Embed Size (px)
Citation preview



Ghent University Faculty of Pharmaceutical Sciences
Biodegradable dextran nanogels as functional carriers for the intracellular delivery of small interfering RNA
Biodegradeerbare dextraan nanogelen als functionele dragers voor de intracellulaire afgifte van small interfering RNA
Koen Raemdonck Pharmacist
Thesis submitted to obtain the degree of Doctor in Pharmaceutical Sciences
Proefschrift voorgedragen tot het bekomen van de graad van Doctor in de Farmaceutische Wetenschappen
2009
Dean
Prof. dr. apr. Jean Paul Remon
Promoters
Prof. dr. apr. Stefaan C. De Smedt Prof. dr. apr. Jo Demeester
Laboratory of General Biochemistry and
Physical Pharmacy


The author and the promoters give the authorization to consult and to copy parts of this
thesis for personal use only. Any other use is limited by the Laws of Copyright, especially the
obligation to refer to the source whenever results from this thesis are cited.
De auteur en de promotoren geven de toelating dit proefschrift voor consultering
beschikbaar te stellen en delen ervan te kopiëren voor persoonlijk gebruik. Elk ander gebruik
valt onder de beperkingen van het auteursrecht, in het bijzonder met betrekking tot de
verplichting uitdrukkelijk de bron te vermelden bij het aanhalen van resultaten uit dit
proefschrift.
Ghent, October 2nd 2009
Promoters: Author:
Prof. dr. apr. Stefaan C. De Smedt Koen Raemdonck
Prof. dr. apr. Jo Demeester




table of contents
TABLE OF CONTENTS
Table of contents 9
List of abbreviations & symbols 11
General introduction & thesis outline 15
CHAPTER 1 | Maintaining the silence: reflections on long‐term RNAi 21 CHAPTER 2 | In situ analysis of single‐stranded and duplex siRNA integrity in living cells 59
CHAPTER 3 | Dextran microgels for time‐controlled delivery of siRNA
85
CHAPTER 4 | Advanced nanogel engineering for drug delivery 109
CHAPTER 5 | Biodegradable dextran nanogels for RNAi: focusing on endosomal escape
and intracellular siRNA delivery 133 CHAPTER 6 | Evaluation of gene silencing kinetics with siRNA loaded dextran nanogels 163 CHAPTER 7 | Liposomal template‐assisted synthesis of dextran based nanogels for
siRNA delivery 183
CHAPTER 8 | Summary & general conclusions 205
APPENDIX A | Samenvatting & algemene besluiten 215
APPENDIX B | Curriculum vitae 223
DANKWOORD | Acknowledgment 233
9

table of contents
10

list of abbreviations & symbols
LIST OF ABBREVIATIONS
AAV adeno‐associated virus
AF488 Alexa Fluor 488
AFM atomic force microscopy
Ago argonaute
agRNA antigene RNA
AMD age‐related macular degeneration
AON antisense oligonucleotide
ATRP atomic transfer radical polymerization
bp basepair
BSA bovine serum albumin
CaF carboxyfluorescein
CDP cyclodextrin polycation
CLSM confocal laser scanning microscopy
coRNAi combinatorial RNA interference
CPP cell penetrating peptide
CRP controlled radical polymerization
DEAEMA 2‐(diethylamino)ethyl methacrylate
DEPC diethyl pyrocarbonate
Dex‐HEMA dextran hydroxyethyl methacrylate
Dex‐MA dextran methacrylate
DGCR8 DiGeorge syndrome chromosomal region 8
DIC differential interference contrast
diINF‐7 dimer of influenza‐derived fusogenic peptide‐7
DLS dynamic light scattering
DMAEMA 2‐(dimethylamino)ethyl methacrylate
DMEM Dulbecco’s modified Eagle’s medium
DNA deoxyribonucleic acid
DNase deoxyribonuclease
DOPE 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine
DOPC 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine
DS degree of substitution
DsiRNA dicer substrate RNA
dsRNA double stranded RNA
DTT dithiothreitol
ECM extracellular matrix
EDTA ethylenediaminetetraacetic acid
11

list of abbreviations & symbols
EE encapsulation efficiency
EGFP enhanced green fluorescent protein
eri‐1 enhanced RNAi‐1
FANA 2’‐fluoro‐β‐D‐arabino nucleic acid
FA/FD ratio of acceptor to donor fluorescence upon donor excitation
FBS fetal bovine serum
FFS fluorescence fluctuation spectroscopy
FRET fluorescence resonance energy transfer
GPC gel permeation chromatography
HBV hepatitis B virus
HDL high density lipoprotein
HEPES 4‐(2‐hydroxyethyl)‐1‐piperazine ethanesulfonic acid
HIV‐1 human immunodeficiency virus‐1
HPH high pressure homogenization
Huh‐7 human hepatoma‐7
kDa kilodalton
KPS potassium peroxodisulfate
LCST lower critical solution temperature
LDL low density lipoprotein
LEN lipid enveloped nanogel
LF lipofectamine
LMV large multilamellar vesicle
LNA locked nucleic acid
LPX lipoplex
MEND multifunctional envelope‐type nano device
miRNA microRNA
MOE 2‐methoxyethyl
mRNA messenger RNA
MW molecular weight
MWCO molecular weight cut‐off
nt nucleotide
NPC nuclear pore complex
OD optical density
OTE off‐target effect
PAGE polyacrylamide gel electrophoresis
PbAE poly(β‐amino ester)
PBS phosphate buffered saline
PCI photochemical internalization
12

list of abbreviations & symbols
PDI polydispersity index
pDNA plasmid DNA
PEG poly(ethylene glycol)
PEI poly(ethylene imine)
PNIPAAm poly(N‐isopropylacrylamide)
PRINT particle replication in nonwetting templates
PS photosensitizer
P/S penicillin/streptomycin
PTGS post‐transcriptional gene silencing
RdDM RNA dependent DNA methylation
RdRP RNA dependent RNA polymerase
RhoGr rhodamine green
RISC RNA induced silencing complex
RLU relative light units
RNA ribonucleic acid
RNAi RNA interference
RNase ribonuclease
RT room temperature
SD standard deviation
SDS sodium dodecylsulfate
SEM scanning electron microscopy
shRNA short hairpin RNA
siRNA small interfering RNA
SNALP stable nucleic acid lipid particle
ssRNA single stranded RNA
SUV small unilamellar vesicle
TBE tris, boric acid and EDTA buffer
TEMED N,N,N’,N’‐tetramethylenediamine
TGS transcriptional gene silencing
TLR toll‐like receptor
TMAEMA [2‐(methacryloyloxy)ethyl] trimethylammonium chloride
TPPS2a meso‐tetraphenylporphine disulfonate
TRBP TAR RNA binding protein
TX100 Triton®X‐100
UV ultraviolet
VPG vesicular phospholipid gel
W/O water‐in‐oil emulsion
w/v weight/volume
13

list of abbreviations & symbols
14
LIST OF SYMBOLS
ε extinction coefficient
λex excitation wavelength
λem emission wavelength
ζ zeta potential

general introduction & thesis outline
GENERAL INTRODUCTION &
THESIS OUTLINE
15

general introduction & thesis outline
16

general introduction & thesis outline
GENERAL INTRODUCTION
RNA interference and siRNAs
Just slightly over a decade ago, Andrew Fire and Craig Mello described in their Nature publication the
peculiar observation that the exogenous introduction of double‐stranded RNA (dsRNA) into the
nematode C. elegans resulted in the suppression of the complementary gene rather than enhancing
its expression.[1] These authors were the first to launch the term RNA interference (RNAi), a discovery
that was awarded in 2006 with the Nobel Prize for Medicine and Physiology.
At present, RNAi is defined essentially as a naturally occurring and evolutionary conserved
mechanism by which short dsRNA triggers the sequence‐specific inhibition of gene expression. The
knowledge that this gene silencing mechanism is operational in mammalian (and therefore also
human) cells, revolutionized our understanding of endogenous gene regulation by small RNAs and
sparked an explosion of research to harness RNAi as a novel therapeutic modality.[2] RNAi can be
implemented for the treatment of a variety of human diseases with a known genetic origin. Now
with the sequencing of the entire human genome, RNAi may lead to a plethora of drug molecules
against any chosen clinical target.[3] The importance of the discovery of RNAi is further emphasized
by the fast transition from lab to clinic with several clinical trials already in progress and several more
that are scheduled for the coming years.
Intensive investigation on the RNAi mechanism identified small interfering RNAs (siRNAs) as the
genuine effectors of the genetic interference.[4] Synthetic siRNAs can easily be produced through
large‐scale chemical synthesis and the majority of reports on RNAi therapeutics have focused on
siRNA for the activation of gene silencing. However, to trigger RNAi, siRNAs need to be delivered into
the cytoplasm of the target cell and many extra‐ and intracellular barriers are encountered before
the site of action is reached. It is therefore imperative for the widespread development of siRNA
therapeutics to focus on the design of novel nucleic acid delivery systems to overcome these.[5] As
virus‐mediated nucleic acid delivery is still associated with insuperable safety concerns, the use of
non‐viral delivery strategies has gained a lot of interest. The majority of reports on siRNA delivery
describe the application of lipid or polymer based carriers to enhance the fraction of the
administered siRNA dose that effectively reaches the target cell cytoplasm.[6]
17

general introduction & thesis outline
Hydrogels for siRNA delivery
Hydrogels can be defined as three‐dimensional networks of hydrophilic polymers that are capable of
absorbing large quantities of water or biological fluid. Both synthetic and natural polymers can be
used as core material for their construction. Since their introduction for biological use almost 50
years ago,[7] at present hydrogels are favoured for many biomedical and pharmaceutical purposes,
including controlled drug delivery.[8‐10] Hydrogels may exist in many geometries such as macroscopic
gels (e.g. scaffolds, cylinders,...), microscopic gels and nanogels. Up to now, literature has mainly
focused on macro‐ and microscopic hydrogels for extracellular drug delivery applications but during
the last decade nanogels have gained momentum as drug delivery vehicles. Since siRNAs are needed
in the cell cytoplasm to be effective, as a consequence hydrogels with nanoscopic dimensions are
required to allow intracellular siRNA release.
In essence, due to the relatively recent discovery of siRNA therapeutics, there is a lack of scientific
studies describing nanogels as siRNA carriers for their controlled delivery across the cellular barriers,
which is therefore the major topic of investigation in this thesis.
THESIS OUTLINE
CHAPTER 1 provides insight into the mechanism of RNA interference and discusses the predominant
barriers for siRNA delivery. Moreover, in this chapter an overview can be found of the most
important parameters that govern the silencing efficiency and influence the duration of the gene
silencing effect.
In CHAPTER 2 we aimed to develop a method, based on fluorescence resonance energy transfer (FRET)
to assess the intracellular siRNA stability. Both double stranded (ds) and single stranded (ss) siRNA
are included in this investigation. In addition to chapter 1, we discuss the significance of intracellular
siRNA stability on the RNAi outcome.
In the context of controlled siRNA delivery, CHAPTER 3 investigates if cationic and biodegradable
dextran microgels are suited for siRNA complexation and subsequent degradation controlled release.
This chapter also provides a first indication that these cationic gel particles are able to deliver active
siRNA across the cellular barriers.
Before we investigate intracellular siRNA delivery with our dextran hydrogel particles in more detail,
CHAPTER 4 reviews the state‐of‐the‐art of nanogel design for drug delivery applications.
18

general introduction & thesis outline
Through mini‐emulsion photopolymerization, in CHAPTER 5 we succeeded in the production of cationic
dextran nanogels, based on the same core material as was used in chapter 3. Their siRNA
complexation potential, intracellular siRNA delivery and eventual gene silencing activity are
evaluated. Additionally, the influence of endosomolytic tools on the RNAi effect is investigated.
Besides the maximal gene suppression, it is equally important to assess the duration of the gene
silencing effect when evaluating a novel siRNA delivery strategy. For this reason, CHAPTER 6 deals with
the kinetics of the RNAi effect obtained following transfection with cationic nanogels of varying
crosslink density.
Finally, in CHAPTER 7, a second approach for the preparation of dextran nanogels is demonstrated.
Here, we apply lipid vesicles as nanoreactors for the selective crosslinking of functionalized dextran
in their aqueous interior. Thus, nanogels are obtained that are surrounded by a lipid shell. This
chapter contains pilot experiments investigating the use of these lipid‐enveloped nanogels (LENs) as
carriers for intracellular siRNA delivery and discusses potential advantages this hybrid nanogel
delivery system may offer.
REFERENCE LIST
1. Fire A., Xu S., Montgomery M.K., Kostas S.A. et al., Potent and specific genetic interference by double‐stranded RNA in Caenorhabditis elegans, Nature 1998, 391: 806‐811.
2. Elbashir S.M., Harborth J., Lendeckel W., Yalcin A. et al., Duplexes of 21‐nucleotide RNAs mediate RNA interference in cultured mammalian cells, Nature 2001, 411: 494‐498.
3. de Fougerolles A.R., Delivery vehicles for small interfering RNA in vivo, Hum. Gene Ther. 2008, 19: 125‐132. 4. Elbashir S.M., Lendeckel W., and Tuschl T., RNA interference is mediated by 21‐ and 22‐nucleotide RNAs,
Genes Dev. 2001, 15: 188‐200. 5. Whitehead K.A., Langer R., and Anderson D.G., Knocking down barriers: advances in siRNA delivery, Nat.
Rev. Drug Discov. 2009, 8: 129‐138. 6. Gao K. and Huang L., Nonviral Methods for siRNA Delivery, Mol. Pharm. 2009, 6: 651‐658. 7. Wichterle O. and Lim D., Hydrophilic gels for biological use, Nature 1960, 185: 117‐118. 8. Gupta P., Vermani K., and Garg S., Hydrogels: from controlled release to pH‐responsive drug delivery, Drug
Discovery Today 2002, 7: 569‐579. 9. Peppas N.A., Bures P., Leobandung W., and Ichikawa H., Hydrogels in pharmaceutical formulations,
European Journal of Pharmaceutics and Biopharmaceutics 2000, 50: 27‐46. 10. Peppas N.A., Hilt J.Z., Khademhosseini A., and Langer R., Hydrogels in biology and medicine: From
molecular principles to bionanotechnology, Advanced Materials 2006, 18: 1345‐1360.
19

general introduction & thesis outline
20

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
CHAPTER 1 | Maintaining the silence: reflections on long‐
term RNAi
Parts of this chapter are published:
Koen Raemdonck1, Roosmarijn E. Vandenbroucke1, Niek N. Sanders1,2, Joseph Demeester1 and
Stefaan C. De Smedt1, Drug Discovery Today 2008, 13: 917‐931.
1Laboratory of General Biochemistry and Physical Pharmacy, Department of Pharmaceutics, Faculty of Pharmaceutical
Sciences, Ghent University, Harelbekestraat 72, B‐9000 Ghent, Belgium. 2Laboratory of Gene Therapy, Department of Nutrition, Genetics and Ethology, Faculty of Veterinary Medicine, Ghent
University, Heidestraat 19, B‐9820 Merelbeke, Belgium.
21

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
22
ABSTRACT
Since the demonstration of RNA interference (RNAi) in mammalian cells, considerable research and
financial effort has gone towards implementing RNAi as a viable therapeutic platform. RNAi is
without doubt the most promising strategy for the treatment of human genetic disorders. As many
of the targets proposed for RNAi therapy require chronic treatment, researchers agree that the
emphasis now must be placed on the safe and long‐term application of RNAi drugs to finally reap the
benefits.

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
1 OVERVIEW CHAPTER CONTENTS
Chapter 1 provides:
• a brief introduction to the post‐transcriptional gene silencing mechanism termed RNA
interference (RNAi)
• a summary of the most important parameters that govern the silencing efficiency and
duration of the RNAi effect
• a discussion of the predominant barriers in the context of siRNA delivery
• an introduction to RNAi mediated transcriptional gene silencing, mathematical modelling in
RNAi therapy and combinatorial RNAi
Key terms:
siRNA – miRNA – shRNA – mathematical modelling – transcriptional gene silencing – coRNAi – siRNA
delivery – chemical modification
INTRODUCTION
RNA interference (RNAi) represents a powerful and versatile gene silencing process in which double‐
stranded RNA (dsRNA) triggers the sequence‐specific cleavage of mRNA transcripts. An explosion of
research that followed its discovery in Caenorhabditis elegans in 1998,[1] has recently resulted in both
RNAi based protocols for analysis of gene function and therapeutic applications in humans.[2,3]
Following pioneering research in plants [4,5] and nematodes,[1,6] RNAi was demonstrated in
mammalian cells in 2001 by Tuschl and colleagues, who were the first to apply small interfering RNAs
(siRNAs) to guide the sequence‐specific suppression of gene expression.[7,8]
23

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
In theory every gene is amenable to RNAi based silencing, and therefore these key papers have led to
a surge of excitement amongst researchers in the medical field. For about a decade now, the quest
for efficient RNAi therapeutics to treat a wide variety of pathologies has been ongoing and has
already made remarkable progress, but we must emphasize that further improvements are still
needed to pave the way for RNAi to develop into a viable therapeutic approach. Obviously, the
translation of RNAi into a broadly applicable therapeutic platform needs appropriate pharmaceutical
considerations at different levels in clinical drug development like e.g. siRNA design [9,10] and siRNA
formulation.[11] In straightforward terms it can be stated that the most efficient delivery of the most
potent siRNA is expected to result in maximal suppression of a target gene.
To extrapolate RNAi from bench to bedside, it is imperative to consider not only the gene knockdown
intensity, but also the dynamics of gene silencing.[12] On one hand, precise control over the duration
of RNAi gene knockdown can provide new information on complex cellular pathways implying
interactions between multiple genes, without having to rely on more elaborate transgene
technologies. On the other hand, from a therapeutic point of view, it may be relevant to silence a
pathogenic gene for a longer period of time, to improve the clinical outcome. Prolongation of the
gene silencing effect of a specific siRNA formulation may be beneficial for chronic patient adherence
if the administration frequency in the dosing schedule could be significantly scaled down.
Although many in vivo studies have already highlighted the enormous therapeutic potential of RNAi
therapeutic strategies,[13] recent studies have shown unintended off target effects (OTEs), activation
of immune and inflammatory pathways and perturbation of endogenous cellular pathways by small
dsRNA drugs. Especially when placing the emphasis on long‐term treatment of chronic diseases,
toxicity issues become increasingly important and should be dealt with accordingly. However, at
present, few data are available on the long‐term toxicity of RNAi triggers but appropriate attention
should be paid to these issues in the context of prolonging RNAi silencing.
In this chapter we will primarily focus on important remarks and strategies in the light of prolonged
RNAi gene silencing for therapeutic purposes. In the different sections we attempt to describe
several topics from the available literature on RNAi that are of particular relevance to the central
theme of this thesis.
INTRINSIC RNAi GENE SILENCING EFFICIENCY AND LONGEVITY
Since the discovery of RNAi, researchers have revealed the basic steps of the RNAi pathway, but
many critical aspects remain to be unveiled. The RNAi effector molecules, termed small interfering
24

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
RNAs (siRNAs), are RNA duplexes ~21‐23 nucleotides (nt) in length consisting of a duplex stem ~19 nt
and both strands bearing a 5’‐phosphate and 2 nt overhangs at the 3’‐ends (Figure 1.1). These small
RNA fragments are defined as the products of longer dsRNA processing by an RNase III type enzyme,
called Dicer. In primitive organisms, long dsRNA can effectively trigger RNAi. However, in higher
organisms, these long dsRNAs (> 30 nt) also trigger innate immune responses resulting in severe
toxicity. Therefore, by preference synthetic small siRNAs are usually applied to experimentally
activate RNAi.
Once introduced in the cell’s cytoplasm, the heterodimer complex Dicer/TRBP processes dsRNAs into
siRNAs that are subsequently incorporated into a protein complex termed RISC (RNA induced
silencing complex). RISC also contains the Argonaute 2 (Ago‐2) protein.[14] Only one strand of the RNA
duplex is retained inside the RISC, which is the antisense strand or ‘guide’ strand. The sense strand or
‘passenger’ strand is nicked by Ago‐2, unwound and discarded.[15] Subsequently, the activated RISC
(RISC*) uses the guide strand to bind to the complementary region on the target mRNA. Ago‐2
contains an RNase H like domain that is responsible for directed transcript cleavage (also called
‘slicing’) in the RNA:RNA duplex region opposite the phosphate linkage between bases 10 and 11
with respect to the 5’ end of the guide strand.[16] The cleavage fragments are then further processed
by cellular RNases.
Our current knowledge of the RNAi pathway is sufficient to recognize the intrinsic gene silencing
potential of RNAi. The fact that siRNAs employ endogenous cellular machinery is imperative in this
regard. SiRNA activated RISC (RISC*) is defined as a multiple‐turnover enzyme that follows Michaelis‐
Menten kinetics. Once the mRNA target is cleaved, RISC* is recycled in the RNAi pathway to identify
and destroy other mRNAs. This implies that a single enzyme complex can bind and cleave multiple
mRNA transcripts.[17] Moreover, RISC can provide additional intracellular stability to the siRNA guide
strand and protect it against an armada of single‐strand specific RNases. The entrapment of siRNA
into RISC and its catalytic action are key parameters explaining the inherent gene silencing efficiency.
On the other hand, when siRNAs are not incorporated into RISC, their double‐stranded nature makes
them more resistant to intracellular degradation. In contrast to single‐stranded RNA, duplex siRNA
has the structural advantage of being better protected against ubiquitous single‐strand specific
nucleases. This feature makes duplex siRNA significantly more stable intracellularly, which can
already be observed on a short time scale.[18] This inherent stability also has to be taken into account
especially when comparing the gene silencing efficiency and duration of siRNAs and unmodified
antisense oligonucleotides, the latter being rapidly degraded in the cell cytoplasm.[19]
25

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
FIGURE 1.1 | Main cellular mechanism of RNA interference (RNAi), mediated by small interfering RNAs (siRNAs). The natural RNAi pathway is initiated by the cleavage of long double‐stranded RNA (dsRNA) by the Dicer/TRBP heterodimer complex into siRNAs. These siRNAs are subsequently incorporated into RISC (RNA Induced Silencing Complex). RISC only maintains one strand (antisense strand or guide strand) while Ago‐2, the catalytic unit of RISC, cleaves the passenger strand (sense strand). The siRNA guide strand in active RISC* then recognizes complementary target sites to direct mRNA cleavage, which is again catalyzed by Ago‐2. Further degradation of the unprotected mRNA fragments is carried out by intracellular ribonucleases. TRBP = TAR RNA binding protein, Ago‐2 = Argonaute 2.
Efforts with the aim of improving siRNA potency led e.g. to the development of longer siRNAs (25‐
27mers) that show superior silencing efficiency at certain target sites when compared to the
conventional 21 mers directed against the same region in the mRNA transcript.[10,20,21] With these
types of duplexes, maximal inhibition and longer gene silencing persistence was observed at
concentrations lower than those required for conventional 21mer siRNAs.[10,20‐22] The improved RNAi
silencing is thought to originate from their recognition and cleavage by Dicer (hence the annotation
Dicer substrate RNAs or DsiRNAs), which could facilitate their incorporation into RISC (Figure 1.1) as
Dicer is believed to participate in the early steps of RISC assembly.[23]
Nature holds several examples of convenient RNAi amplification mechanisms, e.g. in nematodes and
plants. One of the known amplification mechanisms is ‘systemic spreading’ of the RNAi silencing
effect. This effect is attributed to a dsRNA‐receptor, denoted as SID‐1 in C. elegans, which is
26

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
responsible for passive dsRNA translocation over the cell membrane and intercellular dsRNA
transport between neighbouring cells.[24,25] The lack of gene silencing when incubating mammalian
cells with ‘naked’ (i.e. unformulated) siRNA makes it likely that in most mammalian cell types normal
levels of SID‐1 homologue expression can be neglected.[26,27] However, a recent study conducted by
Wolfrum et al. revealed that SID‐1 is at least partially responsible for the in vitro hepatocellular
uptake of lipophilic siRNAs when they are incorporated in lipoprotein complexes such as HDL and LDL
(high and low density lipoprotein respectively) .[28] Specific RNAi silencing of SID‐1 expression in
HepG2 cells and blocking of extracellular SID‐1 epitopes with SID‐1 antibodies decreased the cellular
internalization and subsequent silencing effect of lipophilic siRNAs targeted against apolipoprotein B
(apoB) .[28]
Plants, fungi and worms contain an endogenous RNA dependent RNA polymerase (RdRP) that
produces secondary dsRNAs from an mRNA transcript targeted by a primary siRNA. The resulting
dsRNA can again be recognized and cleaved by Dicer, increasing the intracellular siRNA
concentration. Through this amplification process a small amount of ‘initiator’ dsRNA can lead to
persistent gene silencing.[29‐31] As mammalian cells most likely lack RNA dependent polymerase
activity, one must rely on alternative strategies to achieve prolonged gene silencing.[29,32,33]
STABLE RNAi GENE SILENCING
Twenty‐one mer siRNAs, mimicking Dicer cleavage products, can be chemically synthesized and
introduced into the target cell (Figure 1.1) but they can also be produced intracellularly from short
hairpin RNA (shRNA) precursors that can be continuously expressed from RNA polymerase driven
expression cassettes (Figure 1.2).[34,35] ShRNAs are structurally and functionally related to pre‐
microRNAs, intermediates in the biogenesis of endogenously encoded microRNAs (miRNAs). MiRNAs
constitute a highly conserved class of small RNAs that mediate RNAi mainly through translational
inhibition.[36,37] As Figure 1.2 illustrates, the first step in the production of miRNA occurs in the cell
nucleus, where long primary transcripts (called pri‐miRNA) are expressed from endogenous genetic
regions and processed by Drosha (a RNase III enzyme), with the aid of its cofactor DGCR8, into ~60‐
70 nt hairpins with imperfect complementarities in their stems.[38‐40] These precursor miRNAs (pre‐
miRNAs) are shuttled from the nucleus into the cytoplasm through the Exportin‐5/Ran‐GTP
heterodimer complex.[41] In the cytosol, Dicer processes pre‐miRNA into mature ~22 nt miRNA
duplexes that interact with RISC to modulate transcriptome expression.[42]
27
CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
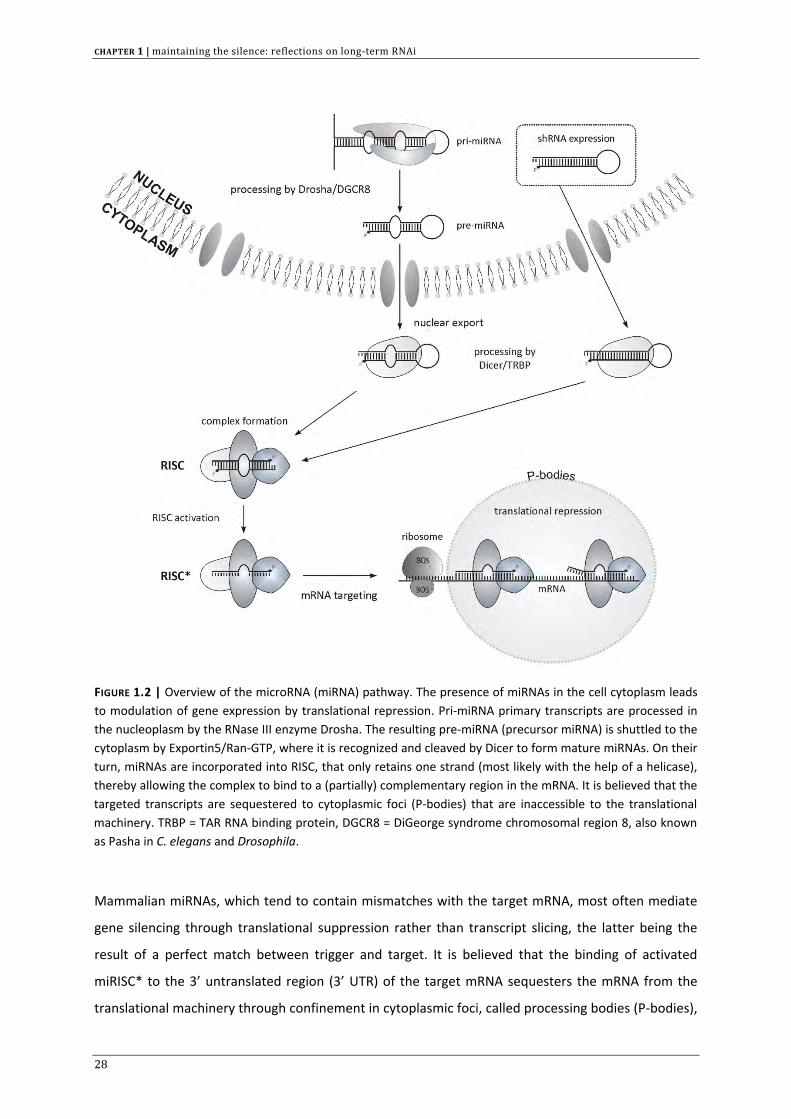
FIGURE 1.2 | Overview of the microRNA (miRNA) pathway. The presence of miRNAs in the cell cytoplasm leads to modulation of gene expression by translational repression. Pri‐miRNA primary transcripts are processed in the nucleoplasm by the RNase III enzyme Drosha. The resulting pre‐miRNA (precursor miRNA) is shuttled to the cytoplasm by Exportin5/Ran‐GTP, where it is recognized and cleaved by Dicer to form mature miRNAs. On their turn, miRNAs are incorporated into RISC, that only retains one strand (most likely with the help of a helicase), thereby allowing the complex to bind to a (partially) complementary region in the mRNA. It is believed that the targeted transcripts are sequestered to cytoplasmic foci (P‐bodies) that are inaccessible to the translational machinery. TRBP = TAR RNA binding protein, DGCR8 = DiGeorge syndrome chromosomal region 8, also known as Pasha in C. elegans and Drosophila.
Mammalian miRNAs, which tend to contain mismatches with the target mRNA, most often mediate
gene silencing through translational suppression rather than transcript slicing, the latter being the
result of a perfect match between trigger and target. It is believed that the binding of activated
miRISC* to the 3’ untranslated region (3’ UTR) of the target mRNA sequesters the mRNA from the
translational machinery through confinement in cytoplasmic foci, called processing bodies (P‐bodies),
28

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
thereby preventing protein synthesis.[43] To date, the exact outcome of imperfect complementarity
with target mRNA is still subject to debate as multiple models have already been proposed.[44]
In analogy with miRNA biogenesis, the intracellular processing of shRNAs originates in the cell
nucleus, where Exportin‐5 is responsible for its nuclear export.[45] In the cytoplasm, these shRNAs are
again cleaved by Dicer to yield the active 21mer siRNAs.[34] In correspondence with Dicer substrate
RNAs (DsiRNAs), plasmid vectors can be designed to produce shRNAs (29 nt stem, 4 nt loop) with
higher RNAi potency over smaller hairpin RNAs (19 nt stem, 4 nt loop) due to improved Dicer
recognition and RISC incorporation.[46] However, in a recent report by Li et al., it was shown that in
the context of a 9 nt loop, the shRNA with a 19 nt stem outperformed the longer 29 nt shRNA.[47] The
explanation for this discrepancy again lies in the ability of the shRNA to be recognized by Dicer.
Indeed, whereas shRNAs with a 19 nt stem and a 4 nt loop bypass Dicer cleavage, increasing the loop
length to 9 nt also turns shRNAs with a 19 nt stem into Dicer substrates.[47,48] Building on our
increased understanding of miRNA processing, second‐generation shRNAs were developed (called
shRNAmir).[49] In contrast to first‐generation shRNA that elicit structural analogy with pre‐miRNA,
shRNAmir are transcribed as pri‐miRNA. Their design is based on the human miRNA, miR‐30, of which
the stem is replaced with a RNA sequence of interest. The advantage of shRNAmir over shRNA lies in
their recognition and processing by both Drosha (nucleoplasm) and Dicer (cytoplasm), leading to a
more efficient intracellular production of mature siRNAs and increased knockdown efficiency.[50]
Important in the context of this chapter is to note that, compared with synthetic siRNAs that induce a
transient knockdown, plasmid vector based interference shows better potential for long‐term gene
silencing.[51] To establish stable RNAi gene silencing in cultured cells, researchers mostly appeal to
viral expression vectors.[34] Lentiviral vectors allow genomic insertion of the shRNA expressing
transgene, while adenoviral and adeno‐associated viral vectors (AAVs) show less successful
integration and viral DNA remains largely episomal.[52] Genomic integration of shRNA expression
cassettes per definition results in stable RNAi mediated gene silencing since the target gene remains
silenced as long as transcription of siRNA precursors proceeds. This RNAi gene therapy approach can
be of interest to study loss‐of‐function phenotypes following prolonged knockdown of a target gene
and for long‐term treatment of chronic diseases (e.g. viral infections such as hepatitis B [53] and
hepatitis C), thereby easing the treatment schedule and improving patient comfort. However, in a
clinical setting, the use of synthetic siRNAs for transient RNAi gene silencing may be advantageous
over continuous shRNA/siRNA production as the dosing schedule (and consequently also the
resulting intracellular siRNA concentrations) in a therapeutic regimen can be more easily adapted
related to therapeutic needs.[54] Some applications will call for a fully‐reversible but long‐duration
gene silencing, without loss of activity following repeated administration. Additionally, transgenes
29

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
that are stably integrated in the host genome can be silenced rapidly by histone modifications and
hypermethylation of CpG islands in the promoter region. Instead of achieving long‐term transgene
expression, this chromatin silencing will result in a gradual extinction of transgene activity.[55]
Recently, Mark Kay and colleagues disclosed that continuous expression of high intracellular levels of
shRNA could result in long‐term toxicity in mice.[56] The observed fatal side effects were primarily
ascribed to saturation of Exportin‐5 leading to interference with nuclear export of miRNA precursors
and miRNA function.[45] Possible solutions to overcome these toxicity issues are (a) lowering the
initial viral load and (b) implementing vector development with inducible or tissue‐specific promoters
that allow more feasible control over intracellular shRNA concentration which may improve the
activity/toxicity ratio.[56‐58] In a mouse model of hyperbilirubinemia, it was shown that the adenoviral
production of shRNAs could silence Abcc2 function (ATP‐binding cassette multidrug resistance
protein 2), involved in liver bilirubin transport, for up to 3 weeks. This effect did not seem to
correlate with changes in the level of endogenous (precursors of) miRNAs.[59]
Nonetheless, it is striking to conclude that the improved RNAi gene silencing of dsRNAs by taking
advantage of the natural miRNA pathway in an earlier stage can have a toxic flip side. In this regard,
synthetic 21mer siRNAs that function further downstream in the RNAi pathway are the better and
safer choice because they bypass Dicer cleavage and do not require nuclear export for their
activity.[45,60] However, when synthetic siRNAs are employed, vigilance is still required, as an
intracellular excess of siRNA may possibly interfere with RISC availability which can also induce
competition with cellular miRNAs.[60‐63] A recent report describing potent and specific knockdown of
hepatocyte‐specific genes in mice and hamsters after systemic delivery of lipid‐formulated siRNAs,
also demonstrated that the treatment schedule did not influence either miRNA biogenesis or miRNA
function.[64] As our knowledge on miRNAs expands, we will be able to better assess any possible
cellular disturbances as a result of siRNA/shRNA treatment. Quantitative data on both the total
number of cellular protein molecules involved in the RNAi pathway and the number of siRNA
molecules that is needed inside a cell for a sufficient silencing effect, could be very helpful towards
optimization of siRNA therapy without the aforementioned toxicity issues.[62]
Another concern that has been raised against the use of (non‐integrating) viral vectors is their
immunogenicity, especially when long‐term use is necessary (e.g. adenoviruses, AAVs) .[3,51]
Furthermore, random genomic insertion of a transgene (e.g. with integrating lentiviral vectors)
coincides with the risk of hazardous in vivo viral recombination and insertional mutagenesis.[65,66]
Although at present viruses are still the most efficient gene delivery vectors, the combination of
these adverse effects could eventually favour a non‐viral approach. Meanwhile, non‐viral strategies
have also been explored for intracellular shRNA production.[51] SiRNA expression plasmids can be
30
maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
introduced in the target cell by complexation with cationic lipids and polymers or by physical
methods like electroporation and microinjection. Unfortunately, the nuclear import of the plasmid
DNA (pDNA) vector remains the predominant bottleneck in the gene delivery process,[67] thereby
giving preference to synthetic siRNAs which function mainly in the cell cytoplasm. Especially non‐
dividing cells are difficult to transfect with non‐viral vectors, since insufficient amounts of the pDNA
can reach the nucleus in the absence of cell division during which the nuclear barrier is temporarily
disassembled.[68]
PERSPECTIVES ON THE DELIVERY OF siRNA: BIOLOGICAL BARRIERS
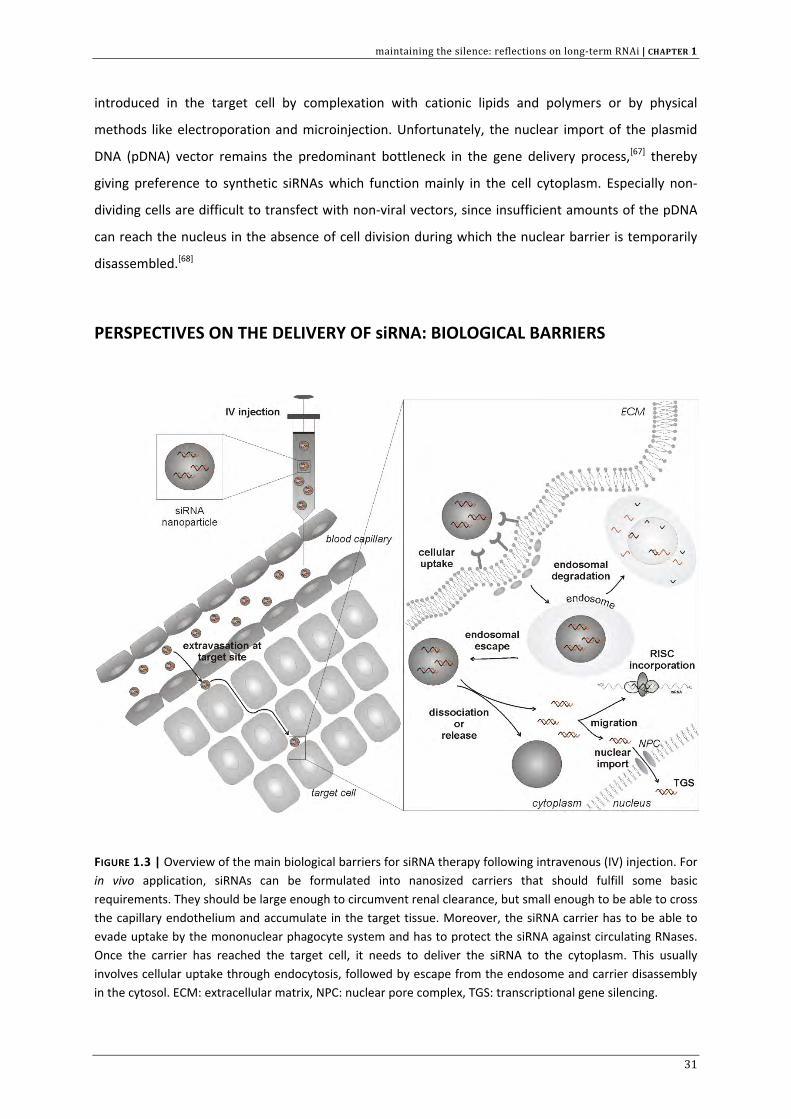
FIGURE 1.3 | Overview of the main biological barriers for siRNA therapy following intravenous (IV) injection. For in vivo application, siRNAs can be formulated into nanosized carriers that should fulfill some basic requirements. They should be large enough to circumvent renal clearance, but small enough to be able to cross the capillary endothelium and accumulate in the target tissue. Moreover, the siRNA carrier has to be able to evade uptake by the mononuclear phagocyte system and has to protect the siRNA against circulating RNases. Once the carrier has reached the target cell, it needs to deliver the siRNA to the cytoplasm. This usually involves cellular uptake through endocytosis, followed by escape from the endosome and carrier disassembly in the cytosol. ECM: extracellular matrix, NPC: nuclear pore complex, TGS: transcriptional gene silencing.
31

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
Although it is generally said that chemically synthesized siRNAs are promising therapeutic candidates
for the treatment of various genetic pathologies, their in vivo drug‐like properties are regarded as
very unfavourable. Before siRNAs can reach the desired intracellular location, many extra‐ and
intracellular barriers have to be overcome (Figure 1.3).[26] Obviously, the efficiency with which siRNAs
can by‐pass these obstacles will have major implications on the extent and duration of gene silencing
and, subsequently, on the determination of a clinically relevant dosing interval. Several concepts and
strategies have been proposed to improve the siRNA pharmacokinetics and eventually their
therapeutic performance.
The extracellular compartment: focus on chemical modification and siRNA formulation
It is well known that siRNAs are rapidly degraded in the extracellular environment. When naked (i.e.
uncomplexed, formulated in saline) siRNA is injected intravenously, it is recognized and cleaved by
RNase A type endonucleases [69‐71] or 3’ exonucleases [72‐74] limiting the serum half‐life to < 30 min.
Therefore, much effort has been undertaken to chemically modify siRNA therapeutics, in order to
reduce their susceptibility to serum RNases.[10,75,76] Chemical modifications include RNA backbone
modifications (e.g. phosphorothioates and boranophosphates), 2’‐ribose modifications, and terminal
3’ and 5’ modifications, amongst others.[77] The most important chemical modifications introduced
into an RNA backbone to date are illustrated in Figure 1.4.[72,78] Initially some concerns were raised
with regard to the possible diminished efficacy of chemically modified siRNAs, due to interference
with RISC incorporation, activation and/or recycling. Since then, however, elaborate research in this
field has proved that chemically modified siRNA can also efficiently lower mRNA levels through a
sequence‐specific RNAi dependent pathway and some modifications even show enhanced gene
silencing compared to the unmodified counterpart.[79,80] An important consideration to make is
whether or not chemical modifications, with the aim of improving the extracellular siRNA stability,
are promising in light of prolonging the knockdown of a target gene. One could expect chemically
modified siRNAs to have a higher bioavailability as they stay intact for a longer period of time in the
blood circulation, thereby also improving the intensity and duration of the RNAi gene silencing effect.
Unexpectedly, Layzer and coworkers were not able to demonstrate a stronger silencing or
persistence of silencing for stabilized 2’‐fluoro pyrimidine siRNAs over the unmodified 2’‐OH siRNAs
after hydrodynamic tail vein injection.[81] Most likely, sufficiently high concentrations of both siRNAs
can reach the intracellular target site after hydrodynamic injection and exert an effect before
nuclease digestion becomes prominent.
32

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
FIGURE 1.4 | Possible chemical modifications that can be incorporated into a siRNA duplex. Modifications include alterations in the phosphodiester backbone (e.g. boranophosphate, methylphosphonate and phosphorothioate) or sugar modifications (LNA, FANA, 4’‐thio and 2’‐OH modifications). FANA = 2’‐fluoro‐β‐D‐arabinonucleic acid, MOE = 2‐methoxyethyl and LNA = locked nucleic acid. Adapted from reference [72].
In contrast to these findings, a modified siRNA duplex, targeted against hepatitis B viral (HBV) RNA
and where all ribose 2’‐OHs were substituted, was shown to be more efficient in decreasing HBV
DNA and HBV surface antigen serum levels when compared to non‐modified siRNA.[76] In the same
report, hydrodynamic injection was used to co‐deliver the siRNA and a replication competent HBV
vector. Surprisingly, the difference in activity between modified and unmodified siRNA was only
significant at the high‐dose level and almost absent at lower doses.[76] When in vivo siRNA
degradation by nucleases is the main limiting factor, one would expect to observe the opposite
trend. This also indicates that factors other than increased nuclease resistance have to be taken into
account when working with chemically modified siRNAs.
Besides degradation by RNases, the short in vivo half life of siRNAs is also influenced by their rapid
renal clearance. Since siRNAs are highly hydrophilic (~40 negatively charged phosphate groups per
siRNA molecule) and have a molecular weight (~14 kDa) far below the cut‐off for glomerular filtration
(~60 kDa), one can expect renal clearance to be prominent. Although RNase digestion can be slowed
33

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
down by chemically modifying the nucleic acid backbone, renal clearance seems to be the rate
limiting factor governing the in vivo half life of siRNAs.[62,82]
Some chemical modifications (e.g. phosphorothioate internucleotide linkages or 4’‐thio
ribonucleotides) can increase the circulatory half‐life of siRNA by promoting interaction with serum
proteins.[77] A more convenient strategy to modulate and improve the siRNA biodistribution and/or
increase cellular uptake is the more dramatic alteration of siRNA structure by conjugation to small
molecular weight moieties.[77,83‐85] Interesting examples are the coupling of siRNA to heavy‐chain
antibody Fab fragments [86] or lipidic moieties like cholesterol, bile acids and long‐chain fatty
acids.[28,87,88] Lipophilic siRNAs seem to incorporate selectively in lipoprotein particles which are rich
in phospholipids and cholesterol (mainly HDL and LDL). These lipophilic siRNA‐lipoprotein complexes
are able to improve siRNA biodistribution by evading renal clearance and promoting cellular uptake
through HDL and LDL lipoprotein receptors.[28] Chimeric peptide‐siRNA complexes, as an alternative
delivery strategy, have recently been shown to protect mice against infection with Japanese
encephalitis virus by delivering their siRNA payload into the brain after intravenous injection.[89]
Intriguingly, the majority of the siRNA treated mice survived for over 4 weeks, while all animals in the
control group died within 10 days. Rozema et al. recently introduced a novel polymer based siRNA‐
conjugate strategy (termed Dynamic Polyconjugates) for in vivo hepatocytic targeting. These
conjugates (~10 nm in size) consist of a reversibly shielded membrane‐destabilizing polycation,
containing a cleavable targeting ligand, to which siRNA is linked through an intracellularly reducible
disulfide bond. Effective knockdown of two endogenous hepatic genes apoB and peroxisome
proliferators‐activated receptor alpha (ppara) was demonstrated in wild‐type mice following low
pressure i.v. injection.[90] In addition, aptamer‐siRNA conjugates seem promising for cell‐type specific
delivery of siRNA, as demonstrated by two groups using a prostate‐specific membrane antigen
(PSMA) targeted aptamer, either directly conjugated to siRNA or linked through streptavidin.[91,92]
The use of cell penetrating peptides (CPPs) for siRNA delivery to date has also produced some
interesting reports, but the exploration of this area of research is only just commencing.[93,94]
Another convenient way to circumvent renal clearance is by the incorporation of siRNA into gene
silencing nanocomplexes that are large enough to evade glomerular filtration, thereby improving
siRNA pharmacokinetics and biodistribution.[85] Paying proper attention to the “packaging” of siRNAs,
can lead to constructs where the siRNA molecules are shielded from circulating RNases and
destabilizing blood components, eliminating the need for chemical modification.[95‐97] Many inventive
siRNA formulations have already shown to dramatically improve the in vivo potency of
siRNAs.[3,11,62,83,98‐100] A textbook case, underlining the importance of siRNA formulation is given by
Morrissey et al. When administering unformulated backbone stabilized anti‐HBV siRNAs through
34

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
hydrodynamic tail vein injection, an effective inhibition of HBV replication could be achieved, albeit
at a dramatically high dosing regimen of three daily doses of 30 mg kg‐1.[76] However, formulating the
therapeutic siRNAs into stable nucleic acid lipid particles (SNALPs) of ~140 nm in size, these
investigators succeeded in achieving a significant and long‐term reduction of HBV activity by three
daily injections of 3 mg kg‐1 d‐1 siRNA followed by a weekly administered maintenance dose for up to
6 weeks.[101] A more recent report, using SNALP technology to deliver anti‐ApoB siRNA in cynomolgus
monkeys, describes marked reduction in plasma ApoB protein levels for as long as 11 days after a
single IV injection (2.5 mg kg‐1).[102] Mark Davis’ group formulated siRNAs in cationic cyclodextrin
containing polycations (CDPs) which can be equipped with targeting ligands, e.g. transferrin for
delivery to tumour cells expressing the transferrin receptor. The systemic delivery of these targeted
siRNA nanoparticles, twice‐weekly for > 4 weeks resulted in long‐term inhibition of tumour cell
engraftment and tumour growth in a murine therapeutic model for metastatic Ewing’s sarcoma.[103]
Interestingly, the long‐term inhibition was only observed for the formulation where the surface was
modified with transferrin moieties and no positive outcome was demonstrated for the non‐targeted
formulation. This targeted nanoparticle delivery system also has been evaluated recently in non‐
human primates [104] and is currently in a phase I clinical trial.[105] Some of the cited delivery
approaches are schematically drawn in Figure 1.5.
The intracellular compartment: focus on chemical modification and cellular dilution
Besides improvements at the extracellular level, also the intracellular behaviour of chemically
modified siRNAs may play a pivotal role in improving the RNAi gene silencing potency and duration.
Some reports ascribe a prolonged gene silencing effect in cell cultures to the use of stabilized siRNA
duplexes that show higher resistance towards nuclease attack, suggesting that intracellular
degradation of siRNA can indeed be partially responsible for transient RNAi gene silencing.[106‐110] An
interesting recent report by Takabatake and coworkers demonstrates the dependence of RNAi gene
silencing duration on the tissue expression level of eri‐1 (enhanced RNAi‐1) nuclease.[111] Eri‐1 can be
regarded as a type of siRNAse, which has been discovered in C. elegans mutants.[73] Preliminary
experiments in EGFP transgenic rats revealed that a single administration of siEGFP into the kidney
resulted in the reduction of EGFP expression for up to two weeks, while the transfection of siEGFP
into muscle tissue silenced EGFP for up to 90 days. The authors hypothesize that the higher eri‐1
expression level in the kidney when compared to muscle could be responsible for higher intracellular
siRNA degradation and decreased RNAi longevity. They propose that blocking eri‐1 expression or
applying eri‐1‐resistant siRNAs can result in more sustained gene silencing.[111]
35
CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
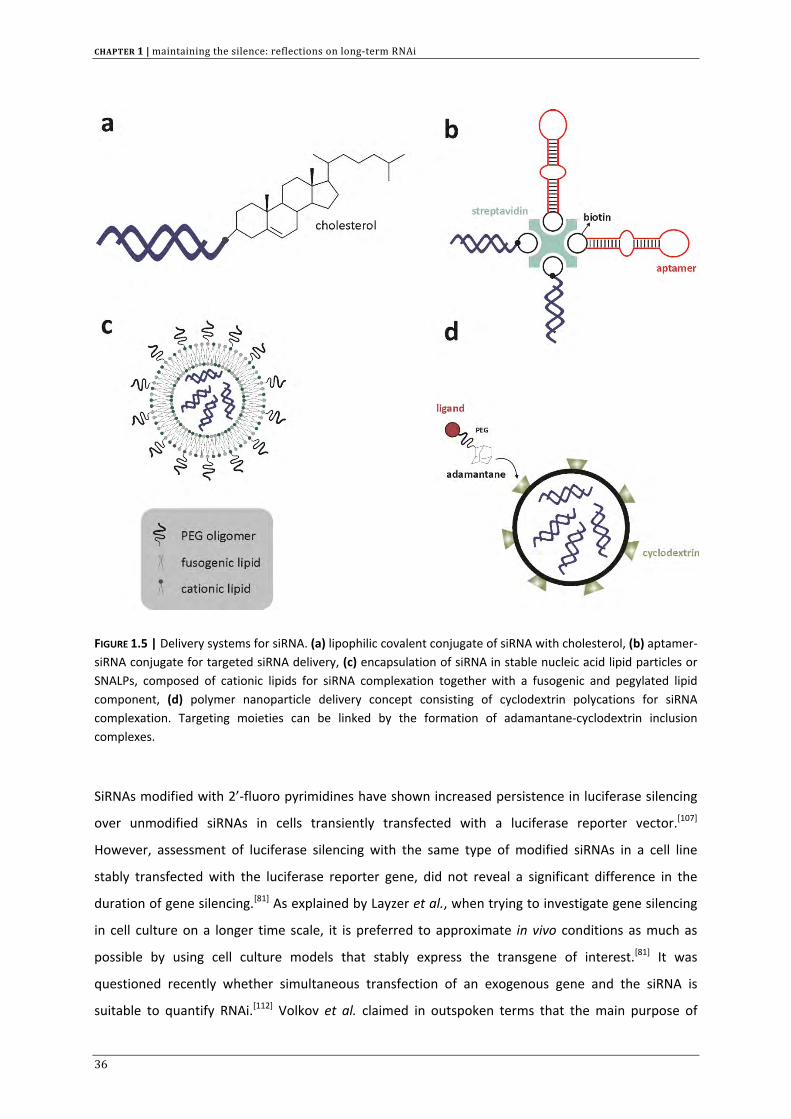
FIGURE 1.5 | Delivery systems for siRNA. (a) lipophilic covalent conjugate of siRNA with cholesterol, (b) aptamer‐siRNA conjugate for targeted siRNA delivery, (c) encapsulation of siRNA in stable nucleic acid lipid particles or SNALPs, composed of cationic lipids for siRNA complexation together with a fusogenic and pegylated lipid component, (d) polymer nanoparticle delivery concept consisting of cyclodextrin polycations for siRNA complexation. Targeting moieties can be linked by the formation of adamantane‐cyclodextrin inclusion complexes.
SiRNAs modified with 2’‐fluoro pyrimidines have shown increased persistence in luciferase silencing
over unmodified siRNAs in cells transiently transfected with a luciferase reporter vector.[107]
However, assessment of luciferase silencing with the same type of modified siRNAs in a cell line
stably transfected with the luciferase reporter gene, did not reveal a significant difference in the
duration of gene silencing.[81] As explained by Layzer et al., when trying to investigate gene silencing
in cell culture on a longer time scale, it is preferred to approximate in vivo conditions as much as
possible by using cell culture models that stably express the transgene of interest.[81] It was
questioned recently whether simultaneous transfection of an exogenous gene and the siRNA is
suitable to quantify RNAi.[112] Volkov et al. claimed in outspoken terms that the main purpose of
36

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
siRNA protection against nuclease attack is the extension of the duration of the siRNA effect.
Interestingly, these authors concluded that only protection of all the nuclease‐sensitive sites in an
siRNA duplex could result in RNAi prolongation, and that partial modification does not induce a more
durable gene silencing when compared to the unmodified duplex.[109]
Fisher et al. investigated the influence of modest altritol (6‐carbon sugar) modifications on siRNA
stability and gene silencing persistence.[113] Although partially modified siRNAs did not show
increased nuclease resistance compared to wild‐type siRNA, the authors did observe enhanced
duration in MDR‐1 gene silencing resulting in more durable suppression of p‐glycoprotein expression.
These data underscore the fact that, at the cellular level, other factors than mere siRNA stability, like
cellular uptake and/or recognition by RNAi machinery, have to be taken into account. Liao and Wang
reported on 2’‐O‐(2,4‐dinitrophenyl) modifications that serve to enhance siRNA lipophilicity.[80] These
chemical adjustments facilitate the intracellular siRNA penetration and improve the siRNA stability
which resulted in a stronger inhibition of insulin‐like growth factor receptor expression, relative to
unmodified siRNA directed against the same target sequence.
Interestingly, it seems that in fast dividing cell lines gene silencing is limited by intracellular dilution
of siRNA (and activated RISC) due to cell division. As a result, in vitro gene silencing usually lasts for
only ~4‐7 days.[54] In slowly dividing and non‐dividing cells the siRNA stability is supposed to be the
main limiting factor for prolonged gene silencing as the intracellular siRNA half‐life is expected to be
shorter than the cell division time.[54] Intracellular persistence of active siRNA has already been
shown to maintain target gene inhibition for several weeks in different types of (non‐dividing)
primary cells.[54,114‐117] In these papers it was assumed that the stability of the RNAi effect is
representative of the physical stability of the intracellular siRNA trigger. Unfortunately, in most
reports this assumption was not experimentally evaluated. Song et al., who did perform a northern
blot analysis for siRNA integrity, suggested that intracellular siRNA survival requires the active
production of the target mRNA transcript.[114] Mantei et al. identified intracellular siRNA stability as
the critical parameter influencing RNAi effect duration in primary T lymphocytes. Whereas target
gene expression was rapidly restored to base levels for unmodified siRNAs upon T cell activation, the
gene inhibition was dramatically lengthened with chemically modified siRNAs. The authors ascribe
this effect to the increased intracellular half‐life of the modified siRNA, rather than dilution resulting
from T cell proliferation.[110]
Also Maliyekkel et al. reported on more stable gene silencing following transient shRNA induction in
growth arrested non‐cycling cells.[115] However, in contrast with other reports on long‐lasting RNAi in
non‐dividing cells,[114] the phenotypic RNAi activity did not coincide with the intracellular siRNA decay
as analyzed by a RNase protection assay. Two possible explanations for this discrepancy were
37

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
suggested. Firstly, it is conceivable that the RNAi phenotype is mainly governed by the fraction of
siRNA associated with RISC*. As already mentioned previously, it can be expected that incorporation
of the guide strand in the RISC* complex provides extra protection against degradation. In this case
quantifying total intracellular siRNA does not represent the effective intracellular RNAi potential.
Secondly, it was proposed that transcriptional silencing of the target gene could be held responsible
for the increased silencing duration ( more information on transcriptional silencing can be found in in
the following section). Recent in vitro and in vivo data on non‐modified and nuclease stabilized siRNA
duplexes have shown that evading nuclease attack by chemically modifying siRNAs does not
significantly prolong the duration of gene silencing once the siRNA has reached the cytosol of the
target cells.[118]
The contradictory findings on the (supposed) advantages of nuclease stabilized siRNAs described in
this section unfortunately hinder drawing clear conclusions in this regard. Nonetheless, although the
benefits of nuclease resistant siRNAs inside the cell cytoplasm still remain questionable, chemical
modifications have proved to be a very useful approach to abrogate off‐target effects and unwanted
stimulation of the mammalian immune system.[119‐122] Several native siRNA sequences are known to
induce a toll‐like receptor (TLR) mediated immune response through endosomal TLR7/8
recognition.[123,124] Cytoplasmic receptors such as PKR (dsRNA‐binding protein kinase receptor), and
the RNA helicases RIG‐1 (retinoic acid‐inducible gene‐I) and MDA‐5 (melanoma differentiation‐
associated protein‐5) are considered to recognize certain structural RNA characteristics, other than
nucleotide sequence.[122] RIG‐1 for instance is activated by uncapped 5’‐triphosphate RNA and longer
blunt‐ended siRNAs.[122,125] Mitigation of immune response is possible by designing siRNAs devoid of
immunostimulatory sequence motifs and by paying proper attention to dsRNA physical structure.
The most robust approach however is the selective incorporation of modified nucleotides (e.g. 2’‐
OMe) to avoid immune receptor activation.[122,126] Kleinman and coworkers showed recently that 21‐
nt or longer siRNAs inhibited choroidal neovascularization (CNV) in mice in a sequence‐ and target
independent manner. The inhibition resulted from the cell‐surface binding of TLR3 by generic
siRNAs.[127] Although this approach could offer perspectives to harness this immune activation for
antiangiogenic effects,[127,128] excessive cytokine release can cause severe toxicity.[129] Modifying the
siRNA duplex to minimize TLR3 activation could reduce any consequential undesired effect and
enhance target specificity. Therefore, chemical modifications can certainly help to optimise siRNA
design towards minimal in vivo toxicity and maximal siRNA tolerance.
38

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
RNAi INDUCED SILENCING AT THE TRANSCRIPTIONAL LEVEL
The majority of reports in the literature dealing with mammalian RNAi gene silencing describe siRNA‐
directed cleavage and destruction of cytoplasmic mRNA transcripts, termed post‐transcriptional gene
silencing (PTGS). Although it was first believed that siRNA only functions in the cell cytoplasm,[130] a
series of recent reports suggest that RNAi may also be active in the cell nucleus. As an example, the
RNAi nuclear function was shown by siRNA‐mediated degradation of 7SK snRNA,[131] an abundant,
well‐characterized RNA that has a highly defined structure and specifically localizes in the nucleus.[132‐
134] Furthermore, it was shown that several siRNAs incorporated in functional RISC existed in the
nucleus and cleaved the target RNA with high efficiency.[131] Irrespective of the fact that siRNA
molecules are small enough to passively diffuse through the nuclear pore complex (NPC), recent data
suggest that nuclear RISC (nRISC) originates from a partial translocation of cytoplasmic RISC (cRISC)
followed by shuttling in the nucleus.[135] Although the cited reports describe RNAi action occurring in
the cell nucleus, this still encompasses a PTGS process, for which essentially the same basic rules
apply as discussed for cytosolic RNAi (see section 2 of this chapter).
To date, accumulating evidence emerges that small interfering RNAs have the ability to silence the
expression of eukaryotic genes by targeting their promoter region, a process which is denoted as
transcriptional gene silencing or TGS. This phenomenon was first observed in lower organisms and
plants, e.g. when doubly transformed tobacco plants exhibited a suppressed phenotype of a
transgene caused by a methylation process.[136] This RNA‐dependent DNA methylation (RdDM)
seemed to be induced by RNA sequences identical to genomic promoter regions, leading to TGS.[137‐
141] While the exact molecular mechanisms of RdDM are still to be fully clarified, it is likely to involve
cytosine methylation, histone modification and chromatin remodelling.
Several independent recent reports also demonstrated transcriptional gene silencing by
siRNAs/shRNAs in mammalian cells.[142‐144] SiRNA‐mediated TGS in mammalian cells appears to be the
result of the siRNA‐directed histone H3K9 and H3K27 methylation at the targeted promoter,[145]
although subsequent DNA methylation has also been observed.[144,146] However, the role of DNA
methylation in TGS remains controversial and highly debated since many reports describe that
potent silencing on the transcriptional level does not necessarily require DNA methylation. A paper
from Janowski et al. describes the inhibition of gene expression by so called antigene RNAs
(agRNAs), complementary to transcription start sites within human chromosomal DNA.[147] In
contrast to other reports, no evidence for a role of DNA methylation in promoter silencing could be
obtained, but the silencing was accompanied by dimethylation of Lys9 in histone H3 (H3K9).[148,149]
Kim et al. and Janowski et al. both investigated the involvement of Argonaute proteins in the TGS
39

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
process. While John Rossi’s group only identified the enrichment of Ago‐1 at the targeted promoter
region,[149] Janowski and co‐workers reported that the siRNA‐mediated silencing of both Ago‐1 and
Ago‐2 seemed to reverse TGS induced by agRNA, suggesting that both Argonautes play a role in the
promoter silencing.[150] Adding to the vast complexity of this field are reports describing TGS by
endogenously encoded miRNAs,[151] or even the transcriptional activation induced by small RNAs
instead of repression.[152,153] It may be clear that the exact molecular mechanisms for TGS or
transcriptional activation still remain rather obscure to date and more data are needed to further
clarify the underlying pathways.
However, evidence on TGS at present is sufficient to envision the use of small RNAs targeting
promoters for therapeutic purposes. An excellent example underlining the therapeutic potential of
TGS can be found in HIV‐1 treatment. To suppress the production of a range of viruses in vitro by
targeting structural and accessory genes, the duration of the PTGS effect is known to be rather
limited, i.e. in HIV‐1 varying from 4 to 7 days.[154] Prolongation of the HIV‐1 silencing up to 14‐25 days
was achieved using adeno‐associated or lentiviral vectors. However, the efficacy of HIV‐1 treatment
based on a PTGS approach is potentially further limited as HIV‐1 is known to adapt to environmental
pressure and rapid selection of siRNA escape mutants has been described in vitro.[155] For this reason,
Suzuki et al. made use of TGS as an alternative approach to prolong the suppressive effect of siRNA
that would be less susceptible to the adaptability of HIV‐1.[156] SiRNA, targeting HIV‐1 LTR, induced a
significant reduction in the reverse transcriptase levels, an effect that was maintained for over one
month. A prolonged effect can be expected as it has been shown that, regardless of the exact
underlying mechanisms, RdDM is long lasting and can be passed on across generations in plant
systems [157‐159] and in C. elegans in the absence of the original RNAi trigger.[160] Therefore,
transcriptional gene silencing through the use of promoter specific siRNAs could be an interesting
method of achieving a more robust gene silencing effect, as recently shown for the first time in
human cells by Kevin Morris’ group, although it has been suggested that this probably needs a
minimal sustained expression of promoter directed siRNA/shRNA in order to increase DNA
methylation events.[161]
MATHEMATICAL MODELLING TO DESCRIBE RNAi GENE SILENCING KINETICS
A broad spectrum of variable parameters can be defined that may influence the outcome of RNAi
gene silencing and the knockdown duration. These parameters are situated on three distinct levels:
(1) the siRNA delivery formulation, (2) the intracellular RNAi pathway and (3) the envisioned target.
40

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
At the level of siRNA delivery one must keep in mind that in vivo the tissue distribution and
pharmacokinetics of the siRNA formulation (e.g. siRNA containing nanoparticles) will have a major
impact on the RNAi effect in the target cells, as discussed earlier. Because the siRNA targets are
located intracellularly, not only the biodistribution in the extracellular space, but also the different
steps in intracellular trafficking have to be considered. Important processes like the cellular uptake
mechanism, the confinement of siRNA carrier to cytosolic vesicles and siRNA release from its carrier
will influence the RNAi effect (Figure 1.3). For example, several studies have shown that the ability of
siRNA or siRNA carrier complexes to escape from the endosomal compartment can be a limiting step
in their gene silencing efficiency.[162,163]
Obviously, RNAi‐specific parameters such as intracellular siRNA stability, RISC activation and siRNA‐
RISC‐mRNA complex kinetics have to be accounted for. The importance of RISC* recycling in the RNAi
pathway towards siRNA gene silencing potency and the potential influence of siRNA chemical
modification was emphasized earlier.
Last but not least, both the turnover of the intracellular mRNA transcript and target protein stability
should be carefully considered. If the protein half‐life extends over several days, even efficient
knockdown of intracellular mRNA levels after a single siRNA dose may fail in altering the cellular
phenotype owing to residual amounts of the stable protein. The rate of cell division is imperative for
the duration of gene silencing in a transient RNAi approach as it determines how transient the RNAi
effect really is. In this regard, both the targeted cell type and the envisioned therapeutic outcome are
of great importance. If a single administration of siRNA is effective in blocking tumour cell growth,
this inherently could imply a prolonged effect since the intracellular siRNA dilution due to cell
division can be disregarded.[164]
A detailed knowledge of the impact of all the parameters listed above, in relation to RNAi gene
silencing kinetics, could expedite the design of therapeutic siRNA strategies. Predictive mathematical
models, illuminating the key factors that govern the duration of gene silencing, could be helpful
instruments in the setup of siRNA treatment regimens. Several reports that describe mathematical
equations to gain more insight into the RNAi silencing pathway are available in the literature.[165,166]
Raab and Stephanopoulos studied the impact of the siRNA concentration and the time of siRNA
transfection relative to reporter plasmid co‐transfection in mouse hepatoma cells.[12] Their model
could be used to select the appropriate time of siRNA transfection as a function of gene induction.
Moment analysis was used by Takahashi and coworkers to evaluate the time‐course of endogenous
protein expression as a function of siRNA concentration.[167] Likewise, an interesting report by Arciero
et al. describes mathematical modelling as a tool to predict tumour‐immune evasion and to study the
41
CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
impact of siRNA administration directed against the immune suppressive cytokine TGF‐β on tumour
growth.[164,168]
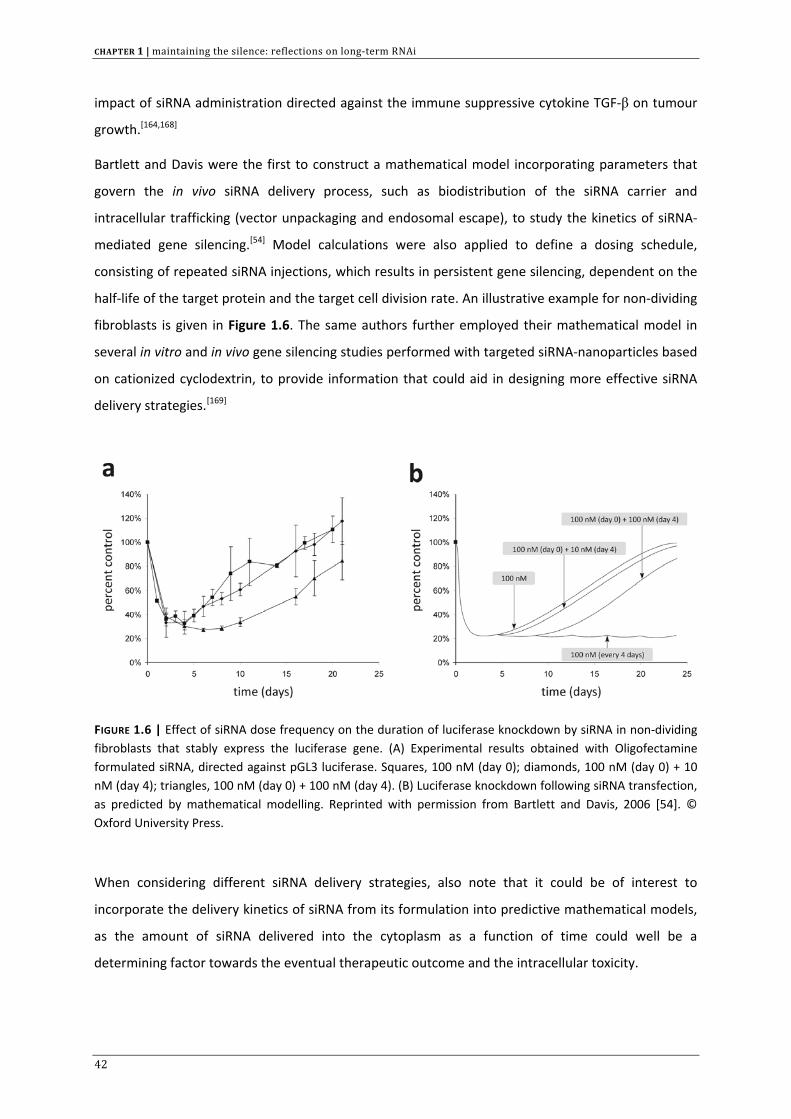
Bartlett and Davis were the first to construct a mathematical model incorporating parameters that
govern the in vivo siRNA delivery process, such as biodistribution of the siRNA carrier and
intracellular trafficking (vector unpackaging and endosomal escape), to study the kinetics of siRNA‐
mediated gene silencing.[54] Model calculations were also applied to define a dosing schedule,
consisting of repeated siRNA injections, which results in persistent gene silencing, dependent on the
half‐life of the target protein and the target cell division rate. An illustrative example for non‐dividing
fibroblasts is given in Figure 1.6. The same authors further employed their mathematical model in
several in vitro and in vivo gene silencing studies performed with targeted siRNA‐nanoparticles based
on cationized cyclodextrin, to provide information that could aid in designing more effective siRNA
delivery strategies.[169]
FIGURE 1.6 | Effect of siRNA dose frequency on the duration of luciferase knockdown by siRNA in non‐dividing fibroblasts that stably express the luciferase gene. (A) Experimental results obtained with Oligofectamine formulated siRNA, directed against pGL3 luciferase. Squares, 100 nM (day 0); diamonds, 100 nM (day 0) + 10 nM (day 4); triangles, 100 nM (day 0) + 100 nM (day 4). (B) Luciferase knockdown following siRNA transfection, as predicted by mathematical modelling. Reprinted with permission from Bartlett and Davis, 2006 [54]. © Oxford University Press.
When considering different siRNA delivery strategies, also note that it could be of interest to
incorporate the delivery kinetics of siRNA from its formulation into predictive mathematical models,
as the amount of siRNA delivered into the cytoplasm as a function of time could well be a
determining factor towards the eventual therapeutic outcome and the intracellular toxicity.
42

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
TIME‐CONTROLLED INTRACELLULAR RELEASE OF siRNA
‘More may not always be better’, it was sharply put by Marsden in a recent edition of the New
England Journal of Medicine,[170] and can be regarded as a basic toxicological paradigm. In theory
every substance is potentially harmful when exceeding a certain concentration level or exposure
time. This is also true for siRNA therapeutics, judging by the concentration dependency of adverse
effects such as off‐target silencing, induction of immune response and saturation of the endogenous
RNAi pathway.[63,122] This obviously stresses that it is imperative to work with the most potent siRNA
designs at the lowest concentration possible. Keeping this in mind, one cannot overemphasize the
need for rigorous control over the intracellular siRNA/shRNA concentrations.
To regulate the number of active siRNA molecules in the cell cytosol, one could consider the use of
conditional shRNA expression from plasmid expression cassettes delivered in the target cells by viral
or non‐viral carriers. However, the regulation of intracellular siRNA concentrations becomes more
complex in the case of (non‐viral) synthetic siRNA delivery where mostly (electrostatic) complexes
between siRNA and (cationic) polymers (i.e. polyplexes) or (cationic) lipids (i.e. lipoplexes) are applied
to the cells. With these poly‐ and lipoplexes a transient RNAi effect lasting for less than one week is
generally observed. For many of these formulations, one can expect that upon escape from the
endosome, they provide a direct release of siRNA in the cytoplasm. The released siRNA is
subsequently prone to dilution through cell division and (possibly) intracellular degradation, leading
to a transient gene silencing. In theory, time‐controlled release of siRNA in the cell cytoplasm could
be interesting to maintain intracellular siRNA concentrations for a longer period of time above the
minimal threshold required for efficient gene silencing, without flooding the cell cytoplasm with
uncomplexed (naked) siRNAs. Controlling the amount of siRNA that is released in the cytosol could
therefore result in a prolonged RNAi effect.
To achieve this goal, there is a need for delivery vehicles that exhibit tailored time‐controlled siRNA
delivery. Our group recently published on cationic biodegradable poly‐β‐amino esters (PbAEs) which
are able to form nanosized electrostatic complexes with the negatively charged siRNA (Figure 1.7).
Following endocytosis of the polyplexes, the hydrolytic degradation of the cationic polymer is
believed to increase the osmotic pressure inside the endosomal vesicles due to an accumulation of
its degradation products.[171] When exceeding a critical pressure threshold, this eventually leads to
endosomal rupture. It is hypothesized that the polymer degradation rate and the number of
polyplexes residing in a single endosome will govern the kinetics of the rise in osmotic pressure over
the endosomal membrane. Therefore, not all endosomes are expected to rupture at the same time.
43
CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
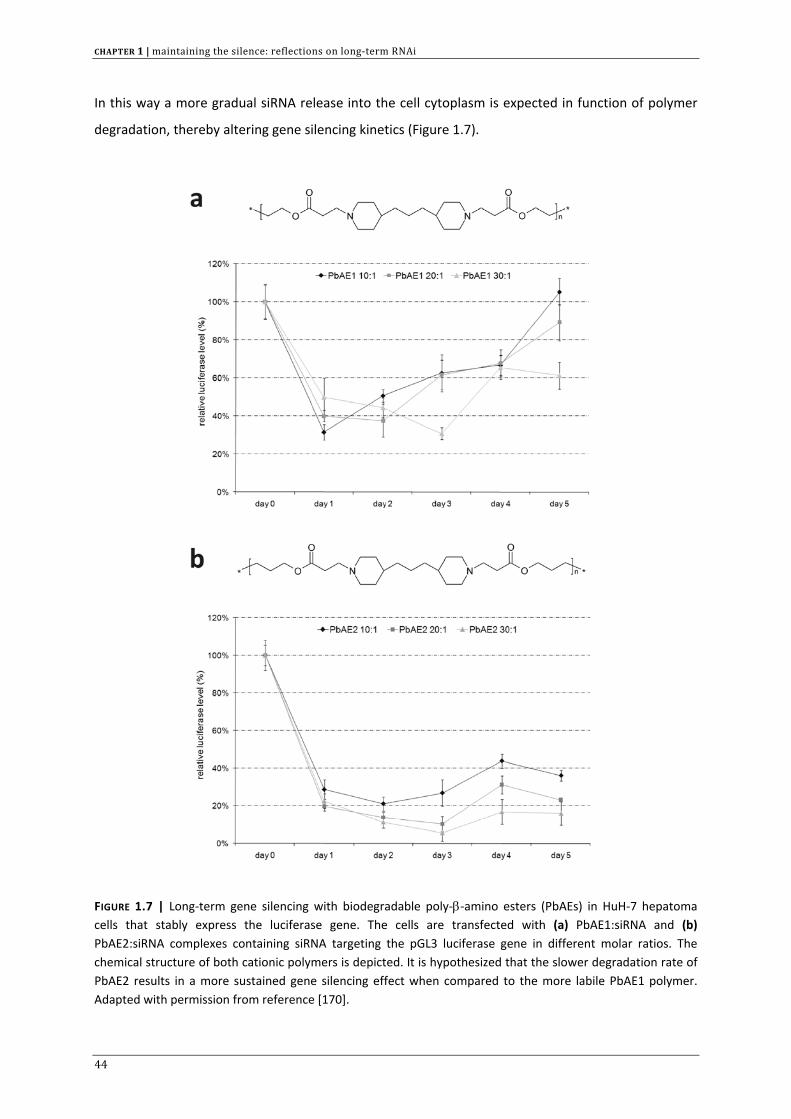
In this way a more gradual siRNA release into the cell cytoplasm is expected in function of polymer
degradation, thereby altering gene silencing kinetics (Figure 1.7).
FIGURE 1.7 | Long‐term gene silencing with biodegradable poly‐β‐amino esters (PbAEs) in HuH‐7 hepatoma cells that stably express the luciferase gene. The cells are transfected with (a) PbAE1:siRNA and (b) PbAE2:siRNA complexes containing siRNA targeting the pGL3 luciferase gene in different molar ratios. The chemical structure of both cationic polymers is depicted. It is hypothesized that the slower degradation rate of PbAE2 results in a more sustained gene silencing effect when compared to the more labile PbAE1 polymer. Adapted with permission from reference [170].
44

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
In a comparable approach, which is explained in more detail in chapter 3, our group is also involved
in the design of biodegradable siRNA impregnated gel beads (microgels) which elicit time‐controlled
release of the incorporated siRNA governed by the hydrolysis of the hydrogel crosslinks.[172]
Depending on the hydrogel characteristics, the siRNA release time can be tailored from hours over
days to several weeks. As such gels can be taken up by target cells they could serve as intracellular
siRNA depots slowly disintegrating and releasing the entrapped siRNA.
Due to the high gene silencing potential of siRNAs, only a small number of active siRNAs per cell
(~several hundred) are needed for activity. Veldhoen et al. provided a detailed quantitative analysis
of cellularly internalised siRNA through a liquid hybridization protocol.[173] Combining these data with
the observed RNAi effect revealed that for a half maximal silencing, ~104 siRNA molecules were
necessary in case of a cell penetrating peptide (CPP) assisted transfection and only ~300 in the case
of transfection with lipofectamine™2000.[173,174] This indicates that the number of siRNA molecules
that becomes available for biological activity may depend strongly on the delivery agent. Data on the
minimal number of siRNA molecules needed to trigger a sufficient RNAi effect and knowledge on the
kinetics of the RNAi effect in relation to the intracellular siRNA concentration are very important
when attempting to achieve a long‐term maximal inhibitory effect.
We emphasised that a number of crucial parameters need to be considered for an understanding of
the RNAi gene silencing kinetics. Similarly, an optimal controlled siRNA release depends on both the
cell type and target gene. For instance, it should be clear that the optimal siRNA release profile to
achieve a more sustained gene silencing will differ significantly for slowly dividing, fast dividing cells
and non‐dividing cells. On the other hand, also the expression level of the target gene will greatly
influence the amount of siRNA needed inside the cell cytoplasm for maximal gene silencing.
COMBINATORIAL APPROACH IN RNAi THERAPY
Because siRNAs/shRNAs can be developed to target viral transcripts in a sequence‐specific manner,
RNAi could prove to be the next best thing to block viral replication.[175] Unfortunately, many reports
have mentioned drug resistance through induced viral mutagenesis under pressure of RNAi mono‐
therapy.[155,175‐177] The perplexing viral genetic flexibility could also entail the emergence of
genetically encoded viral RNAi suppressors.[178] Despite the great potential of RNAi for antiviral
therapy, the issues raised above complicate the achievement of a persistent viral suppression using
RNAi mono‐therapies. Even highly active anti‐retroviral therapy (HAART), where a cocktail of three or
more antiretroviral drugs is used, cannot eradicate viral replication and only delays disease
45

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
progression. Moreover, HAART is notorious for the induction of severe toxicity, putting up an extra
barrier for efficient therapy.[179]
In correlation with HAART, a novel RNAi approach was put forward, called combinatorial RNAi or
coRNAi.[177] This multiplex approach involves the concerted action of different siRNA/shRNA
effectors, directed against multiple viral sequences. Although the majority of antiviral RNAi studies
focus on the targeting of viral transcripts, some studies have also been devoted to suppressing the
expression of cellular genes that are of key importance in the viral life cycle.
The primary goal of coRNAi is therefore to minimize the emergence of viral escape mutants in order
to maintain a long‐term antiviral effect. To give an example, it was shown for HIV‐1 that in response
to a single shRNA inhibitor, mutations inside and even outside the RNAi target sequence provided an
escape route for the virus eventually nullifying the antiviral effect.[180] A combinatorial strategy
applying multiple shRNAs expressed from a single lentiviral vector and targeting a different region in
the HIV‐1 genome, increased the viral inhibitory activity. Moreover, it seemed that a double
expression vector also provided a more durable viral inhibition. Comparable observations are
presented in the literature for Coxsackievirus B3 [181] and SARS‐associated coronavirus, where a clear
synergistic antiviral effect was obtained through combination of siRNAs targeting different functional
genes in the coronaviral genome.[182] Currently, the concept of coRNAi also includes combining RNAi
triggers with non‐RNAi based suppressors of gene expression [183] or even anti‐viral proteins.[184] For
instance, a multiplex strategy resulting in long‐term HIV‐1 inhibition in primary cells was reported
using a triple combination of an anti‐HIV shRNA, a hammerhead ribozyme targeted against the HIV
co‐receptor chemokine receptor 5 (CCR5) and a RNA decoy of HIV‐TAR.[183]
It would be beyond the scope of this review to discuss the plethora of reports available in the
literature with regard to combinatorial RNAi. Recently, an excellent review solely devoted to this
topic has been published by Mark Kay’s group.[177] We would like to refer the reader to this review
(and references herein) for a comprehensive overview of the different strategies already explored for
coRNAi therapy.
In the latter paper, the authors focus mainly on the antiviral potential of coRNAi. Nevertheless, RNAi
can be regarded as a broad therapeutic platform that can additionally be deployed in other
challenging‐to‐treat human pathologies, such as metabolic disorders and cancer, where a
combinatorial approach could be a therapeutic asset in the context of achieving long‐term silencing.
Recent reports established a synergistic effect of an RNAi trigger especially when combined with
conventional low molecular weight chemotherapeutics. Takei et al. employed an effective siRNA
against midkine (MK) together with low (and essentially non‐toxic) doses of paclitaxel in human
46

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
prostate cancer xenografts and found that paclitaxel significantly augmented the antitumour effect
of MK‐siRNA.[185] In this way, tumour growth inhibition could be maintained for several weeks
without the need to switch to higher (and most likely toxic) doses of paclitaxel. Unfortunately, the in
vivo administration of the siRNA‐atelocollagen formulation used in that report was restricted to
intratumoural injection. The same synergistic phenomenon, however, was found for an intravenously
administrated targeted nanoparticle formulation of anti‐EGFR siRNA, in combination with
intraperitoneal cisplatin, in a human lung cancer xenograft model. In contrast to mono‐therapy with
targeted nanoparticles containing anti‐EGFR siRNA, which only partially reduced tumour growth, the
combination of the targeted siRNA‐nanoparticles with cisplatin completely inhibited tumour
proliferation for ~1 week.[186] A third and earlier report describes the packaging of siRNA into neutral
liposomal vesicles for the targeting of the tyrosine kinase receptor EphA2 oncogene that is
overexpressed in ovarian cancer. When liposomal anti‐EphA2 siRNA was administered in addition to
paclitaxel, a significant reduction in tumor growth was observed when compared to paclitaxel in
combination with non‐silencing siRNA.[187] The same neutral liposomal formulation was applied to
target focal adhesion kinase (FAK) and again a synergistic effect could be obtained when combining
therapeutic siRNA with conventional chemotherapeutics such as docetaxel and cisplatin.[188]
Altogether, the studies cited in this section exemplify the tremendous potential of coRNAi in the
battle against various challenging therapeutic targets in human disease. A particular advantage when
combining different RNAi effectors (with or without alternative therapeutics) is the maximization of
the silencing effect, both acute and long‐term. Needless to say that the safety concerns which have
been raised for RNAi monotherapy also hold true for coRNAi. Multiplexing several siRNA/shRNA
triggers could even augment the risks inherently linked to RNAi, such as off‐targeting,
immunostimulation and competition with endogenous miRNAs. Following coadministration of
different types of RNAi drugs, they can even compete with each other for the (limited) amount of
cellular RNAi proteins, thereby diminishing each other’s efficacy.[60,189] On the other hand, combining
RNAi effectors with other non‐RNAi based therapeutics may allow lowering the dosing of both drugs.
In this way, coRNAi can still profit from the synergistic effect along with the benefit of reduced
toxicity.
To date, the concept of coRNAi even has expanded (paradoxically) to single‐molecule approaches,
where distinct functions are synergetically combined in the same molecule. An example of a single‐
molecule combinatorial approach was recently published by Poeck et al. in Nature Medicine, where
they used 5’‐triphosphate siRNAs as a potential therapeutic to combat melanoma.[190] The siRNAs
were targeted against Bcl‐2, a known key tumor survival factor involved in the regulation of
apoptosis. The triphosphate moiety is an activator of RIG‐1, which then leads to the stimulation of
47

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
the innate immune system and cytokine production. The synergy of sequence‐specific Bcl‐2 silencing
and RIG‐1 activation induces tumour apoptosis and tumour growth inhibition, and is a perfect
example of how the immune‐stimulating potential of small interfering RNA may have its therapeutic
benefit.[191] Another original approach is presented by Hossbach et al., who demonstrated the
simultaneous knockdown of two distinct genes with a single asymmetric ‘double‐guide’ siRNA, where
both strands can trigger efficient RNAi on separate targets.[192]
CONCLUSIONS
Advances in biotechnology have led to the emergence of many macromolecular drugs, such as
peptides, proteins and nucleic acids. Researchers are trying ever since to improve the therapeutic
outcome of this new spectrum of drugs using different strategies, related to the drug itself or the
formulation needed to deliver the drug in vivo. RNA interference has the advantage of being able to
profit from lessons learned during preclinical development of antisense oligonucleotides (AONs) and
ribozymes.[193] After several decades of research on AONs, leading experts of the industrial and
academic laboratories still have to acknowledge that inventive engineering strategies to improve the
design of siRNAs/shRNAs and to optimize their in vivo delivery are required to turn the therapeutic
promise of RNAi into clinical reality.
The clinical application of siRNA will call for repeated (often intravenous) administration, so it is
desirable to aim for a durable effect to lengthen treatment intervals. Direct application of synthetic
siRNAs has the disadvantage that the RNAi effect is transient, mainly due to intracellular dilution of
active siRNA dependent on the cell division rate or intracellular siRNA degradation. To maintain a
sufficient silencing of the target gene expression over a prolonged period of time, different strategies
have been proposed. Paying proper attention to their practical implication should eventually lead to
a maximal RNAi effect, both in terms of magnitude and duration. SiRNA duplexes can be designed
and modified to optimally exploit the endogenous RNAi pathway in order to increase their gene
silencing potency. Pursuing optimal siRNA design will lead to siRNA drug candidates with lower IC50
values which should enable effective medical treatment at lower doses. Incorporation of chemically
modified nucleotides into the siRNA sequence has proven to enhance their half‐life in the
bloodstream by protecting them against nuclease activity. However, it is conceivable that the
application of naked (i.e. unformulated or non‐conjugated) siRNAs will be mainly limited to confined
target sites after local administration, such as the eye or the respiratory tract.[189,194,195]
Advances in materials science increased chances of designing new nucleic acid delivery concepts. The
nano‐revolution led to the development of intelligent nanodevices which should enable
48

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
extrapolating drug delivery from the laboratory to a real in vivo situation. Multifunctional delivery
vehicles and siRNA conjugation strategies offer many advantages for the systemic application of
siRNA as they are usually equipped with targeting ligands and carry stabilizing hydrophilic polymers
to avoid aggregation in the bloodstream and to prevent non‐specific uptake by the reticulo‐
endothelial cells.[196] Moreover, delivery vehicles are often modified to improve the intracellular
trafficking (e.g. endosomal escape) which can lead to increased intracellular bioavailability. Including
all these factors in carrier design should enhance the percentage of the administered siRNA dose that
reaches the target site after systemic delivery. The ultimate goal is to define the appropriate dosing
schedule for a given siRNA formulation to maintain the desired therapeutic outcome. Model
predictions can aid in recognizing the main factors that govern the duration of the siRNA effect in
order to adjust the siRNA dose and frequency of administration accordingly. This can provide
researchers with general meaningful insights on systemic siRNA delivery that may apply to a variety
of siRNA carriers.
Depending on the pathological target, a more sustained gene silencing than readily achievable with
synthetic siRNA may be advisable. For this purpose researchers seek solace in the intracellular shRNA
production from plasmid or viral vectors. Although much progress has been made in the
development of viral gene therapy vectors, there are still important safety concerns that remain
troublesome.[197] Moreover, little information is available on the adverse effects that are linked to
sustained shRNA expression in vivo.
Most likely, a safe and long‐term application of RNAi drugs will require rigorous spatiotemporal
control over the intracellular siRNA/shRNA concentrations. Smart nanodevices for controlled
intracellular siRNA delivery or smart plasmids for controllable shRNA expression may fulfil some of
the necessary requirements.
49

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
REFERENCE LIST
1. Fire A., Xu S.Q., Montgomery M.K., Kostas S.A. et al., Potent and specific genetic interference by double‐stranded RNA in Caenorhabditis elegans, Nature 1998, 391: 806‐811.
2. Dorsett Y., siRNAs: applications in functional genomics and potential as therapeutics, Nature Reviews Drug Discovery 2004, 3: 318‐329.
3. Kim D.H. and Rossi J.J., Strategies for silencing human disease using RNA interference, Nat. Rev. Genet. 2007, 8: 173‐184.
4. Hamilton A.J. and Baulcombe D.C., A species of small antisense RNA in posttranscriptional gene silencing in plants, Science 1999, 286: 950‐952.
5. Napoli C., Lemieux C., and Jorgensen R., Introduction of A Chimeric Chalcone Synthase Gene Into Petunia Results in Reversible Co‐Suppression of Homologous Genes in Trans, Plant Cell 1990, 2: 279‐289.
6. Lee R.C., Feinbaum R.L., and Ambros V., The C‐Elegans Heterochronic Gene Lin‐4 Encodes Small Rnas with Antisense Complementarity to Lin‐14, Cell 1993, 75: 843‐854.
7. Elbashir S.M., Harborth J., Lendeckel W., Yalcin A. et al., Duplexes of 21‐nucleotide RNAs mediate RNA interference in cultured mammalian cells, Nature 2001, 411: 494‐498.
8. Elbashir S.M., Lendeckel W., and Tuschl T., RNA interference is mediated by 21‐and 22‐nucleotide RNAs, Genes & Development 2001, 15: 188‐200.
9. Birmingham A., Anderson E., Sullivan K., Reynolds A. et al., A protocol for designing siRNAs with high functionality and specificity, Nat. Protoc. 2007, 2: 2068‐2078.
10. Peek A.S. and Behlke M.A., Design of active small interfering RNAs, Current Opinion in Molecular Therapeutics 2007, 9: 110‐118.
11. de Fougerolles A., Vornlocher H.P., Maraganore J., and Lieberman J., Interfering with disease: a progress report on siRNA‐based therapeutics, Nature Reviews Drug Discovery 2007, 6: 443‐453.
12. Raab R.M. and Stephanopoulos G., Dynamics of gene silencing by RNA interference, Biotechnology and Bioengineering 2004, 88: 121‐132.
13. Behlke M.A., Progress towards in vivo use of siRNAs, Molecular Therapy 2006, 13: 644‐670. 14. Tolia N.H. and Joshua‐Tor L., Slicer and the Argonautes, Nature Chemical Biology 2007, 3: 36‐43. 15. Leuschner P.J.F., Ameres S.L., Kueng S., and Martinez J., Cleavage of the siRNA passenger strand during
RISC assembly in human cells, EMBO Reports 2006, 7: 314‐320. 16. Liu J.D., Carmell M.A., Rivas F.V., Marsden C.G. et al., Argonaute2 is the catalytic engine of mammalian
RNAi, Science 2004, 305: 1437‐1441. 17. Haley B. and Zamore P.D., Kinetic analysis of the RNAi enzyme complex, Nature Structural & Molecular
Biology 2004, 11: 599‐606. 18. Raemdonck K., Remaut K., Lucas B., Sanders N.N. et al., In Situ Analysis of Single‐Stranded and Duplex
siRNA Integrity in Living Cells, Biochemistry 2006, 45: 10614‐10623. 19. Bertrand J.R., Pottier M., Vekris A., Opolon P. et al., Comparison of antisense oligonucleotides and
siRNAs in cell culture and in vivo, Biochemical and Biophysical Research Communications 2002, 296: 1000‐1004.
20. Kim D.H., Behlke M.A., Rose S.D., Chang M.S. et al., Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy, Nature Biotechnology 2005, 23: 222‐226.
21. Amarzguioui M., Rossi J.J., and Kim D., Approaches for chemically synthesized siRNA and vector‐mediated RNAi, FEBS Letters 2005, 579: 5974‐5981.
22. Hefner E., Clark K., Whitman C., Behlke M.A. et al., Increased potency and longevity of gene silencing using validated Dicer substrates, J. Biomol. Tech. 2008, 19: 231‐237.
23. Lee Y.S., Nakahara K., Pham J.W., Kim K. et al., Distinct roles for Drosophila Dicer‐1 and Dicer‐2 in the siRNA/miRNA silencing pathways, Cell 2004, 117: 69‐81.
24. Winston W.M., Molodowitch C., and Hunter C.P., Systemic RNAi in C‐elegans requires the putative transmembrane protein SID‐1, Science 2002, 295: 2456‐2459.
25. Feinberg E.H. and Hunter C.P., Transport of dsRNA into cells by the transmembrane protein SID‐1, Science 2003, 301: 1545‐1547.
26. Oliveira S., Storm G., and Schiffelers R.M., Targeted delivery of siRNA, Journal of Biomedicine and Biotechnology 2006, 63675.
27. Duxbury M.S., Ashley S.W., and Whang E.E., RNA interference: A mammalian SID‐1 homologue enhances siRNA uptake and gene silencing efficacy in human cells, Biochemical and Biophysical Research Communications 2005, 331: 459‐463.
50

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
28. Wolfrum C., Shi S., Jayaprakash K.N., Jayaraman M. et al., Mechanisms and optimization of in vivo delivery of lipophilic siRNAs, Nat. Biotechnol. 2007, 25: 1149‐1157.
29. Baulcombe D.C., Amplified silencing, Science 2007, 315: 199‐200. 30. Sijen T., Steiner F.A., Thijssen K.L., and Plasterk R.H.A., Secondary siRNAs result from unprimed RNA
synthesis and form a distinct class, Science 2007, 315: 244‐247. 31. Pak J. and Fire A., Distinct populations of primary and secondary effectors during RNAi in C. elegans,
Science 2007, 315: 241‐244. 32. Chiu Y.L. and Rana T.M., RNAi in human cells: Basic structural and functional features of small interfering
RNA, Molecular Cell 2002, 10: 549‐561. 33. Stein P., Svoboda P., Anger M., and Schultz R.M., RNAi: Mammalian oocytes do it without RNA‐
dependent RNA polymerase, RNA 2003, 9: 187‐192. 34. Cullen B.R., Induction of stable RNA interference in mammalian cells, Gene Therapy 2006, 13: 503‐508. 35. Brummelkamp T.R., Bernards R., and Agami R., A system for stable expression of short interfering RNAs
in mammalian cells, Science 2002, 296: 550‐553. 36. Barnes M.R., Deharo S., Grocock R.J., Brown J.R. et al., The micro RNA target paradigm: a fundamental
and polymorphic control layer of cellular expression, Expert Opin. Biol. Ther. 2007, 7: 1387‐1399. 37. Bartel D.P., MicroRNAs: Genomics, biogenesis, mechanism, and function, Cell 2004, 116: 281‐297. 38. Zeng Y., Yi R., and Cullen B.R., Recognition and cleavage of primary microRNA precursors by the nuclear
processing enzyme Drosha, EMBO Journal 2005, 24: 138‐148. 39. Han J., Lee Y., Yeom K.H., Nam J.W. et al., Molecular basis for the recognition of primary microRNAs by
the Drosha‐DGCR8 complex, Cell 2006, 125: 887‐901. 40. Yeom K.H., Lee Y., Han J., Suh M.R. et al., Characterization of DGCR8/Pasha, the essential cofactor for
Drosha in primary miRNA processing, Nucleic Acids Res. 2006, 34: 4622‐4629. 41. Lund E., Guttinger S., Calado A., Dahlberg J.E. et al., Nuclear export of microRNA precursors, Science
2004, 303: 95‐98. 42. Kim V.N., MicroRNA biogenesis: coordinated cropping and dicing, Nat. Rev. Mol. Cell Biol. 2005, 6: 376‐
385. 43. Liu J.D., Valencia‐Sanchez M.A., Hannon G.J., and Parker R., MicroRNA‐dependent localization of
targeted mRNAs to mammalian P‐bodies, Nature Cell Biology 2005, 7: 719‐723. 44. Grimm D., Small silencing RNAs: state‐of‐the‐art, Adv. Drug Deliv. Rev. 2009, 61: 672‐703. 45. Yi R., Qin Y., Macara I.G., and Cullen B.R., Exportin‐5 mediates the nuclear export of pre‐microRNAs and
short hairpin RNAs, Genes & Development 2003, 17: 3011‐3016. 46. Siolas D., Lerner C., Burchard J., Ge W. et al., Synthetic shRNAs as potent RNAi triggers, Nature
Biotechnology 2005, 23: 227‐231. 47. Li L.M., Lin X.Y., Khvorova A., Fesik S.W. et al., Defining the optimal parameters for hairpin‐based
knockdown constructs, RNA 2007, 13: 1765‐1774. 48. Vlassov A.V., Korba B., Farrar K., Mukerjee S. et al., shRNAs targeting hepatitis C: Effects of sequence and
structural features, and comparison with siRNA, Oligonucleotides 2007, 17: 223‐236. 49. Fewell G.D. and Schmitt K., Vector‐based RNAi approaches for stable, inducible and genome‐wide
screens, Drug discovery today 2006, 11: 975‐982. 50. Silva J.M., Li M.Z., Chang K., Ge W. et al., Second‐generation shRNA libraries covering the mouse and
human genomes, Nature Genetics 2005, 37: 1281‐1288. 51. Vorhies J.S. and Nemunaitis J., Nonviral delivery vehicles for use in short hairpin RNA‐based cancer
therapies, Expert Review of Anticancer Therapy 2007, 7: 373‐382. 52. Song S.H., Lu Y.Q., Choi Y.K., Han Y.N. et al., DNA‐dependent PK inhibits adeno‐associated virus DNA
integration, Proceedings of the National Academy of Sciences of the United States of America 2004, 101: 2112‐2116.
53. Chen C.C., Ko T.M., Ma H.I., Wu H.L. et al., Long‐term inhibition of hepatitis B virus in transgenic mice by double‐stranded adeno‐associated virus 8‐delivered short hairpin RNA, Gene Therapy 2007, 14: 11‐19.
54. Bartlett D.W. and Davis M.E., Insights into the kinetics of siRNA‐mediated gene silencing from live‐cell and live‐animal bioluminescent imaging, Nucleic Acids Research 2006, 34: 322‐333.
55. Mutskov V. and Felsenfeld G., Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9, EMBO Journal 2004, 23: 138‐149.
56. Grimm D., Streetz K.L., Jopling C.L., Storm T.A. et al., Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways, Nature 2006, 441: 537‐541.
57. Wiznerowicz M., Szulc J., and Trono D., Tuning silence: conditional systems for RNA interference, Nat. Methods 2006, 3: 682‐688.
51

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
58. An D.S., Qin F.X.F., Auyeung V.C., Mao S.H. et al., Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors, Molecular Therapy 2006, 14: 494‐504.
59. Narvaiza I., Aparicio O., Vera M., Razquin N. et al., Effect of adenovirus‐mediated RNA interference on endogenous microRNAs in a mouse model of multidrug resistance protein 2 gene silencing, J. Virol. 2006, 80: 12236‐12247.
60. Castanotto D., Sakurai K., Lingeman R., Li H. et al., Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC, Nucleic Acids Res. 2007, 35: 5154‐5164.
61. Koller E., Propp S., Murray H., Lima W. et al., Competition for RISC binding predicts in vitro potency of siRNA, Nucleic Acids Research 2006, 34: 4467‐4476.
62. Dykxhoorn D.M. and Lieberman J., Knocking down disease with siRNAs, Cell 2006, 126: 231‐235. 63. Snove O., Jr. and Rossi J.J., Toxicity in mice expressing short hairpin RNAs gives new insight into RNAi,
Genome Biol. 2006, 7: 231. 64. John M., Constien R., Akinc A., Goldberg M. et al., Effective RNAi‐mediated gene silencing without
interruption of the endogenous microRNA pathway, Nature 2007, 449: 745‐747. 65. Kohn D.B., Sadelain M., and Glorioso J.C., Occurrence of leukaemia following gene therapy of X‐linked
SCID, Nature Reviews Cancer 2003, 3: 477‐488. 66. Cavazzana‐Calvo M. and Fischer A., Gene therapy for severe combined immunodeficiency: are we there
yet?, Journal of Clinical Investigation 2007, 117: 1456‐1465. 67. Vandenbroucke R.E., Lucas B., Demeester J., De Smedt S.C. et al., Nuclear accumulation of plasmid DNA
can be enhanced by non‐selective gating of the nuclear pore, Nucleic Acids Res. 2007, 35: e86. 68. Munkonge F.M., Dean D.A., Hillery E., Griesenbach U. et al., Emerging significance of plasmid DNA
nuclear import in gene therapy, Advanced Drug Delivery Reviews 2003, 55: 749‐760. 69. Haupenthal J., Baehr C., Kiermayer S., Zeuzem S. et al., Inhibition of RNAse A family enzymes prevents
degradation and loss of silencing activity of siRNAs in serum, Biochemical Pharmacology 2006, 71: 702‐710.
70. Turner J.J., Jones S.W., Moschos S.A., Lindsay M.A. et al., MALDI‐TOF mass spectral analysis of siRNA degradation in serum confirms an RNAse A‐like activity, Mol. Biosyst. 2007, 3: 43‐50.
71. Haupenthal J., Baehr C., Zeuzem S., and Piiper A., RNAse A‐like enzymes in serum inhibit the anti‐neoplastic activity of siRNA targeting polo‐like kinase 1, Int. J. Cancer 2007, 121: 206‐210.
72. Behlke M.A., Chemical modification of siRNAs for in vivo use, Oligonucleotides. 2008, 18: 305‐319. 73. Kennedy S., Wang D., and Ruvkun G., A conserved siRNA‐degrading RNase negatively regulates RNA
interference in C. elegans, Nature 2004, 427: 645‐649. 74. Zou Y., Tiller P., Chen I.W., Beverly M. et al., Metabolite identification of small interfering RNA duplex by
high‐resolution accurate mass spectrometry, Rapid Commun. Mass Spectrom. 2008, 22: 1871‐1881. 75. Choung S., Chemical modification of siRNAs to improve serum stability without loss of efficacy,
Biochemical and Biophysical Research Communications 2006, 342: 919‐927. 76. Morrissey D.V., Blanchard K., Shaw L., Jensen K. et al., Activity of stabilized short interfering RNA in a
mouse model of hepatitis B virus replication, Hepatology 2005, 41: 1349‐1356. 77. Corey D.R., Chemical modification: the key to clinical application of RNA interference?, Journal of Clinical
Investigation 2007, 117: 3615‐3622. 78. Watts J.K., Deleavey G.F., and Damha M.J., Chemically modified siRNA: tools and applications, Drug
Discov. Today 2008, 13: 842‐855. 79. Allerson C.R., Sioufi N., Jarres R., Prakash T.P. et al., Fully 2 '‐modified oligonucleotide duplexes with
improved in vitro potency and stability compared to unmodified small interfering RNA, Journal of Medicinal Chemistry 2005, 48: 901‐904.
80. Liao H.T. and Wang J.H., Biomembrane‐permeable and ribonuclease‐resistant siRNA with enhanced activity, Oligonucleotides 2005, 15: 196‐205.
81. Layzer J.M., McCaffrey A.P., Tanner A.K., Huang Z. et al., In vivo activity of nuclease‐resistant siRNAs, RNA 2004, 10: 766‐771.
82. Dykxhoorn D.M., Palliser D., and Lieberman J., The silent treatment: siRNAs as small molecule drugs, Gene Therapy 2006, 13: 541‐552.
83. Novobrantseva T.I., Akinc A., Borodovsky A., and de Fougerolles A., Delivering silence: Advancements in developing siRNA therapeutics, Current Opinion in Drug Discovery & Development 2008, 11: 217‐224.
84. De Paula D., Bentley M.V., and Mahato R.I., Hydrophobization and bioconjugation for enhanced siRNA delivery and targeting, RNA. 2007, 13: 431‐456.
52

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
85. de Fougerolles A.R., Delivery vehicles for small interfering RNA in vivo, Hum. Gene Ther. 2008, 19: 125‐132.
86. Song E.W., Zhu P.C., Lee S.K., Chowdhury D. et al., Antibody mediated in vivo delivery of small interfering RNAs via cell‐surface receptors, Nature Biotechnology 2005, 23: 709‐717.
87. Soutschek J., Akinc A., Bramlage B., Charisse K. et al., Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs, Nature 2004, 432: 173‐178.
88. Nishina K., Unno T., Uno Y., Kubodera T. et al., Efficient in vivo delivery of siRNA to the liver by conjugation of alpha‐tocopherol, Molecular Therapy 2008, 16: 734‐740.
89. Kumar P., Wu H.Q., McBride J.L., Jung K.E. et al., Transvascular delivery of small interfering RNA to the central nervous system, Nature 2007, 448: 39‐43.
90. Rozema D.B., Lewis D.L., Wakefield D.H., Wong S.C. et al., Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes, Proceedings of the National Academy of Sciences of the United States of America 2007, 104: 12982‐12987.
91. Chu T.C., Twu K.Y., Ellington A.D., and Levy M., Aptamer mediated siRNA delivery, Nucleic Acids Res. 2006, 34: e73.
92. McNamara J.O., Andrechek E.R., Wang Y., Viles K.D. et al., Cell type‐specific delivery of siRNAs with aptamer‐siRNA chimeras, Nat. Biotechnol. 2006, 24: 1005‐1015.
93. Eguchi A., Meade B.R., Chang Y.C., Fredrickson C.T. et al., Efficient siRNA delivery into primary cells by a peptide transduction domain‐dsRNA binding domain fusion protein, Nat. Biotechnol. 2009, doi: 10.1038/nbt.1541.
94. Juliano R., Alam M.R., Dixit V., and Kang H., Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides, Nucleic Acids Res. 2008, 36: 4158‐4171.
95. Sato A., Choi S.W., Hirai M., Yamayoshi A. et al., Polymer brush‐stabilized polyplex for a siRNA carrier with long circulatory half‐life, Journal of controlled release 2007, 122: 209‐216.
96. Urban‐Klein B., RNAi‐mediated gene‐targeting through systemic application of polyethyleneimine (PEI)‐complexed siRNA in vivo, Gene Therapy 2005, 12: 461‐466.
97. Buyens K., Lucas B., Raemdonck K., Braeckmans K. et al., A fast and sensitive method for measuring the integrity of siRNA‐carrier complexes in full human serum, J. Control Release 2007, 126: 67‐76.
98. Howard K.A. and Kjems J., Polycation‐based nanoparticle delivery for improved RNA interference therapeutics, Expert Opinion on Biological Therapy 2007, 7: 1811‐1822.
99. Howard K.A., Rahbek U.L., Liu X.D., Damgaard C.K. et al., RNA interference in vitro and in vivo using a chitosan/siRNA nanoparticle system, Molecular Therapy 2006, 14: 476‐484.
100. Takeshita F., Minakuchi Y., Nagahara S., Honma K. et al., Efficient delivery of small interfering RNA to bone‐metastatic tumors by using atelocollagen in vivo, Proceedings of the National Academy of Sciences of the United States of America 2005, 102: 12177‐12182.
101. Morrissey D.V., Lockridge J.A., Shaw L., Blanchard K. et al., Potent and persistent in vivo anti‐HBV activity of chemically modified siRNAs, Nature Biotechnology 2005, 23: 1002‐1007.
102. Zimmermann T.S., Lee A.C., Akinc A., Bramlage B. et al., RNAi‐mediated gene silencing in non‐human primates, Nature 2006, 441: 111‐114.
103. Hu‐Lieskovan S., Heidel J.D., Bartlett D.W., Davis M.E. et al., Sequence‐specific knockdown of EWS‐FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing's sarcoma, Cancer Research 2005, 65: 8984‐8992.
104. Heidel J.D., Yu Z.P., Liu J.Y.C., Rele S.M. et al., Administration in non‐human primates of escalating intravenous doses of targeted nanoparticles containing ribonucleotide reductase subunit M2 siRNA, Proceedings of the National Academy of Sciences of the United States of America 2007, 104: 5715‐5721.
105. Davis M.E., The First Targeted Delivery of siRNA in Humans via a Self‐Assembling, Cyclodextrin Polymer‐Based Nanoparticle: From Concept to Clinic, Mol. Pharm. 2009, 6: 659‐668.
106. Czauderna F., Structural variations and stabilising modifications of synthetic siRNAs in mammalian cells, Nucleic Acids Research 2003, 31: 2705‐2716.
107. Chiu Y.L. and Rana T.M., siRNA function in RNAi: a chemical modification analysis, RNA. 2003, 9: 1034‐1048.
108. Dowler T., Bergeron D., Tedeschi A.L., Paquet L. et al., Improvements in siRNA properties mediated by 2'‐deoxy‐2'‐fluoro‐beta‐D‐arabinonucleic acid (FANA), Nucleic Acids Res. 2006, 34: 1669‐1675.
109. Volkov A.A., Kruglova N.S., Meschaninova M.I., Venyaminova A.G. et al., Selective protection of nuclease‐sensitive sites in siRNA prolongs silencing effect, Oligonucleotides. 2009, 19: 191‐202.
110. Mantei A., Rutz S., Janke M., Kirchhoff D. et al., siRNA stabilization prolongs gene knockdown in primary T lymphocytes, Eur. J. Immunol. 2008, 38: 2616‐2625.
53

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
111. Takabatake Y., Isaka Y., Mizui M., Kawachi H. et al., Chemically modified siRNA prolonged RNA interference in renal disease, Biochem. Biophys. Res. Commun. 2007, 363: 432‐437.
112. Tagami T., Barichello J.M., Kikuchi H., Ishida T. et al., The gene‐silencing effect of siRNA in cationic lipoplexes is enhanced by incorporating pDNA in the complex, Int. J. Pharm. 2007, 333: 62‐69.
113. Fisher M., Abramov M., Van A.A., Xu D. et al., Inhibition of MDR1 expression with altritol‐modified siRNAs, Nucleic Acids Res. 2007, 35: 1064‐1074.
114. Song E.W., Lee S.K., Dykxhoorn D.M., Novina C. et al., Sustained small interfering RNA‐mediated human immunodeficiency virus type 1 inhibition in primary macrophages, Journal of Virology 2003, 77: 7174‐7181.
115. Maliyekkel A., Davis B.M., and Roninson I.B., Cell cycle arrest drastically extends the duration of gene silencing after transient expression of short hairpin RNA, Cell Cycle 2006, 5: 2390‐2395.
116. Omi K., Tokunaga K., and Hohjoh H., Long‐lasting RNAi activity in mammalian neurons, FEBS Letters 2004, 558: 89‐95.
117. Kakutani K., Nishida K., Uno K., Takada T. et al., Prolonged down regulation of specific gene expression in nucleus pulposus cell mediated by RNA interference in vitro, J. Orthop. Res. 2006, 24: 1271‐1278.
118. Bartlett D.W. and Davis M.E., Effect of siRNA nuclease stability on the in vitro and in vivo kinetics of siRNA‐mediated gene silencing, Biotechnol. Bioeng. 2007, 97: 909‐921.
119. Marques J.T. and Williams B.R., Activation of the mammalian immune system by siRNAs, Nat. Biotechnol. 2005, 23: 1399‐1405.
120. Snove O., Jr. and Rossi J.J., Chemical modifications rescue off‐target effects of RNAi, ACS Chem. Biol. 2006, 1: 274‐276.
121. Jackson A.L., Burchard J., Leake D., Reynolds A. et al., Position‐specific chemical modification of siRNAs reduces "off‐target" transcript silencing, RNA. 2006, 12: 1197‐1205.
122. Judge A. and MacLachlan I., Overcoming the innate immune response to small interfering RNA, Human Gene Therapy 2008, 19: 111‐124.
123. Judge A.D., Sood V., Shaw J.R., Fang D. et al., Sequence‐dependent stimulation of the mammalian innate immune response by synthetic siRNA, Nature Biotechnology 2005, 23: 457‐462.
124. Hornung V., Guenthner‐Biller M., Bourquin C., Ablasser A. et al., Sequence‐specific potent induction of IFN‐alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7, Nature Medicine 2005, 11: 263‐270.
125. Marques J.T., Devosse T., Wang D., Zamanian‐Daryoush M. et al., A structural basis for discriminating between self and nonself double‐stranded RNAs in mammalian cells, Nature Biotechnology 2006, 24: 559‐565.
126. Judge A.D., Bola G., Lee A.C.H., and MacLachlan I., Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo, Molecular Therapy 2006, 13: 494‐505.
127. Kleinman M.E., Yamada K., Takeda A., Chandrasekaran V. et al., Sequence‐ and target‐independent angiogenesis suppression by siRNA via TLR3, Nature 2008, 452: 591‐597.
128. Cho W.G., Albuquerque R.J., Kleinman M.E., Tarallo V. et al., Small interfering RNA‐induced TLR3 activation inhibits blood and lymphatic vessel growth, Proc. Natl. Acad. Sci. U. S. A 2009, 106: 7137‐7142.
129. Robbins M., Judge A., and MacLachlan I., siRNA and innate immunity, Oligonucleotides. 2009, 19: 89‐102. 130. Zeng Y., RNA interference in human cells is restricted to the cytoplasm, RNA 2002, 8: 855‐860. 131. Robb G.B., Brown K.M., Khurana J., and Rana T.M., Specific and potent RNAi in the nucleus of human
cells, Nat. Struct. Mol. Biol. 2005, 12: 133‐137. 132. Gurney T., Jr. and Eliceiri G.L., Intracellular distribution of low molecular weight RNA species in HeLa
cells, J. Cell Biol. 1980, 87: 398‐403. 133. Matera A.G. and Ward D.C., Nucleoplasmic organization of small nuclear ribonucleoproteins in cultured
human cells, J. Cell Biol. 1993, 121: 715‐727. 134. Wassarman D.A. and Steitz J.A., Structural analyses of the 7SK ribonucleoprotein (RNP), the most
abundant human small RNP of unknown function, Mol. Cell Biol. 1991, 11: 3432‐3445. 135. Ohrt T., Mutze J., Staroske W., Weinmann L. et al., Fluorescence correlation spectroscopy and
fluorescence cross‐correlation spectroscopy reveal the cytoplasmic origination of loaded nuclear RISC in vivo in human cells, Nucleic Acids Res. 2008, 36: 6439‐6449.
136. Matzke M.A., Primig M., Trnovsky J., and Matzke A.J., Reversible methylation and inactivation of marker genes in sequentially transformed tobacco plants, EMBO J. 1989, 8: 643‐649.
137. Hamilton A., Voinnet O., Chappell L., and Baulcombe D., Two classes of short interfering RNA in RNA silencing, EMBO J. 2002, 21: 4671‐4679.
54

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
138. Mette M.F., Aufsatz W., van der W.J., Matzke M.A. et al., Transcriptional silencing and promoter methylation triggered by double‐stranded RNA, EMBO J. 2000, 19: 5194‐5201.
139. Wassenegger M., Heimes S., Riedel L., and Sanger H.L., RNA‐directed de novo methylation of genomic sequences in plants, Cell 1994, 76: 567‐576.
140. Zilberman D., Cao X., and Jacobsen S.E., ARGONAUTE4 control of locus‐specific siRNA accumulation and DNA and histone methylation, Science 2003, 299: 716‐719.
141. Zilberman D., Cao X., Johansen L.K., Xie Z. et al., Role of Arabidopsis ARGONAUTE4 in RNA‐directed DNA methylation triggered by inverted repeats, Curr. Biol. 2004, 14: 1214‐1220.
142. Castanotto D., Tommasi S., Li M., Li H. et al., Short hairpin RNA‐directed cytosine (CpG) methylation of the RASSF1A gene promoter in HeLa cells, Mol. Ther. 2005, 12: 179‐183.
143. Kawasaki H. and Taira K., Induction of DNA methylation and gene silencing by short interfering RNAs in human cells, Nature 2004, 431: 211‐217.
144. Morris K.V., Chan S.W., Jacobsen S.E., and Looney D.J., Small interfering RNA‐induced transcriptional gene silencing in human cells, Science 2004, 305: 1289‐1292.
145. Weinberg M.S., Villeneuve L.M., Ehsani A., Amarzguioui M. et al., The antisense strand of small interfering RNAs directs histone methylation and transcriptional gene silencing in human cells, RNA. 2006, 12: 256‐262.
146. Strunnikova M., Schagdarsurengin U., Kehlen A., Garbe J.C. et al., Chromatin inactivation precedes de novo DNA methylation during the progressive epigenetic silencing of the RASSF1A promoter, Mol. Cell Biol. 2005, 25: 3923‐3933.
147. Janowski B.A., Huffman K.E., Schwartz J.C., Ram R. et al., Inhibiting gene expression at transcription start sites in chromosomal DNA with antigene RNAs, Nat. Chem. Biol. 2005, 1: 216‐222.
148. Ting A.H., Schuebel K.E., Herman J.G., and Baylin S.B., Short double‐stranded RNA induces transcriptional gene silencing in human cancer cells in the absence of DNA methylation, Nat. Genet. 2005, 37: 906‐910.
149. Kim D.H., Villeneuve L.M., Morris K.V., and Rossi J.J., Argonaute‐1 directs siRNA‐mediated transcriptional gene silencing in human cells, Nat. Struct. Mol. Biol. 2006, 13: 793‐797.
150. Janowski B.A., Huffman K.E., Schwartz J.C., Ram R. et al., Involvement of AGO1 and AGO‐2 in mammalian transcriptional silencing, Nature Structural & Molecular Biology 2006, 13: 787‐792.
151. Kim D.H., Saetrom P., Snove O., Jr., and Rossi J.J., MicroRNA‐directed transcriptional gene silencing in mammalian cells, Proc. Natl. Acad. Sci. U. S. A 2008, 105: 16230‐16235.
152. Janowski B.A., Younger S.T., Hardy D.B., Ram R. et al., Activating gene expression in mammalian cells with promoter‐targeted duplex RNAs, Nat. Chem. Biol. 2007, 3: 166‐173.
153. Li L.C., Okino S.T., Zhao H., Pookot D. et al., Small dsRNAs induce transcriptional activation in human cells, Proc. Natl. Acad. Sci. U. S. A 2006, 103: 17337‐17342.
154. Capodici J., Kariko K., and Weissman D., Inhibition of HIV‐1 infection by small interfering RNA‐mediated RNA interference, J. Immunol. 2002, 169: 5196‐5201.
155. Boden D., Pusch O., Lee F., Tucker L. et al., Human immunodeficiency virus type 1 escape from RNA interference, J. Virol. 2003, 77: 11531‐11535.
156. Suzuki K., Shijuuku T., Fukamachi T., Zaunders J. et al., Prolonged transcriptional silencing and CpG methylation induced by siRNAs targeted to the HIV‐1 promoter region, J. RNAi Gene Silencing 2005, 1: 66‐78.
157. Robertson K.D., DNA methylation and chromatin ‐ unraveling the tangled web, Oncogene 2002, 21: 5361‐5379.
158. Sijen T., Vijn I., Rebocho A., van B.R. et al., Transcriptional and posttranscriptional gene silencing are mechanistically related, Curr. Biol. 2001, 11: 436‐440.
159. Burgers P.M. and Eckstein F., Absolute configuration of the diastereomers of adenosine 5'‐O‐(1‐thiotriphosphate): consequences for the stereochemistry of polymerization by DNA‐dependent RNA polymerase from Escherichia coli, Proc. Natl. Acad. Sci. U. S. A 1978, 75: 4798‐4800.
160. Vastenhouw N.L., Brunschwig K., Okihara K.L., Muller F. et al., Long‐term gene silencing by RNAi, Nature 2006, 442: 882‐882.
161. Hawkins P.G., Santoso S., Adams C., Anest V. et al., Promoter targeted small RNAs induce long‐term transcriptional gene silencing in human cells, Nucleic Acids Res. 2009, 37: 2984‐2995.
162. Oliveira S., van Rooy I., Kranenburg O., Storm G. et al., Fusogenic peptides enhance endosomal escape improving siRNA‐induced silencing of oncogenes, International Journal of Pharmaceutics 2007, 331: 211‐214.
55

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
163. Oliveira S., Fretz M.M., Hogset A., Storm G. et al., Photochemical internalization enhances silencing of epidermal growth factor receptor through improved endosomal escape of siRNA, Biochimica et Biophysica Acta‐Biomembranes 2007, 1768: 1211‐1217.
164. Bartlett D.W. and Davis M.E., Impact of tumor‐specific targeting and dosing schedule on tumor growth inhibition after intravenous administration of siRNA‐containing nanoparticles, Biotechnol. Bioeng. 2007,
165. Groenenboom M.A., Maree A.F., and Hogeweg P., The RNA silencing pathway: the bits and pieces that matter, PLoS. Comput. Biol. 2005, 1: 155‐165.
166. Bergstrom C.T., McKittrick E., and Antia R., Mathematical models of RNA silencing: unidirectional amplification limits accidental self‐directed reactions, Proc. Natl. Acad. Sci. U. S. A 2003, 100: 11511‐11516.
167. Takahashi Y., Yamaoka K., Nishikawa M., and Takakura Y., Moment analysis for kinetics of gene silencing by RNA interference, Biotechnol. Bioeng. 2006, 93: 816‐819.
168. Arciero J.C., Jackson T.L., and Kirschner D.E., A mathematical model of tumor‐immune evasion and siRNA treatment, Discrete and Continuous Dynamical Systems‐Series B 2004, 4: 39‐58.
169. Bartlett D.W., Su H., Hildebrandt I.J., Weber W.A. et al., Impact of tumor‐specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging, Proc. Natl. Acad. Sci. U. S. A 2007, 104: 15549‐15554.
170. Marsden P.A., RNA interference as potential therapy‐‐not so fast, N. Engl. J. Med. 2006, 355: 953‐954. 171. Vandenbroucke R., De Geest B.G., Bonné S., and Van Haecke T., Prolonged gene silencing in hepatoma
cells and primary hepatocytes after siRNA delivery with biodegradable poly(β‐amino esters), Journal of Gene Medicine 2008, 10: 783‐794.
172. Raemdonck K., Van Thienen T.G., Vandenbroucke R.E., Sanders N.N. et al., Dextran Microgels for Time‐Controlled Delivery of siRNA, Advanced Functional Materials 2008, 18: 993‐1001.
173. Veldhoen S., Laufer S.D., Trampe A., and Restle T., Cellular delivery of small interfering RNA by a non‐covalently attached cell‐penetrating peptide: quantitative analysis of uptake and biological effect, Nucleic Acids Res. 2006, 34: 6561‐6573.
174. Mescalchin A., Detzer A., Wecke M., Overhoff M. et al., Cellular uptake and intracellular release are major obstacles to the therapeutic application of siRNA: novel options by phosphorothioate‐stimulated delivery, Expert. Opin. Biol. Ther. 2007, 7: 1531‐1538.
175. Dykxhoorn D.M. and Lieberman J., Silencing viral infection, PLoS. Med. 2006, 3: e242. 176. Boden D., Pusch O., and Ramratnam B., Overcoming HIV‐1 resistance to RNA interference, Front Biosci.
2007, 12: 3104‐3116. 177. Grimm D. and Kay M.A., Combinatorial RNAi: A winning strategy for the race against evolving targets?,
Molecular Therapy 2007, 15: 878‐888. 178. Zheng Z.M., Tang S.A., and Tao M.F., Development of resistance to RNAi in mammalian cells, Therapeutic
Oligonucleotides: Transcriptional and Translational Strategies for Silencing Gene Expression 2005, 1058: 105‐118.
179. Meadows D.C. and Gervay‐Hague J., Targeting HIV, Chemmedchem 2006, 1: 16‐29. 180. ter Brake O., Konstantinova P., Ceylan M., and Berkhout B., Silencing of HIV‐1 with RNA interference: A
multiple shRNA approach, Molecular Therapy 2006, 14: 883‐892. 181. Schubert S., Grunert H.P., Zeichhardt H., Werk D. et al., Maintaining inhibition: siRNA double expression
vectors against coxsackieviral RNAs, Journal of Molecular Biology 2005, 346: 457‐465. 182. He M.L., Zheng B.J., Chen Y., Wong K.L. et al., Kinetics and synergistic effects of siRNAs targeting
structural and replicase genes of SARS‐associated coronavirus, FEBS Lett. 2006, 580: 2414‐2420. 183. Li M.J., Kim J., Li S., Zaia J. et al., Long‐term inhibition of HIV‐1 infection in primary hematopoietic cells by
lentiviral vector delivery of a triple combination of anti‐HIV shRNA, anti‐CCR5 ribozyme, and a nucleolar‐localizing TAR decoy, Molecular Therapy 2005, 12: 900‐909.
184. Unwalla H.J., Li H.T., Bahner I., Li M.J. et al., Novel Pol II fusion promoter directs human immunodeficiency virus type 1‐inducible coexpression of a short hairpin RNA and protein, J. Virol. 2006, 80: 1863‐1873.
185. Takei Y., Kadomatsu K., Goto T., and Muramatsu T., Combinational antitumor effect of siRNA against midkine and paclitaxel on growth of human prostate cancer xenografts, Cancer 2006, 107: 864‐873.
186. Li S.D., Chen Y.C., Hackett M.J., and Huang L., Tumor‐targeted delivery of siRNA by self‐assembled nanoparticles, Molecular Therapy 2008, 16: 163‐169.
187. Landen C.N., Chavez‐Reyes A., Bucana C., Schmandt R. et al., Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery, Cancer Research 2005, 65: 6910‐6918.
56

maintaining the silence: reflections on long‐term RNAi | CHAPTER 1
188. Halder J., Kamat A.A., Landen C.N., Han L.Y. et al., Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy, Clinical Cancer Research 2006, 12: 4916‐4924.
189. Bitko V., Musiyenko A., Shulyayeva O., and Barik S., Inhibition of respiratory viruses by nasally administered siRNA, Nature Medicine 2005, 11: 50‐55.
190. Poeck H., Besch R., Maihoefer C., Renn M. et al., 5'‐Triphosphate‐siRNA: turning gene silencing and Rig‐I activation against melanoma, Nat. Med. 2008, 14: 1256‐1263.
191. Schlee M., Hornug V., and Hartmann G., siRNA and isRNA: Two edges of one sword, Molecular Therapy 2006, 14: 463‐470.
192. Hossbach M., Gruber J., Osborn M., Weber K. et al., Gene silencing with siRNA duplexes composed of target‐mRNA‐complementary and partially palindromic or partially complementary single‐stranded siRNAs, RNA Biol. 2006, 3: 82‐89.
193. Corey D.R., RNA learns from antisense, Nature Chemical Biology 2007, 3: 8‐11. 194. Campochiaro P.A., Potential applications for RNAi to probe pathogenesis and develop new treatments
for ocular disorders, Gene Therapy 2006, 13: 559‐562. 195. Shen J., Samul R., Silva R.L., Akiyama H. et al., Suppression of ocular neovascularization with siRNA
targeting VEGF receptor 1, Gene Therapy 2006, 13: 225‐234. 196. Remaut K., Sanders N.N., De Geest B.G., Braeckmans K. et al., Nucleic acid delivery: Where material
sciences and bio‐sciences meet, Materials Science & Engineering R‐Reports 2007, 58: 117‐161. 197. Thomas C.E., Ehrhardt A., and Kay M.A., Progress and problems with the use of viral vectors for gene
therapy, Nature Reviews Genetics 2003, 4: 346‐358.
57

CHAPTER 1 | maintaining the silence: reflections on long‐term RNAi
58

in situ analysis of single‐stranded and duplex siRNA integrity in living cells | CHAPTER 2
59
CHAPTER 2 | In situ analysis of single‐stranded and duplex siRNA integrity in
living cells
Parts of this chapter are published:
Koen Raemdonck1, Katrien Remaut1, Bart Lucas1, Niek N. Sanders1,2, Joseph Demeester1 and Stefaan
C. De Smedt1, Biochemistry 2006, 45: 10614‐10623.
1Laboratory of General Biochemistry and Physical Pharmacy, Department of Pharmaceutics, Faculty of Pharmaceutical
Sciences, Ghent University, Harelbekestraat 72, B‐9000 Ghent, Belgium.
2Laboratory of Gene Therapy, Department of Nutrition, Genetics and Ethology, Faculty of Veterinary Medicine, Ghent
University, Heidestraat 19, B‐9820 Merelbeke, Belgium.

CHAPTER 2 | in situ analysis of single‐stranded and duplex siRNA integrity in living cells
60
ABSTRACT
To attain the full therapeutic promise of small interfering RNA (siRNA) it is believed that
improvements, such as increased biostability, are critical. Regrettably, thus far insufficient in situ
data are on hand regarding the intracellular stability of siRNAs. We report on the use of an advanced
fluorescence‐based method to probe nucleolytic decay of double labelled siRNAs which are subject
to fluorescence resonance energy transfer (FRET). In vitro measurements with RNase A and cellular
extracts demonstrate that the ratio of acceptor (5’‐Cy5) to donor (3’‐rhodamine green) fluorescence
can be used to study the degradation of the labelled siRNA substrates upon donor excitation.
Intracellular FRET analysis showed substantial degradation of single‐stranded siRNA, whereas duplex
siRNA stayed intact during the measured time period. These data underline the high intrinsic
nuclease resistance of unmodified duplex siRNA and prove that cellular persistence is much more
critical for the single‐stranded structure. It is for the first time that the stability of siRNA is
investigated in real‐time inside living cells. The presented fluorescence‐based method is a
straightforward technique to gain direct information on siRNA integrity inside living cells and
provides a bright outlook to learn more about the intracellular fate of siRNA therapeutics.

in situ analysis of single‐stranded and duplex siRNA integrity in living cells | CHAPTER 2
2 OVERVIEW CHAPTER CONTENTS
Chapter 2 provides:
• details on the validation and application of a fluorescence based method (FRET‐FFS) to
evaluate siRNA integrity
• a comparison of the stability between ssRNA/dsRNA upon incubation with RNase A and
cellular extracts
• an evaluation of siRNA integrity in living cells with FRET‐FFS following microinjection
Key terms:
FRET – ssRNA – dsRNA – FFS – FA/FD ratio – chemical modification – microinjection
INTRODUCTION
Knockdown of gene expression by RNA interference (RNAi) has evolved as a powerful tool for reverse
genetic analysis and holds a great therapeutic potential.[1,2] RNAi represents an evolutionary
conserved gene silencing mechanism by which ~21 nucleotide double‐stranded RNAs, termed small
interfering RNAs (siRNAs),[3] guide sequence specific cleavage and subsequent degradation of the
targeted mRNA and thus knockdown of the corresponding gene.[4] Intracellularly, duplex siRNA is
recognized by key components of the RNAi pathway which eventually leads to the incorporation of
the antisense (guide) strand in a ribonucleoprotein complex termed RISC (RNA‐induced silencing
complex). This activated RISC (RISC*) subsequently degrades the target mRNA employing the guide
strand as ‘docking site’ to the homologous mRNA sequence.[1‐5] The discovery that siRNAs can also
target mammalian genes without invoking the interferon response lightened the path to the
development of siRNA therapeutics.[5]
61

CHAPTER 2 | in situ analysis of single‐stranded and duplex siRNA integrity in living cells
To effectively use siRNAs as drug molecules, many barriers still need to be overcome.[6] For successful
in vivo application of siRNAs, their biostability and cellular delivery should be improved.[1,2,6‐8] Many
efforts have already been undertaken to chemically modify the siRNA molecules in order to reduce
their susceptibility to RNases.[7,9‐13] Some of the investigated modifications indeed provide a higher
resistance against serum‐derived nucleases.[7,12,13] Moreover, evidence exists that chemical
modifications of siRNAs can lead to a prolonged gene silencing effect in cell culture, suggesting that
the transient and short lived RNAi induced gene knockdown, as usually observed in mammalian cells,
may be partially due to intracellular siRNA degradation.[9,11,12,14,15] However, in an attempt to confirm
this hypothesis, Layzer and coworkers had to conclude that the use of nuclease‐resistant siRNA does
not by definition result in a more potent and/or prolonged RNAi effect in vitro and in vivo.[13] Besides
chemical modification, the packaging of siRNAs into suitable delivery carriers may also protect them
from degradation,[16,17] also in vivo.[8] More detailed information can be found in chapter 1.
The interest of numerous research groups with regard to improvement of siRNA stability emphasizes
the need for useful and straightforward methods to probe siRNA integrity. To the best of our
knowledge, experiments which are able to follow the degradation of siRNA in real‐time in living cells
are not yet reported, mainly due to a lack of suitable non‐invasive methods. It may be clear however
that the development of siRNA as a drug needs an understanding of its intracellular fate. The major
focus of this manuscript is therefore to investigate the intracellular turnover of siRNAs. For this
purpose, we assessed the enzymatic degradation of double fluorescently labelled single‐stranded (ss)
and double‐stranded (ds) siRNA inside living cells by fluorescence resonance energy transfer (FRET;
Figure 2.1a). The intracellular tracking of siRNA duplexes carrying a single fluorophore makes it
impossible to distinguish between intact and degraded siRNA duplexes, strand separation or even
the emergence of free dye. Opposed to this approach, FRET has the advantage that it is based on the
direct and distance‐dependent interaction between two spectrally different fluorescent labels.[17] The
fluorescence intensities of both dyes ‐ i.e. rhodamine green and Cy5, attached to the 3’ and 5’ end of
the sense strand respectively ‐ were monitored using a dual‐colour fluorescence fluctuation
spectroscopy setup (FFS) as depicted in Figure 2.1b. Essentially, FFS measures fluorescence intensity
fluctuations in the excitation volume of a (confocal) microscope (Figure 2.1b ‐ 2.1d). The fluorescence
fluctuations are due to the movement of the fluorophores in and out the confocal volume (Figure
2.1c ‐ 2.1d). In dual‐colour FFS, a sample containing two spectrally different fluorophores (e.g.
rhodamine green and Cy5) is analyzed and their emission light is registered separately by two
detectors monitoring the same excitation volume (Figure 2.1b).[18]
Our results demonstrate that duplex siRNA is more resistant against intracellular nucleases when
compared with single‐stranded siRNA which is degraded within minutes. Additionally, this study
62

in situ analysis of single‐stranded and duplex siRNA integrity in living cells | CHAPTER 2
shows the potential of the delineated fluorescence‐based technique for future research on the cell
biological behaviour of siRNA.
FIGURE 2.1 | (a) Schematical representation of fluorescence resonance energy transfer (FRET) and its applicability to probe the degradation of double labelled siRNA (further information: see experimental section). (b) Setup of the dual‐colour fluorescence fluctuation spectroscopy (dual‐colour FFS) instrument. The excitation light, emerging from a Kr‐Ar ion laser, is reflected by a triple chroic mirror and focused into the objective of a confocal microscope. The fluorescence light, emitted by the fluorophores when diffusing through the excitation volume, passes the same objective and triple chroic mirror to be split by a dichroic mirror into a red and a green component. (c) Animated view of fluorescent molecules diffusing through the excitation volume (typically some femtoliters) which causes fluorescence intensity fluctuations as depicted in (d).
MATERIALS AND METHODS
siRNA oligonucleotides
21 nt single‐stranded (ss) siRNAs were synthesized and purified by Eurogentec (sense strand: 5’‐
GUGCGCUGCUGGUGCCAACdTdT‐3’, antisense strand: 5’‐GUUGGCACCAGCAGCGCACdTdT‐3’). Double
fluorescent labelling occurred at the 3’ and 5’ end of the sense strand with rhodamine green (RhoGr)
and Cy5 respectively. The concentration of the siRNA stock solutions (in DEPC‐treated water) was
calculated from absorption measurements at 260 nm (1 OD260 = 40 µg mL‐1) with a Nanodrop ND‐
1000 spectrophotometer. The absorption of the rhodamine label at 260 nm was taken into account.
The presence of the fluorescent labels was verified by absorption measurements at 500 nm for
RhoGr (εRhoGr, 500 nm = 54 000 L.mol‐1.cm‐1) and 647 nm for Cy5 (εCy5, 647nm = 250 000 L.mol‐1.cm‐1).
Fluorescent siRNA duplexes were prepared through hybridization of non‐labelled antisense and
fluorescently labelled sense RNA strands (either single or double labelled). Equimolar amounts of
63

CHAPTER 2 | in situ analysis of single‐stranded and duplex siRNA integrity in living cells
both RNAs were mixed in annealing buffer (final buffer concentration 50 mM Tris, pH 7.5‐8.0, 100
mM NaCl) and annealing was performed in a thermal cycler (2 min incubation at 94°C followed by
slow cooling to 25°C over a time period of 45 min). Duplex formation was confirmed by 20% non‐
denaturing polyacrylamide gel electrophoresis.
Cell culture and microinjection experiments
VERO cells (African green monkey kidney cells) were maintained in Dulbecco’s modified eagle’s
medium (DMEM) without phenol red (Gibco™ Invitrogen) supplemented with 2 mM glutamine, 10%
heat deactivated fetal bovine serum (FBS) and 1% penicillin‐streptomycin at 37°C in humidified
atmosphere (5% CO2). Cells were prophylactically treated against mycoplasma with Plasmocin
(Invivogen). For intracellular fluorescence measurements, cells were seeded (2.5 x 104 cells per cm2)
on culture dishes with central 150 µm thick glass‐bottomed surface (MatTek Corporation). Cells were
incubated 24 h or 48 h prior to microinjection.
Microinjection experiments were conducted with a Femtojet® microinjector and an Injectman NI 2
micromanipulator (Eppendorf). All injections were performed in the cytoplasm. Directly following
microinjection, intracellular fluorescence measurements were carried out as described below.
Preparation of cytosolic and nuclear cell extracts
Cytosolic and nuclear cell fractions were derived from VERO cells according to a slightly modified BD
Bioscience Transfactor Extraction protocol. Cells were harvested at high confluency by trypsinization
and centrifuged at 450 g for 5 min (4°C). Subsequently, the cell pellets were rinsed twice by
resuspension in cold phosphate buffered saline (PBS), again collected and redispersed in a hypotonic
lysis buffer [10 mM HEPES pH 7.9, 1.5 mM MgCl2 and 10 mM KCl, supplemented with Complete Mini
EDTA‐free Protease Inhibitor Cocktail (Roche)]. Following incubation on ice (15 min) and subsequent
centrifugation (5 min at 420 g, 4°C), the cells were again dispersed in lysis buffer equal to twice the
cell pellet volume. The resulting cell suspension was slowly drawn into a narrow‐gauge syringe and
rapidly ejected to disrupt the outer membrane (disruption step was repeated 10 times). Nuclei were
pelleted by centrifugation of the lysed cells at 11000 g for 20 min. The supernatant (cytosolic extract)
was collected and stored in small aliquots at ‐80°C.
For preparation of the nuclear extract, the remaining nuclear pellet was resuspended in nuclear
extraction buffer (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, 25% (v/v)
glycerol) again supplemented with Complete EDTA‐free Protease Inhibitor Cocktail in appropriate
concentration. Following disruption of the nuclei (using a new syringe), the nuclear suspension was
64

in situ analysis of single‐stranded and duplex siRNA integrity in living cells | CHAPTER 2
shaken gently for 30 min on ice and then centrifuged at 18000 g for 5 min. The supernatant (nuclear
extract) was stored in small aliquots at ‐80°C.
Dual‐colour fluorescence fluctuation spectroscopy setup
Fluorescence measurements were performed on a dual‐colour FFS setup installed on a MRC1024 Bio‐
Rad confocal laser scanning microscope (depicted in Figure 2.1b). An inverted microscope (Eclipse
TE300D, Nikon), equipped with a water‐immersion objective lens (Plan Apo 60x, NA 1.2, collar‐rim
correction, Nikon) was employed. The 488 nm and 647 nm laser lines of a krypton‐argon laser (Bio‐
Rad) were used to excite the RhoGr and Cy5 dye respectively and their emission signal was registered
by ultrasensitive Avalanche photodiode detectors. The fluorescence intensity fluctuations were
recorded on a digital ALV 5000/E correlator. The FFS setup was calibrated as described by Schwille et
al. [19] and summarized by Remaut et al. [20] to optimize the overlap of the excitation and detection
volumes and to determine the size of the latter.
A detailed outlining of the theoretical concept and applications of FFS can be found elsewhere.[21,22]
In this study the dual‐colour FFS setup is mainly employed to measure the fluorescence intensities of
labelled siRNA present at nanomolar concentrations. Additional correlation analysis of the
fluorescence fluctuations to obtain information on the mobility of the siRNA molecules is not
performed. Therefore, the mathematical basics and diffusion models to be applied on FFS data will
not be covered in this chapter.
siRNA stability analysis
Fluorescence resonance energy transfer (FRET) is an important research tool to study inter‐ and
intramolecular processes and is defined as the non‐radiative excitation energy transfer from a donor
fluorophore to an appropriate acceptor fluorophore when both dyes are fixed in close proximity
(typically ~10‐100 Å).[23] The energy transfer efficiency is inversely proportional to the sixth power of
the inter‐dye distance.[23] Hence, virtually any process that induces distance variations between
donor and acceptor on the nanometer scale can be monitored with this technique.[24] In this report,
FRET is used as a tool to study the degradation of double labelled siRNAs (see results section). Intact
nucleic acid molecules, bearing both donor and acceptor, show a significant acceptor emission upon
donor excitation due to energy transfer, as schematically illustrated in Figure 2.1a. When the double
labelled substrates are cleaved into single labelled products, FRET can no longer occur resulting in
negligible acceptor fluorescence. Basically, the ratio of the 5’‐Cy5 fluorescence (acceptor
fluorophore) to the 3’‐RhoGr fluorescence (donor fluorophore) upon donor excitation (termed FA/FD)
65

CHAPTER 2 | in situ analysis of single‐stranded and duplex siRNA integrity in living cells
correlates with siRNA integrity (see results section). For the determination of the FA/FD ratio on
degrading siRNAs, the fluorescence intensities of RhoGr and Cy5 were recorded by the “green” and
“red” detector respectively (Figure 2.1b) during 3 seconds, with laser excitation set at 488 nm. All
samples were analyzed in glass‐bottomed 96‐well plates (Greiner Bio‐One, certified DNase/RNase
free) and the focal volume was placed 50 µm above the bottom of the wells.
Degradation experiments with RNase A (RPA grade, manufactured from bovine pancreas, Ambion)
were performed at ambient temperature by first diluting the siRNA stock solutions with RNase A
buffer (100 mM Tris‐acetate pH 6.5, 1 mM EDTA) to a final concentration of 13 nM. Subsequently, 1
µL of RNase A solution was added and FA/FD was measured as a function of time.
To monitor the degradation of siRNAs in cytosolic and nuclear extracts of VERO cells, a siRNA solution
(400 nM) in “degradation buffer” (20 mM Tris‐HCl, 50 mM Na‐acetate, 2 mM Mg‐acetate, adjusted
with sodium hydroxide to pH 7.4) was mixed with 50% (v/v) of cellular extract and incubated at 37°C
in non‐stick RNase free microfuge tubes (Ambion). Aliquots were taken at different time points and
diluted with sodium citrate buffer to stop degradation (for dsRNA degradation: 10 mM sodium
citrate pH 4, 0.025 % sodium dodecylsulphate (SDS); for ssRNA degradation: 10 mM sodium citrate
pH 6, 20 mM DTT, 0.1 % SDS). Samples (end concentration 13 nM) were analyzed immediately or
incubated on ice prior to FA/FD measurements.
To monitor the stability of siRNA in VERO cells, first confocal images were taken of the cells and the
FFS observation volume was positioned inside the nucleus. Solutions of intact ssRNA (10 µM), intact
dsRNA (5 µM), (RNase A) degraded ssRNA (10 µM) and (RNase A) degraded dsRNA (6 µM) were
prepared in RNase‐free water and loaded in a microinjection needle (Eppendorf II). Subsequently,
cells were microinjected in the cytoplasm. Approximately 30 to 60 sec after injection, the cells were
exposed to 488 nm laser light and the FA/FD ratio was determined in the nucleus. Low laser excitation
power and short measurement time ensured minimal photobleaching and cellular damage. For time‐
dependent stability analysis, the FA/FD ratio was registered inside the nucleus at different time‐points
after injection. At the end of each data series we verified that the nucleus was still in focus.
Polyacrylamide gel electrophoresis combined with dual‐colour FFS
27.5 µL siRNA duplex (8 µM) was added to 0.48 µg RNase A in 12.5 µL RNase A buffer and incubated
at room temperature. At specified times, 5 µL aliquots (for gel electrophoresis, ~ 0.4 µg siRNA) and
0.5 µL aliquots (for dual‐colour FFS) were removed. Electrophoresis aliquots were mixed with 10 µL
formamide (with 0.05% bromophenol blue), snap frozen at ‐80°C and stored afterwards at ‐20°C. A
20% polyacrylamide gel, supplemented with 7 M urea in TBE buffer (0.089 M Tris base, 0.089 M boric
66

in situ analysis of single‐stranded and duplex siRNA integrity in living cells | CHAPTER 2
acid (pH 8.35), 2 mM Na2EDTA), was applied to analyze siRNA degradation (40 min run at 100V).
Following electrophoresis, the gel was stained (40 min) in a 1:104 dilution of SYBR Green II RNA stain
(Molecular Probes) in DEPC‐treated water and samples were visualized on a UV Trans‐illuminator.
The 0.5 µL aliquots were added to 7 µL of RNase A buffer and also snap frozen at ‐80°C. The FA/FD
ratio (upon 488 nm excitation) of every aliquot was calculated.
RESULTS
FRET as a tool to distinguish intact and degraded single‐stranded and duplex siRNA
Figure 2.2 shows representative fluorescence fluctuation profiles as obtained for intact ssRNA (Figure
2.2a) and intact dsRNA (Figure 2.2b) in DEPC‐treated water, upon 488 nm excitation. Although with
these settings only rhodamine green (RhoGr) can be efficiently excited, the Cy5 fluorescence (red
signal) markedly exceeds the RhoGr fluorescence (green signal). This clearly indicates that energy
from the RhoGr excited state (donor) is transferred to and excites the Cy5 acceptor dye (FRET). The
ratio of acceptor fluorescence to donor fluorescence upon 488 nm excitation (FA/FD ratio) is used
here as a measure for energy transfer (see Materials and Methods). Table 2.1 summarizes the FA/FD
values as measured for ssRNA and dsRNA. Both intact ssRNA and dsRNA display a FA/FD ratio >> 1 in
DEPC‐treated water which is mainly due to FRET. Upon annealing with non‐labelled complementary
siRNA, the FA/FD ratio of ssRNA clearly lowers, indicating that the energy transfer is partially inhibited
in the duplex form. This is probably explained by a higher rigidity of the duplex structure which
makes the relative position of both dyes less optimal for FRET. Also, the FA/FD value seems to depend
on the medium in which the siRNA is diluted (Table 2.1). When the siRNA is placed in RNase A buffer
or sodium citrate buffer, a lower FA/FD ratio was measured in comparison with FA/FD in DEPC‐treated
water (Table 2.1), indicating that in the buffer media energy transfer is significantly diminished.
Fluorescence fluctuations (upon 488 nm excitation) of degraded siRNA samples are shown in Figure
2.2c and 2.2d. In contrast with the fluorescence fluctuation profiles of intact siRNA samples, now the
RhoGr emission becomes dominant. This red‐to‐green shift is due to the degradation of the siRNAs
which separates donor and acceptor dye, thus precluding energy transfer (Figure 2.1a). SiRNA
degradation is also clearly reflected in the FA/FD values (Table 2.1). Incubation of siRNA in the
presence of RNase A and cellular extracts (derived from VERO cells) does no longer allow FRET and
significantly lowers the FA/FD ratio (FA/FD << 1).
67
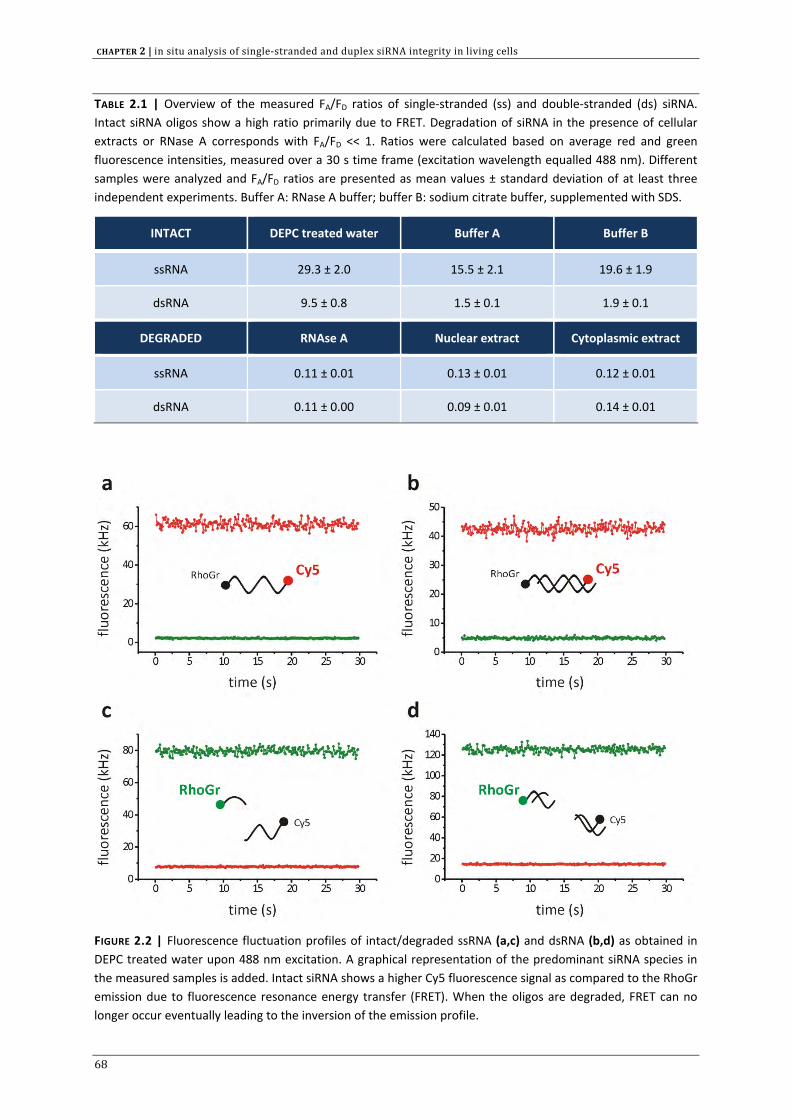
CHAPTER 2 | in situ analysis of single‐stranded and duplex siRNA integrity in living cells
TABLE 2.1 | Overview of the measured FA/FD ratios of single‐stranded (ss) and double‐stranded (ds) siRNA. Intact siRNA oligos show a high ratio primarily due to FRET. Degradation of siRNA in the presence of cellular extracts or RNase A corresponds with FA/FD << 1. Ratios were calculated based on average red and green fluorescence intensities, measured over a 30 s time frame (excitation wavelength equalled 488 nm). Different samples were analyzed and FA/FD ratios are presented as mean values ± standard deviation of at least three independent experiments. Buffer A: RNase A buffer; buffer B: sodium citrate buffer, supplemented with SDS.
INTACT DEPC treated water Buffer A Buffer B
ssRNA 29.3 ± 2.0 15.5 ± 2.1 19.6 ± 1.9
dsRNA 9.5 ± 0.8 1.5 ± 0.1 1.9 ± 0.1
DEGRADED RNAse A Nuclear extract Cytoplasmic extract
ssRNA 0.11 ± 0.01 0.13 ± 0.01 0.12 ± 0.01
dsRNA 0.11 ± 0.00 0.09 ± 0.01 0.14 ± 0.01
FIGURE 2.2 | Fluorescence fluctuation profiles of intact/degraded ssRNA (a,c) and dsRNA (b,d) as obtained in DEPC treated water upon 488 nm excitation. A graphical representation of the predominant siRNA species in the measured samples is added. Intact siRNA shows a higher Cy5 fluorescence signal as compared to the RhoGr emission due to fluorescence resonance energy transfer (FRET). When the oligos are degraded, FRET can no longer occur eventually leading to the inversion of the emission profile.
68
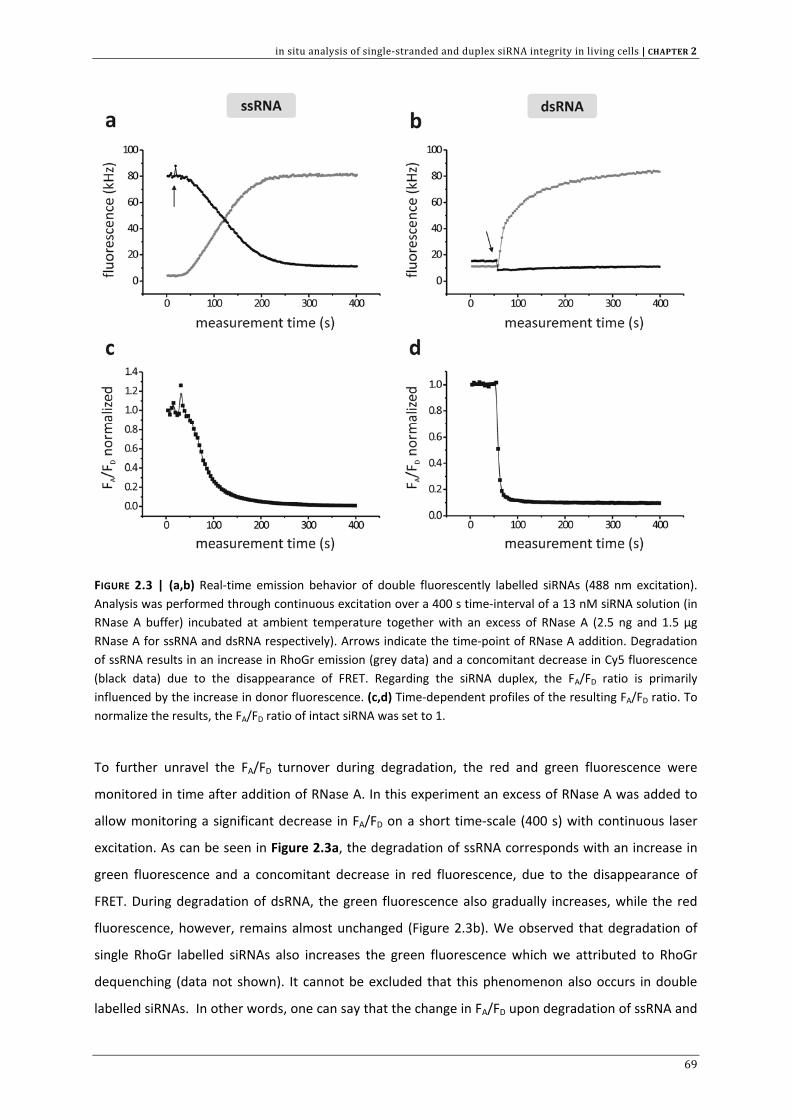
in situ analysis of single‐stranded and duplex siRNA integrity in living cells | CHAPTER 2
FIGURE 2.3 | (a,b) Real‐time emission behavior of double fluorescently labelled siRNAs (488 nm excitation). Analysis was performed through continuous excitation over a 400 s time‐interval of a 13 nM siRNA solution (in RNase A buffer) incubated at ambient temperature together with an excess of RNase A (2.5 ng and 1.5 µg RNase A for ssRNA and dsRNA respectively). Arrows indicate the time‐point of RNase A addition. Degradation of ssRNA results in an increase in RhoGr emission (grey data) and a concomitant decrease in Cy5 fluorescence (black data) due to the disappearance of FRET. Regarding the siRNA duplex, the FA/FD ratio is primarily influenced by the increase in donor fluorescence. (c,d) Time‐dependent profiles of the resulting FA/FD ratio. To normalize the results, the FA/FD ratio of intact siRNA was set to 1.
To further unravel the FA/FD turnover during degradation, the red and green fluorescence were
monitored in time after addition of RNase A. In this experiment an excess of RNase A was added to
allow monitoring a significant decrease in FA/FD on a short time‐scale (400 s) with continuous laser
excitation. As can be seen in Figure 2.3a, the degradation of ssRNA corresponds with an increase in
green fluorescence and a concomitant decrease in red fluorescence, due to the disappearance of
FRET. During degradation of dsRNA, the green fluorescence also gradually increases, while the red
fluorescence, however, remains almost unchanged (Figure 2.3b). We observed that degradation of
single RhoGr labelled siRNAs also increases the green fluorescence which we attributed to RhoGr
dequenching (data not shown). It cannot be excluded that this phenomenon also occurs in double
labelled siRNAs. In other words, one can say that the change in FA/FD upon degradation of ssRNA and
69
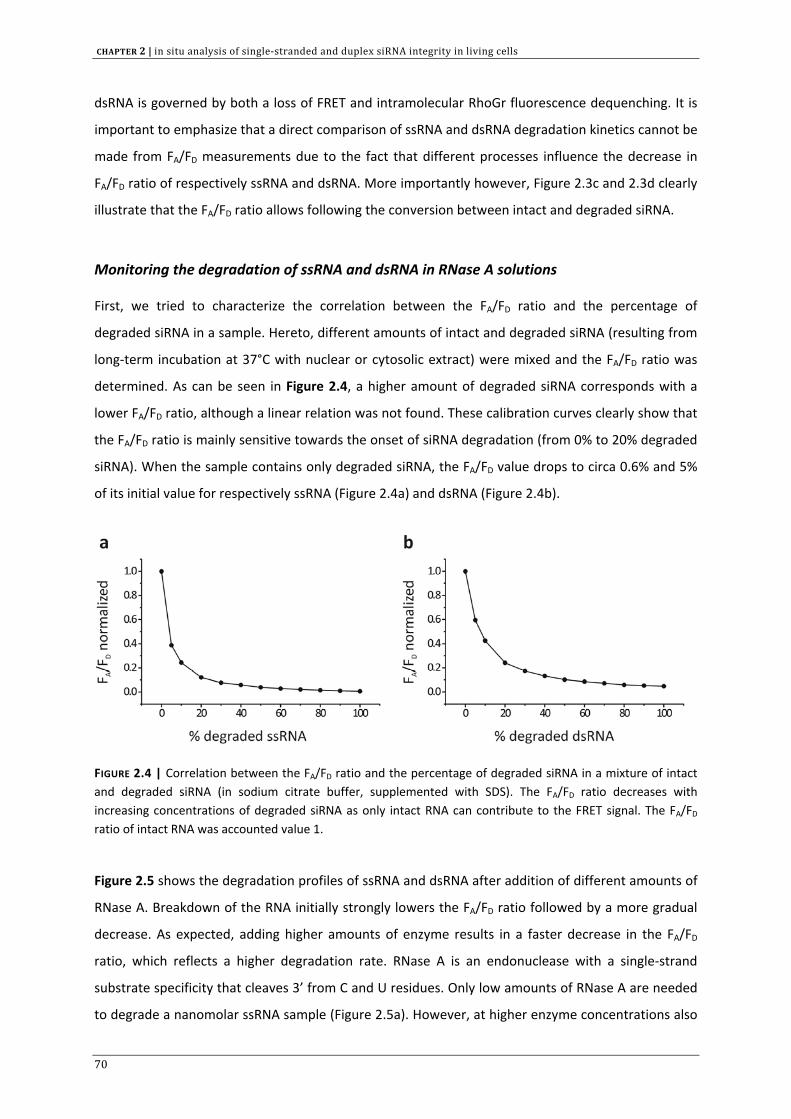
CHAPTER 2 | in situ analysis of single‐stranded and duplex siRNA integrity in living cells
dsRNA is governed by both a loss of FRET and intramolecular RhoGr fluorescence dequenching. It is
important to emphasize that a direct comparison of ssRNA and dsRNA degradation kinetics cannot be
made from FA/FD measurements due to the fact that different processes influence the decrease in
FA/FD ratio of respectively ssRNA and dsRNA. More importantly however, Figure 2.3c and 2.3d clearly
illustrate that the FA/FD ratio allows following the conversion between intact and degraded siRNA.
Monitoring the degradation of ssRNA and dsRNA in RNase A solutions
First, we tried to characterize the correlation between the FA/FD ratio and the percentage of
degraded siRNA in a sample. Hereto, different amounts of intact and degraded siRNA (resulting from
long‐term incubation at 37°C with nuclear or cytosolic extract) were mixed and the FA/FD ratio was
determined. As can be seen in Figure 2.4, a higher amount of degraded siRNA corresponds with a
lower FA/FD ratio, although a linear relation was not found. These calibration curves clearly show that
the FA/FD ratio is mainly sensitive towards the onset of siRNA degradation (from 0% to 20% degraded
siRNA). When the sample contains only degraded siRNA, the FA/FD value drops to circa 0.6% and 5%
of its initial value for respectively ssRNA (Figure 2.4a) and dsRNA (Figure 2.4b).
FIGURE 2.4 | Correlation between the FA/FD ratio and the percentage of degraded siRNA in a mixture of intact and degraded siRNA (in sodium citrate buffer, supplemented with SDS). The FA/FD ratio decreases with increasing concentrations of degraded siRNA as only intact RNA can contribute to the FRET signal. The FA/FD ratio of intact RNA was accounted value 1.
Figure 2.5 shows the degradation profiles of ssRNA and dsRNA after addition of different amounts of
RNase A. Breakdown of the RNA initially strongly lowers the FA/FD ratio followed by a more gradual
decrease. As expected, adding higher amounts of enzyme results in a faster decrease in the FA/FD
ratio, which reflects a higher degradation rate. RNase A is an endonuclease with a single‐strand
substrate specificity that cleaves 3’ from C and U residues. Only low amounts of RNase A are needed
to degrade a nanomolar ssRNA sample (Figure 2.5a). However, at higher enzyme concentrations also
70
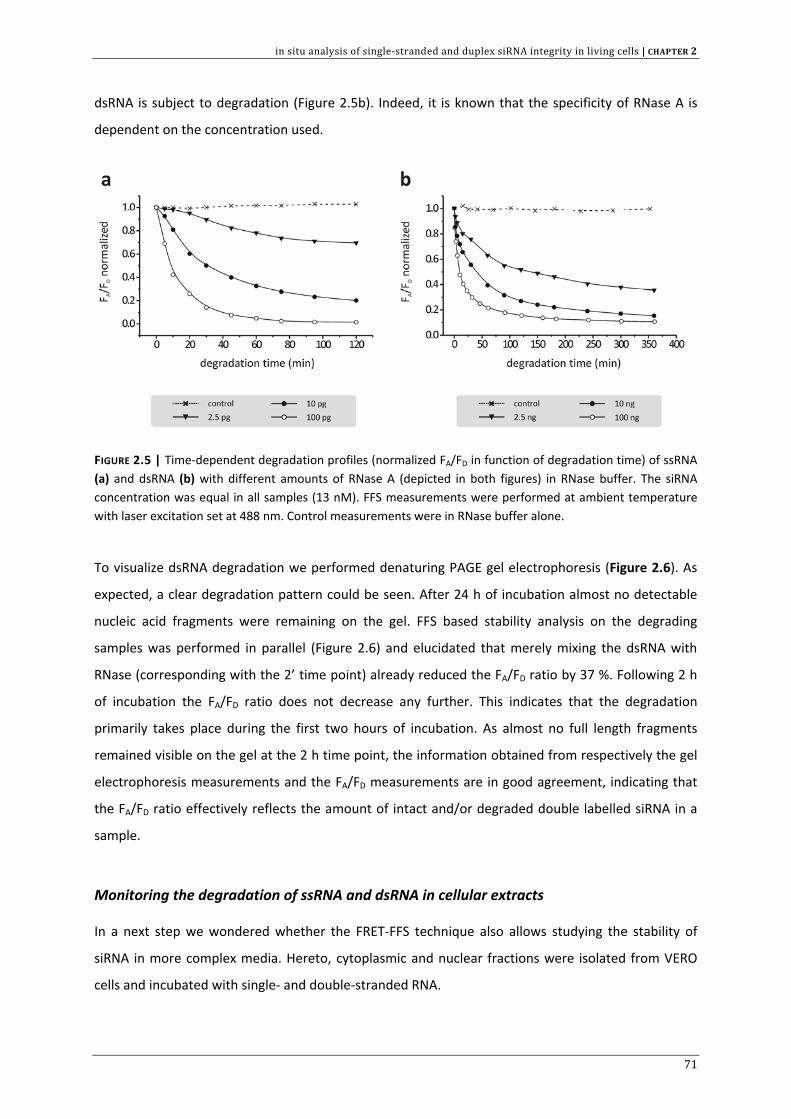
in situ analysis of single‐stranded and duplex siRNA integrity in living cells | CHAPTER 2
dsRNA is subject to degradation (Figure 2.5b). Indeed, it is known that the specificity of RNase A is
dependent on the concentration used.
FIGURE 2.5 | Time‐dependent degradation profiles (normalized FA/FD in function of degradation time) of ssRNA (a) and dsRNA (b) with different amounts of RNase A (depicted in both figures) in RNase buffer. The siRNA concentration was equal in all samples (13 nM). FFS measurements were performed at ambient temperature with laser excitation set at 488 nm. Control measurements were in RNase buffer alone.
To visualize dsRNA degradation we performed denaturing PAGE gel electrophoresis (Figure 2.6). As
expected, a clear degradation pattern could be seen. After 24 h of incubation almost no detectable
nucleic acid fragments were remaining on the gel. FFS based stability analysis on the degrading
samples was performed in parallel (Figure 2.6) and elucidated that merely mixing the dsRNA with
RNase (corresponding with the 2’ time point) already reduced the FA/FD ratio by 37 %. Following 2 h
of incubation the FA/FD ratio does not decrease any further. This indicates that the degradation
primarily takes place during the first two hours of incubation. As almost no full length fragments
remained visible on the gel at the 2 h time point, the information obtained from respectively the gel
electrophoresis measurements and the FA/FD measurements are in good agreement, indicating that
the FA/FD ratio effectively reflects the amount of intact and/or degraded double labelled siRNA in a
sample.
Monitoring the degradation of ssRNA and dsRNA in cellular extracts
In a next step we wondered whether the FRET‐FFS technique also allows studying the stability of
siRNA in more complex media. Hereto, cytoplasmic and nuclear fractions were isolated from VERO
cells and incubated with single‐ and double‐stranded RNA.
71
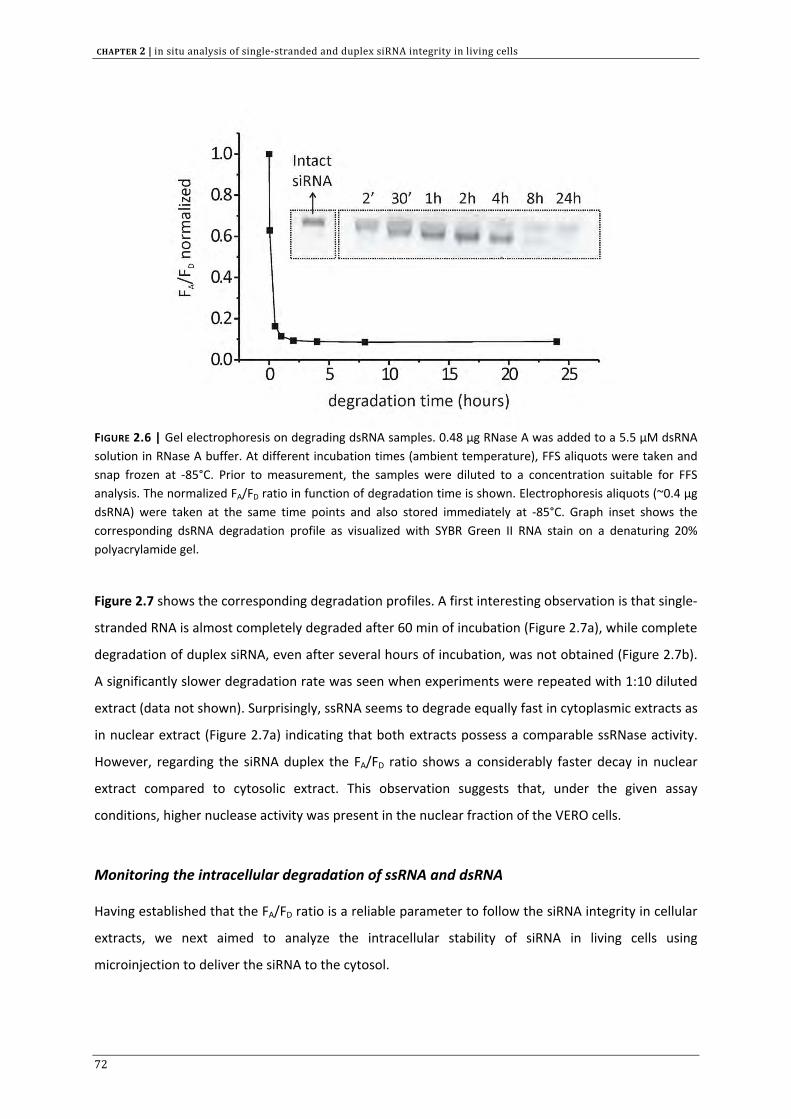
CHAPTER 2 | in situ analysis of single‐stranded and duplex siRNA integrity in living cells
FIGURE 2.6 | Gel electrophoresis on degrading dsRNA samples. 0.48 µg RNase A was added to a 5.5 µM dsRNA solution in RNase A buffer. At different incubation times (ambient temperature), FFS aliquots were taken and snap frozen at ‐85°C. Prior to measurement, the samples were diluted to a concentration suitable for FFS analysis. The normalized FA/FD ratio in function of degradation time is shown. Electrophoresis aliquots (~0.4 µg dsRNA) were taken at the same time points and also stored immediately at ‐85°C. Graph inset shows the corresponding dsRNA degradation profile as visualized with SYBR Green II RNA stain on a denaturing 20% polyacrylamide gel.
Figure 2.7 shows the corresponding degradation profiles. A first interesting observation is that single‐
stranded RNA is almost completely degraded after 60 min of incubation (Figure 2.7a), while complete
degradation of duplex siRNA, even after several hours of incubation, was not obtained (Figure 2.7b).
A significantly slower degradation rate was seen when experiments were repeated with 1:10 diluted
extract (data not shown). Surprisingly, ssRNA seems to degrade equally fast in cytoplasmic extracts as
in nuclear extract (Figure 2.7a) indicating that both extracts possess a comparable ssRNase activity.
However, regarding the siRNA duplex the FA/FD ratio shows a considerably faster decay in nuclear
extract compared to cytosolic extract. This observation suggests that, under the given assay
conditions, higher nuclease activity was present in the nuclear fraction of the VERO cells.
Monitoring the intracellular degradation of ssRNA and dsRNA
Having established that the FA/FD ratio is a reliable parameter to follow the siRNA integrity in cellular
extracts, we next aimed to analyze the intracellular stability of siRNA in living cells using
microinjection to deliver the siRNA to the cytosol.
72
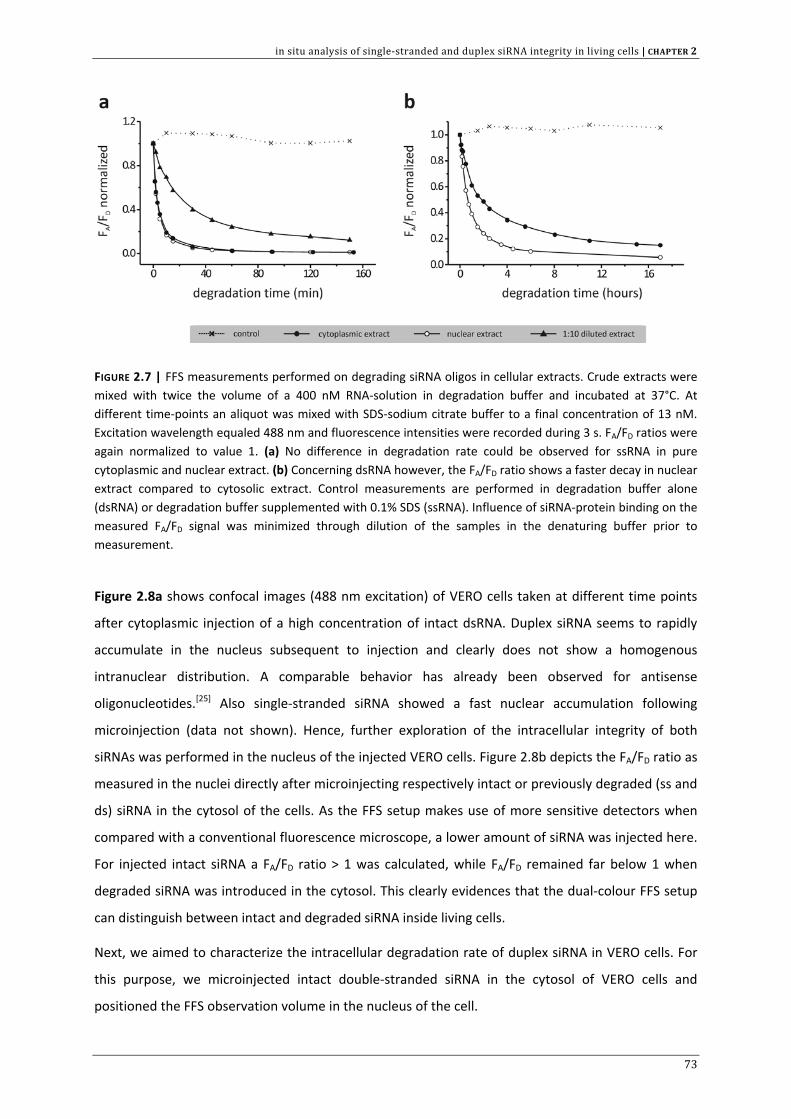
in situ analysis of single‐stranded and duplex siRNA integrity in living cells | CHAPTER 2
FIGURE 2.7 | FFS measurements performed on degrading siRNA oligos in cellular extracts. Crude extracts were mixed with twice the volume of a 400 nM RNA‐solution in degradation buffer and incubated at 37°C. At different time‐points an aliquot was mixed with SDS‐sodium citrate buffer to a final concentration of 13 nM. Excitation wavelength equaled 488 nm and fluorescence intensities were recorded during 3 s. FA/FD ratios were again normalized to value 1. (a) No difference in degradation rate could be observed for ssRNA in pure cytoplasmic and nuclear extract. (b) Concerning dsRNA however, the FA/FD ratio shows a faster decay in nuclear extract compared to cytosolic extract. Control measurements are performed in degradation buffer alone (dsRNA) or degradation buffer supplemented with 0.1% SDS (ssRNA). Influence of siRNA‐protein binding on the measured FA/FD signal was minimized through dilution of the samples in the denaturing buffer prior to measurement.
Figure 2.8a shows confocal images (488 nm excitation) of VERO cells taken at different time points
after cytoplasmic injection of a high concentration of intact dsRNA. Duplex siRNA seems to rapidly
accumulate in the nucleus subsequent to injection and clearly does not show a homogenous
intranuclear distribution. A comparable behavior has already been observed for antisense
oligonucleotides.[25] Also single‐stranded siRNA showed a fast nuclear accumulation following
microinjection (data not shown). Hence, further exploration of the intracellular integrity of both
siRNAs was performed in the nucleus of the injected VERO cells. Figure 2.8b depicts the FA/FD ratio as
measured in the nuclei directly after microinjecting respectively intact or previously degraded (ss and
ds) siRNA in the cytosol of the cells. As the FFS setup makes use of more sensitive detectors when
compared with a conventional fluorescence microscope, a lower amount of siRNA was injected here.
For injected intact siRNA a FA/FD ratio > 1 was calculated, while FA/FD remained far below 1 when
degraded siRNA was introduced in the cytosol. This clearly evidences that the dual‐colour FFS setup
can distinguish between intact and degraded siRNA inside living cells.
Next, we aimed to characterize the intracellular degradation rate of duplex siRNA in VERO cells. For
this purpose, we microinjected intact double‐stranded siRNA in the cytosol of VERO cells and
positioned the FFS observation volume in the nucleus of the cell.
73
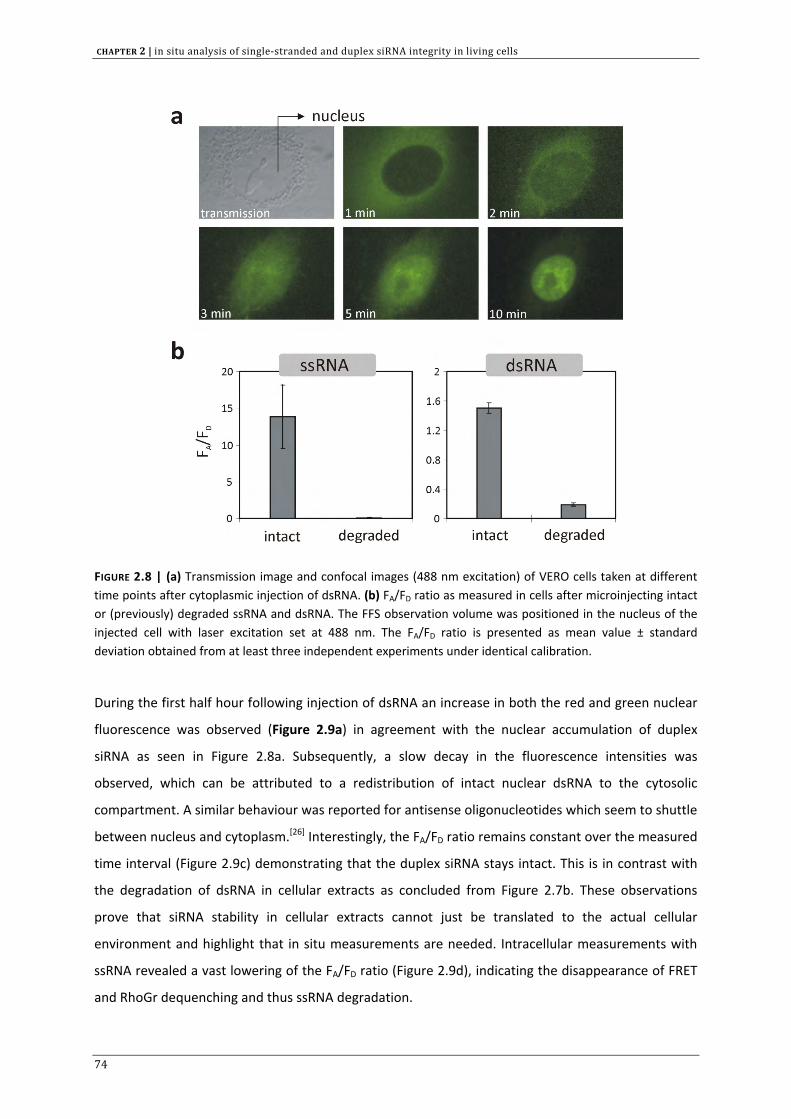
CHAPTER 2 | in situ analysis of single‐stranded and duplex siRNA integrity in living cells
FIGURE 2.8 | (a) Transmission image and confocal images (488 nm excitation) of VERO cells taken at different time points after cytoplasmic injection of dsRNA. (b) FA/FD ratio as measured in cells after microinjecting intact or (previously) degraded ssRNA and dsRNA. The FFS observation volume was positioned in the nucleus of the injected cell with laser excitation set at 488 nm. The FA/FD ratio is presented as mean value ± standard deviation obtained from at least three independent experiments under identical calibration.
During the first half hour following injection of dsRNA an increase in both the red and green nuclear
fluorescence was observed (Figure 2.9a) in agreement with the nuclear accumulation of duplex
siRNA as seen in Figure 2.8a. Subsequently, a slow decay in the fluorescence intensities was
observed, which can be attributed to a redistribution of intact nuclear dsRNA to the cytosolic
compartment. A similar behaviour was reported for antisense oligonucleotides which seem to shuttle
between nucleus and cytoplasm.[26] Interestingly, the FA/FD ratio remains constant over the measured
time interval (Figure 2.9c) demonstrating that the duplex siRNA stays intact. This is in contrast with
the degradation of dsRNA in cellular extracts as concluded from Figure 2.7b. These observations
prove that siRNA stability in cellular extracts cannot just be translated to the actual cellular
environment and highlight that in situ measurements are needed. Intracellular measurements with
ssRNA revealed a vast lowering of the FA/FD ratio (Figure 2.9d), indicating the disappearance of FRET
and RhoGr dequenching and thus ssRNA degradation.
74
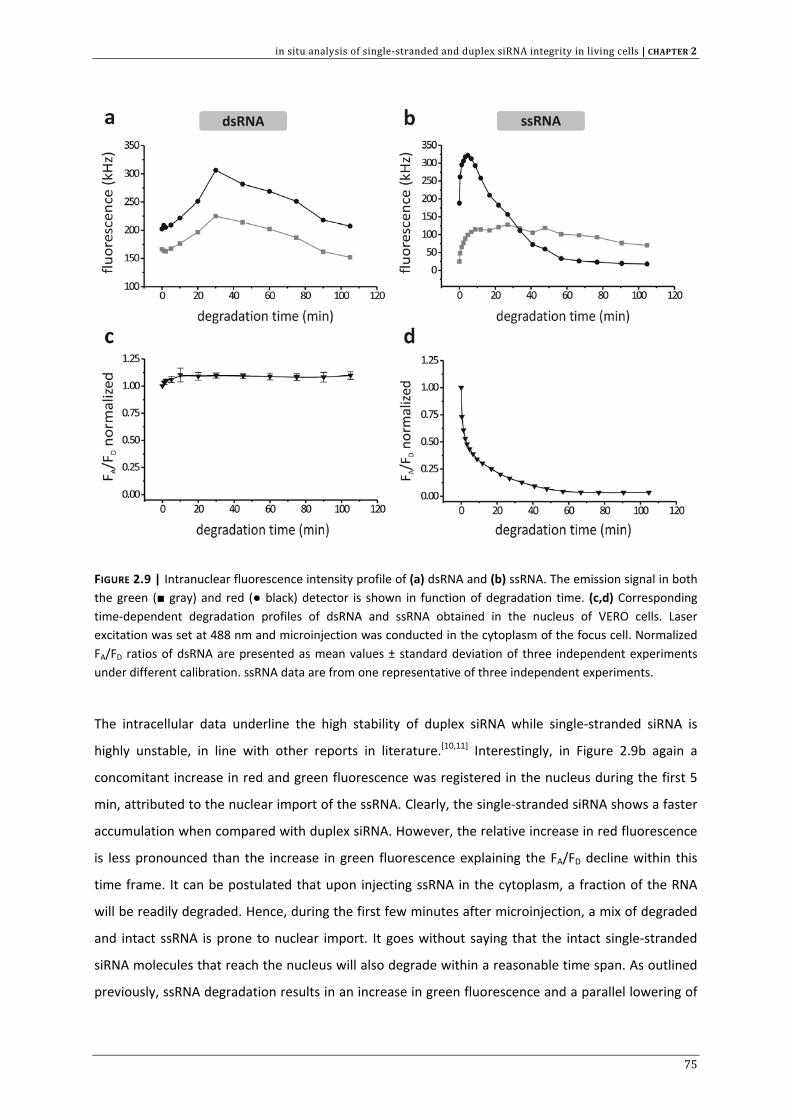
in situ analysis of single‐stranded and duplex siRNA integrity in living cells | CHAPTER 2
FIGURE 2.9 | Intranuclear fluorescence intensity profile of (a) dsRNA and (b) ssRNA. The emission signal in both the green (■ gray) and red (● black) detector is shown in function of degradation time. (c,d) Corresponding time‐dependent degradation profiles of dsRNA and ssRNA obtained in the nucleus of VERO cells. Laser excitation was set at 488 nm and microinjection was conducted in the cytoplasm of the focus cell. Normalized FA/FD ratios of dsRNA are presented as mean values ± standard deviation of three independent experiments under different calibration. ssRNA data are from one representative of three independent experiments.
The intracellular data underline the high stability of duplex siRNA while single‐stranded siRNA is
highly unstable, in line with other reports in literature.[10,11] Interestingly, in Figure 2.9b again a
concomitant increase in red and green fluorescence was registered in the nucleus during the first 5
min, attributed to the nuclear import of the ssRNA. Clearly, the single‐stranded siRNA shows a faster
accumulation when compared with duplex siRNA. However, the relative increase in red fluorescence
is less pronounced than the increase in green fluorescence explaining the FA/FD decline within this
time frame. It can be postulated that upon injecting ssRNA in the cytoplasm, a fraction of the RNA
will be readily degraded. Hence, during the first few minutes after microinjection, a mix of degraded
and intact ssRNA is prone to nuclear import. It goes without saying that the intact single‐stranded
siRNA molecules that reach the nucleus will also degrade within a reasonable time span. As outlined
previously, ssRNA degradation results in an increase in green fluorescence and a parallel lowering of
75

CHAPTER 2 | in situ analysis of single‐stranded and duplex siRNA integrity in living cells
the red fluorescence. As the red emission already drops at 5 min after injection (Figure 2.9b) we
conclude that little intact siRNAs still reach the nucleus, but that the siRNA molecules at this moment
are subject to intranuclear degradation. One can wonder why between 5 min and 50 min following
injection the green fluorescence in the nucleus remains more or less constant. Probably the loss of
intranuclear fluorescence due to nucleocytoplasmic redistribution of (intact and already degraded)
siRNA is countered by the increase in green fluorescence as a result of degradation. Approximately
60 min after injection, the FA/FD does no longer drop (Figure 2.9c), indicating that no further
degradation occurs. The slight decrease in red and green fluorescence is now completely attributed
to diffusion of siRNA fragments from the nucleus into the cytoplasm.
DISCUSSION
Many disease‐causing genes can be silenced efficiently in cell culture with rationally designed small
interfering RNAs under optimized transfection conditions. However, application of siRNA
therapeutics is not yet conceivable as siRNA delivery, biodistribution, target cell selectivity etc.
remain major hurdles to be overcome. Obviously, the therapeutic success of siRNA will also greatly
depend on the biostability of siRNA. Indeed, the siRNA molecules should be sufficiently stable in the
extra‐ and intracellular matrices to be able to exert a biological effect.[1,2,6]
Previous reports in the literature have studied degradation of synthetic RNA/DNA chimera [27] and
antisense oligonucleotides by FRET.[18] Also inside living cells, FRET has been used to evaluate e.g.
ribozyme cleavage of a labelled oligoribonucleotide substrate [28] and for detection of intact and
degraded oligonucleotides.[18,29] In a recent report, FRET was applied to track the degradation of
double‐labelled pDNA in the intracellular environment through confocal time‐lapse imaging.[30] Modi
et al. even describe the design of so called ‘DNA nanomachines’, bearing an appropriate FRET pair, as
intracellular pH sensors that are able to map pH changes during intracellular trafficking.[31] FRET as a
tool to evaluate siRNA integrity in particular is described in three independent reports in the
literature.[17,32,33]
In the presented study we first wanted to evaluate whether our dual‐colour FFS setup (as depicted in
Figure 2.1b) is suited to probe nucleolytic decay of double fluorescently labelled siRNAs which are
subject to FRET. Figures 2.3 to 2.7 clearly show that the FA/FD ratio, as obtained from the
fluorescence fluctuation data (measured through the FFS setup), can be applied to study the
transition from intact to degraded siRNA. As single‐stranded siRNA shows much higher FRET
efficiency than duplex siRNA (Table 2.1), this also allows us to distinguish between dsRNA
degradation and duplex strand denaturation. While dsRNA degradation results in a decrease of the
76

in situ analysis of single‐stranded and duplex siRNA integrity in living cells | CHAPTER 2
FA/FD ratio, destabilization of the intact siRNA duplex will eventually lead to an increase of the FA/FD
ratio. Indeed, release of the more flexible siRNA sense strand from the duplex structure will induce a
higher FRET signal.
Correlation analysis on the fluorescence fluctuation profiles would allow the calculation of the
diffusion coefficient of the fluorescently tagged molecules passing through the observation volume
of the confocal microscope.[19,21,22] However, this strategy is not recommended to monitor the
degradation of siRNA as a function of time.[20] Strictly, FFS can only distinguish between fluorescent
molecules with sufficiently different mobility i.e. when their diffusion coefficients (D) differ for at
least factor 1.6.[34] Considering D ~ 1/(MW)1/3, this implies that FFS could only differentiate between
intact and fully degraded siRNA. In contrast, FRET is sensitive to a single cleavage, irrespective of the
molecular weight (MW) of the degradation products formed.[20] It is also worth highlighting that in
this report end‐labelled siRNAs are used. It could be argued that the labels sterically interfere with
(primarily) exoribonuclease binding to the siRNA substrate thereby limiting the enzymatic activity
which would lead to an underestimation of the degradation rate.
Using a confocal FFS setup to register the fluorescence intensities is in particular advantageous for
quantifying the intracellular fate of siRNAs. Indeed, the confocal excitation volume can easily be
placed inside a defined cellular compartment. As the fluorescence photons emanating from this focal
spot are registered by ultrasensitive avalanche photodiode detectors this enables us to measure low
concentrations of fluorescent molecules which could provide a suitable alternative for confocal
imaging in the case the fluorescence signal becomes too dim to provide satisfactory CLSM images.[35]
Especially for future siRNA research this could be attractive as only low concentrations of highly
active siRNA are needed to show a biological effect. It is also known that fluorescence from
endogenous cellular components (autofluorescence) may interfere with FFS measurements in
mammalian cells, especially when excitation wavelengths in the blue spectral range (< 500 nm) are
used.[36] In particular, the autofluorescence signal may render the interpretation of intracellular data
challenging [37] especially when the fluorescently labelled molecules of interest are present at low
concentration. In this report, the intracellular fluorescence exceeded the autofluorescence at least 5‐
fold at every data point, making sure that the obtained FA/FD ratios really reflect the fate of the
injected siRNAs.
Confocal images showed nuclear accumulation of dsRNA when microinjected in the cytoplasm
(Figure 2.8a). Several reports, however, present evidence for the cytoplasmic nature of RNAi as
activated RISC complexes localize and function in the cytosol where translation of target mRNA takes
place.[38] Transfection of Hela cells with TAT47‐57‐siRNA conjugates, siRNA/lipofectamine nanoparticles
or siRNA/polyamidoamine nanoparticles revealed that the siRNAs preferentially locate in the
77

CHAPTER 2 | in situ analysis of single‐stranded and duplex siRNA integrity in living cells
perinuclear region, thought to contain focal points of RNAi machinery.[39] This specific subcellular
localization showed strong correlation with RNAi activity. A recent report, however, declared that
RNAi activity is also present in the nucleus of HeLa cells as functional RISC complexes were found in
both nuclear and cytoplasmic extracts.[40] Thus, upon transfection, small interfering RNA that is
released from its carrier presumably can enter the nucleus and form RISC* complexes on site to
cleave homologous nuclear RNA transcripts. It has also been shown in the literature that siRNAs can
induce transcriptional gene silencing in mammalian cells through DNA methylation, a process that
occurs in the nucleus, which is explained in more detail in the previous chapter.[41] Järve et al. further
substantiate our observations since these authors also mention the intranuclear transport of
microinjected siRNAs. Moreover, nuclear siRNA was detected following liposomal transfection as
well.[17] The siRNA duplexes that reach the nucleus are however quickly (~hours) relocated to the
cytoplasm through the RanGTP/Exportin 5 pathway [17,42], where they can localize to cytoplasmic P‐
bodies.[33,39] Many uncertainties on the intracellular fate of siRNA remain to exist and further
underline the need for advanced methods to investigate them. On the other hand, one should keep
in mind that the intracellular distribution of siRNAs will probably depend on how they are delivered
to the cytosol. It is indeed possible that the intranuclear accumulation of siRNA is influenced by
microinjection or transfection procedures.[17]
Intracellular FFS measurements showed substantial degradation of the single‐stranded RNA. In
contrast, the FA/FD ratio as measured for dsRNA remained unchanged for up to 2 h, emphasizing the
higher intrinsic stability of dsRNA (Figure 2.9c). Probably the low dsRNase activity in both the
cytoplasmic and nuclear compartment, as compared to the presence of single‐strand specific
nucleases, explains the discrepancy in the stability of ssRNA and dsRNA. A recent article on the
stability of native siRNAs in serum revealed that duplex siRNA is mainly degraded by enzymes of the
(ss)RNase A family.[43] Some of these enzymes can be termed single‐strand preferring, rather than
single‐strand specific, because they are also capable of degrading double‐stranded RNA, although
through a much lesser extent. The biochemical mechanism of this process has already been
described in the literature.[44] Inside living cells, there are numerous types of ribonucleases (exo‐ and
endoribonucleases) with different structures and catalytic function.[45] Because of the high
complexity and diversity of this class of molecules, it is difficult to predict the predominant
ribonuclease pathway involved in the intracellular degradation of siRNA. Because only one siRNA
sequence was studied in this chapter, it is not possible to draw any conclusions with regard to the
sequence‐dependent degradation of siRNA. However, on the basis of the fact that different articles in
literature suggest that single‐stranded siRNA degrades substantially faster than a siRNA
duplex,[3,10,11,46] we are quite confident that in general for native siRNAs the duplex structure will
78

in situ analysis of single‐stranded and duplex siRNA integrity in living cells | CHAPTER 2
show a higher stability in a biological environment compared to the stability of the single‐stranded
counterpart.
The observations in Figure 2.9 also raise the interesting question of how RISC incorporation of the
double‐labelled siRNA duplex would influence the observed FA/FD profile. Assembly of the
(unlabelled) guide strand into RISC, followed by unwinding of the duplex structure and release of the
(double labelled) passenger strand, would imply degradation of the latter.[47,48] As a consequence,
single labelled RNA fragments would emerge and cause a decrease in the FA/FD ratio, which is not
observed here. As blocking of the 5’‐end of the antisense strand can abolish RNAi activity and a free
terminal 5’‐phosphate is believed to be essential for RNAi activity,[42,49‐52] it is very unlikely that the 3’‐
5’ double labelled siRNA strand will act as guide strand. From this we eventually conclude that RISC
activation does not occur under these experimental conditions, thereby also partially explaining the
significant nuclear accumulation as we observed in our confocal microscopy experiments. It is a
possibility that the relatively high intracellular siRNA concentrations can lead to saturation of the
cellular compartments and the RNAi machinery, thereby masking the interaction of siRNA with RISC.
It could be hypothesized that the lower stability of ssRNA is partially responsible for less effective
gene silencing activity of antisense siRNA compared to duplex siRNA.[3,46] It has been suggested that
single‐stranded antisense siRNA can also directly enter the mammalian RNAi pathway with
subsequent depletion of endogenous gene expression [9,46,52] but still the duplex siRNA potency
generally exceeds the single‐stranded RNAi activity. Also, in most cases duplex siRNA shows a more
persistent gene silencing than antisense DNA oligonucleotides [1,2,12] which is, according to Bertrand
et al.,[53] primarily attributed to their difference in susceptibility for nuclease attack.
The stability data we present here have been gathered inside living cells after microinjection of the
siRNA molecules in the cytosol. Because no transfection procedure was applied, the intracellular
environment to which the microinjected siRNAs are exposed resembles that of siRNAs released from
a pharmaceutical nanocarrier (e.g. liposomes and polymeric nanoparticles) into the cytoplasm. As the
applied delivery system, together with incorporation of the antisense siRNA strand into RISC, could
provide extra protection of the siRNA against nuclease attack, especially the time frame between
release from its carrier and interaction with RISC will be crucial for the siRNA integrity inside the
intracellular matrix.[46] Our results suggest that intracellular degradation is much more critical for
ssRNA than for duplex siRNA. The observation that modified siRNA duplexes with enhanced nuclease
resistance do not always show longer lasting RNAi effect [13] supports the hypothesis that intracellular
stability is not the key issue for effective application of siRNA duplexes. It can be postulated that
upon delivery to the cytosol, non‐modified duplex siRNA is able to induce a response before
significant degradation occurs.[13] Bartlett et al. provided data on the anti‐luciferase activity of
79

CHAPTER 2 | in situ analysis of single‐stranded and duplex siRNA integrity in living cells
unmodified and nuclease‐stabilized siRNA duplexes after electroporation and observed no
differences in the amplitude nor the duration of the gene silencing effect.[54] These results strongly
suggest that once the siRNA duplexes reach the cell cytoplasm, nuclease degradation is absent or at
least not the limiting factor. Hence, further research into functional in vivo delivery systems that can
safely guide active siRNA to the cytosol of the target cell should be stimulated.
ACKNOWLEDGMENT
N. Opitz (Max Planck Institute for Molecular Physiology, Dortmund, Germany) is thanked with
gratitude for the installation of the FFS‐module on the MRC‐1024.
80

in situ analysis of single‐stranded and duplex siRNA integrity in living cells | CHAPTER 2
REFERENCE LIST
1. Dorsett Y. and Tuschl T., siRNAs: applications in functional genomics and potential as therapeutics, Nat. Rev. Drug Discov. 2004, 3: 318‐329.
2. Dykxhoorn D.M. and Lieberman J., The silent revolution: RNA interference as basic biology, research tool, and therapeutic, Annu. Rev. Med. 2005, 56: 401‐423.
3. Elbashir S.M., Lendeckel W., and Tuschl T., RNA interference is mediated by 21‐and 22‐nucleotide RNAs, Genes & Development 2001, 15: 188‐200.
4. Zamore P.D., Tuschl T., Sharp P.A., and Bartel D.P., RNAi: Double‐stranded RNA directs the ATP‐dependent cleavage of mRNA at 21 to 23 nucleotide intervals, Cell 2000, 101: 25‐33.
5. Elbashir S.M., Harborth J., Lendeckel W., Yalcin A. et al., Duplexes of 21‐nucleotide RNAs mediate RNA interference in cultured mammalian cells, Nature 2001, 411: 494‐498.
6. Schiffelers R.M., Woodle M.C., and Scaria P., Pharmaceutical prospects for RNA interference, Pharm. Res. 2004, 21: 1‐7.
7. Elmen J., Thonberg H., Ljungberg K., Frieden M. et al., Locked nucleic acid (LNA) mediated improvements in siRNA stability and functionality, Nucleic Acids Res. 2005, 33: 439‐447.
8. Urban‐Klein B., Werth S., Abuharbeid S., Czubayko F. et al., RNAi‐mediated gene‐targeting through systemic application of polyethylenimine (PEI)‐complexed siRNA in vivo, Gene Ther. 2005, 12: 461‐466.
9. Amarzguioui M., Holen T., Babaie E., and Prydz H., Tolerance for mutations and chemical modifications in a siRNA, Nucleic Acids Res. 2003, 31: 589‐595.
10. Braasch D.A., Jensen S., Liu Y., Kaur K. et al., RNA interference in mammalian cells by chemically‐modified RNA, Biochemistry 2003, 42: 7967‐7975.
11. Chiu Y.L. and Rana T.M., siRNA function in RNAi: a chemical modification analysis, RNA 2003, 9: 1034‐1048.
12. Czauderna F., Fechtner M., Dames S., Aygun H. et al., Structural variations and stabilising modifications of synthetic siRNAs in mammalian cells, Nucleic Acids Res. 2003, 31: 2705‐2716.
13. Layzer J.M., McCaffrey A.P., Tanner A.K., Huang Z. et al., In vivo activity of nuclease‐resistant siRNAs, RNA‐A Publication of the Rna Society 2004, 10: 766‐771.
14. Volkov A.A., Kruglova N.S., Meschaninova M.I., Venyaminova A.G. et al., Selective protection of nuclease‐sensitive sites in siRNA prolongs silencing effect, Oligonucleotides. 2009, 19: 191‐202.
15. Mantei A., Rutz S., Janke M., Kirchhoff D. et al., siRNA stabilization prolongs gene knockdown in primary T lymphocytes, Eur. J. Immunol. 2008, 38: 2616‐2625.
16. Remaut K., Lucas B., Braeckmans K., Sanders N.N. et al., Protection of oligonucleotides against nucleases by pegylated and non‐pegylated liposomes as studied by fluorescence correlation spectroscopy, J. Control Release 2005, 110: 212‐226.
17. Jarve A., Muller J., Kim I.H., Rohr K. et al., Surveillance of siRNA integrity by FRET imaging, Nucleic Acids Res. 2007, 35: e124.
18. De Smedt S.C., Remaut K., Lucas B., Braeckmans K. et al., Studying biophysical barriers to DNA delivery by advanced light microscopy, Adv. Drug Deliv. Rev. 2005, 57: 191‐210.
19. Schwille P., Meyer‐Almes F.J., and Rigler R., Dual‐color fluorescence cross‐correlation spectroscopy for multicomponent diffusional analysis in solution, Biophys. J. 1997, 72: 1878‐1886.
20. Remaut K., Lucas B., Braeckmans K., Sanders N.N. et al., FRET‐FCS as a tool to evaluate the stability of oligonucleotide drugs after intracellular delivery, J. Control Release 2005, 103: 259‐271.
21. Elson E.S. and Magde D., Fluorescence correlation spectroscopy: I. Conceptual basis and theory, Biopolymers 1974, 13: 1‐27.
22. Fluorescence Correlation Spectroscopy: Theory and Applications (2001) (Rigler, R. and Elson, E.L., eds.), Springer‐Verlag Berlin‐Heidelberg, Germany.
23. Clegg R.M., Fluorescence resonance energy transfer and nucleic acids, Methods Enzymol. 1992, 211: 353‐388.
24. Selvin P.R., The renaissance of fluorescence resonance energy transfer, Nature Structural Biology 2000, 7: 730‐734.
25. Leonetti J.P., Mechti N., Degols G., Gagnor C. et al., Intracellular‐Distribution of Microinjected Antisense Oligonucleotides, Proceedings of the National Academy of Sciences of the United States of America 1991, 88: 2702‐2706.
26. Lorenz P., Misteli T., Baker B.F., Bennett C.F. et al., Nucleocytoplasmic shuttling: a novel in vivo property of antisense phosphorothioate oligodeoxynucleotides, Nucleic Acids Research 2000, 28: 582‐592.
81

CHAPTER 2 | in situ analysis of single‐stranded and duplex siRNA integrity in living cells
27. Uhler S.A., Cai D., Man Y., Figge C. et al., RNA degradation in cell extracts: real‐time monitoring by fluorescence resonance energy transfer, J. AM. CHEM. SOC. 2003, 125: 14230‐14231.
28. Vitiello D., Pecchia D.B., and Burke J.M., Intracellular ribozyme‐catalyzed trans‐cleavage of RNA monitored by fluorescence resonance energy transfer, RNA 2000, 6: 628‐637.
29. Uchiyama H., Hirano K., Kashiwasake‐Jibu M., and Taira K., Detection of undegraded oligonucleotides in vivo by fluorescence resonance energy transfer. Nuclease activities in living sea urchin eggs, J. Biol. Chem. 1996, 271: 380‐384.
30. Srinivasan C., Siddiqui S., Silbart L.K., Papadimitrakopoulos F. et al., Dual fluorescent labeling method to visualize plasmid DNA degradation, Bioconjug. Chem. 2009, 20: 163‐169.
31. Modi S., M G S, Goswami D., Gupta G.D. et al., A DNA nanomachine that maps spatial and temporal pH changes inside living cells, Nat. Nanotechnol. 2009, 4: 325‐330.
32. Chiu Y.L., Dinesh C.U., Chu C.Y., Ali A. et al., Dissecting RNA‐interference pathway with small molecules, Chem. Biol. 2005, 12: 643‐648.
33. Jagannath A. and Wood M.J., Localization of double‐stranded small interfering RNA to cytoplasmic processing bodies is Ago2 dependent and results in up‐regulation of GW182 and Argonaute‐2, Mol. Biol. Cell 2009, 20: 521‐529.
34. Meseth U., Wohland T., Rigler R., and Vogel H., Resolution of fluorescence correlation measurements, Biophys. J. 1999, 76: 1619‐1631.
35. Remaut K., Lucas B., Raemdonck K., Braeckmans K. et al., Can we better understand the intracellular behavior of DNA nanoparticles by fluorescence correlation spectroscopy?, J. Control Release 2007, 121: 49‐63.
36. Schwille P., Fluorescence correlation spectroscopy and its potential for intracellular applications, Cell Biochem. Biophys. 2001, 34: 383‐408.
37. Brock R., Hink M.A., and Jovin T.M., Fluorescence correlation microscopy of cells in the presence of autofluorescence, Biophys. J. 1998, 75: 2547‐2557.
38. Zeng Y. and Cullen B.R., RNA interference in human cells is restricted to the cytoplasm, RNA. 2002, 8: 855‐860.
39. Chiu Y.L., Ali A., Chu C.Y., Cao H. et al., Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells, Chem. Biol. 2004, 11: 1165‐1175.
40. Robb G.B., Brown K.M., Khurana J., and Rana T.M., Specific and potent RNAi in the nucleus of human cells, Nat. Struct. Mol. Biol. 2005, 12: 133‐137.
41. Matzke M.A. and Birchler J.A., RNAi‐mediated pathways in the nucleus, Nature Reviews Genetics 2005, 6: 24‐35.
42. Ohrt T., Merkle D., Birkenfeld K., Echeverri C.J. et al., In situ fluorescence analysis demonstrates active siRNA exclusion from the nucleus by Exportin 5, Nucleic Acids Research 2006, 34: 1369‐1380.
43. Haupenthal J., Baehr C., Kiermayer S., Zeuzem S. et al., Inhibition of RNAse A family enzymes prevents degradation and loss of silencing activity of siRNAs in serum, Biochemical Pharmacology 2006, 71: 702‐710.
44. Sorrentino S., Naddeo M., Russo A., and D'Alessio G., Degradation of double‐stranded RNA by human pancreatic ribonuclease: Crucial role of noncatalytic basic amino acid residues, Biochemistry 2003, 42: 10182‐10190.
45. Deutscher M.P. and Li Z.W., Exoribonucleases and their multiple roles in RNA metabolism, Prog. Nucleic Acid Res. Mol. Biol. 2001, 67‐105.
46. Holen T., Amarzguioui M., Babaie E., and Prydz H., Similar behaviour of single‐strand and double‐strand siRNAs suggests they act through a common RNAi pathway, Nucleic Acids Res. 2003, 31: 2401‐2407.
47. Leuschner P.J.F., Ameres S.L., Kueng S., and Martinez J., Cleavage of the siRNA passenger strand during RISC assembly in human cells, EMBO Reports 2006, 7: 314‐320.
48. Schwarz D.S., Hutvagner G., Du T., Xu Z.S. et al., Asymmetry in the assembly of the RNAi enzyme complex, Cell 2003, 115: 199‐208.
49. Nykanen A., Haley B., and Zamore P.D., ATP requirements and small interfering RNA structure in the RNA interference pathway, Cell 2001, 107: 309‐321.
50. Harborth J., Elbashir S.M., Vandenburgh K., Manninga H. et al., Sequence, chemical, and structural variation of small interfering RNAs and short hairpin RNAs and the effect on mammalian gene silencing, Antisense & Nucleic Acid Drug Development 2003, 13: 83‐105.
51. Chiu Y.L. and Rana T.M., RNAi in human cells: Basic structural and functional features of small interfering RNA, Molecular Cell 2002, 10: 549‐561.
82

in situ analysis of single‐stranded and duplex siRNA integrity in living cells | CHAPTER 2
52. Martinez J., Patkaniowska A., Urlaub H., Luhrmann R. et al., Single‐stranded antisense siRNAs guide target RNA cleavage in RNAi, Cell 2002, 110: 563‐574.
53. Bertrand J.R., Pottier M., Vekris A., Opolon P. et al., Comparison of antisense oligonucleotides and siRNAs in cell culture and in vivo, Biochemical and Biophysical Research Communications 2002, 296: 1000‐1004.
54. Bartlett D.W. and Davis M.E., Effect of siRNA nuclease stability on the in vitro and in vivo kinetics of siRNA‐mediated gene silencing, Biotechnol. Bioeng. 2007, 97: 909‐921.
83

CHAPTER 2 | in situ analysis of single‐stranded and duplex siRNA integrity in living cells
84

dextran microgels for time‐controlled delivery of siRNA | CHAPTER 3
CHAPTER 3 | Dextran microgels for time‐
controlled delivery of siRNA
Parts of this chapter are published:
Koen Raemdonck1, Tinneke G. Van Thienen1, Roosmarijn E. Vandenbroucke1, Niek N. Sanders1,2,
Joseph Demeester1 and Stefaan C. De Smedt1, Advanced Functional Materials 2008, 18: 993‐1001.
1Laboratory of General Biochemistry and Physical Pharmacy, Department of Pharmaceutics, Faculty of Pharmaceutical
Sciences, Ghent University, Harelbekestraat 72, B‐9000 Ghent, Belgium.
2Laboratory of Gene Therapy, Department of Nutrition, Genetics and Ethology, Faculty of Veterinary Medicine, Ghent
University, Heidestraat 19, B‐9820 Merelbeke, Belgium.
85

CHAPTER 3 | dextran microgels for time‐controlled delivery of siRNA
86
ABSTRACT
To apply siRNA as a therapeutic agent, appropriate attention should be paid to the optimization of
the siRNA gene silencing effect, both in terms of magnitude and duration. Intracellular time‐
controlled siRNA delivery could aid in tailoring the kinetics of siRNA gene knockdown. However,
materials with easily tunable siRNA release properties have not been subjected to thorough
investigation thus far. This report describes cationic biodegradable dextran microgels which can be
loaded with siRNA posterior to gel formation. Even though the siRNAs are incorporated in the
hydrogel network based on electrostatic interaction, still a time‐controlled release can be achieved
by varying the initial network density of the microgels. To demonstrate the biological functionality of
the siRNA loaded gels, we studied their cellular internalization and enhanced green fluorescent
protein (EGFP) gene silencing potential in Huh‐7 human hepatoma cells.

dextran microgels for time‐controlled delivery of siRNA | CHAPTER 3
3 OVERVIEW CHAPTER CONTENTS
Chapter 3 provides:
• details on the formulation of biodegradable dextran hydrogel particles based on
methacrylate functionalized dextran
• information on the applicability of dextran microgels for siRNA encapsulation and siRNA
release in a degradation‐controlled manner
• a first indication that siRNA loaded dextran hydrogel particles show promise towards gene
silencing via the process of RNAi
Key terms:
Microgel – dextran – cationic – siRNA – controlled release – biodegradation – EGFP
INTRODUCTION
Since its discovery in C. elegans [1] and the confirmation of its applicability in mammalian cells,[2] RNA
interference (RNAi) has become the method of choice to suppress gene expression for therapeutic
intervention.[3] RNAi comprises a complex, evolutionary conserved pathway, where ~21 bp RNA
duplexes (termed small interfering RNAs or siRNAs) function as the effector molecules for sequence
specific downregulation of gene expression.[4] Although siRNAs are considered to be more powerful
when compared to antisense oligonucleotides,[2,5] the same barriers exist for successful in vivo
application.[6,7] Indeed, small interfering RNAs have already demonstrated their in vivo potential as
therapeutic agents,[8] but there still is a great need for efficient delivery vehicles that can safely guide
their siRNA payload to the desired target tissue.[3,4,7] Recent reviews critically discuss therapeutic
siRNA delivery, which is still regarded as the main obstacle towards clinical application of siRNA.
87

CHAPTER 3 | dextran microgels for time‐controlled delivery of siRNA
Several strategies for in vivo siRNA delivery have already been proposed, including the application of
siRNA bioconjugates or the incorporation of siRNA into multifunctional polymeric and lipidic
nanodevices, which are also briefly discussed in chapter 1.[4,7‐11]
To apply siRNA as a therapeutic, both the intensity and duration of the gene silencing effect will have
to be controlled and optimized according to the selected disease target. The kinetics of gene
silencing by (unmodified and nuclease stabilized) siRNAs has been recently described in both an in
vitro and an in vivo experimental setting by Bartlett et al.. [12,13] In fast dividing cells, the transient
gene silencing is mainly due to the intracellular dilution of siRNA and activated RISC (RNA Induced
Silencing Complex) upon cell division,[12] while the intracellular siRNA stability has little or no
influence on the magnitude and duration of the gene knockdown effect once the siRNA has reached
the cytosol.[13]
We anticipate that an intracellular time‐controlled release of siRNA could be an interesting approach
to maintain the cytosolic siRNA concentration above a critical threshold for a longer period of time.
The packaging of siRNA in carriers that shield the siRNA from its direct environment and slowly
releases it into the cytosol could be advantageous to prolong their silencing effect. Moreover,
adverse effects originating from saturation of the RNAi pathway or unwanted immune activation may
be minimized when obtaining better control over the intracellular siRNA concentrations. To achieve
such goals it is evident that carriers with ‘flexible’ siRNA release properties are needed. However, the
literature to date contains only few reports on carriers which should be able to deliver siRNA
intracellularly in a time‐controlled manner.[14] Several papers describe the cytosolic release of siRNA
based on an intracellular trigger,[15‐17] but detailed studies on intracellular time‐controlled siRNA
delivery based on the characteristics of the siRNA carrier are not yet available at present.
The siRNA carrier that is envisioned in the present chapter is hydrogel based. Hydrogels can be
defined as three‐dimensional, hydrophilic matrices that are able to retain large amounts of water or
biological fluid.[18] In general, as a consequence of this specific feature, hydrogels can be regarded as
biocompatible and are therefore suitable for a wide range of pharmaceutical and biomedical
applications.[19] In particular, hydrogels offer excellent opportunities for development of controlled
drug delivery devices for different types of drugs.[18‐21] Our research is particularly directed towards
biodegradable hydrogels for controlled nucleic acid delivery. In this application, the ability to tailor
the drug release time depending on the physicochemical properties of the hydrogel matrix will be the
main focus.
This chapter reports on the preparation of siRNA loaded (bio)degradable microgels based on dextran.
Dextran is one the most promising polysaccharides, produced either chemically or by bacteria from
88

dextran microgels for time‐controlled delivery of siRNA | CHAPTER 3
sucrose, for the preparation of hydrogels for controlled drug delivery purposes.[20,22] Bacterial dextran
mainly consists of linear α‐1,6‐linked glucopyranose units, with some degree of 1,3‐branching.
Several coupling reactions have already been described for the modification of the numerous OH‐
groups present along the dextran chain.[22]
We especially wished to know whether the composition of the dextran gel network allows controlling
the release kinetics of the loaded siRNA. We also describe the collection of smaller sized microgels
with the aim to conduct a series of preliminary cell culture experiments. We verified their cellular
uptake in a human hepatoma cell line (Huh‐7) with confocal microscopy. Finally, the gene silencing
potential of these microgels, loaded with siRNA targeted against enhanced green fluorescent protein
(EGFP) is demonstrated in Huh‐7 cells that stably express the EGFP gene (Huh‐7_EGFP cells).
EXPERIMENTAL SECTION
siRNA oligonucleotides.
Twenty‐one nucleotide siRNA duplexes, targeted against enhanced green fluorescent protein
(siEGFP), were purchased from Dharmacon (Lafayette, CO). siCONTROL is a negative control
sequence, synthesized by Eurogentec (Seraing, Belgium), which does not match to any known
sequence in the human genome. siRNA batches were diluted in RNase free water to a final
concentration of 20‐50 µM and stored at ‐85°C. For fluorescence experiments, a siCONTROL duplex
5’‐labeled (sense strand) with atto488 or atto647 dye was used. Fluorescent modifications were
done by Eurogentec.
Synthesis of dex‐HEMA and dex‐MA.
Dextran hydroxyethyl methacrylate (dex‐HEMA; Figure 3.1a) and dextran methacrylate (dex‐MA;
Figure 3.1a) were prepared and characterized according to a method described previously.[23]
Dextran with a number average molecular weight of 19 kDa was used. The degree of substitution
(DS, i.e. the number of HEMA (or MA) groups per 100 glucopyranose residues of dextran) was
determined by proton nuclear magnetic resonance spectroscopy (1H NMR) in D2O with a Gemini 300
spectrometer (Varian). As reported before, the carbonate ester in dex‐HEMA (Figure 3.1a) hydrolyzes
at 37°C, pH 7.4 (further referred to as physiological conditions).
89

CHAPTER 3 | dextran microgels for time‐controlled delivery of siRNA
Preparation of dextran microgels.
To prepare dex‐HEMA and dex‐MA microgels, an emulsion photopolymerization method in mineral
oil was applied. A dex‐HEMA (or dex‐MA) solution (20% w/w) with ~0.1% (w/v) photoinitiator
(irgacure 2959, Ciba Specialty Chemicals) was briefly flushed with N2 and subsequently emulsified in
mineral oil (Sigma) supplemented with 10% (v/v) of surfactant (ABIL EM 90, Goldschmidt) by
vortexing (2 min). To obtain cationic microgels, 2‐(dimethylamino)ethyl methacrylate (DMAEMA, pKa
8.4, Figure 3.1) was added to the dex‐HEMA (or dex‐MA) solution. Within 60 s after emulsification,
the emulsion droplets were polymerized by UV irradiation (450 s) at room temperature (Bluepoint
2.1 UV source, Dr. Hönle UV technology). Subsequently, the continuous oil phase was diluted in
hexane (HPLC grade), followed by short centrifugation (525 g, 5 min) to collect the dextran microgels.
The supernatant was discarded and the washing step was repeated three consecutive times with an
excess of hexane before redispersing the dextran microgels in 20 mM N‐2‐hydroxyethylpiperazine‐
N’‐2‐ethanesulfonic acid (HEPES) buffer (adjusted to pH 7.4 with 1 N NaOH) or in RNase free water.
Remaining hexane was removed by vacuum evaporation. Contrary to dex‐HEMA hydrogels,
hydrogels prepared from dex‐MA are essentially non‐degradable at physiological pH as they lack the
presence of a labile carbonate ester bond.[24,25]
Penetration of siRNA into dextran microgels.
Neutral and cationic dextran microgels were prepared as described above. Intact dex‐HEMA
microgels were immersed in a 2 µM solution of atto488‐siRNA (24 h at 4°C) or a 5 µM solution of
carboxyfluorescein (CaF) (10 min at room temperature) and subsequently confocal micrographs were
taken. Confocal time lapse microscopy (1 frame/25 s) was applied to visualize siRNA penetration in
degrading neutral dextran microgels, following addition of 1 µL of NaOH (0.05 M) to 5 µL of the
siRNA‐microgel dispersion. Non‐degradable dex‐MA microgels were used as a control. To investigate
the interaction of siRNA with cationic gels, dex‐HEMA‐co‐DMAEMA microgels were incubated with
increasing atto488‐siRNA concentrations and subsequently visualized with confocal microscopy.
siRNA release measurements from cationic dextran microgels.
The dextran microgels used in the siRNA release experiments were prepared as described below, this
time using a water‐in‐water emulsion method based on the immiscibility of aqueous solutions of
dextran and PEG. Briefly, 0.025 g dex‐HEMA (or dex‐MA), 0.012 g dextran (19 kDa) and 25 µL
DMAEMA were dissolved in 2.167 mL RNase free water and emulsified in 2.7 mL of a 30% (w/w)
aqueous polyethyleneglycol (PEG; 20 kDa) solution by vortexing (2 min).
90
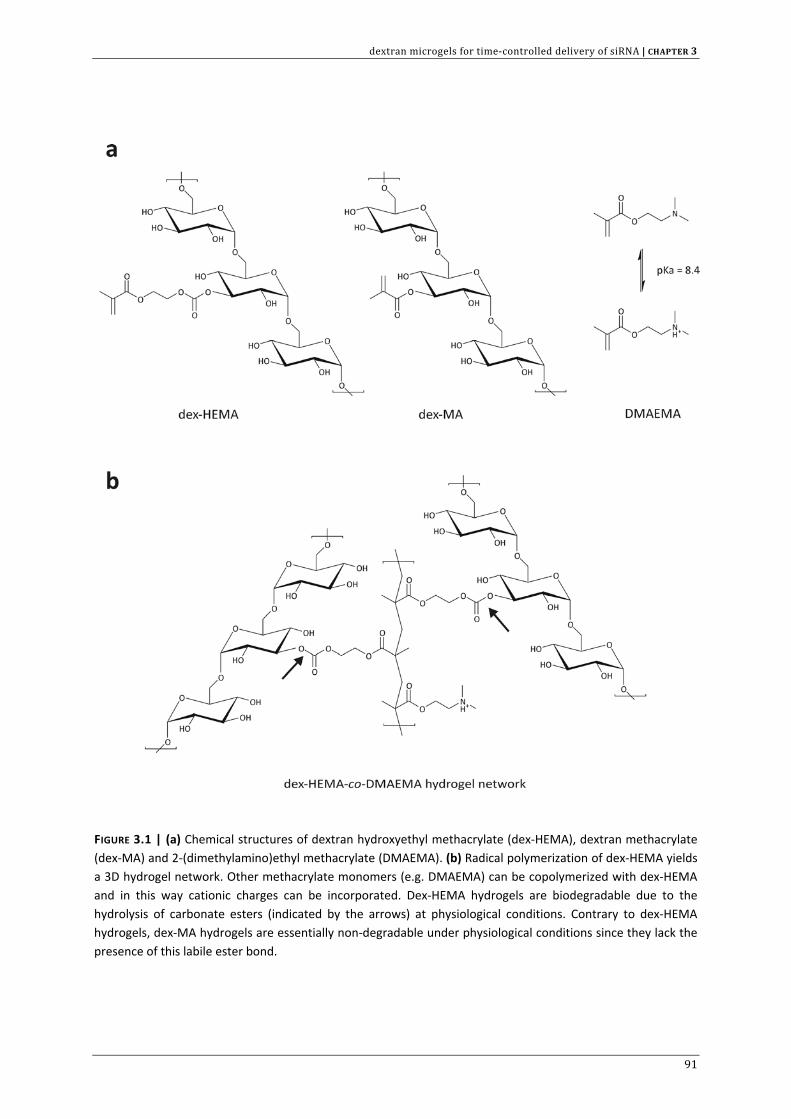
dextran microgels for time‐controlled delivery of siRNA | CHAPTER 3
FIGURE 3.1 | (a) Chemical structures of dextran hydroxyethyl methacrylate (dex‐HEMA), dextran methacrylate (dex‐MA) and 2‐(dimethylamino)ethyl methacrylate (DMAEMA). (b) Radical polymerization of dex‐HEMA yields a 3D hydrogel network. Other methacrylate monomers (e.g. DMAEMA) can be copolymerized with dex‐HEMA and in this way cationic charges can be incorporated. Dex‐HEMA hydrogels are biodegradable due to the hydrolysis of carbonate esters (indicated by the arrows) at physiological conditions. Contrary to dex‐HEMA hydrogels, dex‐MA hydrogels are essentially non‐degradable under physiological conditions since they lack the presence of this labile ester bond.
91

CHAPTER 3 | dextran microgels for time‐controlled delivery of siRNA
Radical polymerization of the dextran/dex‐HEMA (or dextran/dex‐MA) droplets was initiated by 100
µL of a 20% (v/v) N,N,N’,N’‐tetramethylethylenediamine (TEMED) solution (neutralized with 2N HCl)
and 180 µL potassium peroxodisulphate (KPS, 50 mg mL‐1). The reaction mixture was left to
polymerize at room temperature for at least 30 minutes. Microgels of lower crosslink density were
obtained by decreasing the amount of polymerizable dextran while keeping the total amount of
dextran constant (0.0185 g dex‐HEMA (or dex‐MA) + 0.0185 g dextran). To remove remaining KPS,
TEMED, PEG and unpolymerized DMAEMA, the microgels were washed three times with RNase free
water, collected through centrifugation (750 g, 5 min) and finally redispersed in 5 mL HEPES buffer.
To load the microgels with siRNA, a 0.9 mL aliquot of the microgel dispersion was incubated with 100
µL siRNA (50 µM) during 30 min at room temperature in non‐stick RNase free 2 mL eppendorfs
(Ambion). Subsequently, the samples were kept at 37°C under continuous shaking. At defined time
points, the microgels were pelleted down by centrifugation (750g, 5 min). A 50 µL aliquot of the
supernatant was then withdrawn and replaced by 50 µL fresh HEPES buffer. The siRNA concentration
in each aliquot was quantified by measuring the absorbance at 260 nm (Nanodrop ND1000
Spectrophotometer) or with the Quant‐it™ Ribogreen® assay (Molecular Probes). It was verified that
the absorbance and fluorescence values lied within the linear range of detection. The siRNA
concentration measured when the microgels were completely degraded (as verified through optical
microscopy) was set to 100% in the release graphs.
Collection of smaller sized microgels.
Microgels were prepared as described in the previous paragraph. After polymerization of the
microgels, the dispersion (~5 mL) was diluted 1:3 with RNase free water and centrifuged during 30
min at 1000 g. The supernatant was collected and smaller sized microgels were obtained through a
second centrifugation (12000 g, 10 min). The microgels were washed three times with an excess of
RNase free water to remove any impurities and finally redispersed in 667 µL of HEPES buffer. The size
distribution of the microgels was measured by fluorescence microscopy. Therefore, fluorescein O‐
methacrylate (Sigma) was copolymerized in the microgels. Fluorescence images at 488 nm excitation
were recorded with a SPOT Insight CCD camera (Diagnostic Instruments, Sterling Heigths, MI)
attached to a Nikon TE300 inverted epi‐fluorescence microscope with a Nikon Plan Fluor 20x
objective lens. To perform accurate size measurements, the optical system was calibrated using a
stage grid consisting of 10 µm by 10 µm squares (Edmund Optics, York, UK). The fluorescence images
were analyzed with custom built software to obtain size distributions of the microgels.
92

dextran microgels for time‐controlled delivery of siRNA | CHAPTER 3
Scanning electron microscopy (SEM).
Scanning electron microscopy was carried out using a Quanta 200F FEG (Field Emission Gun) SEM (FEI
company) operating in High VacMode at an accelerating voltage of 25 kV. For sample preparation, a
drop of the microgel dispersion was dried under low vacuum on a silica wafer and sputtered with
gold/palladium before visualization.
Atomic Force Microscopy (AFM).
Atomic Force Microscopy images in air were acquired using a PicoPlus instrument (Molecular
Imaging) in tapping mode at room temperature. For sample preparation, a drop of the microgel
dispersion was left to dry overnight on a silica wafer. Height images were flattened for visualization
using WSxM scanning probe microscopy software (Nanotec Electronica).
Confocal Laser Scanning Microscopy.
Confocal micrographs of the dex‐HEMA and dex‐HEMA‐co‐DMAEMA microgels incubated with
atto488‐siRNA were taken using a Nikon EZC1‐si confocal scanning module installed on a motorized
Nikon TE2000‐E inverted microscope (Nikon Benelux, Brussels, Belgium), equipped with an Argon ion
laser (488 nm) and a 60x water‐immersion objective lens (Plan Apo 60x, numerical aperture 1.2,
collar rim correction, Nikon).
Cellular uptake of dextran microgels in Huh‐7 cells.
The human hepatoma cell line Huh‐7 was cultured (37°C, 5% CO2) in DMEM:F12, supplemented with
2 mM glutamine, 10% heat‐inactivated Fetal Bovine Serum (FBS) and 100 U mL‐1 penicillin‐
streptomycin. Huh‐7 cells were seeded in sterile glass‐bottomed culture dishes (MatTek Corporation,
Ashland, MA) at a density of 3 x 104 cells per cm2 and allowed to attach overnight. The cells were
incubated (37°C, 5% CO2) with 50 µL of a dex‐HEMA‐co‐DMAEMA microgel dispersion (18.5 mg
dextran, DS 2.5), loaded with atto647‐siRNA, in a total volume of 250 µL OptiMEM (Invitrogen,
Merelbeke, Belgium) during 20 h (37°C). Subsequently, cells were washed with Phosphate Buffered
Saline (PBS) and prewarmed culture medium was added. The intracellular distribution of the
microgels was visualized with a confocal microscope (see previous section). For lysosomal staining,
cells were incubated for 15 min with 1:104 diluted Lysotracker Green® (Molecular Probes) prior to
image recording. Images were captured with a Nikon Plan Apochromat 60x oil immersion objective
(numerical aperture 1.4) using a 488 nm and 639 nm laser line for the excitation of Lysotracker
Green® and atto647‐siRNA respectively.
93

CHAPTER 3 | dextran microgels for time‐controlled delivery of siRNA
EGFP silencing in Huh‐7_EGFP cells by siRNA loaded dextran microgels.
Huh‐7_EGFP cells stably expressing EGFP were generated by transfecting Huh‐7 cells with the pEGFP‐
N2 plasmid (Clontech, Palo Alto, USA). The pDNA was linearized using the restriction enzyme XbaI
and transfected by complexation with linear polyethylenimine (PEI) 22 kDa. Transfected cells were
incubated in fresh medium for 48 h and then selected with 400 µg mL‐1 G418 (geneticine, Invitrogen).
After two weeks, clones were isolated and expanded. Subsequently, the generated clones were
analyzed by CLSM and a 100% EGFP positive clone was selected. Huh‐7_EGFP cells and Huh‐7 cells
were cultured under identical conditions. To assess the EGFP gene silencing potential of siRNA loaded
dextran microgels, Huh‐7_EGFP cells were seeded at a density of 3 x 104 cells per cm2 in Lab‐Tek™ II
chambered coverglasses (Nalge Nunc International, Rochester, NY). Two days after seeding, cells
were washed with PBS and 30 µL dex‐HEMA‐co‐DMAEMA microgel dispersion (18.5 mg dex‐HEMA,
DS 2.5), loaded with 0.2 nmol siEGFP or siCONTROL, was added per well in a total volume of 400 µL
OptiMEM. After 20 h incubation (37°C, 5% CO2), the transfection medium was removed and replaced
with complete cell culture medium. Cells were left to grow to 100% confluence (~48 h).
Subsequently, the cells were trypsinized and replated (1:3 diluted in culture medium) in a glass‐
bottomed 96‐well plate (Greiner Bio‐One). Huh‐7_EGFP cells were visualized 8 days after transfection
with confocal microscopy using a Nikon Plan Fluor 20x objective (numerical aperture 0.5). The 488
nm laser line was selected for EGFP excitation.
RESULTS AND DISCUSSION
Interaction of siRNA with neutral dextran microgels
Dextran microgels were prepared by emulsion photopolymerization of dextran hydroxyethyl
methacrylate (dex‐HEMA, Figure 3.1a) in mineral oil. In this way, a microscopic gel network is
obtained, consisting of dextran polymer chains coupled by inter‐ and intramolecular
oligomethacrylate crosslinks.[23,25] First, we aimed to investigate to which extent siRNA can diffuse
into intact neutral dextran hydrogel networks with variable crosslinking density. The microgel
crosslink density can be altered by varying the concentration of methacrylated dextran or by using
dex‐HEMA with a different degree of substitution (DS, i.e. the number of HEMA groups per 100
glucopyranose residues of dextran). In the following experiment, dex‐HEMA microgels (20% w/w)
were prepared with different DS values. The microgels were incubated with green fluorescent siRNA
(atto488‐siRNA) during 24 h at 4°C (to avoid excessive hydrolysis of the carbonate esters) and the
samples were analyzed by confocal microscopy. As shown in the left and middle image of Figure 3.2,
94
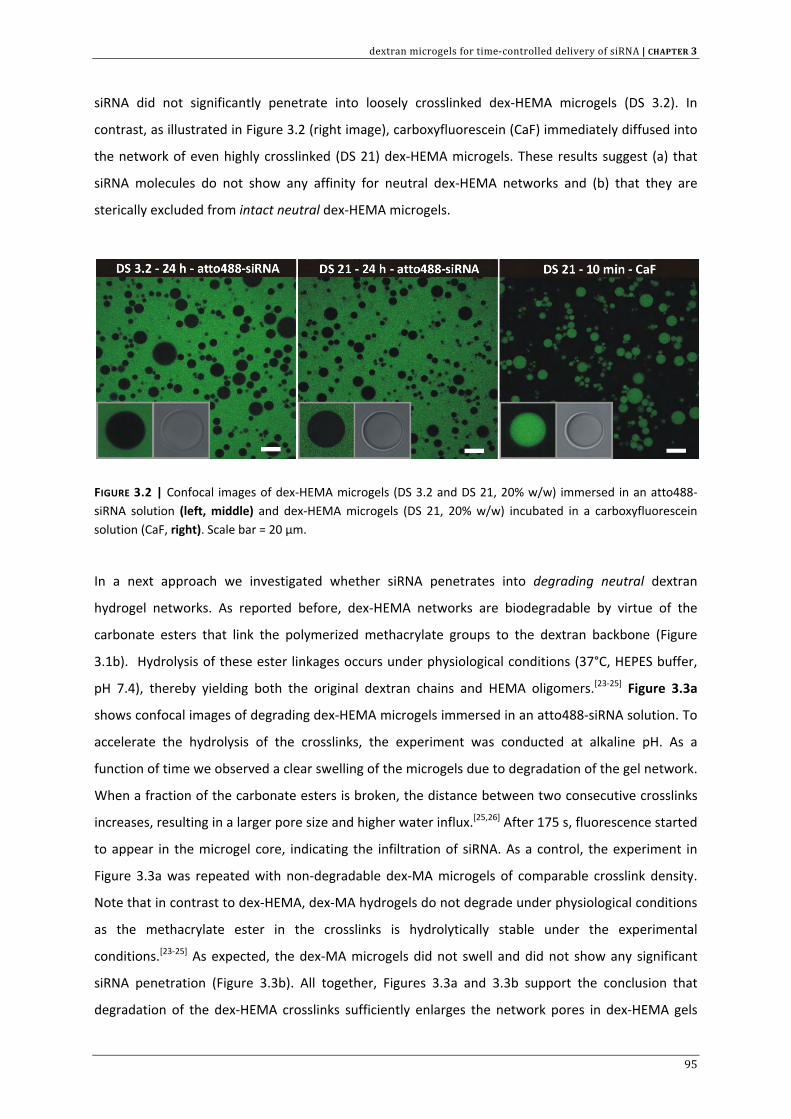
dextran microgels for time‐controlled delivery of siRNA | CHAPTER 3
siRNA did not significantly penetrate into loosely crosslinked dex‐HEMA microgels (DS 3.2). In
contrast, as illustrated in Figure 3.2 (right image), carboxyfluorescein (CaF) immediately diffused into
the network of even highly crosslinked (DS 21) dex‐HEMA microgels. These results suggest (a) that
siRNA molecules do not show any affinity for neutral dex‐HEMA networks and (b) that they are
sterically excluded from intact neutral dex‐HEMA microgels.
FIGURE 3.2 | Confocal images of dex‐HEMA microgels (DS 3.2 and DS 21, 20% w/w) immersed in an atto488‐siRNA solution (left, middle) and dex‐HEMA microgels (DS 21, 20% w/w) incubated in a carboxyfluorescein solution (CaF, right). Scale bar = 20 µm.
In a next approach we investigated whether siRNA penetrates into degrading neutral dextran
hydrogel networks. As reported before, dex‐HEMA networks are biodegradable by virtue of the
carbonate esters that link the polymerized methacrylate groups to the dextran backbone (Figure
3.1b). Hydrolysis of these ester linkages occurs under physiological conditions (37°C, HEPES buffer,
pH 7.4), thereby yielding both the original dextran chains and HEMA oligomers.[23‐25] Figure 3.3a
shows confocal images of degrading dex‐HEMA microgels immersed in an atto488‐siRNA solution. To
accelerate the hydrolysis of the crosslinks, the experiment was conducted at alkaline pH. As a
function of time we observed a clear swelling of the microgels due to degradation of the gel network.
When a fraction of the carbonate esters is broken, the distance between two consecutive crosslinks
increases, resulting in a larger pore size and higher water influx.[25,26] After 175 s, fluorescence started
to appear in the microgel core, indicating the infiltration of siRNA. As a control, the experiment in
Figure 3.3a was repeated with non‐degradable dex‐MA microgels of comparable crosslink density.
Note that in contrast to dex‐HEMA, dex‐MA hydrogels do not degrade under physiological conditions
as the methacrylate ester in the crosslinks is hydrolytically stable under the experimental
conditions.[23‐25] As expected, the dex‐MA microgels did not swell and did not show any significant
siRNA penetration (Figure 3.3b). All together, Figures 3.3a and 3.3b support the conclusion that
degradation of the dex‐HEMA crosslinks sufficiently enlarges the network pores in dex‐HEMA gels
95
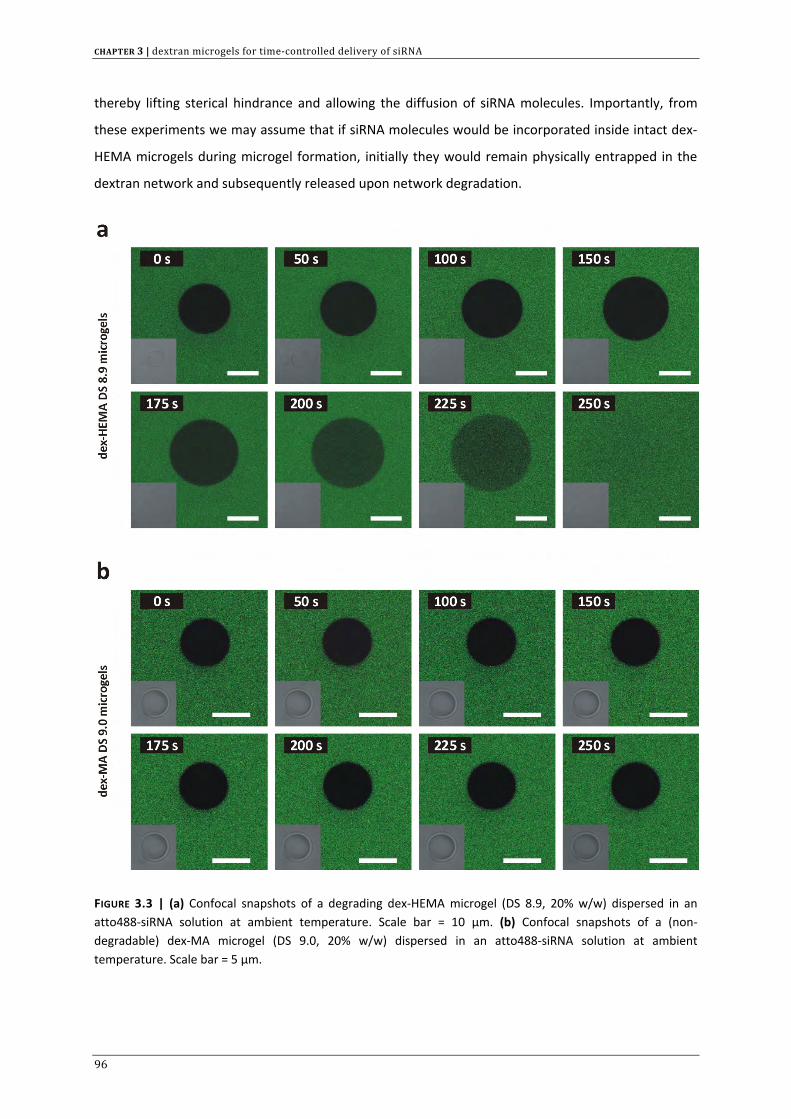
CHAPTER 3 | dextran microgels for time‐controlled delivery of siRNA
thereby lifting sterical hindrance and allowing the diffusion of siRNA molecules. Importantly, from
these experiments we may assume that if siRNA molecules would be incorporated inside intact dex‐
HEMA microgels during microgel formation, initially they would remain physically entrapped in the
dextran network and subsequently released upon network degradation.
FIGURE 3.3 | (a) Confocal snapshots of a degrading dex‐HEMA microgel (DS 8.9, 20% w/w) dispersed in an atto488‐siRNA solution at ambient temperature. Scale bar = 10 µm. (b) Confocal snapshots of a (non‐degradable) dex‐MA microgel (DS 9.0, 20% w/w) dispersed in an atto488‐siRNA solution at ambient temperature. Scale bar = 5 µm.
96

dextran microgels for time‐controlled delivery of siRNA | CHAPTER 3
In the experiments described above, the dextran microgels were prepared by emulsion
polymerization in mineral oil. As siRNA is not soluble in the continuous phase we anticipate a high
loading efficiency when the siRNA is added to the dextran phase, prior to emulsification. However,
this preparation protocol implies that the siRNA molecules would be brought into contact with free
radicals and UV light during the photopolymerization step. To avoid this, we sought for an alternative
preparation protocol which allows (a) high siRNA loading of the dextran microgels and (b) siRNA
loading of the microgels after their preparation.
Penetration of siRNA into cationic dextran microgels
Cationic microgels were prepared by copolymerization of 2‐(dimethylamino)ethyl methacrylate
(DMAEMA, Figure 3.1a, pKa 8.4) with dex‐HEMA or dex‐MA.[27,28] The incorporation of positively
charged groups in the microgel network was verified by measuring the zeta‐potential of the
microgels, which was indeed positive (~20‐30 mV) when DMAEMA was added during microgel
preparation.
Figure 3.4 shows confocal fluorescence images of cationic dextran microgels which were incubated
with increasing amounts of atto488‐siRNA. When the microgels were incubated with a low amount
of siRNA (Figure 3.4a), a bright fluorescent ring was observed, suggesting the selective binding of
negatively charged siRNAs to the cationic DMAEMA groups near the surface of the microgels.
However, in more concentrated siRNA solutions (Figures 3.4b and 3.4c), not only the surface but also
the interior of the microgels became loaded with siRNA. It seems that the siRNA molecules first
neutralize the charges at the surface of the microgels after which remaining siRNA molecules are
able to penetrate deeper into the microgel network. When a sufficient amount of siRNA was added,
a homogeneous loading of the cationic dextran microgels could be achieved (Figure 3.4c and 3.4f).
Interestingly, while the pores in neutral dextran gels (DS 3.2, 20% w/w) are too small to allow siRNA
diffusion into the network (Figure 3.2), the incorporation of cationic groups in these gels seems to aid
the diffusion (Figure 3.4), thereby loading the microgels with siRNA based on electrostatic attraction.
The exact reason for this remains unclear. Possibly, the incorporation of protonable amines into the
gel network leads to electrostatic repulsion between positive charges which may expand the pores in
the network. For this reason, a direct comparison between microgels composed of dex‐HEMA alone
and dex‐HEMA microgels with copolymerization of DMAEMA is not feasible.
The previous experiments demonstrate that loading of positively charged dextran‐co‐DMAEMA gels
with siRNA after gel formation (“post‐loading”) is possible. Post‐loading of siRNA may have major
advantages as (1) exposure of siRNA to UV light and/or free radicals during the gelation process is
97
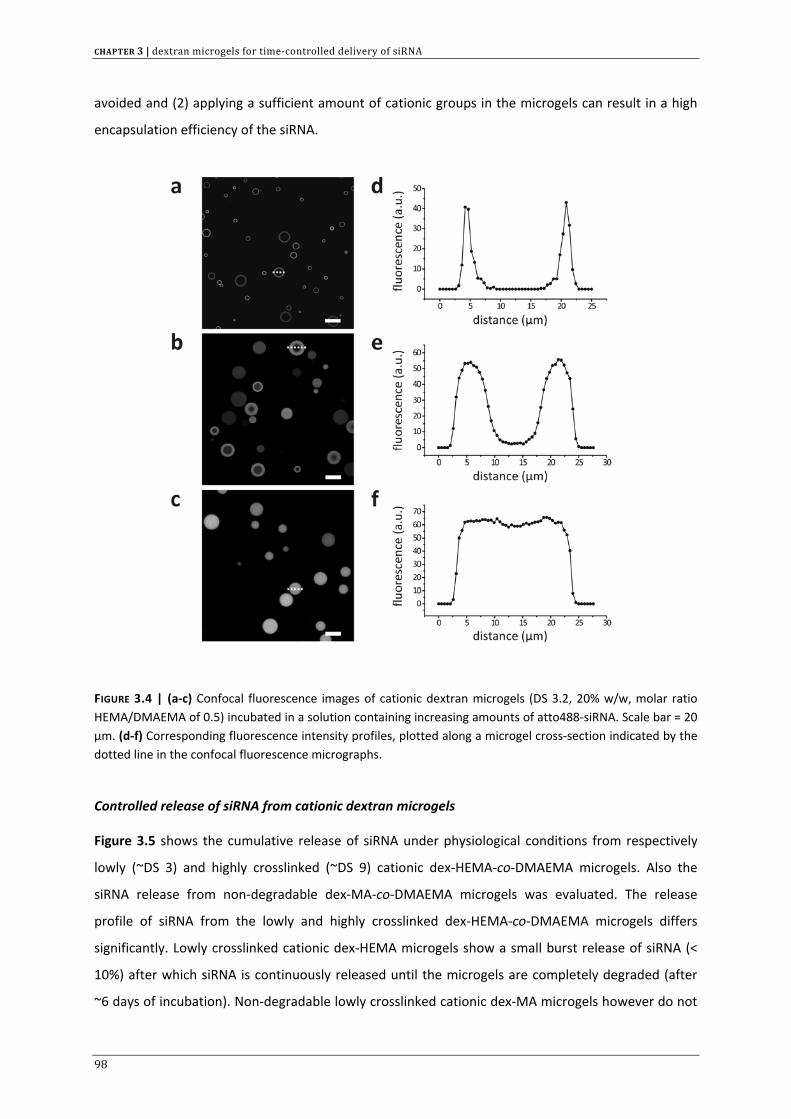
CHAPTER 3 | dextran microgels for time‐controlled delivery of siRNA
avoided and (2) applying a sufficient amount of cationic groups in the microgels can result in a high
encapsulation efficiency of the siRNA.
FIGURE 3.4 | (a‐c) Confocal fluorescence images of cationic dextran microgels (DS 3.2, 20% w/w, molar ratio HEMA/DMAEMA of 0.5) incubated in a solution containing increasing amounts of atto488‐siRNA. Scale bar = 20 µm. (d‐f) Corresponding fluorescence intensity profiles, plotted along a microgel cross‐section indicated by the dotted line in the confocal fluorescence micrographs.
Controlled release of siRNA from cationic dextran microgels
Figure 3.5 shows the cumulative release of siRNA under physiological conditions from respectively
lowly (~DS 3) and highly crosslinked (~DS 9) cationic dex‐HEMA‐co‐DMAEMA microgels. Also the
siRNA release from non‐degradable dex‐MA‐co‐DMAEMA microgels was evaluated. The release
profile of siRNA from the lowly and highly crosslinked dex‐HEMA‐co‐DMAEMA microgels differs
significantly. Lowly crosslinked cationic dex‐HEMA microgels show a small burst release of siRNA (<
10%) after which siRNA is continuously released until the microgels are completely degraded (after
~6 days of incubation). Non‐degradable lowly crosslinked cationic dex‐MA microgels however do not
98
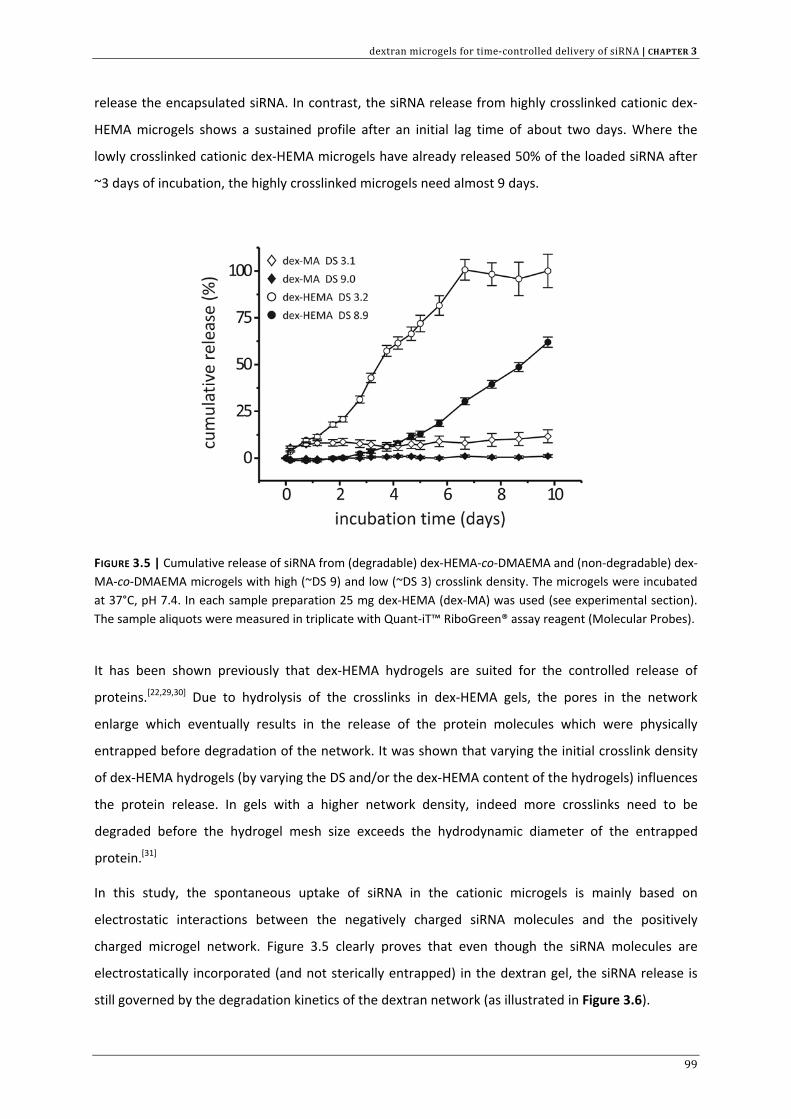
dextran microgels for time‐controlled delivery of siRNA | CHAPTER 3
release the encapsulated siRNA. In contrast, the siRNA release from highly crosslinked cationic dex‐
HEMA microgels shows a sustained profile after an initial lag time of about two days. Where the
lowly crosslinked cationic dex‐HEMA microgels have already released 50% of the loaded siRNA after
~3 days of incubation, the highly crosslinked microgels need almost 9 days.
FIGURE 3.5 | Cumulative release of siRNA from (degradable) dex‐HEMA‐co‐DMAEMA and (non‐degradable) dex‐MA‐co‐DMAEMA microgels with high (~DS 9) and low (~DS 3) crosslink density. The microgels were incubated at 37°C, pH 7.4. In each sample preparation 25 mg dex‐HEMA (dex‐MA) was used (see experimental section). The sample aliquots were measured in triplicate with Quant‐iT™ RiboGreen® assay reagent (Molecular Probes).
It has been shown previously that dex‐HEMA hydrogels are suited for the controlled release of
proteins.[22,29,30] Due to hydrolysis of the crosslinks in dex‐HEMA gels, the pores in the network
enlarge which eventually results in the release of the protein molecules which were physically
entrapped before degradation of the network. It was shown that varying the initial crosslink density
of dex‐HEMA hydrogels (by varying the DS and/or the dex‐HEMA content of the hydrogels) influences
the protein release. In gels with a higher network density, indeed more crosslinks need to be
degraded before the hydrogel mesh size exceeds the hydrodynamic diameter of the entrapped
protein.[31]
In this study, the spontaneous uptake of siRNA in the cationic microgels is mainly based on
electrostatic interactions between the negatively charged siRNA molecules and the positively
charged microgel network. Figure 3.5 clearly proves that even though the siRNA molecules are
electrostatically incorporated (and not sterically entrapped) in the dextran gel, the siRNA release is
still governed by the degradation kinetics of the dextran network (as illustrated in Figure 3.6).
99
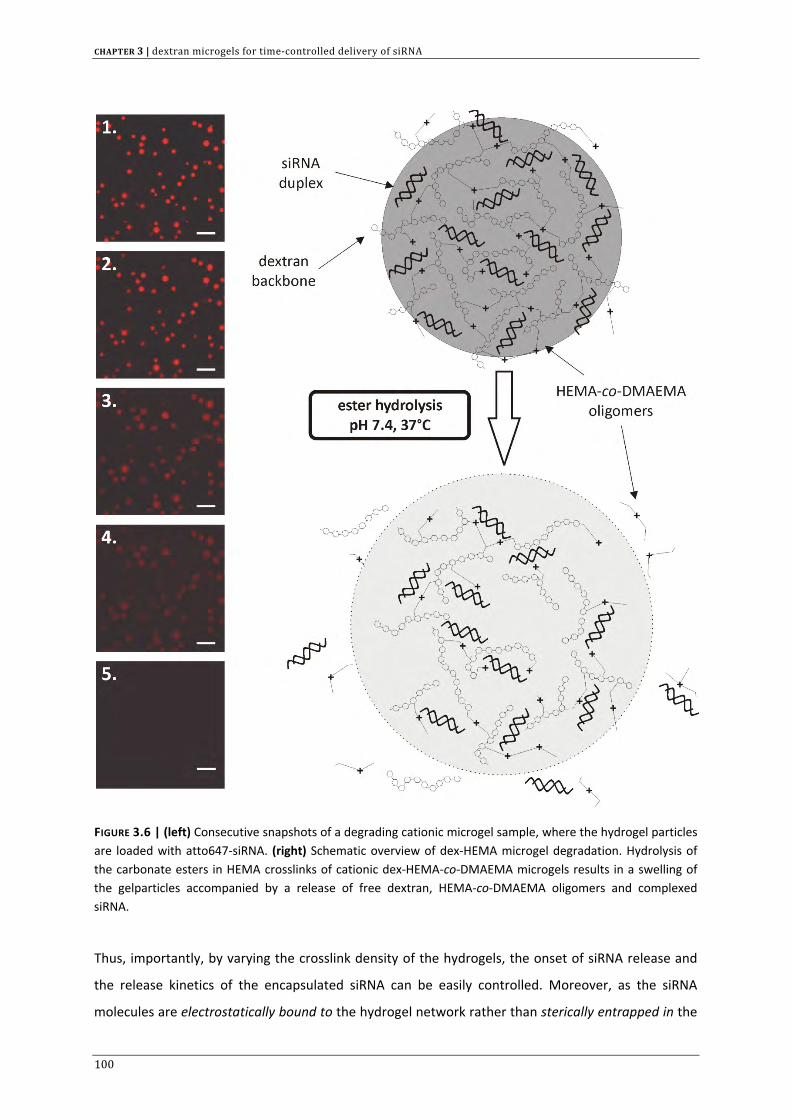
CHAPTER 3 | dextran microgels for time‐controlled delivery of siRNA
FIGURE 3.6 | (left) Consecutive snapshots of a degrading cationic microgel sample, where the hydrogel particles are loaded with atto647‐siRNA. (right) Schematic overview of dex‐HEMA microgel degradation. Hydrolysis of the carbonate esters in HEMA crosslinks of cationic dex‐HEMA‐co‐DMAEMA microgels results in a swelling of the gelparticles accompanied by a release of free dextran, HEMA‐co‐DMAEMA oligomers and complexed siRNA.
Thus, importantly, by varying the crosslink density of the hydrogels, the onset of siRNA release and
the release kinetics of the encapsulated siRNA can be easily controlled. Moreover, as the siRNA
molecules are electrostatically bound to the hydrogel network rather than sterically entrapped in the
100
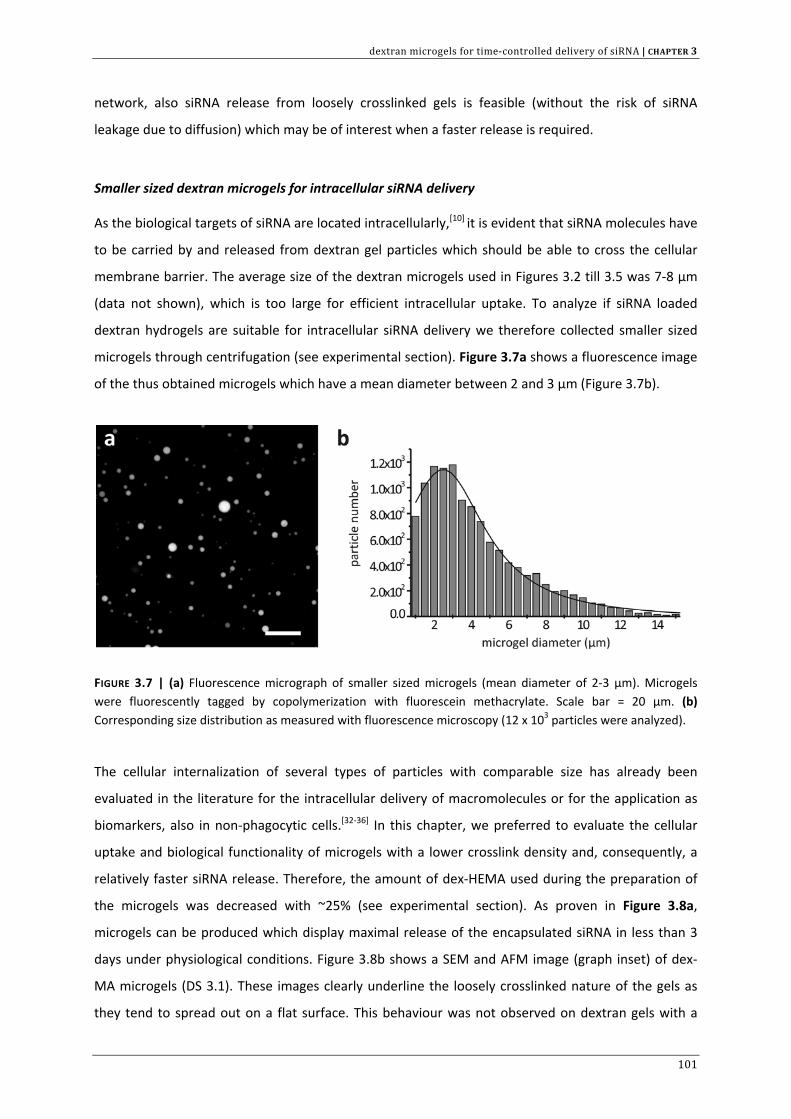
dextran microgels for time‐controlled delivery of siRNA | CHAPTER 3
network, also siRNA release from loosely crosslinked gels is feasible (without the risk of siRNA
leakage due to diffusion) which may be of interest when a faster release is required.
Smaller sized dextran microgels for intracellular siRNA delivery
As the biological targets of siRNA are located intracellularly,[10] it is evident that siRNA molecules have
to be carried by and released from dextran gel particles which should be able to cross the cellular
membrane barrier. The average size of the dextran microgels used in Figures 3.2 till 3.5 was 7‐8 µm
(data not shown), which is too large for efficient intracellular uptake. To analyze if siRNA loaded
dextran hydrogels are suitable for intracellular siRNA delivery we therefore collected smaller sized
microgels through centrifugation (see experimental section). Figure 3.7a shows a fluorescence image
of the thus obtained microgels which have a mean diameter between 2 and 3 µm (Figure 3.7b).
FIGURE 3.7 | (a) Fluorescence micrograph of smaller sized microgels (mean diameter of 2‐3 µm). Microgels were fluorescently tagged by copolymerization with fluorescein methacrylate. Scale bar = 20 µm. (b) Corresponding size distribution as measured with fluorescence microscopy (12 x 103 particles were analyzed).
The cellular internalization of several types of particles with comparable size has already been
evaluated in the literature for the intracellular delivery of macromolecules or for the application as
biomarkers, also in non‐phagocytic cells.[32‐36] In this chapter, we preferred to evaluate the cellular
uptake and biological functionality of microgels with a lower crosslink density and, consequently, a
relatively faster siRNA release. Therefore, the amount of dex‐HEMA used during the preparation of
the microgels was decreased with ~25% (see experimental section). As proven in Figure 3.8a,
microgels can be produced which display maximal release of the encapsulated siRNA in less than 3
days under physiological conditions. Figure 3.8b shows a SEM and AFM image (graph inset) of dex‐
MA microgels (DS 3.1). These images clearly underline the loosely crosslinked nature of the gels as
they tend to spread out on a flat surface. This behaviour was not observed on dextran gels with a
101
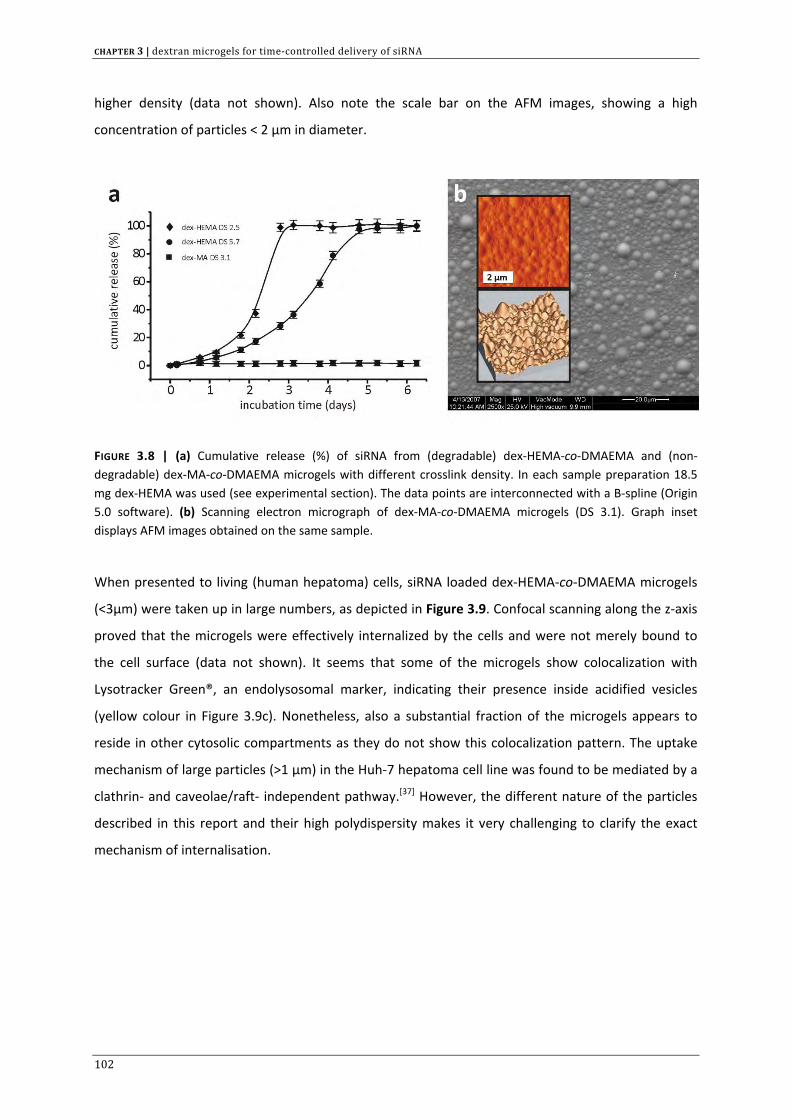
CHAPTER 3 | dextran microgels for time‐controlled delivery of siRNA
higher density (data not shown). Also note the scale bar on the AFM images, showing a high
concentration of particles < 2 µm in diameter.
FIGURE 3.8 | (a) Cumulative release (%) of siRNA from (degradable) dex‐HEMA‐co‐DMAEMA and (non‐degradable) dex‐MA‐co‐DMAEMA microgels with different crosslink density. In each sample preparation 18.5 mg dex‐HEMA was used (see experimental section). The data points are interconnected with a B‐spline (Origin 5.0 software). (b) Scanning electron micrograph of dex‐MA‐co‐DMAEMA microgels (DS 3.1). Graph inset displays AFM images obtained on the same sample.
When presented to living (human hepatoma) cells, siRNA loaded dex‐HEMA‐co‐DMAEMA microgels
(<3µm) were taken up in large numbers, as depicted in Figure 3.9. Confocal scanning along the z‐axis
proved that the microgels were effectively internalized by the cells and were not merely bound to
the cell surface (data not shown). It seems that some of the microgels show colocalization with
Lysotracker Green®, an endolysosomal marker, indicating their presence inside acidified vesicles
(yellow colour in Figure 3.9c). Nonetheless, also a substantial fraction of the microgels appears to
reside in other cytosolic compartments as they do not show this colocalization pattern. The uptake
mechanism of large particles (>1 µm) in the Huh‐7 hepatoma cell line was found to be mediated by a
clathrin‐ and caveolae/raft‐ independent pathway.[37] However, the different nature of the particles
described in this report and their high polydispersity makes it very challenging to clarify the exact
mechanism of internalisation.
102
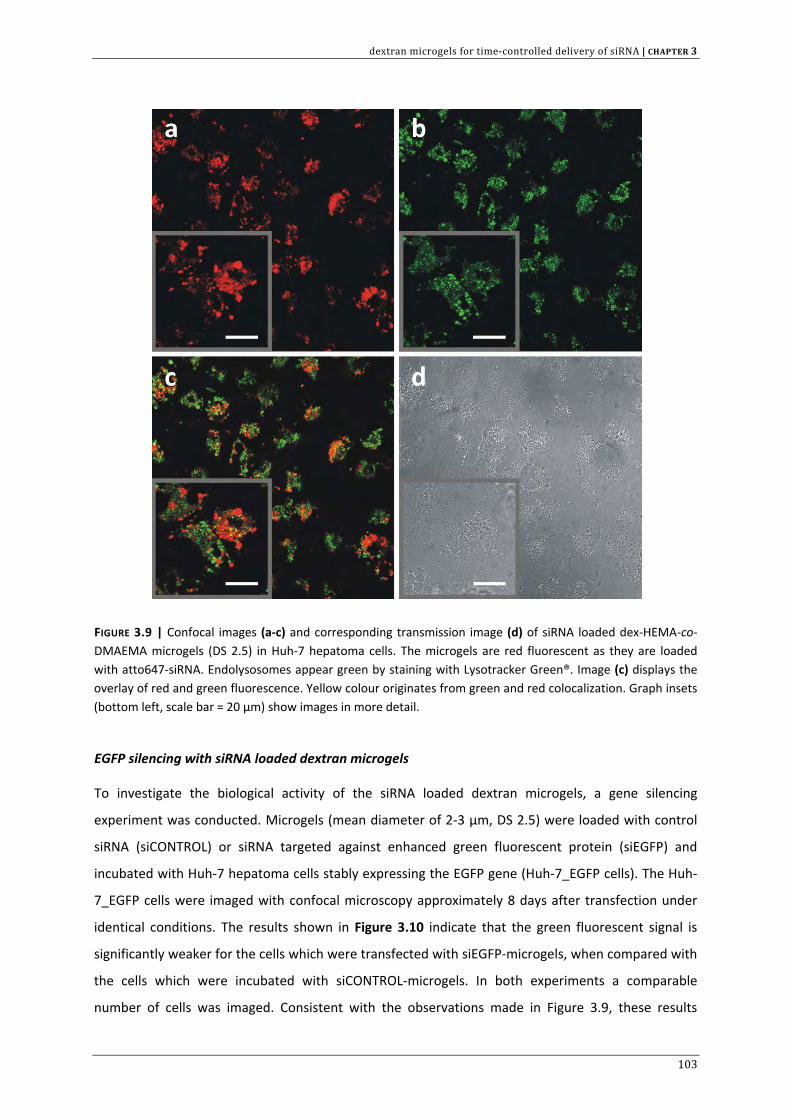
dextran microgels for time‐controlled delivery of siRNA | CHAPTER 3
FIGURE 3.9 | Confocal images (a‐c) and corresponding transmission image (d) of siRNA loaded dex‐HEMA‐co‐DMAEMA microgels (DS 2.5) in Huh‐7 hepatoma cells. The microgels are red fluorescent as they are loaded with atto647‐siRNA. Endolysosomes appear green by staining with Lysotracker Green®. Image (c) displays the overlay of red and green fluorescence. Yellow colour originates from green and red colocalization. Graph insets (bottom left, scale bar = 20 µm) show images in more detail.
EGFP silencing with siRNA loaded dextran microgels
To investigate the biological activity of the siRNA loaded dextran microgels, a gene silencing
experiment was conducted. Microgels (mean diameter of 2‐3 µm, DS 2.5) were loaded with control
siRNA (siCONTROL) or siRNA targeted against enhanced green fluorescent protein (siEGFP) and
incubated with Huh‐7 hepatoma cells stably expressing the EGFP gene (Huh‐7_EGFP cells). The Huh‐
7_EGFP cells were imaged with confocal microscopy approximately 8 days after transfection under
identical conditions. The results shown in Figure 3.10 indicate that the green fluorescent signal is
significantly weaker for the cells which were transfected with siEGFP‐microgels, when compared with
the cells which were incubated with siCONTROL‐microgels. In both experiments a comparable
number of cells was imaged. Consistent with the observations made in Figure 3.9, these results
103
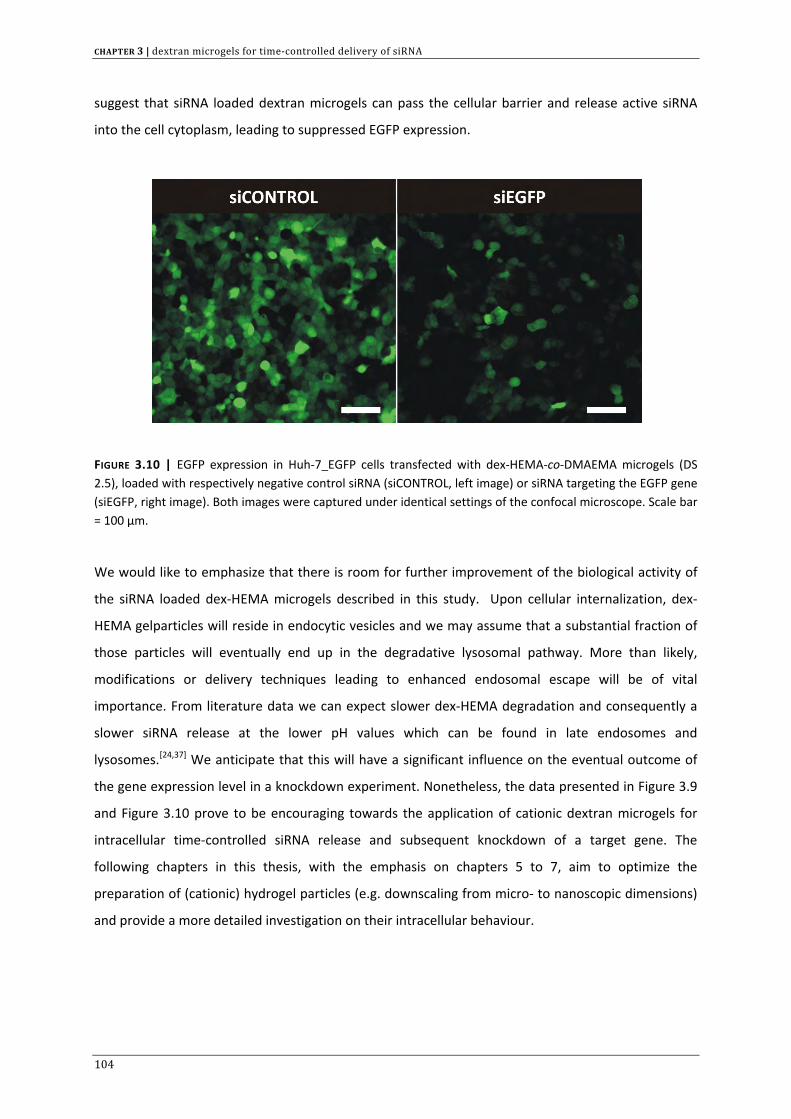
CHAPTER 3 | dextran microgels for time‐controlled delivery of siRNA
suggest that siRNA loaded dextran microgels can pass the cellular barrier and release active siRNA
into the cell cytoplasm, leading to suppressed EGFP expression.
FIGURE 3.10 | EGFP expression in Huh‐7_EGFP cells transfected with dex‐HEMA‐co‐DMAEMA microgels (DS 2.5), loaded with respectively negative control siRNA (siCONTROL, left image) or siRNA targeting the EGFP gene (siEGFP, right image). Both images were captured under identical settings of the confocal microscope. Scale bar = 100 µm.
We would like to emphasize that there is room for further improvement of the biological activity of
the siRNA loaded dex‐HEMA microgels described in this study. Upon cellular internalization, dex‐
HEMA gelparticles will reside in endocytic vesicles and we may assume that a substantial fraction of
those particles will eventually end up in the degradative lysosomal pathway. More than likely,
modifications or delivery techniques leading to enhanced endosomal escape will be of vital
importance. From literature data we can expect slower dex‐HEMA degradation and consequently a
slower siRNA release at the lower pH values which can be found in late endosomes and
lysosomes.[24,37] We anticipate that this will have a significant influence on the eventual outcome of
the gene expression level in a knockdown experiment. Nonetheless, the data presented in Figure 3.9
and Figure 3.10 prove to be encouraging towards the application of cationic dextran microgels for
intracellular time‐controlled siRNA release and subsequent knockdown of a target gene. The
following chapters in this thesis, with the emphasis on chapters 5 to 7, aim to optimize the
preparation of (cationic) hydrogel particles (e.g. downscaling from micro‐ to nanoscopic dimensions)
and provide a more detailed investigation on their intracellular behaviour.
104

dextran microgels for time‐controlled delivery of siRNA | CHAPTER 3
CONCLUSIONS
The results presented here indicate that cationic biodegradable dextran microgels can be produced
through emulsion polymerization and that they can be subsequently loaded with siRNA. The uptake
of siRNA by the microgels is mainly based on electrostatic interaction. This enables the ‘post‐loading’
of siRNA in preformed cationic gels, which offers several advantages: (1) the siRNA integrity is not
affected by the presence of free radicals during the emulsion polymerization, (2) depending on the
number of cationic groups incorporated in the gel network, a high siRNA encapsulation efficiency can
be achieved and (3) loosely crosslinked gels can be loaded with siRNA, without unwanted leakage of
siRNA due to diffusion. Irrespective of the electrostatic nature of the siRNA incorporation in the
microgel network, the siRNA release is still governed by the degradation of the crosslinks in the gel
network. Using a Huh‐7 cell line that stably expresses the EGFP gene, we were able to demonstrate
the biological functionality of the siRNA loaded microgels. We would like to remark that the siRNA
microgels show promise to investigate the influence of siRNA release kinetics on both the magnitude
and duration of gene knockdown, which has not been studied so far.
ACKNOWLEDGMENTS
Financial support from IWT (Institute for the Promotion of Innovation through Science and
Technology in Flanders) and FWO‐Flanders (Fund for Scientific Research‐Flanders) is acknowledged
with gratitude. Prof. Dr. K. Braeckmans is thanked for his help with the size distribution
measurements. Olivier Janssens and Karel Lambert are acknowledged for conducting the SEM and
AFM measurements respectively. The EU (FP6) is acknowledged for financially supporting MediTrans,
an Integrated Project on Targeted Delivery of Nanomedicines.
105

CHAPTER 3 | dextran microgels for time‐controlled delivery of siRNA
REFERENCE LIST
1. Fire A., Xu S.Q., Montgomery M.K., Kostas S.A. et al., Potent and specific genetic interference by double‐stranded RNA in Caenorhabditis elegans, Nature 1998, 391: 806‐811.
2. Elbashir S.M., Harborth J., Lendeckel W., Yalcin A. et al., Duplexes of 21‐nucleotide RNAs mediate RNA interference in cultured mammalian cells, Nature 2001, 411: 494‐498.
3. Bumcrot D., Manoharan M., Koteliansky V., and Sah D.W.Y., RNAi therapeutics: a potential new class of pharmaceutical drugs, Nature Chemical Biology 2006, 2: 711‐719.
4. Kim D.H. and Rossi J.J., Strategies for silencing human disease using RNA interference, Nat. Rev. Genet. 2007, 8: 173‐184.
5. Bertrand J.R., Pottier M., Vekris A., Opolon P. et al., Comparison of antisense oligonucleotides and siRNAs in cell culture and in vivo, Biochemical and Biophysical Research Communications 2002, 296: 1000‐1004.
6. Corey D.R., RNA learns from antisense, Nature Chemical Biology 2007, 3: 8‐11. 7. de Fougerolles A., Vornlocher H.P., Maraganore J., and Lieberman J., Interfering with disease: a progress
report on siRNA‐based therapeutics, Nature Reviews Drug Discovery 2007, 6: 443‐453. 8. Behlke M.A., Progress towards in vivo use of siRNAs, Molecular Therapy 2006, 13: 644‐670. 9. De Paula D., Bentley M.V., and Mahato R.I., Hydrophobization and bioconjugation for enhanced siRNA
delivery and targeting, RNA 2007, 13: 431‐456. 10. Dykxhoorn D.M. and Lieberman J., Knocking down disease with siRNAs, Cell 2006, 126: 231‐235. 11. Oliveira S., Storm G., and Schiffelers R.M., Targeted delivery of siRNA, Journal of Biomedicine and
Biotechnology 2006, 12. Bartlett D.W. and Davis M.E., Insights into the kinetics of siRNA‐mediated gene silencing from live‐cell and
live‐animal bioluminescent imaging, Nucleic Acids Research 2006, 34: 322‐333. 13. Bartlett D.W. and Davis M.E., Effect of siRNA nuclease stability on the in vitro and in vivo kinetics of siRNA‐
mediated gene silencing, Biotechnol. Bioeng. 2007, 97: 909‐921. 14. Kunisawa J., Masuda T., Katayama K., Yoshikawa T. et al., Fusogenic liposome delivers encapsulated
nanoparticles for cytosolic controlled gene release, Journal of controlled release 2005, 105: 344‐353. 15. Jeong J.H., Christensen L.V., Yockman J.W., Zhong Z.Y. et al., Reducible poly(amido ethylenimine) directed
to enhance RNA interference, Biomaterials 2007, 28: 1912‐1917. 16. Kakizawa Y., Furukawa S., and Kataoka K., Block copolymer‐coated calcium phosphate nanoparticles
sensing intracellular environment for oligodeoxynucleotide and siRNA delivery, Journal of controlled release 2004, 97: 345‐356.
17. Read M.L., Singh S., Ahmed Z., Stevenson M. et al., A versatile reducible polycation‐based system for efficient delivery of a broad range of nucleic acids, Nucleic Acids Research 2005, 33: e86.
18. Peppas N.A., Bures P., Leobandung W., and Ichikawa H., Hydrogels in pharmaceutical formulations, Eur. J. Pharm. Biopharm. 2000, 50: 27‐46.
19. Peppas N.A., Hilt J.Z., Khademhosseini A., and Langer R., Hydrogels in biology and medicine: From molecular principles to bionanotechnology, Advanced Materials 2006, 18: 1345‐1360.
20. Coviello T., Matricardi P., Marianecci C., and Alhaique F., Polysaccharide hydrogels for modified release formulations, Journal of controlled release 2007, 119: 5‐24.
21. Lin C.C. and Metters A.T., Hydrogels in controlled release formulations: Network design and mathematical modeling, Advanced Drug Delivery Reviews 2006, 58: 1379‐1408.
22. Van Tomme S.R. and Hennink W.E., Biodegradable dextran hydrogels for protein delivery applications, Expert Review of Medical Devices 2007, 4: 147‐164.
23. van Dijk‐Wolthuis W.N.E., Tsang S.K.Y., Kettenes‐vandenBosch J.J., and Hennink W.E., A new class of polymerizable dextrans with hydrolyzable groups: hydroxyethyl methacrylated dextran with and without oligolactate spacer, Polymer 1997, 38: 6235‐6242.
24. van Dijk‐Wolthuis W.N.E., van Steenbergen M.J., Underberg W.J.M., and Hennink W.E., Degradation kinetics of methacrylated dextrans in aqueous solution, Journal of Pharmaceutical Sciences 1997, 86: 413‐417.
25. van Dijk‐Wolthuis W.N.E., Hoogeboom J.A.M., van Steenbergen M.J., Tsang S.K.Y. et al., Degradation and release behavior of dextran‐based hydrogels, Macromolecules 1997, 30: 4639‐4645.
26. Stubbe B.G., Horkay F., Amsden B., Hennink W.E. et al., Tailoring the swelling pressure of degrading dextran hydroxyethyl methacrylate hydrogels, Biomacromolecules 2003, 4: 691‐695.
27. De Geest B.G., Dejugnat C., Verhoeven E., Sukhorukov G.B. et al., Layer‐by‐layer coating of degradable microgels for pulsed drug delivery, Journal of controlled release 2006, 116: 159‐169.
106

dextran microgels for time‐controlled delivery of siRNA | CHAPTER 3
28. Van Tomme S.R., van Steenbergen M.J., De Smedt S.C., van Nostrum C.F. et al., Self‐gelling hydrogels based on oppositely charged dextran microspheres, Biomaterials 2005, 26: 2129‐2135.
29. Cadee J.A., de Groot C.J., Jiskoot W., den Otter W. et al., Release of recombinant human interleukin‐2 from dextran‐based hydrogels, Journal of controlled release 2002, 78: 1‐13.
30. Vlugt‐Wensink K.D.F., Vlugt T.J.H., Jiskoot W., Crommelin D.J.A. et al., Modeling the release of proteins from degrading crosslinked dextran microspheres using kinetic Monte Carlo simulations, Journal of controlled release 2006, 111: 117‐127.
31. Franssen O., Vandervennet L., Roders P., and Hennink W.E., Degradable dextran hydrogels: controlled release of a model protein from cylinders and microspheres, Journal of controlled release 1999, 60: 211‐221.
32. De Geest B.G., Vandenbroucke R.E., Guenther A.M., Sukhorukov G.B. et al., Intracellularly degradable polyelectrolyte microcapsules, Advanced Materials 2006, 18: 1005.
33. Javier A.M., Kreft O., Alberola A.P., Kirchner C. et al., Combined atomic force microscopy and optical microscopy measurements as a method to investigate particle uptake by cells, Small 2006, 2: 394‐400.
34. Sanchez‐Martin R.M., Muzerelle M., Chitkul N., How S.E. et al., Bead‐based cellular analysis, sorting and multiplexing, Chembiochem 2005, 6: 1341‐1345.
35. Skirtach A.G., Javier A.M., Kreft O., Kohler K. et al., Laser‐induced release of encapsulated materials inside living cells, Angewandte Chemie‐International Edition 2006, 45: 4612‐4617.
36. Sukhorukov G.B., Rogach A.L., Zebli B., Liedl T. et al., Nanoengineered polymer capsules: Tools for detection, controlled delivery, and site‐specific manipulation, Small 2005, 1: 194‐200.
37. von Gersdorff K., Sanders N.N., Vandenbroucke R., De Smedt S.C. et al., The internalization route resulting in successful gene expression depends on polyethylenimine both cell line and polyplex type, Molecular Therapy 2006, 14: 745‐753.
107

CHAPTER 3 | dextran microgels for time‐controlled delivery of siRNA
108

advanced nanogel engineering for drug delivery | CHAPTER 4
109
CHAPTER 4 | Advanced nanogel engineering for drug delivery
Parts of this chapter are published:
Koen Raemdonck, Joseph Demeester and Stefaan C. De Smedt, Soft Matter 2009, 5: 707‐715
1Laboratory of General Biochemistry and Physical Pharmacy, Department of Pharmaceutics, Faculty of Pharmaceutical
Sciences, Ghent University, Harelbekestraat 72, B‐9000 Ghent, Belgium.

CHAPTER 4 | advanced nanogel engineering for drug delivery
110
ABSTRACT
Nanosized hydrogels (nanogels) have attracted considerable attention as multifunctional polymer‐
based drug delivery systems. Their versatility is demonstrated both in drug encapsulation and drug
release. Nanogels can be designed to facilitate the encapsulation of diverse classes of bioactive
compounds. With optimization of their molecular composition, size and morphology, nanogels can
be tailor‐made to sense and respond to environmental changes in order to ensure spatial and
stimuli‐controlled drug release in vivo. This chapter aims to highlight recent advances on the
interface between biology and nanomedicine with the emphasis on nanogels as carriers for
controlled drug delivery.

advanced nanogel engineering for drug delivery | CHAPTER 4
4 OVERVIEW CHAPTER CONTENTS
Chapter 4 provides:
• information on the design of nanogels and highlights on recent advanced preparation
protocols
• details on how nanogels can be used for the encapsulation and subsequent controlled
release of different classes of therapeutics, with the emphasis on the intracellular
environment
• insights in the possible applications of nanogels as drug delivery carriers and their ‘fine‐
tuning’ for in vivo delivery purposes
Key terms:
Crosslinking – hydrogel – dextran – stimuli‐responsive – PEGylation – controlled release
INTRODUCTION
In the past decade the quest for intelligent biomaterials in pharmaceutical applications has moved
into second gear. More than ever, the design of efficient drug delivery carriers has evolved as a
multidisciplinary effort where the interplay between material science, medicine and biology provides
innovative biofunctional materials.[1] Amongst the available biomaterials already described in the
literature, hydrogels have already proved their value in diverse biomedical applications. Hydrogels
are described as hydrophilic three‐dimensional polymer networks that are able to take up large
amounts of water or physiological fluid, while maintaining their internal network structure.[2,3] Their
high water content and low surface tension contribute to their biocompatibility. Some hydrogels
encompass a high loading capacity for bioactive compounds and are able to release their therapeutic
111

CHAPTER 4 | advanced nanogel engineering for drug delivery
payload in a controlled fashion. These unique features explain their widespread application in tissue
engineering, drug delivery and diagnostics.[4]
Although research has mainly focused on macroscopic hydrogels, there is now an increasing interest
in hydrogels confined to micro‐ and especially nanoscopic dimensions (termed micro‐ and
nanogels).[5,6] This is easily proven by a simple literature search on ‘nanogels’ and ‘drug delivery’ that
reveals an exponential growth in the number of publications during the last 10 years (Figure 4.1).
Colloidally stable nanogel particles have properties similar to those of their macrogel counterparts,
but they have the added value that e.g. upon intravenous injection, they can be deployed in areas of
the body that are not easily accessed by macroscopic hydrogels and microgels.[7] As most nanogels
are able to be taken up by cells they are ideal candidates for intracellular drug delivery. This is of
particular interest for therapeutic agents that need to be safely delivered into the cytoplasm of the
target cell, such as antisense oligonucleotides, siRNAs, peptides and various low molecular weight
chemotherapeutics. In addition, nanogel dispersions have a large surface area that can function as a
template for multivalent conjugations which is important to optimize the particles towards in vivo
applications. Moreover, their nanoscaled dimensions ensure that they respond very rapidly to
environmental stimuli, which is attractive when aiming for triggered drug delivery. Hydrogels, and
nanogels in particular, hold all the cards to be versatile drug delivery carriers for exploitation in
biomedicine, as already discussed in many review articles.[2‐6,8,9] In this paper, we especially aim to
highlight some of the most recent advances made in this emerging field of nanomaterials, with the
emphasis on nanogel design towards efficient application for drug delivery purposes.
DESIGNING NANOSIZED HYDROGELS
Hydrogel structures can be formed by introducing chemical or physical crosslinks between
hydrophilic natural or synthetic polymers.[10] These crosslinks are essential for the structural stability
of the hydrogel as they prevent dissolution of the polymer chains in the aqueous environment.
Chemical crosslinking involves the formation of covalent bonds, leading to an insoluble polymer
network. To obtain nanogels instead of macro‐ or microgels by chemical crosslinking, mostly (inverse)
emulsion polymerization[11] and precipitation polymerization[12] methods are applied. Alternatively,
also physical interactions between polymer chains (e.g. hydrogen bonds, chain entanglements, Van
der Waals forces, electrostatic interactions,…) can result in stable hydrogels. Physical self‐assembly
implies the controlled formation of nanosized aggregates as a function of polymer concentration[13‐15]
or environmental parameters such as temperature and pH.[9,16]
112
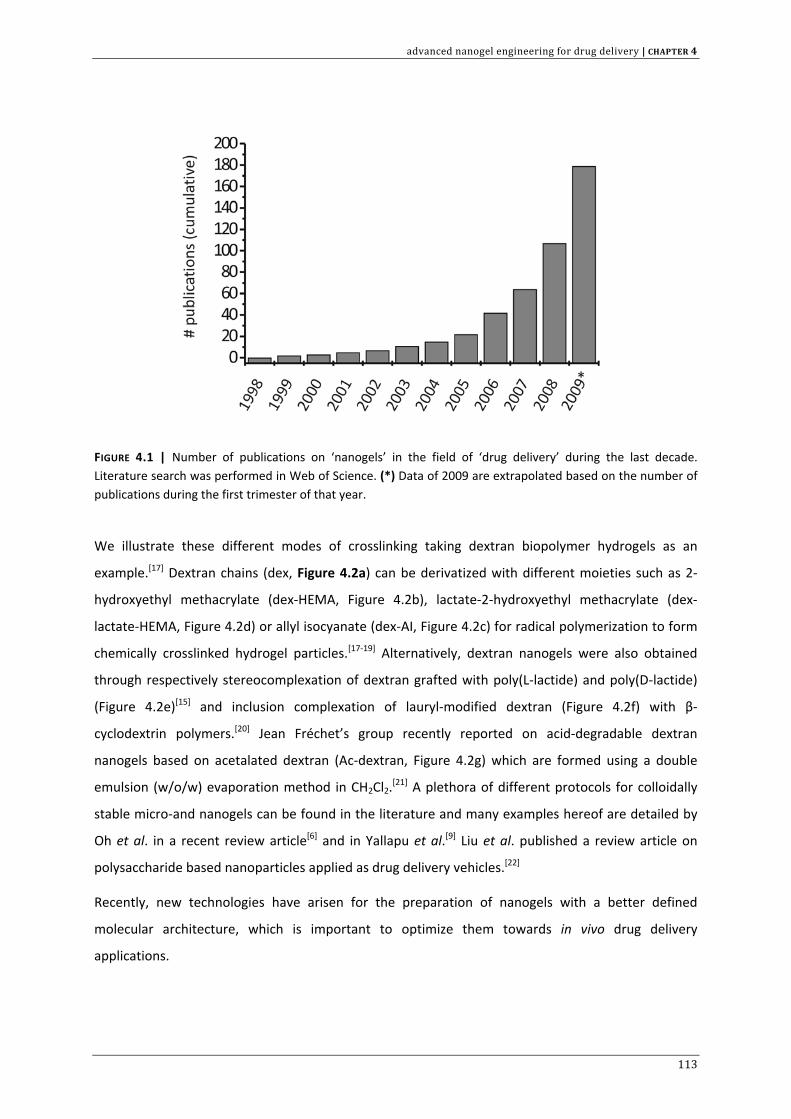
advanced nanogel engineering for drug delivery | CHAPTER 4
FIGURE 4.1 | Number of publications on ‘nanogels’ in the field of ‘drug delivery’ during the last decade. Literature search was performed in Web of Science. (*) Data of 2009 are extrapolated based on the number of publications during the first trimester of that year.
We illustrate these different modes of crosslinking taking dextran biopolymer hydrogels as an
example.[17] Dextran chains (dex, Figure 4.2a) can be derivatized with different moieties such as 2‐
hydroxyethyl methacrylate (dex‐HEMA, Figure 4.2b), lactate‐2‐hydroxyethyl methacrylate (dex‐
lactate‐HEMA, Figure 4.2d) or allyl isocyanate (dex‐AI, Figure 4.2c) for radical polymerization to form
chemically crosslinked hydrogel particles.[17‐19] Alternatively, dextran nanogels were also obtained
through respectively stereocomplexation of dextran grafted with poly(L‐lactide) and poly(D‐lactide)
(Figure 4.2e)[15] and inclusion complexation of lauryl‐modified dextran (Figure 4.2f) with β‐
cyclodextrin polymers.[20] Jean Fréchet’s group recently reported on acid‐degradable dextran
nanogels based on acetalated dextran (Ac‐dextran, Figure 4.2g) which are formed using a double
emulsion (w/o/w) evaporation method in CH2Cl2.[21] A plethora of different protocols for colloidally
stable micro‐and nanogels can be found in the literature and many examples hereof are detailed by
Oh et al. in a recent review article[6] and in Yallapu et al.[9] Liu et al. published a review article on
polysaccharide based nanoparticles applied as drug delivery vehicles.[22]
Recently, new technologies have arisen for the preparation of nanogels with a better defined
molecular architecture, which is important to optimize them towards in vivo drug delivery
applications.
113
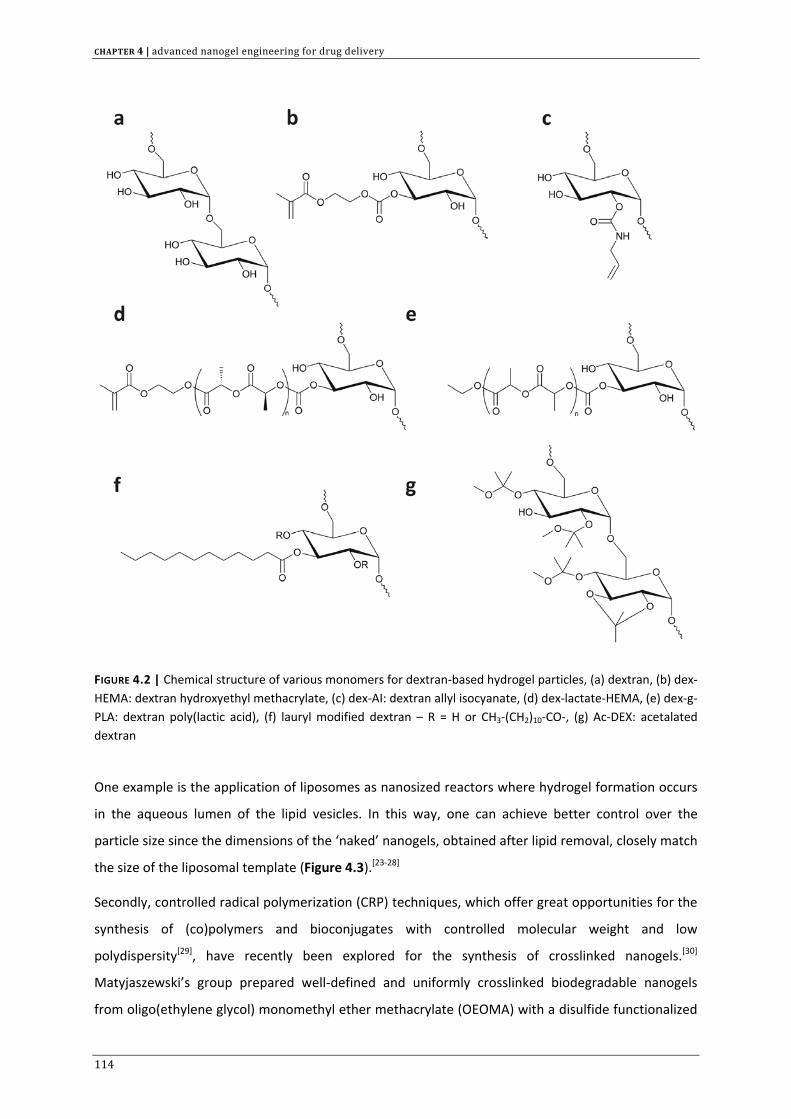
CHAPTER 4 | advanced nanogel engineering for drug delivery
FIGURE 4.2 | Chemical structure of various monomers for dextran‐based hydrogel particles, (a) dextran, (b) dex‐HEMA: dextran hydroxyethyl methacrylate, (c) dex‐AI: dextran allyl isocyanate, (d) dex‐lactate‐HEMA, (e) dex‐g‐PLA: dextran poly(lactic acid), (f) lauryl modified dextran – R = H or CH3‐(CH2)10‐CO‐, (g) Ac‐DEX: acetalated dextran
One example is the application of liposomes as nanosized reactors where hydrogel formation occurs
in the aqueous lumen of the lipid vesicles. In this way, one can achieve better control over the
particle size since the dimensions of the ‘naked’ nanogels, obtained after lipid removal, closely match
the size of the liposomal template (Figure 4.3).[23‐28]
Secondly, controlled radical polymerization (CRP) techniques, which offer great opportunities for the
synthesis of (co)polymers and bioconjugates with controlled molecular weight and low
polydispersity[29], have recently been explored for the synthesis of crosslinked nanogels.[30]
Matyjaszewski’s group prepared well‐defined and uniformly crosslinked biodegradable nanogels
from oligo(ethylene glycol) monomethyl ether methacrylate (OEOMA) with a disulfide functionalized
114

advanced nanogel engineering for drug delivery | CHAPTER 4
crosslinker using inverse mini‐emulsion atom transfer radical polymerization (ATRP).[31] These
nanogels showed potential as drug delivery carriers in their studies.[32,33] Another interesting example
is the photolithographic fabrication of sub‐micrometer particles using a technique called PRINT
(Particle Replication in Nonwetting Templates). PRINT applies photocurable perfluoropolyether
molds for imprint lithography to form uniform nanoparticles of well‐controlled size, shape and
composition.[34‐36] Gratton and coworkers used PRINT nano‐ and microgels to investigate the
influence of particle size, shape and surface chemistry on their cellular internalization. The
experimental data point out that cationic rod‐shaped particles are internalized at higher rates,
underlining the importance of particle design when intracellular targets are envisioned.[37] An
alternative nanoimprint photo‐lithographic approach (Step and Flash Imprint Lithography or S‐FIL) for
the preparation of crosslinked peptide nanogels was recently presented by Glangchai et al. (Figure
4.4).[38]
FIGURE 4.3 | Synthesis of nanogels using a liposomal template. Liposomes are prepared in the presence of a polymer solution. Non‐encapsulated polymer is removed or diluted below the critical polymerization concentration. Crosslinking of the polymers inside the lipid vesicles leads to the formation of lipid coated nanogels. Removal of the lipid coat by e.g. the addition of a detergent, finally results in naked nanogels with a size comparable to that of the liposomal template.
Rigorous control over the size of nanocolloids is essential for their in vivo biodistribution. While
microgels are often designed to release their payload in the extracellular space or in the cytoplasm of
dendritic cells following subcutaneous or intramuscular injection, nanogels may also be applied for
intravenous administration where they need to extravasate through the capillary endothelium to
reach the target tissue. The morphology of the capillary wall will set the size limits for particle
extravasation into the surrounding tissue. Discontinuous or sinusoidal capillaries can be found in
liver, spleen and bone marrow. This ‘leaky’ vasculature is also observed in inflammation sites and
tumours which allows extravasation of particles even up to 400 nm in diameter.[1,39] Since size is a
critical determinant for nanogels to reach the intended target following intravenous administration,
115
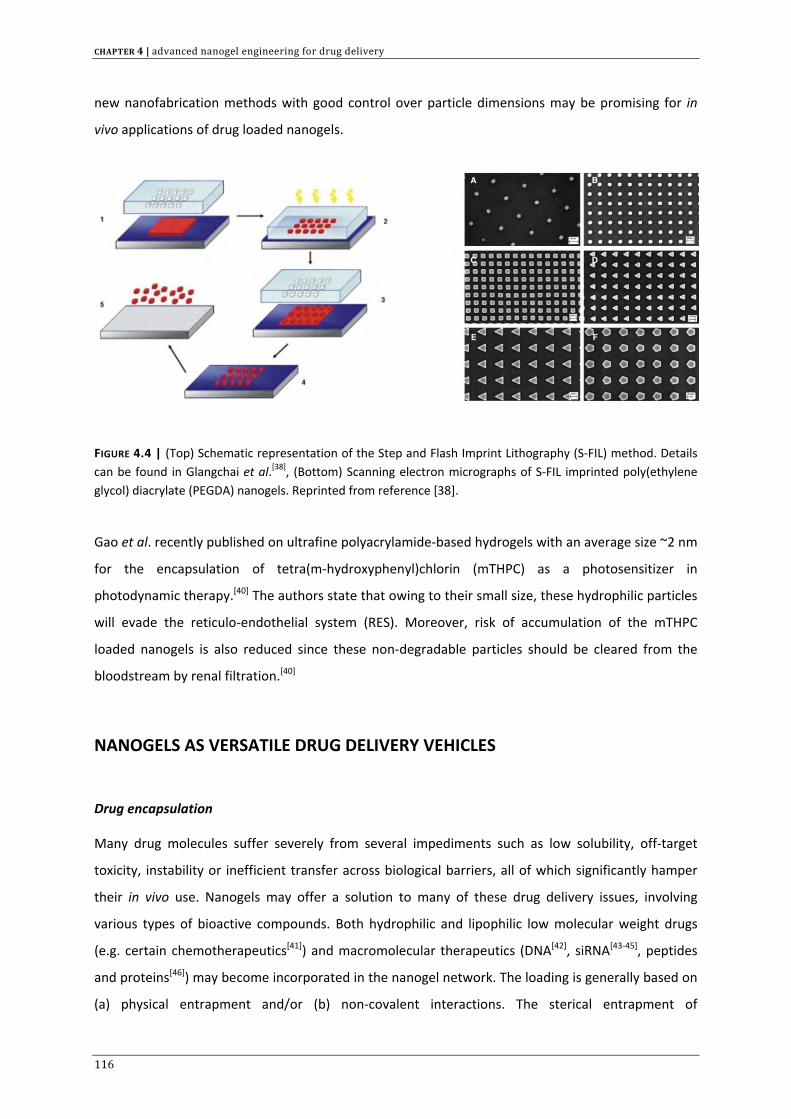
CHAPTER 4 | advanced nanogel engineering for drug delivery
new nanofabrication methods with good control over particle dimensions may be promising for in
vivo applications of drug loaded nanogels.
FIGURE 4.4 | (Top) Schematic representation of the Step and Flash Imprint Lithography (S‐FIL) method. Details can be found in Glangchai et al.[38], (Bottom) Scanning electron micrographs of S‐FIL imprinted poly(ethylene glycol) diacrylate (PEGDA) nanogels. Reprinted from reference [38].
Gao et al. recently published on ultrafine polyacrylamide‐based hydrogels with an average size ~2 nm
for the encapsulation of tetra(m‐hydroxyphenyl)chlorin (mTHPC) as a photosensitizer in
photodynamic therapy.[40] The authors state that owing to their small size, these hydrophilic particles
will evade the reticulo‐endothelial system (RES). Moreover, risk of accumulation of the mTHPC
loaded nanogels is also reduced since these non‐degradable particles should be cleared from the
bloodstream by renal filtration.[40]
NANOGELS AS VERSATILE DRUG DELIVERY VEHICLES
Drug encapsulation
Many drug molecules suffer severely from several impediments such as low solubility, off‐target
toxicity, instability or inefficient transfer across biological barriers, all of which significantly hamper
their in vivo use. Nanogels may offer a solution to many of these drug delivery issues, involving
various types of bioactive compounds. Both hydrophilic and lipophilic low molecular weight drugs
(e.g. certain chemotherapeutics[41]) and macromolecular therapeutics (DNA[42], siRNA[43‐45], peptides
and proteins[46]) may become incorporated in the nanogel network. The loading is generally based on
(a) physical entrapment and/or (b) non‐covalent interactions. The sterical entrapment of
116

advanced nanogel engineering for drug delivery | CHAPTER 4
therapeutics inside a hydrogel network implies in most cases that the compounds are present in the
polymer solution during the gelation process. However, it is conceivable that polymerization may
occur under conditions that are possibly detrimental for the therapeutic cargo. Therefore this
encapsulation technique obviously requires rigorous control over the gelation process to ensure drug
stability.[38,42,47] When the dimensions of the encapsulated therapeutics exceed the mesh size of the
hydrogel network, their diffusional leaching can be prevented. This encapsulation method is
therefore most feasible for macromolecular therapeutics,[38,42,43,48,49] especially when loosely
crosslinked or porous hydrogels are formed.
In contrast, non‐covalent interactions between the biological agent and the polymer matrix are the
basis of the research of Akiyoshi and co‐workers who focus on amphiphilic cholesterol‐modified
pullulan (CHP) nanogels of typically 20‐50 nm in diameter, mainly for the encapsulation of different
proteins.[50] Already in 1993, Akiyoshi disclosed that polysaccharides partially modified with
hydrophobic moieties could form nanoparticles through a self‐assembly process in an aqueous
environment.[51] The main driving force for protein encapsulation is the hydrophobic attraction
between the cholesteryl moieties in the nanogels and hydrophobic nanodomains in the protein of
interest. Recently, phase I clinical trials were conducted with CHP nanogels encapsulating truncated
HER2 protein for cancer vaccination. Patients received repetitive subcutaneous injections of the CHP‐
HER2 vaccine and results showed HER2 specific antibody and T‐cell immune responses.[52,53] A
drawback of this nanogel platform is their limitation for intravenous administration since abundant
serum proteins, like albumin, can destabilize the nanogel complex.[50] Especially when an intracellular
drug target is envisioned, the stability of drug delivery systems in the extracellular matrix should be
routinely tested to avoid premature drug release before the target cells have been reached.[54] For
the intracellular delivery of membrane‐impermeable compounds, amino‐functionalized CHP
nanogels were proposed to improve the efficiency of interactions with the cellular membrane and
their subsequent internalization. Once the cellular barrier is overcome, the authors assume that the
aforementioned protein exchange mechanism is also partially responsible for the intracellular release
of the encapsulated therapeutic protein.[46]
An alternative non‐covalent approach for the incorporation of therapeutics in charged nanogels is
based on electrostatic interactions between the biological agent and the ionized polymer matrix. This
approach has proved valuable for e.g. the complexation of low molecular weight anionic compounds
like nucleoside analog 5’‐triphosphates (NTPs) in crosslinked poly(ethylene glycol)‐poly(ethylene
imine) (PEG‐g‐PEI) nanogels (Figure 4.5)[55] Electrostatic complexation can also be exploited for
encapsulation of polynucleotides in cationic nanogels if the pore size of the nanogels is large enough
to allow penetration of the polynucleotides into the interior of the gel particle. In this way, antisense
117

CHAPTER 4 | advanced nanogel engineering for drug delivery
oligonucleotides[56‐58] and short interfering RNA (siRNA)[18,57] can be loaded in preformed cationic
hydrogel networks. On the other hand, Hu et al. reported on nanogels for the surface binding of
proteins, inactivated influenza virions (for the purpose of antigen delivery to dendritic cells) and
siRNA duplexes (for gene silencing activity). These (predominantly) anionic cargos are adsorbed onto
preformed core‐shell particles, based on electrostatic interactions with the positively charged outer
layer.[45]
FIGURE 4.5 | Schematic representation of PEG‐g‐PEI nanogels, electrostatically loaded with 5’‐ nucleoside triphosphate anticancer agents (5’‐NTPs). The nanogels consist of a PEI core for 5’‐NTP binding and a PEG envelope, containing targeting ligands (●). Reprinted and modified from Vinogradov et al. [55].
Exceptionally, bioactive compounds are incorporated into the nanogel interior through covalent
conjugation. One example is given by Standley and co‐workers who linked a methacrylate‐modified
CpG oligonucleotide ligand into acid‐degradable nanogels prepared through an inverse emulsion
radical polymerization technique.[49] In addition to the oligonucleotide strand, also ovalbumin (OVA)
as an antigen model was encapsulated. The incorporation of CpG oligonucleotides resulted in a more
potent production of interleukin‐12 and enhanced OVA‐specific CD8 T‐cell immunity in vivo due to
activation of Toll‐like receptor 9 (TLR‐9). However, covalent attachment is not feasible for every type
of drug or application as it may also alter the drug’s effectiveness.
Triggered drug release
Hydrogels tend to swell when they are brought into a polymer compatible medium due to solvent
penetration into free spaces between the macromolecular chains. The resulting volumetric
118

advanced nanogel engineering for drug delivery | CHAPTER 4
expansion of the hydrogel network is governed by the balance between the internal osmotic
pressure and its elastic deformability.[59] An attractive feature of hydrogels is that their swelling
behaviour can be influenced by a diversity of external triggers, such as changes in environmental pH,
ionic strength or temperature and the application of light or a magnetic or electric field.[2,5,60] A
plethora of stimuli‐responsive nanocarriers is described in the literature, e.g. polymer‐drug
conjugates, liposomes and micelles, which all have their strengths and weaknesses.[61] The latter two
examples are of particular interest in drug delivery, but they suffer from drawbacks such as a lack of
controlled loadability, limitations in material composition, limitations in the type of biological cargo
that can be loaded/released and/or physical instability.[7,9,35] For these reasons, many nanogel
platforms are often regarded as a better alternative. The responsiveness of ‘intelligent’ hydrogels is
largely dependent on the type of polymer used in making the gel.[62,63] Control over the swelling‐
deswelling transitions or other physical changes on a nanoscopic scale are especially of interest for
intracellular controlled drug delivery.
Among the class of ‘intelligent nanogels’, those responding to changes in temperature and pH are
most extensively investigated. Nanogels constructed from polymers bearing pendant basic or acidic
groups (ionic nanogels) are generally regarded as pH responsive.[62] When they are dispersed in an
aqueous medium of appropriate pH and ionic strength, ionization of pendant groups occurs, thereby
introducing fixed charges in the polymer network. By electrostatic repulsion the pores in the
nanogels enlarge, allowing excessive solvent influx, which eventually leads to their swelling. Cationic
nanogels swell at a pH below the pKa of the polymer network, while anionic nanogels are ionized at
higher pH. Small changes in the external pH may have a significant influence on the swelling
behaviour.[2] Na and Bae reported on self‐assembled pH‐sensitive nanogels, composed of a
sulphonamide‐pullulan acetate conjugate[64] and hydrophobized pullulan‐Na‐Boc‐L‐histidine[65] for
the tumor specific release of doxorubicin. Most solid tumours elicit a lower extracellular pH (pH ~6.8)
when compared with the pH in other tissues or the blood circulation (pH 7.4).[61,66,67] The particles
proposed by Na and Bae were stable at pH 7.4. However, at pH 6.8 the sulphonamide (SA) tagged
nanogels shrank and aggregated due to SA deionization while the histidine tagged nanogels swelled
as a result of imidazole protonation in the histidine moieties. In both cases drug release occurred
faster in response to these physical changes resulting into a more specific deposition of the drug in
the tumour microenvironment.
In the intracellular microenvironment, such pH responsive carriers may also prove beneficial. It is
known that nanocarriers are internalized mostly through an endocytotic mechanism upon interaction
with the cellular membrane. Endocytosis eventually confines the particles in an acidic environment
as the pH in the late endosomal and lysosomal lumen gradually lowers from neutral to ~pH 5.[68]
119

CHAPTER 4 | advanced nanogel engineering for drug delivery
Indeed, pH sensitive nanogels that undergo structural changes upon endocytosis could enhance the
intracellular bioavailability of the encapsulated drug. Chitosan nanogels around 180 nm in diameter
were described by Zhang and colleagues for the encapsulation of the anticancer agent methotrexate
disodium (MTX).[69] The nanogels originated from the ionic crosslinking of N‐[2‐hydroxy‐3‐
(trimethylammonium)propyl] chitosan chloride (HTCC) with sodium tripolyphosphate (TPP)
counterions. MTX is negatively charged and could be incorporated in the cationic HTCC nanogels at
physiological pH through electrostatic binding. Following efficient cellular internalization, these
nanogels showed a 2.2 fold increase in hydrodynamic diameter at endolysosomal pH due to higher
protonation of primary and secondary amines in HTCC and stronger electrostatic repulsion. As a
result, a faster diffusion driven release of MTX was achieved.[69] Shen et al. prepared chitosan
nanogels ~70‐80 nm through the crosslinking of chitosan with ethylenediamine tetraacetic acid
(EDTA). These nanogels exhibit a reversible change in surface charge and surface structure depending
on the environmental pH. The authors attempted to exploit this behaviour for the encapsulation and
release of the water‐insoluble anticancer drug camptothecin.[70] Similarly, Nagasaki’s lab showed pH‐
triggered release of doxorubicin due to protonation and subsequent swelling of ionic PEG‐poly[2‐
(N,N‐(diethylamino)ethyl methacrylate) nanogels at endolysosomal pH.[71,72] In particular for
biomacromolecules like protein antigens, pDNA and siRNA, an efficient transfer from the endosomal
compartment into the cytosol is crucial since otherwise they become degraded by endolysosomal
proteases and nucleases. Employing pH‐sensitive nanogels could enhance this endosomal escape
significantly. Indeed, it is thought that when nanogels are constructed from polyelectrolytes with
endosomal buffering capacity, they can disrupt endosomes via an osmotic pressure buildup (proton
sponge hypothesis). In addition, the swelling of the particles, due to endosomal protonation, may
facilitate endosomal destabilization.[45,73,74]
The pharmaceutical relevance of thermosensitive nanogels may lie within the treatment of local
infections (with elevated environmental temperature), tumour hyperthermia or by artificially
increasing temperature in the affected tissue to tailor local drug delivery.[7,61] Accumulation of a drug
loaded nanoparticle at the disease site, followed by a local temperature increase can trigger drug
release due to thermally triggered volume transitions. Poly(N‐isopropylacrylamide) or PNIPAAm is
the most widely investigated thermoresponsive polymer for biomedical applications.[63] PNIPAAm
nanogels can exhibit significant volume change in response to thermal fluctuations.[75‐78] In aqueous
solution the polymer has a lower critical solution temperature (LCST) around 32°C.[44] Below the LCST,
the polymer network is hydrated by hydrogen bonding of water to the amide side groups. When the
temperature increases towards or above the LCST, the polymer‐polymer hydrophobic interactions
become more dominant. As the hydrogen bonds are weakened, phase separation occurs and the
120

advanced nanogel engineering for drug delivery | CHAPTER 4
polymer chains collapse. The resulting expulsion of water and nanogel aggregation can be
accompanied with the release of both hydrophilic and hydrophobic drugs in the surrounding
medium.[79] A slightly different therapeutic concept, but also based on thermally triggered volume
transition, was recently proposed by Lee et al.. The authors prepared complex degradable nanogels
that exhibit a size increase from nano‐ to microscale when lowering the environmental temperature
from 37°C to 15°C. These nanogels are subsequently employed as ‘nanobombs’ to induce necrotic
cell death after cold‐shock treatment of cells that had internalized the nanogels in deswollen state.[80]
Modification of NIPAAm with different comonomers or post‐polymerization modifications on
PNIPAAm nanogels may significantly alter the hydrogel characteristics and shift the LCST. This feature
enables the application of PNIPAAm nanogels over a wide temperature range.[76] As an example,
incorporation of ionic and hydrophilic comonomers, such as acrylic acid (AAc) or methacrylic acid
(MAAc), increases the LCST and makes the nanogels sensitive both to changes in temperature as well
as changes in pH.[74,81‐86] A plethora of reports, describing different modifications of NIPAAm, can be
found in the literature.[5,6,63,83,86,87] A particularly interesting example of nanogel engineering is the
development of amphoteric, glucose‐responsive PNIPAAm based nanogels for triggered insulin
release.[88] Grafting of phenylboronic acid (PBA) to cationic‐anionic nanogels introduces glucose‐
responsiveness in the nanogels. The swelling‐deswelling behaviour of the nanogels as a function of
physiological glucose concentrations can be fine‐tuned by varying the relative amount of PBA,
cationic and anionic groups in the PNIPAAm hydrogel particles.[88] Besides pH and temperature
sensitive nanogels, also nanogels which are sensitive simultaneously to multiple triggers have already
been prepared. For instance, Bhattacharya et al. engineered hybrid nanogels that contain
ferromagnetic nanoparticles and which are responsive to both temperature, pH and magnetic
fields.[89]
Depending on the elastic properties of the polymer network, the deformation of hydrogels can be
reversible. In this way, hydrogels can regain their original shape and structure when the stimulus,
triggering the swelling or shrinking, is removed. This ‘shape memory effect’ can also have its
implications on drug delivery as recently disclosed by You Han Bae’s group. They prepared nanogels
consisting of a pH‐sensitive hydrophobic core of poly(L‐histidine‐co‐phenylalanine), held together by
a double hydrophilic shell.[90] These nanogels are colloidally stable at neutral pH, but swell
substantially at endosomal pH (< pH 6.8) explaining the faster release rate of encapsulated
doxorubicin at lower pH values. After endocytosis, the swelling of the nanogels together with the
buffering effect of poly(L‐histidine) is considered to disrupt the endosomal membranes, thereby
facilitating the transfer of the nanogels and the released doxorubicin to the cytoplasm. There the
nanogels shrink again to their original size, imparting further release of doxorubicin. The doxorubicin
121

CHAPTER 4 | advanced nanogel engineering for drug delivery
released intracellularly induces apoptosis that allows the nanogels, still containing sufficient amounts
of doxorubicin, to be taken up by neighbouring cells. In this way, doxorubicin loaded nanogels may
‘infect’ several cells and release the anticancer agent in a pH triggered pulsatile fashion (Figure
4.6).[90]
Different types of degradable nanogels are currently explored with the aim of enhancing the
intracellular delivery of encapsulated drugs. Moreover, degradability of the nanogel carrier is
essential in drug delivery to lower toxicity and avoid accumulation in the body upon repeated
administration. Labile bonds can be incorporated in the polymer backbone or in the crosslinks of the
hydrogel network. This class of nanogels releases its payload upon degradation in response to
specific intracellular stimuli such as pH[47], reducing agents or enzymatic activity. As a first example,
the Fréchet group reported on DNA and protein antigen encapsulation into acrylamide nanogels
copolymerized with an acid cleavable bisacrylamide acetal crosslinker.[42,48,49] These nanogels are
designed to degrade under the acidic conditions found in the phagolysosomes of Antigen Presenting
Cell’s (APCs). Upon lysosomal degradation of the nanogels, the encapsulated DNA can be released
and interact with TLR‐9 for enhanced cytokine production.[42,49]
Moreover, the accumulation of degradation fragments in the lysosomal lumen induces a rise in
osmotic pressure which is believed to destabilize the endosomal membrane and enables the transfer
of the encapsulated antigen to the cytoplasm for MHC class I processing.[48,49] Second, disulfide
crosslinked nanogels employ the intracellular reductive potential to deliver their therapeutic
payload.[91] It is known that disulfide linkages can be selectively cleaved inside the target cell by the
reductive environment in the endo/lysosomes and by cytoplasmic glutathione.[92] Lee et al.
succeeded in the fabrication of hyaluronic acid (HA) based nanogels around 200‐500 nm in size.[43]
For this purpose, thiol modified HA was crosslinked by ultrasonic treatment through the formation of
disulfide bonds in an inverse water‐in‐oil emulsification process. SiRNA was added to the HA solution
during the emulsification process which led to their physical entrapment inside the resulting
nanogels. Upon cellular internalisation, disulfide reduction disassembled the nanogels which resulted
in siRNA release. In a comparable approach, cationic PEI‐cl‐PEG/Pluronic nanogels[93] were made
degradable by the use of branched PEI polycations in which the 2 kDa PEI segments were linked via
disulfide bridges.[92] Very recently, Morimoto et al. reported on the self‐assembly of PNIPAAm‐g‐
pullulan chains carrying thiol end groups, resulting in nanogels responding to changes in
environmental temperature and redox potential. Below the LCST, only disulfide bridges secure
nanogel stability, while above the LCST the hydrophobic interactions between the PNIPAAm side
chains introduce additional physical crosslinks in the hydrogel network.[94]
122
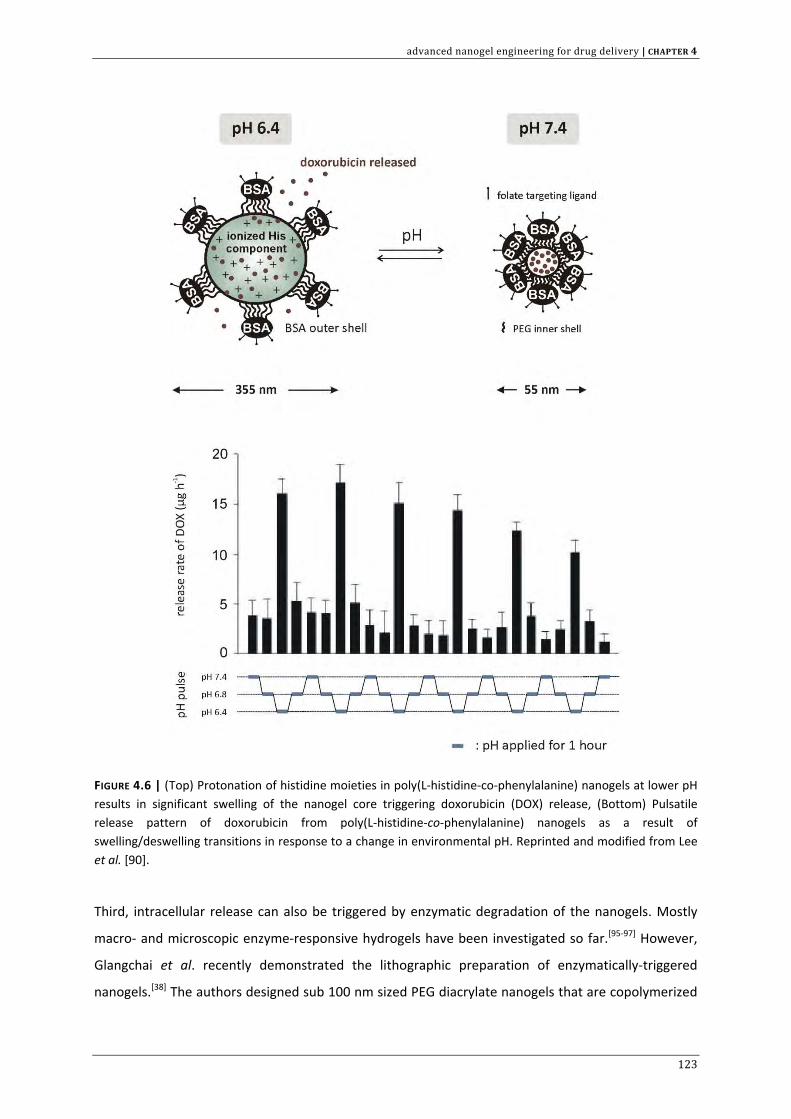
advanced nanogel engineering for drug delivery | CHAPTER 4
FIGURE 4.6 | (Top) Protonation of histidine moieties in poly(L‐histidine‐co‐phenylalanine) nanogels at lower pH results in significant swelling of the nanogel core triggering doxorubicin (DOX) release, (Bottom) Pulsatile release pattern of doxorubicin from poly(L‐histidine‐co‐phenylalanine) nanogels as a result of swelling/deswelling transitions in response to a change in environmental pH. Reprinted and modified from Lee et al. [90].
Third, intracellular release can also be triggered by enzymatic degradation of the nanogels. Mostly
macro‐ and microscopic enzyme‐responsive hydrogels have been investigated so far.[95‐97] However,
Glangchai et al. recently demonstrated the lithographic preparation of enzymatically‐triggered
nanogels.[38] The authors designed sub 100 nm sized PEG diacrylate nanogels that are copolymerized
123

CHAPTER 4 | advanced nanogel engineering for drug delivery
with an acrylated GFLGK peptide crosslinker. Enzymatic cleavage of the crosslinker by thiol proteases,
e.g. cathepsin B, releases the therapeutic payload which was shown for antibodies and pDNA. As this
class of enzymes is mainly located in the extracellular matrix of tumours and inside lysosomes, the
drug will be mainly released locally in these confined compartments.
Optimizing nanogel architecture for in vivo application
Ideally, drug delivery vehicles have to safely guide their therapeutic payload to the desired target
tissue. However, to reach the target tissue, several extracellular barriers need to be overcome.[1]
Shielding of the nanoparticle surface with inert, hydrophilic polymers and introducing targeting
ligands may significantly improve in vivo delivery and therapeutic efficacy (Figure 4.7). The
hydrophilic modification of nanoparticles can (a) prevent particle uptake by the mononuclear
phagocytic system, (b) decrease the recognition by the immune system and (c) enhance their
circulation time in the bloodstream. Indeed, the presence of a hydrophilic surface is known to reduce
non‐specific interactions with serum proteins and thereby also lowers the risk of phagocytosis by
immune cells as opsonisation is prevented.[98] Longer circulation times offer nanogels a better chance
of targeting the site of interest and may increase passive accumulation of drug loaded nanogels into
distant tumours due to the enhanced permeability and retention (EPR) effect. It has been described
that the leaky discontinuous vasculature often found in tumour tissue, together with the slower
efflux, can result in high local deposition of nanoparticles.[61,99,100] In addition, targeting of cell‐surface
receptors is a very attractive concept to achieve cell specific binding and subsequent internalization.
In this way, the fraction of the administered dose that accumulates at the target site can be
enhanced thereby also minimizing the risk of adverse effects. Many recent investigations have
evaluated both strategies for drug loaded nanogels.
With regard to the shielding of nanogels, most research is focused on PEG based nanogels or the
surface modification of nanogels with a hydrophilic PEG shell (also termed PEGylation). Several
groups constructed stimuli‐responsive core‐crosslinked polymeric micelles with a PEG corona.[101‐104]
However, some controversy exists on whether or not this type of nanocarriers can be regarded as
nanogels. Nagasaki and co‐workers copolymerized PEG, carrying a vinylbenzyl group, with 2‐(N,N‐
diethylamino)ethyl methacrylate to obtain cationic nanogels with a hydrophilic PEG shell.[71,72,105] Lee
et al. fabricated a double hydrophilic shell, consisting of PEG and bovine serum albumin (BSA), at the
surface of polypeptide nanogels.[90] Besides PEG, also other hydrophilic polymers have been
investigated for their ‘stealth’ properties, such as various polysaccharides.[106‐109]
124

advanced nanogel engineering for drug delivery | CHAPTER 4
FIGURE 4.7 | Biofunctionalization of stimuli‐responsive nanogels through the introduction of a hydrophilic shell and the coupling of targeting ligands may result in nanogel carriers that are of interest for clinical applications.
With regard to the active targeting of nanoparticles, the iron transporting plasma protein transferrin
is frequently used as a targeting ligand for tumour targeting since the expression of the transferrin
receptor is often upregulated in fast‐dividing tissues.[110] Chitosan[69], poly(NIPAAm) based[82,87] and
PEG‐cl‐PEI nanogels[93] were modified with transferrin to improve their cellular uptake and
intracellular drug delivery. The folate receptor is also overexpressed in many tumours[111], making
folic acid a prime candidate for active targeting of nanogels.[55,74,75,90,112] Other valuable approaches
include incorporating ligands for the asialoglycoprotein receptor, used in hepatocyte targeting[81,113],
or the conjugation of monoclonal antibodies for specific cell‐surface markers.[57,114] A recent
approach by the group of Andrew Lyon involves the coupling of a peptide shell to PNIPAAm core
nanogels for receptor mediated siRNA targeting towards carcinoma cells overexpressing the
erythropoietin producing hepatocellular A2 receptor (EphA2).[44]
CONCLUSIONS AND FUTURE PERSPECTIVES
Owing to our increasing knowledge on the biological barriers that nanomaterials encounter in vivo,
research in the field of drug delivery has gradually evolved to a top‐to‐bottom approach.[1] This
125

CHAPTER 4 | advanced nanogel engineering for drug delivery
approach implies that scientists now aim to design nanoparticles with appropriate drug delivery
properties in relation to the envisioned biological target. Nanogels fit extremely well into this
strategy, since they offer the possibility for stimuli‐controlled release of therapeutic agents. Several
proof‐of‐concept studies have highlighted the potential of such ‘intelligent’ nanogels that are able to
sense and respond to environmental changes. In this way, drug release can be controlled in a spatial
and temporal fashion. Especially attractive so far has been the design of nanogels that retain their
therapeutic payload in the extracellular environment but release it once they are internalized by
target cells, in response to an intracellular trigger. However, like for all types of nanoparticles under
investigation for drug delivery, the in vivo biodistribution of nanogels is mainly governed by their size
and surface properties which should also be taken into careful consideration in the engineering of
nanogels for clinical applications.
Despite the progress made in this field of nanomaterials there still is a long way to go before drug
loaded nanogels will reach the clinic. More than a decade of intensive research provided the
technological basis for the development of ‘intelligent’ soft nanomaterials. Nonetheless, new
developments both in engineering stimuli‐responsive materials, bioconjugation methods as in
controlled colloid synthesis are key considerations to further improve the design of advanced
hydrogel nanomaterials. Moreover, it must be emphasized that there is an urgent need for in vivo
data from nanogels in preclinical development. To translate the nanogel concept into a viable
therapeutic approach, it will indeed become increasingly important to study toxicity, immunogenicity
and pharmacokinetics together with drug effects in validated in vivo models.
Amongst the different classes of polymeric materials in drug delivery, polysaccharides are very
frequently used to prepare nanoparticles and nanogels. As abundant natural biomaterials, they can
be regarded as safe, biocompatible and they usually have a low processing cost.[22] Chapter 3 already
mentioned the use of methacrylate functionalized dextran (dex‐HEMA) for the production of
hydrogels and microgels. In the next chapter, this material will be investigated for the preparation of
nanosized hydrogels that are to be deployed as carriers of small interfering RNA (siRNA).
ACKNOWLEDGMENTS
Koen Raemdonck is a doctoral fellow of the Institute for the Promotion of Innovation through
Science and Technology in Flanders (IWT‐Vlaanderen). The authors would like to acknowledge the EU
(FP6) for funding of MediTrans, an integrated project on nanomedicines.
126

advanced nanogel engineering for drug delivery | CHAPTER 4
REFERENCE LIST
1. Remaut K., Sanders N.N., De Geest B.G., Braeckmans K. et al., Nucleic acid delivery: Where material sciences and bio‐sciences meet, Materials Science & Engineering R‐Reports 2007, 58: 117‐161.
2. Gupta P., Vermani K., and Garg S., Hydrogels: from controlled release to pH‐responsive drug delivery, Drug Discovery Today 2002, 7: 569‐579.
3. Peppas N.A., Bures P., Leobandung W., and Ichikawa H., Hydrogels in pharmaceutical formulations, European Journal of Pharmaceutics and Biopharmaceutics 2000, 50: 27‐46.
4. Peppas N.A., Hilt J.Z., Khademhosseini A., and Langer R., Hydrogels in biology and medicine: From molecular principles to bionanotechnology, Advanced Materials 2006, 18: 1345‐1360.
5. Nayak S. and Lyon L.A., Soft nanotechnology with soft nanoparticles, Angewandte Chemie‐International Edition 2005, 44: 7686‐7708.
6. Oh J.K., Drumright R., Siegwart D.J., and Matyjaszewski K., The development of microgels/nanogels for drug delivery applications, Progress in Polymer Science 2008, 33: 448‐477.
7. Malmsten M., Soft drug delivery systems, Soft Matter 2006, 2: 760‐769. 8. Ulanski P. and Rosiak J.M., polymeric nano/microgels, encyclopedia of nanoscience and nanotechnology
2008, 8: 845‐871. 9. Yallapu M.M., Reddy M.K., and Labhasetwar V., Nanogels: chemistry to drug delivery, Biomedical
applications of nanotechnology 2008, 131‐171. 10. Hennink W.E. and van Nostrum C.F., Novel crosslinking methods to design hydrogels, Advanced Drug
Delivery Reviews 2002, 54: 13‐36. 11. Landfester K., Synthesis of colloidal particles in miniemulsions, Annual Review of Materials Research
2006, 36: 231‐279. 12. Pelton R., Temperature‐sensitive aqueous microgels, Advances in Colloid and Interface Science 2000, 85:
1‐33. 13. Akiyama E., Morimoto N., Kujawa P., Ozawa Y. et al., Self‐assembled nanogels of cholesteryl‐modified
polysaccharides: Effect of the polysaccharide structure on their association characteristics in the dilute and semidilute regimes, Biomacromolecules 2007, 8: 2366‐2373.
14. Kuroda K., Fujimoto K., Sunamoto J., and Akiyoshi K., Hierarchical self‐assembly of hydrophobically modified pullulan in water: Gelation by networks of nanoparticles, Langmuir 2002, 18: 3780‐3786.
15. Nagahama K., Mori Y., Ohya Y., and Ouchi T., Biodegradable nanogel formation of polylactide‐grafted dextran copolymer in dilute aqueous solution and enhancement of its stability by stereocomplexation, Biomacromolecules 2007, 8: 2135‐2141.
16. Yu S.Y., Hu J.H., Pan X.Y., Yao P. et al., Stable and pH‐sensitive nanogels prepared by self‐assembly of chitosan and ovalbumin, Langmuir 2006, 22: 2754‐2759.
17. Van Tomme S.R. and Hennink W.E., Biodegradable dextran hydrogels for protein delivery applications, Expert Review of Medical Devices 2007, 4: 147‐164.
18. Raemdonck K., Van Thienen T.G., Vandenbroucke R.E., Sanders N.N. et al., Dextran microgels for time‐controlled delivery of siRNA, Advanced Functional Materials 2008, 18: 993‐1001.
19. Huang X., Misra G.P., Vaish A., Flanagan J.M. et al., Novel Nanogels with Both Thermoresponsive and Hydrolytically Degradable Properties, Macromolecules 2008, 41: 8339‐8345.
20. Daoud‐Mahammed S., Ringard‐Lefebvre C., Razzouq N., Rosilio V. et al., Spontaneous association of hydrophobized dextran and poly‐[beta]‐cyclodextrin into nanoassemblies.: Formation and interaction with a hydrophobic drug, Journal of Colloid and Interface Science 2007, 307: 83‐93.
21. Broaders K.E., Cohen J.A., Beaudette T.T., Bachelder E.M. et al., Acetalated dextran is a chemically and biologically tunable material for particulate immunotherapy, Proc. Natl. Acad. Sci. U. S. A 2009, 106: 5497‐5502.
22. Liu Z.H., Jiao Y.P., Wang Y.F., Zhou C.R. et al., Polysaccharides‐based nanoparticles as drug delivery systems, Advanced Drug Delivery Reviews 2008, 60: 1650‐1662.
23. Hong J.S., Vreeland W.N., Paoli Lacerda S.H., Locascio L.E. et al., Liposome‐templated supramolecular assembly of responsive alginate nanogels, Langmuir 2008, 24: 4092‐4096.
24. Kazakov S., Kaholek M., Teraoka I., and Levon K., UV‐induced gelation on nanometer scale using liposome reactor, Macromolecules 2002, 35: 1911‐1920.
25. Van Thienen T.G., Lucas B., Flesch F.M., van Nostrum C.F. et al., On the synthesis and characterization of biodegradable dextran nanogels with tunable degradation properties, Macromolecules 2005, 38: 8503‐8511.
127

CHAPTER 4 | advanced nanogel engineering for drug delivery
26. Van Thienen T.G., Raemdonck K., Demeester J., and De Smedt S.C., Protein release from biodegradable dextran nanogels, Langmuir 2007, 23: 9794‐9801.
27. Van Thienen T.G., Demeester J., and De Smedt S.C., Screening poly(ethyleneglycol) micro‐ and nanogels for drug delivery purposes, International Journal of Pharmaceutics 2008, 351: 174‐185.
28. An S.Y., Bui M.P., Nam Y.J., Han K.N. et al., Preparation of monodisperse and size‐controlled poly(ethylene glycol) hydrogel nanoparticles using liposome templates, J. Colloid Interface Sci. 2009, 331: 98‐103.
29. Braunecker W.A. and Matyjaszewski K., Controlled/living radical polymerization: Features, developments, and perspectives, Progress in Polymer Science 2007, 32: 93‐146.
30. Kim K.H., Kim J., and Jo W.H., Preparation of hydrogel nanoparticles by atom transfer radical polymerization of N‐isopropylacrylamide in aqueous media using PEG macro‐initiator, Polymer 2005, 46: 2836‐2840.
31. Oh J.K., Tang C.B., Gao H.F., Tsarevsky N.V. et al., Inverse miniemulsion ATRP: A new method for synthesis and functionalization of well‐defined water‐soluble/cross‐linked polymeric particles, Journal of the American Chemical Society 2006, 128: 5578‐5584.
32. Oh J.K., Siegwart D.J., Lee H.I., Sherwood G. et al., Biodegradable nanogels prepared by atom transfer radical polymerization as potential drug delivery carriers: Synthesis, biodegradation, in vitro release, and bioconjugation, Journal of the American Chemical Society 2007, 129: 5939‐5945.
33. Oh J.K., Siegwart D.J., and Matyjaszewski K., Synthesis and biodegradation of nanogels as delivery carriers for carbohydrate drugs, Biomacromolecules 2007, 8: 3326‐3331.
34. Euliss L.E., Dupont J.A., Gratton S., and DeSimone J., Imparting size, shape, and composition control of materials for nanomedicine, Chemical Society Reviews 2006, 35: 1095‐1104.
35. Napier M.E. and Desimone J.M., Nanoparticle drug delivery platform, Polymer Reviews 2007, 47: 321‐327.
36. Rolland J.P., Maynor B.W., Euliss L.E., Exner A.E. et al., Direct fabrication and harvesting of monodisperse, shape‐specific nanobiomaterials, Journal of the American Chemical Society 2005, 127: 10096‐10100.
37. Gratton S.E.A., Ropp P.A., Pohlhaus P.D., Luft J.C. et al., The effect of particle design on cellular internalization pathways, Proceedings of the National Academy of Sciences of the United States of America 2008, 105: 11613‐11618.
38. Glangchai L.C., Caldorera‐Moore M., Shi L., and Roy K., Nanoimprint lithography based fabrication of shape‐specific, enzymatically‐triggered smart nanoparticles, Journal of Controlled Release 2008, 125: 263‐272.
39. Brown J.M. and Giaccia A.J., The unique physiology of solid tumors: Opportunities (and problems) for cancer therapy, Cancer Research 1998, 58: 1408‐1416.
40. Gao D., Xu H., Philbert M.A., and Kopelman R., Ultrafine hydrogel nanoparticles: Synthetic approach and therapeutic application in living cells, Angewandte Chemie‐International Edition 2007, 46: 2224‐2227.
41. Missirlis D., Kawamura R., Tirelli N., and Hubbell J.A., Doxorubicin encapsulation and diffusional release from stable, polymeric, hydrogel nanoparticles, European Journal of Pharmaceutical Sciences 2006, 29: 120‐129.
42. Goh S.L., Murthy N., Xu M.C., and Frechet J.M.J., Cross‐linked microparticles as carriers for the delivery of plasmid DNA for vaccine development, Bioconjugate Chemistry 2004, 15: 467‐474.
43. Lee H., Mok H., Lee S., Oh Y.K. et al., Target‐specific intracellular delivery of siRNA using degradable hyaluronic acid nanogels, Journal of Controlled Release 2007, 119: 245‐252.
44. Blackburn W.H., Dickerson E.B., Smith M.H., McDonald J.F. et al., Peptide‐Functionalized Nanogels for Targeted siRNA Delivery, Bioconjug. Chem. 2009, 286: 563‐569.
45. Hu Y., Atukorale P.U., Lu J.J., Moon J.J. et al., Cytosolic delivery mediated via electrostatic surface binding of protein, virus, or siRNA cargos to pH‐responsive core‐shell gel particles, Biomacromolecules 2009, 10: 756‐765.
46. Ayame H., Morimoto N., and Akiyoshi K., Self‐assembled cationic nanogels for intracellular protein delivery, Bioconjugate Chemistry 2008, 19: 882‐890.
47. Shi L.J., Khondee S., Linz T.H., and Berkland C., Poly(N‐vinylformamide) nanogels capable of pH‐sensitive protein release, Macromolecules 2008, 41: 6546‐6554.
48. Kwon Y.J., Standley S.M., Goh S.L., and Fréchet J.M.J., Enhanced antigen presentation and immunostimulation of dendritic cells using acid‐degradable cationic nanoparticles, Journal of Controlled Release 2005, 105: 199‐212.
128

advanced nanogel engineering for drug delivery | CHAPTER 4
49. Standley S.M., Mende I., Goh S.L., Kwon Y.J. et al., Incorporation of CpG oligonucleotide ligand into protein‐loaded particle vaccines promotes antigen‐specific CD8 T‐cell immunity, Bioconjugate Chemistry 2007, 18: 77‐83.
50. Shimizu T., Kishida T., Hasegawa U., Ueda Y. et al., Nanogel DDS enables sustained release of IL‐12 for tumor immunotherapy, Biochemical and Biophysical Research Communications 2008, 367: 330‐335.
51. Akiyoshi K., Deguchi S., Moriguchi N., Yamaguchi S. et al., Self‐Aggregates of Hydrophobized Polysaccharides in Water ‐ Formation and Characteristics of Nanoparticles, Macromolecules 1993, 26: 3062‐3068.
52. Kageyama S., Kitano S., Hirayama M., Nagata Y. et al., Humoral immune responses in patients vaccinated with 1‐146 HER2 protein complexed with cholesteryl pullulan nanogel, Cancer Science 2008, 99: 601‐607.
53. Kitano S., Kageyama S., Nagata Y., Miyahara Y. et al., HER2‐specificT‐cell immune responses in patients vaccinated with truncated HER2 protein complexed with nanogels of cholesteryl pullulan, Clinical Cancer Research 2006, 12: 7397‐7405.
54. Buyens K., Lucas B., Raemdonck K., Braeckmans K. et al., A fast and sensitive method for measuring the integrity of siRNA‐carrier complexes in full human serum, Journal of Controlled Release 2008, 126: 67‐76.
55. Vinogradov S.V., Zeman A.D., Batrakova E.V., and Kabanov A.V., Polyplex Nanogel formulations for drug delivery of cytotoxic nucleoside analogs, Journal of Controlled Release 2005, 107: 143‐157.
56. McAllister K., Sazani P., Adam M., Cho M.J. et al., Polymeric nanogels produced via inverse microemulsion polymerization as potential gene and antisense delivery agents, Journal of the American Chemical Society 2002, 124: 15198‐15207.
57. Vinogradov S.V., Colloidal microgels in drug delivery applications, Current Pharmaceutical Design 2006, 12: 4703‐4712.
58. Vinogradov S., Batrakova E., and Kabanov A., Poly(ethylene glycol)‐polyethyleneimine NanoGel(TM) particles: novel drug delivery systems for antisense oligonucleotides, Colloids and Surfaces B: Biointerfaces 1999, 16: 291‐304.
59. Eichenbaum G.M., Kiser P.F., Simon S.A., and Needham D., pH and ion‐triggered volume response of anionic hydrogel microspheres, Macromolecules 1998, 31: 5084‐5093.
60. Ahn S.K., Kasi R.M., Kim S.C., Sharma N. et al., Stimuli‐responsive polymer gels, Soft Matter 2008, 4: 1151‐1157.
61. Ganta S., Devalapally H., Shahiwala A., and Amiji M., A review of stimuli‐responsive nanocarriers for drug and gene delivery, Journal of Controlled Release 2008, 126: 187‐204.
62. Dai S., Ravi P., and Tam K.C., pH‐Responsive polymers: synthesis, properties and applications, Soft Matter 2008, 4: 435‐449.
63. Rzaev Z.M.O., Dincer S., and Piskin E., Functional copolymers of N‐isopropylacrylamide for bioengineering applications, Progress in Polymer Science 2007, 32: 534‐595.
64. Na K. and Bae Y.H., Self‐assembled hydrogel nanoparticles responsive to tumor extracellular pH from pullulan derivative/sulfonamide conjugate: Characterization, aggregation, and adriamycin release in vitro, Pharmaceutical Research 2002, 19: 681‐688.
65. Na K., Lee E.S., and Bae Y.H., Self‐organized nanogels responding to tumor extracellular pH: pH‐dependent drug release and in vitro cytotoxicity against MCF‐7 cells, Bioconjugate Chemistry 2007, 18: 1568‐1574.
66. Gerweck L.E. and Seetharaman K., Cellular pH gradient in tumor versus normal tissue: Potential exploitation for the treatment of cancer, Cancer Research 1996, 56: 1194‐1198.
67. Stubbs M., McSheehy P.M.J., Griffiths J.R., and Bashford C.L., Causes and consequences of tumour acidity and implications for treatment, Molecular Medicine Today 2000, 6: 15‐19.
68. Van Dyke R.W., Acidification of lysosomes and endosomes, Subcell. Biochem. 1996, 27: 331‐360. 69. Zhang H., Mardyani S., Chan W.C.W., and Kumacheva E., Design of biocompatible chitosan microgels for
targeted pH‐mediated intracellular release of cancer therapeutics, Biomacromolecules 2006, 7: 1568‐1572.
70. Shen X.C., Zhang L.Y., Jiang X.Q., Hu Y. et al., Reversible surface switching of nanogel triggered by external stimuli, Angewandte Chemie‐International Edition 2007, 46: 7104‐7107.
71. Hayashi H., Iijima M., Kataoka K., and Nagasaki Y., pH‐sensitive nanogel possessing reactive PEG tethered chains on the surface, Macromolecules 2004, 37: 5389‐5396.
72. Oishi M., Hayashi H., Michihiro I.D., and Nagasaki Y., Endosomal release and intracellular delivery of anticancer drugs using pH‐sensitive PEGylated nanogels, Journal of Materials Chemistry 2007, 17: 3720‐3725.
129

CHAPTER 4 | advanced nanogel engineering for drug delivery
73. Hu Y., Litwin T., Nagaraja A.R., Kwong B. et al., Cytosolic delivery of membrane‐impermeable molecules in dendritic cells using pH‐Responsive core‐shell nanoparticles, Nano Letters 2007, 7: 3056‐3064.
74. Soppimath K.S., Liu L.H., Seow W.Y., Liu S.Q. et al., Multifunctional core/shell nanoparticles self‐assembled from pH‐induced thermosensitive polymers for targeted intracellular anticancer drug delivery, Advanced Functional Materials 2007, 17: 355‐362.
75. Blackburn W.H. and Lyon L.A., Size‐controlled synthesis of monodisperse core/shell nanogels, Colloid and Polymer Science 2008, 286: 563‐569.
76. Debord J.D. and Lyon L.A., Synthesis and characterization of pH‐responsive copolymer microgels with tunable volume phase transition temperatures, Langmuir 2003, 19: 7662‐7664.
77. Gan D.J. and Lyon L.A., Tunable swelling kinetics in core‐shell hydrogel nanoparticles, Journal of the American Chemical Society 2001, 123: 7511‐7517.
78. Kuckling D., Vo C.D., Adler H.J.P., Volkel A. et al., Preparation and characterization of photo‐cross‐linked thermosensitive PNIPAAm nanogels, Macromolecules 2006, 39: 1585‐1591.
79. Shin Y.S., Chang J.H., Liu J., Williford R. et al., Hybrid nanogels for sustainable positive thermosensitive drug release, Journal of Controlled Release 2001, 73: 1‐6.
80. Lee Y., Park S.Y., Kim C., and Park T.G., Thermally triggered intracellular explosion of volume transition nanogels for necrotic cell death, J. Control Release 2009, 135: 89‐95.
81. Choi S.H., Yoon J.J., and Park T.G., Galactosylated poly(N‐isopropylacrylamide) hydrogel submicrometer particles for specific cellular uptake within hepatocytes, Journal of Colloid and Interface Science 2002, 251: 57‐63.
82. Das M., Mardyani S., Chan W.C.W., and Kumacheva E., Biofunctionalized pH‐responsive microgels for cancer cell targeting: Rational design, Advanced Materials 2006, 18: 80‐83.
83. Soppimath K.S., Tan D.C.W., and Yang Y.Y., pH‐triggered thermally responsive polymer core‐shell nanoparticles for drug delivery, Advanced Materials 2005, 17: 318.
84. Teng D.Y., Hou J.L., Zhang X.G., Wang X. et al., Glucosamine‐carrying temperature‐ and pH‐sensitive microgels: Preparation, characterization, and in vitro drug release studies, Journal of Colloid and Interface Science 2008, 322: 333‐341.
85. Tian P., Wu Q.L., and Lian K., Preparation of temperature‐ and pH‐sensitive, stimuli‐responsive poly(N‐isopropylacrylamide‐co‐methacrylic acid) nanoparticles, Journal of Applied Polymer Science 2008, 108: 2226‐2232.
86. Zhang L.Y., Guo R., Yang M., Jiang X.Q. et al., Thermo and pH dual‐responsive nanoparticles for anti‐cancer drug delivery, Advanced Materials 2007, 19: 2988‐+.
87. Quan C.Y., Sun Y.X., Cheng H., Cheng S.X. et al., Thermosensitive P(NIPAAm‐co‐PAAc‐co‐HEMA) nanogels conjugated with transferrin for tumor cell targeting delivery, Nanotechnology 2008, 19.
88. Hoare T. and Pelton R., Charge‐switching, amphoteric glucose‐responsive microgels with physiological swelling activity, Biomacromolecules 2008, 9: 733‐740.
89. Bhattacharya S., Eckert F., Boyko V., and Pich A., Temperature‐, pH‐, and magnetic‐field‐sensitive hybrid microgels, Small 2007, 3: 650‐657.
90. Lee E.S., Kim D., Youn Y.S., Oh K.T. et al., A virus‐mimetic nanogel vehicle, Angewandte Chemie‐International Edition 2008, 47: 2418‐2421.
91. Bae K.H., Mok H., and Park T.G., Synthesis, characterization, and intracellular delivery of reducible heparin nanogels for apoptotic cell death, Biomaterials 2008, 29: 3376‐3383.
92. Kohli E., Han H.Y., Zeman A.D., and Vinogradov S.V., Formulations of biodegradable Nanogel carriers with 5'‐triphosphates of nucleoside analogs that display a reduced cytotoxicity and enhanced drug activity, Journal of Controlled Release 2007, 121: 19‐27.
93. Vinogradov S.V., Kohli E., and Zeman A.D., Comparison of nanogel drug carriers and their formulations with nucleoside 5 '‐triphosphates, Pharmaceutical Research 2006, 23: 920‐930.
94. Morinloto N., Qiu X.P., Winnik F.M., and Akiyoshi K., Dual stimuli‐responsive nanogels by self‐assembly of polysaccharides lightly grafted with thiol‐terminated poly(N‐isopropylacrylamide) chains, Macromolecules 2008, 41: 5985‐5987.
95. Thornton P.D., Mart R.J., and Ulijn R.V., Enzyme‐responsive polymer hydrogel particles for controlled release, Advanced Materials 2007, 19: 1252..
96. Thornton P.D., Mart R.J., Webb S.J., and Ulijn R.V., Enzyme‐responsive hydrogel particles for the controlled release of proteins: designing peptide actuators to match payload, Soft Matter 2008, 4: 821‐827.
97. Ulijn R.V., Enzyme‐responsive materials: a new class of smart biomaterials, Journal of Materials Chemistry 2006, 16: 2217‐2225.
130

advanced nanogel engineering for drug delivery | CHAPTER 4
98. Owens D.E. and Peppas N.A., Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles, International Journal of Pharmaceutics 2006, 307: 93‐102.
99. Cho K.J., Wang X., Nie S.M., Chen Z. et al., Therapeutic nanoparticles for drug delivery in cancer, Clinical Cancer Research 2008, 14: 1310‐1316.
100. Maeda H., The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor‐selective macromolecular drug targeting, 2001, 189‐207.
101. Bontha S., Kabanov A.V., and Bronich T.K., Polymer micelles with cross‐linked ionic cores for delivery of anticancer drugs, Journal of Controlled Release 2006, 114: 163‐174.
102. Rijcken C.J., Snel C.J., Schiffelers R.M., van Nostrum C.F. et al., Hydrolysable core‐crosslinked thermosensitive polymeric micelles: Synthesis, characterisation and in vivo studies, Biomaterials 2007, 28: 5581‐5593.
103. Tamura A., Oishi M., and Nagasaki Y., Enhanced Cytoplasmic Delivery of siRNA Using a Stabilized Polyion Complex Based on PEGylated Nanogels with a Cross‐Linked Polyamine Structure, Biomacromolecules 2009, doi: 10.1021/bm900252d.
104. Yusa S., Sugahara M., Endo T., and Morishima Y., Preparation and characterization of a pH‐responsive nanogel based on a photo‐cross‐linked micelle formed from block copolymers with controlled structure, Langmuir 2009, 25: 5258‐5265.
105. Oishi M. and Nagasaki Y., Synthesis, characterization, and biomedical applications of core‐shell‐type stimuli‐responsive nanogels ‐ Nanogel composed of poly[2‐(N,N‐diethylamino)ethyl methacrylate] core and PEG tethered chains, Reactive & Functional Polymers 2007, 67: 1311‐1329.
106. Lemarchand C., Gref R., and Couvreur P., Polysaccharide‐decorated nanoparticles, European Journal of Pharmaceutics and Biopharmaceutics 2004, 58: 327‐341.
107. Lemarchand C., Gref R., Passirani C., Garcion E. et al., Influence of polysaccharide coating on the interactions of nanoparticles with biological systems, Biomaterials 2006, 27: 108‐118.
108. Li J., Yu S.Y., Yao P., and Jiang M., Lysozyme‐dextran core‐shell nanogels prepared via a green process, Langmuir 2008, 24: 3486‐3492.
109. Labarre D., Vauthier C., Chauvierre C., Petri B. et al., Interactions of blood proteins with poly(isobutylcyanoacrylate) nanoparticles decorated with a polysaccharidic brush, Biomaterials 2005, 26: 5075‐5084.
110. Ogris M. and Wagner E., Targeting tumors with non‐viral gene delivery systems, Drug Discovery Today 2002, 7: 479‐485.
111. Leamon C.P. and Low P.S., Folate‐mediated targeting: from diagnostics to drug and gene delivery, Drug Discovery Today 2001, 6: 44‐51.
112. Nayak S., Lee H., Chmielewski J., and Lyon L.A., Folate‐mediated cell targeting and cytotoxicity using thermoresponsive microgels, Journal of the American Chemical Society 2004, 126: 10258‐10259.
113. Na K., Park K.H., Kim S.W., and Bae Y.H., Self‐assembled hydrogel nanoparticles from curdlan derivatives: characterization, anti‐cancer drug release and interaction with a hepatoma cell line (HepG2), Journal of Controlled Release 2000, 69: 225‐236.
114. Kwon Y.J., James E., Shastri N., and Frechet J.M.J., In vivo targeting of dendritic cells for activation of cellular immunity using vaccine carriers based on pH‐responsive microparticles, Proceedings of the National Academy of Sciences of the United States of America 2005, 102: 18264‐18268.
131

CHAPTER 4 | advanced nanogel engineering for drug delivery
132

biodegradable dextran nanogels for RNAi | CHAPTER 5
CHAPTER 5 | Biodegradable dextran nanogels for RNAi: focusing on endosomal escape and intracellular
siRNA delivery
Parts of this chapter are published:
Koen Raemdonck1, Broes Naeye1, Kevin Buyens1, Roosmarijn E. Vandenbroucke1, Anders Høgset2,
Joseph Demeester1 and Stefaan C. De Smedt1, Advanced Functional Materials 2009, 19: 1406‐1415.
1Laboratory of General Biochemistry and Physical Pharmacy, Department of Pharmaceutics, Faculty of Pharmaceutical
Sciences, Ghent University, Harelbekestraat 72, B‐9000 Ghent, Belgium.
2PCI Biotech, Hoffsveien 48, N‐0377, Oslo, Norway
133

CHAPTER 5 | biodegradable dextran nanogels for RNAi
134
ABSTRACT
The successful therapeutic application of small interfering RNA (siRNA) largely relies on the
development of safe and effective delivery systems that are able to guide the siRNA therapeutics to
the cytoplasm of the target cell. In this report, biodegradable cationic dextran nanogels are
engineered by inverse emulsion photopolymerization and their potential as siRNA carriers is
evaluated. The nanogels are able to entrap siRNA with a high loading capacity, based on electrostatic
interaction. Once inside the cells, the degradation of the nanogel core by ester hydrolysis again
releases the siRNA cargo. Confocal microscopy and flow cytometry analysis revealed that high
amounts of siRNA loaded nanogels can be internalized by Huh‐7 human hepatoma cells, without
significant cytotoxicity. Following their endocytic uptake, it is found that they are mainly trafficked
towards the endolysosomes. Since endosomal confinement of siRNA carrying nanoparticles is known
to be one of the most important intracellular barriers for efficient gene silencing, we investigated the
influence of two different strategies to enhance endosomal escape on the extent of gene silencing.
We found that both the application of photochemical internalization (PCI) and the use of an
influenza‐derived fusogenic peptide (diINF‐7), could significantly improve the silencing efficiency of
our siRNA loaded nanogels. Furthermore, it is shown that an efficient gene silencing requires the
degradation of the nanogels. As the degradation kinetics of the nanogels can easily be tailored, these
particles show potential for intracellular controlled release of small interfering RNA.

biodegradable dextran nanogels for RNAi | CHAPTER 5
5 OVERVIEW CHAPTER CONTENTS
Chapter 5 provides:
• a protocol for the production of nanosized dextran hydrogel particles (nanogels) and their
subsequent loading with siRNA
• an evaluation of their cellular internalization and the subsequent intracellular siRNA release
• details regarding the improvement of intracellular siRNA delivery by application of validated
endosomolytic tools
Key terms:
Nanogel – siRNA – cationic – flow cytometry – PCI – fusogenic peptide ‐ luciferase
INTRODUCTION
RNAi is now generally regarded as a promising strategy to modulate aberrant expression of disease‐
causing genes in the treatment of among others, cancer, viral infections, inflammatory diseases, and
hypercholesterolemia.[1‐3] One approach to evoking RNAi is by the intracellular delivery of chemically
synthesized siRNAs. However, the relatively large size of siRNA duplexes (approximately 14 kDa; 1 Da
equals 1.66 x 10‐27 kg) and their negative charge (~40 ionized phosphate groups at physiological pH)
do not allow them to spontaneously cross cellular membranes. As a consequence, clinical
applications of siRNA therapeutics require the development of delivery strategies that can guide
intact siRNA into the cytoplasm of the target cell, where the RNAi machinery resides.[4,5] Both viral
and non‐viral carriers have been used for the intracellular delivery of RNAi molecules. Although viral
carriers are beyond doubt the most efficient delivery systems for nucleic acids, they still suffer from
135

CHAPTER 5 | biodegradable dextran nanogels for RNAi
important disadvantages, such as the risk of insertional mutagenesis [6], undesired immunogenicity [7]
and challenging and expensive large‐scale production. For these reasons, many research groups now
focus on non‐viral siRNA delivery. The use of non‐viral carriers is often based on electrostatic
complexation of the negatively charged siRNA with cationic lipids [8] (to form lipoplexes) or polymers
(to form polyplexes).[9‐11]
We previously reported cationic microgels (mean diameter ~2‐3 µm) that consist of dextran
hydroxyethyl methacrylate (dex‐HEMA, Figure 5.1a), which was copolymerized with cationic
methacrylate monomers. The functionalization of dextran with methacrylate moieties, as discussed
in Chapter 3, makes it feasible to incorporate different functionalities in the dextran hydrogel
network by copolymerization with varying methacrylate monomers. In this way, the physicochemical
properties of the hydrogel and its behaviour in a biological system may be adjusted according to the
final needs. HEMA is coupled to the dextran backbone through a carbonate ester that is subject to
hydrolysis under physiological conditions, which makes the cationic hydrogel network
biodegradable.[12,13] We showed in Chapter 3 that the micron‐sized dex‐HEMA gel particles allow a
slow release of encapsulated siRNA over several days.[14] In the present chapter, we aim to translate
this concept to the nanoscale by designing cationic dex‐HEMA nanogels in order to achieve time‐
controlled siRNA delivery in an intracellular environment. Cationic biodegradable dextran nanogels
were synthesized and their ability to complex and subsequently release the entrapped siRNA was
evaluated. Data on cellular internalization and intracellular trafficking were linked to the eventual
biological RNAi effect in a human hepatoma cell line. Since cationic nanogels are most likely
internalized through endocytosis, the effect of validated endosomolytic tools on the extent of gene
silencing was evaluated. Finally, to gain more insight into the exact mechanism of intracellular siRNA
delivery, we conducted control experiments with non‐degradable cationic dextran methacrylate
nanogels (dex‐MA, Figure 5.1a).
EXPERIMENTAL SECTION
Preparation of cationic dextran nanogels
For the preparation of positively charged dextran nanogels, a mini‐emulsion photo‐polymerization
method was applied (Figure 5.1b). Briefly, 100 mg dextran hydroxyethyl methacrylate or dextran
methacrylate (dex‐HEMA or dex‐MA, Figure 5.1a) was dissolved in 20 mM HEPES buffer pH 7.4 and
supplemented with a known amount of the cationic methacrylate monomer [2‐(methacryloyloxy)‐
ethyl] trimethylammonium chloride (TMAEMA, Figure 5.1a, Sigma) to a final volume of 340 µL.
136
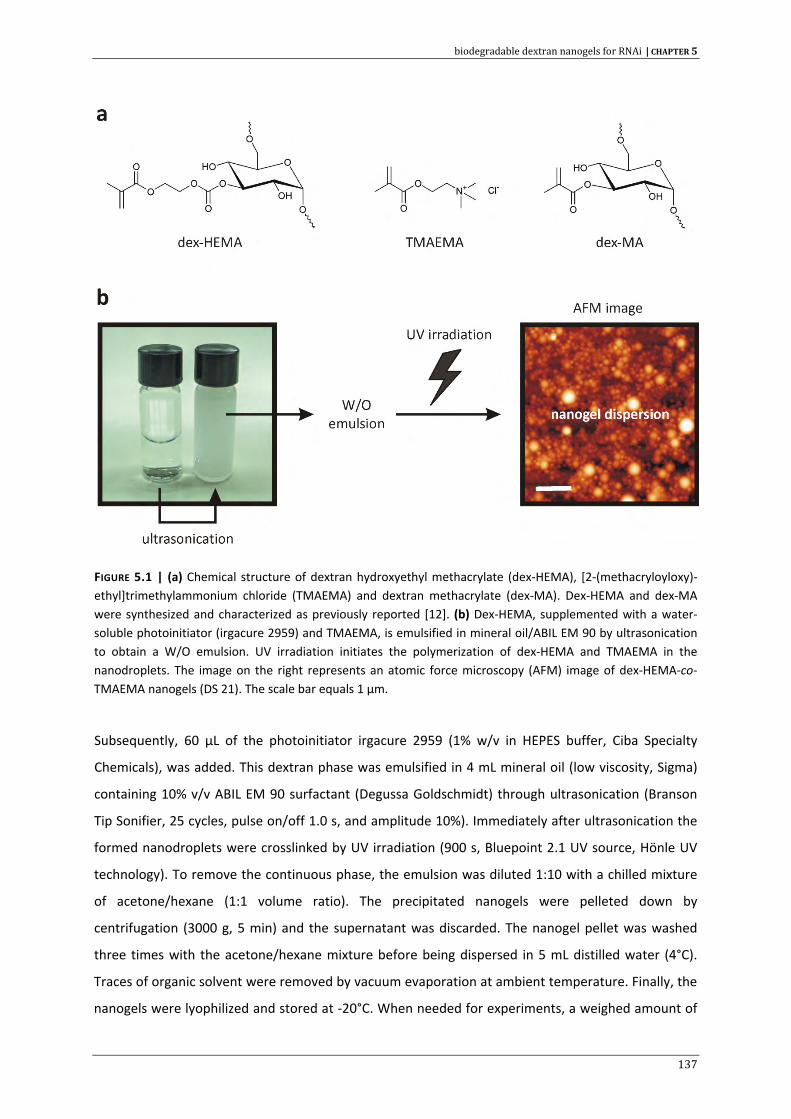
biodegradable dextran nanogels for RNAi | CHAPTER 5
FIGURE 5.1 | (a) Chemical structure of dextran hydroxyethyl methacrylate (dex‐HEMA), [2‐(methacryloyloxy)‐ethyl]trimethylammonium chloride (TMAEMA) and dextran methacrylate (dex‐MA). Dex‐HEMA and dex‐MA were synthesized and characterized as previously reported [12]. (b) Dex‐HEMA, supplemented with a water‐soluble photoinitiator (irgacure 2959) and TMAEMA, is emulsified in mineral oil/ABIL EM 90 by ultrasonication to obtain a W/O emulsion. UV irradiation initiates the polymerization of dex‐HEMA and TMAEMA in the nanodroplets. The image on the right represents an atomic force microscopy (AFM) image of dex‐HEMA‐co‐TMAEMA nanogels (DS 21). The scale bar equals 1 µm.
Subsequently, 60 µL of the photoinitiator irgacure 2959 (1% w/v in HEPES buffer, Ciba Specialty
Chemicals), was added. This dextran phase was emulsified in 4 mL mineral oil (low viscosity, Sigma)
containing 10% v/v ABIL EM 90 surfactant (Degussa Goldschmidt) through ultrasonication (Branson
Tip Sonifier, 25 cycles, pulse on/off 1.0 s, and amplitude 10%). Immediately after ultrasonication the
formed nanodroplets were crosslinked by UV irradiation (900 s, Bluepoint 2.1 UV source, Hönle UV
technology). To remove the continuous phase, the emulsion was diluted 1:10 with a chilled mixture
of acetone/hexane (1:1 volume ratio). The precipitated nanogels were pelleted down by
centrifugation (3000 g, 5 min) and the supernatant was discarded. The nanogel pellet was washed
three times with the acetone/hexane mixture before being dispersed in 5 mL distilled water (4°C).
Traces of organic solvent were removed by vacuum evaporation at ambient temperature. Finally, the
nanogels were lyophilized and stored at ‐20°C. When needed for experiments, a weighed amount of
137

CHAPTER 5 | biodegradable dextran nanogels for RNAi
the lyophilized nanogels was dispersed in a known volume of buffer. Throughout this paper the
concentration of the nanogel suspensions is denoted as µg nanogel per mL buffer.
Atomic Force Microscopy (AFM)
AFM images were acquired on a PicoPlus instrument (Molecular Imaging) operating in tapping mode
(open air, ambient temperature). For sample preparation, a drop of nanogel dispersion was dried on
a silica slide overnight before imaging. Height images were flattened using WSxM Scanning Probe
Microscopy software (Nanotec electronica).
Dynamic Light Scattering (DLS)
Nanogel sizes, size distributions and surface charge were determined by dynamic light scattering
(DLS) and zeta‐potential measurements respectively, using a Zetasizer Nano ZS (Malvern,
Worcestershire, UK), equipped with Dispersion Technology Software (DTS). The samples were
equilibrated at 25°C prior to measurement and measurement settings were put on automatic. To
screen the degradation of the nanogels, nanogel suspensions (500 µg mL‐1) were prepared in
respectively 20 mM HEPES buffer (pH 7.4 or pH 6.0) and 20 mM sodium acetate buffer (pH 5.0) in a
low volume cuvette that was subsequently sealed with parafilm. The nanogel suspensions were kept
at 37°C and the intensity of the scattered light was measured every 5 minutes. Each measurement
time point consisted of 6 runs, with 5 s per run.
Small Interfering RNA (siRNA)
Two 21 mer siRNA duplexes from different providers, targeting the pGL3 firefly luciferase, were used
in our experiments: anti‐luc siRNA‐1 from Dharmacon (Lafayette, CO, USA) and pGL3 luciferase siRNA
from Eurogentec (Seraing, Belgium), respectively denoted as siLUCD and siLUCE. Alexa fluor 488
(AF488) labelled pGL3 luciferase siRNA and a non‐targeting negative control siRNA duplex
(siCONTROL) were also purchased from Eurogentec and siGLO® green fluorescent Transfection
Indicator was from Dharmacon (Lafayette, CO).
Preparation and characterization of siRNA loaded nanogels
In a first step, a nanogel suspension (1 mg mL‐1) was prepared by dispersing a weighed amount of
lyophilized particles in RNase free water or buffer, followed by short sonication (Branson sonifier,
30”‐10% amplitude). From this 1 mg mL‐1 stock solution, dilutions were prepared according to the
138

biodegradable dextran nanogels for RNAi | CHAPTER 5
experimental needs. Nanogels were loaded with siRNA by mixing equal volumes of a diluted nanogel
dispersion and a siRNA solution at room temperature. The mixture was incubated for at least 15 min
at ambient temperature before initiating further experiments. The average size and zeta‐potential of
the nanogels before and after siRNA impregnation were measured by dynamic light scattering as
described above.
Fluorescence Fluctuation Spectroscopy (FFS)
A fluorescence fluctuation spectroscopy instrument was used as described previously.[15] FFS
monitors fluorescence intensity fluctuations in the excitation volume of a confocal microscope and
can be used to evaluate the complexation of fluorescently labelled molecules to cationic lipids or
polymers.[16‐19] The fluorescence fluctuations originate from the movement of (poorly concentrated)
fluorescent molecules in and out the fixed confocal excitation volume. In this study the 488 nm laser
line of a Krypton‐Argon ion laser (BioRad, Cheshire, UK) was used to excite the samples that contain
AF488‐siRNA. Typically, for FFS experiments, 50 µL of a nanogel dispersion was mixed with 50 µL of
an AF488‐siRNA solution (200 pmol mL‐1) in RNase free eppendorf tubes. After 20 min incubation, the
sample was transferred to the wells of a glass‐bottomed 96‐well plate (Greiner Bio‐one, certified
DNase/RNase free). Subsequently, the focal volume of the microscope was positioned 50 µm above
the bottom of the wells and the fluorescence fluctuations were recorded during a 30 s time interval.
All samples were prepared in triplicate and fluorescence measurements on every well were
performed in duplicate. The average fluorescence intensity of freely diffusing and complexed AF488‐
siRNA in the fluorescence fluctuation profile was determined as described previously.[16,20]
Cell lines and culture conditions
All cell experiments in this study were conducted on a human hepatoma cell line (Huh‐7). The wild
type cells are denoted as Huh‐7_wt. For luciferase gene silencing experiments, Huh‐7_eGFPLuc_rLuc
cells, stably expressing both the firefly and renilla luciferase (hereafter abbreviated as Huh‐7_LUC
cells), were generated by stably transfecting Huh‐7_eGFPLuc cells (gift from E. Wagner, Munchen)
with the pGL4.76_CMV plasmid. The pGL4.76_CMV plasmid was generated by ligating the PCR
amplified (forward primer AATAGTCGACTAGTTATTAATAGTAATCAA and reversed primer
AATAGGATCCGATCTGACG‐GTTCACTAAAC) and SalI/BamHI double digested CMV promotor into the
XhoI/BglII double digested pGL4.76 plasmid (Promega). The resulting pGL4.76_CMV plasmid was
linearized with the BamHI restriction enzyme and transfected using linear polyethylenimine (PEI) 22
kDa. Transfected cells were incubated in fresh medium for 48 h and then selected with 250 µg mL‐1
139

CHAPTER 5 | biodegradable dextran nanogels for RNAi
hygromycin. After two weeks, clones were isolated and expanded. Subsequently, the generated
clones were analyzed and a firefly and renilla luciferase positive clone was selected. Huh‐7_LUC cells
are grown in DMEM:F12, supplemented with 2 mM glutamine, 10% heat‐inactivated fetal bovine
serum (FBS) and 100 U mL‐1 P/S at 37°C in a humidified atmosphere containing 5% CO2.
Cytotoxicity assay
The influence of the nanogels on cell viability was analyzed using the CellTiter‐Glo® Assay (Promega).
Huh‐7_LUC cells (7 x 104 cells per cm2) were seeded in 24‐well plates and allowed to adhere. After 48
h, the cell culture medium was removed and 200 µL of a nanogel dispersion in OptiMEM was brought
onto the cells. Following 4 h of incubation (37°C, 5% CO2), nanogels that were not internalized were
removed from the cells and replaced with 200 µL of preheated culture medium. The 24‐well plates
were incubated for another 24 h at 37°C before cell viability was analyzed. Therefore, the cell plates
were incubated at room temperature during 30 min and 200 µL CellTiter‐Glo® reagent was added to
each well. After shaking the plate for 2 min (to lyse the cells) and 10 min incubation at room
temperature (to stabilize the luminescence signal), 100 µL of each well was transferred to a white‐
opaque 96‐well plate. Luminescence was measured on a GloMax™ 96 luminometer with 0.6 s
integration time. Cells treated with OptiMEM only and cells incubated with phenol (10 mg mL‐1)
served as negative and positive controls.
Confocal Laser Scanning Microscopy (CLSM)
Huh‐7_LUC cells were seeded in sterile glass‐bottomed culture dishes (MatTek Corporation, Ashland,
MA) at a density of 7 x 104 cells per cm2 and allowed to attach overnight. Nanogels loaded with
AF488‐siRNA were incubated with the cells in 200 µL OptiMEM during 4h (37°C, 5% CO2).
Subsequently, the cells were washed with phosphate buffered saline (PBS) and preheated culture
medium was added. The intracellular fluorescence distribution was visualized with a Nikon EZC1‐si
confocal scanning module installed on a motorized Nikon TE2000‐E inverted microscope (Nikon
Benelux, Brussels, Belgium). For lysosomal staining, cells were incubated for 20 min with 1:104
diluted Lysotracker Red® (Molecular Probes). Prior to image recording, the cells were fixed with a 4%
paraformaldehyde solution in PBS for 20 minutes at room temperature. After fixation, the cells were
washed twice with PBS and mounted with VectaShield® Mounting Medium (Vector Labs). Confocal
images were captured with a Nikon Plan Apochromat 60x oil immersion objective (numerical
aperture 1.4) using a 488 nm and 561 nm laser line for the excitation of AF488‐siRNA and
Lysotracker Red® respectively. A non‐confocal diascopic DIC (differential interference contrast) image
140

biodegradable dextran nanogels for RNAi | CHAPTER 5
was collected simultaneously. An object based co‐localization algorithm was used to quantify the
number of nanogel particles that are located inside acidified endosomes. First, the AF488‐siRNA
loaded nanogels were identified in the green image by intensity thresholding. Only “objects”
consisting of at least 8 pixels were retained as nanogels. The thresholded image was then binarized in
order to generate a mask for superposition on the corresponding red image (containing the
Lysotracker Red® stained endosomes). This allows to calculate the red (endosomal) intensity R for
each gel particle. Next, the background intensity (mean value BG and standard deviation S) of the cell
in the image was determined within a rectangular area not containing any endosomes. Finally, a gel
particle is decided to be co‐localized with an acidified endosome if its red intensity R > BG+S.
Flow cytometry
Huh‐7_LUC cells were plated in 6‐well plates (7 x 104 cells per cm2) and treated two days later with
AF488‐siRNA loaded nanogels (200 pmol mL‐1 AF488‐siRNA), prepared as described above. After the
incubation period, the cells were washed twice with PBS, trypsinized (750 µL trypsin/EDTA 0.25% per
well) and diluted with 7 mL cell culture medium. Following centrifugation (7 min, 1200 rpm – 4°C),
the cell pellet was resuspended in 400 µL flow buffer (PBS supplemented with 1% BSA and 0.1%
sodium azide) and placed on ice until flow analysis. The cells were analyzed using a Beckman Coulter
Cytomics™ FC500 flow cytometer. Generally, a minimum of ~104 cells was analyzed in each
measurement. Dead cells were discriminated from viable cells by the addition of propidium iodide
(Sigma). The data were analyzed using Beckman Coulter CXP analysis software.
Luciferase silencing in human hepatoma cells
For gene silencing experiments, Huh‐7_LUC cells were seeded in 24‐well plates at a density of 7 x 104
cells per cm2 and allowed to attach during 48 h. Two hundred microliter of a siRNA loaded nanogel
dispersion (in OptiMEM) was added to the cells in each well (50 pmol of siRNA per well). After 4h
incubation (37°C), the nanogels were removed from the cells and replaced with 400 µL prewarmed
cell culture medium. Forty eight hours later, the cells were lysed with 85 µL passive lysis buffer
(Promega, Leiden, The Netherlands). Firefly and Renilla luciferase activities were determined with the
Promega Dual‐Luciferase® Reporter Assay according to the manufacturer’s instructions. For
luciferase analysis we used a GloMax™ 96 luminometer with two injectors. Briefly, 10 µL of cell
lysate was mixed with 50 µL firefly luciferase substrate and after a 2 sec delay, firefly luminescence
(relative light units, RLU) was integrated during 10 s. Subsequently, 50 µL of renilla luciferase
substrate in Stop & Glo Buffer (Promega), was added and again after 2 s renilla luminescence (RLU)
141

CHAPTER 5 | biodegradable dextran nanogels for RNAi
was measured during a 10 s interval. For each sample, the ratio of firefly luminescence to renilla
luminescence was calculated. The ratios obtained with siLUC nanogels were compared to the ratios
obtained with siCONTROL nanogels and expressed as remaining luciferase expression (%). As a
positive control, siRNA complexes based on Lipofectamine™RNAiMAX (Invitrogen) were used and
prepared according to the manufacturer’s instructions. The time‐dependent gene silencing
experiment was conducted as described above albeit that 5 x 104 cells per cm2 were plated and the
transfection was performed in 5‐fold. Every day one 24‐well plate was analyzed for luciferase
expression.
Fusogenic peptide diINF‐7
The influenza‐derived fusogenic peptide diINF‐7 [21,22] was a kind gift from S. Oliveira (Utrecht
University). For luciferase gene silencing, Huh‐7_LUC cells were plated in 24‐well plates (7 x 104 cells
per cm2). After 48 h, a dispersion of siRNA loaded nanogels was prepared as described above (with
15 min incubation) and subsequently mixed with the fusogenic peptide (15 µg mL‐1). The mixture was
left at room temperature for an additional 15 minutes before incubation with the cells (200 µL per
well). After 4h incubation (37°C), the nanogels were removed from the cells and replaced with 400 µL
prewarmed cell culture medium. Luciferase silencing was analyzed two days later as described
above.
Photochemical internalization (PCI)
The photosensitizer (PS) meso‐tetraphenylporphine disulfonate (TPPS2a) was kindly provided by Dr.
A. Høgset (PCI Biotech, Oslo, Norway). The PS was protected from light and stored at 4°C until use.
Huh‐7_LUC cells were seeded in 24‐well plates at a density of 7 x 104 cells per cm2. One day after
seeding, the cells were incubated overnight with 250 µL PS (0.4 µg mL‐1 in complete cell culture
medium) at 37°C and protected from light. The next day, the cells were washed with PBS and 200 µL
siRNA loaded nanogels was added to the cells. Following 4 h of incubation, the nanogel complexes
were replaced with 400 µL fresh cell culture medium and the cells were exposed to blue light (375
nm – 450 nm) emitted by LumiSource® (Osram 18W/67, 13 mW per cm2, PCI Biotech, Oslo, Norway).
Cells were incubated for another 48 h in the dark before luciferase activity was quantified as
described above.
Fluorescence microscopy before and after PCI, was evaluated by seeding 12.5 x 104 Huh‐7_wt cells in
Lab‐Tek™ II chambered coverglasses (Nalge Nunc International, Rochester, NY), two days before
transfection. One day pre‐transfection, PS was added to half of the wells (0.4 µg mL‐1). At the day of
142

biodegradable dextran nanogels for RNAi | CHAPTER 5
transfection, the PS was removed and the cells were washed with PBS. Cationic nanogels were
loaded with fluorescent siGLO® in HEPES buffer as indicated earlier and incubated with the cells in
400 µl OptiMEM. The final concentration of nanogels and siGLO® was 25 µg mL‐1 and 200 pmol mL‐1
respectively. After 3 h incubation, the remaining complexes were removed and replaced with
complete cell culture medium (37°C). One hour of incubation later, the culture chambers were
illuminated with LumiSource® (45s). The cells were placed at 37°C for another 36 h, protected from
light. Afterwards, the cell‐bound siGLO® fluorescence was quenched by treatment with 0.4% trypan
blue (8 min) and after washing with PBS the cells were fixed with 4% paraformaldehyde (20 min, RT).
Following 3 washing steps with PBS, the cells were mounted with Vectashield® DAPI Mounting
Medium (Vector Labs, UK) and kept at 4°C (dark) until fluorescence imaging.
RESULTS AND DISCUSSION
Preparation of cationic nanogels
Several methods for the engineering of nanogels are described in the literature.[23] We synthesized
dextran nanogels by using an inverse mini‐emulsion photopolymerization method, as schematically
depicted (Figure 5.1b). In this process, the internal aqueous dex‐HEMA phase, supplemented with
TMAEMA (Figure 5.1a) and photoinitiator, is dispersed in the continuous mineral oil phase with the
aid of an oil‐soluble surfactant (ABIL EM 90, HLB value ~5). Sonification provides the energy that is
necessary to form nanoscopic droplets.[23] Exposure to UV light initiates the radical polymerization
process within the dex‐HEMA droplets, yielding stable crosslinked dextran nanogels. Using this
procedure, we obtained dextran nanogels with a hydrodynamic diameter ~200‐300 nm with an
acceptable polydispersity index (Table 5.1).
We also visualized the nanogels by atomic force microscopy (Figure 5.1b, scale bar = 1 µm) showing
particles with a size in good agreement with the values obtained through dynamic light scattering.
Following polymerization, the particles were purified through centrifugation/washing steps with
organic solvent. Finally, the obtained aqueous dispersion of nanogels was lyophilized. We observed
that lyophilization had no significant influence on nanogel size and surface charge (data not shown).
Adding TMAEMA to the aqueous dex‐HEMA solution before emulsification enables copolymerization
of this methacrylate monomer with dex‐HEMA in the emulsion droplets, leading to nanogels with a
cationic surface charge, as revealed from zeta‐potential measurements (Table 5.1). Increasing the
amount of TMAEMA increased the surface charge of the nanogels, indicating that more TMAEMA
units get incorporated in the nanogel network.
143
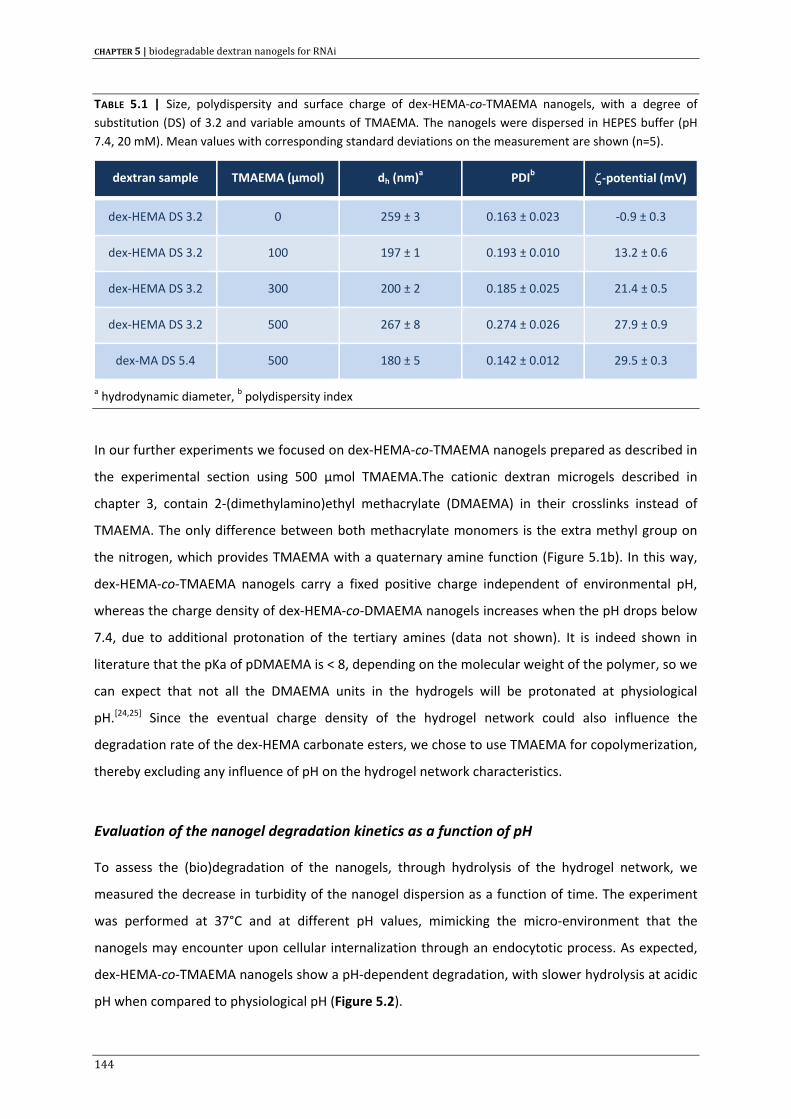
CHAPTER 5 | biodegradable dextran nanogels for RNAi
TABLE 5.1 | Size, polydispersity and surface charge of dex‐HEMA‐co‐TMAEMA nanogels, with a degree of substitution (DS) of 3.2 and variable amounts of TMAEMA. The nanogels were dispersed in HEPES buffer (pH 7.4, 20 mM). Mean values with corresponding standard deviations on the measurement are shown (n=5).
dextran sample TMAEMA (µmol) dh (nm)a PDIb ζ‐potential (mV)
dex‐HEMA DS 3.2 0 259 ± 3 0.163 ± 0.023 ‐0.9 ± 0.3
dex‐HEMA DS 3.2 100 197 ± 1 0.193 ± 0.010 13.2 ± 0.6
dex‐HEMA DS 3.2 300 200 ± 2 0.185 ± 0.025 21.4 ± 0.5
dex‐HEMA DS 3.2 500 267 ± 8 0.274 ± 0.026 27.9 ± 0.9
dex‐MA DS 5.4 500 180 ± 5 0.142 ± 0.012 29.5 ± 0.3
a hydrodynamic diameter, b polydispersity index
In our further experiments we focused on dex‐HEMA‐co‐TMAEMA nanogels prepared as described in
the experimental section using 500 µmol TMAEMA.The cationic dextran microgels described in
chapter 3, contain 2‐(dimethylamino)ethyl methacrylate (DMAEMA) in their crosslinks instead of
TMAEMA. The only difference between both methacrylate monomers is the extra methyl group on
the nitrogen, which provides TMAEMA with a quaternary amine function (Figure 5.1b). In this way,
dex‐HEMA‐co‐TMAEMA nanogels carry a fixed positive charge independent of environmental pH,
whereas the charge density of dex‐HEMA‐co‐DMAEMA nanogels increases when the pH drops below
7.4, due to additional protonation of the tertiary amines (data not shown). It is indeed shown in
literature that the pKa of pDMAEMA is < 8, depending on the molecular weight of the polymer, so we
can expect that not all the DMAEMA units in the hydrogels will be protonated at physiological
pH.[24,25] Since the eventual charge density of the hydrogel network could also influence the
degradation rate of the dex‐HEMA carbonate esters, we chose to use TMAEMA for copolymerization,
thereby excluding any influence of pH on the hydrogel network characteristics.
Evaluation of the nanogel degradation kinetics as a function of pH
To assess the (bio)degradation of the nanogels, through hydrolysis of the hydrogel network, we
measured the decrease in turbidity of the nanogel dispersion as a function of time. The experiment
was performed at 37°C and at different pH values, mimicking the micro‐environment that the
nanogels may encounter upon cellular internalization through an endocytotic process. As expected,
dex‐HEMA‐co‐TMAEMA nanogels show a pH‐dependent degradation, with slower hydrolysis at acidic
pH when compared to physiological pH (Figure 5.2).
144
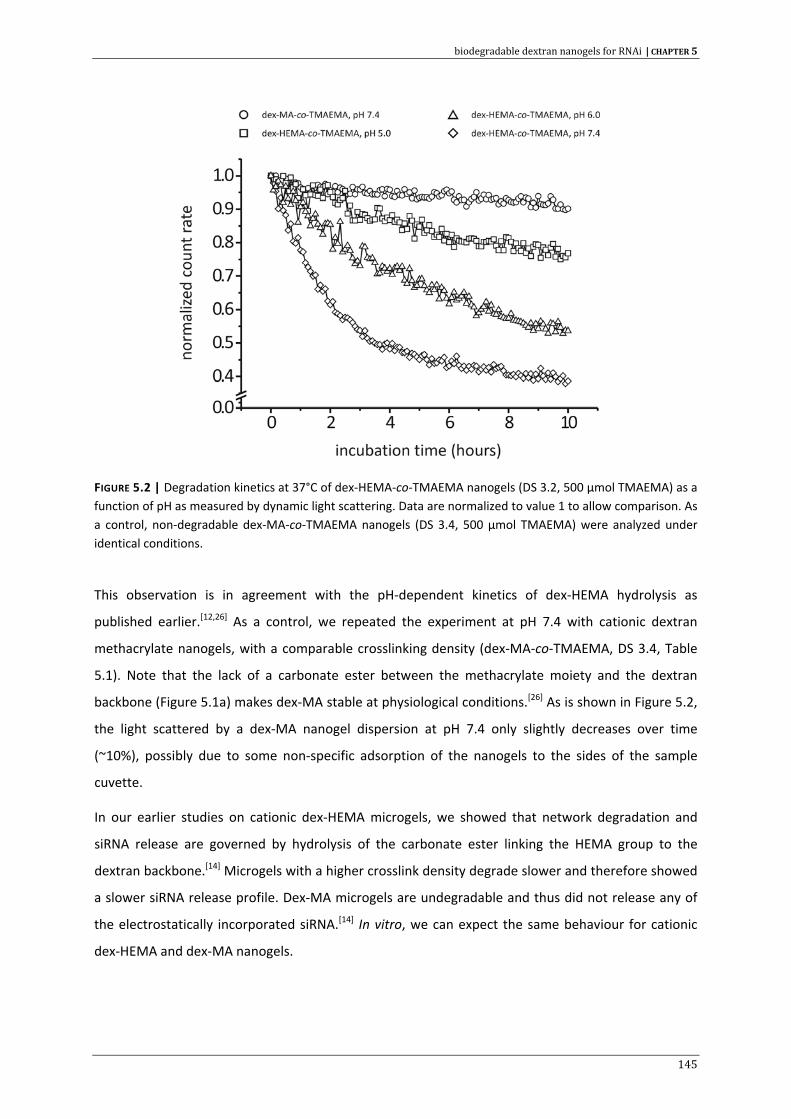
biodegradable dextran nanogels for RNAi | CHAPTER 5
FIGURE 5.2 | Degradation kinetics at 37°C of dex‐HEMA‐co‐TMAEMA nanogels (DS 3.2, 500 µmol TMAEMA) as a function of pH as measured by dynamic light scattering. Data are normalized to value 1 to allow comparison. As a control, non‐degradable dex‐MA‐co‐TMAEMA nanogels (DS 3.4, 500 µmol TMAEMA) were analyzed under identical conditions.
This observation is in agreement with the pH‐dependent kinetics of dex‐HEMA hydrolysis as
published earlier.[12,26] As a control, we repeated the experiment at pH 7.4 with cationic dextran
methacrylate nanogels, with a comparable crosslinking density (dex‐MA‐co‐TMAEMA, DS 3.4, Table
5.1). Note that the lack of a carbonate ester between the methacrylate moiety and the dextran
backbone (Figure 5.1a) makes dex‐MA stable at physiological conditions.[26] As is shown in Figure 5.2,
the light scattered by a dex‐MA nanogel dispersion at pH 7.4 only slightly decreases over time
(~10%), possibly due to some non‐specific adsorption of the nanogels to the sides of the sample
cuvette.
In our earlier studies on cationic dex‐HEMA microgels, we showed that network degradation and
siRNA release are governed by hydrolysis of the carbonate ester linking the HEMA group to the
dextran backbone.[14] Microgels with a higher crosslink density degrade slower and therefore showed
a slower siRNA release profile. Dex‐MA microgels are undegradable and thus did not release any of
the electrostatically incorporated siRNA.[14] In vitro, we can expect the same behaviour for cationic
dex‐HEMA and dex‐MA nanogels.
145

CHAPTER 5 | biodegradable dextran nanogels for RNAi
Quantification of siRNA complexation to cationic dextran nanogels
The data in chapter 3 demonstrated that cationic dextran microgels could be loaded with siRNA after
microgel formation, based on electrostatic interactions.[14] This ‘post‐loading’ step has the advantage
that the siRNA molecules are not exposed to free radicals and UV light during the
photopolymerization process, thereby avoiding potential interference with the siRNA integrity.[14]
Similarly, cationic dex‐HEMA‐co‐TMAEMA nanogels could be post‐loaded with siRNA by dispersing
the nanogels in an siRNA solution.
We quantified the amount of siRNA that can be complexed by dex‐HEMA‐co‐TMAEMA nanogels by
fluorescence fluctuation spectroscopy (see experimental section), as described earlier by Buyens et
al.[16] In a solution of free (thus non‐complexed) AF488 labelled siRNA, the fluorescence signal in the
confocal excitation volume of the microscope fluctuates around an average value that is proportional
to the AF488‐siRNA concentration (Figure 5.3a). Upon complexation of the siRNA to cationic
nanogels, the fluorescence intensity of the ‘baseline’, which is representative for the concentration
of free siRNA, lowers and peaks of high fluorescence appear (Figure 5.3b). The latter originate from
the movement of highly fluorescent nanogel particles that complex many fluorescent siRNA
molecules, through the excitation volume. By determination of the free siRNA concentration,
through analysis of the average fluorescence intensity of the baseline, the percentage of siRNA in a
complexed state can be quantified.[16] Figure 5.3c shows the degree of siRNA complexation to
different concentrations of dex‐HEMA‐co‐TMAEMA nanogels, dispersed in respectively HEPES buffer
and OptiMEM. In every FFS experiment, the final siRNA concentration was 100 pmol mL‐1. We
estimated the maximal siRNA loading for the nanogels to be between 50 and 100 pmol siRNA per µg
nanogel in HEPES buffer which corresponds to >60 wt% loading. A closer look to Figure 5.2c shows
that in OptiMEM the maximal siRNA loading of the nanogels is slightly lower than in HEPES buffer,
probably due to partial screening of the cationic charges on the nanogels by the higher ionic strength
of OptiMEM.
Influence of siRNA complexation on nanogel size and surface charge
Besides information on the degree of siRNA complexation, it is equally important to evaluate
whether the loading of the nanogels with siRNA influences their size and surface charge. Indeed,
many studies have reported that these physicochemical parameters significantly influence the extent
and method of cellular internalization of nanoparticulate matter.[9,27,28] Negatively charged siRNA
duplexes will bind electrostatically to the charged ammonium groups incorporated in the nanogel
network.
146
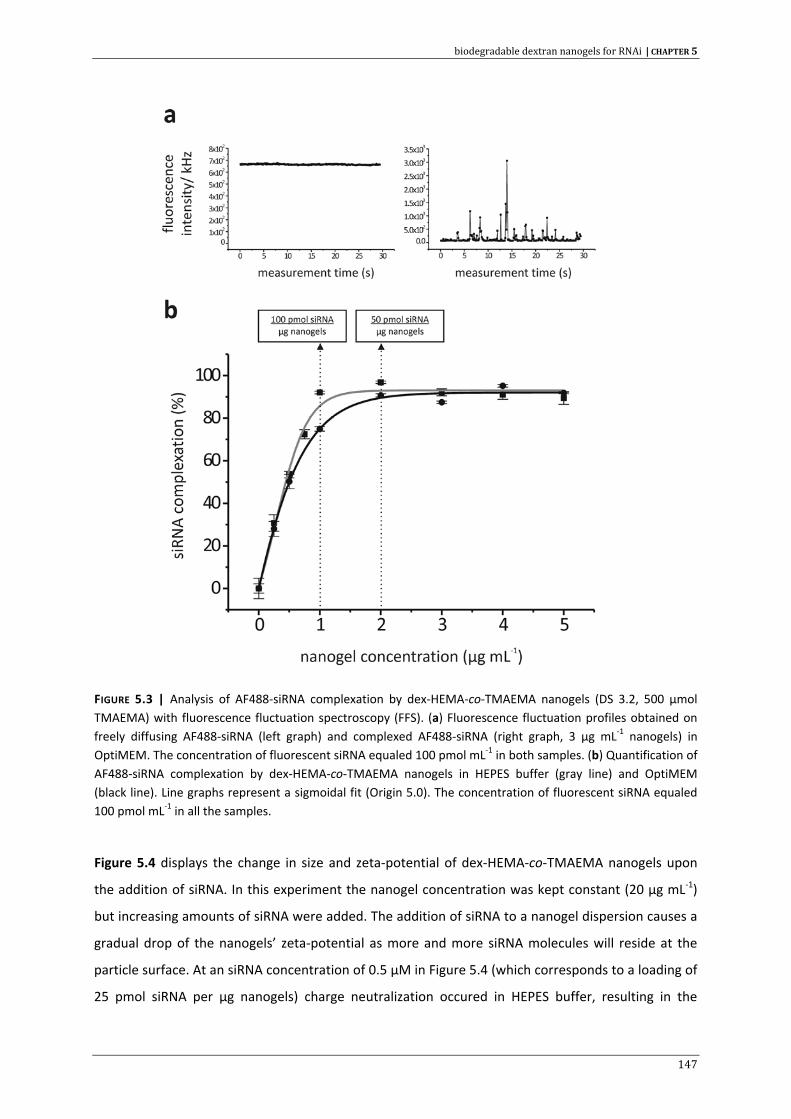
biodegradable dextran nanogels for RNAi | CHAPTER 5
FIGURE 5.3 | Analysis of AF488‐siRNA complexation by dex‐HEMA‐co‐TMAEMA nanogels (DS 3.2, 500 µmol TMAEMA) with fluorescence fluctuation spectroscopy (FFS). (a) Fluorescence fluctuation profiles obtained on freely diffusing AF488‐siRNA (left graph) and complexed AF488‐siRNA (right graph, 3 µg mL‐1 nanogels) in OptiMEM. The concentration of fluorescent siRNA equaled 100 pmol mL‐1 in both samples. (b) Quantification of AF488‐siRNA complexation by dex‐HEMA‐co‐TMAEMA nanogels in HEPES buffer (gray line) and OptiMEM (black line). Line graphs represent a sigmoidal fit (Origin 5.0). The concentration of fluorescent siRNA equaled 100 pmol mL‐1 in all the samples.
Figure 5.4 displays the change in size and zeta‐potential of dex‐HEMA‐co‐TMAEMA nanogels upon
the addition of siRNA. In this experiment the nanogel concentration was kept constant (20 µg mL‐1)
but increasing amounts of siRNA were added. The addition of siRNA to a nanogel dispersion causes a
gradual drop of the nanogels’ zeta‐potential as more and more siRNA molecules will reside at the
particle surface. At an siRNA concentration of 0.5 µM in Figure 5.4 (which corresponds to a loading of
25 pmol siRNA per µg nanogels) charge neutralization occured in HEPES buffer, resulting in the
147
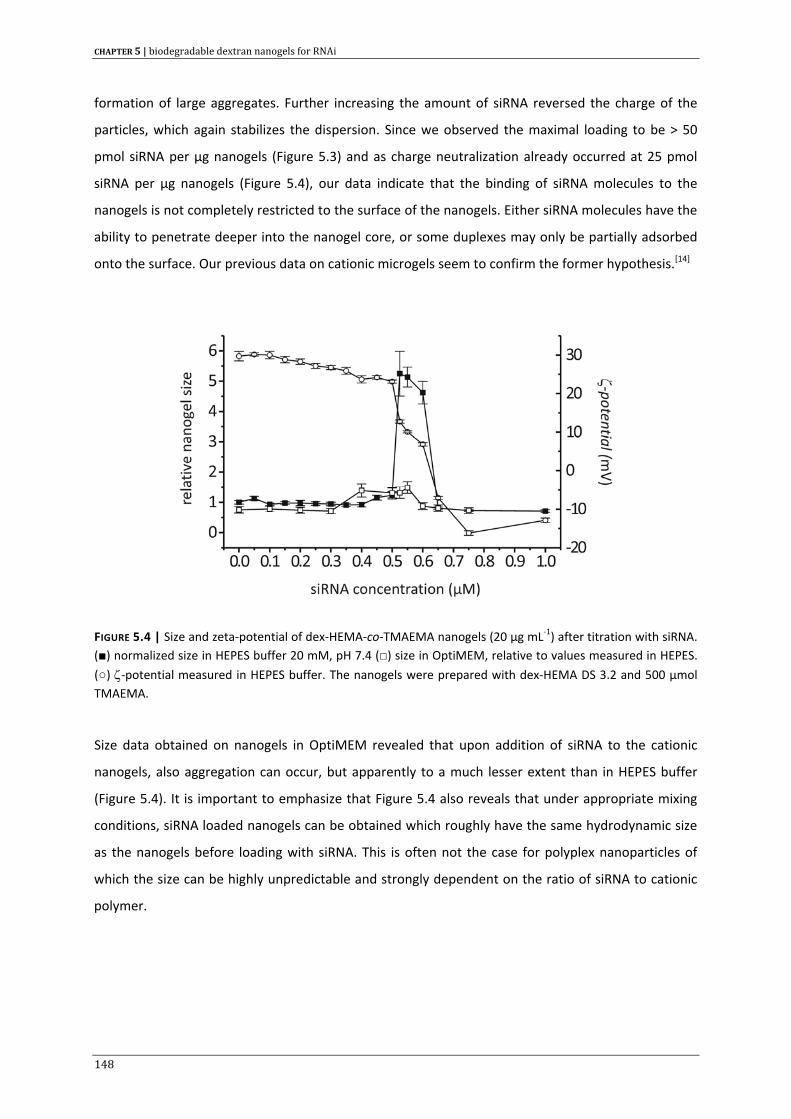
CHAPTER 5 | biodegradable dextran nanogels for RNAi
formation of large aggregates. Further increasing the amount of siRNA reversed the charge of the
particles, which again stabilizes the dispersion. Since we observed the maximal loading to be > 50
pmol siRNA per µg nanogels (Figure 5.3) and as charge neutralization already occurred at 25 pmol
siRNA per µg nanogels (Figure 5.4), our data indicate that the binding of siRNA molecules to the
nanogels is not completely restricted to the surface of the nanogels. Either siRNA molecules have the
ability to penetrate deeper into the nanogel core, or some duplexes may only be partially adsorbed
onto the surface. Our previous data on cationic microgels seem to confirm the former hypothesis.[14]
FIGURE 5.4 | Size and zeta‐potential of dex‐HEMA‐co‐TMAEMA nanogels (20 µg mL‐1) after titration with siRNA. (■) normalized size in HEPES buffer 20 mM, pH 7.4 (□) size in OptiMEM, relative to values measured in HEPES.
(○) ζ‐potential measured in HEPES buffer. The nanogels were prepared with dex‐HEMA DS 3.2 and 500 µmol TMAEMA.
Size data obtained on nanogels in OptiMEM revealed that upon addition of siRNA to the cationic
nanogels, also aggregation can occur, but apparently to a much lesser extent than in HEPES buffer
(Figure 5.4). It is important to emphasize that Figure 5.4 also reveals that under appropriate mixing
conditions, siRNA loaded nanogels can be obtained which roughly have the same hydrodynamic size
as the nanogels before loading with siRNA. This is often not the case for polyplex nanoparticles of
which the size can be highly unpredictable and strongly dependent on the ratio of siRNA to cationic
polymer.
148
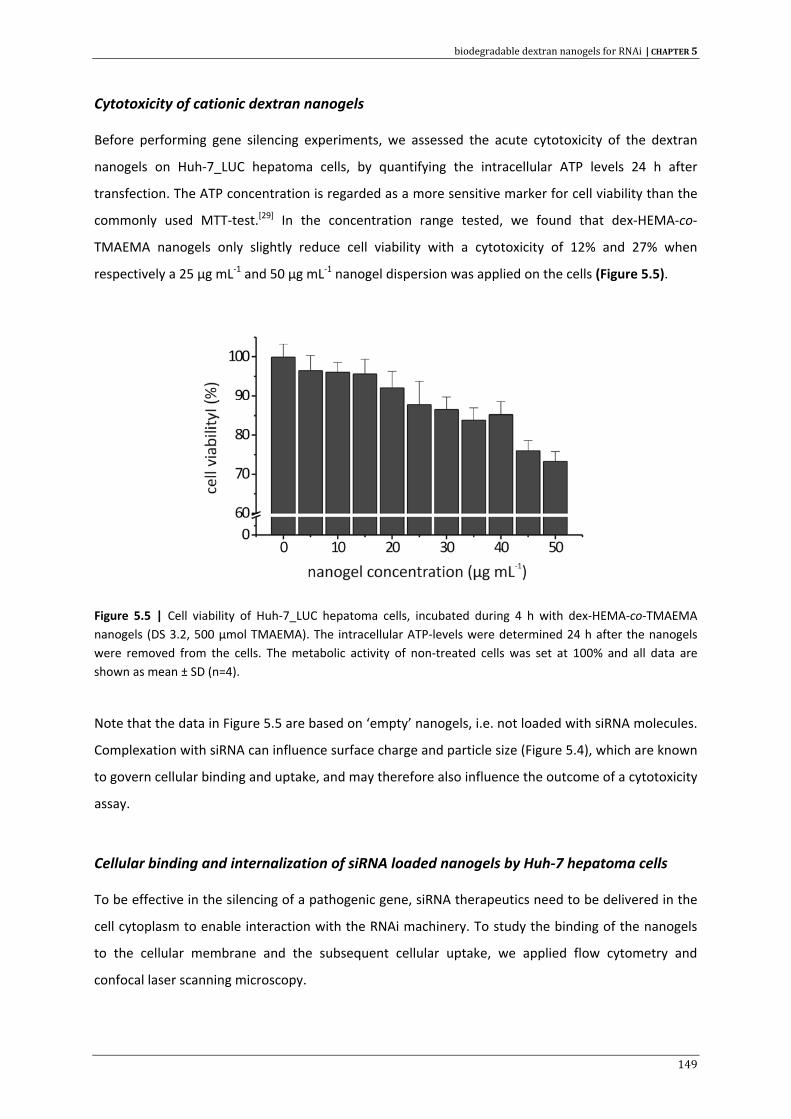
biodegradable dextran nanogels for RNAi | CHAPTER 5
Cytotoxicity of cationic dextran nanogels
Before performing gene silencing experiments, we assessed the acute cytotoxicity of the dextran
nanogels on Huh‐7_LUC hepatoma cells, by quantifying the intracellular ATP levels 24 h after
transfection. The ATP concentration is regarded as a more sensitive marker for cell viability than the
commonly used MTT‐test.[29] In the concentration range tested, we found that dex‐HEMA‐co‐
TMAEMA nanogels only slightly reduce cell viability with a cytotoxicity of 12% and 27% when
respectively a 25 µg mL‐1 and 50 µg mL‐1 nanogel dispersion was applied on the cells (Figure 5.5).
Figure 5.5 | Cell viability of Huh‐7_LUC hepatoma cells, incubated during 4 h with dex‐HEMA‐co‐TMAEMA nanogels (DS 3.2, 500 µmol TMAEMA). The intracellular ATP‐levels were determined 24 h after the nanogels were removed from the cells. The metabolic activity of non‐treated cells was set at 100% and all data are shown as mean ± SD (n=4).
Note that the data in Figure 5.5 are based on ‘empty’ nanogels, i.e. not loaded with siRNA molecules.
Complexation with siRNA can influence surface charge and particle size (Figure 5.4), which are known
to govern cellular binding and uptake, and may therefore also influence the outcome of a cytotoxicity
assay.
Cellular binding and internalization of siRNA loaded nanogels by Huh‐7 hepatoma cells
To be effective in the silencing of a pathogenic gene, siRNA therapeutics need to be delivered in the
cell cytoplasm to enable interaction with the RNAi machinery. To study the binding of the nanogels
to the cellular membrane and the subsequent cellular uptake, we applied flow cytometry and
confocal laser scanning microscopy.
149
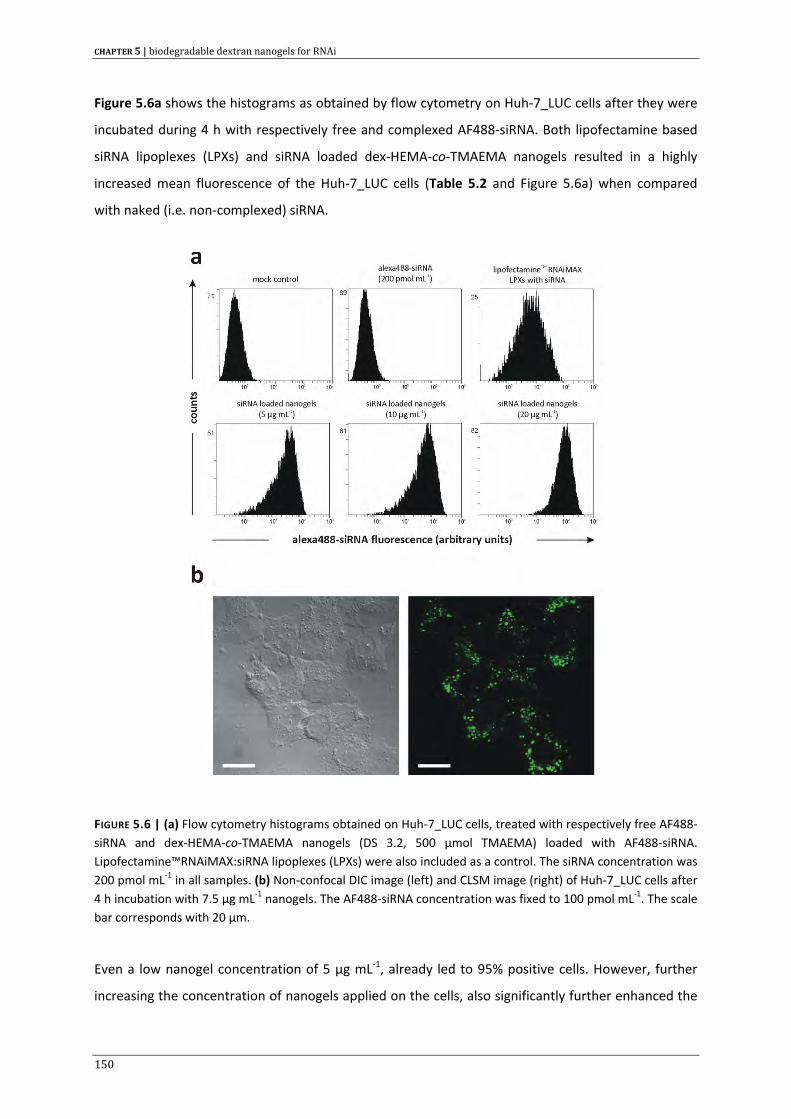
CHAPTER 5 | biodegradable dextran nanogels for RNAi
Figure 5.6a shows the histograms as obtained by flow cytometry on Huh‐7_LUC cells after they were
incubated during 4 h with respectively free and complexed AF488‐siRNA. Both lipofectamine based
siRNA lipoplexes (LPXs) and siRNA loaded dex‐HEMA‐co‐TMAEMA nanogels resulted in a highly
increased mean fluorescence of the Huh‐7_LUC cells (Table 5.2 and Figure 5.6a) when compared
with naked (i.e. non‐complexed) siRNA.
FIGURE 5.6 | (a) Flow cytometry histograms obtained on Huh‐7_LUC cells, treated with respectively free AF488‐siRNA and dex‐HEMA‐co‐TMAEMA nanogels (DS 3.2, 500 µmol TMAEMA) loaded with AF488‐siRNA. Lipofectamine™RNAiMAX:siRNA lipoplexes (LPXs) were also included as a control. The siRNA concentration was 200 pmol mL‐1 in all samples. (b) Non‐confocal DIC image (left) and CLSM image (right) of Huh‐7_LUC cells after 4 h incubation with 7.5 µg mL‐1 nanogels. The AF488‐siRNA concentration was fixed to 100 pmol mL‐1. The scale bar corresponds with 20 µm.
Even a low nanogel concentration of 5 µg mL‐1, already led to 95% positive cells. However, further
increasing the concentration of nanogels applied on the cells, also significantly further enhanced the
150
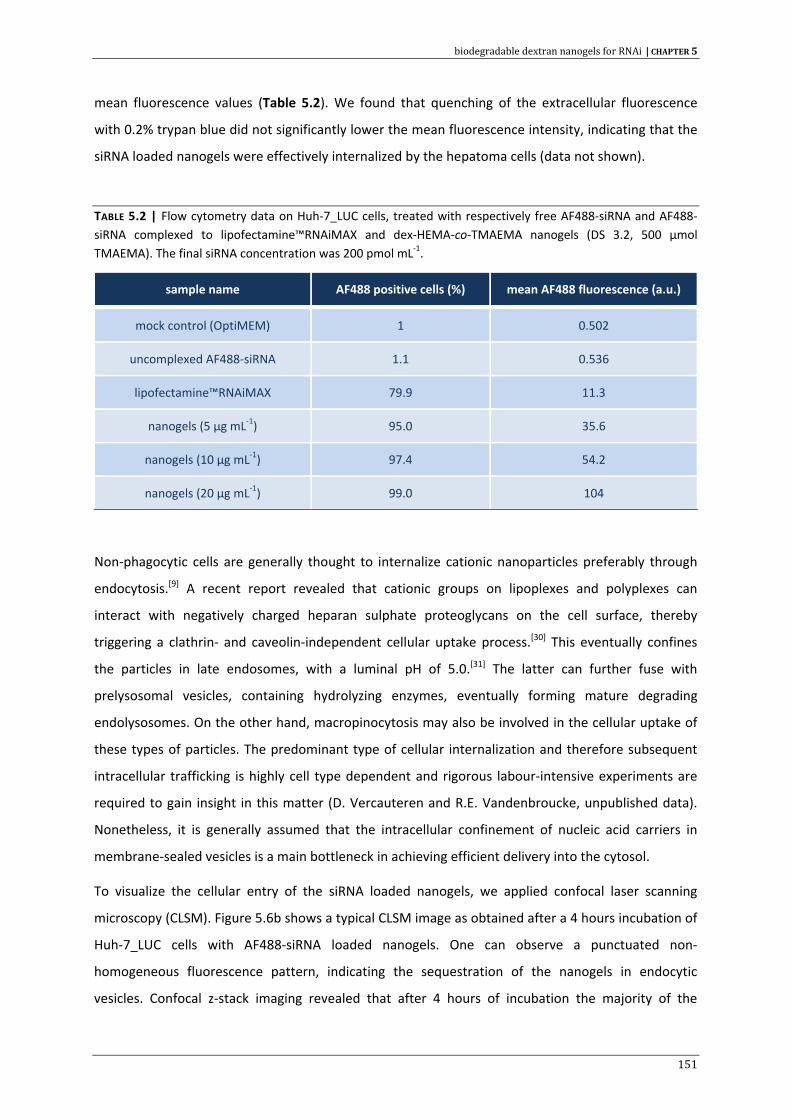
biodegradable dextran nanogels for RNAi | CHAPTER 5
mean fluorescence values (Table 5.2). We found that quenching of the extracellular fluorescence
with 0.2% trypan blue did not significantly lower the mean fluorescence intensity, indicating that the
siRNA loaded nanogels were effectively internalized by the hepatoma cells (data not shown).
TABLE 5.2 | Flow cytometry data on Huh‐7_LUC cells, treated with respectively free AF488‐siRNA and AF488‐siRNA complexed to lipofectamine™RNAiMAX and dex‐HEMA‐co‐TMAEMA nanogels (DS 3.2, 500 µmol TMAEMA). The final siRNA concentration was 200 pmol mL‐1.
sample name AF488 positive cells (%) mean AF488 fluorescence (a.u.)
mock control (OptiMEM) 1 0.502
uncomplexed AF488‐siRNA 1.1 0.536
lipofectamine™RNAiMAX 79.9 11.3
nanogels (5 µg mL‐1) 95.0 35.6
nanogels (10 µg mL‐1) 97.4 54.2
nanogels (20 µg mL‐1) 99.0 104
Non‐phagocytic cells are generally thought to internalize cationic nanoparticles preferably through
endocytosis.[9] A recent report revealed that cationic groups on lipoplexes and polyplexes can
interact with negatively charged heparan sulphate proteoglycans on the cell surface, thereby
triggering a clathrin‐ and caveolin‐independent cellular uptake process.[30] This eventually confines
the particles in late endosomes, with a luminal pH of 5.0.[31] The latter can further fuse with
prelysosomal vesicles, containing hydrolyzing enzymes, eventually forming mature degrading
endolysosomes. On the other hand, macropinocytosis may also be involved in the cellular uptake of
these types of particles. The predominant type of cellular internalization and therefore subsequent
intracellular trafficking is highly cell type dependent and rigorous labour‐intensive experiments are
required to gain insight in this matter (D. Vercauteren and R.E. Vandenbroucke, unpublished data).
Nonetheless, it is generally assumed that the intracellular confinement of nucleic acid carriers in
membrane‐sealed vesicles is a main bottleneck in achieving efficient delivery into the cytosol.
To visualize the cellular entry of the siRNA loaded nanogels, we applied confocal laser scanning
microscopy (CLSM). Figure 5.6b shows a typical CLSM image as obtained after a 4 hours incubation of
Huh‐7_LUC cells with AF488‐siRNA loaded nanogels. One can observe a punctuated non‐
homogeneous fluorescence pattern, indicating the sequestration of the nanogels in endocytic
vesicles. Confocal z‐stack imaging revealed that after 4 hours of incubation the majority of the
151
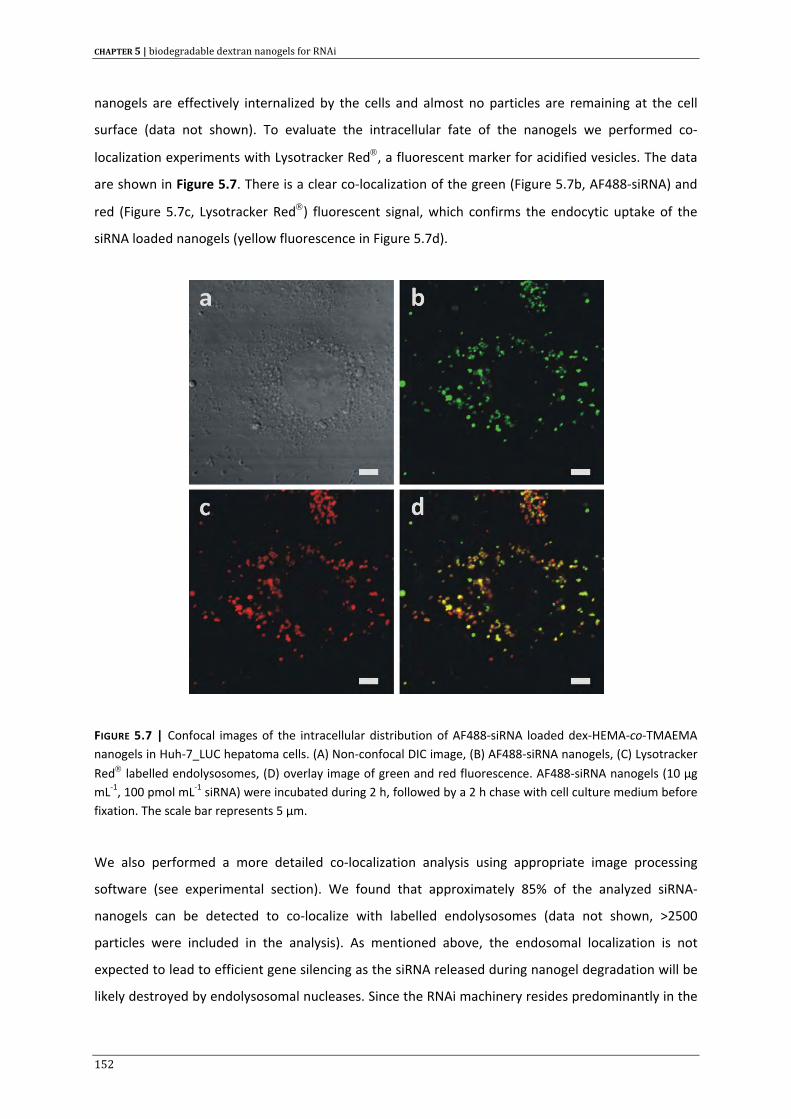
CHAPTER 5 | biodegradable dextran nanogels for RNAi
nanogels are effectively internalized by the cells and almost no particles are remaining at the cell
surface (data not shown). To evaluate the intracellular fate of the nanogels we performed co‐
localization experiments with Lysotracker Red®, a fluorescent marker for acidified vesicles. The data
are shown in Figure 5.7. There is a clear co‐localization of the green (Figure 5.7b, AF488‐siRNA) and
red (Figure 5.7c, Lysotracker Red®) fluorescent signal, which confirms the endocytic uptake of the
siRNA loaded nanogels (yellow fluorescence in Figure 5.7d).
FIGURE 5.7 | Confocal images of the intracellular distribution of AF488‐siRNA loaded dex‐HEMA‐co‐TMAEMA nanogels in Huh‐7_LUC hepatoma cells. (A) Non‐confocal DIC image, (B) AF488‐siRNA nanogels, (C) Lysotracker
Red® labelled endolysosomes, (D) overlay image of green and red fluorescence. AF488‐siRNA nanogels (10 µg mL‐1, 100 pmol mL‐1 siRNA) were incubated during 2 h, followed by a 2 h chase with cell culture medium before fixation. The scale bar represents 5 µm.
We also performed a more detailed co‐localization analysis using appropriate image processing
software (see experimental section). We found that approximately 85% of the analyzed siRNA‐
nanogels can be detected to co‐localize with labelled endolysosomes (data not shown, >2500
particles were included in the analysis). As mentioned above, the endosomal localization is not
expected to lead to efficient gene silencing as the siRNA released during nanogel degradation will be
likely destroyed by endolysosomal nucleases. Since the RNAi machinery resides predominantly in the
152
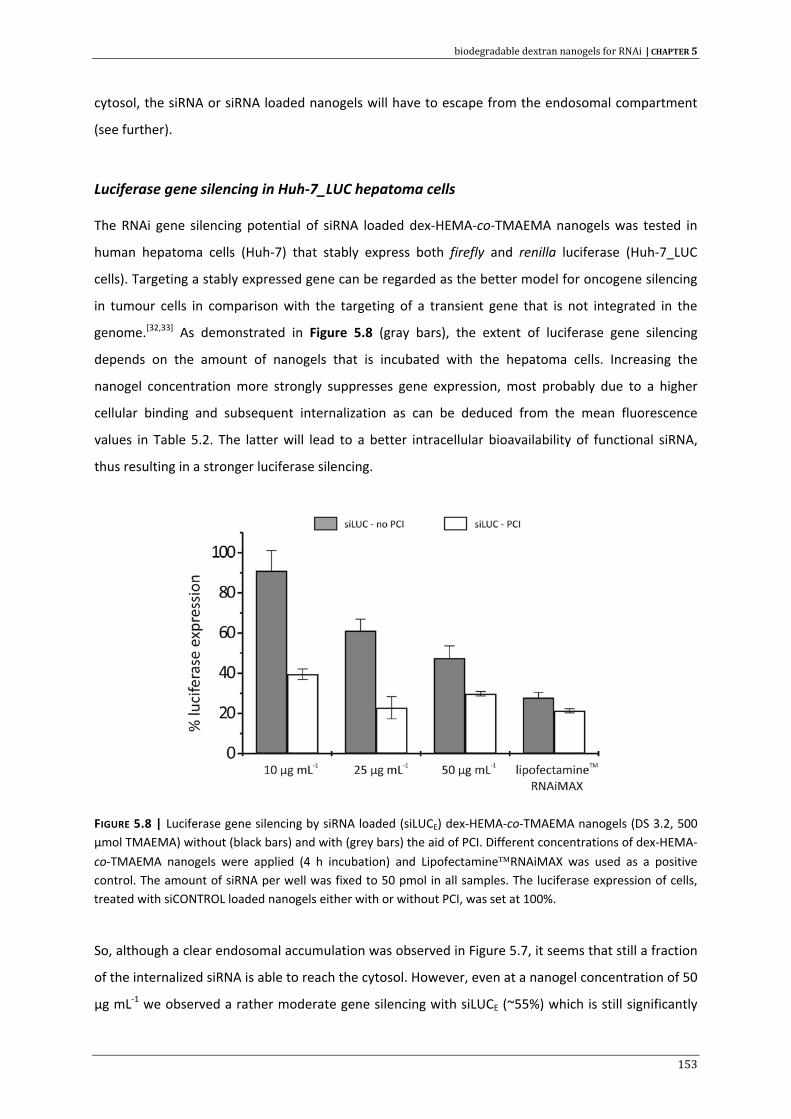
biodegradable dextran nanogels for RNAi | CHAPTER 5
cytosol, the siRNA or siRNA loaded nanogels will have to escape from the endosomal compartment
(see further).
Luciferase gene silencing in Huh‐7_LUC hepatoma cells
The RNAi gene silencing potential of siRNA loaded dex‐HEMA‐co‐TMAEMA nanogels was tested in
human hepatoma cells (Huh‐7) that stably express both firefly and renilla luciferase (Huh‐7_LUC
cells). Targeting a stably expressed gene can be regarded as the better model for oncogene silencing
in tumour cells in comparison with the targeting of a transient gene that is not integrated in the
genome.[32,33] As demonstrated in Figure 5.8 (gray bars), the extent of luciferase gene silencing
depends on the amount of nanogels that is incubated with the hepatoma cells. Increasing the
nanogel concentration more strongly suppresses gene expression, most probably due to a higher
cellular binding and subsequent internalization as can be deduced from the mean fluorescence
values in Table 5.2. The latter will lead to a better intracellular bioavailability of functional siRNA,
thus resulting in a stronger luciferase silencing.
FIGURE 5.8 | Luciferase gene silencing by siRNA loaded (siLUCE) dex‐HEMA‐co‐TMAEMA nanogels (DS 3.2, 500 µmol TMAEMA) without (black bars) and with (grey bars) the aid of PCI. Different concentrations of dex‐HEMA‐
co‐TMAEMA nanogels were applied (4 h incubation) and Lipofectamine™RNAiMAX was used as a positive control. The amount of siRNA per well was fixed to 50 pmol in all samples. The luciferase expression of cells, treated with siCONTROL loaded nanogels either with or without PCI, was set at 100%.
So, although a clear endosomal accumulation was observed in Figure 5.7, it seems that still a fraction
of the internalized siRNA is able to reach the cytosol. However, even at a nanogel concentration of 50
µg mL‐1 we observed a rather moderate gene silencing with siLUCE (~55%) which is still significantly
153

CHAPTER 5 | biodegradable dextran nanogels for RNAi
lower than that achieved with the positive control (lipofectamine based siRNA complexes) which
show ~75% silencing. Moreover, at a nanogel concentration of 50 µg mL‐1, cytotoxicity concerns arise
(Figure 5.5) and therefore it is imperative that appropriate measures are taken to achieve sufficient
gene silencing without interfering too much with cell viability.
Since the data on cellular uptake, presented in Figure 5.7, suggest that the nanogels are trafficked
toward the endolysosomes, it is likely that this intracellular fate poses a major barrier for efficient
RNAi. For this reason, we investigated the influence of photochemical internalization (PCI), a
technique known to enhance endosomal escape, on the luciferase gene silencing efficiency achieved
with dex‐HEMA‐co‐TMAEMA nanogels. PCI involves the use of amphiphilic photosensitizers (PS) that
accumulate in the membranes of endocytic vesicles.[34,35] In this study, meso‐tetraphenylporphine
carrying two sulfonate groups on adjacent phenyl rings (TPPS2a) was used. Upon illumination with a
specific light source (blue light in our experiments), excitation of the PS compound induces the
formation of reactive oxygen species (ROS), primarily singlet oxygen. This highly reactive
intermediate can damage cellular components, but this effect is mainly confined to the local
production site owing to its short range of action and short life‐time.[34] This localized effect will
therefore selectively disrupt the endosomal membranes, releasing the entrapped macromolecules or
nanoparticles into the cytosol (Figure 5.9). Since its discovery in 1999 [36], PCI has been successfully
applied to stimulate the cytosolic delivery of several types of macromolecules (peptides, proteins,
nucleic acids), incorporated in non‐viral carrier systems.[34,35,37‐39]
PCI experiments were conducted under conditions that induce moderate cytotoxicity on adherent
cells, i.e. 0.4 µg mL‐1 PS and 50s illumination time (data not shown and reference [37]). The effect of
PCI on gene silencing is shown in Figure 5.8 (white bars). Upon application of PCI a 60% and 78%
knockdown of luciferase expression was achieved, with 10 µg mL‐1 and 25 µg mL‐1 nanogels
respectively, in comparison with a 10% and 40% knockdown when the cells were not treated with
PCI. This large difference in gene silencing is a strong indication that the endosomal accumulation of
the siRNA loaded nanogels is a significant barrier for RNAi gene silencing. PCI had almost no influence
on the gene silencing by Lipofectamine™ RNAiMAX:siRNA complexes, suggesting that this carrier is
able to overcome the endosomal barrier by itself. Comparable results for Lipofectamine™2000 were
described by Bøe et al.[40]
To visually confirm the improvement in intracellular siRNA delivery after PCI treatment, we
conducted fluorescence microscopy experiments in which our cationic dextran nanogels were loaded
with siGLO® green transfection indicator.
154

biodegradable dextran nanogels for RNAi | CHAPTER 5
FIGURE 5.9 | Schematic overview of photochemical internalization (PCI). In a typical PCI experiment, the amphiphilic photosensitizer TPPS2a accumulates in the endosomal membranes of the target cells (I). Excitation with blue light induces the local production of mainly singlet oxygen, that disrupts the endocytic vesicles (II). This rupture of the endosomal barrier allows internalized nanosized matter to be released into the cytoplasm (III).
This green fluorescent (FAM‐labelled) siRNA duplex is chemically modified to accumulate
predominantly in the cell nucleus following successful delivery in the cell cytoplasm. Figure 5.10 (top
row) shows fluorescence micrographs of Huh‐7_wt cells that have internalized a large amount of
siGLO‐nanogels before PCI treatment. However, the punctuated fluorescence pattern suggest
vesicular confinement, as already pointed out above, and as a result none of the cells show
fluorescent nuclei. In contrast, applying PCI on this cell population leads to a detectable fluorescence
in the cytoplasm and nuclei of a fraction of cells which are signs of successful endosomal escape
(Figure 5.10, bottom row, indicated by the white arrows). Contrast enhancement further accentuates
the difference in cytoplasmic and nuclear siGLO fluorescence between the control cells and the cells
treated with PCI. Clearly, PCI promotes the delivery of siRNA to the cytoplasm of the cell and hence
this observation further supports the enhancement of the RNAi effect we observed in Figure 5.8.
To additionally validate the conclusions drawn from the PCI data, we also tested a different approach
to enhance the endosomal escape, i.e. by adding a fusogenic peptide to the siRNA loaded dex‐HEMA‐
co‐TMAEMA nanogel dispersion. We used a dimeric influenza‐derived peptide (diINF‐7) [22], which has
previously been shown to enhance the oncogene silencing by non‐viral siRNA complexes.[21] Other
investigators also successfully applied the INF‐7 peptide or its dimeric form to enhance intracellular
gene delivery and gene expression by improving the endosomal escape of polyplexes or liposomal
gene carriers.[41‐43]
155
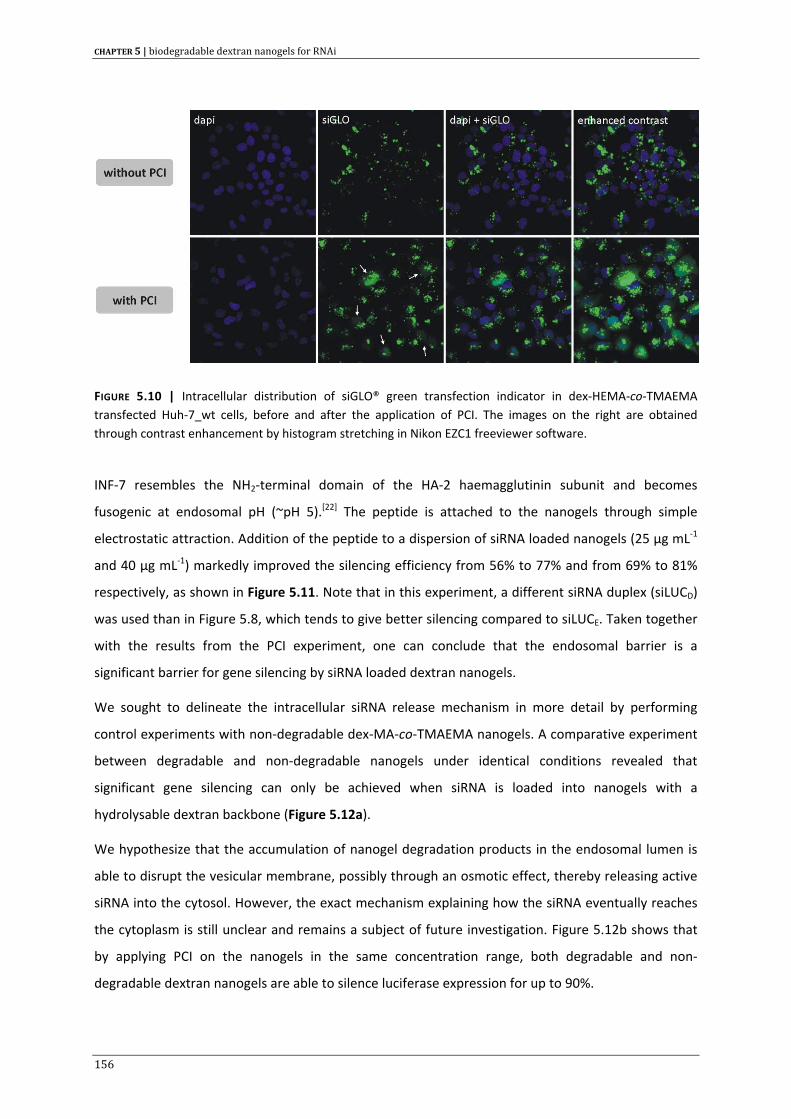
CHAPTER 5 | biodegradable dextran nanogels for RNAi
FIGURE 5.10 | Intracellular distribution of siGLO® green transfection indicator in dex‐HEMA‐co‐TMAEMA transfected Huh‐7_wt cells, before and after the application of PCI. The images on the right are obtained through contrast enhancement by histogram stretching in Nikon EZC1 freeviewer software.
INF‐7 resembles the NH2‐terminal domain of the HA‐2 haemagglutinin subunit and becomes
fusogenic at endosomal pH (~pH 5).[22] The peptide is attached to the nanogels through simple
electrostatic attraction. Addition of the peptide to a dispersion of siRNA loaded nanogels (25 µg mL‐1
and 40 µg mL‐1) markedly improved the silencing efficiency from 56% to 77% and from 69% to 81%
respectively, as shown in Figure 5.11. Note that in this experiment, a different siRNA duplex (siLUCD)
was used than in Figure 5.8, which tends to give better silencing compared to siLUCE. Taken together
with the results from the PCI experiment, one can conclude that the endosomal barrier is a
significant barrier for gene silencing by siRNA loaded dextran nanogels.
We sought to delineate the intracellular siRNA release mechanism in more detail by performing
control experiments with non‐degradable dex‐MA‐co‐TMAEMA nanogels. A comparative experiment
between degradable and non‐degradable nanogels under identical conditions revealed that
significant gene silencing can only be achieved when siRNA is loaded into nanogels with a
hydrolysable dextran backbone (Figure 5.12a).
We hypothesize that the accumulation of nanogel degradation products in the endosomal lumen is
able to disrupt the vesicular membrane, possibly through an osmotic effect, thereby releasing active
siRNA into the cytosol. However, the exact mechanism explaining how the siRNA eventually reaches
the cytoplasm is still unclear and remains a subject of future investigation. Figure 5.12b shows that
by applying PCI on the nanogels in the same concentration range, both degradable and non‐
degradable dextran nanogels are able to silence luciferase expression for up to 90%.
156
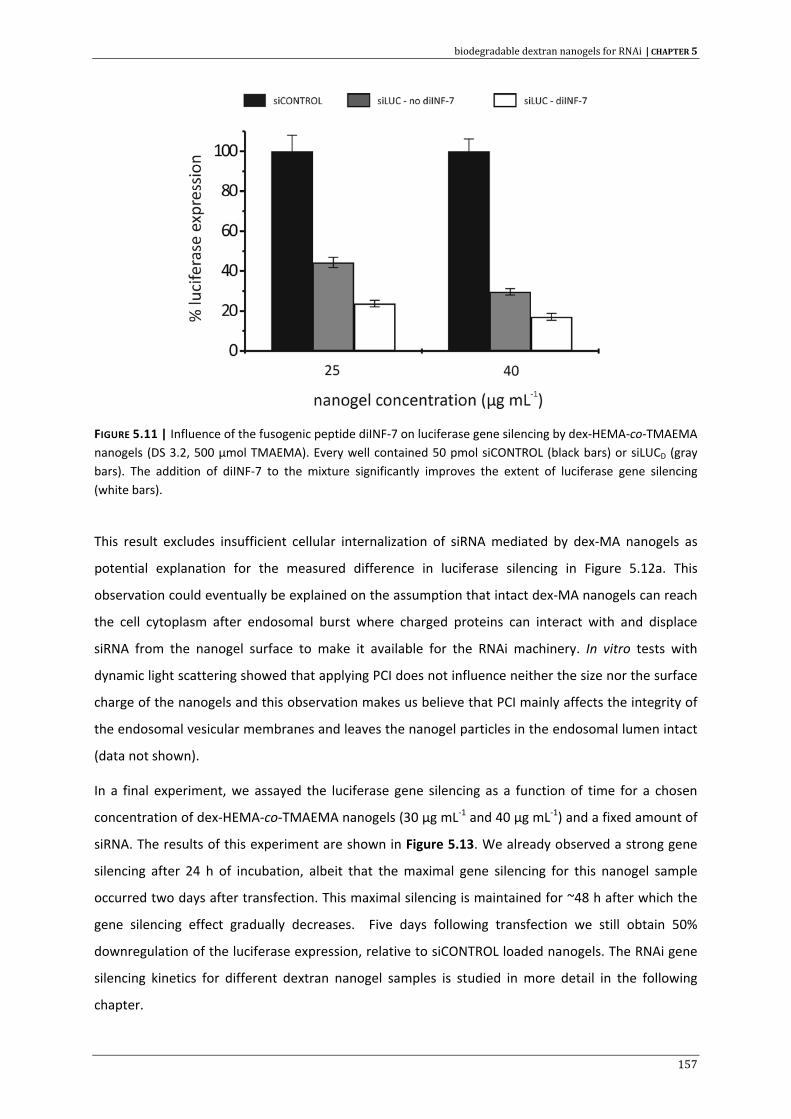
biodegradable dextran nanogels for RNAi | CHAPTER 5
FIGURE 5.11 | Influence of the fusogenic peptide diINF‐7 on luciferase gene silencing by dex‐HEMA‐co‐TMAEMA nanogels (DS 3.2, 500 µmol TMAEMA). Every well contained 50 pmol siCONTROL (black bars) or siLUCD (gray bars). The addition of diINF‐7 to the mixture significantly improves the extent of luciferase gene silencing (white bars).
This result excludes insufficient cellular internalization of siRNA mediated by dex‐MA nanogels as
potential explanation for the measured difference in luciferase silencing in Figure 5.12a. This
observation could eventually be explained on the assumption that intact dex‐MA nanogels can reach
the cell cytoplasm after endosomal burst where charged proteins can interact with and displace
siRNA from the nanogel surface to make it available for the RNAi machinery. In vitro tests with
dynamic light scattering showed that applying PCI does not influence neither the size nor the surface
charge of the nanogels and this observation makes us believe that PCI mainly affects the integrity of
the endosomal vesicular membranes and leaves the nanogel particles in the endosomal lumen intact
(data not shown).
In a final experiment, we assayed the luciferase gene silencing as a function of time for a chosen
concentration of dex‐HEMA‐co‐TMAEMA nanogels (30 µg mL‐1 and 40 µg mL‐1) and a fixed amount of
siRNA. The results of this experiment are shown in Figure 5.13. We already observed a strong gene
silencing after 24 h of incubation, albeit that the maximal gene silencing for this nanogel sample
occurred two days after transfection. This maximal silencing is maintained for ~48 h after which the
gene silencing effect gradually decreases. Five days following transfection we still obtain 50%
downregulation of the luciferase expression, relative to siCONTROL loaded nanogels. The RNAi gene
silencing kinetics for different dextran nanogel samples is studied in more detail in the following
chapter.
157
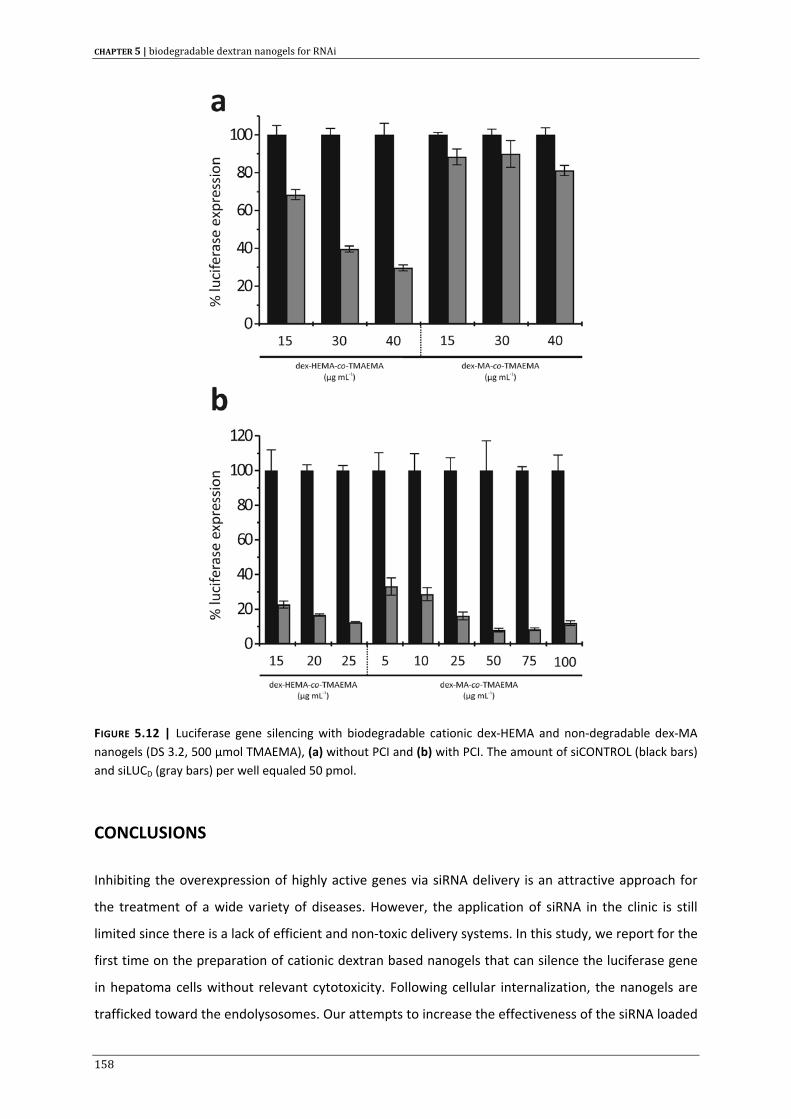
CHAPTER 5 | biodegradable dextran nanogels for RNAi
FIGURE 5.12 | Luciferase gene silencing with biodegradable cationic dex‐HEMA and non‐degradable dex‐MA nanogels (DS 3.2, 500 µmol TMAEMA), (a) without PCI and (b) with PCI. The amount of siCONTROL (black bars) and siLUCD (gray bars) per well equaled 50 pmol.
CONCLUSIONS
Inhibiting the overexpression of highly active genes via siRNA delivery is an attractive approach for
the treatment of a wide variety of diseases. However, the application of siRNA in the clinic is still
limited since there is a lack of efficient and non‐toxic delivery systems. In this study, we report for the
first time on the preparation of cationic dextran based nanogels that can silence the luciferase gene
in hepatoma cells without relevant cytotoxicity. Following cellular internalization, the nanogels are
trafficked toward the endolysosomes. Our attempts to increase the effectiveness of the siRNA loaded
158
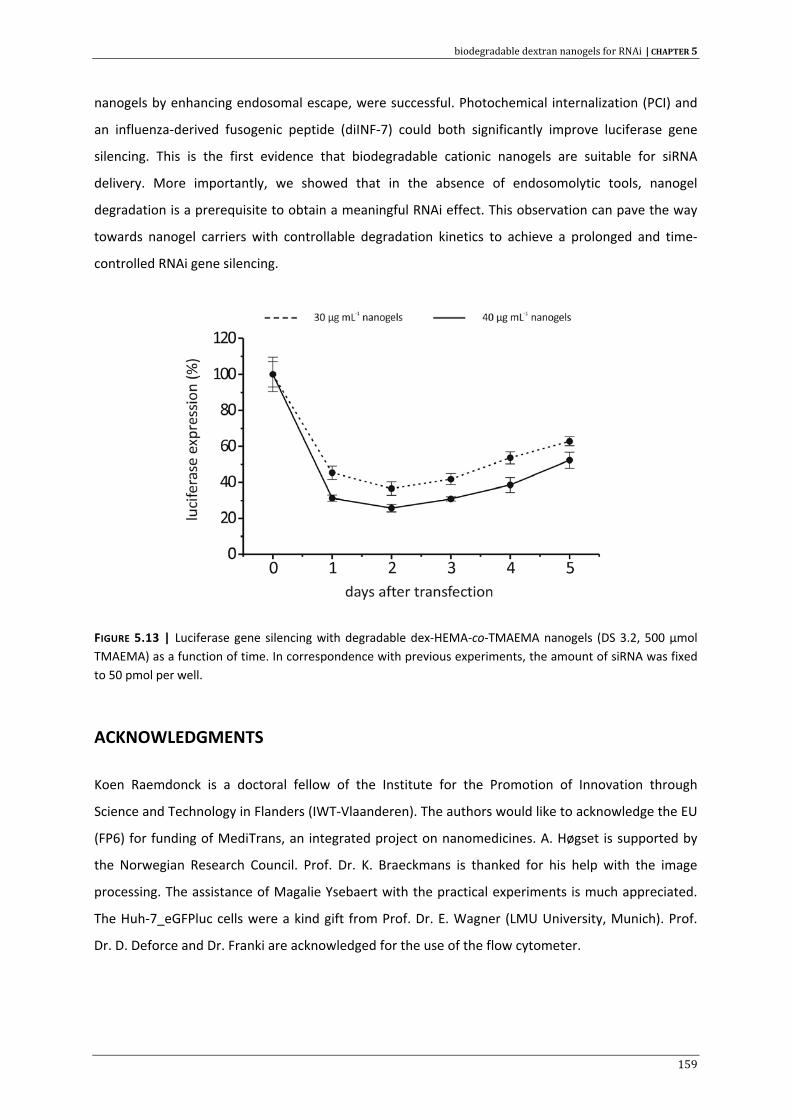
biodegradable dextran nanogels for RNAi | CHAPTER 5
nanogels by enhancing endosomal escape, were successful. Photochemical internalization (PCI) and
an influenza‐derived fusogenic peptide (diINF‐7) could both significantly improve luciferase gene
silencing. This is the first evidence that biodegradable cationic nanogels are suitable for siRNA
delivery. More importantly, we showed that in the absence of endosomolytic tools, nanogel
degradation is a prerequisite to obtain a meaningful RNAi effect. This observation can pave the way
towards nanogel carriers with controllable degradation kinetics to achieve a prolonged and time‐
controlled RNAi gene silencing.
FIGURE 5.13 | Luciferase gene silencing with degradable dex‐HEMA‐co‐TMAEMA nanogels (DS 3.2, 500 µmol TMAEMA) as a function of time. In correspondence with previous experiments, the amount of siRNA was fixed to 50 pmol per well.
ACKNOWLEDGMENTS
Koen Raemdonck is a doctoral fellow of the Institute for the Promotion of Innovation through
Science and Technology in Flanders (IWT‐Vlaanderen). The authors would like to acknowledge the EU
(FP6) for funding of MediTrans, an integrated project on nanomedicines. A. Høgset is supported by
the Norwegian Research Council. Prof. Dr. K. Braeckmans is thanked for his help with the image
processing. The assistance of Magalie Ysebaert with the practical experiments is much appreciated.
The Huh‐7_eGFPluc cells were a kind gift from Prof. Dr. E. Wagner (LMU University, Munich). Prof.
Dr. D. Deforce and Dr. Franki are acknowledged for the use of the flow cytometer.
159

CHAPTER 5 | biodegradable dextran nanogels for RNAi
REFERENCE LIST
1. Aagaard L. and Rossi J.J., RNAi therapeutics: Principles, prospects and challenges, Advanced Drug Delivery Reviews 2007, 59: 75‐86.
2. Kim D.H. and Rossi J.J., Strategies for silencing human disease using RNA interference, Nature Reviews Genetics 2007, 8: 173‐184.
3. Grimm D. and Kay M.A., Therapeutic application of RNAi: is mRNA targeting finally ready for prime time?, J. Clin. Invest 2007, 117: 3633‐3641.
4. Novobrantseva T.I., Akinc A., Borodovsky A., and de Fougerolles A., Delivering silence: Advancements in developing siRNA therapeutics, Current Opinion in Drug Discovery & Development 2008, 11: 217‐224.
5. de Fougerolles A., Vornlocher H.P., Maraganore J., and Lieberman J., Interfering with disease: a progress report on siRNA‐based therapeutics, Nature Reviews Drug Discovery 2007, 6: 443‐453.
6. Bohne J. and Cathomen T., Genotoxicity in gene therapy: An account of vector integration and designer nucleases, Current Opinion in Molecular Therapeutics 2008, 10: 214‐223.
7. Zaiss A.K. and Muruve D.A., Immunity to adeno‐associated virus vectors in animals and humans: a continued challenge, Gene Therapy 2008, 15: 808‐816.
8. Akinc A., Zumbuehl A., Goldberg M., Leshchiner E.S. et al., A combinatorial library of lipid‐like materials for delivery of RNAi therapeutics, Nature Biotechnology 2008, 26: 561‐569.
9. Remaut K., Sanders N.N., De Geest B.G., Braeckmans K. et al., Nucleic acid delivery: Where material sciences and bio‐sciences meet, Materials Science & Engineering R‐Reports 2007, 58: 117‐161.
10. Juliano R., Alam M.R., Dixit V., and Kang H., Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides, Nucleic Acids Research 2008, 36: 4158‐4171.
11. Raemdonck K., Vandenbroucke R.E., Demeester J., Sanders N.N. et al., Maintaining the silence: reflections on long‐term RNAi, Drug Discov. Today 2008, 13: 917‐931.
12. van Dijk‐Wolthuis W.N.E., Tsang S.K.Y., Kettenes‐vandenBosch J.J., and Hennink W.E., A new class of polymerizable dextrans with hydrolyzable groups: hydroxyethyl methacrylated dextran with and without oligolactate spacer, Polymer 1997, 38: 6235‐6242.
13. Van Tomme S.R. and Hennink W.E., Biodegradable dextran hydrogels for protein delivery applications, Expert Review of Medical Devices 2007, 4: 147‐164.
14. Raemdonck K., Van Thienen T.G., Vandenbroucke R.E., Sanders N.N. et al., Dextran microgels for time‐controlled delivery of siRNA, Advanced Functional Materials 2008, 18: 993‐1001.
15. Raemdonck K., Remaut K., Lucas B., Sanders N.N. et al., In situ analysis of single‐stranded and duplex siRNA integrity in living cells, Biochemistry 2006, 45: 10614‐10623.
16. Buyens K., Lucas B., Raemdonck K., Braeckmans K. et al., A fast and sensitive method for measuring the integrity of siRNA‐carrier complexes in full human serum, Journal of Controlled Release 2008, 126: 67‐76.
17. Remaut K., Lucas B., Raemdonck K., Braeckmans K. et al., Can we better understand the intracellular behavior of DNA nanoparticles by fluorescence correlation spectroscopy?, Journal of Controlled Release 2007, 121: 49‐63.
18. Lucas B., Remaut K., Braeckmans K., Haustraete J. et al., Studying pegylated DNA complexes by dual color fluorescence fluctuation spectroscopy, Macromolecules 2004, 37: 3832‐3840.
19. Lucas B., Van Rompaey E., De Smedt S.C., Demeester J. et al., Dual‐color fluorescence fluctuation spectroscopy to study the complexation between poly‐l‐lysine and oligonucleotides, Macromolecules 2002, 35: 8152‐8160.
20. Van Craenenbroeck E., Matthys G., Beirlant J., and Engelborghs Y., A statistical analysis of fluorescence correlation data, Journal of Fluorescence 1999, 9: 325‐331.
21. Oliveira S., van Rooy I., Kranenburg O., Storm G. et al., Fusogenic peptides enhance endosomal escape improving siRNA‐induced silencing of oncogenes, International Journal of Pharmaceutics 2007, 331: 211‐214.
22. Mastrobattista E., Koning G.A., van Bloois L., Filipe A.C.S. et al., Functional characterization of an endosome‐disruptive peptide and its application in cytosolic delivery of immunoliposome‐entrapped proteins, Journal of Biological Chemistry 2002, 277: 27135‐27143.
23. Oh J.K., Drumright R., Siegwart D.J., and Matyjaszewski K., The development of microgels/nanogels for drug delivery applications, Progress in Polymer Science 2008, 33: 448‐477.
24. van de Wetering P., Zuidam N.J., van Steenbergen M.J., van der Houwen O.A.G.J. et al., A mechanistic study of the hydrolytic stability of poly(2‐(dimethylamino)ethyl methacrylate), Macromolecules 1998, 31: 8063‐8068.
160

biodegradable dextran nanogels for RNAi | CHAPTER 5
25. Van Tomme S.R., van Nostrum C.F., Dijkstra M., De Smedt S.C. et al., Effect of particle size and charge on the network properties of microsphere‐based hydrogels, European Journal of Pharmaceutics and Biopharmaceutics 2008, 70: 522‐530.
26. van Dijk‐Wolthuis W.N., van Steenbergen M.J., Underberg W.J., and Hennink W.E., Degradation kinetics of methacrylated dextrans in aqueous solution, J. Pharm. Sci. 1997, 86: 413‐417.
27. Rejman J., Oberle V., Zuhorn I.S., and Hoekstra D., Size‐dependent internalization of particles via the pathways of clathrin‐and caveolae‐mediated endocytosis, Biochemical Journal 2004, 377: 159‐169.
28. Gratton S.E.A., Ropp P.A., Pohlhaus P.D., Luft J.C. et al., The effect of particle design on cellular internalization pathways, Proceedings of the National Academy of Sciences of the United States of America 2008, 105: 11613‐11618.
29. Vandenbroucke R., De Geest B., Demeester J., De Smedt S. et al., Prolonged gene silencing in hepatoma cells and primary hepatocytes after siRNA delivery with biodegradable poly(alpha‐amino ester[s]), Human Gene Therapy 2007, 18: 1056‐1056.
30. Payne C.K., Jones S.A., Chen C., and Zhuang X.W., Internalization and trafficking of cell surface proteoglycans and proteoglycan‐binding ligands, Traffic 2007, 8: 389‐401.
31. Van Dyke R.W., Acidification of lysosomes and endosomes, Subcell. Biochem. 1996, 27: 331‐360. 32. Vandenbroucke R.E., De Geest B.G., Bonne S., Vinken M. et al., Prolonged gene silencing in hepatoma cells
and primary hepatocytes after small interfering RNA delivery with biodegradable poly(beta‐amino esters), J. Gene Med. 2008, 10: 783‐794.
33. Grayson A.C.R., Ma J., and Putnam D., Kinetic and efficacy analysis of RNA interference in stably and transiently expressing cell lines, Molecular Pharmaceutics 2006, 3: 601‐613.
34. Berg K., Folini M., Prasmickaite L., Selbo P.K. et al., Photochemical internalization: A new tool for drug delivery, Current Pharmaceutical Biotechnology 2007, 8: 362‐372.
35. Fretz M.M., Hogset A., Koning G.A., Jiskoot W. et al., Cytosolic delivery of liposomally targeted proteins induced by photochemical internalization, Pharm. Res. 2007, 24: 2040‐2047.
36. Berg K., Selbo P.K., Prasmickaite L., Tjelle T.E. et al., Photochemical internalization: A novel technology for delivery of macromolecules into cytosol, Cancer Research 1999, 59: 1180‐1183.
37. Oliveira S., Fretz M.M., Hogset A., Storm G. et al., Photochemical internalization enhances silencing of epidermal growth factor receptor through improved endosomal escape of siRNA, Biochimica et Biophysica Acta‐Biomembranes 2007, 1768: 1211‐1217.
38. Kloeckner J., Prasmickaite L., Hogset A., Berg K. et al., Photochemically enhanced gene delivery of EGF receptor‐targeted DNA polyplexes, Journal of Drug Targeting 2004, 12: 205‐213.
39. Oliveira S., Hogset A., Storm G., and Schiffelers R.M., Delivery of siRNA to the target cell cytoplasm: photochemical internalization facilitates endosomal escape and improves silencing efficiency, in vitro and in vivo, Curr. Pharm. Des 2008, 14: 3686‐3697.
40. Boe S., Longva A.S., and Hovig E., Photochemically induced gene silencing using small interfering RNA molecules in combination with lipid carriers, Oligonucleotides 2007, 17: 166‐173.
41. El‐Sayed A., Masuda T., Khalil I., Akita H. et al., Enhanced gene expression by a novel stearylated INF7 peptide derivative through fusion independent endosomal escape, J. Control Release 2009, doi: 10.1016/j.jconrel.2009.05.18.
42. Moore N.M., Sheppard C.L., Barbour T.R., and Sakiyama‐Elbert S.E., The effect of endosomal escape peptides on in vitro gene delivery of polyethylene glycol‐based vehicles, J. Gene Med. 2008, 10: 1134‐1149.
43. Jiang X., Lok M.C., and Hennink W.E., Degradable‐brushed pHEMA‐pDMAEMA synthesized via ATRP and click chemistry for gene delivery, Bioconjug. Chem. 2007, 18: 2077‐2084.
161

CHAPTER 5 | biodegradable dextran nanogels for RNAi
162

evaluation of gene silencing kinetics with siRNA loaded dextran nanogels | CHAPTER 6
CHAPTER 6 | Evaluation of gene silencing kinetics with
siRNA loaded dextran nanogels
Manuscript of this chapter is in preparation:
Koen Raemdonck1, A. Høgset2, Joseph Demeester1 and Stefaan C. De Smedt1
1Laboratory of General Biochemistry and Physical Pharmacy, Department of Pharmaceutics, Faculty of Pharmaceutical
Sciences, Ghent University, Harelbekestraat 72, B‐9000 Ghent, Belgium.
2PCI Biotech, Hoffsveien 48, N‐0377, Oslo, Norway
163

CHAPTER 6 | evaluation of gene silencing kinetics with siRNA loaded dextran nanogels
164

evaluation of gene silencing kinetics with siRNA loaded dextran nanogels | CHAPTER 6
6 OVERVIEW CHAPTER CONTENTS
Chapter 6 provides:
• preparation and characterization of cationic dextran nanogels with varying gel network
density
• dose response suppression of EGFP with cationic nanogels
• the screening of RNAi knockdown kinetics with the aforementioned nanogels, in hepatoma
cells that stably express (1) firefly luciferase and (2) EGFP
• influence of photochemical internalization on the RNAi kinetics outcome
Key terms:
RNAi kinetics – nanogels – siRNA – EGFP – luciferase – degradation – PCI
INTRODUCTION
Since the discovery of RNA interference (RNAi) at the end of the 20th century,[1] intensive research on
RNAi therapeutics and their potential applications has led to an astonishing progress in just over a
single decade. The research investments resulted in a fast transition of promising lead siRNA
therapeutics from in vitro tests to clinical trials.[2‐4] Advances are made, both in the area of siRNA
design[5‐9] e.g. aiming for improved intrinsic siRNA activity as in the field of siRNA delivery. New
formulation strategies emerge, designed to overcome intra‐ and extracellular hurdles and to improve
the fraction of the administered siRNA dose that eventually reaches the target cell cytoplasm.[3,10,11]
165

CHAPTER 6 | evaluation of gene silencing kinetics with siRNA loaded dextran nanogels
In the majority of in vitro and in vivo assays to screen different siRNA delivery strategies, the maximal
gene suppression for a given siRNA dose is regarded as the main parameter to determine what
strategies should be taken into consideration. As already mentioned in previous chapters, not only
the maximal gene silencing effect at a fixed time‐point, but also and in our opinion more importantly,
the longevity of this effect must be routinely evaluated. While the magnitude of target gene (or
protein) suppression, relative to a control sample, may be a good indicator for the success of an
siRNA treatment, looking at the time‐window that the siRNA therapeutic intervention is able to keep
the intracellular protein titers below a certain threshold, is perhaps a better parameter to decide on
the efficacy of the treatment, even in vitro, and may also be used to compare different treatment
regimens and siRNA delivery strategies.[12] The duration of the silencing effect will eventually strongly
determine the dosage frequency in clinical therapy. Especially when long‐term RNAi activity is
necessary to obtain a desired downstream phenotype,[13] it should be common practice to check
upon the long‐term target gene suppression.
In the present chapter, dex‐HEMA‐co‐TMAEMA nanogels were prepared with dex‐HEMA of varying
methacrylate substitution and therefore also different nanogel crosslink densities.[14] The nanogel
batches were carefully characterised with regard to size, surface charge and siRNA interaction. In the
light of the comments made above, their gene silencing effect was analyzed as a function of time in
both luciferase and EGFP expressing human hepatoma cells. Finally, the influence of photochemical
internalization (PCI, see also chapter 5) on the gene silencing duration was assessed.
EXPERIMENTAL SECTION
Preparation of dextran nanogels and loading with siRNA
Cationic dextran nanogels (dex‐HEMA‐co‐TMAEMA) were prepared using the UV induced emulsion
photopolymerization as described in chapter 5, with minor adjustments. Dex‐HEMA (65 mg) was
dissolved in a solution containing 40 µL irgacure 1% (w/v), 80 µL TMAEMA (75% w/v) and 80 µL
HEPES buffer (pH 7.4, 20 mM). The obtained dextran solution was emulsified in 5 mL pre‐cooled
mineral oil, supplemented with 5% (v/v) of ABIL EM 90 as emulsifier. Following sonification (Branson
sonifier, 30 s ‐ 20%), the emulsion was exposed to UV light during 15 min under cooling (~4°C). The
prepared nanogels were obtained in pure form by precipitation with acetone:hexane (1:1) and
centrifugation. After washing, the precipitate was redispersed in 5 mL distilled water and traces of
organic solvent were removed by vacuum evaporation. To assure long‐term stability, the nanogels
were lyophilized and stored exsiccated. To load the nanogels with siRNA, a stock dispersion of
166

evaluation of gene silencing kinetics with siRNA loaded dextran nanogels | CHAPTER 6
nanogels (~2 mg mL‐1) was prepared in ice‐cooled HEPES buffer and sonicated to loosen the gel
particles (20 s, 10%). From this stock dispersion, dilutions are prepared according to the experimental
needs. Subsequently, equal volumes of nanogels and siRNA are mixed at room temperature and left
to stand at RT for ≥ 15 min to allow complexation.
Dynamic light scattering and zeta‐potential
The size and surface charge of the different nanogel dispersions, listed in Table 6.1, was analyzed
with DLS and zeta‐potential measurements respectively, as described earlier in this thesis. For more
details on the analysis of nanogel degradation, using the DLS instrument, we would like to refer to
the experimental section of chapter 5.
siRNA duplexes
Twenty‐one nt siRNAs targeting the pGL3 firefly luciferase gene (siLUC), the enhanced green
fluorescent protein (siEGFP) and a universal negative control duplex (siCONTROL) were purchased
from Eurogentec and stored in 20 µM aliquots in DEPC treated water (‐20°C). For flow cytometry and
fluorescence microscopy experiments, a green fluorescent siRNA duplex was ordered from
Dharmacon (siGLO® green transfection indicator).
Cell lines and culture conditions
All cell experiments were performed on Huh‐7 hepatoma cells of which the cell culture conditions
can be found in previous chapters. Huh‐7_LUC cells are cells that stably express the firefly and renilla
luciferase gene (see also chapter 5). Huh‐7_EGFP cells stably produce the enhanced green
fluorescent protein (see also chapter 3).
Luciferase gene silencing kinetics
Two days before transfection, Huh‐7_LUC cells were seeded in 96‐well plates (1 x 103 cells per well).
For transfection, first 90 µL of pre‐heated OptiMEM (37°C) was pipetted on the cells and
subsequently 10 µL of a siRNA loaded nanogel dispersion (in HEPES buffer) was added to the cells in
each well (20 pmol of siRNA). After 4 h incubation (37°C), the nanogels were removed from the cells
and replaced with 150 µL pre‐heated cell culture medium. This transfection protocol was performed
in duplicate (plate 1 and plate 2 respectively) for every sample tested. Twenty‐four hours later, the
167

CHAPTER 6 | evaluation of gene silencing kinetics with siRNA loaded dextran nanogels
cells on plate 1 were lysed with 20 µL passive lysis buffer (Promega, Leiden, The Netherlands). Firefly
and Renilla luciferase activities were determined with the Promega Dual‐Luciferase® Reporter Assay
as explained in chapter 5. The cells on plate 2 were trypsinized with 100 µL trypsin/EDTA (0.25%) and
replated in 5 separate 96‐well plates. Four of these plates are analyzed for luciferase expression on
day 2 till day 5 following transfection. On day 5, the trypsinization step was repeated on the
remaining 96‐well plate in order to avoid overgrowth of the cells and to maintain the cells in a state
of active cell division. Luciferase expression was additionally analyzed at day 6 and day 7.
Silencing EGFP with DS 2.5 cationic dextran nanogels
Two days before transfection, 7.5 x 104 Huh‐7_EGFP cells were seeded in 12‐well plates. At the day of
transfection, the culture medium was replaced with 900 µL pre‐heated OptiMEM. DS 2.5 siEGFP
nanogel complexes were prepared in HEPES buffer (~20 min incubation at room temperature) and
added to the wells to obtain a final volume of 1 mL. The nanogel and siEGFP concentration on the
cells eventually equalled 60 µg mL‐1 and 75 pmol mL‐1 respectively. As controls, empty nanogels or
nanogels loaded with siCONTROL and siLUC were included in the transfection experiment. After 5 h
incubation at 37°C, the transfection medium was replaced with 2 mL of complete cell culture
medium. Five days post‐transfection, the cells were washed with PBS and collected for flow
cytometry by trypsinization (500 µL trypsin/EDTA 0.25% per well, followed by dilution in 5 mL cell
culture medium). After centrifugation (1200 rpm, 7 min) the cell pellet was resuspended in 300 µL
flow buffer and kept on ice until flow analysis. The cells were analyzed using a Beckman Coulter
Cytomics™ FC500 flow cytometer. Dead cells were discriminated from viable cells by the addition of
propidium iodide (Invitrogen). The data were analyzed using Beckman Coulter CXP analysis software.
Silencing EGFP with DS 5.2 cationic dextran nanogels – fluorescence microscopy
1.5 x 104 Huh‐7_EGFP cells were grown on Lab‐Tek™ II chambered coverglasses (Nalge Nunc
International, Rochester, NY) two days pre‐transfection. As described above, the siRNA‐nanogel
complexes were prepared in HEPES buffer as a 10x stock dispersion. The transfection was performed
by incubating the cells during 4 h with 50 µg mL‐1 DS 5.2 nanogels, loaded with 40 pmol siEGFP in a
final volume of 400 µL OptiMEM and after this incubation period, the remaining complexes were
removed and 400 µL of fresh cell culture medium was added per well. The same controls as
indicated in the above paragraph were used. After 96 h, the cells were washed with PBS and fixed in
4% paraformaldehyde solution (20 min incubation at RT). Subsequently, after three additional
washing steps with PBS, the coverglasses were mounted with Vectashield® DAPI mounting medium
168

evaluation of gene silencing kinetics with siRNA loaded dextran nanogels | CHAPTER 6
(Vector Labs). EGFP expression was visualized with fluorescence microscopy using a Nikon EZC1‐si
confocal scanning module installed on a motorized Nikon TE2000‐E inverted microscope (Nikon
Benelux, Brussels, Belgium ‐ see previous chapters) with open pinhole setting.
Dose response EGFP silencing with DS 5.2 cationic nanogels
Huh‐7_EGFP cells were seeded in 24‐well plates (3 x 104 cells per well). Two days after seeding, the
cells were transfected with 50 µg mL‐1 nanogels (500 µL in OptiMEM) during 4 h after which the
medium was replaced with 500 µL of fresh complete cell culture medium. We verified for the DS 5.2
nanogels that 4 h incubation with the cells was the optimal incubation time. Longer incubation did
not improve the RNAi activity but rather induced more cytotoxic effects (data not shown). The
amount of siEGFP per well was varied between 0.025 pmol and 125 pmol. Nanogels that complex
siLUC are used as a control. At day 3 post‐transfection, the cells were prepared for flow cytometry by
trypsinization (250 µL trypsin/EDTA 0.25% per well, followed by dilution in 1 mL cell culture medium).
After centrifugation (1200 rpm, 7 min) the cell pellet was resuspended in 200 µL flow buffer and kept
on ice until flow analysis as explained above.
EGFP gene silencing kinetics
Huh‐7_EGFP cells were seeded in 24‐well plates at a density of 5 x 104 cells per well. Twenty‐four
hours later, the cells were incubated during 4 h with DS 2.5, DS 5.2 and DS 8.9 nanogels (50 µg mL‐1),
loaded with siEGFP or siLUC (50 pmol per well) in a final volume of 500 µL (OptiMEM).
Lipofectamine™RNAiMAX complexes were prepared in OptiMEM and included in the transfection
experiment (1.56 µL RNAiMAX stock per well). For all samples, the transfection was performed in
duplicate. Subsequently, the transfection medium was replaced with 500 µL cell culture medium.
EGFP expression was quantified at different time‐points post‐transfection. At every time‐point, the
cells in the duplicate transfection were again replated in two new 24‐well plates (1/3 diluted) to be
analysed at the next time‐point.
Influence of photochemical internalization on EGFP gene silencing kinetics
The effect of PCI on the silencing duration was evaluated in two distinct protocols where PCI was
conducted at the day of transfection (PCI t0) or two days post‐transfection (PCI t2). More details on
the protocol time‐scale can be found in Figure 6.8. For the transfection, 5 x 104 cells are seeded in 24‐
well plates and left to grow for 48 h. Transfection of the cells (in 500 µL OptiMEM) was performed
169
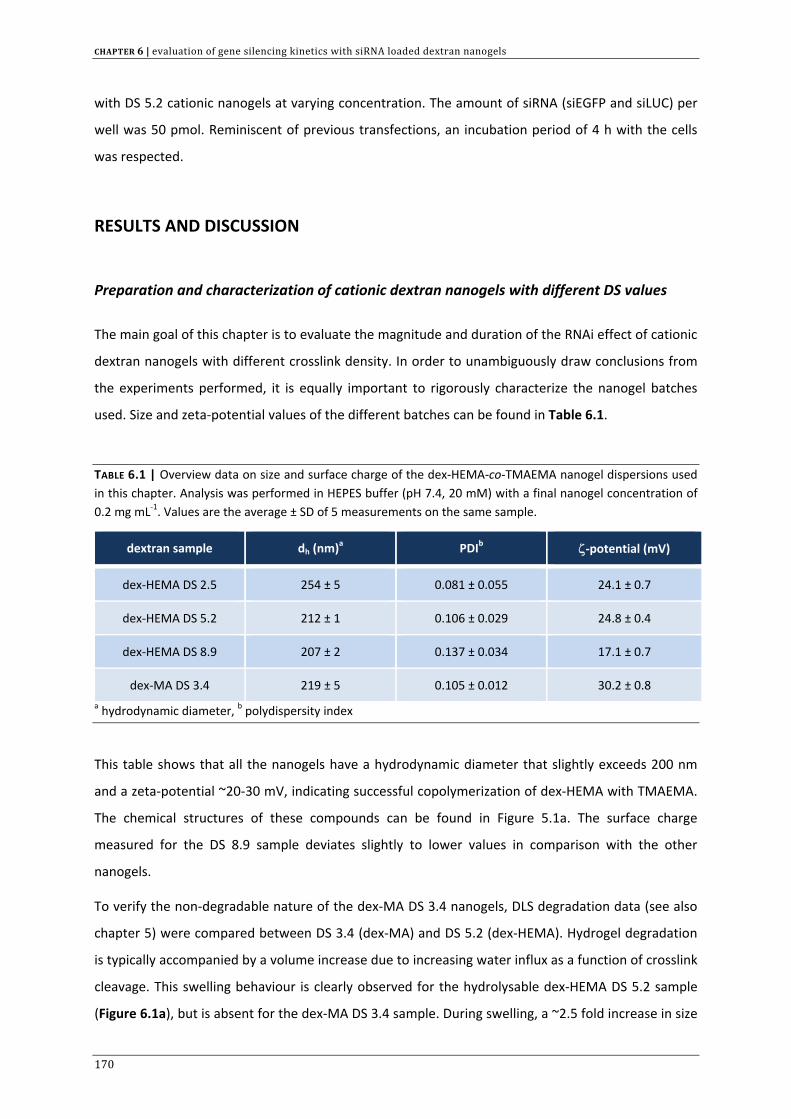
CHAPTER 6 | evaluation of gene silencing kinetics with siRNA loaded dextran nanogels
with DS 5.2 cationic nanogels at varying concentration. The amount of siRNA (siEGFP and siLUC) per
well was 50 pmol. Reminiscent of previous transfections, an incubation period of 4 h with the cells
was respected.
RESULTS AND DISCUSSION
Preparation and characterization of cationic dextran nanogels with different DS values
The main goal of this chapter is to evaluate the magnitude and duration of the RNAi effect of cationic
dextran nanogels with different crosslink density. In order to unambiguously draw conclusions from
the experiments performed, it is equally important to rigorously characterize the nanogel batches
used. Size and zeta‐potential values of the different batches can be found in Table 6.1.
TABLE 6.1 | Overview data on size and surface charge of the dex‐HEMA‐co‐TMAEMA nanogel dispersions used in this chapter. Analysis was performed in HEPES buffer (pH 7.4, 20 mM) with a final nanogel concentration of 0.2 mg mL‐1. Values are the average ± SD of 5 measurements on the same sample.
dextran sample dh (nm)a PDIb ζ‐potential (mV)
dex‐HEMA DS 2.5 254 ± 5 0.081 ± 0.055 24.1 ± 0.7
dex‐HEMA DS 5.2 212 ± 1 0.106 ± 0.029 24.8 ± 0.4
dex‐HEMA DS 8.9 207 ± 2 0.137 ± 0.034 17.1 ± 0.7
dex‐MA DS 3.4 219 ± 5 0.105 ± 0.012 30.2 ± 0.8 a hydrodynamic diameter, b polydispersity index
This table shows that all the nanogels have a hydrodynamic diameter that slightly exceeds 200 nm
and a zeta‐potential ~20‐30 mV, indicating successful copolymerization of dex‐HEMA with TMAEMA.
The chemical structures of these compounds can be found in Figure 5.1a. The surface charge
measured for the DS 8.9 sample deviates slightly to lower values in comparison with the other
nanogels.
To verify the non‐degradable nature of the dex‐MA DS 3.4 nanogels, DLS degradation data (see also
chapter 5) were compared between DS 3.4 (dex‐MA) and DS 5.2 (dex‐HEMA). Hydrogel degradation
is typically accompanied by a volume increase due to increasing water influx as a function of crosslink
cleavage. This swelling behaviour is clearly observed for the hydrolysable dex‐HEMA DS 5.2 sample
(Figure 6.1a), but is absent for the dex‐MA DS 3.4 sample. During swelling, a ~2.5 fold increase in size
170
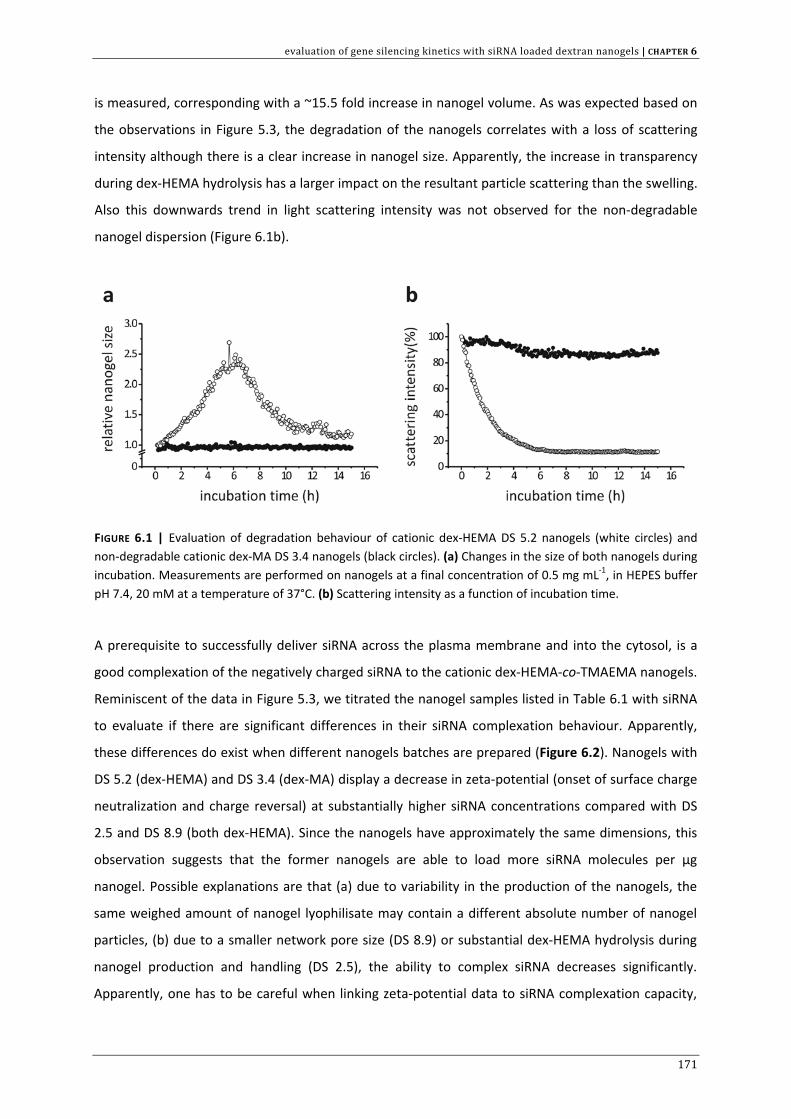
evaluation of gene silencing kinetics with siRNA loaded dextran nanogels | CHAPTER 6
is measured, corresponding with a ~15.5 fold increase in nanogel volume. As was expected based on
the observations in Figure 5.3, the degradation of the nanogels correlates with a loss of scattering
intensity although there is a clear increase in nanogel size. Apparently, the increase in transparency
during dex‐HEMA hydrolysis has a larger impact on the resultant particle scattering than the swelling.
Also this downwards trend in light scattering intensity was not observed for the non‐degradable
nanogel dispersion (Figure 6.1b).
FIGURE 6.1 | Evaluation of degradation behaviour of cationic dex‐HEMA DS 5.2 nanogels (white circles) and non‐degradable cationic dex‐MA DS 3.4 nanogels (black circles). (a) Changes in the size of both nanogels during incubation. Measurements are performed on nanogels at a final concentration of 0.5 mg mL‐1, in HEPES buffer pH 7.4, 20 mM at a temperature of 37°C. (b) Scattering intensity as a function of incubation time.
A prerequisite to successfully deliver siRNA across the plasma membrane and into the cytosol, is a
good complexation of the negatively charged siRNA to the cationic dex‐HEMA‐co‐TMAEMA nanogels.
Reminiscent of the data in Figure 5.3, we titrated the nanogel samples listed in Table 6.1 with siRNA
to evaluate if there are significant differences in their siRNA complexation behaviour. Apparently,
these differences do exist when different nanogels batches are prepared (Figure 6.2). Nanogels with
DS 5.2 (dex‐HEMA) and DS 3.4 (dex‐MA) display a decrease in zeta‐potential (onset of surface charge
neutralization and charge reversal) at substantially higher siRNA concentrations compared with DS
2.5 and DS 8.9 (both dex‐HEMA). Since the nanogels have approximately the same dimensions, this
observation suggests that the former nanogels are able to load more siRNA molecules per µg
nanogel. Possible explanations are that (a) due to variability in the production of the nanogels, the
same weighed amount of nanogel lyophilisate may contain a different absolute number of nanogel
particles, (b) due to a smaller network pore size (DS 8.9) or substantial dex‐HEMA hydrolysis during
nanogel production and handling (DS 2.5), the ability to complex siRNA decreases significantly.
Apparently, one has to be careful when linking zeta‐potential data to siRNA complexation capacity,
171
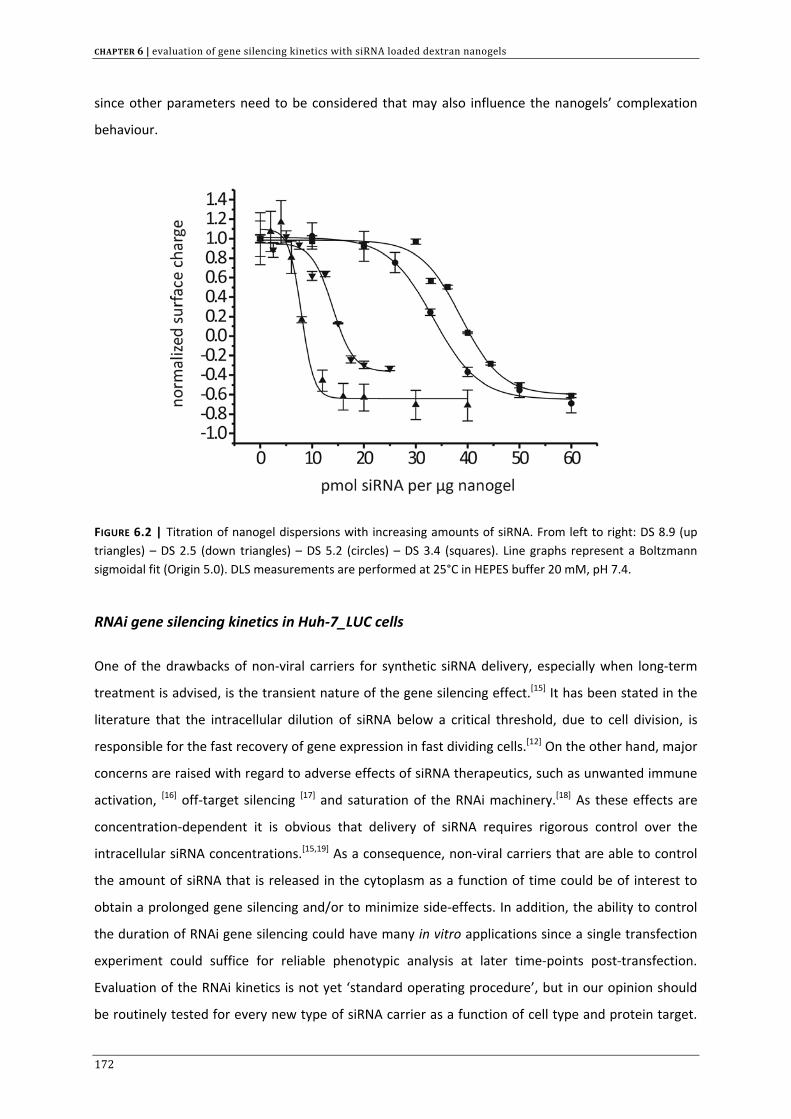
CHAPTER 6 | evaluation of gene silencing kinetics with siRNA loaded dextran nanogels
since other parameters need to be considered that may also influence the nanogels’ complexation
behaviour.
FIGURE 6.2 | Titration of nanogel dispersions with increasing amounts of siRNA. From left to right: DS 8.9 (up triangles) – DS 2.5 (down triangles) – DS 5.2 (circles) – DS 3.4 (squares). Line graphs represent a Boltzmann sigmoidal fit (Origin 5.0). DLS measurements are performed at 25°C in HEPES buffer 20 mM, pH 7.4.
RNAi gene silencing kinetics in Huh‐7_LUC cells
One of the drawbacks of non‐viral carriers for synthetic siRNA delivery, especially when long‐term
treatment is advised, is the transient nature of the gene silencing effect.[15] It has been stated in the
literature that the intracellular dilution of siRNA below a critical threshold, due to cell division, is
responsible for the fast recovery of gene expression in fast dividing cells.[12] On the other hand, major
concerns are raised with regard to adverse effects of siRNA therapeutics, such as unwanted immune
activation, [16] off‐target silencing [17] and saturation of the RNAi machinery.[18] As these effects are
concentration‐dependent it is obvious that delivery of siRNA requires rigorous control over the
intracellular siRNA concentrations.[15,19] As a consequence, non‐viral carriers that are able to control
the amount of siRNA that is released in the cytoplasm as a function of time could be of interest to
obtain a prolonged gene silencing and/or to minimize side‐effects. In addition, the ability to control
the duration of RNAi gene silencing could have many in vitro applications since a single transfection
experiment could suffice for reliable phenotypic analysis at later time‐points post‐transfection.
Evaluation of the RNAi kinetics is not yet ‘standard operating procedure’, but in our opinion should
be routinely tested for every new type of siRNA carrier as a function of cell type and protein target.
172

evaluation of gene silencing kinetics with siRNA loaded dextran nanogels | CHAPTER 6
Dependent on the degree of protein suppression that is needed to observe the desired phenotype,
these kinetic data could provide the basis for the outline of a clinical treatment schedule.
In this chapter we aimed to investigate the kinetics of RNAi gene silencing for our various nanogel
dispersions, to identify possible variation in the RNAi outcome induced by differences in DS value and
aiming to gather more information to further optimize our concept of siRNA delivery through
biodegradable nanogels.
For every nanogel sample, first the expression of both firefly and renilla luciferase was followed for
the course of one week. To maintain the cells in a state of active cell division and prevent cellular
overgrowth, we replated the cells at day 1 and day 5 post‐transfection (Figure 6.3). As a positive
control, the RNAi gene silencing kinetics for lipofectamine™RNAiMAX was evaluated using exactly the
same protocol. Since the cellular uptake of siRNA varies as a function of the nanogel concentration
that is incubated with the cells, we screened different concentrations for every nanogel type, ranging
from 10 µg mL‐1 to 70 or 80 µg mL‐ 1.
Figure 6.3 shows that none of the siRNA carriers investigated was able to maintain sufficient gene
silencing up to 7 days. Although the DS 2.5, DS 5.2 and Lipofectamine™RNAiMAX achieve a maximal
gene silencing of > 75% two days post‐transfection, this level of gene suppression is only maintained
for an additional 24 h at most. Five days post‐transfection, on average gene expression has recovered
to ~50% compared to control (reminiscent of the luciferase silencing kinetics presented in Figure
5.13), and seven days post‐transfection, in most cases little gene suppression remains. Another
interesting observation is the lack of substantial gene silencing (< 50% knockdown) when performing
experiments with slowly degrading nanogels (DS 8.9). DS 8.9 nanogels reach their maximal effect at a
concentration of 60 µg mL‐1 after which no further improvement is observed. Also non‐degradable
nanogels (DS 3.4, dex‐MA) were not able to silence the luciferase gene to an appreciable extent
(Figure 6.4).
173
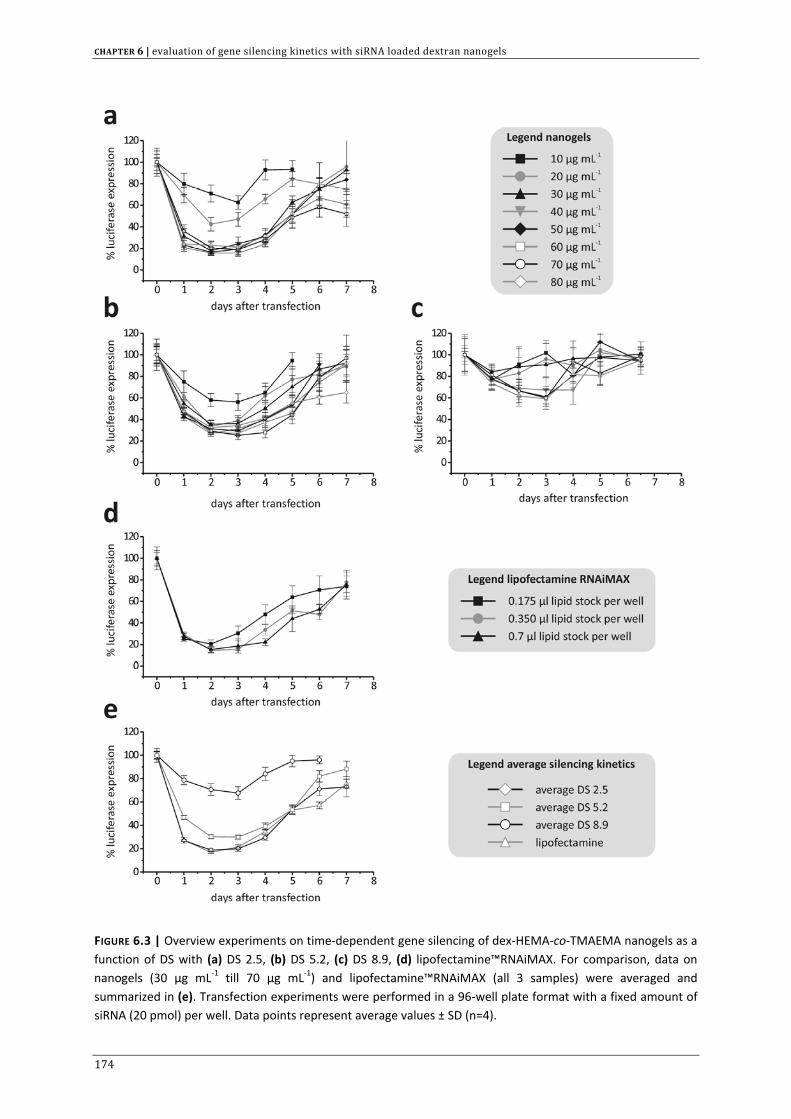
CHAPTER 6 | evaluation of gene silencing kinetics with siRNA loaded dextran nanogels
FIGURE 6.3 | Overview experiments on time‐dependent gene silencing of dex‐HEMA‐co‐TMAEMA nanogels as a function of DS with (a) DS 2.5, (b) DS 5.2, (c) DS 8.9, (d) lipofectamine™RNAiMAX. For comparison, data on nanogels (30 µg mL‐1 till 70 µg mL‐1) and lipofectamine™RNAiMAX (all 3 samples) were averaged and summarized in (e). Transfection experiments were performed in a 96‐well plate format with a fixed amount of siRNA (20 pmol) per well. Data points represent average values ± SD (n=4).
174
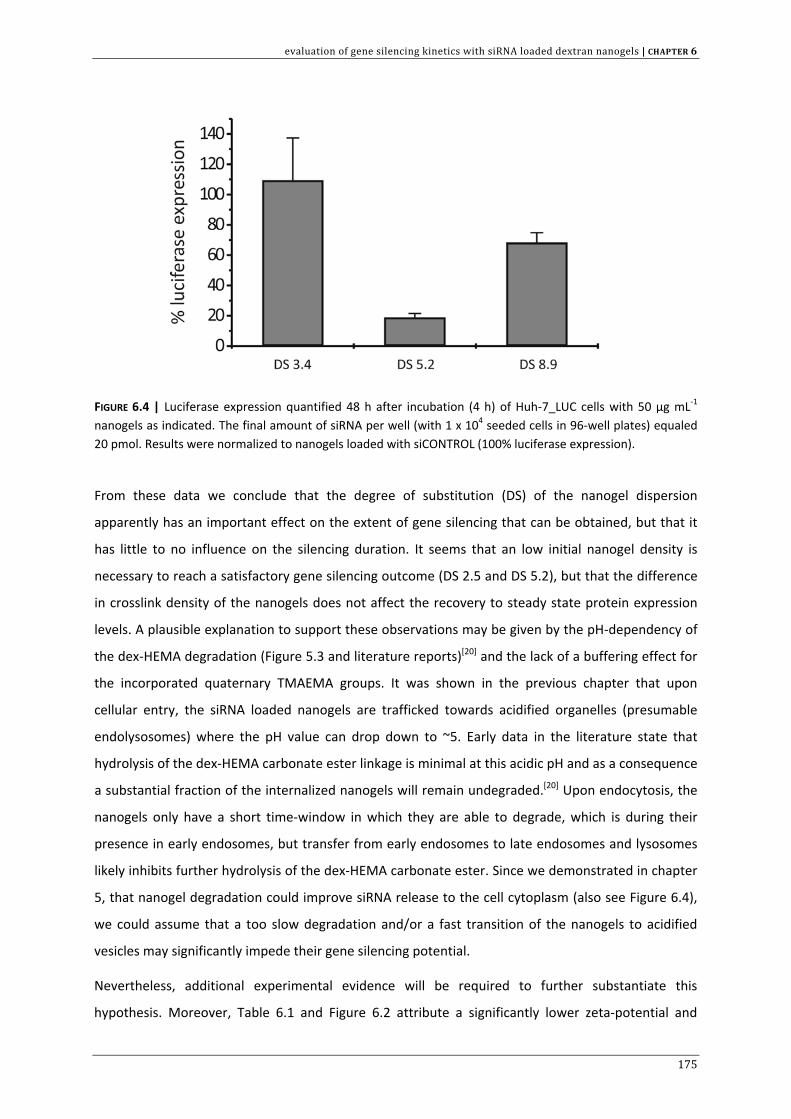
evaluation of gene silencing kinetics with siRNA loaded dextran nanogels | CHAPTER 6
FIGURE 6.4 | Luciferase expression quantified 48 h after incubation (4 h) of Huh‐7_LUC cells with 50 µg mL‐1 nanogels as indicated. The final amount of siRNA per well (with 1 x 104 seeded cells in 96‐well plates) equaled 20 pmol. Results were normalized to nanogels loaded with siCONTROL (100% luciferase expression).
From these data we conclude that the degree of substitution (DS) of the nanogel dispersion
apparently has an important effect on the extent of gene silencing that can be obtained, but that it
has little to no influence on the silencing duration. It seems that an low initial nanogel density is
necessary to reach a satisfactory gene silencing outcome (DS 2.5 and DS 5.2), but that the difference
in crosslink density of the nanogels does not affect the recovery to steady state protein expression
levels. A plausible explanation to support these observations may be given by the pH‐dependency of
the dex‐HEMA degradation (Figure 5.3 and literature reports)[20] and the lack of a buffering effect for
the incorporated quaternary TMAEMA groups. It was shown in the previous chapter that upon
cellular entry, the siRNA loaded nanogels are trafficked towards acidified organelles (presumable
endolysosomes) where the pH value can drop down to ~5. Early data in the literature state that
hydrolysis of the dex‐HEMA carbonate ester linkage is minimal at this acidic pH and as a consequence
a substantial fraction of the internalized nanogels will remain undegraded.[20] Upon endocytosis, the
nanogels only have a short time‐window in which they are able to degrade, which is during their
presence in early endosomes, but transfer from early endosomes to late endosomes and lysosomes
likely inhibits further hydrolysis of the dex‐HEMA carbonate ester. Since we demonstrated in chapter
5, that nanogel degradation could improve siRNA release to the cell cytoplasm (also see Figure 6.4),
we could assume that a too slow degradation and/or a fast transition of the nanogels to acidified
vesicles may significantly impede their gene silencing potential.
Nevertheless, additional experimental evidence will be required to further substantiate this
hypothesis. Moreover, Table 6.1 and Figure 6.2 attribute a significantly lower zeta‐potential and
175
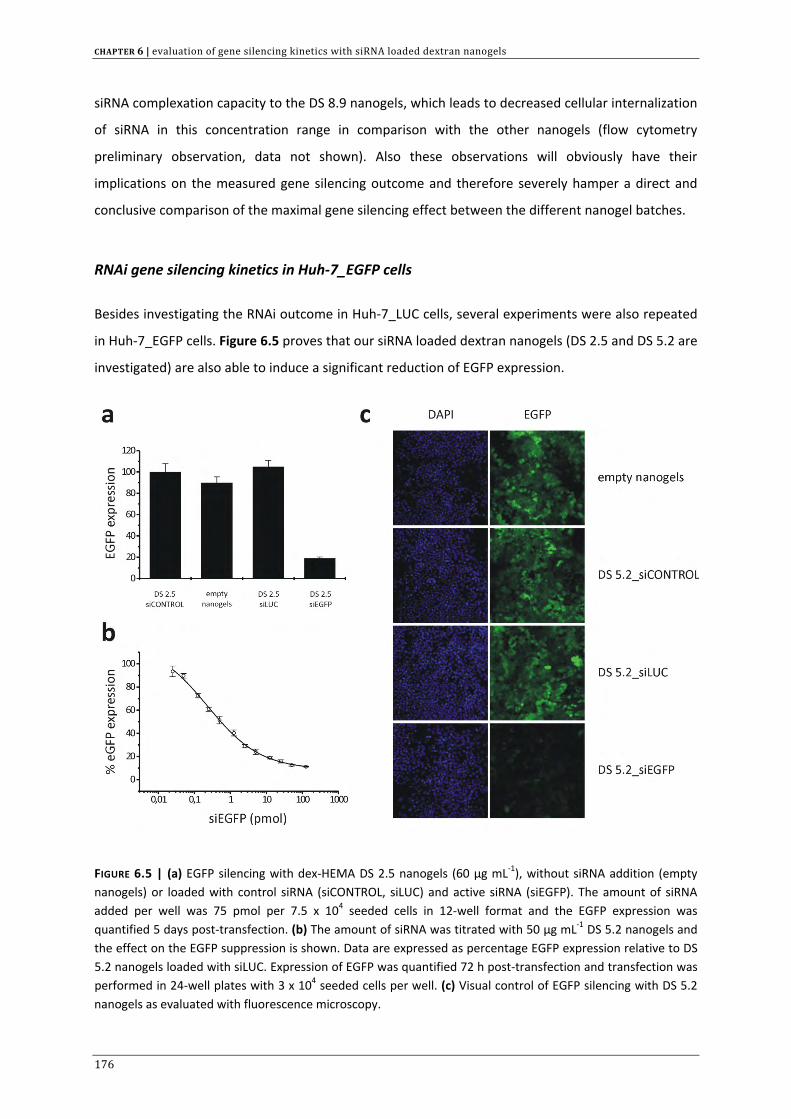
CHAPTER 6 | evaluation of gene silencing kinetics with siRNA loaded dextran nanogels
siRNA complexation capacity to the DS 8.9 nanogels, which leads to decreased cellular internalization
of siRNA in this concentration range in comparison with the other nanogels (flow cytometry
preliminary observation, data not shown). Also these observations will obviously have their
implications on the measured gene silencing outcome and therefore severely hamper a direct and
conclusive comparison of the maximal gene silencing effect between the different nanogel batches.
RNAi gene silencing kinetics in Huh‐7_EGFP cells
Besides investigating the RNAi outcome in Huh‐7_LUC cells, several experiments were also repeated
in Huh‐7_EGFP cells. Figure 6.5 proves that our siRNA loaded dextran nanogels (DS 2.5 and DS 5.2 are
investigated) are also able to induce a significant reduction of EGFP expression.
FIGURE 6.5 | (a) EGFP silencing with dex‐HEMA DS 2.5 nanogels (60 µg mL‐1), without siRNA addition (empty nanogels) or loaded with control siRNA (siCONTROL, siLUC) and active siRNA (siEGFP). The amount of siRNA added per well was 75 pmol per 7.5 x 104 seeded cells in 12‐well format and the EGFP expression was quantified 5 days post‐transfection. (b) The amount of siRNA was titrated with 50 µg mL‐1 DS 5.2 nanogels and the effect on the EGFP suppression is shown. Data are expressed as percentage EGFP expression relative to DS 5.2 nanogels loaded with siLUC. Expression of EGFP was quantified 72 h post‐transfection and transfection was performed in 24‐well plates with 3 x 104 seeded cells per well. (c) Visual control of EGFP silencing with DS 5.2 nanogels as evaluated with fluorescence microscopy.
176
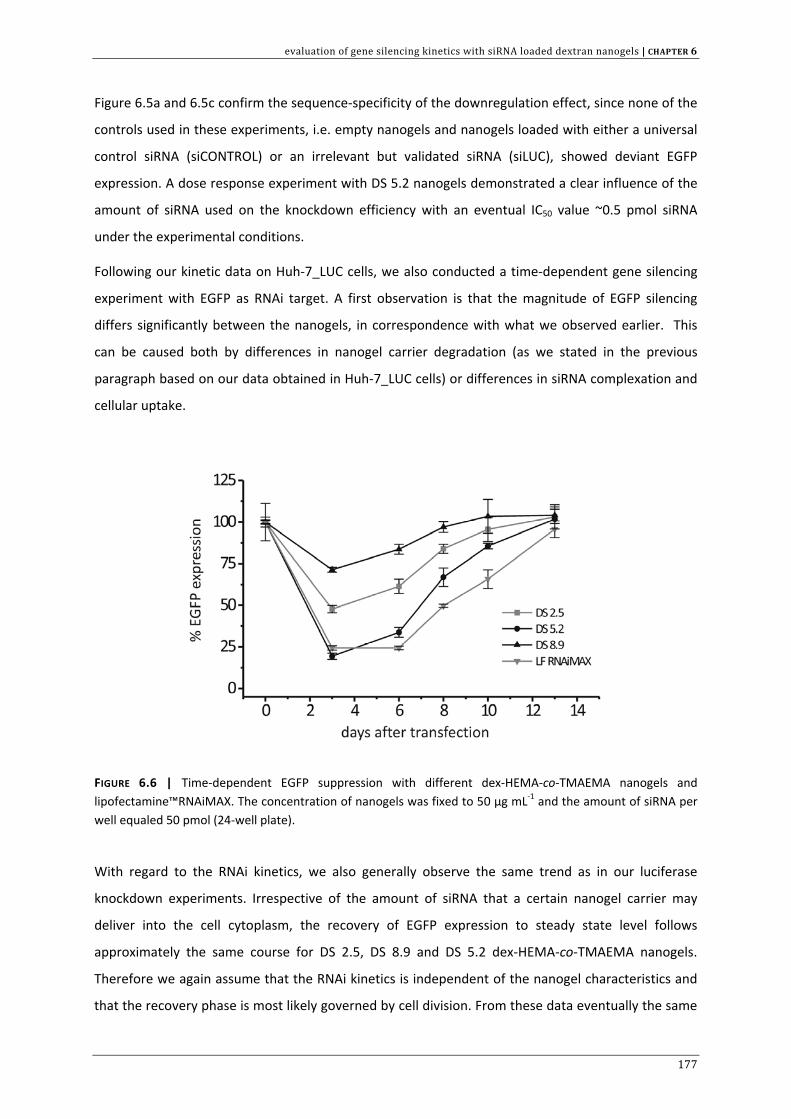
evaluation of gene silencing kinetics with siRNA loaded dextran nanogels | CHAPTER 6
Figure 6.5a and 6.5c confirm the sequence‐specificity of the downregulation effect, since none of the
controls used in these experiments, i.e. empty nanogels and nanogels loaded with either a universal
control siRNA (siCONTROL) or an irrelevant but validated siRNA (siLUC), showed deviant EGFP
expression. A dose response experiment with DS 5.2 nanogels demonstrated a clear influence of the
amount of siRNA used on the knockdown efficiency with an eventual IC50 value ~0.5 pmol siRNA
under the experimental conditions.
Following our kinetic data on Huh‐7_LUC cells, we also conducted a time‐dependent gene silencing
experiment with EGFP as RNAi target. A first observation is that the magnitude of EGFP silencing
differs significantly between the nanogels, in correspondence with what we observed earlier. This
can be caused both by differences in nanogel carrier degradation (as we stated in the previous
paragraph based on our data obtained in Huh‐7_LUC cells) or differences in siRNA complexation and
cellular uptake.
FIGURE 6.6 | Time‐dependent EGFP suppression with different dex‐HEMA‐co‐TMAEMA nanogels and lipofectamine™RNAiMAX. The concentration of nanogels was fixed to 50 µg mL‐1 and the amount of siRNA per well equaled 50 pmol (24‐well plate).
With regard to the RNAi kinetics, we also generally observe the same trend as in our luciferase
knockdown experiments. Irrespective of the amount of siRNA that a certain nanogel carrier may
deliver into the cell cytoplasm, the recovery of EGFP expression to steady state level follows
approximately the same course for DS 2.5, DS 8.9 and DS 5.2 dex‐HEMA‐co‐TMAEMA nanogels.
Therefore we again assume that the RNAi kinetics is independent of the nanogel characteristics and
that the recovery phase is most likely governed by cell division. From these data eventually the same
177

CHAPTER 6 | evaluation of gene silencing kinetics with siRNA loaded dextran nanogels
hypothesis arises, namely that during a short time‐window following cellular internalization, a small
fraction of the complexed siRNA may be delivered in the cell cytoplasm, but that a genuine
intracellular time‐controlled delivery based upon the characteristics of the dex‐HEMA nanogels is
hampered by the endolysosomal barrier. Other reports in the literature also state that only a minor
fraction of the internalized siRNA is eventually functionally active in the cytosol.[21‐24] Since most
cationic nanoparticles are effectively endocytosed by the target cells, exploiting possibilities to
overcome their resulting vesicular confinement will dramatically increase the biological availability of
siRNA and the RNAi activity.
RNAi gene silencing kinetics in Huh‐7_EGFP cells: influence of PCI
To achieve an intracellular degradation controlled release of siRNA with our cationic dextran
nanogels in order to improve the biological availability of siRNA in the target cell, it is imperative to
induce a transition of siRNA loaded nanogels from the endosomal compartment towards the
cytoplasm of the cell. Chapter 5 showed that photochemical internalization (PCI) was able to
enhance the RNAi effect of both degradable dex‐HEMA as non‐degradable dex‐MA nanogels (Figure
5.9 and 5.10).[25] For this reason, we again turned to PCI to test its effect on the nanogel samples
used in this chapter in order to implement a PCI driven time‐dependent gene silencing. In agreement
with the results in the previous chapter, we observed a significant improvement in EGFP gene
silencing for all nanogel samples used in this chapter, also for the non‐degradable dex‐MA DS 3.4
nanogels (data not shown). Improvement in cellular siRNA delivery with non‐degradable dex‐MA
nanogels is best explained by the fact that charged anionic macromolecules that the particles
encounter in the cell cytoplasm (e.g. proteins, mRNA, negatively charged phospholipids) are able to
displace siRNA molecules from the surface of the nanogels (see also PCI data in chapter 5). Currently,
we cannot be sure whether the observed improvement in the siRNA effect is a result of quantitative
siRNA displacement from the nanogel surface or that a fraction of the internalized siRNA remains
complexed after endosomal escape. In the latter case, this siRNA fraction may still be released in a
degradation controlled fashion from our dex‐HEMA‐co‐TMAEMA nanogels. Unfortunately, up to now
we have insufficient data at our disposal to unambiguously address this question.
As a proof of concept of how photochemical internalization may be applied not only to enhance the
intracellular release of siRNA but also to induce a more sustained gene silencing effect, we followed
an experimental gene silencing protocol as schematically depicted in Figure 6.7. In these
experiments, photochemical internalization was not only induced during transfection as is normally
178

evaluation of gene silencing kinetics with siRNA loaded dextran nanogels | CHAPTER 6
done (Figure 6.7a – PCI t0), but also the influence of a two days delay in the photochemical treatment
was investigated (Figure 6.7b – PCI t2).
PCI t0 on dex‐HEMA‐co‐TMAEMA nanogels (DS 5.2, 25 µg mL‐1) already improved the EGFP expression
on day 3 post‐transfection (i.e. also 3 days following PCI treatment), an effect that shows even more
obvious at the next analysis point (day 6) (Figure 6.8a). Nevertheless, the improvement in the effect
is from this point on gradually lost as can be seen at day 9 and day 12 post‐transfection. As expected,
promoting endosomal escape by PCI, eventually enlarges the fraction of functional siRNA molecules
in the cell cytoplasm, resulting in an improvement of target gene suppression, lasting for at least 6
days under the experimental conditions. Surprisingly, when applied in a transfection with 50 µg mL‐1
as nanogel concentration, the PCI t0 protocol was not able to enhance the eventual gene silencing
outcome (data not shown).
FIGURE 6.7 | Schematic overview of the experimental protocols used with the application of PCI. (a) PCI t0: one day after seeding of the Huh‐7_EGFP cells, photosensitizer (PS) was added and incubation proceeded overnight. At day 0, PS was removed from the cells and the transfection was performed in PS‐free OptiMEM (4 h) after which the cells were illuminated. (b) PCI t2: 48 h after seeding the cells, they were transfected with siRNA loaded nanogels. One day post‐transfection, PS was added and left to incubate with the cells overnight. At day 2 post‐transfection, also a 4 h PS‐free incubation period was respected before illumination. In both protocols, EGFP expression was analyzed day 3 – day 6 – day 9 and day 12 post‐transfection.
The results on the delayed PCI treatment (PCI t2) are displayed in Figure 6.8b‐d. At day 3 post‐
transfection, being 24 h after photochemical treatment, already a significant difference is observed
between the treated and non‐treated cells for the lower nanogel concentrations (10 µg mL‐1, Figure
6.8b and 25 µg mL‐1, Figure 6.8c), but not when 50 µg mL‐1 nanogels were applied on the cells. More
interestingly, for the remainder of the investigated time course, this difference in gene silencing is at
179
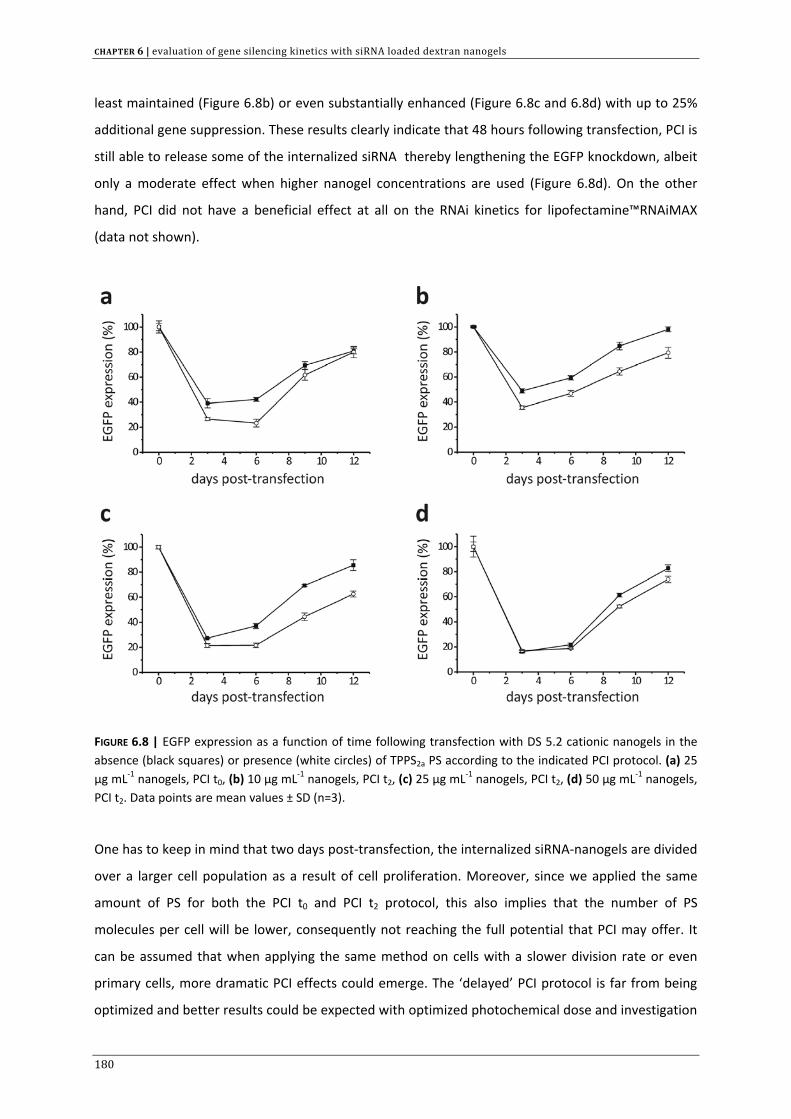
CHAPTER 6 | evaluation of gene silencing kinetics with siRNA loaded dextran nanogels
least maintained (Figure 6.8b) or even substantially enhanced (Figure 6.8c and 6.8d) with up to 25%
additional gene suppression. These results clearly indicate that 48 hours following transfection, PCI is
still able to release some of the internalized siRNA thereby lengthening the EGFP knockdown, albeit
only a moderate effect when higher nanogel concentrations are used (Figure 6.8d). On the other
hand, PCI did not have a beneficial effect at all on the RNAi kinetics for lipofectamine™RNAiMAX
(data not shown).
FIGURE 6.8 | EGFP expression as a function of time following transfection with DS 5.2 cationic nanogels in the absence (black squares) or presence (white circles) of TPPS2a PS according to the indicated PCI protocol. (a) 25 µg mL‐1 nanogels, PCI t0, (b) 10 µg mL‐1 nanogels, PCI t2, (c) 25 µg mL‐1 nanogels, PCI t2, (d) 50 µg mL‐1 nanogels, PCI t2. Data points are mean values ± SD (n=3).
One has to keep in mind that two days post‐transfection, the internalized siRNA‐nanogels are divided
over a larger cell population as a result of cell proliferation. Moreover, since we applied the same
amount of PS for both the PCI t0 and PCI t2 protocol, this also implies that the number of PS
molecules per cell will be lower, consequently not reaching the full potential that PCI may offer. It
can be assumed that when applying the same method on cells with a slower division rate or even
primary cells, more dramatic PCI effects could emerge. The ‘delayed’ PCI protocol is far from being
optimized and better results could be expected with optimized photochemical dose and investigation
180

evaluation of gene silencing kinetics with siRNA loaded dextran nanogels | CHAPTER 6
of the ideal illumination time‐point. In conclusion, this PCI strategy indicates that delayed application
of PS to transfected cells is still able to liberate a fraction of the siRNA‐nanogels trapped in endocytic
vesicles, which is a potential strategy to prolong the therapeutic effect in the case when endosomal
escape is the effect‐limiting step. Furthermore, the proposed data suggest that the prolonged
residence time in intracellular vesicles (e.g. endolysosomes) is not detrimental at least for some of
the internalized siRNA duplexes. Still this is one of the major concerns when applying a delayed PCI
treatment.[26] PCI on nanogels complexing nuclease‐stabilized siRNA could therefore also be an
interesting alternative.
CONCLUSIONS
In the present chapter, we screened the RNAi gene silencing kinetics on Huh‐7_LUC and Huh‐7_EGFP
cells with dex‐HEMA‐co‐TMAEMA nanogels of varying crosslink density. We first verified that our
cationic nanogels could also induce a sequence‐specific knockdown of the EGFP expression, which
was not yet studied in this thesis. Although the maximal gene silencing effect differed for the nanogel
dispersions investigated in both screening experiments, no marked deviations could be noted in the
recovery to steady state expression levels of the target gene. Photochemical internalization (PCI) was
able to substantially enhance the extent and duration of EGFP knockdown, even when applied 48
hours post‐transfection. This opens up new possibilities to use PCI as a tool to prolong the siRNA
silencing effect when identifying the optimal time‐point for PCI application to maximize the effect of
a single administration.
ACKNOWLEDGMENTS
Koen Raemdonck is a doctoral fellow of the Institute for the Promotion of Innovation through
Science and Technology in Flanders (IWT‐Vlaanderen). The authors would like to acknowledge the EU
(FP6) for funding of MediTrans, an integrated project on nanomedicines. A. Høgset is supported by
the Norwegian Research Council. Saartje Steppe is acknowledged for her help with the practical
experiments.
181

CHAPTER 6 | evaluation of gene silencing kinetics with siRNA loaded dextran nanogels
182
REFERENCE LIST
1. Fire A., Xu S., Montgomery M.K., Kostas S.A. et al., Potent and specific genetic interference by double‐stranded RNA in Caenorhabditis elegans, Nature 1998, 391: 806‐811.
2. Whitehead K.A., Langer R., and Anderson D.G., Knocking down barriers: advances in siRNA delivery, Nature Reviews Drug Discovery 2009, 8: 129‐138.
3. de Fougerolles A.R., Delivery vehicles for small interfering RNA in vivo, Hum. Gene Ther. 2008, 19: 125‐132. 4. Davis M.E., The First Targeted Delivery of siRNA in Humans via a Self‐Assembling, Cyclodextrin Polymer‐
Based Nanoparticle: From Concept to Clinic, Mol. Pharm. 2009, 6: 659‐668. 5. Behlke M.A., Chemical modification of siRNAs for in vivo use, Oligonucleotides. 2008, 18: 305‐319. 6. Hefner E., Clark K., Whitman C., Behlke M.A. et al., Increased potency and longevity of gene silencing using
validated Dicer substrates, J. Biomol. Tech. 2008, 19: 231‐237. 7. Amarzguioui M. and Rossi J.J., Principles of Dicer substrate (D‐siRNA) design and function, Methods Mol.
Biol. 2008, 442: 3‐10. 8. Chu C.Y. and Rana T.M., Potent RNAi by short RNA triggers, RNA 2008, 14: 1714‐1719. 9. Sano M., Sierant M., Miyagishi M., Nakanishi M. et al., Effect of asymmetric terminal structures of short
RNA duplexes on the RNA interference activity and strand selection, Nucleic Acids Res. 2008, 36: 5812‐5821.
10. Gao K. and Huang L., Nonviral Methods for siRNA Delivery, Mol. Pharm. 2009, 6: 651‐658. 11. Novobrantseva T.I., Akinc A., Borodovsky A., and de F.A., Delivering silence: advancements in developing
siRNA therapeutics, Curr. Opin. Drug Discov. Devel. 2008, 11: 217‐224. 12. Bartlett D.W. and Davis M.E., Insights into the kinetics of siRNA‐mediated gene silencing from live‐cell and
live‐animal bioluminescent imaging, Nucleic Acids Research 2006, 34: 322‐333. 13. Mantei A., Rutz S., Janke M., Kirchhoff D. et al., siRNA stabilization prolongs gene knockdown in primary T
lymphocytes, Eur. J. Immunol. 2008, 38: 2616‐2625. 14. Raemdonck K., Van Thienen T.G., Vandenbroucke R.E., Sanders N.N. et al., Dextran microgels for time‐
controlled delivery of siRNA, Advanced Functional Materials 2008, 18: 993‐1001. 15. Raemdonck K., Vandenbroucke R.E., Demeester J., Sanders N.N. et al., Maintaining the silence: reflections
on long‐term RNAi, Drug Discovery Today 2008, 13: 917‐931. 16. Judge A. and Maclachlan I., Overcoming the innate immune response to small interfering RNA, Human
Gene Therapy 2008, 19: 111‐124. 17. Svoboda P., Off‐targeting and other non‐specific effects of RNAi experiments in mammalian cells, Current
Opinion in Molecular Therapeutics 2007, 9: 248‐257. 18. Grimm D., Streetz K.L., Jopling C.L., Storm T.A. et al., Fatality in mice due to oversaturation of cellular
microRNA/short hairpin RNA pathways, Nature 2006, 441: 537‐541. 19. Dykxhoorn D.M. and Lieberman J., Knocking down disease with siRNAs, Cell 2006, 126: 231‐235. 20. Van Dijk‐Wolthuis W.N., van Steenbergen M.J., Underberg W.J., and Hennink W.E., Degradation kinetics of
methacrylated dextrans in aqueous solution, J. Pharm. Sci. 1997, 86: 413‐417. 21. Detzer A., Overhoff M., Wunsche W., Rompf M. et al., Increased RNAi is related to intracellular release of
siRNA via a covalently attached signal peptide, RNA. 2009, 15: 627‐636. 22. Mescalchin A., Detzer A., Wecke M., Overhoff M. et al., Cellular uptake and intracellular release are major
obstacles to the therapeutic application of siRNA: novel options by phosphorothioate‐stimulated delivery, Expert. Opin. Biol. Ther. 2007, 7: 1531‐1538.
23. Veldhoen S., Laufer S.D., Trampe A., and Restle T., Cellular delivery of small interfering RNA by a non‐covalently attached cell‐penetrating peptide: quantitative analysis of uptake and biological effect, Nucleic Acids Res. 2006, 34: 6561‐6573.
24. Lu J.J., Langer R., and Chen J., A Novel Mechanism Is Involved in Cationic Lipid‐Mediated Functional siRNA Delivery, Mol. Pharm. 2009, 6: 763‐771.
25. Berg K., Folini M., Prasmickaite L., Selbo P.K. et al., Photochemical internalization: a new tool for drug delivery, Curr. Pharm. Biotechnol. 2007, 8: 362‐372.
26. Prasmickaite L., Hogset A., Selbo P.K., Engesaeter B.O. et al., Photochemical disruption of endocytic vesicles before delivery of drugs: a new strategy for cancer therapy, Br. J. Cancer 2002, 86: 652‐657.

liposomal template‐assisted synthesis of dextran based nanogels | CHAPTER 7
CHAPTER 7 | Liposomal
template‐assisted synthesis of dextran based nanogels
Manuscript of this chapter is in preparation:
Koen Raemdonck1, Joseph Demeester1, Stefaan C. De Smedt1 (2009)
1Laboratory of General Biochemistry and Physical Pharmacy, Department of Pharmaceutics, Faculty of Pharmaceutical
Sciences, Ghent University, Harelbekestraat 72, B‐9000 Ghent, Belgium.
183

CHAPTER 7 | liposomal template‐assisted synthesis of dextran based nanogels
184

liposomal template‐assisted synthesis of dextran based nanogels | CHAPTER 7
7 OVERVIEW CHAPTER CONTENTS
Chapter 7 provides:
• details on the creation of hybrid lipid‐enveloped (cationic) nanogels (LENs) using liposomes
as nanoreactor
• pilot experiments to evaluate if these nanoparticles are suited for siRNA encapsulation
• A brief overview of other literature reports describing lipid‐coated nanoparticles and a
discussion on the additional value of these nanocomposites for siRNA delivery
Key terms:
Liposomes – nanogels – siRNA – charge neutral – phospholipid gel – encapsulation
INTRODUCTION
The successful intracellular delivery of nucleic acid therapeutics, such as siRNA and pDNA, depends
on the efficient formulation into pharmaceutical nanocarriers that can guide their therapeutic
payload to the target cells. However, as already discussed in chapters 1 and 4, extracellular and
intracellular barriers may prevent efficient accumulation of the drug loaded particles at the site of
action.[1‐3] We provided evidence in chapter 5 and chapter 6 that the presented cationic dextran
nanogels are able to carry high amounts of siRNA therapeutics and transport them across the cellular
membrane. The designated chapters, however, also revealed that the endosomal sequestration of
the siRNA loaded nanogels, significantly limited the full RNAi potential. To achieve our goal of
intracellular controlled release of siRNA, a transition of intact siRNA loaded nanogels from the
endosomal lumen into the cytoplasm needs to occur.
185

CHAPTER 7 | liposomal template‐assisted synthesis of dextran based nanogels
Besides polymeric nanoparticles, liposomes have also been widely investigated as pharmaceutical
nanocarriers.[1,4‐6] Liposomes are defined as self‐assembled spherical vesicles consisting of (a) lipid
bilayer(s) that enclose an aqueous core. Like nanogels, liposomes can be tailored to respond to
environmental changes e.g. pH, redox potential and temperature,[6‐10] but they usually lack the ability
to provide a time‐controlled cytosolic release of macromolecular therapeutics. In this chapter, we
aim to combine the advantages of our biodegradable dextran nanogels for cytosolic controlled siRNA
release with the multifunctionality of liposomes into a hybrid nanogel‐lipid core‐shell composite.
Several reports in the literature describe the design of lipid coated nanoparticles.[11‐16] As an example,
Harashima’s group published several papers on multifunctional envelope‐type nanodevices (MENDs),
that consist of a condensed core containing the complexed nucleic acids (siRNA or pDNA) surrounded
by a multifunctional lipid envelope.[17,18] The MEND particles are usually obtained by an optimized
lipid hydration method, where a preformed lipid film is hydrated with an aqueous solution containing
the nucleic acid‐polycation core complexes.[19,20] Alternatively, encapsulation in a lipid bilayer may
also be achieved through fusion of anionic small unilamellar vesicles (SUVs) on the surface of
positively charged nanoparticles.[21] The authors state that during these processes the nucleic acid
core and the lipid bilayers exist as separate structures. It was shown that the packaging of a pDNA or
siRNA complex core into a pH‐responsive fusogenic lipid envelope may lead to efficient endosomal
escape of the nucleic acid complexes due to fusion triggered decoating upon interaction with the
endosomal membrane.[21] Alternatively, Jana et al. prepared polyvinylpyrrolidone nanoparticles that
were subsequently encapsulated in fusogenic Sendai virosomes. The authors indicate that the viral
envelope may trigger fusion with the plasma membrane of (hepatic) cells, thereby injecting the
nanoparticles directly into the cytosol.[22] A comparable approach is presented by Kunisawa et al.
who incorporated poly(vinyl)amine nanoparticles into Sendai viral envelopes.[23]
A potential drawback of the described preparation methods is that they generally consist of several
consecutive steps that need separate optimization (nanoparticle preparation, hydration of lipid coat,
sonication or extrusion,...) which makes the preparation process laborious and difficult to control. It
seems not so straightforward to incorporate the necessary functionalities in a hybrid nanosized
structure by a ‘one‐step’ production process.
However, in the literature one can find some examples of the single‐step preparation of lipid coated
particles, by using liposomes as nanoscaled reactors for particle formation.[24‐33] Hereby, the aqueous
core of the liposomal template is employed as a reaction vessel for the synthesis of nanostructures,
that are thereby automatically equipped with a lipid shell. In this chapter we aimed to evaluate if
hybrid lipid‐enveloped dextran nanogels (LENs) could be prepared for siRNA delivery, by using a
186

liposomal template‐assisted synthesis of dextran based nanogels | CHAPTER 7
liposomal template‐assisted synthesis and we further discuss the potential advantages this delivery
system may provide.
EXPERIMENTAL SECTION
Phospholipids
1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC, Tm=‐20°C, 20 mg mL‐1 in chloroform); 1,2‐dioleoyl‐
sn‐glycero‐3‐phosphoethanolamine (DOPE, Tm=‐16°C, 10 mg mL‐1 in chloroform); 1,2‐dioleoyl‐sn‐
glycero‐3‐phosphoethanolamine‐N‐(lissamine rhodamine‐B sulfonyl) ammonium salt (Rho‐B‐DOPE,
λex/λem = 557 nm/571 nm) and 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(7‐nitro‐2‐1,3‐
benzoxadiazol‐4‐yl) (ammonium salt) (NBD‐DOPE, λex/λem = 460 nm/534 nm) were purchased from
Avanti Polar Lipids (Alabama, USA). Lyophilized DOPC and DOPE were obtained from Sigma Aldrich.
Cholesteryl Bodipy FLC12 (cholesteryl 4,4‐difluoro‐5,7‐dimethyl‐4‐bora‐3a,4a‐diaza‐s‐indacene‐3‐
dodecanoate) was from Molecular probes (Eugene Oregon, USA).
siRNA oligonucleotides
Twenty‐one nucleotide siRNA duplexes, targeted against pGL3 luciferase, were purchased from
Eurogentec (Seraing, Belgium). SiRNA batches were diluted in RNase free water and placed at ‐85°C.
For fluorescence experiments, a pGL3 siRNA duplex 5’‐labeled (sense strand) with Alexa Fluor 488
(AF488‐siRNA) was used.
Preparation of lipid‐enveloped dextran nanogels through extrusion
Dextran nanogels were prepared using liposomes as nanosized reactor. Appropriate amounts of
DOPC or DOPC and DOPE (molar ratio 1:1), dissolved in chloroform, were mixed in a glass vial. For
fluorescently labelled liposomes, rho‐B DOPE, NBD‐DOPE or cholesteryl Bodipy were added in 0.1‐0.2
mol% of total phospholipid. To obtain a lipid film, the chloroform was evaporated under a nitrogen
flow, while rotating the vial gently. The lipid film is then placed under a vigorous stream of nitrogen
for at least 1h to remove traces of chloroform and stored at ‐20°C. A dex‐HEMA solution (20% (w/w)
in 20 mM HEPES buffer pH 7.4, containing irgacure 2959 as photoinitiator (Ciba Specialty Chemicals),
is used to hydrate the lipids by vortexing at room temperature (2 min). To obtain lipid coated cationic
dextran nanogels, dimethyl aminoethyl methacrylate (DMAEMA, also see chapter 3) was added to
the dextran solution. The resulting dispersion of large multilamellar vesicles (LMVs) was allowed to
187

CHAPTER 7 | liposomal template‐assisted synthesis of dextran based nanogels
stabilize for 5 min and subsequently extruded through a double stacking of 0.4 or 0.2 µm
polycarbonate filters using a hand‐held extruder (Avanti polar lipids). To prevent the polymerization
of the dex‐HEMA which was not encapsulated in the liposome interior, the liposome dispersion was
diluted 1:10 in 20 mM HEPES buffer so that the dex‐HEMA concentration of the solution surrounding
the liposomes became too low to form a hydrogel. Subsequently the sample was UV irradiated at
room temperature for 450 s under N2 atmosphere (365 nm from a Bluepoint 2.1 UV source, Honle
UV Technology) thereby selectively crosslinking the dex‐HEMA inside the lipid bilayer which results in
the formation of lipid‐enveloped dextran nanogels. To obtain naked dextran nanogels, the lipid layer
was solubilized by addition of TX100 (10% w/v) at a molar ratio ≥ 10 relative to total phospholipid
content. In one experiment, the lipid coat was removed by subsequent washing steps with diluted
isopropanol. Briefly, a dispersion of LENs (containing ~1.3 mM total phospholipid) is filtrated on a
Vivaspin 6 ultrafiltration device (300.000 MWCO, polyethersulfone, Vivascience – Sartorius
instruments) by centrifugation (2000 g, 20 min). The remaining volume (~100 µL) is diluted with 1 mL
of distilled water followed by the addition of 700 µL isopropanol (35% final concentration) and again
subjected to a washing step (1000 g, 20 min). This washing step is repeated a second time. After
every centrifugation‐dilution step with distilled water, an aliquot is taken for zeta‐potential
measurement and phosphate analysis according to Rouser et al. after destruction of the lipids using
perchloric acid.[31,34]
Preparation of lipid coated dextran nanogels through ultrasonication
Lyophilized lipids (total amount 150 mg), i.e. DOPC or DOPC:DOPE (1:1), are weighed in a non‐stick
RNase free 2 mL eppendorf (Ambion, Austin, TX) and 500 µL of chloroform is added to dissolve the
lipids. Subsequently, the chloroform is removed under nitrogen flow followed by additional vacuum
evaporation (Büchi Rotavapor R‐200). The dried lipid constituents were hydrated with a dex‐MA
solution (25 wt% in HEPES buffer) with a CapMix™ (3*30s) and left to hydrate during 30 min (4°C). To
further size the formed lipid vesicles, the dispersion was sonicated using a probe sonicator (Branson
digital sonifier) in subsequent steps of maximum 25 s. After every cycle, the resulting vesicular
phospholipid gel (VPG) was briefly placed on ice before starting the next sonication step. To evaluate
the size of the liposomes in the VPG, ~1 mg of VPG was dispersed in 2 mL HEPES buffer (CapMix™
during 15 s) and analyzed with DLS at 25°C.
188

liposomal template‐assisted synthesis of dextran based nanogels | CHAPTER 7
siRNA loading during preparation of lipid‐enveloped dextran nanogels
The loading of dex‐HEMA nanogels with siRNA was obtained through hydration of the DOPC:DOPE
lipid film with a siRNA containing dex‐HEMA solution (passive loading). To estimate the siRNA uptake
inside the liposomes formed by extrusion (final lipid concentration ~30 mM) in a preliminary
experiment, the non‐encapsulated siRNA was separated from the liposomes by gel permeation
chromatography (GPC) using a Sephadex CL‐4B column (Sigma), equilibrated with Tris/EDTA (10
mM/1 mM) buffer at pH 7.4. The elution of the liposomes was detected by turbidity measurements
with a Pharmacia LKB Biochrom UV/VIS spectrophotometer (λ = 633 nm). The siRNA concentration in
each elution fraction was quantified (after the addition of TX100) using the Quant‐it™ Ribogreen®
RNA Reagent (Molecular Probes, Eugene, Oregon, USA). To quantify the encapsulation efficiency of
siRNA in liposomes formed by ultrasonication, DOPC:DOPE lipids were hydrated with a 25% dex‐MA
solution in HEPES buffer, supplemented with siRNA (final lipid concentration 500 mg mL‐1, final siRNA
concentration 40 µM). The encapsulation efficiency was quantified by measuring the fluorescence of
Quant‐it™ Ribogreen® RNA Reagent before and after TX100 addition (0.25%) to the lipid dispersion.
In the absence of detergent, the signal comes from accessible (i.e. unencapsulated) siRNA only, while
the signal after detergent addition corresponds with total siRNA.[35] Calibration curves of siRNA were
first constructed with and without TX100 (R2= 0.998). Ribogreen fluorescence (λex= 500 nm, λem=525
nm) was measured using a Wallac Victor2 fluorescence plate reader (Perkin Elmer, Waltham, MA).
Dynamic Light Scattering (DLS) and zeta‐potential
The particle size of the nanogels was measured by dynamic light scattering using a Malvern Autosizer
4700, consisting of a He‐Ne laser (633 nm, 25 mW output power). Polystyrene nanospheres (220 ± 6
nm, Duke Scientific Corporation) were used as calibration standard. The mean hydrodynamic
diameter of the particles (dh) was obtained from the intensity fluctuations of the scattered light using
the software package provided by Malvern based on the Stokes‐Einstein equation: D = kbT.(3πηdh)‐1
where D is the diffusion coefficient, kb is de boltzmann constant, T is the temperature of the water
bath (298 K) and η is the solvent viscosity. Scattering was measured at a fixed angle of 90°. HEPES
buffer, used to dilute the nanoparticle dispersions, was filtered through 0.22 µm membrane before
use. Some of the size measurements were repeated and/or performed on the Malvern Zetasizer
Nano ZS, as described in previous chapters.
Zeta potential measurements in this chapter were performed on a Zetasizer 2000, Malvern,
Worcestershire, UK.
189

CHAPTER 7 | liposomal template‐assisted synthesis of dextran based nanogels
Scanning electron microscopy
Scanning electron microscopy on naked nanogels was carried out using a Zeiss DSM 40 instrument
operating at an accelerating voltage of 5 kV. For sample preparation, a drop of the dispersion of
naked nanogels (obtained through washing of the lipid coated nanogels with isopropanol) was dried
under low vacuum on a glass slide and subsequently sputtered with gold/palladium.
Fluorescence Fluctuation Spectroscopy (FFS)
A dual‐colour fluorescence fluctuation spectroscopy instrument (dual‐colour FFS) was used as
described previously.[36] FFS monitors fluorescence intensity fluctuations in the excitation volume of a
confocal microscope. In dual‐colour FFS measurements, two spectrally different fluorophores
(respectively AF488 and lissamine‐rhodamine B in this study) are used. The fluorescence of the two
fluorophores is registered by two separate detectors monitoring the same excitation volume. In this
study the 488 nm and 561 nm laser lines of a Krypton‐Argon ion laser (BioRad, Cheshire, UK) were
used to excite the samples and the fluorescence fluctuations were recorded during a 50 s time
interval. For a profound theoretical background on FFS we would like to refer to the indicated
references.[37‐39]
FFS experiments were carried out on dex‐HEMA‐co‐DMAEMA containing DOPC:DOPE liposomes
respectively before and after UV irradiation. Therefore 50 µL of the liposomal dispersions (50 nM
phospholipids) was applied in the wells of a glass‐bottomed 96‐well plate (Greiner Bio‐one, certified
DNase/RNase free). The focal volume was placed 50 µm above the bottom of the wells. The lipid
layer was fluorescently labelled by addition of 0.1 mol% of rhodamine B‐DOPE to the lipid mixture.
TX100 was added to solubilize the lipid layer (molar ratio TX100/phospholipids = 10). After
solubilization, AF488‐siRNA (final concentration = 50 nM) was mixed with the samples in the wells
and fluorescence fluctuations were again recorded.
Cellular uptake of lipid coated cationic nanogels in Huh‐7 cells
The wild‐type human hepatoma cell line Huh‐7_wt was cultured (37°C, 5% CO2) in DMEM:F12,
supplemented with 2 mM glutamine, 10 % heat‐inactivated FBS and 100 U mL‐1 P/S. Huh‐7 cells were
seeded in sterile glass‐bottomed culture dishes (MatTek Corporation, Ashland, MA) at a density of 3 x
104 cells per cm2 and allowed to attach overnight. DOPC:DOPE (1:1) coated cationic nanogels (20 %
w/w dex‐HEMA DS 5.7, molar ratio HEMA/DMAEMA = 0.25, 25 nmol lipids) containing 0.2 mol% of
cholesteryl Bodipy were incubated with the cells (37°C, 5% CO2) in OptiMEM during 4 h.
Subsequently, cells were washed with PBS and preheated culture medium was added. After another
190

liposomal template‐assisted synthesis of dextran based nanogels | CHAPTER 7
20 h of incubation, the intracellular distribution of the lipid coated nanogels was visualized with a
Nikon EZC1‐si confocal scanning module installed on a motorized Nikon TE2000‐E inverted
microscope (Nikon Benelux, Brussels, Belgium). For lysosomal staining, cells were incubated for 15
min with 1:104 diluted Lysotracker Red® (Molecular Probes) prior to image recording. Nuclei were
stained through 10 min incubation with a 1:103 dilution of DraQ5™ (Alexis Biochemicals,
Switzerland). Extracellular fluorescence was quenched by short incubation with 0.4% trypan blue
(Sigma). Images were captured with a Nikon Plan Apochromat 60x oil immersion objective (numerical
aperture 1.4) using a 488 nm, 561 nm and 639 nm laser line for the excitation of cholesteryl Bodipy,
Lysotracker Red® and DraQ5™ respectively.
Quantification of cellular uptake of DOPC:DOPE LENs was analyzed using flow cytometry. Briefly, 6 x
104 cells were seeded in 24‐well plates and 48 h after seeding, they were incubated for 4 h with
increasing concentrations of fluorescently labelled LENs (DOPC:DOPE containing 0.2 mol% NBD‐
DOPE) in complete cell culture medium (0.5 mL final volume). Following incubation, the cells were
washed with PBS, quenched with 0.4% trypan blue (8 min, 37°C) and prepared for flow cytometry as
already explained in previous chapters.
RESULTS AND DISCUSSION
Formation of biodegradable nanogels inside a liposome reactor
In this manuscript we aimed to design dextran nanogels using liposomes as nanoscaled reactor for
the selective photopolymerization of dextran hydroxyethyl methacrylate (dex‐HEMA), as described
earlier by Van Thienen et al..[32] Figure 7.1 schematically depicts the preparation procedure. In a first
step, a lipid film is hydrated with a buffered dex‐HEMA solution, that is supplemented with irgacure
2959 as a photoinitiator. In this way, polydisperse multilamellar liposomal vesicles are formed with
dex‐HEMA encapsulated in their aqueous core. To obtain a more homogeneous dispersion of
(predominantly) unilamellar vesicles, the sample is subsequently extruded through a polycarbonate
filter with a defined pore size. A final step is the UV induced photopolymerization of dex‐HEMA inside
the liposomal template, to obtain dextran nanogels surrounded by a lipid bilayer. In order to prevent
the polymerization of dex‐HEMA in between the liposomal vesicles, the liposome dispersion is
diluted at least ten‐fold. In this way, the dex‐HEMA crosslinking will be restricted to the liposome
inner compartment as the concentration of unencapsulated dex‐HEMA will be below the critical
threshold that is needed for hydrogel formation. Alternatively, the external dex‐HEMA may also be
removed by size exclusion chromatography or dialysis, which are however more time‐consuming and
191

CHAPTER 7 | liposomal template‐assisted synthesis of dextran based nanogels
labour intensive methods. Schillemans et al. reported on the use of ascorbic acid to inhibit the free
radical polymerization process in between the liposomal vesicles to ensure selective crosslinking in
the liposome interior.[31]
FIGURE 7.1 | Schematic overview of the preparation protocol to obtain dextran nanogels surrounded by a lipid coat. Hydration of a thin lipid film with a dex‐HEMA solution leads to a dispersion of large and polydisperse multilamellar vesicles (LMVs). Extrusion of the LMV dispersion through a polycarbonate membrane is used to ‘size’ the obtained multilamellar vesicles into monodisperse large unilamellar vesicles or LUVs. These liposomes carry dex‐HEMA chains in their aqueous core that can be polymerized during UV irradiation to form a dex‐HEMA gel. Redrawn from reference [24].
Figure 7.2a shows dynamic light scattering (DLS) data on a dispersion of DOPC:DOPE (1:1) liposomes
filled with dex‐HEMA respectively before and after UV treatment. In both cases vesicles of ~200 nm
were detected. To solubilise the lipid bilayer, TX100 was added to the dispersions. The removal of the
lipid coat was evidenced by the lowering of the scattering intensity (the removal of the lipid bilayer
lowers the difference in refractive index between the nanoparticles and the aqueous dispersion
buffer)[32] and the appearance of detergent‐phospholipid mixed micelles (~15‐20 nm in size; see
Figure 7.2c). Upon addition of TX100 to the liposome dispersion which was not exposed to UV light
only micelles remained (monomodal distribution, data not shown). In contrast, both micelles and
uncoated nanogels were observed after addition of TX100 to the UV irradiated liposome dispersions
(Figure 7.2c, bimodal distribution). Figure 7.2d displays a scanning electron micrograph of ‘naked’
nanogels, obtained after removal of the lipid coat. The DLS and electron microscopy data in Figure
7.2 clearly indicate that UV irradiation of the dex‐HEMA filled liposomes results in nanogel particles
formed within the liposomal reactor.
We also monitored the size of these lipid coated dex‐HEMA nanogels as a function of time at 37°C by
DLS. In agreement with the observations by Van Thienen et al.,[32] even after > 2 weeks of incubation,
still vesicles of about 200 nm in size were detected (Figure 7.2a ‐ UV degraded, white bar).
192
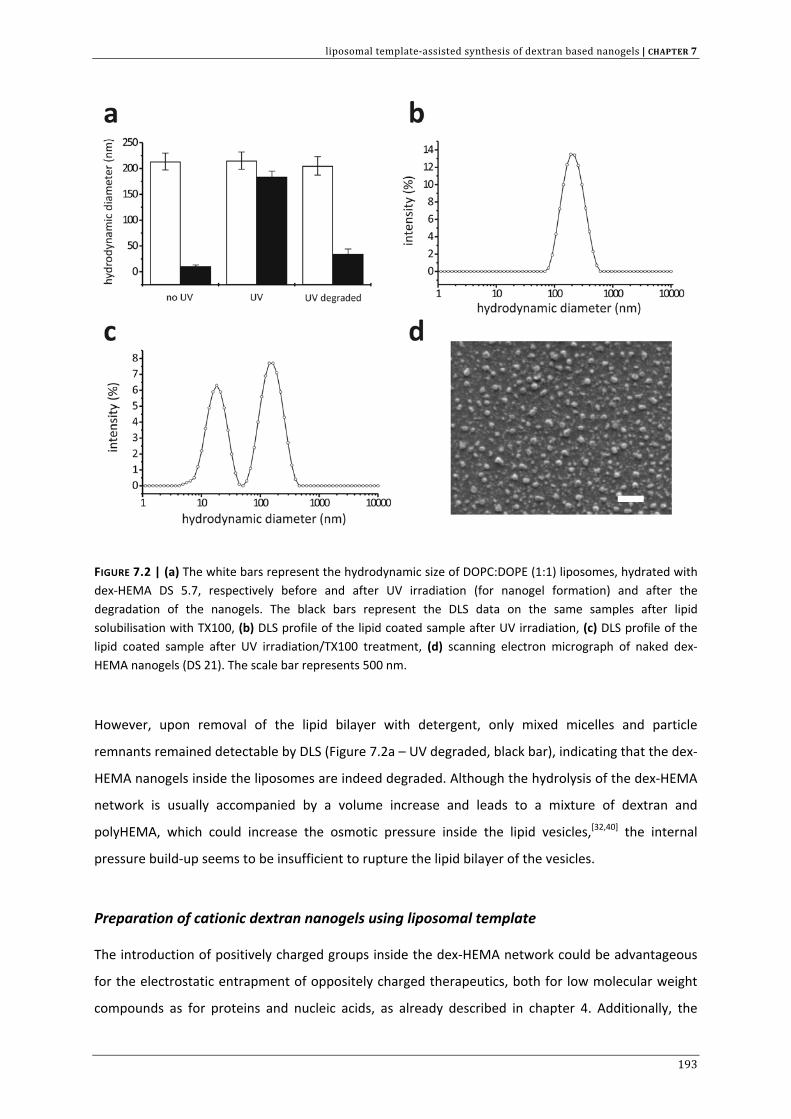
liposomal template‐assisted synthesis of dextran based nanogels | CHAPTER 7
FIGURE 7.2 | (a) The white bars represent the hydrodynamic size of DOPC:DOPE (1:1) liposomes, hydrated with dex‐HEMA DS 5.7, respectively before and after UV irradiation (for nanogel formation) and after the degradation of the nanogels. The black bars represent the DLS data on the same samples after lipid solubilisation with TX100, (b) DLS profile of the lipid coated sample after UV irradiation, (c) DLS profile of the lipid coated sample after UV irradiation/TX100 treatment, (d) scanning electron micrograph of naked dex‐HEMA nanogels (DS 21). The scale bar represents 500 nm.
However, upon removal of the lipid bilayer with detergent, only mixed micelles and particle
remnants remained detectable by DLS (Figure 7.2a – UV degraded, black bar), indicating that the dex‐
HEMA nanogels inside the liposomes are indeed degraded. Although the hydrolysis of the dex‐HEMA
network is usually accompanied by a volume increase and leads to a mixture of dextran and
polyHEMA, which could increase the osmotic pressure inside the lipid vesicles,[32,40] the internal
pressure build‐up seems to be insufficient to rupture the lipid bilayer of the vesicles.
Preparation of cationic dextran nanogels using liposomal template
The introduction of positively charged groups inside the dex‐HEMA network could be advantageous
for the electrostatic entrapment of oppositely charged therapeutics, both for low molecular weight
compounds as for proteins and nucleic acids, as already described in chapter 4. Additionally, the
193
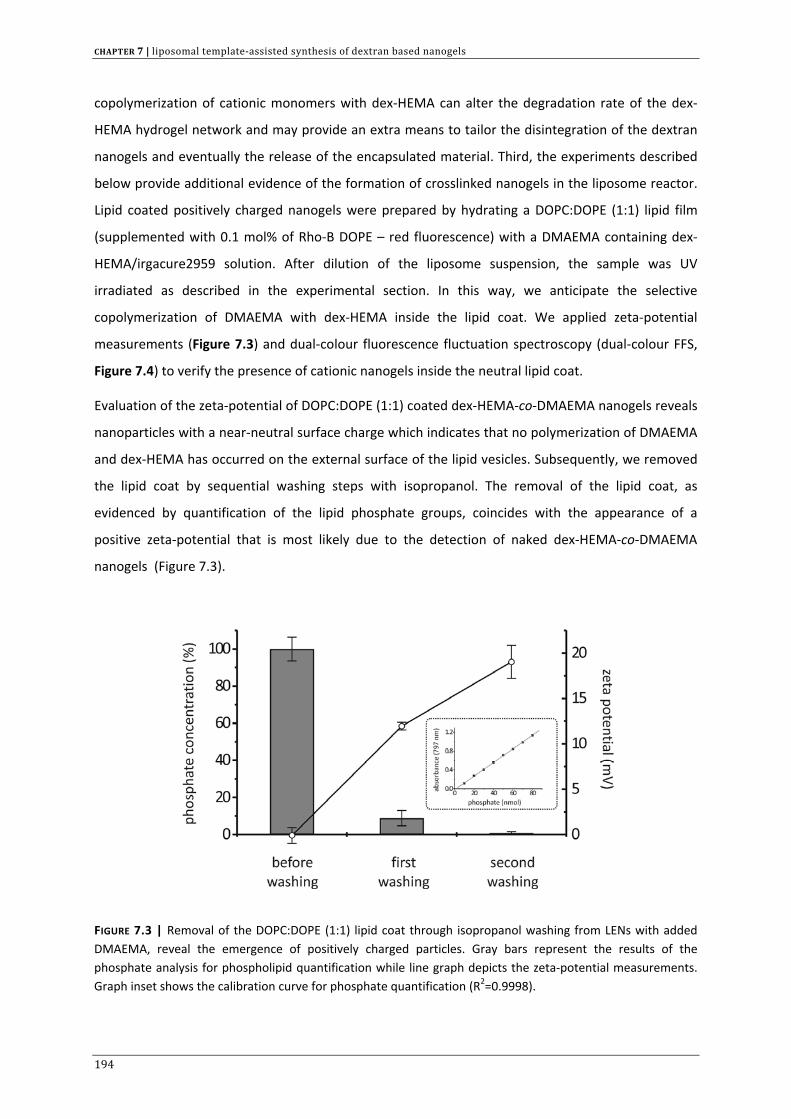
CHAPTER 7 | liposomal template‐assisted synthesis of dextran based nanogels
copolymerization of cationic monomers with dex‐HEMA can alter the degradation rate of the dex‐
HEMA hydrogel network and may provide an extra means to tailor the disintegration of the dextran
nanogels and eventually the release of the encapsulated material. Third, the experiments described
below provide additional evidence of the formation of crosslinked nanogels in the liposome reactor.
Lipid coated positively charged nanogels were prepared by hydrating a DOPC:DOPE (1:1) lipid film
(supplemented with 0.1 mol% of Rho‐B DOPE – red fluorescence) with a DMAEMA containing dex‐
HEMA/irgacure2959 solution. After dilution of the liposome suspension, the sample was UV
irradiated as described in the experimental section. In this way, we anticipate the selective
copolymerization of DMAEMA with dex‐HEMA inside the lipid coat. We applied zeta‐potential
measurements (Figure 7.3) and dual‐colour fluorescence fluctuation spectroscopy (dual‐colour FFS,
Figure 7.4) to verify the presence of cationic nanogels inside the neutral lipid coat.
Evaluation of the zeta‐potential of DOPC:DOPE (1:1) coated dex‐HEMA‐co‐DMAEMA nanogels reveals
nanoparticles with a near‐neutral surface charge which indicates that no polymerization of DMAEMA
and dex‐HEMA has occurred on the external surface of the lipid vesicles. Subsequently, we removed
the lipid coat by sequential washing steps with isopropanol. The removal of the lipid coat, as
evidenced by quantification of the lipid phosphate groups, coincides with the appearance of a
positive zeta‐potential that is most likely due to the detection of naked dex‐HEMA‐co‐DMAEMA
nanogels (Figure 7.3).
FIGURE 7.3 | Removal of the DOPC:DOPE (1:1) lipid coat through isopropanol washing from LENs with added DMAEMA, reveal the emergence of positively charged particles. Gray bars represent the results of the phosphate analysis for phospholipid quantification while line graph depicts the zeta‐potential measurements. Graph inset shows the calibration curve for phosphate quantification (R2=0.9998).
194
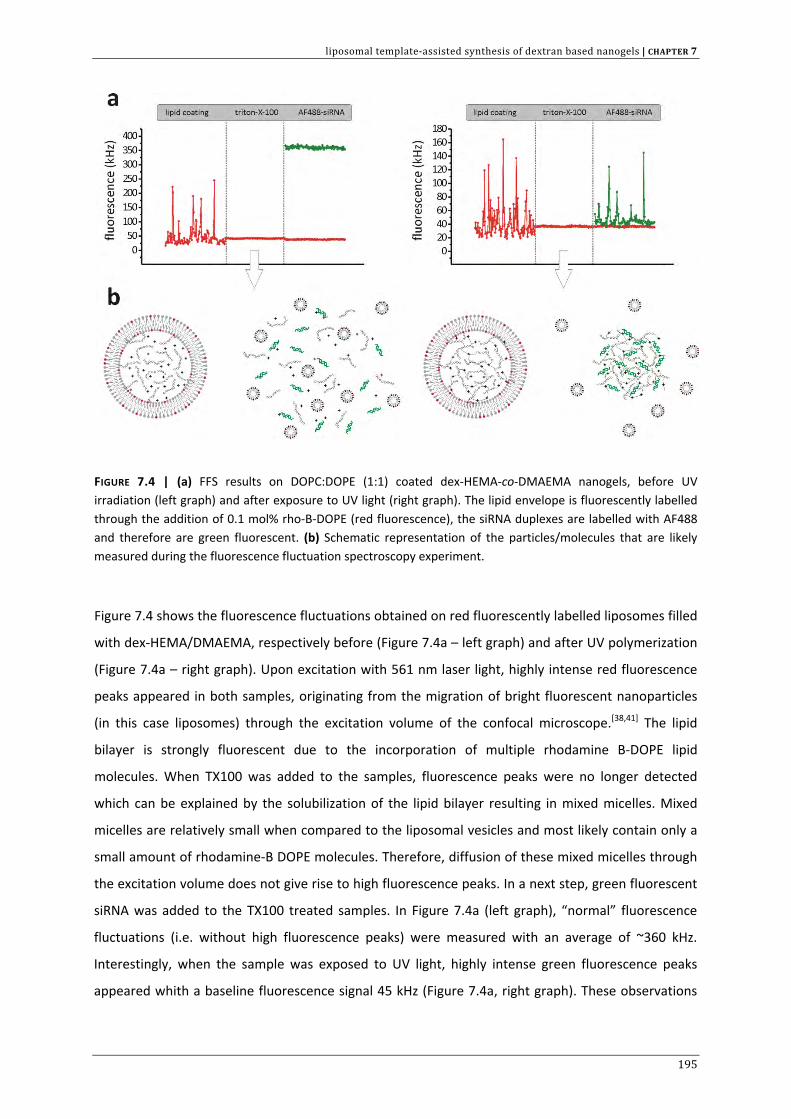
liposomal template‐assisted synthesis of dextran based nanogels | CHAPTER 7
FIGURE 7.4 | (a) FFS results on DOPC:DOPE (1:1) coated dex‐HEMA‐co‐DMAEMA nanogels, before UV irradiation (left graph) and after exposure to UV light (right graph). The lipid envelope is fluorescently labelled through the addition of 0.1 mol% rho‐B‐DOPE (red fluorescence), the siRNA duplexes are labelled with AF488 and therefore are green fluorescent. (b) Schematic representation of the particles/molecules that are likely measured during the fluorescence fluctuation spectroscopy experiment.
Figure 7.4 shows the fluorescence fluctuations obtained on red fluorescently labelled liposomes filled
with dex‐HEMA/DMAEMA, respectively before (Figure 7.4a – left graph) and after UV polymerization
(Figure 7.4a – right graph). Upon excitation with 561 nm laser light, highly intense red fluorescence
peaks appeared in both samples, originating from the migration of bright fluorescent nanoparticles
(in this case liposomes) through the excitation volume of the confocal microscope.[38,41] The lipid
bilayer is strongly fluorescent due to the incorporation of multiple rhodamine B‐DOPE lipid
molecules. When TX100 was added to the samples, fluorescence peaks were no longer detected
which can be explained by the solubilization of the lipid bilayer resulting in mixed micelles. Mixed
micelles are relatively small when compared to the liposomal vesicles and most likely contain only a
small amount of rhodamine‐B DOPE molecules. Therefore, diffusion of these mixed micelles through
the excitation volume does not give rise to high fluorescence peaks. In a next step, green fluorescent
siRNA was added to the TX100 treated samples. In Figure 7.4a (left graph), “normal” fluorescence
fluctuations (i.e. without high fluorescence peaks) were measured with an average of ~360 kHz.
Interestingly, when the sample was exposed to UV light, highly intense green fluorescence peaks
appeared whith a baseline fluorescence signal 45 kHz (Figure 7.4a, right graph). These observations
195

CHAPTER 7 | liposomal template‐assisted synthesis of dextran based nanogels
provide additional evidence for the formation of cationic dex‐HEMA‐co‐DMAEMA nanogels inside the
neutral lipid coat. Indeed, when the liposomes are not exposed to UV light, no polymerization of the
dex‐HEMA/DMAEMA can occur. After removal of the lipid coating with TX100, a solution of
unpolymerized dex‐HEMA/DMAEMA together with neutral mixed micelles remains, that cannot
complex the added fluorescent siRNA (Figure 7.4b). As a result “normal” fluorescence fluctuations
are detected, corresponding to freely diffusing AF488‐siRNA, and high fluorescence peaks are absent.
On the other hand, UV irradiation results in the formation of DMAEMA containing dextran nanogels
within the lipid bilayer. After addition of TX100, naked cationic nanogels emerge that are able to
complex the added fluorescent siRNA (Figure 7.4b). The diffusion of nanogels, loaded with AF488‐
siRNA, explains (a) the emergence of green fluorescence peaks and (b) the lowering of the baseline
fluorescence signal.
Cellular internalization of neutral lipid‐enveloped dextran nanogels
The cellular internalisation of nanoparticles often depends on the interaction of positively charged
groups with the oppositely charged heparan sulphate proteoglycans spanning the plasma membrane
of eukaryotic cells.[42] Since the lipid nanoreactor that is envisioned in this chapter, in accordance
with our experimental needs, does not contain charged components, we aimed to verify if this
characteristic significantly impedes cellular uptake of our LENs. Flow cytometry on DOPC:DOPE (1:1)
coated dex‐HEMA DS 5.2 nanogels (fluorescently tagged by 0.2 mol% NBD‐DOPE), still indicated
efficient cellular binding and internalization in Huh‐7_wt cells from a ratio of ~10 nmol total lipid per
6 x 104 seeded cells when a 4 h incubation time with the cells was respected (Figure 7.5a). The LENs
were characterized by DLS and zeta‐potential (198.6 ± 1.7 nm; PDI 0.093 ± 0.022; ‐6.1 ± 0.4 mV) .
During this incubation period, even at the highest amount of lipid tested (1 µmol), no signs of
cytotoxicity or morphological aberrations were observed. The uptake of fluorescently tagged LENs
was also visualized with confocal microscopy (Figure 7.5b).
In this section we chose to use DOPC:DOPE (1:1) neutral liposomes for several reasons: (1) neutral
liposomes will not interfere with the incorporation of charged methacrylate monomers in the dex‐
HEMA nanogels, (2) neutral liposomes generally have low cytotoxicity, especially when compared
with cationic lipid formulations,[4] and (3) DOPC, carrying two mono‐unsaturated acyl chains, forms
stable vesicles at ambient and physiological temperature and the ability to fuse (e.g. with endosomal
vesicular membranes) can be enhanced by incorporation of DOPE, which enhances the
hydrophobicity of the lipid bilayer.[43,44]
196
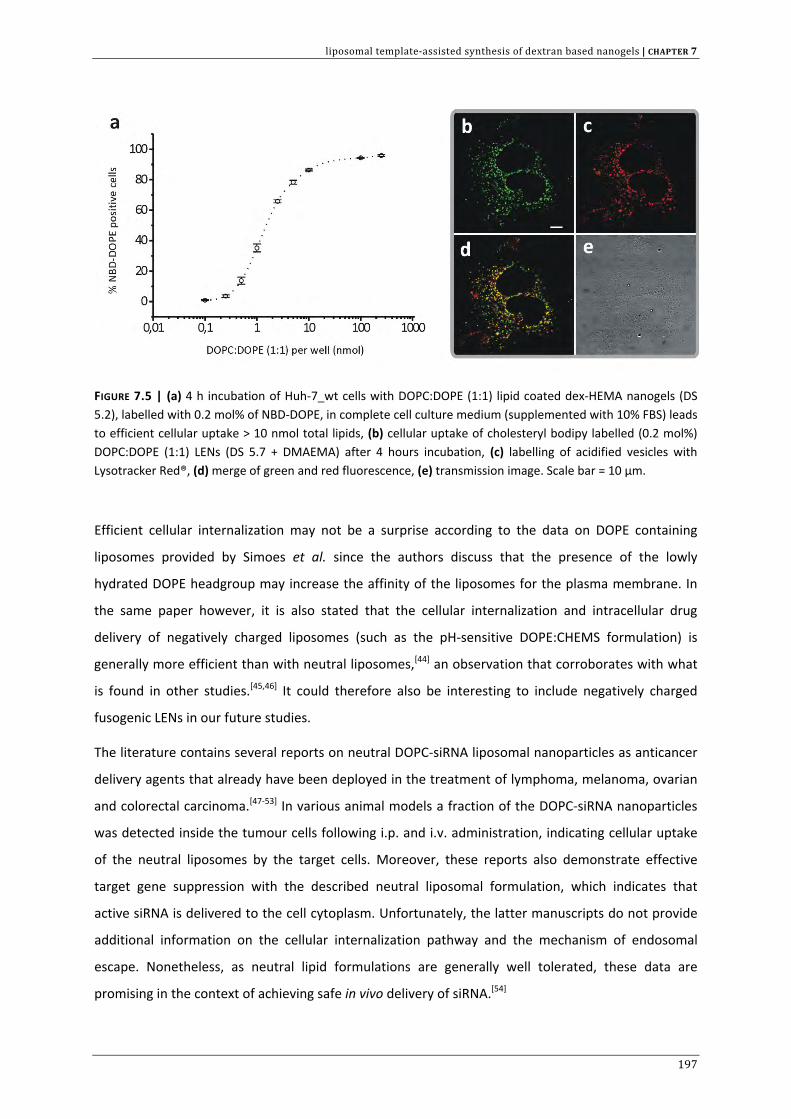
liposomal template‐assisted synthesis of dextran based nanogels | CHAPTER 7
FIGURE 7.5 | (a) 4 h incubation of Huh‐7_wt cells with DOPC:DOPE (1:1) lipid coated dex‐HEMA nanogels (DS 5.2), labelled with 0.2 mol% of NBD‐DOPE, in complete cell culture medium (supplemented with 10% FBS) leads to efficient cellular uptake > 10 nmol total lipids, (b) cellular uptake of cholesteryl bodipy labelled (0.2 mol%) DOPC:DOPE (1:1) LENs (DS 5.7 + DMAEMA) after 4 hours incubation, (c) labelling of acidified vesicles with Lysotracker Red®, (d) merge of green and red fluorescence, (e) transmission image. Scale bar = 10 µm.
Efficient cellular internalization may not be a surprise according to the data on DOPE containing
liposomes provided by Simoes et al. since the authors discuss that the presence of the lowly
hydrated DOPE headgroup may increase the affinity of the liposomes for the plasma membrane. In
the same paper however, it is also stated that the cellular internalization and intracellular drug
delivery of negatively charged liposomes (such as the pH‐sensitive DOPE:CHEMS formulation) is
generally more efficient than with neutral liposomes,[44] an observation that corroborates with what
is found in other studies.[45,46] It could therefore also be interesting to include negatively charged
fusogenic LENs in our future studies.
The literature contains several reports on neutral DOPC‐siRNA liposomal nanoparticles as anticancer
delivery agents that already have been deployed in the treatment of lymphoma, melanoma, ovarian
and colorectal carcinoma.[47‐53] In various animal models a fraction of the DOPC‐siRNA nanoparticles
was detected inside the tumour cells following i.p. and i.v. administration, indicating cellular uptake
of the neutral liposomes by the target cells. Moreover, these reports also demonstrate effective
target gene suppression with the described neutral liposomal formulation, which indicates that
active siRNA is delivered to the cell cytoplasm. Unfortunately, the latter manuscripts do not provide
additional information on the cellular internalization pathway and the mechanism of endosomal
escape. Nonetheless, as neutral lipid formulations are generally well tolerated, these data are
promising in the context of achieving safe in vivo delivery of siRNA.[54]
197
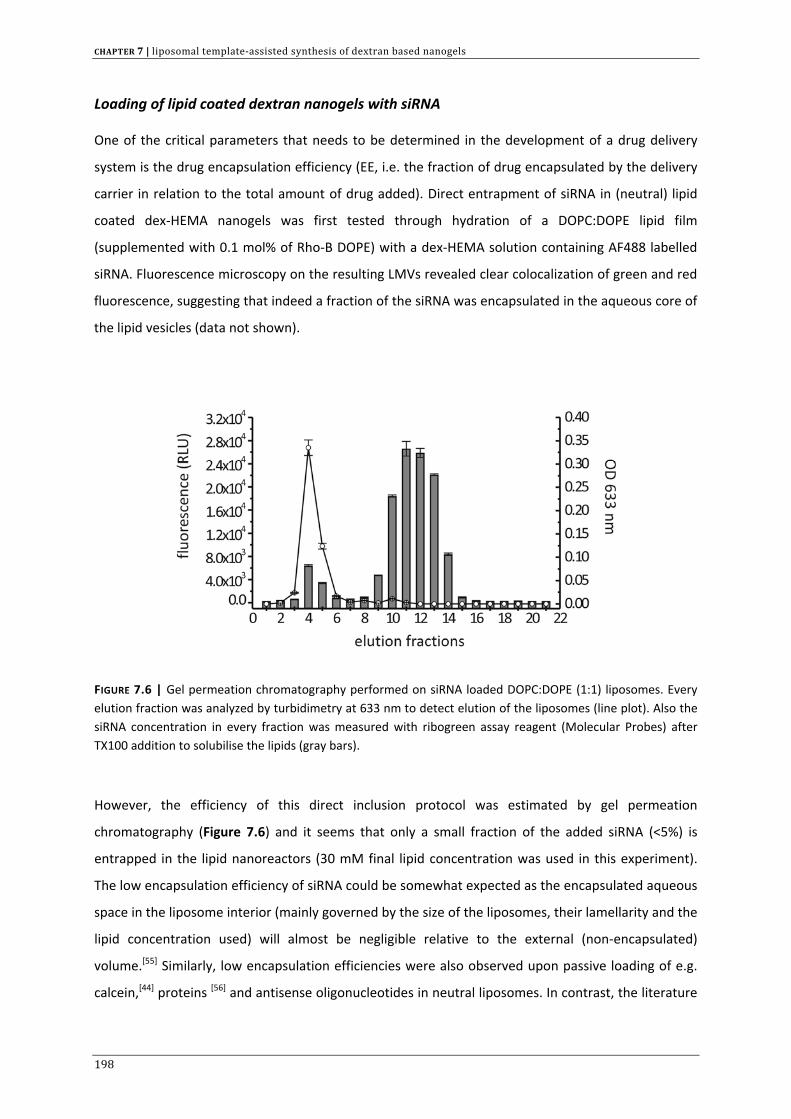
CHAPTER 7 | liposomal template‐assisted synthesis of dextran based nanogels
Loading of lipid coated dextran nanogels with siRNA
One of the critical parameters that needs to be determined in the development of a drug delivery
system is the drug encapsulation efficiency (EE, i.e. the fraction of drug encapsulated by the delivery
carrier in relation to the total amount of drug added). Direct entrapment of siRNA in (neutral) lipid
coated dex‐HEMA nanogels was first tested through hydration of a DOPC:DOPE lipid film
(supplemented with 0.1 mol% of Rho‐B DOPE) with a dex‐HEMA solution containing AF488 labelled
siRNA. Fluorescence microscopy on the resulting LMVs revealed clear colocalization of green and red
fluorescence, suggesting that indeed a fraction of the siRNA was encapsulated in the aqueous core of
the lipid vesicles (data not shown).
FIGURE 7.6 | Gel permeation chromatography performed on siRNA loaded DOPC:DOPE (1:1) liposomes. Every elution fraction was analyzed by turbidimetry at 633 nm to detect elution of the liposomes (line plot). Also the siRNA concentration in every fraction was measured with ribogreen assay reagent (Molecular Probes) after TX100 addition to solubilise the lipids (gray bars).
However, the efficiency of this direct inclusion protocol was estimated by gel permeation
chromatography (Figure 7.6) and it seems that only a small fraction of the added siRNA (<5%) is
entrapped in the lipid nanoreactors (30 mM final lipid concentration was used in this experiment).
The low encapsulation efficiency of siRNA could be somewhat expected as the encapsulated aqueous
space in the liposome interior (mainly governed by the size of the liposomes, their lamellarity and the
lipid concentration used) will almost be negligible relative to the external (non‐encapsulated)
volume.[55] Similarly, low encapsulation efficiencies were also observed upon passive loading of e.g.
calcein,[44] proteins [56] and antisense oligonucleotides in neutral liposomes. In contrast, the literature
198

liposomal template‐assisted synthesis of dextran based nanogels | CHAPTER 7
also describes protocols for the preparation of neutral and/or negatively charged liposomal
formulations to achieve higher loading efficiency for pDNA [4,43], DNA oligonucleotides [57] and
siRNA.[50] The DOPC lipid nanoparticles from A.K. Sood’s group that were described earlier in this
chapter, have an estimated ~65% loading efficiency (based on centrifugational ultrafiltration to
separate encapsulated from non‐encapsulated material), but unfortunately the physicochemical
characteristics of the lipid formulation remain uncertain.[50] A common feature of the latter reports is
the use of (water‐miscible) organic solvents during the process of liposome formation, which can
cause a precipitation of the added dextran during LENs formation. For this reason the proposed
methods are not the first choice when aiming to optimize siRNA loading inside our lipid coated
dextran nanogels.
The theoretical encapsulation efficiency of hydrophilic compounds is directly proportional to the
total lipid concentration.[55] Increasing the amount of lipids used during liposome preparation
augments the total internal liposome volume and thus also the potential entrapment of hydrophilic
(macro)molecules. At highly concentrated lipid mixtures, exceeding 300 mM of total phospholipid,
viscous vesicle dispersions are formed defined as a vesicular phospholipid gel or VPG. In this capacity,
the liposomal vesicles are so closely packed that the aqueous space in between the vesicles is
minimal and intervesicle interactions give rise to a semi‐solid consistency. Subsequent dilution of a
VPG eventually leads to a conventional liposomal dispersion. Due to the high viscosity of the VPG
sample, conventional liposomal preparation methods (e.g. the thin film hydration method follow by
extrusion) are not the best choice. Usually, high pressure homogenization (HPH) is considered to be
the gold standard for VPG production.[58] Recently, Massing and co‐workers introduced a different
homogenization technique called differential asymmetric centrifugation or DAC, that can be applied
to homogenize concentrated phospholipid mixtures [59] and this technique led to high encapsulation
of siRNA up to 70%.[60]
In analogy with these reports, we aimed to prepare lipid coated dextran nanogels at higher lipid
concentrations than used earlier, in order to achieve higher entrapment efficiency of our siRNA
duplexes. In a first experiment, it needed to be evaluated if such high lipid concentrations could be
hydrated and transformed into vesicles of monodisperse size. Lyophilized DOPC was hydrated with a
dex‐MA 20 wt% solution to reach a final lipid concentration of 500 mg mL‐1 (equal to 636 mM). To
homogenize the lipid dispersion at this high concentration, we tested probe sonication (for more
details see experimental section) and the results are summarized in Figure 7.7. Merely mixing the
lipids with a dex‐HEMA solution produces LMVs (average size > 700 nm) with high polydispersity.
Clearly, probe sonication provides enough shear force to ‘size’ the liposomal vesicles in the obtained
vesicular phospholipid gel down to ~110 nm with low polydispersity. The probe sonication has the
199
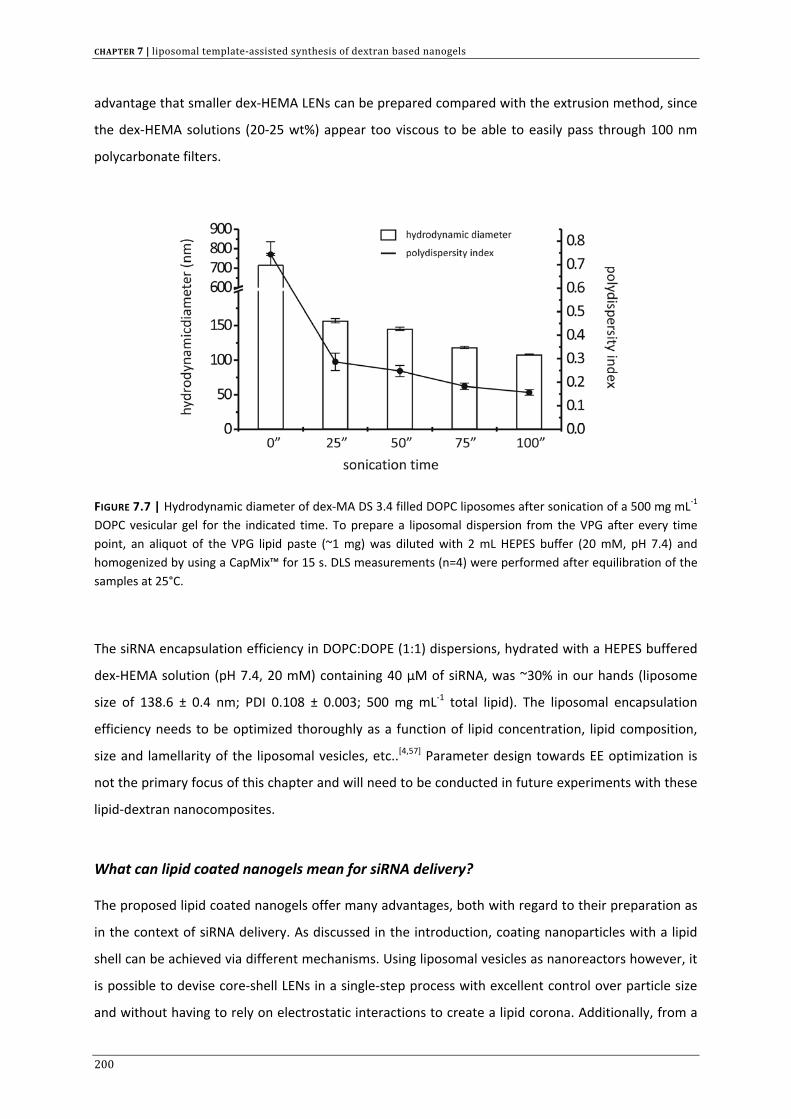
CHAPTER 7 | liposomal template‐assisted synthesis of dextran based nanogels
advantage that smaller dex‐HEMA LENs can be prepared compared with the extrusion method, since
the dex‐HEMA solutions (20‐25 wt%) appear too viscous to be able to easily pass through 100 nm
polycarbonate filters.
FIGURE 7.7 | Hydrodynamic diameter of dex‐MA DS 3.4 filled DOPC liposomes after sonication of a 500 mg mL‐1 DOPC vesicular gel for the indicated time. To prepare a liposomal dispersion from the VPG after every time point, an aliquot of the VPG lipid paste (~1 mg) was diluted with 2 mL HEPES buffer (20 mM, pH 7.4) and homogenized by using a CapMix™ for 15 s. DLS measurements (n=4) were performed after equilibration of the samples at 25°C.
The siRNA encapsulation efficiency in DOPC:DOPE (1:1) dispersions, hydrated with a HEPES buffered
dex‐HEMA solution (pH 7.4, 20 mM) containing 40 µM of siRNA, was ~30% in our hands (liposome
size of 138.6 ± 0.4 nm; PDI 0.108 ± 0.003; 500 mg mL‐1 total lipid). The liposomal encapsulation
efficiency needs to be optimized thoroughly as a function of lipid concentration, lipid composition,
size and lamellarity of the liposomal vesicles, etc..[4,57] Parameter design towards EE optimization is
not the primary focus of this chapter and will need to be conducted in future experiments with these
lipid‐dextran nanocomposites.
What can lipid coated nanogels mean for siRNA delivery?
The proposed lipid coated nanogels offer many advantages, both with regard to their preparation as
in the context of siRNA delivery. As discussed in the introduction, coating nanoparticles with a lipid
shell can be achieved via different mechanisms. Using liposomal vesicles as nanoreactors however, it
is possible to devise core‐shell LENs in a single‐step process with excellent control over particle size
and without having to rely on electrostatic interactions to create a lipid corona. Additionally, from a
200

liposomal template‐assisted synthesis of dextran based nanogels | CHAPTER 7
combinatorial point of view, merging a liposomal and nanogel carrier into one single nanocomposite,
combines the controlled release properties of the nanogel matrix with the favourable membrane
properties of phospholipid vesicles.[61]
The multifunctionality of the lipid coat is of significant importance in overcoming the most prominent
barriers in siRNA delivery. The choice of lipids is imperative in tailoring the surface chemistry of the
LENs, that will eventually influence to a large extent their behaviour in the extra‐ and intracellular
environment. The mechanisms with which our lipid coated nanogels could improve siRNA delivery
are therefore in agreement with what literature reports on how liposomes can be functionalized for
improved in vivo performance.[1,5,8,62] First, the preparation of stealth LENs by the incorporation of
PEGylated lipids could be used to prevent (1) aggregation by interaction with serum proteins and (2)
massive non‐specific phagocytosis, mainly by Kupffer cells in the liver and/or macrophages in the
spleen. As a result, the increased residence time in the blood circulation should favour accumulation
in leaky tumours through the Enhanced Permeation and Retention (EPR) effect or could augment the
uptake in other tissues and organs. Nonetheless, this is mainly a prerequisite for particles carrying
charged lipid bilayers and is of less importance for neutral liposomes or the LENs we envision. Once
the LENs gain access to the target cells, in a next step they should cross the cell membrane and
deliver intact siRNA into the cytoplasm for RNAi activation. PEGylation is usually accompanied by
coupling of a targeting moiety to the PEG distal end to improve cell surface receptor binding and
subsequent internalization. Following cell uptake, lipid‐based nanoparticles generally end up being
trapped in (acidified) endosomal vesicles. The preparation of LENs with pH‐sensitive lipid bilayers
that become fusogenic at endosomal pH (pH 5‐6.5) could facilitate the intracytoplasmic injection of
the encapsulated siRNA loaded nanogels.[5,8,44] From these examples it should be clear that the
predominant motivation to equip nanogels with a lipid‐shell is to overcome the major extracellular
barriers and allow disposition of biodegradable siRNA loaded nanogels in the target cell cytoplasm. It
would be interesting to assess the effect of lipid composition on siRNA loading, intracellular siRNA
delivery and the eventual RNAi outcome (both maximal effect as gene silencing duration) in future
experiments.
201

CHAPTER 7 | liposomal template‐assisted synthesis of dextran based nanogels
REFERENCE LIST
1. Remaut K., Sanders N.N., De Geest B.G., Braeckmans K. et al., Nucleic acid delivery: Where material sciences and bio‐sciences meet, Materials Science & Engineering R‐Reports 2007, 58: 117‐161.
2. Raemdonck K., Demeester J., and De Smedt S., Advanced nanogel engineering for drug delivery, Soft Matter 2009, 5: 707‐715.
3. Raemdonck K., Vandenbroucke R.E., Demeester J., Sanders N.N. et al., Maintaining the silence: reflections on long‐term RNAi, Drug Discovery Today 2008, 13: 917‐931.
4. Li W. and Szoka F.C., Jr., Lipid‐based nanoparticles for nucleic acid delivery, Pharm. Res. 2007, 24: 438‐449. 5. Torchilin V.P., Recent advances with liposomes as pharmaceutical carriers, Nature Reviews Drug Discovery
2005, 4: 145‐160. 6. Ganta S., Devalapally H., Shahiwala A., and Amiji M., A review of stimuli‐responsive nanocarriers for drug
and gene delivery, Journal of Controlled Release 2008, 126: 187‐204. 7. Ong W., Yang Y.M., Cruciano A.C., and McCarley R.L., Redox‐Triggered Contents Release from Liposomes,
Journal of the American Chemical Society 2008, 130: 14739‐14744. 8. Simoes S., Moreira J.N., Fonseca C., Duzgunes N. et al., On the formulation of pH‐sensitive liposomes with
long circulation times, Adv. Drug Deliv. Rev. 2004, 56: 947‐965. 9. Kullberg M., Mann K., and Owens J.L., A two‐component drug delivery system using Her‐2‐targeting
thermosensitive liposomes, J. Drug Target 2009, 17: 98‐107. 10. Auguste D.T., Furman K., Wong A., Fuller J. et al., Triggered release of siRNA from poly(ethylene glycol)‐
protected, pH‐dependent liposomes, J. Control Release 2008, 130: 266‐274. 11. Sengupta S., Eavarone D., Capila I., Zhao G.L. et al., Temporal targeting of tumour cells and neovasculature
with a nanoscale delivery system, Nature 2005, 436: 568‐572. 12. Messerschmidt S.K., Musyanovych A., Altvater M., Scheurich P. et al., Targeted lipid‐coated nanoparticles:
Delivery of tumor necrosis factor‐functionalized particles to tumor cells, J. Control Release 2009, doi: 10.1016/j.jconrel.2009.03.010.
13. Li S.D. and Huang L., Surface‐modified LPD nanoparticles for tumor targeting, Oligonucleotide Therapeutics 2006, 1082: 1‐8.
14. Li S.D., Chen Y.C., Hackett M.J., and Huang L., Tumor‐targeted delivery of siRNA by self‐assembled nanoparticles, Molecular Therapy 2008, 16: 163‐169.
15. De Miguel I, Imbertie L., Rieumajou V., Major M. et al., Proofs of the structure of lipid coated nanoparticles (SMBV) used as drug carriers, Pharm. Res. 2000, 17: 817‐824.
16. Mastrobattista E., Kapel R.H., Eggenhuisen M.H., Roholl P.J. et al., Lipid‐coated polyplexes for targeted gene delivery to ovarian carcinoma cells, Cancer Gene Ther. 2001, 8: 405‐413.
17. Hatakeyama H., Akita H., Kogure K., and Harashima H., A Novel Nonviral Gene Delivery System: Multifunctional Envelope‐Type Nano Device, Adv. Biochem. Eng Biotechnol. 2009, doi: 10.1007/10_2008_40.
18. Sakurai Y., Hatakeyama H., Akita H., Oishi M. et al., Efficient short interference RNA delivery to tumor cells using a combination of octaarginine, GALA and tumor‐specific, cleavable polyethylene glycol system, Biol. Pharm. Bull. 2009, 32: 928‐932.
19. Kogure K., Akita H., and Harashima H., Multifunctional envelope‐type nano device for non‐viral gene delivery: Concept and application of Programmed Packaging, Journal of Controlled Release 2007, 122: 246‐251.
20. El‐Sayed A., Khalil I.A., Kogure K., Futaki S. et al., Octaarginine‐ and octalysine‐modified nanoparticles have different modes of endosomal escape, J. Biol. Chem. 2008, 283: 23450‐23461.
21. Akita H., Kudo A., Minoura A., Yamaguti M. et al., Multi‐layered nanoparticles for penetrating the endosome and nuclear membrane via a step‐wise membrane fusion process, Biomaterials 2009, 30: 2940‐2949.
22. Jana S.S., Bharali D.J., Mani P., Maitra A. et al., Targeted cytosolic delivery of hydrogel nanoparticles into HepG2 cells through engineered Sendai viral envelopes, FEBS Letters 2002, 515: 184‐188.
23. Kunisawa J., Masuda T., Katayama K., Yoshikawa T. et al., Fusogenic liposome delivers encapsulated nanoparticles for cytosolic controlled gene release, Journal of Controlled Release 2005, 105: 344‐353.
24. An S.Y., Bui M.P., Nam Y.J., Han K.N. et al., Preparation of monodisperse and size‐controlled poly(ethylene glycol) hydrogel nanoparticles using liposome templates, J. Colloid Interface Sci. 2009, 331: 98‐103.
25. Hong J.S., Vreeland W.N., Lacerda S.H., Locascio L.E. et al., Liposome‐templated supramolecular assembly of responsive alginate nanogels, Langmuir 2008, 24: 4092‐4096.
202

liposomal template‐assisted synthesis of dextran based nanogels | CHAPTER 7
26. Kazakov S., Kaholek M., Teraoka I., and Levon K., UV‐induced gelation on nanometer scale using liposome reactor, Macromolecules 2002, 35: 1911‐1920.
27. Kazakov S., Kaholek M., Kudasheva D., Teraoka I. et al., Poly(N‐isopropylacrylamide‐co‐1‐vinylimidazole) hydrogel nanoparticles prepared and hydrophobically modified in liposome reactors: Atomic force microscopy and dynamic light scattering study, Langmuir 2003, 19: 8086‐8093.
28. Kazakov S. and Levon K., Liposome‐nanogel structures for future pharmaceutical applications, Curr. Pharm. Des 2006, 12: 4713‐4728.
29. Patton J.N. and Palmer A.F., Engineering temperature‐sensitive hydrogel nanoparticles entrapping hemoglobin as a novel type of oxygen carrier, Biomacromolecules 2005, 6: 2204‐2212.
30. Patton J.N. and Palmer A.F., Photopolymerization of bovine hemoglobin entrapped nanoscale hydrogel particles within liposomal reactors for use as an artificial blood substitute, Biomacromolecules 2005, 6: 414‐424.
31. Schillemans J.P., Flesch F.M., Hennink W.E., and van Nostrum C.F., Synthesis of bilayer‐coated nanogels by selective cross‐linking of monomers inside liposomes, Macromolecules 2006, 39: 5885‐5890.
32. Van Thienen T.G., Lucas B., Flesch F.M., van Nostrum C.F. et al., On the synthesis and characterization of biodegradable dextran nanogels with tunable degradation properties, Macromolecules 2005, 38: 8503‐8511.
33. Van Thienen T.G., Demeester J., and De Smedt S.C., Screening poly(ethyleneglycol) micro‐ and nanogels for drug delivery purposes, International Journal of Pharmaceutics 2008, 351: 174‐185.
34. Rouser G., Fkeischer S., and Yamamoto A., Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots, Lipids 1970, 5: 494‐496.
35. Akinc A., Goldberg M., Qin J., Dorkin J.R. et al., Development of Lipidoid‐siRNA Formulations for Systemic Delivery to the Liver, Molecular Therapy 2009, 17: 872‐879.
36. Raemdonck K., Remaut K., Lucas B., Sanders N.N. et al., In situ analysis of single‐stranded and duplex siRNA integrity in living cells, Biochemistry 2006, 45: 10614‐10623.
37. Buyens K., Lucas B., Raemdonck K., Braeckmans K. et al., A fast and sensitive method for measuring the integrity of siRNA‐carrier complexes in full human serum, Journal of Controlled Release 2008, 126: 67‐76.
38. Lucas B., Remaut K., Braeckmans K., Haustraete J. et al., Studying pegylated DNA complexes by dual color fluorescence fluctuation spectroscopy, Macromolecules 2004, 37: 3832‐3840.
39. Remaut K., Lucas B., Raemdonck K., Braeckmans K. et al., Can we better understand the intracellular behavior of DNA nanoparticles by fluorescence correlation spectroscopy?, Journal of Controlled Release 2007, 121: 49‐63.
40. Stubbe B.G., Horkay F., Amsden B., Hennink W.E. et al., Tailoring the swelling pressure of degrading dextran hydroxyethyl methacrylate hydrogels, Biomacromolecules 2003, 4: 691‐695.
41. Lucas B., Van Rompaey E., De Smedt S.C., Demeester J. et al., Dual‐color fluorescence fluctuation spectroscopy to study the complexation between poly‐l‐lysine and oligonucleotides, Macromolecules 2002, 35: 8152‐8160.
42. Payne C.K., Jones S.A., Chen C., and Zhuang X., Internalization and trafficking of cell surface proteoglycans and proteoglycan‐binding ligands, Traffic 2007, 8: 389‐401.
43. Bailey A.L. and Sullivan S.M., Efficient encapsulation of DNA plasmids in small neutral liposomes induced by ethanol and calcium, Biochim. Biophys. Acta 2000, 1468: 239‐252.
44. Simoes S., Slepushkin V., Duzgunes N., and Pedroso de Lima M.C., On the mechanisms of internalization and intracellular delivery mediated by pH‐sensitive liposomes, Biochim. Biophys. Acta 2001, 1515: 23‐37.
45. Skalko‐Basnet N., Tohda M., and Watanabe H., Uptake of liposomally entrapped fluorescent antisense oligonucleotides in NG108‐15 cells: conventional versus pH‐sensitive, Biol. Pharm. Bull. 2002, 25: 1583‐1587.
46. Malaekeh‐Nikouei B., Jaafari M.R., Tabassi S.A., and Samiei A., The enhancement of immunosuppressive effects of cyclosporine A on human T‐cells using fusogenic liposomes, Colloids Surf. B Biointerfaces. 2008, 67: 238‐244.
47. Gray M.J., Dallas N.A., Van B.G., Xia L. et al., Therapeutic targeting of Id2 reduces growth of human colorectal carcinoma in the murine liver, Oncogene 2008, 27: 7192‐7200.
48. Halder J., Kamat A.A., Landen C.N., Jr., Han L.Y. et al., Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy, Clin. Cancer Res. 2006, 12: 4916‐4924.
49. Landen C.N., Merritt W.M., Mangala L.S., Sanguino A.M. et al., Intraperitoneal delivery of liposomal siRNA for therapy of advanced ovarian cancer, Cancer Biol. Ther. 2006, 5: 1708‐1713.
203

CHAPTER 7 | liposomal template‐assisted synthesis of dextran based nanogels
204
50. Landen C.N., Jr., Chavez‐Reyes A., Bucana C., Schmandt R. et al., Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery, Cancer Res. 2005, 65: 6910‐6918.
51. Lin Y.G., Immaneni A., Merritt W.M., Mangala L.S. et al., Targeting aurora kinase with MK‐0457 inhibits ovarian cancer growth, Clin. Cancer Res. 2008, 14: 5437‐5446.
52. Merritt W.M., Lin Y.G., Spannuth W.A., Fletcher M.S. et al., Effect of interleukin‐8 gene silencing with liposome‐encapsulated small interfering RNA on ovarian cancer cell growth, J. Natl. Cancer Inst. 2008, 100: 359‐372.
53. Villares G.J., Zigler M., Wang H., Melnikova V.O. et al., Targeting melanoma growth and metastasis with systemic delivery of liposome‐incorporated protease‐activated receptor‐1 small interfering RNA, Cancer Res. 2008, 68: 9078‐9086.
54. de Fougerolles A.R., Delivery vehicles for small interfering RNA in vivo, Hum. Gene Ther. 2008, 19: 125‐132. 55. Brandl M., Drechsler M., Bachmann D., Tardi C. et al., Preparation and characterization of semi‐solid
phospholipid dispersions and dilutions thereof, International Journal of Pharmaceutics 1998, 170: 187‐199. 56. Fretz M.M., Hogset A., Koning G.A., Jiskoot W. et al., Cytosolic delivery of liposomally targeted proteins
induced by photochemical internalization, Pharm. Res. 2007, 24: 2040‐2047. 57. Edwards K.A. and Baeumner A.J., DNA‐oligonucleotide encapsulating liposomes as a secondary signal
amplification means, Anal. Chem. 2007, 79: 1806‐1815. 58. Brandl M., Vesicular phospholipid gels: A technology platform, Journal of Liposome Research 2007, 17: 15‐
26. 59. Massing U., Cicko S., and Ziroli V., Dual asymmetric centrifugation (DAC) ‐ A new technique for liposome
preparation, Journal of Controlled Release 2008, 125: 16‐24. 60. Hirsch M., Ziroli V., Helm M., and Massing U., Preparation of small amounts of sterile siRNA‐liposomes with
high entrapping efficiency by dual asymmetric centrifugation (DAC), Journal of Controlled Release 2009, 135: 80‐88.
61. Soussan E., Cassel S., Blanzat M., and Rico‐Lattes I., Drug delivery by soft matter: matrix and vesicular carriers, Angew. Chem. Int. Ed Engl. 2009, 48: 274‐288.
62. Tseng Y.C., Mozumdar S., and Huang L., Lipid‐based systemic delivery of siRNA, Adv. Drug Deliv. Rev. 2009, doi: 10.1016/j.addr.2009.03.003.

summary & general conclusions | CHAPTER 8
205
CHAPTER 8 | Summary & general conclusions
Koen Raemdonck, J. Demeester and Stefaan C. De Smedt
Laboratory of General Biochemistry and Physical Pharmacy, Department of Pharmaceutics, Faculty of Pharmaceutical
Sciences, Ghent University, Harelbekestraat 72, B‐9000 Ghent, Belgium.

CHAPTER 8 | summary & general conclusions
206

summary & general conclusions | CHAPTER 8
8 SUMMARY
RNA interference (RNAi) certainly is a hot topic among the scientific community, judging by the broad
financial investments and the tremendous research output to date.[1] This statement is further
illustrated through a PubMed search on the term ‘RNA interference’ (performed on June 15th 2009),
which yielded ~16.500 entries. RNAi is a naturally conserved gene silencing mechanism functioning in
eukaryotic cells and is activated by small interfering RNAs (siRNAs) that trigger the degradation of
mRNA in a sequence‐specific manner (CHAPTER 1). Besides its use as a laboratory tool in functional
genomics and drug target discovery, the therapeutic potential of RNAi by blocking the production of
disease‐causing proteins has also long been recognized. RNAi therapeutics have the advantage to act
(post)transcriptionally, while small molecular drugs are generally limited to interference on the
protein level. Thus, siRNAs may be designed to downregulate almost every gene transcript and hopes
are high for the future to make RNAi a therapeutic reality. In just over 10 years since the discovery of
RNAi, we already witnessed the transition of small interfering RNAs into the clinic and an overview of
siRNA compounds in active and already completed clinical trials is presented in Table 8.1.[1‐4]
The siRNAs that are in the most advanced stages of clinical evaluation are delivered locally at the site
of disease as a conventional saline formulation. Scientists however agree that to broaden the
therapeutic potential of RNAi, the principal challenge that remains is the hurdle of efficient siRNA
delivery in the human body. As summarized in chapter 1, many extra‐ and intracellular barriers are
encountered before siRNAs can be safely delivered to their site of action, which is the cytoplasm of
the target cells. Advanced RNAi delivery systems, either viral or non‐viral, are desperately needed
and constitute the main focus of the research on RNAi therapeutics. Since the RNAi machinery is
located in the cytoplasm, siRNA therapeutics need to be guided across the cellular membrane for
biological activity. However, owing to their high negative charge and relatively large size (~14.000
Da), naked siRNAs are not readily internalized by cells. Although the viral delivery systems are
generally the most effective to date, their clinical use is limited mainly due to safety issues. For this
reason, non‐viral delivery approaches have emerged as the safe(r) alternative. The experimental
207
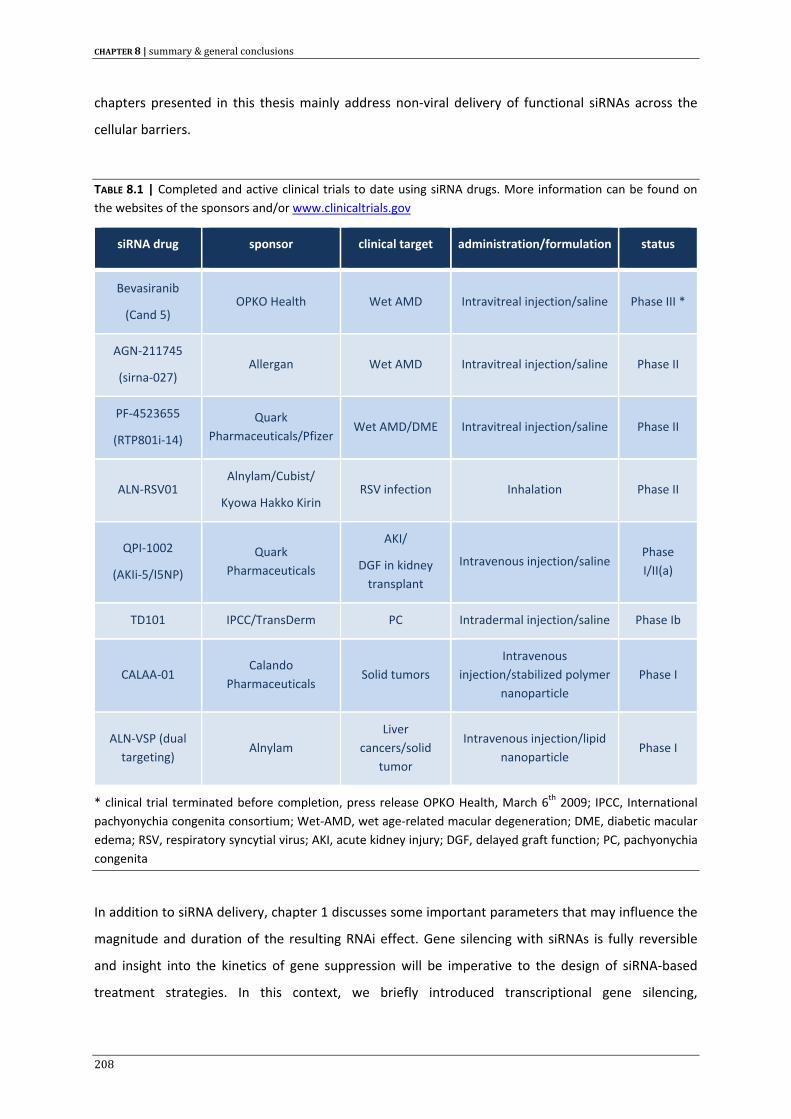
CHAPTER 8 | summary & general conclusions
chapters presented in this thesis mainly address non‐viral delivery of functional siRNAs across the
cellular barriers.
TABLE 8.1 | Completed and active clinical trials to date using siRNA drugs. More information can be found on the websites of the sponsors and/or www.clinicaltrials.gov
siRNA drug sponsor clinical target administration/formulation status
Bevasiranib
(Cand 5) OPKO Health Wet AMD Intravitreal injection/saline Phase III *
AGN‐211745
(sirna‐027) Allergan Wet AMD Intravitreal injection/saline Phase II
PF‐4523655
(RTP801i‐14)
Quark Pharmaceuticals/Pfizer
Wet AMD/DME Intravitreal injection/saline Phase II
ALN‐RSV01 Alnylam/Cubist/
Kyowa Hakko Kirin RSV infection Inhalation Phase II
QPI‐1002
(AKIi‐5/I5NP)
Quark Pharmaceuticals
AKI/
DGF in kidney transplant
Intravenous injection/saline Phase I/II(a)
TD101 IPCC/TransDerm PC Intradermal injection/saline Phase Ib
CALAA‐01 Calando
Pharmaceuticals Solid tumors
Intravenous injection/stabilized polymer
nanoparticle Phase I
ALN‐VSP (dual targeting)
Alnylam Liver
cancers/solid tumor
Intravenous injection/lipid nanoparticle
Phase I
* clinical trial terminated before completion, press release OPKO Health, March 6th 2009; IPCC, International pachyonychia congenita consortium; Wet‐AMD, wet age‐related macular degeneration; DME, diabetic macular edema; RSV, respiratory syncytial virus; AKI, acute kidney injury; DGF, delayed graft function; PC, pachyonychia congenita
In addition to siRNA delivery, chapter 1 discusses some important parameters that may influence the
magnitude and duration of the resulting RNAi effect. Gene silencing with siRNAs is fully reversible
and insight into the kinetics of gene suppression will be imperative to the design of siRNA‐based
treatment strategies. In this context, we briefly introduced transcriptional gene silencing,
208

summary & general conclusions | CHAPTER 8
mathematical modelling and combinatorial RNAi as new tools for the further optimization of genetic
interference.
In the exponentially growing field of RNAi, besides siRNA delivery also stability and degradation of
siRNA receives increasing attention. An essential issue that needs to be taken into account when
applying siRNAs, is their susceptibility to nuclease attack in biological fluids. Naked siRNA is highly
vulnerable to exo‐and endonucleases, leading to a short half‐life in serum and a wealth of reports in
the literature describe chemical modifications and formulation strategies to improve siRNA stability
and pharmacokinetics. With regard to the intracellular siRNA persistence, some conflicting studies
exist on whether or not the siRNA integrity significantly influences RNAi kinetics and therefore a
direct correlation between siRNA stability and RNAi duration is not yet conclusive. Techniques for the
investigation of the siRNA integrity after cellular internalization are therefore of substantial interest
to address the intracellular fate of siRNA. In CHAPTER 2, we describe a method based on fluorescence
resonance energy transfer (FRET) to assess the stability of siRNA in the intracellular
microenvironment. The method was validated by analyzing the loss of FRET as a function of
degradation by RNase A or through incubation in cellular extracts. In these experiments we used the
ratio of acceptor fluorescence to donor fluorescence upon donor excitation (FA/FD ratio) as a
parameter to evaluate the cleavage of both single‐stranded (ss) as double‐stranded (ds) siRNAs. Such
techniques may prove useful to explore the correlation between the intracellular fate of delivered
nucleic acids and their biological effectiveness, as already partially demonstrated in the literature.[5‐7]
A concept to induce a more robust RNAi response, as first mentioned in chapter 1, is through a
controlled release of siRNA in the cytoplasm of the target cell. The design of carriers that are able to
function as an siRNA depot across the cellular barriers, may induce a more sustained gene silencing
effect by maintaining the intracellular siRNA concentrations above the level required for activity for a
longer period of time. Following the broad applicability of hydrogels as controlled drug delivery
matrices and their experience in a biopharmaceutical context, we focused in this thesis on dextran
based hydrogels as carriers for siRNA. We first evaluated if biodegradable dextran microgels could be
produced to achieve a controlled release of the encapsulated siRNA (CHAPTER 3). For the preparation
of the microgels, both a water‐in‐water emulsion as well as a water‐in‐oil emulsion were tested.
Preformed microscopic gel particles (microgels), composed of dextran hydroxyethyl methacrylate
(dex‐HEMA) and further functionalized with cationic methacrylate monomers (2‐
(dimethylamino)ethyl methacrylate or DMAEMA), were able to bind siRNA through electrostatic
interaction. Confocal microscopy revealed that the siRNA duplexes were mainly adsorbed onto the
microgel surface but that they could also penetrate deeper in the pores of the gel network at higher
concentrations. The HEMA moieties are coupled to the dextran backbone via a labile carbonate ester
209

CHAPTER 8 | summary & general conclusions
linkage. Hydrolysis of this ester function, cleaves the crosslinks in between the dextran chains,
thereby again releasing the complexed siRNA. The degradation time of the dex‐HEMA microgels is
governed by their initial crosslink density and we showed in chapter 3 that by tailoring the network
properties we are able to control the release kinetics of the encapsulated siRNA over several days.
The vast majority of non‐viral delivery approaches makes use of (cationic) polymer or lipid‐based
nanosized particles to transfer complexed or encapsulated siRNA across the cellular membrane. A
principal requirement to assess the effectiveness of our siRNA delivery concept, was therefore to
scale down our cationic hydrogel particles to nanoscopic dimensions, in order to ensure efficient
cellular internalization. CHAPTER 4 briefly describes some interesting drug delivery applications based
on nanogels as the drug carrier. This chapter highlights several eye‐catching examples of nanogel
design and illustrates nanogels in terms of versatile stimuli‐responsive drug delivery systems.
Although the number of publications addressing this issue is growing exponentially since the
beginning of this century, to this date only a very limited number of reports has evaluated nanogels
for RNAi. In CHAPTER 5, we employed an inverse miniemulsion photopolymerization method for the
production of hydrolysable cationic dex‐HEMA nanogels. In contrast to chapter 3, this time we used a
methacrylate monomer carrying a quaternary ammonium group (trimethylammonium ethyl
methacrylate or TMAEMA) for copolymerization to incorporate positively charged amines into the
hydrogel network. Owing to the positive charge, the nanogels are able to complex siRNA with a high
loading capacity exceeding 70 wt%. Confocal microscopy and flow cytometry analysis revealed that
large numbers of siRNA loaded nanogels can effectively be internalized by human hepatoma cells
(Huh‐7 ) without relevant cytotoxicity. It is generally accepted that the majority of non‐viral carriers is
taken up through an endocytic process where most particles end up being trapped in acidified
(endo)lysosomes. We found that also our nanogel particles accumulated to a high extent in these
degradative organelles. Nevertheless, dependent on the amount of nanogels applied on the cells, still
a substantial suppression of luciferase expression could be obtained, indicating that a fraction of the
complexed siRNA reaches the cell cytoplasm where it is recognized by the RNAi machinery.
Moreover, this RNAi effect was almost absent for non‐degradable cationic dextran nanogels,
suggesting an influence of dex‐HEMA hydrolysis on the delivery of siRNA. On the other hand, the
extent of gene silencing could be markedly enhanced by applying validated endosomolytic tools, such
as photochemical internalization (PCI) and the co‐complexation of an influenza‐derived fusogenic
peptide (diINF‐7). These findings provide evidence that the endosomal sequestration of our siRNA
loaded nanogels partially abrogates the potential RNAi outcome.
While the magnitude of target gene suppression may be a good indicator for the success of an siRNA
treatment, evaluation of the kinetics of the RNAi effect is perhaps a more favourable parameter to
210

summary & general conclusions | CHAPTER 8
decide on the efficacy of the treatment. The time‐window that the siRNA therapy is able to keep the
intracellular protein titers below the threshold that is necessary to obtain the desired downstream
phenotype, will strongly determine the dosage frequency in clinical therapy. In CHAPTER 6, we
therefore analyzed the kinetics of gene inhibition for dex‐HEMA‐co‐TMAEMA nanogels with varying
crosslink density and compared the results with a commercially available lipid‐based transfection
reagent, i.e. lipofectamine™RNAiMAX. The transfection experiments revealed that the duration of
gene silencing (as assessed in Huh‐7 cells with firefly luciferase and EGFP as target respectively) is
largely independent on the type of carrier used. The magnitude of downregulation does however
differ for the investigated nanogel formulations with in general lower gene silencing efficiency for
higher crosslinked nanogels. Most likely, the extent of gene silencing is governed by the amount of
siRNA that is initially delivered in the cell cytoplasm while the recovery of gene expression to its
steady state level is caused by sequential cell divisions that dilute the intracellular siRNA pool until
insufficient active drug resides in the cytoplasm. In addition, we found that application of PCI, even
48 hours post‐transfection, was able to improve and lengthen the RNAi outcome for our dextran
nanogels, with up to 25% additional gene suppression. In conclusion, this result indicates that PCI is
able to liberate a fraction of the siRNA (‐loaded nanogels) that remain trapped in intracellular
compartments, which can be regarded as a potential strategy to prolong the therapeutic response
when endosomal escape is effect‐limiting, albeit that further optimization is required.
From the previous chapters it may be clear that to achieve an intracellular time‐controlled siRNA
delivery and to evaluate to what extent this may contribute to a more durable gene inhibition, a
transition of siRNA loaded nanogels from the endosomal lumen into the cytoplasm is imperative.
Following pioneering work by Van Thienen et al.,[8] CHAPTER 7 discusses the design of hybrid lipid‐
enveloped nanogels (LENs), where a multifunctional lipid shell is entrusted with the task to bypass
the major extracellular barriers to allow disposition of biodegradable siRNA loaded nanogels in the
target cell cytoplasm. These hybrid nanocomposites can be prepared by using liposomes as
nanoreactors for selective UV polymerization of dex‐HEMA in their aqueous core. SiRNA can be
loaded into LENs by passive hydration of a preformed lipid film with an encapsulation efficiency (non‐
optimized) of >30%. Both extrusion and probe sonication are successfully applied for LENs
production, as evidenced by dynamic light scattering. Although a majority of liposomal drug delivery
systems for RNAi typically comprises a cationic lipid, we chose to use a charge neutral lipid
combination for LENs development instead, consisting of an equimolar mixture of DOPC and DOPE.
In spite of this, preliminary cellular experiments suggested that Huh‐7 cells still quantitatively
internalize DOPC:DOPE coated LENs. Future experiments on LENs will be oriented toward a further
optimization of the siRNA encapsulation efficiency and the assessment of their RNAi potential.
211

CHAPTER 8 | summary & general conclusions
212
GENERAL CONCLUSIONS
In conclusion, this thesis comprises a novel FRET based approach for the intracellular assessment of
small interfering RNA (siRNA) integrity, which could aid in clarifying the correlation between
intracellular siRNA fate and the eventual RNAi outcome. Besides siRNA stability, this thesis also
describes the design of cationic and biodegradable nanogels for the time‐controlled delivery of
siRNA. We provide evidence that these nanogels can effectively deliver active siRNA across the
cellular barrier, leading to substantial and durable gene silencing. Endosomal escape is identified as
the predominant barrier confining the full RNAi potential, opening up new opportunities to further
improve this delivery concept. It is conceivable that, although RNAi can generally be applied to
interfere with the expression of virtually any gene, several distinct in vivo delivery agents will be
needed depending on the disease target and the chosen route of administration. Our nanogels may
well claim a future position in this ensemble of siRNA delivery systems.
REFERENCE LIST
1. Haussecker D., The business of RNAi therapeutics, Hum. Gene Ther. 2008, 19: 451‐462. 2. Behlke M.A., Chemical modification of siRNAs for in vivo use, Oligonucleotides. 2008, 18: 305‐319. 3. de Fougerolles A.R., Delivery vehicles for small interfering RNA in vivo, Hum. Gene Ther. 2008, 19: 125‐132. 4. Whelan J., First clinical data on RNAi, Drug Discov. Today 2005, 10: 1014‐1015. 5. Jagannath A. and Wood M.J., Localization of double‐stranded small interfering RNA to cytoplasmic
processing bodies is Ago2 dependent and results in up‐regulation of GW182 and Argonaute‐2, Mol. Biol. Cell 2009, 20: 521‐529.
6. Jarve A., Muller J., Kim I.H., Rohr K. et al., Surveillance of siRNA integrity by FRET imaging, Nucleic Acids Research 2007, 1‐13.
7. Remaut K., Lucas B., Raemdonck K., Braeckmans K. et al., Can we better understand the intracellular behavior of DNA nanoparticles by fluorescence correlation spectroscopy?, Journal of Controlled Release 2007, 121: 49‐63.
8. Van Thienen T.G., Lucas B., Flesch F.M., van Nostrum C.F. et al., On the synthesis and characterization of biodegradable dextran nanogels with tunable degradation properties, Macromolecules 2005, 38: 8503‐8511.

213

214

samenvatting en algemene besluiten | APPENDIX A
215
APPENDIX A | SAMENVATTING EN
ALGEMENE BESLUITEN

APPENDIX A | samenvatting en algemene besluiten
216

samenvatting en algemene besluiten | APPENDIX A
A SAMENVATTING VAN HET DOCTORAAL PROEFSCHRIFT
Het is slechts 11 jaar geleden dat Andrew Fire and Craig Mello in hun Nature publicatie de vreemde
observatie beschreven dat de exogene toediening van dubbelstrengig RNA (dsRNA) aan de nematode
C. elegans resulteerde in de onderdrukking van het complementaire gen. Zij waren de eersten die de
term RNA interference (RNAi) hanteerden en werden voor hun ontdekking beloond met de Nobelprijs
voor Geneeskunde en Fysiologie in 2006. Dit doet reeds vermoeden dat het hier een belangrijke
wetenschappelijke doorbraak betreft. RNAi is dan ook daadwerkelijk een ‘hot topic’ in de exacte
wetenschap, hetgeen we verder ook kunnen afleiden uit de immense financiële investeringen van de
afgelopen jaren en de uitgebreide wetenschappelijke output omtrent RNAi die men vandaag reeds in
de vakliteratuur kan terugvinden.
In essentie is RNAi een intracellulair mechanisme dat actief is in eukaryote cellen en waarvan het
biochemisch raderwerk in de loop van de evolutie bewaard is gebleven. De RNAi machine wordt
geactiveerd door de introductie van dsRNA in het cytoplasma van de doelwitcel, hetgeen uiteindelijk
leidt tot de sequentie‐specifieke onderdrukking van een doelwitgen op het posttranscriptioneel
niveau (HOOFDSTUK 1). Naar aanleiding van de ontdekking in 2001 dat RNAi ook functioneel zou zijn in
menselijke cellen, nam het onderzoek naar hoe dit mechanisme zou kunnen gemanipuleerd worden
voor therapeutische doeleinden een hoge vlucht. RNAi biedt immers vele mogelijkheden voor de
behandeling van ziekten met een gekende genetische oorzaak, waaronder diverse kankers,
neurondegeneratieve aandoeningen, maar ook virale infecties. RNAi therapeutica bieden
daarenboven het voordeel messenger RNA (mRNA) als doelwit te hebben, in tegenstelling tot vele
conventionele geneesmiddelen waarvan de werking meestal beperkt is tot het eiwitniveau. Met het
ontrafelen van het menselijk genoom en onze toenemende kennis omtrent de functionaliteit van
menselijke genen, zou RNAi kunnen leiden tot een brede waaier van geneesmiddelen tegen eender
welk (genetisch) klinisch doelwit.
Intensief onderzoek heeft kleine intermediaire dsRNA fragmenten, afgekort als siRNAs (small
interfering RNAs), geïdentificeerd als de authentieke effectoren van de genetische interferentie. Deze
RNA moleculen kunnen eenvoudig op grote schaal aangemaakt worden via chemische synthese en in
het merendeel van de publicaties tot op heden omtrent RNAi therapeutica, staan siRNAs centraal.
217

APPENDIX A | samenvatting en algemene besluiten
Uiteindelijk is men nu in staat siRNAs te ontwerpen die, in functie van hun nucleotide sequentie,
mogelijks elk gentranscript kunnen onderdrukken. In iets meer dan 10 jaar sinds de ontdekking van
RNAi waren we reeds getuige van de overgang van siRNA naar de klinische testfase, hetgeen
nogmaals het belang en de mogelijkheden van RNAi onderstreept. Tabel 8.1 (zie hoofdstuk 8, pagina
208) toont een overzicht van de siRNA therapeutica die tot op heden reeds werden geëvalueerd in
klinische testen of in de toekomst aan evaluatie zullen onderworpen worden.
Deze data tonen aan dat de siRNAs in de meer gevorderde stadia van klinische evaluatie vooral lokaal
worden toegediend, geformuleerd als een 0.9% NaCl zoutoplossing. Wetenschappers zijn het er
echter over eens dat, om het therapeutisch potentieel van RNAi in de breedte te verzilveren, een
efficiënte afgifte van siRNA in het menselijk lichaam het belangrijkste obstakel blijft. Zoals
samengevat in HOOFDSTUK 1 bestaan er vele extra‐ en intracellulaire barrières voor nucleïnezuren als
geneesmiddel die overwonnen dienen te worden om hen een veilige doortocht naar het cytoplasma
van de doelwitcel te garanderen. In dit proefschrift wordt voornamelijk de focus geplaatst op de
hindernissen op cellulair niveau. Aangezien het RNAi raderwerk hoofdzakelijk is gelokaliseerd in het
cytoplasma, moeten siRNAs immers intact overheen de cellulaire membraan worden geloodst. Het
sterk negatief geladen karakter van siRNA (≥ 40 negatieve ladingen per molecule) en hun relatief
grote afmetingen (~14 kDa) laten niet zomaar toe dat deze spontaan door cellen worden
opgenomen. Onder meer om deze reden vormt het evalueren van nanoscopische dragers voor de
formulatie en afgifte van small RNAs een belangrijk segment in het onderzoek rond RNAi. Zowel
virale als niet‐virale afgifteystemen (of vectoren) werden reeds ontwikkeld voor de intracellulaire
vrijgave van RNAi effectoren, waarvan de virale vectoren ontegensprekelijk de meest efficiënte zijn.
Ondanks hun efficiëntie is het klinisch gebruik van virale vectoren gelimiteerd door tot op heden
onoverkoombare veiligheidsproblemen, hetgeen het onderzoek voedt naar niet‐virale alternatieven.
De meerderheid van wetenschappelijke publicaties beschrijft hierin het gebruik van synthetische
vetten en polymeren voor de complexatie en intracellulaire afgifte van siRNA, waarvan enkele
belangrijke voorbeelden ook in HOOFDSTUK 1 worden aangehaald.
Behalve siRNA afgifte, bespreekt dit hoofdstuk eveneens de meest voorname parameters die de
grootteorde en levensduur van het RNAi effect beïnvloeden. Genonderdrukking via siRNAs is volledig
reversibel en meer inzichten in de kinetiek van dit effect zullen cruciaal zijn voor het opstellen van
een geschikt doseringsschema. In de context van een meer duurzaam RNAi effect worden
genonderdrukking op het transcriptioneel niveau (transcriptional gene silencing of TGS), het gebruik
van mathematische modellen en combinatoriële RNAi (coRNAi) kort besproken als nieuwe
strategieën voor de verdere optimalisatie van genetische interferentie.
218

samenvatting en algemene besluiten | APPENDIX A
In het exponentieel groeiende veld van RNAi, gaat er behalve naar methodes voor verbeterde siRNA
afgifte ook aandacht uit naar de stabiliteit en afbraak van siRNA. Het is essentieel bij het gebruik van
siRNA als geneesmiddel dat de stabiliteit van de siRNA duplex en zijn vatbaarheid voor afbraak door
nucleasen in biologische media wordt nagegaan. Naakt siRNA (i.e. niet geformuleerd in virale of niet‐
virale nanoscopische dragers) wordt snel afgebroken door exo‐ en endonucleasen, hetgeen leidt tot
een zeer korte halfwaardetijd in de algemene circulatie. De literatuur bevat daarom ook vele
publicaties die chemische modificaties en formulatie strategieën bespreken met als doel de siRNA
stabiliteit en farmacokinetiek te verbeteren. Met betrekking tot het belang van de intracellulaire
siRNA stabiliteit zijn de meningen echter verdeeld. Tegenstrijdige rapporten of de siRNA integriteit al
dan niet een invloed zou hebben op het levensduur van het RNAi effect maakt de correlatie tussen
beide nog niet beslissend. Technieken om de siRNA integriteit na te gaan, volgend op cellulaire
opname, zijn daarom van belang om het intracellulair lot van siRNA te verifiëren. In HOOFDSTUK 2
wordt een methode voorgesteld, gebaseerd op fluorescentie resonantie energie transfer (FRET), om
de stabiliteit van dubbel fluorescent gemerkt siRNA te evalueren in de cellulaire micro‐omgeving. De
methode werd gevalideerd door het wegvallen van FRET te detecteren in functie van siRNA
degradatie door RNase A of door incubatie in celextracten. In deze experimenten werd de
verhouding van acceptor fluorescentie tot donor fluorescentie bij donor excitatie (FA/FD ratio)
gebruikt als parameter voor de stabiliteit van zowel enkelstrengig (ssRNA) als dubbelstrengig RNA
(dsRNA). Dergelijke technieken kunnen bruikbaar zijn om de correlatie tussen het intracellulair
gedrag van siRNA en het uiteindelijke biologische effect meer in detail te bestuderen, zoals reeds
gedeeltelijk aangetoond in de literatuur.
Zoals reeds aangehaald is het transiënte effect een mogelijk nadeel van het gebruik van siRNA als
therapeuticum, waardoor meerdere toedieningen noodzakelijk zullen zijn om het therapeutisch
effect gedurende langere tijd aan te houden. Een concept om een meer robust RNAi effect te
verkrijgen is via de gecontroleerde vrijstelling van siRNA in het cytoplasma van de doelwitcel. Het
ontwerpen van nanoscopische dragers die fungeren als een intracellulair depot, zou een verlengde
genonderdrukking kunnen verwezenlijken door de intracytoplasmatische siRNA concentratie
gedurende langere tijd in het therapeutisch venster te houden. In navolging van de brede
toepasbaarheid van hydrogelen als matrices voor gecontroleerde afgifte van geneesmiddelen en hun
veelvuldig gebruik in een biofarmaceutische context, werd in deze thesis de focus geplaatst op
dextraan hydrogelen als dragers voor siRNA. Hydrogelen kunnen gedefinieerd worden als 3D
netwerken, opgebouwd uit hydrofiele polymeren, die in staat zijn om grote hoeveelheden water of
fysiologische vloeistof op te nemen. Sinds hun introductie voor biologisch gebruik bijna 50 jaar
219

APPENDIX A | samenvatting en algemene besluiten
geleden, nemen hydrogelen een belangrijke plaats in voor allerhande biomedische en
farmaceutische toepassingen.
Hydrogelen kunnen worden aangemaakt in diverse geometrieën. HOOFDSTUK 3 beschrijft de aanmaak
van microscopische biodegradeerbare dextraan hydrogelen (i.e. microgelen) via zowel een w/w
emulsie in polyethyleenglycol als een w/o emulsie in minerale olie. Voorgevormde microgelen,
bestaande uit dextraan hydroxyethyl methacrylaat (dex‐HEMA) en verder gefunctionaliseerd met
kationische methacrylaat monomeren (2‐(dimethylamino)ethyl methacrylaat of DMAEMA), bleken in
staat siRNA op te nemen via elektrostatische aantrekking. De HEMA groepen zijn gekoppeld aan
dextraan via een onstabiele ester binding. Hydrolyse van deze ester functie, klieft de verbinding
tussen de dextraan ketens, waardoor het gelnetwerk langzaam uit elkaar valt en het gecomplexeerde
siRNA wordt vrijgesteld. Aangezien de degradatie snelheid van dex‐HEMA microgelen bepaald wordt
door de initiële dichtheid aan crosslinks, kan de vrijstellingskinetiek van siRNA gecontroleerd worden
door de dichtheid van het hydrogel netwerk te laten variëren.
Een primaire vereiste om de efficiëntie van het voorgestelde siRNA afgifte systeem te bepalen, is het
vertalen ervan naar nanoscopische dimensies om cellulaire internalisatie mogelijk te maken.
HOOFDSTUK 4 presenteert een overzicht van enkele opvallende applicaties waarin nanoscopische
hydrogelen (nanogelen) gebruikt worden als geneesmiddeldragers voor de gecontroleerde vrijstelling
van diverse therapeutica. Ondanks het toenemend aantal publicaties omtrent nanogelen for
farmaceutische toepassingen het voorbije decennium, zijn de rapporten die nanogelen beschrijven
voor siRNA afgifte tot op heden nog op één hand te tellen. De aanmaak van positief geladen en
hydrolyseerbare dex‐HEMA nanogelen via een inverse mini‐emulsie fotopolymerisatie staat
beschreven in HOOFDSTUK 5. In vergelijking met hoofstuk 3 werd ditmaal een methacrylaat
monomeer met een kwaternaire stikstofgroep (2‐(trimethylammonium)ethyl methacrylaat of
TMAEMA) gekozen voor copolymerisatie met dex‐HEMA. Dankzij de incorporatie van positieve
ladingen in het nanogel netwerk, hebben deze een grote ladingscapaciteit voor siRNA. Confocale
microscopie en flow cytometrie leverden het bewijs dat grote hoeveelheden siRNA beladen
nanogelen efficiënt konden opgenomen worden door humane hepatoma cellen (Huh‐7) onder
behoud van de celviabiliteit. Het wordt algemeen aangenomen dat voor het merendeel van de niet‐
virale dragers de cellulaire internalisatie gebeurt via endocytose, waarbij een grote fractie van de
opgenomen partikels uiteindelijk eindigt in verzuurde (endo)lysosomen. Dit bleek ook voor onze
kationische nanogelen het geval. Ongeacht deze nadelige intracellulaire eindbestemming, toonden
onze experimenten toch een substantiële en sequentie‐specifieke onderdrukking aan van het
luciferase gen na transfectie met afbreekbare dex‐HEMA nanogelen. Dit is een onrechtstreekse
aanwijzing dat een deel van het gecomplexeerd siRNA het cytoplasma bereikt alwaar het opgenomen
220

samenvatting en algemene besluiten | APPENDIX A
wordt in het RNAi raderwerk. Bovendien bleek het RNAi effect bijna afwezig wanneer de transfectie
werd uitgevoerd met stabiele (niet afbreekbaar door hydrolyse bij fysiologische pH) dextraan
nanogelen, hetgeen doet vermoeden dat dex‐HEMA hydrolyse een invloed heeft op de efficiëntie
van siRNA afgifte. Anderzijds kon het genonderdrukkende effect markant verbeterd worden door het
toepassen van gevalideerde endosomolytische hulpmiddelen, zoals fotochemische internalisatie
(PCI) en de co‐complexatie van siRNA beladen nanogelen met een fusogeen peptide afgeleid van de
hemagluttinine HA2 subeenheid van het influenza virus. Deze data leveren het bewijs aan dat de
endolysosomale accumulatie van de siRNA nanogelen nefast is voor een optimale siRNA afgifte in het
cytoplasma van de doelwitcel.
Terwijl de omvang van de genonderdrukking een goede indicator kan zijn voor het succes van een
siRNA behandeling, is de kinetiek van het RNAi effect hiervoor misschien beter geschikt. De tijd dat
de siRNA behandeling de intracellulaire concentratie van het doelwit proteïne beneden de drempel
kan houden die nodig is om het gewenste fenotype te bekomen, zal in belangrijke mate de
doseringsfrequentie bepalen in klinische therapie. In HOOFDSTUK 6 wordt o.a. om deze reden de
kinetiek van genonderdrukking nagegaan voor dex‐HEMA‐co‐TMAEMA nanogelen met variërende
crosslink densiteit. De transfectie experimenten toonden aan dat de kinetiek van genonderdrukking
grotendeels onafhankelijk is van de gebruikte nanogel formulatie. De grootteorde van het
onderdrukkend effect daarentegen bleek wel te verschillen met in het algemeen een kleiner effect
voor sterker gecrosslinkte nanogelen. Het lijkt aannemelijk te veronderstellen dat het maximaal
biologisch effect bepaald wordt door de hoeveelheid siRNA dat initieel het cytoplasma bereikt terwijl
het herstel van de genexpressie naar steady‐state gedicteerd wordt door sequentiële celdelingen die
de intracellulaire pool van actief siRNA gestaag reduceren. Daarbij werd eveneens aangetoond dat de
toepassing van PCI, zelfs 48 h post‐transfectie, het RNAi effect in belangrijke mate kon verbeteren
(tot 25% bijkomende genonderdrukking) en verlengen. Dit resultaat geeft aan dat PCI in staat is om
een fractie van de siRNA beladen nanogelen, die in een vesiculaire micro‐omgeving gestrand waren,
vrij te stellen. Dit vormt een mogelijke strategie om de therapeutische respons te verlengen in het
geval dat de endosomale barriere limiterend is, alhoewel deze strategie verdere optimalisatie vereist.
Uit de vorige hoofdstukken blijkt duidelijk dat om een intracellulaire gecontroleerde vrijstelling van
siRNA mogelijk te maken en de implicaties na te gaan in de context van een meer duurzame
genonderdrukking, een verbeterde overgang van siRNA beladen nanogelen van het endosomale
lumen naar het cytoplasma cruciaal is. In navolging van vernieuwend onderzoek, uitgevoerd door
Van Thienen et al. in 2005 bediscussieert HOOFDSTUK 7 het ontwerp van nanogelen omgeven door
een vetlaagje (lipid‐enveloped nanogels or LENs). De multifunctionele lipide mantel wordt in dit
concept belast met de opdracht om de belangrijkste extracellulaire barrières te overwinnen (i.e.
221

APPENDIX A | samenvatting en algemene besluiten
222
stabiliteit in de bloedstroom, vermijden van opsonizatie en een verbeterde mobiliteit in de
extracellulaire matrix). Bovendien anticiperen we op basis van literatuurgegevens een positieve
invloed op de accumulatie van intacte nanogelen in het cytoplasma, die daar als siRNA depot kunnen
fungeren. Hiertoe kan beroep gedaan worden op fusogene lipiden, die de eigenschap hebben te
versmelten met de endosomale membraan. Deze vorm van hybride nanopartikels werd aangemaakt
door liposomen in te schakelen als nanoreactor voor de selectieve UV polymerisatie van dex‐HEMA
in hun waterig lumen. Het opladen met siRNA gebeurt door passieve hydratatie van een
voorgevormde lipide film waarbij een encapsulatie efficiëntie werd bereikt van 30%. Zowel extrusie
als ultrasonicatie werden succesvol ingezet voor de aanmaak van LENs, zoals aangetoond via
dynamische lichtverstrooiing. In contrast met de vele liposomale formulaties voor RNAi die meestal
een kationisch lipide bevatten voor complexatie met het anionisch siRNA, werd in dit hoofdstuk
geopteerd voor een equimolaire mix van neutrale lipiden. Ondanks hun ladingsneutraliteit, kon via
flow cytometrie toch een duidelijke dosis afhankelijke Huh‐7 cellulaire opname waargenomen
worden in preliminaire experimenten. Naar de toekomst toe zullen LENs experimenten zich
hoofdzakelijk concentreren op de verdere optimalisatie van siRNA encapsulatie en het evalueren van
hun RNAi potentieel.
ALGEMENE BESLUITEN
Samenvattend beschrijft dit proefschrift een nieuwe methode, gebaseerd op FRET, met als
voornaamste doel de in situ evaluatie van siRNA stabiliteit in levende cellen. Deze methode moet het
mogelijk maken om bijkomende informatie te verkrijgen omtrent de correlatie tussen de
intracellulaire siRNA integriteit en het uiteindelijke RNAi effect. Daarnaast werd een nieuw concept
voorgesteld voor de intracellulaire gecontroleerde vrijstelling van siRNA. Hiertoe werd siRNA
gecomplexeerd in biodegradeerbare nanoscopische hydrogelen (i.e. nanogelen). Significante
onderdrukking van een reportergen vormde een onrechtstreekse aanwijzing dat deze nanogelen in
staat zijn actief siRNA in het cytoplasma van de doelwitcel af te leveren. Het ontsnappen van
geïnternaliseerde nanogelen uit de endosomen werd echter geïdentificeerd als de voornaamste
cellulaire barrière, hetgeen het RNAi potentieel gedeeltelijk beperkt. Tegelijkertijd biedt deze
observatie nieuwe opportuniteiten om het nanogel concept verder te verfijnen. Het is aannemelijk
dat afhankelijk van de te behandelen pathologie en de toedieningswijze, een grote verscheidenheid
aan siRNA dragers nodig zal zijn wanneer siRNAs de stap zetten naar nieuwe klinische toepassingen.
Het is best mogelijk dat ook nanogelen hierin hun plaats zullen opeisen.

curriculum vitae | APPENDIX B
223
APPENDIX B | CURRICULUM VITAE

APPENDIX B | curriculum vitae
224

curriculum vitae | APPENDIX B
B PERSONALIA
Name RAEMDONCK
First name KOEN
Nationality Belgian
Place and date of birth Dendermonde, 28‐08‐1981
Marital status Married
Private address Nieuwbrugkaai 70
9000 Gent
Telephone + 32 (0)485 28 08 12
Professional address Lab of General Biochemistry and Physical Pharmacy
Faculty of Pharmaceutical Sciences – Ghent University
Harelbekestraat 72
9000 Ghent
Telephone +32 (0)9 264 80 95
Fax +32 (0)9 264 81 89
E‐mail: [email protected]
URL http://www.biofys.ugent.be
225

APPENDIX B | curriculum vitae
LANGUAGES
Dutch mother tongue
English fluent
French good
DEGREES
June, 2004 pharmacist with great distinction
Faculty of Pharmaceutical Sciences, Ghent University
INTERNATIONAL PEER REVIEWED PUBLICATIONS
Raemdonck K., Remaut K., Lucas B., Sanders N.N., Demeester J., and De Smedt S.C., In situ analysis of
single‐stranded and duplex siRNA integrity in living cells. Biochemistry 2006, 45: 10614‐10623.
Remaut K., Lucas B., Raemdonck K., Braeckmans K., Demeester J., and De Smedt S.C., Can we better
understand the intracellular behavior of DNA nanoparticles by fluorescence correlation
spectroscopy?. Journal of Controlled Release 2007, 121: 49‐63.
Remaut K., Lucas B., Raemdonck K., Braeckmans K., Demeester J., and De Smedt S.C., Protection of
oligonucleotides against enzymatic degradation by pegylated and nonpegylated branched
polyethyleneimine. Biomacromolecules 2007, 8: 1333‐1340.
Van Thienen T.G., Raemdonck K., Demeester J., and De Smedt S.C., Protein release from
biodegradable dextran nanogels. Langmuir 2007, 23: 9794‐9801.
Buyens K., Lucas B., Raemdonck K., Braeckmans K., Vercammen J., Hendrix J., Engelborghs Y., De
Smedt S.C., and Sanders N.N., A fast and sensitive method for measuring the integrity of siRNA‐
carrier complexes in full human serum. Journal of Controlled Release 2008, 126: 67‐76.
226

curriculum vitae | APPENDIX B
Raemdonck K., Van Thienen T.G., Vandenbroucke R.E., Sanders N.N., Demeester J., and De Smedt
S.C., Dextran microgels for time‐controlled delivery of siRNA. Advanced Functional Materials 2008,
18: 993‐1001.
Raemdonck K., Vandenbroucke R.E., Demeester J., Sanders N.N., and De Smedt S.C., Maintaining the
silence: reflections on long‐term RNAi. Drug Discovery Today 2008, 13: 917‐931.
Raemdonck K., Naeye B., Buyens K., Vandenbroucke R.E., Høgset A., Demeester J., De Smedt S.C.,
Biodegradable dextran nanogels for RNAi: focusing on endosomal escape and intracellular siRNA
delivery, Advanced Functional Materials 2009, 19: 1406‐1415. Press release (2009‐05‐28) on
www.pcibiotech.com.
Raemdonck K., Demeester J., De Smedt S.C., Advanced nanogel engineering for drug delivery, Soft
Matter 2009, 5: 707‐715. Invited.
Naeye B., Raemdonck K., Demeester J., and De Smedt S.C., PEGylation strategies for cationic dextran
nanogels as siRNA delivery agents. Manuscript in preparation.
CONFERENCE PROCEEDINGS
Raemdonck K., Remaut K., Lucas B., Sanders N.N., Demeester J., De Smedt S.C., Real‐time Monitoring
of Single‐Stranded and Duplex Short Interfering RNA Integrity inside living cells. Proceedings of the 9th
European Symposium on Controlled Drug Delivery 2006, 241‐243.
Raemdonck K., Remaut K., Lucas B., Sanders N.N., Demeester J., De Smedt S.C., Real‐time Monitoring
of Single‐Stranded and Duplex Short Interfering RNA Integrity inside living cells. Molecular Therapy
2006, 13: S273.
Raemdonck K., Demeester J., De Smedt S.C., Intracellular time‐controlled delivery of siRNA from
cationic nanogel depots. Molecular Therapy 2008, 16: S245.
227

APPENDIX B | curriculum vitae
ORAL PRESENTATIONS (presenting author)
Raemdonck K., Remaut K., Lucas B., Sanders N.N., Demeester J., De Smedt S.C., In situ analysis of
single‐stranded and duplex siRNA integrity in living cells. Spring Meeting of the Belgian‐Dutch
Biopharmaceutical Society, Janssen Pharmaceutica, Beerse (Belgium), May 22th 2006.
De Smedt S.C., Lucas B., Remaut K., Raemdonck K., Braeckmans K., Sanders N.N., Demeester J., Can
we better understand the intracellular behaviour of nucleic acid containing nanoparticles by
fluorescence correlation spectroscopy? Nanomedicine and drug delivery symposium, Omaha,
Nebraska (USA), October 8th‐10th 2006.
Raemdonck K., Demeester J., De Smedt S.C., Dextran micro‐and nanogels for controlled release of
siRNA. Pre‐satellite Meeting of the Pharmaceutical Sciences World Congres, Amsterdam (The
Netherlands), April 20th‐21st 2007.
Naeye B., Raemdonck K., Demeester J. and De Smedt S.C., Optimization of biodegradable dextran
nanogels for controlled siRNA delivery. MediTrans 2nd annual meeting, Rehovot (Israel), March 26th‐
29th 2009.
Raemdonck K., Naeye B., Demeester J., De Smedt S.C., Evaluation of a dextran nanogel concept for
intracellular siRNA delivery. EuroNanoMedicine, Bled (Slovenia), September 28th‐30th 2009.
POSTER PRESENTATIONS (presenting author)
Raemdonck K., Remaut K., De Smedt S.C., Demeester J., Studying the degradation of single‐ and
double‐stranded short interfering RNA in living cells by means of FCS‐FRET
• 1st Young Scientist Day of the Belgian Biophysical Society, Ghent (Belgium), May 19th 2005.
• Biopharmacy Day, Spring Meeting of the Belgian‐Dutch Biopharmaceutical Society, Ghent
(Belgium), May 20th 2005.
228

curriculum vitae | APPENDIX B
Raemdonck K., Remaut K., De Smedt S.C., Demeester J., Real‐time monitoring of single‐stranded and
uplex short interfering RNA integrity inside living cells
• RNAi Europe, Amsterdam (The Netherlands), September 28th‐29th 2005.
• Biopharmacy Day, Autumn Meeting of the Belgian‐Dutch Biopharmaceutical Society, Utrecht
(The Netherlands), December 2nd 2005.
Raemdonck K., Remaut K., Lucas B., Sanders N.N., Demeester J., De Smedt S.C.*, Real‐time
Monitoring of Single‐Stranded and Duplex Short Interfering RNA Integrity inside Living Cells
• 9th European Symposium on Controlled Drug Delivery, Noordwijk aan Zee (The Netherlands),
April 5th‐7th 2006.
• American Society of Gene Therapy annual meeting, Baltimore, MD (USA) May 31st‐June 4th
2006.
• 33rd Annual meeting of the Controlled Release Society, Vienna (Austria) July 22nd‐July 26th
2006 (*presenting author).
• 13th International symposium on recent advances in drug delivery systems, Salt Lake City, UT
(USA), February 26th‐28th 2007.
Raemdonck K., Demeester J., and De Smedt S.C., Towards dextran based nanogels for the
intracellular delivery of siRNA
• 13th International symposium on recent advances in drug delivery systems, Salt Lake City, UT
(USA), February 26th‐28th 2007.
Raemdonck K., Demeester J., and De Smedt S.C.*, Cytosolic nanogel depots for time‐controlled
siRNA delivery
• 11th annual Meeting of the American Society of Gene Therapy (ASGT), Boston, MA (USA) May
28th‐June 1st 2008
• 2nd International Symposium on Cellular Delivery of Therapeutic Macromolecules, Cardiff
University (Wales), June 22nd‐June 25th 2008
• RNAi Europe, Stockholm (Sweden), September 16th‐18th 2008
229

APPENDIX B | curriculum vitae
230
• Right Symposium “RNA interference technology as Human Therapeutic Tool”, Brussels
(Belgium), November 3rd‐5th 2008 (* presenting author)
Raemdonck K., Demeester J., and De Smedt S.C., Biodegradable dextran nanogels for RNAi
• 14th international symposium on recent advances in drug delivery systems, Salt Lake City, UT
(USA), February 15th‐18th 2009.
Naeye B., Raemdonck K., Demeester J., and De Smedt S.C., Pegylation of biodegradable dextran
nanogels for controlled siRNA release
• Nanomedicine, Sant Feliu de Guixols (Spain), September 20th‐24th 2008
Naeye B., Raemdonck K., Demeester J. and De Smedt S.C., Optimization of biodegradable dextran
nanogels for controlled siRNA delivery
• MediTrans 2nd annual meeting, Rehovot (Israel), March 26th‐29th 2009.
SCIENTIFIC COURSES
Training course ‐ Particle characterization & Innovation (organized by Analis), Sint‐Martens‐Latem
(Belgium), September 13th, 2007.
Flow cytometry course – learning and knowledge, BD Biosciences, Erembodegem (Belgium), February
10th, 2009.
Biogazelle course on qPCR experiment design and data‐analysis, Vlerick Business Center, Ghent
(Belgium), March 5th‐6th, 2009.

231

232

| DANKWOORD
DANKWOORD | ACKNOWLEDGMENT
Ik ga proberen het kort te houden. Proberen dus…
Eerst en vooral wil ik even melden dat ik een dankwoord beschouw als een nawoord, dat je hoort te
schrijven met een goed en volledig overzicht van (en inzicht in) de voorbije jaren en wie daar
allemaal een voorname rol in heeft gespeeld. Vandaar dus ook de keuze om het dankwoord
desgevallend een plaats te geven op hetgeen uiteindelijk pagina 233 is geworden. Desalniettemin
moet ik iedereen gelijk geven die in zijn of haar dankwoord vermeldt dat het niet vanzelfsprekend is
een dergelijk stukje tekst op papier te zetten. “Schrijven is schrappen”, vertrouwde Stefaan mij al
allitererend toe in de periode dat ik op het FFW het schrijven van mijn masterthesis aanvatte, en
deze stelling geldt ongetwijfeld ook voor een dankwoord, zo weet ik nu.
Een eerste woord van dank ben ik uiteraard verschuldigd aan Stefaan & Jo, promotoren van het
proefschrift dat u op dit punt volledig gelezen zou moeten hebben. Een wetenschappelijk labo steunt
grotendeels op een goed uitgeruste (infra)structuur die gevoed wordt vanuit gezonde financiële
fundamenten. Als directeur van het labo vervult Jo hierin een belangrijke en creatieve rol, waarvoor
onzer appreciatie! Deeltijds wetenschappelijk bezieler en deeltijds filosoof vat ongeveer het profiel
van Stefaan samen. Bedankt voor de professionele steun, motivatie en de extra dosis zelfvertrouwen
wanneer dat nodig was. Eveneens bedankt voor de vele anekdotes waarin je zelf een hoofdrol speelt
en die iedereen van ons nog regelmatig met plezier doorvertelt (carport en dergelijke, u weet het wel
;)). Sinds kort horen in deze paragraaf ook Niek en Kevin Br. thuis. Ik wens jullie beiden nog veel
succes met het verder uitbouwen van een wetenschappelijke carrière (dit geldt voor iedereen
trouwens…).
De laatste jaren is het labo exponentieel gegroeid. Dit vertaalde zich in een grote ‘voorraad’ aan
collega’s waaruit kon geput worden in geval van wetenschappelijke nood. Een welgekomen
extralegaal voordeel aangezien een doctoraat vervolmaken ook grotendeels steunt op advies van en
discussies met scientific peers. Bedankt allemaal (Bart L., Farzaneh, Roos, Lies, Tinneke, Barbara,
Stefaan D., Bruno DG, Joanna, Katrien, Ine, Sofie T., Elien, Marie‐Luce, Geertrui, Oliwia, Chaobo,
Broes, Hendrik, Dries, Nathalie, Kevin Bu. en Bart G.)! Er arriveerde zowaar een nanogel‐partner‐in‐
science (NPiS) onder de vorm van Broes. Met ons tweeën gaan we nu resoluut voor die Nature
publicatie, zeker nu we uit goede bron hebben vernomen dat die garant staat voor een fles
champagne. Het labo Algemene Biochemie en Fysische Farmacie is op meerdere vlakken uniek. Naar
mijn bescheiden mening vormen we een hechte groep gemotiveerde onderzoekers (beetje reclame
maken mag wel) die op en naast het werk zeer goed met elkaar kunnen opschieten. Moge het GRGN
Social Committee nog vele activiteiten op de kalender plaatsen!
233

DANKWOORD |
234
In ieder dankwoord wordt (en dit is meer dan terecht!) ook een paragraaf gereserveerd voor
Katharine en Bruno VdB, de beste secretariaatstandem die een onderzoekslabo zich kan wensen. Een
kort literatuuronderzoek in menig doctoraatsdankwoord leverde lyrische lofzangen op met als
voornaamste kernwoorden ‘werkijver’, ‘onmisbaar’, ‘glimlach’ en ‘grapjes’. Ik sluit mij hier volledig bij
aan. Misschien nog even opmerken dat Bruno VdB zich bovendien nuttig maakte als mijn
persoonlijke ‘ping‐pong’ coach. Volgend jaar gaan we voor de sportieve doorbraak!
Naast het wetenschappelijk werk (ping‐pongen werd onder de middag gepland red.) was er nog tijd
voor ontspanning na de uren en in de weekends onder de vorm van tennis, mountainbike, menig
cafébezoek, BBQ, wijndegustaties, festivals, weekendjes Ardennen, limericks en vooral veel gezellige
babbels! Special thanks to Bart W., Klaar, Tim, Bart L., Jo, Ine, Philippe, Sofie S., Olivier, Patricia,
Katrien, Björn, Birger, Line, Wim, Eva, Lieven, Julie, Griet, Nathalie, Steven, Annelies, Ruben,
Kathleen, Bert… .
Tot slot een overweldigende merci aan ma en pa, my brother and sister‐in‐law, ons meter en mijn
Meetjeslandse schoonfamilie voor vanalles en eigenlijk teveel om op te noemen (wij weten het!).
Details kan u lezen in mijn autobiografie die misschien ooit zal verschijnen. Iemand die hierin ook een
niet te onderschatten rol zal spelen is m’n vraaiken, Evelien, mijn steun en toeverlaat, die nu
ongetwijfeld ook tevreden zal zijn dat het doctoraat achter de rug is… ;). Dikke kus.
Oh ja, ook een woordje van dank aan iedereen die ik ben vergeten…
Gent, oktober 2009
Koen Raemdonck

| DANKWOORD
A PhD is a real way of life
That takes you beyond nine to five
But the effort’s in vain
If you try to explain
It’s ‘no pain, no gain’ to your wife
:)
(A PhD limerick© by Koen Raemdonck)
235

DANKWOORD |
236































