStabilizer® continuing infusion
for the intervention group (Section 5.1.2, (4). 4. If a glucose check is due at nearly the same time as the meal, check the
glucose first and adjust the infusion prior to giving the meal.

SHINE Manual of Procedures 41 Version 6– December 19, 2016
(c) Bolus tube feeds
(i) For patients receiving bolus tube feeds, a 60 grams/meal carbohydrate bolus feed is recommended.
(ii) Bolus tube feeds will also be encouraged to be given at around 06:00, 12:00 and 18:00.
(iii) The nurse will estimate the proportion of the bolus given. 1. 50-60 grams carbohydrates/bolus Enter 60 grams carbohydrates in
GlucoStabilizer® 2. 0-9 grams carbohydrates/bolus No entry in GlucoStabilizer® is
indicated. 3. 10-49 grams carbohydrates/bolus Enter 30 grams carbohydrates in
GlucoStabilizer® (iv) If a glucose check is due at nearly the same time as the bolus tube feed,
check glucose first and adjust the infusion prior to giving the bolus.
(d) NPO or Continuous tube feeds (i) Continuous tube feeds - For patients receiving continuous tube feeds, no
meal insulin is given. Patients who are receiving continuous tube feeds will receive placebo saline subcutaneous injections TWO times per day. Continuous tube feeds should include approximately 180 grams of carbohydrates per day. Special instructions are provided on the study protocol when there are interruptions in continuous tube feeds (see Section 9 - Procedure for interruptions in continuous tube feeds below).
(ii) NPO – For patients who are NPO, no meal insulin is given. Patients who are NPO will receive placebo saline subcutaneous injections TWO times per day.
(iii) Subcutaneous saline injections (only for patients who are not eating AND in intervention group of study) Patients who are not eating or are receiving continuous tube feeds will receive placebo saline subcutaneous injections TWO times per day (at approximately 9:00 and 21:00 associated with a glucose check).
FAQ Q: Should meal insulin be given if a study patient was hypoglycemic at the beginning of the meal? A: When the blood glucose is < 80mg/dL, the hypoglycemia prevention and management protocol indicates that the insulin infusion should be stopped and all subcutaneous insulin injections should be held. Any time that any portion of the meal occurs at the same time that the BG < 80mg/dL and the hypoglycemia protocol has been initiated, the meal insulin should not be administered for that meal. An example of the way that this could occur is that if a glucose check happens to be due at the same time the meal is delivered. If the glucose is < 80mg/dL, the hypoglycemia protocol will be initiated. If the patient starts eating while the glucose is known to be < 80mg/dL, no meal insulin should be given. Because no meal insulin should be administered, no entry in GlucoStabilizer should be made, and no injection is given. Consumption should be documented on the study CRF (Form 20: Daily Care Log). The questions, 'Was meal insulin given?' should be answered "No", and the reason should be listed as "Other". Specify that the reason that no meal insulin was given and "Other" was selected was due to the "Hypoglycemia protocol". The D50 should always be given per the recommendation of GlucoStabilizer - even if the patient is just starting to eat at the same time. Questions about the hypoglycemia protocol and meal insulin dosing can be directed to the SHINE PI on call, and concerns about safety should be directed to the Independent Safety Monitor.
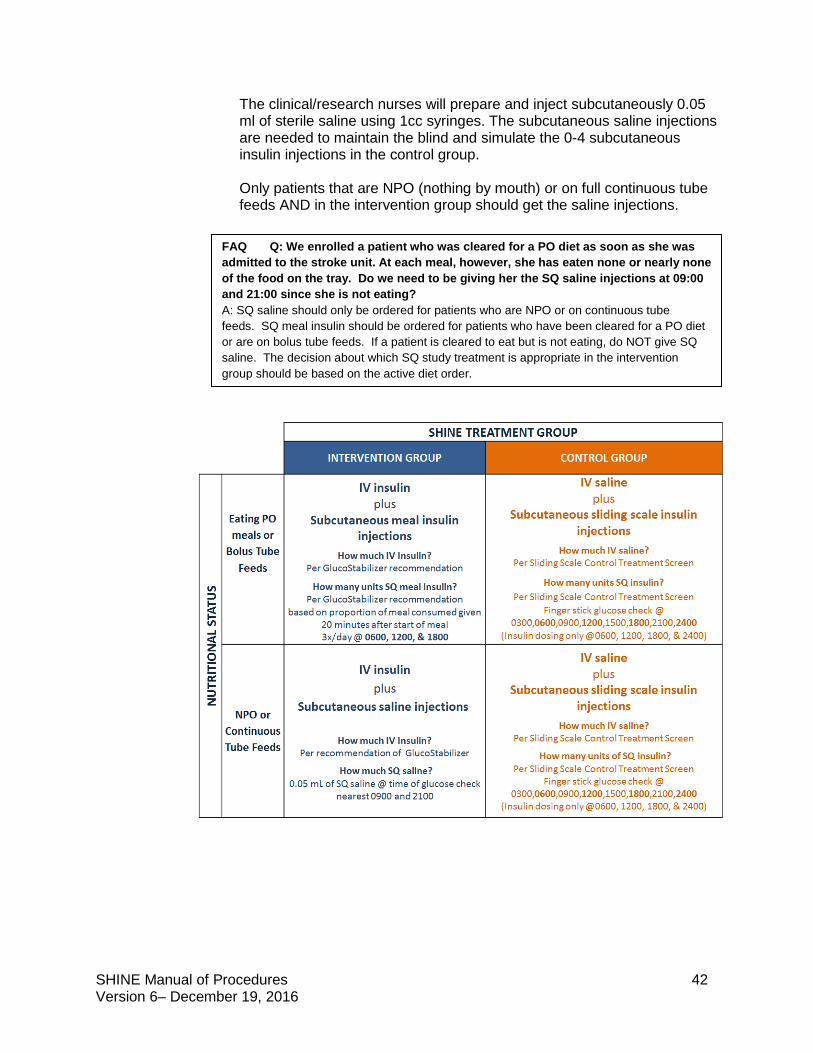
SHINE Manual of Procedures 42 Version 6– December 19, 2016
The clinical/research nurses will prepare and inject subcutaneously 0.05 ml of sterile saline using 1cc syringes. The subcutaneous saline injections are needed to maintain the blind and simulate the 0-4 subcutaneous insulin injections in the control group. Only patients that are NPO (nothing by mouth) or on full continuous tube feeds AND in the intervention group should get the saline injections.
FAQ Q: We enrolled a patient who was cleared for a PO diet as soon as she was admitted to the stroke unit. At each meal, however, she has eaten none or nearly none of the food on the tray. Do we need to be giving her the SQ saline injections at 09:00 and 21:00 since she is not eating? A: SQ saline should only be ordered for patients who are NPO or on continuous tube feeds. SQ meal insulin should be ordered for patients who have been cleared for a PO diet or are on bolus tube feeds. If a patient is cleared to eat but is not eating, do NOT give SQ saline. The decision about which SQ study treatment is appropriate in the intervention group should be based on the active diet order.
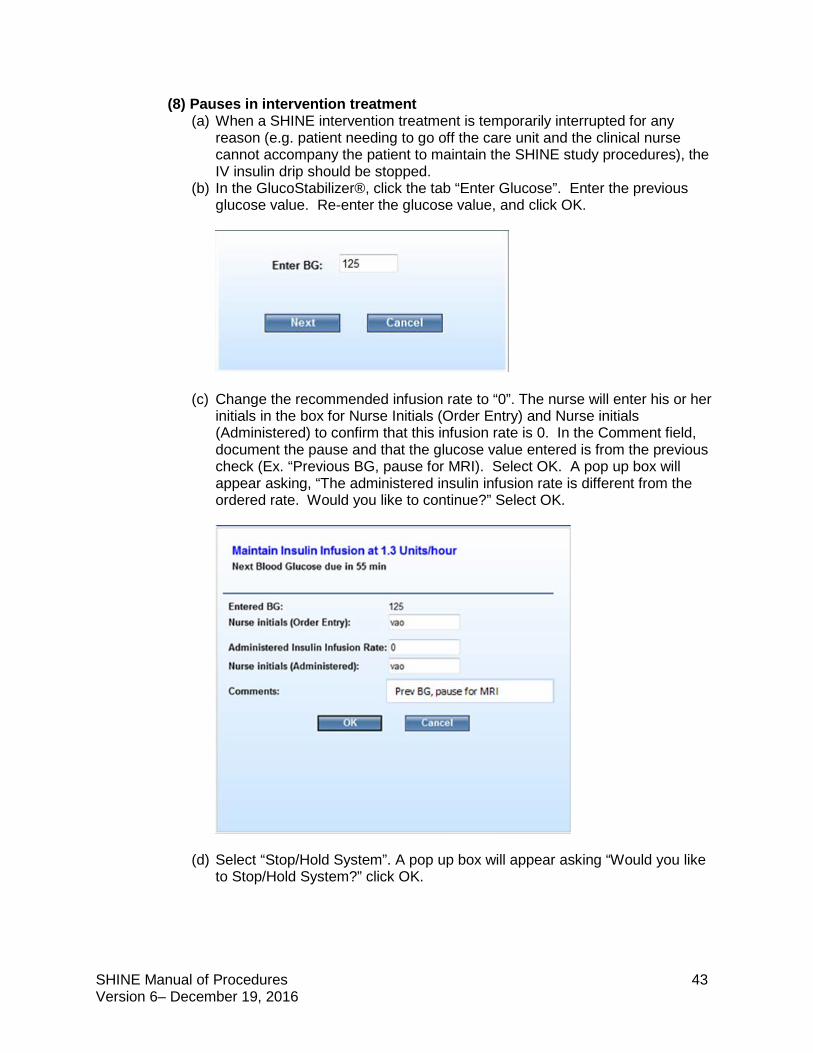
SHINE Manual of Procedures 43 Version 6– December 19, 2016
(8) Pauses in intervention treatment (a) When a SHINE intervention treatment is temporarily interrupted for any
reason (e.g. patient needing to go off the care unit and the clinical nurse cannot accompany the patient to maintain the SHINE study procedures), the IV insulin drip should be stopped.
(b) In the GlucoStabilizer®, click the tab “Enter Glucose”. Enter the previous glucose value. Re-enter the glucose value, and click OK.
(c) Change the recommended infusion rate to “0”. The nurse will enter his or her initials in the box for Nurse Initials (Order Entry) and Nurse initials (Administered) to confirm that this infusion rate is 0. In the Comment field, document the pause and that the glucose value entered is from the previous check (Ex. “Previous BG, pause for MRI). Select OK. A pop up box will appear asking, “The administered insulin infusion rate is different from the ordered rate. Would you like to continue?” Select OK.
(d) Select “Stop/Hold System”. A pop up box will appear asking “Would you like to Stop/Hold System?” click OK.
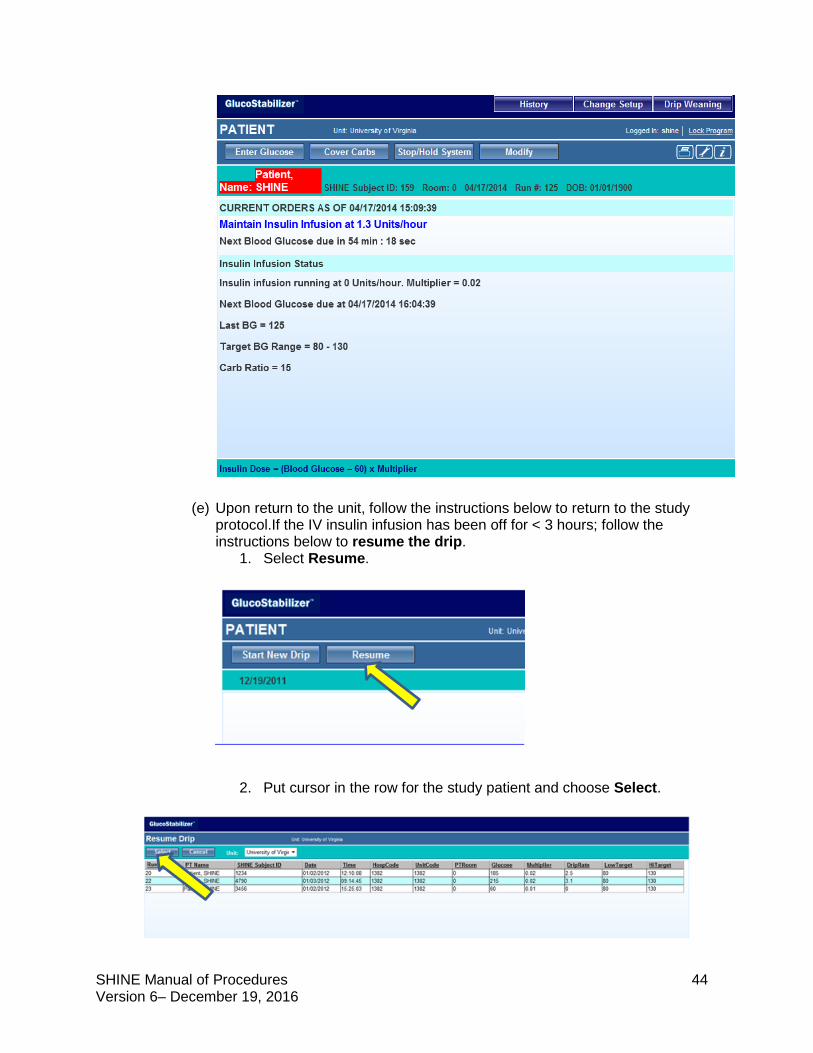
SHINE Manual of Procedures 44 Version 6– December 19, 2016
(e) Upon return to the unit, follow the instructions below to return to the study protocol.If the IV insulin infusion has been off for < 3 hours; follow the instructions below to resume the drip.
1. Select Resume.
2. Put cursor in the row for the study patient and choose Select.

SHINE Manual of Procedures 45 Version 6– December 19, 2016
3. Review the Drip Setup, confirming the SHINE Subject ID. Select Save.
4. Select No for changing to advanced setup.
5. The home screen of the GlucoStabilizer® will display with the current
countdown ongoing. 6. Follow instructions in Section 5.1.2 (4)-GlucoStabilizer® Continuing
Infusion to repeat the glucose check and insulin dosing adjustments per protocol.
(i) If the IV insulin infusion has been off for ≥ 3 hours, follow the instructions as described above to start a new drip (Section 5.1.2 (3) GlucoStabilizer® and IV insulin initiation).
(ii) For patients that are NPO or on continuous tube feeds, if a scheduled SQ saline injection was missed during the pause, give the dose immediately after the glucose check to restart the insulin infusion.
(9) Procedure for interruptions in continuous tube feeds

SHINE Manual of Procedures 46 Version 6– December 19, 2016
(a) When there is a pause in the continuous tube feeds for patients in the intervention group, subcutaneous saline injections will continue per protocol unless the patient is cleared for PO diet or bolus tube feeds.
(b) When there is an interruption in continuous tube feedings in the intervention group, the insulin drip will be stopped, and the option to stop/hold GlucoStabilizer® will be selected.
(c) If the continuous tube feeds are restarted within 1 hour after the drip was stopped, the nurse will select the GlucoStabilizer® option to ‘Resume’ and check the glucose and re-start the insulin drip per recommendation of GlucoStabilizer®. If the continuous tube feeds have not been restarted within 1 hour, the nurse will select the GlucoStabilizer® option to ‘Start a New Drip’, check the glucose and re-start the insulin drip at the rate recommended by GlucoStabilizer®.
(10) Locking the GlucoStabilizer® Program When the nurse leaves the bedside, the GlucoStabilizer® should be locked. (i) Choose lock program

SHINE Manual of Procedures 47 Version 6– December 19, 2016
(i) Upon return to the bedside, unlock the GlucoStabilizer® by entering the username and password (shine/shine). The Lock Program feature is designed to help maintain the blind.
(12) Stopping study treatment when there is a change in diagnosis of ischemic stroke/stroke mimic (a) In the event that there is a change in the diagnosis of ischemic stroke during
the 72 hour treatment period, all study treatments should be stopped at the time that the study team receives knowledge of the new diagnosis.
(b) The clinical care team will determine glucose control therapy after the SHINE treatment protocol is discontinued.
(c) Instructions for completing CRFs when the study treatment is stopped due to a change in diagnosis of ischemic stroke (i) The Day 1-Day 3 visits should be completed only during the period of
time that the patient was receiving study treatment. (ii) The final diagnosis will be captured on the Study Treatment CRF (Form
15) as: Migraine, Seizure, Brain tumor/mass, Psychogenic, Anamnestic syndrome, CNS Infection (abscess, meningitis, encephalitis), Toxic/metabolic (encephalopathy) and Other: specify).
(iii) A brief narrative explaining the change in diagnosis is required. This narrative should include sufficient information to demonstrate that the new diagnosis is most likely including appropriate imaging and laboratory data. A brief narrative template is included in WebDCU: [On ________(full date), the diagnosis was changed to ________. This diagnosis was based on ________(lab/imaging results) and ________(clinical information).]
(iv) On the Study Treatment CRF (Form 15), the study treatment status should be captured as: Started but discontinued prior to 72 hours of study treatment

SHINE Manual of Procedures 48 Version 6– December 19, 2016
(d) The End of Study Visit must be completed. Unless consent is withdrawn, patients whose treatment ends early are still in the study and the 6 week and 90 day follow up visits must be completed in addition to the End of Study Visit.
(13) Transition from study treatment
(a) The start time for the study treatment period is the time of randomization. The treatment period will be 72 hours or until the time of discharge from the hospital, whichever comes first.
(b) At the end of the treatment period, the clinical nurse should chart that the study infusion was stopped in the medical record and follow these steps to document the end of study treatment in GlucoStabilizer®. (i) In the GlucoStabilizer®, click the tab “Enter Glucose”. Enter the previous
glucose value. Re-enter the same value, and click OK.
(ii) Change the recommended infusion rate to “0”. The nurse will enter his or
her initials in the box for Nurse initials (Order Entry) and Nurse initials (Administered) to confirm that this infusion rate is 0. In the Comment field, document the pause and that the glucose value entered is from the previous check (Ex. “Previous BG, pause for MRI). Select OK. A pop up box will appear asking, “The administered insulin infusion rate is different from the ordered rate. Would you like to continue?” Select OK.

SHINE Manual of Procedures 49 Version 6– December 19, 2016
(iii) Select “Stop/Hold System” in GlucoStabilizer®.
(iv) Select the option to “Log out” and shut down the study laptop.
(c) Patients who discontinue study treatment are required to do so 6 hours prior to actual discharge from the hospital.
(d) After the SHINE protocol ends, the clinical care team will determine inpatient and outpatient glucose control therapy. The treatment should be based upon on ADA/AACE guidelines and individual patient needs.28
(e) The table below offers information to consider for hospitalized patients requiring glucose control. These guidelines are not part of the SHINE protocol and are not required actions.
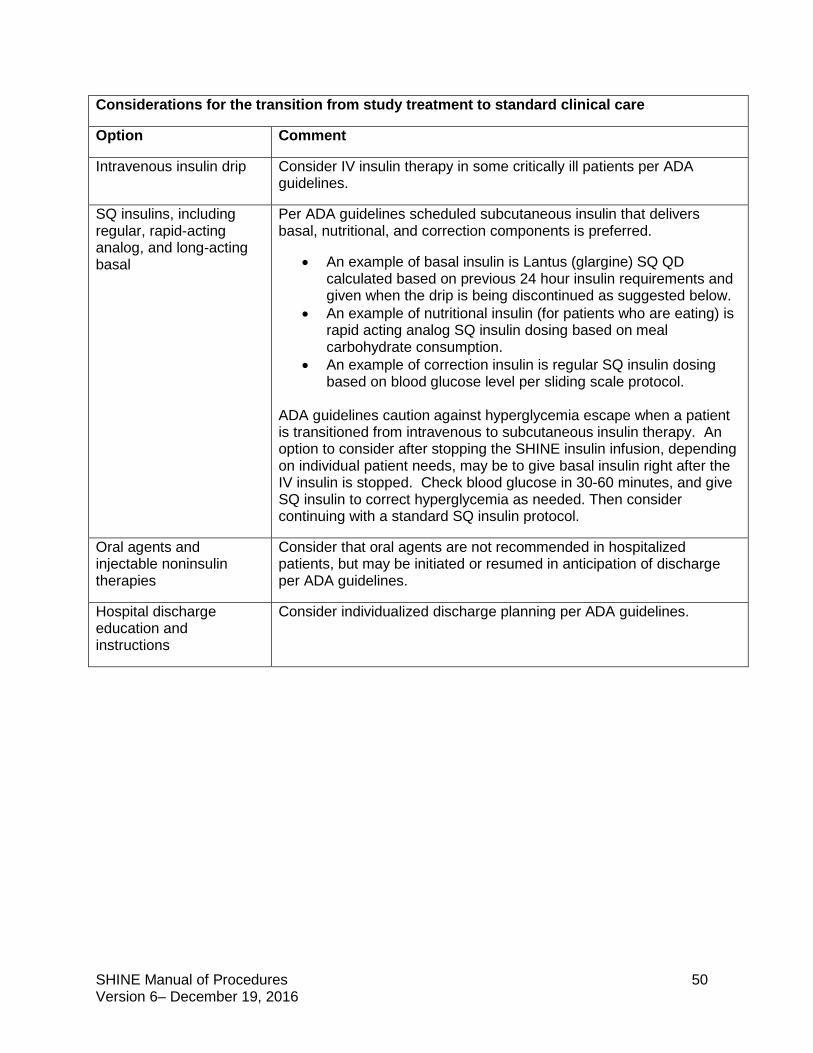
SHINE Manual of Procedures 50 Version 6– December 19, 2016
Considerations for the transition from study treatment to standard clinical care
Option Comment
Intravenous insulin drip Consider IV insulin therapy in some critically ill patients per ADA guidelines.
SQ insulins, including regular, rapid-acting analog, and long-acting basal
Per ADA guidelines scheduled subcutaneous insulin that delivers basal, nutritional, and correction components is preferred.
• An example of basal insulin is Lantus (glargine) SQ QD calculated based on previous 24 hour insulin requirements and given when the drip is being discontinued as suggested below.
• An example of nutritional insulin (for patients who are eating) is rapid acting analog SQ insulin dosing based on meal carbohydrate consumption.
• An example of correction insulin is regular SQ insulin dosing based on blood glucose level per sliding scale protocol.
ADA guidelines caution against hyperglycemia escape when a patient is transitioned from intravenous to subcutaneous insulin therapy. An option to consider after stopping the SHINE insulin infusion, depending on individual patient needs, may be to give basal insulin right after the IV insulin is stopped. Check blood glucose in 30-60 minutes, and give SQ insulin to correct hyperglycemia as needed. Then consider continuing with a standard SQ insulin protocol.
Oral agents and injectable noninsulin therapies
Consider that oral agents are not recommended in hospitalized patients, but may be initiated or resumed in anticipation of discharge per ADA guidelines.
Hospital discharge education and instructions
Consider individualized discharge planning per ADA guidelines.

SHINE Manual of Procedures 51 Version 6– December 19, 2016
(ii) Drip Weaning Report sample
5.1.3 Control Group Protocol Study treatment: IV saline + subcutaneous insulin injections Target blood glucose: < 180 mg/dL Once randomized to the control group (saline infusion + subcutaneous sliding scale insulin injections with a target blood glucose of < 180 mg/dL), the control treatment screen on the study laptop will be activated. A sliding scale for determination of the IV saline rate and subcutaneous insulin dosing will be provided on the control treatment screen. A schedule for the required timing of the glucose checks will also be displayed on the control treatment screen. Patients will continue on study treatment for 72 hours. Discontinuation prior to 72 hour is allowed when discharge is clinically indicated and requires that all study protocol treatment must be stopped at least 6 hours in advance of scheduled discharge.
(1) Study treatment preparation
(a) Drug preparation (i) Upon randomization the SHINE study team will contact the pharmacy to
communicate the randomization allocation. Study treatment must begin as soon as possible following randomization.
(ii) Orders for the study treatments are to be entered by the study investigator per site procedures.
(iii)The pharmacy will provide the study IV saline per site procedures.
(b) Study patient preparation (i) The clinical nurse and/or study team will gather supplies for the IV saline
infusion (catheter, tubing, infusion pump),) and will follow site procedures for obtaining supplies for the subcutaneous insulin injections.
(ii) Human Regular Insulin (Humulin R or Novolin R) must be used for the subcutaneous insulin injections in the control group.

SHINE Manual of Procedures 52 Version 6– December 19, 2016
(iii) If the patient advances to Level 3 on the sliding scale, a one-time dose of basal insulin will be given at 48 hours. Glargine (Lantus) must be used for this one-time subcutaneous basal insulin injection at a dose equal to 40% of previous day’s (24 h) entire insulin dose.
(iv) The D50 will be stored to allow immediate availability at the bedside. If a patient in the control group becomes hypoglycemic (< 80 mg/dL), IV D50 25 mL (1/2 amp D50) will be given.
(v) The nurse will place an IV for study medication only. All other IV medications should be administered separately from the IV for study saline infusion.
(vi) Upon receipt of the IV saline bag from the pharmacy, the clinical nurse will hang it with an infusion pump. The IV pump should be programmed to maintain the blind (e.g. This should read SHINE Study Drug rather than saline).
(vii) The IV saline bag will be labeled with an ORANGE sticker that says SHINE study drug. The label MUST only be applied by the pharmacist who is preparing the IV infusion.
(viii) The background for the study laptop screens will be ORANGE for the control group. The nurse should verify that the screen and sticker match to assure that the correct study drug has been provided.
(ix) In the control arm of the study, the initial IV saline infusion rate and
subsequent rate adjustments will be based on POC glucose levels and displayed on the control treatment screen of the study laptop tool. The subcutaneous insulin dose will be determined using the sliding scale also displayed on the control treatment screen.
(2) Blood glucose monitoring (a) Blood glucose will be monitored using POC glucose checks. The Accu-
Chek System is the SHINE study preferred system. Other blood glucose monitoring systems can also be used per site procedures.
(b) The SHINE protocol recommends the use of capillary blood for the POC glucose tests However, if there is concern about continuing participation in the treatment protocol due to the number of finger stick blood glucose checks or if the number of checks exceeds the number listed in the consent form, non-capillary blood can be used for the POC test.
(c) For patients who are in the control arm of the study, the control protocol will require POC glucose checks at least every 1 hour for the first 4 hours.

SHINE Manual of Procedures 53 Version 6– December 19, 2016
Complete one POC glucose test to start the infusion and then check hourly for the next four hours (for a total of 5 checks before transitioning to the schedule for checks approximately every 3 hours; Check to start study infusion:_ _:_ _, ___mg/dL, 1hr:_ _:_ _, ___mg/dL, 2hr: :_ _:_ _, ___mg/dL, 3hr:_ _:_ _, ___mg/dL,4hr:_ _:_ _, ___mg/dL). After the hourly checks for the first four hours on the study infusion, transition to q3hr checks. The schedule for the q3 hour checks is 03:00, 06:00, 09:00, 12:00, 15:00, 18:00, 21:00 and 24:00 and should be maintained for the remainder of the study treatment period.
(d) Glucose must be checked with 15 minutes of the scheduled time for a regular check and within 5 minutes of the scheduled time when a patient is hypoglycemic.
(e) Only the results of POC glucose should be recorded in the control treatment screen of study laptop. Lab glucose levels should NOT be entered!
(f) In the control group, the timing of POC glucose monitoring is displayed on the control treatment screen of the study laptop for the duration of the study treatment period.
(g) If the blood glucose drops < 80 mg/dL, the hypoglycemia prevention and management protocol will be initiated.
(3) Control treatment protocol
(a) Overview (i) SHINE study laptops will be supplied to all participating sites and must be
used to access the control treatment screen. The icon for the SHINE portal will be located on the desktop of the laptop. Study laptops are to remain powered on and open throughout the treatment period. The screens for the control group will allow the clinical nurses to see the sliding scale table and timing for glucose checks. The glucose levels, subcutaneous insulin administered and saline infusion rates will be entered in the control treatment screen. The sliding scale table will have three levels of dosing (Level 1, Level 2 and Level 3). Changes in the level of dosing will occur only if indicated and only at 24 and 48 hours after the initiation of study treatment (time of randomization).
(ii) Administrative set up instructions 1. Retrieve the study laptop and ensure internet connectivity per site
procedures. 2. The initial setup for each subject will be done by the site study team. 3. The login for the computer is the SHINE user profile, and the
password is shine. 4. The GlucoStabilizer® login will be User ID: shine, Password: shine
for all personnel and all sites. Contact the study team with questions about the login.
5. The SHINE subject ID is a unique identification number for each randomized patient. This will be provided by the study team after the patient has been randomized in WebDCUTM.
(iii) Initiating IV saline/subcutaneous insulin
1. Select the icon for the SHINE Trial Portal on the desktop.
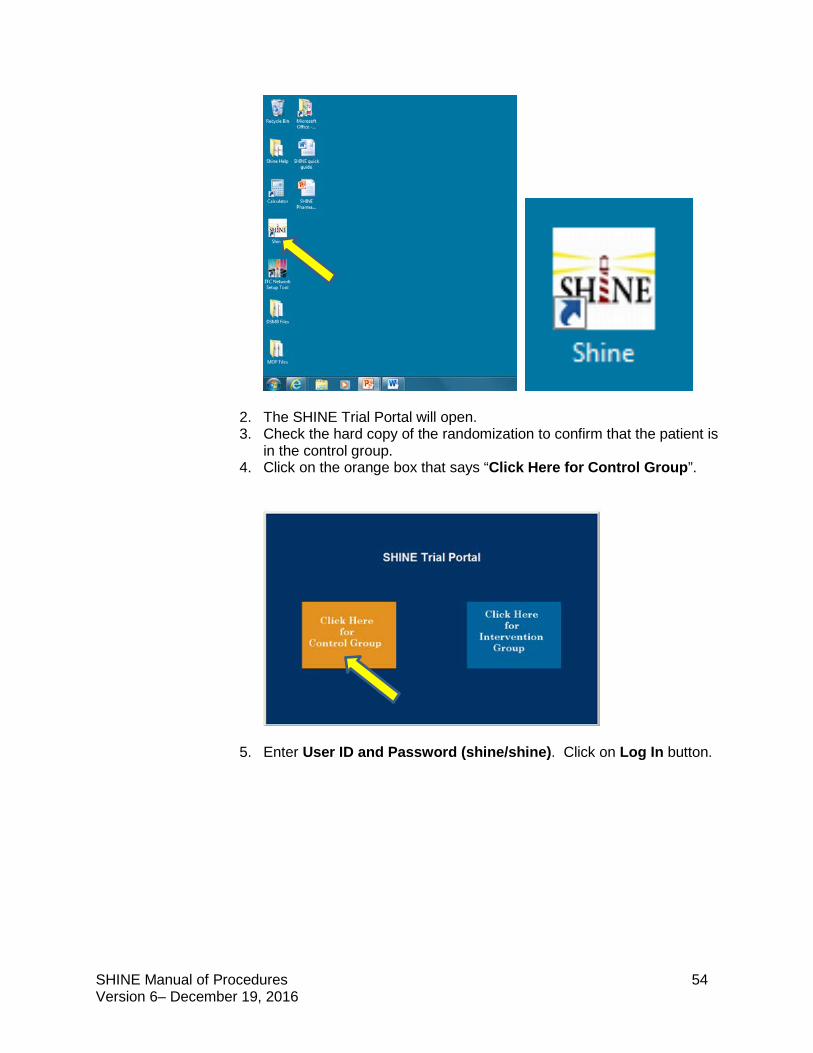
SHINE Manual of Procedures 54 Version 6– December 19, 2016
2. The SHINE Trial Portal will open. 3. Check the hard copy of the randomization to confirm that the patient is
in the control group. 4. Click on the orange box that says “Click Here for Control Group”.
5. Enter User ID and Password (shine/shine). Click on Log In button.
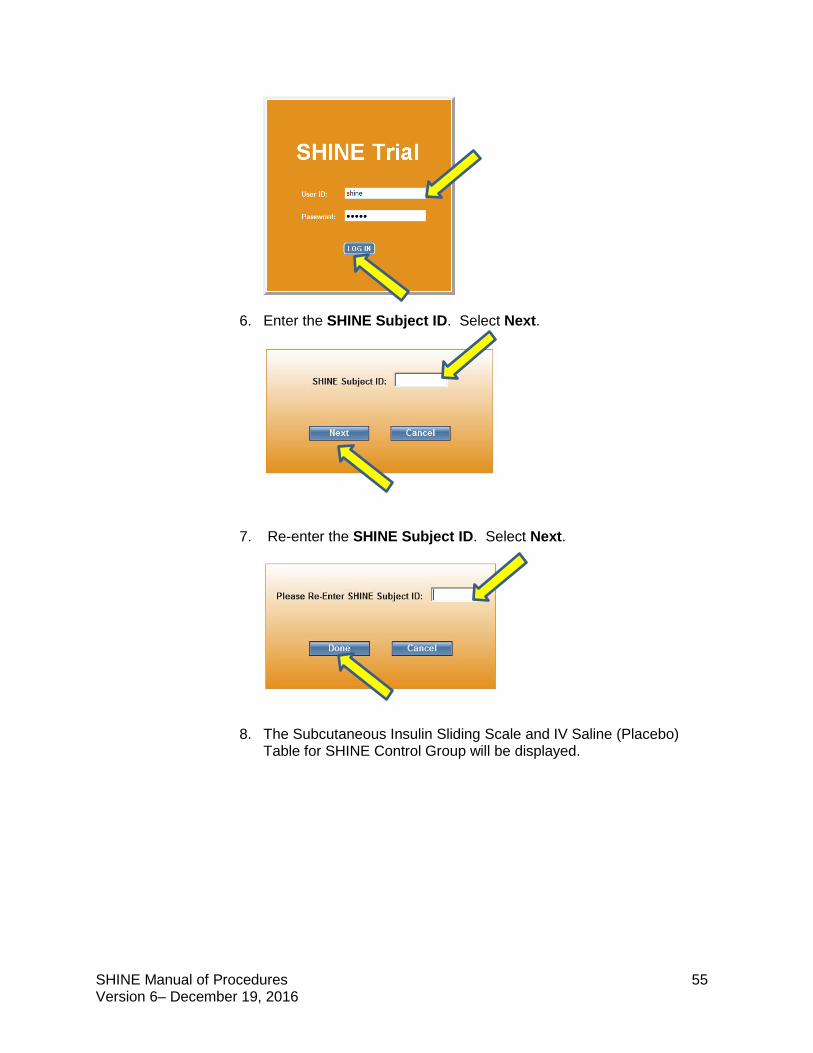
SHINE Manual of Procedures 55 Version 6– December 19, 2016
6. Enter the SHINE Subject ID. Select Next.
7. Re-enter the SHINE Subject ID. Select Next.
8. The Subcutaneous Insulin Sliding Scale and IV Saline (Placebo)
Table for SHINE Control Group will be displayed.
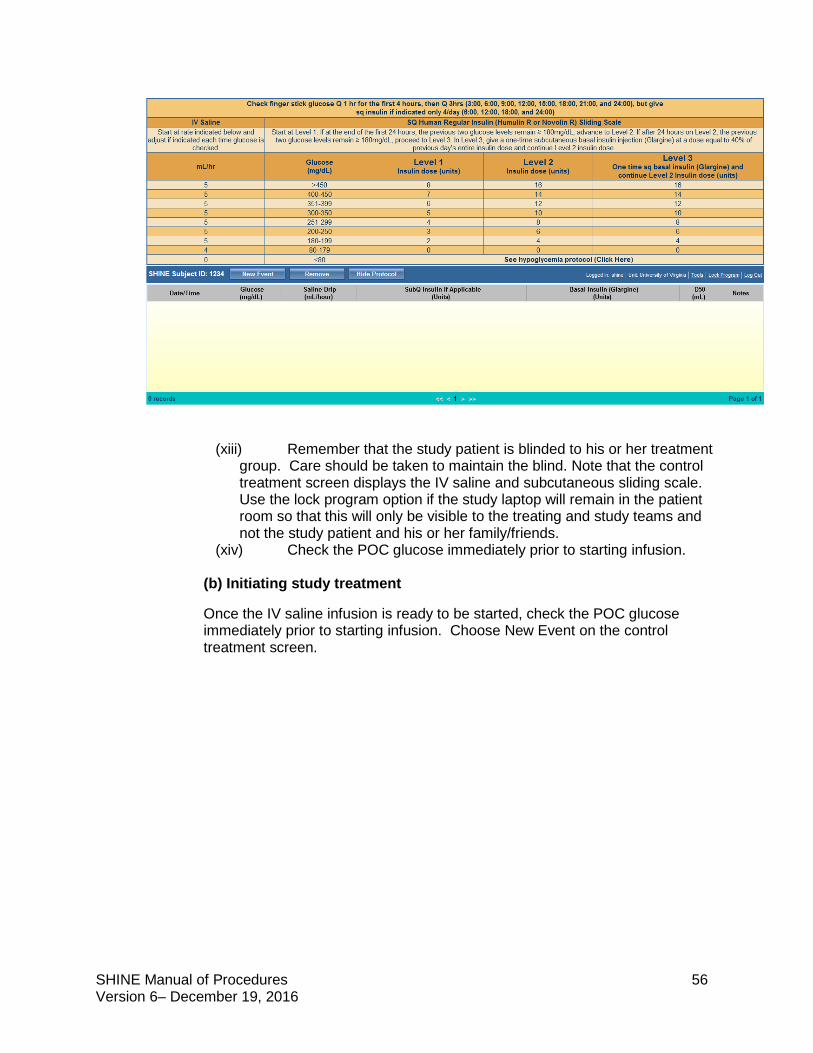
SHINE Manual of Procedures 56 Version 6– December 19, 2016
(xiii) Remember that the study patient is blinded to his or her treatment group. Care should be taken to maintain the blind. Note that the control treatment screen displays the IV saline and subcutaneous sliding scale. Use the lock program option if the study laptop will remain in the patient room so that this will only be visible to the treating and study teams and not the study patient and his or her family/friends.
(xiv) Check the POC glucose immediately prior to starting infusion.
(b) Initiating study treatment
Once the IV saline infusion is ready to be started, check the POC glucose immediately prior to starting infusion. Choose New Event on the control treatment screen.
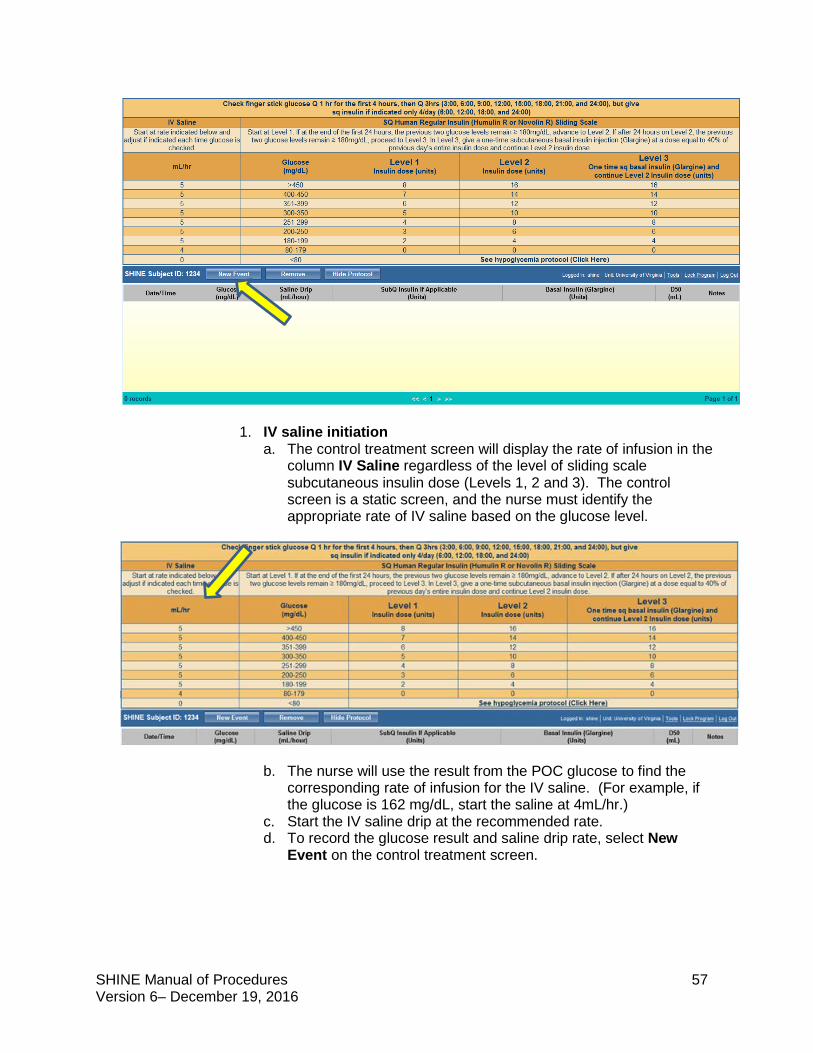
SHINE Manual of Procedures 57 Version 6– December 19, 2016
1. IV saline initiation a. The control treatment screen will display the rate of infusion in the
column IV Saline regardless of the level of sliding scale subcutaneous insulin dose (Levels 1, 2 and 3). The control screen is a static screen, and the nurse must identify the appropriate rate of IV saline based on the glucose level.
b. The nurse will use the result from the POC glucose to find the
corresponding rate of infusion for the IV saline. (For example, if the glucose is 162 mg/dL, start the saline at 4mL/hr.)
c. Start the IV saline drip at the recommended rate. d. To record the glucose result and saline drip rate, select New
Event on the control treatment screen.
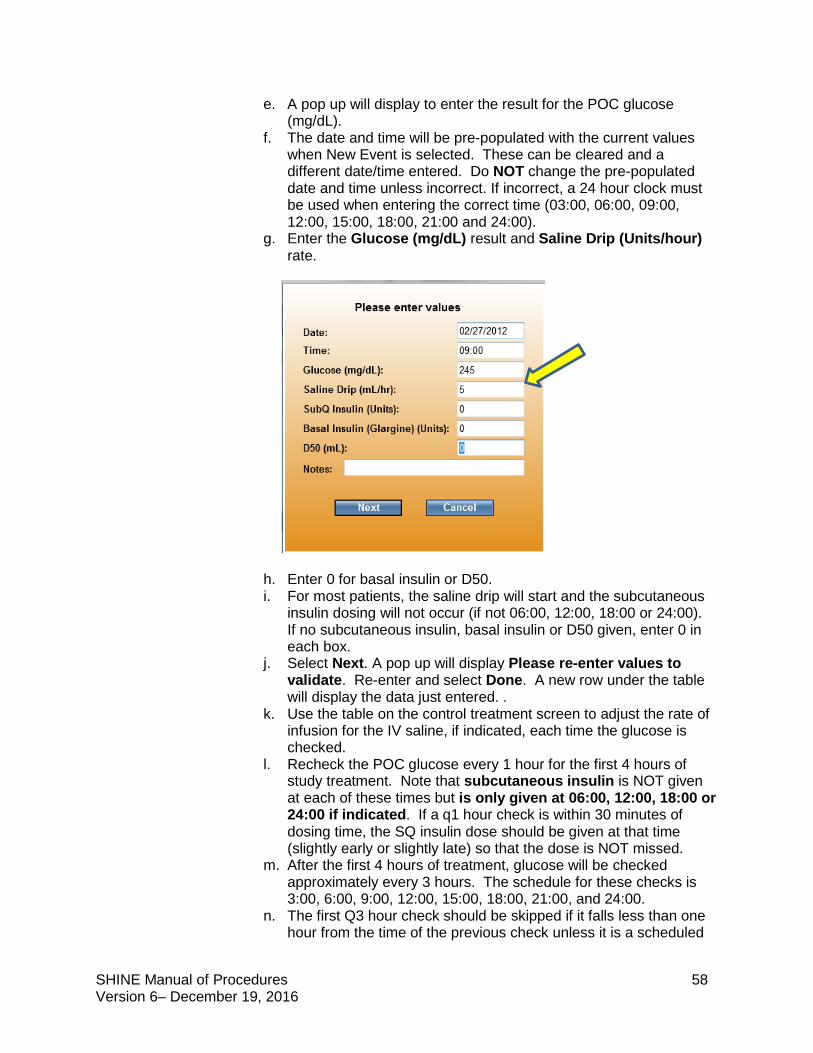
SHINE Manual of Procedures 58 Version 6– December 19, 2016
e. A pop up will display to enter the result for the POC glucose (mg/dL).
f. The date and time will be pre-populated with the current values when New Event is selected. These can be cleared and a different date/time entered. Do NOT change the pre-populated date and time unless incorrect. If incorrect, a 24 hour clock must be used when entering the correct time (03:00, 06:00, 09:00, 12:00, 15:00, 18:00, 21:00 and 24:00).
g. Enter the Glucose (mg/dL) result and Saline Drip (Units/hour) rate.
h. Enter 0 for basal insulin or D50. i. For most patients, the saline drip will start and the subcutaneous
insulin dosing will not occur (if not 06:00, 12:00, 18:00 or 24:00). If no subcutaneous insulin, basal insulin or D50 given, enter 0 in each box.
j. Select Next. A pop up will display Please re-enter values to validate. Re-enter and select Done. A new row under the table will display the data just entered. .
k. Use the table on the control treatment screen to adjust the rate of infusion for the IV saline, if indicated, each time the glucose is checked.
l. Recheck the POC glucose every 1 hour for the first 4 hours of study treatment. Note that subcutaneous insulin is NOT given at each of these times but is only given at 06:00, 12:00, 18:00 or 24:00 if indicated. If a q1 hour check is within 30 minutes of dosing time, the SQ insulin dose should be given at that time (slightly early or slightly late) so that the dose is NOT missed.
m. After the first 4 hours of treatment, glucose will be checked approximately every 3 hours. The schedule for these checks is 3:00, 6:00, 9:00, 12:00, 15:00, 18:00, 21:00, and 24:00.
n. The first Q3 hour check should be skipped if it falls less than one hour from the time of the previous check unless it is a scheduled

SHINE Manual of Procedures 59 Version 6– December 19, 2016
dosing time. Also, if one of the scheduled dosing times (06:00, 12:00, 18:00 or 24:00) falls within one hour of the previous glucose check due to a pause, the POC test should be done and SQ insulin dosed per protocol
o. The saline drip instructions remain the same regardless of Level 1, 2 or 3 subcutaneous insulin dosing.
2. Subcutaneous insulin initiation a. The sliding scale table that is displayed on the control treatment
screen to determine subcutaneous insulin dosing for patients in the control arm of the study.
b. The control screen is a static screen used to determine the appropriate dose of subcutaneous insulin based on the glucose level.
c. A POC glucose check is required to start the study infusion, and then the glucose should be checked at least every 1 hour for the first 4 hours. After the first four hours on the study infusion, the glucose will be checked approximately every 3 hours for the remainder of the study treatment period. The schedule for the checks after the first 4 hours is 3:00, 6:00, 9:00, 12:00, 15:00, 18:00, 21:00, and 24:00.
d. The subcutaneous insulin should only be given (if indicated based on the POC glucose result) four times per day at 6:00, 12:00, 18:00, and 24:00.
e. At each POC glucose check the glucose level needs to be entered into the SHINE study portal. When the glucose is > 80 mg/dL there is a +/- 15 minute window to enter the data, when glucose is < 80 mg/dL there is a +/- 5 minute window to enter the data to remain protocol compliant.
f. Select New Event on the control treatment screen.

SHINE Manual of Procedures 60 Version 6– December 19, 2016
g. A pop up will display to enter the result for the POC glucose
(mg/dL). h. The date and time will be pre-populated with the current values
when New Event is selected. These can be cleared and a different date/time entered. Do NOT change the pre-populated date and time unless they are incorrect. If incorrect, a 24 hour clock must be used when entering the correct time (03:00, 06:00, 09:00, 12:00, 15:00, 18:00, 21:00 or 24:00).
i. Type in the glucose level. j. Enter 0 for basal insulin or D50. k. For most patients, the saline drip will start and the subcutaneous
insulin dosing will not occur (if not 06:00, 12:00, 18:00 or 24:00). If no subcutaneous insulin, basal insulin or D50 given, enter 0 in each box.
l. Select Next. m. At the first scheduled time for subcutaneous insulin dosing (6:00,
12:00, 18:00, and 24:00), use the sliding scale to determine the insulin dose.
n. All patients will start at Level 1 on the sliding scale at initiation. Level adjustment decisions will only be made at 24 and 48 hours in consultation with the study team.
o. The nurse will use the sliding scale to determine the insulin dose in units in Level 1 that corresponds to the POC glucose level.
p. If an insulin dose is indicated, Human Regular Insulin (Humulin R or Novolin R) should be used for the subcutaneous insulin injections. DO NOT USE ANALOG RAPID ACTING INSULIN (HUMALOG/NOVOLOG/APIDRA) IN THE CONTROL GROUP.
q. The nurse will administer the indicated dose of subcutaneous Human Regular Insulin and record in control treatment screen.
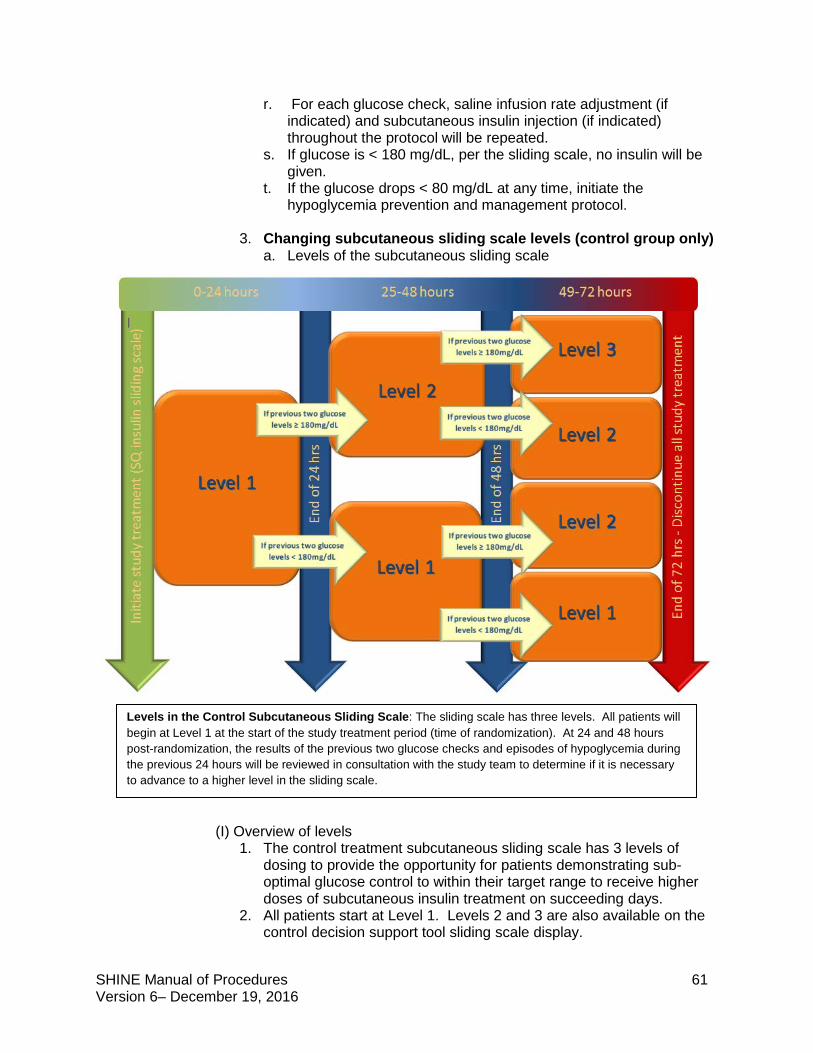
SHINE Manual of Procedures 61 Version 6– December 19, 2016
r. For each glucose check, saline infusion rate adjustment (if indicated) and subcutaneous insulin injection (if indicated) throughout the protocol will be repeated.
s. If glucose is < 180 mg/dL, per the sliding scale, no insulin will be given.
t. If the glucose drops < 80 mg/dL at any time, initiate the hypoglycemia prevention and management protocol.
3. Changing subcutaneous sliding scale levels (control group only) a. Levels of the subcutaneous sliding scale
(I) Overview of levels
1. The control treatment subcutaneous sliding scale has 3 levels of dosing to provide the opportunity for patients demonstrating sub-optimal glucose control to within their target range to receive higher doses of subcutaneous insulin treatment on succeeding days.
2. All patients start at Level 1. Levels 2 and 3 are also available on the control decision support tool sliding scale display.
Levels in the Control Subcutaneous Sliding Scale: The sliding scale has three levels. All patients will begin at Level 1 at the start of the study treatment period (time of randomization). At 24 and 48 hours post-randomization, the results of the previous two glucose checks and episodes of hypoglycemia during the previous 24 hours will be reviewed in consultation with the study team to determine if it is necessary to advance to a higher level in the sliding scale.

SHINE Manual of Procedures 62 Version 6– December 19, 2016
3. The opportunity to consider advancing by 1 level occurs only twice during the study treatment: at the time points of 24 and 48 hours of treatment based on time of randomization.
4. At 24 and 48 hours, the results of the previous two glucose checks and episodes of hypoglycemia during the previous 24 hours will be reviewed in consultation with the study team to determine if it is necessary to advance to a higher level in the sliding scale.
5. For any study patient in the control group who experiences 3 or more episodes of hypoglycemia (glucose concentration of < 70 mg/dL in a 24 hour period, contact the Shine Safety Hotline. At the 24 and 48 hour intervals, regardless of the previous two glucose measurements, do NOT advance to a higher level on the subcutaneous sliding scale.
(c) Navigating the levels of control subcutaneous sliding scale
(i) Level 1 1. All patients will be initiated at Level 1.
(ii) 24 hours from the time of randomization:
1. If at the end of the first 24 hours either of the previous two glucose levels are < 180 mg/dL, continue at Level 1.
2. If at the end of the first 24 hours, both of the previous two glucose levels are ≥ 180 mg/dL, advance to Level 2.

SHINE Manual of Procedures 63 Version 6– December 19, 2016
3. Follow instructions above for glucose checks, saline infusion and subcutaneous insulin injections. 4. No basal insulin should be given at this point.
(ii) 48 hours after randomization: 1. If on Level 1:
a. If at the end of 48 hours, either of the previous two glucose levels are < 180 mg/dL, continue on Level 1.
b. If at the end of 48 hours, both of the previous two glucose levels are ≥ 180 mg/dL, advance to Level 2.
2. If on Level 2: a. If at the end of 48 hours and either of the previous two glucose
levels are < 180 mg/dL, continue on Level 2. b. If at the end of 48 hours, both of the previous two glucose
levels remain ≥ 180 mg/dL, proceed to Level 3.

SHINE Manual of Procedures 64 Version 6– December 19, 2016
c. Follow instructions above for glucose checks, saline infusion and subcutaneous insulin injections.
(iii) If a patient advances to Level 3:
1. Give a one-time subcutaneous basal insulin injection Glargine (Lantus) at a dose of 40% of previous 24 hours entire insulin dose given.
2. Calculate the dose of basal insulin. a. Sum the total insulin given over the past 24 hours. b. Multiply this sum by 0.40 to calculate the dose. c. Round down if < 0.4 and round up if > 0.5
3. Basal insulin (Glargine) will be requested from pharmacy per site procedures.
4. This dose of basal insulin should be given approximately at the 48 hour point (from time of randomization) regardless of time of day.
5. Record the dose of basal insulin by entering New Event. The pop up will display. a. If not a dosing time, enter the following values.
i. Time: actual time given (do not change unless incorrect) ii. Glucose: enter the previous glucose value (because this
cannot be blank or 0) and note that this is the previous glucose value
iii. SQ insulin: If it is not a SQ dosing time, enter 0 as no SQ insulin dose should be administered.
iv. D50: Enter 0 as no D50 should be administered. v. Basal: Enter the dose given. vi. Note: Since not a scheduled glucose check time, type
‘previous value entered because not a scheduled check time’. vii. Select next and re-enter values and select OK.
b. If it is a dosing time, enter the following values: i. If the 48 hour point is within 15 minutes of a glucose check,
check POC, give SQ insulin per protocol. ii. Time: actual time given (do not change unless incorrect). iii. Glucose: enter the POC glucose value. iv. SQ insulin: enter the SQ regular insulin dose based on sliding
scale if one of the scheduled dosing times (06:00, 12:00, 18:00 or 21:00).
v. D50: Enter 0 as no D50 should be administered. vi. Basal: Enter the dose given. vii. Note: Note that this was a scheduled glucose check time as
well as the time of the 48 hour/glargine (Lantus) dose time.

SHINE Manual of Procedures 65 Version 6– December 19, 2016
viii. Select next and re-enter values and select OK. 6. Continue Level 3 of the sliding scale insulin dose. 7. Follow instructions above for glucose checks, saline infusion and
subcutaneous insulin injections.
(iv) After 72 hours on study protocol, study treatment should be discontinued.
(4 ) Hypoglycemia prevention and management protocol (a) Hypoglycemia prevention and management (hypoglycemia protocol)
(i) The hypoglycemia protocol will be initiated for any patient whose glucose level drops below 80 mg/dL.
(ii) Definitions 1. Blood glucose < 80 mg/dL initiate hypoglycemia prevention and
management protocol 2. Hypoglycemia: defined as blood glucose < 70 mg/dL (will require
additional steps) 3. Severe hypoglycemia: defined as blood glucose < 40 mg/dL (will
require SAE reporting)
(iii) Hypoglycemia protocol for control group 1. If the blood glucose drops to < 80 mg/dL, select the link on the control
treatment screen to view the hypoglycemia protocol. a. Stop the saline infusion and hold all subcutaneous insulin
injections when the glucose is < 80 mg/dL! b. For all POC glucose < 80 mg/dL, the nurse will prepare a dose of
IV D50 25 ml (1/2 amp). c. D50 will be stored to allow immediate availability at the bedside. d. Give 25mL of D50, slow IV push (over 1-2 minutes). e. Check glucose again in 15 minutes. f. To record the glucose measurement and D50 administration,
select New Event.

SHINE Manual of Procedures 66 Version 6– December 19, 2016
g. A pop up will display to enter the result for the POC glucose (mg/dL).
h. Enter the Glucose result and D50 administered. i. As long as the blood glucose is < 80 mg/dL, all insulin treatments
and the IV saline will be held. j. Enter 0 for SubQ Insulin and 0 for Saline Drip. k. Select Save. l. After 15 minutes, the nurse will repeat the POC glucose. Check
POC glucose every 15 minutes until glucose is ≥ 80 mg/dL. m. These steps will be repeated for every glucose measurement that is < 80 mg/dL. n. If the glucose drops below 70 mg/dL, additional steps are required (described below).
(iv) Additional steps for blood glucose < 70 mg/dL If the blood glucose drops < 70 mg/dL, the following additional actions are required: 1. Continue to use the hypoglycemia protocol above (e.g. hold saline
and SQ insulin, give D50, repeat glucose checks every 15 minutes). 2. Draw a STAT laboratory serum glucose measurement but do not
delay treatment with D50 by waiting for this blood draw result. Only the results POC glucose checks should be used for insulin dosing. The study coordinator will document the result of the serum glucose measurement.
3. Screen the patient for hypoglycemia symptoms using Hypoglycemia Symptomatic Questionnaire. a. The worksheet and instructions will be available on paper and on
the desktop of the laptops. b. The Hypoglycemia Symptomatic Questionnaire must be repeated
every 15 minutes when glucose is < 70 mg/dL.

SHINE Manual of Procedures 67 Version 6– December 19, 2016
c. Once the glucose is ≥ 70 mg/dL, no additional assessment with the Hypoglycemia Symptomatic Questionnaire is required.
d. Once the glucose is ≥ 80 mg/dL, the timing of glucose checks and insulin infusion rate will again be determined by the control treatment screen.
4. Screen the patient for neurological worsening. a. An assessment of neurological change is required per site
standard care (“neuro check”) every 15 minutes when glucose is < 70 mg/dL even if there are no new neurological findings.
b. The SHINE study definition of neurological worsening is any clinical change that is associated with a ≥ 4 point increase from baseline on the NIHSS score. (Baseline is considered to be the most recent daily NIHSS..)
c. Any patient with neurological worsening will be assessed for hypoglycemia and for relatedness of the hypoglycemia to neurological worsening if present.
d. If the patient has not returned to neurological baseline within 24 hours, a NIHSS assessment is required within 24(+/-4) hours from onset of hypoglycemic event (< 70 mg/dL). If the patient has returned to neurological baseline at any point in less than 24 hours, the NIHSS is not required at 24 hours.
e. If neurological worsening persists for greater than or equal to 24 hours and is associated with a glucose concentration of < 55 mg/dL, an SAE form is required. If the glucose falls below 40 mg/dL at any time, an SAE form is required.
5. For any study patient in the control group who experiences 3 or more episodes of hypoglycemia (glucose concentration of < 70 mg/dL in a 24 hour period), contact the SHINE Safety Hotline (800-915-7320 ext 2). At the 24 and 48 hour intervals, regardless of the previous two glucose measurements, do NOT advance to a higher level on the subcutaneous sliding scale.
(5) Meals
(a) Patients who are PO (i) SHINE protocol-recommended diet for the control group
All patients in the control group who pass a swallow test and are cleared to eat will have a 60 gram carbohydrate diet. The 60 gram carbohydrate diet should be ordered for breakfast, lunch and dinner as a part of standard care. Meals should be delivered at about 06:00, 12:00 and 18:00. Meals will be withheld until subcutaneous insulin is given per the study schedule for glucose checks and sliding scale for insulin dosing occurring at meal time. Meal insulin will not be given in this group.
(ii) Protocol-approved snacks
SHINE patients may also consume up to 2 low carbohydrate snacks (< 5 grams carbohydrates per serving) from the list below between meals (up to 6 low carbohydrate snacks daily). The study protocol diet does not limit the consumption of sugar free foods and drinks listed in the unlimited category below. Patients should only consume food included on the meal tray from the hospital kitchen or the protocol approved snacks during the 72 hour treatment period.

SHINE Manual of Procedures 68 Version 6– December 19, 2016
Low carbohydrate snack options (up to 2 between meals) 5 celery sticks + Tablespoon peanut butter 5 baby carrots 5 cherry tomatoes + 1 Tablespoon ranch 1 hard-boiled egg ½ cup raw broccoli + 1 Tablespoon ranch 1 cup cucumber slices + 1 Tablespoon ranch dressing ¼ cup of fresh blueberries 1 cup of salad greens, 1/2 cup of diced cucumber, and with vinegar and oil 2 saltine crackers 1 piece of string cheese stick ½ cup of egg salad, tuna salad or chicken salad 3 oz of deli ham, chicken or turkey slices 1 serving of cubed or sliced cheese (1 oz) ½ cup cottage cheese ½ cup tofu 1 slice deli ham, chicken or turkey + 1 slice cheese Unlimited Bouillon and broth Club soda, unsweetened Coffee, unsweetened or artificial sweeteners only Diet soft drinks Flavoring extracts Horseradish Mineral water Mustard Pickles Soy sauce Spices Sugar-free drink mixes Sugar-free gum Sugar-free Jell-O Tabasco or hot sauce Unsweetened lemon or lime juice Unsweetened tea Vinegar
(iii) Patients receiving tube feedings may receive either bolus or continuous tube feedings per standard care. Bolus tube feedings will be encouraged to be given at around 06:00, 12:00 and 18:00 and will also be 60 gram carbohydrate per feed.

SHINE Manual of Procedures 69 Version 6– December 19, 2016
(6) Special Situations with the Control Treatment Protocol (a) Pauses in control treatment protocol
(i) When the SHINE control group treatment protocol is temporarily interrupted for any reason (e.g. patient needing to go off the care unit), and the clinical nurse or study team cannot accompany the patient, the IV saline drip should be stopped. Document the pause following the steps below. 1. Select “New Event” on the control treatment screen. Enter the
previous glucose value. Enter “0” for Saline Drip, SubQ Insulin, Basal Insulin, and D50. In the Notes field, enter a comment documenting the pause and that the glucose entered was a previous value (Ex. Prev BG, pause for MRI).
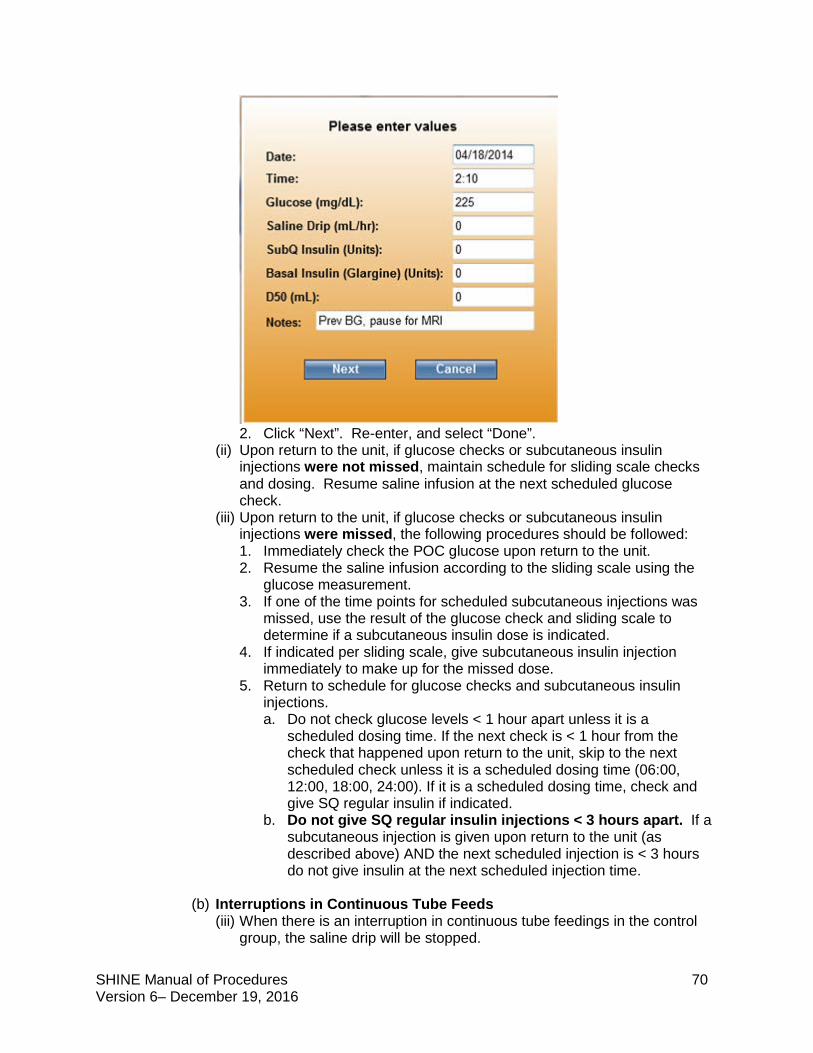
SHINE Manual of Procedures 70 Version 6– December 19, 2016
2. Click “Next”. Re-enter, and select “Done”.
(ii) Upon return to the unit, if glucose checks or subcutaneous insulin injections were not missed, maintain schedule for sliding scale checks and dosing. Resume saline infusion at the next scheduled glucose check.
(iii) Upon return to the unit, if glucose checks or subcutaneous insulin injections were missed, the following procedures should be followed: 1. Immediately check the POC glucose upon return to the unit. 2. Resume the saline infusion according to the sliding scale using the
glucose measurement. 3. If one of the time points for scheduled subcutaneous injections was
missed, use the result of the glucose check and sliding scale to determine if a subcutaneous insulin dose is indicated.
4. If indicated per sliding scale, give subcutaneous insulin injection immediately to make up for the missed dose.
5. Return to schedule for glucose checks and subcutaneous insulin injections. a. Do not check glucose levels < 1 hour apart unless it is a
scheduled dosing time. If the next check is < 1 hour from the check that happened upon return to the unit, skip to the next scheduled check unless it is a scheduled dosing time (06:00, 12:00, 18:00, 24:00). If it is a scheduled dosing time, check and give SQ regular insulin if indicated.
b. Do not give SQ regular insulin injections < 3 hours apart. If a subcutaneous injection is given upon return to the unit (as described above) AND the next scheduled injection is < 3 hours do not give insulin at the next scheduled injection time.
(b) Interruptions in Continuous Tube Feeds (iii) When there is an interruption in continuous tube feedings in the control
group, the saline drip will be stopped.

SHINE Manual of Procedures 71 Version 6– December 19, 2016
(iv) The control protocol for subcutaneous insulin dosing does not change if continuous tube feeds are interrupted and the schedule for glucose checks and subcutaneous insulin dosing should be followed.
(v) If the continuous tube feeds are restarted less than 1 hour from the time that the saline drip was stopped, the nurse will check the glucose at the time that the continuous tube feeds are restarted. The glucose measurement will be used to restart the saline drip if indicated per the saline sliding scale.
(vi) If the continuous tube feeds are not restarted within 1 hour, the nurse will re-start the saline drip, at the time of the next scheduled glucose check based on the saline sliding scale (0, 4, or 5 ml/hr).
(7) Locking the GlucoStabilizer® Program
When the nurse leaves the bedside, the GlucoStabilizer® should be locked. (ii) Choose lock program
(c) When the control program is locked, a blank screen displays. (d) The User ID is shine, and the Password is shine.
(8) Stopping study treatment when there is a change in diagnosis of ischemic stroke/stroke mimic (a) In the event that there is a change in the diagnosis of ischemic stroke during
the 72 hour treatment period, all study treatments should be stopped at the time that the study team receives knowledge of the new diagnosis.
(b) The clinical care team will determine glucose control therapy after the SHINE treatment protocol is discontinued.
(c) Instructions for completing CRFs when the study treatment is stopped due to a change in diagnosis of ischemic stroke

SHINE Manual of Procedures 72 Version 6– December 19, 2016
(i) The Day 1-Day 3 visits should be completed only during the period of time that the patient was receiving study treatment.
(ii) The final diagnosis will be captured on the Study Treatment CRF (Form 15) as: Migraine, Seizure, Brain tumor/mass, Psychogenic, Anamnestic syndrome, CNS Infection (abscess, meningitis, encephalitis), Toxic/metabolic (encephalopathy) and Other: specify).
(iii) A brief narrative explaining the change in diagnosis is required. This narrative should include sufficient information to demonstrate that the new diagnosis is most likely including appropriate imaging and laboratory data. A brief narrative template is included in WebDCU: [On ________(full date), the diagnosis was changed to ________. This diagnosis was based on ________(lab/imaging results) and ________(clinical information).]
(iv) On the Study Treatment CRF (Form 15), the study treatment status should be captured as: Started but discontinued prior to 72 hours of study treatment
(d) The End of Study Visit must be completed. Unless consent is withdrawn, patients whose treatment ends early are still in the study and the 6 week and 90 day follow up visits must be completed in addition to the End of Study Visit.
(9) Transition from study treatment (a) The start time for the study treatment period is the time of randomization.
The treatment period will be 72 hours or until the time of discharge from the hospital, whichever comes first.
(b) Patients who discontinue study treatment are required to do so 6 hours prior to actual discharge from the hospital.
(c) To document stopping study treatment, select “New Event” on the control treatment screen. Enter the previous glucose value. Enter “0” for Saline Drip, SubQ Insulin, Basal Insulin, and D50. In the Notes field, document that the glucose entered was a previous value and that study treatment was stopped.
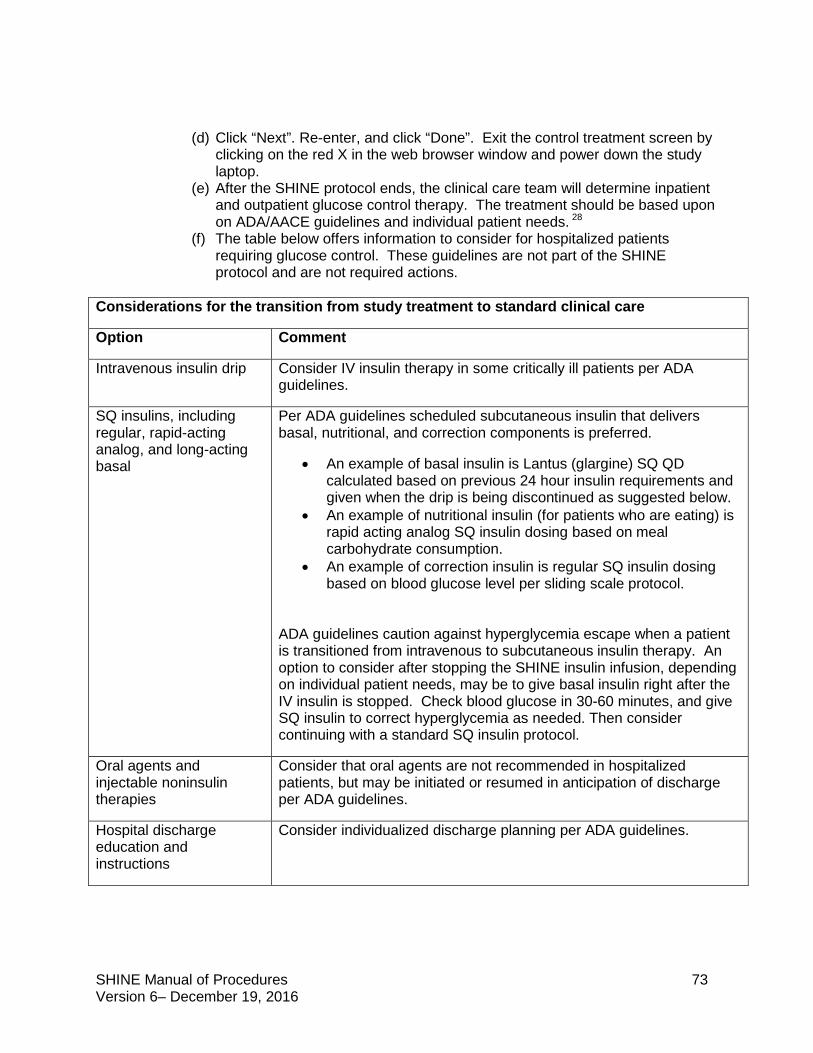
SHINE Manual of Procedures 73 Version 6– December 19, 2016
(d) Click “Next”. Re-enter, and click “Done”. Exit the control treatment screen by clicking on the red X in the web browser window and power down the study laptop.
(e) After the SHINE protocol ends, the clinical care team will determine inpatient and outpatient glucose control therapy. The treatment should be based upon on ADA/AACE guidelines and individual patient needs. 28
(f) The table below offers information to consider for hospitalized patients requiring glucose control. These guidelines are not part of the SHINE protocol and are not required actions.
Considerations for the transition from study treatment to standard clinical care
Option Comment
Intravenous insulin drip Consider IV insulin therapy in some critically ill patients per ADA guidelines.
SQ insulins, including regular, rapid-acting analog, and long-acting basal
Per ADA guidelines scheduled subcutaneous insulin that delivers basal, nutritional, and correction components is preferred.
• An example of basal insulin is Lantus (glargine) SQ QD calculated based on previous 24 hour insulin requirements and given when the drip is being discontinued as suggested below.
• An example of nutritional insulin (for patients who are eating) is rapid acting analog SQ insulin dosing based on meal carbohydrate consumption.
• An example of correction insulin is regular SQ insulin dosing based on blood glucose level per sliding scale protocol.
ADA guidelines caution against hyperglycemia escape when a patient is transitioned from intravenous to subcutaneous insulin therapy. An option to consider after stopping the SHINE insulin infusion, depending on individual patient needs, may be to give basal insulin right after the IV insulin is stopped. Check blood glucose in 30-60 minutes, and give SQ insulin to correct hyperglycemia as needed. Then consider continuing with a standard SQ insulin protocol.
Oral agents and injectable noninsulin therapies
Consider that oral agents are not recommended in hospitalized patients, but may be initiated or resumed in anticipation of discharge per ADA guidelines.
Hospital discharge education and instructions
Consider individualized discharge planning per ADA guidelines.

SHINE Manual of Procedures 74 Version 6– December 19, 2016
5.1.4 Considerations for Study Coordinators during the Treatment Period
Study team should: 1. Communicate with nurses at shift change, meals, end of each 24 hour period
during the treatment period 2. Establish site procedures to ensure that study coordinator is notified if any
hypoglycemia, severe hyperglycemia or neurologic changes occur. 3. When a study patient experiences study defined neurological worsening,
capture the NIHSS score 24 hours (+/-4 hours) after the start of the hypoglycemic event (blood glucose < 70 mg/dL).
4. Be notified if patient planned for discharge. This is particularly important if the patient will be discharged before 72 hours as the study treatment must be discontinued at least 6 hours prior to discharge.
5. Confirm that the NIHSS score is captured each day. 6. Review the Daily Care Log and ensure that data are being captured accurately
and that meals are being ordered and recorded per protocol. 7. Control group – assist with transition from hourly checks to q3 hour scheduled
check times and 8. Assist in planning the transition to standard care.
5.1.5 Concomitant or Ancillary Therapy Throughout the study period, all AHA guidelines for acute stroke care will be followed. This includes standard care for acute ischemic stroke patients such as swallowing evaluation prior to initiation of PO intake, DVT prophylaxis and secondary stroke prevention therapies. Blood pressure and temperature recommendations also will be followed. Rehabilitation evaluation should be considered for every study patient.
FAQ Q: What happens if you check a POC finger stick glucose when it is not due? Do you enter it into the study laptop?
A: If POC glucose is measured when not scheduled (e.g. concern for hypoglycemia, other standard care protocol or test, etc.), only enter in GlucoStabilizer/ control treatment screen if the result is <80mg/dL.
The reason that values that are <80mg/dL should be entered is to document the POC glucose levels as they relate to the initiation of the hypoglycemia prevention and management in both treatment groups. Also, importantly, GlucoStabilizer will not know to prompt the hypoglycemia protocol for study patients in the intervention group if a level of <80mg/dL is not entered in the study laptop.
The reason that POC glucose values should NOT be entered if >80mg/dL is that this can change the timing of next glucose check and the multiplier which is used to calculated future insulin dosing for study patients in the intervention group. GlucoStabilizer has been programmed and validated based on entering glucose values when due per the programmed schedule for checks recommended by the decision support tool.
All POC glucose values should be documented in the clinical chart per institutional policies.
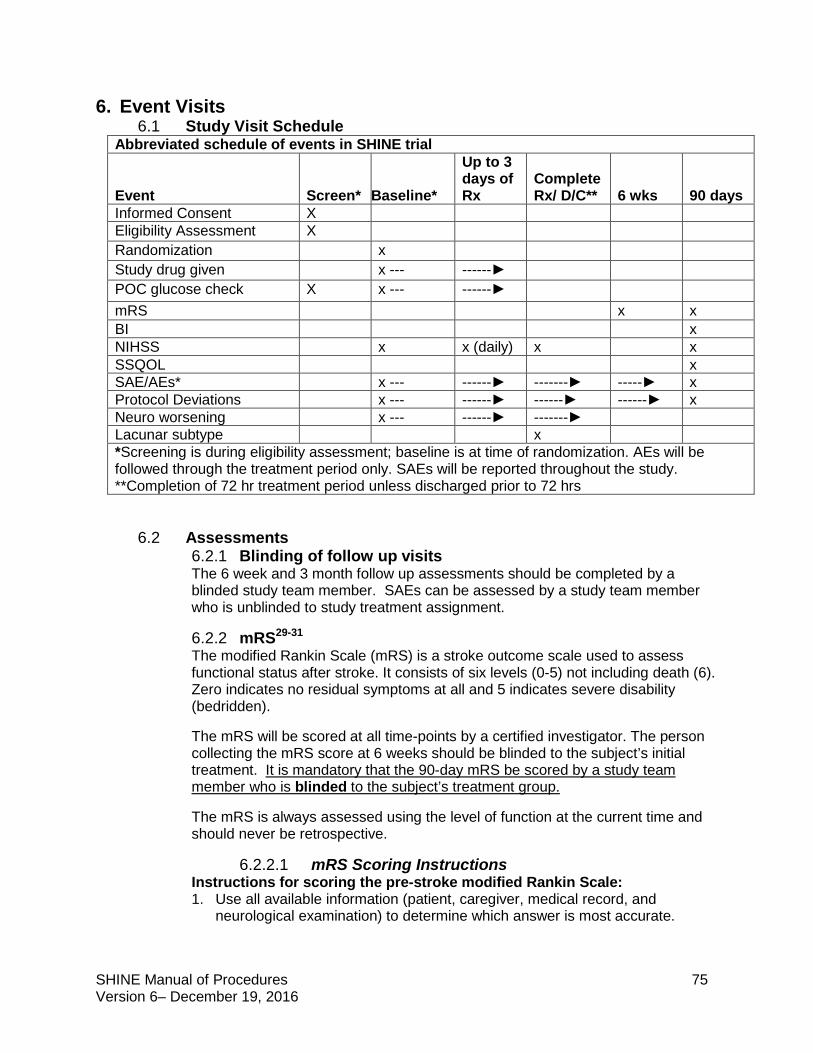
SHINE Manual of Procedures 75 Version 6– December 19, 2016
6. Event Visits 6.1 Study Visit Schedule
Abbreviated schedule of events in SHINE trial
Event
Screen*
Baseline*
Up to 3 days of Rx
Complete Rx/ D/C**
6 wks
90 days
Informed Consent X Eligibility Assessment X Randomization x Study drug given x --- ------► POC glucose check X x --- ------► mRS x x BI x NIHSS x x (daily) x x SSQOL x SAE/AEs* x --- ------► -------► -----► x Protocol Deviations x --- ------► ------► ------► x Neuro worsening x --- ------► -------► Lacunar subtype x *Screening is during eligibility assessment; baseline is at time of randomization. AEs will be followed through the treatment period only. SAEs will be reported throughout the study. **Completion of 72 hr treatment period unless discharged prior to 72 hrs
6.2 Assessments
6.2.1 Blinding of follow up visits The 6 week and 3 month follow up assessments should be completed by a blinded study team member. SAEs can be assessed by a study team member who is unblinded to study treatment assignment.
6.2.2 mRS29-31 The modified Rankin Scale (mRS) is a stroke outcome scale used to assess functional status after stroke. It consists of six levels (0-5) not including death (6). Zero indicates no residual symptoms at all and 5 indicates severe disability (bedridden).
The mRS will be scored at all time-points by a certified investigator. The person collecting the mRS score at 6 weeks should be blinded to the subject’s initial treatment. It is mandatory that the 90-day mRS be scored by a study team member who is blinded to the subject’s treatment group.
The mRS is always assessed using the level of function at the current time and should never be retrospective.
6.2.2.1 mRS Scoring Instructions Instructions for scoring the pre-stroke modified Rankin Scale: 1. Use all available information (patient, caregiver, medical record, and
neurological examination) to determine which answer is most accurate.

SHINE Manual of Procedures 76 Version 6– December 19, 2016
2. If the patient had a stroke before the currently acute stroke, score zero only if all signs and symptoms from the previous stroke have resolved. Otherwise, score 1-5 according to the mRS definitions. Note that only patients with a prior stroke can have a mRS score of 1.
3. If the patient has never had a stroke before, in order to score 0 they must have been capable of living independently and walking without assistance from another person. (Necessary devices for walking are not considered assistance.)
Instructions for scoring the modified Rankin Scale during follow up: 1. Use all available information (patient, caregiver, medical record, and
neurological examination) to determine which answer is most accurate. 2. Follow the mRS definitions found: 1) on CRF form 00 and 2) in the NETT
mRS training video located onthe NETT SHINE trial website under education.
6.2.2.2 Modified Rankin Scale (mRS) Training and Certification
The NETT CCC will provide mRS training and certification* to SHINE study team members. The training and certification will be located on the SHINE Education page (https://nett.umich.edu/clinical-trials/shine/education-and-training). A training video will contain two training vignettes. The certification will consist of correctly scoring a set of vignettes. The certification will be valid for two years.
* The NETT mRS training and certification was informed by the following publications:
van Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, van GJ. Interobserver agreement for the assessment of handicap in stroke patients. Stroke 1988;19:604–607.

SHINE Manual of Procedures 77 Version 6– December 19, 2016
Quinn TJ, Dawson J, Walters MR, Lees KR. Variability in modified Rankin scoring across a large cohort of international observers. Stroke 2008;39:2975–2979.
Bruno A, Akinwuntan AE, Lin C, Close B, Davis K, Baute V, et al. Simplified Modified Rankin Scale Questionnaire: Reproducibility Over the Telephone and Validation With Quality of life. Stroke 2011;42:2276-2279.
6.2.3 BI32 The Barthel Index (BI) is a scale that measures a person’s ability to perform Activities of Daily Living (ADLs). Higher scores indicate a greater chance of living independently.
The BI must be assessed at the Day 90 visit by a blinded assessor.
6.2.4 NIHSS33-35 The National Institutes of Health Stroke Scale (NIHSS) is an assessment tool that quantitatively measures stroke-related neurologic deficits. The NIHSS will be obtained prior to randomization (baseline) by a certified investigator. The NIHSS score used to determine eligibility must be the score closest to the time of randomization and must have been done within 30 minutes of the time of randomization.
Based on the NIHSS scoring instructions, untestable is only considered for the following: Question 5: Motor Arm (UN: Amputation or joint fusion) Question 6: Motor Leg (UN: Amputation or joint fusion) Question 7: Limb Ataxia (UN: Amputation or joint fusion) Question 10: Dysarthria (UN: Intubation or other physical barrier) This only includes patients who are intubated or who have other physical barriers to assess the NIHSS during the entire screening period. If the NIHSS cannot be scored for any of these reasons within 30 minutes prior to randomization, the patient is not eligible. Intubation during the screening period will be exclusionary if the NIHSS cannot be assessed with 30 minutes of randomization or if medications or neurologic worsening are confounding neurological assessment. The NIHSS will be repeated at least once daily during the treatment period, and it is strongly recommended that this be completed by a certified investigator. The daily NIHSS should be documented in the medical record or other source documentation but is not captured in WebDCUTM unless there is an adverse event with neurological worsening. Per the study protocol, the full NIHSS must be assessed daily; however, in cases when this has not been captured, the documented neurological exam from that day can be converted to attain a retrospective NIHSS score.34, 35
It is required that the Day 90 NIHSS is scored by an NIHSS-certified study team member who is blinded to the treatment assignment. Based on the NIHSS scoring instructions, untestable is only considered for the following: Question 5: Motor Arm (UN: Amputation or joint fusion), Question 6: Motor Leg (UN: Amputation or joint fusion), Question 7: Limb Ataxia (UN: Amputation or joint fusion), Question 10: Dysarthria (UN: Intubation or other physical barrier).

SHINE Manual of Procedures 78 Version 6– December 19, 2016
If any item is untestable at follow up, select UN for the response in WebDCUTM, leave the total field blank and make a note in the general comments.
6.2.5 SSQOL36 The SSQOL must be assessed at the Day 90 Visit by a blinded assessor or the subject/blinded family or LAR may self-complete.
The Stroke Specific Quality of Life (SS-QOL) Scale is a validated 49-item, 12-domain (mobility, energy, upper extremity function, work/productivity, mood, self-care, social roles, family roles, vision, language, thinking, and personality) scale. It was designed specifically for stroke patients with input from such patients. Scores range 1.0-5.0 with 5.0 being best. Scores tend to follow a normal distribution and can be analyzed as continuous variables. The total score and the individual SS-QOL domains can be analyzed separately. This scale takes 10-15 minutes to administer and caregivers can answer the scale questions for patient who are unable to do so.
6.2.6 SSQOL Administration The SSQOL has been validated for face-to-face and telephone administration. Both modes of administration require about 12 minutes. In interviewer-administering the questionnaire, several basic guidelines apply:
• Read all introductory text verbatim. This helps to frame the questions for the patient and introduces the reference time frame (the past week).
• Read each item verbatim.
• Read each response option clearly and without any influence, such as expression, tone of voice or eye contact changes. You may wish to give the patient a laminated sheet with the two different response sets for ease of reference during the interview.
• Repeat the time reference (“During the past week…” every 4-5 items to remind the patient that you are asking about how they were in that specific time frame.
• If a patient responds that they are unsure, remind them that there are no right or wrong answers, read all the response choices again and then ask them to choose the answer that seems best.
• If a patient responds that s/he doesn’t do a particular task and so can’t answer that (e.g. says that they don’t use stairs because they live in a one-level house) ask them to imagine how they would do if they had to perform the specific task in question.
6.2.7 Neuro Checks and Neurological Worsening Any clinically relevant neurological change or decline will trigger a clinical assessment (“neuro check”) and glucose measurement.
Any blood glucose < 70 mg/dL will also trigger a neuro check. Neuro checks must be repeated every 15 minutes when the glucose is < 70 mg/dL. Any patient

SHINE Manual of Procedures 79 Version 6– December 19, 2016
with neurological worsening will be assessed for hypoglycemia and for relatedness of the hypoglycemia to neurological worsening if present.
The SHINE study definition of neurological worsening is any clinical change lasting greater than or equal to 24 hours that is associated with a ≥4 point increase on the NIHSS score from baseline. (Baseline is considered to be the most recent daily NIHSS.) The SHINE study definition of transient neurological decline will be defined as an increase in the NIHSS score of >4 (from baseline or most recent) that lasts less than 24 hours.
Neurological worsening (lasting greater than or equal to 24 hours) and associated with glucose concentration of < 55 mg/dL is a required SAE. Transient neurological decline may or may not be an SAE per the judgment of the site PI. If a patient is intubated, it is recommended that an NIHSS score is completed immediately prior to intubation. An NIHSS should also be assessed immediately after intubation and this most recent daily score should be used to determine if there is a ≥ 4 point increase. 6.2.8 Lacunar Stroke Etiology The small vessel occlusion (lacunar) subtype is a common data element and the definition for scoring is traceable to the TOAST criteria37 and the Causative Classification System38. Small vessel occlusive stroke (lacune) is defined for the SHINE study as: 1. Lacunar syndrome with or without evidence of ischemic lesion less than
1.5cm in diameter in the brainstem or subcortical white matter (TOAST criteria) OR
2. Imaging evidence of a single and clinically relevant acute infarction less than 20mm in greatest diameter within the territory of basal or brainstem penetrating arteries in the absence of any focal pathology in the parent artery at the site of origin of the penetrating artery (focal atheroma, parent vessel dissection, vasculitis, vasospasm, etc.) (CCS criteria).
Lacunar Stroke Etiology is assessed at the End of Treatment Visit.
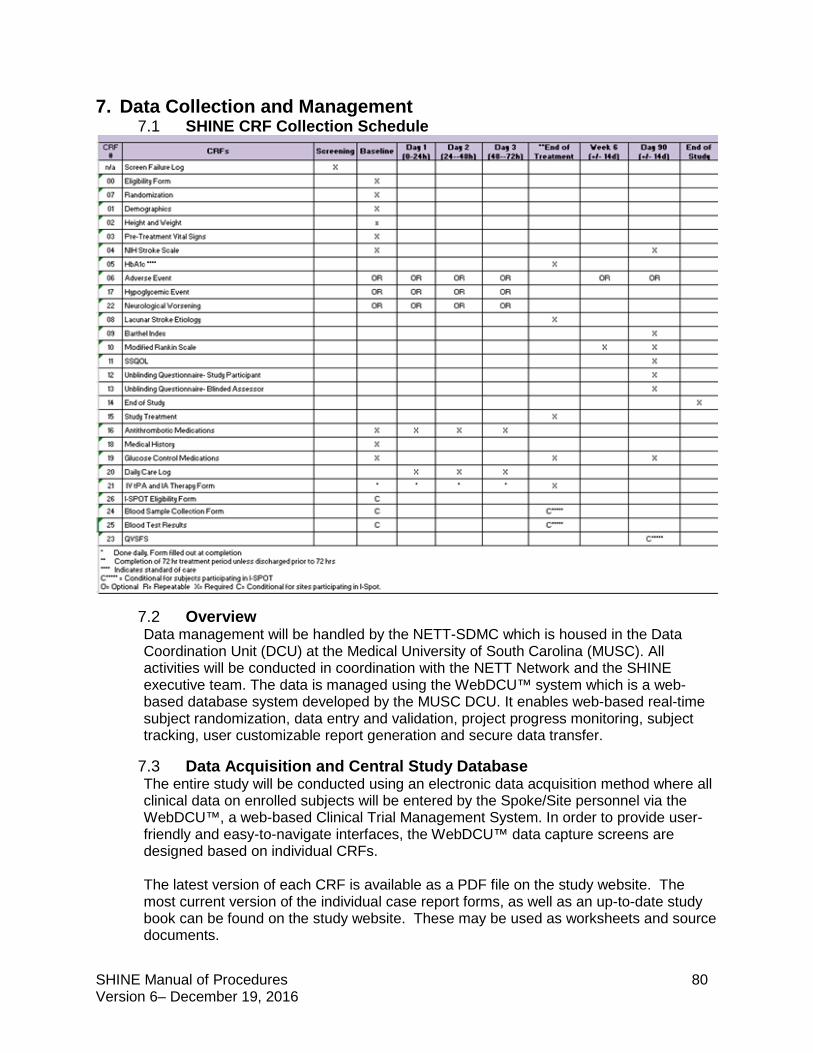
SHINE Manual of Procedures 80 Version 6– December 19, 2016
7. Data Collection and Management 7.1 SHINE CRF Collection Schedule
7.2 Overview Data management will be handled by the NETT-SDMC which is housed in the Data Coordination Unit (DCU) at the Medical University of South Carolina (MUSC). All activities will be conducted in coordination with the NETT Network and the SHINE executive team. The data is managed using the WebDCU™ system which is a web-based database system developed by the MUSC DCU. It enables web-based real-time subject randomization, data entry and validation, project progress monitoring, subject tracking, user customizable report generation and secure data transfer.
7.3 Data Acquisition and Central Study Database The entire study will be conducted using an electronic data acquisition method where all clinical data on enrolled subjects will be entered by the Spoke/Site personnel via the WebDCU™, a web-based Clinical Trial Management System. In order to provide user-friendly and easy-to-navigate interfaces, the WebDCU™ data capture screens are designed based on individual CRFs. The latest version of each CRF is available as a PDF file on the study website. The most current version of the individual case report forms, as well as an up-to-date study book can be found on the study website. These may be used as worksheets and source documents.

SHINE Manual of Procedures 81 Version 6– December 19, 2016
The data validation procedure is implemented in two stages. The study database has extensive consistency checks programmed into the forms during the development of the database. These checks are in place to flag potential data entry errors and protocol violations, including missing required data, data out of a pre-specified range, data conflicts and disparities within each CRF and across different CRFs. When data that violates the consistency check is entered, a rule violation message appears on the data entry screen alerting the data entry person to address it. The choices are:
(1) to correct the entry immediately; (2) to correct the entry at a later time; or (3) to dismiss the rule with an explanation if the entered data is confirmed to be
correct. This may be done for warnings and protocol deviations. To do so, click the red rule violation to go to the ‘View Record: Rule Violation’ page. Click ‘Data Confirmed’ to dismiss the violation. Provide a reason before saving.
Secondly, for some checks that are more complicated, additional consistency checks are periodically run after data entry occurs at the site. All data items that fail these secondary consistency checks are queried via Data Clarification Requests (DCRs) within the database, by NETT-SDMC data managers. Site monitors are also able to generate Data Clarification Requests when discrepancies are found during source to database verification. The DCRs will be generated, communicated to the Spokes/Sites, and resolved on the secure study website. Any changes made in the system have a full audit trail.
7.4 Case Report Forms and Worksheets
7.4.1 Overview of Forms and Requirements • Although it is not a requirement that you use paper worksheets for data
collection, all data defined on the worksheets must be collected and entered into WebDCUTM.
• If paper worksheets are used as source documents, they must be retained at the Clinical Site according to local and federal regulations.
• No data should be missing unless allowed by a skip pattern. • If data for a numerical field is unknown or missing, please leave that field blank.
Do not enter 0 (zero). • Circles or radio buttons O indicate that you should choose only one answer. • Boxes indica te tha t you s hould ‘che ck a ll tha t • Use the following format for all date fields: DD-MMM-YYYY (e.g., 31-JAN-2010) • Complete dates should be entered, whenever possible, for all date fields. If the
complete date isn’t known, partial dates are allowed for select data points. • In WebDCUTM, use the following format for all time fields: 24 hour clock hh:mm
Please note: 24:00 is not an allowable response. 24 hour clock time goes from 00:00 to 23:59. Midnight should be entered as 00:00.

SHINE Manual of Procedures 82 Version 6– December 19, 2016
Time on Clock 24 Hour Clock Time 12:00 AM 00:00 01:00 AM 01:00 02:00 AM 02:00 03:00 AM 03:00 04:00 AM 04:00 05:00 AM 05:00 06:00 AM 06:00 07:00 AM 07:00 08:00 AM 08:00 09:00 AM 09:00 10:00 AM 10:00 11:00 AM 11:00 12:00 PM 12:00 01:00 PM 13:00 02:00 PM 14:00 03:00 PM 15:00 04:00 PM 16:00 05:00 PM 17:00 06:00 PM 18:00 07:00 PM 19:00 08:00 PM 20:00 09:00 PM 21:00 10:00 PM 22:00 11:00 PM 23:00
• Name of person who collected the CRF data must be entered on the bottom of the paper worksheet, when the paper worksheet is used as a source document. This field will not be data entered but is required for monitoring purposes and should be dated and signed.
• Data Entry Timelines: • Screen Failure Log - The Clinical Site staff should update the Screen
Failure Log forms in WebDCUTM by the 10th of the following month. • Baseline through End of Study CRFs - Within 5 days of collection. • SAEs – Within 24 hours from the time the study team received
knowledge of the event. • Please note that site payments are dependent upon the subject’s data
being entered and submitted.
• Data Clarification Request (DCR) Timelines: All responses to DCRs must be submitted within 5 days of query generation with the exception of DCRs for SAEs, which must be submitted within 24 hours of query generation.

SHINE Manual of Procedures 83 Version 6– December 19, 2016
7.5 Guidelines for Completion of Specific CRFs (See Appendix 8) A comprehensive view of the SHINE CRF Completion Guidelines can be found in WebDCU by clicking on SHINE → project documents. A list of FAQs regarding the SHINE trial are also located in WebDCU project documents under the tab ‘Help Instructions’. An example of an FAQ off ‘Help Instructions’ Q: How do I remove a team member in SHINE? A: Follow the instructions below: 1. Click on [User Management] tab, and then [User Permission Request]. 2. On the list record page, click on the blue link in the # column to the left of the study team member name you are looking for. 3. Click on “Edit Record” located on the top right corner of the page. Remove all user group permissions by selecting the blank option in each drop down box on question 7. Once completed, click “Save Record.” Please be sure to add an end date for this user on the eDOA Log under section 5: Active Team Member. 4. The data manager will then review and approve this request.
8. Adverse Events 8.1 Definitions Adverse events will be defined and severity graded according to the Common Terminology Criteria for Adverse Events (CTCAE) version 4.0, found at http://evs.nci.nih.gov/ftp1/CTCAE/About.html.
8.1.1 Adverse Event An adverse event (AE) is any unfavorable and unintended sign (including an abnormal laboratory finding), symptom, or disease temporally associated with the use of a medical treatment or procedure that may or may not be considered related to the medical treatment or procedure. An AE is a term that is a unique representation of a specific event used for medical documentation and scientific analyses. Non-serious adverse events will be reported from the time of randomization until the end of the study treatment period. The end of the study treatment period is defined as the infusion stop time, which may be prior to 72 hours from randomization. The only time that a non-serious AE is to be reported after the end of the study treatment period is a hypoglycemic event that occurs within 8 hours of stopping the study infusion and the study investigator determines to be possibly, probably or definitely related to the study treatment. See Section 8.2.2 for guidelines for assessing relatedness of hypoglycemic events that occur after the end of the study treatment period.
8.1.2 Serious Adverse Event A serious adverse event (SAE) is any untoward medical occurrence that results in death, is life-threatening, requires or prolongs hospitalization, causes persistent or significant disability/incapacity, results in congenital anomalies/birth defects, or in the opinion of the investigators represents other significant hazards or potentially serious harm to research subjects or others.

SHINE Manual of Procedures 84 Version 6– December 19, 2016
Serious adverse events (SAE) must be reported and submitted within 24 hours of discovery. SAEs will be captured from the time of randomization until the end of the study. End of study may be past 90 days from randomization if the final study visit is conducted late however SAEs will not be captured past 120 days from randomization or end of study, whichever occurs first.
8.2 Anticipated Adverse Events The following adverse events are anticipated and should be reported; however, seriousness should be determined by the principal investigator or designee:
• Mild Hypoglycemia (glucose 56-69) • Moderate Hypoglycemia (glucose 40-54)
8.2.1 Clinically Important Adverse Events The following adverse events should be treated as serious. • Severe Hypoglycemia (glucose < 40 mg/dL). • Neurological worsening lasting greater than or equal to 24 hours and
associated with glucose concentration of < 55 mg/dL.
8.2.1.1 Severe Hypoglycemia (glucose < 40 mg/dL) Severe hypoglycemia is a serious adverse event. For treatment of hypoglycemia, refer to Section 10.
8.2.1.2 Neurological Worsening Associated with Hypoglycemia
For the SHINE study, neurological worsening is defined as a decline in neurological function associated with an increase in NIHSS score of ≥ 4 (from baseline or most recent) lasting for ≥ 24 hours.
For the SHINE study, transient neurological decline will be defined as an increase in the NIHSS score of > 4 (from baseline or most recent) that lasts less than 24 hours.
Neurological worsening or transient neurological decline in the presence of the above symptoms during a period of glucose < 55 mg/dL will be presumed to represent symptomatic hypoglycemia. This must be categorized as an SAE and neurological worsening if symptoms persist for > 24 hours. Transient neurological decline in the setting of a glucose < 55 mg/dL may be categorized as an SAE per the judgment of the site PI. Any patient with neurological worsening in the presence of hypoglycemia (< 70 mg/dL) will be assessed for relatedness of the hypoglycemia to neurological worsening.
SAEs must be reported in WebDCU™ within 24 hours of knowledge that the subject meets criteria for neurological worsening. In the event that the transient neurological decline is determined to be an SAE, the study team has 24 hours from that determination to report the event. If the event persists for greater than or equal to 24 hours, the CRF can be updated from transient neurological decline to neurological worsening.
For treatment of hypoglycemia, refer to Section 10.
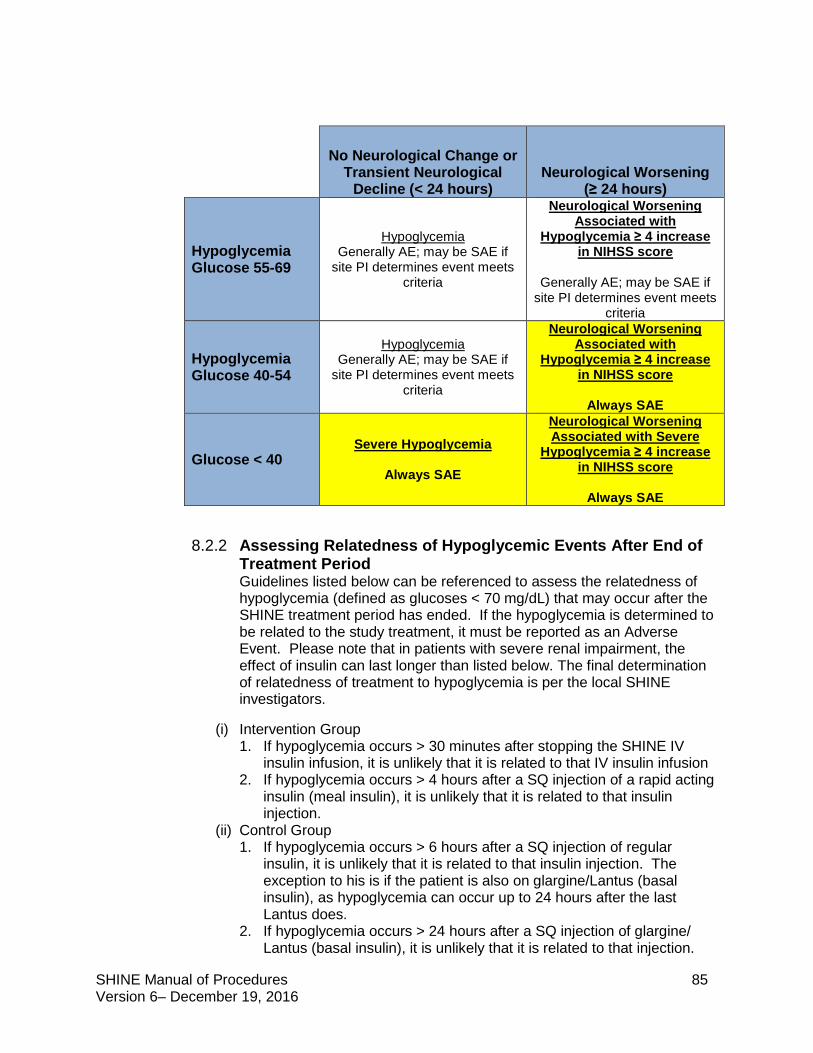
SHINE Manual of Procedures 85 Version 6– December 19, 2016
No Neurological Change or
Transient Neurological Decline (< 24 hours)
Neurological Worsening (≥ 24 hours)
Hypoglycemia Glucose 55-69
Hypoglycemia Generally AE; may be SAE if
site PI determines event meets criteria
Neurological Worsening Associated with
Hypoglycemia ≥ 4 increase in NIHSS score
Generally AE; may be SAE if
site PI determines event meets criteria
Hypoglycemia Glucose 40-54
Hypoglycemia Generally AE; may be SAE if
site PI determines event meets criteria
Neurological Worsening Associated with
Hypoglycemia ≥ 4 increase in NIHSS score
Always SAE
Glucose < 40 Severe Hypoglycemia
Always SAE
Neurological Worsening Associated with Severe
Hypoglycemia ≥ 4 increase in NIHSS score
Always SAE
8.2.2 Assessing Relatedness of Hypoglycemic Events After End of Treatment Period Guidelines listed below can be referenced to assess the relatedness of hypoglycemia (defined as glucoses < 70 mg/dL) that may occur after the SHINE treatment period has ended. If the hypoglycemia is determined to be related to the study treatment, it must be reported as an Adverse Event. Please note that in patients with severe renal impairment, the effect of insulin can last longer than listed below. The final determination of relatedness of treatment to hypoglycemia is per the local SHINE investigators.
(i) Intervention Group 1. If hypoglycemia occurs > 30 minutes after stopping the SHINE IV
insulin infusion, it is unlikely that it is related to that IV insulin infusion 2. If hypoglycemia occurs > 4 hours after a SQ injection of a rapid acting
insulin (meal insulin), it is unlikely that it is related to that insulin injection.
(ii) Control Group 1. If hypoglycemia occurs > 6 hours after a SQ injection of regular
insulin, it is unlikely that it is related to that insulin injection. The exception to his is if the patient is also on glargine/Lantus (basal insulin), as hypoglycemia can occur up to 24 hours after the last Lantus does.
2. If hypoglycemia occurs > 24 hours after a SQ injection of glargine/ Lantus (basal insulin), it is unlikely that it is related to that injection.

SHINE Manual of Procedures 86 Version 6– December 19, 2016
8.3 Adverse Event Exceptions Death due to the natural history of ischemic stroke will be recorded as a non-related serious adverse event. Additionally, all serious but known complications of stroke (i.e. malignant brain edema) will be recorded as non-related serious adverse events.
8.4 Algorithm to Determine Relatedness (from http://www.nihtraining.com/ohsrsite/irb/Attachments/5-10_Serious_ Adverse_Event_Rep.htm) The best estimate of the PI at the time of reporting of the causal relationship between an experimental intervention and an adverse event; the degree of certainty about causality is graded as follows: Unrelated: Adverse event is clearly due to extraneous causes (e.g., underlying disease, environment) Unlikely (must have 2): Adverse event:
• does not have temporal relationship to intervention, • could readily have been produced by the subject’s clinical state, • could have been due to environmental or other interventions, • does not follow known pattern of response to intervention, • does not reappear or worsen with reintroduction of intervention
Possible (must have 2): Adverse event:
• has a reasonable temporal relationship to intervention, • could not readily have been produced by the subject’s clinical state, • could not readily have been due to environmental or other interventions, • follows a known pattern of response to intervention
Probable (must have 3): Adverse event:
• has a reasonable temporal relationship to intervention, • could not readily have been produced by the subject’s clinical state or
have been due to environmental or other interventions, • follows a known pattern of response to intervention, • disappears or decreases with reduction in dose or cessation of
intervention Definite (must have all 4): Adverse event:
• has a reasonable temporal relationship to intervention, • could not readily have been produced by the subject’s clinical state or
have been due to environmental or other interventions, • follows a known pattern of response to intervention,

SHINE Manual of Procedures 87 Version 6– December 19, 2016
• disappears or decreases with reduction in dose or cessation of intervention and recurs with re-exposure
8.5 Adverse Event Collection and Reporting
8.5.1 Study Site AE Screening Adverse events will be captured by the treating team and the study team based on history, physical exam, medical records and laboratory findings.
8.5.2 Adverse Event Reporting In order to assure prompt and complete reporting of adverse events or complications of intervention, the following general guidelines are to be observed.
AEs will be submitted online through WebDCU™. All adverse events, whether serious or not, will be collected from the time of randomization through completion of study treatment. Serious adverse events will be assessed, recorded and followed through end of study. End of study may be past 90 days from randomization if the final study visit is conducted late however SAEs will not be captured past 120 days from randomization or end of study, whichever occurs first. The site PI or Study Coordinator is responsible for entering AEs and SAEs into the database and updating the information (e.g., date of resolution, action taken) as needed. For serious adverse events (SAEs) that occur between the End of Study Treatment and the Six Week visit, complete Form 06 - Adverse Event under the last treatment day visit in WebDCU. If the study team learns of an SAE that occurred between the Six Week and 90 Day visit, the AE form that posts under the 6 week visit in WebDCU can be used to document the event. Non-serious AEs must be reported and submitted within 5 days from time of discovery by site personnel, and SAEs must be reported and submitted within 24 hours of the time that the study team receives knowledge of the event.
Serious adverse events are defined as meeting one of the following criteria:
Death: All deaths occurring during the course of the study must be reported.
Life-threatening: Report if suspected that the patient was at substantial risk of dying at the time of the adverse event, or use or continued use of the study treatment might have resulted in the death of the patient.
Hospitalization (initial or prolonged): Report if admission to the hospital or prolongation of hospitalization was a result of the adverse event. Emergency room visits that do not result in admission to the hospital should be evaluated for one of the other serious outcomes (e.g., life-threatening; required intervention to prevent permanent impairment or damage; other serious medically important event). Hospitalization that is due to a planned or elective admission should be evaluated for one of the other serious outcomes (e.g., life-threatening; required intervention to prevent permanent impairment or damage; other serious medically important event).
Disability or Permanent Damage: Report if the adverse event resulted in a substantial disruption of a person's ability to conduct normal life functions, i.e., the adverse event resulted in a significant, persistent or permanent change,

SHINE Manual of Procedures 88 Version 6– December 19, 2016
impairment, damage or disruption in the patient's body function/structure, physical activities and/or quality of life.
Congenital Anomaly/Birth Defect: Report if you suspect that exposure to study treatment prior to conception or during pregnancy may have resulted in an adverse outcome in the child.
Required Intervention to Prevent Permanent Impairment or Damage: Report if you believe that medical or surgical intervention was necessary to preclude permanent impairment of a body function, or prevent permanent damage to a body structure, either situation suspected to be due to the use of study treatment.
Other Serious (Important Medical Events): Report when the event does not fit the other outcomes, but the event may jeopardize the patient and may require medical or surgical intervention (treatment) to prevent one of the other outcomes. Examples include allergic bronchospasm (a serious problem with breathing) requiring treatment in an emergency room, serious blood dyscrasias (blood disorders) or seizures/convulsions that do not result in hospitalization.
The submission of an SAE will generate an automatic email notification to the SHINE Site Manager (SM). The SM will review the SAE for completeness of information in WebDCUTM. If the information is insufficient, the SM will send a query to the site on WebDCU™ indicating what additional information is required. The process triggers an automatic email notification that will be sent to the site PI and coordinator requesting the additional information. Once the SM feels that SAE has been properly entered and is ready for clinical review, they will indicate this in WebDCU™ which will prompt review by the Internal Quality and Safety Reviewer (IQSR). If the information is insufficient, the IQSR will either issue a DCR to request additional information OR will send this SAE for review back to the SM after indication on the CRF that the “information is clinically accurate = No” and contacts the SM with additional information required for review; based on this, SM issues DCR to site. SAE review process restarts requiring SM review when site responds to query and submits CRF. When the IQSR completes his or her clinical review, they will indicate this, prompting an automatic email notification to be forwarded to the Independent Medical Safety Monitor. The Independent Medical Safety Monitor will review SAEs to determine relatedness and whether expected or unexpected. This review must be completed and entered within 48 hours of notification.
8.5.3 SAE Narrative and Templates In order to assure prompt and complete review of Serious Adverse Events or complications of intervention, kindly use the SHINE narrative template to provide us with as much information regarding the patient, SHINE Enrollment and treatment following general event reporting guidelines.
It is important for narratives to describe the event, chronologically and contextually. To help sites with the essential information that is needed to review an SAE, templates have been drafted and are located under “SAE Templates” in the SHINE Website under Toolbox.
https://nett.umich.edu/clinical-trials/shine/toolbox

SHINE Manual of Procedures 89 Version 6– December 19, 2016
9. Protocol Adherence
9.1 Protocol Deviations Protocol deviations must be assessed throughout each subject’s participation in the study.
Protocol deviations must be reported in WebDCU™ when applicable. Protocol deviations must also be reported to the IRB per local site requirements. Once a deviation has been acknowledged by the IRB, it must be uploaded to WebDCUTM.
Serious or repeated protocol deviations will require the development of a Corrective Action/Preventative Action (CAPA) plan (see Section 9.2).
9.2 Corrective Action/Preventative Action Plans Development of a CAPA plan may be initiated by the site study team, local IRB, or the NETT-CCC. Potential triggers include protocol deviations, data quality problems, or systematic problems identified by study teams or monitors.
CAPA plans must be reviewed by the site study team, NETT-CCC site manager and monitor. The NETT-CCC site manager, NETT-CCC project monitors, or the SHINE project director may approve CAPA plans.
Once a CAPA plan has been approved and enacted, data must be collected as agreed upon in the plan until the study team demonstrates that the issue has been resolved. The criteria for determining this point will vary depending upon the frequency and severity of the issue. After it has been demonstrated that the deficiency has been corrected, the CAPA plan will be closed by the NETT-CCC site manager, NETT-CCC site monitors, or the SHINE project director.
9.3 Protocol Adherence Monitoring Procedures The overall goal of site monitoring and aggregate protocol adherence monitoring in the SHINE trial is to identify patterns suggestive of overall study protocol issues or individual site issues that may require support and/or retraining on study treatment procedures.
Details of the SHINE Protocol Adherence Monitoring Procedures can be found in Appendix 11.
Q: Our patient had an episode of hypoglycemia that occurred immediately after the end of the treatment period. Is this an event that we should report?
A: Non-serious adverse events will be reported from the time of randomization until the end of the study treatment period. The end of the study treatment period is defined as the infusion stop time, which may be prior to 72 hours from randomization. The only time that a non-serious AE is to be reported after the end of the study treatment period is a hypoglycemic event that occurs within 8 hours of stopping the study infusion and the study investigator determines to be possibly, probably or definitely related to the study treatment.

SHINE Manual of Procedures 90 Version 6– December 19, 2016
10. Management of Hypoglycemia 10.1 Hypoglycemia Protocols
10.1.1 Initiating hypoglycemia protocol (Glucose concentration < 80 mg/dL)
All insulin therapy will be stopped in the event that glucose drops below 80 mg/dL and the following protocol will be initiated:
(1) Stop all IV infusions and hold all subcutaneous injections. (2) D50 will be stored to allow immediate availability at the bedside. (3) Glucose administration
(a) Control Group – A dose of IV D50 25 ml (1/2 amp) will be given (slow IV push over 1-2 minutes) every 15 minutes until blood glucose is ≥ 80 mg/dL. Repeat POC glucose checks and treatment every 15 minutes if needed until glucose is ≥ 80 mg/dL.
(b) Intervention Group – An individualized dose of IV D50 will be given (slow IV push over 1-2 minutes). The specific dose will be determined by GlucoStabilizer® based on the glucose concentration. Recheck blood glucose every 15 minutes as directed by GlucoStabilizer®. Repeat treatment every 15 minutes as directed by GlucoStabilizer® until glucose is ≥ 80 mg/dL.
(c) Glucose level entry into the SHINE Trial Portal needs to occur q15 minutes +/- 5 minute window to remain protocol compliant.
(4) Once glucose is ≥ 80 mg/dL: (a) Restart IV insulin or saline per protocol (b) Restart SQ insulin or saline per protocol
10.1.2 Extra steps for hypoglycemia < 70 mg/dL Continue to use the hypoglycemia protocol above (e.g. hold insulin, give D50, repeat glucose checks every 15 minutes).
• Draw a STAT laboratory serum glucose measurement but do not delay treatment with D50 by waiting for this blood draw result. Only the results from POC checks should be used for study protocol dosing. The result of the serum glucose measurement will be captured in the Hypoglycemic Event CRF (Form 17).
• Screen the patient for hypoglycemia symptoms using Hypoglycemia Symptomatic Questionnaire (downloadable from NETT SHINE website).
o The worksheet and instructions will be available on paper and on the desktop of the laptops.
o The Hypoglycemia Symptomatic Questionnaire must be repeated every 15 minutes when glucose is < 70 mg/dL.
o Once the glucose is ≥ 70 mg/dL, one final assessment with the Hypoglycemia Symptomatic Questionnaire is required.
o Once the glucose is ≥ 80 mg/dL, the timing of glucose checks and insulin infusion rate will again be determined by the study computer.
• Screen the patient for neurological worsening. o An assessment of neurological change is required per site standard
care (“neuro check”) every 15 minutes when glucose is < 70mg/dL.

SHINE Manual of Procedures 91 Version 6– December 19, 2016
o The SHINE study definition of neurological worsening is any clinical change that is associated with a ≥ 4 point increase from most recent NIHSS score that lasts ≥ 24 hours.
o Any patient with neurological worsening will be assessed for hypoglycemia and for relatedness of the hypoglycemia to neurological worsening if present.
o If the patient has not returned to neurological baseline within 24 hours, a NIHSS assessment is required at 24 hours(+/- 4 hours) from onset of hypoglycemic event (< 70 mg/dL). If the patient has returned to neurological baseline at any point in less than 24 hours, the NIHSS is not required at 24 hours.
o If neurological worsening persists for ≥ 24 hours and is associated with a glucose concentration < 55 mg/dL, an SAE form is required.
• If the glucose falls below 40 mg/dL at any time, an SAE form is required. The SAE must be entered into WebDCU within 24 h of discovery. In addition, this SAE must be filed with your local IRB per their guidelines.
10.1.3 Severe Hypoglycemia (Glucose concentration < 40 mg/dL) All glucose measurements < 40 mg/dL will be captured as severe hypoglycemia. Each
event will be characterized as symptomatic or asymptomatic based on symptoms and signs. All spontaneous symptomatic complaints will trigger a stat glucose measure per standard care. Nonverbal patients will be assessed for only the physiologic signs of symptomatic hypoglycemia.
10.1.4 Determining Symptomatic Hypoglycemia
The Hypoglycemia Symptomatic Questionnaire will be used to assess symptomatic hypoglycemia in patients with glucose levels below 70 mg/dL. Symptomatic hypoglycemia, which consistently differentiates hypoglycemia from euglycemia, will be assessed using questions of: shaky, heart pounding, nervous, sweaty, hungry, tingly, warm, weak, confused and drowsy. Of these symptoms, shakiness, heart pounding and nervousness may be reduced in patients on medication that result in adrenergic blockade though the other symptoms persist despite such medications.
In addition, spontaneous complaints of these symptoms will trigger an immediate glucose level assessment. Standard care orders should specify PRN glucose level check for patient complaints of these symptoms or others suggestive of hypoglycemia. Aphasic patients, unresponsive patients or patients who cannot otherwise participate in the hypoglycemic questionnaire will be evaluated for signs of a physiologic response to hypoglycemia only and not symptoms on the questionnaire (See Appendix 6).
10.1.5 Additional steps for ≥ 3 episodes of hypoglycemia (Glucose concentration < 70 mg/dL) in 24 hour period
• Intervention Group o For any study patient in the intervention group who experiences 3 or
more episodes of hypoglycemia (glucose concentration of < 70 mg/dL)

SHINE Manual of Procedures 92 Version 6– December 19, 2016
in a 24 hour period, the SHINE Safety Hotline must be notified (800-915-7320 ext 2).
• Control Group o For any study patient in the control group who experiences 3 or more
episodes of hypoglycemia (glucose concentration of < 70 mg/dL) in a 24 hour period, the SHINE Safety Hotline must be notified (800-915-7320 ext 2).
o Also, at the 24 and 48 hour intervals, regardless of the previous two glucose measurements, do NOT advance to a higher level on the subcutaneous sliding scale.
10.1.6 Additional steps for severe hyperglycemia (Glucose concentration ≥ 500 mg/dL)
If a POC meter shows that the blood glucose is too high to provide an exact measurement (generally greater than 500-600 mg/dL depending on the meter) or the glucose reads ≥ 500 mg/dL
1. Draw a STAT laboratory serum glucose measurement 2. Call the SHINE Independent Safety Monitor (800-915-7320 ext 2).
11. Safety Monitoring 11.1 Safety Reporting The Independent Safety Monitor and DSMB will receive periodic safety reports of all adverse events and serious adverse events. The DSMB will have the ability to review all serious AEs in real time. Statistical monitoring for safety will be limited to severe hypoglycemia (< 40 mg/dL) during the treatment period, death rate within 90 days post randomization and neurological worsening lasting > 24 hours and associated with blood glucose < 55 mg/dL. All adverse events and serious adverse events will be summarized by MedDRA code in terms of frequency of the event, number of subjects having the event, severity, and relatedness to the study treatment.
11.2 Safety Review Process The project Site Manager (SM), Internal Quality and Safety Reviewer (IQSR), Independent Safety Monitor (ISM) and/or the designated backup personnel participate in the Safety Review process.
When an SAE occurs, the clinical research staff at the respective site enters the SAE into WebDCU™ via the AE case report form (CRF). The study site team will determine and enter: the seriousness, severity, the relatedness to the study protocol, action taken as a result of the SAE, the outcome and date of resolution (if applicable or known) and a narrative of the event. The submission of the CRF triggers an automatic e-mail that will be sent via the WebDCU™ system to the SM. This e-mail will alert the SM that an SAE has occurred, and the date and time will be included in this e-mail. The SM will access the event information within 48 hours via the password protected WebDCU™, reviewing the SAE for completeness of information. Once the SM determines that SAE has been properly entered and is ready for clinical review, this will be indicated in WebDCU™ which will prompt an automated email to be sent to the IQSR as a notification of the pending review. The IQSR will review the SAE within 48 hours for clinical accuracy and completeness in WebDCU™. When the IQSR completes the review process, an automatic e-mail notification will be triggered to the ISM. The ISM will access the event information, subsequently making a designation of causality, severity and

SHINE Manual of Procedures 93 Version 6– December 19, 2016
expectedness. This review must be completed and entered within 48 hours of notified. After 48 hours, WebDCU™ will close the review process. Should the ISM, IQSR or SM require changes and/or additional information about the SAE, a data clarification request will be entered alerting the site of the query. If additional information is required by a site in order to make a designation of causality and severity, the 2 day data entry deadline will be extended in order to gain the necessary information.
All AEs, regardless of seriousness, will be coded centrally via MedDRA. All AEs, regardless of seriousness, will be aggregated and provided via quarterly safety reports via the unblinded statistician to the ISM for review. These reports will present all SAEs and AEs by treatment arm as well as by treatment relation and severity. Relative risks with 95% confidence intervals will be reported for treatment related SAEs.
If any safety concerns arise, the ISM will report these concerns to the DSMB chair via the DSMB liaison.
11.3 Blinding This is a single blinded trial with double blinded outcome assessments. Patients will remain blinded throughout the trial as they all will receive an IV infusion and SQ injections. The treating team (physicians/nurses) will be unblinded to treatment to assure appropriate patient safety. The outcome assessments will be performed by blinded investigators who did not participate in the acute care. The subjects will not be told of their actual treatment until the trial completion. The protocol adherence team monitors unblinding questionnaire responses in aggregate and by site. The DSMB is partially unblinded for the closed reports. However, if it so wishes, it may be completely unblinded at any time during the trial. If the DSMB wishes to be unblinded on a particular subject only, the NINDS Liaison to the DSMB should email the request to the unblinded SDMC biostatistician. At the time of the scheduled follow-up assessments, the blinded assessor and subject each complete the blinding assessment CRF.
11.4 Emergency Unblinding No emergency unblinding is anticipated. Any emergency unblinding requests must be directed to the SHINE Study Hotline (800-915-7320).
12. Pharmacy Manual
12.1 Study Design SHINE is a Phase III, randomized, single-blinded, controlled trial of approximately 1400 patients at approximately 60 US sites. The study is composed of two treatment groups:
Intervention Group: continuous IV insulin + subcutaneous meal insulin or saline injections (Target blood glucose: 80-130 mg/dL)
Control Group: IV saline + subcutaneous sliding scale insulin (Target blood glucose: < 180 mg/dL)
Study drugs are NOT supplied by the sponsor. Study-specific accountability is not required. Sites must establish and adhere to pharmacy procedures for the preparation and storage of study medications required for the SHINE treatment protocols.

SHINE Manual of Procedures 94 Version 6– December 19, 2016
12.2 Study Drug Descriptions The following drugs will be used for the intervention and control group protocols.
i) Intravenous Insulin (1) Human regular insulin (Humulin R or Novolin R) will be the only insulin used
intravenously. The concentration of the insulin infusion will be 1:1 (e.g. 100 Units of human regular insulin in 100 mL of normal saline (0.9% NaCl)) unless a site-specific exception is approved.
ii) Subcutaneous Insulin (1) Intervention Group
(a) Only rapid acting analog insulin will be used for the subcutaneous insulin (meal insulin) in the intervention group. These insulins are Lispro (Humalog), Aspart (Novolog) or Glulisine (Apidra), and the choice of the insulin will be determined by the local hospital formulary.
(b) Human regular insulin (Humulin R and Novolin R) is NOT to be used for subcutaneous injections in the intervention group.
(2) Control Group (a) Only Human Regular Insulin (Humulin R or Novolin R) is to be used in the
control group. Choice of this insulin type is determined by local hospital formulary.
(b) Basal Insulin [Glargine (Lantus)] at a dose of 40% of the total insulin requirement in the previous 24 hours for patients who progress to Level 3.
(c) Rapid Acting Analog Insulins (Humalog/Novolog/Apidra) are NOT to be used in the control group.
iii) Saline (1) Intervention Group
(a) Saline will be stored according to local site procedures and drawn up from multidose vials/single dose ampoules ONLY for study patients in the intervention group who are NOT eating or are receiving continuous tube feeds. Subcutaneous saline injections are to be administered for these study patients around 09:00 and 21:00 and associated with a glucose check.
(2) Control Group (a) The saline infusion (normal saline (0.9% NaCl)) will be prepared according to
local site procedures.
iv) Dextrose 50% in Water (D50) (for both the Intervention and Control Groups) Each clinical unit where a randomized subject is being treated should – per hospital policy- have ready access to D50. If such hospital policy does not exist, the site study team will ensure that D50 is stored to allow immediate availability at the bedside in the units in which randomized subjects are to be admitted. D50 will be stored at an appropriate secure and readily accessible location as per local site procedures.
12.3 Method for Assigning Patients to the Treatment Group After the site study team has identified a patient as eligible for the study and informed consent has been obtained, the patient will be randomized using WebDCUTM. Site pharmacists are not required to access the WebDCUTM. A copy of the randomization assignment with the SHINE study ID and treatment allocation will be provided to the pharmacist according to site procedures (deliver/fax/email).

SHINE Manual of Procedures 95 Version 6– December 19, 2016
12.4 Drug Labeling The IV infusion bag for insulin (intervention group) and saline (control group) should be labeled SHINE Study Drug.
A sticker with a blue background and the text SHINE Study Drug will be provided for the IV insulin bag. A sticker with an orange background and the text SHINE Study Drug will be provided for the IV saline bag. Stickers will be supplied at the time of site activation, and the site study team will provide to the pharmacy. The label MUST only be applied by the pharmacist who is preparing the IV infusion. Contact site study coordinator to request additional stickers.
See Appendix 10 for the SHINE Pharmacy Guidance and Instructions for Developing Study Orders.
13. Source Documentation and Monitoring 13.1 Source Documentation
Source documents are any documents on which study data are recorded for the first time. Source documents include, but are not limited to, medical records (inpatient and outpatient), worksheets developed for study use, standardized test forms and laboratory reports. The source documents are necessary to validate the information that has been entered into WebDCU™. The source documents also will need to be readily available during site monitoring visits. These documents should be in good order and should be placed in a study-titled binder or clearly marked file.
13.2 Site Monitoring Monitoring visits will be conducted to ensure that the conduct of the trial is in compliance with the current version of the IRB approved protocol, the GCP/ICH Guidelines and all applicable regulatory requirements. It is paramount that the trial is performed in accordance with all regulatory requirements and protocol criteria and that the rights and well-being of human subjects are protected. A project monitor from the NETT-CCC will perform the on-site monitoring for all SHINE study sites. Project monitors are selected and managed by the NETT-CCC. In accordance with the GCP Guidelines, monitoring of clinical sites will take place on a periodic basis – at least once each year for sites that have enrolled within the year, and a final close-out visit upon completion of the trial. More frequent monitoring visits will be determined by the site manager and the project monitor based upon factors such as rate of enrollment, findings of previous monitoring reports, etc. The Site Monitor will coordinate/perform the on-site monitoring for the SHINE study sites. At each monitoring visit, the investigator(s), study coordinator(s),

SHINE Manual of Procedures 96 Version 6– December 19, 2016
and other study staff members should be available to meet with the Site Monitor. The Site Monitor will provide a minimum of two weeks’ notice before the on-site visit. During a monitoring visit, Site Monitors will:
• Conduct meetings with the study investigators and/or study staff to ensure that the obligations of the investigator are being fulfilled.
• Discuss enrollment strategies, reason(s) for early subject discontinuation, and identify any challenges at the site.
• Review Screen Failure Logs with investigator and/or study staff. • Examine the Regulatory Binder for identified regulatory materials not
uploaded to the WebDCU™ database. • Verify CRF data against source documents. Review relevant
documentation (CRFs, source documents, shadow documents etc.) to confirm adherence to the study protocol.
• Ensure current version of IRB approved informed consent documents are available and signed appropriately for each enrolled subject.
• Verify that enrolled subjects were eligible at time of enrollment, and that study procedures or interventions were initiated based on the timeline and enrollment procedures described in the study protocol.
• Review SAEs/AEs and verify to source/shadow document. • Review source/shadow documents to identify S/AEs that have not been
previously reported. • Review evidence that SAEs have been appropriately reported to the IRB. • Verify adequacy of facility (records storage, visit pharmacy and review
pharmacy log). • Ensure the adequacy of clinical supplies. • Review findings with the site personnel. • Resolve outstanding queries.
The monitors will verify to source documents 100% of the data on the first two subjects enrolled. A target of at least 50% of the clinical data submitted to the database is verified to source documents at the clinical sites for each subsequent subject. Examples of the data monitored for all subjects include all serious adverse events, subject deaths, randomization information, NIHSS and mRS assessments and study drug administration data. In addition, all cases are monitored for the presence of a signed informed consent and adequate documentation of the informed consent process. A Monitoring Log should be signed and dated by the monitor and a member of the clinical site staff at each visit and maintained in the MOP and/or Regulatory Binder. See Appendix 7 for a sample Monitoring Log. Following the monitoring visit, the findings will be reported to the site. If major or recurrent violations are identified during a monitoring visit, a Corrective Action/Preventative Action (CAPA) plan will be required. The site will be notified in the Monitoring Report. For more information on developing CAPA plans, please see Section 9.2.
13.3 Queries/Data Clarification Requests (DCRs)

SHINE Manual of Procedures 97 Version 6– December 19, 2016
Data clarification requests (DCRs) may be initiated either by the SDMC, the Site Monitor, or the Site Manager. Notification of DCRs will be found on the WebDCU™ SHINE home page. DCRs must be responded to within 5 days.
13.4 Regulatory Documentation and E-Binder All required regulatory documents, as found in the SHINE Regulatory Parameters Document (found on WebDCU™ under Project Management/Project Documents), must be uploaded into WebDCU™.
When a document is nearing expiration, the primary study coordinator will receive 30- and 7-day notices via e-mail. A weekly email will be sent if a site has any missing/rejected/expired regulatory documents outstanding.
13.5 Change in Study Personnel 13.5.1 Change in Site PI Any changes in site PI must be approved by SHINE study leadership and the NIH.
13.5.2 Change in Other Study Personnel Changes to site personnel during the study need to be updated in WebDCU™. The SHINE site manager or project director should be made aware of changes in the primary study coordinator to facilitate training. The following information is required to be uploaded into WebDCU™ for all additions/changes/deletions:
• All required regulatory documents • Updated Delegation of Authority Log • IRB approval for change in study team
13.5.3 Notifying SDMC about Changes in Study Personnel The SDMC must be notified when study personnel are no longer affiliated with the trial. The Study Team Member table and User Permission Request tables located under the User Management tab must be updated, and the SDMC team will adjust WebDCU™ permissions.
14. Reports
14.1 Monthly Report Reports can be generated from WebDCU™ to monitor trial progress and patient recruitment at each site. These reports will count the patients recruited overall and at each clinical site. They will also provide site-specific information on the number of subjects with missing or incomplete data in the WebDCU™ system (based on subject registration data), number of errors, follow-up visits completed and required procedures completed.
14.2 Enrollment Reports Enrollment reports will be sent to NINDS annually summarizing data on subject accrual (overall and by site), date of first randomization and participant retention.
14.3 Newsletter A newsletter may be sent to all site PI’s and Study Coordinators. This newsletter will contain information on trial recruitment, updates and/or changes in the trial,

SHINE Manual of Procedures 98 Version 6– December 19, 2016
as well as any additional information, suggestions, reminders, or advice that may be appropriate or necessary.
14.4 DSMB Reports A NIH-NINDS appointed independent DSMB will monitor all aspects of safety in this trial. Interim reports containing summary tables describing patient enrollment, baseline characteristics of patients, safety information, losses to follow up, outcome information, data quality information and any other information requested related to data integrity, continued relevance of the research questions, or patient safety will be provided every 6 months or as requested to the DSMB. The DSMB will follow the guidelines as described in the National Institutes of Health (NIH) issued policy on data and safety monitoring. The DSMB may pause or halt the trial at any time based on data from this or other trials. The DSMB will meet twice yearly unless otherwise determined. The results of all DSMB meetings will be summarized to the study PI and forwarded to site study teams for IRB submission at the enrolling institutions.
14.5 Recruitment and Retention Reports The SHINE trial procedures include standards for monitoring sites for recruitment/enrollment performance. At the request of the trial’s Data Safety Monitoring Board, the SHINE recruitment team reviews enrollments and reports findings to the SHINE Executive Committee regularly. On a quarterly basis, all NETT hubs and SHINE Ancillary sites are provided with a site-specific recruitment report.
15. Site Recruitment, Training and Initiation
15.1 Site Start-Up and Initiation Prior to the enrollment of the first subject, site initiation will occur. Site initiation will take place by telephone (Readiness Call) after the site is deemed “Regulatory Ready”. Sites will not be allowed to enroll subjects until all regulatory documents are complete. All regulatory documents must remain current throughout the course of the trial. It is the responsibility of the hub and/or ancillary site to ensure regulatory compliance is maintained at the enrolling site/spoke. The NETT-CCC will routinely monitor the WebDCU™ SHINE database for regulatory compliance. Regulatory documents specific to the SHINE trial include:
• Investigator’s Agreement Form • Documentation of Full Study IRB Application Submittal • IRB approval of SHINE Protocol/Full Study IRB approval • IRB Approved Informed Consent Form • Institutional Federalwide Assurance (FWA) • Human Subjects Protection Policy • Electronic Delegation of Authority Log (eDOA) • CLIA • Current Medical/Professional license • Current CV • NIHSS Certification • mRS Certification

SHINE Manual of Procedures 99 Version 6– December 19, 2016
• Human Subjects Training Certification or Waiver • HIPAA Training Certification or Waiver • SHINE Protocol Training Certification • SHINE Data Training Certification • SHINE Pharmacy Plan • SHINE Recruitment Plan • SHINE Nurse Inservice Sign-in Sheets
A description of each requirement and what needs to be uploaded into WebDCU™ SHINE (Regulatory Parameters Document) can be found on the WebDCU™ SHINE Regulatory database under Toolbox/Project Documents. Once these documents are uploaded into the WebDCU™ SHINE Regulatory database and accepted, a Readiness Call will be scheduled. In preparation for the Readiness Call, each study team will be asked to complete a Readiness Checklist documenting regulatory readiness and study logistics; a checklist can be downloaded from the SHINE Website under Toolbox for sites to complete. The Readiness Checklist, Recruitment Plan and Pharmacy Plan will be reviewed during the readiness call.. Following the Readiness Call, a Readiness Report will be entered in WebDCU™ by the NETT-CCC. The NETT-CCC will activate the site to begin enrollment.
15.2 Training for Study Personnel All training is located on the SHINE website (www.shinetrial.com). Study team members may be required to complete the following certifications:
• Modified Rankin Scale (mRS): Required for all study team members who will be completing this assessment.
• NIH Stroke Scale (NIHSS): Required for all study team members who will be completing this assessment.
• Database Training: Required for all study team members who will be entering data in WebDCUTM.
• Randomization Database Training: Required for all study team members who will be randomizing subjects.
• Health Insurance Portability and Accountability Act (HIPAA): Required for all study team members. This may be taken at your own institution.
• Human Subjects Protection (HSP): Required for all study team members. This may be taken at your own institution.
Instructions to create an account to access and complete the SHINE trial trainings follow: The training modules are located on the SHINE Education and Training page on the SHINE website. In order to access this page you will need to create a UM Friend Account. If you've already created one then follow the instructions below. If you have not created one please go to http://www.itcs.umich.edu/itcsdocs/s4316/#how to create an account.

SHINE Manual of Procedures 100 Version 6– December 19, 2016
Instructions to access SHINE Training and Resources:
1. Go to http://nett.umich.edu
2. Click on NETT LOGIN at the top of the homepage, and enter your UM Friend Account.
3. Click on the SHINE tile on the homepage, then the Education and Training link.
Once you’ve accessed the SHINE Education and Training page, click on the Regulatory Parameters Document at the top of the page. This will be your guide to what is required for each role in the study.
Certificates of completion should be saved electronically. The study personnel at the site responsible for regulatory document management will upload these certifications to the WebDCU™ SHINE. It is the responsibility of the site PI to train or update the personnel site study team members with regard to the SHINE protocol and trial procedures.
Study team members that will randomize subject and/or enter CRF data will require access to the SHINE database. To receive access to the SHINE database, study personnel must view the SHINE Data Training which is found on the SHINE trial website under ‘SHINE Education and Training’. After viewing, the electronic Data Training Certificate must be completed and uploaded to the WebDCU™ SHINE database. Once uploaded, the CCC will approve the certification. Once approved, a Data Manager at SDMC will grant SHINE database permissions. When access has been granted, an email notification will be sent to each user, with a username and password.
15.3 Training for Site Clinical Personnel (Non-Study Team Members) Site study investigators, coordinators and lead clinical nurses will attend SHINE Investigator Meetings as appropriate. Each site study team will then use a train-the-trainer approach to train site clinical nurses. Training content will include SHINE protocol and study procedures with a focus on GlucoStabilizer® and study laptops. In response to staff nurse turnover, there will need to be a continuous education plan at each site. There will not be any sponsor-required individual certifications required for the nurses caring for subjects. An informal review on the use of the study laptops should also take place between nursing shifts by trained personnel on an as needed basis.
Protocol Changes: Any protocol changes in the study impacting the GlucoStabilizer® or control treatment screen will mandate re-training. Such training will be planned as face-to-face training, but may also include on-line or other options as deemed appropriate by the PI based on the nature of the protocol change.
15.4 Training Investigator Meeting Clinical site Principal Investigators and Study Coordinators will receive training in the protocol, trial procedures (e.g., assessments), CRF completion, WebDCU™ procedures, etc. at the SHINE Investigator Meeting, and at subsequent SHINE Investigator Meetings. At the meetings, SHINE investigators and personnel from the DCU will present training material on the current protocol, the Manual of Procedures, and use of the WebDCU™ data entry system (generally with a Power Point or comparable presentation or other forms and instructions, as necessary). A goal of these training sessions is to ensure that all clinical site personnel who are able to attend receive the same information; and with

SHINE Manual of Procedures 101 Version 6– December 19, 2016
regard to data collection, to try to standardize the methods of data collection to help ensure comparability of data across sites.
Specific elements of the Investigator Meeting include the following:
Protocol-Specific Training: Prior to the initiation of SHINE at any participating site, Principal Investigators, co-Investigators, Study Coordinators, and other study personnel will participate and complete the Protocol-Specific Training. The training includes protocol details, GlucoStabilizer® procedures, WebDCU™ procedures, and other forms and instructions as necessary. To promote adherence with the study protocol, aggressive training, reminders, and encouragement will be provided to each participating site. The CCC can mandate re-training or additional training as needed for specific sites based on site performance.
Study Laptop Training: Hands-on GlucoStabilizer® and control treatment screen training will be conducted during the Investigator Meeting. Each site will be provided a laptop to use at the meeting during the training. The laptop will be returned to the site for training other study team members and for use in the trial.
Data Training/WebDCU™: Primary Study Coordinators will be trained (train-the-trainer) in study procedures, subject enrollment, and data entry procedures at the SHINE Investigator Meeting. The goal of the training is to standardize the methods of data collection to help ensure comparability of data across sites. Once the Primary Study Coordinator has received his/her training, it is the responsibility of that person to train other site personnel as needed.
The SDMC will conduct data training via webcasts on an as needed basis for study personnel wishing to receive additional training. Additionally, the WebDCU™ User Manual (See Appendix 4), which contains step-by-step entry instructions, is also available online, and NETT-SDMC personnel are available during working hours to answer questions regarding data collection and entry.
15.5 Site Re-Training (Due to Lack of Subject Enrollments in 6 Months) Trial-specific refresher training will be mandated for Hub Complexes and SHINE Ancillary sites that do not enroll a subject in the specified trial for six (6) consecutive months. Retraining will include:
1. Nursing Inservices for nursing staff or provide with a just-in-time nursing training plan. 2. Protocol training in the form of reviewing the protocol training video OR review the recruitment, protocol adherence quizzes and FAQs for investigators and study coordinators 3. Review of Data/Randomization Training Series by team member as appropriate to their role. 4. For sites participating in ISPOT study, review ISPOT blood collection and processing procedures and expiration dates for blue tops in their collection kits. 5. Ensure study laptop is updated and there are no internet connectivity issues.
Once the above are completed, upload the all training certifications and Hub PI attestation into the SHINE Regulatory Database. For more details regarding procedures, refer to the NETT SOP for Hub Performance Assessment or contact SHINE site manager.

SHINE Manual of Procedures 102 Version 6– December 19, 2016
16. Patient Recruitment and Retention 16.1 Recruitment Recruitment is the dialogue which takes place between an investigator and a potential participant prior to the initiation of the consent process. It begins with the identification, targeting and enlistment of participants for the research study. It involves providing information to the potential participants and generating their interest in the proposed study. To successfully enroll patients into the trial, the study team must be able to explain in an enthusiastic, informative and convincing manner that the trial is something in which they would want to participate. Subject recruitment includes frequent collaboration with the hospital stroke team, radiologist(s), ED team and house staff as a primary referral mechanism. It is, however, important to have multiple recruitment strategies to reach all potential patients at all times. The site PI, coordinator, and study team will agree to a screening plan before initiating patient enrollment. Each site will provide a recruitment plan including estimates of volumes for patients to be screened and enrolled. Other strategies aimed at improving recruitment include identifying site-specific barriers by telephone or in person and the implementation of a recognition system for excellent recruitment, retention and data quality.
16.2 Retention 16.2.1 Retention and Study Participation
Before leaving the hospital, the coordinator will obtain multiple forms of contact information in order to avoid loss to follow-up.
The participant/LAR will be called at 6 weeks (+/- 14 days) to evaluate the participant’s progress thus far. The coordinator will make every effort to begin making this contact early in the window of opportunity.
The 90-day post follow-up visit will include the mRS, NIHSS, BI, SSQOL, assessment of SAEs and unblinding questionnaires. The target for this visit will be (+/-) 14 days. Late data will be collected and analyzed for the 90 day visit with a window of (+30 days/-14 days) for the primary outcome. In the event that the study team cannot make contact with the participant/LAR, the mRS may be assessed using information provided by caregivers or other individuals with current knowledge of the patient’s condition. Late data may be collected and analyzed for the 90 day visit with a window of (+90 days/-14 days) for secondary analyses. In the event that a subject is not lost to follow up but has missed the target for the primary analysis, it is recommended that the 90 day outcomes are captured whenever possible. SAEs will be captured from the time of randomization until the end of the study. End of study may be past 90days from randomization if the final study visit is conducted late however SAEs will not be captured past 120days from randomization or end of study, whichever occurs first.
16.2.2 Lost to Follow Up A subject can be classified as lost to follow-up at the clinical site only when the study team has tried numerous ways to contact the subject and has been unsuccessful.
All attempts must be documented and include the following, but are not limited to:
• Repeated phone calls are made to the subject and documented on a log • Contact is attempted with all individuals listed on the Patient Information page
within the medical record

SHINE Manual of Procedures 103 Version 6– December 19, 2016
• A certified letter is sent to the subject’s address, with a return envelope included, asking the subject if they wish to continue participation in the trial
• Contact is made with the subject’s primary care physician and a message is left for the subject to contact the Coordinator
• The local appointment scheduler is consulted to determine when the subject is returning for a clinic visit and where they could be reached, even if the appointment is in a different specialty
• The use of the people locating service was considered (and used), as appropriate. The outcome of the search using the people locating service will be documented, if applicable.
• If all methods of contact have been attempted and are unsuccessful, the Coordinator should indicate that the subject is lost to follow up on the End of Study CRF.
• The site study team member(s) (PI/coordinator will be asked to present the case on a scheduled NETT Operations Committee call in order to adjudicate the case.
16.3 Use of People Locating Service (OmniTrace) The SHINE trial has partnered with a licensed people search company, OmniTrace, to assist in locating contact information for subjects that are at risk of being lost to follow up. The service will serve to obtain current contact information (address, phone number) for participants who have become unable to be located. Only sites that have the Omnitrace language approved within their consent forms are able to use this service.
The SHINE trial recruitment team will work with enrolling sites to determine whether use of the locating service is warranted for subjects who cannot be located for completion of scheduled follow up. When a subject is not able to be located and the SHINE recruitment team determines use of the service is indicated, this information will be communicated to the site. The enrolling site will complete the Patient Information Form located in the SHINE Toolbox and provide directly to OmniTrace. OmniTrace will complete a search for updated contact information and will provide directly to the enrolling site.
OmniTrace maintains strict patient privacy guidelines and data security policies. The University of Virginia has a business agreement with OmniTrace and will pay OmniTrace directly for all completed searches. OmniTrace does not access patient medical records and does not electronically store patient data.
The Patient Information Form, OmniTrace FAQs, and an introductory memo are found in Appendix 12.
16.4 Discontinuation of Study Participation The treatment period for the study is 72 hours or until discharged, whichever comes first. If discharge is clinically indicated, the treating team can stop study treatment before 72 hours. Subjects who are discharged prior to 72 hours are considered to have completed the study treatment and will be followed for the duration of the study. This is not considered discontinuation from participation and is distinct from discontinuation that is initiated by the subject/LAR or the treating or study team for safety reasons.

SHINE Manual of Procedures 104 Version 6– December 19, 2016
There are two categories of discontinuation of study participation: A subject can have the treatment protocol discontinued but remain in the study until completion of the 90-day follow up. Alternatively, the subject or LAR may withdraw informed consent and terminate all further participation in the study. Procedures on how to process and document these discontinuations follows.
A distinction will be made between subjects who fail to complete all visits on schedule or who miss some follow up assessments and the withdrawal of consent. Missed or rescheduled visits will be documented, but the subject will continue to be followed in the future according to protocol requirements.
16.4.1 Discontinuation of Study Participation
16.4.1.1 Initiated by Subject or LAR As participation in the SHINE trial is voluntary, a subject or LAR can request that treatment be discontinued at any time. Study team members should attempt to understand the subject or LAR’s reasons for desiring discontinuation. Further discussion with the subject/LAR about their concerns and the subject’s treatment and treatment alternatives may enhance retention. Study team members should also discern whether the subject/LAR would like the treatment ceased or if they would like to withdraw informed consent. If a subject or LAR decides to discontinue study treatment but is willing to remain in the study, the reason for discontinuation should be documented in the research file and reported on the Study Treatment CRF (Form 15).
If a subject/LAR wishes to discontinue treatment unless they are unblinded to which treatment the subject is receiving, they may be unblinded. However, these requests must first be presented to the SHINE Study Hotline and may be minimized by thorough discussion by the study team of the rationale for blinding and the subject/LAR’s specific concerns about the differences in treatment groups.
16.4.1.2 Initiated by Treating Investigator or Treating Physician
A treating investigator or treating physician may discontinue the study treatment if it is determined that continued participation is not in the subject’s best interests. Prior to discontinuing for this reason, the treating investigator or treating physician is encouraged to contact the SHINE Study Hotline to discuss discontinuation with a SHINE study Principal Investigator. If a subject is discontinued for clinical or safety reasons, this should be clearly documented in the research file and reported on the Study Treatment CRF (Form 15).
16.4.1.3 Discontinuation of Treatment for Other Reasons If treatment is discontinued for other reasons (e.g., death, discharge, subject is transferred to a floor where IV insulin cannot be given, etc.), it should be reported on the Study Treatment CRF (Form 15). Early discontinuation may also need to be reported as a protocol deviation and reported to the IRB.
16.4.2 Procedures for Withdrawal of Informed Consent A subject or LAR may withdraw informed consent at any time, and will not be contacted for any further study data collection. However, any data already collected on the subject up to the point of withdrawal will continue to be used in the study. In

SHINE Manual of Procedures 105 Version 6– December 19, 2016
instances where a subject wishes to withdraw consent, the date and reason for consent withdrawal should be documented in the research file and documented on the End of Study CRF (Form 14). Additionally, if a subject withdraws consent during the treatment period, this should be documented on the Study Treatment CRF (Form 15). Subject data will be included in the analysis up to the date of the consent withdrawal. Notify the SHINE Study Hotline when a subject has withdrawn consent to participate in all trial activities by calling 800-915-7320 (ext 1).
If a subject/LAR wishes to withdraw consent unless they are unblinded to which treatment the subject is receiving, they may be unblinded. However, these requests may be minimized by thorough discussion by the study team of the rationale for blinding and the subject/LAR’s specific concerns about the differences in treatment groups. Every measure should be taken to ensure that blinded study team members remain blind, even in the event the patient becomes unblinded.
16.4.2.1 Required Documents
Subjects wishing to withdraw informed consent should provide written documentation of his or her withdrawal to the extent possible.
16.4.2.2 Procedures If a subject/LAR wishes to withdraw consent, the following procedure should be followed:
• Check for the development of adverse events. • Complete the End-of-Study CRF (Form 14) and include an explanation of why
the subject/LAR is withdrawing. If the subject is receiving study treatment, complete the Study Treatment CRF (Form 15), indicating that the subject has withdrawn consent.
• Subject or LAR will be asked to document in writing his or her desire to withdraw.
16.5 Milestones and Subject Payments to Clinical Sites SHINE sites will receive milestone-based payments prior to enrollment of their first patient. A summary of these payments is provided below.
16.5.1 Milestone MILESTONE 1
• Enter Hub, spoke(s) or ancillary site into WebDCU™, assigning the site to the SHINE trial
• If the site IRB requires review of the Informed Consent at this point, submit site‐specific informed consent to the NETT CCC for review/approval (if not, see Milestone 2)
• Upload copy of above submission into WebDCU™ (document should be titled, “SHINE IRB Submittal”)
MILESTONE 2
• Reconcile regulatory documents in WebDCU™ • Confirm that all contracts (site, investigational pharmacy, etc.) and required
training are in place • Complete site readiness call • Upload IRB approval • Obtain approval to begin subject enrollment

SHINE Manual of Procedures 106 Version 6– December 19, 2016
MILESTONE 3
• Continuation of enrollment at the Hub
16.5.2 Per Subject Payments SHINE sites will receive two per-subject payments based on the schedule outlined below. In order to qualify for payment for an individual subject, all associated CRFs must be complete and query free to show as ready for payment. The site is then eligible to request payment by submitting an institutional acceptable invoice document to the subcontractor for payment. Figures provided represent “total payments”, and thus include any Indirect Cost recovery.
Payment 1 Payment 1 is provided when the following criteria are met:
• Eligible subject is enrolled and completes initial study visit • All data for eligible visit is entered into WebDCU™ • All queries are resolved for the visit • Subject visit reads “Ready” in WebDCU™
Payment 2 Payment 2 is provided when the following criteria are met:
• Subject is not lost to follow up • Eligible subject completes all required components of 3-month outcomes
visit • All required data for second visit is entered into WebDCU™ • All queries are resolved for the visit • Subject visit reads “Ready” in WebDCU™
Contact Information for Milestones and Payments For NETT Hubs and Spoke Sites: All email correspondence regarding milestone payments should be sent to the SHINE study team at [email protected]. For SHINE Ancillary Sites: All email correspondence regarding milestone payments should be sent to the SHINE Grants and Contracts Administrator: Emily Gray at [email protected].
17. Close-Out and Termination Stages 17.1 Site Closeout Site Closeout procedures will be distributed and coordinated by the NETT-CCC Site Manager to ensure regulatory compliance. Contact site manager with questions.
17.2 Retention of Study Records Study records will be retained for a minimum of 5 years from the approval date of the sponsor’s final study report in accordance with contract or grant stipulations or until you are otherwise notified by NIH. These include, but are not limited to: 1. Patient CRF binders and informed consent forms 2. Patient medical charts containing progress notes, laboratory reports, etc.

SHINE Manual of Procedures 107 Version 6– December 19, 2016
3. Regulatory Binders 4. Study Reference Manual 5. All other personal/site files and documents related to the trial
18. Study Policies and Research Conduct 18.1 Protection of Human Subjects Participating sites must maintain a human subjects protection program compliant with 45 CFR 46 and 21CFR 50 and 56 and with state, local or institutional requirements related to the protection of human subjects, an approved Assurance for human subjects research and an IRB registration number. Enrolling local institutions must also ensure the safe and appropriate performance of the research at its institution. This includes, but is not limited to, monitoring protocol compliance, managing any major protocol violations, managing any serious adverse events occurring at the institution, ensuring qualifications of research staff and providing a mechanism by which complaints about the research can be made by local study participants or others. Prior to enrolling subjects in SHINE, each site must submit documentation that the study has been approved by the local IRB, including locally approved informed consent forms. Written, valid informed consent must be obtained from each SHINE participant as part of the subject enrollment process only after the investigator is satisfied that the participant understands the potential risks and benefits of participation in the study. See also:
https://nett.umich.edu/sites/default/files/docs/sop_certification_for_protection_of_human_subjects_final01.pdf 18.2 The HIPAA Privacy Rule Under the Health Insurance Portability and Accountability Act of 1996 (HIPAA) Privacy Rule, SHINE investigators are required by the Department of Health and Human Services (HHS) or the Food and Drug Administration (FDA) Protection of Human Subjects Regulations (45 CFR part 46 or 21 CFR parts 50 and 56, respectively) to take measures to protect personal health information (PHI) from inappropriate use or disclosure. PHI includes identifiable health information about subjects of clinical research gathered by a researcher who is a covered health care provider. Compliance with HIPAA regulations is considered a local context issue and remains the purview of the local institution and local IRB. The HIPAA Privacy Rule is concerned with the risk to the subject's privacy associated with the use and disclosure of the subject's PHI, and permits researchers, as health care providers and therefore covered entities, to use or disclose PHI for research under certain circumstances and conditions, including if the subject of the PHI has granted specific written permission through an Authorization that satisfies section 164.508 and if the PHI has been de-identified in accordance with the standards set by the Privacy Rule at section 164.514(a)-(c) in which case, the health information is no longer PHI. The individual SHINE IRBs will act as Privacy Boards (required by HIPAA) to review the use and disclosure of PHI and to determine whether subjects should sign a Subject Authorization for Release of PHI for Research in addition to the consent to participate in research, or if a Waiver of Authorization may be granted analogous to a Waiver of Consent under the Common Rule.

SHINE Manual of Procedures 108 Version 6– December 19, 2016
For a more detailed discussion of permitted uses or disclosures of PHI for clinical research under the Privacy Rule, refer to Protecting Personal Health Information in Research: Understanding the HIPAA Privacy Rule; Research Repositories, Databases, and the HIPAA Privacy Rule; Institutional Review Boards and the HIPAA Privacy Rule; and Privacy Boards and the HIPAA Privacy
18.3 Protocol Amendments Full protocol amendments are prepared to incorporate significant changes, those involving more than minimal impact on participant safety and risk-to-benefit ratio of participation in SHINE, and will result in the generation of a new protocol version with a new version number. When amendments are prepared, any prior protocol modifications specified in a contract or agreement are also incorporated into the amendment.
In accordance with 45 Code of Federal Regulations (CFR) 46.103(b) (4) (iii) and 21 CFR 56.108(a) (4), changes to the SHINE protocol or its related consent document must be approved by the IRB prior to implementation except where necessary to eliminate apparent immediate hazards to participants.
Once finalized by the NIH/DSMB, the amendment is distributed to all team members and participating study sites by the NETT-CCC Site Manager/staff. Sites must seek local IRB approval of the protocol and other associated documents for the amended version of the protocol. Revised procedures specified in the amendment may not be conducted until after protocol approval is obtained. Participants enrolled in a study after approval of a protocol amendment must be consented to the study using the revised informed consent form associated with the amended version of the protocol.
For participants enrolled prior to approval and registration of an amendment, guidance on whether re-consenting is required (using the revised informed consent form associated with the amendment) will be provided by the CCC, typically in the summary of changes that accompanies the amended protocol.
Regardless of protocol team’s recommendations, site IRBs may require re-consenting of previously enrolled participants; in such cases, IRB requirements must be followed.
18.4 Ancillary Studies Proposals, budgets and personnel for ancillary studies will be reviewed by the Executive Committee. The committee will assure that all such studies are hypothesis driven, methodologically robust and contain complete and accurate data. Approval will follow the ancillary study approval process which defines the standard procedures for proposing, reviewing, and approving ancillary studies and/or sub-studies conducted within the trial. Approval is conditional on the following: 1. Collaboration with the SHINE investigators and other SHINE ancillary study teams as
necessary to support all the research being done under the auspices of the SHINE trial.
2. The use SHINE data and resources is permitted only for the agreed upon reasons. Additional use of SHINE data or resources requires additional review and approval by the SHINE executive team.
3. The ancillary study team must abide by all SHINE policies and procedures including the data sharing policies, publication policies.

SHINE Manual of Procedures 109 Version 6– December 19, 2016
4. No results that relate to the SHINE trial outcomes can be initiated until all outcomes are complete and validated in the study data set and approved for release to the ancillary teams by the SHINE executive committee and the SDMC.
5. Review and approval to publish or present SHINE data is required from the SHINE executive team. All publications must include at least 1 SHINE PI as well as the clause “for the SHINE investigators” in addition to an appendix of SHINE principal investigators (PIs) and coordinators. If the clause “for the SHINE investigators” is not allowed by the journal then the executive team should be considered for authorship and all SHINE PIs and coordinators must be listed in an appendix.
6. For ancillary studies that are funded, a representative from the ancillary study team is expected to participate on SHINE executive team meetings at least once monthly.
7. For ancillary studies that are funded, a representative from the ancillary study team must attend investigator meetings and all requested face to face meetings with the SHINE executive team.
8. The SHINE executive team and SHINE DSMB will monitor the execution of the ancillary study and reserve the right to alter the study should it impede the ability of the parent SHINE trial to succeed.
9. The conditions of this agreement should be included in applications for NIH or other funding.
Data will be controlled by the Executive Committee, which will review requests for access and specific analyses. Monitoring during the trial will be dictated by safety and scientific concerns rather than regulatory requirements. Publication of the results of these studies will be governed by the policies and procedures developed by the Executive Committee. Sites will not be required to participate in any ancillary study that requires additional data collection, but they will be encouraged to participate in accepted studies.
18.5 Publications and Presentations Policies and Procedures The goal of the SHINE Trial Publications Policy is to provide guidelines for preparing, reviewing, submitting and maximizing productivity of high quality peer-reviewed publications. In addition to overseeing the performance of the trial, the Executive Committee is responsible for encouraging paper production, ensuring timely publication of data, maintaining a high standard for the quality of papers produced for SHINE, and determining appropriate authorship. When the Committee is discussing manuscripts associated with ancillary studies, the PI of the ancillary study and His/her designee will also join the Executive Committee for that discussion. Manuscript proposals will be submitted to the Executive Committee via a website based form at https://nett.umich.edu/clinical-trials/shine/data-analysis-publication-application-form. These proposals will include the type (primary, secondary, tertiary and quaternary), list of authors and their qualifications for authorship, a statement that no others deserving authorship have been omitted, the scientific rationale for the paper, the data needed and a description of the proposed analyses and any deadlines for submission of abstracts or presentation dates if applicable.
18.6 Data Sharing Policy and Procedures Within one year of the primary publication or within two years of the last study visit of the last subject, a cleaned dataset and any supporting documentation required for the analysis of the data will be submitted to NINDS Office of Clinical Research.
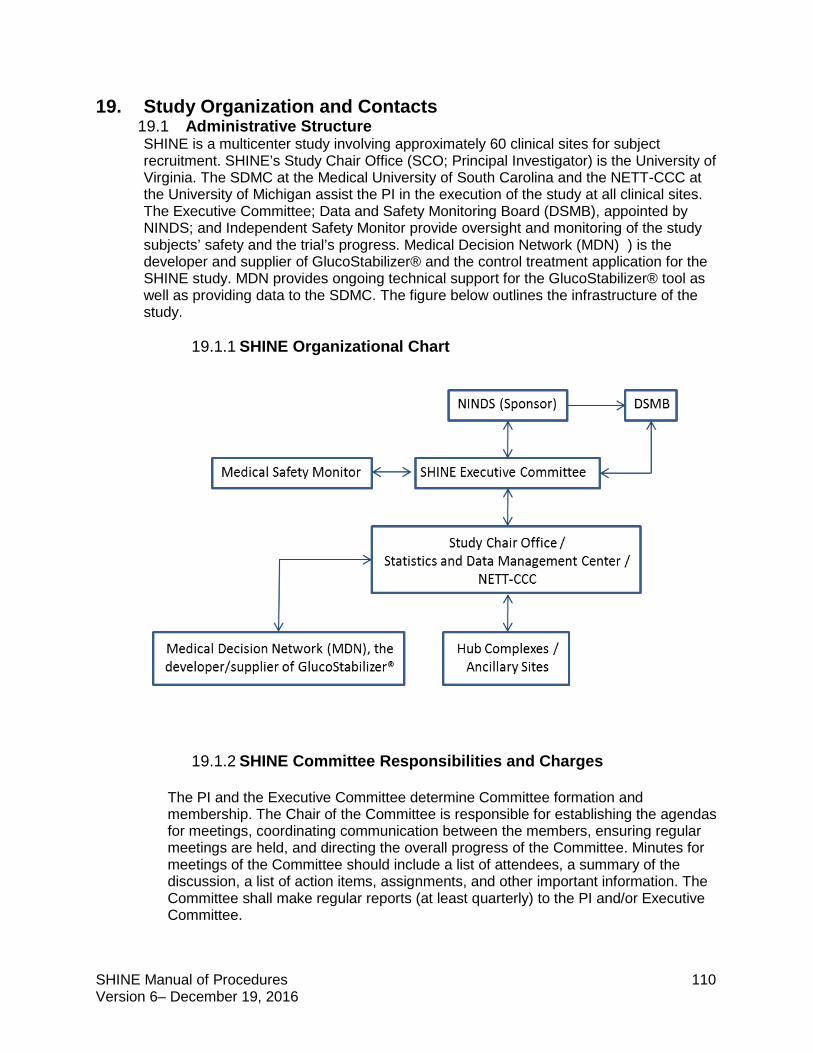
SHINE Manual of Procedures 110 Version 6– December 19, 2016
19. Study Organization and Contacts 19.1 Administrative Structure SHINE is a multicenter study involving approximately 60 clinical sites for subject recruitment. SHINE’s Study Chair Office (SCO; Principal Investigator) is the University of Virginia. The SDMC at the Medical University of South Carolina and the NETT-CCC at the University of Michigan assist the PI in the execution of the study at all clinical sites. The Executive Committee; Data and Safety Monitoring Board (DSMB), appointed by NINDS; and Independent Safety Monitor provide oversight and monitoring of the study subjects’ safety and the trial’s progress. Medical Decision Network (MDN) ) is the developer and supplier of GlucoStabilizer® and the control treatment application for the SHINE study. MDN provides ongoing technical support for the GlucoStabilizer® tool as well as providing data to the SDMC. The figure below outlines the infrastructure of the study.
19.1.1 SHINE Organizational Chart
19.1.2 SHINE Committee Responsibilities and Charges
The PI and the Executive Committee determine Committee formation and membership. The Chair of the Committee is responsible for establishing the agendas for meetings, coordinating communication between the members, ensuring regular meetings are held, and directing the overall progress of the Committee. Minutes for meetings of the Committee should include a list of attendees, a summary of the discussion, a list of action items, assignments, and other important information. The Committee shall make regular reports (at least quarterly) to the PI and/or Executive Committee.

SHINE Manual of Procedures 111 Version 6– December 19, 2016
19.1.2.1 Executive Committee The SHINE Executive Committee (EC) is a small study leadership group that guides study implementation and operations. The EC is responsible for reviewing study progress and for identifying and resolving critical issues that arise. The EC will develop and maintain the MOP, trial-specific SOPs, and other important trial documents. The EC members are charged with operational decisions that require more expeditious action and complex implementation than can be achieved through meetings of the Operational Committee. The EC meets monthly or more frequently if needed.
19.1.2.2 Publications Committee
The Publications Committee (PC) is responsible for authorizing any publication using the SHINE database prior to public disclosure. The PC will meet on an as-needed basis.
19.1.2.3 WebDCUTM The entire study will be conducted using an electronic data acquisition method where all clinical data on enrolled subjects will be entered by the Hub/Spoke and SHINE Ancillary site personnel in real time. In order to provide user-friendly and easy-to-navigate interfaces, the WebDCU™ SHINE data capture screens are designed based on individual CRFs. The latest version of each CRF is available as a PDF file on the study website for use as worksheets and source documents by study personnel. A full study casebook is entered into the system to ensure the data entry screens mirror the CRFs and that the pre-programmed data rules appropriately detect incorrect data. This process not only tests the format and use of the forms but also tests the database data entry screens and rules. The latest version of each CRF will be available as a PDF file on the study website for use as worksheets and source documents by study personnel. This process facilitates version control of all study related documents, particularly since documents may evolve over the course of the study. The data will be captured and data entered (single-keyed) at the Hubs/Spokes and Affiliate Hubs via the study website interface. The data will be managed (including data queries) by the SDMC using the WebDCUTM system. This user-friendly web-based database system, developed and validated by the SDMC, will be used for subject randomization, data entry, data validation, project progress monitoring, subject payment, subject tracking, drug shipment tracking, user customizable report generation and secure data transfer.
19.1.2.4 Neurological Emergencies Treatment Trials (NETT) Network
The NIH created a Neurological Emergencies Treatment Trials (NETT) Network for conducting large simple trials to reduce the burden of very acute injuries and illnesses affecting the brain, spinal cord, and peripheral nervous system. The network recognizes and seeks to explore the special narrow window of opportunity that seems to exist in treating neurologic damage from a variety of pathologies ranging from stroke to traumatic brain injury, and seizures to meningitis. The study of very rapid interventions will have to be implemented by paramedics in the field, or by physicians in the Emergency

SHINE Manual of Procedures 112 Version 6– December 19, 2016
Department. This network provides the basis for conducting efficient studies in these environments.
The mission of the network is to improve outcomes of patients with acute neurologic problems through innovative research focused on the emergent phase of patient care. The NETT network consists of 22 regional Hub Complexes, each with several community hospital Spokes, a Statistical & Data Management Center (SDMC), and a Clinical Coordinating Center (CCC). Oversight of the network is provided by an NINDS appointed Advisory Group (NAG), the NINDS NETT Scientific Program Director and the NINDS NETT Administrative Program Director.
19.1.2.5 DSMB
The NINDS-appointed Data Safety Monitoring Board (DSMB) will plan to meet semi-annually with interim conference calls as needed, to review interim data relating to the trial. The functions of the DSMB will include: (1) Review of all adverse effects or complications related to the trial
interventions; (2) Review of interim data on the primary and secondary outcomes according
to prespecified monitoring guidelines; (3) Monitor subject accrual; (4) Review summary reports relating to performance of centers relative to
compliance with protocol requirements; (5) Recommend to the Executive Committee that:
a) Trial continue as planned; b) Trial be modified with justification of modification as DSMB deems
appropriate; c) Trial should be stopped with disclosure of basis for recommendation.
In general, clinical investigators will not be present at the Closed Sessions of the DSMB, but may on occasion be requested to present or provide information to assist the DSMB in carrying out its functions appropriately. DSMB Roster Michael D. Hill, M.D. Professor Department of Clinical Neurosciences Hotchkiss Brain Institute University of Calgary, Foothills Hospital 1403 29th Street NW, Room 1242A Calgary, AB, T2N 2T9, Canada Susan Shapiro Braithwaite, M.D. Visiting Clinical Professor University of Illinois―Chicago Endocrine Consults and Care, S.C. West Tower Office Building, Suite 605 800 Austin Street Evanston, IL 60202

SHINE Manual of Procedures 113 Version 6– December 19, 2016
William R. Clarke, Ph.D. Professor of Biostatistics Director, Clinical Trials Statistical and Data Management Center 2450 University Capitol Centre Iowa City, Iowa 52242 Jennifer Frontera, M.D. Cleveland Clinic Main Campus, Cerebrovascular Center Mail Code S80 9500 Euclid Avenue Cleveland, OH 44195 Kama Guluma, M.D. Assistant Professor Department of Emergency Medicine University of California San Diego 200 West Arbor Drive San Diego, CA 92103-8676 Rema Raman, Ph.D. Associate Professor Division of Biostatistics and Bioinformatics Department of Family and Preventive Medicine and Department of Neurosciences University of California, San Diego 9500 Gilman Drive, M/C 0717 La Jolla, CA 92093-0717 19.1.3 Medical Decision Network (GlucoStabilizer®) Medical Decision Network (MDN) is the developer and supplier of GlucoStabilizer® and the control treatment application for the SHINE study. MDN specializes in information management for POC testing. For the SHINE trial, MDN will also facilitate data transfer to SDMC, training, server installation and hosting, laptop installation and hosting.

SHINE Manual of Procedures 114 Version 6– December 19, 2016
19.2 SHINE Trial Contacts Karen C. Johnston, MD, MSc Principal Investigator
University of Virginia Department of Neurology P.O. Box #800394 Charlottesville, VA Telephone: 434-924-5323 Fax: 434-982-1726 Email: [email protected] Pager: 434-982-3500 # 2820
Askiel Bruno, MD, MS Co-Principal Investigator
Askiel Bruno, MS, MD Department of Neurology Medical College of Georgia at Augusta UniversityGeorgia Regents University 1120 15th St., BI 3076 Augusta, GA 30912 Email: [email protected] Telephone: 706-721-6050
Christiana E. Hall, MD, MS Co-Principal Investigator
University of Texas Southwestern Dept. of Neurology & Neurotherapeutics 5323 Harry Hines Blvd; MC 8855 Dallas, TX 75390-8855 Telephone: (214) 648-8511 Fax: (214) 648-0341 Email: [email protected] Pager: 214-786-0120
Kevin Barrett, MD, MSc Co-Principal Investigator
Vice-Chair, Department of Neurology Associate Professor of Neurology Director, Neurohospitalist Fellowship Program Mayo Clinic 4500 San Pablo Road Jacksonville, FL 32224 904-953-7102 [email protected]
Mark Conaway, PhD Blinded Statistician
University of Virginia Department of Public Health Sciences P.O. Box 800717 Telephone: 434 924-8510 Fax: 434 924-8437 Email: [email protected]
Valerie Durkalski, PhD NETT SDMC Principal Investigator and Study Unblinded Statistician
Medical University of South Carolina Division of Biostatistics and Epidemiology 135 Cannon St, Rm 305J, MSC 835 Charleston, SC 29425-8350 Telephone: 843- 876-1911 Fax: 843- 876-1923 Email: [email protected]
SHINE Manual of Procedures 115 Version 6– December 19, 2016
Yuko Palesch, PhD NETT SDMC Principal Investigator
Medical University of South Carolina Division of Biostatistics and Epidemiology 135 Cannon St, Rm 303Q Charleston, SC 29425-8350 Telephone: 843-876-1917 Fax: 843-876-1923 Email: [email protected]
William Barsan, MD NETT CCC Principal Investigator
University of Michigan Health System Department of Emergency Medicine 1500 East Medical Center Dr. B1354-TC, Box 0303 Ann Arbor, MI 48109 Telephone: 734-936-6020 Fax: 734-763-7228 Email: [email protected]
William Meurer, MD, MS University of Michigan Health System Department of Emergency Medicine 1500 East Medical Center Dr. B1354-TC, Box 0303 Ann Arbor, MI 48109 Telephone: 734-936-1616 Email: [email protected]
Cemal Sozener, MD NETT CCC Co-Investigator Internal Quality and Safety Reviewer
University of Michigan Health System Emergency Medicine University Hospital Floor B1 Reception Emergency 1500 E Medical Center Dr SPC 5301 Ann Arbor, MI 48109 Telephone: 734-936-6666 Fax: 734-936-8763 Email: [email protected]
Thomas Bleck, MD Independent Safety Monitor
Rush University Medical Center 1725 W. Harrison, Ste 529 Chicago, IL 60612 Telephone: 312-942-6706 Cell: 434-227-0279 Email: [email protected]
Mercedes Falciglia, MD Study Endocrinologist
UC Medical Center Stetson Center 260 Stetson Street, Ste 4200, ML 0547 Cincinnati, OH 45219 Phone: 513-558-4444 Email: [email protected]
Heather M Haughey, PhD SHINE Project Director
University of Virginia Department of Neurology P.O. Box #800394 Charlottesville, VA Telephone: (434) 243-8065 Fax: (434) 243-4691 Email: [email protected]
SHINE Manual of Procedures 116 Version 6– December 19, 2016
Katrina van de Bruinhorst SHINE Recruitment Specialist
UT Southwestern Medical Center at Dallas Division of Neurocritical Care Department of Neurology and Neurotherapeutics 5323 Harry Hines Blvd., CS7.522, MC 8855 Dallas, TX 75390 Telephone: 214-648-9248 E: [email protected]
Arthi Ramakrishnan Site Manager, NETT CCC
University of Michigan Department of Emergency Medicine 24 Frank Lloyd Wright Drive Lobby H, Suite 3100, P.O. Box 381 Ann Arbor, MI 48106 Telephone: 734-936-2454 Email: [email protected]
Carol Van Huysen Project Monitor, NETT CCC
University of Michigan Department of Emergency Medicine 24 Frank Lloyd Wright Drive Lobby H, Suite 3100, P.O. Box 381 Ann Arbor, MI 48106 Telephone: 734-764-9254 Email: [email protected]
Donna Harsh Project Monitor, NETT CCC
University of Michigan Department of Emergency Medicine 24 Frank Lloyd Wright Drive Lobby H, Suite 3100, P.O. Box 381 Ann Arbor, MI 48106 Ph. 734-232-2136 Email: [email protected]
Valerie Stevenson Administrative Director, NETT CCC
University of Michigan Department of Emergency Medicine 24 Frank Lloyd Wright Drive Lobby H, Suite 3100, P.O. Box 381 Ann Arbor, MI 48106 Telephone: 734-232-2131 Email: [email protected]
Emily Gray Senior Grants and Contracts Administrator, UVA SHINE
University of Virginia Department of Neurology P.O. Box #800394 Charlottesville, VA Telephone: 434-982-6773 Fax: (434) 243-4691 Email: [email protected]
Joy Pinkerton, Education Coordinator, NETT CCC
University of Michigan Department of Emergency Medicine 24 Frank Lloyd Wright Drive Lobby H, Suite 3100, P.O. Box 381 Ann Arbor, MI 48106 Telephone: 734-232-2138 Email: [email protected]
SHINE Manual of Procedures 117 Version 6– December 19, 2016
Angela Pauls RA/Statistical Programmer, NETT SDMC
Medical University of South Carolina Division of Biostatistics and Epidemiology 135 Cannon St, Rm 305R, MSC 835 Charleston, SC 29425-8350 Telephone: 843-792-4192 Fax: 843-876-1923 Email: [email protected]
Wenle Zhao, PhD Co-Investigator/ Associate Director of Information Systems, NETT SDMC
Medical University of South Carolina Division of Biostatistics and Epidemiology 135 Cannon St, Rm 305P, MSC 835 Charleston, SC 29425-8350 Telephone: 843- 876-1937 Fax: 843- 876-1923 Email: [email protected]
Keith Pauls Senior Database Programmer, NETT SDMC
Medical University of South Carolina Division of Biostatistics and Epidemiology 135 Cannon St, Rm 305U, MSC 835 Charleston, SC 29425-8350 Telephone: 843-876-1889 Fax: 843-876-1923 Email: [email protected]
Catherine Dillon Associate Director of Data Management, NETT SDMC
Medical University of South Carolina Division of Biostatistics and Epidemiology 135 Cannon St, Rm 305S, MSC 835 Charleston, SC 29425-8350 Telephone: 843-876-1261 Fax: 843-876-1923 Email: [email protected]
Kavita Patel Data Manager, NETT SDMC
Medical University of South Carolina Division of Biostatistics and Epidemiology 135 Cannon St, Rm 305R, MSC 835 Charleston, SC 29425-8350 Telephone: 843-876-1167 Fax: 843-876-1923 Email: [email protected]
Scott Janis, PhD NINDS Program Official
NIH/NINDS Neuroscience Center Room 2191 6001 Executive Blvd MSC 9520 Bethesda, MD 20892 Telephone: (301) 496-9135 Email: [email protected]
Sei Kim
Principal Engineer/Architect
Medical Decision Network 2220 Ivy Road, Suite 403 Charlottesville VA 22903 Telephone: 434-202-7280 Toll Free: 866-791-6108 x306 Email: [email protected]
SHINE Manual of Procedures 118 Version 6– December 19, 2016
19.3 Quick Reference Table The following contact sheet provides guidance to investigators and their staff regarding who to contact regarding specific study related questions or issues.
Category Contact Name Business Hours Number
24 Hour Contact
Protocol questions
Karen Johnston, MD, MSc Christiana Hall, MD, MS Askiel Bruno, MD, MS Kevin Barrett, MD, MSc
800-915-7320 ext 1
Eligibility criteria Karen Johnston, MD, MSc Christiana Hall, MD, MS Askiel Bruno, MD, MS Kevin Barrett, MD, MSc
800-915-7320 ext 1
Randomization Catherine Dillon Kavita Patel
843-876-1942 843-876-1167
8:00AM-5:00PM ET
866-450-2016 (For technical randomization
emergencies only) WebDCUTM support and passwords
Catherine Dillon Kavita Patel
843-876-1942 843-876-1167
8:00AM-5:00PM ET
CRF questions Kavita Patel
843-876-1167
Contracts and invoicing
Valerie Stevenson (NETT sites) Emily Gray (Affiliate sites)
734-232-2131
434-982-6773 Laptop & GlucoStabilizer®
Heather M Haughey 434-243-8065 800-915-7320 ext 1
Protocol training Heather M Haughey 434-243-8065
Recruitment questions
Katrina van de Bruinhorst 214-648-9248
Safety questions Karen Johnston, MD, MSc Christiana Hall, MD, MS Askiel Bruno, MD, MS Kevin Barrett, MD, MSc
800-915-7320 ext 2
SAE reporting Arthi Ramakrishnan Catherine Dillon Kavita Patel
734-936-2454
843-876-1942 843-876-1167
8:00AM-5:00PM EST Site status, readiness, and regulatory questions
Arthi Ramakrishnan 734-936-2454

SHINE Manual of Procedures 119 Version 6– December 19, 2016
20. References
1. Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K, Haase N, Hailpern S, Ho M, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott M, Meigs J, Mozaffarian D, Nichol G, O'Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R, Steinberger J, Thom T, Wasserthiel-Smoller S, Wong N, Wylie-Rosett J, Hong Y. Heart disease and stroke statistics--2009 update: A report from the American Heart Association statistics committee and stroke statistics subcommittee. Circulation. 2009 Jan 27; 119(3): 480-6.
2. Gentile NT, Seftchick MW, Huynh T, Kruus LK, Gaughan J. Decreased mortality by normalizing blood glucose after acute ischemic stroke. Acad Emerg Med. 2006 Feb; 13(2): 174-80.
3. Williams LS, Rotich J, Qi R, Fineberg N, Espay A, Bruno A, Fineberg SE, Tierney WR. Effects of admission hyperglycemia on mortality and costs in acute ischemic stroke. Neurology. 2002 Jul 9; 59(1): 67-71.
4. Capes SE, Hunt D, Malmberg K, Pathak P, Gerstein HC. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview. Stroke. 2001 Oct; 32(10): 2426-32.
5. McCormick MT, Muir KW, Gray CS, Walters MR. Management of hyperglycemia in acute stroke: How, when, and for whom? Stroke. 2008 Jul; 39(7): 2177-85.
6. Malmberg K, Ryden L, Efendic S, Herlitz J, Nicol P, Waldenstrom A, Wedel H, Welin L. Randomized trial of insulin-glucose infusion followed by subcutaneous insulin treatment in diabetic patients with acute myocardial infarction (DIGAMI study): Effects on mortality at 1 year. J Am Coll Cardiol. 1995 Jul; 26(1): 57-65.
7. van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001 Nov 8; 345(19): 1359-67.
8. Adams HP Jr, del Zoppo G, Alberts MJ, Bhatt DL, Brass L, Furlan A, Grubb RL, Higashida RT, Jauch EC, Kidwell C, Lyden PD, Morgenstern LB, Qureshi AI, Rosenwasser RH, Scott PA, Wijdicks EF. Guidelines for the early management of adults with ischemic stroke: A guideline from the American Heart Association/American Stroke Association stroke council, clinical cardiology council, cardiovascular radiology and intervention council, and the atherosclerotic peripheral vascular disease and quality of care outcomes in research interdisciplinary working groups: The American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007 May; 38(5): 1655-711.
9. Adams HP Jr, Leclerc JR, Bluhmki E, Clarke W, Hansen MD, Hacke W. Measuring outcomes as a function of baseline severity of ischemic stroke. Cerebrovasc Dis. 2004; 18(2): 124-9.
10. Saver JL, Yafeh B. Confirmation of tPA treatment effect by baseline severity-adjusted end point reanalysis of the NINDS-tPA stroke trials. Stroke. 2007 Feb; 38(2): 414-6.

SHINE Manual of Procedures 120 Version 6– December 19, 2016
11. Young FB, Lees KR, Weir CJ. Improving trial power through use of prognosis-adjusted end points. Stroke. 2005 Mar; 36(3): 597-601.
12. Lyden P, Lu M, Jackson C, Marler J, Kothari R, Brott T, Zivin J. Underlying structure of the national institutes of health stroke scale: Results of a factor analysis. NINDS tPA stroke trial investigators. Stroke. 1999 Nov; 30(11): 2347-54.
13. Wade DT, Collin C. The barthel ADL index: A standard measure of physical disability? Int Disabil Stud. 1988; 10(2): 64-7.
14. Williams LS, Weinberger M, Harris LE, Clark DO, Biller J. Development of a stroke-specific quality of life scale. Stroke. 1999 Jul; 30(7): 1362-9.
15. Gray CS, Hildreth AJ, Sandercock PA, O'Connell JE, Johnston DE, Cartlidge NE, Bamford JM, James OF, Alberti KG. Glucose-potassium-insulin infusions in the management of post-stroke hyperglycaemia: The UK glucose insulin in stroke trial (GIST-UK). Lancet Neurol. 2007 May; 6(5): 397-406.
16. Bruno A, Kent TA, Coull BM, Shankar RR, Saha C, Becker KJ, Kissela BM, Williams LS. Treatment of hyperglycemia in ischemic stroke (THIS): A randomized pilot trial. Stroke. 2008 Feb; 39(2): 384-9.
17. Johnston KC, Hall CE, Kissela BM, Bleck TP, Conaway MR. Glucose regulation in acute stroke patients (GRASP) trial: A randomized pilot trial. Stroke. 2009 Dec; 40(12): 3804-9.
18. Walters MR, Weir CJ, Lees KR. A randomised, controlled pilot study to investigate the potential benefit of intervention with insulin in hyperglycaemic acute ischaemic stroke patients. Cerebrovasc Dis. 2006; 22(2-3): 116-22.
19. Kreisel SH, Berschin UM, Hammes HP, Leweling H, Bertsch T, Hennerici MG, Schwarz S. Pragmatic management of hyperglycaemia in acute ischaemic stroke: Safety and feasibility of intensive intravenous insulin treatment. Cerebrovasc Dis. 2009; 27(2): 167-75.
20. Johnston KC, Wagner DP, Wang XQ, Newman GC, Thijs V, Sen S, Warach S. Validation of an acute ischemic stroke model: Does diffusion-weighted imaging lesion volume offer a clinically significant improvement in prediction of outcome? Stroke. 2007 Jun; 38(6): 1820-5.
21. Matz K, Keresztes K, Tatschl C, Nowotny M, Dachenhausenm A, Brainin M, Tuomilehto J. Disorders of glucose metabolism in acute stroke patients: An underrecognized problem. Diabetes Care. 2006 Apr; 29(4): 792-7.
22. Vancheri F, Curcio M, Burgio A, Salvaggio S, Gruttadauria G, Lunetta MC, Dovico R, Alletto M. Impaired glucose metabolism in patients with acute stroke and no previous diagnosis of diabetes mellitus. QJM. 2005 Dec; 98(12): 871-8.
23. Kernan WN, Inzucchi SE, Viscoli CM, Brass LM, Bravata DM, Shulman GI, McVeety JC, Horwitz RI. Impaired insulin sensitivity among nondiabetic patients with a recent TIA or ischemic stroke. Neurology. 2003 May 13; 60(9): 1447-51.

SHINE Manual of Procedures 121 Version 6– December 19, 2016
24. Adams HP Jr, del Zoppo G, Alberts MJ, Bhatt DL, Brass L, Furlan A, Grubb RL, Higashida RT, Jauch EC, Kidwell C, Lyden PD, Morgenstern LB, Qureshi AI, Rosenwasser RH, Scott PA, Wijdicks EF. Guidelines for the early management of adults with ischemic stroke: A guideline from the American Heart Association/American Stroke Association stroke council, clinical cardiology council, cardiovascular radiology and intervention council, and the atherosclerotic peripheral vascular disease and quality of care outcomes in research interdisciplinary working groups: The American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007 May; 38(5): 1655-711.
25. Demchuk AM, Tanne D, Hill MD, Kasner SE, Hanson S, Grond M, Levine SR. Predictors of good outcome after intravenous tPA for acute ischemic stroke. Neurology. 2001 Aug 14; 57(3): 474-80.
26. Bruno A, Levine SR, Frankel MR, Brott TG, Lin Y, Tilley BC, Lyden PD, Broderick JP, Kwiatkowski TG, Fineberg SE. Admission glucose level and clinical outcomes in the NINDS rt-PA stroke trial. Neurology. 2002 Sep 10; 59(5): 669-74.
27. Cucchiara B, Tanne D, Levine SR, Demchuk AM, Kasner S. A risk score to predict intracranial hemorrhage after recombinant tissue plasminogen activator for acute ischemic stroke. J Stroke Cerebrovasc Dis. 2008 Nov-Dec; 17(6): 331-3.
28. American Diabetes Association. Standards of medical care in diabetes - 2011. Diabetes Care. 2011;34 Suppl 1:S11-61.
29. van Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, van GJ. Interobserver agreement for the assessment of handicap in stroke patients. Stroke 1988;19:604–607.
30. Quinn TJ, Dawson J, Walters MR, Lees KR. Variability in modified Rankin scoring across a large cohort of international observers. Stroke 2008;39:2975–2979.
31. Bruno A, Akinwuntan AE, Lin C, Close B, Davis K, Baute V, et al. Simplified Modified Rankin Scale Questionnaire: Reproducibility Over the Telephone and Validation With Quality of life. Stroke 2011;42:2276-2279.
32. Wade DT, Collin C. The barthel ADL index: A standard measure of physical disability? Int Disabil Stud. 1988; 10(2): 64-7.
33. Lyden P, Lu M, Jackson C, Marler J, Kothari R, Brott T, Zivin J. Underlying structure of the national institutes of health stroke scale: Results of a factor analysis. NINDS tPA stroke trial investigators. Stroke. 1999 Nov; 30(11): 2347-54.
34. Williams LS, Yilmaz EY, Lopez-Yunez AM. Retrospective assessment of initial stroke severity with the NIH Stroke Scale. Stroke. 2000 Apr;31(4):858-62. 35. Kasner SE, Chalela JA, Luciano JM, Cucchiara BL, Raps EC, McGarvey ML, Conroy MB, Localio AR. Reliability and validity of estimating the NIH stroke scale score from medical records. Stroke. 1999 Aug;30(8):1534-7.
36. Williams LS, Weinberger M, Harris LE, Clark DO, Biller J. Development of a stroke-specific quality of life scale. Stroke. 1999 Jul; 30(7): 1362-9.

SHINE Manual of Procedures 122 Version 6– December 19, 2016
37. Adams HP, Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, Marsh EE. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993; 24: 35-41. 38. Ay H, Benner T, Arsava M, Furie K, Singhal A, Jensen M, Ayata C, Towfighi A, Smith E, Chong J, Koroshetz W, Sorensen A. A computerized algorithm for etiologic classification of ischemic stroke: The causative classification of stroke system. Stroke. 2007: 2979-2984.

SHINE Manual of Procedures 123 Version 6– December 19, 2016
21. Appendices Appendix 1: SHINE Protocol Appendix 2: SHINE Screen Failure Log and Decision Tree Tool Appendix 3: SHINE Consent Appendix 4: WebDCU™ User Manual Appendix 5: Daily Care Log Appendix 6: Hypoglycemia Symptomatic Questionnaire Appendix 7: Sample Monitoring Log Appendix 8: Guidelines for Completion of Specific CRFs Appendix 9: Site Recruitment Performance and Milestone Plan and Recruitment Milestones Appendix 10: SHINE Pharmacy Guidance and Instructions for Developing Study Orders Appendix 11: SHINE Protocol Adherence Monitoring Procedures Appendix 12: OmniTrace Documents

SHINE CONFIDENTIAL Version 2 – 10/23/2012
Stroke Hyperglycemia Insulin Network Effort (SHINE) Trial Protocol
STUDY GROUP CHAIR
Karen C. Johnston, MD, MSc
CLINICAL PRINCIPAL INVESTIGATORS
Askiel Bruno, MD, MS; Christiana E. Hall, MD, MS; Karen C. Johnston, MD, MSc
Co-INVESTIGATORS
Rattan Juneja, MD; Mark Conaway, PhD
The NETT Statistical and Data Management Center
PRINCIPAL INVESTIGATORS
Valerie Durkalski, PhD; Yuko Palesch, PhD
The NETT Clinical Coordinating Center
PRINCIPAL INVESTIGATORS
William Barsan, MD; William Meurer, MD, MS
GLUCOSE MONITORING TEAM
Medical Automation Systems an Alere Company
Denise R. Zito
SPONSOR
NIH - National Institutes of Neurological Disease and Stroke
U01 NS069498

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-1-
Investigator's Agreement
I have read the attached clinical protocol titled Stroke Hyperglycemia Insulin Network Effort
(SHINE) dated 10/23/2012 and agree to conduct the protocol as written in this document.
I agree to comply with the Declaration of Helsinki/Tokyo/Venice on Experimentation in Humans
as required by the United States Food and Drug Administration regulations; the Code of Federal
Regulations Title 21 parts 50, 56, 312, 800, as applicable; the Code of Federal Regulations Title
45 part 46; International Conference on Harmonisation Good Clinical Practice Guidelines; and
all other applicable guidelines.
I understand this document contains confidential information of SHINE Executive Committee,
the NETT CCC and SDMC and cannot be disclosed to anyone other than members of my staff
conducting this trial and members of my Institutional Review Board or Ethical Committee.
I agree to ensure that this information will not be used for any purpose other than the evaluation
or conduct of this clinical trial without the prior written permission of the SHINE Executive
Committee.
_________________________________ _____________________________
Signature of Site Principal Investigator Date
_________________________________
Printed name of Site Principal Investigator
_________________________________ ______________________________
Signature of Co-Principal Investigator Date
(When applicable)
_________________________________
Printed name of Co-Principal Investigator
(When applicable)

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-2-
1. SUMMARY ............................................................................................................................ 6
2. OBJECTIVES ......................................................................................................................... 6
2.1 Specific Aim 1 ................................................................................................................ 6
2.2 Specific Aim 2 ................................................................................................................ 7
3. BACKGROUND AND RATIONALE ................................................................................... 7
3.1 Background ..................................................................................................................... 7
3.2 Significance..................................................................................................................... 7
3.3 Hyperglycemia Correction Trials: Acute Ischemic Stroke ............................................. 9
3.4 THIS and GRASP Preliminary Data............................................................................. 10
3.5 Rationale ....................................................................................................................... 15
4. STUDY PLAN ...................................................................................................................... 15
4.1 Study Design ................................................................................................................. 15
4.2 Study Population ........................................................................................................... 15
4.3 Study Therapy (insulin versus saline) ........................................................................... 16
4.4 Study Decision Support Tool for the Intervention Group ............................................ 16
4.5 Study Sites .................................................................................................................... 16
4.6 Estimated Study and Enrollment Duration ................................................................... 17
5. ELIGIBILITY CRITERIA .................................................................................................... 17
5.1 Inclusion Criteria .......................................................................................................... 17
5.2 Exclusion Criteria ......................................................................................................... 17
5.3 Prohibited Therapy During Study Period ..................................................................... 19
6. SUBJECT RECRUITMENT ................................................................................................ 19
6.1 Methods......................................................................................................................... 19
6.2 Screen Failure Logs ...................................................................................................... 20
7. SUBJECT ENROLLMENT ................................................................................................. 20
7.1 Eligibility Assessment .................................................................................................. 20
7.2 Presentation of Informed Consent ................................................................................ 20
7.3 Randomization .............................................................................................................. 21
7.3.1 Central Randomization Procedure ............................................................................ 21
8. STUDY PROCEDURE ........................................................................................................ 21
8.1 Baseline Assessments ................................................................................................... 21
8.1.1 Point of Care Finger Stick Blood glucose level ........................................................ 21
8.1.2 Concomitant medications documentation ................................................................. 21
8.1.3 Vital signs ................................................................................................................. 21
8.1.4 NIHSS ....................................................................................................................... 22
8.1.5 Neuro Worsening Assessment .................................................................................. 22
8.1.6 Protocol Deviation Documentation........................................................................... 22
8.2 Treatment Procedures ................................................................................................... 22
8.2.1 Decision Support Tool set up .................................................................................... 22
8.2.2 Blinding Set Up......................................................................................................... 22
8.2.3 Drug Dosage/ Drug Administration .......................................................................... 22
8.2.4 Concomitant or Ancillary Therapy ........................................................................... 26
8.3 Clinical Guidelines ........................................................................................................ 26
8.4 Follow-up Procedure ..................................................................................................... 26
8.5 Notification of Death .................................................................................................... 26

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-3-
8.6 Procedure for Unblinding ............................................................................................. 27
8.7 Schedule of Events ........................................................................................................ 27
9. DISCONTINUATION OF PARTICIPATION .................................................................... 27
9.1 Subject Removal from Therapy .................................................................................... 27
9.2 Subject Withdrawal ....................................................................................................... 28
9.3 Procedure for Discontinuation ...................................................................................... 28
9.4 Subject Lost to Follow-Up ............................................................................................ 28
9.5 Subject Transfers .......................................................................................................... 28
10. OUTCOMES DEFINTIONS ............................................................................................ 28
10.1 Primary .......................................................................................................................... 28
10.2 Secondary ...................................................................................................................... 29
11. DATA MANAGEMENT.................................................................................................. 29
11.1 Data Processing ............................................................................................................. 29
11.2 Data Security and Confidentiality ................................................................................. 29
11.3 Data Quality Assurance ................................................................................................ 30
12. STATISTICAL CONSIDERATIONS.............................................................................. 30
12.1 Sample Size and Power Estimation .............................................................................. 30
12.2 Statistical Analyses ....................................................................................................... 31
12.2.1 Interim Analysis .................................................................................................... 31
12.2.2 Interim Safety Analysis......................................................................................... 31
12.2.3 Primary Efficacy Analysis .................................................................................... 31
12.2.4 Secondary Analyses .............................................................................................. 31
12.2.5 Safety Outcome Analyses ..................................................................................... 32
13. ADVERSE EVENTS ........................................................................................................ 32
13.1 Adverse Events ............................................................................................................. 32
13.2 Clinically Important Adverse Events ............................................................................ 32
13.3 Adverse Event Exceptions ............................................................................................ 32
13.4 Obligation of Investigator ............................................................................................. 32
13.5 Reporting Procedures .................................................................................................... 33
14. INVESTIGATIONAL DRUG DESCRIPTION ............................................................... 33
15. REGULATORY AND ETHICAL OBLIGATIONS ........................................................ 33
15.1 Informed Consent.......................................................................................................... 33
15.2 Institutional Review Board (IRB) ................................................................................. 34
15.2.1 Initial Review and Approval ................................................................................. 34
15.2.2 Amendments ......................................................................................................... 34
15.2.3 Annual Renewal .................................................................................................... 35
16. STUDY ORGANIZATION .............................................................................................. 35
16.1 Executive Committee .................................................................................................... 35
16.2 Data and Safety Monitoring Board ............................................................................... 35
16.3 Ancillary Studies ........................................................................................................... 36
16.3.1 Optional Insights on Selected Procoagulation Markers and Outcomes in Stroke
Trial (I-SPOT) Ancillary Study ............................................................................................ 36
17. References ......................................................................................................................... 37

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-4-
ABBREVIATIONS
Abbreviation Description
ADA American Diabetes Association
AE Adverse Event
AHA American Heart Association
ALIAS High-Dose Albumin Therapy for Neuroprotection in Acute
Ischemic Stroke
ASAP Acute Stroke Accurate Prediction
ATLANTIS Alteplase Thrombolysis for Acute Noninterventional Therapy in
Ischemic Stroke
BG baseline glucose
BI Barthel Index
CCC Clinical Coordinating Center
CI confidence interval
CRF case report form
D/C discharge
DSMB Data and Safety Monitoring Board
DVT deep venous thrombosis
EC Executive Committee
ED Emergency Department
FDA Food and Drug Administration
GIST-UK Glucose Insulin in Stroke Trial – United Kingdom
GRASP Glucose Regulation in Acute Stroke Patients
IA intra-arterial
ICH intracranial hemorrhage
IND Investigational New Drug
IRB Institutional Review Board
IV intravenous
LAR Legally Authorized Representative
MSM Medical Safety Monitor
MOP Manual of Procedures
NETT Neurological Emergency Treatment Trials
NIH National Institutes of Health
NIHSS NIH Stroke Scale
NINDS National Institute of Neurological Disorders and Stroke
mRS modified Rankin Scale
PI Principal Investigator
PO by mouth
q every
RX treatment
SAP Statistical Analysis Plan
SAE Serious Adverse Event
SDMC Statistical Data Management Center
SHINE Stroke Hyperglycemia Insulin Network Effort
SQ subcutaneous

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-5-
ABBREVIATIONS
Abbreviation Description
SSQOL Stroke Specific Quality Of Life
THIS Treatment of Hyperglycemia in Ischemic Stroke
TOAST Trial of ORG 10172 in Acute Stroke Treatment
tPA Tissue Plasminogen Activator

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-6-
1. SUMMARY
There is an increasing need for improved treatments for stroke patients as stroke is the
most common cause of serious long term adult disability and the third most common
cause of death in the United States.1 Hyperglycemia is seen in approximately 40% of
acute ischemic stroke patients2,3
and has been associated with worse clinical outcomes.4,5
Intravenous (IV) insulin therapy with tight glucose control has been found to improve
clinical outcomes in some non-stroke acute illness trials.6,7
Current stroke guidelines
emphasize the need for definitive clinical trials to determine best practice for managing
hyperglycemia in acute stroke patients.8 A clear determination of the risk and benefit of
glucose control with IV insulin would have a dramatic impact on acute ischemic stroke
patient therapy.
This Phase III multicenter, randomized, controlled trial will determine the efficacy and
provide further safety data on glycemic control in stroke patients. The hyperglycemic
acute ischemic stroke patients that meet all eligibility criteria will receive up to 72 hours
of hyperglycemia control with IV insulin therapy or control therapy with subcutaneous
(SQ) insulin. Treatment will be given within 12 hours of symptom onset and is
recommended, but not required, to begin within 3 hours of arrival to the emergency
department (ED). The primary efficacy outcome to be assessed at 90 days will be the
severity adjusted difference in favorable outcome between the groups. Favorable
outcome will be defined by a previously described baseline severity adjusted
dichotomized modified Rankin scale (mRS).9-11
Outcome success will depend on the
severity of the initial stroke (per NIH Stroke Scale Score (NIHSS)). The primary safety
outcome will be the hypoglycemic event rate. Secondary outcomes will assess additional
neurological and functional status using stroke severity, functional and quality scales12-14
as well as glucose control success and adherence to the protocol dosing recommendations
of the computerized decision support tool. This trial launches a highly collaborative
model for stroke research providing a foundation for maximally generalizable results
based on performance at academic, community, urban, rural, large and small hospitals
throughout North America to produce a highly representative national population sample.
A validated computer decision support tool will guide delivery of IV insulin therapy. A
baseline severity-adjusted dichotomized outcome analysis (responder analysis)9 will
adjust for variability of individual patient characteristics to allow detection of the true
clinically relevant treatment effect. In this setting an absolute 7% treatment effect is
recognized as a threshold at or above which a profound effect on a large stroke
population would be realized.
2. OBJECTIVES
2.1 Specific Aim 1
To determine the efficacy of tight glucose control to a target range of 80-130
mg/dL with IV insulin infusion in hyperglycemic acute ischemic stroke patients
within 12 hours of symptom onset as measured by mRS at 90 days after stroke.

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-7-
o Hypothesis 1: Tight glucose control (target 80-130 mg/dL) with IV insulin
infusion therapy using a validated computerized decision support tool, will
increase the severity adjusted 90 day favorable outcome on the mRS by an
absolute 7% or more, as compared to the control group.
2.2 Specific Aim 2
To determine the safety of tight glucose control with IV insulin infusion in
hyperglycemic acute ischemic stroke patients treated for up to 72 hrs.
o Hypothesis 1: Tight glucose control with IV insulin infusion therapy using a
decision support tool is safe as determined by a severe hypoglycemia (<40
mg/dL) rate that does not exceed that of the control group by more than 4%.
3. BACKGROUND AND RATIONALE
3.1 Background
Ischemic Stroke represents a large burden to society with only a single proven
acute treatment. IV Thrombolytic therapy has proven applicable to only a small
minority of stroke patients who present within a narrow time window. Even for
those patients receiving this therapy, the chances of recovery to normal or near
normal are only increased by approximately 30%. Sorely needed are additional
new stroke treatments applicable to a larger universe of early acute stroke patients
which are safe and efficacious when delivered over an expanded time window.
Hyperglycemia, seen in large numbers of acute stroke patients and well associated
with poorer clinical outcomes, provides a compelling target for intervention.
3.2 Significance
Stroke remains the third leading cause of death and leading cause of adult
disability in the U.S. The total cost of stroke for 2009 is estimated at nearly $69
billion.1 Stroke occurs in nearly 800,000 people annually in the United States
1
with approximately 85% (637,500) of them being ischemic and approximately
40% (250,000) of the ischemic strokes being hyperglycemic (≥130 mg/dL) at
presentation to the hospital.2,3
An efficacious and effective treatment for
hyperglycemia in this population would have an enormous impact. Preliminary
data have demonstrated the safety and feasibility of insulin infusion therapy in
acute ischemic stroke patients, but efficacy remains unknown.15-21
Hyperglycemia
is associated with worse outcomes and yet hypoglycemia is bad for ischemic
brain. Treatment for hyperglycemia with a very low hypoglycemia rate is highly
likely to be beneficial. The stroke community has struggled with uncertainty
regarding how hyperglycemic stroke patients should be managed and the most
recent American Heart Association (AHA) guidelines suggest that the
management uncertainty in acute stroke patients will require clarification by
clinical trials.8 Health care providers are currently making clinical decisions
regarding hyperglycemic management without adequate data. The guidelines
classify the current evidence as C (consensus of experts) only. The information
gained from this efficacy trial will guide clinical practice and provide answers
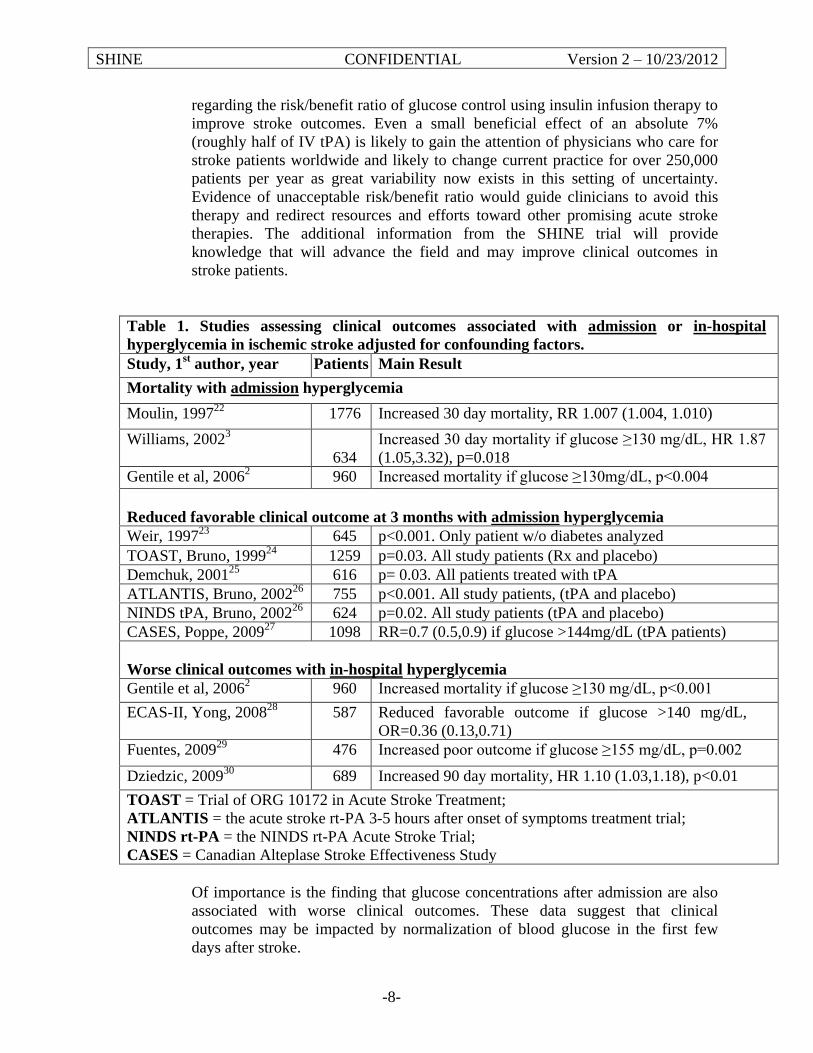
SHINE CONFIDENTIAL Version 2 – 10/23/2012
-8-
regarding the risk/benefit ratio of glucose control using insulin infusion therapy to
improve stroke outcomes. Even a small beneficial effect of an absolute 7%
(roughly half of IV tPA) is likely to gain the attention of physicians who care for
stroke patients worldwide and likely to change current practice for over 250,000
patients per year as great variability now exists in this setting of uncertainty.
Evidence of unacceptable risk/benefit ratio would guide clinicians to avoid this
therapy and redirect resources and efforts toward other promising acute stroke
therapies. The additional information from the SHINE trial will provide
knowledge that will advance the field and may improve clinical outcomes in
stroke patients.
Table 1. Studies assessing clinical outcomes associated with admission or in-hospital
hyperglycemia in ischemic stroke adjusted for confounding factors.
Study, 1st author, year Patients Main Result
Mortality with admission hyperglycemia
Moulin, 199722
1776 Increased 30 day mortality, RR 1.007 (1.004, 1.010)
Williams, 20023
634
Increased 30 day mortality if glucose ≥130 mg/dL, HR 1.87
(1.05,3.32), p=0.018
Gentile et al, 20062 960 Increased mortality if glucose ≥130mg/dL, p<0.004
Reduced favorable clinical outcome at 3 months with admission hyperglycemia
Weir, 199723
645 p<0.001. Only patient w/o diabetes analyzed
TOAST, Bruno, 199924
1259 p=0.03. All study patients (Rx and placebo)
Demchuk, 200125
616 p= 0.03. All patients treated with tPA
ATLANTIS, Bruno, 200226
755 p<0.001. All study patients, (tPA and placebo)
NINDS tPA, Bruno, 200226
624 p=0.02. All study patients (tPA and placebo)
CASES, Poppe, 200927
1098 RR=0.7 (0.5,0.9) if glucose >144mg/dL (tPA patients)
Worse clinical outcomes with in-hospital hyperglycemia
Gentile et al, 20062 960 Increased mortality if glucose ≥130 mg/dL, p<0.001
ECAS-II, Yong, 200828
587 Reduced favorable outcome if glucose >140 mg/dL,
OR=0.36 (0.13,0.71)
Fuentes, 200929
476 Increased poor outcome if glucose ≥155 mg/dL, p=0.002
Dziedzic, 200930
689 Increased 90 day mortality, HR 1.10 (1.03,1.18), p<0.01
TOAST = Trial of ORG 10172 in Acute Stroke Treatment;
ATLANTIS = the acute stroke rt-PA 3-5 hours after onset of symptoms treatment trial;
NINDS rt-PA = the NINDS rt-PA Acute Stroke Trial;
CASES = Canadian Alteplase Stroke Effectiveness Study
Of importance is the finding that glucose concentrations after admission are also
associated with worse clinical outcomes. These data suggest that clinical
outcomes may be impacted by normalization of blood glucose in the first few
days after stroke.
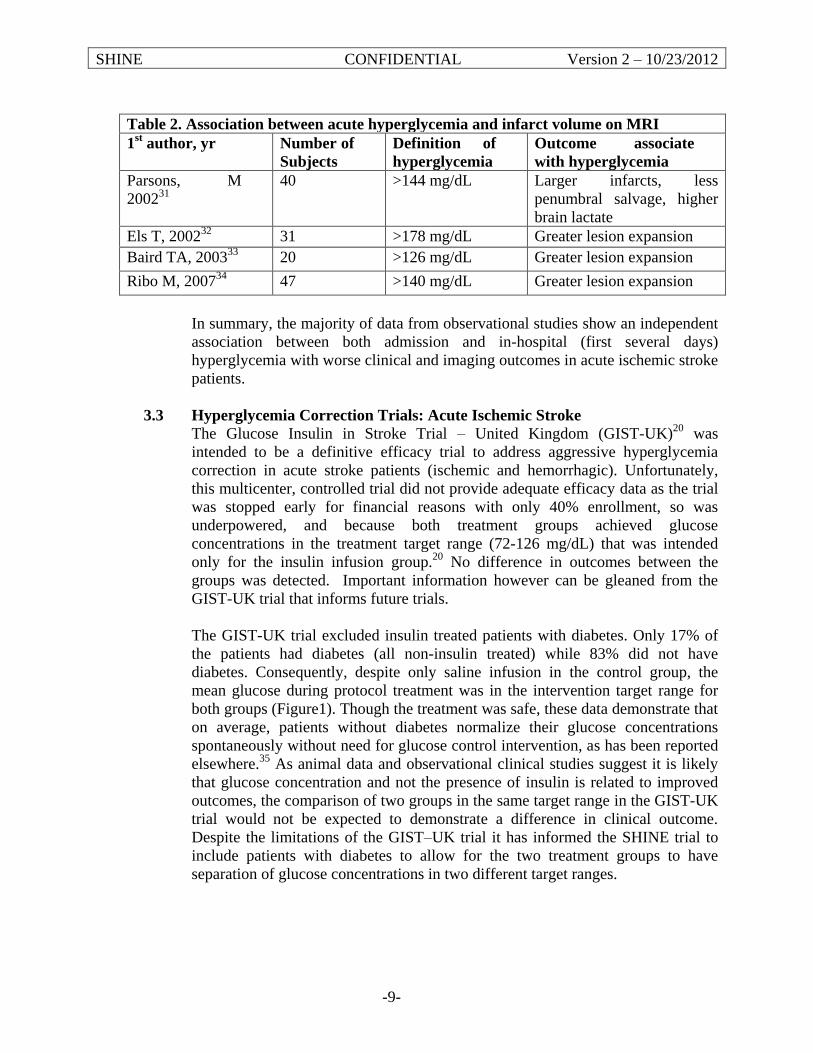
SHINE CONFIDENTIAL Version 2 – 10/23/2012
-9-
Table 2. Association between acute hyperglycemia and infarct volume on MRI
1st author, yr Number of
Subjects
Definition of
hyperglycemia
Outcome associate
with hyperglycemia
Parsons, M
200231
40 >144 mg/dL Larger infarcts, less
penumbral salvage, higher
brain lactate
Els T, 200232
31 >178 mg/dL Greater lesion expansion
Baird TA, 200333
20 >126 mg/dL Greater lesion expansion
Ribo M, 200734
47 >140 mg/dL Greater lesion expansion
In summary, the majority of data from observational studies show an independent
association between both admission and in-hospital (first several days)
hyperglycemia with worse clinical and imaging outcomes in acute ischemic stroke
patients.
3.3 Hyperglycemia Correction Trials: Acute Ischemic Stroke
The Glucose Insulin in Stroke Trial – United Kingdom (GIST-UK)20
was
intended to be a definitive efficacy trial to address aggressive hyperglycemia
correction in acute stroke patients (ischemic and hemorrhagic). Unfortunately,
this multicenter, controlled trial did not provide adequate efficacy data as the trial
was stopped early for financial reasons with only 40% enrollment, so was
underpowered, and because both treatment groups achieved glucose
concentrations in the treatment target range (72-126 mg/dL) that was intended
only for the insulin infusion group.20
No difference in outcomes between the
groups was detected. Important information however can be gleaned from the
GIST-UK trial that informs future trials.
The GIST-UK trial excluded insulin treated patients with diabetes. Only 17% of
the patients had diabetes (all non-insulin treated) while 83% did not have
diabetes. Consequently, despite only saline infusion in the control group, the
mean glucose during protocol treatment was in the intervention target range for
both groups (Figure1). Though the treatment was safe, these data demonstrate that
on average, patients without diabetes normalize their glucose concentrations
spontaneously without need for glucose control intervention, as has been reported
elsewhere.35
As animal data and observational clinical studies suggest it is likely
that glucose concentration and not the presence of insulin is related to improved
outcomes, the comparison of two groups in the same target range in the GIST-UK
trial would not be expected to demonstrate a difference in clinical outcome.
Despite the limitations of the GIST–UK trial it has informed the SHINE trial to
include patients with diabetes to allow for the two treatment groups to have
separation of glucose concentrations in two different target ranges.
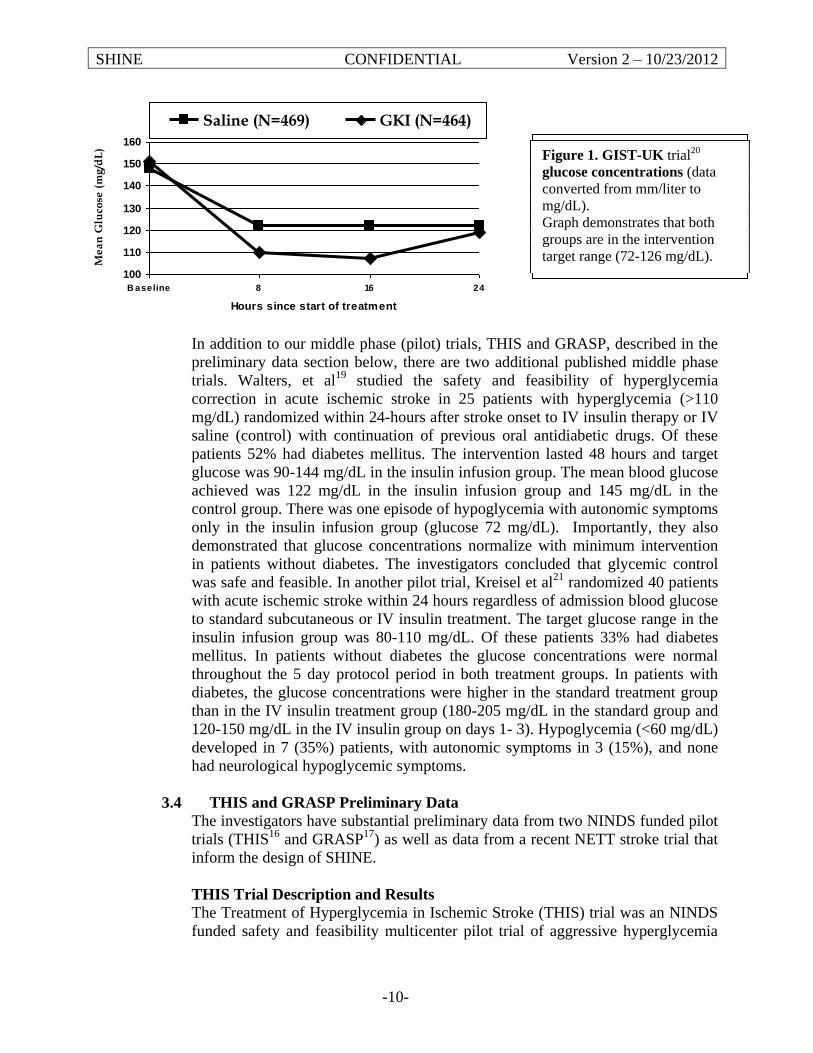
SHINE CONFIDENTIAL Version 2 – 10/23/2012
-10-
In addition to our middle phase (pilot) trials, THIS and GRASP, described in the
preliminary data section below, there are two additional published middle phase
trials. Walters, et al19
studied the safety and feasibility of hyperglycemia
correction in acute ischemic stroke in 25 patients with hyperglycemia (>110
mg/dL) randomized within 24-hours after stroke onset to IV insulin therapy or IV
saline (control) with continuation of previous oral antidiabetic drugs. Of these
patients 52% had diabetes mellitus. The intervention lasted 48 hours and target
glucose was 90-144 mg/dL in the insulin infusion group. The mean blood glucose
achieved was 122 mg/dL in the insulin infusion group and 145 mg/dL in the
control group. There was one episode of hypoglycemia with autonomic symptoms
only in the insulin infusion group (glucose 72 mg/dL). Importantly, they also
demonstrated that glucose concentrations normalize with minimum intervention
in patients without diabetes. The investigators concluded that glycemic control
was safe and feasible. In another pilot trial, Kreisel et al21
randomized 40 patients
with acute ischemic stroke within 24 hours regardless of admission blood glucose
to standard subcutaneous or IV insulin treatment. The target glucose range in the
insulin infusion group was 80-110 mg/dL. Of these patients 33% had diabetes
mellitus. In patients without diabetes the glucose concentrations were normal
throughout the 5 day protocol period in both treatment groups. In patients with
diabetes, the glucose concentrations were higher in the standard treatment group
than in the IV insulin treatment group (180-205 mg/dL in the standard group and
120-150 mg/dL in the IV insulin group on days 1- 3). Hypoglycemia (<60 mg/dL)
developed in 7 (35%) patients, with autonomic symptoms in 3 (15%), and none
had neurological hypoglycemic symptoms.
3.4 THIS and GRASP Preliminary Data
The investigators have substantial preliminary data from two NINDS funded pilot
trials (THIS16
and GRASP17
) as well as data from a recent NETT stroke trial that
inform the design of SHINE.
THIS Trial Description and Results
The Treatment of Hyperglycemia in Ischemic Stroke (THIS) trial was an NINDS
funded safety and feasibility multicenter pilot trial of aggressive hyperglycemia
100
110
120
130
140
150
160
B aseline 8 16 24
Hours since start of treatment
Me
an
Glu
cose
(m
g/d
L)
Saline (N=469) GKI (N=464)
Figure 1. GIST-UK trial20
glucose concentrations (data
converted from mm/liter to
mg/dL).
Graph demonstrates that both
groups are in the intervention
target range (72-126 mg/dL).

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-11-
reduction in acute ischemic stroke.16
Patients (N=46) were randomized within 12
hours of stroke onset to continuous IV insulin infusion (N=31) (target 80-130
mg/dL) or sliding scale SQ insulin injections up to four times daily if needed
(control N=15). The baseline glucose threshold for inclusion was 150 mg/dL and
91% were diabetic.
The main findings in THIS were safety and feasibility. Elevated glucose levels
were lowered into target range with mean glucose nearly 60 mg/dL (31%) lower
in the IV insulin group than the SQ insulin group (133 vs. 190 mg/dL) within four
hours of starting therapy.
THIS Safety Results
There were no serious adverse events related to the IV insulin intervention.
Overall, episodic hypoglycemia (<60 mg/dL) occurred in 11 patients (11/31,
35%) in the IV insulin group. Seven of these hypoglycemic patients were
asymptomatic. There was only one patient who had transient neurologic
symptomatic hypoglycemia. No patient had severe (<40 mg/dL) or prolonged
hypoglycemia. Ten patients (22%) were treated with standard IV tPA within three
hours of stroke onset and no treatment related adverse events were seen in this
subgroup. Glucose concentrations were consistently and significantly lower
throughout the trial in the IV insulin group. The adjusted mean glucose difference
between the groups during treatment was 66 mg/dL. Non-diabetic patients had
glucose levels return to normal without aggressive treatment.
THIS Clinical Outcomes
There were no statistically significant differences in clinical outcomes between
the two treatment groups. The primary exploratory analysis compared mRS ≤2 at
3 months. Favorable outcome was more common in the aggressive treatment
group, by an absolute 5%. Secondary favorable clinical outcomes were also more
favorable in the IV insulin group. The largest difference was in the NIH Stroke
Scale ≤2, with p=0.09 in favor of the IV insulin after adjusting for baseline
inequalities. No patient was lost to follow up.
GRASP Trial Description and Results
The GRASP trial (N=74) was a multicenter randomized safety, feasibility, “dose
finding” trial that included 3 treatment groups (usual, loose and tight control).17
Patients were enrolled within 24 hours of stroke and treated for up to 5 days.
Patients with diabetes in the trial had a mean glucose of 185 mg/dL and patients
without diabetes had a mean glucose of 149 mg/dL at randomization. These data
support a lesser need for insulin therapy in stroke patients without diabetes.
GRASP Protocol Insulin Treatments
1. Insulin Infusion Protocol (eProtocol-insulin)
eProtocol-insulin is a computerized decision support tool used in the GRASP trial.
Both loose and tight control groups received continuous insulin infusion based on
eProtocol-insulin. Overall, there was a 97% adherence to eProtocol

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-12-
recommendations suggesting that nurses agreed with the computer
recommendation and followed the study protocol.
2. Control Group
The control group was a community control with the intent to observe standard
care and to inform the control group in future trials. A total of 88% of control
patients were treated with SQ insulin.
GRASP Safety
Total hypoglycemia rates (<55 mg/dL) were 4% in the control group, 4% in the
loose control group, and 30% in the tight control group. One patient in the loose
control group had transient symptomatic hypoglycemia with a glucose of 50 and
52 mg/dL that was rapidly corrected. A total of 26 patients (36% of enrolled
subjects) received standard IV thrombolysis within 3 hours. The Data Safety
Monitoring Board (DSMB) and the safety monitor had no safety concerns in this
group. There were 10 (14%) deaths in this trial. No patient had severe or
prolonged hypoglycemia.
GRASP Glucose Concentrations and Feasibility
The median glucose during treatment in the control group was 151 mg/dL, in the
loose group was 151 mg/dL, and in the tight group was 111 mg/dL. Target
glucose levels were achieved. They also demonstrate that IV insulin is not
necessary for the loose target as the same glucose control can be achieved with
SQ insulin.
GRASP Recruitment/Enrollment
Recruitment and enrollment were highly successful with completion of
enrollment of 74 subjects within 18 months and ahead of schedule. The
recruitment specialist was critically important to the recruitment success and
because of her expertise and commitment she will oversee recruitment in the
SHINE trial.
GRASP/THIS Target Glucose Concentration (“dose”) Selection
Both GRASP IV insulin doses are safe and feasible, but the tight control approach
is appropriate to study in the phase III trial. GRASP data, combined with THIS
trial data (target 80-130 mg/dL), the AHA guidelines (treat glucose over ~180
mg/dL), the ADA guidelines (treat over180mg/dL) and the literature reviewed
above support an intervention group target glucose of 80–130 mg/dL and a
control group treated with SQ insulin for glucose >180 mg/dL. Separation will
be achieved as diabetics will be the study population and our preliminary data
demonstrate we can adequately separate levels.
GRASP/THIS Time Window
GRASP had a 24 hour window for enrollment, but 58% of patients were enrolled
<12 hours from stroke onset and 45% <9 hours. These data with the THIS trial
demonstrate that patients can be enrolled within 12 hours to an acute IV insulin
trial. A 12 hour window will allow tPA therapy for eligible patients, allow “wake

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-13-
up stroke” enrollment and is supported by the literature. Data suggest that both
inflammation and excitotoxicity likely play a major role in the injury
pathophysiology that tight glucose control is interrupting.36
TNF alpha may be a
major component of the injury pathway and is expressed by 4 hours but doesn’t
peak until 12 hours.37
A 12 hour window therefore may have greater ability to
capture patients that will benefit from this therapy. The rapid sequence of
evaluation and study enrollment for acute stroke patients will assure that patients
will be enrolled quickly and many will likely be early in the time window (as
shown in GRASP). This rapid enrollment will be maximized in our collaboration
with the NETT and will allow a secondary analysis of time to treatment as we are
likely to have a broad distribution of enrollment times.
GRASP/THIS Treatment Period
The GRASP trial treated patients for 5 days or until discharge with the intent of
establishing the time period for inpatient therapy. The GRASP data clearly
demonstrate that 5 days was not feasible and that 4 days was too long for most
patients due to current acute stroke length of stay. Three days was the longest
feasible treatment window and was successful in both trials. Additionally, human
data suggest better outcomes in patients with glucose control for first 3 days
compared to those with hyperglycemia.38
Pathophysiologic mechanisms that may
be involved include protein synthesis dependent protection mechanisms that
continue for up to 3 days.39
Additionally, inflammatory mechanisms (suspected
to be involved) are known to continue for several days. Animal model data
suggest that 3 days of anti-inflammatory treatment can reduce infarct volume.40
These mechanistic data plus our pilot feasibility data strongly suggest that a 3 day
treatment period is ideal. Mild stroke patients will be discharged as clinically
indicated which may be <3 days.
GRASP/THIS Meal Insulin
The GRASP and THIS trials used a simple algorithm for estimating carbohydrate
consumption and providing meal insulin. One unit/15 grams carbohydrate
consumed will be used in SHINE.
GRASP Exploratory Efficacy Outcomes (N=73 patients with 3 month
outcomes)
None of the efficacy outcome relationships reached statistical significance, but
have been used to estimate the control group outcome rates and predict the
population most likely to benefit from treatment. The primary exploratory
analysis analyzed modified Rankin score ≤1 at 3 months and the tight group had
the most favorable outcomes. Three months outcomes were captured on 99% of
patients.
Responder Analysis
Based on the well-known statistical principle of loss of information with the use
of dichotomized outcomes and the recent literature suggesting that more
innovative and powerful analyses may be necessary for “neuroprotection” trials,41-
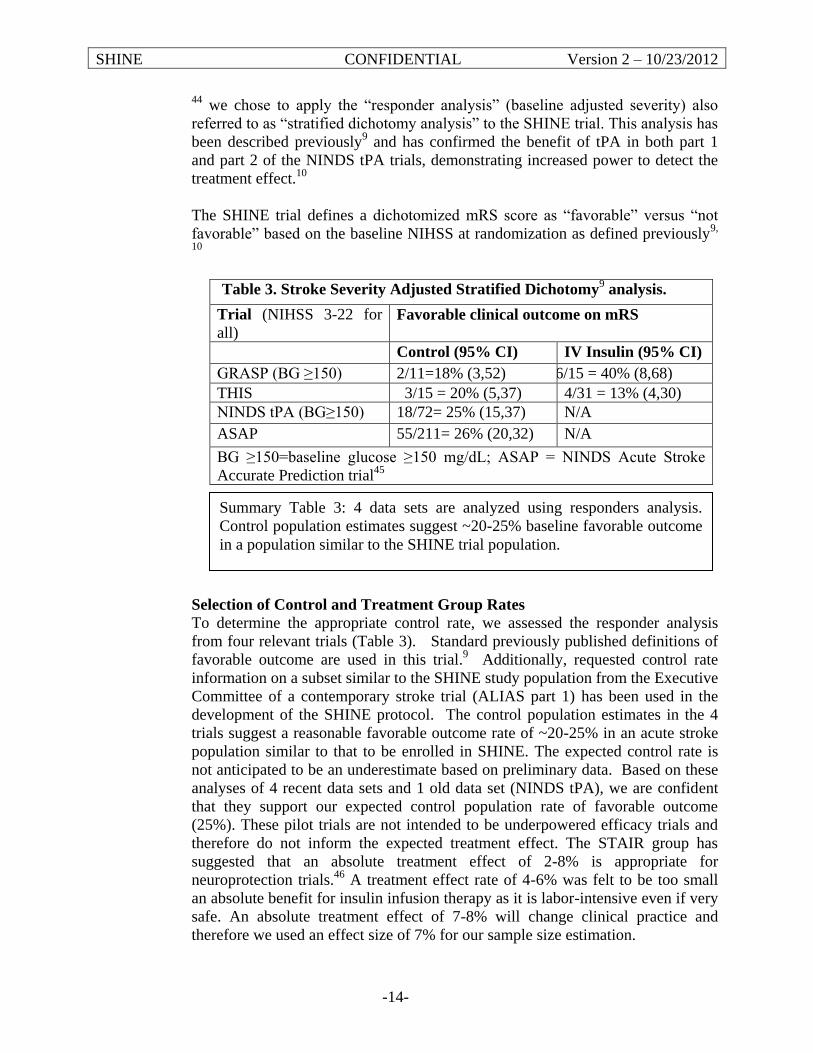
SHINE CONFIDENTIAL Version 2 – 10/23/2012
-14-
44 we chose to apply the “responder analysis” (baseline adjusted severity) also
referred to as “stratified dichotomy analysis” to the SHINE trial. This analysis has
been described previously9 and has confirmed the benefit of tPA in both part 1
and part 2 of the NINDS tPA trials, demonstrating increased power to detect the
treatment effect.10
The SHINE trial defines a dichotomized mRS score as “favorable” versus “not
favorable” based on the baseline NIHSS at randomization as defined previously9,
10
Table 3. Stroke Severity Adjusted Stratified Dichotomy9 analysis.
Trial (NIHSS 3-22 for
all)
Favorable clinical outcome on mRS
Control (95% CI) IV Insulin (95% CI)
GRASP (BG ≥150) 2/11=18% (3,52) 6/15 = 40% (8,68)
THIS 3/15 = 20% (5,37) 4/31 = 13% (4,30)
NINDS tPA (BG≥150) 18/72= 25% (15,37) N/A
ASAP 55/211= 26% (20,32) N/A
BG ≥150=baseline glucose ≥150 mg/dL; ASAP = NINDS Acute Stroke
Accurate Prediction trial45
Selection of Control and Treatment Group Rates
To determine the appropriate control rate, we assessed the responder analysis
from four relevant trials (Table 3). Standard previously published definitions of
favorable outcome are used in this trial.9 Additionally, requested control rate
information on a subset similar to the SHINE study population from the Executive
Committee of a contemporary stroke trial (ALIAS part 1) has been used in the
development of the SHINE protocol. The control population estimates in the 4
trials suggest a reasonable favorable outcome rate of ~20-25% in an acute stroke
population similar to that to be enrolled in SHINE. The expected control rate is
not anticipated to be an underestimate based on preliminary data. Based on these
analyses of 4 recent data sets and 1 old data set (NINDS tPA), we are confident
that they support our expected control population rate of favorable outcome
(25%). These pilot trials are not intended to be underpowered efficacy trials and
therefore do not inform the expected treatment effect. The STAIR group has
suggested that an absolute treatment effect of 2-8% is appropriate for
neuroprotection trials.46
A treatment effect rate of 4-6% was felt to be too small
an absolute benefit for insulin infusion therapy as it is labor-intensive even if very
safe. An absolute treatment effect of 7-8% will change clinical practice and
therefore we used an effect size of 7% for our sample size estimation.
Summary Table 3: 4 data sets are analyzed using responders analysis.
Control population estimates suggest ~20-25% baseline favorable outcome
in a population similar to the SHINE trial population.

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-15-
Decision Support Tool
The decision support tool, GlucoStabilizer, is an FDA cleared device that has
been utilized in over 50,000 patients. The tool has demonstrated extremely low
rates of severe hypoglycemia (<40 mg/dL). The severe hypoglycemia rate during
use is 1.67% of patients and 0.07% of total glucose readings.
Decision support tools have been used in previous studies, and the insulin
infusion patients in the GRASP Trial used eProtocol-insulin, a similar decision
support tool. Adherence to the recommendations using decision support tool
devices has been reported to be 91-98%,47, 48
and it was 97% in the GRASP trial
using eProtocol17
.
Summary Preliminary Results
The GRASP and THIS trials have shown that insulin infusion therapy for tight
glucose control is safe and is feasible in hyperglycemic acute ischemic stroke
patients. Additionally, these trials have identified the appropriate glucose target,
timing, treatment period, patient population for enrollment, expected loss to
follow up rate, expected recruitment rate, and expected control group outcome
rates. These pilot data ideally inform the design of the SHINE Trial.
3.5 Rationale
The goal in the treatment group is glucose levels within the target range of 80-130
mg/dL. This requires that the target range be rapidly established and maintained
with minimal variability for up to 72 hours. IV insulin infusion due to its rapid
onset and offset of action and its superior degree of frequent titratability provides
the ideal intervention to achieve and tightly maintain the target range goal across
the study period. The use of computerized decision support tools is now part of
standard care.
4. STUDY PLAN
4.1 Study Design
SHINE is a multicenter, randomized, controlled clinical trial of approximately
1400 patients to be conducted at 17 Neurological Emergencies Treatment Trials
(NETT) hubs and their spoke hospitals as well as approximately 10 non-NETT
sites.
4.2 Study Population
The study population will include hyperglycemic acute ischemic stroke patients of
either gender who are 18 years of age or older. They will have a history of type 2
diabetes and hyperglycemia or in the absence of diabetes, must have a baseline
glucose levels ≥150 mg/dL. To be eligible, treatment must begin within 12 hours
of stroke symptom onset and is recommended, but not required, to begin within 3
hours of arrival to the Emergency Department. Participation is limited to those
with baseline NIHSS scores 3-22.

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-16-
4.3 Study Therapy (insulin versus saline)
Insulin is indicated for the treatment of hyperglycemic conditions and the
maintenance of euglycemia in the setting of diabetes mellitus. It is also known to
be effective in normalizing blood glucose when hyperglycemia results from other
conditions. It is frequently used for both the inpatient and outpatient management
of patients with diabetes. Insulin given by IV or subcutaneous (SQ) route
stimulates carbohydrate metabolism and facilitates transfer of glucose into cardiac
muscle, skeletal muscle and adipose tissue, and glucose is converted to glycogen.
In addition, lipogenesis is stimulated with inhibition of lipolysis from adipose
cells; protein synthesis is stimulated. An Investigational New Drug (IND)
approval is not required for this protocol.
Major adverse reactions
Common – Skin site reactions (SQ), Lypodystrophy, Mild hypoglycemia
Serious – Moderate to Severe Hypoglycemia
The SHINE trial will use a validated computerized electronic decision support
tool for the IV insulin group only. IV insulin or saline infusions will be adjusted
by the nurses to maintain the assigned glucose target. The nurses will consider
the recommendation made by the computer for the IV insulin group and will
follow the protocol for the saline adjustments.
4.4 Study Decision Support Tool for the Intervention Group
The decision support system, GlucoStabilizer, has been developed by Medical
Automation Systems an Alere Company and cleared for use by the FDA. The
system provides an alert to the nurse for the next required glucose check. Other
decision support tool rules moderate the insulin dose recommendation depending
upon previous glucose readings or steep drops in glucose values. Nurses will have
the option of accepting or declining the recommended insulin rates. Nurses are
encouraged to use clinical judgment and reject any recommendation that seems
inappropriate but will be required to stop the insulin drip when notified to do so
by the decision support tool.
For blood glucose <80 mg/dL, the decision support tool will instruct the treating
team to discontinue the insulin infusion, and investigators will be required to stop
the insulin infusion at that time. Sites will be monitored for rates of adherence to
the decision support tool recommendation for those patients receiving IV insulin
infusion. All glucose measurements, insulin dosing, times of glucose checks and
acceptance of recommendations are captured in the electronic system.
4.5 Study Sites
Study sites will include The Neurological Emergency Treatment Trials (NETT)
sites and non NETT sites. Details of study sites are listed in the Manual of
Procedures (MOP).

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-17-
4.6 Estimated Study and Enrollment Duration
The project period will begin in late 2011. Enrollment is expected to begin in
2012. Enrollment is anticipated to take 3.5 – 4 years and follow up is 3 months.
Final data analysis and close out activities will occur after completion of
enrollment and patient follow up.
5. ELIGIBILITY CRITERIA
5.1 Inclusion Criteria
(1) Age 18 years or older
(2) Clinical diagnosis of ischemic stroke defined as acute neurological deficit
occurring in one or more cerebral vascular territories. Neuroimaging must
be done to exclude intracranial hemorrhage (ICH).
(3) Protocol treatment must begin within 12 hours after stroke symptom onset
and is recommended, but not required, to begin within 3 hours after
hospital arrival. If time of symptom onset is unclear or patient is
awakening with stroke symptoms, the time of onset will be the time the
patient was last known to be normal.
(4) Known history of type 2 diabetes mellitus and glucose >110 mg/dL OR
admission glucose ≥150 mg/dL in those w/o known diabetes mellitus
(5) Baseline NIHSS score of 3-22
(6) Pre-stroke modified Rankin Scale score = 0 for patients with an NIHSS
score of 3-7. Pre-stroke modified Rankin Scale score = 0 or 1 for patients
with an NIHSS score of 8-22.
(7) Able to provide a valid informed consent to be in the study (self or their
authorized legally accepted representative). The approved consent form
must be signed and dated in accordance with federal and institutional
guidelines.
5.2 Exclusion Criteria
(1) Known history of type1 diabetes mellitus
(2) Substantial pre-existing neurological or psychiatric illness that would
confound the neurological assessment or other outcome assessment
(3) Having received experimental therapy for the enrollment stroke. IV tPA
(up to 4.5 hrs) or IA tPA are allowed as are IA therapies including use of

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-18-
FDA cleared devices. Non FDA cleared devices are considered
experimental and are excluded.
(4) Known to be pregnant or breast-feeding at the time of study entry
(5) Other serious conditions that make the patient unlikely to survive 90 days
(6) Inability to follow the protocol or return for the 90 day follow up
(7) Renal dialysis (including hemo or peritoneal dialysis)
Justification for Eligibility Criteria
Only ≥18 year old ischemic stroke subjects will be included since ischemic
strokes in the pediatric population are substantially different from adult strokes.
The population under age 18 is excluded to avoid confounding the results.
The 12-hour eligibility requirement was chosen as this is almost universally
before the development of maximum edema in acute ischemic stroke patients, but
is a wide enough time window to be inclusive of most patients allowing
generalizable results and is supported by the preliminary data. Treatment within 3
hours of arrival to the Emergency Department will assure the avoidance of
treatment delays in hopes of maximizing treatment effect as suggested by much of
the animal and human data in acute ischemic stroke. This will also allow the
patients to be treated with standard IV tPA as per published eligibility criteria and
then enrolled in the trial. An enrollment blood glucose >110 mg/dL in patients
with type 2 diabetes or ≥150 mg/dL in patients without diabetes was based on
our preliminary data and other data suggesting this group is most likely to benefit.
As demonstrated by GIST-UK20
, THIS16
, GRASP17
, Walters19
and Kriesel21
hyperglycemia frequently resolves spontaneously in most patients without
diabetes. Thus, patients without diabetes mellitus or admission hyperglycemia
will be excluded. Both THIS16
and GRASP17
trials and an observational study45
demonstrated that most patients enrolled with hyperglycemia ≥150 mg/dL
remained hyperglycemic during hospitalization unless they received intravenous
insulin. The vast majority of patients with admission glucose ≥150 mg/dL ha
undiagnosed diabetes or impaired glucose metabolism (insulin resistance) as has
been reported,49-51
thus making them good subjects for this trial. Patients with an
NIHSS score of 3-7 will be required to have a pre-stroke mRS score of 0 to be
eligible. This is intended to allow patients to reach the success criteria defined by
the stratified dichotomy outcome. Patients with an NIHSS of 8-22 will be eligible
if they have a pre-stroke mRS score of 0 or 1. Patients with type I diabetes
mellitus are excluded for safety reasons. Usual care for type I diabetes patients
during acute illness usually includes intravenous insulin infusion accompanied by
dextrose otherwise these patients would be at risk of diabetic ketoacidosis. Since
withholding standard care is unacceptable, these patients are excluded. Patients

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-19-
with type I diabetes will be identified based on medical history. Study
definitions of type 1 and type 2 diabetes are provided in the MOP.
Patients with a neurological or psychiatric illness likely to confound the final
outcome assessment will be excluded since their baseline deficits and outcomes
cannot be accurately obtained. Any patient deemed by the enrolling physician to
have a condition that confounds the enrollment neurological exam will be
excluded. Patients receiving experimental stroke therapies will be excluded due
to uncertain effects of such therapies on outcomes. Standard care IV tPA or IA
tPA according to the AHA/ASA guidelines will be allowed.8 The pilot trials
demonstrated safety in the population treated with IV tPA. The increased risk of
symptomatic hemorrhagic transformation of infarcts observed in IV tPA treated
stroke patients with hyperglycemia25, 26, 52
may be reduced with glucose control.
Intra-arterial (IA) treatments that are standard care, including the use of FDA
cleared devices, will be allowed. FDA cleared devices must be employed
according to their Instructions for Use. Non FDA cleared devices or other
experimental interventions will not be allowed. No clear data are available on the
risk/benefit ratio of these interventions and they could confound the results.
Pregnant women will be excluded since the standard care for this population
often includes IV insulin treatment for hyperglycemia. Patients unlikely to
return at 90 days will be excluded since the primary efficacy outcome is
measured at that time. Renal dialysis patients will be excluded including those
requiring hemodialysis and peritoneal dialysis due to inability to accurately follow
glucose levels and variability in insulin requirements that would put patients at
risk.
5.3 Prohibited Therapy During Study Period
No other diabetes treatment medication besides the assigned protocol treatment
will be allowed during the treatment period because such additional medications
would confound the study. All oral agents that are used for the treatment of
hyperglycemia are unauthorized. The use of non FDA cleared devices for IA
therapy is not allowed. Patients taking PO diets must eat protocol specified diets
as defined in the MOP.
6. SUBJECT RECRUITMENT
6.1 Methods
The methods used for recruitment of subjects in the study will be devoid of any
procedures that may be construed as coercive. The recruitment process will not
involve any restrictions on sociodemographic factors including gender or ethnic
characteristics of the patient population. However, the composition of the study
population will depend on patient sources available to the enrolling sites.
Patients will be recruited by members of the research teams at enrolling sites.
Stroke teams and Emergency Medicine teams involved in the immediate
evaluation and management of acute stroke patients will be trained to recognize

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-20-
potentially eligible candidates and to rapidly refer them for formal screening by
appropriate study team personal. When a patient is found eligible, they or their
Legally Authorize Representative (LAR) as appropriate to the situation will be
approached for discussion of the trial and informed consent.
Recruitment and enrollment will usually occur at the acute portal of entry to the
enrolling site. Most often this will be in the Emergency Department, but this
could also occur in a hospital inpatient unit directly receiving an acute stroke
patient in transfer from another facility as long as arrival is within the specified
enrollment window.
6.2 Screen Failure Logs
Screen failure logs at each site must be entered into the study database listing all
patients with acute ischemic stroke who have presented within 12 hours of onset
and are hyperglycemic but not randomized into the SHINE Trial. Documentation
will include reasons for non-enrollment. Screening data will be reviewed to
assure that full efforts are being made with respect to recruitment and enrollment
and to identify any patterns with regard to ineligibility or reasons for non-
enrollment.
7. SUBJECT ENROLLMENT
7.1 Eligibility Assessment
All patients who present within 12 hours of onset of an acute ischemic stroke will
be screened for eligibility for entry into the study. Rapid referral to the study
team should be made immediately upon recognition of potentially eligible
candidates. Hyperglycemia as defined in the eligibility criteria will be a major
trigger for consideration for enrollment.
As in all trials, the goal is to achieve a high level of compliance with protocol
requirements by assuring during the eligibility assessment that the potential
subject or LAR is fully informed and agrees to the protocol requirements. In
addition, patients with a strong likelihood of non-adherence as described in the
inclusion and exclusion criteria should not knowingly be randomized. Careful
assessment of the patient’s understanding of the trial is required prior to
enrollment.
7.2 Presentation of Informed Consent
Consent will be obtained by either the site Principal Investigator or by a member
of the study team as described in the MOP. The consent should be the IRB-
approved version corresponding to the version of the protocol approved when the
screening was initiated. Informed consent is to be obtained from the patient or
patient’s LAR. For the SHINE study, there is no activity required in the screening
process that is not typically included within the reasonable scope of standard care
evaluation for acute stroke patients; therefore, unless called for by local regulatory

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-21-
request, patients would be approached for consent only after the clinical screening
process had established eligibility.
7.3 Randomization
7.3.1 Central Randomization Procedure
A web-based central randomization system developed by the NETT
Statistical Data Management Center (SDMC) and installed on the
WebDCUTM
SHINE study website.
Subjects will be assigned to one of the treatment groups according to the
randomization scheme developed at the SDMC. Randomization must be
done centrally using the WebDCUTM
for all patients entered in the trial.
In the event that the site cannot access the randomization module of
WebDCUTM
, emergency randomization procedures as defined in the study
MOP should be followed.
8. STUDY PROCEDURE
8.1 Baseline Assessments
8.1.1 Point of Care Finger Stick Blood glucose level
Accu-Chek Comfort Curve Test Strip System is a glucose dehydrogenase
based system of quantitatively measuring the concentration of glucose in
whole blood. A minimum blood sample of 4μL is the required volume on
the strip. The reportable range of glucose concentration is 10 – 600
mg/dL. This is the SHINE study preferred test strip. Strips must be stored
at < 90◦ F or 32◦ C. Strips should be used at temperatures between 57◦ -
104◦ F and less than 85% humidity. Hematocrit must be between 20-55%
for glucose measurements >200mg/dL. Avoid using test strips during
xylose absorption testing, in patients receiving maltose infusion, at
altitudes above 10,150 feet, in situations of decreased peripheral blood
flow to the fingers, galactose level >10 mg/dL, maltose >16 mg/dL,
bilirubin (unconjugated) >20 mg/dL, lipemia >5,000 mg/dL,
acetaminophen levels >8 mg/dL, uric acid >10 mg/dL.
8.1.2 Concomitant medications documentation
Concomitant medications will not be systematically collected on the case
report form; however some relevant medications taken just before and
during the treatment period will be captured in the study database.
Additionally, medications that relate to glucose control will be captured
from just prior to treatment through completion of the study period.
8.1.3 Vital signs
Routine vital signs per AHA guidelines for acute stroke patients will be
followed.

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-22-
8.1.4 NIHSS
The NIHSS will be obtained prior to randomization (baseline) by an
investigator who is NIHSS certified. The NIHSS will be repeated at least
once daily during the treatment period, and it is strongly recommended
that this be completed by a certified investigator. It is required that the
Day 90 NIHSS is scored by an NIHSS certified investigator.
8.1.5 Neuro Worsening Assessment
Any clinically relevant neurological worsening will trigger a clinical
assessment including an NIHSS score. The SHINE study definition of
neurological worsening will be considered any clinical change that is
associated with a ≥4 point increase on the NIHSS score.
8.1.6 Protocol Deviation Documentation
Protocol deviations will be assessed throughout the study period and will
be captured in WebDCUTM
. The details of identifying and reporting
protocol deviations are described in the MOP.
8.2 Treatment Procedures
8.2.1 Decision Support Tool set up
Study laptops capable of internet connection will be supplied to all
participating sites. All appropriate site personnel will be trained in the use
of the tool. The decision support tool and control accounts are housed and
run on a central dedicated secure server and are not resident on the laptop.
The laptop serves as a convenient portal to access the central system. The
FDA cleared computerized decision support tool will simplify and
streamline study procedures both for bedside nurses and local study teams,
and thereby substantially minimize possibilities for protocol deviations.
Details of the decision support tool can be found in the MOP.
8.2.2 Blinding Set Up
o The SHINE trial is single blinded to the subject throughout the study
(the treating team will be unblinded to treatment) and then is double
blinded to include the examiner for the 90 day efficacy outcome
assessment.
o Patients in both groups will receive an IV treatment (insulin or saline)
and subcutaneous treatment (insulin or saline).
8.2.3 Drug Dosage/ Drug Administration
8.2.3.1 Control group protocol
IV Saline with Subcutaneous Insulin Injections Target Blood
Glucose <180 mg/dL
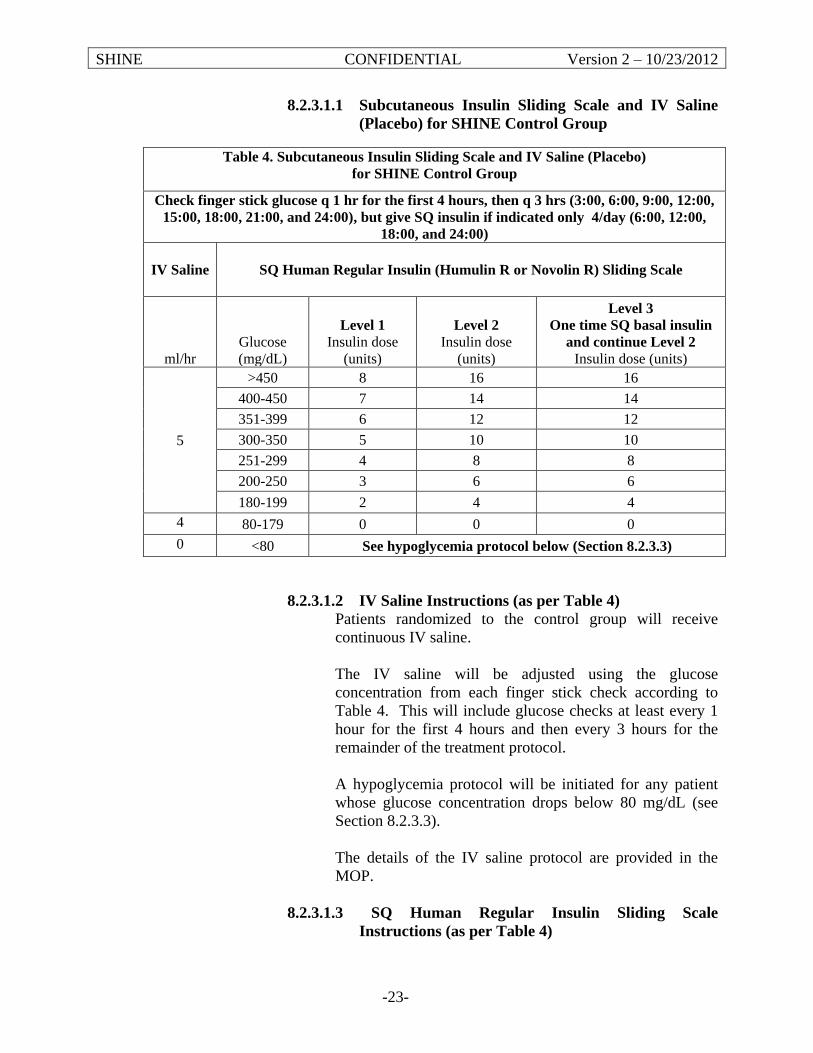
SHINE CONFIDENTIAL Version 2 – 10/23/2012
-23-
8.2.3.1.1 Subcutaneous Insulin Sliding Scale and IV Saline
(Placebo) for SHINE Control Group
8.2.3.1.2 IV Saline Instructions (as per Table 4)
Patients randomized to the control group will receive
continuous IV saline.
The IV saline will be adjusted using the glucose
concentration from each finger stick check according to
Table 4. This will include glucose checks at least every 1
hour for the first 4 hours and then every 3 hours for the
remainder of the treatment protocol.
A hypoglycemia protocol will be initiated for any patient
whose glucose concentration drops below 80 mg/dL (see
Section 8.2.3.3).
The details of the IV saline protocol are provided in the
MOP.
8.2.3.1.3 SQ Human Regular Insulin Sliding Scale
Instructions (as per Table 4)
Table 4. Subcutaneous Insulin Sliding Scale and IV Saline (Placebo)
for SHINE Control Group
Check finger stick glucose q 1 hr for the first 4 hours, then q 3 hrs (3:00, 6:00, 9:00, 12:00,
15:00, 18:00, 21:00, and 24:00), but give SQ insulin if indicated only 4/day (6:00, 12:00,
18:00, and 24:00)
IV Saline
SQ Human Regular Insulin (Humulin R or Novolin R) Sliding Scale
ml/hr
Glucose
(mg/dL)
Level 1 Insulin dose
(units)
Level 2 Insulin dose
(units)
Level 3
One time SQ basal insulin
and continue Level 2 Insulin dose (units)
5
>450 8 16 16
400-450 7 14 14
351-399 6 12 12
300-350 5 10 10
251-299 4 8 8
200-250 3 6 6
180-199 2 4 4
4 80-179 0 0 0
0 <80 See hypoglycemia protocol below (Section 8.2.3.3)

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-24-
Patients randomized to the control group will receive
subcutaneous insulin only up to four times a day as needed
according the sliding scale in Table 4.
The glucose concentration will be checked using finger
stick testing at least every 1 hour for the first 4 hours and
then every 3 hours for the remaining treatment period as
per Table 4. The subcutaneous insulin will only be given at
6:00, 12:00, 18:00 and 24:00 as needed.
All patients will start at Level 1 sliding scale dosing as
shown in Table 4. If glucose concentrations on that dosing
schedule are not adequately controlled (<180 mg/dL),
patients will begin Level 2 on the second day. If they are
still not adequately controlled at Level 2, they will advance
to Level 3 on the third day. Level 3 treatment will include
basal insulin (glargine, Lantus) at a dose of 40% of
previous day’s entire insulin dose and then continuation of
the Level 2 sliding scale dosing as shown in Table 4.
The details of the subcutaneous protocol are provided in the
MOP.
8.2.3.2 Intervention group
IV Insulin with Subcutaneous Meal Insulin or Saline Injections
Target Glucose Concentration 80-130 mg/dL
8.2.3.2.1 IV Insulin Instructions
Patients randomized to the intervention group will receive
continuous IV insulin.
The decision support tool will require finger stick glucose
checks at least every 1 hour for the first 4 hours and then
approximately every 1 to 2 hours for the remainder of the
treatment period.
The dosing will be recommended by the decision support
tool. The required timing of the glucose check will also be
indicated by the decision support tool. The decision
support tool uses information about the specific patient to
make the recommendation of best dosing to keep the
glucose concentration in target.
A hypoglycemia protocol will be initiated for any patient
whose glucose concentration drops below 80 mg/dL (see
Section 8.2.3.3).

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-25-
The details of the decision support tool and the insulin
infusion dosing are provided in the MOP.
8.2.3.2.2 SQ Insulin and Saline Instructions
Subcutaneous injections will be given at meal time up to
three times a day for patients who are eating.
Patients who are not eating or are receiving continuous tube
feeds will receive placebo saline subcutaneous injections
2/day at 9:00 and 21:00 to simulate the 0-4 SQ insulin
injections in the control group.
The details of the subcutaneous insulin/saline dosing are
provided in the MOP.
8.2.3.3 Hypoglycemia Protocols for Control and Intervention
Groups
Hypoglycemia protocol (Glucose concentration <80 mg/dL)
All insulin therapy will be stopped in the event that glucose
falls below 80 mg/dL and the following protocol will be
initiated:
Stop all IV insulin and hold all subcutaneous insulin
(stop all IV saline and saline subcutaneous
injections as well).
Stat draw of a serum sample is to be sent to the
laboratory for confirmation if glucose <70 mg/dL.
Glucose administration
Control Group – A dose of IV D50 25 ml
(1/2 amp D50) will be given until blood
glucose is ≥80 mg/dL.
Intervention Group – A dose of IV D50 will
be given. The specific dose will be
determined by the decision support tool
based on the glucose concentration.
Recheck blood glucose in 15 minutes, and repeat
treatment every 15 minutes if needed until glucose
is ≥80 mg/dL.
Once glucose is ≥80 mg/dL:
Restart IV insulin or saline per protocol
Restart SQ insulin or saline per protocol
The details of the hypoglycemia protocol are provided in
the MOP.

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-26-
Severe Hypoglycemia (Glucose <40 mg/dL)
All glucose measurements <40 mg/dL will be captured as
severe hypoglycemia, and measurements <70 mg/dL will be
captured as hypoglycemia. Each event will be characterized
as symptomatic or asymptomatic based on symptoms and
signs determined by the hypoglycemia assessment tool
described in the MOP. All spontaneous symptomatic
complaints will trigger a stat glucose measure.
Nonverbal patients will be assessed for only the physiologic
signs of hypoglycemia.
8.2.3.4 Suggested Care Post Study Treatment (based on ADA
guidelines)53,54
At the completion of study treatment, the hospital treating team
(not the study team) will determine the best long-term glucose
control therapy for each patient. Investigators will be encouraged
to follow the most recent ADA guidelines.
8.2.4 Concomitant or Ancillary Therapy
Throughout the study period, all AHA guidelines for acute stroke care8
will be followed. This includes standard care for acute ischemic stroke
patients such as swallowing evaluation prior to initiation of PO intake,
DVT prophylaxis and early secondary stroke prevention therapy. Blood
pressure and temperature recommendations also will be followed.
Rehabilitation evaluation will be considered for every subject.
8.3 Clinical Guidelines
As above, the AHA acute ischemic stroke guidelines for standard clinical care
will be followed for all subjects.
8.4 Follow-up Procedure
The goal of the study is to achieve complete, accurate follow-up for the three
month study period. Subjects will be contacted at 6 weeks by phone for reports of
SAEs, to capture early follow up information and to make arrangements for the 3
month study follow up visit during which time the primary clinical outcome
(mRS) and secondary outcomes will be captured. Any subject unable to return in
person will undergo a 3 month study follow up visit by phone.
8.5 Notification of Death
For each death, an AE form must be completed documenting the cause of death.
An event that leads to death is always an SAE, although it may not be related or
unexpected. The underlying cause of death will be required to be reported.
Concurrent disease processes such as sepsis, pneumonia, etc. should be
investigated. Within 24 hours of the discovery of a treatment related death, the
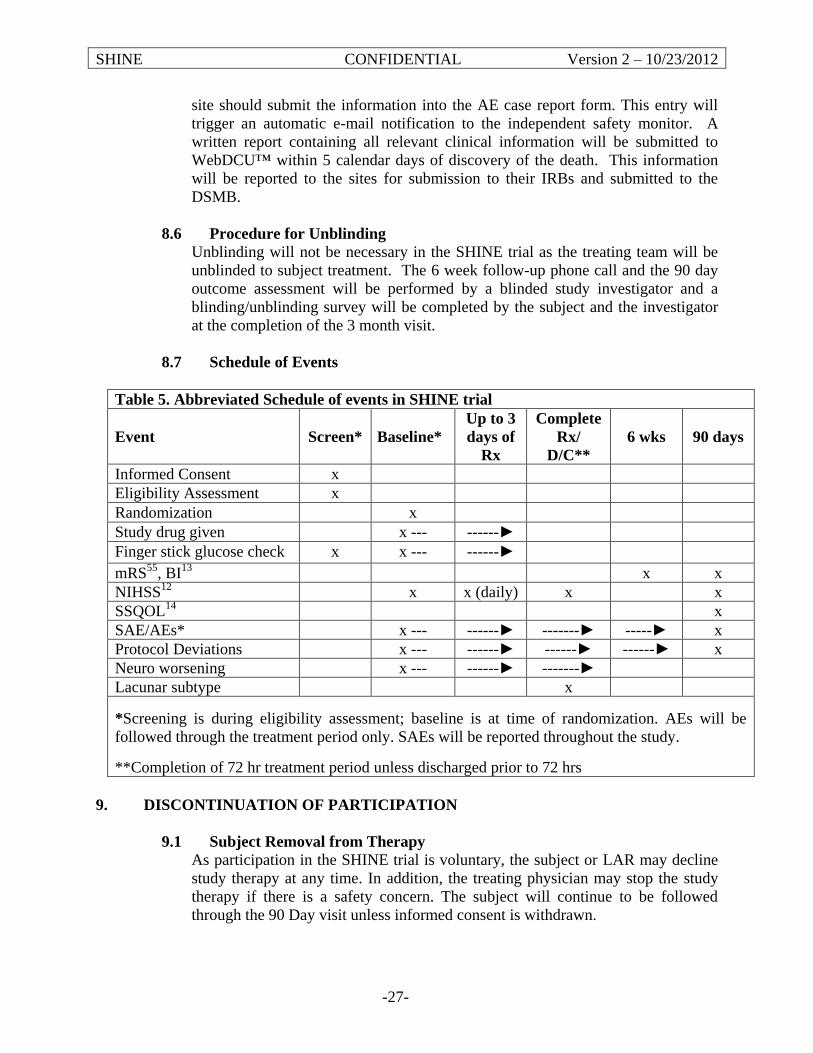
SHINE CONFIDENTIAL Version 2 – 10/23/2012
-27-
site should submit the information into the AE case report form. This entry will
trigger an automatic e-mail notification to the independent safety monitor. A
written report containing all relevant clinical information will be submitted to
WebDCU™ within 5 calendar days of discovery of the death. This information
will be reported to the sites for submission to their IRBs and submitted to the
DSMB.
8.6 Procedure for Unblinding
Unblinding will not be necessary in the SHINE trial as the treating team will be
unblinded to subject treatment. The 6 week follow-up phone call and the 90 day
outcome assessment will be performed by a blinded study investigator and a
blinding/unblinding survey will be completed by the subject and the investigator
at the completion of the 3 month visit.
8.7 Schedule of Events
Table 5. Abbreviated Schedule of events in SHINE trial
Event
Screen*
Baseline*
Up to 3
days of
Rx
Complete
Rx/
D/C**
6 wks
90 days
Informed Consent x
Eligibility Assessment x
Randomization x
Study drug given x --- ------►
Finger stick glucose check x x --- ------►
mRS55
, BI13
x x
NIHSS12
x x (daily) x x
SSQOL14
x
SAE/AEs* x --- ------► -------► -----► x
Protocol Deviations x --- ------► ------► ------► x
Neuro worsening x --- ------► -------►
Lacunar subtype x
*Screening is during eligibility assessment; baseline is at time of randomization. AEs will be
followed through the treatment period only. SAEs will be reported throughout the study.
**Completion of 72 hr treatment period unless discharged prior to 72 hrs
9. DISCONTINUATION OF PARTICIPATION
9.1 Subject Removal from Therapy
As participation in the SHINE trial is voluntary, the subject or LAR may decline
study therapy at any time. In addition, the treating physician may stop the study
therapy if there is a safety concern. The subject will continue to be followed
through the 90 Day visit unless informed consent is withdrawn.

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-28-
9.2 Subject Withdrawal
The subject has the right to voluntarily withdraw from the study at any time for
any reason without prejudice to his/her future medical care by the physician or at
the institution.
For the occasional subject who withdraws consent, the date and reason for
consent withdrawal should be documented. Subject data will be included in the
analysis up to the date of the consent withdrawal.
A distinction will be made between subjects who fail to complete all forms on
schedule or who miss some follow up assessments and the withdrawal of consent.
Missed or rescheduled visits will be documented, but the subject will continue to
be followed in the future according to protocol requirements, and all follow-up
data will be included in the analysis.
9.3 Procedure for Discontinuation
The procedure to be followed at the time a subject/LAR withdraws consent from
the trial:
(1) Check for the development of adverse events.
(2) Complete the End-of-Study form and include an explanation of why the
subject is withdrawing.
(3) Subject or LAR will be required to document in writing his or her desire to
withdraw.
9.4 Subject Lost to Follow-Up
In the event that all possible attempts to locate the subject have failed, a formal
letter will be submitted to the Executive Committee (EC). The letter must
document all efforts made by the investigator to contact the subject. When the
EC is satisfied that all methods have been tried and have failed, the subject will be
coded as lost to follow-up and the delinquency status will be removed.
9.5 Subject Transfers
Whenever a subject's medical care transfers to another clinical setting, every
attempt must be made to obtain continued follow-up data and information on self-
administered forms. The study coordinator should be notified immediately when
this occurs, so that appropriate arrangements can be made, wherever possible for
the subject to continue to participate in the study.
10. OUTCOMES DEFINTIONS
10.1 Primary

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-29-
The primary outcome is the severity adjusted 90-day mRS. “Favorable” outcome
is defined as mRS score of 0 in subjects with baseline NIHSS of 3-7, mRS of 0-1
in subjects with baseline NIHSS of 8-14, and mRS of 0-2 in subjects with
baseline NIHSS of 15-22.9, 10
All NETT and non-NETT investigators will be
trained and certified in the same manner.
10.2 Secondary
o NIHSS
o Barthel Index (BI)
o Stroke Specific Quality Of Life (SSQOL) scale
Favorable outcomes for the NIHSS and BI are defined as: a score of 0-1 on the
NIHSS and 95-100 on the BI. The SSQOL will be analyzed as a continuous
outcome.
11. DATA MANAGEMENT
11.1 Data Processing
Data management will be handled by the NETT SDMC which is housed in the
Data Coordination Unit (DCU) in the Division of Biostatistics and Epidemiology
at the Medical University of South Carolina (MUSC). All study activities will be
conducted in coordination with the study co-PIs, the hubs/spokes, and the NETT
CCC and SDMC, and will use an electronic data acquisition method where all
clinical data on enrolled subjects will be entered by the hub/spoke personnel in
real time. The latest version of each CRF will be available as a PDF file on the
study website for use as worksheets and source documents by study personnel.
The study data will be managed (including data queries) by the SDMC using the
WebDCU™ system. This user-friendly web-based database system, developed by
the SDMC, will be used for regulatory document management, subject
randomization, data entry, data validation, project progress monitoring, subject
tracking, user customizable report generation and secure data transfer.
11.2 Data Security and Confidentiality
During the course of the trial, user access to the files with subject identifiers,
treatment assignment and files with study outcomes will be restricted to core staff
with any exceptions to be approved by the Executive Committee.
In addition to use of passwords and other security measures, all documents
containing identifying information on individuals or physicians are considered
confidential materials and will be safeguarded to the greatest possible extent. No
information, which identifies a specific person, hospital, or physician, will be
released to, or discussed with anyone other than study staff members.
Because the SDMC uses a web-based system, source documents and CRFs will
remain at the participating sites. The study database only identifies study subjects

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-30-
by unique study identification codes. All data will be stored in a manner that is
HIPAA compliant, without the ability to track the information back to a specific
subject except through a password protected system. All collected information
about a subject will be stored by a unique identification code. All SDMC
personnel are certified by the NIH Office of Human Subjects Research in the
Protection of Human Research Subjects course.
11.3 Data Quality Assurance
Upon entry of CRFs into the study database, quality control procedures will be
applied at each stage of data handling in order to ensure compliance with GCP
guidelines, integrity of the study data and document processing system reliability.
All sites will be monitored by the CCC and monitors will review source
documents and case report form information. An quality assurance record audit
will be implemented. Audit findings will be used to identify and correct problems
in data collection.
12. STATISTICAL CONSIDERATIONS
12.1 Sample Size and Power Estimation
The primary outcome variable is the overall proportion of subjects experiencing a
favorable outcome 90-days post randomization, where favorable is defined by the
dichotomized mRS score as adjusted to the baseline NIHSS (stroke severity). Our
preliminary data suggest a 25% rate of favorable outcome in the control group at
90 days is reasonable. A clinically relevant absolute difference in success rates
between the two interventions is chosen as 7% (see preliminary data) (success
ratecontrol=25%; success rateinfusion=32%). If the IV insulin group does not have at
least a 7% or higher success rate than the control group, then IV insulin within 12
hours of symptom onset will not be considered a worthwhile therapy for
hyperglycemic acute ischemic stroke subjects.
Based on the above information and taking into consideration the planned interim
analyses, the study is powered to assure 80% likelihood of identifying a
difference in success rates greater than or equal to 7%. Sample size estimation is
based on the comparison of independent proportions. This estimation approach is
conservative as the proposed regression analysis is generally more powerful than
chi square test on individual outcomes. The maximum sample size required for
randomization is 1314 subjects (657/treatment group). Although every attempt
will be made to avoid drop outs and losses to follow up, the required sample size
is inflated for a 3% non-adherence rate. The 3% rate is consistent with pilot data
and the ALIAS study rates. The estimated total of subjects required is a
maximum of 1400. The analysis plan does include a sample size re-estimation so
this number could potentially change. Details of the re-estimation are in the
Statistical Analysis Plan (SAP).

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-31-
12.2 Statistical Analyses
12.2.1 Interim Analysis
Details of all planned statistical analyses including the interim analyses
will be provided in the study SAP. In summary, four interim analyses for
futility and overwhelming efficacy are planned. These analyses will use
the error-spending function method with stopping guidelines that are
similar to the O'Brien and Fleming (OBF) type stopping boundaries. The
proposed timelines can be altered based on the input from the DSMB. The
SDMC will conduct these analyses and compile the reports for the DSMB.
12.2.2 Interim Safety Analysis
The safety monitor and DSMB will receive periodic safety reports of all
adverse events and serious adverse events. Statistical monitoring for
safety will be limited to severe hypoglycemia (<40 mg/dL) during the
treatment period and death rate within 90 days post randomization.
Details on this monitoring plan are in the SAP.
12.2.3 Primary Efficacy Analysis
The primary outcome analysis of the 90-day mRS will use a stratified
dichotomy methodology for assessing improvement as defined in the
Primary Outcome section above. Outcome differences will be analyzed
under the intention-to-treat principle, therefore all randomized subjects
will be included in the primary analysis sample. To assess efficacy, the
treatment groups will be compared with respect to the proportion with
favorable outcome 90 days post randomization after controlling for the
prognostic variables included in the randomization scheme. A Wald chi-
square test will be performed to compare the treatment group proportions
using a two-tailed significance level of 0.05. Adjusted relative risks will
be reported with two-sided 95% confidence intervals.
Additional analyses will identify potential confounding (prognostic)
variables to be used as covariates in subsequent secondary analyses of the
primary outcome using generalized linear modeling techniques. Specific
covariates include age, gender, race, ethnicity, admission blood glucose,
previous stroke, time between stroke onset and treatment and lacunar
subtype. If statistically significant differences are evident, post-hoc
analyses will be conducted to determine if differences had an effect on the
conclusions from the pre-specified primary analysis.
12.2.4 Secondary Analyses
This study is designed to test the primary hypothesis. However, it also
offers the opportunity to conduct analyses to evaluate important additional
neurological and functional outcomes using the NIHSS, BI and SSQOL.
The secondary analyses will be conducted using the intention to treat
population. In addition to these secondary clinical outcomes, the analysis

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-32-
will include time to randomization from symptom onset and protocol
conduct metrics between treatment arms: time to target, time in target,
time to treatment and adherence to the decision support tool.
12.2.5 Safety Outcome Analyses
In addition to the continual monitoring of adverse events by the safety
monitor and DSMB and the planned statistical monitoring for safety, final
analyses of specified safety outcomes will be conducted as specified in the
SAP.
13. ADVERSE EVENTS
13.1 Adverse Events
Adverse event reporting and procedures are described in detail in the MOP.
Adverse events will be defined and severity graded according to the Common
Terminology Criteria for Adverse Events, (CTCAE), found in the MOP. AEs will
be submitted online through WebDCU™ and coded centrally using MedDRA.
Guidelines for report content and structure will be provided in the MOP. All
adverse events occurring during treatment and all serious adverse events
occurring during the study will be recorded. The site PI or Study Coordinator is
responsible for entering any and all AEs and SAEs into the database and updating
the information (e.g., date of resolution, action taken) as needed.
Study personnel will evaluate subjects while in the hospital and at each telephone
communication and follow up for the presence of AEs (during treatment only) and
SAEs.
13.2 Clinically Important Adverse Events
The following adverse events should be handled as serious.
o Neurological worsening lasting greater than 24 hours and associated with
glucose concentration of <55 mg/dL.
o Severe Hypoglycemia (glucose <40 mg/dL).
13.3 Adverse Event Exceptions
Death due to the natural history of ischemic stroke will be recorded as a non-
related serious adverse event. Additionally, all serious but known complications
of stroke (i.e. malignant brain edema) will be recorded as non-related serious
adverse events.
13.4 Obligation of Investigator
At the time of enrollment, an assessment of ongoing medical conditions and/or
signs or symptoms will be recorded. Upon initiation of study protocol, all adverse
events, whether or not attributed to the study intervention, observed by the
Investigator or reported by the subject, will be recorded on the appropriate Case

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-33-
Report Forms. Attributes will include a description, date of onset and resolution,
duration, severity, assessment of relatedness to the intervention, action taken, and
outcome. All adverse events must be followed to resolution or until the end of the
study treatment period, whichever comes first.
If the adverse event is sufficiently severe, the investigator is obligated to conduct
a termination assessment, and if appropriate, should halt study treatment. The
subject should be provided with medical supervision and appropriate treatment
until symptoms cease.
13.5 Reporting Procedures
In order to assure prompt and complete reporting of adverse events or
complications of intervention, the following general guidelines are to be observed.
All adverse events, whether serious or not, will be collected through completion
of study therapy. Adverse events will be captured by the treating team and the
study team based on history, physical exam, medical records and laboratory
findings. The independent safety monitor will review non serious AEs regularly.
Serious adverse events (SAE) will be captured throughout the 3 month study
period. All SAEs, including those that are judged to be related to study therapy,
and especially severe hypoglycemia, will be data entered by the site personnel
into the study database within timelines defined in the MOP. Upon data entry, the
system will trigger an automatic e-mail notification to the independent Medical
Safety Monitor (MSM) stating that an SAE has occurred. The MSM will access
the information via the password protected web based system for review and
report safety concerns to the DSMB.
14. INVESTIGATIONAL DRUG DESCRIPTION
The investigational drug is insulin which is FDA approved for the treatment of
hyperglycemia. Therefore, it will be labeled, stored, prepared and distributed through the
hospital pharmacy as it otherwise would. Blinding procedures (to maintain the subject
blind) will take place and are described in detail in the MOP.
15. REGULATORY AND ETHICAL OBLIGATIONS
15.1 Informed Consent
In accordance with US FDA regulations (21 CFR 50) and guidelines (Federal
Register, May 9, 1997, Vol. 62, Number 90 - ICH Good Clinical Practice
Consolidated Guideline) it is the investigator’s responsibility to ensure that
informed consent is obtained from the subject or subject’s LAR before
participating in an investigational study, after an adequate explanation of the
purpose, methods, risks, potential benefits and subject responsibilities of the
study. Procedures that are to be performed as part of the practice of medicine and
which would be done whether or not study entry was contemplated, such as for

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-34-
diagnosis or treatment of a disease or medical condition, may be performed and
the results subsequently used for determining study eligibility without first
obtaining consent. On the other hand, informed consent must be obtained prior to
initiation of any screening procedures that are performed solely for the purpose of
determining eligibility for research.
Each subject/LAR must be given a copy of the informed consent. The original
signed consent must be retained in the institution’s records and is subject to
review by the sponsor, DCU, the FDA or representative from another agency that
performs the same function, and the IRB responsible for the conduct of the
institution. All elements listed in the International Conference on Harmonisation
Good Clinical Practice guidelines must be included in the informed consent.
Informed consent will be obtained by either the site Principal Investigator or by
individuals approved by the site Principal Investigator and whose names have
been submitted to the NETT Regulatory Database. Informed consent will be
obtained from the subject or subject’s LAR after the details of the protocol have
been reviewed. The individual responsible for obtaining consent will assure, prior
to signing of the informed consent, that the subject has had all questions regarding
therapy and the protocol answered.
15.2 Institutional Review Board (IRB)
In accordance with US FDA regulations (21 CFR 56) and guidelines (Federal
Register, May 9, 1997 Vol. 62 Number 90 - International Conference on
Harmonisation Good Clinical Practice Consolidated Guideline) all research
involving human subjects and changes to the research plan must be reviewed and
approved by an IRB.
15.2.1 Initial Review and Approval
A copy of the protocol, proposed informed consent form, other written
subject information, and any proposed advertising material must be
submitted to the enrolling site’s IRB for written approval. A copy of the
IRB approval of the protocol and informed consent form must be
submitted to the NETT Regulatory database and approved before
recruitment of subjects into the study.
15.2.2 Amendments
Protocol amendments may only be made with the prior approval of
Executive Committee. The site Principal Investigator must agree to, and
obtain approval from the IRB, for all protocol amendments and revisions
to the informed consent document. The site Principal Investigator should
notify the IRB of serious adverse events occurring at the site and other
adverse event reports recorded in the study database, in accordance with
local procedures and Section 13.5 of this protocol. Copies of all approvals
and approved versions of the informed consent must be submitted to the
central regulatory document database housed within WebDCU™.

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-35-
15.2.3 Annual Renewal
The site Principal Investigator will be responsible for obtaining annual
IRB approval renewal throughout the duration of the study. Copies of the
site Principal Investigator’s reports and the IRB’s continuance of approval
must be submitted electronically to the central regulatory document
database.
16. STUDY ORGANIZATION
16.1 Executive Committee
In addition to the SHINE leadership, the Executive Committee (EC) has overall
responsibility for assuring the scientific, clinical and ethical integrity of the study.
The executive committee will include the NINDS Project Officer, administrative
core leadership, the NETT CCC leadership and SDMC leadership.
The details of the executive committee functions and membership are detailed in
the MOP.
16.2 Data and Safety Monitoring Board
The NINDS-appointed DSMB will plan to meet semi-annually with interim
conference calls as needed, to review interim data relating to the trial. The
functions of the DSMB will include:
(1) Review of all adverse effects or complications related to the trial
interventions;
(2) Review of interim data on the primary and secondary outcomes according
to prespecified monitoring guidelines;
(3) Monitor accrual;
(4) Review summary reports relating to performance of centers relative to
compliance with protocol requirements;
(5) Recommend to the Executive Committee that:
a) Trial continue as planned;
b) Trial be modified with justification of modification as DSMB deems
appropriate;
c) Trial should be stopped with disclosure of basis for recommendation.

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-36-
In general, clinical investigators will not be present at the Closed Sessions of the
DSMB, but may on occasion be requested to present or provide information to
assist the DSMB in carrying out its functions appropriately.
16.3 Ancillary Studies
General information on proposals for SHINE ancillary studies and detailed
procedures for approved and funded ancillary studies can be found in the MOP.
Sites will not be required to participate in any ancillary study that requires
additional data collection, but they will be encouraged to participate in accepted
studies.
16.3.1 Optional Insights on Selected Procoagulation Markers and
Outcomes in Stroke Trial (I-SPOT) Ancillary Study
Participants who consent to the SHINE trial may be asked to consent to an
optional ancillary study. Subjects who consent to I-SPOT will provide blood
samples at 2 time-points (baseline and 48 hours). Those patients who decline
the ancillary study may still participate in the SHINE trial.
The I-SPOT trial will recruit 315 SHINE patients. Blood coagulation marker
levels will be measured before and at 48 hours after the start of treatment.
Baseline and temporal changes in biomarkers levels will be compared between
SHINE treatment groups. Details of I-SPOT can be found in the ancillary trial
protocol and laboratory manual.

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-37-
17. REFERENCES
1. Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, Ford
E, Furie K, Go A, Greenlund K, Haase N, Hailpern S, Ho M, Howard V, Kissela B,
Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott M, Meigs J, Mozaffarian D,
Nichol G, O'Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R,
Steinberger J, Thom T, Wasserthiel-Smoller S, Wong N, Wylie-Rosett J, Hong Y. Heart
disease and stroke statistics--2009 update: A report from the American Heart Association
statistics committee and stroke statistics subcommittee. Circulation. 2009 Jan 27; 119(3):
480-6.
2. Gentile NT, Seftchick MW, Huynh T, Kruus LK, Gaughan J. Decreased mortality by
normalizing blood glucose after acute ischemic stroke. Acad Emerg Med. 2006 Feb;
13(2): 174-80.
3. Williams LS, Rotich J, Qi R, Fineberg N, Espay A, Bruno A, Fineberg SE, Tierney
WR. Effects of admission hyperglycemia on mortality and costs in acute ischemic stroke.
Neurology. 2002 Jul 9; 59(1): 67-71.
4. Capes SE, Hunt D, Malmberg K, Pathak P, Gerstein HC. Stress hyperglycemia and
prognosis of stroke in nondiabetic and diabetic patients: A systematic overview. Stroke.
2001 Oct; 32(10): 2426-32.
5. McCormick MT, Muir KW, Gray CS, Walters MR. Management of hyperglycemia in
acute stroke: How, when, and for whom? Stroke. 2008 Jul; 39(7): 2177-85.
6. Malmberg K, Ryden L, Efendic S, Herlitz J, Nicol P, Waldenstrom A, Wedel H, Welin
L. Randomized trial of insulin-glucose infusion followed by subcutaneous insulin
treatment in diabetic patients with acute myocardial infarction (DIGAMI study): Effects
on mortality at 1 year. J Am Coll Cardiol. 1995 Jul; 26(1): 57-65.
7. van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M,
Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R. Intensive insulin therapy in the
critically ill patients. N Engl J Med. 2001 Nov 8; 345(19): 1359-67.
8. Adams HP Jr, del Zoppo G, Alberts MJ, Bhatt DL, Brass L, Furlan A, Grubb RL,
Higashida RT, Jauch EC, Kidwell C, Lyden PD, Morgenstern LB, Qureshi AI,
Rosenwasser RH, Scott PA, Wijdicks EF. Guidelines for the early management of adults
with ischemic stroke: A guideline from the American Heart Association/American Stroke
Association stroke council, clinical cardiology council, cardiovascular radiology and
intervention council, and the atherosclerotic peripheral vascular disease and quality of
care outcomes in research interdisciplinary working groups: The American Academy of
Neurology affirms the value of this guideline as an educational tool for neurologists.
Stroke. 2007 May; 38(5): 1655-711.
9. Adams HP Jr, Leclerc JR, Bluhmki E, Clarke W, Hansen MD, Hacke W. Measuring
outcomes as a function of baseline severity of ischemic stroke. Cerebrovasc Dis. 2004;
18(2): 124-9.

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-38-
10. Saver JL, Yafeh B. Confirmation of tPA treatment effect by baseline severity-
adjusted end point reanalysis of the NINDS-tPA stroke trials. Stroke. 2007 Feb; 38(2):
414-6.
11. Young FB, Lees KR, Weir CJ. Improving trial power through use of prognosis-
adjusted end points. Stroke. 2005 Mar; 36(3): 597-601.
12. Lyden P, Lu M, Jackson C, Marler J, Kothari R, Brott T, Zivin J. Underlying
structure of the national institutes of health stroke scale: Results of a factor analysis.
NINDS tPA stroke trial investigators. Stroke. 1999 Nov; 30(11): 2347-54.
13. Wade DT, Collin C. The barthel ADL index: A standard measure of physical
disability? Int Disabil Stud. 1988; 10(2): 64-7.
14. Williams LS, Weinberger M, Harris LE, Clark DO, Biller J. Development of a stroke-
specific quality of life scale. Stroke. 1999 Jul; 30(7): 1362-9.
15. Kanji S, Jones E, Goddard R, Meggison HE, Neilipovitz D. Efficiency and safety of
a standardized protocol for intravenous insulin therapy in ICU patients with
neurovascular or head injury. Neurocritical Care. 2010 Feb;12(1):43-9
16. Bruno A, Kent TA, Coull BM, Shankar RR, Saha C, Becker KJ, Kissela BM,
Williams LS. Treatment of hyperglycemia in ischemic stroke (THIS): A randomized pilot
trial. Stroke. 2008 Feb; 39(2): 384-9.
17. Johnston KC, Hall CE, Kissela BM, Bleck TP, Conaway MR. Glucose regulation in
acute stroke patients (GRASP) trial: A randomized pilot trial. Stroke. 2009 Dec; 40(12):
3804-9.
18. Bruno A, Saha C, Williams LS, Shankar R. IV insulin during acute cerebral infarction
in diabetic patients. Neurology. 2004 Apr 27; 62(8): 1441-2.
19. Walters MR, Weir CJ, Lees KR. A randomised, controlled pilot study to investigate
the potential benefit of intervention with insulin in hyperglycaemic acute ischaemic
stroke patients. Cerebrovasc Dis. 2006; 22(2-3): 116-22.
20. Gray CS, Hildreth AJ, Sandercock PA, O'Connell JE, Johnston DE, Cartlidge NE,
Bamford JM, James OF, Alberti KG. Glucose-potassium-insulin infusions in the
management of post-stroke hyperglycaemia: The UK glucose insulin in stroke trial
(GIST-UK). Lancet Neurol. 2007 May; 6(5): 397-406.
21. Kreisel SH, Berschin UM, Hammes HP, Leweling H, Bertsch T, Hennerici MG,
Schwarz S. Pragmatic management of hyperglycaemia in acute ischaemic stroke: Safety
and feasibility of intensive intravenous insulin treatment. Cerebrovasc Dis. 2009; 27(2):
167-75.
22. Moulin T, Tatu L, Crepin-Leblond T, Chavot D, Berges S, Rumbach T. The besancon
stroke registry: An acute stroke registry of 2,500 consecutive patients. Eur Neurol. 1997;
38(1): 10-20.

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-39-
23. Weir CJ, Murray GD, Dyker AG, Lees KR. Is hyperglycaemia an independent
predictor of poor outcome after acute stroke? results of a long-term follow up study.
BMJ. 1997 May 3; 314(7090): 1303-6.
24. Bruno A, Biller J, P. AH,Jr, Clarke WR, Woolson RF, Williams LS, Hansen MD.
Acute blood glucose level and outcome from ischemic stroke. trial of ORG 10172 in
acute stroke treatment (TOAST) investigators. Neurology. 1999 Jan 15; 52(2): 280-4.
25. Demchuk AM, Tanne D, Hill MD, Kasner SE, Hanson S, Grond M, Levine SR.
Predictors of good outcome after intravenous tPA for acute ischemic stroke. Neurology.
2001 Aug 14; 57(3): 474-80.
26. Bruno A, Levine SR, Frankel MR, Brott TG, Lin Y, Tilley BC, Lyden PD, Broderick
JP, Kwiatkowski TG, Fineberg SE. Admission glucose level and clinical outcomes in the
NINDS rt-PA stroke trial. Neurology. 2002 Sep 10; 59(5): 669-74.
27. Poppe AY, Majumdar SR, Jeerakathil T, Ghali W, Buchan AM, Hill MD. Admission
hyperglycemia predicts a worse outcome in stroke patients treated with intravenous
thrombolysis. Diabetes Care. 2009 Apr; 32(4): 617-22.
28. Yong M, Kaste M. Dynamic of hyperglycemia as a predictor of stroke outcome in the
ECASS-II trial. Stroke. 2008 Oct; 39(10): 2749-55.
29. Fuentes B, Castillo J, San Jose B, Leira R, Serena J, Vivancos J, Davalos A, Nunez
AG, Egido J, Diez-Tejedor E. The prognostic value of capillary glucose levels in acute
stroke: The GLycemia in acute stroke (GLIAS) study. Stroke. 2009 Feb; 40(2): 562-8.
30. Dziedzic T, Slowik A, Pera J, Szczudlik A. Association between hyperglycemia,
heart failure and mortality in stroke patients. Eur J Neurol. 2009 Feb; 16(2): 251-6.
31. Parsons MW, Barber PA, Desmond PM, Baird TA, Darby DG, Byrnes G, Tress BM,
Davis SM. Acute hyperglycemia adversely affects stroke outcome: A magnetic resonance
imaging and spectroscopy study. Ann Neurol. 2002 Jul; 52(1): 20-8.
32. Els T, Klisch J, Orszagh M, Hetzel A, Schulte-Monting J, Schumacher M, Lucking
CH. Hyperglycemia in patients with focal cerebral ischemia after intravenous
thrombolysis: Influence on clinical outcome and infarct size. Cerebrovasc Dis. 2002;
13(2): 89-94.
33. Baird TA, Parsons MW, Phanh T, Butcher KS, Desmond PM, Tress BM, Colman PG,
Chambers BR, Davis SM. Persistent poststroke hyperglycemia is independently
associated with infarct expansion and worse clinical outcome. Stroke. 2003 Sep; 34(9):
2208-14.
34. Ribo M, Molina CA, Delgado P, Rubiera M, Delgado-Mederos R, Rovira A,
Munuera J, Alvarez-Sabin J. Hyperglycemia during ischemia rapidly accelerates brain
damage in stroke patients treated with tPA. J Cereb Blood Flow Metab. 2007 Sep; 27(9):
1616-22.

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-40-
35. Wong AA, Schluter PJ, Henderson RD, O'Sullivan JD, Read SJ. Natural history of
blood glucose within the first 48 hours after ischemic stroke. Neurology. 2008 Mar 25;
70(13): 1036-41.
36. Garg R, Chaudhuri A, Munschauer F, Dandona P. Hyperglycemia, insulin, and acute
ischemic stroke: A mechanistic justification for a trial of insulin infusion therapy. Stroke.
2006 Jan; 37(1): 267-73.
37. Becker KJ. Targeting the central nervous system inflammatory response in ischemic
stroke. Curr Opin Neurol. 2001 Jun; 14(3): 349-353.
38. Gentile NT, Siren K. Glycemic control and the injured brain. Emerg Med Clin North
Am. 2009 Feb; 27(1): 151-69, x.
39. Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral
ischaemia: From experimental strategies to clinical use. Lancet Neurol. 2009 Apr; 8(4):
398-412.
40. Prass K, Dirnagl U. Glutamate antagonists in therapy of stroke. Restor Neurol
Neurosci. 1998; 13(1-2): 3-10.
41. Fisher M, Hanley DF, Howard G, Jauch EC, Warach S. Recommendations from the
STAIR V meeting on acute stroke trials, technology and outcomes. Stroke. 2007 Feb;
38(2): 245-8.
42. Saver JL. Novel end point analytic techniques and interpreting shifts across the entire
range of outcome scales in acute stroke trials. Stroke. 2007 Nov; 38(11): 3055-62.
43. Saver JL, Gornbein J. Treatment effects for which shift or binary analyses are
advantageous in acute stroke trials. Neurology. 2009 Apr 14; 72(15): 1310-5.
44. Brown DL, Coffey CS. Stroke trials: A shift to shift analysis? Neurology. 2009 Apr
14; 72(15): 1292-3.
45. Johnston KC, Wagner DP, Wang XQ, Newman GC, Thijs V, Sen S, Warach S.
Validation of an acute ischemic stroke model: Does diffusion-weighted imaging lesion
volume offer a clinically significant improvement in prediction of outcome? Stroke. 2007
Jun; 38(6): 1820-5.
46. Fisher M, Albers GW, Donnan GA, Furlan AJ, Grotta JC, Kidwell CS, Sacco RL,
Wechsler LR. Enhancing the development and approval of acute stroke therapies: Stroke
therapy academic industry roundtable. Stroke. 2005 Aug; 36(8): 1808-13.
47. Morris AH, Orme J, Rocha BH, Holmen J, Clemmer T, Nelson N, Allen J, Jephson
A, Sorenson D, Sward K, Warner H. An electronic protocol for translation of research
results to clinical practice: A preliminary report. J Diabetes Sci Technol. 2008
Sep;2(5):802-8.
48. Thompson BT, Orme JF, Zheng H, Lucktt PM, Truwit JD, Wilson DF, Duncan Hite
R, Brower RG, Bernard GR, Curley MA, Steingrub JS, Sorenson DK, Sward K,

SHINE CONFIDENTIAL Version 2 – 10/23/2012
-41-
Hirshberg E, Morris AH. Multicenter validation of a computer based clinical decision
support tool for glucose control in adult and pedicatric intensive care units. J Diabetes
Sci Technol. 2008 May;2(3):357-68.
49. Matz K, Keresztes K, Tatschl C, Nowotny M, Dachenhausenm A, Brainin M,
Tuomilehto J. Disorders of glucose metabolism in acute stroke patients: An
underrecognized problem. Diabetes Care. 2006 Apr; 29(4): 792-7.
50. Vancheri F, Curcio M, Burgio A, Salvaggio S, Gruttadauria G, Lunetta MC, Dovico
R, Alletto M. Impaired glucose metabolism in patients with acute stroke and no previous
diagnosis of diabetes mellitus. QJM. 2005 Dec; 98(12): 871-8.
51. Kernan WN, Inzucchi SE, Viscoli CM, Brass LM, Bravata DM, Shulman GI,
McVeety JC, Horwitz RI. Impaired insulin sensitivity among nondiabetic patients with a
recent TIA or ischemic stroke. Neurology. 2003 May 13; 60(9): 1447-51.
52. Cucchiara B, Tanne D, Levine SR, Demchuk AM, Kasner S. A risk score to predict
intracranial hemorrhage after recombinant tissue plasminogen activator for acute
ischemic stroke. J Stroke Cerebrovasc Dis. 2008 Nov-Dec; 17(6): 331-3.
53. Moghissi ES, Korytkowski MT, DiNardo M, Einhorn D, Hellman R, Hirsch IB,
Inzucchi SE, Ismail-Beigi F, Kirkman MS, Umpierrez GE. American association of
clinical endocrinologists and american diabetes association consensus statement on
inpatient glycemic control. Endocr Pract. 2009 Jul-Aug; 15(4): 353-69.
54. American Diabetes Association. Standards of Medical Care in Diabetes - 2010.
Diabetes Care. 2010 Jan;33:S11-S61.
55. van Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, van Gijn J. Interobserver
agreement for the assessment of handicap in stroke patients. Stroke. 1988 May; 19(5):
604-7.
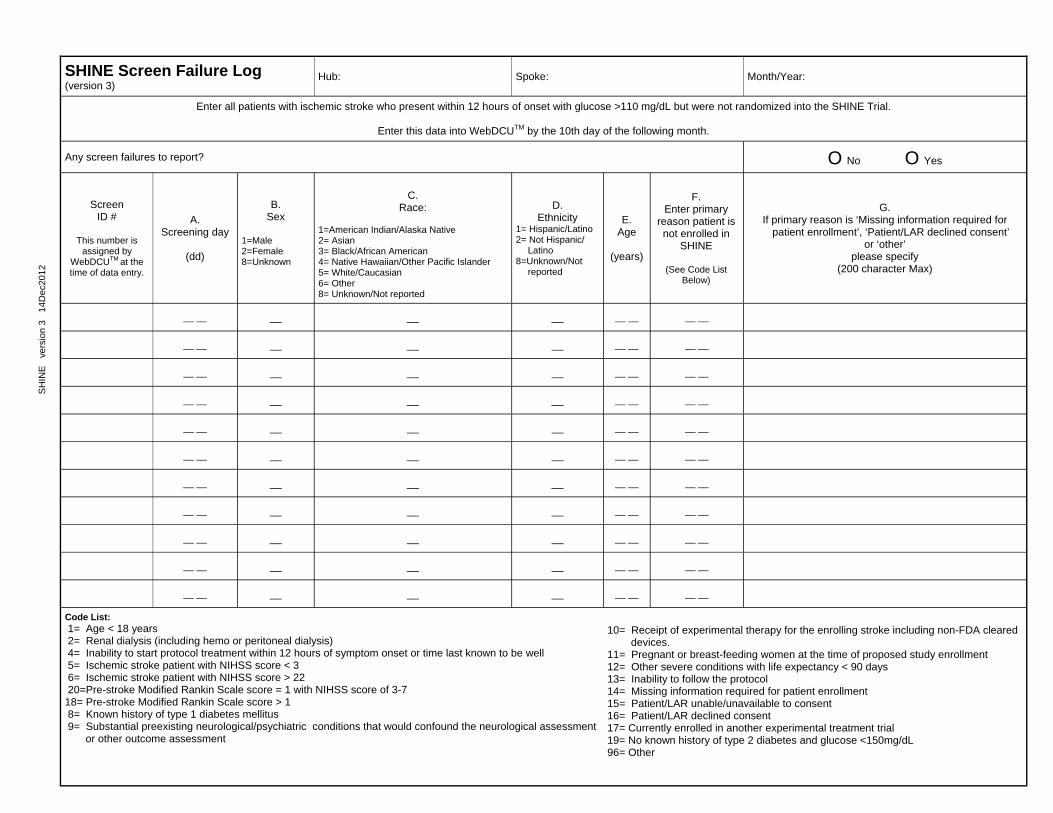
SH
INE
ve
rsio
n 3
14D
ec20
12
SHINE Screen Failure Log (version 3)
Hub: Spoke: Month/Year:
Enter all patients with ischemic stroke who present within 12 hours of onset with glucose >110 mg/dL but were not randomized into the SHINE Trial.
Enter this data into WebDCUTM by the 10th day of the following month.
Any screen failures to report? O No O Yes
Screen ID #
This number is
assigned by WebDCUTM at the time of data entry.
A. Screening day
(dd)
B. Sex
1=Male 2=Female 8=Unknown
C.
Race:
1=American Indian/Alaska Native 2= Asian 3= Black/African American 4= Native Hawaiian/Other Pacific Islander 5= White/Caucasian 6= Other 8= Unknown/Not reported
D. Ethnicity
1= Hispanic/Latino 2= Not Hispanic/
Latino 8=Unknown/Not
reported
E. Age
(years)
F. Enter primary
reason patient is not enrolled in
SHINE
(See Code List Below)
G. If primary reason is ‘Missing information required for
patient enrollment’, ‘Patient/LAR declined consent’ or ‘other’
please specify (200 character Max)
__ __ __ __ __ __ __ __ __
__ __ __ __ __ __ __ __ __
__ __ __ __ __ __ __ __ __
__ __ __ __ __ __ __ __ __
__ __ __ __ __ __ __ __ __
__ __ __ __ __ __ __ __ __
__ __ __ __ __ __ __ __ __
__ __ __ __ __ __ __ __ __
__ __ __ __ __ __ __ __ __
__ __ __ __ __ __ __ __ __
__ __ __ __ __ __ __ __ __
Code List: 1= Age < 18 years 2= Renal dialysis (including hemo or peritoneal dialysis) 4= Inability to start protocol treatment within 12 hours of symptom onset or time last known to be well 5= Ischemic stroke patient with NIHSS score < 3 6= Ischemic stroke patient with NIHSS score > 22 20=Pre-stroke Modified Rankin Scale score = 1 with NIHSS score of 3-7 18= Pre-stroke Modified Rankin Scale score > 1 8= Known history of type 1 diabetes mellitus 9= Substantial preexisting neurological/psychiatric conditions that would confound the neurological assessment or other outcome assessment
10= Receipt of experimental therapy for the enrolling stroke including non-FDA cleared devices. 11= Pregnant or breast-feeding women at the time of proposed study enrollment 12= Other severe conditions with life expectancy < 90 days 13= Inability to follow the protocol 14= Missing information required for patient enrollment 15= Patient/LAR unable/unavailable to consent 16= Patient/LAR declined consent 17= Currently enrolled in another experimental treatment trial 19= No known history of type 2 diabetes and glucose <150mg/dL 96= Other

Has the patient had an acute ischemic stroke?
If yes, did they arrive to hospital within 12h of symptom
onset/LKW?
If no, do not add to screen failure log (SFL).
If no, do not add to SFL.
If no, do not add to SFL.
If yes, add patient to SFL.
Screen Failure Log Entries
If yes, does patient have BG > 110
Screen failure logs must be entered monthly into WebDCU by the 10th of the month for the previous month.

SHINE Trial Consent Template Version Date: 01/13/2016
Page 1 of 13
INSTITUTION NAME
CONSENT TO BE PART OF A RESEARCH STUDY
INFORMATION ABOUT THIS FORM
You, or your family member, may be eligible to take part in a research study. This form gives you important information about the study. It describes the purpose of the study, and the risks and possible benefits of participating in the study. Legally authorized representatives who are giving permission for a family member, please note: in the sections that follow the word “you” refers to your family member.
Please take time to review this information carefully. After you have finished, you should talk to the researchers about the study and ask them any questions you have. You may also wish to talk to others (for example, your friends, family, or other doctors) about your participation in this study. If you decide to take part in the study, you will be asked to sign this form. Before you sign this form, be sure you understand what the study is about, including the risks and possible benefits to you.
1. GENERAL INFORMATION ABOUT THIS STUDY AND THE RESEARCHERS
1.1 Study title: Stroke Hyperglycemia Insulin Network Effort (SHINE) Trial
1.2 Company or agency sponsoring the study: This study is sponsored by the University of Virginia and funded by the National Institutes of Health – National Institute of Neurological Disorders and Stroke (NIH-NINDS).
1.3 Names, degrees, and affiliations of the researchers conducting the study:
Include local PI, local Co-Is
2. PURPOSE OF THIS STUDY
2.1 Study purpose:
Laboratory and human studies have shown that high blood sugar levels during a stroke can be associated with more damage to the brain than normal blood sugar levels. Blood sugar levels can be lowered with a hormone called insulin, which can either be given as a shot under your skin (subcutaneously) or into a vein in your arm through a tube called an IV.
The purpose of this research study is to find out if treating high blood sugar in stroke patients with IV insulin shows better recoveries than the current standard care for treating high blood sugar.
IV insulin for blood sugar control has been shown to be a safe treatment in previous studies of acute stroke patients. The next step is to learn if this treatment can improve recovery after stroke.

SHINE Trial Consent Template Version Date: 01/13/2016
Page 2 of 13
3. INFORMATION ABOUT STUDY PARTICIPANTS (SUBJECTS)
Taking part in this study is completely voluntary. You do not have to participate if you don't want to. You may also leave the study at any time. If you leave the study before it is finished, there will be no penalty to you, and you will not lose any benefits to which you are otherwise entitled.
3.1 Who can take part in this study?
To participate in this study, you must:
Have recently had a stroke caused by a blood clot in your brain
Have either: o a history of type 2 diabetes and a blood sugar level above 110 mg/dL,
or o no history of type 2 diabetes and a blood sugar level at or above 150 mg/dL
Be over 18 years old
Be able to start treatment within 12 hours of the start of your stroke symptoms or the last time you were known to be well Have had no significant disability before your stroke
Meet all study inclusion and exclusion criteria Please tell the study doctor if you have any of the following:
Any history of type 1 diabetes
Any history of severe neurological or psychiatric illness
Current pregnancy or currently breast feeding
Current kidney dialysis treatment If you have any of the conditions listed above, you will not be able to participate in this study because it may not be safe for you. 3.2 How many people (subjects) are expected to take part in this study?
About 1400 stroke patients are expected to take part in this study at about 65 medical facilities around the country. You will be one of ________ enrolled at _____________ Hospital.
4. INFORMATION ABOUT STUDY PARTICIPATION
4.1 What will happen to me in this study?
Baseline and Enrollment Procedures
You will be given a full medical and neurological exam, including a CT scan which takes a picture of your brain, baseline blood pressure, blood tests, and electrocardiogram (ECG). Though these are all part of standard care for stroke patients, we also record the results for the study. The study will also record information about your age, gender, and ethnic origin.
The study will use random assignment (like the flip of a coin) to place each patient into one of two groups. One group will be given IV insulin and subcutaneous insulin shots to control high blood sugar. The other group will be given standard subcutaneous insulin shots to control high blood

SHINE Trial Consent Template Version Date: 01/13/2016
Page 3 of 13
sugar. Both groups will be getting IV solutions and subcutaneous shots so that you will not be able to tell which group you are in.
As the trial goes on, your chance of being placed in one treatment group or the other will be based on the recovery of the patients enrolled before you. At the time you are randomized, if one treatment group is doing better than the other group then you will have a better chance of being placed in the group with the patients who have had better recovery.
Study Activities - First 3 Days in Hospital
During the first 3 days, you will receive medical and neurological exams at least once a day. These are standard care for stroke patients. In addition to these exams, a special neurological exam (National Institutes of Health Stroke Scale - NIHSS) will be done at the beginning of the study, each of the first 3 days of the study and/or at the end of the treatment if you are ready to be discharged from the hospital earlier than the third day. The NIHSS will also be done if at any time your symptoms should worsen during the first 3 days.
An IV catheter will be placed in your arm for the IV study solution. During the first 3 days in the hospital, you will receive both an IV study solution (approximately 1 teaspoon per hour consisting of either insulin or sterile salt water) and up to four subcutaneous shots per day (about 1-4 drops of either insulin or sterile salt water) based on the study insulin dosage plan. You will have your blood sugar level checked by a finger stick about every hour for the first four hours and then about every 1-3 hours. IV solutions or subcutaneous shots will be adjusted by your nurse to keep your blood sugar at the right levels.
As part of your hospital stay, you should expect about 6 finger stick checks per day as part of your standard care. Extra finger stick checks will be done as part of the study. In total you should expect about 8-20 checks per day as this is required for the safe management of insulin therapy. If your sugars are easy to control you may receive fewer checks, or if difficult, more checks per day may be needed.
For the first 3 days during study treatment in the hospital, you will not be allowed to take any other medicines to lower your blood sugar, such as pills you may usually take for diabetes, that are not part of the study treatments. You will also need to follow a special study diet during this time, and will be allowed to have only study-approved snacks. At the end of the first 3 days of the study or before leaving the hospital, whichever comes first, the plan to restart your home medicines for high blood sugar or to start a new treatment program will be decided between you and your treating doctor and is not part of this research study.
Study Activities – 6-week Interview
You will receive a telephone call from the study team about 6 weeks after your stroke that will last about 20 minutes. You will be asked about any new symptoms or illnesses, what medicines you are taking, how your recovery is going and how well you are able to do regular daily activities. If you are not able to speak on the phone, we may ask a family member or caregiver questions about how you are doing.
Study Activities – 3-month Visit

SHINE Trial Consent Template Version Date: 01/13/2016
Page 4 of 13
You will see a study doctor and/or study nurse, who has special interest and training in caring for stroke patients, about 3 months after your stroke. During this outpatient follow-up visit at __________________________________, you will be given a neurological exam including the NIH Stroke Scale. You will also be asked a series of questions that will measure how well you have recovered in your physical, thinking, and speaking abilities and how much help you need (if any) with usual daily activities. You will also be asked about your general quality of life and recovery using standard scales with simple questions which are answered by you and/or your family members (in case their help is needed). You will be asked what medications you are currently taking and whether you have had any serious illness or any hospitalizations since the 6-week telephone interview.
If for some reason we are unable to locate you for the 3 month follow up visit, we will employ an people locating service to help us locate you. The firm will not have access to your health records or study results, but will be able to access other personal information such as phone number, address, etc. We will keep all records that we produce private to the extent we are required to do so by law.
4.2 How much of my time will be needed to take part in this study?
Neurological examinations, including the study-related NIH Stroke Scale, will take 5-15 minutes each. Each blood sugar check will take less than one minute, approximately 8-20 times per day for 3 days. The 6-week phone call will take approximately 20 minutes, and the 3-month visit will take approximately two hours.
4.3 When will my participation in the study be over?
Your participation in the study will be over about 3 months after your stroke.
5. INFORMATION ABOUT RISKS AND BENEFITS
5.1 What risks will I face by taking part in the study? What will the researchers do to protect me against these risks?
The known or expected risks are:
Low blood sugar. If your blood sugar is low enough, you might feel:
o Confused
o Nervous
o Hungry
o Tingly or weak or
o Show signs like fast heart rate, sweatiness, sleepiness.
Your nurse will ask you about these and record them in case your blood sugar is low, but you should also report these feelings to your nurse if you are having them because they can

SHINE Trial Consent Template Version Date: 01/13/2016
Page 5 of 13
mean your blood sugar may be low and needs to be checked. If your blood sugar stays very low for a long time, this can be serious and result in worsened neurological symptoms, seizures or even death, though this is extremely rare. If your blood sugar goes low but is brought back to normal quickly it is unlikely to result in injury.
The researchers will try to minimize this risk in these ways:
o Your blood sugar levels will be checked frequently to prevent low blood sugar from occurring.
o If you have a finger stick blood sugar of less than 80, the study drug infusion will be temporarily stopped and you will be given glucose (sugar) therapy right away so that your blood sugar will not go too low.
o Your blood sugar level will be checked every 15 minutes until your blood sugar level reaches at least 80 mg/dL. The study drug infusion will be restarted when it is safe to do so.
Problems associated with blood drawing, finger stick blood sugar checks, and IV placement:
o Pain from inserting a needle under the skin surface (common)
o Fainting at or about the time of blood drawing (infrequent)
o Bruising at the site (infrequent)
o Infection at the site (rare).
To allow the study IV solution to be given safely and adequately, during this study, you may occasionally need to have the first IV catheter replaced.
There is a slight risk that your personal information may be accidentally released for reasons other than the ones listed in Section 9 below. The researchers will try to minimize this risk by handling your information carefully, storing it securely, and sharing it only with authorized persons.
As with any research study, there may be additional risks that are unknown or unexpected.
5.2 What happens if I get hurt, become sick, or have other problems as a result of this research?
The researchers have taken steps to minimize the risks of this study. Even so, you may still have problems or side effects, despite the best efforts of the researchers to avoid them. Please tell your regular doctor(s) and/or the researchers listed in Section 10 about any injuries, side effects, or other problems that you have during this study.
Compensation for an injury resulting from your participation in this research is not available from ___________________ Hospital or the sponsor for the study. By signing this form, you do not give up your right to seek payment if you are harmed as a result of being in this study.

SHINE Trial Consent Template Version Date: 01/13/2016
Page 6 of 13
5.3 If I take part in this study, can I also participate in other studies?
Being in more than one research study at the same time, or even at different times, may increase the risks to you. It may also affect the results of the studies. You should not take part in more than one study without approval from the researchers involved in each study.
5.4 How could I benefit if I take part in this study? How could others benefit?
You may not receive any personal benefits from being in this study. The researchers hope the information learned from your participation in the study will increase our knowledge about which way is best to treat patients like you who have suffered a stroke and also have high blood sugar at the hospital. This knowledge will help make it possible to provide the best type of treatment for stroke patients in the future. While you may or may not personally benefit from being in the study, your participation will provide a benefit to others with stroke and to society.
5.5 Will the researchers tell me if they learn of new information that could change my willingness to stay in this study?
Yes, the researchers will tell you if they learn of important new information that may change your willingness to stay in this study. If new information is provided to you after you have joined the study, it is possible that you may be asked to sign a new consent form that includes the new information. This information may be shared with you at the time of the 6-week phone call or the scheduled follow-up study visit.
6. OTHER OPTIONS
6.1 If I decide not to take part in this study, what other options do I have?
You do not have to join this study or sign this Consent to Participate in Research. By not signing you would not have the opportunity to receive the study treatments. If you choose not to participate, during your hospitalization, you will receive standard care for stroke patients, which usually would include about 6 finger sticks daily and subcutaneous insulin shots to treat your blood sugar if it is high.
7. ENDING THE STUDY
7.1 If I want to stop participating in the study, what should I do?
You are free to leave the study at any time. If you leave the study before it is finished, there will be no penalty to you. You will not lose any benefits to which you may otherwise be entitled. If you choose to tell the researchers why you are leaving the study, your reasons for leaving may be kept

SHINE Trial Consent Template Version Date: 01/13/2016
Page 7 of 13
as part of the study record. If you decide to leave the study before it is finished, please tell one of the persons listed in Section 10 “Contact Information” (below).
7.2 Could there be any harm to me if I decide to leave the study before it is finished?
There is no harm to you if you decide to leave the study before it is finished. You will still be treated for high blood sugar as part of your standard care.
7.3 Could the researchers take me out of the study even if I want to continue to participate?
Yes. There are many reasons why the researchers may need to end your participation in the study. Some examples are:
The researcher believes that it is not in your best interest to stay in the study.
You become ineligible to participate.
Your condition changes and you need treatment that is not allowed while you are taking part in the study.
You do not follow instructions from the researchers.
The study is suspended or canceled.
8. FINANCIAL INFORMATION
8.1 Who will pay for the costs of the study? Will I or my health plan be billed for any costs of the study?
You will not be required to pay any extra money because of this study, but you will be responsible for the costs of the standard care for your stroke. The study will pay for the study treatment medicine that is not standard care and for the materials for the extra research finger stick blood sugar tests needed.
The normal costs of taking care of you for your stroke will have to be paid for by you or your insurance company. These costs are the same as you would be responsible for if you were not in this research study. The 3-month clinical follow up with your doctor for continuing stroke treatment and care which is not part of the scheduled research follow up activities will be not be paid for as part of the study.
8.2 Will I be paid or given anything for taking part in this study?
[Select from the following two statements (and delete the other) as appropriate for your local procedure:]
You will not receive payment for participating in this study.

SHINE Trial Consent Template Version Date: 01/13/2016
Page 8 of 13
You will receive reimbursement to compensate for transportation costs.
8.3 Who could profit or financially benefit from the study results?
Dr. Rattan Juneja, the study endocrinologist from the Indiana University, may profit from the commercial sales of the GlucoStabilizer. The terms of this arrangement have been reviewed and approved by Indiana University and a management plan is in place in accordance with its conflict of interest policies.
9. CONFIDENTIALITY OF SUBJECT RECORDS AND AUTHORIZATION TO RELEASE YOUR PROTECTED HEALTH INFORMATION
The information below describes how your privacy and the confidentiality of your research records will be protected in this study.
9.1 How will the researchers protect my privacy?
Any time information about you is collected there is a potential risk for loss of confidentiality. Every effort will be made to keep your information confidential; however, this cannot be guaranteed. Information about you collected for this research study will remain confidential, unless you give your permission to share it with others or if we are required by law to release it.
9.2 What information about me could be seen by the researchers or by other people? Why? Who might see it?
Signing this form gives the researchers your permission to obtain, use, and share information about you for this study, and is required in order for you to take part in the study. Information about you may be obtained from any hospital, doctor, and other health care provider involved in your care, including:
Hospital/doctor's office records, including test results (X-rays, blood tests, urine tests, etc.)
Mental health care records (except psychotherapy notes not kept with your medical records)
Alcohol/substance abuse treatment records
Your AIDS/HIV status
All records relating to your stroke, the treatment you have received, and your response to the treatment
Billing information
There are many reasons why information about you may be used or seen by the researchers or others during or after this study. Examples include:
The researchers may need the information to make sure you can take part in the study.
The researchers may need the information to check your test results or look for side effects.

SHINE Trial Consent Template Version Date: 01/13/2016
Page 9 of 13
University, Food and Drug Administration (FDA), and/or other government officials may need the information to make sure that the study is done in a safe and proper manner.
Study sponsors or funders, or safety monitors or committees, may need the information to: o Make sure the study is done safely and properly
o Learn more about side effects
o Analyze the results of the study
Insurance companies or other organizations may need the information in order to pay your medical bills or other costs of your participation in the study.
The researchers may need to use the information to create a databank of information about your condition or its treatment.
Information about your study participation may be included in your regular medical record.
Federal or State law may require the study team to give information to government agencies. For example, to prevent harm to you or others, or for public health reasons.
The following groups may access your information as part of your participation in this study:
Members of the SHINE Trial and the Neurological Emergency Trials Network Leadership Teams who are located at the following centers: University of Virginia, University of Texas Southwestern Medical Center, Georgia Health Sciences University, Medical University of South Carolina, University of Michigan, Indiana University, Rush University Medical Center-Chicago.
The results of this study could be published in an article, but would not include any information that would let others know who you are.
A description of this clinical trial will be available on http://www.clinicaltrials.gov, as required by U.S. Law. This web site will not include information that can identify you. At most, the web site will include a summary of the results. You can search the web site at any time.
9.3 What happens to information about me after the study is over or if I cancel my permission?
As a rule, the researchers will not continue to use or disclose information about you, but will keep it secure until it is destroyed. Sometimes, it may be necessary for information about you to continue to be used or disclosed, even after you have canceled your permission or the study is over. Examples of reasons for this include:
To avoid losing study results that have already included your information
To provide limited information for research, education, or other activities (This information would not include your name, social security number, or anything else that could let others know who you are.)
To help University and government officials make sure that the study was conducted properly
9.4 When does my permission expire?

SHINE Trial Consent Template Version Date: 01/13/2016
Page 10 of 13
Your permission will not expire unless you cancel it. You may cancel your permission at any time by writing to the researchers listed in Section 10 "Contact Information" (below). If you decide not to continue participating or if you withdraw consent, any information already collected about you while you were in the study will be used in this study.
The collected clinical information will be available indefinitely. This gives the investigators information to be used for medical, scientific, educational or research purposes.
10. CONTACT INFORMATION
10.1 Who can I contact about this study?
Please contact the researchers listed below to:
Obtain more information about the study
Ask a question about the study procedures or treatments
Talk about study-related costs to you or your health plan
Report an illness, injury, or other problem (you may also need to tell your regular doctors)
Leave the study before it is finished
Express a concern about the study
Principal Investigator: Mailing Address: Telephone:
Study Coordinator: Mailing Address: Telephone:
You may also express a concern about a study by contacting the Institutional Review Board listed below.
IRB Name: Mailing Address: Telephone:
11. RECORD OF INFORMATION PROVIDED
11.1 What documents will be given to me?
Your signature in the next section means that you have received copies of all of the following documents:
This "Consent to be Part of a Research Study" document. (Note: In addition to the copy you receive, copies of this document will be stored in a separate confidential research file and may be entered into your regular medical record.)

SHINE Trial Consent Template Version Date: 01/13/2016
Page 11 of 13

SHINE Trial Consent Template Version Date: 01/13/2016
Page 12 of 13
Optional SHINE Sub-Study: I-SPOT (Insight on Selected Procoagulation markers and Outcomes in stroke Trial)
------------------------ Use the following page only if applicable ----------------------
In addition to the main research study, there may be an optional sub-study in which you may qualify to participate. Participating in this optional sub-study is important to the research; however you may participate in SHINE without agreeing to take part in this sub-study. The purpose of the sub-study is to gather information about the effects of high blood sugar and insulin treatment on blood clotting. The blood samples collected will be used for this purpose only. If you decide to participate, we will collect two blood samples, one before the SHINE study treatment is started, and the second will be collected about two days later. Each blood sample will be approximately one tablespoon of blood. With blood drawing, you may experience local pain and, rarely, infection or bruising. Your blood samples will be labeled with your SHINE study identification number and the visit date. No personal information will be on the tubes used to store the blood samples. Your blood samples will be stored at Temple University for about 3 years after the completion of the SHINE study or up to 10 years. The results of these tests will not have an effect on your care, and neither you nor your doctor will receive the results. You are free to choose not to participate. If you do participate, you are free to withdraw at any time up until the samples are de-identified, which means the samples are not linked to you. If you have any questions, or you would like to withdraw your blood sample, you should contact the principal investigator listed in this form. If you choose not to participate or if you withdraw, there will be no penalty to you. You will not lose any benefits to which you may otherwise be entitled. It will not affect your ability to participate in the SHINE trial. Please initial whether you permit the collection of a repository blood sample as described above: ______ YES, I give my permission for blood samples to be collected for the sub-study I-SPOT ______ NO, I do not give my permission for blood samples to be collected the sub-study I-SPOT

SHINE Trial Consent Template Version Date: 01/13/2016
Page 13 of 13
12. SIGNATURES
Research Subject:
I understand the information printed on this form. I have discussed this study, its risks and potential benefits, and my
other choices with ____________________. My questions so far have been answered. I understand that if I have
more questions or concerns about the study or my participation as a research subject, I may contact one of the people
listed in Section 10 (above). I understand that I will receive a copy of this form at the time I sign it and later upon
request. I understand that if my ability to consent for myself changes, either I or my legal representative may be asked
to re-consent prior to my continued participation in this study.
Signature of Subject: Date: ______ Time:__________
Name (Print legal name):
Patient ID: ___________________ Date of Birth: ________________________________
Legal Representative (if applicable): Signature of Person Legally
Authorized to Give Consent Date: ______ Time:__________
Name (Print legal name): ____________ __ Phone: _______________________
Address:
Check Relationship to Subject:
Parent Spouse Child Sibling Legal Guardian Other:
Reason subject is unable to sign for self:___________________________ _____________________
__________________________________________________________________ ______________
Principal Investigator (or Designee):
I have given this research subject (or his/her legally authorized representative, if applicable) information about this
study that I believe is accurate and complete. The subject has indicated that he or she understands the nature of the
study and the risks and benefits of participating.
Name: Title:
Signature: Date: Time:________
Witness (optional):
I observed the above subject (or his/her legally authorized representative, if applicable) sign this consent document.
Name:
Signature: Date of Signature: Time:__________

NNEETTTTWWeebbDDCCUU™™
UUsseerr MMaannuuaall
NETT: Neurological Emergency
Treatment Trials Data Coordination Unit
Medical University of South Carolina 24Apr2015 Version 19

2
TABLE OF CONTENTS
GENERAL ............................................................................................................ 6
WebDCU™ General Overview............................................................................ 6
Data Manager Contact Information ................................................................... 6
System Requirements for Users ........................................................................ 7
Suggested Settings ............................................................................................ 7
System Security and Password Protection ...................................................... 7
User Accounts ..................................................................................................... 8
System Login ...................................................................................................... 9
Main Menu .........................................................................................................11
General Operations Guide ...............................................................................12
Adding a Record to a Table .............................................................................12
Viewing/Sorting/Filtering Records ..................................................................13
Editing Records in a Table ...............................................................................18
‘My Lists’ Bookmarks .......................................................................................19
Basic Summary Tool ........................................................................................21
NETT REGULATORY DOCUMENT DATABASE ..............................................21
[Network Management] Button ........................................................................22
[Project Management] Button ..........................................................................23
[Regulatory Documents] Button ......................................................................25
Viewing Reports of Required Documents ......................................................26
Submitting Regulatory Documents ................................................................. 26
Sharing Documents ..........................................................................................30
Editing Rejected Documents ...........................................................................30

3
Adding from Existing Documents ...................................................................30
Changing the Status of Submitted Regulatory Documents ..........................31
STUDY DATABASES ........................................................................................31
Locating Project Documents ...........................................................................32
Printing Study Casebooks or Individual Worksheets ....................................32
Screen Failure Log ............................................................................................33
Adding and Deleting a Subject ........................................................................34
Adding and Deleting a Subject Visit ................................................................35
Entering/Viewing CRF Data ..............................................................................36
Dismissing Protocol Violations and Warnings ...............................................38
Submitting CRF Data ........................................................................................39
Deleting CRFs ...................................................................................................39
Adding Extra CRFs ...........................................................................................40
Interpreting the CRF Collection Table .............................................................40
Creating and Closing Data Clarification Requests (DCRs) ...........................41
Responding to Data Clarification Requests (DCRs) ......................................43
Monitor Verification (For Monitors) ................................................................. 45
Audit Trail ..........................................................................................................45
Study Calendar ..................................................................................................46
Entering Community Consultation Summaries ..............................................46
Entering Public Disclosure Summaries ..........................................................46
STUDY-SPECIFIC INFORMATION....................................................................47
POINT SPECIFICS .............................................................................................47
Data Entry Timelines ........................................................................................47
POINT Study Drug Tracking Module ...............................................................48

4
Study Drug Labeling .........................................................................................48
Study Drug Shipping ........................................................................................48
Study Drug Receipt ...........................................................................................48
Study Drug Damage ..........................................................................................49
Study Drug Expired ..........................................................................................49
Randomization ..................................................................................................49
Unblinding .........................................................................................................50
POINT Medical Safety Monitoring ....................................................................51
Clinical Event/SAE Site Manager Review (For Site Managers Only) ............51
Clinical Event Coordinator Review ..................................................................52
Adjudication Review .........................................................................................52
SHINE SPECIFICS .............................................................................................53
Data Entry Timelines ........................................................................................53
Randomization ..................................................................................................54
Glucose Stabilizer/Study Computer ................................................................55
Stroke Mimic Review ........................................................................................55
SHINE SAE Safety Monitoring .........................................................................55
Site Manager Review (For the Site Manager Only) .........................................55
Internal Quality and Safety Reviewer Review ................................................. 56
Independent Medical Safety Monitor Review .................................................57
Blinded Assessors ............................................................................................58
Emergency Unblinding .....................................................................................58
ESETT SPECIFICS ............................................................................................58
Obtaining a User Account and Defining Study Team Members ...................58
Adding Study Team Member User Accounts .................................................59

5
Adding/Editing Study Team Members on Electronic DOA Log ....................60
Regulatory Documents .....................................................................................62
Viewing Status of Required Documents .........................................................63
Submitting Regulatory Documents ................................................................. 64
Editing Rejected Documents ...........................................................................66
Approving Electronic DOA Logs .....................................................................66
Approving/Rejecting Documents ....................................................................67

6
GENERAL
WEBDCU™ GENERAL OVERVIEWSeveral hundreds of clinical trials are being conducted at any one time in the United States. Investigators of these trials invest vast amounts of resources and energy into conducting these studies and often face daily challenges with data management and data quality control. The management, transfer and storage of data generated from these studies create several pitfalls to conducting successful clinical trials including lack of real-time data reporting, lack of resources for recording the data, endless amounts of paper and lack of document storage space.
The WebDCU™ system combines study tools required for these trials into one user-friendly system. Data is directly entered into the database via a secure internet connection at each clinical site. Pre-programmed logic checks allow for automatic notification of rule violations (i.e., out of range values, inclusion/exclusion criteria deviations, dates) which are displayed on the data entry screen for quick and efficient resolution at the site. This allows the clinical sites and principal investigators access to real time data.
DATA MANAGER CONTACT INFORMATION
NETT Regulatory Database: Cassidy Conner, [email protected], 843-876-1105Catherine Dillon, [email protected], 843-876-1942
POINT Database: Aaron Perlmutter, [email protected], 843-876-1261Adam Henry, [email protected], 843-792-3980 Kristina Hill, [email protected], 843-792-1453
SHINE Database: Kavita Patel, [email protected], 843-876-1167
ESETT Database: Cassidy Conner, [email protected], 843-876-1105Kristina Hill, [email protected], 843-792-1453

7
SYSTEM REQUIREMENTS FOR USERSAccess to WebDCU™ requires a computer with high speed internet access. (Please note that JavaScript must be enabled.)
As required by 21 CFR part 11, any WebDCU™ user who leaves the workstation must log off the system. Additionally, automatic system protection (screen saver and password protection) must be enabled for SHORT periods of inactivity to protect against unauthorized data entry.
Suggested Settings
Website layout is optimized for Internet Explorer 7 and a screen resolution of 1280 x 1024. A minimum resolution of 1024 x 768 is strongly recommended.
Officially supported browsers: Internet Explorer 6 or greater; Firefox 2.0 or greater; Opera 9.0 or greater.
SYSTEM SECURITY AND PASSWORD PROTECTION
Security of the database is dependent on maintaining individual password security.
Each user is issued an individual password which is not to be shared with any other user. Recording of passwords which might expose them to viewing by others is strictly forbidden.
Keep your password secure at all times. It is imperative that you do not share your user account information with others. The audit trail that is maintained in the study database is directly linked to your username. Sharing account information falsifies the audit trail and is contrary to Good Clinical Practice. If you feel the security of your password has been compromised, immediately change your password. To do so, click on [Toolbox] on the main menu page, then click ‘Change Password’ and follow the instructions.
An acceptable password will consist of at least 6 characters, including at least one letter and at least one number. If a valid username was used, that account will be locked, and can only be reactivated by request to DCU.
Due to security and technical issues, please refrain from using any auto-complete setting that store passwords.

8
When a user is logged in to WebDCU™ but is inactive for 20 minutes, the screen will be blocked. To activate the screen, the user will need to correctly re-enter their password. If the password is entered correctly, the screen will be activated, and all data previously entered will be present. If the user fails to enter their username/password combination correctly 3 times, they will be automatically logged out and all data will be lost. After 90 minutes of inactivity the user will be logged off from the website and will have to log back in, and any unsaved data will be lost. Any activity which involves sending or receiving data to or from the website will maintain an active connection. Scrolling through a report, reading or typing an extended narrative will not be detected as activity.
To prevent loss of newly entered CRF data, the form should be saved before leaving the system inactive. Any data not saved will be lost if the system times out.
USER ACCOUNTSUser accounts are activated by DCU data managers, and each account is assigned different user permissions based on the roles and responsibilities of study team members. See below for descriptions of each type of user account and the steps involved in activating an account.
STUDY SPECIFIC USER ACCOUNTS
Before WebDCUTM user accounts can be activated, Hub Project Managers or designees must add the study team member to the [People] table and the [Project Spoke Team Member] table in the NETT regulatory database.After this has been done, accounts will be set up based on the following permissions:
Users that will need to enter data or randomize subjects: These users must be assigned a role in the [Project Spoke Team Member] and the user’s data training certificate must then be uploaded to the NETT regulatory database. After the data training certification has been uploaded and accepted, accounts will be automatically activated (within 3 days) by DCU. Because of automatic activation, it is not necessary to contact DCU to activate accounts with data entry permissions.
Users that will need to enter/manage pharmacy data: After being added to the [Project Spoke Team Member] table with a site

9
pharmacy role, pharmacy data training will need to be completed (as applicable) and a certificate uploaded to the NETT regulatory database by the Hub Project Manager or designee. After this has been uploaded and accepted, accounts will be automatically activated (within 3 days). Because of automatic activation, it is not necessary to contact WebDCUTM to activate these accounts.
Users that only need to view data: After the Hub Project Managers or designee has added a team member to the [Project Spoke Team Member] table, users who will not need to enter data or randomize subjects can request data view permissions for WebDCUTM. It will be necessary to contact DCU to activate these types of accounts.
NETT REGULATORY DATABASE USER ACCOUNTS
Contact DCU in order to activate user accounts with permissions to access the NETT regulatory database. There are two types of permissions that can be granted in the NETT regulatory database:
Hub Project Manager Permissions: includes permissions to upload/edit regulatory documents and to manage project management tables.
View Permissions: includes permissions to view regulatory documents and project management tables.
DCU will notify the user of his/her user name and temporary password once the account has been created. The username will always be the email address on record in the [People] table of the database. If an email address is changed in the database, the username is similarly changed.
NOTE: It is critical that Hub personnel immediately notify DCU of personnel who are no longer working on NETT and users whose
permissions have changed, so that account access can be terminated and/or adjusted accordingly. Notification in advance is preferred.
SYSTEM LOGINLogging in to WebDCUTM
If you do not have a user name or password and need access to the website, the primary study coordinator should refer to the previous section “User Accounts”.

10
Turn on your computer, click in your internet browser (Internet Explorer 7 or higher is recommended) and type https://webdcu.musc.edu/login.asp into the address bar at the top of the page.
Type in your user name (email address) and your password and click [Sign In]. Once you are logged in, icons for all of the studies in which you are an active member will be displayed.
Select the project icon to go to the main menu page of each project. To return to the project selection page, click on the WebDCUTM icon in the upper left corner.The first time you log in you will automatically be directed to the ‘Change password’ page. Enter your current or temporary password into the first box. Enter the new password you have chosen into the 2 boxes below and click on [Save]. Please note that your password must be 6-35characters long and must only contain letters, numbers, and spaces. At least one upper case letter, one lower case letter and one number must be included. Upon changing your password you will automatically exit from WebDCU™. You can then log in using your new password.
The email address entered will be that person’s username. For that reason, email addresses cannot be edited after they are saved. To change an email address that has already been entered, contact the DCU data manager.
Logging out of WebDCUTM
In order to protect the integrity of the trial data always log out prior to leaving your workstation. To log out, either click on [Sign Out] in the upper

11
right hand corner of the main menu page or shut down the application by closing your internet browser.
MAIN MENU
Features on the Main Menu Page
Note: Icons that display on the homepage are specific to each user and the permissions that they are assigned. Therefore, the icons on the home page for each study team member may vary.
[Subject CRF Binder] Allows you to add Visits to existing Subjects and enter or edit Case Report Form (CRF) data.
[Study Progress] Allows you to view subject enrollment and enter monthly Screen Failure Logs.
[Data Management] Allows you to view data management information such as CRF, rule, and DCR status.
[Safety Monitoring] Allows medical safety monitor to review serious adverse events.
[Site Management] Allows you to review site status.
[Drug Tracking] Allows you to access drug accountability (for site pharmacist only).
[CRF Data List] This icon contains a separate icon for each CRF. Selecting an individual CRF icon populates a list of the forms that have been entered specific to that CRF. The selection can be further refined by applying the filters available to each column.
[Project Setup] Allows you to review the Data Collection Schedule and Study Design.

12
[User Management] Allows you to request user accounts for study team members, view a listing of your study team members, and submit your site’s Delegation of Authority (DOA) Log.
[Regulatory Document] Allows you to submit and view regulatory documents.
[Help & Toolbox] Allows you to change your WebDCUTM password and access help instructions/site navigation tools. This is also where you will find ‘Project Documents.’ ‘Project Documents’ contains documents that have trial-wide importance, such as the protocol, study book sections, and the Manual of Procedures (MOP).
[Project Management] (Not Pictured) Where you will find CIRB minutes and a module for reporting Unanticipated Events to the CIRB.
[Data Monitoring] (Not Pictured) Allows on site monitors to schedule monitoring visits and submit monitoring reports.
GENERAL OPERATIONS GUIDE The WebDCUTM data management system has a generic design. This means that the menu functions and database format will be similar throughout the system, even though the tables and questions may differ.
Adding a Record to a Table
From any List Record page, click the [Add New] button located at the top right-hand corner of the table.
13
This will show the form. Questions numbered in red are required. Questions numbered in black are optional. After entering the required information, click the [Save Record] button at the bottom of the page.
The browser will be forwarded to the ‘Record View’ page, which allows the user to view the information that was just entered. To return to the List Record page, click the [List Record] button in the bottom-right hand corner of the page.
Viewing/Sorting/Filtering Records

14
The sorting and filtering features are available for all List Record pages.
To view additional details about a record, click the blue number link in the first column of the ‘List Record’ table.
To sort any column in ascending order, click the column header once. To sort any column in descending order, click the column header twice. Clicking the header a third time will remove sorting order for that column. Please note that you can sort more than one column at a time.
To filter records, select the criteria you want to filter by clicking on a value within any cell in the column. When a cell is clicked, it will be highlighted in a light orange color. You may deselect a cell by clicking on it again or by clicking on another cell.
The field that was selected will display in the upper left hand corner of the screen. You may make any changes to the selection by either typing in a different selection or making another choice from the drop down box. Then click the appropriate filter operator button (< >,=, like, etc.) to apply the filter. Different operators are provided based on the field type selected. The records will be filtered by the parameters selected.
15

16
The Filter feature will allow users to save filters currently being applied to a table. There are two types of filters, System filters and Personal filters.
System Filters are filters that are provided to all users and are setup and maintained by the DCU. Each table will have specific system filters based on the records found in the table.
The ‘Page Action’ Dropdown Box is located in the upper right-handcorner of the table, just below the menu tabs. Click on the dropdown box and select the filter you wish to apply. The listing will then show only those records which meet the filter criteria.
To remove a single filter, click on the in the upper left hand corner of the screen adjacent to the filter you would like to remove. Note: This button will be viewable only when a filter has been applied.
To remove the entire filter (after multiple filters have been applied), click on the in the upper left hand corner of the screen. Note: This button will be viewable only when multiple filters have been applied.
17
Personal Filters are filters created by specific users and are unique to each specific user. To save a personal filter, apply the sorting/filtering criteria, and then click the button in the top left hand corner of the table.

18
You will be forwarded to the Edit Record page for the ‘My Lists’ table where you can name your query. Then click [Save Record].
To return to the page you were working on, click on the Link icon.Personal queries are accessible by clicking on the ‘Page Actions’ drop down box in the upper left hand corner of any ‘List Record’ page.
To edit or delete personal queries go to ‘Project Management’ and click on ‘My Lists’. Click on the blue number link in the first column of the ‘List Record’ table adjacent to the record you would like to edit/delete. Click [Edit Record] or [Delete Record] at the bottom left-hand side of the page.
You must confirm the deletion of any personal query by entering the reason for deletion and your password, then click [Confirm Delete].
Editing Records in a Table
If a record needs to be updated/edited, click on the blue number link in the first column of the ‘List Record’ table.
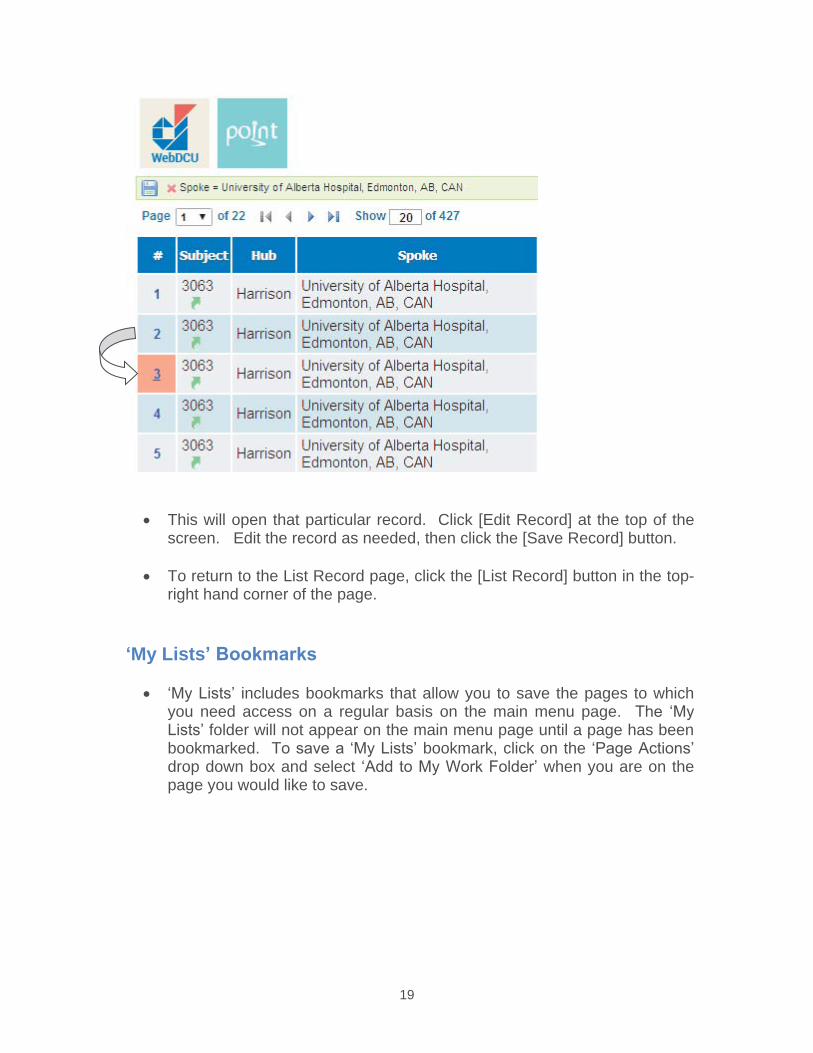
19
This will open that particular record. Click [Edit Record] at the top of the screen. Edit the record as needed, then click the [Save Record] button.
To return to the List Record page, click the [List Record] button in the top-right hand corner of the page.
‘My Lists’ Bookmarks
‘My Lists’ includes bookmarks that allow you to save the pages to which you need access on a regular basis on the main menu page. The ‘My Lists’ folder will not appear on the main menu page until a page has been bookmarked. To save a ‘My Lists’ bookmark, click on the ‘Page Actions’ drop down box and select ‘Add to My Work Folder’ when you are on the page you would like to save.

20
You will be forwarded to the Edit Record page for the ‘My Lists’ table where you can name your bookmark. Then click [Save Record].
To return to the page you were working on, click on the Link icon.
To edit or delete ‘My Lists’ bookmarks go to ‘Toolbox’ and click on ‘My Lists’. Click on the blue number link in the first column of the ‘List Record’ table adjacent to the record you would like to edit/delete. Click [Edit Record] or [Delete Record] at the top right-hand side of the page.
You must confirm the deletion of any personal bookmark by entering the reason for deletion and your password, then click [Confirm Delete].

21
Basic Summary Tool
The basic summary tool allows authorized users to summarize the data to which they have access.
From any list record view, select ‘Basic Summary’ from the Page Actions drop down box located in the top right-hand corner of the table.
To return to the page you were working on, click ‘back’ in the upper right hand corner of the summary report.
NETT REGULATORY DOCUMENT DATABASETo get to the NETT Regulatory Document Database, Select ‘NETT’ from the project selection screen.
The ‘Project Management’ and ‘Network Management’ buttons containinterfaces for managing the operations of NETT projects. The ‘Regulatory Document’ button contains interfaces for managing the regulatory documents required for NETT projects.

22
[Network Management] Button
Hub Any hub participating in any NETT study must be added to the [Hub] table. To add a Hub to this table, CCC should contact the appropriate data manager for the study (See previous section ‘Contact Information of Data Managers’). A unique Hub ID number will be assigned when the Hub is added to the Hub table. No Hubs should ever be deleted from this table.
Spoke Any spoke participating in any NETT study must be added to the [Spoke] table. To add a spoke to this table, CCC should contact the appropriate data manager for the study (See previous section ‘Contact Information of Data Managers’). A unique Spoke ID number will be assigned when the Spoke is added to the Spoke table. No Spokes should ever be deleted from this table.
People Anyone participating in NETT (across all hubs and projects) must be entered into the [People] table. Entering a person in the People table will allow that person to be later linked to specific projects, spokes, and documents. Hub Project Managers or designees can add people to this table by clicking on ‘Project Management’, then ‘People’. Hub personnel can then enter the person’s name and contact information. No people should ever be deleted from this table. The email address entered will be that person’s username. For that reason, email address cannot be edited by the Hub Project Manager or CCC after they are saved. To change a person’s email address, Hub personnel should contact the appropriate data manager for the study (See previous section ‘Contact Information of Data Managers’). If someone in the [People] table needs to be associated with more than one hub, the site should contact the appropriate data manager (See previous section ‘Contact Information of Data Managers’).
Regulatory Documents Any regulatory document that will be collected for any NETT study must be added to the [Regulatory Documents] table. CCC can add a regulatory document to the Regulatory Documents table by clicking on ‘Project Management’, then ‘Regulatory Documents’. CCC will enter the information required. If a document is project specific, CCC will include the project name in the naming convention (i.e. ALIAS 1572, RAMPART 1572, etc.). If a document is not project specific, CCC will not include the project name in the naming convention (i.e. medical license, CV). The CCC will indicate if this is a people, spoke, or EMS document. Please note that 1572’s are considered to be people documents.

23
Strict version control must be maintained in this table. If a document has been updated from its previous version, CCC must add the revised document as a new document. Documents should not be deleted or renamed. For example: If an amendment is made to a protocol, ‘ALIAS version 1 IRB approval’ SHOULD NOT be changed to ‘ALIAS Version 2 IRB approval’. ‘ALIAS version 1 IRB approval’ must remain the Regulatory Documents table and ‘ALIAS Version 2 IRB approval’ should be added to it. No regulatory documents should ever be deleted from this table.
[Project Management] Button
Project Hub Each Hub participating in a particular project/study must be linked in the [Project Hub] table. CCC can link a Hub to a project by clicking on ‘Project Management’, then ‘Project Hub’. CCC will enter the information required.
No Project Hubs should ever be deleted from this table. Hubs that are no longer participating in a project should be changed to a status of ‘inactive’ in the Project Hub table.
Project Spoke Each spoke participating in a particular project/study must be linked in the [Project Spoke] table. Contact the appropriate data manager or CCC project manager to request that a spoke be linked to the project/study in the [Project Spoke] table. After the spoke has been added to the project, Hub Project Managers or designees can edit the project spoke to add the spoke address and set the project status to either ‘preparing- high priority or ‘preparing- low priority’ until the spoke is regulatory ready and is released to enroll subjects by CCC. After the Project Spoke is released to enroll subjects, the status can only be changed by CCC Project Managers.
The drug shipping address for each project spoke is collected in this table. Hub personnel will indicate whether the spoke will be receiving study drug directly from the Central Pharmacy or not. If the spoke will be receiving study drug directly from the Central Pharmacy, Hub personnel will select ‘yes,’ select the Drug Recipient’s name from the drop down box (this person will first need to entered into the Project Spoke Team Member Table) and enter the shipping address. If the spoke will not be receiving study drug directly from the Central Pharmacy, Hub personnel will select ‘no’ and indicate which project spoke will be supplying study drug by selecting the spoke from the drop down box. Please note that the supplying spoke’s Drug Shipping information must be entered into the Project Spoke table before the receiving spoke can receive study drug. If, at the time of data entry, the drug recipient and shipping address is unknown, click on the response indicating that the site will receive drug

24
directly from the Central Pharmacy, leave the Drug Recipient field blank, and enter ‘To be determined’ in the Drug Shipping Address. CCC Project Managers will review the drug shipping information, prior to releasing a site for enrollment, to make sure the information is accurate and complete.
Once a Project Spoke is regulatory ready, the CCC will edit the Project Spoke status accordingly. The Project Spoke status should be listed as ‘actively enrolling’ once that spoke is released to enroll subjects into the project. If the Project Spoke does not have a status of ‘actively enrolling’, the spoke will not be able to add subjects to the database. The Project Spoke status should be listed as ‘enrollment suspended’ if subject enrollment is temporarily halted at that spoke. (Note: Required regulatory documents will continue to be posted in the in the appropriate Regulatory Document tables for Project spokes that have a status of ‘enrollment suspended’). The Project Spoke status should be listed as ‘study activity suspended’ when the project is completed at that particular spoke, for example, the study is complete or the spoke is no longer participating in the project. (Note: Required regulatory documents will no longer be posted in the Regulatory Document tables for Project spokes that have a status of ‘study activity suspended’). No Project Spokes should ever be deleted from this table.
Project Spoke Team Member Table Each person participating in a particular project/study at a particular spoke must be linked in the [Project Spoke Team Member] table. Hub Project Managers or designees can link a person to a project spoke by clicking on ‘Project Management’, then ‘Project Spoke Team Member’. Hubpersonnel will enter the information required.
No Project Spoke Team Members should ever be deleted from this table. Project Spoke Team Members who are no longer participating should be changed to a status of ‘inactive’. (Note: Required regulatory documents will no longer be posted in the Regulatory Document tables for Project Spoke Team Members that have a status of ‘inactive’).
Reg Doc Requirements Each regulatory document to be collected for a particular project must be linked to the [Reg Doc Requirements] table. CCC can link a required regulatory document to a project by clicking on ‘Project Management’, then ‘Reg Doc Requirements’. CCC will enter the information required.
Reg Doc Requirements should not be deleted and should only be changed from ‘currently required’ to ‘not currently required’ after much thought. For example: If an amendment is made to a protocol, ‘ALIAS version 1 IRB approval’ may be changed to ‘not currently required’ provided there are no outstanding ‘ALIAS version 1 IRB approvals’ at the

25
spokes. ‘ALIAS Version 2 IRB approval’ should be added to this table at that time as ‘currently required’.
[Regulatory Documents] Button
Required Doc Status Report This table is a view only report that allows users to view the status of required spoke/people/EMS documents. Only requirements for active entities (spoke/people/EMS) are listed here. Deferment information is also listed here. Documents cannot be uploaded here. Documents must be uploaded via the ‘Submit & Update Docs’ interface. Clicking the icon will take the user directly to the ‘Submit & Update Docs’ interface.
Required Doc Status Report CCC This table is allows the CCC to view the status of all required spoke/people/EMS documents across sites. Only requirements for active entities (spoke/people/EMS) are listed here. Deferment information is also listed here. Documents cannot be uploaded here. Documents must be uploaded via the ‘Submit & Update Docs' interface. Clicking the icon will take the user directly to the ‘Submit & Update Docs’ interface.
Document Collection Report This table is a view only report that allows users to view all documents that have been uploaded. Each document is listed as either being current or not to allow for ease of filtering for the most recent document collected. Deferment information is also listed here. Clicking the icon will take the user directly to the ‘Submit & Update Docs’ interface.
Document Collection Report CCC This table is a view only report that allows the CCC to view all documents that have been uploaded across sites. Each document is listed as either being current or not to allow for ease of filtering for the most recent document collected. Deferment information is also listed here. Clicking the icon will take the user directly to the ‘Submit & Update Docs’ interface.
Submit & Update Docs This table is where Hub Project Managers or designees can submit new spoke/people/EMS documents or update rejected documents.
Submit & Update Docs- CCCThis table is where CCC can review and accept/reject documents that are uploaded by Hub Project Managers.

26
Viewing Reports of Required Documents
To view a listing of the documents that are required at your Hub, click on ‘Regulatory Document’ from the main menu page. Then click on ‘Required Doc Status Report’.
This will provide you with a listing of the documents required to be
collected at your Hub (for all projects) as well as the submission status of each document. System queries are provided to allow the user to easily filter for missing, expired, pending, and rejected documents. A status that is empty/missing indicates that the document has not yet been submitted by the Hub Project Manager. A status of ‘pending’ indicates that the document has been uploaded by the Hub Project Manager but not yet reviewed by the CCC. While the status is ‘pending’, Hub ProjectManagers have the option of editing their entries. A status of ‘accepted’ indicates that the document has been uploaded by the Hub Project Manager and been accepted by the CCC. Once ‘accepted’, Hub Project Managers will be unable to edit the entry. A status of ‘rejected’ indicates that the document has been uploaded by the Hub Project Manager,rejected by the CCC, and requires correction from the Hub Project Manager. Hub Project Managers should edit a document when it has a status of ‘rejected’ rather than creating a new document. When the rejected document has been edited, click ‘save record.’ This will move the document back to pending status for CCC review.
Submitting Regulatory Documents
To submit regulatory documents, the Hub Project Manager or designees can click on [Submit & Update Docs] under [Regulatory Document] located in the NETT study database on the Project Selection page.
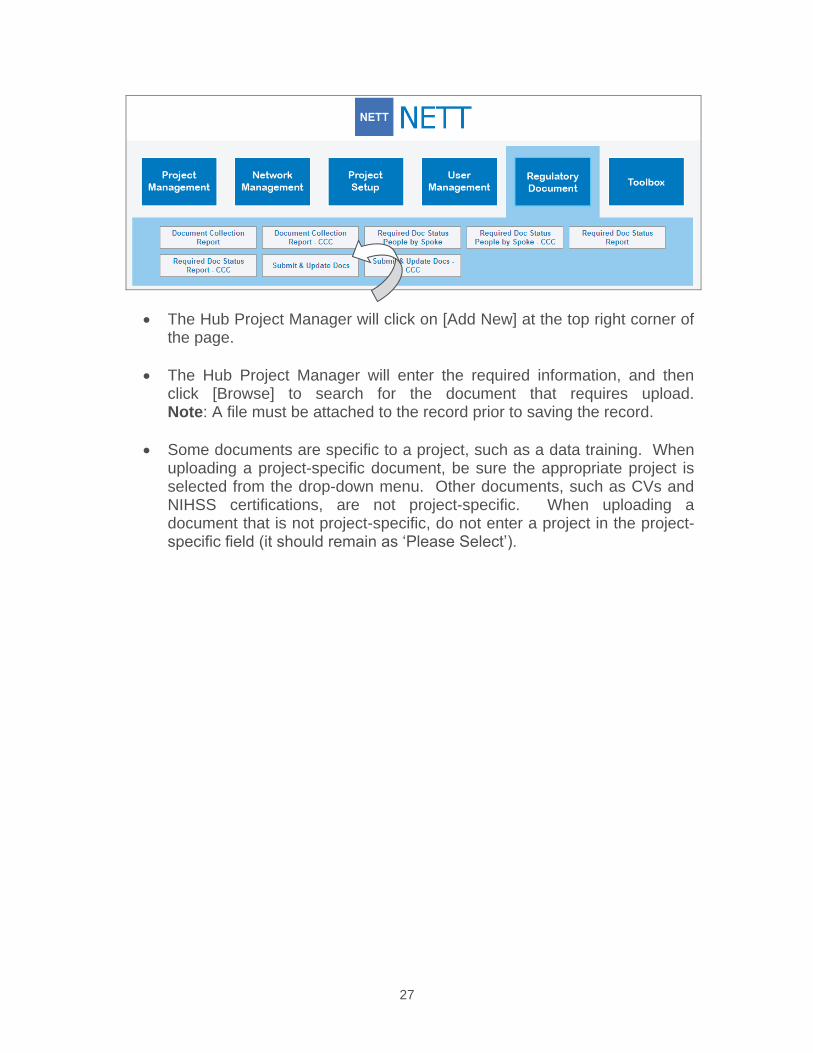
27
The Hub Project Manager will click on [Add New] at the top right corner of the page.
The Hub Project Manager will enter the required information, and then click [Browse] to search for the document that requires upload. Note: A file must be attached to the record prior to saving the record.
Some documents are specific to a project, such as a data training. When uploading a project-specific document, be sure the appropriate project is selected from the drop-down menu. Other documents, such as CVs and NIHSS certifications, are not project-specific. When uploading a document that is not project-specific, do not enter a project in the project-specific field (it should remain as ‘Please Select’).

28
Once the desired file is selected, the Hub Project Manager will click on [Upload File] to upload the document. File Upload Restrictions: Only Adobe PDF files less than 2MB can beuploaded. If you are using Adobe Acrobat Professional, set your options to ‘higher compression’ (as opposed to ‘higher quality’).
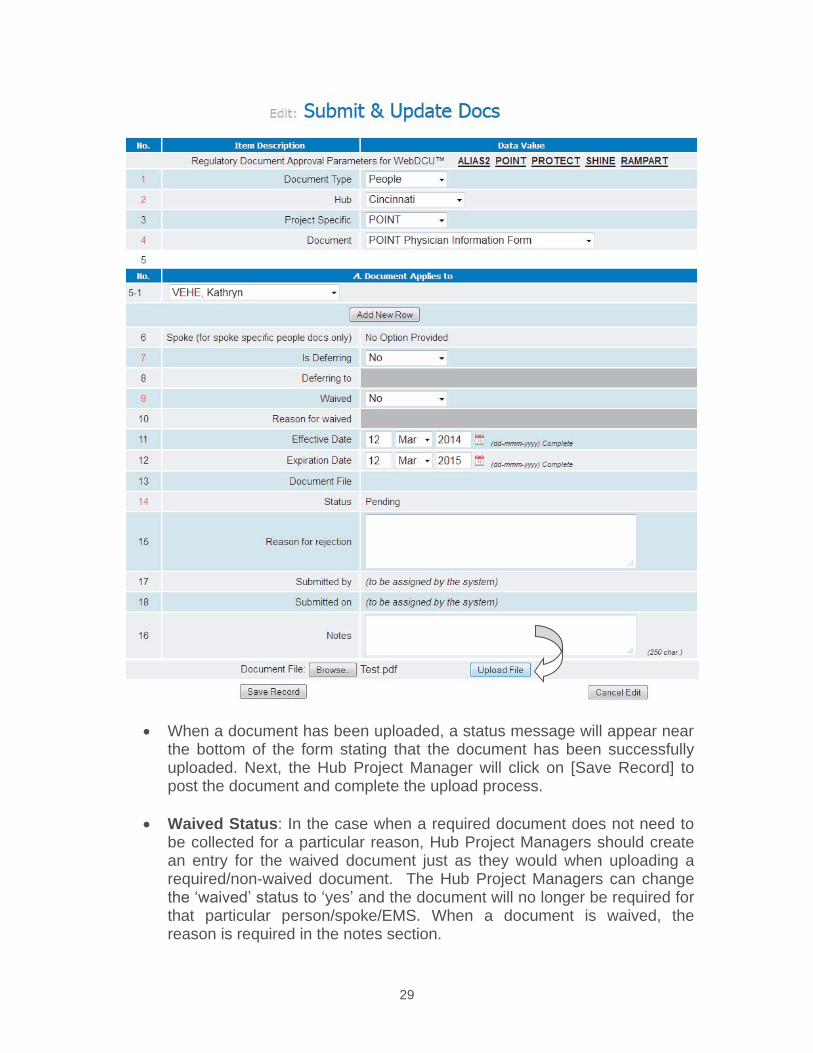
29
When a document has been uploaded, a status message will appear near the bottom of the form stating that the document has been successfully uploaded. Next, the Hub Project Manager will click on [Save Record] to post the document and complete the upload process.
Waived Status: In the case when a required document does not need to be collected for a particular reason, Hub Project Managers should create an entry for the waived document just as they would when uploading a required/non-waived document. The Hub Project Managers can change the ‘waived’ status to ‘yes’ and the document will no longer be required forthat particular person/spoke/EMS. When a document is waived, the reason is required in the notes section.

30
Sharing Documents
Occasionally, one document may need to be uploaded for more than one person (i.e. RAMPART 1572). Instead of uploading a new document, the user should enter all entities which are ‘sharing’ the document in field 5. More rows can be added as needed by clicking on [Add new row].
Editing Rejected Documents
To edit a document that has been rejected by CCC, the Hub Project Manager should click on ‘Regulatory Document’ from the main menu page, and then on ‘Submit & Update Docs’.
Filter for ‘Rejected’ documents (or selected the ‘Rejected Documents’ system query), click the blue number link in the first column of the ‘List Record Table’ table adjacent to the record that requires editing, click on [Edit Record], make the required changes, and then click [Save]. This will change the status back to pending for CCC review.
Adding from Existing Documents
It may be time saving to use a previously entered regulatory document as a template for a new document. This is called ‘Add from Existing’. This is different from editing a document in that editing a document merely changes the record, while ‘Add from Existing’ creates a new record.
To ‘Add from an existing’ record, click on ‘Regulatory Document’ from the main menu page, and then on ‘Submit & Update Docs’.
Select the record you would like to copy, click the blue number link in the first column of the ‘List Record Table’ table adjacent to the record that you wish to copy, click on [Add from Existing], edit the record accordingly, and then click [Save].

31
Changing the Status of Submitted Regulatory Documents
Once a document is submitted, it will be reviewed by the CCC who will update the document status.
To change the status of a document that has been submitted by a Hub Project Manager , CCC should go to the [Submit & Update Docs- CCC] table, filter for ‘Pending’ documents (or selected the ‘Pending Documents’ system query), and click the blue number link in the first column of the ‘List Record Table’ table adjacent to the document requiring review. CCC will then change the status as required.
STUDY DATABASES The study databases contain interfaces for collecting and processing a study’s CRFs, medical safety monitoring, Community Consultation/Public Disclosure Summaries, and drug accountability. Regulatory documents for all studies are collected in the NETT Regulatory Document Database.
To get to a study specific database, select the database name (e.g. ‘SHINE’) from the WebDCUTM Project Selection page seen below.
Once you are in a project database (e.g. SHINE), you can return to the Project Selection page by clicking the WebDCUTM icon located in the top left-handcorner of a specific study database.

32
Locating Project Documents
Study specific documents can be found by clicking on the [Toolbox] tab, and then clicking on [Project Documents]. To view or print a project document click on icon next to the document. You may then print the document by selecting the appropriate print function from your browser toolbar.
Printing Study Casebooks or Individual Worksheets
Prior to enrolling a subject into a study, you may wish to print the subject’s study book. This is a collection of worksheets which defines the data that is required to be collected for the protocol.
To print a study book for a subject, click on [Toolbox] tab, and then click on ‘Project Documents’.
Click on the icon adjacent to the document to open the PDF file. The PDF file can then be printed by selecting the appropriate print function from your browser toolbar
To print an individual worksheet, from the main menu page, click on ‘CRF Collection Schedule’ in the upper left hand corner of the screen and click on the ‘PDF’ link adjacent to the CRF name you would like to print. The PDF file can be printed by selecting the appropriate print function from your browser toolbar.
Note: If any change is made to the study casebook during the conduct of the study, you will be notified via e-mail and the revised worksheet and

33
study book will be posted. Due to the possibility of revisions to the worksheets, it is recommended that you print only a few study books at a time.
Screen Failure Log
To print a copy of the Screen Failure Log worksheets, click on the [Toolbox] tab, and then click on ‘Project Documents’.
Click on ‘PDF’ icon adjacent to the Screen Failure Log to open the PDF file. The PDF file can be printed by selecting the appropriate print function from your browser toolbar.
Screen failure logs are posted on the 1st of every month. The previous month screen failure log should be submitted in WebDCUTM by the 10th of the following month. If a log is not entered by this date, your site will receive a late Screen Failure Log email notification, which is automatically generated by the WebDCUTM database. Even if a site does not have any screen failures, the log still must be submitted.
To data enter the Screen Failure Log, click on ‘[Study Progress] tab, and then click on the [Screen Failure Log]

34
A screen failure log will be automatically posted on the first of the month. Click on the number cell in the left hand column corresponding to the correct log, then select ‘edit record’ and data enter the required information. Once finished, click on [Save record]. Users will be unable to add more than one screen failure log per month. To update a pre-existing log, open the pre-existing log, and then click [Edit Record]. Instructions for completing the Screen Failure Log can be found in the study’s CRF Guidelines.
Adding and Deleting a Subject
From the main menu page, click on ‘Add Subject’ after selecting the spoke and baseline/randomization date. If you only have permissions for one spoke, the spoke will be populated automatically.
The subject will be assigned the next available subject ID number across all spokes. This means that there will be gaps in the ID numbers for the subjects enrolled at your spoke.
For detailed instructions about randomizing a subject, visit the Randomization section specific to your project.
If you added a subject in error, you can remove it (provided you have not entered any CRF data for this subject) by clicking [Delete Subject ####] at the top of the screen.

35
Adding and Deleting a Subject Visit
The first study visit will be posted automatically when a subject is added to WebDCUTM.
To add a subsequent visit (and therefore make the CRFs available for that particular visit), click on ‘Subject CRF’ link at the top of the main menu page. Select the subject you would like to work on from the ‘Subject’ drop down box, select the visit from the Next Visit drop down box (if applicable), enter the date of the visit, and click the [Add Visit] button.
The forms for that visit will be posted in the CRF Collection Table in the middle of your screen for that subject. Note: You should not move a subject to a visit until that visit has occurred. Failure to follow these instructions will result in incorrect late data reminders being sent to yourhub.
You can delete a visit (provided no CRFs for that visit have been data entered) by clicking on [Delete Visit] in header on the CRF Collection Table.

36
Entering/Viewing CRF Data
From the main menu page, click the [Subject CRF Binder] tab. Now select the subject you would like to work on from the ‘Subject’ drop down box. The CRFs for that subject are posted on the CRF Collection Table in the middle of the screen.
Click on the appropriate icon for the CRF you would like to work on (An example of the icon is in the figure below).
The CRF will appear on screen. Enter the data, then click [Save Record] located at the bottom left of the page.
If required data is missing or contains formatting errors, you will receive a small pop up window in the middle of your screen listing your errors. You are required to fix the errors before you can save the form.

37
After the data is saved, you will receive notification of any rule violationsadjacent to the offending data. You can view the violation message by mousing over the red rule violation number.
The different types of rule violations are listed below:
Rejections are signified by a red “R” preceding the violation number. You can view the rejection message by mousing over it (or clicking on it).
These types of errors involve significant logical data errors. The only way to remove a rejection is to correct the data by editing the CRF.
Protocol violations are signified by a red “P” preceding the violation number. You can view the protocol violation message by mousing over it (or clicking on it).
These types of rule violations are put in place to protect against typographical errors and to notify users of protocol violations occurring at the spoke. Protocol Violations may be addressed in two ways.
1.) If the data was incorrectly entered, the data may be edited. Click on [Edit CRF] located at the top right-hand corner of the screen. Edit the data as needed, enter the ‘reason for change’ at the bottom of the screen, and click on [Save Record].
Or
2.) If the entered data is correct as is and a protocol violation truly occurred, the site may dismiss the protocol violation (see Dismissing Protocol Violations or Warnings)
Warnings are signified by a red “W” preceding the violation number. You can view the warning message by mousing over violation number.

38
These types of rule violations are put in place to reduce the number of typographical errors and missing data. Warnings may be addressed in two ways.
1.) If the data was incorrectly entered, the data may be edited. Click on [Edit CRF] located at the top right-hand corner of the screen. Edit the data as needed, enter the ‘reason for change’ at the bottom of the screen, and click on [Save Record].
Or
2.) If the entered data is correct as is, the site may dismiss the warning (see Dismissing Protocol Violations or Warnings).
Notifications are signified by the icon. Notifications do not need to be dismissed prior to CRF submission but are put in place to remind users of pertinent information.
Dismissing Protocol Violations and Warnings
To dismiss a warning or protocol violation, click on the red warning/protocol violation link next to the offending data. This will take you to the View Record page for that specific rule violation.
Click on the [Edit Record] at the top right-hand corner of the page, change the violation status to ‘Data Confirmed’, enter your reason for dismissing the rule violation, and then click [Save Record].
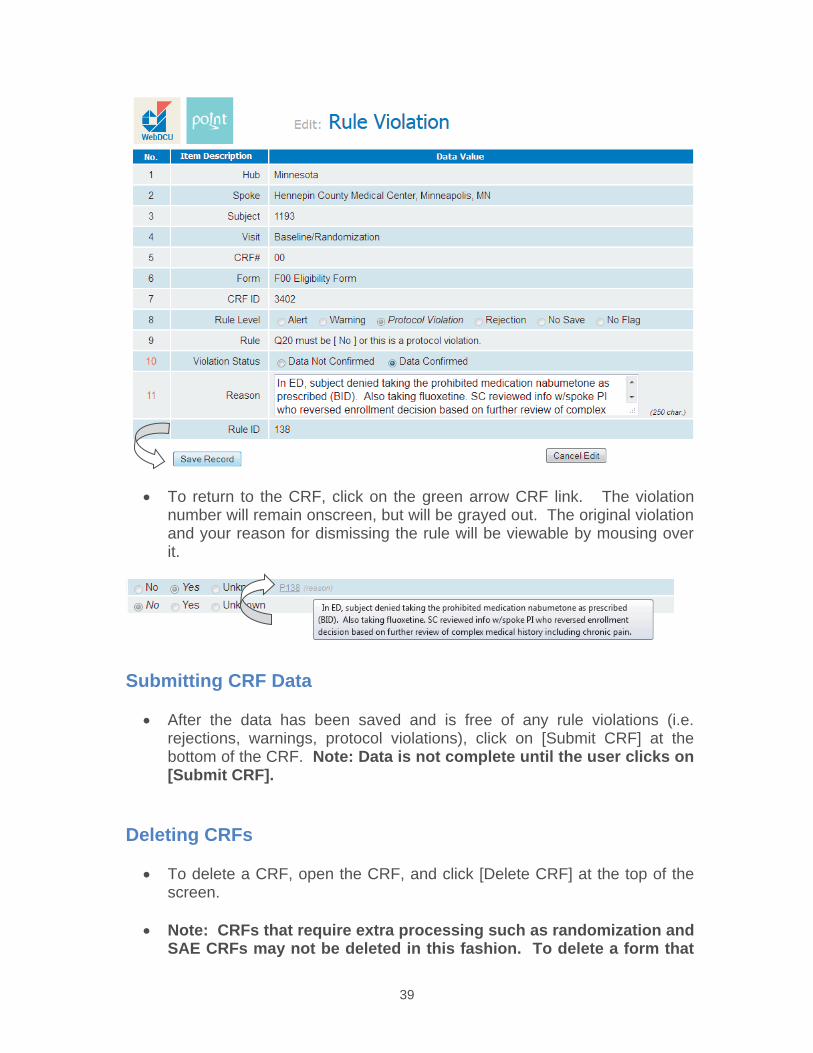
39
To return to the CRF, click on the green arrow CRF link. The violation number will remain onscreen, but will be grayed out. The original violation and your reason for dismissing the rule will be viewable by mousing over it.
Submitting CRF Data
After the data has been saved and is free of any rule violations (i.e. rejections, warnings, protocol violations), click on [Submit CRF] at the bottom of the CRF. Note: Data is not complete until the user clicks on [Submit CRF].
Deleting CRFs
To delete a CRF, open the CRF, and click [Delete CRF] at the top of the screen.
Note: CRFs that require extra processing such as randomization and SAE CRFs may not be deleted in this fashion. To delete a form that

40
requires extra processing, please contact the appropriate DCU Data Manager.
Adding Extra CRFs
Certain additional CRFs (such as AE CRFs) may need to be completed; however, these extra forms are not automatically included on the CRF Collection Page.
To add an extra CRF, open any previously entered form for that subject visit and click [Add Repeat Form] in the header of the CRF. This will post an extra form on the CRF collection table for that visit.
Note: This function is available only for forms that may need to be repeated per the study protocol. Not all CRFs are repeatable.
Interpreting the CRF Collection Table
CRF ID Mouse Over Pop-up Notification Each CRF is assigned a CRF ID number when it is saved. This number will appear while mousing over the document icon in the CRF Collection Table. Mousing over the document icon will also display the status of the CRF validation (errors, rule violations, protocol violations).
These icons will appear in the CRF collection table and represent CRFs in various stages of data processing. Mousing over an icon in the CRF collection table will provide additional details.
Icon Description
A CRF that has not yet been data entered
A CRF that been data entered and has no violations or DCRs, but is not submitted
A CRF with an open rule violation or DCR

41
A CRF that contains no clinical datawhich has been submitted
A CRF that contains clinical data whichhas been submitted
Yellow/Tan Cell background colors are used to indicate to the current user that a CRF needs attention.
Blue Cell background color is used to indicate to the current user that a CRF does not need attention.
Creating and Closing Data Clarification Requests (DCRs) (For Monitors and Data Managers)
To generate a DCR, open the CRF in need of review by clicking on the document icon of the Subject’s CRF Collection Table. Please note that you can only create DCRs for CRFs after they have been submitted.
At the top of the page, click [Create New Monitor/Data Manager DCR]. This will open the New Data Clarification Request screen at the bottom of the page.

42
Enter the required information and click [Save].
You may edit your query by clicking on [Edit Query]. Please note that you will not be able to edit your query once a response is submitted by the site. To delete your query, click on [Delete Query].
Once the site submits a response and resubmits the CRF, you will have the option of closing the DCR or generating a new query. If the data and response are satisfactory upon review, specify what action was taken by the site: If the site confirmed the accuracy of the data and no change was needed, select ‘Data confirmed’. If the site corrected an error on the CRF, select ‘Error corrected’. If the site added missing data or more information to the CRF or if a new CRF was data entered in response to the DCR, select ‘Missing item/CRF entered’. If the site was responding to a query based on a change that was made to the database by DCU, select
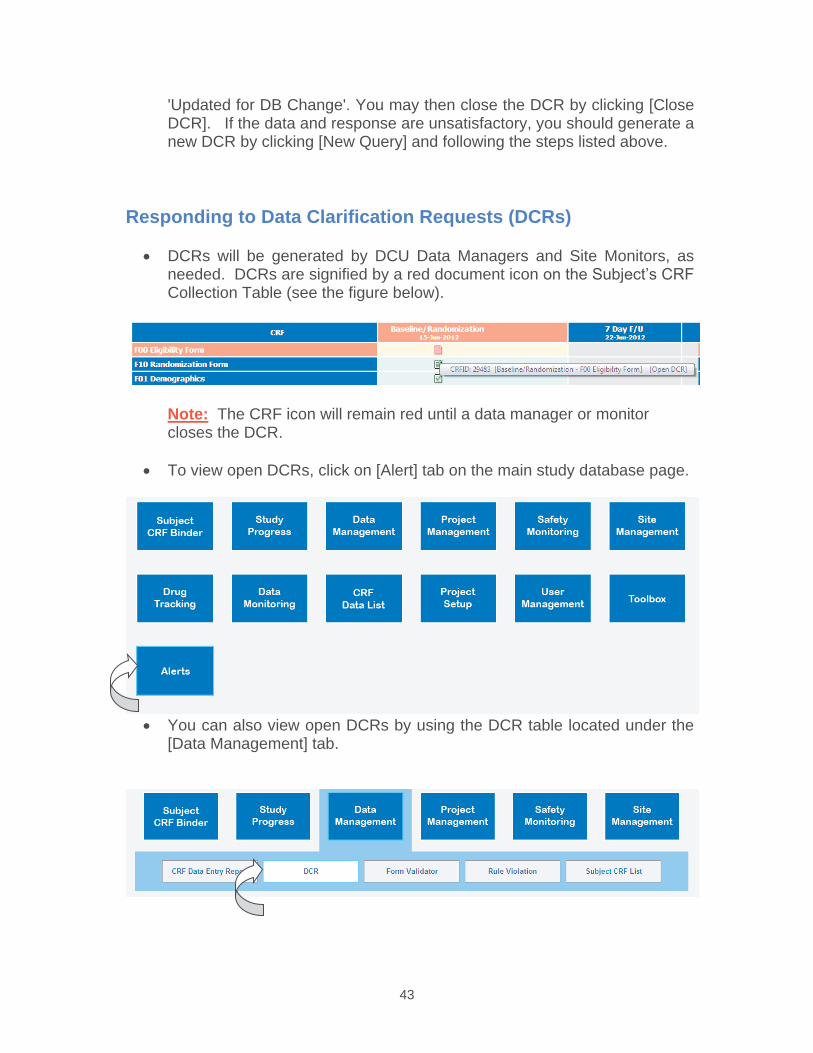
43
'Updated for DB Change'. You may then close the DCR by clicking [Close DCR]. If the data and response are unsatisfactory, you should generate a new DCR by clicking [New Query] and following the steps listed above.
Responding to Data Clarification Requests (DCRs)
DCRs will be generated by DCU Data Managers and Site Monitors, as needed. DCRs are signified by a red document icon on the Subject’s CRF Collection Table (see the figure below).
Note: The CRF icon will remain red until a data manager or monitor closes the DCR.
To view open DCRs, click on [Alert] tab on the main study database page.
You can also view open DCRs by using the DCR table located under the [Data Management] tab.

44
To view CRFs with DCRs, select the ‘Open DCR’ system query from the ‘Page Actions’ drop down box.
Click on the green arrow under the CRF ID column for the query you want to review. This will take you to the CRF page that has been queried.
The DCR will be posted at the bottom of the CRF page. Click on [Add Response] next to the query and enter a response. Then click [Save Response] at the bottom of the page.
If the CRF requires editing, click [Edit CRF] and correct the CRF data as needed. Then click [Save Record] and then [Submit CRF] at the bottom of the page.

45
Monitor Verification (For Monitors)
Site Monitors will be performing source to database verification.
Once a CRF passes monitor verification, the Site Monitor will document this by clicking on [CRF passed verification] at the top of the CRF page.Please note that you can only pass a CRF as verified after it has been submitted.
Audit Trail
The audit trail will display data points on a CRF have been that have been updated/edited.
To view the audit trail, open the CRF, and click on [View Audit Trail] at the top of the CRF page.
The audit trail will be displayed in chronological order with changes highlighted in yellow. Note: If no changes have been made to the form, you will receive a message indicating that 'no audit record’ is available.

46
Study Calendar
Once a subject has been added to the study database, the study calendar will be populated with target and actual visit dates. To view the study calendar, click on Study Calendar under the [Study Progress] tab the main menu page of the study database.
Actual visit dates are shown in green. Projected/target visit dates are shown in pink.
To filter the calendar for a particular subject, click on the ‘Data Management’ tab at the top of the main menu page, then click on ‘Subject Progress’. Filter for the appropriate subject, then select ‘Subject Visit/Projected Subject Visits’ from the page actions drop down box at the top right corner of the page.
Entering Community Consultation Summaries
Hub Project Managers can data enter Community Consultation Summaries by clicking on ‘Project Management’, and then Community Consultation Summary.
Then click on [Add Record] and enter the required information.
Section 1 consists of demographic questions. Section 2 is for closed ended questions (question with definite answers) asked by the Community Consultant(s). Section 3 is for open ended and unsolicited questions. Section 4 is for survey questions for information distribution and advertising effectiveness. Once all responses are entered and confirmed, click [Save Record] at the bottom of the page.
Entering Public Disclosure Summaries
Hub Project Managers can data enter Public Disclosure Summaries by clicking on ‘Project Management’, and then Public Disclosure Summary.
Then click on [Add Record] and enter the required information.
MedDRA Coding
Central AE coders can code AEs using MedDRA by clicking on ‘Medical Safety Monitoring’, and then AE MedDRA Coding.

47
Click on the Link icon adjacent to any AE name. This will initiate auto coding and generate a coding status report.
To interactively code AEs that did not automatically code, click on [Interactive MedDRA], select the AE to code from the drop down box, and enter your search criteria. Select the correct code for each term and then click [Code AE].
NOTE: If the coder would prefer not to propagate this code to other identically named adverse events, uncheck the box adjacent to these instructions prior to clicking [Code AE]: ‘Check this box to always use this coding path to autocode any current or future AEs with matching AE Name. If this box is unchecked, this record will not be used for autocoding.’
Any AE name that is changed will return to a pending AE coding status.
STUDY-SPECIFIC INFORMATION
POINT SPECIFICS Data Entry Timelines
Screening Failure Log - By the 10th day of the following month.
Eligibility and Randomization Forms - Must be data entered and submitted with all eligibility criteria met before randomization number will be assigned.
All other CRFs - Within 5 days of collection.
In the In the event of a Serious Adverse Event (SAE) or Clinical Outcome, the SAE/Clinical Outcome Reporting Form must be submitted within 24 hours of first knowledge of the event. In addition, any pertinent subject data should be data entered within 24 hours.
If there will be a delay in data entry, please contact DCU. Please note that site payments are dependent upon the subject’s data being entered and submitted.
Data Clarification Request (DCR) Timelines: All responses to DCRs must be submitted within 5 days of query generation with the exception of

48
DCRs for SAEs/Clinical Outcomes, which must be submitted within 24 hours of query generation.
POINT Study Drug Tracking Module
Study Drug Labeling
Central Pharmacy (DPSL) must confirm labeling of study drug in WebDCUTM. From the main menu page, click on ‘Drug Tracking’, then on ‘Drug Labeling Clopidogrel/Placebo’.
Click the blue number link in the first column of the ‘List Record’ table adjacent to the study drug number you wish to pack. Click [Edit Record] at the top of the screen and enter the required information. Then, click [Save Record].
Study Drug Shipping
Central Pharmacy (DPSL) must document shipping of study drug in WebDCUTM. From the main menu page, click on ‘Drug Tracking’, then on ‘Drug Shipping’. Note: Only study drug that has been requested to be shipped will appear in this table.
Click the blue number link in the first column of the ‘List Record’ table adjacent to the study drug number you wish to ship. Click [Edit Record] at the top of the screen and indicate that the study drug has been shipped.Then, click [Save Record].
To print the packing slip, click the icon in the ‘packing slip’ column on the list record page.
Study Drug Receipt
Study Coordinator or designee must confirm receipt of study drug in WebDCUTM. From the main menu page, click on ‘Drug Tracking’, then on‘Drug Receiving’. Note: You must receive the study drug before it will be available for randomization.
Click the blue number link in the first column of the ‘List Record’ table adjacent to the bottle number you wish to receive. Click [Edit Record] at the top of the screen and enter the required information. Then, click [Save Record].

49
Study Drug Damage
Study Coordinator or designee must document damage of study drug in WebDCUTM. From the main menu page, click on ‘Drug Tracking’, then on ‘Drug Report Damage’. Note: You must receive the study drug before it will be available to report as damaged.
Click the blue number link in the first column of the ‘List Record’ tableadjacent to the bottle number for which you wish to report damage. Click [Edit Record] at the top of the screen and indicate that the study drug has been removed from inventory. Then, click [Save Record].
Study Drug Expired
Study Coordinator or designee must document expiration of study drug in WebDCUTM. From the main menu page, click on ‘Drug Tracking’, then on ‘Drug Expired’. Note: Only drug that has been received and will expire within the next 100 days will be listed.
Click the blue number link in the first column of the ‘List Record’ table adjacent to the bottle number for which you wish to report as expired. Click [Edit Record] at the top of the screen and indicate that the study drug has been removed from inventory. Then, click [Save Record].
Randomization
On the main menu page, select the appropriate spoke from the spoke drop-down box, if applicable. Enter the date of enrollment, and click [Add Subject].
The subject ID will be assigned, the enrollment/randomization forms will be posted, and the subject’s CRF collection grid will display.
Click on the CRF icon adjacent to Form 00: Eligibility Form. Enter the required information and click [Save Record]. Then click [Submit CRF].
Click on the ‘Subject CRF’ tab at the top of the screen, and then click the CRF icon adjacent to Form 10: Randomization Form. Enter the required information and click [Save Record]. Then click [Submit CRF].
The computer will generate the randomization assignment and will display the bottle number to be used for that subject on the screen. A screen shot of this form can be printed using the print function in the browser by selecting the appropriate print function from your browser toolbar.

50
Click the link to the Randomization Verification Form (RVF). This form should be printed and signed verifying that the study drug bottle assignedby WebDCU™ matches the study drug bottle taken from the pharmacy shelf for the subject. After this form is completed, it should be filed in the subject record.
If you experience any difficulties when trying to randomize a subject, call the 24 hour randomization hotline at 1-866-450-2016.
Because this is an intent to treat (ITT) trial, all randomized subjects should remain in the study and should be followed, even if he/she does do not receive study drug.
Unblinding
In general, unblinding to treatment assignment should be avoided. If the site investigator feels the need to unblind, he/she must first call 1-866-94POINT. If the person manning the call line agrees that unblinding is required, he/she can initiate this request in WebDCUTM.
To unblind the treatment assignment of the subject, the on-call investigator who receives the request to unblind will click on [Project Management], then ‘Unblinding’ from the main menu page. All subjects randomized will be listed on the screen.
Click the blue number link in the first column of the ‘List Record’ table adjacent to the subject number for which unblind is requested. Click [Edit Record] at the top of the screen, enter the required information, and then click [Save Record]. NOTE: The treatment assignment will not beindicated on this screen.
The on-call investigator should instruct the site investigator to open the Randomization CRF for this subject. The treatment assignment will be listed at the bottom of the screen. The treatment assignment is revealed FOR 30 MINUTES ONLY. After 30 minutes, the treatment assignment is concealed. After the unblinding, the Randomization CRF will have a

51
message stating that the subject’s treatment assignment has been unblinded due to site request.
NOTE: At the time of unblinding and automatic email notification will be triggered to the POINT Executive Committee.
POINT Medical Safety Monitoring
Clinical Event/SAE Site Manager Review (For Site Managers Only)
From the main menu page, under ‘Safety Monitoring’ click on ‘SAE Review ’. The submitted SAEs/Clinical Outcomes for all subjects will be posted on the List Record Table in the middle of the screen.
To view records that require review, select the ‘Pending Site Manager Review’ system query from the ‘Page Actions’ drop down box. This query includes records that have never been Site Manager reviewed as well as those that require re-review due to data changes.
Click on adjacent to Adverse Event record requiring review and review the record.
Then click on [Add New Site Manager review] at the bottom of the page. Enter the required information and click [Save Record]. If the event requires review by the Clinical Event Coordinator, an email notification will be sent to the Clinical Event Coordinator indicating that they should add their review.
If the information is insufficient according to the Site Manager, click on [Create new DM DCR] to generate a Data Clarification Request. Enter a description of the missing information and then click [Save]. An email notification will be sent to the site requesting more information (see Generating DCRs).
To view the audit trail which shows all revisions/updates to the CRF, click on [View Audit Trail] at the top of the screen (see Audit Trail).

52
Clinical Event Coordinator Review (For Clinical Event Coordinators Only)
From the main menu page, under ‘Safety Monitoring’ click on ‘SAE Review ’. The submitted Clinical Events/SAEs for all subjects will be posted on the List Record Table in the middle of the screen.
To view records that require review, select the ‘Pending CEC Review’ system query from the ‘Page Actions’ drop down box. This query includes records that have never been CEC reviewed as well as those that require re-review due to data changes.
Click on adjacent to the record requiring review and review the record.
Click on [Add New CEC review] at the bottom of the page. Enter the required information and click [Save Record]. As appropriate, anautomatic email notification will be sent to the Adjudicators indicating that CEC review is complete and adjudication is required.
To view the audit trail which shows all revisions/ updates to the CRF, click on [View Audit Trail] at the top of the screen (see Audit Trail).
Adjudication Review (for Adjudicators Only)
From the main menu page under ‘AE Alerts,’ click on ‘Pending Adjudicator Review.’ This link will bring you to the List Records: SAE Adjudication Table, which will be limited to those events pending adjudicator review that have been assigned to you.
Click on adjacent to the record requiring adjudication and review the record.
After you have reviewed the record including all of the files in the event packet, click on [Add New Adjudicated Outcome]. Enter the required information and click [Save Record].
NOTE: If you do not have enough information for adjudication, you may request additional information by contacting the Clinical Event Coordinator (CEC), who will work with the Site Manager and the site to provide theinformation being requested, whether it is in the form itself or the event packet. This event will be removed from your list of events requiring adjudication while the form/event packet is being updated but will return to your list once the additional information is added and reviewed by the Site Manager/CEC.

53
There may be cases where you are asked to submit an adjudication for an event you have already reviewed and adjudicated. You will be able to see your previous adjudication review, and in order to know the changes that have been made to the SAE/ Clinical Outcome Reporting Form since your last review, click on [View Audit Trail] at the top of the screen (see Audit Trail).
The flow of the adjudication process is as follows: If the first two adjudicators assigned to an event agree, no further action is required. If there are discrepancies between the first and second adjudicators, a third adjudicator is asked to adjudicate. If the third adjudicator agrees with either of the first two adjudicators, no further action is required. If all three adjudicators disagree, the Adjudicator Chair reviews the event and attempts to gain consensus among the other adjudicators, though ultimately it is his/her adjudication that is assigned to the event.
SHINE SPECIFICS Data Entry Timelines
Screening Failure Log - By the 10th day of the following month.
Randomization Form – The Eligibility and NIH Stroke Scale forms must be data entered, saved and submitted with all eligibility criteria met and no rule violations before the Randomization form may be submitted to generate a subject ID number and treatment assignment.
All other CRFs - Within 5 days of collection.
In the event of a Serious Adverse Event (SAE), the Adverse Event Form must be submitted within 24 hours of first knowledge of the event. In addition, any pertinent subject data should be data entered within 24 hours. If the Serious Adverse Event included Hypoglycemia, the Hypoglycemic Event Form must be data entered and associated with theAE form in question within 24 hours of first knowledge of the event. If the Serious Adverse Event included Neurological Worsening, the Neurological Worsening Form must be data entered and associated with the AE form in question within 24 hours of first knowledge of the event.
If there will be a delay in data entry, please contact DCU. Please note that site payments are dependent upon the subject’s data being entered and submitted.

54
Data Clarification Request (DCR) Timelines: All responses to DCRs must be submitted within 5 days of query generation with the exception of DCRs for SAEs, which must be submitted within 24 hours of query generation.
Randomization
On the main menu page, select the appropriate spoke from the spoke drop-down box, if applicable. Enter the date of enrollment, and click [Add Subject].
The enrollment/randomization forms will be posted, and the subject’s CRF collection grid will display.
Click on the CRF icon adjacent to Form 00: Eligibility Form. Enter the required information and click [Save Record]. Then click [Submit CRF].
Click on the CRF icon adjacent to Form 04: NIHSS. Enter the required information and click [Save Record]. Then click [Submit CRF].
Click on the ‘Subject CRF’ tab at the top of the screen, and then click the CRF icon adjacent to Form 07: Randomization Form. Enter the required information and click [Save Record]. Then click [Submit CRF].
The computer will generate the subject ID and will display the randomization assignment for the subject on the screen. A screen shot of this form can be printed using the print function in the browser by selecting the appropriate print function from your browser toolbar.
If you experience any difficulties when trying to randomize a subject, call the 24 hour randomization hotline at 1-866-450-2016.
Because this is an intent to treat (ITT) trial, all randomized subjects should remain in the study and should be followed, even if he/she does do not receive study drug.

55
Glucose Stabilizer/Study Computer
After a subject has been randomized, please refer to the printed copy or screen shot of the submitted randomization CRF.
Refer to the subject’s unique Subject ID number and the treatment assignment printed on this form while entering subject information into GlucoseStabilizer/Study Computer.
Enter the subject’s treatment assignment. If the subject is randomized to the Control group, once indicated as such in GlucoseStabilizer/Study Computer, the computer screen will be orange. If it is indicated that the subject was randomized to the Intervention group, the computer screenwill be blue.
Please double check that you are entering the correct Subject ID number and treatment assignment into GlucoseStabilizer.
Stroke Mimic Review
The Stroke Mimic Review Functionality is triggered when any final diagnosis of enrolling event, EXCEPT ‘Acute cerebral ischemia: ischemic Stroke’ or ‘Acute cerebral ischemia: TIA’, is data entered on ‘Form 15: Study Treatment’.
When this occurs, the Primary Reviewer will receiving an e-mail notification to make a primary review. Only the site narrative related to the final diagnosis will be visible to the primary reviewer.
Should the site and Primary Reviewer’s diagnoses differ, an e-mail will be sent to the Secondary Reviewer, to make a final review.
Only the site narrative related to the final diagnosis will be visible to the secondary reviewer. Neither reviewer will be able to see the other’s diagnosis.
Only when the secondary reviewer completes their diagnosis, this processis complete.
SHINE SAE Safety Monitoring
Site Manager Review (For the Site Manager Only)
From the main menu page, click the [Project Management] tab, followed by ‘SAE Review ’. The submitted SAEs for all subjects will be posted on the List Record Table in the middle of the screen.

56
To view records that require review, select the ‘Site Manager Review On’ system query from the ‘Page Actions’ drop down box. This query includes records that have never been Project Safety Manager reviewed as well as those that require re-review due to data changes.
Click on adjacent to Adverse Event CRF ID record requiring review, scroll to the bottom of the form and click [Add New Site Manager Review]to begin reviewing the record.
If the information is sufficient and ready for clinical review, click ‘Yes’ in response to “Requires review by Internal Quality and Safety Reviewer’ and then [Save]. After indicating that the SAE should be reviewed by the IQSR and clicking [Save], an automatic email notification will be sent to the Internal Quality and Safety Reviewer indicating that they should add their review.
If the information is insufficient according to the Site Manager, exit the review by clicking ‘Cancel’. To gain appropriate information, query the site by clicking on [Create New Monitor DCR] at the top of the page, to generate a Data Clarification Request. Enter a description of the missing information and then click [Save]. An email notification will be sent to the site requesting more information (see Generating DCRs). When the site provides necessary information and/or updates the SAE form, return to the bottom of the form and click [Add New Site Manager Review] to review the record.
To view the audit trail which shows all revisions/updates to the CRF, click on [View Audit Trail] at the top of the screen (see Audit Trail).
Internal Quality and Safety Reviewer Review (For the Internal Quality and Safety Reviewer)
From the main menu page, click the [Project Management] tab, followed by ‘SAE Review ’. The submitted SAEs for all subjects will be posted on the List Record Table in the middle of the screen.
To view records that require review, select the ‘Pending Internal Quality and Safety Review’ system query from the ‘Page Actions’ drop down box. This query includes records that have never been Internal Quality and Safety Reviewer reviewed as well as those that require re-review due to data changes.
Click on adjacent to Adverse Event CRF ID record requiring review, scroll to the bottom of the form and click [Add New Internal Quality and Safety Review] to begin reviewing the record.

57
If the information is clinically accurate and complete and ready for the Independent Medical Safety Monitor Review, click ‘Yes’ and then [Save].After clicking ‘Yes’ and [Save], an automatic email notification will be sent to the Independent Medical Safety Monitor indicating that they should add their review.
If the information is insufficient according to the Internal Quality and Safety Review, click ‘No’ and [Save]. To gain appropriate information, query the site by clicking on [Create New Monitor DCR] at the top of the page, to generate a Data Clarification Request. Enter a description of the missing information and then click [Save]. An email notification will be sent to the site requesting more information (see Generating DCRs). When the site provides necessary information and/or updates the SAE form, return to the bottom of the form and click [Edit] to adjust your review.
To view the audit trail which shows all revisions/ updates to the CRF, click on [View Audit Trail] at the top of the screen (see Audit Trail).
Independent Medical Safety Monitor Review (For the Independent Medical Safety Monitor Only)
From the main menu page, click the [Project Management] tab, followed by ‘SAE Review ’. The submitted SAEs for all subjects will be posted on the List Record Table in the middle of the screen.
To view records that require review, select the ‘Pending Independent MSM Review’ system query from the ‘Page Actions’ drop down box. This query includes records that have never been Independent MSM reviewed as well as those that require re-review due to data changes.
Click on adjacent to Adverse Event CRF ID record requiring review, scroll to the bottom of the form and click [Add New Independent Medical Safety Monitor Review] to begin reviewing the record.
If the information is clinically accurate and complete enough to complete, complete the review and click [Save]. [Save], an automatic email notification of the SAE review will be sent to the DSMB liaison and Chair, as per their request.
If the information is insufficient according to the Independent Medical Safety Monitor, exit the review by clicking ‘Cancel’. To gain appropriate information, query the site by clicking on [Create New Monitor DCR] at the top of the page, to generate a Data Clarification Request. Enter a description of the missing information and then click [Save]. An email

58
notification will be sent to the site requesting more information (see Generating DCRs). When the site provides necessary information and/or updates the SAE form, return to the bottom of the form and click [Add New Independent Medical Safety Monitor Review] to review the record.
To view the audit trail which shows all revisions/ updates to the CRF, click on [View Audit Trail] at the top of the screen (see Audit Trail).
Blinded Assessors
The 6 week follow up phone call and 90 day outcome assessment will be performed by a blinded study investigator.
When attempting to view a CRF potentially containing unblinding information for the first time, a pop-up window with a warning will appear. The warning will alert users that the form contains potentially unblinding information.
For each subject, pop-ups will appear only once, per each potentially unblinding form.
When presented with this pop-up warning, the user must confirm whether he/she would like view the form or cancel the view. The history of users who have been potential unblinded will be stored in the database.
Emergency Unblinding
No emergency unblinding is anticipated. Any emergency unblinding requests must be directed to the PI on call (800-915-7320). If a subject/LAR wishes to discontinue treatment- unless they are unblinded to which treatment the subject is receiving- they may be unblinded. However, these requests must first be presented to the SHINE PI on call and may be minimized by thorough discussion by the study team of the rationale for blinding and the subject/LAR’s specific concerns about the differences in treatment groups.
ESETT SPECIFICS Obtaining a User Account and Defining Study Team Members
In the WebDCU™ database, the User Account governs many of the functions available to study team members. Regardless of whether the study team

59
member will need to gain access to the WebDCU™ database, each study team member must have a WebDCU™ User Account in order to track the required regulatory documents for that person.
To start the process, a user account for the Site Study/Regulatory Coordinator at each site will be created by the Data Manager, or designee, at the DCU. An email notification will be sent from WebDCU™ with the user’s account information.
Adding Study Team Member User Accounts(For Site Study/Regulatory Coordinators)
To obtain a user account for the remainder of the study team, the Study Coordinator should enter a study team member request. From the study’s main menu page, the Site Study/Regulatory Coordinator should click on [User Management], and then [Study Team Member Request]. Click on [Add New] in the upper right hand corner of the screen. Complete all information on the ‘Study Team Member Request’ page, and then click ‘Save Record’. If the team member is already a WebDCUTM
user, choose “Select from existing WebDCU member.” If not, select “Add a new WebDCU member.”
If the ‘Study Team Member Request’ was approved (see the Request Process Status column in list view), return to the study’s main menu page. Click on [User Management] and then [User Permission Request]. On the list record page, click on the blue number link to the left of the study team member name you are looking for.
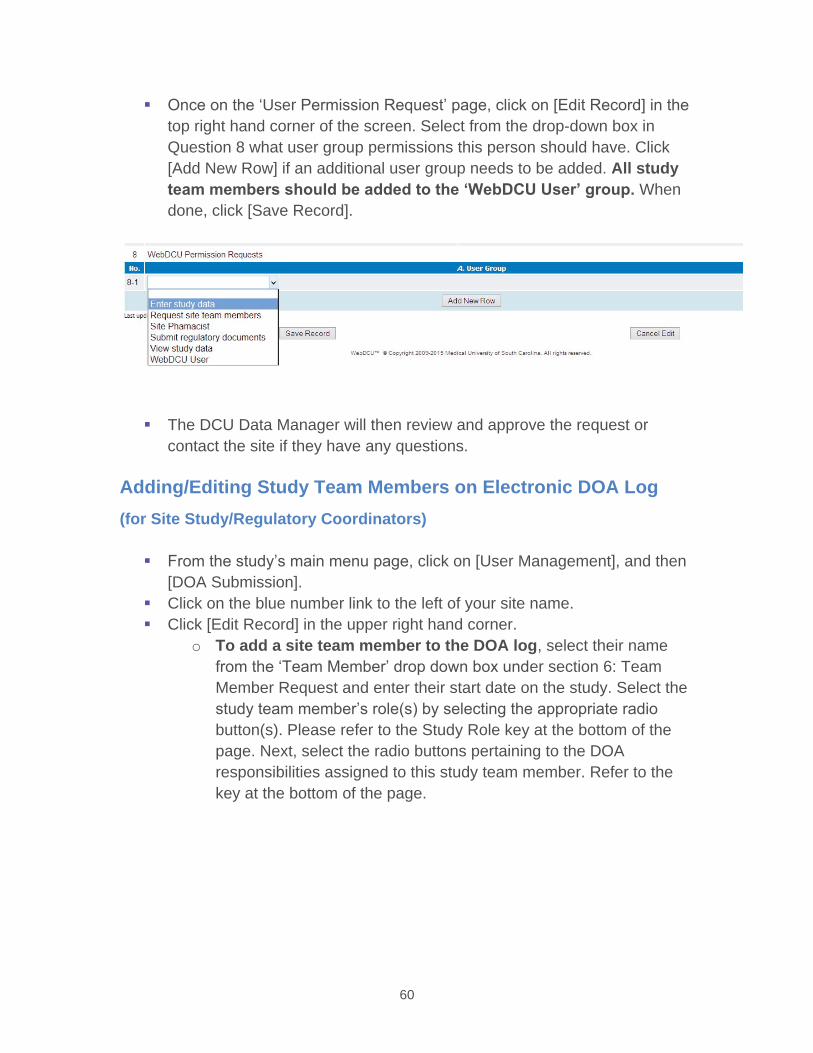
60
Once on the ‘User Permission Request’ page, click on [Edit Record] in the top right hand corner of the screen. Select from the drop-down box in Question 8 what user group permissions this person should have. Click [Add New Row] if an additional user group needs to be added. All study team members should be added to the ‘WebDCU User’ group. When done, click [Save Record].
The DCU Data Manager will then review and approve the request or contact the site if they have any questions.
Adding/Editing Study Team Members on Electronic DOA Log (for Site Study/Regulatory Coordinators)
From the study’s main menu page, click on [User Management], and then [DOA Submission].Click on the blue number link to the left of your site name. Click [Edit Record] in the upper right hand corner.
o To add a site team member to the DOA log, select their name from the ‘Team Member’ drop down box under section 6: Team Member Request and enter their start date on the study. Select the study team member’s role(s) by selecting the appropriate radio button(s). Please refer to the Study Role key at the bottom of the page. Next, select the radio buttons pertaining to the DOA responsibilities assigned to this study team member. Refer to the key at the bottom of the page.
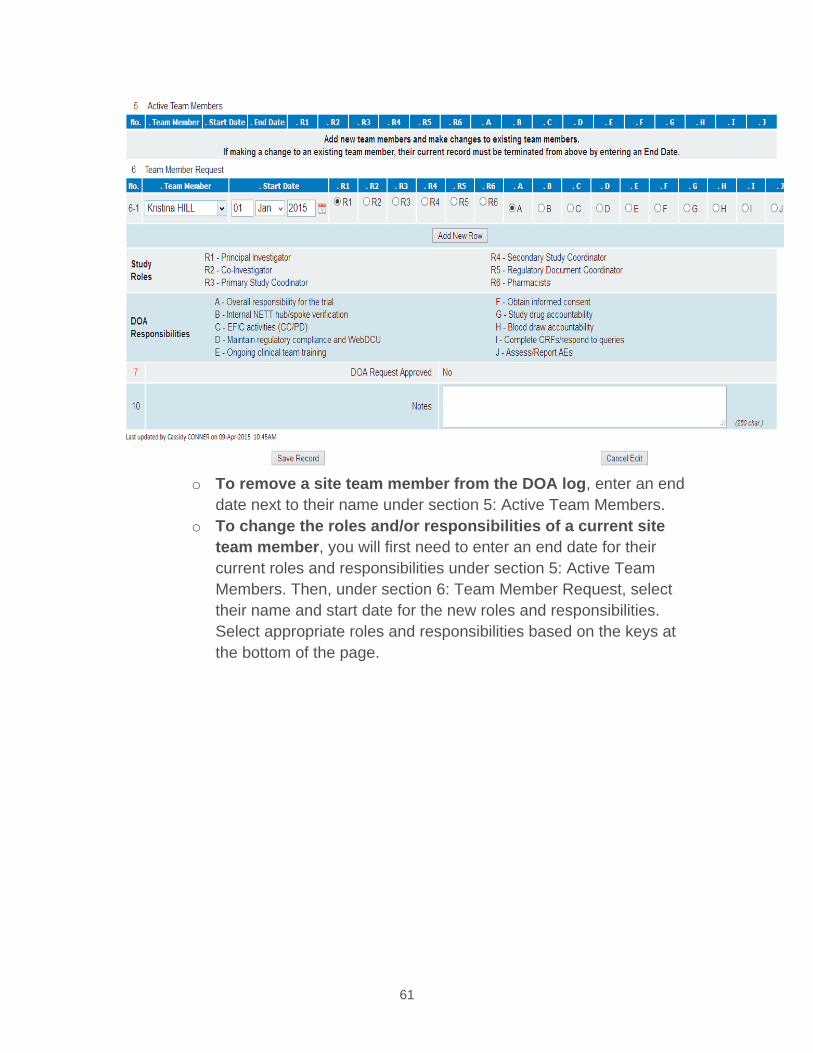
61
o To remove a site team member from the DOA log, enter an end date next to their name under section 5: Active Team Members.
o To change the roles and/or responsibilities of a current site team member, you will first need to enter an end date for their current roles and responsibilities under section 5: Active Team Members. Then, under section 6: Team Member Request, select their name and start date for the new roles and responsibilities. Select appropriate roles and responsibilities based on the keys at the bottom of the page.

62
Once all study team members have been added/edited along with their roles and responsibilities, click on [Save Record]. The DOA will then be sent to the CCC Site Manager for review and approval. The regulatory document requirements for the newly added or edited study team members will not populate in the regulatory database until the DOA is approved. If the DOA log submitted by the Site Study/Regulatory Coordinator is deemed unacceptable (e.g. because it contains errors or requires further clarification prior to approval), the Project Manager will respond “No” in the “DOA Request Approved” field and explain the reason the DOA was not approved in the text field below it. The Site Study/Regulatory Coordinator will be notified by email of the decision and can then make changes and resubmit for approval.
REGULATORY DOCUMENTS
The Site Study/Regulatory Coordinator will be able to submit regulatory documents through WebDCUTM based on the pre-specified regulatory document collection requirements. Regulatory documents are divided into two groups: “SiteDocuments,” such as IRB Approval, FWA, CAP/CLIA, and “People Documents,”such as CV and medical licenses.
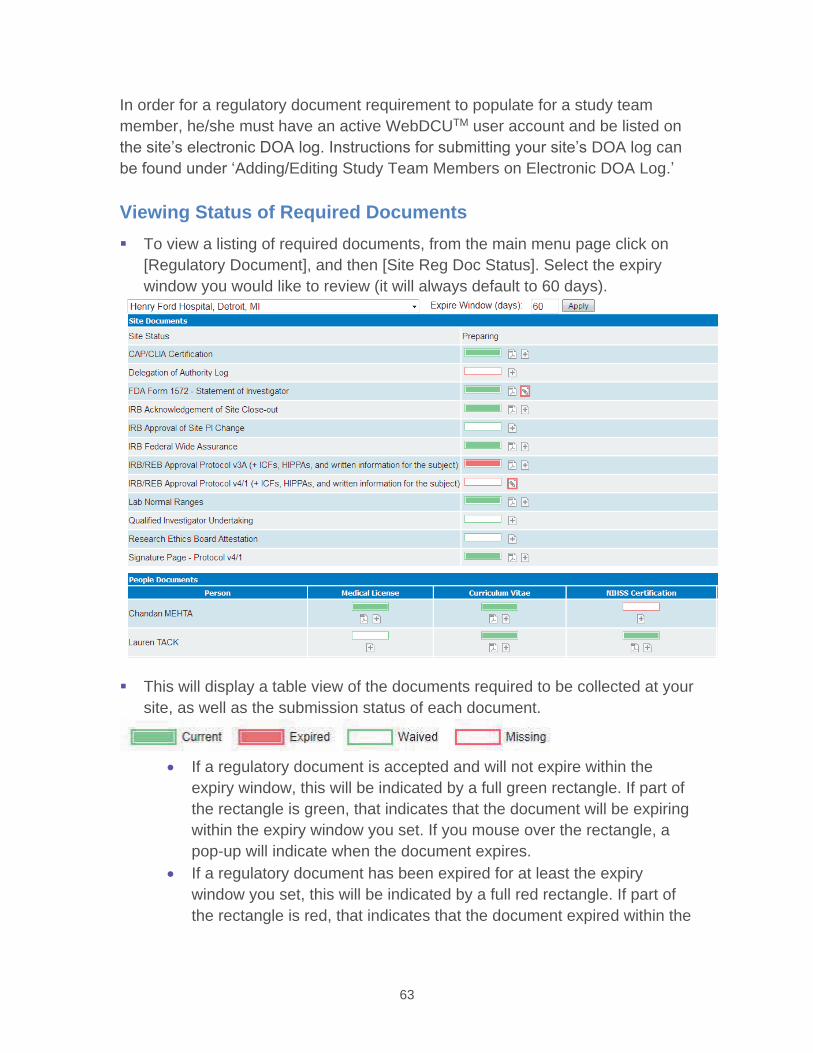
63
In order for a regulatory document requirement to populate for a study team member, he/she must have an active WebDCUTM user account and be listed on the site’s electronic DOA log. Instructions for submitting your site’s DOA log can be found under ‘Adding/Editing Study Team Members on Electronic DOA Log.’
Viewing Status of Required DocumentsTo view a listing of required documents, from the main menu page click on [Regulatory Document], and then [Site Reg Doc Status]. Select the expiry window you would like to review (it will always default to 60 days).
This will display a table view of the documents required to be collected at your site, as well as the submission status of each document.
If a regulatory document is accepted and will not expire within the expiry window, this will be indicated by a full green rectangle. If part of the rectangle is green, that indicates that the document will be expiring within the expiry window you set. If you mouse over the rectangle, a pop-up will indicate when the document expires. If a regulatory document has been expired for at least the expiry window you set, this will be indicated by a full red rectangle. If part of the rectangle is red, that indicates that the document expired within the

64
expiry window you set. If you mouse over the rectangle, a pop-up will indicate when the document expired.
To upload a new document, click on the link and this will take you to [Reg Doc Submission] (see ‘Submitting Regulatory Documents’ below for more information).If a regulatory document is waived and therefore not required, this will be indicated by an empty green rectangle If a regulatory document is missing or has not been submitted yet, this will be indicated by an empty red rectangle.
Submitting Regulatory Documents (for Site Study/Regulatory Coordinators)
From the main menu page, click on [Regulatory Document], and then [Site Reg Doc Submission] or [People Reg Doc Submission] depending on the type of regulatory document you will be submitting. You can also get to this
view by clicking on a document’s link in the [Site Reg Doc Status] table.
This will take you to a ‘List Record’ page of all the regulatory documents at your site.
Click on the ‘Add New’ green arrow link adjacent to the document you would like to upload or edit.
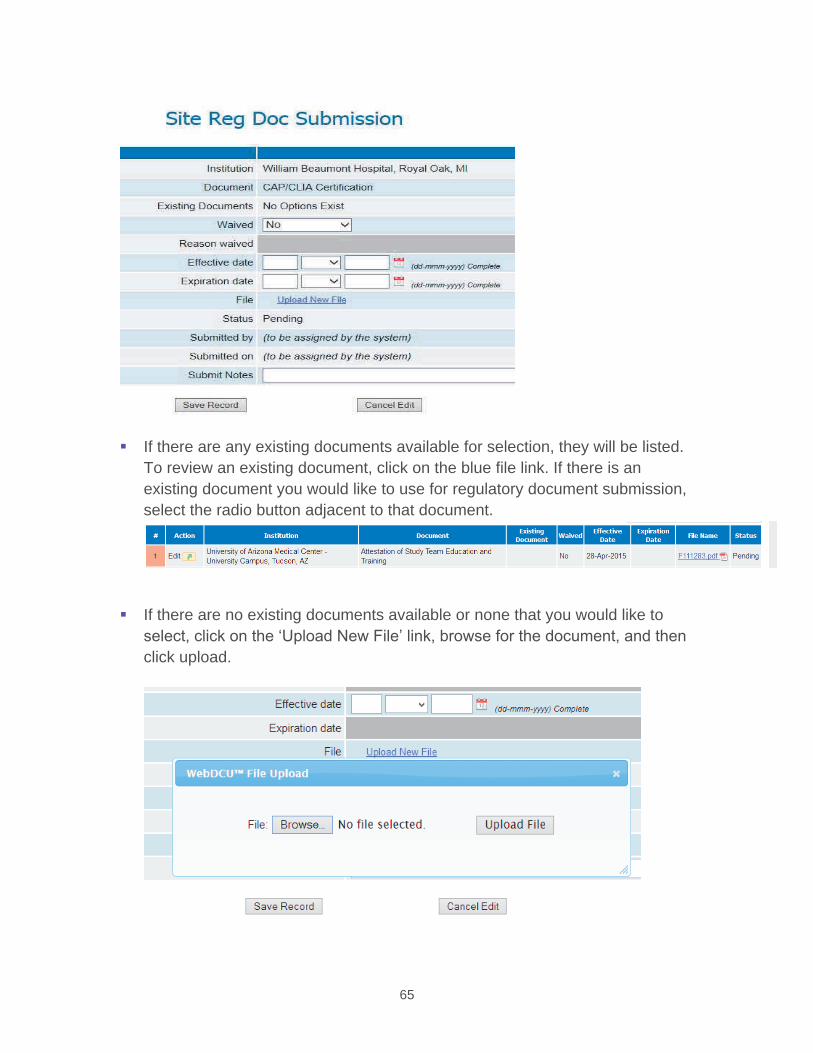
65
If there are any existing documents available for selection, they will be listed. To review an existing document, click on the blue file link. If there is an existing document you would like to use for regulatory document submission, select the radio button adjacent to that document.
If there are no existing documents available or none that you would like to select, click on the ‘Upload New File’ link, browse for the document, and then click upload.

66
Enter the required information, and then click ‘Save Record.’
The document will then be in a pending status until the trial’s Project Manager verifies the information and approves/accepts the document.
Editing Rejected Documents (for Site Study/Regulatory Coordinators)
Documents that have been rejected should not be re-entered. The original entry should be edited instead.
To edit a document that has been rejected, from the main menu page, click on [Regulatory Document], and then [Site Reg Doc Submission] or [People Reg Doc Submission]. Click the ‘Edit’ green arrow link adjacent to the record you would like to edit. Update the new file and/or information, and then click ‘Save Record.’
File Upload Restrictions: Only Adobe PDF files less than 3MB can be uploaded. If you are using Adobe Acrobat Professional, set your options to ‘higher compression’ (as opposed to ‘higher quality’). If you are not using Adobe or experiencing difficulties with the file size limit, you may contact the appropriate DCU data manager. If the file size is too large, WebDCUTM will show an error message and will not allow you to upload the document until it has been resized.
Approving Electronic DOA Logs (for CCC Site Managers)
From the study’s main menu page, click on [User Management] and then [DOA Review]. Click on the blue number link to the left of the site you would like to review. Once on the site’s ‘DOA Review’ page, click [Edit Record] in the upper right hand corner of the page.

67
Review all changes made to the DOA. All changes made by the site will be listed under section 6. If the DOA log submitted by the Site Study/Regulatory Coordinator is deemed unacceptable (e.g. because it contains errors or requires further clarification prior to approval), the Project Manager will respond “No” in the “DOA Request Approved” field and explain the reason the DOA was not approved in the text field below it. The Site Study/Regulatory Coordinator will be notified by email of the decision and can then make changes and resubmit for approval. Approve the DOA by selecting ‘Yes’ for Q7 and then [Save Record].
Approving/Rejecting Documents (for CCC Site Managers)
To review uploaded documents for approval/rejection, click on the [Regulatory Document] tab on the study-specific main menu page. Next, click on [Site Reg Doc Review] or [People Reg Doc Review], as appropriate. This will take you to a ‘List Record’ page of all submitted regulatory documents.
To filter for the documents awaiting approval, select “Pending Docs” from the drop-down box in the upper right hand corner of the page. Click on the blue number link adjacent to the document you would like to review. This will take you to [Reg Doc Review].
Click on the document link to review the document for accuracy. Select the ‘Edit Record’ button in the top right hand corner of the page. If the documentis correct, select “Accepted.” If the document is incorrect, select “Rejected” and enter a reason for rejection. The site will receive an automatic email notification regarding the rejected regulatory document.
�
General Comments:
Name of person who collected these data (not for data entry):
Data Coordination Unit—MUSC SHINE Study book version 5-08-Nov-2012
SHINE Visit :
___ ___
Site ID
___ ___ ___ ___
Subject ID
Data Collected?
O No O Yes
___ ___ - ___ ___ ___ - ___ ___ ___ ___
(dd-mmm-yyyy) Date of assessment
Page 40
PO
INT
ver
sion
1
24S
EP2
008
Page 1 of 1 Form 20: Daily Care Log (version 2)
SH
INE
ve
rsio
n 2
19J
uly2
012
Page 2 of 2
If the treatment assignment is Control, complete ONLY columns A and F for each meal, regardless of whether it was ordered or not. If the treatment assignment is ‘Intervention’, complete all rows for each meal, regardless of whether it was ordered or not.
A. Meal
F. Meal status
If meal was not ordered,
skip to question C.
B.
What was the estimated amount of meal consumption?
C.
Was meal insulin given?
D. If ‘no’, indicate reasons:
E. If ‘staff error’ or ‘other’, specify:
�
10-1
O Breakfast
O Lunch
O Dinner
O Meal was not ordered, NPO
O Meal was not ordered for another reason
O A 60 gram carbohydrate meal was ordered
O Bolus tube feeding
O Continuous tube feeding
O Another diet was ordered
O Full or nearly full
O Partial
O None or nearly none
O No
O Yes
O No consumption or NPO
O Continuous tube feeding
O Staff error
O Other
10-2
O Breakfast
O Lunch
O Dinner
O Meal was not ordered, NPO
O Meal was not ordered for another reason
O A 60 gram carbohydrate meal was ordered
O Bolus tube feeding
O Continuous tube feeding
O Another diet was ordered
O Full or nearly full
O Partial
O None or nearly none
O No
O Yes
O No consumption or NPO
O Continuous tube feeding
O Staff error
O Other
10-3
O Breakfast
O Lunch
O Dinner
O Meal was not ordered, NPO
O Meal was not ordered for another reason
O A 60 gram carbohydrate meal was ordered
O Bolus tube feeding
O Continuous tube feeding
O Another diet was ordered
O Full or nearly full
O Partial
O None or nearly none
O No
O Yes
O No consumption or NPO
O Continuous tube feeding
O Staff error
O Other
Randomization assignment: Day 1 (0-24h)

�
General Comments:
Name of person who collected these data (not for data entry):
Data Coordination Unit—MUSC SHINE Study book version 5-08-Nov-2012
SHINE Visit :
___ ___
Site ID
___ ___ ___ ___
Subject ID
Data Collected?
O No O Yes
___ ___ - ___ ___ ___ - ___ ___ ___ ___
(dd-mmm-yyyy) Date of assessment
Page 41
PO
INT
ver
sion
1
24S
EP2
008
Page 1 of 1 Form 06: Adverse Event (version 2)
SH
INE
ve
rsio
n 2
08F
eb20
12
Page 1 of 4
All AEs must be reported through completion of study treatment. SAEs must be reported through End of Study.
1 Name of the adverse event: Please avoid colloquialisms and abbreviations and use correct medical terminology. (100 character max)
�
2
Severity: (Please refer to NCI Common Terminology Criteria for Adverse Events. See http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_40) If finger stick or confirmatory serum glucose is <40 mg/dl, Question 2 must be severe, life threatening/disabling, or fatal. If the AE was not fatal, skip to question 4.
O Mild
O Moderate
O Severe
O Life threatening / Disabling
O Fatal
3 Date of death: ___ ___ - ___ ___ ___ - ___ ___ ___ ___ (dd-mmm-yyyy)
4
Serious adverse event? Required Serious Adverse Events include, but are not limited to,
Events associated with finger stick or confirmatory serum glucose of <40 mg/dl are serious. Events associated with neurological worsening lasting longer than 24 hours and associated with glucose concentrations of <=55 mg/dL, are serious.
O No O Yes
5 Start date: ___ ___ - ___ ___ ___ - ___ ___ ___ ___ (dd-mmm-yyyy)
6 Start time: __ __ : __ __ (hh:mm, 24 hour clock)
7
Outcome: If outcome is ‘Continuing’, skip to question 9.
O Resolved
O Resolved w/ sequellae
O Continuing– Follow up is required
O Continuing– No follow up is required (SAE ongoing at end of study or AE ongoing at end of treatment)
8 End date: ___ ___ - ___ ___ ___ - ___ ___ ___ ___ (dd-mmm-yyyy)
�Day 2 (24-48h)
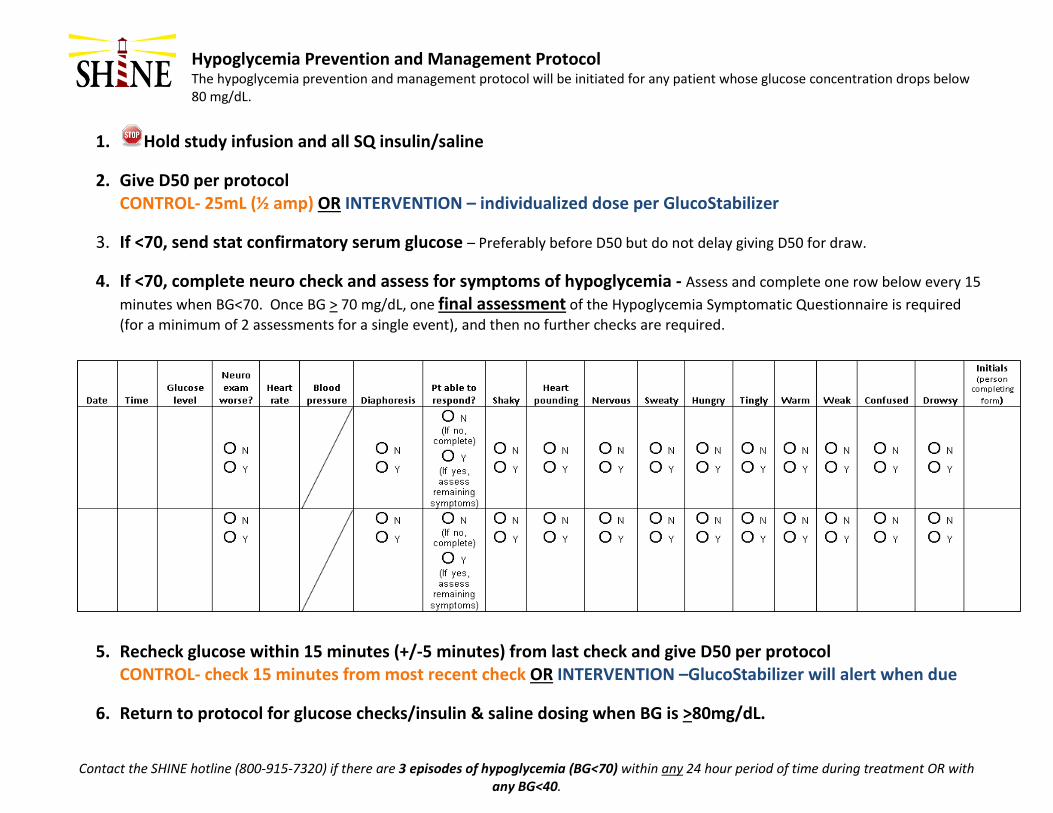
Hypoglycemia Prevention and Management Protocol The hypoglycemia prevention and management protocol will be initiated for any patient whose glucose concentration drops below
80 mg/dL.
Contact the SHINE hotline (800-915-7320) if there are 3 episodes of hypoglycemia (BG<70) within any 24 hour period of time during treatment OR with any BG<40.
1. Hold study infusion and all SQ insulin/saline
2. Give D50 per protocol CONTROL- 25mL (½ amp) OR INTERVENTION – individualized dose per GlucoStabilizer
3. If <70, send stat confirmatory serum glucose – Preferably before D50 but do not delay giving D50 for draw.
4. If <70, complete neuro check and assess for symptoms of hypoglycemia - Assess and complete one row below every 15 minutes when BG<70. Once BG > 70 mg/dL, one final assessment of the Hypoglycemia Symptomatic Questionnaire is required (for a minimum of 2 assessments for a single event), and then no further checks are required.
5. Recheck glucose within 15 minutes (+/-5 minutes) from last check and give D50 per protocol CONTROL- check 15 minutes from most recent check OR INTERVENTION –GlucoStabilizer will alert when due
6. Return to protocol for glucose checks/insulin & saline dosing when BG is >80mg/dL.
Hypoglycemia Prevention and Management Protocol The hypoglycemia prevention and management protocol will be initiated for any patient whose glucose concentration drops below
80 mg/dL.
Contact the SHINE hotline (800-915-7320) if there are 3 episodes of hypoglycemia (BG<70) within any 24 hour period of time during treatment OR with any BG<40.

Site Visit Report Template
STUDY/SITE INFORMATION
Protocol Name: SHINE
Stroke Hyperglycemia Insulin Network Effort
Visit conducted by:
Date of this Visit:
Previous Visit Date:
Investigator Name:
Location:
Report Number
Monitors- please note that tips and reminders specific to monitoring requirements have been
embodied in this report in italic text for your ease of reference. Also, please comment on anything that
is not done (N/D)
A. STUDY PERSONNEL: List all personnel that participated in the visit
NAME STUDY ROLE
COMMENTS
B. FOLLOW-UP FROM PREVIOUS VISIT:
YES NO
1. Are there any follow-up issues from the last visit? If yes, list below and include resolution or
status.
Comments:
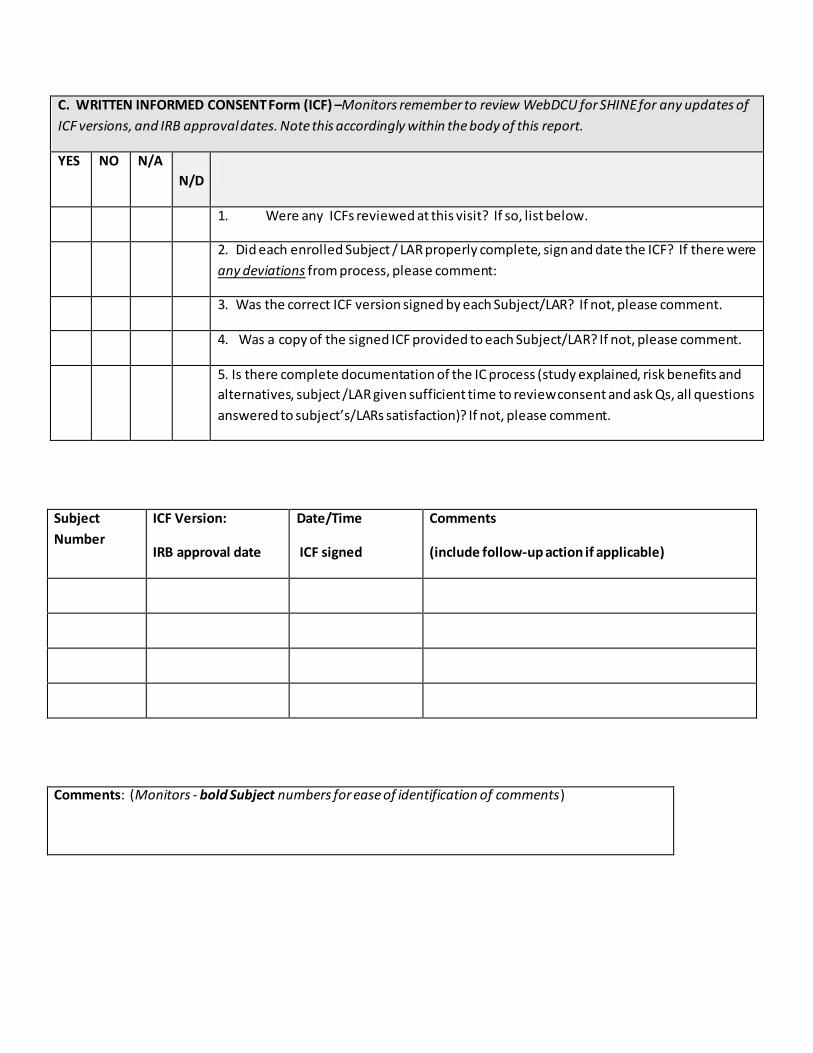
C. WRITTEN INFORMED CONSENT Form (ICF) –Monitors remember to review WebDCU for SHINE for any updates of
ICF versions, and IRB approval dates. Note this accordingly within the body of this report.
YES NO N/A
N/D
1. Were any ICFs reviewed at this visit? If so, list below.
2. Did each enrolled Subject / LAR properly complete, sign and date the ICF? If there were
any deviations from process, please comment:
3. Was the correct ICF version signed by each Subject/LAR? If not, please comment.
4. Was a copy of the signed ICF provided to each Subject/LAR? If not, please comment.
5. Is there complete documentation of the IC process (study explained, risk benefits and
alternatives, subject /LAR given sufficient time to review consent and ask Qs, all questions
answered to subject’s/LARs satisfaction)? If not, please comment.
Subject
Number
ICF Version:
IRB approval date
Date/Time
ICF signed
Comments
(include follow-up action if applicable)
Comments: (Monitors - bold Subject numbers for ease of identification of comments)

D. ELECTRONIC CASE REPORT FORMS (CRFs) & SOURCE DOCUMENTATION
YES NO N/A N/D
1. Were any eCRFs and Source Documents reviewed at this Visit? If so, then list
below
2. Are eCRFs being completed correctly and were source documents available to
support data reviewed? If no, comment:
3. Are subjects’ source documents and study records stored in a double - locked
location? If no, comment.
4. Are queries being addressed in a timely manner? If no, comment
Subject ID
Number
eCRFs Reviewed
(Visit and Forms)
Comments
Comments: (Monitors - bold Subject numbers for ease of identification of comments) Include # of
DCRs written, resolved during visit, remaining
E. PROTOCOL VIOLATIONS / DEVIATIONS
YES NO N/A
1. Were any Protocol Deviations noted at this visit? If yes, please comment below.
Subject ID
Number List Deviation Describe Deviation and document Action Taken

F. SERIOUS ADVERSE EVENTS (SAEs)
YES NO N/A N/D Monitors, review 100% SDV of all SAEs. Sites are to enter SAE into WebDCU within 24
hours of first knowledge. Monitors are advised to take a copy of the Adverse Event List
from the WebDCU for reference.
1. Were any SAEs noted since the last visit?
2. If yes, were SAEs reported to WebDCU and the IRB in the appropriate timeframe If
no, comment:
3. 3. Were any unreported SAEs identified during the visit?
4. . Were there SAEs from previous visits that required follow-up? If yes, comment:
5. Have all AEs/SAEs been appropriately documented & causality assessed by the
investigator?
Subject No. Event
Date
Serious Adverse Event Term Comments
ADDITIONAL COMMENTS:
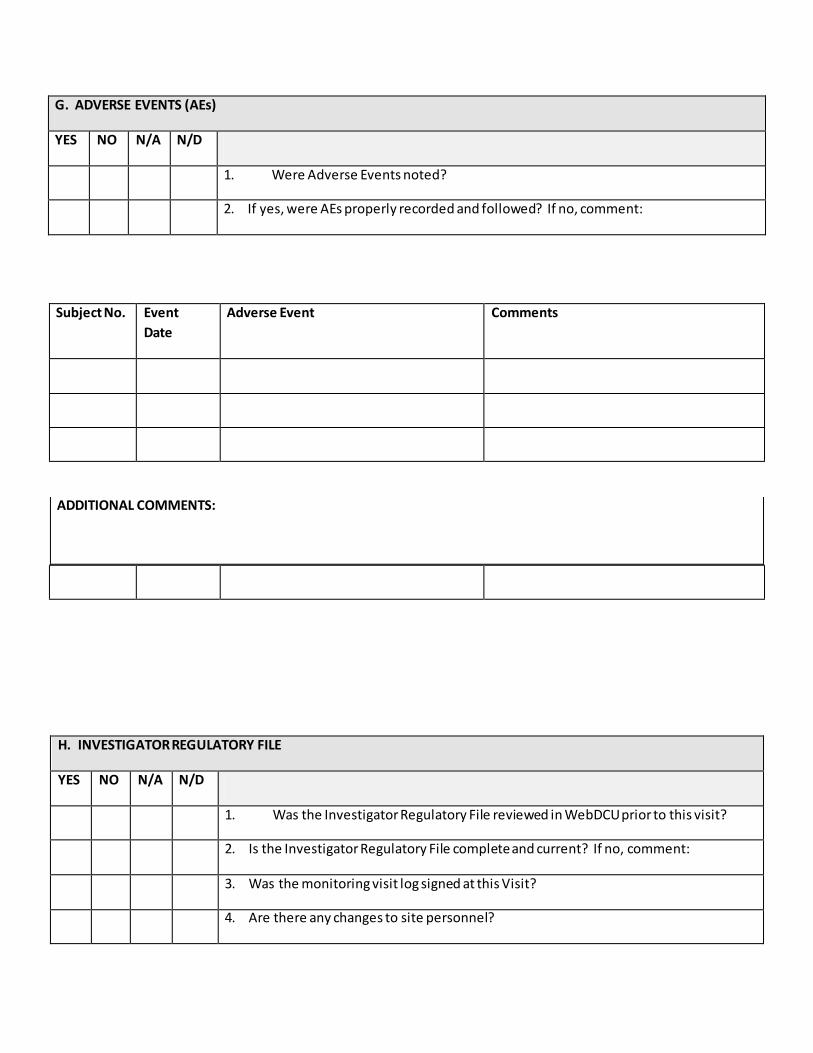
G. ADVERSE EVENTS (AEs)
YES NO N/A N/D
1. Were Adverse Events noted?
2. If yes, were AEs properly recorded and followed? If no, comment:
Subject No. Event
Date
Adverse Event Comments
H. INVESTIGATOR REGULATORY FILE
YES NO N/A N/D
1. Was the Investigator Regulatory File reviewed in WebDCU prior to this visit?
2. Is the Investigator Regulatory File complete and current? If no, comment:
3. Was the monitoring visit log signed at this Visit?
4. Are there any changes to site personnel?
ADDITIONAL COMMENTS:
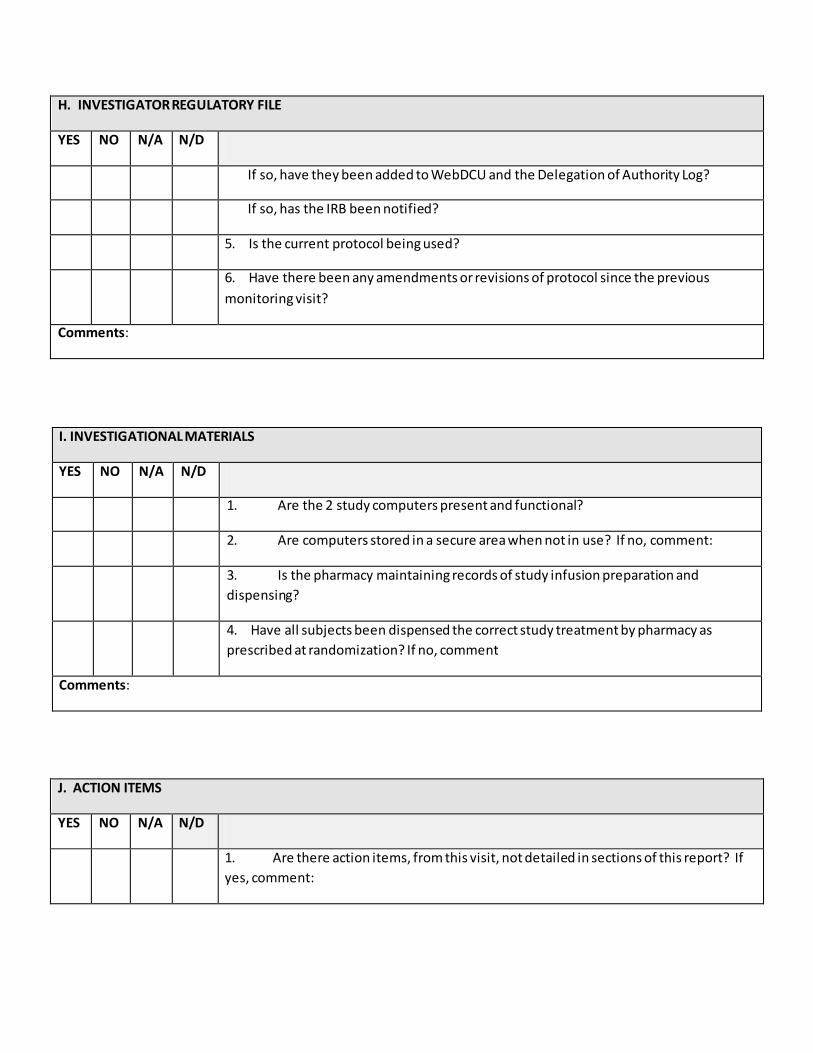
H. INVESTIGATOR REGULATORY FILE
YES NO N/A N/D
If so, have they been added to WebDCU and the Delegation of Authority Log?
If so, has the IRB been notified?
5. Is the current protocol being used?
6. Have there been any amendments or revisions of protocol since the previous
monitoring visit?
Comments:
I. INVESTIGATIONAL MATERIALS
YES NO N/A N/D
1. Are the 2 study computers present and functional?
2. Are computers stored in a secure area when not in use? If no, comment:
3. Is the pharmacy maintaining records of study infusion preparation and
dispensing?
4. Have all subjects been dispensed the correct study treatment by pharmacy as
prescribed at randomization? If no, comment
Comments:
J. ACTION ITEMS
YES NO N/A N/D
1. Are there action items, from this visit, not detailed in sections of this report? If
yes, comment:
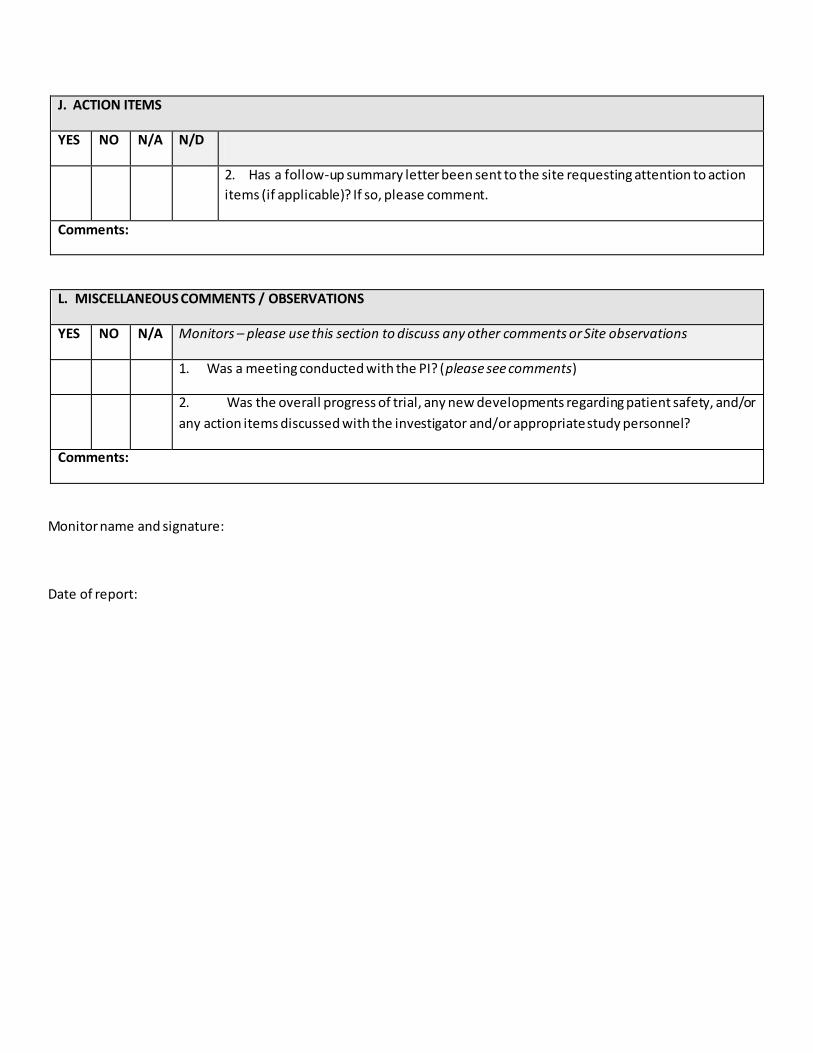
J. ACTION ITEMS
YES NO N/A N/D
2. Has a follow-up summary letter been sent to the site requesting attention to action
items (if applicable)? If so, please comment.
Comments:
L. MISCELLANEOUS COMMENTS / OBSERVATIONS
YES NO N/A Monitors – please use this section to discuss any other comments or Site observations
1. Was a meeting conducted with the PI? (please see comments)
2. Was the overall progress of trial, any new developments regarding patient safety, and/or
any action items discussed with the investigator and/or appropriate study personnel?
Comments:
Monitor name and signature:
Date of report:
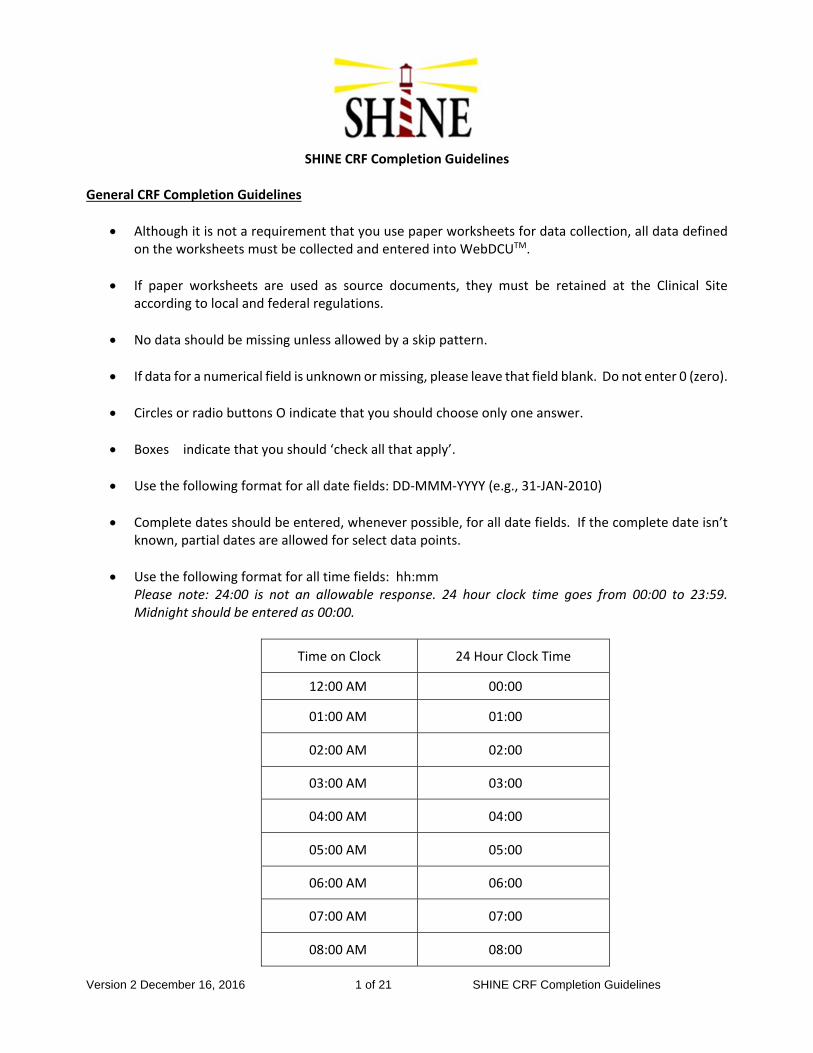
Version 2 December 16, 2016 1 of 21 SHINE CRF Completion Guidelines
SHINE CRF Completion Guidelines General CRF Completion Guidelines
Although it is not a requirement that you use paper worksheets for data collection, all data defined on the worksheets must be collected and entered into WebDCUTM.
If paper worksheets are used as source documents, they must be retained at the Clinical Site according to local and federal regulations.
No data should be missing unless allowed by a skip pattern.
If data for a numerical field is unknown or missing, please leave that field blank. Do not enter 0 (zero).
Circles or radio buttons O indicate that you should choose only one answer.
Boxes � indicate that you should ‘check all that apply’.
Use the following format for all date fields: DD‐MMM‐YYYY (e.g., 31‐JAN‐2010)
Complete dates should be entered, whenever possible, for all date fields. If the complete date isn’t known, partial dates are allowed for select data points.
Use the following format for all time fields: hh:mm Please note: 24:00 is not an allowable response. 24 hour clock time goes from 00:00 to 23:59. Midnight should be entered as 00:00.
Time on Clock 24 Hour Clock Time
12:00 AM 00:00
01:00 AM 01:00
02:00 AM 02:00
03:00 AM 03:00
04:00 AM 04:00
05:00 AM 05:00
06:00 AM 06:00
07:00 AM 07:00
08:00 AM 08:00

Version 2 December 16, 2016 2 of 21 SHINE CRF Completion Guidelines
09:00 AM 09:00
10:00 AM 10:00
11:00 AM 11:00
12:00 PM 12:00
01:00 PM 13:00
02:00 PM 14:00
03:00 PM 15:00
04:00 PM 16:00
05:00 PM 17:00
06:00 PM 18:00
07:00 PM 19:00
08:00 PM 20:00
09:00 PM 21:00
10:00 PM 22:00
11:00 PM 23:00
Name of person who collected the CRF data must be entered on the bottom of the paper worksheet, when the paper worksheet is used as a source document. This field will not be data entered but is required for monitoring purposes.
Data Entry Timelines:
Screen Failure Log ‐ The Clinical Site staff should update the Screen Failure Log forms in WebDCU by the 10th of the following month.
Baseline through End of Study CRFs ‐ Within 5 days of collection.
In the event of a Serious Adverse Event (SAE), the Adverse Event Form must be submitted within 24 hours of first knowledge of the event.

Version 2 December 16, 2016 3 of 21 SHINE CRF Completion Guidelines
Please note that site payments are dependent upon the subject’s data being entered and submitted.
Data Clarification Request (DCR) Timelines: All responses to DCRs must be submitted within 5 days of query generation with the exception of DCRs for SAEs, which must be submitted within 24 hours of query generation.
Screen Failure Log The Screen Failure Log is used to help identify the number of potential SHINE subjects who flow through a Clinical Site’s Emergency Department. Patients that are actively screened for the SHINE study by your study team but not randomized at your site should be included on the log. A screen failure log will be completed for all NON RANDOMIZED patients who were screened for the SHINE study. All patient diagnosed with ischemic stroke who present within 12 hours of onset with glucose >110 md/dl but were not randomized into the trial should be entered on the log. All potentially eligible subjects that were not randomized into the SHINE trial will be recorded on the log. Within the SHINE database, The Screen Failure Log is located under [Study Progress] Click on the number cell in the left hand column corresponding to the correct log, then select ‘edit record’ and data enter the required information. Once finished, click on [Save record]. To update a pre‐existing log, open the pre‐existing log, and then click [Edit Record]. The monthly Screen Failure Logs for actively enrolling sites will be posted automatically on the 1st of every month by WebDCU™. Screen failures for the previous month must be reported by the 10th of the following month. If a log is not entered by this date, your site will receive a late Screen Failure Log email notification, which is automatically generated by the WebDCU™ database. Even if a site does not have any screen failures, the log still must be submitted. Any screen failures to report? Answer ‘No’ or ‘Yes’. If ‘No’, no further information needs to be entered. If ‘Yes’, enter all screen failures that month, as designated on the form. Column F (Primary reason patient is not enrolled in SHINE): Select the code that corresponds with the primary reason for non‐enrollment. Additional reasons must be entered in general comments. Column G (Specify): If primary reason is “Missing information required for patient enrollment” ‘’Patient/LAR declined consent” or “other,” it must be specified in this column.

Version 2 December 16, 2016 4 of 21 SHINE CRF Completion Guidelines
Screen failures can be defined by reviewing the code list located on the bottom of the Screen Failure Log. Screen Failure Code List: 1= Age < 18 years 2= Renal dialysis (including hemo or peritoneal dialysis) 4= Inability to start protocol treatment within 12 hours of symptom onset or time last known to be well 5= Ischemic stroke patient with NIHSS score < 3 6= Ischemic stroke patient with NIHSS score > 22 20=Pre‐stroke Modified Rankin Scale score = 1 with NIHSS score of 3‐7 18= Pre‐stroke Modified Rankin Scale score > 1 8= Known history of type 1 diabetes mellitus 9= Substantial preexisting neurological/psychiatric conditions that would confound the neurological
assessment or other outcome assessment 10= Receipt of experimental therapy for the enrolling stroke including non‐FDA cleared devices. 11= Pregnant or breast‐feeding women at the time of proposed study enrollment 12= Other severe conditions with life expectancy < 90 days 13= Inability to follow the protocol 14= Missing information required for patient enrollment 15= Patient/LAR unable/unavailable to consent 16= Patient/LAR declined consent 17= Currently enrolled in another experimental treatment trial 19= No known history of type 2 diabetes and glucose <150mg/dL 96= Other
Version 2 December 16, 2016 5 of 21 SHINE CRF Completion Guidelines
Data Collection Schedule
Form 00: Eligibility Form This form is intended to document the subject’s eligibility prior to randomization. This form must be data entered and submitted into WebDCUTM. Note: All eligibility criteria must be met or randomization will be blocked. In a situation where an eligibility violation is discovered after randomization, the eligibility violation must be documented on the Eligibility Form. Eligibility criteria must be reviewed by a physician investigator before the form can be submitted. The reviewing physician investigator must be listed on the Physician Information form and the Delegation of Authority log.

Version 2 December 16, 2016 6 of 21 SHINE CRF Completion Guidelines
If you are unable to access the study database due to technical connectivity issues, please call the DCU Technical Emergency Randomization Hotline (1‐866‐450‐2016) to have a treatment assignment given to this subject. Remember, the Hotline is to be used only for emergency questions relating to enrollment. Form 04: NIH Stroke Scale The NIHSS is a well‐validated clinical tool to score the stroke neurological examination. The scale is scored from a minimum of 0, indicating no measurable neurological deficit, to a maximum score of 42. In practice, a score of <5 is a mild stroke, 6‐15 is a moderate to severe stroke, and >15 is a severe stroke. The scale can be administered in about 10 minutes. All health care personnel can be certified in the use of the scale. The NIHSS must be assessed in person by a clinical investigator at the site who has a current NIHSS certification and is included on the Delegation of Authority log. Certification is available through the American Stroke Association. The NIHSS will be completed at Baseline and the 90‐day visit. Administer stroke scale items in the order listed. Record performance in each category after each subscale exam. Do not go back and change scores. Follow directions provided for each exam technique. Scores should reflect what the patient does, not what the clinician thinks the patient can do. The clinician should record answers while administering the exam and work quickly. Except where indicated, the patient should not be coached (i.e., repeated requests to patient to make a special effort). Please remember that if the subject is in a coma, use coma scoring. WebDCU will check to ensure that coma scoring is data entered properly. Once a final score is determined, WebDCU will check to make sure that all questions have been answered, and summed properly. Subjects who are intubated, or have other physical barriers to speech, or have an amputation or joint fusion, requiring an ‘UN’ answer choice to be chosen, should NOT be enrolled in the study. 1a) Level of Consciousness The investigator must choose a response, even if a full evaluation is prevented by such obstacles as an endotracheal tube, language barrier, orotracheal trauma/bandages. A 3 is scored only if the patient makes no movement (other than reflexive posturing) in response to noxious stimulation. Coma score “3”. 0=Alert; keenly responsive. 1=Not alert, but arousable by minor stimulation to obey, answer or respond. 2=Not alert, requires repeated stimulation to attend, or is obtunded and requires strong or painful stimulation to make movements (not stereotyped). 3=Responds only with reflex motor or autonomic effects or totally unresponsive, flaccid, and flexic. (Complete form using coma scoring)

Version 2 December 16, 2016 7 of 21 SHINE CRF Completion Guidelines
1b) LOC Questions The patient is asked the month and his/her age. The answer must be correct ‐ there is no partial credit for being close. Aphasic and stuporous patients who do not comprehend the questions will score 2. Patients unable to speak because of endotracheal intubation, orotracheal trauma, and severe dysarthria from any cause, language barrier or any other problem not secondary to aphasia are given a 1. It is important that only the initial answer be graded and that the examiner not "help" the patient with verbal or non‐verbal cues. Coma score “2”. 0=Answers both questions correctly. 1=Answers one question correctly. 2=Answers neither question correctly. 1c) LOC Commands The patient is asked to open and close the eyes and then to grip and release the non‐paretic hand. Substitute another one step command if the hands cannot be used. Credit is given if an unequivocal attempt is made but not completed due to weakness. If the patient does not respond to command, the task should be demonstrated to him or her (pantomime) and the result scored (i.e., follows none, one or two commands). Patients with trauma, amputation, or other physical impediments should be given suitable one‐step commands. Only the first attempt is scored. Coma score “2”. 0=Performs both tasks correctly. 1=Performs one task correctly. 2=Performs neither task correctly. (2) Best Gaze Only horizontal eye movements will be tested. Voluntary or reflexive (oculocephalic) eye movements will be scored but caloric testing is not done. If the patient has a conjugate deviation of the eyes that can be overcome by voluntary or reflexive activity, the score will be 1. If a patient has an isolated peripheral nerve paresis (CN III, IV or VI), score a 1. Gaze is testable in all aphasic patients. Patients with ocular trauma, bandages, pre‐existing blindness or other disorder of visual acuity or fields should be tested with reflexive movements and a choice made by the investigator. Establishing eye contact and then moving about the patient from side to side will occasionally clarify the presence of a partial gaze palsy. Coma score as examined. 0=Normal. 1=Partial gaze palsy; gaze is abnormal in one or both eyes, but forced deviation or total gaze paresis is not
present. 2=Forced deviation, or total gaze paresis not overcome by the oculocephalic maneuver.

Version 2 December 16, 2016 8 of 21 SHINE CRF Completion Guidelines
(3) Visual Visual fields (upper and lower quadrants) are tested by confrontation, using finger counting or visual threat, as appropriate. Patient must be encouraged, but if they look at the side of the moving fingers appropriately, this can be scored as normal. If there is unilateral blindness or enucleation, visual fields in the remaining eye are scored. Score 1 only if a clear‐cut asymmetry, including quadrantanopia, is found. If patient is blind from any cause, score 3. Double simultaneous stimulation is performed at this point. If there is extinction, patient receives a 1, and the results are used to respond to question 22. Score as examined, using bilateral threat. 0=No visual loss. 1=Partial hemianopia. 2=Complete hemianopia. 3=Bilateral hemianopia (blind including cortical blindness). (4) Facial Palsy Ask—or use pantomime to encourage—the patient to show teeth or raise eyebrows and close eyes. Score symmetry of grimace in response to noxious stimuli in the poorly responsive or non‐comprehending patient. If facial trauma/bandages, orotracheal tube, tape or other physical barrier obscures the face, these should be removed to the extent possible. Coma Score “3”. 0=Normal symmetrical movement. 1=Minor paralysis (flattened nasolabial fold, asymmetry on smiling). 2=Partial paralysis (total or near total paralysis of lower face). 3=Complete paralysis of one or both sides (absence of facial movement in the upper and lower face). 5a) Motor Arm Left The limb is placed in the appropriate position: extend the arms (palms down) 90 degrees (if sitting) or 45 degrees (if supine). Drift is scored if the arm falls before 10 seconds. The aphasic patient is encouraged using urgency in the voice and pantomime, but not noxious stimulation. Each limb is tested in turn, beginning with the non‐paretic arm. Only in case of amputation or joint fusion at the shoulder, the examiner should record the score as untestable (UN), and clearly write the explanation for this choice. Coma Score “4”. 0=No drift, limb holds 90 (or 45) degrees for full 10 seconds. 1=Drift, limb holds 90 (or 45) degrees, but drifts down before full 10 seconds; does not hit bed or other
support. 2=Some effort against gravity, limb cannot get to or maintain (if cued) 90 (or 45) degrees, drifts down to
bed, but has some effort against gravity. 3=No effort against gravity, limb falls. 4=No movement. UN=Amputation, joint fusion.

Version 2 December 16, 2016 9 of 21 SHINE CRF Completion Guidelines
(5b) Motor Arm Right The limb is placed in the appropriate position: extend the arms (palms down) 90 degrees (if sitting) or 45 degrees (if supine). Drift is scored if the arm falls before 10 seconds. The aphasic patient is encouraged using urgency in the voice and pantomime, but not noxious stimulation. Each limb is tested in turn, beginning with the non‐paretic arm. Only in case of amputation or joint fusion at the shoulder, the examiner should record the score as untestable (UN), and clearly write the explanation for this choice. Coma Score “4”. 0=No drift, limb holds 90 (or 45) degrees for full 10 seconds. 1=Drift, limb holds 90 (or 45) degrees, but drifts down before full 10 seconds; does not hit bed or other
support. 2=Some effort against gravity, limb cannot get to or maintain (if cued) 90 (or 45) degrees, drifts down to
bed, but has some effort against gravity. 3=No effort against gravity, limb falls. 4=No movement. UN=Amputation, joint fusion. 6a) Motor Leg Left The limb is placed in the appropriate position: hold the leg 30 degrees (always tested supine). Drift is scored if the leg falls before 5 seconds. The aphasic patient is encouraged using urgency in the voice and pantomime, but not noxious stimulation. Each limb is tested in turn, beginning with the non‐paretic leg. Only in case of amputation or joint fusion at the hip, the examiner should record the score as untestable (UN), and clearly write the explanation for this choice. Coma score “4”. 0=No drift, leg holds 30 degrees position for full 5 seconds. 1=Drift, leg falls by the end of the 5 second period but does not hit bed. 2=Some effort against gravity; leg falls to bed by 5 seconds, but has some effort against gravity. 3=No effort against gravity; leg falls to bed immediately. 4=No movement. UN=Amputation, joint fusion. (6b) Motor Leg Right The limb is placed in the appropriate position: hold the leg 30 degrees (always tested supine). Drift is scored if the leg falls before 5 seconds. The aphasic patient is encouraged using urgency in the voice and pantomime, but not noxious stimulation. Each limb is tested in turn, beginning with the non‐paretic leg. Only in case of amputation or joint fusion at the hip, the examiner should record the score as untestable (UN), and clearly write the explanation for this choice. Coma score “4”.

Version 2 December 16, 2016 10 of 21 SHINE CRF Completion Guidelines
0=No drift, leg holds 30 degrees position for full 5 seconds. 1=Drift, leg falls by the end of the 5 second period but does not hit bed. 2=Some effort against gravity; leg falls to bed by 5 seconds, but has some effort against gravity. 3=No effort against gravity; leg falls to bed immediately. 4=No movement. UN=Amputation, joint fusion. (7) Limb Ataxia This item is aimed at finding evidence of a unilateral cerebellar lesion. Test with eyes open. In case of visual defect, ensure testing is done in intact visual field. The finger‐nose‐finger and heel‐shin tests are performed on both sides, and ataxia is scored only if present out of proportion to weakness. Ataxia is absent in the patient who cannot understand or is paralyzed. Only in the case of amputation or joint fusion, the examiner should record the score as untestable (UN), and clearly write the explanation for this choice. In case of blindness, test by having the patient touch nose from extended arm position. Coma score “0”. 0=Absent 1=Present in one limb 2=Present in two limbs UN=amputation or joint fusion 8) Sensory Sensation or grimace to pin prick when tested, or withdrawal from noxious stimulus in the obtunded or aphasic patient. Only sensory loss attributed to stroke is scored as abnormal and the examiner should test as many body areas [arms (not hands), legs, trunk, face] as needed to accurately check for hemisensory loss. A score of 2, “severe or total sensory loss,” should only be given when a severe or total loss of sensation can be clearly demonstrated. Stuporous and aphasic patients will, therefore, probably score 1 or 0. The patient with brainstem stroke who has bilateral loss of sensation is scored 2. If the patient does not respond and is quadriplegic score 2. Patients in coma (item 1a=3) are automatically given a 2 on this item. Coma Score “2”. 0=Normal; no sensory loss. 1=Mild to moderate sensory loss; patient feels pinprick is less sharp or is dull on the affected side; or there
is a loss of superficial pain with pinprick, but patient is aware of being touched. 2=Severe to total sensory loss; patient is not aware of being touched in the face, arm & leg. (9) Best Language A great deal of information about comprehension will be obtained during the preceding sections of the examination. For this scale item, the patient is asked to describe what is happening in the attached picture, to name the items on the attached naming sheet and to read from the attached list of sentences. Comprehension is judged from responses here, as well as to all of the commands in the preceding general neurological exam. If visual loss interferes with the tests, ask the patient to identify objects placed in the

Version 2 December 16, 2016 11 of 21 SHINE CRF Completion Guidelines
hand, repeat, and produce speech. The intubated patient should be asked to write. The patient in a coma (question 1a=3) will automatically score 3 on this item. The examiner must choose a score for the patient with stupor or limited cooperation, but a score of 3 should be used only if the patient is mute and follows no one‐step commands. Coma Score “3”. 0=No aphasia, normal. 1=Mild to moderate aphasia; some obvious loss of fluency or facility of comprehension, without significant
limitation on ideas expressed or form of expression. Reduction of speech and/or comprehension, however, makes conversation about provided materials difficult or impossible. For example, in conversation about provided materials, examiner can identify picture or naming card content from patient’s response.
2=Severe aphasia; all communication is through fragmentary expression; great need for inference, questioning, and guessing by the listener. Range of information that can be exchanged is limited; listener carries burden of communication. Examiner cannot identify materials provided from patient response.
3=Mute, global aphasia; no usable speech or auditory comprehension. (10) Dysarthria If patient is thought to be normal, an adequate sample of speech must be obtained by asking patient to read or repeat words from the attached list. If the patient has severe aphasia, the clarity of articulation of spontaneous speech can be rated. Only if the patient is intubated or has other physical barriers to producing speech, the examiner should record the score as untestable (UN), and clearly write an explanation for this choice. Do not tell the patient why he or she is being tested. Coma Score “2”. 0=Normal. 1=Mild to moderate dysarthria; patient slurs at least some words and, at worst, can be understood with
some difficulty. 2=Severe dysarthria; patient’s speech is so slurred as to be unintelligible in the absence of or out of
proportion to any dysphasia, or is mute/anarthric. UN=Intubated or other physical barrier. 11) Extinction and Inattention (formerly Neglect) Sufficient information to identify neglect may be obtained during the prior testing. If the patient has a severe visual loss preventing visual double simultaneous stimulation, and the cutaneous stimuli are normal, the score is normal. If the patient has aphasia but does appear to attend to both sides, the score is normal. The presence of visual spatial neglect or anosagnosia may also be taken as evidence of abnormality. Since the abnormality is scored only if present, the item is never untestable. Coma Score “2”. 0=No abnormality. 1=Visual, tactile, auditory, spatial, or personal inattention or extinction to bilateral simultaneous
stimulation in one of the sensory modalities. 2=Profound hemi‐inattention or extinction to more than one modality; does not recognize own hand or

Version 2 December 16, 2016 12 of 21 SHINE CRF Completion Guidelines
orients to only one side of space. For NIHSS certification, or to review the training information, please visit: http://nett.umich.edu/clinical‐trials/shine/education‐and‐training The assessor must be a study team member who has completed the NIHSS certification, whose certification is current and who is listed on the Delegation of Authority log. Form 07: Randomization To successfully randomize a patient, both the Eligibility and NIHSS forms must be saved and submitted without warning, protocol deviation or rejection. If there are errors or rule violations on either of these forms, randomization will be blocked. In the event that this occurs, an error message will display. Links will be provided to edit forms with rule violations that prevent randomization. In the event that a subject is not eligible to be randomized, please delete CRF data for the Eligibility and NIHSS Forms by opening the form and clicking [Delete CRF Data]. You will need to enter a reason for the deletion. Please call Kavita Patel at the data coordination unit during business hours at 843‐876‐1167 to have the Randomization form deleted. When a subject is eligible for randomization and the Eligibility and NIHSS forms are successfully saved and submitted, open and data enter the Randomization Form. Click [Save record] AND [Submit CRF]. Do not forget to click [Submit CRF] or randomization will not be complete. Once randomization is complete, the Subject ID, Treatment arm to which the subject is randomized, the color of the screen in Glucose Stabilizer based on the subject’s treatment assignment, Local Randomization Time and Date will be displayed. Please print, email or fax a copy of your randomization screen to your pharmacy and save a copy for your records. Once randomization is complete, it cannot be un‐done. Once the randomization number is assigned, this subject is enrolled in the trial and must be followed until the 90 day visit or withdrawal of consent. Form 01: Demographics This form is intended to capture basic demographic information. Several of the items on Form 01 are Common Data Element (CDE) questions.
It is important that all demographic information be verified by either subject self‐report, medical records, or a reliable individual accompanying the subject.
Ethnicity is a self‐reported or self‐identified data field that is required by the NIH. This field should be marked Unknown unless the subject/family members/medical records can provide the information.

Version 2 December 16, 2016 13 of 21 SHINE CRF Completion Guidelines
Sex is also a field required by the NIH. In cases of transgender and/or transsexual subjects, preference will be given to the subject’s birth sex. Form 02: Height and Weight The Height and Weight CRF is used to collect the actual or estimated height and weight of subjects. These measurements should be measured prior and closest to randomization. Form 03: Pre‐Treatment Vitals The Pre‐Treatment Vitals CRF contains items recommended as core stroke CDEs. Those items not identified as core stroke CDEs were not included in the Pre‐Treatment Vitals CRF. Form 05: HbA1c The HbA1c Form is used to capture the amount of sugar in the subject’s blood. The SHINE trial is collecting the HbA1c test that is performed within +/‐ 30 days from the randomization date. If more than one is collected, report the HbA1c collected closest to baseline. Form 06: Adverse Events The Adverse Event form is completed when an adverse event occurs and is used for documenting all AEs and SAEs. An adverse event (AE) is any unfavorable and unintended sign (including a clinically significant abnormal laboratory finding), symptom, or disease temporally associated with the use of a medical treatment or procedure regardless of whether it is considered related to the medical treatment or procedure (attribution of unrelated, unlikely, possible, probable, or definite). Each AE is a unique representation of a specific event used for medical documentation and scientific analysis. All AEs must be reported from the time of randomization through completion of the 72 h study treatment period. SAE’s must be reported through End of Study on the online AE case report form (CRF) through the WebDCU™. For serious adverse events (SAEs) that occur between the End of Study Treatment and the Six Week visit, complete Form 06 ‐ Adverse Event under the last treatment day visit in WebDCU. If the study team learns of an SAE that occurred between the Six Week and 90 Day visit, the AE form that posts under the 6 week visit in WebDCU can be used to document the event. Several SAE narrative templates are available at: https://nett.umich.edu/clinical‐trials/shine/toolbox#SAE Serious Adverse Events, must be data entered and submitted in WebDCUTM within 24 hours of the first knowledge of the event. The PI at each Clinical Site is responsible for the review, monitoring and follow‐up of all AEs until resolution (or end of study for that subject), ensuring the submission of AE data into the study database within the

Version 2 December 16, 2016 14 of 21 SHINE CRF Completion Guidelines
required timelines, adding appropriate documentation in the subject research record and submitting follow up data in a timely manner. In addition to performing protocol‐specified follow‐up, the participating PI must review all previously reported ongoing AEs to evaluate the current status. Upon completion of the study protocol by the subject, premature withdrawal from the study by the subject, or subject’s death, all information regarding each AE must be completed, if not done so earlier. If an AE changes in severity or frequency, it is considered a separate AE and must be reported on a new Adverse Events CRF. In this case, the outcome date of the first AE and the onset date of the new AE will both be the date upon which the severity or frequency changed. If an AE fully resolves and then recurs at a later date, the second occurrence is considered a new AE and a new AE CRF must be completed. Resolution is the normalization or return to baseline (of laboratory values, clinical signs or symptoms). If an AE occurs before the end of study treatment and involves neurological worsening, after completing the Adverse Event Form 06, the Neurological Worsening Form 22, must be completed. Choose the Adverse Event CRF ID# from Form 22’s drop down box in question 1. If an AE occurs before the end of study treatment and involves Hypoglycemia, after completing the Adverse Event Form 06, the Hypoglycemic Event Form 17, must be completed. Choose the Adverse Event CRF ID# from Form 17’s drop down box in question 1. Name of AE Please note that when reporting an AE, you should report the diagnosis and not each individual symptom. For example, it would be incorrect to report pneumonia as 4 separate events (fever, cough, chest pain, crackles). Pneumonia should be reported as one AE with the AE name being ‘pneumonia’. Death, surgery, intubation, etc. are not adverse events. They are outcomes of adverse events. When a subject dies, has surgery, is intubated, etc. please enter the reason for the death, surgery, intubation, etc. in the response. Severity Severity is often used to describe the intensity (severity) of a specific event (as in mild, moderate, severe myocardial infarction). However, the event itself may be of relatively minor medical significance (such as severe headache). Seriousness (not severity) serves as a guide for defining regulatory reporting obligations. Adverse events will be documented using the NCI Common Terminology Criteria for Adverse Events Version 4.0 (CTCAE) (see MoP). The CTCAE provides a grading (severity) scale for each AE term and AEs are listed alphabetically within categories based on anatomy or pathophysiology. The CTCAE (v 4.0) displays Grades 1‐5 with unique clinical descriptions of severity for each AE based on this general guidance:

Version 2 December 16, 2016 15 of 21 SHINE CRF Completion Guidelines
Grade 1: Mild AE Grade 2: Moderate AE Grade 3: Severe AE Grade 4: Life‐Threatening or Disabling AE Grade 5: Fatal Serious The seriousness of a Clinical Outcome is based on subject/event outcome or action (i.e., usually associated with events that pose a threat to a subject’s life or functioning). Serious Adverse Events are:
Fatal
Life‐Threatening
Result in hospitalization (or prolonged hospitalization)
Result in disability/congenital anomaly or
Require intervention to prevent permanent impairment or damage
Events associated with finger stick or confirmatory serum glucose of <40 mg/dl are serious.
Events associated with neurological worsening lasting longer than 24 hours and associated with glucose concentrations of <=55 mg/dL, are serious.
Outcome Any AE that is not resolved must be followed until the Day 90 visit, whichever comes first. Relatedness One of the most important components of AE reporting is determining the cause of the AE. It is imperative that the investigator assess AE causality in terms of overall study participation and make an independent determination as to whether the AE was thought to be related to any study‐related activity (i.e., study intervention, test article administration, study‐related tests or procedures). For each Adverse Event, the relationship to the study treatment must be recorded as one of the choices on the following scale: Not Related The temporal relationship between treatment exposure and the serious adverse event is unreasonable or incompatible and/or adverse event is clearly due to extraneous causes (e.g., underlying disease, environment)

Version 2 December 16, 2016 16 of 21 SHINE CRF Completion Guidelines
Unlikely (must have 2)
∙ May have reasonable or only tenuous temporal relationship to intervention. ∙ Could readily have been produced by the subject’s clinical state, or environmental or other interventions.
∙ Does not follow known pattern of response to intervention. ∙ Does not reappear or worsen with reintroduction of intervention.
Possibly (must have 2)
∙ Has a reasonable temporal relationship to intervention. ∙ Could not readily have been produced by the subject’s clinical state or environmental or other interventions.
∙ Follows a known pattern of response to intervention.
Probably (must have all 3)
∙ Has a reasonable temporal relationship to intervention. ∙ Could not readily have been produced by the subject’s clinical state or have been due to environmental or other interventions.
∙ Follows a known pattern of response to intervention. ∙ Disappears or decreases with reduction in dose or cessation of intervention.
Definitely (must have all 4) ∙ Has a reasonable temporal relationship to intervention. ∙ Could not readily have been produced by the subject’s clinical state or have been due to environmental or other interventions.
∙ Follows a known pattern of response to intervention. ∙ Disappears or decreases with reduction in dose or cessation of intervention and recurs with re‐exposure.
SAE Narratives These sections are used to provide additional relevant details about SAEs. This section should be as complete as possible, but only include information pertinent to the SAE. The Site Manager will utilize an outcome specific checklist to ensure that the event packet is sufficient for the medical monitor’s review. These narratives should not include any patient identifying information.

Version 2 December 16, 2016 17 of 21 SHINE CRF Completion Guidelines
To assist in the review of all SAEs, certain information is required for each SAE entered. Describe the event in detail. DO NOT identify any study participant, physician, or institution by name. The following are specific items to include in the SAE narrative:
1. Provide age, race, gender, most pertinent history, and time and date of enrollment. 2. Indicate whether subject previously experienced a stroke. 3. Include dates and times for the event and relevant procedures/clinical assessments. 4. Include a description of what happened and a summary of all relevant clinical information
(medical status prior to the event, signs and or symptoms. 5. Provide differential diagnosis for the event in question. 6. Provide complete clinical course information (relevant test/laboratory data: both positive and
negative results with corresponding dates. 7. Include all treatment outcomes. 8. Provide the discharge summary at length (if applicable).
Medical Safety Monitoring All SHINE SAEs will be reviewed by a Site Manager and Internal Quality and Safety Reviewer for data accuracy and completeness. Once the data is deemed accurate and complete, SAEs will be reviewed by an Independent Medical Safety Monitor for a determination of relatedness and expectedness. Once reviewed, SAEs will be sent to the DSMB liaison and Chair as per their request. At any time, any one of these parties may submit a Data Clarification Request (DCR), requesting more information about the SAE. Investigator Review Each SAE must be reviewed by a Site Investigator prior to data submission. Form 08: Lacunar Stroke Etiology This form should be filled out at End of Treatment and serves to measure whether the stroke was Lacunar. Form 09: Barthel Index The Barthel Index is a scale to evaluate performance in activities of daily living. This form should be collected at Day 90 visit. Answer all questions as correctly as possible. All questions on Form 09 are Common Data Elements (CDEs).

Version 2 December 16, 2016 18 of 21 SHINE CRF Completion Guidelines
Form 10: Modified Rankin Scale The mRS should reflect the subject’s current status. The Modified Rankin Scale (mRS) is a functional disability scale heavily weighted toward neurological disability. It is widely used and has strong face validity worldwide. The scale is best scored by medical personnel in person. However, a structured interview has been shown to have good reproducibility by telephone. The mRS will be administered at Week 6 and Day 90 visits. The assessor must be a blinded study team member who has completed the mRS certification, and one who is listed on the Delegation of Authority log. For more information about mRS certification, visit: http://www.rankinscale.org/links.shtml Form 11: SSQOL The SSQOL is an outcome measure intended to provide an assessment of health related, quality of life, specific to patients with stroke. This form should be collected at the 90 day visit. All questions on Form 11 are Common Data Elements (CDEs). Form 12: Unblinding Questionnaire ‐ Study Participant
This assessment is performed to document whether the study participant during the Day 90 visit is blinded to their treatment assignment. The study participant should make his/her best guess as to which treatment they received. If study participant marks ‘very sure’ to which treatment assignment they have been given, find out reasoning from participant and add the findings on the general comments portion of the form. Form 13: Unblinding Questionnaire ‐ Blinded Assessor
This assessment is performed to document whether the Investigator performing the Day 90 visit is blinded to the subject’s treatment assignment. The Investigator should make his/her best guess as to which treatment the subject received. If assessment is completed by an unblinded assessor, attempt to contact the blinded assessor to complete assessment within 120 days from Randomization window. Form 14: End of Study This form should be completed once a subject has completed the study (i.e., after 90 days, withdrawal of consent, lost to follow up, or death). The site PI, listed on the Delegation of Authority log, must review and affirm (by providing a signature and date the forms were reviewed) the accuracy of the information reflected in ALL of the case report forms for the study subject.
Form 15: Study Treatment This form captures information regarding the final diagnosis, the status of IV insulin and saline administration, sliding scale insulin levels and associated protocol deviations. The information on this form

Version 2 December 16, 2016 19 of 21 SHINE CRF Completion Guidelines
should match values entered into Glucose Stabilizer. This form should be collected at End of Treatment visit. Form 16: Current Antithrombotic Medications This form serves to capture all antithrombotic medications taken by the study participant prior to randomization and during treatment. This form should be collected at Baseline, Day 1, Day 2 and Day 3 of treatment. For the baseline visit, indicate which antithrombotic medications were taken by the study participant in the 48 hours prior to randomization. Include medications taken or given in the ED. For subsequent visits, indicate which antithrombotic medications were taken by the study participant in the previous 24 hours. If an antiplatelet or anticoagulant medication is not listed as an item on the CRF, it should be described in the “specify any other antiplatelet” or “specify any other anticoagulant” fields. Form 21: IV tPA and IA Therapy Form This form serves to capture all IV tPA and IA Therapy interventions from onset through the end of study treatment. Form 21 should be collected at End of Treatment (defined as the completion of a 72 hour treatment period unless discharged prior to 72 hours). Form 18: Medical History Form This form is collected at Baseline and is intended to document both the data obtained from the patient and/or his/her family, the medical record while screening the patient and pre‐existing conditions that are discovered after randomization into the trial.
For pre‐existing conditions discovered after a patient is randomized and after the Medical History CRF has been submitted, the Study Coordinator, or data entry staff, should edit the CRF accordingly AND enter a notation in the General Comments section at the bottom of the CRF. The General Comments notation should indicate the date the information became available and a brief description of the circumstances (e.g., dd‐mmm‐yyy at Hospital x.) Form 19: Glucose Control Medications This form should be collected at Baseline, End of Treatment and the 90 Day Visit. Its purpose is to track all Glucose Control medications taken by the subject prior to randomization, during treatment and after discharge. Form 20: Daily Care Log The Dietary Form should be collected at Day, 1 Day 2 and Day 3 of Treatment. This form captures the type of unit in which the subject was treated. If the subject was randomized to the intervention group, the glucose

Version 2 December 16, 2016 20 of 21 SHINE CRF Completion Guidelines
monitoring system used, whether the NIHSS was collected, and meal consumption and meal insulin is collected.
Form 17: Hypoglycemic Event The Hypoglycemic form is completed only if question 19 on Form 06: Adverse Event is ’yes’ (the reported adverse event is ‘hypoglycemia’ defined as blood glucose <70 mg/dL). Assess and complete one row every 15 minutes when BG<70. Once BG > =70 mg/dL, one final assessment of the Hypoglycemia Symptomatic Questionnaire is required (for a minimum of 2 assessments for a single event), and then no further checks are required. Form 22: Neurological Worsening The Neurological Worsening form is completed only if question 18 on Form 06: Adverse Event is ’yes’ which means that the reported adverse event meets the SHINE study definition of neurological worsening which is a clinical change associated with a ≥ 4 point NIHSS score increase from the most recent daily NIHSS score or baseline (whichever is most recent), that persists for 24± 4 hour. ****************************Forms and Visit Checklist ************************************ Baseline Please submit the following forms for this visit within 5 days of collection (unless otherwise indicated): Height and Weight (Form 02) Demographics (Form 01) Medical History (Form 18) Pre‐Treatment Vital Signs (Form 03) Glucose Control Medications (Form 19) Antithrombotic Medications (Form 16)
*The Eligibility (Form 00) and NIH Stroke Scale (Form 04) forms must be completed prior to Randomization (Form 07) or else randomization will be blocked. Please complete all AE forms with an SAE within 1 day of first knowledge of the event. Day 1 Please submit the following forms for this visit within 5 days of collection:
Antithrombotic Medications (Form 16) Daily Care Log (Form 20)
Day 2 Please submit the following forms for this visit within 5 days of collection:
Antithrombotic Medications (Form 16) Daily Care Log (Form 20)

Version 2 December 16, 2016 21 of 21 SHINE CRF Completion Guidelines
Day 3 Please submit the following forms for this visit within 5 days of collection:
Antithrombotic Medications (Form 16) Daily Care Log (Form 20)
End of Treatment Visit Please submit the following forms for this visit within 5 days of collection: HbA1c (Form 05) IV tPA and IA Therapy (Form 21) Glucose Control Medications (Form 19) Lacunar Stroke Etiology (Form 08) Study Treatment (Form 15)
Week 6 (+/‐ 14 days) Please submit the following forms for this visit within 5 days collection: Modified Rankin Scale (Form 10)
90 Day Visit (+/‐ 14 days) late data (+30 days) Please submit the following forms for this visit within 5 days of collection: NIH Stroke Scale (Form 04) Glucose Control Medications (Form 19) Barthel Index (Form 09) mRS (Form 10) SSQOL (Form 11) Unblinding Questionnaire‐Blinded Assessor (Form 13) Unblinding Questionnaires‐Study Participant (Form 12)
End of Study Visit (‐14/+30 days) Please submit the following forms for this visit within 5 days of collection: End of Study (Form 14)

Stroke Hyperglycemia Insulin Network Effort Trial NIH-NINDS Sponsored Trial In collaboration with Neurological Emergencies Treatment Trials (NETT) Network
SHINE Recruitment and Site Performance Monitoring Procedures
The SHINE trial procedures include standards for monitoring sites for recruitment/enrollment performance. Sites found to have major and/or ongoing deficiencies will be required to comply with the following at the discretion of the SHINE Executive Committee (EC).
1. Meeting with the SHINE leadership and project staff either in person or via telelconference.
2. Additional and/or refresher protocol training will be mandated for sites that do not
enroll a subject in the SHINE Trial for six (6) consecutive months.
3. Other actions deemed appropriate and necessary by the SHINE EC.
In addition, the SHINE EC has developed the following recruitment monitoring program plan:
The SHINE recruitment PIs (Drs. Kevin Barrett and Chris Hall) and the recruitment specialist (Katrina van de Bruinhorst) will review expected versus actual enrollment for each site, and reports to the EC and to the sites will be generated on a regular basis. The NETT performance PI (Dr. Bill Barsan) will participate in all communications to NETT sites. Sites performing below recruitment expectations for 6 months will be contacted by phone by the recruitment team to discuss recruitment obstacles and develop plans for improvement. Continued underperformance will be reported to the EC and may result in a site visit by a member of the EC and/or recruitment team to explore additional options. Termination of a site for poor recruitment is unlikely as continued efforts to improve recruitment may be beneficial to the SHINE trial.
Recruitment Milestones- Overall Study Recruitment Real-time overall recruitment reports will be generated and reviewed by the SHINE project team on a weekly basis. Individual site recruitment reports will be generated and reviewed by the SHINE project team on a monthly basis. Drs. Barrett and Hall and the recruitment specialist will be provided the recruitment reports for review on a routine basis. The overall recruitment progress will be benchmarked against expected recruitment and reported to the executive committee weekly along with a summary of which sites account for any variances from the 100% benchmark noted. Recruitment Milestones- Site Specific Recruitment Similar to the overall study Milestone benchmarking report, SHINE quarterly data quality and performance reports will be generated every 3 months as to take into account that stroke patient presentation may ebb & flow but will average over time. The recruitment team will conduct annual teleconferences with the sites to discuss enrollment. The minimum enrollment goal for each site is to enroll at least 1 subject every other month. The minimum annual enrollment goal for each site is to enroll 4 subjects per year. If a site fails to meet the minimum expected goals for enrollment two quarters in a row the recruitment team will schedule a teleconference with the site PI’s and PSCs to discuss enrollment barriers and if possible to devise an enrollment remediation plan. Version date 16DEC2016 Page 1 of 1

Stroke Hyperglycemia Insulin Network Effort Trial
NIH-NINDS Sponsored Trial In collaboration with the Neurological Emergencies Treatment Trials network (NETT)
Contact Askiel Bruno ([email protected]) with questions about study orders and pharmacy plan.
Pharmacy Plan Guidance
The pharmacy plan is a tool used during the site readiness process to develop and document the site-specific procedures for study drug ordering, labeling and dispensing for the SHINE trial. Please include a ½-1 page summary that details the following:
STUDY DRUG: List the study drugs that will be used at your site based on your hospital formulary
DRUG SUPPLY & STORAGE: Detail how study drugs will be supplied and where they will be stored
RANDOMIZATION: Describe the randomization process as it pertains to your site pharmacy (how is the
randomization assignment provided to pharmacy and documentation of this maintained)
DRUG ORDERING & DISPENSING: Describe the process for ordering/dispensing study-required treatments;
Detail the system used to place orders (electronic, hard copy orders, etc.)
STUDY DRUG NAMING & LABELS: Describe how the study drug will appear in the medical record and how the
actual treatments (vials, infusion bags) are labeled
PHARMACY CONTACT INFORMATION: Include name & contact info for pharmacist who will be working with SHINE
In addition to the Pharmacy Plan summary, please attach study order sets (screenshots of electronic orders, actual hard copy orders or instructions for manual order entry into an electronic system), pharmacy instructions and labels as available and upload the compiled document in WebDCU.
Please reference the instructions that follow on developing study orders for SHINE as you begin to draft the study order set, or contact Dr. Askiel Bruno for more information or for sample plans and order sets.

Stroke Hyperglycemia Insulin Network Effort Trial
NIH-NINDS Sponsored Trial In collaboration with the Neurological Emergencies Treatment Trials network (NETT)
Contact Askiel Bruno ([email protected]) with questions about study orders and pharmacy plan.
Developing study orders for SHINE
We have compiled the information included below as a resource for developing the SHINE study order set. We would encourage you to adapt the template language organized by treatment group on the following pages to draft your study orders. Additionally, several enrolling sites have shared copies of their site pharmacy plan and orders, and these are posted on the SHINE study website (https://www.nett.umich.edu/nett/shine_toolbox) for your reference.
We are available to work with your study team and pharmacy to develop the study orders for your site and recommend starting to work on this early. Because of the complexity of the protocol and dosing regimens, prior to the readiness call, we will review copies of your site study order sets. If corrections or updates to the orders are required, this can delay overall site readiness, so again we encourage you to start early. When complete, study orders are compiled and uploaded in WebDCU with the ½ to 1 page pharmacy plan that addresses the process, labeling and pharmacy contact information.
Pharmacy/Medication orders and Physician to Nurse/Nursing communication orders Please plan to incorporate both the orders for the study treatments as well as the necessary communication orders to manage the study protocol during the 72 hour treatment period. Orders should be implemented using the standard practice for research at your institution. Hard copy or paper orders, templates for manual order entry or an electronic order set are all acceptable formats for the orders.
Labels
Consider what will print on the labels.
The IV infusion should be blinded since it will be hanging in the patient’s room.
The labels for the SQ treatments do not have to be blinded as these can be drawn up outside of the view of the patient/family.
SHINE study drug stickers o These are supplied by UVA to be applied by the pharmacy on the study infusion bag. (Orange –
control group; Blue – intervention group) o Pharmacy preparation instructions should detail the application of the study drug sticker on the
study infusion bag. o These study drug stickers must only be applied by the pharmacy. o The MOP specifies that the study team retains one study drug sticker per subject for monitoring
purposes. This can be removed from the infusion bag and retained, or a color-copy can be made of the bag/sticker with the subject ID visible, and the bag can be discarded.
o Please contact Heather M. Haughey ([email protected]) for resupply, allowing 2 weeks for these to arrive.
Documentation in the medical record/charting
Consider how the study treatments will be charted in the medical record
The study monitors will review source documentation to confirm the name of the actual study treatment administered with time and dose.

Stroke Hyperglycemia Insulin Network Effort Trial
NIH-NINDS Sponsored Trial In collaboration with the Neurological Emergencies Treatment Trials network (NETT)
Contact Askiel Bruno ([email protected]) with questions about study orders and pharmacy plan.
Naming conventions
GlucoStabilizer is the name for the commercially available decision support tool. The company that has developed the decision support tool has asked that we do not refer to the programming that they have done for the control protocol and data entry as GlucoStabilizer.
For the control group, you may refer to the control protocol, control treatment screen, study sliding scale, study laptop, etc. (e.g. Rate per study laptop)
For the intervention group, you may refer to GlucoStabilizer, intervention protocol, study laptop, etc. (e.g. Timing of glucose checks: q1 to q2 hours per GlucoStabilizer)
Dosing calculations for the intervention group The GlucoStabilizer calculations for the insulin infusion, meal insulin and D50 follow.
Regular insulin infusion: There is not a hard maximum cutoff to the IV regular insulin dosage recommendation. The recommendation is calculated based on the formula "(BG - 60 ) * multiplier". However if the insulin dose is > 32, a warning message is displayed to check with the physician before proceeding.
For the rapid acting analog (meal) insulin dosing, the insulin to carbohydrate ratio is 1:15. One unit of insulin will be recommended for every 15 grams of carbs consumed. Based on the study protocol diet and instructions for estimating consumption, carbohydrate intake per meal will be estimated to be either 30 or 60 grams. Two units of rapid acting analog insulin will be recommended for patients that partially consume the meal (entry of 30 into Cover Carbs option in GlucoStabilizer). Four units of SQ rapid acting analog insulin will be recommended for patients that consume all or nearly all of the meal (entry of 60 into Cover Carbs option in GlucoStabilizer). If none or nearly none of the meal is consumed, there should be no entry in GlucoStabilizer, and no rapid acting analog insulin given for that meal.
For dextrose 50% in Water (D50), when the BG < 80mg/dL, an individualized dose is calculated per
GlucoStabilizer based on the following formula: (100 - BG) * 0.4 The lowest value that is allowed for the BG is 1, so the highest theoretical value for D50 in the intervention group is (100 - 1) * 0.4 = 40mL. (As a reminder in the control group, IV D50 25 mL (1/2 amp D50) will be given every 15 minutes when the BG <80mg/dL.)

Stroke Hyperglycemia Insulin Network Effort Trial
NIH-NINDS Sponsored Trial In collaboration with the Neurological Emergencies Treatment Trials network (NETT)
Contact Askiel Bruno ([email protected]) with questions about study orders and pharmacy plan.
CONTROL GROUP
1) Glucose checks
POC glucose tests
i) Check glucose levels using finger stick testing every hour for the first 4 hours, then every 3 hours for the remaining treatment period (03:00, 06:00, 09:00, 12:00, 15:00, 18:00, 21:00, & 24:00). Use capillary blood only unless otherwise directed by study team. POC testing ONLY Adjust saline infusion with each glucose check.
OR
POC glucose checks i) Blood Glucose Monitoring POCT
(1) Frequency: Q1hours (2) Duration: 4 hours (3) Special instructions: Use capillary blood only unless otherwise directed by study team. POC
testing ONLY. Complete on POC test to start the study infusion and then hourly for four hours (POC to start study infusion mg/dL _ _:_ _; H1 _ mg/dL _ _:_ _; H2 _mg/dL _ _:_ _; H3 mg/dL _ _:_ _; and H4 mg/dL _ _:_ _; then transition to the schedule for Q3hour checks
ii) Blood Glucose Monitoring POCT (1) Frequency: Q3hours (2) Duration: Up to 66 hours (3) Special Instructions: Time testing 0300; 0600; 0900; 1200; 1500; 1800; 2100; 24:00 (4) Special instructions: Use capillary blood only unless otherwise directed by study team. POC
testing ONLY
2) IV saline - SHINE Study Drug in Normal Saline _ _ _mL infusion (1:1); Insulin regular human or placebo in NS _ _ _mL infusion (1:1)
IV continuous
Adjusted with each glucose check
Rate determined per control treatment screen
o 4mL/hr for glucose of 80-179 o 5mL/hr for glucose of ≥180mg/dL
If glucose is < 80 mg/dL, administer NO saline or SQ insulin and initiate study hypoglycemia protocol
3) Regular Insulin SQ (Humulin R or Novolin R)
Route: SQ Insulin regular subcutaneous
Frequency: Q6 hours, up to four times a day -- ONLY at 06:00, 12:00, 18:00, & 24:00
Dose per Level 1, 2 or 3 on study sliding scale
Duration: Up to 72 hours
Special instructions: Level will be determined by study team every 24 hours period on study protocol based on the last two POCT BG results
4) Dextrose 50%
Route: IV
Frequency: Every 15 MIN PRN (Blood glucose < 80mg/dL)
Dose: 25mL
Special instructions: Recheck blood glucose every 15 minutes and repeat treatment until blood glucose > 80. When blood glucose > 80, restart study saline infusion and insulin SQ per protocol.
5) Basal insulin SQ (Glargine (Lantus))
Route: SQ

Stroke Hyperglycemia Insulin Network Effort Trial
NIH-NINDS Sponsored Trial In collaboration with the Neurological Emergencies Treatment Trials network (NETT)
Contact Askiel Bruno ([email protected]) with questions about study orders and pharmacy plan.
Dose: To be determined by study team per special instructions
Frequency: Once
Special Instructions: For Level 3, one-time SQ basal insulin injection at a dose of 40% of previous 24 hours entire insulin requirement (>.5 round up; <.5, round down). This dose of basal insulin should be given as close as possible to the 48 hour point regardless of time of day.
6) Hypoglycemia prevention and management
Hypoglycemia protocol for BG < 80 mg/dL i) Stop the saline infusion and hold all subcutaneous insulin injections ii) A dose of IV D50 25 ml (1/2 amp) will be given (slow IV push over 1-2 minutes) q 15 min until blood
glucose is ≥80 mg/dL. iii) Repeat finger stick glucose checks and treatment q 15 min until glucose is ≥80 mg/dL.
b) Additional steps for blood glucose <70mg/dL i) Draw a STAT laboratory serum glucose measurement but do not delay treatment with D50. ii) Screen the patient for hypoglycemia symptoms using Hypoglycemia Symptomatic Questionnaire.
(1) Repeat q15 min when glucose is <70mg/dL. (2) Once the glucose is ≥ 70mg/dL, no further assessment with the Hypoglycemia Symptomatic
Questionnaire is required. iii) Screen the patient for neurological worsening. iv) Once the glucose is ≥80 mg/dL, the timing of glucose checks and saline infusion rate will again be
determined by the control treatment screen.
6) Diet/Nutrition Orders – Control group a) 60 gram carbohydrate diet for PO patients for breakfast lunch and dinner & protocol approved snacks
i) Timing of meals (1) Breakfast after 06:00 check (2) Lunch after 12:00 check (3) Dinner after 18:00 check
b) Patients should not consume additional food not included on the meal tray from the hospital kitchen. c) Family, friends and visitors should be instructed not to consume food from the patient’s trays unless
approved by the nurses, after patients finish eating.
7) Procedures for pauses/interruptions in study protocol
a) Notify the study team OR
b) When the study protocol needs to be paused, stop the IV saline infusion. c) Upon return to the unit, if glucose checks or subcutaneous insulin injections were not missed, maintain
schedule for sliding scale checks and dosing. Resume saline infusion at the next scheduled glucose check.
d) Upon return to the unit, if glucose checks or subcutaneous insulin injections were missed, the following procedures should be followed: i) Immediately check the finger stick point of care glucose upon return to the unit. ii) Resume the saline infusion according to the sliding scale using the glucose measurement. iii) If one of the time points for scheduled subcutaneous injections was missed, use the result of the
glucose check and sliding scale to determine if a subcutaneous insulin dose is indicated. iv) If indicated per sliding scale, give subcutaneous insulin injection immediately (rather than waiting for
next scheduled dose). v) Return to schedule for glucose checks and subcutaneous insulin injections.
(1) Do not check glucose levels <1 hour apart unless it is a check associated with a scheduled insulin dose. If the next check is <1 hour from the check that happened upon return to the unit, skip to the next scheduled check.

Stroke Hyperglycemia Insulin Network Effort Trial
NIH-NINDS Sponsored Trial In collaboration with the Neurological Emergencies Treatment Trials network (NETT)
Contact Askiel Bruno ([email protected]) with questions about study orders and pharmacy plan.
(2) Do not give SQ regular insulin injections <3 hours apart. If a subcutaneous injection is given upon return to the unit (as described above) AND the next scheduled injection is <3 hours do not give insulin at the next scheduled injection time.
8) Steps for >3 episodes of hypoglycemia (Glucose < 70mg/dL) in 24 hour period
a) Notify the study team (Study team contacts safety monitor) b) At the 24 and 48 hour intervals, regardless of the previous two glucose measurements, do NOT advance
to a higher level on the subcutaneous sliding scale.
9) Severe hyperglycemia (Glucose > 500mg/dL)
a) Draw a STAT laboratory serum glucose measurement b) Notify the study team (Study team contacts safety monitor)
10) Discharge prep instructions
a) Notify study team as study treatment must be discontinued 6 hours in advance of discharge
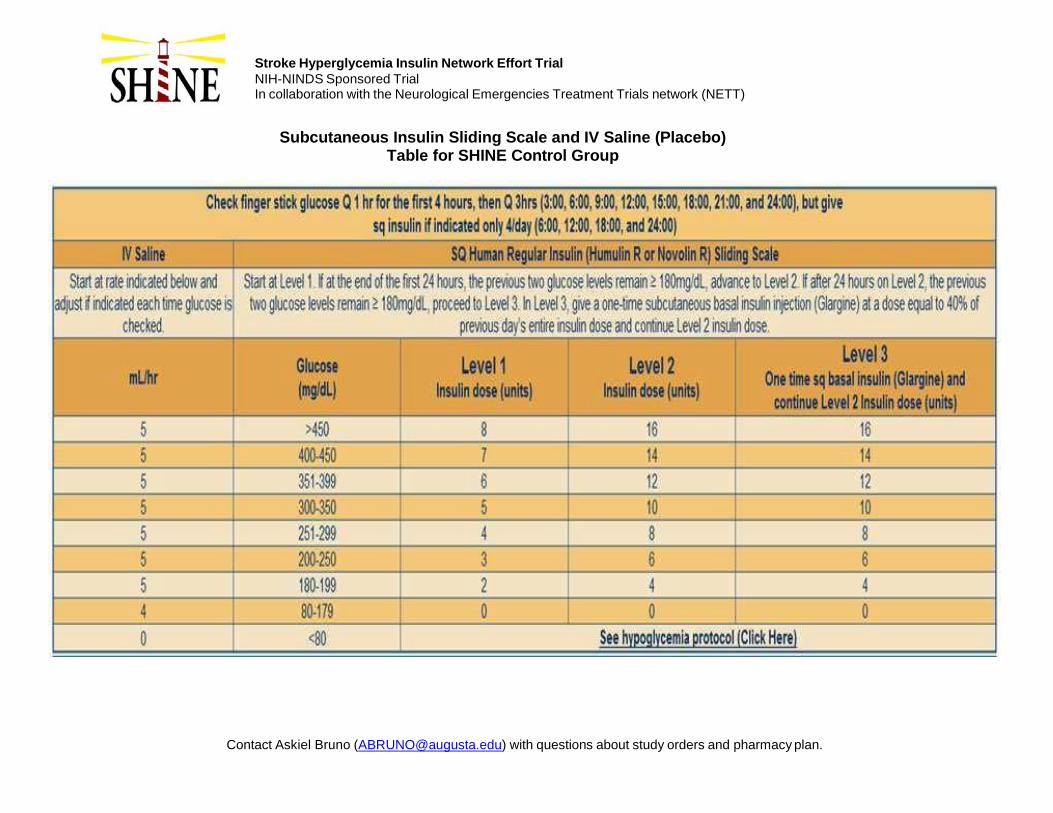
Stroke Hyperglycemia Insulin Network Effort Trial
NIH-NINDS Sponsored Trial In collaboration with the Neurological Emergencies Treatment Trials network (NETT)
Subcutaneous Insulin Sliding Scale and IV Saline (Placebo) Table for SHINE Control Group
Contact Askiel Bruno ([email protected]) with questions about study orders and pharmacy plan.

Stroke Hyperglycemia Insulin Network Effort Trial
NIH-NINDS Sponsored Trial In collaboration with the Neurological Emergencies Treatment Trials network (NETT)
Contact Askiel Bruno ([email protected]) with questions about study orders and pharmacy plan.
INTERVENTION GROUP
1) Glucose checks-Intervention group
POC glucose tests
Check glucose levels using finger stick testing as instructed by GlucoStabilizer Use capillary blood only unless otherwise directed by study team. POC testing ONLY
OR
POC glucose checks i) Blood Glucose Monitoring POCT
(1) Frequency: Q1hours per GlucoStabilizer (2) Duration: 4 hours (3) Special instructions: Use capillary blood only unless otherwise directed by study team. POC
testing ONLY
vi) Blood Glucose Monitoring POCT (1) Frequency: Q1-2 hours per GlucoStabilizer (2) Duration: Up to 68 hours (3) Special instructions: Use capillary blood only unless otherwise directed by study team. POC
testing ONLY
2) IV insulin (human insulin regular- Humulin R or Novolin R) - SHINE Study Drug in Normal Saline _ _ _mL
infusion (1:1); Insulin regular human or placebo in NS _ _ _mL infusion (1:1)
Route: IV continuous
Frequency: Adjusted with each glucose check
Rate: Rate determined per GlucoStabilizer
Special instructions: If glucose is < 80 mg/dL, administer NO IV insulin or SQ insulin or saline and initiate study hypoglycemia protocol
3) Meal insulin (Rapid acting analog insulin SQ - Lispro (Humalog), Aspart (Novolog) or Glulisine (Apidra)
Route: SQ
Frequency: Up to three times daily with meals
Duration: Up to 72 hours
Dose: To be determined by carbohydrate count using GlucoStabilizer tool
Special instructions: Approximately 20 minutes after patient begins eating, estimate meal consumption. o If none or nearly none, DO NOT enter carbohydrate amount in GlucoStabilizer and do not give
meal insulin SQ o If partial meal consumption, enter 30gm carbohydrate in GlucoStabilizer and give SQ rapid acting
insulin according to recommendation by GlucoStabilizer tool. o If all or nearly all, enter 60gm carbohydrate in GlucoStabilizer give SQ rapid acting insulin
according to recommendation by GlucoStabilizer tool.
4) Saline SQ
Dose: 0.05mL (equivalent to 5 Units)
Route: SQ
Frequency: BIS (0900,2100)
Duration: Up to 72 hours
Special Instructions: Administer for patient who are NPO or on continuous tube feeds to maintain the blind. Administer after the glucose checks closest to 09:00am and 21:00pm.

Stroke Hyperglycemia Insulin Network Effort Trial
NIH-NINDS Sponsored Trial In collaboration with the Neurological Emergencies Treatment Trials network (NETT)
Contact Askiel Bruno ([email protected]) with questions about study orders and pharmacy plan.
5) Dextrose 50% - Intervention group
Route: IV
Frequency: Every 15 MIN PRN (Blood glucose < 80mg/dL)
Dose: Dose determined by GlucoStabilizer tool
Duration: Up to 72 hours
Special instructions: Recheck blood glucose every 15 minutes and repeat treatment as directed by GlucoStabilizer until blood glucose > 80. When blood glucose > 80, restart study insulin infusion and insulin SQ or the saline SQ per protocol.
6) Hypoglycemia prevention and management- Intervention group
Hypoglycemia protocol for BG < 80 mg/dL v) Stop the insulin infusion and hold all subcutaneous insulin and saline injections vi) An individualized dose of IV D50 will be recommended by GlucoStabilizer and should be
administered slow IV push (over 1-2 minutes). vii) Repeat finger stick glucose checks and treatment q 15 min per GlucoStabilizer until BG is ≥80 mg/dL.
c) Additional steps for blood glucose <70mg/dL i) Draw a STAT laboratory serum glucose measurement but do not delay treatment with D50. ii) Screen the patient for hypoglycemia symptoms using Hypoglycemia Symptomatic Questionnaire.
(1) Repeat q15 min when glucose is <70mg/dL. (2) Once the glucose is ≥ 70mg/dL, no further assessment with the Hypoglycemia Symptomatic
Questionnaire is required. iii) Screen the patient for neurological worsening. iv) Once the glucose is ≥80 mg/dL, the timing of glucose checks and insulin infusion rate will again be
determined by the GlucoStabilizer.
11) Diet/Nutrition Orders – Intervention group
a) 60 gram carbohydrate diet for PO patients for breakfast lunch and dinner & protocol approved snacks Estimate the proportion of the meal consumed approximately 20 minutes after the start of the meal i) Full or near full consumption 60 grams carbohydrates ii) No or nearly no consumption 0 grams carbohydrates iii) Partial consumption 30 grams carbohydrates
b) Patients should not consume additional food not included on the meal tray from the hospital kitchen. c) Family, friends and visitors should be instructed not to consume food from the patient’s trays unless
approved by the nurses, after patients finish eating.
12) Procedures for pauses/interruptions in study protocol- Intervention group a) Notify the study team
OR e) When the study protocol needs to be paused, stop the IV insulin infusion.
i) Upon return to the unit, if the IV insulin infusion has been off for <3 hours, follow the protocol to resume the drip.
ii) Upon return to the unit, if the IV insulin infusion has been off for ≥ 3 hours, follow the protocol to start new drip).
13) Steps for >3 episodes of hypoglycemia (Glucose < 70mg/dL) in 24 hour period
a) Notify the study team (Study team contacts safety monitor)
14) Severe hyperglycemia (Glucose > 500mg/dL)
a) Draw a STAT laboratory serum glucose measurement b) Notify the study team (Study team contacts safety monitor)
15) Discharge prep instructions
a) Notify study team as study treatment must be discontinued 6 hours in advance of discharge

Stroke Hyperglycemia Insulin Network Effort Trial NIH-NINDS Sponsored Trial In collaboration with Neurological Emergencies Treatment Trials (NETT) Network
SHINE Protocol Adherence Monitoring Procedures
The overall goal of site monitoring and aggregate protocol adherence monitoring in the SHINE trial is to identify patterns suggestive of overall study protocol issues or individual site issues that may require support and/or retraining on study treatment procedures. The SHINE trial procedures and NETT Standard Operating Procedures (SOPs) include individual site monitoring for protocol adherence and corrective action plans for sites in need of improvement. Sites found to have major and/or ongoing deficiencies will be required to comply with the following at the discretion of the SHINE executive committee (EC).
1. Meeting with the SHINE leadership and project staff either in person or via telelconference. 2. Additional and/or refresher protocol training will be required as deemed appropriate based on the
nature of the deficiency. 3. Interruption in study related activities including suspension of enrollment in the SHINE trial. 4. Other actions deemed appropriate and necessary by the SHINE EC.
Below is the process for review of protocol adherence monitoring reports by the protocol PI (Dr. Askiel Bruno). Due to the different treatment protocols, the protocol PI needs to view certain adherence items by treatment group. The protocol PI is the only member of the SHINE EC, except for the unblinded statistician (Valerie Durkalski), who views these reports. The information is reported in aggregate by treatment group. The protocol PI remains blind to the total number of patients in each group and to all patient outcomes.
1. The unblinded SHINE statistical team will generate a protocol monitoring adherence report
(template attached) every 50 randomized patients. A site-specific report will be generated once a site reaches 5 randomizations and then after every additional 5 randomizations. These reports will be emailed to the protocol PI for review.
2. The primary adherence data point is percent of late blood glucose checks. Late checks are defined as more than 15 minutes past the scheduled check. This threshold has been deemed appropriate for identifying delayed glucose control that may be associated with hypoglycemia. The adherence threshold for late checks for each treatment group is defined as 10% of all glucose checks. If this threshold is exceeded in either the aggregate report or site-specific report, the protocol PI will work with the statistical team and/or site monitors to gather information on reasons for the late checks and to determine if the EC should be notified.
3. Upon notification, the SHINE EC and the NETT performance PI (Dr. Bill Barsan) may work with all sites or individual sites on a corrective or preventive action plan as appropriate.
4. The adherence reports include additional protocol adherence data that the protocol PI will be reviewing including: median time interval between randomization and infusion start, prolonged pauses, early glucose checks, correct administration of D50, control group level changes, correct insulin dosing and timing in the control group and carbs consumed/correct meal insulin dosing in the intervention group. The protocol PI will be monitoring these data overall and by individual sites and will notify the SHINE EC in the event of unexpected results. Individual site performance will determine the need for individual site corrective action plans.
5. Communication about site monitoring and adherence may be through the protocol PI, the NETT performance PI, SHINE trial monitors or any other appropriate study team member.
This process allows the SHINE EC to consider study issues that may be problematic for all sites as well as to identify and work with individual sites on any necessary protocol-specific issues without becoming unblinded.
Version date 16DEC2016 Page 1 of 1

Page 1 of 1
Stroke Hyperglycemia Insulin Network Effort Trial NIH-NINDS Sponsored Trial In collaboration with Neurological Emergencies Treatment Trials (NETT) Network
To: SHINE Study Teams
From: SHINE Recruitment Leadership
Date: January 21, 2016
Re: Use of OmniTrace People Locating Service in SHINE
Thank you for taking a moment to review this letter of instruction in regards to the SHINE clinical trial sponsored
by the NIH-NINDS and the University of Virginia. As you may be aware, OmniTrace Corp. has been selected to
assist in locating Lost to Follow-Up (LTFU) patients who have participated in the SHINE trial. OmniTrace
specializes in LTFU searching and has assisted hundreds of studies since 2003.
This letter describes the process and transfer of information between the enrolling site and OmniTrace.
1) When a subject is at risk for being lost to follow up and contact information is not up to date, the clinical
enrolling site will work with the SHINE Recruitment Leadership team to determine whether use of
OmniTrace is indicated. Please note that OmniTrace can only be used at sites that have IRB approval
and for subjects that have signed a consent form that includes details about the people locating service.
2) The University of Virginia has a business associate agreement with OmniTrace and will centrally pay for
the cost of the service. The use of OmniTrace must be approved by the SHINE Recruitment Team for
each subject and will be initiated by the University of Virginia.
3) Upon approval to use the service, the enrolling site will complete the attached Information Form. The
form collects contact information from the enrolling site (Site name, Site number, contact person,
phone, fax and email) as well as the patient’s name, their last known address, last known phone and
date of birth. Please note that OmniTrace does not access patient medical records and does not
electronically store patient data. The enrolling site will directly fax the completed Information Form to
OmniTrace.
4) Once OmniTrace has completed the search, they will fax a completed report containing all results to the
enrolling site. For compliance reasons, OmniTrace does not make contact with any subjects of the
search in order to verify results. As such, there will be occasions where their information may not be
accurate. Please notify OmniTrace immediately if this is the case as they may have to run secondary
searches. If they are not notified of incorrect results, they will consider them to be accurate. If
OmniTrace determines the patient is deceased, they will provide the date of death and a source
reference.

Primary Contact: Ashley Watkins
Direct Phone: +(1) 561-470-8937
Toll Free: 800-799-1911
E-Mail: [email protected]
Secondary Contact: Heather Groeneveld
Direct Phone: +(1) 561-470-8989
Fax: + (1) 561-470-8905 Toll Free: 800-799-1911
E-Mail: [email protected]
Study Name: SHINE
City: State: Zip Code:
Phone: Fax:
Email:
Date of Birth: Gender:
Street:
City:
State/Province:
Zip/Postal Code:
Date?:
Date Received:
Last Known Address
Additional Information?:
Citizenship,Marital Status
Misc.
Last Known Phone Number:
Investigator Information
(Last)Subject Name:
Additional Information
Omnitrace Use Only
Date Completed:
(First) (Middle)
200 Congress Park Drive Suite 206
Delray Beach, FL 33445
Toll Free: 800-799-1911
Subject Information
Site/Hospital/Clinic Name:
Contact Person:
Address:
PLEASE COMPLETE ALL THE FIELDS BELOW AND FAX THIS FORM TO OMNITRACE AT: +(1) 561 470 8905
Country
Subject ID:
1/1 LTFU Request Form_DEC2007

OmniTrace FAQ
OmniTrace FAQs
What primary function does OmniTrace provide to a clinical study?
OmniTrace provides the administrative function of updating current contact information for
subjects identified as LTFU or at risk of LTFU. Our second primary area of involvement is
assisting to obtain the vital status data of designated subjects. These two key functions are
required to maintain study integrity by significantly reducing missing data.
Does OmniTrace provide additional clinical trial support services?
OmniTrace provides comprehensive project support including ICF language review,
LTM/CRA/Site LTFU training, IRB and EC response and rebuttals, death certificate retrieval,
MAE monitoring, EDC assistance, Site communication, metrics development as well as other
custom services.
What is OmniTrace’s experience with clinical trials?
OmniTrace has assisted clinical trials since 2003, worked on hundreds of domestic and global
studies and has successfully handled over twenty thousand requests. OmniTrace is the premier
provider of LTFU services and is the primary option when a firm has an issue with Lost to
Follow-Up patients (LTFU) or is simply establishing an LTFU contingency plan.
Who are OmniTrace’s typical clients?
OmniTrace’s clients consist of Pharmaceutical Companies, CROs, AROs, Hospitals, public
research institutions and government agencies such as the National Institutes of Health.
Is OmniTrace licensed and insured?
OmniTrace is licensed (A2200304) by the State of Florida as a Private Investigative Agency.
OmniTrace’s current coverages include General Liability and Errors and Omissions of
$2,000,000.00 per Occurrence and $2,000,000.00 Aggregate.
What countries does OmniTrace provide services to?
OmniTrace’s LTFU search capabilities are global in scope. We also work with our clients, ethics
committees and IRBs to determine if any local regulations prevent the use of our service.
OmniTrace offers multiple international programs to select from or can customize a program to
suit EC requirements.

OmniTrace FAQ
How long does an average search last?
For USA searches average turnaround time is 24 - 48 hours. Internationally, the length of time for
a search will vary by the country and program selection. Turnaround time can typically range from
3 days to 6 weeks depending on the type of search we conduct.
How does OmniTrace collect information from Sites?
OmniTrace prefers not to transmit subject data electronically as part of its standard operating
procedures. Information is received and transmitted to Sites via facsimile. If faxing is not an
option, we can verbally transmit information or utilize other secure means.
Does OmniTrace utilize independent contractors?
In the USA, all searches are conducted by OmniTrace employees. Globally, all searches are
initially conducted by OmniTrace employees. If the results of an international search are not
successful, we can utilize the assistance of a local, in-country investigator but only when
authorized by the client.
Can OmniTrace assist us in scheduling a patient visit or collect medical data?
OmniTrace does not get involved with patient medical issues, data collection or patient interviews.
We do not access medical records as part of our search. We only are providing the administrative
function of updating contact information or providing vital status.
What information does OmniTrace need to conduct a search?
We typically only require the patient’s name, last known address, previous phone number and date
of birth.
Can you tell us about OmniTrace’s compliance?
OmniTrace maintains strict patient privacy guidelines and data security policies. HIPAA,
HITECH, GCP, EU Data Directives, Graham-Leach Bliley and other associated regulations are
embedded in our processes. OmniTrace’s records and processes are open for audit and assessment.
Has OmniTrace been subject to any FDA Audits?
OmniTrace has never been subject to an FDA or regulatory audit, but our facilities and policies
have been assessed and audited on multiple occasions by numerous clients.
How long does OmniTrace archive subject information?
OmniTrace only maintains hard copy records for a period of two years. No electronic records are
maintained.
Does OmniTrace communicate directly with IRBs and Ethics Committees?
OmniTrace works with the Sponsor to respond to IRB/EC questions regarding our services but
typically works at arm’s length versus directly. OmniTrace will assist in answering any questions
an IRB or EC may have, provide policy documents pertaining to our services, assist in preparing
amendment letters and otherwise work with the sponsor, Site and EC/IRB to gain approval.

OmniTrace FAQ
Why Use OmniTrace?
o Significantly reduce the number of LTFU patients
o To provide 100% vital status on all subjects
o Monitor Major Adverse Events
o Reduce FDA scrutiny
o Maximize clinical trial success
o Eliminate missing trial data
o Allow for a faster clinical trial endpoint
o Enhance regulatory compliance and post study surveillance
o Greatly reduce clinical trial costs
Can you tell us more about OmniTrace’s vital status programs?
Domestically, OmniTrace is often requested to conduct vital status determinations on large subject
populations as various endpoint and study outcome dates approach. OmniTrace provides an “alive
as of date” or “date of death” along with a supporting reference. These vital status runs can be
scheduled in advance or on an as needed basis. Internationally, there is a lack of searchable death
records and therefore vital status determinations are often predicated on locating the subject alive.





























