Embed Size (px)
Citation preview

ORIGINAL PAPER
Lineage pattern, trans-species polymorphism, and selectionpressure among the major lineages of feline Mhc-DRBpeptide-binding region
Kun Wei & Zhihe Zhang & Xiaofang Wang &
Wenping Zhang & Xiao Xu & Fujun Shen & Bisong Yue
Received: 21 September 2009 /Accepted: 16 March 2010 /Published online: 7 April 2010# Springer-Verlag 2010
Abstract The long-term evolution of major histocompati-bility complex (MHC) involves the birth-and-death processand independent divergence of loci during episodespunctuated by natural selection. Here, we investigated themolecular signatures of natural selection at exon-2 of MHCclass II DRB gene which includes a part of the peptide-binding region (PBR) in seven of eight putative extantFelidae lineages. The DRB alleles in felids can be mainlydivided into five lineages. Signatures of trans-speciespolymorphism among major allelic lineages indicate thatbalancing selection has maintained the MHC polymorphismfor a long evolutionary time. Analysis based on maximumlikelihood models of codon substitution revealed overallpurifying selection acting on the feline DRB. Sites that haveundergone positive selection and those that are underdivergent selective pressure among lineages were detectedand found to fall within the putative PBR. This studyincreased our understanding of the nature of selective forcesacting on DRB during feline radiation.
Keywords Allelic lineages .DRB . Felid .MHC .
Positive selection . Trans-species polymorphism
Introduction
The major histocompatibility complex (MHC) is a largemultigene family that plays a pivotal role in the adaptiveimmune system of jawed vertebrates. MHC genes can bedivided in two major classes on the basis of molecularstructure and function of the polypeptide encoded: class I,which is responsible for the recognition and binding ofintra-cellular antigens, and class II, which corresponds toextracellular antigens (Klein and Sato 1998). In the typicalpattern of MHC evolution, class II genes usually exhibitslower rate of birth-and-death evolution, higher longevity,and higher polymorphism than class I genes (Takahashiet al. 2000; Bos and Waldman 2006). The highest levels ofpolymorphism are often exhibited in the functionallyimportant peptide-binding region (PBR), in which thegenetic variation alters the peptide-binding site of theencoded proteins, enabling them to bind a variety of foreignpeptides (Furlong and Yang 2008). Therefore, positiveselection is commonly expected to act intensively on PBR,making PBR to be the focus of studying adaptive selectionin MHC molecules. For long-term evolution of mammalianMHC genes, those polymorphisms are thought to have beenmaintained by balancing selection (a form of positiveselection; Hughes and Yeager 1998; Hedrick 2007). Underbalancing selection, some allelic lineages exhibit unusuallongevity which predates speciation events, leading to aproper name “trans-species polymorphism (TSP)” which isused to describe polymorphism in such case (Klein et al.1998, 2007). In this way, pattern of TSP produces a genetree that can differ markedly from the species relation-ships revealed by neutral molecular markers, that is,some alleles from different species would cluster together
K. Wei :X. Wang :X. Xu : B. Yue (*)Sichuan Key Laboratory of Conservation Biology on EndangeredWildlife, College of Life Sciences, Sichuan University,Sichuan 610064, People’s Republic of Chinae-mail: [email protected]
Z. Zhang :W. Zhang : F. ShenChengdu Research Base of Giant Panda Breeding,Sichuan 610081, People’s Republic of China
Immunogenetics (2010) 62:307–317DOI 10.1007/s00251-010-0440-5

in the inferred phylogenetic tree rather than all intra-specific alleles clustering together.
Felid species that possess a prominent position atop thefood chain in natural habitats are of keen concern abouttheir radiation and the processes of adaptive evolution(Janczewski et al. 1995; Johnson et al. 2006). Anunderstanding about the evolution of feline MHC mayprovide insights into the variation of immunologicalfunction, the evolutionary constraints, and the drivingforces in feline radiation. Yuhki and O’Brien (1997)examined the variation of Mhc-DRB exon-2 that containsa large part of PBR in the domestic cat (Felis catus), whichrevealed a high level of polymorphism, with an observationof positive selection for PBR and negative selection fornon-PBR. In addition, a phylogenetic analysis on acombined set of the domestic cat and other three felidspecies revealed a sharing pattern of allelic lineages,suggesting a trans-species mode of evolution. In theirsubsequent review, O'Brien and Yuhki (1999) presented aprospect of monitoring the selective forces that influencethe feline genome and the MHC evolutionary processes.Since then, the Immuno Polymorphism Database projectfor feline MHC (FLA) was built, aiming at integratingand facilitating advanced MHC research on felid species(see http://www.ebi.ac.uk/ipd/mhc; Kennedy et al. 2002;Robinson et al. 2003). A recent study on the MHC genomeorganization spanning the domestic cat classical andextended class II region confirmed that there are exactlythree functional DRB loci (DRB 1, 3, and 4) and apseudogene (DRB2) which lacks the full complement ofexons (Yuhki et al. 2007). Following these previous work,we continue to use the method of clonal sequence analysis(Yuhki and O'Brien 1997; Kennedy et al. 2002) to collectMhc-DRB exon-2 alleles from felid species and examine theselective pressure among the allelic lineages, with the purposeof providing a synthetic understanding of molecular signa-tures of natural selection in feline MHC evolution.
Materials and methods
DNA specimen, molecular cloning, and nomenclature
Species from the Panthera lineage included the cloudedleopard (Neofelis nebulosa; number of individuals = 3), thelion (Panthera leo; n=2), the snow leopard (Panthera uncia;n=3), the leopard (Panthera pardus; n=7), and the tiger(Panthera tigris; n=47). Representatives from the leopardcat lineage were the Asian leopard cat (Prionailurusbengalensis; n=5) and the Pallas cat (Otocolobus manul;n=13). The domestic cat lineage was represented by theChinese desert cat (Felis bieti; n=4) and the European wildcat (Felis silvestris; n=2). The Asian golden cat (Pardofelis
temminckii; n=3) and the Eurasian lynx (Lynx lynx; n=20)represented the bay cat lineage and lynx lineage, respectively.Most of the animals belonging to these species we sampledwere wild-caught and then kept in 20 zoos (including parksand museums) in China, except for those “big cats” such astigers that were captive-born within a captive history of nomore than 60 years, according to the studbook established bythe Chinese Association of Zoological Gardens.
Genomic DNA from peripheral blood was isolated withQIAamp DNA Mini Kit (Qiagen Inc.), and DNA from hairswas extracted with DNeasy Tissue Kit (Qiagen Inc.),according to the manufacturer’s protocols. The primersDRB219m (5′-CCACACAGCACGTTTC(C/T)T-3′) andDRB61a (5′-CCGCTGCACTGTGAAGCT-3′) which hadbeen successfully applied to several felid species (Drakeet al. 2004; Kennedy et al. 2002, 2003; Luo et al. 2004;Yuhki and O'Brien 1997) were used in our study. In order tominimize the PCR-induced substitutions, all PCRs wereperformed with a proofreading Pfu DNA polymerase(Fermentas Inc.). Each PCR was carried out with 100 ngof each primer and 1 μl of template DNA in a final volumeof 50 μl, and the cycling consisted of 35 cycles at 94°C for1 min, 55°C for 1 min, and 72°C for 1 min, followed by afinal extension stage at 72°C for 10 min. All blunt-endedamplicons were 5′-phosphorylated (T4 Polynucleotide Kinase[Fermentas Inc.]) and cloned (Digested & DephosphorylatedpUC19/SamI Vector and Rapid DNA Ligation & Transfor-mation Kit [Fermentas Inc.]), and the recombinant plasmidswere selected and purified. Ten to 15 clones of each ampliconwere sequenced on ABI 3730 DNA analyzer (AppliedBiosytems, USA).
A sequence with variants was considered as a uniqueallele only when it met the criteria summarized by Kennedyet al. (2003). The criteria are, briefly, that there have to beat least three identical clones, identified in either twoseparate PCRs from the same individual or from PCRs fromat least two different individuals. The obtained 57 DRBalleles of felid species in our test were of 237 bp in lengthwithout stop codons. Among these, 36 were identified asnew sequences by the BLAST search again GenBank. Wedesignated the local name of the new alleles by followingthe proposed nomenclature for MHC in non-human species(Klein et al. 1990) with lineage-specific series numberattached (Table 1). These were sequences were deposited inGenBank with accession numbers of FJ210683–FJ210718.
Some available feline DRB exon-2 alleles were includedin our analysis. We chose 24 domestic cat alleles fromrepeatable results (Kennedy et al. 2002; Kuwahara et al.2000; Yuhki and O'Brien 1997) and from the EMBL-EBIImmuno Polymorphism Database (http://www.ebi.ac.uk/ipd/mhc; Robinson et al. 2003). Retrieved from the resultsof Yuhki and O'Brien (1997), one allele (U51548) ofGeoffroy’s cat (Oncifelis geoffroyi) and three alleles
308 Immunogenetics (2010) 62:307–317

(U51543–U51545) of Tigrina (Leopardus tigrinus) werechosen to represent the ocelot lineage; two alleles (U51546and U51547) of Iriomote cat (Mayailurus iriomorensis),which was once considered to be a subspecies of leopardcat but later suggested to be a separate species (Leyhausen
and Pfleiderer 1999), were also used. In addition, five alleles(GenBank accession numbers AY312960–AY312964) ofcheetah (Acinonyx jubatus) generated by the RSCA technol-ogy (Drake et al. 2004) were used to represent the pumalineage. We also included one allele of tiger, three alleles
Table 1 The 57 unique feline DRB alleles detected in this study
New identified alleles (n=36) Previously reported alleles (n=21)
Species and the number of samples Local name NoA References Local name NoA
Clouded leopard (n=3) Nene-DRB*201 3 Wang et al. 2008 Nene-DRB*1 1
Nene-DRB*202 3 Nene-DRB*2 3
Lion (n=2) Pale-DRB*201 2 Nene-DRB*3 1
Pale-DRB*202 2 Nene-DRB*4 2
Snow leopard (n=3) Paun-DRB*201 3 Pati-DRB*1 21
Leopard (n=7) Papa-DRB*201 a 2 Papa-DRB*1 1
Papa-DRB*301 2 Papa-DRB*2 1
Papa-DRB*302 6 Papa-DRB*3 1
Tiger (n=47) Pati-DRB*201 5 Wang et al. 2009 Lyly-DRB*01 7
Pati-DRB*202 2 Lyly-DRB*02 13
Pati-DRB*203 21 Lyly-DRB*03 6
Pati-DRB*204 5 Lyly-DRB*04 2
Pati-DRB*205 17 Lyly-DRB*05 2
Pati-DRB*206 b 6 Lyly-DRB*06 3
Pati-DRB*207 a 2 Lyly-DRB*07 9
Pati-DRB*301 4 Lyly-DRB*08 2
Pati-DRB*302 5 Lyly-DRB*09 3
Pati-DRB*303 2 Lyly-DRB*10 7
Pati-DRB*304 27 Lyly-DRB*11 2
Pati-DRB*305 c 3 Lyly-DRB*12 2
Pati-DRB*306 2 Lyly-DRB*13 2
Asian leopard cat (n=5) Prbe-DRB*101 3
Prbe-DRB*201 2
Prbe-DRB*202 2
Prbe-DRB*203 2
Prbe-DRB*401 4
Prbe-DRB*501 3
Pallas cat (n=13) Otma-DRB*201 b 13
Otma-DRB*301 c 13
Chinese desert cat (n=4) Febi-DRB*101 4
Febi-DRB*502a 3
Febi-DRB*503 2
European wild cat (n=2) Fesi-DRB*101b 2
Asian golden cat (n=3) Pate-DRB*201 b 2
Pate-DRB*501 2
Pate-DRB*502 2
Alleles with the same letters (a, b, and c) are identical
NoA no. of animals containing the assigned allele in our studya Febi-DRB*502 is identical with Feca-DRB*0504b Fesi-DRB*101 is identical with Feca-DRB*0103
Immunogenetics (2010) 62:307–317 309

of leopard, four alleles of clouded leopard (DQ189257–DQ189264; Wang et al. 2008) and 13 alleles of lynx(EU918395–EU918407; Wang et al. 2009) which were alsoconfirmed in our test. The large-insert BAC/PAC clones224e2 (AY152833), g7/160a17 (AY152835–AY152836),and f2 (AY152834) which correspond to DRB4, DRB3,and DRB1 of domestic cat (Yuhki et al. 2007), respectively,were also used. Taken together, 96 sequences were includedin our analysis, which contains seven of the extant eightFelidae lineages (except the caracal lineage; Johnson et al.2006; O'Brien and Johnson 2007). The alleles of sheep(Ovar-DRB1*0101) and swine (SLA-DRB1*0101) from theEMBL-EBI Immuno Polymorphism Database were used asoutgroups in phylogenetic analysis.
Data analysis
All nucleotide sequences were aligned with ClustalX 2.0.9(Larkin et al. 2007). Initial sequence comparisons and mea-sures of variation were performed using MEGA 4 (Tamura etal. 2007). The program RDP3 Beta 28 (Martin et al. 2005)was used to detect recombination with default selections ofRDP, GENECONV, and MaxChi methods. The Iss and Iss.cstatistics (Xia et al. 2003) was carried out by using DAMBEVersion 5.0.23 (Xia and Xie 2001) to measure substitutionsaturation which may affect the phylogenetic inference.
In the phylogenetic analysis, in order to avoid relying onsingle best-fit substitution model, the model averagedphylogenies were constructed by using jModelTest v0.1.1(Posada 2008). In brief, a maximum likelihood (ML)optimized tree for each candidate model was specified andthen a weighted major rule consensus tree (Felsenstein2005) was built from all candidate trees using modelweights as tree weights (Posada and Buckley 2004; Posada2008). The obtained tree was rebooted from outgroup withPHYLIP 3.67 (Felsenstein 2005). Under the 24-modelscheme, both the AICc (due to n/K<40 in our dataset)and BIC-weighted consensus trees showed a consistenttopology (see “Results” section) which was close toprevious five-lineage pattern in domestic cat (Yuhki andO'Brien 1997) and was then used in subsequent analysis. Toquantify the degree of trans-specificity observed within asingle allelic lineage, the number of “deep coalescent”events that necessary to make the DRB allelic tree andneutral-marker tree concordant was estimate by using themethod of reconciled phylogenetic trees (Page and Charleston1997) implemented in GeneTree 1.3 (Page 1998).
The topology of feline DRB was extracted (with out-groups deleted) and analyzed with CODEML implementedin PAML 4 (Yang 2007). The branch lengths were re-estimated. Under the “random-site” models, the model M0was used to calculated the average ratio of the non-synonymous substitution rate to the synonymous substitution
rate (ω=dN/dS) among all aligned sequences; the M1a(neutral) vs. M2a (selection) and M7 (beta) vs. M8 (beta&ω;Nielsen and Yang 1998; Yang et al. 2000, 2005) were usedto form two pairs of likelihood ratio tests (LRTs) for whichthe twice log likelihood difference (2ΔInL) was used tocompare with a χ2 distribution to determine the better-fitmodels.
To test whether the inferred positive selection sites arerobust to tree topology, a ML tree based on a smaller dataset was built to get comparative bootstrap values as inYuhki and O'Brien (1997). This smaller data set contained73 of 96 sequences above and was subjected to jModelTestv0.1.1 (Posada 2008) for calculating a best-fit model (GTR +I + G). The phylogeny reconstruction was performed usingRAxML-7.0.3 (Stamatakis 2006) under the GTRMIXImodel and 1,000 bootstraps, and the topology was subjectedagain to “random-site” models in PAML 4 as describedabove. Besides, in the PAML 4 analysis, the five lineageswhich got reasonable bootstrap support in the ML tree(Fig. 2) were also used in the “branch-site” model (test 2,model A1 vs. model A; Zhang et al. 2005) and the “clade”model C (Bielawski and Yang 2004; Yang et al. 2005) fordetecting positive selection sites along a given foregroundlineage and sites evolving under divergent selective pressure,respectively. In this scenario, due to the absence of biologicalhypothesis to designate so-called foreground, eachMhc-DRBlineage was specified in turn as the foreground lineage,and the family-wise error rate in such multiple testing wascontrolled under the Bonferroni’s correction (Anisimova andYang 2007).
Results
Fifty-seven unique alleles were found among the collectedanimals in this study (Table 1): six in the clouded leopard(N. nebulosa), one in the snow leopard (P. uncia), six in theAsian leopard cat (P. bengalensis), 14 in the tiger (P. tigris),three in the Asian golden cat (P. temminckii), two in thePallas cat (O. manul), two in the lion (P. leo), six in theleopard (P. pardus), one in the European wild cat(F. silvestris), three in the Chinese desert cat (F. bieti),and 13 in the Eurasian lynx (L. lynx). Of these 57 alleles, 36were new identified sequences and 21 were previouslyreported and were confirmed again in our test. The range ofthe numbers of alleles found in single individuals was fromone to six.
For all the 96 feline alleles involved in data analysis, 111of the 237 (47%) nucleotide sites were polymorphic and 47of the 79 (59%) amino acids were variable. The number ofnucleotide substitutions per site between two sequences,corrected by the Jukes–Cantor method for multiple sub-stitutions, ranged from 0.000 to 0.264 (mean of 0.137), and
310 Immunogenetics (2010) 62:307–317
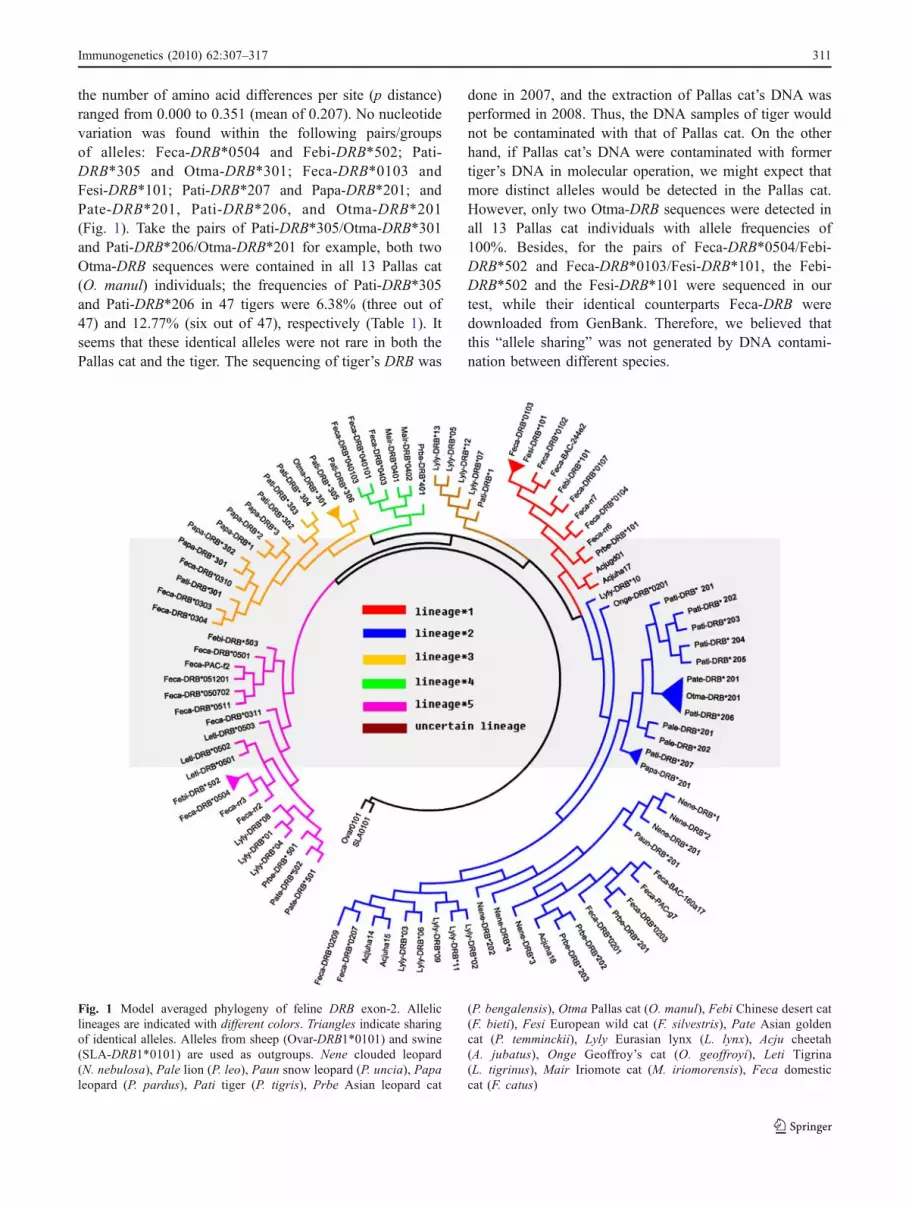
the number of amino acid differences per site (p distance)ranged from 0.000 to 0.351 (mean of 0.207). No nucleotidevariation was found within the following pairs/groupsof alleles: Feca-DRB*0504 and Febi-DRB*502; Pati-DRB*305 and Otma-DRB*301; Feca-DRB*0103 andFesi-DRB*101; Pati-DRB*207 and Papa-DRB*201; andPate-DRB*201, Pati-DRB*206, and Otma-DRB*201(Fig. 1). Take the pairs of Pati-DRB*305/Otma-DRB*301and Pati-DRB*206/Otma-DRB*201 for example, both twoOtma-DRB sequences were contained in all 13 Pallas cat(O. manul) individuals; the frequencies of Pati-DRB*305and Pati-DRB*206 in 47 tigers were 6.38% (three out of47) and 12.77% (six out of 47), respectively (Table 1). Itseems that these identical alleles were not rare in both thePallas cat and the tiger. The sequencing of tiger’s DRB was
done in 2007, and the extraction of Pallas cat’s DNA wasperformed in 2008. Thus, the DNA samples of tiger wouldnot be contaminated with that of Pallas cat. On the otherhand, if Pallas cat’s DNA were contaminated with formertiger’s DNA in molecular operation, we might expect thatmore distinct alleles would be detected in the Pallas cat.However, only two Otma-DRB sequences were detected inall 13 Pallas cat individuals with allele frequencies of100%. Besides, for the pairs of Feca-DRB*0504/Febi-DRB*502 and Feca-DRB*0103/Fesi-DRB*101, the Febi-DRB*502 and the Fesi-DRB*101 were sequenced in ourtest, while their identical counterparts Feca-DRB weredownloaded from GenBank. Therefore, we believed thatthis “allele sharing” was not generated by DNA contami-nation between different species.
Fig. 1 Model averaged phylogeny of feline DRB exon-2. Alleliclineages are indicated with different colors. Triangles indicate sharingof identical alleles. Alleles from sheep (Ovar-DRB1*0101) and swine(SLA-DRB1*0101) are used as outgroups. Nene clouded leopard(N. nebulosa), Pale lion (P. leo), Paun snow leopard (P. uncia), Papaleopard (P. pardus), Pati tiger (P. tigris), Prbe Asian leopard cat
(P. bengalensis), Otma Pallas cat (O. manul), Febi Chinese desert cat(F. bieti), Fesi European wild cat (F. silvestris), Pate Asian goldencat (P. temminckii), Lyly Eurasian lynx (L. lynx), Acju cheetah(A. jubatus), Onge Geoffroy’s cat (O. geoffroyi), Leti Tigrina(L. tigrinus), Mair Iriomote cat (M. iriomorensis), Feca domesticcat (F. catus)
Immunogenetics (2010) 62:307–317 311

No recombination event was detected in the recombina-tion analysis. Little substitution saturation was found toaffect phylogenetic inference because, for codon positions1, 2, 3 and the whole length sequence, the Iss.c was alwayssignificantly larger than Iss in both symmetrical andasymmetrical topology (P<0.05). Topology inferred bythe ML-based model averaged consensus method is shownin Fig. 1. Highly similar to a previous report on domesticcat (Yuhki and O'Brien 1997), the topology of feline DRB
showed five distinct allelic lineages with the exception ofan additional uncertain lineage. The ML tree of the smallerdata set (Fig. 2) also clearly showed the five-lineage patternof feline MHC-DRB with reasonable bootstrap values(>50%) that comparative as in Yuhki and O’Brien (1997).Therefore, we used the definition of allelic lineages suggestedby Yuhki and O'Brien (1997) to specify the name of lineagesin our study. The lineage*2, *5, *1, *3, and *4 contained 12,six, five, four, and three species, respectively. The lineage*2
Feca-DRB*0103 Fesi-DRB*101 Feca-DRB*0102 Feca-BAC-244e2 Feca-DRB*0107 Febi-DRB*101 Feca-rr7 Feca-DRB*0104
lineage*1 (98%)
Prbe-DRB*0402 Prbe-DRB*401 Feca-DRB*040101 Feca-DRB*040103 Feca-DRB*0403
lineage*4 (67%)
Feca-DRB*0304 Feca-DRB*0303 Pati-DRB*301 Papa-DRB*301 Papa-DRB*302 Feca-DRB*0310
lineage*3 (90%)
(57%)
(57%)
Nene-DRB*3 Acjuha16 Prbe-DRB*203 Prbe-DRB*202 Feca-BAC-160a17 Feca-PAC-g7 Prbe-DRB*201 Feca-DRB*0203 Feca-DRB*0201 Paun-DRB*201 Nene-DRB*201 Nene-DRB*2 Nene-DRB*1 Pale-DRB*201 Pale-DRB*202 Pati-DRB*204 Pati-DRB*205 Pati-DRB*201 Pati-DRB*203 Pati-DRB*202 Pate-DRB*201 Otma-DRB*201 Pati-DRB*206 Papa-DRB*201 Pati-DRB*207 Lyly-DRB*02 Lyly-DRB*11 Lyly-DRB*09 Feca-DRB*0207 Feca-DRB*0209 Acjuha15 Acjuha14 Lyly-DRB*06 Lyly-DRB*03 Nene-DRB*202 Nene-DRB*4
lineage*2 (51%)
(49%)
Feca-DRB*0504 Febi-DRB*502 Feca-rr3 Feca-rr2 Leti-DRB*0501 Leti-DRB*0502 Lyly-DRB*08 Lyly-DRB*04 Lyly-DRB*01 Leti-DRB*0503 Pate-DRB*502 Pate-DRB*501 Prbe-DRB*501 Feca-DRB*051201 Feca-PAC-f2 Feca-DRB*0511 Feca-DRB*050702 Feca-DRB*0501
lineage*5 (55%)
(97%)
SLA0101 Ovar0101 outgroups (97%)
Fig. 2 The ML tree of thesmaller data set (73 alleles) withcomparative bootstrap values asin Yuhki and O’Brien (1997).The bootstrap values of nodesare indicated in numbers ordescribed in brackets
312 Immunogenetics (2010) 62:307–317

of Fig. 1, which was assumed to be the oldest lineage (see“Discussion” section), was involved in reconciliation withthe accepted neutral-marker tree (Fig. 3), and 45 “deepcoalescent” events were calculated to account for theincongruence between the two trees.
When the topology of Fig. 1 was subjected to the“random-site” models implemented in PAML 4, the modelM0 gave a result of ω (dN/dS)=0.416, revealing overallpurifying selection acting over feline MHC-DRB. However,the null models of neutrality were significantly rejected(P<0.001, df=2) in both LRTs (M1a vs. M2a and M7 vs.M8), indicating that signals of positive selection weredetected. Three codon sites (9th, 57th, and 86th) wereinferred to be subject to positive selection with high ωvalues under the Bayes empirical Bayes (BEB) analysis(Table 2).
When the smaller data set (topology of Fig. 2) was used,similar results were obtained under the “random-site”models (Table 3), with the exception that only two sites(57th and 86th) were inferred to be under positive selection.Results of the “test 2 (model A1 vs. model A)” showed thatthe null model A1 (with ω2=1 fixed) could not be rejectedwhen Bonferroni’s correction was involved. However,under the clade model, sites 57th and 86th were detectedto be subject to divergent pressure between foregroundlineages and background lineages, no matter which lineagewas assigned as the foreground (Table 4).
Discussion
Lineage pattern and signature of trans-speciespolymorphism
Yuhki and O’Brien (1997) have defined five DRB exon-2allelic lineages in the domestic cat and three other felids, sothe alleles from the domestic cat can be used to indicatefeline allelic lineages when more felids are involved. In ourtest, we chose 24 alleles of the domestic cat which has beenfound to be repeatable in different labs and alleles fromthe other 15 felid species (described in “Materials andmethods” section) to set up our analysis. The data hereincome from seven of eight putative extant Felidae lineages(except the caracal lineage), according to Johnson et al.(2006). The ML-based model averaged consensus treeclearly showed a pattern of five distinct allelic lineages(Fig. 1), to which we specified the lineage name accordingto Yuhki and O'Brien’s (1997) definition.
Since we followed a strict cloning criterion, singleindividuals in our test possessed up to six distinctsequences, revealing that there are at least three DRB lociexisting in the felid genome. Similar results have beenfound in some species such like the domestic cat and thecheetah, in which up to three separate FLA-DRB loci werefound (Drake et al. 2004). In fact, a complete nucleotidesequence spanning the domestic cat classical and extended
Fig. 3 Topology of lineage*2 extracted from Fig. 1 (right) isreconciled with the accepted neutral-marker tree (left). The neutral-marker tree is extracted from Johnson et al. (2006). Separation of linesradiating from the tips of the species tree into different branches of the
gene tree indicates extensive trans-species evolution of DRB alleles. Atotal of 45 “deep coalescent” events are detected to be required forreconciling the two topologies, according to the method of Page andCharleston (1997)
Immunogenetics (2010) 62:307–317 313

class II region has recently confirmed that there are exactlythree functional DRB loci (DRB 1, 3, and 4); the DRB2lacks the full complement of exons (with only a remnant ofexon-1) and thus represents a pseudogene (Yuhki et al.2007). However, different level of gene’s birth and deathleads to a new locus (DRB5) adjacent to DRB1 in the DRhaplotype 2 of domestic cat (Yuhki et al. 2008). Thiscomplexity of gene duplication makes it hard to specifyeach lineage to any specific locus.
Balancing selection maintains TSP and leads to alleliclineage sharing (Klein et al. 2007). An extreme case of TSPis that different species share identical alleles. Five allele-sharing events (no variation between alleles) were foundin our test (Fig. 1), revealing that balancing selectionmaintains ancient alleles across speciation.
Assuming in a diploid population, allele “A” is wild typeand allele “a” is mutant type, then for the fitness (F) of thealleles: (1) if FAA=FAa=Faa, the neutrality is expected; (2)if FAA<FAa<Faa, “a” is expected to be an advantageousmutant and the directional positive selection will increasethe fixation of the advantageous mutants; (3) if FAA>FAa>Faa, “a” is a deleterious mutant and the purifying selectionwill eliminate the deleterious mutants; and (4) if FAA<FAa>Faa, namely the heterozygote shows more advantage than
homozygote, then the balancing selection is proposed notonly to maintain the advantageous mutants for a long timebut also to diversify the genetic variance to ensure adequateheterozygosis (Dobzhansky 1970). By examining the allelicdistribution in our tree, we found the obtained alleles fromthe Panthera lineage (clouded leopard N. nebulosa, lionP. leo, snow leopard P. uncia, leopard P. pardus, and tigerP. tigris) were fastened on lineages *2 and *3, especially allsix clouded leopard’s alleles appeared only in lineage*2.Considering that the expected balancing selection maintainsthe ancient allelic lineage for a long history (even throughmany speciation events) and such ancient lineage would beshared by many extant species, we inferred that lineage*2,which contains all Panthera lineage species and possesses12 species in our investigation, is the oldest allelic lineage.The maintenance of lineage*2 could even be traced back atleast to 10.8 MY, to which point the Panthera lineagediverged from the other lineages of felid species (Johnsonet al. 2006).
Regardless of the recent duplication event, we assumedthat all alleles in lineage*2 are orthologs from the samelocus because in such ancient lineage, some orthologs areexpected to have been maintained for a long history; if anancient duplication has occurred, the paralogous sequences
Table 2 Log-likelihood values and parameter estimates under random-site models for feline DRB alleles
Model NFP ℓ Estimates of parameters Positively selected sitesa,b
M0: one ratio 1 −3,810.46 ω=0.416 None
M1a: neutral 2 −3,511.69 p0=0.645 (p1=0.355), ω0=0.047 (ω1=1) Not allowed
M2a: selection 4 −3,435.73 p0=0.609, p1=0.353 (p2=0.038), ω0=0.044 (ω1=1), ω2=7.309 9th, 57th, and 86th
M3: discrete 5 −3,435.19 p0=0.619, p1=0.342 (p2=0.038), ω0=0.052, ω1=1.126, ω2=7.812 9th, 57th, and 86th
M7: beta 2 −3,490.31 p=0.164, q=0.335 Not allowed
M8: beta&ω 4 −3,425.59 p0=0.962 (p1=0.038), p=0.160, q=0.307, ω=6.230 9th, 57th, and 86th
NFP number of free parametersa Sites are identified with ω>6 at cutoff p>99% (BEB)b Number of sites is corresponding to that of HLA-DRB specified by Stern et al. (1994)
Table 3 Log-likelihood values and parameter estimates under random-site models for the smaller data set of feline DRB alleles
Model NFP ℓ Estimates of parameters Positively selected sitesa,b
M0: one ratio 1 −2,663.67 ω=0.368 None
M1a: neutral 2 −2,490.14 p0=0.652 (p1=0.348), ω0=0.049 (ω1=1) Not allowed
M2a: selection 4 −2,450.07 p0=0.622, p1=0.353 (p2=0.025), ω0=0.045 (ω1=1), ω2=10.429 57th, 86th
M3: discrete 5 −2,450.04 p0=0.619, p1=0.356 (p2=0.025), ω0=0.043, ω1=0.971, ω2=10.261 57th, 86th
M7: beta 2 −2,479.64 p=0.166, q=0.377 Not allowed
M8: beta&ω 4 −2,442.54 p0=0.975 (p1=0.025), p=0.159, q=0.337, ω=8.828 57th, 86th
NFP number of free parametersa Sites are identified with ω>8 at cutoff p>99% (BEB)b Number of sites is corresponding to that of HLA-DRB specified by Stern et al. (1994)
314 Immunogenetics (2010) 62:307–317

should have diverged enough to be distinguished andformed different lineages in the phylogenetic tree; if a recentduplication has occurred, the paralogs (even identical) wouldcluster very closely together, and this would not substantiallyaffect the calculation of “deep coalescent”. To quantify thedegree of trans-species evolution, topology of lineage*2 wasused to reconcile with the accepted neutral-marker tree, towhich 45 “deep coalescent” events were detected to accountfor the incongruence between the trees (Fig. 3). A “deepcoalescent” event in such a case, as described by Gutierrez-Espeleta et al. (2001), can be defined as polymorphism in theancestral species being retained in each of the descendantspecies, a phenomenon also known as trans-species evolu-tion. Under the simplest models of neutrality and drift, thereshould be complete congruence between gene and speciestrees, and a set of species separated by >4Ne generationsshould have (n−1) inter-specific coalescent events, where nis the number of species sampled (Edwards et al. 1997;Garrigan and Hedrick 2003). Therefore, the 45 “deepcoalescent” events detected in lineage*2 is about 4-foldhigher than that expected from inter-specific coalescentevents when there is complete congruence between the geneand species trees (n−1=11), which confirms that balancingselection drives the evolution of genes in lineage*2.
Selective pressure indicated by dN/dS
Besides TSP, from the long-term evolutionary point ofview, another common method for detecting positiveselection (including both balancing and directional selec-tion) is to calculate the ratio of the non-synonymoussubstitution rate to the synonymous substitution rate(ω=dN/dS) among aligned sequences, where the ratio >1,<1, and =1 indicate positive selection, negative (purifying)selection, and neutral evolution, respectively (Li andGojobori 1983; Nei 2005).
In our test with maximum likelihood estimates, the one-ratio model (M0) which averages dN/dS over all sites and
over all braches gave results of ω<1, revealing overallpurifying selection acting over feline MHC-DRB. However,because purifying selection is ubiquitous and tends todominate in evolution, positive selection most likely affectsonly a few sites at a few time points (Nielsen 2005; Yang2002). In both the 96-allele and the smaller data sets, sites57th and 86th were confirmed by M2a and M8 to besubject to positive selection with high ω values under theBEB analysis. In the analysis of the HLA-DR1 crystalstructure, Stern et al. (1994) listed 20 codon sites thatcorresponding to peptide binding: 9W, 11L, 13F, 28E, 47Y,56P, 57D, 60Y, 61W, 67L, 70Q, 71R, 74A, 78Y, 81H, 82N,85V, 86G, 89F, and 90T. Comparing with this description,these two codon sites suggested under positive selectionacross feline radiation are all within the PBR. Furlong andYang (2008) investigated selective pressure on DRB acrossseveral mammals and found that several sites (11L, 13F,57D, 74A, and 86G) were in common subject to positiveselection in at least five mammals. Of these, the 57th and86th sites are also confirmed in felids in our test. It seemsthat variation at these two sites favor amino acid alterna-tions for adapting to environmental change and variousantigens, and this property can be traced back even to alonger evolutionary history of mammals rather than just thefeline radiation.
Under the “branch-site” models (Table 4), the multiple“test 2 (model A1 vs. model A)” with each of the fivelineages in turn as foreground failed to significantly rejectthe null model A1 in all LRTs, indicating that no positivelyselective sites were specific to any foreground lineage.Instead, if we did not require the condition of ω2≥1 but justallowed sites with different ω between the foreground andbackground lineages, then these sites were expected toevolve under so-called divergent selective pressure. In thisscenario, multiple LRTs (M1a vs. model C) with singlelineage in turn as foreground provided significant rejectionof null M1a, after Bonferroni’s correction (P<0.001, df=3).We notice that the two sites (57th and 86th) detected to be
Table 4 Log-likelihood values and suggested sites under branch-site and clade models with different lineage as foreground
Model Foreground lineage
Lineage*1 Lineage*2 Lineage*3 Lineage*4 Lineage*5
ℓ Sitesa ℓ Sitesa ℓ Sitesa ℓ Sitesa ℓ Sitesa
LRTs of test 2 (df=1)
Model A1 −2,488.99 – −2,492.54 – −2,488.72 – −2,491.32 – −2,492.00 –
Model A −2,488.32 62th −2,491.12 86th −2,487.68 None −2,490.35 None −2,491.66 86th
LRTs of clade model (df=3)
M1a −2,490.14 – −2,490.14 – −2,490.14 – −2,490.14 – −2,490.14 –
Model C −2,476.47 57th, 86th −2,473.89 57th, 86th −2,477.33 57th, 86th −2,475.78 57th, 86th −2,476.36 57th, 86th
a Sites are identified under positive selection or divergent selective pressure with BEB probability >95%
Immunogenetics (2010) 62:307–317 315

under divergent selective pressure are those suggestedunder positive selection in “random-site” models. However,these sites are not lineage specific because the multipletesting with any lineage as the foreground can result insignificant rejection of the null model.
From the long-term evolutionary point of view, positiveselection acting on MHC is expected to both retainmutations longer than normal expectations under a neutralmodel and increase the apparent rate of nucleotidesubstitution relative to neutral expectations. As discussedabove, both trans-species polymorphism and dN/dS methodsproperly reflect the features of long-term evolution of theMhc-DRB genes in feline radiation. This monitoring willhelp to understand the formation of the allelic lineagepattern and the exiguous changes among lineages andfurther help to get insights into the nature of selective forcesat molecular level in feline MHC evolutionary processes.
Acknowledgments We thank Beijing Zoo, Changsha Zoo, ChengduZoo, Chongqing Zoo, Fuzhou Zoo, Guiyang Zoo, Jiujiang Zoo,Kunming Zoo, Lanzhou Zoo, Luoyang Zoo, Nanchang Zoo, GuangzhouZoo, Shanghai Zoo, Suzhou Zoo, Urumchi Zoo, Wuhan Zoo, XiamenZoo, Xining Zoo, Guizhou Museum, and Heilongjiang Siberia TigerPark for providing felid samples in this study. This study was supportedby National Basic Research Program of China (973 Project:2007CB411605), Chengdu Giant Panda Research Fund (CPF06001),and China Scholarship Council (CSC2007102736).
References
Anisimova A, Yang Z (2007) Multiple hypothesis testing to detectlineages under positive selection that affects only a few sites. MolBiol Evol 24:1219–1228
Bielawski JP, Yang Z (2004) A maximum likelihood method fordetecting functional divergence at individual codon sites, withapplication to gene family evolution. J Mol Evol 59:121–132
Bos DH, Waldman B (2006) Evolution by recombination andtransspecies polymorphism in the MHC class I gene of Xenopuslaevis. Mol Biol Evol 23:137–143
Dobzhansky T (1970) Genetics of the evolutionary process. ColumbiaUniversity Press, New York
Drake GJ, Kennedy LJ, Auty HK et al (2004) The use of referencestrand-mediated conformational analysis for the study of cheetah(Acinonyx jubatus) feline leucocyte antigen class II DRB poly-morphisms. Mol Ecol 13:221–229
Edwards SV, Chesnut K, Satta Y, Wakeland EK (1997) Ancestralpolymorphism of Mhc class II genes in mice: implications forbalancing selection and the mammalian molecular clock. Genetics146:655–668
Felsenstein J (2005) PHYLIP (Phylogeny Inference Package) version3.6. Distributed by the author. Department of Genome Sciences,University of Washington, Seattle (USA)
Furlong RF, Yang Z (2008) Diversifying and purifying selection in thepeptide binding region of DRB in mammals. J Mol Evol 66:384–394
Garrigan D, Hedrick PW (2003) Perspective: detecting adaptivemolecular polymorphism: lessons from the MHC. Evolution57:1707–1722
Gutierrez-Espeleta GA, Hedrick PW, Kalinowski ST, Garrigan D,Boyce WM (2001) Is the decline of desert bighorn sheep frominfectious disease the result of low MHC variation? Heredity86:439–450
Hedrick PW (2007) Balancing selection. Curr Biol 17:R230–R231Hughes AL, Yeager M (1998) Natural selection at major histocompat-
ibility complex loci of vertebrates. Annu Rev Genet 32:415–435Janczewski DN, Modi WS, Stephens JC, O'Brien SJ (1995) Molecular
evolution of mitochondrial 12S RNA and cytochrome bsequences in the pantherine lineage of Felidae. Mol Biol Evol12:690–707
Johnson WE, Eizirik E, Pecon-Slattery J et al (2006) The late Mioceneradiation of modern Felidae: a genetic assessment. Science311:73–77
Kennedy LJ, Ryvar R, Gaskell RM et al (2002) Sequence analysis ofMHC DRB alleles in domestic cats from the United Kingdom.Immunogenetics 54:348–352
Kennedy LJ, Ryvar R, Brown JJ, Ollier WE, Radford AD (2003)Resolution of complex feline leukocyte antigen DRB loci byreference strand-mediated conformational analysis (RSCA).Tissue Antigens 62:313–323
Klein J, Sato A (1998) Birth of the major histocompatibility complex.Scand J Immunol 47:199–209
Klein J, Bontrop RE, Dawkins RL et al (1990) Nomenclature for themajor histocompatibility complexes of different species: aproposal. Immunogenetics 31:217–219
Klein J, Sato A, Nagl S, O'hUigin C (1998) Molecular trans-speciespolymorphism. Annu Rev Ecol Syst 29:1–21
Klein J, Sato A, Nikolaidis N (2007) MHC, TSP, and the origin ofspecies: from immunogenetics to evolutionary genetics. AnnuRev Genet 41:281–304
Kuwahara Y, Kitoh K, Kobayashi R et al (2000) Genotyping of felineMHC (FLA) class II DRB by PCR-RFLP method using group-specific primers. J Vet Med Sci 62:1283–1289
Larkin MA, Blackshields G, Brown NP et al (2007) Clustal W andClustal X version 2.0. Bioinformatics 23:2947–2948
Leyhausen P, Pfleiderer M (1999) The systematic status of theIriomote cat (Prionailurus iriomotensis Imaizumi 1967) and thesubspecies of the leopard cat (Prionailurus bengalensis Kerr1792). J Zool Syst Evol Res 37:121–131
Li W-H, Gojobori T (1983) Rapid evolution of goat and sheep globingenes following gene duplication. Mol Biol Evol 1:94–108
Luo SJ, Kim JH, Johnson WE et al (2004) Phylogeography and geneticancestry of tigers (Panthera tigris). PLoS Biol 2:2275–2293
Martin DP, Williamson C, Posada D (2005) RDP2: recombinationdetection and analysis from sequence alignments. Bioinformatics21:260–262
Nei M (2005) Selectionism and neutralism in molecular evolution.Mol Biol Evol 22:2318–2342
Nielsen R (2005) Molecular signatures of natural selection. Annu RevGenet 39:197–218
Nielsen R, Yang Z (1998) Likelihood models for detecting positivelyselected amino acid sites and applications to the HIV-1 envelopegene. Genetics 148:929–936
O'Brien SJ, Yuhki N (1999) Comparative genome organization of themajor histocompatibility complex: lessons from the Felidae.Immunol Rev 167:133–144
O'Brien SJ, Johnson WE (2007) The evolution of cats. Sci Am297:68–75
Page RDM (1998) GeneTree: comparing gene and species phyloge-nies using reconciled trees. Bioinformatics 14:819–820
Page RDM, Charleston MA (1997) From gene to organismalphylogeny: reconciled trees and the gene tree/species treeproblem. Mol Phylogenet Evol 7:231–240
Posada D (2008) jModelTest: phylogenetic model averaging. Mol BiolEvol 25:1253–1256
316 Immunogenetics (2010) 62:307–317

Posada D, Buckley TR (2004) Model selection and model averagingin phylogenetics: advantages of the AIC and Bayesianapproaches over likelihood ratio tests. Syst Biol 53:793–808
Robinson J, Waller MJ, Parham P et al (2003) IMGT/HLA and IMGT/MHC: sequence databases for the study of the major histocom-patibility complex. Nucleic Acids Res 31:311–314
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-basedphylogenetic analyses with thousands of taxa and mixed models.Bioinformatics 22:2688–2690
Stern LJ, Brown JH, Jardetzky TS et al (1994) Crystal structure of thehuman class II MHC protein HLA-DR1 complexed with aninfluenza virus peptide. Nature 368:215–221
Takahashi K, Rooney AP, Nei M (2000) Origins and divergence timesof mammalian class II MHC gene clusters. J Hered 19:198–204
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: MolecularEvolutionary Genetics Analysis (MEGA) software version 4.0.Mol Biol Evol 24:1596–1599
Wang Q, Wu X, Yan P, Zheng S (2008) Sequence variability analysisof major histocompatibility complex class II DRB alleles in threefelines. Front Biol China 3:55–62
Wang X, Wei K, Zhang Z, Xu X, Zhang W, Shen F, Zhang L, Yue B(2009) MHC Class II DRB exon-2 diversity of the Eurasian lynx(Lynx lynx) in China. J Nat Hist 43:245–257
Xia X, Xie Z (2001) DAMBE: data analysis in molecular biology andevolution. J Hered 92:371–373
Xia X, Xie Z, SalemiM, Chen L,WangY (2003) An index of substitutionsaturation and its application. Mol Phylogenet Evol 26:1–7
Yang Z (2002) Inference of selection from multiple species align-ments. Curr Opin Genet Dev 12:688–694
Yang Z (2007) PAML 4: a program package for phylogenetic analysisby maximum likelihood. Mol Biol Evol 24:1586–1591
Yang Z, Nielsen R, Goldman N, Pedersen A-MK (2000) Codon-substitution models for heterogeneous selection pressure at aminoacid sites. Genetics 155:431–449
Yang Z, Wong WSW, Nielsen R (2005) Bayes empirical Bayesinference of amino acid sites under positive selection. Mol BiolEvol 22:1107–1118
Yuhki N, O’Brien SJ (1997) Nature and origin of polymorphism infeline MHC class II DRA and DRB genes. J Immunol 158:2822–2833
Yuhki N, Beck T, Stephens RM, Nishigaki Y, Newmann K, O'Brien SJ(2007) Comparative genome organization of human, murine, andfeline MHC class II region. Genome Res 13:1169–1179
Yuhki N, Mullikin JC, Beck T, Stephens R, O'Brien SJ (2008)Sequences, annotation and single nucleotide polymorphism ofthe major histocompatibility complex in the domestic cat. PLoSONE 3:e2674
Zhang J, Nielsen R, Yang Z (2005) Evaluation of an improved branch-site likelihood method for detecting positive selection at themolecular level. Mol Biol Evol 22:2472–2479
Immunogenetics (2010) 62:307–317 317





























