Embed Size (px)
Citation preview

Applied Catalysis A: General 234 (2002) 191–205
Low temperature use of SiC-supported NiS2-based catalystsfor selective H2S oxidation
Role of SiC surface heterogeneity and nature of the active phase
Nicolas Kellera,∗, Cuong Pham-Huua, Claude Estournèsb, Marc J. Ledouxaa Laboratoire des Matériaux, Surfaces et Procédés pour la Catalyse, UMR 7515 du CNRS, ECPM,
Université Louis Pasteur, 25 rue Becquerel, BP08, 67087 Strasbourg Cedex 2, Franceb Groupe des Matériaux Inorganiques, IPCMS-ECPM-ULP, UMR 7504 du CNRS, 23 rue du Loess, 67037 Strasbourg Cedex, France
Received 4 February 2002; received in revised form 13 March 2002; accepted 27 March 2002
Abstract
High activity for the direct oxidation of H2S into elemental sulfur at low reaction temperature (40–60◦C) on medium surfacearea silicon carbide-supported nickel sulfide (NiS2/SiC) catalyst was attributed to the formation of a highly active superficialnickel oxysulfide. The hypothesis of the superficial formation of either nickel oxide or nickel sulfate was rejected. Thesuperiority of the SiC support in terms of performance as compared to silica, high surface area alumina and activated charcoalwas evidenced. The high stability of the SiC-supported catalyst as a function of time and solid sulfur loading in the presenceof water was explained by a peculiar mode of sulfur deposition, involving the role of water and the hydrophilic/hydrophobicduality of the SiC support surface. Water acts as a conveyor belt, continuously cleaning the active phase particles, locatedon the hydrophilic oxygen-containing surfaces of the support. Hydrophobic pure SiC, located outside the mesoporosity andexempt of active phase, remains as an available surface for the storage of high amounts of solid sulfur.© 2002 Elsevier Science B.V. All rights reserved.
Keywords:Silicon carbide; Nickel sulfide; Hydrophilic; Hydrophobic; Hydrogen sulfide oxidation; Sulfur formation
1. Introduction
Removal of hydrogen sulfide from waste gasescoming from petrochemical refineries or from naturalgas processing is an important aspect of air pollutioncontrol, due to the high toxicity of H2S. The generaltrend is to selectively transform H2S into elementalsulfur which is a valuable product, by the modifiedClaus process[1,2]. Thermodynamic limitations ofthe Claus equilibrium reaction led to the develop-
∗ Corresponding author. Tel.:+33-3-90-24-26-75;fax: +33-3-90-24-26-74.E-mail address:[email protected] (N. Keller).
ment of new processes to deal with the Claus tail-gas,based on the direct oxidation of H2S by oxygen andH2S absorption/recycling technologies, in order tomeet ever newer and stricter legislation requirements.All these processes were summarized during the lastdecade in a series of reviews published in the litera-ture [3–6]. Up to now, two main catalytic processesdealing with the selective oxidation of H2S by oxygeninto elemental sulfur have been developed: the Doxo-sulfreen (Elf–Lurgi) process working around 100◦Cin a discontinuous mode of reaction/regenerationand the Superclaus (Comprimo) process workingcontinuously over the dew point of sulfur. Liquidphase processes involving homogeneous catalysts
0926-860X/02/$ – see front matter © 2002 Elsevier Science B.V. All rights reserved.PII: S0926-860X(02)00226-0

192 N. Keller et al. / Applied Catalysis A: General 234 (2002) 191–205
were also studied at lower temperatures and usedvanadium- and iron-based catalysts, respectively,in the case of the Stretford and Lo-Cat processes[3].
Trovarelli and co-workers[7,8] have reported theuse of high surface area activated charcoal (AC) as cat-alyst for deep oxidation of H2S at room temperature.Total selectivity into sulfur was obtained, according tothe literature which reported that SO2 was not formedat such a low reaction temperature[9–12]. Deactiva-tion rapidly occurred as a function of the sulfur depo-sition, due to pore plugging of the catalyst. In addition,regeneration in order to remove the sulfur depositfrom the catalyst surface led to a slow decrease in thesulfur storage capacity and in the initial desulfuriza-tion activity [7,8]. The loss of catalyst performanceas a function of the reaction/regeneration cycles wasattributed to an irreversible accumulation of adsorbedsolid sulfur inside the micropores of the support. Ithas been recently reported by Ledoux and co-workers[13–16] that selective oxidation of H2S into elemen-tal sulfur could be efficiently performed on a siliconcarbide-supported nickel sulfide (NiS2/SiC) catalystat low reaction temperatures (40–60◦C). The use of anon-microporous support at a low temperature allowedto obtain a total selectivity into sulfur. The forma-tion on stream of a new and very active nickel-basedphase and a peculiar mode of sulfur deposition wereboth suggested to explain how a very stable H2S con-version of 100%, even at high sulfur loadings on thecatalyst (≥80 wt.%) was obtained. Such results arethe highest results which have ever been reported in thefield. The macroporosity of the SiC support played animportant role and contributed to the high stability ofthe catalyst, by allowing the storage of high amountsof sulfur inside the material. The determinant pres-ence of water in the feed was evidenced, as the sameNiS2/SiC catalyst rapidly deactivated as a function oftime on stream in dry reaction conditions with onlya small sulfur loading of a few percent on its surface[16,17].
The aim of the present article is to provide fur-ther insight into the mode of sulfur deposition andthe nature of the active phase for the selective oxi-dation of H2S into elemental sulfur at low tempera-ture. The influence of the type of support and of thesupported nickel-based phase on the desulfurizationactivity and the catalyst stability at 60◦C was, there-
fore, studied. The nickel sulfide catalyst was supportedon SiC, SiC oxidized in air at 1000◦C, alumina andAC in order to show the role of the sulfur depositionmode on the desulfurization stability of the catalysts.A model of the sulfur deposition mode involving therole of water and the dual nature of the support sur-face will be presented in order to explain the highperformances obtained on the SiC-supported catalystas compared to other supports. The possible natureof the active phase stabilized on stream will also bediscussed.
2. Experimental
2.1. Supports and catalysts
Medium surface area SiC (grains 0.4–1 mm) usedas support was synthesized at the CRV-Pechineyaccording to the shape memory synthesis developedby Ledoux and co-workers, involving the gas–solidreaction between SiO vapor and solid carbon underdynamic vacuum around 1200–1300◦C [18–20]. Acalcination was then performed at 700◦C for 3 h inorder to stabilize the textural characteristics and toburn off the remaining unreacted carbon. OxidizedSiC was prepared by subjecting the fresh supportto an oxidative treatment in air at 1000◦C for 2 hin order to cover the SiC surface with a thick layerof silica and to keep the same overall morphol-ogy. High surface area alumina (�-Al2O3, KetjenCK-300B, Akzo) and high surface area AC (Calgon,puriss grade), used without any pre-treatment, wereground and subsequently sieved in order to obtain thesame gross morphology as the previously describedsupports.
The nickel-based catalysts were prepared by in-cipient wetness impregnation of the correspondingsupports with an aqueous solution of Ni(NO3)2·H2O(Merck). The dried catalysts (at 120◦C for 14 h)were then calcined at 350◦C for 2 h in order to de-compose the nickel salt into its oxide form. Thenickel oxide-based catalysts were then sulfided insitu by reaction with a H2S/He flow at 300◦C for2 h to form the corresponding NiS2 phase. Thenickel concentration of the supported catalysts, mea-sured by atomic absorption spectroscopy (AAS) was5 ± 0.2 wt.%.

N. Keller et al. / Applied Catalysis A: General 234 (2002) 191–205 193
2.2. Characterization techniques
The metal loading was analyzed by AAS performedat the Service Central d’Analyse of the CNRS, Ver-naison, France.
Structural characterization of the samples was doneby powder X-ray diffraction (XRD) measurements,carried out with a Siemens diffractometer modelD-5000, using a Cu K� radiation and operating at40 kV and 20 mA. The measurements were madeusing a long time scan (10 s) and a small step scan(2θ = 0.02◦). The nature of the crystalline phase inthe sample was checked using the data base of theJoint Committee on Powder Diffraction Standards(JCPDS).
The pore size and surface area measurements wereperformed on a Coulter SA-3100 porosimeter usingN2 as adsorbant. Before each measurement the sam-ple was transferred into the BET cell via a glove-boxunder dry nitrogen. The cell was equipped with agreaseless valve in order to avoid air exposure of thesample during the transfer to the porosimeter. Beforethe N2 adsorption, samples were not heated under dy-namic vacuum in order to avoid any modification of thedifferent phases on the surface.SBET is the surface areaof the sample calculated from the nitrogen isothermusing the BET method. The desorption branch of thenitrogen isotherm allowed to obtain the pore size dis-tribution following the Barrett et al. method[21] andthe surface area of all the pores, except micropores(SBJH). The micropore surface area and volume werecalculated using thet-plot method developed by DeBoer [22]. The t-plot consists of the analysis of thevl–t plot curve, wherevl is the volume of nitrogen ad-sorbed as liquid at a given pressureP/P0 by the BETsurface andt the statistical thickness obtained by di-viding the volume of nitrogen adsorbed as liquid at agiven pressureP/P0 by the BET surface. A more de-tailed study has been published by Mikhail et al.[23]concerning the correctness of the different parametersused in this method.
The morphology of the catalyst was observed byscanning electron microscopy (SEM) using a Jeol mi-croscope model JMS-840 operating at 20 mA with anaccelerating voltage of 20 kV. Before observation thesamples were coated with a thin layer of carbon in or-der to avoid a charge effect phenomenon. Great carewas taken during SEM observations in order to avoid
any modifications of the sulfur deposit from the elec-tron beam under vacuum.
2.3. Selective oxidation of H2S and micropilot
Selective oxidation of H2S was carried out in theapparatus working at atmospheric pressure alreadydescribed in detail in previous work[17,24]. The cat-alysts were tested in a fixed-bed mode, with a reac-tant feed containing 2000 ppm of H2S, 3200 ppm ofO2, H2O at various concentrations and balance He,corresponding to a O2/H2S molar ratio of 1.6 and gashourly space velocities (GHSVs) ranged between 500and 1000 h−1. The steam was provided by a saturatorkept at the required temperature allowing variation ofthe water partial pressure from 0 to 30 vol.%. In orderto avoid condensation, all the lines were maintainedat 120◦C with heating tapes. The analysis of the in-let and the outlet gas was performed online using aVarian CX-3400 gas chromatograph equipped with aChrompack JSQ capillary column allowing the sepa-ration of O2, H2S, H2O and SO2, a catharometer de-tector and a calibrated six port loop (500�l). SO2 wasalso measured by means of a specific detector (Drägermodel 81-01531) allowing the detection of SO2 in arange of concentrations between 10 and 5000 ppm andpreviously calibrated using a known amount of SO2.
Before the reaction the reactor was purged withHe at room temperature until no trace of oxygen wasdetected by gas chromatography at the exit of the reac-tor, then the dry helium flow was replaced by the onecontaining steam. The catalyst was heated from roomtemperature to the reaction temperature (40–60◦C)with a heating rate of 1◦C min−1 and the wet heliumflow was replaced by the reactant flow. At such lowtemperatures, a large fraction of the steam condensedat the head of the reactor and trickled down throughthe catalyst. Sulfur mass balance was calculated bothfrom the continuous chromatographic analysis and byweighing, at the end of the test, the elemental sulfurdeposited on the catalyst during the reaction becausethe sulfur deposit represented the sole increase in thecatalyst weight. It has already been reported that to-tally efficient regeneration of the used NiS2/SiC cata-lyst could be performed under flowing helium at about250–300◦C for 2 h [14,15,17]. In this regenerativeprocess, the elemental sulfur deposited on the catalystduring the desulfurization reaction was evaporated and

194 N. Keller et al. / Applied Catalysis A: General 234 (2002) 191–205
condensed in the cold part of the reactor. Confirma-tion of the sulfur mass balance was thus obtained byweighing the regenerated catalyst.
3. Results and discussion
3.1. Support and catalyst characteristics
The XRD pattern of the SiC only exhibited diffrac-tion lines corresponding to the carbide, crystallized ina cubic system (Fig. 1). Traces of other compounds,such as Si or SiO2 were not detected, meaning thatsuch species if present were only in an amorphousform or in very small amounts. The SiC has a surfacearea of 29 m2 g−1 with no microporosity and moredetailed characterizations are reported in previouslypublished work[20,25,26]. Compared to the freshSiC, the XRD pattern of the oxidized SiC showed aslight increase in the background level between 2θ =18 and 25◦ which could be related to the formationof a badly crystallized silica phase, thick enough tobegin to be observed by the XRD technique (Fig. 1).It is well known that SiC is not stable in air, in a ther-modynamic sense, because it reacts with oxygen witha negative change in free energy and the SiO2 coatingacts as a diffusion barrier for SiC oxidation. The super-ficial oxidation of the SiC has already been reported
Fig. 1. XRD pattern of the SiC support. The SiO2–SiC support was formed by oxidizing the SiC in air at 1000◦C for 2 h. The SiO2formed on the SiC surface could be evidenced in inset by the presence of a small broad peak at low angle.
by Moene and co-workers according to the equationSiC(s)+ (3/2)O2(g) → SiO2(s)+CO(g) [27,28]andthermogravimetric analysis of SiC showed a smallincrease in weight starting over 800◦C [25,26]. Thesurface area of the material (SiO2–SiC) after oxida-tion was 18 m2 g−1 without any microporosity. Thisdecrease in surface area was attributed to a slightsintering of the material, due to surface diffusion phe-nomena. For the alumina, after grinding and sieving,the surface area was 223 m2 g−1 (about 10 m2 g−1 ofmicropores), whereas the AC had a mainly microp-orous surface area of about 1490 m2 g−1.
Table 1summarizes the specific surface areas of thefresh supports and sulfided catalysts. The XRD patternof the NiS2 catalysts supported on SiC and SiO2–SiC(not shown) show sharp diffraction lines correspond-ing to the sulfide phase along with the SiC peaks. Onthe other hand, only diffraction lines attributed to thesupports were observed on the XRD patterns of thecatalysts supported on alumina and AC (not shown),meaning that the nickel sulfide phase was highly dis-persed on both supports. This was easily explainedby the high surface area of each support 223 and1490 m2 g−1, respectively. The sulfidation of the oxidephase was accompanied by a decrease in the surfacearea of the catalysts, more or less strong according tothe nature of the support. Such phenomena were at-tributed to a sintering of the supported phase during
N. Keller et al. / Applied Catalysis A: General 234 (2002) 191–205 195
Table 1Specific surface area of fresh and sulfur-loaded nickel sulfide-based catalysts, period of total activity, sulfur loading of the catalyst after32 h of time on stream and catalyst density of the fresh catalysts and weights used for test
Support Surface of thefresh support(m2 g−1)
Surface area ofthe fresh catalyst(m2 g−1)
Catalyst density(g cm−3) [weightused for test (g)]
Period oftotalactivity (h)
Sulfur loading on thecatalyst after 32 h ofreaction (wt.%)
Surface area of thesulfur-loaded catalyst after32 h of reaction (m2 g−1)
SiC 29 25 0.70 [3.52] >32 21 13Al2O3 223 163 0.74 [3.68] 7 15 97SiO2–SiC 18 16 0.72 [3.58] 7 7 13AC 1490 700 0.60 [3.02] 3 13 1
the sulfidation, detailed in previous work[14,17]. Thecleavage and formation of atomic bonds occurring dur-ing sulfidation might temporarily lead to the formationof a more mobile phase, strongly prone to sinter, ashas been reported in the literature[29]. For the AC, thedecrease in the surface area was strongly enhanced bythe high microporosity of the support, easily blockedby the nickel sulfide particles. Moreover, one couldimagine that the hydrophobic nature of the support didnot favor the resistance to this sintering, due to theweak interaction between the supported phase and thesupport[30]. On the contrary, the hydrophilic nature(or partly hydrophilic, see later) of the other supportsallowed to limit the sintering and only a slight de-crease in surface area during the sulfidation treatmentwas evidenced, due to stronger interactions with thesupport.
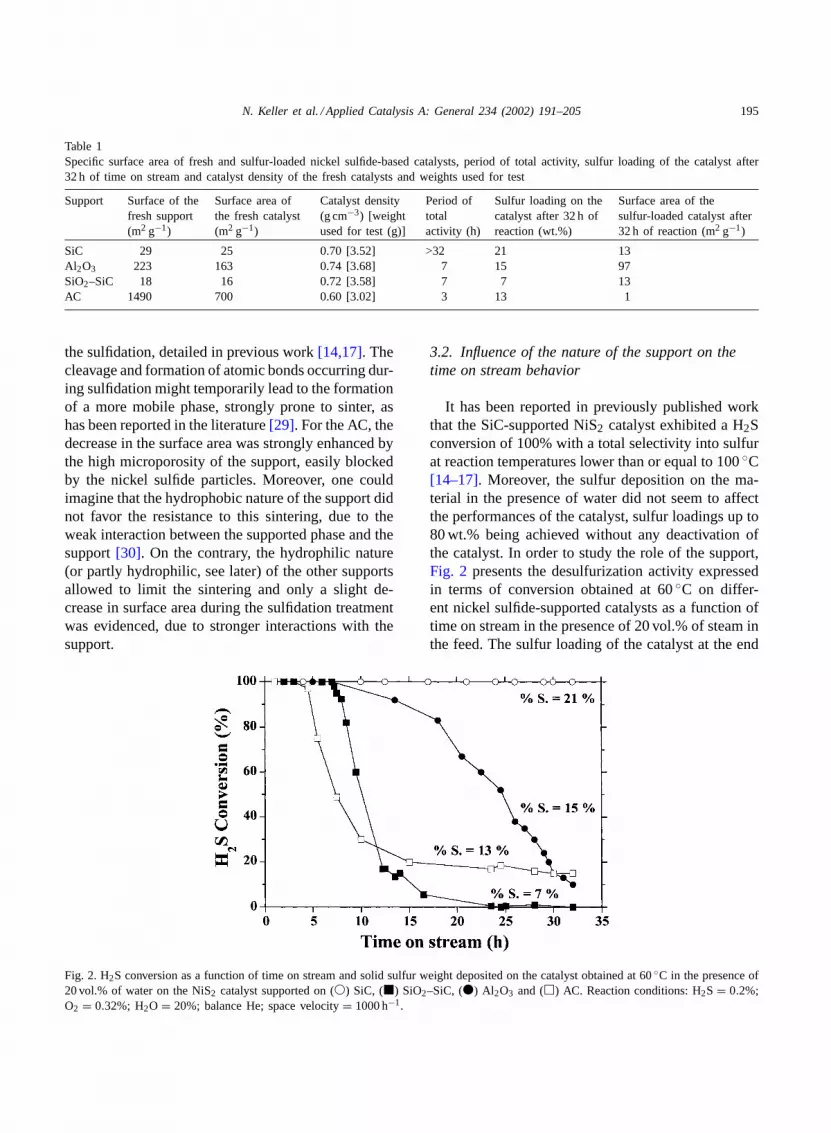
Fig. 2. H2S conversion as a function of time on stream and solid sulfur weight deposited on the catalyst obtained at 60◦C in the presence of20 vol.% of water on the NiS2 catalyst supported on (�) SiC, (�) SiO2–SiC, (�) Al2O3 and (�) AC. Reaction conditions: H2S = 0.2%;O2 = 0.32%; H2O = 20%; balance He; space velocity= 1000 h−1.
3.2. Influence of the nature of the support on thetime on stream behavior
It has been reported in previously published workthat the SiC-supported NiS2 catalyst exhibited a H2Sconversion of 100% with a total selectivity into sulfurat reaction temperatures lower than or equal to 100◦C[14–17]. Moreover, the sulfur deposition on the ma-terial in the presence of water did not seem to affectthe performances of the catalyst, sulfur loadings up to80 wt.% being achieved without any deactivation ofthe catalyst. In order to study the role of the support,Fig. 2 presents the desulfurization activity expressedin terms of conversion obtained at 60◦C on differ-ent nickel sulfide-supported catalysts as a function oftime on stream in the presence of 20 vol.% of steam inthe feed. The sulfur loading of the catalyst at the end

196 N. Keller et al. / Applied Catalysis A: General 234 (2002) 191–205
of the test was also reported. Whatever the support,the selectivity into elemental sulfur (not reported) re-mained total and no trace of SO2 was ever detectedat the outlet of the reactor. The high selectivity wasattributed to the use of a low reaction temperature inclose agreement with the literature, which reportedthat SO2 formation only occurred at higher tempera-tures[8,10–12]. Previous results published by Steijnset al. showed that between 100 and 200◦C, the rate offormation of SO2 by the successive oxidation of sulfurby oxygen was 100 times lower than the rate of oxida-tion of H2S, due to the very large difference betweenthe corresponding experimental activation energies for
Fig. 3. SEM micrographs of the sulfur-loaded SiC-supported NiS2 catalyst: (a) overall morphology; (b) sulfur aggregate in formationcorresponding to the catalyst after few hours of test and thus with a low sulfur loading; (c) back-scattering mode image of a sulfur aggregate;(d) high resolution image of a sulfur aggregate, showing the presence of chimneys and small sulfur particles located on the larger ones.
oxidation, in good agreement with those reported inthe literature ([9] and numerous references therein).One could nevertheless note that at a very low temper-ature, the use of a highly microporous support, suchas AC did not decrease the total selectivity to sulfur,despite the artificial increase in the contact time.
Fig. 3shows SEM micrographs of the sulfur-loadedSiC-supported catalyst. Big aggregates of solid sul-fur on the support surface can be observed, whereasa large part of the surface still remains accessiblefor the reactants (Fig. 3a). Higher magnification im-ages (Fig. 3b–d) show the high porosity of the sul-fur deposit, which could be described as a network of

N. Keller et al. / Applied Catalysis A: General 234 (2002) 191–205 197
interconnected chimneys, with a mean pore diameterof about 1–4�m. This porosity was probably due tothe presence of water during the agglomeration pro-cess of the sulfur particles. It could allow some of theactive sites covered by the sulfur deposit to remainaccessible for the reactants. Small sulfur particles lo-cated on the larger sulfur aggregates were also evi-denced, with a very homogeneous size distribution ofabout 0.2–0.3�m.
The catalysts showed a total conversion during thefirst hours of test whatever the support used, but sev-eral kinds of deactivation occurred, depending on thenature of the support (Fig. 2). The NiS2/Al2O3 catalystbegan to deactivate after 7 h of time on stream and ex-hibited a H2S conversion lower than 20% with a sulfurloading of about 15 wt.% after 32 h of time on stream.Conversely, a drastic deactivation was observed onthe catalysts supported on SiO2–SiC and AC, respec-tively, occurring after 7 and 3 h of test. It was signifi-cant to note that the deactivation occurred after aboutthe same time on stream whatever the nature and thesurface area of the support (except for the SiC cata-lyst). The specific surface area of the support had noinfluence on the mode of sulfur deposition and thusthe desulfurization activity.Table 1summarizes thesedifferent observations. Whatever the sulfur loading onthe catalyst, the solid sulfur deposit did not displayany increase in the micropore contents. Previous workreported that the supported NiS2 phase was stable asa function of time on stream at a temperature of 60◦Cin the reaction conditions used in this study. Deacti-vation was thus attributed to the progressive block-ade by the sulfur deposit of the nickel sulfide sites, ashas been reported by several authors in the literature[7,8,10].
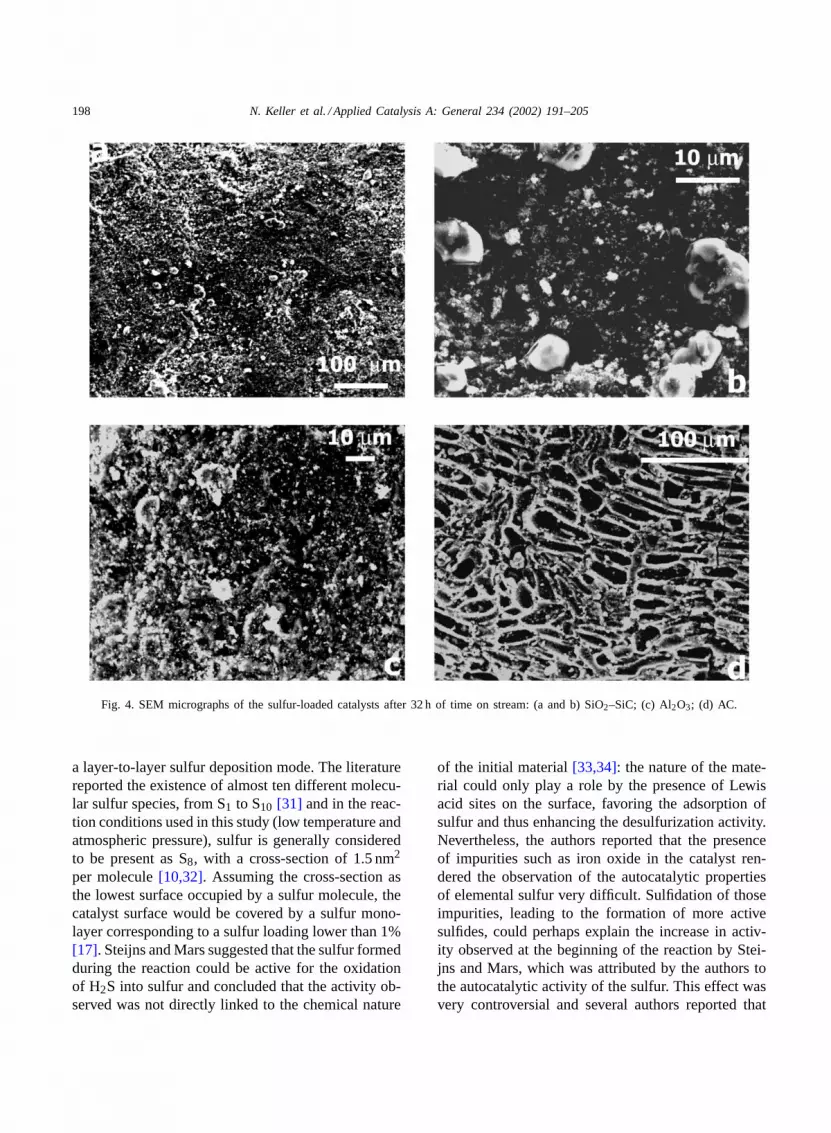
Fig. 4 shows the SEM images of the deactivatedcatalysts supported on AC, SiO2–SiC and Al2O3 after32 h of time on stream. On the SiO2–SiC-supportedcatalyst, only few small sulfur aggregates were ob-served. The solid sulfur deposited on the catalyst was7 wt.% (Fig. 4a and b). On the other hand, it was diffi-cult to observe by SEM sulfur particles on the surfaceof the alumina-supported catalyst after reaction, eventhough 14 wt.% of sulfur was deposited. SEM obser-vation only showed few sulfur particles on the catalystsurface and indicated that the sulfur formed was prob-ably well dispersed throughout the catalyst surface orwas hidden inside the support porosity (Fig. 4c). The
rapid deactivation observed on the AC-supported cat-alyst was explained by the high microporosity of thesupport, which could be rapidly blocked by the firstsulfur deposits, in close agreement with the strong de-crease in surface area of the catalyst from 700 m2 g−1
for the fresh NiS2 catalyst to 1 m2 g−1 for the 13 wt.%sulfur-loaded material. Similar results have been re-ported by Trovarelli and co-workers during the use ofAC, the surface area of the material decreasing from944 m2 g−1 to few square meters for the deactivatedcatalyst[7,8]. This was confirmed by the SEM micro-graph, which only showed few small sulfur particleson the AC surface, nevertheless loaded with 14 wt.%of sulfur (Fig. 4d). The residual activity after 32 h oftime on stream, quite surprising for a material withsuch a low surface area, could be attributed to: (i) theactivity of the biggest NiS2 particles located in largepores, requiring a long time to be completely coveredby the sulfur deposit; or (ii) the activity of non coveredAC active centers, as has been reported at such a lowtemperature by Trovarelli and co-workers. The lowerrate of deactivation observed on the alumina-supportedcatalyst as compared to those observed on the ACand SiO2–SiC supports was probably due to the su-perficial sulfation of the alumina support in the pres-ence of the reactant mixture along with the exothermicnature of the reaction. Such a exothermicity togetherwith the low thermal conductibility of the aluminacould allow the reaction of a fraction of H2S in pres-ence of water with the alumina surface parallely withthe reaction of H2S with oxygen to give elementalsulfur.
3.3. Proposed mechanism for the sulfur deposition
The results obtained in the present study evidencedthe high and stable activity of the NiS2/SiC catalystat 60◦C. The desulfurization activity remained un-changed even with a sulfur loading greater than 60% ofthe weight of the starting catalyst. Such performancefor a medium surface area catalyst was quite surpris-ing, as the literature reported that a high surface areacatalyst is required to successfully perform the reac-tion at low temperature, due to the sulfur depositionduring the reaction, which blocks the access to theactive sites[7,8,10].
The high amount of sulfur which could be stored bythe catalyst allowed the rejection of the hypothesis of
198 N. Keller et al. / Applied Catalysis A: General 234 (2002) 191–205
Fig. 4. SEM micrographs of the sulfur-loaded catalysts after 32 h of time on stream: (a and b) SiO2–SiC; (c) Al2O3; (d) AC.
a layer-to-layer sulfur deposition mode. The literaturereported the existence of almost ten different molecu-lar sulfur species, from S1 to S10 [31] and in the reac-tion conditions used in this study (low temperature andatmospheric pressure), sulfur is generally consideredto be present as S8, with a cross-section of 1.5 nm2
per molecule[10,32]. Assuming the cross-section asthe lowest surface occupied by a sulfur molecule, thecatalyst surface would be covered by a sulfur mono-layer corresponding to a sulfur loading lower than 1%[17]. Steijns and Mars suggested that the sulfur formedduring the reaction could be active for the oxidationof H2S into sulfur and concluded that the activity ob-served was not directly linked to the chemical nature
of the initial material[33,34]: the nature of the mate-rial could only play a role by the presence of Lewisacid sites on the surface, favoring the adsorption ofsulfur and thus enhancing the desulfurization activity.Nevertheless, the authors reported that the presenceof impurities such as iron oxide in the catalyst ren-dered the observation of the autocatalytic propertiesof elemental sulfur very difficult. Sulfidation of thoseimpurities, leading to the formation of more activesulfides, could perhaps explain the increase in activ-ity observed at the beginning of the reaction by Stei-jns and Mars, which was attributed by the authors tothe autocatalytic activity of the sulfur. This effect wasvery controversial and several authors reported that

N. Keller et al. / Applied Catalysis A: General 234 (2002) 191–205 199
the sulfur deposit blocked the access to the active cen-ters located in the pores of the catalyst, whereas nocorrelation was observed between the amount of solidsulfur and the activity measured[12,35–37]. Catalytictests performed with different amounts of sulfur rang-ing from 5 to 50 wt.% stored in SiC in the absence ofnickel catalyst, allowed to state that the autocatalyti-cal effect of sulfur, if any, was really insignificant inthe reaction conditions used[17].
The formation of sulfur via a vapor–liquid–solid ortip growth mechanism, as usually reported in the caseof carbon nanofiber or -tube growth[38], could al-low the nickel sulfide phase to be pushed away fromthe support surface and also to remain accessible tothe reactants. However, the low reaction temperatureused in the present study should strongly inhibit thediffusion rate of the elemental sulfur on or throughthe NiS2 particle. In addition, TEM analysis of thesulfur-loaded catalyst showed no evidence of the for-mation of filamentous sulfur on the material[17,39].
In order to explain the results observed, a peculiarmode of sulfur deposition on the SiC-supported nickelsulfide catalyst was proposed in this study, involv-ing the role of water and the dual nature of the cata-lyst surface. Previous characterizations by XPS/Augermapping evidenced that the surface of SiC preparedaccording to the “shape-memory” gas–solid synthe-sis was heterogeneous[17,19,25,26,40]. A fractionof the surface is composed of a mixture of silicaand oxycarbide of silicon, expected to present a hy-drophilic character due to the presence of oxygen onthe surface, whereas the remaining surface is pure SiC,oxygen-free, expected to be hydrophobic in nature,due to the absence of any oxygen bonds. This dual na-ture of SiC has already been successfully used to per-form the specific impregnation of supported phases.The change from hydrophobic to hydrophilic naturewhen creating oxygenated groups on carbon nanofibersurfaces was reported by de Jong and Geus in their ex-tended review on carbon nanofiber catalytic synthesis[41]. An oxidative treatment under reflux in boilingnitric acid was applied to introduce polarity in the sur-face, thus giving hydrophilic properties to hydropho-bic carbon nanofibers.
It was proposed that the hydrophilic part of thesupport covered the internal surface of pores, due tothe very high density of crystal defects in these ar-eas, formed of planes with high Miller indexes. The
hydrophobic part would be located on the outside ofthe pores, consisting in stable and low Miller indexplanes. During the preparation of the catalyst, per-formed by aqueous impregnation, one could easilyimagine that the interaction between the precursor saltand the hydrophilic oxygenated phases led to the dis-persion and anchorage of the supported nickel oxidephase on these surfaces in the pores of the catalyst.After the sulfidation pre-treatment, the active NiS2phase would thus be located inside the catalyst pores.At such a low reaction temperature, the presence ofwater in the feed led to the formation of a trickle-bed,a large fraction of water condensing at the head of thereactor during the reaction. The formation of a waterfilm on the hydrophilic parts of the SiC surface al-lowed the removal of sulfur from the active sites tothe hydrophobic parts, at the edge of the liquid film.Acting as a conveyor belt, the water film was mechan-ically and continuously cleaning the active sites, keep-ing them free and accessible for the reactants, what-ever the amount of sulfur deposited on its surface. Thesulfur deposition thus occurred on the pure SiC sur-face, with no active phase and also where very highamounts of sulfur could be stored without affectingthe performance of the SiC-supported NiS2 catalyst.Fig. 5aindicates the proposed process of sulfur depo-sition when the reaction is performed in the presenceof water in the feed.
In dry reaction conditions, the rapid deactivation ofthe SiC-based catalyst after only few hours of time onstream, even in the presence of a very low amount ofthe solid sulfur deposit, proved that presence of wateris required to perform the reaction. The absence ofany liquid film on the surface did not allow the freshlygenerated sulfur to be mechanically removed from theactives sites. The sulfur deposition, therefore, directlyoccurred on the hydrophilic parts of the catalyst wherethe active particles are located, leading thus to theblockade of the active sites, as shown inFig. 5b.
In order to check if the solid sulfur formed couldhave escaped from the catalyst, the wet reaction wasperformed at 60◦C on the NiS2/SiC catalyst, mechan-ically mixed with fresh and inactive SiC extrudates(4–6 mm length and 2–3 mm diameter). This SiC wasprepared at the CRV-Pechiney according to the sameshape memory synthesis as the grains. This mixing hadno effect on the catalytic results and both total H2Sconversion and selectivity into sulfur were achieved

200 N. Keller et al. / Applied Catalysis A: General 234 (2002) 191–205
Fig. 5. Proposed mode of sulfur deposition on the surface of theSiC-based catalyst: (a) in the presence of water in the feed; (b)in the absence of water in the feed.
(not reported). The surface of the SiC extrudate af-ter the test, stopped at a theoretical sulfur loading of30 wt.%, only exhibited a very small amount of sulfur(Fig. 6). The weight gain of the extrudates after testwas negligible and the sulfur-loaded NiS2/SiC mate-rial contained 98% of the total solid sulfur formed dur-ing the course of the reaction. The intra-grain transferof the sulfur particles was thus evidenced and con-firmed the hypothesis of the non-homogeneous natureof the liquid film at the surface of the material dur-ing the reaction in the presence of water. Indeed, ifan homogeneous film was formed on the catalyst sur-face due to the water condensation, it would allowan inter-grain transfer to occur and higher amounts ofsulfur would be observed on the fresh SiC extrudates.The aqueous solution condensed at the outlet of the
Fig. 6. SEM micrograph of a SiC extrudate after the test performedwith the mechanical mixing of the NiS2/SiC grains and fresh SiCextrudate (total sulfur loading of 30 wt.%).
reactor and frequently analyzed should furthermorecontain sulfur particles, which were never observed.The results well confirmed the ability of the SiC sup-port to retain the total amount of sulfur formed on itssurface during the test.
3.4. On the nature of the active phase supportedon SiC
Whereas the NiS2/SiC catalyst exhibited a total H2Sconversion since the beginning of the test at a tempera-ture of 60◦C, previous work reported that at the lowertemperature of 40◦C the catalyst required a few hoursactivation period on stream before reaching 100% con-version[16]. A slight oxidative pre-treatment with anoxygen flow at the reaction temperature for 1 h allowedto avoid this activation period and superficially trans-formed the sulfide starting phase into the stable fi-nal active phase. The same transformation probablyoccurred at 60◦C, but could not be observed due tothe higher reactivity at 60◦C as compared to 40◦Cwhich accelerated the formation of the stable activephase.Fig. 7reports the influence of the GHSV on theevolution with time on stream of the H2S conversionobtained on the NiS2/SiC at a temperature of 40◦C.The selectivity into sulfur remained 100% at this lowtemperature whatever the GHSV and thus was not re-ported. One could observe that higher GHSV led toa shorter activation period, which decreased from 4

N. Keller et al. / Applied Catalysis A: General 234 (2002) 191–205 201
Fig. 7. H2S conversion as a function of time on stream obtained at 40◦C in the presence of 20 vol.% of water on the NiS2/SiC catalystwith different HSVs between 500 and 1000 h−1. Reaction conditions: H2S = 0.2%; O2 = 0.32%; H2O = 20%; balance He.
to 2 h when the velocity was increased from 500 to1000 h−1. Different transformations of the NiS2 phaseduring the oxidative pre-treatment were thus investi-gated at a reaction temperature of 40◦C.
The NiS2 phase could be superficially oxidized intoits corresponding oxide. The H2S conversion obtainedon the SiC-supported NiO catalyst at 40◦C in the pres-ence of 20 vol.% of water in the feed is reported inFig. 8 (the total sulfur selectivity is not drawn). The
Fig. 8. H2S conversion as a function of time on stream obtained at 40◦C in the presence of 20 vol.% of water on the NiO/SiC catalyst.Reaction conditions: H2S = 0.2%; O2 = 0.32%; H2O = 20%; balance He; space velocity= 500 h−1.
catalyst rapidly deactivated with time on stream be-fore reaching a stable H2S conversion of about 10%after 3 h of test. The presence of 20% of water in thefeed and the low sulfur loading on the catalyst afteronly a few hours on stream allowed the exclusion ofdeactivation by site blockade. It confirmed the low ac-tivity of the oxide phase already evidenced at a tem-perature of 100◦C [17]. The high affinity between theNiO phase and H2S could lead to a superficial and

202 N. Keller et al. / Applied Catalysis A: General 234 (2002) 191–205
also probably partial sulfidation of the oxide phasewith time on stream. Previous work showed that theNiO-supported phase was transformed during the re-action at a temperature of 100◦C into a partially sul-fided NiSx phase[17], in close agreement with theliterature which showed in the case of Co, Mo or Nioxides that the lower the temperatures, the more su-perficial and partial the phenomenon[42–45]. Thislow temperature sulfidation has been particularly re-ported by several authors during the study of H2S ad-sorption on NiO/NaY and NiO/�-Al2O3 materials atambient temperature[46,47]. The formation of sucha superficial phase could explain the residual activityobtained on the NiO/SiC catalyst after a few hoursof time on stream. The phase formed at 40◦C shouldhave a much less sulfidic nature than the NiSx phaseobserved at 100◦C, leading thus to a very low residualactivity, as compared to the conversion of 93% thatcould be obtained at 100◦C with the NiSx phase[17].The XRD diagram of the tested catalyst (not shown)did not evidence any modifications of the NiO phaseand confirmed the superficial nature of the transfor-mation. Results obtained on the NiO/SiC catalyst thusallowed to definitively reject the hypothesis of a su-perficial transformation of the NiS2 phase into its cor-responding oxide at a temperature of 40◦C.
The superficial oxidation of NiS2 by oxygen orliquid water into sulfate could be proposed, becauseof the known affinity of sulfide phases for oxygen
Fig. 9. H2S conversion as a function of time on stream obtained at 40◦C in the presence of 20 vol.% of water on the NiS2/SiC catalystwith a O2/H2S molar ratio of 12.5 and an inlet H2S concentration of 1.1 vol.% (balance He; space velocity= 500 h−1).
to form their corresponding sulfates. The H2S con-version obtained at 40◦C on the NiS2/SiC catalystwith simultaneously a high O2 to H2S molar ratioof 12.5 (instead of 2.5) and a H2S concentration in-creased from 0.2 to 0.9% is reported inFig. 9 (thesulfur selectivity remaining 100% is not reported).These reaction conditions (both high oxygen and H2Spartial pressures) were chosen in order to favor thesulfation of the nickel sulfide phase, by using the highaffinity between sulfides and oxygen and the knownhigh exothermicity of the direct oxidation of H2S intosulfur (H = −222 kJ mol−1 [48]). Indeed, highH2S concentrations to be treated induced a significantexothermicity during the reaction which could leadto an oxidation or a sulfation of the supported phase:industrial tests revealed that an increase in tempera-ture of 70◦C occurred for the transformation of a H2Sconcentration of 1% in the total feed. Other reactionparameters remained unchanged. The catalyst rapidlydeactivated with time on stream, whereas a green soliddeposit was observed after reaction inside the silicawool which supported the catalyst bed. InFig. 10arereported the XRD diagrams of the deactivated cata-lyst and of this solid deposit. The deposit diffractionpattern exhibited high intensity diffraction peaks cor-responding to the sulfate phase NiSO4·2H2O, in closeagreement with the green color observed. In addition,the XRD diagram of the catalyst after test revealed thepresence of�-sulfur, together with this sulfate phase

N. Keller et al. / Applied Catalysis A: General 234 (2002) 191–205 203
Fig. 10. XRD diagrams of: (a) the deactivated NiS2/SiC catalystafter the test inFig. 9; (b) the solid deposit located inside the silicawool which supported the catalyst bed during the reaction. (�)NiS2 phase and (�) �-sulfur. Non-attributed peaks correspond tothe NiSO4·2H2O sulfate phase.
and the NiS2 phase was only observed in a very smallamount (at 32◦). The high solubility of the nickel sul-fate in water is well known from 0.3 to 0.8 g cm−3 atambient temperature, depending on the nature of thesulfate and increasing with water temperature[49].It is very likely that the sulfation of the NiS2 phaseoccurred during the reaction and the nickel sulfateformed was rapidly dissolved by the trickling water,which continuously cleaned the catalyst surface. Thedissolved sulfate phase then recrystallized in the silicawool. This phenomenon would also explain the signif-icant decrease in the intensity of the diffraction peakcorresponding to the NiS2 phase on the deactivatedcatalyst. If such a sulfation process occurred during thecatalyst test with a H2S concentration of 2000 ppm,the nickel sulfate phase formed would be rapidlydissolved by water and the catalyst should deactivatewith time on stream. The stability of the NiS2/SiCcatalyst as a function of time on stream and the ab-sence of any green solid deposit inside the silica woolafter test thus allowed the rejection of the hypothesis
of the superficial formation of a nickel sulfate phasewhich would be the stable active phase on stream.
The superficial oxidation on stream of the nickelsulfide phase into a new nickel oxysulfide ornon-stochiometric sulfate phase was then proposed inorder to explain the activation period observed at atemperature of 40◦C on the NiS2/SiC catalyst. Theformation of this new phase should remain superficial,because of the relatively rapid transformation of thesulfide (between 2 and 4 h depending on the GHSV)despite the very low reaction temperature. The oxy-sulfide phase would be formed by exchange betweenthe sulfur atoms of the network and the oxygen atomsof the feed, present on the catalyst surface as O2molecules or O∗ activated radicals[8,10,36,50]. Thelarger the oxygen amount, corresponding to a highervelocity, the shorter was the activation period of thecatalyst. The formation of such an oxysulfide phasehas been already reported by de Jong et al. during theexposure of MoO3 to a H2S containing flow[43]. Theauthors showed that the formation of a molybdenumoxysulfide phase occurred at ambient temperatureafter the first minutes of exposure. The nickel oxysul-fide phase was probably formed at a temperature of60◦C, but the too rapid S→ O atom exchange wouldlead to a H2S conversion of 100% from the beginningof the test and no activation period was observed.Intensive work is continuing in order to evidence andcharacterize this superficial nickel-based phase andthese results will be separately published later.
4. Conclusions
The use of medium surface area SiC as catalystsupport allowed a very active, but also highly stablecatalyst as a function of time on stream and solid sul-fur loading, to be obtained compared to other sup-ports such as silica and both high surface area aluminaand AC. This exceptional stability was attributed toa peculiar mode of sulfur deposition, occurring whenSiC was used as support, involving water present inthe feed and the SiC surface heterogeneity. The activephase, located on the hydrophilic oxygen-containingareas of the SiC (mixture of silica and silicon oxy-carbide), was continuously cleaned by water duringthe reaction. The solid sulfur particles formed thus es-caped from the active phase up to the next hydropho-

204 N. Keller et al. / Applied Catalysis A: General 234 (2002) 191–205
bic/hydrophilic interface, where the liquid film of wa-ter stopped. The solid sulfur particles could conse-quently be stored in high amounts on the hydrophobicpure SiC surfaces, which are free from nickel, withoutmodifying the activity of the catalyst.
The nature of the very active phase stabilized onstream during the direct oxidation of H2S by oxy-gen into elemental sulfur at low reaction temperatures(40–60◦C) was investigated. Hypotheses concerningthe formation of nickel oxide and nickel sulfate wereboth rejected. Superficial transformation on stream ofthe starting NiS2 into a very active nickel oxysulfideor non-stochiometric phase was proposed to explainthe high activity for H2S oxidation.
Acknowledgements
This work was supported by the Pechiney and ELFAquitaine Production Companies.
References
[1] H. Baehr, Refiner Nat. Gasoline Manufacturer 17 (1938) 237.[2] Sulphur, no. 252, 1997.[3] Sulphur, no. 231, 1994.[4] J. Wieckowska, Catal. Today 24 (1995) 405.[5] Sulphur, no. 257, 1998.[6] A. Piéplu, O. Saur, J.-C. Lavalley, O. Legendre, C. Nedez,
Catal. Rev. Sci. Eng. 43 (4) (1998) 409.[7] A. Trovarelli, F. Felluga, C. De Leitenburg, P. Andreussi, G.
Dolcetti, in: G. Centi, et al. (Eds.), Proceedings of the FirstWorld Congress on Environmental Catalysis, Pisa, Italy, 1–5May 1995, p. 667.
[8] A. Primavera, A. Trovarelli, P. Andreussi, G. Dolcetti, Appl.Catal. A Gen. 173 (1998) 185.
[9] M. Steijns, F. Derks, A. Verloop, P. Mars, J. Catal. 42 (1976)87.
[10] J. Klein, H.-D. Henning, Fuel 63 (1984) 1064.[11] A.I. Chowdhury, E.L. Tollefson, Can. J. Chem. Eng. 68 (1990)
449.[12] A.K. Dalai, A. Majumdar, A. Chowdhury, E.L. Tollefson,
Can. J. Chem. Eng. 71 (1993) 75.[13] M.J. Ledoux, J.-B. Nougayrède, S. Savin-Poncet, C.
Pham-Huu, N. Keller, C. Crouzet, French Patent 97-16617(1997), assigned to Elf Aquitaine Production.
[14] N. Keller, C. Pham-Huu, C. Estournès, M.J. Ledoux, Catal.Lett. 61 (3/4) (1999) 151.
[15] N. Keller, C. Pham-Huu, C. Crouzet, M.J. Ledoux, S.Savin-Poncet, J.-B. Nougayrède, J. Bousquet, Catal. Today53 (4) (1999) 535.
[16] M.J. Ledoux, C. Pham-Huu, N. Keller, J.-B. Nougayrède,S. Savin-Poncet, J. Bousquet, Catal. Today 61 (2000)157.
[17] N. Keller, Thesis, Louis Pasteur University, Strasbourg,France, 1999.
[18] M.J. Ledoux, S. Hantzer, C. Pham-Huu, J. Guille, M.P.Desaneaux, J. Catal. 114 (1988) 176.
[19] M.J. Ledoux, S. Hantzer, J.L. Guille, D. Dubots, US Patent4,914,070 (1990), assigned to Pechiney Recherche.
[20] E. Peschiera, Thesis, Louis Pasteur University, Strasbourg,France, 1993.
[21] E.P. Barrett, L.G. Joyner, P.H. Halenda, J. Am. Chem. Soc.73 (1951) 373.
[22] J.H. De Boer, in: D.H. Everett (Ed.), The Structure andProperties of Porous Materials, 1958.
[23] R.S.H. Mikhail, S. Brunauer, E.E. Bodor, J. Coll. Int. Sci. 26(1968) 45.
[24] N. Keller, C. Pham-Huu, M.J. Ledoux, Appl. Catal. A Gen.217 (2001) 205.
[25] N. Keller, C. Pham-Huu, M.J. Ledoux, C. Estournès, G. Ehret,Appl. Catal. A Gen. 187 (1999) 255.
[26] N. Keller, C. Pham-Huu, S. Roy, M.J. Ledoux, C. Estournès,J. Guille, J. Mater. Sci. 34 (1999) 3189.
[27] R. Moene, Ph.D. dissertation, Delft University of Technology,The Netherlands, 1995.
[28] R. Moene, M. Makkee, J.A. Moulijn, Appl. Catal. A Gen.167 (1998) 321.
[29] C.K. Groot, Ph.D. dissertation, Delft University ofTechnology, The Netherlands, 1984.
[30] C. Pham-Huu, N. Keller, V.V. Roddatis, G. Mestl, R. Schlögl,M.J. Ledoux, Phys. Chem. Chem. Phys. 4 (3) (2002)514.
[31] H.G. Paskall, Capability of the Modified Claus Process,Western Research, Calgary, 1979.
[32] B.W. Gamson, R.H. Elkins, Chem. Eng. Prod. 49 (1953)203.
[33] M. Steijns, P. Mars, J. Catal. 35 (1974) 11.[34] M. Steijns, P. Mars, Ind. Ing. Chem. Prod. Res. Dev. 16 (1)
(1977) 35.[35] R. Sreeramamurthy, Ph.D. dissertation, University of Andhra,
India, 1972.[36] I. Coskun, E.L. Tollefson, Can. J. Chem. Eng. 58 (1980)
72.[37] T.K. Ghosh, E.L. Tollefson, Can. J. Chem. Eng. 64 (1986)
969.[38] N.M. Rodriguez, J. Mater. Res. 8 (1993) 3233.[39] M.J. Ledoux, C. Pham-Huu, N. Keller, J.-B. Nougayrède,
S. Savin-Poncet, J. Bousquet, in: A. Corma, F.V. Melo, S.Mendioroz, J.L.G. Fierro (Eds.), Proceedings of the 12thInternational Congress on Catalysis, Stud. Surf. Sci. Catal.130D (2000) 2891.
[40] Pechiney Recherche, unpublished results.[41] K.P. de Jong, J.W. Geus, Catal. Rev. Sci. Eng. 42 (4) (2000)
481.[42] P.J. Mangnus, Ph.D. dissertation, Delft University of
Technology, The Netherlands, 1991.

N. Keller et al. / Applied Catalysis A: General 234 (2002) 191–205 205
[43] A.M. de Jong, H.J. Borg, L.Z. van Ijzendoorn, V.G.M.F.Soudant, V.H.J. de Beer, J.A.R. van Veen, J.W. Niemants-verdriert, J. Phys. Chem. 97 (24) (1993) 6477.
[44] P. de Bont, Ph.D. dissertation, Delft University of Technology,The Netherlands, 1998.
[45] M.J. Vissenberg, Ph.D. dissertation, Eindhoven University ofTechnology, The Netherlands, 1999.
[46] B. Scheffer, P. Mangnus, J.A. Moulijn, J. Catal. 121 (1990)18.
[47] W.J.J. Welters, Ph.D. dissertation, Eindhoven University ofTechnology, The Netherlands, 1994.
[48] Dupuy, M. Graulier, Informations chimie, Spécial Catalyse33 (1969).
[49] Handbook of Chemistry and Physics, 54th Edition, R.C. Weast(Ed.), CRC Press, Cleveland, Ohio, 1973.
[50] K. Hedden, L. Huber, B.R. Rao, Adsorptive Reinigung vonSchwefelwasserstoffhaltigen Abgasen, VDI-Bericht no. 253,VDI-Verlag, Düsseldorf, 1976, p. 37.









![Guillelmi Caonrsin [sic] Rhodiorum Uicecancellarij: obsidionis](https://img.dokumen.tips/doc/110x75/633da63291fe0b691903a6ca/guillelmi-caonrsin-sic-rhodiorum-uicecancellarij-obsidionis-.jpg)



















