Embed Size (px)
Citation preview

KDIGO CLINICAL PRACTICE GUIDELINE FOR GLOMERULONEPHRITIS
Public Review Draft
March 2011
CONFIDENTIAL: DO NOT DISTRIBUTE

KDIGO® GN Guideline March 2011 Public Review Draft
ii
Table of Contents
Disclaimer ...................................................................................................................................... iv Work Group Membership ............................................................................................................... v KDIGO Board Members ............................................................................................................... vii Reference Keys ............................................................................................................................ viii Abbreviations and Acronyms ........................................................................................................ ix Recommendation Statements ......................................................................................................... xi
Chapter 1: Introduction ................................................................................................................... 1 Chapter 2: General Principles in the Management of Glomerular Disease .................................... 3 Chapter 3: Treatment of Steroid-Sensitive Nephrotic Syndrome in Children .............................. 15 Chapter 4: Steroid-Resistant Nephrotic Syndrome in Children .................................................... 33 Chapter 5: Minimal-Change Disease in Adults ............................................................................ 43 Chapter 6: Treatment of Adult Patients with Idiopathic Focal Segmental Glomerulosclerosis ... 51 Chapter 7: Idiopathic Membranous Nephropathy ......................................................................... 62 Chapter 8: Membranoproliferative Glomerulonephritis ............................................................... 89 Chapter 9: Infection-Related Glomerulonephritis ........................................................................ 93 Chapter 10: Immunoglobulin A Nephropathy ........................................................................... 116 Chapter 11: Henoch-Schönlein Purpura Nephritis .................................................................... 135 Chapter 12: Treatment of Lupus Nephritis ................................................................................. 142 Chapter 13: Treatment of Pauci-immune Focal and Segmental Necrotizing
Glomerulonephritis ............................................................................................................ 168 Chapter 14: Treatment of Anti-Glomerular Basement Membrane Antibody
Glomerulonephritis ............................................................................................................ 182
Appendix: Methods for Guideline Development ........................................................................ 188 Biographic and Disclosure Information ...................................................................................... 206

KDIGO® GN Guideline March 2011 Public Review Draft
iv
DISCLAIMER
SECTION I: USE OF THE CLINICAL PRACTICE GUIDELINE
This Clinical Practice Guideline document is based upon the best information available as of January 2011. It is designed to provide information and assist decision-making. It is not intended to define a standard of care, and should not be construed as one, nor should it be interpreted as prescribing an exclusive course of management. Variations in practice will inevitably and appropriately occur when clinicians take into account the needs of individual patients, available resources, and limitations unique to an institution or type of practice. Every health-care professional making use of these recommendations is responsible for evaluating the appropriateness of applying them in the setting of any particular clinical situation. The recommendations for research contained within this document are general and do not imply a specific protocol.
SECTION II: DISCLOSURE
Kidney Disease: Improving Global Outcomes (KDIGO) makes every effort to avoid any actual or reasonably perceived conflicts of interest that may arise as a result of an outside relationship or a personal, professional, or business interest of a member of the Work Group. All members of the Work Group are required to complete, sign, and submit a disclosure and attestation form showing all such relationships that might be perceived or actual conflicts of interest. This document is updated annually and information is adjusted accordingly. All reported information will be printed in the final publication and are on file at the National Kidney Foundation (NKF), Managing Agent for KDIGO.
Note: KDIGO Clinical Practice Guideline for Glomerulonephritis is not final. Please do not quote or reproduce any part of this document.

KDIGO® GN Guideline March 2011 Public Review Draft
v
WORK GROUP MEMBERSHIP
Work Group Co-Chairs
Daniel C. Cattran, MD, FRCPC Toronto General Hospital Toronto, Canada
John Feehally, DM, FRCP University Hospitals of Leicester Leicester, United Kingdom
Work Group
H. Terence Cook, MBBS, MRCP, MRCPath, FRCPath, FMedSci Imperial College London London, United Kingdom Fernando C. Fervenza, MD, PhD Mayo Clinic Rochester, MN Jürgen Floege, MD University hospital, RWTH Aachen Aachen, Germany Debbie Gipson, MD, MS University of Michigan Ann Arbor, MI Richard J. Glassock, MD, MACP The Geffen School of Medicine at UCLA Laguna Niguel, CA Elisabeth M. Hodson, MBBS, FRACP The Children's Hospital at Westmead Westmead, Australia Vivekanand Jha, MD, FRCP, FRCP, FAMS Postgraduate Institute of Medical Education Chandigarh, India Philip K. T. Li, MD, FRCP, FACP Chinese University of Hong Kong Hong Kong, China
Zhi-Hong Liu, MD Nanjing University School of Medicine Nanjing, China Sergio A. Mezzano, MD, FASN, FACP Universidad Austral Valdivia, Chile
Patrick H. Nachman, MD University of North Carolina Chapel Hill, NC Manuel Praga, MD, PhD Hospital 12 de Octubre Madrid, Spain Jai Radhakrishnan, MD, MS, MRCP, FACC, FASNNew York Presbyterian-Columbia New York, NY Brad H. Rovin, MD, FACP, FASN The Ohio State University College of Medicine Columbus, OH Stéphan Troyanov, MD Hôpital du Sacré-Cœur Montreal, Canada Jack Wetzels, MD, PhD Radboud University Nijmegen Medical Center Nijmegen, Netherlands

KDIGO® GN Guideline March 2011 Public Review Draft
vi
EVIDENCE REVIEW TEAM
Tufts Center for Kidney Disease Guideline Development and Implementation Tufts Medical Center, Boston, MA, USA:
Ethan M. Balk, MD, MPH, Project Director; Program Director, Evidence Based Medicine Gowri Raman, MD, MS, Scientific Staff
Dana C. Miskulin, MD, MS, Staff Nephrologist Aneet Deo, MD, MS, Nephrology Fellow
Amy Earley, BS, Project Coordinator Shana Haynes, MS, DHSc, Research Assistant
In addition, support and supervision were provided by: Katrin Uhlig, MD, MS, Director, Guideline Development

KDIGO® GN Guideline March 2011 Public Review Draft
vii
KDIGO BOARD MEMBERS
Garabed Eknoyan, MD Norbert Lameire, MD, PhD
Founding KDIGO Co-Chairs
Kai-Uwe Eckardt, MD KDIGO Co-Chair
Bertram L. Kasiske, MD KDIGO Co-Chair
Omar I. Abboud, MD, FRCP Sharon Adler, MD, FASN Rajiv Agarwal, MD Sharon P. Andreoli, MD Robert Atkins, MD Gavin J. Becker, MD, FRACP Fred Brown, MBA, FACHE Daniel Cattran, MD, FRCPC Allan J. Collins, MD, FACP Josef Coresh, MD, PhD Ricardo Correa-Rotter, MD Adrian Covic, MD, PhD Jonathan C. Craig, MBChB, MM (Clin Epi), DCH, FRACP, PhD Angel de Francisco, MD Paul de Jong, MD, PhD Tilman B. Drüeke, MD, FRCP Ana Figueiredo, RN, MSc, PhD Mohammed Benghanem Gharbi, MD Gordon Guyatt, MD, MSc, BSc, FRCPC Philip Halloran, MD, PhD David Harris, MD Lai Seong Hooi, MD
Enyu Imai, MD, PhD Michel Jadoul, MD Simon Jenkins, MBE, FRCGP Vivekanand Jha, MD, FRCP Suhnggwon Kim, MD, PhD Martin K. Kuhlmann, MD Nathan W. Levin, MD, FACP Philip K.T. Li, MD, FRCP, FACP Zhi-Hong Liu, MD Pablo Massari, MD Peter A. McCullough, MD, MPH, FACC, FACP Rafique Moosa, MD Miguel C. Riella, MD Bernardo Rodriquez-Iturbe, MD Robert Schrier, MD Justin Silver, MD, PhD Lesley A. Stevens, MD, MS, FRCP Marcello Tonelli, MD, SM, FRCPC Yusuke Tsukamoto, MD Theodor Vogels, MSW David C. Wheeler, MD, FRCP Elena Zakharova, MD, PhD
NKF-KDIGO Guideline Development Staff Kerry Willis, PhD, Senior Vice-President for Scientific Activities
Michael Cheung, MA, Guideline Development Director Sean Slifer, BA, Guideline Development Manager

KDIGO® GN Guideline March 2011 Public Review Draft
viii
REFERENCE KEYS
NOMENCLATURE AND DESCRIPTION FOR RATING GUIDELINE RECOMMENDATIONS
Within each recommendation, the strength of recommendation is indicated as Level 1, Level 2, or Not Graded, and the quality of the supporting evidence is shown as A, B, C, or D.
Grade* Implications
Patients Clinicians Policy
Level 1 “We recommend”
Most people in your situation would want the recommended course of action and only a small proportion would not.
Most patients should receive the recommended course of action.
The recommendation can be evaluated as a candidate for developing a policy or a performance measure.
Level 2 “We suggest”
The majority of people in your situation would want the recommended course of action, but many would not.
Different choices will be appropriate for different patients. Each patient needs help to arrive at a management decision consistent with her or his values and preferences.
The recommendation is likely to require substantial debate and involvement of stakeholders before policy can be determined
* The additional category “Not Graded” was used, typically, to provide guidance based on common sense or where the topic does not allow adequate application of evidence. The most common examples include recommendations regarding monitoring intervals, counseling, and referral to other clinical specialists. The ungraded recommendations are generally written as simple declarative statements, but are not meant to be interpreted as being stronger recommendations than Level 1 or 2 recommendations.
Grade Quality of Evidence Meaning
A High We are confident that the true effect lies close to that of the estimate of the effect. B Moderate The true effect is likely to be close to the estimate of the effect, but there is a possibility that
it is substantially different. C Low The true effect may be substantially different from the estimate of the effect. D Very low The estimate of effect is very uncertain, and often will be far from the truth.

KDIGO® GN Guideline March 2011 Public Review Draft
ix
ABBREVIATIONS AND ACRONYMS
(in progress) ACEi Angiotensin-converting enzyme inhibitor(s) ACTH Adrenocorticotropic hormone AKI Acute kidney injury ALMS Aspreva Lupus Management Study ANCA Anti-neutrophil cytoplasmic antibody APS Antiphospholipid antibody syndrome ARB Angiotensin receptor blocker ATN Acute tubular necrosis BMI Body mass index CI Confidence interval CKD Chronic kidney disease CNI Calcineurin inhibitor CrCl Creatinine clearance eGFR Estimated glomerular filtration rate ERT Evidence Review Team ESRD End-stage renal disease FR Frequently relapsing FRNS Frequently relapsing nephrotic syndrome FSGS Focal segmental glomerulosclerosis GBM Glomerular basement membrane GFR Glomerular filtration rate GN Glomerulonephritis GRADE Grading of Recommendations Assessment, Development and Evaluation HAART Highly active antiretroviral therapy HBV Hepatitis B virus HCV Hepatitis C virus HIVAN Human immunodeficiency virus–associated nephropathy HR Hazards ratio HSP Henoch-Schönlein purpura HSV Herpes simplex virus i.v. Intravenous IFN Interferon IgAN Immunoglobulin A nephropathy IMN Idiopathic membranous nephropathy INR International normalized ratio ISKDC International Study of Kidney Disease in Children IU International units KDIGO Kidney Disease: Improving Global Outcomes LN Lupus Nephritis MCD Minimal-change disease MDRD Modification of Diet in Renal Disease MEPEX Methylprednisolone or Plasma Exchange MMF Mycophenolate mofetil MN Membranous nephropathy MPGN Mesangial proliferative glomerulonephritis MPO Myeloperoxidase NCGN Necrotizing and crescentic glomerulonephritis NS Not significant OR Odds ratio

KDIGO® GN Guideline March 2011 Public Review Draft
x
p.o. Oral(ly) PR3. Proteinase 3 RAS Renin-angiotensin system RAVE Rituximab for the Treatment of Wegener’s Granulomatosis and Microscopic Polyangiitis RCT Randomized controlled trial RR Relative risk RRT Renal replacement therapy SCr Serum creatinine SD Steroid-dependent SLE Systemic lupus erythematosus SRNS Steroid-resistant nephrotic syndrome SSNS Steroid-sensitive nephrotic syndrome TMA Thrombotic microangiopathies TTP Thrombotic thrombocytopenic purpura uPCR Urine protein:creatinine ratio

KDIGO® GN Guideline March 2011 Public Review Draft
xi
RECOMMENDATION STATEMENTS
CHAPTER 3: TREATMENT OF STEROID-SENSITIVE NEPHROTIC SYNDROME IN CHILDREN
3.1: Treatment of the initial episode of SSNS 3.1.1: We recommend that corticosteroid therapy (prednisone or
prednisolone)* be given for at least 12 weeks. (1B) 3.1.1.1: We recommend that oral prednisone be administered as a
single daily dose (1B) starting at 60 mg/m2/d or 2 mg/kg/d to a maximum 60 mg/d. (1D)
3.1.1.2: We recommend that daily oral prednisone be given for at least 4 weeks (1C) followed by alternate-day oral medication as a single daily dose starting at 40 mg/m2 or 1.5 mg/kg (maximum 40 mg on alternate days) (1D) to be continued for 2-5 months. (1B)
3.2: Treatment of relapsing SSNS with corticosteroids 3.2.1: Corticosteroid therapy for children with infrequent relapses of SSNS:
3.2.1.1: We suggest that infrequent relapses of SSNS in children be treated with a single-daily dose of prednisone 60 mg/m2 or 2 mg/kg (maximum of 60 mg/d) until the child has been in complete remission for at least 3 days. (2D)
3.2.1.2: We suggest that, after achieving complete remission, children be given prednisone as a single dose on alternate days (40 mg/m2 per dose or 1.5 mg/kg per dose: maximum 40 mg on alternate days) for at least 4 weeks. (2C)
3.2.2: Corticosteroid therapy for frequently relapsing (FR) and steroid-dependent (SD) SSNS: 3.2.2.1: We suggest that relapses in children with FR or SD SSNS be
treated with daily prednisone until the child has been in remission for at least 3 days, followed by alternate-day prednisone for at least 3 months. (2C)
3.2.2.2: We suggest that prednisone be given on alternate days in the lowest dose to maintain remission without major adverse effects in children with SD SSNS. (2D)
3.2.2.3: We suggest that daily prednisone at the lowest dose be given to maintain remission without major adverse effects in children with SD SSNS where alternate-day prednisone therapy is not effective. (2D)
3.2.2.4: We suggest that daily prednisone be given during episodes of upper respiratory tract infection to reduce the risk for relapse in children with SD SSNS already on alternate-day prednisone. (2C)
*Prednisone and prednisolone are equivalent, used in the same dosage, and have both been used in RCTs depending on the country of origin. All later references to prednisone refer to prednisone or prednisolone. All later references to oral corticosteroids refer to prednisone or prednisolone.

KDIGO® GN Guideline March 2011 Public Review Draft
xii
3.3: Treatment of FR and SD SSNS with corticosteroid-sparing agents 3.3.1: We recommend that corticosteroid-sparing agents be prescribed for
children with FR SSNS and SD SSNS, who develop steroid-related adverse effects. (1B)
3.3.2: We recommend that alkylating agents, cyclophosphamide or chlorambucil, be given as corticosteroid-sparing agents for FR SSNS. (1B) We suggest that alkylating agents, cyclophosphamide or chlorambucil, be given as corticosteroid-sparing agents for SD SSNS. (2C) 3.3.2.1: We suggest that cyclophosphamide (2 mg/kg/d) be given for 8-
12 weeks (maximum cumulative dose 168 mg/kg). (2C) 3.3.2.2: We suggest that cyclophosphamide not be started until the
child has achieved remission with corticosteroids. (2D) 3.3.2.3: We suggest that chlorambucil (0.1-0.2 mg/kg/d) may be given
for 8 weeks (maximum cumulative dose 11.2 mg/kg) as an alternative to cyclophosphamide. (2C)
3.3.2.4: We suggest that second courses of alkylating agents not be given. (2D)
3.3.3: We recommend that levamisole be given as a corticosteroid-sparing agent. (1D) 3.3.3.1: We suggest that levamisole be given at a dose of 2.5 mg/kg on
alternate days (2B) for at least 12 months (2C) as most children will relapse when levamisole is stopped.
3.3.4: We recommend that the calcineurin inhibitors, cyclosporine or tacrolimus, be given as corticosteroid-sparing agents. (1C) 3.3.4.1: We suggest that cyclosporine be administered at a dose of 4-
5 mg/kg/d (starting dose) in two divided doses. (2C) 3.3.4.2: We suggest that tacrolimus 0.1 mg/kg/d (starting dose) in two
divided doses be used instead of cyclosporine when the cosmetic side-effects of cyclosporine are unacceptable. (2D)
3.3.4.3: Monitor CNI levels during therapy to limit toxicity. (Not Graded)
3.3.4.4: We suggest that CNIs be given for at least 12 months as most children will relapse when CNIs are stopped. (2C)
3.3.5: We suggest that MMF be given as a corticosteroid-sparing agent. (2C) 3.3.5.1: We suggest that MMF (starting dose 1200 mg/m2/d) be given in
two divided doses for at least 12 months, as most children will relapse when MMF is stopped. (2C)
3.3.6: We suggest that rituximab be considered only in children with SD SSNS, who have continuing frequent relapses despite optimal combinations of prednisone and corticosteroid-sparing agents, and/or who have serious adverse effects of therapy. (2D)
3.3.7: We suggest that mizoribine not be used as a corticosteroid-sparing agent in FR and SD SSNS. (2C)
3.3.8: We recommend that azathioprine not be used as a corticosteroid-sparing agent in FR and SD SSNS. (1B)

KDIGO® GN Guideline March 2011 Public Review Draft
xiii
3.4: Indication for kidney biopsy 3.4.1: Indications for kidney biopsy in children with SSNS are (Not Graded):
• late failure to respond following initial response to corticosteroids; • a high index of suspicion for a different underlying pathology; • decreasing kidney function in children receiving CNIs.
3.5: Immunizations in children with SSNS 3.5.1: To reduce the risk of serious infections in children with SSNS (Not
Graded): • Children should receive pneumococcal vaccination. • Children and their household contacts should receive influenza
vaccination annually. • Vaccination with live vaccines should be deferred until prednisone
dose is below either 1 mg/kg daily or 2 mg/kg on alternate days. • Live vaccines are contraindicated in children receiving
corticosteroid-sparing immunosuppressive agents. • Healthy household contacts should receive live vaccines to minimize
the risk of transfer of infection to the immunosuppressed child. • Following close contact with Varicella infection, nonimmune
children on immunosuppressive agents should receive zoster immune globulin if available.
CHAPTER 4: STEROID-RESISTANT NEPHROTIC SYNDROME IN CHILDREN
4.1: Evaluation of children with SRNS 4.1.1: We suggest a minimum of 8 weeks treatment with corticosteroids to
define steroid resistance. (2D) 4.1.2: The following are required to evaluate the child with SRNS (Not Graded):
• a diagnostic kidney biopsy; • evaluation of kidney function by GFR or eGFR; • quantitation of urine protein excretion.
4.2: Treatment recommendations for SRNS 4.2.1: We recommend using a calcineurin inhibitor (CNI) as first-line therapy.
(1B) 4.2.1.1: We suggest that CNI therapy be continued for a minimum of 6
months and then stopped if a partial or complete remission of proteinuria is not achieved. (2C)
4.2.1.2: We suggest CNIs be continued for a minimum of 12 months when at least a partial remission is achieved by 6 months. (2C)

KDIGO® GN Guideline March 2011 Public Review Draft
xiv
4.2.1.3: We suggest that low-dose corticosteroid therapy be combined with CNI therapy. (2D)
4.2.2: We recommend treatment with ACEi or ARBs for children with SRNS. (1B)
4.2.3: In children who fail to achieve remission with CNI therapy: 4.2.3.1: We suggest that mycophenolate mofetil (2D), high-dose
corticosteroids (2D), or a combination of these agents (2D) be considered in children who fail to achieve complete or partial remission with CNIs and corticosteroids.
4.2.3.2: We suggest that cyclophosphamide not be given to children with SRNS. (2B)
4.2.4: In patients with a relapse of nephrotic syndrome after complete remission, we suggest that therapy be restarted using one of the following options: (2C) • oral corticosteroids (2D); • return to previous successful immunosuppressive agent (2D); • an alternative immunosuppressive agent to avoid cumulative
potential toxicity (2D).
CHAPTER 5: MINIMAL-CHANGE DISEASE IN ADULTS
5.1: Treatment of initial episode of adult MCD 5.1.1: We recommend that corticosteroids be given for initial treatment of
nephrotic syndrome. (1C) 5.1.2: We suggest prednisone or prednisolone* be given at a daily single dose of
1 mg/kg (maximum 80 mg) or alternate-day single dose of 2 mg/kg (maximum 120 mg). (2C)
5.1.3: We suggest the initial high dose of corticosteroids, if tolerated, be maintained for a minimum period of 4 weeks if complete remission is achieved, and for a maximum period of 16 weeks if complete remission is not achieved. (2C)
5.1.4: In patients who remit, we suggest that corticosteroids be tapered slowly over a total period of 6 months. (2D)
5.1.5: For patients with relative contraindications or intolerance to high-dose corticosteroids (e.g., uncontrolled diabetes, psychiatric conditions, severe osteoporosis), we suggest oral cyclophosphamide or CNIs as discussed in frequently relapsing MCD. (2D)
5.1.6: We suggest using the same initial dose of corticosteroids for infrequent relapses as in Recommendation 5.1.2 until remission is achieved, followed by at least 2 months of tapering steroids. (2D)
*Prednisone and prednisolone are equivalent, used in the same dosage, and have both been used in RCTs depending on the country of origin. All later references to prednisone refer to prednisone or prednisolone. All later references to oral corticosteroids refer to prednisone or prednisolone.

KDIGO® GN Guideline March 2011 Public Review Draft
xv
5.2: FR/SD MCD 5.2.1: We suggest oral cyclophosphamide 2-2.5 mg/kg/d for 8 weeks. (2C) 5.2.2: We suggest CNI (cyclosporine 3-5 mg/kg/d or tacrolimus 0.05-0.1 mg/kg/d
in divided doses) for FR/SD MCD patients who have relapsed despite cyclophosphamide, and for people who wish to preserve fertility. (2C)
5.2.3: We suggest MMF 750-1000 mg twice daily for patients who are intolerant of corticosteroids, cyclophosphamide, and CNIs. (2D)
5.3: Corticosteroid-resistant MCD 5.3.1: Re-evaluate patients who are corticosteroid-resistant for other causes of
nephrotic syndrome. (Not Graded) A repeat kidney biopsy (if performed) commonly shows FSGS pathology (see Chapter 6 for treatment guideline for FSGS).
5.4: Supportive therapy 5.4.1: We suggest that MCD patients who have AKI be treated with renal
replacement therapy as indicated, but together with corticosteroids, as for a first episode of MCD. (2D)
5.4.2: We suggest that, for the initial episode of nephrotic syndrome associated with MCD, statins not be used to treat hyperlipidemia, and ACEi or ARBs not be used in normotensive patients to lower proteinuria. (2D)
CHAPTER 6: TREATMENT OF ADULT PATIENTS WITH IDIOPATHIC FOCAL SEGMENTAL GLOMERULOSCLEROSIS
6.1: Initial evaluation of FSGS 6.1.1: Undertake thorough evaluation to exclude secondary forms of FSGS. (Not
Graded) 6.1.2: Do not routinely perform genetic testing. (Not Graded)
6.2: Initial treatment of FSGS 6.2.1: We recommend that corticosteroid and immunosuppressive therapy be
considered only in idiopathic FSGS associated with clinical features of the nephrotic syndrome. (1C)
6.2.2: We suggest prednisone* be given at a daily single dose of 1 mg/kg (maximum 80 mg) or alternate-day dose of 2 mg/kg (maximum 120 mg). (2C)
*Prednisone and prednisolone are equivalent, used in the same dosage, and have both been used in RCTs depending on the country of origin. All later references to prednisone refer to prednisone or prednisolone. All later references to oral corticosteroids refer to prednisone or prednisolone.

KDIGO® GN Guideline March 2011 Public Review Draft
xvi
6.2.3: We suggest the initial high dose of corticosteroids be given for a minimum of 4 weeks; continue high-dose corticosteroids up to a maximum of 16 weeks, as tolerated, or until complete remission has been achieved, whichever is earlier. (2D)
6.2.4: We suggest corticosteroids be tapered slowly over a period of 6 months after achieving complete remission. (2D)
6.2.5: We suggest CNIs be considered as first-line therapy for patients with relative contraindications or intolerance to high-dose corticosteroids (e.g., uncontrolled diabetes, psychiatric conditions, severe osteoporosis). (2D)
6.3: Treatment for relapse 6.3.1: We suggest that a relapse of nephrotic syndrome is treated as per the
recommendations for relapsing MCD in adults (see Chapter 5.2). (2D)
6.4: Treatment for steroid-resistant FSGS 6.4.1: We suggest that in steroid-resistant FSGS, cyclosporine at 3-5 mg/kg/d in
divided doses be given for at least 4-6 months. (2B) 6.4.2: If there is a partial or complete remission, we suggest continuing
cyclosporine treatment for at least 12 months, followed by a slow taper. (2D)
CHAPTER 7: IDIOPATHIC MEMBRANOUS NEPHROPATHY
7.1: Evaluation of IMN 7.1.1: Perform appropriate investigations to exclude secondary causes in all
cases of idiopathic membranous nephropathy. (Not Graded)
7.2: Selection of patients with IMN to be considered for treatment with corticosteroids and immunosuppressive agents 7.2.1: We recommend that initial therapy be started ONLY in patients with
nephrotic syndrome AND when at least ONE of the following conditions are met: • urinary protein excretion persistently exceeds 4 g/d, remains >50% of
baseline value, AND does not show progressive decline, during antihypertensive and antiproteinuric therapy (see Chapter 1) during an observation period of at least 6 months; (1B)
• the presence of severe, disabling, or life-threatening symptoms related to the nephrotic syndrome; (1C)
• SCr has risen by 30% or more within 6 to 12 months from the time of diagnosis but the eGFR is not less than 25-30 ml/min per 1.73 m2 AND this change is not explained by superimposed complications. (2C)

KDIGO® GN Guideline March 2011 Public Review Draft
xvii
7.2.2: Patients with a SCr persistently >3.5 mg/dl (>320 µmol/l) or eGFR <30 ml/min per 1.73 m2 and those with marked reduction of kidney size on ultrasound should not be exposed to immunosuppressive therapy. (Not Graded)
7.3: Initial therapy of IMN 7.3.1: We recommend that initial therapy consist of a 6-month course of
alternating monthly cycles of oral and i.v. corticosteroids, and oral alkylating agents. (1B)
7.3.2: We suggest using cyclophosphamide rather than chlorambucil for initial therapy. (2B)
7.3.3: We recommend patients be managed conservatively for at least 6 months following the completion of the initial regimen before being evaluated for remission, unless kidney function is deteriorating. (1C)
7.3.4: Perform a repeat kidney biopsy only if the patient has rapidly deteriorating kidney function (doubling of SCr over 1-2 months of observation), in the absence of massive proteinuria (>15 g/d). (Not Graded)
7.3.5: Adjust the dose of cyclophosphamide or chlorambucil according to age of the patient and eGFR. (Not Graded)
7.3.6: We suggest that continuous (noncyclical) use of alkylating agents may also be effective, but is associated with greater risk of toxicity, particularly when administered for >6 months. (2C)
7.4: Alternative regimens for the initial therapy of IMN: CNIs 7.4.1: We recommend that cyclosporine or tacrolimus be used for a period of at
least 6 months in patients who meet the criteria for initial therapy (as described in Recommendation 7.1.2), but who choose not to receive the cyclical corticosteroid/alkylating-agent regimen or who have contraindications to this regimen. (See Table 7.7 for specific recommendations for dosage during therapy.) (1C)
7.4.2: We suggest that CNIs be discontinued in patients who do not achieve complete or partial remission after 6 months of treatment. (2C)
7.4.3: We suggest that the dosage of CNI be reduced at intervals of 4-8 weeks to a level of about 50% of the starting dosage and continued for at least 12 months if a complete or partial remission of proteinuria occurs, and no treatment-limiting CNI-related nephrotoxicity occurs. (2C)
7.4.4: Monitor CNI blood levels regularly during the initial treatment period, and when there is an unexplained rise in SCr (>20%) during therapy. (Not Graded). (See Table 7.7 for specific CNI-based regimen dosage recommendations.)
7.5: Regimens not recommended or suggested for initial therapy of IMN 7.5.1: We recommend that corticosteroid monotherapy not be used for initial
therapy of IMN. (1B)

KDIGO® GN Guideline March 2011 Public Review Draft
xviii
7.5.2: We suggest that monotherapy with MMF not be used for initial therapy of IMN. (2C)
7.5.3: We suggest that rituximab not be used for initial therapy of IMN. (2D) 7.5.4: We suggest that ACTH not be used for initial therapy of IMN. (2C)
7.6: Treatment of IMN resistant to recommended initial therapy 7.6.1: We suggest that patients with IMN resistant to alkylating agent–based
initial therapy be treated with a CNI. (2C) 7.6.2: We suggest that patients with IMN resistant to CNI-based initial therapy
be treated with an alkylating agent. (2C)
7.7: Treatment for relapses of nephrotic syndrome in IMN 7.7.1: We suggest that relapses of nephrotic syndrome in IMN be treated by
reinstitution of the same therapy that resulted in the initial remission. (2D)
7.7.2: We suggest that, if a 6-month cyclical corticosteroid/alkylating-agent regimen was used for initial therapy (see Recommendation 7.2.1), it be repeated only once for treatment of a relapse. (2B)
7.8: Treatment of IMN in children 7.8.1: We suggest that treatment of IMN in children follows the
recommendations for treatment of IMN in adults. (2C) (See Recommendations 7.2.1, 7.3.1.)
7.8.2 We suggest that no more than one course of the cyclical corticosteroid/alkylating-agent regimen be given in children. (2D)
7.9: Prophylactic anticoagulants in IMN 7.9.1: We suggest that patients with IMN and nephrotic syndrome, with
marked reduction in serum albumin (<2.5 g/dl [<25 g/l]) and additional risks for thrombosis, be considered for prophylactic anticoagulant therapy, using oral warfarin. (2C)
CHAPTER 8: MEMBRANOPROLIFERATIVE GLOMERULONEPHRITIS
8.1: Evaluation of MPGN 8.1.1: Evaluate patients with the histological (light microscopic) pattern of
MPGN for underlying diseases before considering a specific treatment regimen (see Table 8.1). (Not Graded)

KDIGO® GN Guideline March 2011 Public Review Draft
xix
8.2: Treatment of MPGN 8.2.1: We suggest that adults or children with idiopathic type I MPGN
accompanied by nephrotic syndrome and/or progressive decline of kidney function receive oral cyclophosphamide or MMF plus low-dose alternate-day or daily corticosteroids. (2D)
CHAPTER 9: INFECTION-RELATED GLOMERULONEPHRITIS
9.1: For the following infection-related GN, we suggest appropriate treatment of the infectious disease and standard approaches to management of the kidney manifestations: (2D) • post-streptococcal GN; • infective endocarditis-related GN; • shunt nephritis.
9.2: Hepatitis C virus (HCV) infection–related GN (Please also refer to the published KDIGO Clinical Practice Guidelines for the
Prevention, Diagnosis, Evaluation, and Treatment of Hepatitis C in Chronic Kidney Disease [14].) 9.2.1: We suggest, for HCV-infected patients with CKD Stages 1 and 2,
combined antiviral treatment using pegylated interferon and ribavirin as in the general population. (2C) [based on KDIGO HCV Recommendation 2.2.1] 9.2.1.1: Titrate ribavirin dose according to patient tolerance. (Not
Graded) 9.2.2: We suggest, for HCV-infected patients with CKD Stages 3, 4, and 5 not
yet on dialysis, monotherapy with pegylated interferon, with doses adjusted to the level of kidney function. (2D) [based on KDIGO HCV Recommendation 2.2.2]
9.2.3: We suggest that patients with HCV and mixed cryoglobulinemia (IgG/IgM) with nephrotic proteinuria or evidence of progressive kidney disease or an acute flare of cryoglobulinemia be treated with one of the following options (not necessarily in the order below), in conjunction with i.v. methylprednisolone and with antiviral therapy: (2D) • cyclophosphamide; • plasma exchange; • rituximab.
9.3: Hepatitis B virus (HBV) infection–related GN 9.3.1: We recommend that patients with HBV infection and GN receive
treatment with IFN-α or with nucleoside analogues as recommended for the general population by standard clinical practice guidelines for HBV infection (see Table 9.3). (1C)

KDIGO® GN Guideline March 2011 Public Review Draft
xx
9.3.2: We recommend that the dosing of these antiviral agents be adjusted to the degree of kidney function. (1C)
9.4: Human Immunodeficiency virus (HIV) infection–related glomerular disorders 9.4.1: We recommend that antiretroviral therapy be initiated in all patients
with biopsy-proven HIV-associated nephropathy, regardless of CD4 count. (1B)
9.5: Schistosomal, filarial, and malarial nephropathies 9.5.1: We suggest that patients with GN and concomitant malarial,
schistosomal, or filarial infection be treated with an appropriate antiparasitic agent in sufficient dosage and duration to eradicate the organism. (Not Graded)
9.5.2: We suggest that corticosteroids or immunosuppressive agents are not used for treatment of schistosomal GN. (2D)
9.5.3: We suggest that blood culture for Salmonella be performed in all patients with hepatosplenic schistosomiasis who show urinary abnormality and/or reduced GFR. (2C) 9.5.3.1: We suggest that all patients who show a positive blood culture
for Salmonella receive anti-Salmonella therapy. (2C)
CHAPTER 10: IMMUNOGLOBULIN A NEPHROPATHY
10.1: Initial evaluation including assessment of risk of progressive kidney disease 10.1.1: Assess all patients with biopsy-proven IgAN for secondary causes of
IgAN. (Not Graded) 10.1.2: Assess the risk of progression in all cases by evaluation of proteinuria,
blood pressure, and eGFR at the time of diagnosis and during follow-up. (Not Graded)
10.1.3: Pathological features may be used to assess prognosis. (Not Graded)
10.2: Antiproteinuric and antihypertensive therapy 10.2.1: We recommend long-term ACEi or ARB treatment when proteinuria is
>1 g/d. (1B) 10.2.2: We suggest ACEi or ARB treatment if proteinuria is between 0.5 to 1 g/d
(in children, between 0.5 to 1 g/d per 1.73 m2). (2D) 10.2.3: We suggest the ACEi or ARB be titrated upwards as far as tolerated to
achieve proteinuria <1 g/d. (2C) 10.2.4: The goal of blood pressure treatment in IgAN should be <130/80 mmHg
in patients with proteinuria <1 g/d, and <125/75 mmHg when initial proteinuria is >1 g/d (see Chapter 2). (Not Graded)

KDIGO® GN Guideline March 2011 Public Review Draft
xxi
10.3: Corticosteroids 10.3.1: We suggest that patients with persistent proteinuria ≥1 g/d, despite 3-6
months of optimized supportive care (including ACEi or ARBs and blood pressure control), and GFR >50 ml/min, receive a 6-month course of corticosteroid therapy. (2C)
10.4: Immunosuppressive agents (cyclophosphamide, azathioprine, MMF, cyclosporine) 10.4.1: We suggest not treating with corticosteroids combined with
cyclophosphamide or azathioprine in IgAN patients (unless there is crescentic IgAN with rapidly deteriorating kidney function; see Recommendation 10.6.3). (2D)
10.4.2: We suggest not using immunosuppressive therapy in patients with GFR <30 ml/min unless there is crescentic IgAN with rapidly deteriorating kidney function (see Section 10.6). (2C)
10.4.3: We suggest not using MMF in IgAN. (2C)
10.5: Other treatments 10.5.1: Fish oil treatment
10.5.1.1: We suggest using fish oil in the treatment of IgAN. (2D) 10.5.2: Antiplatelet agents
10.5.2.1: We suggest not using antiplatelet agents to treat IgAN. (2C) 10.5.3: Tonsillectomy
10.5.3.1: We suggest that tonsillectomy not be performed for IgAN. (2C)
10.6: Atypical forms of IgAN 10.6.1: MCD with mesangial IgA deposits
10.6.1.1: We recommend treatment as for MCD (see Chapter 5) in nephrotic patients showing pathological findings of MCD with mesangial IgA deposits on kidney biopsy. (2B)
10.6.2: AKI associated with macroscopic hematuria 10.6.2.1: Perform a repeat kidney biopsy in IgAN patients with AKI
associated with macroscopic hematuria if, after 5 days from the onset of kidney function worsening, there is no improvement. (Not Graded)
10.6.2.2: We suggest general supportive care for AKI in IgAN, with a kidney biopsy performed during an episode of macroscopic hematuria showing only ATN and intratubular erythrocyte casts. (2C)
10.6.3: Crescentic IgAN 10.6.3.1: We suggest the use of steroids and cyclophosphamide in
patients with IgAN and rapidly progressive crescentic IgAN, analogous to the treatment of ANCA vasculitis (see Chapter 13). (2D)

KDIGO® GN Guideline March 2011 Public Review Draft
xxii
CHAPTER 11: HENOCH-SCHÖNLEIN PURPURA NEPHRITIS
11.1: Treatment of HSP nephritis in children 11.1.1: We suggest that children with HSP nephritis and persistent proteinuria,
>0.5-1 g/d per 1.73 m2, are treated with ACEi or ARBs. (2D) 11.1.2: We suggest that children with persistent proteinuria, >1 g/d per 1.73 m2,
after a trial of ACEi or ARBs, and GFR >50 ml/min per 1.73 m2, be treated the same as for IgAN with a 6-month course of corticosteroid therapy (see Chapter 10). (2D)
11.2: Treatment of crescentic HSP nephritis in children 11.2.1: We suggest that children with crescentic HSP with nephrotic syndrome
and/or deteriorating kidney function are treated the same as for crescentic IgAN (see Recommendation 10.6.3). (2D)
11.3: Prevention of HSP nephritis in children 11.3.1: We recommend not using corticosteroids to prevent HSP nephritis. (1B)
11.4: HSP nephritis in adults 11.4.1: We suggest that HSP nephritis in adults be treated the same as in
children. (2D)
CHAPTER 12: TREATMENT OF LUPUS NEPHRITIS
12.1: Class I LN (minimal-mesangial LN) 12.1.1: Treat patients with class I LN with corticosteroids and
immunosuppressives only as dictated by the extrarenal clinical manifestations of lupus. (Not Graded)
12.2: Class II LN (mesangial-proliferative LN) 12.2.1: Treat patients with class II LN and proteinuria <3 g/d with
corticosteroids and immunosuppressives only as dictated by the extrarenal clinical manifestations of lupus. (Not Graded)
12.2.2: We suggest that class II LN with proteinuria >3 g/d be treated with corticosteroids or CNIs as described for MCD (see Chapter 5). (2D)
12.3: Class III LN (focal LN) and class IV LN (diffuse LN)—initial therapy 12.3.1: We recommend initial therapy with corticosteroids (1A), combined with
either cyclophosphamide (1B) or MMF (1B). 12.3.2: We suggest that, if patients have worsening LN (rising SCr, worsening
proteinuria) during their initial treatment, a change to the alternative recommended initial therapy be instituted. (2D)

KDIGO® GN Guideline March 2011 Public Review Draft
xxiii
12.4: Class III LN (focal LN) and class IV LN (diffuse LN)—maintenance therapy 12.4.1: We recommend that, after initial therapy is complete, patients with class
III and IV LN receive maintenance therapy with azathioprine (1.5-2.5 mg/kg/d) or MMF (1-3 g/d in divided doses), and low-dose oral corticosteroids (≤10 mg/d prednisone equivalent). (1B)
12.4.2: We suggest that CNIs with low-dose corticosteroids can be used for maintenance therapy in patients who are intolerant of MMF and azathioprine. (2C)
12.4.3: We suggest that, after complete remission is achieved, maintenance therapy be continued for at least 1 year before tapering the immunosuppression. (2D)
12.4.4: If complete remission has not been achieved after 12 months of maintenance therapy, consider performing a repeat kidney biopsy before determining if a change in therapy is indicated. (Not Graded)
12.4.5: While maintenance therapy is being tapered, if kidney function deteriorates and/or proteinuria worsens, we suggest that treatment be increased to the previous level of immunosuppression that controlled the LN. (2D)
12.5: Class V LN (membranous LN) 12.5.1: We recommend that patients with class V LN and non-nephrotic range
proteinuria be treated with antiproteinuria and antihypertensive medications, and only receive corticosteroids and immunosuppressives as dictated by the extrarenal manifestations of systemic lupus. (1B)
12.5.2: We suggest that patients with pure class V LN and persistent nephrotic proteinuria be treated with corticosteroids plus an additional immunosuppressive agent: cyclophosphamide (2C), or CNIs (2C), or MMF (2D), or azathioprine (2D).
12.6: General treatment of LN 12.6.1: We suggest that all patients with LN of any class be treated with
hydroxychloroquine unless they have a specific contraindication to this drug. (2C)
12.7: Class VI LN (advanced sclerosis LN) 12.7.1: We recommend that patients with class VI LN be treated with
corticosteroids and immunosuppressives only as dictated by the extrarenal manifestations of systemic lupus. (2D)

KDIGO® GN Guideline March 2011 Public Review Draft
xxiv
12.8: Relapse of LN 12.8.1: We suggest that a relapse of LN after complete or partial remission be
treated with the initial therapy followed by the maintenance therapy that was effective in inducing the original remission. (2B) 12.8.1.1: If resuming the original therapy would put the patient at risk
for excessive lifetime cyclophosphamide exposure, then we suggest a non-cyclophosphamide-based initial regimen be used (Regimen D). (2B)
12.8.2: Consider a repeat kidney biopsy during relapse if there is suspicion that the histologic class of LN has changed, or there is uncertainty whether a rising SCr and/or worsening proteinuria represents disease activity or chronicity. (Not Graded)
12.9: Treatment of resistant disease 12.9.1: For patients with worsening SCr and/or proteinuria after completing one
of the initial treatment regimens, consider a repeat kidney biopsy to distinguish active LN from scarring. (Not Graded)
12.9.2: Treat patients with worsening SCr and/or proteinuria who continue to have active LN on biopsy with one of the alternative initial treatment regimens (see Section 12.4). (Not Graded)
12.9.3: We suggest that nonresponders who have failed more than one of the recommended initial regimens (see Section 12.4) may be considered for treatment with i.v. immunoglobulin, CNIs, or rituximab. (2D)
12.10: Systemic lupus and thrombotic microangiopathy
12.10.1: We suggest that the antiphospholipid antibody syndrome (APS) involving the kidney in systemic lupus patients, with or without LN, be treated by anticoagulation (target INR 2-3). (2D)
12.10.2: We suggest that thrombotic thrombocytopenic purpura (TTP) in systemic lupus be treated as for TTP without systemic lupus. (2D)
12.11: Systemic lupus and pregnancy 12.11.1: We suggest that women be counseled to delay pregnancy until a complete
remission of LN has been achieved. (2D) 12.11.2: We recommend that cyclophosphamide, MMF, ACEi, and ARBs not be
used during pregnancy. (1A) 12.11.3: We suggest that hydroxychloroquine be continued during pregnancy.
(2B) 12.11.4: We recommend that LN patients who become pregnant while being
treated with MMF be switched to azathioprine. (1B) 12.11.5: We recommend that, if LN patients relapse during pregnancy, they
receive treatment with corticosteroids and, depending on the severity of the relapse, azathioprine. (1B)

KDIGO® GN Guideline March 2011 Public Review Draft
xxv
12.11.6: If pregnant patients are receiving corticosteroids or azathioprine, do not taper these drugs during pregnancy or for at least 3 months after delivery. (Not Graded)
12.11.7: We suggest administration of low-dose aspirin during pregnancy to decrease the risk of fetal loss. (2C)
12.12: LN in children 12.12.1: We suggest that children with LN receive the same therapies as adults
with LN, with dosing based on patient size and GFR. (2D)
CHAPTER 13: TREATMENT OF PAUCI-IMMUNE FOCAL AND SEGMENTAL NECROTIZING GLOMERULONEPHRITIS
13.1: Initial treatment of pauci-immune focal and segmental necrotizing GN 13.1.1: We recommend that cyclophosphamide and corticosteroids be used as
initial treatment for ANCA vasculitis. (1A) 13.1.2: We suggest that rituximab and corticosteroids be used as an alternative
initial treatment in patients. (1A)
13.2: Special patient populations 13.2.1: We recommend the addition of plasmapheresis for patients requiring
dialysis or with rapidly increasing SCr. (1C) 13.2.2: We suggest the addition of plasmapheresis for patients with diffuse
pulmonary hemorrhage. (2C) 13.2.3: We suggest the addition of plasmapheresis for patients with overlap
syndrome of ANCA vasculitis and anti-GBM, according to proposed criteria and regimen for anti-GBM GN (see Chapter 14). (2D)
13.2.4: We suggest discontinuing cyclophosphamide therapy after 3 months in patients who remain dialysis-dependent and who do not have any extrarenal manifestations of disease. (2C)
13.3: Maintenance therapy 13.3.1: We recommend maintenance therapy in patients who have achieved
remission. (1B) 13.3.2: We suggest continuing maintenance therapy for 12-18 months in patients
who remain in complete remission. (2D) 13.3.3: We recommend no maintenance therapy in patients who are dialysis-
dependent and have no extrarenal manifestations of disease. (1C)
13.4: Choice of agent for maintenance therapy 13.4.1: We recommend azathioprine 1-2 mg/kg/d orally as maintenance therapy.
(1B)

KDIGO® GN Guideline March 2011 Public Review Draft
xxvi
13.4.2: We suggest that MMF 1 g twice daily be used for maintenance therapy in patients who are allergic to, or intolerant of, azathioprine. (2C)
13.4.3: We suggest trimethoprim-sulfamethoxazole as an adjunct to maintenance therapy in patients with upper respiratory tract disease. (2B)
13.4.4: We suggest methotrexate (initially 0.3 mg/kg/wk, maximum 25 mg/wk) for maintenance therapy in patients intolerant of azathioprine and MMF, but not if GFR is <60 ml/min. (1C)
13.4.5: We recommend not using etanercept as adjunctive therapy. (1A)
13.5: Treatment of relapse 13.5.1: We recommend treating patients with severe relapse of ANCA vasculitis
(life- or organ-threatening) according to the same guidelines as for the initial therapy (see Section 13.1). (1C)
13.5.2: We suggest treating other relapses of ANCA vasculitis by reinstituting immunosuppressive therapy or increasing its intensity with agents other than cyclophosphamide, including instituting or increasing dose of corticosteroids, with or without azathioprine or MMF. (2C)
13.6: Treatment of resistant disease 13.6.1: In ANCA GN resistant to induction therapy with cyclophosphamide and
corticosteroids, we suggest the addition of i.v. immunoglobulin (2C) or rituximab (2D), or plasmapheresis (2D).
13.7: Monitoring 13.7.1: We recommend not changing immunosuppression based on changes in
ANCA titer alone. (2D)
13.8: Transplantation 13.8.1: We recommend delaying transplantation until patients are in complete
extrarenal remission for 12 months. (1C) 13.8.2: We recommend not delaying transplantation for patients who are in
complete remission but are still ANCA-positive. (1C)

KDIGO® GN Guideline March 2011 Public Review Draft
xxvii
CHAPTER 14: TREATMENT OF ANTI-GLOMERULAR BASEMENT MEMBRANE ANTIBODY GLOMERULONEPHRITIS
14.1: Treatment of anti-GBM GN 14.1.1: We recommend initiating immunosuppression using plasmapheresis,
cyclophosphamide, and corticosteroids (see Table 14.1) in all patients with anti-GBM GN except those who are dialysis-dependent at presentation and have 100% crescents in an adequate biopsy sample, and do not have pulmonary hemorrhage. (1B)
14.1.2: We recommend no maintenance immunosuppressive therapy for anti-GBM GN. (1C)
14.1.3: Start treatment for anti-GBM GN without delay once the diagnosis is confirmed. If the diagnosis is highly suspected, it would be appropriate to begin high-dose corticosteroids (Table 14.1) while waiting for confirmation. (Not Graded)
14.1.4: Defer kidney transplantation after anti-GBM GN until anti-GBM antibodies have been undetectable for a minimum of 6 months. (Not Graded)

KDIGO® GN Guideline March 2011 Public Review Draft
1
CHAPTER 1: INTRODUCTION
SCOPE
This clinical practice guideline has been developed to provide recommendations for the treatment of patients already diagnosed with glomerulonephritis (GN). The emphasis is on the more common forms of immune-mediated glomerular disease in both children and adults. The scope includes histologic variants of GN restricted to the kidney, as well as the most common ones associated with systemic immune-mediated disease. This guideline does not cover diagnosis or prevention of GN.
The guideline addresses the following forms of GN:
• Steroid-sensitive nephrotic syndrome (SSNS) and steroid-resistant nephrotic syndrome (SRNS) in children;
• Minimal-change disease (MCD) and idiopathic focal segmental glomerulosclerosis (FSGS) in children and adults;
• Idiopathic membranous nephropathy (IMN); • Idiopathic membranoproliferative GN; • GN associated with infections; • Immunoglobulin A (IgA) nephropathy and Henoch-Schönlein purpura (HSP)
nephritis; • Lupus nephritis (LN); • Renal vasculitis; • Antiglomerular basement membrane (anti-GBM) GN.
METHODOLOGY
The Work Group members defined the overall topics and goals for the guideline. Then, in collaboration with the evidence review team (ERT), the Work group further developed and refined each systematic review topic, specified screening criteria, literature search strategies, and data extraction forms.
The ERT performed literature searches, organized the abstracts and article screening, coordinated the methodological and analytic processes of the report, defined and standardized the methodology relating to these searches and data extraction, and produced summaries of the evidence. Using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach, they created preliminary evidence profiles (described in the appendices) that were subsequently reviewed and completed by the Work Group members. The ERT searches were updated to January 2011 and supplemented with additional studies known to the Work Group members through December 2010. Through an iterative process that involved all Work Group members, the chairs of the Work Group, and the ERT, the individual chapters were refined, reviewed, and finalized. All the details in the multiple steps involved in the assessment of grade and strength of the evidence are detailed fully in the appendices. The Work Group made

KDIGO® GN Guideline March 2011 Public Review Draft
2
two levels of recommendations (1 or 2) based on the strength of the evidence supporting the recommendation, the net medical benefit, values and preferences, and costs. Recommendations were also graded based on the overall quality of the evidence (A to D). Recommendations that provided general guidance about routine medical care (and related issues) were not graded.
The recommendations made in this guideline are directed by the available evidence to support the specific treatment options listed. When the published evidence is very weak or nonexistent no recommendations are made, although the reasons for such omissions are explained in the rationale in each chapter. There are, therefore, a number of circumstances in this guideline where treatments in wide use in current clinical practice are given only level 2 recommendations (i.e., suggested) or not included for lack of evidence.
The starting point for this guideline is that a morphological characterization of the glomerular lesion has been established by kidney biopsy or, in the case of some children with nephrotic syndrome, by characteristic clinical features. An important corollary is that the guideline does not provide recommendations on how to evaluate patients presenting with suspected glomerular disease nor when or in whom to perform a diagnostic kidney biopsy. We recognize these are relevant management issues in these patients but have chosen to begin the guideline at the point of an established diagnosis based on an adequate biopsy reviewed by a knowledgeable nephropathologist. This has dictated the starting point of our evidence-based systematic reviews and subsequent recommendations.
INTENDED USERS
This guideline was written primarily for nephrologists, although it should also be useful for other physicians, nurses, pharmacists, and health-care professionals who care for patients with GN. It was not developed for health-care administrators or regulators per se, and no attempts were made to develop clinical performance measures. This guideline was also not written directly for patients or caregivers, though appropriately drafted explanations of guideline recommendations could potentially provide useful information for these groups.

KDIGO® GN Guideline March 2011 Public Review Draft
3
CHAPTER 2: GENERAL PRINCIPLES IN THE MANAGEMENT OF GLOMERULAR DISEASE
There are a number of general principles in the management of glomerular injury which apply to most or all of the histologic variants of GN covered by this guideline. In this chapter, we discuss these general principles to minimize repetition in the guideline. Where there are specific applications or exceptions to these general statements, an expansion and rationale for these variations and/or recommendations are made in each chapter.
Kidney Biopsy Kidney biopsy is mandatory for diagnosis. It defines the morphologic patterns of GN that
will be reviewed in this guideline. The single exception to this rule is SSNS in children. This entity has an operational clinical definition that is sufficiently robust to direct initial treatment, with the kidney biopsy reserved for identifying pathology only when the clinical response is atypical.
Adequacy of kidney biopsy There are two components in terms of assessing adequacy of the tissue sample. The first
relates to the size of biopsy necessary to diagnose or exclude a specific histopathologic pattern with a reasonable level of confidence, and the second concerns the amount of tissue needed for an adequate assessment of the amount of acute or chronic damage present.
In some cases a diagnosis may be possible from examination of a single glomerulus (e.g., membranous nephropathy), but generally a substantially larger specimen is required to ensure that the material reviewed by the nephropathologist adequately represents the glomerular, tubular, interstitial, and vascular compartments of the kidney. In addition, sufficient tissue is needed to perform not only an examination by light microscopy, but also immunohistochemical staining to detect immune reactants (including immunoglobulins and complement components), and electron microscopy to define precisely the location, extent and, potentially, the specific characteristics of the immune deposits. We recognize that electron microscopy is not routinely available in many parts of the world, but the additional information defined by this technique can influence therapeutic decisions and hence it is recommended whenever possible.
In some diseases, for example necrotizing glomerulonephritis and FSGS associated with antineutrophilic cytoplasmic antibodies (ANCA), lesions are only seen in some segments of some glomeruli. In these cases, it is important that the biopsy is examined by light microscopy at several levels if lesions are not to be missed. If a lesion that affects only 5% of glomeruli is to be detected or excluded with 95% confidence, then over 20 glomeruli are needed in the biopsy [1]. Although many biopsies will have fewer glomeruli, it is important to realize that this limits diagnostic accuracy, especially when the diagnostic lesions are focal and/or segmental.
An important component of kidney biopsy examination is the assessment of “activity”, that is lesions which are acute and potentially responsive to specific therapy, and “chronicity”, where they are not reversible or treatable. As glomeruli become scarred there is consequent atrophy of the rest of the nephron with interstitial fibrosis, and it is usually the case in GN that the degree of

KDIGO® GN Guideline March 2011 Public Review Draft
4
chronic irreversible damage is most easily assessed from the amount of tubular atrophy. The accuracy of this assessment is increased with larger biopsies. The assessment of chronic damage from the biopsy must always be interpreted together with the clinical data to avoid misinterpretation if the biopsy is taken from a focal cortical scar. The amount of information that can be derived from kidney pathology varies substantially in the different GN types; when of particular relevance, this is addressed specifically within the appropriate chapters.
Repeat kidney biopsy Repeat kidney biopsy during therapy or following a relapse may be informative. There is no
systematic evidence to support recommendations for when or how often a repeat biopsy is necessary, but given the invasive nature of the procedure and the low but unavoidable risks involved, it should be used sparingly. In general, a decision about the value of a repeat biopsy should be driven by whether a change in therapy is being considered. More specifically, a repeat biopsy should be considered:
• when an unexpected deterioration in kidney function occurs (not compatible with the natural history) that suggests there may be a change or addition to the primary diagnosis (e.g., crescentic GN developing in known membranous nephropathy or interstitial nephritis secondary to the drugs being used in the disease management);
• when changes in clinical or laboratory parameters suggest a change of injury pattern within the same diagnosis (e.g., conversion of membranous to diffuse proliferative LN);
• when the relative contributions to the clinical picture of disease activity and chronicity are unknown, creating therapeutic uncertainty in regards to intensifying, maintaining, or reducing therapy;
• to assist in defining a “point of no return” and to help define therapeutic futility (i.e., such extensive and irreversible kidney scarring that no response to available therapies can be expected).
Assessment of Kidney Function Key outcome measures for the management of GN include assessment of kidney function,
particularly measurement of proteinuria and glomerular filtration rate (GFR).
Proteinuria Whether urine albumin or urine protein excretion is the preferred measurement to assess
glomerular injury continues to be debated. However, 24-hour protein excretion remains the reference (“gold standard”) method for quantification of proteinuria in patients with GN. It averages the variation of proteinuria due to the circadian rhythm, physical activity, and posture. Almost all of the published clinical trials used in the development of this guideline utilized 24-hour measurement of proteinuria to assess responses. Although this method is subject to error due to over- or under-collection, the simultaneous measurement of urine creatinine helps to standardize the collection in terms of completeness, thereby improving its reliability.
Protein-creatinine or albumin-creatinine ratio on a random (“spot”) urine sample is a practical alternative to 24-hour urine collection [2]. It is increasingly used in clinical practice

KDIGO® GN Guideline March 2011 Public Review Draft
5
because the sample is easy to obtain, is not influenced by variation in water intake or by urinary flow rate. There may still be gender and racial variations that are not accounted for, given these factors may modify creatinine generation. There is a correlation between the protein-creatinine ratio in a random urine sample and 24-hour protein excretion. Although the reliability of protein-creatinine ratio for the monitoring of proteinuria during treatment is still not proven, it has practical clinical utility, especially in children. In some recent studies, urine samples have been collected over a longer period (e.g., 4 hours) to address the limitations of “spot” urine samples that can be influenced by activity and circadian rhythm, but without the problems associated with a 24-hour urine collections [3]. The correlation with a 24-hour urine collection does improve steadily as the collection period is lengthened. However, there is currently insufficient evidence to preferentially recommend for this guideline 24-hour, shorter-timed, or spot urine collections in the management of GN.
The conventional definition of nephrotic syndrome in the published literature is proteinuria >3.5 g per 24 hours (in children, urine protein:creatinine ratio [uPCR] >200 mg/mmol [>2 g/g] or >300 mg/dl or 3+ on urine dipstick) plus hypoalbuminemia and edema. Nephrotic-range proteinuria is nearly always arbitrarily defined as proteinuria >3.5 g per 24 hours [uPCR >200 mg/mmol [>2 g/g] in children) in the absence of clinically overt nephrotic syndrome. Asymptomatic proteinuria, by definition without clinical symptoms, has variable levels of proteinuria in the range of 0.3-1.5 g per 24 hours (or equivalent). Treatment trials even within the same pattern of GN have used a variety of entry criteria based on severity of proteinuria. This is only one of the issues that make direct comparison of trial outcomes difficult. Nevertheless, quantifying proteinuria (and perhaps even assessing its qualitative nature) is an important measure in the assessment of the patient with GN. This is relevant in almost all the primary and secondary glomerular diseases in this guideline. It is also important and necessary to define, within each of the specific GN types in the subsequent chapters, what levels and changes in proteinuria have been used to categorize both the risk of progression and the definition of response. These parameters are not uniform and vary widely across the spectrum of GN. There is insufficient evidence currently to recommend basing treatment decisions on more detailed qualitative analysis of proteinuria, such as measurement of fractional urinary excretion of immunoglobulin G (IgG), β-2 microglobulin, and α-1 macroglobulin.
Estimation of GFR Most of the available evidence for treatment of GN has been based on estimations of
excretory kidney function using serum creatinine (SCr) or creatinine clearance (CrCl) requiring a 24-hour urine collection. Very few studies have reported gold standard measurements of GFR using inulin or radioisotope clearance techniques. Other techniques used in past studies include adjustment of SCr for age, weight, and sex using the Cockcroft-Gault formula and reciprocal or log transformation of SCr. All these methods have limitations [ref] but are informative when sequential measurements are made in each subject.
Recently, estimation of GFR using the Modification of Diet in Renal Disease (MDRD) 4 variable equation has gained increasing acceptance. Another estimating equation, CKD Epi has recently been proposed, which may be more accurate than the MDRD equation. Ethnicity may also influence estimated glomerular filtration rate (eGFR). There is no robust evidence to recommend the superiority of any of the available methods for estimating GFR in the management of GN. One particular limitation is that eGFR using creatinine-based formulas

KDIGO® GN Guideline March 2011 Public Review Draft
6
should be interpreted with caution in nephrotic syndrome, since tubular creatinine handling is altered in this condition. As a result, CrCl and eGFR may overestimate true GFR in nephrotic syndrome by 50% or more [4].
Outcome Measures
Complete remission, ESRD, mortality A definitive assessment of the efficacy of a treatment for GN requires the demonstration that
end-stage renal disease (ESRD) has been prevented, and mortality reduced. Very few studies in GN have been of sufficient duration or have analyzed sufficient numbers of patients to accurately assess these outcomes. This is not surprising, given the slow natural history of many of the histologic variants of GN in this guideline. The other accepted outcome measure for many of these disorders is complete remission, assessed by the complete disappearance of abnormal proteinuria (<300 mg per 24 hours). However, most studies rely on other surrogates as predictors of clinical outcomes. These surrogate outcome measures include changes in proteinuria, e.g., partial remission of proteinuria, change in kidney function, “point of no return”, quality of life, and quality of health.
Changes in proteinuria A quantitative change in proteinuria is presented in most studies. This is often categorized as
complete remission, defined as proteinuria <0.3 g per 24 hours (uPCR <30 mg/mmol) or partial remission defined as proteinuria >0.3 to <3.5 g per 24 hours or a decrease in proteinuria by at least 50% from the initial value and to <3.5 g per 24 hours. However, definitions vary and are not used consistently even within a specific GN pattern. The variations in these definitions will be discussed in each chapter.
Changes in kidney function Changes in kidney function are usually measured by changes in SCr or CrCl. These need to
be substantial to indicate true disease progression, e.g., doubling of SCr, or halving of CrCl or eGFR. This is because most patients with GN have gradual changes in function and there are many factors that may modify the SCr value besides progression of kidney disease. These factors include changes in intravascular volume, intercurrent illness, comorbid conditions, and many drugs. In addition, there are specific issues related to the SCr value independent of the disease, such as the method used for its measurement, changes in muscle mass, and alterations in urine flow and level of kidney function that both alter the tubular secretion of creatinine. In more recent studies, changes over time in eGFR have been reported. In the absence of ESRD as a defined adverse outcome, slope of CrCl or slope of eGFR may be an adequate and reliable marker of change in kidney function, provided that sufficient data at sequential time points are available, and that the slope is sufficiently linear [5].
Changes in GFR are often described qualitatively as “deteriorating” or “rapidly deteriorating” kidney function. Although these terms have no precise definitions, they are in common usage especially in certain histologic categories such as vasculitis and lupus nephritis. These are descriptive terms, and the value of a particular therapy can be properly evaluated only when compared to another group with similar clinical and histologic characterizations and in the

KDIGO® GN Guideline March 2011 Public Review Draft
7
setting of a randomized controlled trial (RCT). Where available, these will be presented in each chapter.
“Point of no return” This concept has no precise definition, but describes a situation in the natural history of a
chronic glomerular disease where loss of kidney function is accompanied by such extensive and irreversible kidney injury that any therapeutic strategy being tested cannot reasonably be expected to alter the natural history of progressive deterioration in kidney function (therapeutic futility). The presumption is that such patients should be excluded from clinical trials, since they are expected to be “nonresponders” and therefore may dilute any treatment effect, and adversely affect the power of the study. Furthermore these subjects with reduced kidney function may be at higher risk of adverse effects of the therapies being tested. In the absence of precise definitions of the ‘point of no return” it is not possible to know, in most of the published trials, whether the inclusion of such patients may have masked any therapeutic benefit.
Quality of life and quality of health Patients’ own perceptions of their quality of life and quality of health, and their preferences
is an extremely important element of the assessment of therapy, but is often an underappreciated and/or unmeasured parameter in the evaluation of many of the clinical trials reviewed in this guideline. This is particularly relevant when considering the risk-benefit analysis of interventions, which includes the short- and long-term risks of immunosuppressive treatments but, even here, does not account for the patient's perspective in relationship to real or perceived impact on their quality of life. The lack of such data is a substantial evidence gap in the evaluation of studies relating to the management of GN.
Impact of Age, Sex, Ethnicity, and Genetic Background Published RCTs of treatment for GN remain few, and many are small, short in duration of
follow-up, and of variable quality. This has resulted in uncertainty about generalizability, i.e., whether the demonstrated benefits (or lack of efficacy) of any treatments will still emerge if patients are then treated who come from different ethnic groups, and/or are of different age or sex, compared to those included in the published studies. The specific limitations of studies in this regard are discussed in later chapters but the following are examples of this issue: whether it is reasonable to extrapolate treatment recommendations from children to adults with MCD, and vice versa; whether the effectiveness of regimens for LN proven in Caucasians can be extended to those of other ethnicities; and whether the safety observed with a course of immunosuppression in the young applies equally to the elderly.
Furthermore few available RCTs are statistically powered to examine less-common adverse effects of therapy. It is not yet clear if new insights into these and other issues will emerge from a better understanding of the pharmacogenetic variations that can substantially alter the pharmacokinetics and/or pharmacodynamics of immunosuppressive and other agents. Although early evidence is suggestive that such genetic traits may alter clinical outcome [6], the cost of such pharmacogenetic testing also needs consideration and, as yet, there is little robust evidence that these factors should modify the treatment of GN.

KDIGO® GN Guideline March 2011 Public Review Draft
8
Management of Complications of Glomerular Disease A number of complications of glomerular disease are a consequence of the clinical
presentation rather than the specific histolopathologic pattern. Active management of such complications—although not subject to evidence review in this guideline—should always be considered and may have a significant positive impact on the natural history of the disease. These include measures to treat blood pressure, reduce proteinuria, control edema, and address other metabolic and thrombophilic consequences of nephrotic syndrome, which can result in significant morbidity and even mortality. If successful, these relatively nontoxic therapies may prevent—or at least modulate—the need for immunosuppressive drugs with their potential adverse effects. Such supportive therapy is usually not necessary in steroid-sensitive MCD with rapid remission, or in patients with GN and only microscopic hematuria, preserved GFR, and neither proteinuria nor hypertension. The latter is a common scenario, for instance, in IgA nephropathy.
Hypertension As in all chronic kidney disease (CKD), the aim of blood pressure control is both to protect
against the cardiovascular risks of hypertension and to delay progressive loss of GFR. Lifestyle modification (salt restriction, weight normalization, regular exercise, and smoking cessation) should be an integral part of the therapy for blood pressure control.
The ideal goal for blood pressure is not firmly established but current recommendations suggest that 130/80 mm Hg should be the treatment goal, and 125/75 mm Hg if there is proteinuria >1 g/d) [7,7A,7B,7C]. There is no specific evidence in GN on which to base a recommendation about the preferential importance of systolic or diastolic blood pressure , or about timing of blood pressure measurements. There are strong theoretical and experimental reasons for angiotensin-converting enzyme inhibitors (ACEi) and angiotensin-receptor blockers (ARB) to be first-choice therapy; this is now well-documented in clinical studies [8]. Children with GN should have blood pressure controlled to below the 50th percentile for systolic and diastolic pressure for age and sex using published [9,10] or locally available standards.
The evidence for blood pressure goals and choice of antihypertensive therapy in GN and other CKD has not been systematically evaluated for this guideline; it will be the subject of a forthcoming KDIGO Clinical Practice Guideline [10A].
Proteinuria Most studies suggest that the progressive loss of kidney function observed in many
glomerular diseases can largely be prevented if proteinuria can be reduced to levels below 0.5 g/d. Reduction in proteinuria is important, as it reflects control of the primary disease, reduction of glomerular hypertension, and also reduction of podocyte damage, which is probably a major factor in glomerular scarring. Proteinuria or factors present in proteinuric urine may also be toxic to the tubulointerstitium. In nephrotic syndrome, a reduction of proteinuria to a non-nephrotic range often results in an elevation to normal of serum proteins (particularly albumin). This elevation, in turn, alleviates many of the patient's symptoms as well as the metabolic complications of the nephrotic syndrome, thus improving quality of life.

KDIGO® GN Guideline March 2011 Public Review Draft
9
The antiproteinuric agents of choice are ACEi or ARB, which may reduce proteinuria by up to 40-50% in a dose-dependent manner, particularly if the patient complies with dietary salt restriction. There is little evidence to suggest that ACEi differ from ARBs in this respect. However, the combination of the two may result in additive antiproteinuric activity, although there is conflicting evidence as to the risk-benefit ratio of this strategy, especially if GFR is significantly reduced. Since ACEi and ARBs lower GFR, a 10-20% increase in SCr is often observed. Unless SCr continues to rise, this moderate increase reflects their effect on kidney hemodynamics and not worsening disease, and should not prompt withdrawal of the medication.
Adequate dietary protein should be ensured in the proteinuric patient (0.8-1.0g/kg daily) with a high carbohydrate intake to maximize utilization of that protein. When there is very heavy proteinuria, the amount of urinary protein loss should be added to dietary protein intake on a gram for gram basis.
Recommendations on the dosing of these agents and the target levels of proteinuria are outside the scope of this introduction, but are addressed when there is available evidence for specific forms of GN in subsequent chapters.
The evidence for the benefit of reducing proteinuria in CKD in general, and the choice of specific agents, has not been systematically evaluated for this guideline with the exception of the value of partial remission discussed in the relevant chapters. The evidence for renal protective therapy will be the subject of a forthcoming KDIGO Clinical Practice Guideline.
Hyperlipidemia Treatment of hyperlipidemia in patients with glomerular disease should usually follow the
guidelines that apply to those at high risk for the development of cardiovascular disease. This is most relevant in the patients where the manifestations of the disease cannot be completely ameliorated, and when other risk factors for cardiovascular disease coexist, most commonly hypertension and proteinuria. Dietary restriction of fats and cholesterol alone has only modest effects on hyperlipidemia in glomerular disease, in particular in nephrotic syndrome. Statins (HMG CoA reductase inhibitors) are well tolerated and effective in correcting the lipid profile, although not proven to reduce cardiovascular events in nephrotic syndrome. It may also be that statin therapy protects from a decline in GFR, although this is not established. Care is needed when statins are used in combinations with other drugs, notably an increased risk of myalgia/myositis when combined with calcineurin inhibitors.
Nephrotic edema The mainstay of treatment is diuretics accompanied by moderate dietary sodium restriction
(60-80 mmol [1.5-2 g] sodium per 24 hours). Nephrotic patients are often diuretic-resistant even if GFR is normal: oral loop diuretics with once- or twice-daily administration are usually preferred, given the ease of administration and longer therapeutic effect compared to i.v. therapy. However, in severe nephrotic syndrome, gastrointestinal absorption of the diuretic may be uncertain because of intestinal-wall edema, and i.v. diuretic, by bolus injection or infusion, may be necessary to provoke an effective diuresis. Alternatively, combining a loop diuretic with a thiazide diuretic or with metolazone is often an effective oral regimen that may overcome “diuretic resistance”. i.v. albumin infusions may be combined with diuretics to treat diuretic

KDIGO® GN Guideline March 2011 Public Review Draft
10
resistance, but are of unproven benefit. Occasionally, mechanical ultrafiltration is required for resistant edema.
Significant hypovolemia is not often a clinical problem, provided that fluid removal is controlled and gradual, but the pediatric and the elderly populations are at more risk of this complication. In the elderly, associated conditions such as diabetes mellitus and hypertension may increase the likelihood of hypovolemic shock and acute ischemic kidney injury.
Hypercoagulability The risk of thrombotic events becomes progressively more likely as serum albumin values
fall below 2.5 g/dl (25 g/l). Immobility as a consequence of edema, obesity, intercurrent illness or admission to hospital for surgery can further aggravate the risk. Prophylactic low-dose anticoagulation (e.g., heparin 5000 units subcutaneously twice daily) is common practice at times of high risk. Full-dose prophylactic anticoagulation with low-molecular-weight heparin or warfarin is mandatory if an arterial or venous thrombosis, or pulmonary embolism, is documented. It should also be considered if serum albumin drops below 2.0-2.5 g/dl (20-25 g/l) with one or more of the following: proteinuria >10 g/d; body mass index (BMI) >35 kg/m2; family history of thromboembolism with documented genetic predisposition; New York Heart Association class III or IV congestive heart failure; recent abdominal or orthopedic surgery; or prolonged immobilization. Contraindications to prophylactic anticoagulation are: an uncooperative patient; a bleeding disorder; prior gastrointestinal bleeding; a central nervous lesion prone to hemorrhage (brain tumor, aneurysms); or a genetic abnormality influencing warfarin metabolism or efficacy.
During treatment initiation, a significantly higher than average dose may be required because part of the action of heparin depends on antithrombin III, which may be lost in the urine in the nephrotic patient, resulting in reduced plasma concentrations. Warfarin is the long-term treatment of choice but should be monitored with special care because of potential alterations in the protein binding of the drug with fluctuations in serum albumin in the nephrotic patient. A target international normalized ratio (INR) of 2-3 is usually recommended, although not supported by specific evidence.
Risk of infection A high order of clinical vigilance for bacterial infection is vital in nephrotic patients. This is
particularly important in nephrotic children with ascites, in whom the fluid should be examined microscopically and cultured for spontaneous bacterial peritonitis. Bacteremia can occur even if clinical signs are localized to the abdomen. Erythrocyte sedimentation rate is unhelpful, but an elevated C-reactive protein may be informative. Parenteral antibiotics should be started once cultures are taken and the regimen should include benzylpenicillin (to treat pneumococcal infection). If repeated infections occur, serum immunoglobulins should be measured. If serum IgG is less than 600 mg/dl (6 g/l), there is limited evidence that infection risk is reduced by monthly administration of i.v. immunoglobulin 10-15 g to keep serum IgG >600 mg/dl (>6 g/l) [11].
Those with GN and nephrotic syndrome are at increased risk of invasive pneumococcal infection and should receive pneumococcal vaccination with the heptavalent conjugate vaccine (7vPCV) and the 23-valent polysaccharide vaccine (23vPPV) as well as the annual influenza

KDIGO® GN Guideline March 2011 Public Review Draft
11
vaccination. The response does not seem to be impaired by concurrent corticosteroid therapy. Vaccination with live vaccines (measles, mumps, rubella, varicella, rotavirus) is contraindicated while on immunosuppressive or cytotoxic agents, and should be deferred until prednisone is <20 mg/d and/or immunosuppressive agents have been stopped for at least 1-3 months. Exposure to varicella can be life-threatening, especially in children. Treatment should be given with zoster immune globulin if exposure does occur and antiviral therapy with acyclovir or valaciclovir begun at the first sign of chicken pox lesions [12] (See chapter 3, SSNS, for additional details on management in children).
Use of Corticosteroids and Immunosuppressive Therapy The chapters that follow will focus on the effectiveness of therapy based on current evidence
in the most common histologic variants of GN.
The therapeutic decisions of the physician are predicated on the continuing need to balance the risks and benefits of treatment. Nothing stated in this guideline replaces the physician’s assessment in this regard. The physician ideally seeks a treatment regimen that reduces immunosuppressive therapy exposure to the minimum, minimizes immediate morbidity (e.g., achieving remission of nephrotic syndrome), and prevents disease progression. However, physicians must also recognize that more prolonged treatment may be required, given the long-term threat that failure to prevent ESRD will shorten life expectancy and may only delay prolonged immunosuppressive drug exposure that would be required after kidney transplantation.
The focus in the management of chronic patterns of GN has shifted from cure to control, exemplified by recognition of the short- and long-term benefits of a reduction in proteinuria (in addition to the benefits known to accrue with complete remission). This paradigm has translated into use of more extended (or repeated) treatment regimens with the corollary of more toxic drug exposure.
The specific adverse effects of the recommended immunosuppressive agents and the need for routine prophylactic measures are beyond the scope of this guideline, but are familiar in clinical practice, and have been reviewed [13]. Specific regimens that potentially require prolonged exposure to these immunosuppressive agents are identified in the chapters to follow.
Adverse effects The potential adverse effects of immunosuppressive therapy must always be discussed with
the patient and family before treatment is initiated. This part of the management cannot be overemphasized. The risks of treatment with many of the agents are significant and may have a substantial latent period (e.g., cyclophosphamide). A balance must be struck between the potential risks of immunosuppressive treatment for GN, and the seriousness of the patient’s condition. It is sometimes difficult to reconcile the immediate risks of immunosuppression, in the otherwise clinically well patient, vs. the potential for progression to ESRD. However, given that advanced CKD—and, particularly, ESRD—is associated with a significant shortening of life expectancy even with dialysis or transplantation, the balancing of risks and benefits over time must be considered. The physician must be aware of this conundrum and where the evidence for treatment is weak (but potentially life-altering) and the risk for harm strong, a full disclosure is

KDIGO® GN Guideline March 2011 Public Review Draft
12
mandatory. Individual patient perceptions of the acceptability of any adverse effect may strongly influence the decision (e.g., the possibility of hirsutism with cyclosporin therapy may be perceived as less tolerable in a young female than in an older male). What might be seen as an acceptable trade-off by the physician may not be viewed similarly by the patient, leading to an issue over compliance with therapy.
With more intensive immunosuppressive regimens, prophylaxis may be required to minimize possible adverse effects. Specific recommendations are beyond the scope of this guideline, and are without an evidence base specific to treatment of GN, but better evidenced when immunosuppression is used in kidney transplantation. Common examples are the use of prophylactic antimicrobials to minimize opportunistic infection, and H2-receptor antagonists or proton pump inhibitors to prevent peptic ulceration. Two other important and more drug-specific examples are the use of bisphosphonates (except in the presence of kidney failure) to minimize loss of bone density during prolonged treatment with corticosteroids, and the need to offer the opportunity for sperm or ovum storage/preservation before treatment with the gonadotoxic agents, cyclophosphamide and chlorambucil.
Drug monitoring Immunosuppressive agents with a narrow therapeutic index include the calcineurin
inhibitors, cyclosporin and tacrolimus. There are no RCTs that compare response to treatment in GN and different achieved blood levels of these agents. Dosing and target blood levels are based on established practice in kidney transplantation. The main goal of blood level monitoring is to avoid toxicity due to high drug levels, while still maintaining efficacy. The latter can often be assessed by proteinuria reduction, which can sometimes be achieved with trough blood levels of calcineurin inhibitors that would be considered subtherapeutic for solid-organ transplantation. There is no evidence for the value of monitoring mycophenolic acid levels to guide dosing of mycophenolate in the treatment of GN.
Pregnancy in Women with GN In women of child-bearing potential, the risks of pregnancy must be considered. Issues
include the toxicity, especially in the first trimester, of immunosuppressive agents, ACEi, and ARBs, and also the hazards to fetal and maternal outcome of pregnancy with uncontrolled proteinuric conditions. There is also a risk of relapse of LN both during and after pregnancy.
Treatment Costs and Related Issues
These guidelines have been developed with the goal of providing evidence-based treatment recommendations for GN that can be used by physicians in all parts of the world. Most of the medications recommended are available at low cost in many parts of the world. These include prednisone, azathioprine, and cyclophosphamide tablets. Monitoring (e.g., by regular checks of blood count) is also cheap and widely available.
The cost of some agents (e.g., calcineurin inhibitors and mycophenolate) remains high, but the development and marketing of generic agents and biosimilars is now rapidly reducing costs. However, care must be taken to ensure that variations in bioavailablity with these less expensive generic agents do not compromise effectiveness or safety.

KDIGO® GN Guideline March 2011 Public Review Draft
13
Plasmapheresis remains unavailable in some parts of the world, related not only to the high cost and limited availability of replacement fluids (including human albumin and fresh frozen plasma) but also to the equipment and staffing costs.
Some treatments suggested as potential “rescue” therapies in this guideline (e.g., rituximab) remain prohibitively expensive in most parts of the world. This is another indication of the urgent need for developing trials that will provide robust evidence of their efficacy. Uncertainty about the value of such high-cost agents would also be mitigated if there were comprehensive national or international registries collecting comprehensive observational data on their use, but unfortunately none exist.
Recurrent Post-transplantation GN Virtually all of the histologic variants discussed in this guideline (with the exception of
MCD) may recur after transplantation. Recurrent disease is recognized as the third most common cause of kidney transplant failure. Currently there are no proven strategies to prevent recurrent GN in kidney transplant recipients. Despite the high rate of recurrent disease, long-term graft survival is still very good and transplantation remains the best treatment option for patients with ESRD secondary to GN. Where there are specific recommendations in particular variants of GN that relate to management before transplantation, they will be discussed in each relevant chapter.
RESEARCH RECOMMENDATIONS
The evidence review underpinning this clinical practice guideline has confirmed the paucity of robust data from RCTs to support the treatment recommendations and suggestions that have been made. This raises the question of why there are so few RCTs of good design and sufficient power in GN, compared to many other areas of nephrology and internal medicine. The slowly progressive natural history of many patterns of GN means that trials designed to provide definitive outcome data (using ESRD or mortality) require long follow-up, significantly increasing their cost as well as effort for both the physician and the patient. Studies often employ “composite end-points” in order to enhance event rates. Furthermore, there are two competing elements in GN trial design. On the one hand, there exists the recognition that most GN variants are uncommon; on the other hand, there is a need to acquire an adequate sample size within a reasonable time frame, an essential element for any successful study. This virtually mandates multicenter and multinational trial organization which, in turn, is challenging from both organizational and cost perspectives. These factors have made trials in GN less attractive both to funding agencies and pharmaceutical companies, compared to more common and higher-profile clinical domains such as cardiovascular disease and cancer.
However there is an urgent need for such studies to be carried out. The costs—both to society, and to patients with GN and their families, if disease progression is not prevented—are often grossly underestimated. As an integral part of this guideline, we make recommendations in each chapter about the most pressing areas of uncertainty where RCTs and other areas of research would significantly inform clinical practice.

KDIGO® GN Guideline March 2011 Public Review Draft
14
References
1. Corwin HL, Schwartz MM, Lewis EJ. The importance of sample size in the interpretation of the renal biopsy. Am J Nephrol. 1988; 8(2):85-9
2. Price CP, Newall R, Boyd JC. Use of protein:creatinine ratio measurements on random urine samples for prediction of significant proteinuria: a systematic review. Clin Chem 51: 1577-1586, 2005
3. Fine DM, Ziegenbein M, Petri M, Han EC, McKinley AM, Chellini JW, Nagaraja HN, Carson KA, Rovin BH. A prospective study of protein excretion using short-interval timed urine collections in patients with lupus nephritis. Kidney Int. 2009 Dec; 76(12):1284-8
4. Branten AJ, Vervoort G, Wetzels JF. Serum creatinine is a poor marker of GFR in nephrotic syndrome. Nephrol Dial Transplant. 2005 Apr; 20(4):707-11
5. Botev R, Mallié JP, Couchoud C, Schück O, Fauvel JP, Wetzels JF, Lee N, De Santo NG, Cirillo M. Estimating glomerular filtration rate: Cockcroft-Gault and Modification of Diet in Renal Disease formulas compared to renal inulin clearance. Clin J Am Soc Nephrol. 2009 May; 4(5):899-906.
6. Rovin BH, McKinley AM, Birmingham DJ. Can we personalize treatment for kidney diseases? Clin J Am Soc Nephrol. 2009 Oct;4(10):1670-6
7. Haddad N, Brown C, Hebert LA: Retarding Progression of Kidney Disease. In Johnson R, Fehally J (Eds) Comprehensive Clinical Nephrology, 3rd Ed. Philadelphia, Elsevier, 2007, pp 823-830.
7A. CARI: http://www.cari.org.au/guidelines.php 7B. UK Renal Association: http://www.renal.org/CKDguide/full/UKCKDfull.pdf 7C. Japanese: http://www.nature.com/hr/journal/v32/n1/abs/hr200818a.html 8. Agarwal A, Haddad N, Hebert LA: Progression of kidney disease: diagnosis and management, In: Molony D,
Craig J (Eds) Evidence-Based Nephrology, Hoboken NJ, John Wiley & Sons, 2008, pp 311-322. 9. National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and
Adolescents. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics 2004; 114(2 Suppl 4th Report):555-576.
10. ESCAPE Trial Group, Wuhl E, Trivelli A, Picca S, Litwin M, Peco-Antic A et al. Strict blood-pressure control and progression of renal failure in children. New England Journal of Medicine 2009; 361(17):1639-1650.)
10A. KDIGO Clinical Practice Guideline for Management of Blood Pressure in CKD. In preparation. 11. Ogi M, Yokoyama H, Tomosui N, et al: Risk factors for infection and immunoglobulin replacement therapy
in adult nephrotic syndrome. Am J Kidney Dis 1994; 24: 427–436. 12. Furth SL, Arbus GS, Hogg R, Tarver J, Chan C, Fivush BA et al. Varicella vaccination in children with
nephrotic syndrome: a report of the Southwest Pediatric Nephrology Study Group. Journal of Pediatrics 2003; 142(2):145-148
13. Philibert D, Cattran D. Remission of proteinuria in primary glomerulonephritis: we know the goal but do we know the price? Nat Clin Pract Nephrol. 2008 Oct;4(10):550-9.

KDIGO® GN Guideline March 2011 Public Review Draft
15
CHAPTER 3: TREATMENT OF STEROID-SENSITIVE NEPHROTIC SYNDROME IN CHILDREN
INTRODUCTION
This guideline makes treatment recommendations for children aged 1 to 18 years with nephrotic syndrome, who respond to corticosteroid therapy by achieving complete remission (SSNS). The cost implications for global application of this guideline are addressed in Chapter 2.
3.1: Treatment of the initial episode of SSNS 3.1.1: We recommend that corticosteroid therapy (prednisone or
prednisolone)* be given for at least 12 weeks. (1B) 3.1.1.1: We recommend that oral prednisone be administered as a
single daily dose (1B) starting at 60 mg/m2/d or 2 mg/kg/d to a maximum 60 mg/d. (1D)
3.1.1.2: We recommend that daily oral prednisone be given for at least 4 weeks (1C) followed by alternate-day oral medication as a single daily dose starting at 40 mg/m2 or 1.5 mg/kg (maximum 40 mg on alternate days) (1D) to be continued for 2-5 months. (1B)
*Prednisone and prednisolone are equivalent, used in the same dosage, and have both been used in RCTs. All later references to prednisone in this chapter refer to prednisone or prednisolone. All later references to oral corticosteroids refer to prednisone or prednisolone.
BACKGROUND
Nephrotic syndrome affects 1-3 per 100,000 children below 16 years of age [1]. Eighty percent of children respond to corticosteroid therapy [1]. A kidney biopsy diagnosis is not required routinely at presentation because the International Study of Kidney Disease in Children (ISKDC) demonstrated that, while 93% of children with MCD responded to corticosteroids, 25-50% of children with mesangial proliferative glomerulonephritis (MPGN) or FSGS also responded to corticosteroids [2]. The majority of children who relapse continue to respond completely to corticosteroids throughout their subsequent course, and the long-term prognosis, including maintenance of normal kidney function, is good [3-5]. In contrast, without treatment, nephrotic syndrome in children is associated with high risk of death, particularly from bacterial infection. Before the use of corticosteroids and antibiotics, 40% of children died, with half of these deaths being from infection [6]. A recent study reports only one death (0.7%) associated with nephrotic syndrome among 138 children with SSNS presenting between 1970 and 2003 [7].
The definitions used for nephrotic syndrome, complete remission, initial responder, initial and late steroid nonresponders (steroid resistance), infrequent relapses, frequent relapses and steroid dependence are listed in Table 3.1. The likelihood of initial corticosteroid unresponsiveness is increased with increasing age at presentation [1], in African and African-American children [8], and in children with kidney pathologies other than MCD [2]. The

KDIGO® GN Guideline March 2011 Public Review Draft
16
likelihood of late resistance to corticosteroids is associated with a shorter interval to the first relapse, and relapsing during the initial course of corticosteroid therapy [9].
Table 3.1. Definitions used in nephrotic syndrome in children
Classification Definition Nephrotic syndrome Edema, uPCR ≥200 mg/mmol (≥2 g/g), or ≥300mg/dl, or 3+ protein on urine
dipstick, hypoalbuminaemia ≤25 g/l (≤2.5 mg per 100 ml) Complete remission uPCR <20 mg/mmol (<0.2 g/g) or <1+ of protein on urine dipstick for 3 consecutive
days Partial remission Proteinuria reduction of 50% or greater from the presenting value and absolute
uPCR between 0.2 and 2 mg/mg (20-200 mg/mmol) No remission Failure to reduce urine protein excretion by 50% from baseline or persistent
excretion uPCR >2 mg/mg (>200 mg/mmol) Initial responder Attainment of complete remission within initial 4 weeks of corticosteroid therapy Initial nonresponder/steroid resistance Failure to achieve remission after 8 weeks of corticosteroid therapy Relapse uPCR ≥200 mg/mmol (≥2 g/g) or ≥3+ protein on urine dipstick for 3 consecutive
days Infrequent relapse One relapse within 6 months of initial response, or one to three relapses in any 12-
month period Frequent relapse Two or more relapses within 6 months of initial response, or four or more relapses
in any 12-month period Steroid dependence Two consecutive relapses during corticosteroid therapy, or within 14 days of
ceasing therapy Late nonresponder Persistent proteinuria during 4 or more weeks of corticosteroids following one or
more remissions uPCR, urine protein:creatinine ratio.
RATIONALE
• There is moderate-quality evidence that administering prednisone for three months reduces the risk of relapse in children with the first episode of SSNS, with an increase in benefit seen with up to 6 months of treatment.
• There is moderate-quality evidence that corticosteroid therapy should be given as a single daily dose for at least 4 weeks, followed by alternate-day therapy for 2-5 months.
• The initial dose regimen of corticosteroid therapy is based on recommendations from the ISKDC, and has not been defined in RCTs.
Corticosteroid Use in the First Episode of SSNS in Children With corticosteroid therapy, 80-90% of patients with childhood nephrotic syndrome achieve
complete remission [1,3]. However, 80-90% of these children have one or more relapses [3,5] following the 2- month steroid regimen proposed by the ISKDC [10] and adapted by Arbeitsgemeinschaft für Pädiatrische Nephrologie [11]. Therefore, RCTs have evaluated the benefits of increased duration of therapy for the initial episode of SSNS. A meta-analysis [12] of six RCTs (422 children) demonstrated that the risk of relapse at 12-24 months was reduced by 30% (relative risk [RR] of relapse 0.70; 95% confidence intervals [CI] 0.58-0.84) with 3 months or more of corticosteroid therapy compared to 2 months. There was an increase in benefit seen

KDIGO® GN Guideline March 2011 Public Review Draft
17
when prednisone was given for up to the end of the sixth month (RR = 1.26 - 0.112 duration; r² = 0.56, P = 0.03) [12]. Also a meta-analysis [12] of four RCTs (382 children) identified that treatment for 6 months significantly reduced the risk of relapse at 12-24 months compared to 3 months (RR 0.57; 95% CI 0.45-0.71). No significant differences in the incidence of adverse effects between treatment groups were demonstrated. However, individual trials were not designed specifically to study harm, and so were underpowered for the detection of side-effects of corticosteroids [12].
There are no RCTs examining different initial doses of corticosteroid for the first episode of childhood nephrotic syndrome. A dose of prednisone 60 mg/m2/d was recommended empirically by the ISKDC in 1979; this is roughly equivalent to 2 mg/kg. Although theoretical studies indicate that dosing for body weight results in a lower total dose compared to dosing for surface area, there are no data on whether this is of clinical relevance, so either method of calculating prednisone dose may be utilized [13]. Two RCTs have demonstrated that the mean time to remission did not differ significantly when daily corticosteroid therapy was given as a single daily dose compared to divided doses (weighted mean difference 0.04 days; 95% CI –0.98 to –1.06) [12].
The majority (94%) of children respond to corticosteroids within 4 weeks of daily prednisone therapy [14]. To reduce the risk of relapse, prednisone should be given daily for at least 4 weeks in the initial episode of nephrotic syndrome. In an RCT, the risk of relapse was significantly higher at 6 months and 12 months when prednisone was given for 1 month compared to 2 months (RR 1.46; 95% CI 1.01-2.12 at 12 months) [15]. Prednisone should be given on alternate days after 4 weeks of daily treatment rather than on 3 consecutive days out of 7 days, based on an RCT that showed the former had a lower risk of relapse [16]. Alternate-day (rather than daily) prednisone is suggested to maintain remission, because linear growth is less affected [17]. Although widely used particularly in France [18], there is no evidence to support the administration of high-dose i.v. methylprednisolone to a child with nephrotic syndrome, who has not achieved remission after 4 weeks of daily corticosteroids, before labeling that child as steroid-resistant.
3.2: Treatment of relapsing SSNS with corticosteroids 3.2.1: Corticosteroid therapy for children with infrequent relapses of SSNS:
3.2.1.1: We suggest that infrequent relapses of SSNS in children be treated with a single-daily dose of prednisone 60 mg/m2 or 2 mg/kg (maximum of 60 mg/d) until the child has been in complete remission for at least 3 days. (2D)
3.2.1.2: We suggest that, after achieving complete remission, children be given prednisone as a single dose on alternate days (40 mg/m2 per dose or 1.5 mg/kg per dose: maximum 40 mg on alternate days) for at least 4 weeks. (2C)
3.2.2: Corticosteroid therapy for frequently relapsing (FR) and steroid-
dependent (SD) SSNS:

KDIGO® GN Guideline March 2011 Public Review Draft
18
3.2.2.1: We suggest that relapses in children with FR or SD SSNS be treated with daily prednisone until the child has been in remission for at least 3 days, followed by alternate-day prednisone for at least 3 months. (2C)
3.2.2.2: We suggest that prednisone be given on alternate days in the lowest dose to maintain remission without major adverse effects in children with SD SSNS. (2D)
3.2.2.3: We suggest that daily prednisone at the lowest dose be given to maintain remission without major adverse effects in children with SD SSNS where alternate-day prednisone therapy is not effective. (2D)
3.2.2.4: We suggest that daily prednisone be given during episodes of upper respiratory tract infection to reduce the risk for relapse in children with SD SSNS already on alternate-day prednisone. (2C)
BACKGROUND
Children with nephrotic syndrome who respond to corticosteroids have an 80-90% chance of having one or more relapses [3,5]. Half of those that relapse have infrequent relapses and can be managed with short courses of prednisone. The remaining children have FR or SD SSNS [3,5]. The risks of a child developing frequent relapses or becoming steroid-dependent are increased with shorter time to first relapse [19], the number of relapses in the first 6 months after initial treatment [2,5], younger age at the initial episode [20], prolonged time to first remission [18,21], infection at first relapse [19], and hematuria in first episode [21]. The most consistent indicator for a frequently relapsing course is early relapse after initial treatment. Studies have not assessed whether the other factors are independent risk factors for predicting frequent relapses or steroid dependence. Children with FR or SD SSNS, and children whose first episode of SSNS occurred at a young age, have a longer duration of relapsing or SD nephrotic syndrome compared to children with infrequent relapses or older age of onset [4,20]. Corticosteroids are needed to achieve remission, and low doses given on alternate days may maintain remission in patients with FR SSNS without recourse to corticosteroid-sparing agents. Low-dose daily or alternate-day corticosteroids may still be required to maintain remission in SD SSNS, despite receiving corticosteroid-sparing agents.
RATIONALE
• In children with infrequent relapses of SSNS, corticosteroid therapy regimens are based on empirical recommendations from the ISKDC and an RCT in children with FR SSNS.
• In children with FR and SD SSNS, there is low-quality evidence that increasing the duration of corticosteroid therapy increases the duration of remission.
• In children with SD SSNS, there is low-quality evidence that changing children from alternate-day to daily corticosteroids at onset of upper respiratory infections reduced the risk of relapse.

KDIGO® GN Guideline March 2011 Public Review Draft
19
• In children with FR and SD SSNS, there is very low–quality evidence that low-dose alternate-day or daily corticosteroid therapy reduces the risk of relapse.
Corticosteroid Use in Relapses in Children with Infrequent Relapses of SSNS There are no RCTs examining relapse regimens with corticosteroids in infrequently
relapsing SSNS. In children with frequently relapsing SSNS, the ISKDC demonstrated that the number of relapses in the 7 months after treatment did not differ significantly between children treated with 8 weeks of daily prednisone compared to daily prednisone till remission followed by 4 weeks of prednisone given on 3 consecutive days out of 7 days (further relapse by 9 months RR 1.07; 95% CI 0.77-1.50) [12]. Based on these data we suggest that children with infrequently relapsing SSNS should receive daily corticosteroids only until remission followed by four weeks of alternate day prednisone.
Corticosteroid Therapy in Frequently Relapsing (FR) and Steroid-Dependent (SD) SSNS in Children
Approximately 40% of children with SSNS have FR or SD SSNS. A single RCT in children with relapsing nephrotic syndrome has demonstrated that the risk of relapse at 12 and 24 months was significantly reduced with prednisone treatment for 7 months compared to 2 months of therapy [12]. These data, and the data on prednisone duration in the initial episode of SSNS, suggest that it is reasonable to treat a child with FR or SD SSNS with longer corticosteroid regimens than those suggested for children who relapse infrequently. Two RCTs have demonstrated that daily prednisone dose during URTI reduced the risk for relapse in children with SD SSNS [12,22].
To maintain remission in children with SD SSNS, prednisone may be given on alternate days in the lowest dose possible to maintain remission. An observational study demonstrated that low-dose alternate-day prednisone (mean dose 0.48 mg/kg on alternate days) reduced the risk of relapse in FR SSNS compared to historical controls [23]. Guidelines from the British Association of Paediatric Nephrology recommend that children with SD SSNS receive 0.1-0.5 mg/kg on alternate days for at least 3-6 months before tapering [24]. Guidelines from the Indian Paediatric Nephrology Group recommend that the prednisone dose is tapered to 0.5-0.7 mg/kg on alternate days or lower, and continued for 9-18 months with careful monitoring of corticosteroid toxicity [25]. A nonrandomized comparator study indicated that low-dose daily prednisone (0.25 mg/kg) was more effective in maintaining remission compared to historical controls not treated with low-dose prednisone with a reduction in relapse rate from 2.25 per patient per year to 0.5 per patient per year [26].
3.3: Treatment of FR and SD SSNS with corticosteroid-sparing agents 3.3.1: We recommend that corticosteroid-sparing agents be prescribed for
children with FR SSNS and SD SSNS, who develop steroid-related adverse effects. (1B)
3.3.2: We recommend that alkylating agents, cyclophosphamide or chlorambucil, be given as corticosteroid-sparing agents for FR SSNS. (1B) We suggest that alkylating agents, cyclophosphamide or

KDIGO® GN Guideline March 2011 Public Review Draft
20
chlorambucil, be given as corticosteroid-sparing agents for SD SSNS. (2C) 3.3.2.1: We suggest that cyclophosphamide (2 mg/kg/d) be given for 8-
12 weeks (maximum cumulative dose 168 mg/kg). (2C) 3.3.2.2: We suggest that cyclophosphamide not be started until the
child has achieved remission with corticosteroids. (2D) 3.3.2.3: We suggest that chlorambucil (0.1-0.2 mg/kg/d) may be given
for 8 weeks (maximum cumulative dose 11.2 mg/kg) as an alternative to cyclophosphamide. (2C)
3.3.2.4: We suggest that second courses of alkylating agents not be given. (2D)
3.3.3: We recommend that levamisole be given as a corticosteroid-sparing agent. (1D) 3.3.3.1: We suggest that levamisole be given at a dose of 2.5 mg/kg on
alternate days (2B) for at least 12 months (2C) as most children will relapse when levamisole is stopped.
3.3.4: We recommend that the calcineurin inhibitors, cyclosporine or tacrolimus, be given as corticosteroid-sparing agents. (1C) 3.3.4.1: We suggest that cyclosporine be administered at a dose of 4-5
mg/kg/d (starting dose) in two divided doses. (2C) 3.3.4.2: We suggest that tacrolimus 0.1 mg/kg/d (starting dose) in two
divided doses be used instead of cyclosporine when the cosmetic side-effects of cyclosporine are unacceptable. (2D)
3.3.4.3: Monitor CNI levels during therapy to limit toxicity. (Not Graded)
3.3.4.4: We suggest that CNIs be given for at least 12 months as most children will relapse when CNIs are stopped. (2C)
3.3.5: We suggest that MMF be given as a corticosteroid-sparing agent. (2C) 3.3.5.1: We suggest that MMF (starting dose 1200 mg/m2/d) be given in
two divided doses for at least 12 months, as most children will relapse when MMF is stopped. (2C)
3.3.6: We suggest that rituximab be considered only in children with SD SSNS, who have continuing frequent relapses despite optimal combinations of prednisone and corticosteroid-sparing agents, and/or who have serious adverse effects of therapy. (2D)
3.3.7: We suggest that mizoribine not be used as a corticosteroid-sparing agent in FR and SD SSNS. (2C)
3.3.8: We recommend that azathioprine not be used as a corticosteroid-sparing agent in FR and SD SSNS. (1B)
BACKGROUND
About half of the children with SSNS who relapse will have FR or SD SSNS [3,5]. The long-term prognosis for most children with SSNS is for complete resolution of their disease over time and maintenance of normal kidney function. Therefore limiting the long-term adverse effects of treatment is an important objective. Children with FR or SD SSNS require prolonged

KDIGO® GN Guideline March 2011 Public Review Draft
21
corticosteroid therapy, which is associated with significant adverse effects. including impaired linear growth, behavioral changes, obesity, Cushing’s syndrome, hypertension, ophthalmological disorders, impaired glucose tolerance, and reduced bone mineral density. Adverse effects may persist into adult life in young people, who continue to relapse after puberty [27]. To reduce the risk of corticosteroid-related adverse effects, children with FR or SD SSNS may require other agents, including alkylating agents (cyclophosphamide, chlorambucil) and CNI (cyclosporine, tacrolimus). Adverse effects of these agents include increased risk of infection and reduced fertility (alkylating agents) [27,28] and kidney dysfunction and hypertension (CNI) [29]. CNIs and MMF are expensive, further limiting their use in many countries.
RATIONALE
In children with FR and SD SSNS:
• There is moderate-quality evidence to support the use of alkylating agents (cyclophosphamide, chlorambucil), levamisole, and CNI (cyclosporine, tacrolimus).
• There is low-quality evidence to support the use of mycophenolate mofetil (MMF). • There is very low–quality evidence to support the efficacy of rituximab. • There is moderate-quality evidence to demonstrate that mizoribine and azathioprine
are not effective.
Children with FR or SD SSNS often continue to relapse into adolescence or adulthood, and require prednisone in variable doses for long periods of time to achieve and maintain remission. Patients successfully treated with corticosteroid-sparing therapy have improved growth rates, reduced body mass index, reduction of Cushingoid features, and improvement in other corticosteroid-related adverse effects [30-33]. In all cases when contemplating corticosteroid-sparing therapy, the adverse effects of such therapy must be assessed against the benefits for that child in terms of reduced risk of relapse and reduced adverse effects of corticosteroids.
Fourteen RCTs in children have compared cyclophosphamide (three trials), chlorambucil (two trials), levamisole (six trials), mizoribine (one trial), and azathioprine (two trials) to placebo, no specific treatment, or prednisone in children with FR and/or SD SSNS. Trials either did not differentiate between FR and SD SSNS, or included only SD SSNS patients. Cyclophosphamide, chlorambucil, and levamisole reduced the risk of relapse during short term follow up (6-12 months) by more than 50% (Table 3.2). Two RCTs demonstrated no significant differences in the risk of relapse between cyclosporine and cyclophosphamide, or between cyclosporine and chlorambucil during cyclosporine treatment. RCTs have identified no significant differences in the risk for relapse between levamisole and i.v. cyclophosphamide, and between oral cyclophosphamide and oral chlorambucil [34].
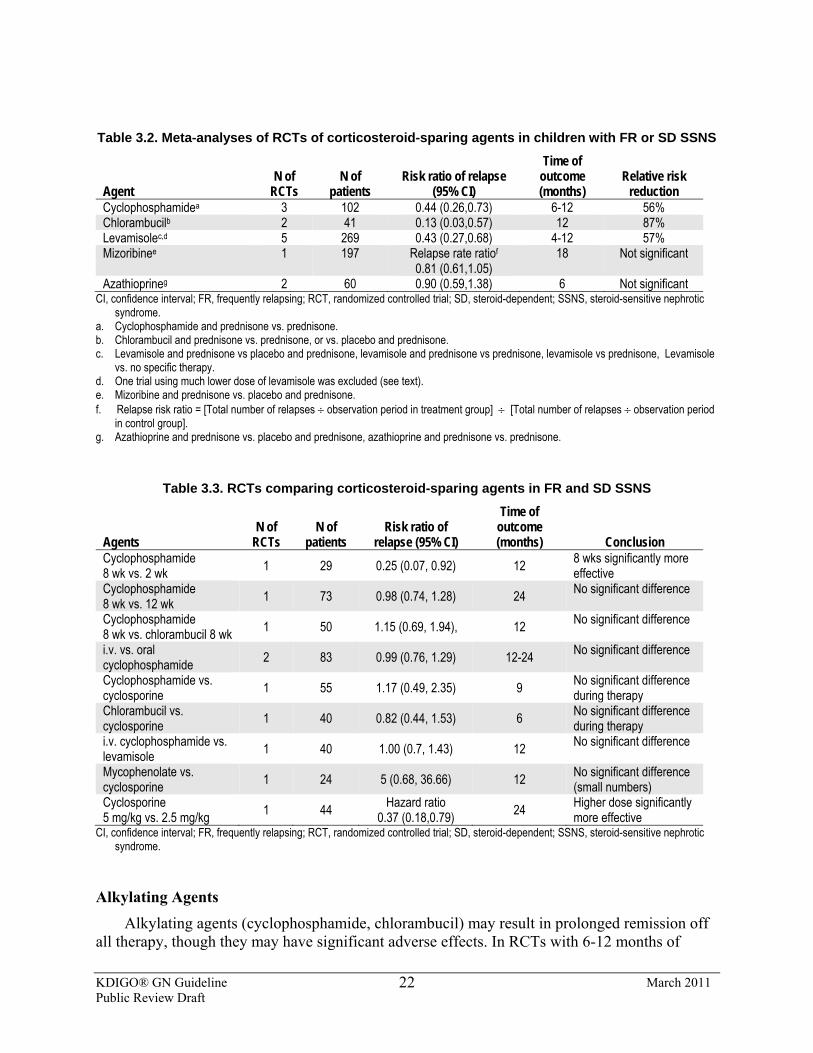
KDIGO® GN Guideline March 2011 Public Review Draft
22
Table 3.2. Meta-analyses of RCTs of corticosteroid-sparing agents in children with FR or SD SSNS
Agent N of
RCTs N of
patients Risk ratio of relapse
(95% CI)
Time of outcome (months)
Relative risk reduction
Cyclophosphamidea 3 102 0.44 (0.26,0.73) 6-12 56% Chlorambucilb 2 41 0.13 (0.03,0.57) 12 87% Levamisolec,d 5 269 0.43 (0.27,0.68) 4-12 57% Mizoribinee 1 197 Relapse rate ratiof
0.81 (0.61,1.05) 18 Not significant
Azathioprineg 2 60 0.90 (0.59,1.38) 6 Not significant CI, confidence interval; FR, frequently relapsing; RCT, randomized controlled trial; SD, steroid-dependent; SSNS, steroid-sensitive nephrotic
syndrome. a. Cyclophosphamide and prednisone vs. prednisone. b. Chlorambucil and prednisone vs. prednisone, or vs. placebo and prednisone. c. Levamisole and prednisone vs placebo and prednisone, levamisole and prednisone vs prednisone, levamisole vs prednisone, Levamisole
vs. no specific therapy. d. One trial using much lower dose of levamisole was excluded (see text). e. Mizoribine and prednisone vs. placebo and prednisone. f. Relapse risk ratio = [Total number of relapses ÷ observation period in treatment group] ÷ [Total number of relapses ÷ observation period
in control group]. g. Azathioprine and prednisone vs. placebo and prednisone, azathioprine and prednisone vs. prednisone.
Table 3.3. RCTs comparing corticosteroid-sparing agents in FR and SD SSNS
Agents N of
RCTs N of
patients Risk ratio of
relapse (95% CI)
Time of outcome (months) Conclusion
Cyclophosphamide 8 wk vs. 2 wk 1 29 0.25 (0.07, 0.92) 12 8 wks significantly more
effective Cyclophosphamide 8 wk vs. 12 wk 1 73 0.98 (0.74, 1.28) 24 No significant difference
Cyclophosphamide 8 wk vs. chlorambucil 8 wk 1 50 1.15 (0.69, 1.94), 12 No significant difference
i.v. vs. oral cyclophosphamide 2 83 0.99 (0.76, 1.29) 12-24 No significant difference
Cyclophosphamide vs. cyclosporine 1 55 1.17 (0.49, 2.35) 9 No significant difference
during therapy Chlorambucil vs. cyclosporine 1 40 0.82 (0.44, 1.53) 6 No significant difference
during therapy i.v. cyclophosphamide vs. levamisole 1 40 1.00 (0.7, 1.43) 12 No significant difference
Mycophenolate vs. cyclosporine 1 24 5 (0.68, 36.66) 12 No significant difference
(small numbers) Cyclosporine 5 mg/kg vs. 2.5 mg/kg 1 44 Hazard ratio
0.37 (0.18,0.79) 24 Higher dose significantly more effective
CI, confidence interval; FR, frequently relapsing; RCT, randomized controlled trial; SD, steroid-dependent; SSNS, steroid-sensitive nephrotic syndrome.
Alkylating Agents Alkylating agents (cyclophosphamide, chlorambucil) may result in prolonged remission off
all therapy, though they may have significant adverse effects. In RCTs with 6-12 months of

KDIGO® GN Guideline March 2011 Public Review Draft
23
follow-up, alkylating agents reduced the risk of relapse compared to prednisone, placebo, or no specific treatment by about 65% (RR 0.34; 95% CI 0.18-0.63) [34] (Table 3.2; Suppl Tables 14-16). In a systematic review of observational studies and RCTs, alkylating agents in FR SSNS resulted in remission rates of 72% after 2 years and 36% after 5 years, but were less effective in SD SSNS with remission rates of 40% and 24% after 2 and 5 years, respectively [28]. Eight weeks of cyclophosphamide therapy was significantly more effective in reducing the risk for relapse compared to 2 weeks (Table 3.3). In SD SSNS patients, there was no significant difference in the risk of relapse between 8 and 12 weeks of cyclophosphamide therapy in one RCT (Table 3.3). However the Arbeitsgemeinschaft für Pädiatrische Nephrologie concluded that 12 weeks of cyclophosphamide was more effective compared to historical controls treated for 8 weeks. Cyclophosphamide is associated with hemorrhagic cystitis but this rarely occurs at the doses used [34A]. Nevertheless, where possible, cyclophosphamide should be administered when the child is in remission, with a good urine output, and can receive a high fluid intake. The i.v. route may be considered where nonadherence to therapy is likely. Two RCTs found no significant difference in the risk of relapse between oral and i.v. cyclophosphamide at 12-24 months follow-up. However, at 6 months, significantly more children treated with monthly pulses of i.v. cyclophosphamide for 6 months were in remission, compared to oral treatment for 8-12 weeks (Table 3.3; Suppl Tables 1-3). Studies have demonstrated the efficacy of chlorambucil at doses of 0.1-0.2 mg/kg/d given for 8 weeks (cumulative dose 11.2 mg/kg) (Table 3.2). Higher doses did not increase efficacy and resulted in increased risks, particularly of hematological and infectious adverse effects [35].
It is suggested that second courses of alkylating agents not be given. Gonadal toxicity with alkylating agents is well documented, with males more affected than females. There is a dose-dependent relationship between the total dose of cyclophosphamide and probability of sperm counts below 106/ml. A “safe” dose of cyclophosphamide remains unclear, but a maximum cumulative dose of 168 mg/kg (2 mg/kg/d for 12 weeks) in boys is below the total dose (>200-300 mg/kg) at which azoospermia has generally been reported [28,36]. There are fewer data available on chlorambucil, but studies in patients treated for lymphoma found that azoospermia was associated with total doses of 10-17 mg/kg, suggesting that the margin between efficacy and toxicity is narrow for chlorambucil [37]. Studies have reported a higher risk of malignancy following chlorambucil use compared to cyclophosphamide [28].
Levamisole
Five of six RCTs have demonstrated a significant reduction in the risk for relapse during levamisole treatment compared to prednisone, placebo, or no specific treatment (RR 0.50; 95% CI 0.25-0.99) [34] (Table 3.2). In four of these five RCTs that involved children with FR or SD SSNS, levamisole was given at a dose of 2.5 mg/kg on alternate days. In the sixth trial, a smaller dose (2.5 mg/kg of levamisole on 2 consecutive days of 7 days) did not reduce the risk of relapse compared to placebo [38]. Most children relapse when levamisole was discontinued. Observational studies have documented a more prolonged reduction in relapse frequency when it is used for 12-24 months [39-41]. Adverse effects of levamisole are uncommon and minor, with mild leucopenia and gastrointestinal upsets described. Rare cases of cutaneous vasculitis have been described with levamisole therapy [42].

KDIGO® GN Guideline March 2011 Public Review Draft
24
CNIs Two RCTs have demonstrated, no significant differences in the risk of relapse between
cyclosporine during treatment and cyclophosphamide or chlorambucil during treatment [43,44]. However, cyclosporine has a high relapse rate compared to alkylating agents when assessed at 12-24 months after treatment. An RCT from Japan reported that 50% of 49 children treated with cyclosporine relapsed compared to 70% of 59 children treated with placebo during the 24 weeks of therapy [45]. In observational studies, cyclosporine maintains remission in 60-90% of children with SD SSNS, who had relapsed after alkylating-agent therapy [46-48]. However, in children with SD SSNS due to biopsy-proven MCD, only 40% remained in remission after 2 years of therapy and all relapsed within a median of 26 days when cyclosporine was discontinued [46]. Most studies have used cyclosporine at 3-6 mg/kg/d in two divided doses targeting 12-hour trough levels of 80-150 ng/ml with maintenance of lower levels after a child has been in stable remission for 3-6 months, aiming to minimize cyclosporine nephrotoxicity. In an RCT the sustained remission rate was significantly higher in children maintaining a 12-hour cyclosporine trough level of 60-80 ng/ml (mean dose 4.7 mg/kg/d) compared to children treated with 2.5 mg/kg/d [49]. Limited data suggest peak (C2) levels rather than trough (C0) levels can be used for monitoring [50].
Tacrolimus has not been studied in RCTs in children with SSNS. Tacrolimus is widely used in North America in children with FR and SD SSNS, because of the cosmetic side effects of cyclosporine. There are few data to support its use, though its efficacy would appear to be similar to that of cyclosporine based on an observational study in SD SSNS [51]. The tacrolimus dose is adjusted according to 12-hour trough levels of 5-10 ng/ml initially based on data from kidney transplant studies.
The principal side-effects of cyclosporine are kidney dysfunction, hypertension, gum hypertrophy, and hypertrichosis. Hypertension and kidney dysfunction are reported in 5-10% of children [34,47,48]. Hypertrichosis and gum hypertrophy develop in 70% and 30%, respectively, in children treated with cyclosporine for more than 1 year [47]. Tacrolimus also causes kidney dysfunction and hypertension, but significantly less hypertrichosis; tacrolimus-associated diabetes mellitus has been described in children with nephrotic syndrome [52].
In children receiving cyclosporine for 12 months or more, tubulointerstitial lesions on kidney biopsy are reported in 30-40% of cases. This increases to 80% after 4 or more years of treatment [53]. Cyclosporine-associated arteriopathy is uncommon. The duration of safe therapy is controversial, with some authors suggesting that CNI therapy should be restricted to 2 years [53], while others have suggested that longer courses of cyclosporine may be well tolerated [54].
Coadministration of ketoconazole with cyclosporine in children with SD SSNS resulted in a 48% reduction in mean dose of cyclosporine, equivalent to a net cost saving of 38% with no reduction in efficacy, in a nonrandomized comparator study [55].
MMF To date all studies of mycophenolic acid prodrugs in nephrotic syndrome have used MMF.
In a small RCT, five of 12 children treated for 1 year with MMF relapsed compared to one of 12 treated with cyclosporine. Although this difference was not statistically significant, the patient

KDIGO® GN Guideline March 2011 Public Review Draft
25
numbers were too small to determine the relative efficacies of MMF and cyclosporine [56]. GFR remained stable during MMF treatment but fell during cyclosporine treatment. In a prospective study of 33 children (26 with FR SSNS) treated for 6 months, 24 (75%) children remained in remission during therapy, with 12 remaining relapse-free for 6 months after MMF was ceased; eight of these 12 patients continued in remission during 18-30 months of follow-up [57]. In a retrospective study of SD SSNS in 42 children, who were treated for at least 6 months, mean reduction in relapse rate was 3.8 per year [58]. MMF was generally well tolerated, with small numbers of children developing leucopenia and abdominal pain. In observational studies, MMF has been used for up to 45 months and has been well tolerated [58]. In most studies, MMF has been given in a dose of 1200 mg/m2/d or about 30 mg/kg/d in two divided doses. MMF has been used with cyclosporine in children with poorly controlled SD SSNS and has allowed reduction in cyclosporine dose [59]. Mycophenolate sodium may be an alternative if MMF is not tolerated because of adverse effects, but there are no data to support its use in nephrotic syndrome. In pediatric kidney transplant patients on cyclosporine, a single-dose pharmacokinetic study has demonstrated that 450 mg/m2 mycophenolate sodium and 600 mg/m2 of MMF provide similar mycophenolic acid exposure [60].
Choice of First Agent for FR or SD SSNS There are no data from RCTs to determine which corticosteroid-sparing agent should be
used as the first agent in a child with FR or SD SSNS. In Table 3.4, the advantages and disadvantages of alkylating agents, levamisole, CNIs, and MMF are presented.This table should allow the clinician and families to determine which agent a child with FR or SD SSNS should receive as their first corticosteroid-sparing agent.
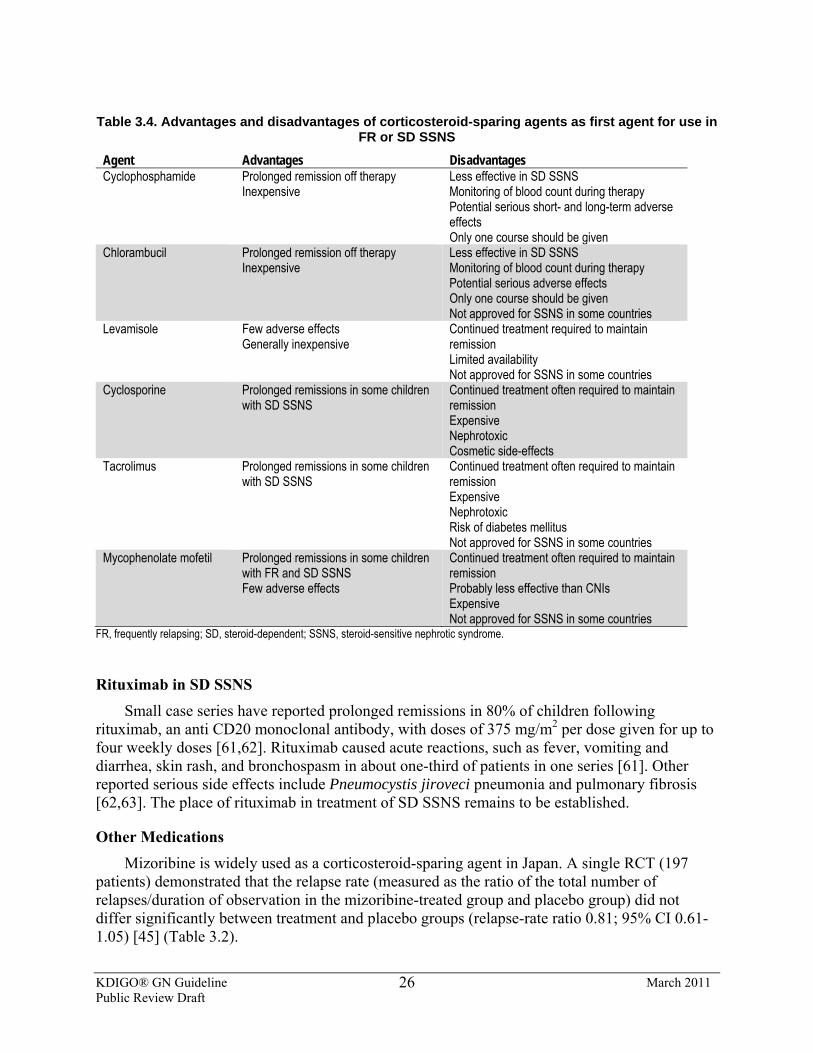
KDIGO® GN Guideline March 2011 Public Review Draft
26
Table 3.4. Advantages and disadvantages of corticosteroid-sparing agents as first agent for use in
FR or SD SSNS
Agent Advantages Disadvantages Cyclophosphamide Prolonged remission off therapy
Inexpensive Less effective in SD SSNS Monitoring of blood count during therapy Potential serious short- and long-term adverse effects Only one course should be given
Chlorambucil Prolonged remission off therapy Inexpensive
Less effective in SD SSNS Monitoring of blood count during therapy Potential serious adverse effects Only one course should be given Not approved for SSNS in some countries
Levamisole Few adverse effects Generally inexpensive
Continued treatment required to maintain remission Limited availability Not approved for SSNS in some countries
Cyclosporine Prolonged remissions in some children with SD SSNS
Continued treatment often required to maintain remission Expensive Nephrotoxic Cosmetic side-effects
Tacrolimus Prolonged remissions in some children with SD SSNS
Continued treatment often required to maintain remission Expensive Nephrotoxic Risk of diabetes mellitus Not approved for SSNS in some countries
Mycophenolate mofetil Prolonged remissions in some children with FR and SD SSNS Few adverse effects
Continued treatment often required to maintain remission Probably less effective than CNIs Expensive Not approved for SSNS in some countries
FR, frequently relapsing; SD, steroid-dependent; SSNS, steroid-sensitive nephrotic syndrome.
Rituximab in SD SSNS Small case series have reported prolonged remissions in 80% of children following
rituximab, an anti CD20 monoclonal antibody, with doses of 375 mg/m2 per dose given for up to four weekly doses [61,62]. Rituximab caused acute reactions, such as fever, vomiting and diarrhea, skin rash, and bronchospasm in about one-third of patients in one series [61]. Other reported serious side effects include Pneumocystis jiroveci pneumonia and pulmonary fibrosis [62,63]. The place of rituximab in treatment of SD SSNS remains to be established.
Other Medications Mizoribine is widely used as a corticosteroid-sparing agent in Japan. A single RCT (197
patients) demonstrated that the relapse rate (measured as the ratio of the total number of relapses/duration of observation in the mizoribine-treated group and placebo group) did not differ significantly between treatment and placebo groups (relapse-rate ratio 0.81; 95% CI 0.61-1.05) [45] (Table 3.2).

KDIGO® GN Guideline March 2011 Public Review Draft
27
It is recommended that azathioprine not be used as a corticosteroid-sparing agent in FR and SD SSNS, since two RCTs have demonstrated no significant difference in the risk of relapse between azathioprine and placebo (RR 0.90; 95% CI 0.59-1.38) [34] (Table 3.2).
3.4: Indication for kidney biopsy 3.4.1: Indications for kidney biopsy in children with SSNS are (Not Graded):
• late failure to respond following initial response to corticosteroids; • a high index of suspicion for a different underlying pathology; • decreasing kidney function in children receiving CNIs.
RATIONALE
Kidney biopsy is indicated in children with nephrotic syndrome who fail to respond to corticosteroids after one or more remissions (late nonresponder) to determine kidney pathology. There is no fixed upper age limit for treating children with nephrotic syndrome without prior kidney biopsy, particularly in Northern Europe and India where 40-50% adolescents have MCD [1,64,65]. However, in populations with a much higher prevalence of FSGS and other pathologies, particularly African or African-American populations, it is reasonable to consider routine biopsy at the time of onset of nephrotic syndrome diagnosis before treatment [66]. While it is sometimes recommended that children with SSNS should undergo annual kidney biopsy if CNI therapy is continued beyond 2 years [53], there are no data to determine whether the benefits of regular biopsies exceed the harm. Biopsies should be considered in children with deteriorating kidney function, particularly if this persists when CNI doses are reduced. Routine biopsies of children with FR or SD SSNS before using corticosteroid-sparing therapy are not indicated. Studies show that the most important predictor for kidney survival in childhood nephrotic syndrome is not kidney pathology, but the achievement and maintenance of remission following any therapy [67].
3.5: Immunizations in children with SSNS 3.5.1: To reduce the risk of serious infections in children with SSNS (Not
Graded): • Children should receive pneumococcal vaccination. • Children and their household contacts should receive influenza
vaccination annually. • Vaccination with live vaccines should be deferred until prednisone
dose is below either 1 mg/kg daily or 2 mg/kg on alternate days. • Live vaccines are contraindicated in children receiving
corticosteroid-sparing immunosuppressive agents. • Healthy household contacts should receive live vaccines to minimize
the risk of transfer of infection to the immunosuppressed child. • Following close contact with Varicella infection, nonimmune
children on immunosuppressive agents should receive zoster immune globulin if available.

KDIGO® GN Guideline March 2011 Public Review Draft
28
RATIONALE
Children with nephrotic syndrome are at increased risk of invasive pneumococcal disease [68] and should receive pneumococcal immunization with the heptavalent conjugate vaccine (7vPCV) and the 23-valent polysaccharide vaccine (23vPPV) according to local recommendations. Adequacy of response to the 7vPCV vaccine has not been studied in children with nephrotic syndrome. Serological response to 23vPPV was not different in children with active nephrotic syndrome on high-dose prednisone (60 mg/m2/d) compared to children who received the vaccine while on low-dose alternate day prednisone [69]. Children with SSNS and their household contacts should receive annual influenza vaccination [70,71].
Live Vaccines Live vaccines (measles, mumps, rubella, varicella, rotavirus) are contraindicated in children
on immunosuppressive or cytotoxic agents [70,71] and should be deferred until:
• Prednisone dose is below 1 mg/kg/d (below 20 mg/d) or below 2 mg/kg on alternate days (below 40 mg on alternate days).
• The child has been off cytotoxic agents (cyclophosphamide, chlorambucil) for more than 3 months.
• The child has been off other immunosuppressive agents (CNIs, levamisole, MMF) for more than 1 month.
Healthy siblings and household contacts of children with impaired immunity should be
vaccinated with measles, mumps, rubella, varicella, and rotavirus vaccines (where indicated) to prevent them from infecting children with impaired immunity [71].
Varicella Immunization Varicella infection may lead to life-threatening disease in children receiving
immunosuppressive medications. Varicella immunization is safe and effective in children with nephrotic syndrome, including children on low-dose alternate-day prednisone [72].
• Children with SSNS, who are not receiving immunosuppressive or cytotoxic agents other than low-dose daily or alternate-day prednisone, should be offered varicella immunization if nonimmune [70,71].
• Families of nonimmune children with SSNS, who are receiving immunosuppressive agents, should be asked to contact their physician as soon as possible if the child comes into close contact with another child with chicken pox, or an adult with herpes zoster, so that the child can receive zoster immune globulin (if available) within 72 hours of exposure [71].
• Aciclovir or valaciclovir should be administered to immunosuppressed children at the onset of chicken pox lesions.
RESEARCH RECOMMENDATIONS
Further information from RCTs is required:

KDIGO® GN Guideline March 2011 Public Review Draft
29
• To determine the relative efficacies of alkylating agents, levamisole, MMF, CNIs in FR and SD SSNS.
• To determine the relative benefits and adverse effects of cyclosporine and tacrolimus in FR and SD SSNS.
• To determine the additional benefits and risks of mycophenolic acid when added to CNIs in SD SSNS.
• To determine the additional benefits and risks of rituximab in addition to other corticosteroid-sparing agents in SD SSNS.
Please refer to Supplemental Tables 1-7 for additional data related to Chapter 3.
References 1. McKinney PA, Feltbower RG, Brocklebank JT, Fitzpatrick MM. Time trends and ethnic patterns of
childhood nephrotic syndrome in Yorkshire, UK. Pediatric Nephrology 16(12):1040-4, 2001. 2. Primary nephrotic syndrome in children: clinical significance of histopathologic variants of minimal change
and of diffuse mesangial hypercellularity. A Report of the International Study of Kidney Disease in Children. Kidney International 1981; 20(6):765-771.
3. Koskimies O, Vilska J, Rapola J, Hallman N. Long-term outcome of primary nephrotic syndrome. Archives of Disease in Childhood 57(7):544-8, 1982.
4. Fakhouri F, Bocquet N, Taupin P, Presne C, Gagnadoux MF, Landais P et al. Steroid-sensitive nephrotic syndrome: from childhood to adulthood. American Journal of Kidney Diseases 41(3):550-7, 2003.
5. Tarshish P, Tobin JN, Bernstein J, Edelmann CM, Jr. Prognostic significance of the early course of minimal change nephrotic syndrome: report of the International Study of Kidney Disease in Children. Journal of the American Society of Nephrology 1997; 8(5):769-776.
6. Arneil GC. 164 children with nephrosis. Lancet 1961; 2:1103-1110. 7. Ruth EM, Kemper MJ, Leumann EP, Laube GF, Neuhaus TJ. Children with steroid-sensitive nephrotic
syndrome come of age: long-term outcome. Journal of Pediatrics 2005; 147(2):202-207. 8. Bhimma R, Coovadia HM, Adhikari M. Nephrotic syndrome in South African children: changing
perspectives over 20 years. Pediatric Nephrology 11(4):429-34, 1997. 9. Kim JS, Bellew CA, Silverstein DM, Aviles DH, Boineau FG, Vehaskari VM. High incidence of initial and
late steroid resistance in childhood nephrotic syndrome. Kidney International 68(3):1275-81, 2005. 10. Arneil GC. The nephrotic syndrome. Pediatric Clinics of North America 18(2):547-59, 1971. 11. Alternate-day versus intermittent prednisone in frequently relapsing nephrotic syndrome. A report of
"Arbetsgemeinschaft fur Padiatrische Nephrologie". Lancet 1(8113):401-3, 1979. 12. Hodson EM, Willis NS, Craig JC. Corticosteroid therapy for nephrotic syndrome in children. Update of
Cochrane Database Syst Rev. 2005;(1):CD001533; PMID: 15674881. Cochrane Database of Systematic Reviews 2007;(4):CD001533.
13. Feber J, Al Matrafi J, Farhadi E, Vaillancourt R, Wolfish N. Prednisone dosing per body weight or body surface area in children with nephrotic syndrome: is it equivalent? Pediatric Nephrology 2009; 24(5):1027-1031.
14. The primary nephrotic syndrome in children. Identification of patients with minimal change nephrotic syndrome from initial response to prednisone. A report of the International Study of Kidney Disease in Children. Journal of Pediatrics 98(4):561-4, 1981.
15. Short versus standard prednisone therapy for initial treatment of idiopathic nephrotic syndrome in children. Arbeitsgemeinschaft fur Padiatrische Nephrologie. Lancet 1(8582):380-3, 1988.
16. Alternate-day prednisone is more effective than intermittent prednisone in frequently relapsing nephrotic syndrome. A report of "Arbeitsgemeinschaft fur Padiatrische Nephrologie. European Journal of Pediatrics 135(3):229-37, 1981.

KDIGO® GN Guideline March 2011 Public Review Draft
30
17. Broyer M, Guest G, Gagnadoux MF. Growth rate in children receiving alternate-day corticosteroid treatment after kidney transplantation. Journal of Pediatrics 120(5):721-5, 1992.
18. Letavernier B, Letavernier E, Leroy S, Baudet-Bonneville V, Bensman A, Ulinski T. Prediction of high-degree steroid dependency in pediatric idiopathic nephrotic syndrome. Pediatric Nephrology 2008; 23(12):2221-2226.
19. Noer MS. Predictors of relapse in steroid-sensitive nephrotic syndrome. Southeast Asian Journal of Tropical Medicine & Public Health 2005; 36(5):1313-1320.
20. Lewis MA, Baildom EM, Davis N, Houston IB, Postlethwaite RJ. Nephrotic syndrome: from toddlers to twenties. Lancet 1(8632):255-9, 1989.
21. Constantinescu AR, Shah HB, Foote EF, Weiss LS. Predicting first-year relapses in children with nephrotic syndrome. Pediatrics 105(3 Pt 1):492-5, 2000.
22. Abeyagunawardena AS, Trompeter RS. Increasing the dose of prednisolone during viral infections reduces the risk of relapse in nephrotic syndrome: a randomised controlled trial. Archives of Disease in Childhood 2008; 93(3):226-228.
23. Elzouki AY, Jaiswal OP. Long-term, small dose prednisone therapy in frequently relapsing nephrotic syndrome of childhood. Effect on remission, statural growth, obesity, and infection rate. Clinical Pediatrics 27(8):387-92, 1988.
24. Consensus statement on management and audit potential for steroid responsive nephrotic syndrome. Report of a Workshop by the British Association for Paediatric Nephrology and Research Unit, Royal College of Physicians. Archives of Disease in Childhood 70(2):151-7, 1994.
25. Indian Pediatric Nephrology Group IAoP, Bagga A, Ali U, Banerjee S, Kanitkar M, Phadke KD et al. Management of steroid sensitive nephrotic syndrome: revised guidelines. Indian Pediatrics 2008; 45(3):203-214.
26. Srivastava RN, VasudevAS, Bagga A, Sunderam KR. Long-term, low-dose prednisolone therapy in frequently relapsing nephrotic syndrome. Pediatric Nephrology 1992; 6(3):247-250.
27. Kyrieleis AC, Lowik MM, Pronk I, Cruysberg HRM, Kremer JAM, Oyen WJG et al. Long-term outcome of biopsy-proven, frequently relapsing minimal-change nephrotic syndrome in children. Clinical Journal of American Society of Nephrology 2009; 4:1593-1600.
28. Latta K, von Schnakenburg C, Ehrich JH. A meta-analysis of cytotoxic treatment for frequently relapsing nephrotic syndrome in children. Pediatric Nephrology 16(3):271-82, 2001.
29. Niaudet P, Habib R, Tete MJ, Hinglais N, Broyer M. Cyclosporin in the treatment of idiopathic nephrotic syndrome in children. Pediatric Nephrology 1(4):566-73, 1987.
30. Hino S, Takemura T, Okada M, Murakami K, Yagi K, Fukushima K et al. Follow-up study of children with nephrotic syndrome treated with a long-term moderate dose of cyclosporine. American Journal of Kidney Diseases 31(6):932-9, 1998.
31. Mowry JA, McCarthy ET. Cyclosporine in glomerular disease. Seminars in Nephrology 1996; 16(6):548-554. 32. Boyer O, Moulder JK, Grandin L, Somers MJ. Short- and long-term efficacy of levamisole as adjunctive
therapy in childhood nephrotic syndrome. Pediatric Nephrology 2008; 23(4):575-580. 33. Leroy V, Baudouin V, Alberti C, Guest G, Niaudet P, Loirat C et al. Growth in boys with idiopathic nephrotic
syndrome on long-term cyclosporin and steroid treatment. Pediatric Nephrology 2009; 24(12):2393-2400. 34. Hodson EM, Willis NS, Craig JC. Non-corticosteroid treatment for nephrotic syndrome in children. [Update
of Cochrane Database Syst Rev. 2005;(2):CD002290; PMID: 15846634]. Cochrane Database of Systematic Reviews 2008;(1):CD002290.
34A. Cyclophosphamide treatment of steroid dependent nephrotic syndrome: comparison of eight week with 12 week course. Report of Arbeitsgemeinschaft fur Padiatrische Nephrologie. Archives of Disease in Childhood 62(11):1102-6, 1987
35. Williams SA, Makker SP, Ingelfinger JR, Grupe WE. Long-term evaluation of chlorambucil plus prednisone in the idiopathic nephrotic syndrome of childhood. New England Journal of Medicine 1980; 302(17):929-933.
36. Wetzels JF. Cyclophosphamide-induced gonadal toxicity: a treatment dilemma in patients with lupus nephritis? Netherlands Journal of Medicine 2004; 62(10):347-352.

KDIGO® GN Guideline March 2011 Public Review Draft
31
37. Miller DG. Alkylating agents and human spermatogenesis. JAMA 217(12):1662-5, 1971. 38. Weiss R. Randomized double-blind placebo controlled, multi-center trial of levamisole for children with
frequently relapsing/steroid dependent nephrotic syndrome. Journal of the American Society of Nephrology 1993; 4:289.
39. Hafeez F, Ahmed TM, Samina U. Levamisole in steroid dependent and frequently relapsing nephrotic syndrome. Jcpsp, Journal of the College of Physicians & Surgeons - Pakistan 2006; 16(1):35-37.
40. Fu LS, Shien CY, Chi CS. Levamisole in steroid-sensitive nephrotic syndrome children with frequent relapses and/or steroid dependency: comparison of daily and every-other-day usage. Nephron 97(4):c137-41, 2004.
41. Kemper MJ, Amon O, Timmermann K, Altrogge H, ller-Wiefel DE. [The treatment with levamisole of frequently recurring steroid-sensitive idiopathic nephrotic syndrome in children]. [German]. Deutsche Medizinische Wochenschrift 1998; 123(9):239-243.
42. Palcoux JB, Niaudet P, Goumy P. Side effects of levamisole in children with nephrosis. Pediatric Nephrology 1994; 8(2):263-264.
43. Niaudet P. Comparison of cyclosporin and chlorambucil in the treatment of steroid-dependent idiopathic nephrotic syndrome: a multicentre randomized controlled trial. The French Society of Paediatric Nephrology. Pediatric Nephrology 6(1):1-3, 1992.
44. Ponticelli CE. Cyclosporin versus cyclophosphamide for patients with steroid-dependent and frequently relapsing idiopathic nephrotic syndrome: A multicentre randomized controlled trial. Nephrology Dialysis Transplantation 1993; 8(12):1993.
45. Yoshioka K, Ohashi Y, Sakai T, Ito H, Yoshikawa N, Nakamura H et al. A multicenter trial of mizoribine compared to placebo in children with frequently relapsing nephrotic syndrome. Kidney International 58(1):317-24, 2000.
46. Hulton SA, Neuhaus TJ, Dillon MJ, Barratt TM. Long-term cyclosporin A treatment of minimal-change nephrotic syndrome of childhood. Pediatric Nephrology 8(4):401-3, 1994.
47. El Husseini A, El Basuony F, Mahmoud I, Sheashaa H, Sabry A, Hassan R et al. Long-term effects of cyclosporine in children with idiopathic nephrotic syndrome: a single-centre experience. Nephrology Dialysis Transplantation 2005;(11):2433-2438.
48. Niaudet P, Broyer M, Habib R. Treatment of idiopathic nephrotic syndrome with cyclosporin A in children. Clinical Nephrology 35 Suppl 1:S31-6, 1991.
49. Ishikura K, Ikeda M, Hattori S, Yoshikawa N, Sasaki S, Iijima K et al. Effective and safe treatment with cyclosporine in nephrotic children: a prospective, randomized multicenter trial. Kidney International 2008; 73(10):1167-1173.
50. Filler G. How should microemulsified Cyclosporine A (Neoral) therapy in patients with nephrotic syndrome be monitored? Nephrology Dialysis Transplantation 2005; 20(6):1032-1034.
51. Sinha MD, MacLeod R, Rigby E, Clark AG. Treatment of severe steroid-dependent nephrotic syndrome (SDNS) in children with tacrolimus. Nephrology Dialysis Transplantation 2006; 21(7):1848-1854.
52. Dotsch J, Dittrich K, Plank C, Rascher W. Is tacrolimus for childhood steroid-dependent nephrotic syndrome better than cyclosporin A? Nephrology Dialysis Transplantation 2006; 21:1761-1763.
53. Iijima K, Hamahira K, Tanaka R, Kobayashi A, Nozu K, Nakamura H et al. Risk factors for cyclosporine-induced tubulointerstitial lesions in children with minimal change nephrotic syndrome. Kidney International 61(5):1801-5, 2002.
54. Kranz B, Vester U, scher R, Wingen AM, Hoyer PF. Cyclosporine-A-induced nephrotoxicity in children with minimal-change nephrotic syndrome: long-term treatment up to 10 years. Pediatric Nephrology 2008; 23(4):581-586.
55. El Husseini A, El Basuony F, Mahmoud I, Donia A, Sheashaa H, Sabry A et al. Impact of the cyclosporine-ketoconazole interaction in children with steroid-dependent idiopathic nephrotic syndrome. European Journal of Clinical Pharmacology 2006; 62(1):3-8.
56. Dorresteijn EM, Kist-van Holthe JE, Levtchenko EN, Nauta J, Hop WC, van der Heijden AJ. Mycophenolate mofetil versus cyclosporine for remission maintenance in nephrotic syndrome. Pediatric Nephrology 2008; 23(11):2013-2020.

KDIGO® GN Guideline March 2011 Public Review Draft
32
57. Hogg RJ, Fitzgibbons L, Bruick J, Bunke M, Ault B, Baqi N et al. Clinical trial of mycophenolate mofetil (MMF) for frequent relapsing nephrotic syndrome in children. Pediatric Nephrology 2004; 19(9):C18.
58. Afzal K, Bagga A, Menon S, Hari P, Jordan SC. Treatment with mycophenolate mofetil and prednisolone for steroid-dependent nephrotic syndrome. Pediatric Nephrology 2007; 22(12):2059-2065.
59. Barletta GM, Smoyer WE, Bunchman TE, Flynn JT, Kershaw DB. Use of mycophenolate mofetil in steroid-dependent and -resistant nephrotic syndrome. Pediatric Nephrology 2003; 18(8):833-837.
60. Ettenger R, Bartosh S, Choi L, Zhu W, Niederberger W, Campestrini J et al. Pharmacokinetics of enteric-coated mycophenolate sodium in stable pediatric renal transplant recipients. Pediatric Transplantation 2005; 9(6):780-787.
61. Prytula A, Iijima K, Kamei K, Geary D, Gottlich E, Majeed A et al. Rituximab in refractory nephrotic syndrome. Pediatric Nephrology 2010; 25(3):461-468.
62. Guigonis V, Dallocchio A, Baudouin V, Dehennault M, Hachon-Le Camus C, Afanetti M et al. Rituximab treatment for severe steroid- or cyclosporine-dependent nephrotic syndrome: a multicentric series of 22 cases. Pediatric Nephrology 2008; 23(8):1269-1279.
63. Chaumais MC, Garnier A, Chalard F, Peuchmaur M, Dauger S, Jacqz-Agrain E et al. Fatal pulmonary fibrosis after rituximab administration. Pediatric Nephrology 2009; 24(9):1753-1755.
64. Gulati S, Sural S, Sharma RK, Gupta A, Gupta RK. Spectrum of adolescent-onset nephrotic syndrome in Indian children. Pediatric Nephrology 16(12):1045-8, 2001.
65. Gulati S, Sharma AP, Sharma RK, Gupta A, Gupta RK. Do current recommendations for kidney biopsy in nephrotic syndrome need modifications? Pediatric Nephrology 17(6):404-8, 2002.
66. Baqi N, Singh A, Balachandra S, Ahmad H, Nicastri A, Kytinski S et al. The paucity of minimal change disease in adolescents with primary nephrotic syndrome. Pediatric Nephrology 12(2):105-7, 1998.
67. Gipson DS, Chin H, Presler TP, Jennette C, Massengill S, Gibson K et al. Differential risk of remission and ESRD in childhood FSGS. Pediatric Nephrology 2006; 21(3):344-349.
68. Gorensek MJ, Lebel MH, Nelson JD. Peritonitis in children with nephrotic syndrome. Pediatrics 1988; 81(6):849-856.
69. Ulinski T, Leroy S, Dubrel M, Danon S, Bensman A. High serological response to pneumococcal vaccine in nephrotic children at disease onset on high-dose prednisone. Pediatric Nephrology 2008; 23(7):1107-1113.
70. American Academy of Pediatrics. Active and passive immunization. In: Pickering LK, Baker CJ, Long SS, McMillan JA, editors. Red Book. Elk Grove Village: AmericanAcademy of Pediatrics, 2006: 9-40.
71. The Australian Immunisation Handbook. 9th ed. Australian Government: Department of Health and Ageing, 2008.
72. Furth SL, Arbus GS, Hogg R, Tarver J, Chan C, Fivush BA et al. Varicella vaccination in children with nephrotic syndrome: a report of the Southwest Pediatric Nephrology Study Group. Journal of Pediatrics 2003; 142(2):145-148.

KDIGO® GN Guideline March 2011 Public Review Draft
33
CHAPTER 4: STEROID-RESISTANT NEPHROTIC SYNDROME IN CHILDREN
INTRODUCTION
This chapter makes treatment recommendations for children aged 1 to 18 years with nephrotic syndrome, who do not achieve a complete remission with corticosteroid therapy, i.e., SRNS. This chapter does not apply to children with SRNS under 1 year of age, nor to SRNS due to histologic patterns of glomerular injury other than MCD, MPGN, or FSGS.
The cost implications for global application of this guideline are addressed in Chapter 2.
4.1: Evaluation of children with SRNS 4.1.1: We suggest a minimum of 8 weeks treatment with corticosteroids to
define steroid resistance. (2D) 4.1.2: The following are required to evaluate the child with SRNS (Not Graded):
• a diagnostic kidney biopsy; • evaluation of kidney function by GFR or eGFR; • quantitation of urine protein excretion.
BACKGROUND
SRNS generally, and FSGS specifically, is associated with a 50% risk for ESRD within 5 years of diagnosis if patients do not achieve a partial or complete remission [1]. Persistent nephrotic syndrome is associated with poor patient-reported quality of life, thromboembolic events, hypertension, peritonitis and other serious infections, persistent dyslipidemia, and death [2-5]. Children reaching ESRD have a greatly reduced life expectancy of 19 years on average following initiation of dialysis, and approximately 40 years following transplantation [6].
The cumulative burden of ongoing disease-related complications is measured against potential medication-associated toxicities due to corticosteroids and other immunosuppressive agents. These issues are discussed in Chapter 3, SSNS and in Chapter 1, Introduction.
The potential benefit of therapy includes disease cure, control of nephrotic syndrome, and/or or slowing the progression to ESRD. Unfortunately, there are times when the nephrologist, with the child’s family or caregivers, may have to accept a point of futility has been reached, characterized by unremitting loss of kidney function toward ESRD, resistance to multiple drug therapies, or concern for cumulative drug-associated toxicities.

KDIGO® GN Guideline March 2011 Public Review Draft
34
RATIONALE
• Management of children with SRNS requires confirmation of resistance to corticosteroids, usually defined by unresponsiveness to oral prednisone or prednisolone* 60 mg/m2/d or 2 mg/kg/d to a maximum 60 mg/d for a minimum of 8 weeks.
• Kidney biopsy is necessary to exclude secondary causes of nephrotic syndrome, and assess the extent of interstitial and glomerular fibrosis.
• Kidney function, measured by eGFR, at presentation is associated with the long-term risk for kidney failure.
• Quantification of proteinuria is essential, since this provides the comparison for subsequent treatment responsiveness.
*Prednisone and prednisolone are equivalent, used in the same dosage, and have both been used in RCTs. All later references to prednisone in this chapter refer to prednisone or prednisolone. All later references to oral corticosteroids refer to prednisone or prednisolone.
Steroid Resistance The minimum requirement of corticosteroid exposure to define resistance remains unclear.
Discrepancies in the definition of SRNS create difficulties in comparing therapeutic trials. Based upon the International Study of Kidney Disease in Children (ISKDC), 95% of children with SRNS will demonstrate resolution of proteinuria with 4 weeks of daily corticosteroid therapy and 100% after an additional 3 weeks of alternate-day therapy [7]. Subsequent studies have reported additional remissions after an extended exposure to steroids in low-dose prednisone control arms within RCTs and after high doses of i.v. or oral corticosteroids in observational studies [8,9]. It is not clear if these late responses are due to the extended corticosteroid exposure, a late effect of prior therapy, or natural history of the disease. Consequently, we have elected to utilize one of the commonly used definitions of resistance, i.e., a minimum exposure of 8 weeks of prednisone 2 mg/kg/d or 60 mg/m2/d for 4 weeks followed by 1.5 mg/kg/d or 40 mg/m2 per dose alternate-day for 4 weeks. At this point, steroid resistance dictates the requirement for kidney biopsy to define the histopathology. Steroids may be continued for an additional 4 weeks, totaling 12 weeks, while awaiting histopathology results.
Kidney Biopsy A kidney biopsy in the evaluation of SRNS is generally recommended. This evaluation—
including light microscopy, immunofluorescence, and electron microscopy—may indicate disorders that also result in the clinical features of the nephrotic syndrome, e.g., immunoglobulin A nephropathy (IgAN) or LN. The therapy is subsequently dictated by the underlying diagnosis. It may also show pathologic lesions of FSGS or, despite steroid resistance, may show MCD. In Chapter 2 it was noted that 20 glomeruli are needed in a biopsy to confidently exclude lesions that are affecting only 5% of them; hence, there is a possibility of missing an FSGS lesion in many routine biopsies containing fewer than this number. The kidney biopsy will also provide information regarding the degree of interstitial and glomerular fibrosis, which will be utilized in the assessment of prognosis of children with SRNS. Results of the biopsy are also often used to

KDIGO® GN Guideline March 2011 Public Review Draft
35
explain to both patient and family why there has not been a response to therapy, and that the prognosis is likely to be substantially altered from the initial one.
Laboratory Assessment Kidney function should be measured at the time a diagnosis of SRNS is made to inform
prognosis and assessment of response to subsequent therapy. Kidney function at the time of diagnosis is a predictor of the long-term risk for kidney failure. Proteinuria should be quantified by uPCR to allow subsequent treatment response to be defined as partial, complete, or no remission [1,10-13] (Table 3.1, Chapter 3). The uPCR should be measured in a first morning void to prevent variation based upon orthostatic effects [14]. Measurements of 24-hour urine protein may also be used but such collections are impractical in young children who are not toilet-trained. Observational studies of patients with FSGS demonstrate a 5-year kidney survival of 90% in patients with a complete remission following any single or combination of tested therapies [1,10]. Partial remission has been associated with an intermediate 5-year kidney survival of 80% in adults, although these data are not available for children [10]. Absence of remission predicts a 5-year kidney survival of approximately 50% [1, 10, 13].
Many genetic mutations have been identified in SRNS and FSGS studies. In children with SRNS over 1 year of age, podocin mutations have been reported in 0-30%. The significant variation in the prevalence of SRNS-associated mutations is exemplified by the absence of podocin mutations in an African-American cohort of 18 children with FSGS [15] and the findings of a 28% prevalence of podocin mutations in a European cohort of 25 children published by the same group of investigators [16]. Routine evaluation for genetic mutations is not included in this guideline due to the variability in availability, significant cost, low to absent prevalence observed in some populations, and our current uncertainty about the individual response to therapy and prognosis when one of the many mutations is identified.
4.2: Treatment recommendations for SRNS 4.2.1: We recommend using a calcineurin inhibitor (CNI) as first-line therapy.
(1B) 4.2.1.1: We suggest that CNI therapy be continued for a minimum of 6
months and then stopped if a partial or complete remission of proteinuria is not achieved. (2C)
4.2.1.2: We suggest CNIs be continued for a minimum of 12 months when at least a partial remission is achieved by 6 months. (2C)
4.2.1.3: We suggest that low-dose corticosteroid therapy be combined with CNI therapy. (2D)
4.2.2: We recommend treatment with ACEi or ARBs for children with SRNS. (1B)
4.2.3: In children who fail to achieve remission with CNI therapy: 4.2.3.1: We suggest that mycophenolate mofetil (2D), high-dose
corticosteroids (2D), or a combination of these agents (2D) be considered in children who fail to achieve complete or partial remission with CNIs and corticosteroids.

KDIGO® GN Guideline March 2011 Public Review Draft
36
4.2.3.2: We suggest that cyclophosphamide not be given to children with SRNS. (2B)
4.2.4: In patients with a relapse of nephrotic syndrome after complete remission, we suggest that therapy be restarted using one of the following options: (2C) • oral corticosteroids (2D); • return to previous successful immunosuppressive agent (2D); • an alternative immunosuppressive agent to avoid cumulative
potential toxicity (2D).
BACKGROUND
The risk for kidney failure in patients with persistent nephrotic syndrome provides the rationale for utilizing an alternate therapy once steroid resistance has been established.
Both cyclosporine and corticosteroids have a direct effect on the podocyte cytoskeleton [17], in addition to their immune-modulating properties, indicating these agents may have multiple beneficial mechanisms of action in nephrotic syndrome.
RATIONALE
• There is moderate-quality evidence that cyclosporine induces complete or partial remission in a majority of children with SRNS.
• There is low-quality evidence that tacrolimus has a similar impact on proteinuria control and may improve adherence to treatment, based upon lower risk for hypertrichosis and gingival hyperplasia compared to cyclosporine.
• There is moderate-quality evidence that treatment with renin-angiotensin system (RAS) blockade is associated with a reduction in proteinuria.
• The risk for kidney failure is significantly greater for patients who fail to achieve a partial or complete remission with any single or combination therapy.
CNI Therapy Cyclosporine has been most widely studied for treatment of SRNS. In three RCTs with 49
patients, 26 treated with cyclosporine and 23 with placebo or control therapy [18-20] (Table 4.1), cyclosporine resulted in a complete remission in 31% and partial remission in 38% during 6 months of therapy. The 69% cumulative complete and partial remission was significantly better than the 0-16% remission in the control arms of these randomized studies. Based upon case series, complete and partial remissions are less common in the presence of nephrotic syndrome associated with podocin mutations. However, remissions have been reported, and suggest that a trial of CNI therapy may induce at least a partial remission even in these patients [21].
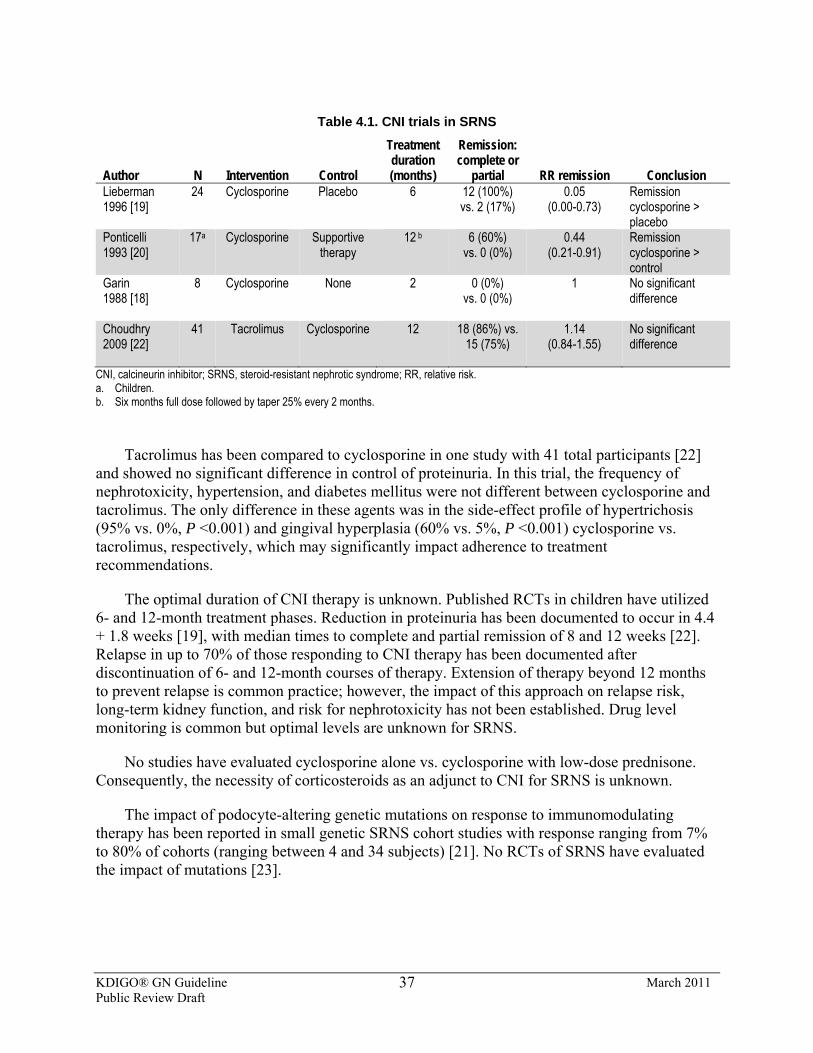
KDIGO® GN Guideline March 2011 Public Review Draft
37
Table 4.1. CNI trials in SRNS
Author N Intervention Control
Treatment duration (months)
Remission: complete or
partial RR remission Conclusion Lieberman 1996 [19]
24 Cyclosporine Placebo 6 12 (100%) vs. 2 (17%)
0.05 (0.00-0.73)
Remission cyclosporine > placebo
Ponticelli 1993 [20]
17a Cyclosporine Supportive therapy
12 b 6 (60%) vs. 0 (0%)
0.44 (0.21-0.91)
Remission cyclosporine > control
Garin 1988 [18]
8 Cyclosporine None 2 0 (0%) vs. 0 (0%)
1 No significant difference
Choudhry 2009 [22]
41 Tacrolimus Cyclosporine 12 18 (86%) vs. 15 (75%)
1.14 (0.84-1.55)
No significant difference
CNI, calcineurin inhibitor; SRNS, steroid-resistant nephrotic syndrome; RR, relative risk. a. Children. b. Six months full dose followed by taper 25% every 2 months.
Tacrolimus has been compared to cyclosporine in one study with 41 total participants [22] and showed no significant difference in control of proteinuria. In this trial, the frequency of nephrotoxicity, hypertension, and diabetes mellitus were not different between cyclosporine and tacrolimus. The only difference in these agents was in the side-effect profile of hypertrichosis (95% vs. 0%, P <0.001) and gingival hyperplasia (60% vs. 5%, P <0.001) cyclosporine vs. tacrolimus, respectively, which may significantly impact adherence to treatment recommendations.
The optimal duration of CNI therapy is unknown. Published RCTs in children have utilized 6- and 12-month treatment phases. Reduction in proteinuria has been documented to occur in 4.4 + 1.8 weeks [19], with median times to complete and partial remission of 8 and 12 weeks [22]. Relapse in up to 70% of those responding to CNI therapy has been documented after discontinuation of 6- and 12-month courses of therapy. Extension of therapy beyond 12 months to prevent relapse is common practice; however, the impact of this approach on relapse risk, long-term kidney function, and risk for nephrotoxicity has not been established. Drug level monitoring is common but optimal levels are unknown for SRNS.
No studies have evaluated cyclosporine alone vs. cyclosporine with low-dose prednisone. Consequently, the necessity of corticosteroids as an adjunct to CNI for SRNS is unknown.
The impact of podocyte-altering genetic mutations on response to immunomodulating therapy has been reported in small genetic SRNS cohort studies with response ranging from 7% to 80% of cohorts (ranging between 4 and 34 subjects) [21]. No RCTs of SRNS have evaluated the impact of mutations [23].
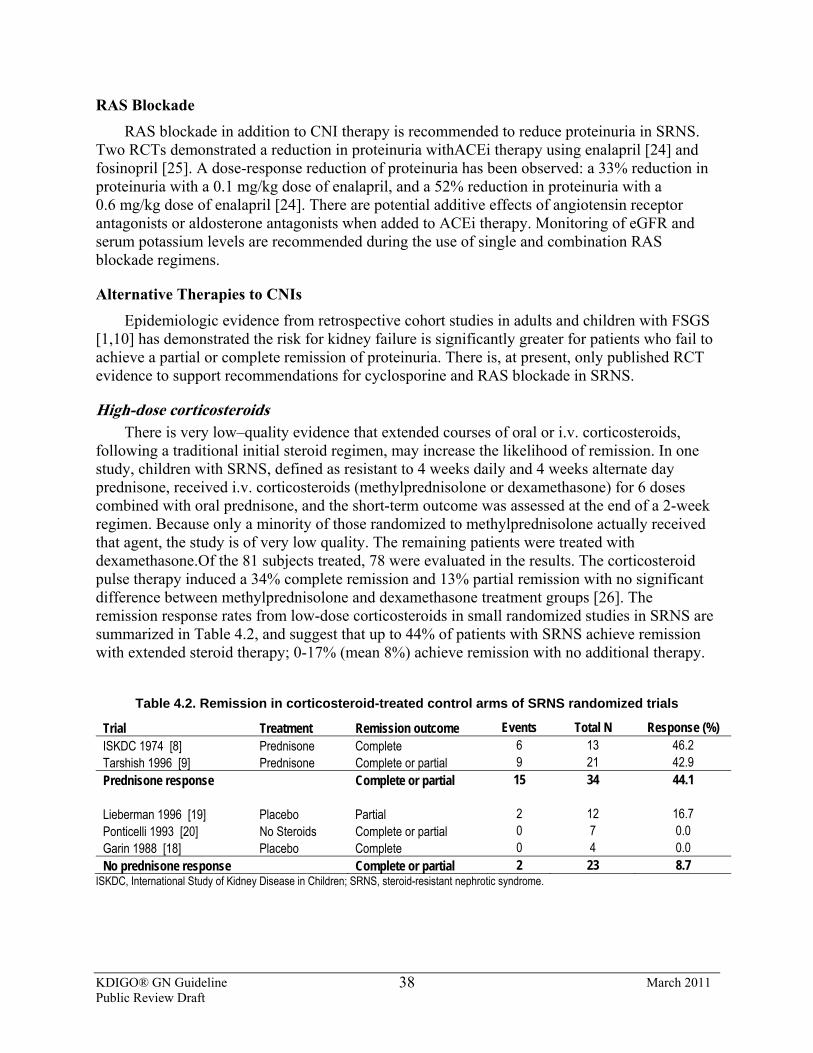
KDIGO® GN Guideline March 2011 Public Review Draft
38
RAS Blockade RAS blockade in addition to CNI therapy is recommended to reduce proteinuria in SRNS.
Two RCTs demonstrated a reduction in proteinuria withACEi therapy using enalapril [24] and fosinopril [25]. A dose-response reduction of proteinuria has been observed: a 33% reduction in proteinuria with a 0.1 mg/kg dose of enalapril, and a 52% reduction in proteinuria with a 0.6 mg/kg dose of enalapril [24]. There are potential additive effects of angiotensin receptor antagonists or aldosterone antagonists when added to ACEi therapy. Monitoring of eGFR and serum potassium levels are recommended during the use of single and combination RAS blockade regimens.
Alternative Therapies to CNIs Epidemiologic evidence from retrospective cohort studies in adults and children with FSGS
[1,10] has demonstrated the risk for kidney failure is significantly greater for patients who fail to achieve a partial or complete remission of proteinuria. There is, at present, only published RCT evidence to support recommendations for cyclosporine and RAS blockade in SRNS.
High-dose corticosteroids There is very low–quality evidence that extended courses of oral or i.v. corticosteroids,
following a traditional initial steroid regimen, may increase the likelihood of remission. In one study, children with SRNS, defined as resistant to 4 weeks daily and 4 weeks alternate day prednisone, received i.v. corticosteroids (methylprednisolone or dexamethasone) for 6 doses combined with oral prednisone, and the short-term outcome was assessed at the end of a 2-week regimen. Because only a minority of those randomized to methylprednisolone actually received that agent, the study is of very low quality. The remaining patients were treated with dexamethasone.Of the 81 subjects treated, 78 were evaluated in the results. The corticosteroid pulse therapy induced a 34% complete remission and 13% partial remission with no significant difference between methylprednisolone and dexamethasone treatment groups [26]. The remission response rates from low-dose corticosteroids in small randomized studies in SRNS are summarized in Table 4.2, and suggest that up to 44% of patients with SRNS achieve remission with extended steroid therapy; 0-17% (mean 8%) achieve remission with no additional therapy.
Table 4.2. Remission in corticosteroid-treated control arms of SRNS randomized trials
Trial Treatment Remission outcome Events Total N Response (%) ISKDC 1974 [8] Prednisone Complete 6 13 46.2 Tarshish 1996 [9] Prednisone Complete or partial 9 21 42.9 Prednisone response Complete or partial 15 34 44.1 Lieberman 1996 [19] Placebo Partial 2 12 16.7 Ponticelli 1993 [20] No Steroids Complete or partial 0 7 0.0 Garin 1988 [18] Placebo Complete 0 4 0.0 No prednisone response Complete or partial 2 23 8.7
ISKDC, International Study of Kidney Disease in Children; SRNS, steroid-resistant nephrotic syndrome.

KDIGO® GN Guideline March 2011 Public Review Draft
39
MMF Observational studies involving children with SRNS who were treated for a minimum of 6
months with mycophenolate have demonstrated a complete remission rate from 23% to 62%, a partial remission rate of 25% to 37% and no remission in 8% to 40% [25,27]. Data from SRNS patients are expected from the North American FSGS Clinical Trial in early 2011 which compares the combination of high-dose oral corticosteroids to MMF to cyclosporine.
Cytotoxic agents There is moderate evidence to suggest that cytotoxic agents in children with SRNS should
not be used, based upon two randomized controlled trials that show no evidence of benefit of these agents combined with prednisone, compared to corticosteroids alone. The evidence is of moderate quality due to the small sample size [8,9] (Table 4.3). In the ISKDC trial, there was no significant difference in achieving a complete remission with cyclophosphamide therapy plus corticosteroids compared to corticosteroids alone with 10/18 versus 6/13 achieving complete remission in the combined-therapy group vs. corticosteroids alone group (RR 1.01; 95% CI 0.74-1.36) and an increase in adverse events [28]. Although imprecision may affect this risk estimate, the RR and CI are centered around 1. In the Tarshish trial comparing cyclophosphamide plus corticosteroids vs. corticosteroids alone, there was also no evidence of benefit with the addition of cyclophosphamide, i.e., 16/32 with combination vs. 12/21 monotherapy (P = NS). One additional randomized trial compared cyclophosphamide plus methylprednisolone (N =17) to cyclosporine (N = 15). The study was halted at week 12 according to predefined stopping rules, due to the significant difference between the combined complete and partial remission rates of 60% in cyclosporine group and 17% in the cyclophosphamide group (P <0.05) [29]. At the present time, the potential harm from cytotoxic agents—including serious infections, increased risk for late onset malignancy, reduced fertility, hemorrhagic cystitis, and alopecia—far exceeds any evidence of benefit [30]
Table 4.3. Cytotoxic therapy in SRNS
Author N Intervention Control
Remission complete or
partial RR Remission Conclusion ISKDC 1974 [8]
31 Cyclophosphamide p.o. + prednisone 3 mo
Prednisone 3 mo
10 (56%) vs. 6 (46%)
0.83 (0.40-1.70) ND
Tarshish 1996 [9] 53 Cyclophosphamide po x 3 mo +
prednisone 12 mo q.o.d. Prednisone 12 mo q.o.d.
16 (50%) vs. 12 (57%)
1.17 (0.64- 2.13) ND
ISKDC, International Study of Kidney Disease in Children; ND, not determined; p.o., orally; q.o.d., every other day; SRNS, steroid-resistant nephrotic syndrome.
Rituximab Rituximab is not recommended as a treatment option for SRNS due to the lack of RCTs and
risk for serious adverse events, which may persist long after the discontinuation of the therapy [31]. Although this may be a promising agent, prospective randomized studies are required.

KDIGO® GN Guideline March 2011 Public Review Draft
40
Relapsing Disease In SRNS patients with relapse after complete remission, we suggest that immunosuppressant
therapy be reinstated. This recommendation is based upon the concern that uncontrolled SRNS is likely to lead both to complications from the persistent nephrotic state as well as a high risk for kidney failure. We have no evidence in the literature to support a specific treatment choice. Options are provided without prioritization, and include oral corticosteroids, a return to the previously effective immunosuppressant agent, or the selection of an alternate immunosuppressant agent to avoid potential toxicity. Assessment of risk vs. benefit needs reassessment and becomes more relevant with each relapse.
RESEARCH RECOMMENDATIONS
• RCTs are needed in resistant nephrotic syndrome comparing CNIs to alternate immunosuppressive and nonimmunosuppressive agents.
• Investigation of treatment options is needed for patients with nephrotic syndrome associated with genetic mutations.
• RCTs are needed examining rituximab therapy for SRNS.
Please refer to Supplemental Tables 8-19 for additional data related to Chapter 4.
References 1. Gipson DS, Chin H, Presler TP, Jennette C, Ferris ME, Massengill S, Gibson K, Thomas DB. Differential
Risk of Remission and ESRD in Childhood FSGS. Pediatr Nephrol. 21:344-9, 2006. 2. Trachtman H, Fine R, Friedman A, Gassman J, Gipson D, Greene T, Hogan S, Hogg R, Kaskel F, Moxey-
Mims M, Radeva M, Siegel N, Watkins S. Quality Of Life In Children With Focal Segmental Glomerulosclerosis: Baseline Findings. Report Of The FSGS Clinical Trial. American Society of Nephrology Annual Meeting, Abstract 551104, 2009
3. Kerlin BA, Blatt NB, Fuh B, Zhao S, Lehman A, Blanchong C, Mahan JD, Smoyer WE. Epidemiology and risk factors for thromboembolic complications of childhood nephrotic syndrome: a Midwest Pediatric Nephrology Consortium (MWPNC) study. J Pediatr 155:105-10, 2009.
4. Uncu N, Bülbül M, Yildiz N, Noyan A, Koşan C, Kavukçu S, Calişkan S, Gündüz Z, Beşbaş N, Gür Güven A. Primary peritonitis in children with nephrotic syndrome: results of a 5-year multicenter study. Eur J Pediatr. 169:73-6, 2010. PMID: 19430812
5. Soeiro EM, Koch VH, Fujimura MD, Okay Y. Influence of nephrotic state on the infectious profile in childhood idiopathic nephrotic syndrome. Rev Hosp Clin Fac Med Sao Paulo. 59:273-8, 2004. PMID: 15543399
6. National Institutes of Health (2003) US renal data system, USRDS 2003 annual data report: atlas of end-stage renal disease in the United States. In: National Institutes of Health (ed) National Institutes of Diabetes and Digestive and Kidney Diseases, Bethesda, MD
7. ISKDC. The primary nephrotic syndrome in children. Identification of patients with minimal change nephrotic syndrome from initial response to prednisone. A report of the International Study of Kidney Disease in Children. J Pediatr 98:561-564, 1981.
8. ISKDC. Prospective controlled trial of cyclophosphamide therapy in children with the nephrotic syndrome Lancet 2:423-7, 1974.
9. Tarshish P, Tobin JN, Bernstein J, Edelmann CM Jr. Cyclophosphamide does not benefit patients with focal segmental glomerulosclerosis. A report of the International Study of Kidney Disease in Children. Pediatr Nephrol 10:590-3, 1996. MEDLINE 8897562

KDIGO® GN Guideline March 2011 Public Review Draft
41
10. Troyanov S, Wall CA, Miller JA, Scholey JW, Cattran DC. Focal and Segmental Glomerulosclerosis: Definition and Relevance of a Partial Remission. J Am Soc Nephrol 16: 1061-1068, 2005
11. Cosio FG, Hernandez RA. Favorable prognostic significance of raised serum C3 concentration in patients with idiopathic focal glomerulosclerosis. Clinical Nephrology 45:146-152, 1996
12. Chitalia VC, Wells JE, Robson RA, Searle M, Lynn KL. Predicting renal survival in primary focal glomerulosclerosis from the time of presentation. Kidney International 56:2236-2242, 1999
13. Abrantes MM, Cardoso LSB, Lima EM, Penido Silva JM, Diniz JS, Bambirra EA, Oliveira EA. Predictive factors of chronic kidney disease in primary focal segmental glomerulosclerosis. Pediatr Nephrol 21:1003-1012, 2006
14. Hogg RJ, Furth S, Lemley KV, Portman R, Schwartz GJ, Coresh J, Balk E, Lau J, Levin A, Kausz AT, Eknoyan G, Levey AS; National Kidney Foundation's Kidney Disease Outcomes Quality Initiative. National Kidney Foundation's Kidney Disease Outcomes Quality Initiative clinical practice guidelines for chronic kidney disease in children and adolescents: evaluation, classification, and stratification. Pediatrics. 111:1416-21, 2003.
15. Chernin G, Heeringa SF, Gbadegesin R, Liu J, Hinkes BG, Vlangos CN, Vega-Warner V, Hildebrandt F. Low prevalence of NPHS2 mutations in African American children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 23:1455-60, 2008. PMID: 18543005
16. Karle SM, Uetz B, Ronner V et al. Novel mutations in NPHS2 detected in both familial and sporadic steroid-resistant nephrotic syndrome. J Am Soc Nephrol 13: 388–393, 2002.
17. Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J and Mundel P. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nature Medicine 14: 931 – 938, 2008
18. Garin EH, Orak JK, Hiott KL, Sutherland SE. Cyclosporine therapy for steroid-resistant nephrotic syndrome. A controlled study. Am J Dis Child. 142:985-8, 1988.PMID: 3046332
19. Lieberman KV, Tejani A. A Randomized Double-Blind Placebo-Controlled Trial of Cyclosporine in Steroid-Resistant Idiopathic Focal Segmental Glomerulosclerosis in Children. J Am Soc Nephrol. 7:56-63, 1996
20. Ponticelli C, Rizzoni G, Edefonti A, Altieri P, Rivolta E, Rinaldi S, et al. A randomized trial of cyclosporine in steroid-resistant idiopathic nephrotic syndrome. Kidney International 43:1377-84, 1993. MEDLINE 8315953
21. Caridi G, Perfumo F, Ghiggeri GM. NPHS2 (Podocin) mutations in nephrotic syndrome. Clinical spectrum and fine mechanisms. Pediatr Res 57:54R–61R, 2005
22. Choudhry S, Bagga A, Hari P, Sharma S, Kalavani M, Dinda A. Efficacy and Safety of Tacrolimus Versus Cyclosporine in Children with Steroid-Resistant Nephrotic Syndrome: A Randomized Controlled Trial. Am J Kidney Dis 53:760-769, 2009
23. Winn MP. Not all in the family: mutations of podocin in sporadic steroid-resistant Nephrotic syndrome. J Am Soc Nephrol. 13:577-9, 2002.
24. Bagga A, Mudigoudar BD, Hari P, Vasudev V. Enalapril dosage in steroid-resistant nephrotic syndrome. Pediatr Nephrol. 19:45-50, 2004.
25. Li Z, Duan C, He J, Wu T, Xun M, Zhang Y, Yin Y. Mycophenlate mofetil therapy for children with steroid-resistant nephritic syndrome. Pediatr Nephrol 2009 Dec 2
26. Hari P Bagga A, Mantan M. Short Term Efficacy of Intravenous Dexamethasone and Methylprednisolone Therapy in Steroid Resistant Nephrotic Syndrome. Indian Pediatrics. 41:993-1000, 2004
27. Raposo de Mello V, Rodrigues M, Mastrocinque T, Martins S, Braga de Andrade O, Guidoni E, Scheffer D, Filho D, Toporovski J, Benini V. Mycophenolate mofetil in children with steroid/cyclophosphamide-resistant nephritic syndrome. Pediatr Nephrol 25: 453-460, 2010
28. Hodson EM, Craig JC. Therapies for steroid-resistant nephrotic syndrome.Pediatr Nephrol. 23:1391-4, 2008. PMID: 18368428
29. Plank C, Kalb V, Hinkes B, Hildebrandt F, Gefeller O, Rascher W. Cyclosporin A is superior to cyclophosphamide in children with steroid-resistant nephrotic syndrome – a randomized controlled multicentre trial by the Arbeitsgemeinschaft fur Padiatrische nephrologie. Pediatr Nephrol 23:1483-1493, 2008

KDIGO® GN Guideline March 2011 Public Review Draft
42
30. Latta K, von S, Ehrich JH. A meta-analysis of cytotoxic treatment for frequently relapsing nephrotic syndrome in children. Pediatr Nephrol 16:271-282, 2001.
31. Chaumais MC, Garnier A, Chalard F, Peuchmaur M, Dauger S, Jacqz-Agrain E, Deschenes G. Fatal pulmonary fibrosis after rituximab administration. Pediatr Nephrol 24:1753-1755, 2009. DOI 10.1007/s00467-009-1195-9

KDIGO® GN Guideline March 2011 Public Review Draft
43
CHAPTER 5: MINIMAL-CHANGE DISEASE IN ADULTS
INTRODUCTION
This chapter refers to the treatment of adults with MCD. The cost implications for global application of this guideline are addressed in Chapter 2.
5.1: Treatment of initial episode of adult MCD 5.1.1: We recommend that corticosteroids be given for initial treatment of
nephrotic syndrome. (1C) 5.1.2: We suggest prednisone or prednisolone* be given at a daily single dose of
1 mg/kg (maximum 80 mg) or alternate-day single dose of 2 mg/kg (maximum 120 mg). (2C)
5.1.3: We suggest the initial high dose of corticosteroids, if tolerated, be maintained for a minimum period of 4 weeks if complete remission is achieved, and for a maximum period of 16 weeks if complete remission is not achieved. (2C)
5.1.4: In patients who remit, we suggest that corticosteroids be tapered slowly over a total period of 6 months. (2D)
5.1.5: For patients with relative contraindications or intolerance to high-dose corticosteroids (e.g., uncontrolled diabetes, psychiatric conditions, severe osteoporosis), we suggest oral cyclophosphamide or CNIs as discussed in frequently relapsing MCD. (2D)
5.1.6: We suggest using the same initial dose of corticosteroids for infrequent relapses as in Recommendation 5.1.2 until remission is achieved, followed by at least 2 months of tapering steroids. (2D)
*Prednisone and prednisolone are equivalent, used in the same dosage, and have both been used in RCTs depending on the country of origin. All later references to prednisone in this chapter refer to prednisone or prednisolone. All later references to oral corticosteroids refer to prednisone or prednisolone.
BACKGROUND
MCD refers to the occurrence of nephrotic syndrome with no glomerular lesions by light microscopy (or only minimal mesangial prominence), no staining on immunofluorescence microscopy (or low-intensity staining for C3 and IgM), and foot process effacement but no electron-dense deposits on electron microscopy [1].
Although spontaneous remission can occur in MCD [2-5], untreated nephrotic syndrome is associated with significant morbidity due to accelerated atherosclerosis, in part due to dyslipidemia [13], infections [5,14], and thromboembolic events [15]. Therefore, specific treatment should be given with the goal of achieving remission. The cornerstone of treatment has been corticosteroids. MCD in children is exquisitely sensitive to corticosteroids; however, adults

KDIGO® GN Guideline March 2011 Public Review Draft
44
tend to respond more slowly, with responses occurring as late as 3-4 months after starting therapy. The response to corticosteroids is also less predictable in adults, as only about 75% of adults with MCD are steroid-responsive (Table 5.1). Also, in contrast to children, there is a paucity of well-designed RCTs investigating the treatment of MCD in adults.
Although acute kidney injury (AKI) is common in adults with MCD (up to 20-25%) [6,7], progressive CKD is not part of the natural history of adults with MCD, and its occurrence suggests underlying FSGS.
More than half of adult MCD patients will experience relapses, and up to a third of patients may become frequent relapsers or corticosteroid-dependent [7-10]. Furthermore, a 40% relapse rate has been reported in adults who had MCD as children [11], and these patients continue to relapse. Secondary etiologies associated with MCD are uncommon, but should be considered. They include Hodgkin’s disease, lithium therapy, and nonsteroidal anti-inflammatory drugs [12].
Corticosteroids are generally well-tolerated, but drug-related adverse effects are common with prolonged/repeated courses in SD or FR patients.
Disease Definitions Definitions of proteinuria outcomes are as listed in Table 6.2 in Chapter 6: Treatment of
adult patients with idiopathic focal segmental glomerulosclerosis (FSGS). Partial remissions in proteinuria are not seen in MCD.
RATIONALE
• There is only low-quality evidence to recommend corticosteroids in the treatment of adult MCD. This recommendation is based largely on extrapolation from RCTs in children, as well as small RCTs and observational studies in adults.
• There is only low-quality evidence to define the optimal dose and duration of corticosteroids in adults, but a high dose until remission is achieved followed by a slow taper to minimize relapse is usually prescribed.
• There is very low–quality evidence suggesting that alternate-day is equivalent to daily corticosteroids in adult MCD.
• MCD in adults may take a longer time to remit compared to MCD in children.

KDIGO® GN Guideline March 2011 Public Review Draft
45
Table 5.1. Dosage regimens in MCD
Drug and dosing scheme Initial treatment Prednisone Daily single dose of 1 mg/kg (maximum 80 mg) or alternate-day single dose of 2 mg/kg (maximum 120 mg) —until complete remission (minimum 4 weeks to a maximum of 16 weeks) —after complete remission, tapered slowly over 6 months FR or SD MCD 1. Cyclophosphamide (oral) single course
2-3 mg/kg/d as tolerated for 8 weeks 2. Relapsed despite cyclophosphamide, or patients of childbearing age
a. Cyclosporine starting dose 3-5 mg/kg/d (in two equally divided doses) b. Tacrolimus 0.05-0.1 mg/kg/d (in two equally divided doses)
Following 3 months of stable remission, tapered to reach the minimum dosage that maintains remission, for 1-2 years 3. Intolerant to corticosteroids and/or CNIs
a. Mycophenolate mofetil 750-1000 mg twice daily for 1-2 years FR, frequently relapsing; MCD, minimal-change disease; SD, steroid-dependent.
Corticosteroids have been studied in several large prospective RCTs in children [16,17] and observational studies in children and adults [6,7,9,10]. In a very early multicenter controlled study of corticosteroids compared to no treatment in 125 nephrotic adults (including 31 MCD patients defined by light microscopy alone), those treated with at least 20 mg/d prednisone for at least 6 months showed an early and rapid decrease in proteinuria compared to the control group. However, by two and a half years, there was no difference in proteinuria or serum albumin in the two groups [2]. Similarly, in one small RCT of 28 adult MCD patients that compared prednisone 125 mg every other day for 2 months with placebo, there was no difference in overall remission rates over 77 months follow-up, although a significant percentage of the placebo arm ended up being treated with prednisone over this time frame. However, patients treated with prednisone went into remission more rapidly; 12 of 14 treated patients were in complete remission before 2 months, compared to 6 of 14 controls. [3,18].
Although there are no controlled trials comparing daily vs. alternate-day corticosteroids in adults, observational studies have not shown any difference in response rates [7]. Corticosteroid therapy leads to complete remission in over 80% of adults with MCD. The time course to a complete remission is delayed compared to children, with 50% responding by 4 weeks but the remaining 10-25% requiring 12-16 weeks of therapy [6,7]. It is known that, in children, 6 months of corticosteroid treatment is associated with a lower relapse rate than 3 months of therapy [16]. The optimal method to taper corticosteroids in adults is not known, but corticosteroids are commonly tapered by 5-10 mg/wk or less after achieving remission, for a total period of corticosteroid exposure of at least 24 weeks [5-7].
Only a few patients have been treated at the time of initial presentation with steroid-free regimens (e.g., cyclophosphamide [10,19,20] or cyclosporine [21]). In this very limited experience, the typical response rate of 75% is comparable to corticosteroids.

KDIGO® GN Guideline March 2011 Public Review Draft
46
For infrequent relapses, repeat courses of corticosteroids may be used as in the first episode of MCD. There are no RCTs to guide the therapy of relapse in adult MCD. Reinstitution of prednisone usually results in a remission.
5.2: FR/SD MCD 5.2.1: We suggest oral cyclophosphamide 2-2.5 mg/kg/d for 8 weeks. (2C) 5.2.2: We suggest CNI (cyclosporine 3-5 mg/kg/d or tacrolimus 0.05-0.1 mg/kg/d
in divided doses) for FR/SD MCD patients who have relapsed despite cyclophosphamide, and for people who wish to preserve fertility. (2C)
5.2.3: We suggest MMF 750-1000 mg twice daily for patients who are intolerant of corticosteroids, cyclophosphamide, and CNIs. (2D)
RATIONALE
• There is low-quality evidence to suggest the value of alkylating agents in adult FR/SD MCD. Support for this approach comes from RCTs in children, and observational studies in adults.
• There is low-quality evidence to suggest that CNIs can induce complete or partial remission in adult MCD, but relapse rates may be higher than with alkylating agents after cessation of CNIs.
• There is very low–quality evidence to suggest the use of MMF as a corticosteroid or CNI-sparing agent.
In observational studies, treatment with cyclophosphamide leads to remission in a
significant number of adults [6,7,10]. The relapse-free interval appears to be longer than with cyclosporine (see below). In an observational study, the initial response rates with cyclophosphamide in SD adults appeared excellent (all nine patients were able to be weaned off steroids in one study) [6]; however, five of these patients relapsed. In this study FR MCD patients appeared to fare better than SD MCD, with 80% of patients showing sustained remission at a mean follow-up of 9.1 years. Similarly, SD children may be less responsive to cyclophosphamide than frequent relapsers [22]. In another study, 21 of 36 adults with FR/SD MCD attained remission within 8 weeks and four more patients (total of 25/31 or 69%) within 16 weeks. The addition of prednisone to cyclophosphamide did not appear to provide added benefit. Remissions appeared to be more durable with cyclophosphamide compared to steroids [10]. In another study, 55% of 20 patients treated with cyclophosphamide (for FR or SD MCD) had a complete or partial remission [7]. There is one report of the effectiveness of regimens using i.v. cyclophosphamide in adults [23].
Many observational studies have reported the efficacy of cyclosporine with remission rates of 70-90% [7,24]. In an RCT of 73 adults and children with FR/SD nephrotic syndrome (31 with MCD; 42 with FSGS), treatment was given with either cyclophosphamide (2.5 mg/kg/d) for 8 weeks or cyclosporine (5 mg/kg/d) for 9 months, followed by a 3-month taper to withdrawal. At 9 months, remission rate did not differ significantly: 64% (18/28) of patients on cyclophosphamide and 74% (26/35) of patients on cyclosporine maintained remission. However,

KDIGO® GN Guideline March 2011 Public Review Draft
47
at 2 years, 25% of patients assigned to cyclosporine vs. 63% of patients assigned to cyclophosphamide were still in remission [25]. Another RCT of 52 patients noted that remission was achieved sooner in patients treated with cyclosporine plus 0.8 mg/kg/d prednisone compared to patients receiving only 1 mg/kg/d prednisone, suggesting an additional benefit of lower exposure to corticosteroids [26].
The optimal dose and duration of cyclosporine therapy is unknown. In an RCT of adults and children with FR/SD nephrotic syndrome, cyclosporine was dosed at 5 mg/kg/d for 9 months followed by a taper over 3 months [25]. The possibility of cyclosporine dependency is high when treatment is abruptly stopped after achieving complete remission. However, prolonged treatment in 36 adult patients for a mean of 26 months, followed by slow withdrawal, led to sustained remissions without steroids in 11 of 14 patients and with low doses of corticosteroids in three patients. In 20% of patients, who remained cyclosporine-dependent, doses of <3 mg/kg/d were sufficient to maintain remission. The cumulative rate of remissions appears to reaches a plateau by 6 months [27, 28].
Tacrolimus, administered for 24 weeks was compared to i.v. cyclophosphamide in a small RCT in SD patients with achieved response rates similar to cyclosporine. All patients in this study were able to discontinue corticosteroids [23].
In children with MCD, MMF has been used as a steroid-sparing agent (see recommendation 3.3.5). The experience with MMF in adults has been limited to case reports [29-31].
5.3: Corticosteroid-resistant MCD 5.3.1: Re-evaluate patients who are corticosteroid-resistant for other causes of
nephrotic syndrome. (Not Graded) A repeat kidney biopsy (if performed) commonly shows FSGS pathology (see Chapter 6 for treatment guideline for FSGS).
RATIONALE
• Corticosteroid-resistant MCD suggests FSGS.
An estimated 10% of adult MCD patients are steroid-resistant (failed 16 weeks of daily or alternate-day corticosteroids as outlined previously). Steroid resistance may be due to undetected FSGS (which may not be seen in a biopsy specimen because it is a focal lesion). A repeat biopsy could be considered and may show FSGS, which is associated with a worse prognosis than MCD. There are no RCTs and very few observational data on treatment strategies of steroid-resistant MCD in adults. Treatment strategy as outlined in Chapter 6 is suggested.
5.4: Supportive therapy 5.4.1: We suggest that MCD patients who have AKI be treated with renal
replacement therapy as indicated, but together with corticosteroids, as for a first episode of MCD. (2D)

KDIGO® GN Guideline March 2011 Public Review Draft
48
5.4.2: We suggest that, for the initial episode of nephrotic syndrome associated with MCD, statins not be used to treat hyperlipidemia, and ACEi or ARBs not be used in normotensive patients to lower proteinuria. (2D)
RATIONALE
• AKI may accompany MCD in adults. This is usually reversible with continued steroid therapy. Supportive care, including renal replacement therapy, may be temporarily required. Proteinuria in adult MCD will typically remit with corticosteroids. As a consequence, the accompanying hyperlipidemia will remit with resolution of proteinuria, negating the need for statin therapy.
• Proteinuria in adult MCD will typically remit with corticosteroids, and RAS blockade to help reduce proteinuria is not necessary.
AKI, sometimes severe enough to require dialysis, can occur in patients with MCD. Risk
factors include older age, hypertension, severe nephrotic syndrome, and underlying arteriosclerosis of the kidney [7, 32]. Kidney function typically recovers even in the most severely affected patients, although patients who have experienced kidney failure may have residual chronic renal impairment [7]. Careful attention to volume status, as well as continued therapy with corticosteroids, and other supportive therapy for AKI are suggested.
There is only one small study of 40 adults who had relapsing nephritic syndrome as children. This study did not show a higher incidence of cardiovascular disease, implying that long-term cardiovascular risk was not increased by intermittent hyperlipidemia during nephrotic relapses in childhood [33]. The use of antihyperlipidemic agents and ACEi or ARBs may be considered on a case-by-case basis in FR/SD MCD adults.
Economic Considerations Prednisone and cyclophosphamide are less costly than CNIs and MMF. Cost factors need to
be considered in patients who are not able to afford or access the more expensive medications [34]. The addition of ketoconazole is safe and can lead to significant reduction in costs associated with CNIs, but drug levels need to be assessed to avoid nephrotoxicity [35].
RESEARCH RECOMMENDATIONS
• RCTs should investigate the use of CNIs or MMF as alternatives to corticosteroids for the first episode of adult MCD.
• RCTs are needed to compare CNIs to cyclophosphamide in FR/SD MCD, and to establish if cyclosporine or tacrolimus should be the preferred CNI.
• RCTs are needed to study the role of rituximab in FR/SD MCD. • Evidence should be collected in these RCTs to evaluate the long-term cardiovascular,
metabolic, infectious, and bone risk of FR/SD MCD, and corresponding treatment.

KDIGO® GN Guideline March 2011 Public Review Draft
49
References
1. Nachman PH, Jennette JC, Falk RJ. Primary glomerular diseases. In: Brenner BM, editors. The Kidney. 8 ed. Philadelphia: W.B. Saunders Company; 2008 p. 987-1279.
2. Black DA, Rose G, Brewer DB. Controlled trial of prednisone in adult patients with the nephrotic syndrome. Br Med J 1970; 3(5720):421-426.
3. Coggins CH. Adult minimal change nephropathy: experience of the collaborative study of glomerular disease. Trans Am Clin Climatol Assoc 1986; 97:18-26.
4. Arneil GC, Lam CN. Long-term assessment of steroid therapy in childhood nephrosis. Lancet 1966; 2(7468):819-821.
5. Huang JJ, Hsu SC, Chen FF, Sung JM, Tseng CC, Wang MC. Adult-onset minimal change disease among Taiwanese: clinical features, therapeutic response, and prognosis. Am J Nephrol 2001; 21(1):28-34.
6. Mak SK, Short CD, Mallick NP. Long-term outcome of adult-onset minimal-change nephropathy. Nephrol Dial Transplant 1996; 11(11):2192-2201.
7. Waldman M, Crew RJ, Valeri A et al. Adult minimal-change disease: clinical characteristics, treatment, and outcomes. Clin J Am Soc Nephrol 2007; 2(3):445-453.
8. Korbet SM, Schwartz MM, Lewis EJ. Minimal-change glomerulopathy of adulthood. Am J Nephrol 1988; 8(4):291-297.
9. Tse KC, Lam MF, Yip PS et al. Idiopathic minimal change nephrotic syndrome in older adults: steroid responsiveness and pattern of relapses. Nephrol Dial Transplant 2003; 18(7):1316-1320.
10. Nolasco F, Cameron JS, Heywood EF, Hicks J, Ogg C, Williams DG. Adult-onset minimal change nephrotic syndrome: a long-term follow-up. Kidney Int 1986; 29(6):1215-1223.
11. Fakhouri F, Bocquet N, Taupin P et al. Steroid-sensitive nephrotic syndrome: from childhood to adulthood. Am J Kidney Dis 2003; 41(3):550-557.
12. Appel GB, Radhakrishnan J, D'Agati V. Secondary Glomerular Disease. In: Brenner BM, editor. BRENNER & RECTOR'S THE KIDNEY. 8 ed. Philadelphia: Saunders; 2008 p. 1067-1126.
13. Radhakrishnan J, Appel AS, Valeri A, Appel GB. The nephrotic syndrome, lipids, and risk factors for cardiovascular disease. Amer J Kid Dis 1993; 22:135-142.
14. McIntyre P, Craig JC. Prevention of serious bacterial infection in children with nephrotic syndrome. J Paediatr Child Health 1998; 34(4):314-317.
15. Mahmoodi BK, ten Kate MK, Waanders F et al. High absolute risks and predictors of venous and arterial thromboembolic events in patients with nephrotic syndrome: results from a large retrospective cohort study. Circulation 2008; 117(2):224-230.
16. Hodson EM, Willis NS, Craig JC. Corticosteroid therapy for nephrotic syndrome in children. Cochrane Database Syst Rev 2007;(4):CD001533.
17. Gipson DS, Massengill SF, Yao L et al. Management of childhood onset nephrotic syndrome. Pediatrics 2009; 124(2):747-757.
18. Palmer SC, Nand K, Strippoli GF. Interventions for minimal change disease in adults with nephrotic syndrome. Cochrane Database Syst Rev 2008;(1):CD001537.
19. Uldall PR, Feest TG, Morley AR, Tomlinson BE, Kerr DN. Cyclophosphamide therapy in adults with minimal-change nephrotic syndrome. Lancet 1972; 1(7763):1250-1253.
20. Al-Khader AA, Lien JW, Aber GM. Cyclophosphamide alone in the treatment of adult patients with minimal change glomerulonephritis. Clin Nephrol 1979; 11(1):26-30.
21. Matsumoto H, Nakao T, Okada T et al. Favorable outcome of low-dose cyclosporine after pulse methylprednisolone in Japanese adult minimal-change nephrotic syndrome. Intern Med 2004; 43(8):668-673.
22. Latta K, von SC, Ehrich JH. A meta-analysis of cytotoxic treatment for frequently relapsing nephrotic syndrome in children. Pediatr Nephrol 2001; 16(3):271-282.
23. Li X, Li H, Chen J et al. Tacrolimus as a steroid-sparing agent for adults with steroid-dependent minimal change nephrotic syndrome. Nephrol Dial Transplant 2008; 23(6):1919-1925.

KDIGO® GN Guideline March 2011 Public Review Draft
50
24. Meyrier A, Condamin MC, Broneer D, and The Collaborative Group of the French Society of Nephrology. Treatment of adult idiopathic nephrotic syndrome with cyclosporin A: Minimal-change disease and focal-segmental glomerulosclerosis. Clin Nephrol 1991; 35(suppl 1):37-42.
25. Ponticelli C, Edefonti A, Ghio L et al. Cyclosporin versus cyclophosphamide for patients with steroid-dependant and frequently relapsing idiopathic nephrotic syndrome: a multicenter randomized controlled trial. Nephrol Dial Transplant 1993; 8:1326-1332.
26. Eguchi A, Takei T, Yoshida T, Tsuchiya K, Nitta K. Combined cyclosporine and prednisolone therapy in adult patients with the first relapse of minimal-change nephrotic syndrome. Nephrol Dial Transplant 2010; 25(1):124-129.
27. Meyrier A, Niaudet P, Brodehl J. Optimal Use of Sandimmun in Nephrotic Syndrome. Springer; 1993. 28. Meyrier A, Noel L-H, Auriche P, Callard P. Long-term renal tolerance of cyclosporin A tretment in adult
idiopathic neprhotic syndrome. Collaborative Group of the Societe de Nephrologie. Kidney Int 1994; 45:1446-1456.
29. Siu YP, Tong MK, Leung K, Kwan TH, Au TC. The use of enteric-coated mycophenolate sodium in the treatment of relapsing and steroid-dependent minimal change disease. J Nephrol 2008; 21(1):127-131.
30. Choi MJ, Eustace JA, Gimenez LF et al. Mycophenolate mofetil treatment for primary glomerular diseases. Kidney Int 2002; 61(3):1098-1114.
31. Fujinaga S, Ohtomo Y, Hirano D et al. Mycophenolate mofetil therapy for childhood-onset steroid dependent nephrotic syndrome after long-term cyclosporine: extended experience in a single center. Clin Nephrol 2009; 72(4):268-273.
32. Jennette JC, Falk RJ. Adult minimal change glomerulopathy with acute renal failure. Am J Kidney Dis 1990; 16(5):432-437.
33. Lechner BL, Bockenhauer D, Iragorri S, Kennedy TL, Siegel NJ. The risk of cardiovascular disease in adults who have had childhood nephrotic syndrome. Pediatr Nephrol 2004; 19(7):744-748.
34. Colquitt JL, Kirby J, Green C, Cooper K, Trompeter RS. The clinical effectiveness and cost-effectiveness of treatments for children with idiopathic steroid-resistant nephrotic syndrome: a systematic review. Health Technol Assess 2007; 11(21):iii-xi, 1.
35. El-Husseini A, El-Basuony F, Mahmoud I et al. Impact of the cyclosporine-ketoconazole interaction in children with steroid-dependent idiopathic nephrotic syndrome. Eur J Clin Pharmacol 2006; 62(1):3-8.

KDIGO® GN Guideline March 2011 Public Review Draft
51
CHAPTER 6: TREATMENT OF ADULT PATIENTS WITH IDIOPATHIC FOCAL SEGMENTAL GLOMERULOSCLEROSIS
INTRODUCTION
This chapter applies to the adult with biopsy-proven, idiopathic FSGS. The cost implications for global application of this guideline are addressed in Chapter 2.
6.1: Initial evaluation of FSGS 6.1.1: Undertake thorough evaluation to exclude secondary forms of FSGS. (Not
Graded) 6.1.2: Do not routinely perform genetic testing. (Not Graded)
BACKGROUND
The classical description of FSGS includes segmental increase of mesangial matrix with obliteration of the capillaries, sclerosis, hyalinosis, foam cells, and segmental scarring, and adhesion between the glomerular tuft and Bowman’s capsule. A recently proposed pathology classification has pointed to the existence of nonsclerotic forms of FSGS [1]. There has been a marked increase in the number of known underlying causes for the lesion of FSGS over the last 10-20 years. Perhaps a consequence of this has been that the incidence, the age of onset, and the clinical presentation have also dramatically altered over this timeframe. FSGS is now one of the most common patterns of glomerular injury encountered in human kidney biopsies [2,3], and it is the most common cause of proteinuria in the African-American and US Hispanic populations.
RATIONALE
• FSGS should be classified as idiopathic (primary) FSGS or secondary FSGS. This is not merely semantic, but has therapeutic implications. Idiopathic FSGS is defined by exclusion of any other identifiable cause of secondary FSGS [4]. Secondary causes of FSGS are listed in Table 6.1, and should be evaluated by detailed examination of the patient, including medical history, physical examination, family history, kidney imaging, and kidney pathology, including electron micoscopy studies [5].
• There are no good data to support genetic testing in adults with FSGS, even in cases of steroid resistance. In the absence of a family history of FSGS, mutations of NPHS1 (nephrin), NPHS2 (podocin), alpha-actinin-4, CD2AP, and TRPC-6 are detected in only 0-3% of adults with FSGS [6-13]. In addition, since some patients with a genetic abnormality have responded to therapy, the results of genetic analysis should not change treatment decisions.

KDIGO® GN Guideline March 2011 Public Review Draft
52
Table 6.1. Causes of FSGS
Idiopathic (primary) FSGS Secondary FSGS 1. Familial
a. Mutations in α-actinin 4 b. Mutations in NPHS1 (nephrin) c. Mutations in NPHS2 (podocin) d. Nutations in WT-1 e. Mutations in TRPC6 f. Mutations in SCARB2 (LIMP2) g. Mutations in INF2 (formin) h. Mutations in CD2-associated protein i. Mitochondrial cytopathies
2. Virus associated
a. HIV-associated nephropathy b. Parvovirus B19
3. Medication
a. Heroin-nephropathy b. Interferon-α c. Lithium d. Pamidronate/alendronate e. Anabolic steroids
4. Adaptive structural-functional responses likely mediated by glomerular hypertrophy or hyperfiltration
4.1 Reduced kidney mass a. Oligomeganephronia b. Unilateral kidney agenesis c. Kidney dysplasia d. Cortical necrosis e. Reflux nephropathy f. Surgical kidney ablation g. Chronic allograft nephropathy h. Any advanced kidney disease with reduction in functioning nephrons
4.2 Initially normal kidney mass a. Diabetes mellitus b. Hypertension c. Obesity d. Cyanotic congenital heart disease e. Sickle cell anemia
5. Malignancy (lymphoma) Nonspecific pattern of FSGS caused by kidney scarring in glomerular disease
a. Focal proliferative glomerulonephritis (IgAN, LN, pauci-immune focal necrotizing and crescentic GN) b. Hereditary nephritis (Alport syndrome) c. Membranous glomerulopathy d. Thrombotic microangiopathy
FSGS, focal segmental glomerulosclerosis; GN, glomerulonephritis; HIV, Human immunodeficiency virus ; IgAN, immunoglobulin A nephropathy; LN, lupus nephritis.
Adapted with permission. [4]

KDIGO® GN Guideline March 2011 Public Review Draft
53
6.2: Initial treatment of FSGS 6.2.1: We recommend that corticosteroid and immunosuppressive therapy be
considered only in idiopathic FSGS associated with clinical features of the nephrotic syndrome. (1C)
6.2.2: We suggest prednisone* be given at a daily single dose of 1 mg/kg (maximum 80 mg) or alternate-day dose of 2 mg/kg (maximum 120 mg). (2C)
6.2.3: We suggest the initial high dose of corticosteroids be given for a minimum of 4 weeks; continue high-dose corticosteroids up to a maximum of 16 weeks, as tolerated, or until complete remission has been achieved, whichever is earlier. (2D)
6.2.4: We suggest corticosteroids be tapered slowly over a period of 6 months after achieving complete remission. (2D)
6.2.5: We suggest CNIs be considered as first-line therapy for patients with relative contraindications or intolerance to high-dose corticosteroids (e.g., uncontrolled diabetes, psychiatric conditions, severe osteoporosis). (2D)
*Prednisone and prednisolone are equivalent, used in the same dosage, and have both been used in RCTs depending on the country of origin. All later references to prednisone in this chapter refer to prednisone or prednisolone. All later references to oral corticosteroids refer to prednisone or prednisolone.
BACKGROUND
Patients with FSGS and persistent proteinuria are at increased risk of progressive CKD and its accompanying cardiovascular morbidity and mortality. Risks are dependent on the level of proteinuria and kidney function.
The potential benefit of therapy includes disease cure, control, and/or slowing the progression to ESRD. In FSGS, outcome parameters can be divided into kidney and proteinuric events. Disease cure and control are defined primarily by changes in proteinuria (see Table 6.2).
In most cases of idiopathic FSGS, the natural history of the disease is prolonged, with even complete remitters having a relapse rate of up to 40%. Those with partial remissions still have a risk of slowly progressive loss of kidney function. There is also a significant minority with no response to therapy; hence, the potential benefits of treatment must be constantly weighed against the risks of the chosen immunosuppressive therapy [14].

KDIGO® GN Guideline March 2011 Public Review Draft
54
Table 6.2. Definitions of proteinuria used in adult nephrotic syndrome (FSGS)
Complete remission Reduction of proteinuria to <0.2 g/d or <20 mg/mmol (<0.2 g/g) creatinine and serum albumin >35 g/l
Partial remission* Reduction of proteinuria to 0.2 -2.0 g/d or 20-200 mg/mmol (0.2-2.0 g/g) creatinine and stable GFR (change in creatinine <25%) or Reduction of proteinuria to 0.2-3.5 g/d and a decrease >50% from baseline, and stable GFR
Relapse Proteinuria >3.5 g/d or >350 mg/mmol creatinine (>3.5 g/g) after complete remission has been obtained
Frequent relapses Not defined in adults Steroid-dependent Two relapses during or within 2 weeks of completing steroid therapy Steroid-resistant Persistence of proteinuria despite prednisone 1 mg/kg/d or 2 mg/kg every other day for
>4 months FSGS, focal segmental glomerulosclerosis; GFR, glomerular filtration rate. *Both definitions of partial remission have been used in the literature.
Prognosis in patients with idiopathic FSGS is predicted by the severity and persistence of proteinuria. Patients with non-nephrotic proteinuria have a good prognosis, with kidney survival rates of more than 95% after a mean follow-up of 6.5 to 9.3 years [15-17], even in older studies when few patients, if any, were treated with RAS blockade. The conclusion still seems to be valid, since a very recent study concluded that even partial remission (reduction to non-nephrotic range proteinuria) was associated with significant improvement in kidney survival (80% vs. 40%) compared to no remission [18].
Many observational studies have demonstrated that remission of proteinuria, whether spontaneous or induced by therapy, is associated with a good outcome [18-22]. Many studies have shown, in univariate and multivariate analyses, that development of a remission was associated with prednisone treatment [18, 23-25].
The natural history of primary FSGS with nephrotic syndrome is quite variable. Important predictors are the magnitude of proteinuria, the level of kidney function, and the amount of tubulo-interstitial injury [15, 26, 27]. Resistance to corticosteroids and immunosuppressive therapy is now considered the strongest predictor of ESRD [16,28]. Prognosis is poor in patients who do not achieve remission, with 5-year kidney survival averaging 65% (60-90%) and 10-year kidney survival 30% (25-56%) [15-17,29].
RATIONALE
• Most patients that progress have persistent nephrotic-range proteinuria; patients with non-nephrotic proteinuria are at low risk for progressive kidney failure and ESRD.
• Those with sustained non-nephrotic proteinuria are at increased risk of cardiovascular morbidity and mortality.Tthose risks should be managed, including treatment of proteinuria with RAS blockade and tight control of blood pressure.
• There is low-quality evidence to recommend corticosteroid or immunosuppressive therapy in primary FSGS when accompanied by nephrotic syndrome.
• There is no evidence to suggest corticosteroid or immunosuppressive therapy in secondary FSGS.

KDIGO® GN Guideline March 2011 Public Review Draft
55
RAS Blockade and Blood Pressure Control Optimal conservative management of patients with FSGS should follow guidelines for
patients with persistent proteinuria (see Chapter 1). RAS blockade should be routine; however, it may be delayed in nephrotic syndrome to see if there is a response to initial corticosteroid therapy. This is particularly relevant if the nephrotic syndrome is severe, since the risk of developingAKI due to hypoperfusion and acute tubular necrosis (ATN) is increased in this setting [30,31].
Corticosteroids Corticosteroid therapy should only be considered for patients with idiopathic FSGS
associated with nephrotic syndrome. There are no data to support treatment with corticosteroids in patients without nephrotic-range proteinuria and, although there are no RCTs, there are numerous observational studies to support the use of corticosteroids in FSGS when associated with nephrotic-range proteinuria.
Prior to 1985, idiopathic FSGS was considered a steroid-resistant disease with poor outcome [15]. In contrast, observational studies conducted after 1985 have reported better outcomes and suggested that this improvement in response was associated with a higher initial dose and longer duration of treatment with corticosteroids.
Treatment routines have varied with durations from 4 to 24 months, and prednisone dosing from 0.3 to 1.5 mg/kg/d, reported complete remission rates range from 28% to 74%, and partial remission rates from 0% to 50%. The average time to complete remission is 3-4 months, with a range up to 8 months [16,19-21].
The timing of prednisone therapy initiation has been debated. Spontaneous remissions do occur, with reported rates varying from 5% to 23%. Spontaneous remissions are more likely to occur in patients with tip lesions, with preserved kidney function, and lower grades of proteinuria [32A]. In such patients, prednisone treatment could be delayed to see if spontaneous remission occurs with RAS blockade and other conservative approaches, but no studies have investigated this approach, or systematically analyzed its risks and benefits.
In the absence of any evidence specific for FSGS, we suggest that the guidelines for adult MCD are used to direct further therapy in steroid-responsive primary FSGS (see Chapter 5).
There is no evidence to support the use of corticosteroids in secondary FSGS and, in current practice, such patients are not treated with immunosuppressive therapy [32].
Other Immunosuppressive Agents Adult patients may tolerate poorly the sustained corticosteroid regimen recommended for
primary FSGS, but there are no RCTs to support the use of alternative immunosuppressive agents as first-line therapy.
A retrospective observational study compared high-dose oral prednisone (1 mg/kg/d) for at least 4 months and tapering thereafter, with low-dose prednisone (0.5 mg/kg/d) in combination with cyclosporine (3 mg/kg/d initial dose, tapering to 50 mg/d) or azathioprine (2 mg/kg/d initial

KDIGO® GN Guideline March 2011 Public Review Draft
56
dose, tapering to 0.5 mg/kg/d). Average duration of treatment was 20 months. Low-dose prednisone was given to 16 patients with obesity, bone disease, or mild diabetes. Remission rates were comparable; 63% for prednisone (n = 9), 80% for prednisone plus azathioprine (n = 6), and 86% for prednisone plus cyclosporine (n = 10) [25]. Another study used tacrolimus as initial therapy in six patients and noted a remission in all [33].
A randomized study in adult patients with FSGS and persistent nephrotic syndrome after 6 months of RAS blockade compared MMF (2 g/d for 6 months) plus low-dose prednisone (0.5 mg/kg/d for 8-12 weeks) to high-dose prednisone (1 mg/kg/d for 12-24 weeks, followed by tapering over 8 weeks). Similar remission rates were observed in the two regimens, 71% (12/17 patients) vs. 69% (11/16 patients) [34]. These limited data suggest that patients who do not tolerate prolonged high-dose prednisone might benefit from alternative immunosuppressive agents, alone or in combination with a lower dose of prednisone. A CNI is favored in view of the evidence derived from studies in patients with steroid-resistant FSGS (see below).
6.3: Treatment for relapse 6.3.1: We suggest that a relapse of nephrotic syndrome is treated as per the
recommendations for relapsing MCD in adults (see Chapter 5.2). (2D)
RATIONALE
• There is very low–quality evidence to guide treatment of relapses in steroid-responsive FSGS. We suggest that the guidelines for relapsing MCD are followed (see Chapter 5.2).
6.4: Treatment for steroid-resistant FSGS 6.4.1: We suggest that in steroid-resistant FSGS, cyclosporine at 3-5 mg/kg/d in
divided doses be given for at least 4-6 months. (2B) 6.4.2: If there is a partial or complete remission, we suggest continuing
cyclosporine treatment for at least 12 months, followed by a slow taper. (2D)
BACKGROUND
There is no agreement in the literature regarding the duration of prednisone therapy that defines steroid-resistance. Some authors advise the use of alternative immunosuppressive therapy after only 4-8 weeks of prednisone, whereas others define resista nce as persistent nephrotic syndrome after 4 months prednisone in a dose of 1 mg/kg/d [22, 35-37]. We suggest that prednisone be given for 4 months before defining resistance to therapy.

KDIGO® GN Guideline March 2011 Public Review Draft
57
RATIONALE
• Cyclosporine is effective in inducing remission of proteinuria in patients with steroid-resistant FSGS. Remissions can develop slowly, and may take 3-6 months after start of therapy.
• A partial remission provides a substantial outcome benefit. • Relapses are very frequent after withdrawal of cyclosporine. More prolonged
treatment may lead to more persistent remissions. Relapses occur frequently when using cyclosporine for a 6-month period. Longer duration of therapy and slow tapering is advised in cyclosporine-responsive patients with MCD. A similar strategy can be used in FSGS (Table 6.3).
• There is no evidence to support the efficacy of other regimens in patient with steroid-resistant proteinuria.
CNIs Two RCTs have shown that cyclosporine is more effective than no treatment in inducing
remission of proteinuria in FSGS with SRNS [38-40]. In one of the two studies, cyclosporine was combined with low-dose prednisone. These are summarized in Supplemental Table 14. Remission in the two studies occurred in 60% and 69%, but relapse after cyclosporine withdrawal occurred in 69% and 61%, respectively. An additional benefit to cyclosporine treatment was an attenuated deterioration of kidney function in one study, with doubling of SCr in 25% of treated vs. 52% of control patients. An additional, but low-quality, controlled trial (Suppl Table 14) as well as various uncontrolled studies have confirmed that treatment with cyclosporine reduces proteinuria in patients with FSGS [41-45]. These observational studies reported remission rates of 10-75%. The variation in reported remission rates may depend on the definition of steroid resistance, the prior use of alkylating agents, and the concomitant use of low-dose prednisone. Remissions usually develop within 2-3 months, but may take longer (4-6 months). All studies report high relapse rates (60-80%). Patients who respond within 6 months to cyclosporine can sometimes be maintained for periods of years without untoward effects on kidney function; however, deterioration of kidney function may occur, even if proteinuria has remitted [42]. Deterioration of kidney function is more likely in patients who use high-dose cyclosporine (>5.5 mg/kg/d), in patients with pre-existing reduced GFR (<60 ml/min per 1.73 m2) and pre-existent tubulo-interstitial fibrosis [36].
There are no RCTs using tacrolimus. Uncontrolled studies suggest that tacrolimus may be an alternative to cyclosporine [33, 46]. Segarra et al. [46] treated 25 patients with cyclosporine-resistant or cyclosporine-dependent FSGS. Tacrolimus was used in a dose of 0.15 mg/kg/d and targeted to trough levels of 5-10 µg/l; there was a 100% remission rate in the cyclosporine-dependent patients, 100% in patients who had developed resistance to cyclosporine, and 62% in patients with resistance to the initial treatment with cyclosporine. These limited observational studies suggest tacrolimus may be an alternative in patients intolerant of cyclosporine.
Other Immunosuppressive Agents Case reports and small observational studies have reported response to alkylating agents,
MMF [47], sirolimus, and rituximab, but there is insufficient evidence to support the use of any of these agents in patients with steroid-resistant FSGS.

KDIGO® GN Guideline March 2011 Public Review Draft
58
Table 6.3. Treatment schedules
Drug and dosing scheme Initial treatment Prednisone* 1 mg/kg/d in patients (up to a maximum of 80 mg/d) or alternate-day prednisone 2 mg/kg (up to 120 mg) for at least 4 weeks and for a maximum of 4 months; in case of a complete remission, taper prednisone: e.g., reduce dose by 10 mg per 2 weeks down to 0.15 mg/kg/d, then taper dose every 2-4 weeks by 2.5 mg. In SR patients, taper off prednisone over 6 weeks. Therapy for SR FSGS Cyclosporine 3-5 mg/kg/d: in two divided doses (initial target levels 125-175 ng/ml); in case of a remission continue treatment for 1 year then try to slowly taper cyclosporine: reduce cyclosporine dose by 25% every 2 months. If no remission by 6 months, discontinue cyclosporine treatment. Or Tacrolimus 0.1-0.2 mg/kg/d in two divided doses (initial target levels 5-10 ng/ml); in case of remission see advice for cyclosporine. And Prednisone 0.15 mg/kg/d for 4-6 months, then taper off over 4-8 weeks. FSGS, Focal segmental glomerulosclerosis; SR, steroid-resistant.
RESEARCH RECOMMENDATIONS
• An RCT is needed of corticosteroid therapy at presentation compared to delayed corticosteroid therapy.
• An RCT is needed to evaluate the comparative efficacy of CNIs, alkylating agents, and MMF in steroid-resistant FSGS.
• Validation studiesare needed of the most recent classification of FSGS [1] to test its reproducibility, impact on outcome, and capacity to predict response to corticosteroids and immunosuppressive agents.
References 1. D'Agati V. Pathologic classification of focal segmental glomerulosclerosis. Semin Nephrol. 2003
Mar;23(2):117-34. Review. PubMed PMID: 12704572. 2. Haas M, Meehan SM, Karrison TG, Spargo BH. Changing etiologies of unexplained adult nephrotic
syndrome: a comparison of renal biopsy findings from 1976-1979 and 1995-1997. Am J Kidney Dis 1997; 30: 621 -631
3. Braden GL, Mulhern JG, O’Shea MH, et al. Changing incidence of glomerular disease in adults. Am J Kidney Dis 2000; 35: 878-883

KDIGO® GN Guideline March 2011 Public Review Draft
59
4. Deegens JK, Steenbergen EJ, Wetzels JF. Review on diagnosis and treatment of focal segmental glomerulosclerosis. Neth J Med. 2008 Jan;66(1):3-12. Review.
5. Deegens JK, Dijkman HB, Borm GF, Steenbergen EJ, van den Berg JG, Weening JJ,Wetzels JF.Podocyte foot process effacement as a diagnostic tool in focal segmentalGlomerulosclerosis. Kidney Int. 2008 Dec;74(12):1568-76. Epub 2008 Aug 27.
6. He N, Zahirieh A, Mei Y, et al. Recessive NPHS2 (Podocin) mutations are rare in adult-onset idiopathic focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2007 Jan;2(1):31-7. Epub 2006 Oct 25. PubMed PMID: 17699384.
7. Caridi G, Bertelli R, Scolari F, Sanna-Cherchi S, Di Duca M, Ghiggeri GM. Podocin mutations in sporadic focal-segmental glomerulosclerosis occurring in adulthood. Kidney Int. 2003 Jul;64(1):365. PubMed PMID: 12787432.
8. Aucella F, De Bonis P, Gatta G, et al. Molecular analysis of NPHS2 and ACTN4 genes in a series of 33 Italian patients affected by adult-onset nonfamilial focal segmental glomerulosclerosis. Nephron Clin Pract.2005;99(2):c31-6. 2004 Dec 21. PubMed PMID: 15627790.
9. Santín S, Ars E, Rossetti S, et al. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2009 Oct;24(10):3089-96. Epub 2009 May 20. PubMed PMID: 19458060.
10. Santín S, García-Maset R, Ruíz P, et al. Nephrin mutations cause childhood- and adult-onset focal segmental glomerulosclerosis. Kidney Int. 2009 Dec;76(12):1268-76. Epub 2009 Oct 7. PubMed PMID: 19812541.
11. Gigante M, Pontrelli P, Montemurno E. CD2AP mutations are associated with sporadic nephrotic syndrome and focal segmental glomerulosclerosis (FSGS). Nephrol Dial Transplant. 2009 Jun;24(6):1858-64. Epub 2009 Jan 7. PubMed PMID: 19131354.
12. Chernin G, Heeringa SF, Gbadegesin R, et al. Low prevalence of NPHS2 mutations in African American children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2008 Sep;23(9):1455-60. Epub 2008 Jun 10. PubMed PMID: 18543005.
13. Machuca E, Hummel A, Nevo F, et al. Clinical and epidemiological assessment of steroid-resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney Int. 2009 Apr;75(7):727-35. Epub 2009 Jan 14. PubMed PMID: 19145239.
14. Philibert D, Cattran D. Remission of proteinuria in primary glomerulonephritis: we know the goal but do we know the price? Nat Clin Pract Nephrol 2008; 4: 550-559
15. Cameron JS, Turner DR, Ogg CS, Chantler C, Williams DG. The long-term prognosis of patients with focal segmental glomerulosclerosis. Clin Nephrol. 1978 Dec;10(6):213-8. PubMed PMID: 365407.
16. Korbet SM, Schwartz MM, Lewis EJ. Primary focal segmental glomerulosclerosis: clinical course and response to therapy. Am J Kidney Dis. 1994 Jun;23(6):773-83. Review. PubMed PMID: 8203357.
17. Velosa JA, Donadio JV Jr, Holley KE. Focal sclerosing glomerulonephropathy: a clinicopathologic study. Mayo Clin Proc. 1975 Mar;50(3):121-33. PubMed PMID: 1090790.
18. Troyanov S, Wall CA, Miller JA, Scholey JW, Cattran DC; Toronto Glomerulonephritis Registry Group. Focal and segmental glomerulosclerosis: definition and relevance of a partial remission. J Am Soc Nephrol. 2005 Apr;16(4):1061-8. Epub 2005 Feb 16. PubMed PMID: 15716334.
19. Banfi G, Moriggi M, Sabadini E, et al. The impact of prolonged immunosuppression on the outcome of idiopathic focal-segmental glomerulosclerosis with nephrotic syndrome in adults. A collaborative retrospective study. Clin Nephrol. 1991 Aug;36(2):53-9. PubMed PMID: 1934660.
20. Cattran DC, Rao P. Long-term outcome in children and adults with classic focal segmental glomerulosclerosis. Am J Kidney Dis. 1998 Jul;32(1):72-9. PubMed PMID: 9669427.
21. Rydel JJ, Korbet SM, Borok RZ, Schwartz MM. Focal segmental glomerular sclerosis in adults: presentation, course, and response to treatment. Am J Kidney Dis. 1995 Apr;25(4):534-42. PubMed PMID: 7702047.
22. Korbet SM. Treatment of primary focal segmental glomerulosclerosis. Kidney Int. 2002 Dec;62(6):2301-10. PubMed PMID: 12427162.
23. Pei Y, Cattran D, Delmore T, Katz A, Lang A, Rance P. Evidence suggesting under-treatment in adults with idiopathic focal segmental glomerulosclerosis. Regional Glomerulonephritis Registry Study. Am J Med. 1987 May;82(5):938-44. PubMed PMID: 3578362.

KDIGO® GN Guideline March 2011 Public Review Draft
60
24. Stirling CM, Mathieson P, Boulton-Jones JM, et al. Treatment and outcome of adult patients with primary focal segmental glomerulosclerosis in five UK renal units. QJM. 2005 Jun;98(6):443-9. Epub 2005 May 6. PubMed PMID: 15879445.
25. Goumenos DS, Tsagalis G, El Nahas AM, et al. Immunosuppressive treatment of idiopathic focal segmental glomerulosclerosis: a five-year follow-up study. Nephron Clin Pract. 2006;104(2):c75-82. Epub 2006 Jun 19. PubMed PMID: 16785738.
26. Wehrmann M, Bohle A, Held H, Schumm G, Kendziorra H, Pressler H. Long-term prognosis of focal sclerosing glomerulonephritis. An analysis of 250 cases with particular regard to tubulointerstitial changes. Clin Nephrol. 1990 Mar;33(3):115-22. PubMed PMID: 2323110.
27. Chitalia VC, Wells JE, Robson RA, Searle M, Lynn KL. Predicting renal survival in primary focal glomerulosclerosis from the time of presentation. Kidney Int. 1999 Dec;56(6):2236-42. PubMed PMID: 10594800.
28. Caridi G, Gigante M, Ravani P, et al. Clinical features and long-term outcome of nephrotic syndrome associated with heterozygous NPHS1 and NPHS2 mutations. Clin J Am Soc Nephrol. 2009 Jun;4(6):1065-72. Epub 2009 Apr 30. PubMed PMID: 19406966; PubMed Central PMCID: PMC2689885.
29. Beaufils H, Alphonse JC, Guedon J, Legrain M. Focal glomerulosclerosis: natural history and treatment. A report of 70 cases. Nephron. 1978;21(2):75-85. PubMed PMID: 673092.
30. Praga M, MartinezMA, Andres A, et al. Acute renal failure and tubular dysfunction associated with minimal change nephrotic syndrome. Nephrol Dial Transplant. 1989;4(10):914-6. PubMed PMID: 2515498.
31. Jennette JC, Falk RJ. Adult minimal change glomerulopathy with acute renal failure. Am J Kidney Dis. 1990 Nov;16(5):432-7. PubMed PMID: 2239933.
32. Gupta K, Iskandar SS, Daeihagh P et al. Ditribution of pathologic findings in individuals with nephrotic proteinuria according to serum albumin Nephrol Dial Transplant 2008; 23: 1595-1599
32A. Idiopathic focal segmental glomerulosclerosis: a favourable prognosis in untreated patients? Deegens JK, Assmann KJ, Steenbergen EJ, Hilbrands LB, Gerlag PG, Jansen JL, Wetzels JF. Neth J Med. 2005 Nov;63(10):393-8
33. Duncan N, Dhaygude A, Owen J, et al. Treatment of focal and segmental glomerulosclerosis in adults with tacrolimus monotherapy. Nephrol Dial Transplant. 2004 Dec;19(12):3062-7. Epub 2004 Oct 26. PubMed PMID: 15507477.
34. Nayagam LS, Ganguli A, Rathi M., et al. Mycophenolate mofetil or standard therapy for membranous nephropathy and focal segmental glomerulosclerosis: a pilot study. Nephrol. Dial. Transplant. (2008) 23(6): 1926-1930
35. Shiiki H, Nishino T, Uyama H, et al. Clinical and morphological predictors of renal outcome in adult patients with focal and segmental glomerulosclerosis (FSGS). Clin Nephrol. 1996 Dec;46(6):362-8. PubMed PMID: 8982551.
36. Meyrier A, Noël LH, Auriche P, Callard P. Long-term renal tolerance of cyclosporin A treatment in adult idiopathic nephrotic syndrome. Collaborative Group of the Société de Néphrologie. Kidney Int. 1994 May;45(5):1446-56. PubMed PMID: 8072258.
37. Cattran DC, Alexopoulos E, Heering P, et al. Cyclosporin in idiopathic glomerular disease associated with the nephrotic syndrome: workshop recommendations. Kidney Int. 2007 Dec;72(12):1429-47. Epub 2007 Sep 26. PubMed PMID: 17898700.
38. Ponticelli C, Rizzoni G, Edefonti A, et al. A randomized trial of cyclosporine in steroid-resistant idiopathic nephrotic syndrome. Kidney Int. 1993 Jun;43(6):1377-84. PubMed PMID: 8315953.
39. Cattran DC, Appel GB, Hebert LA, et al. A randomized trial of cyclosporine in patients with steroid-resistant focal segmental glomerulosclerosis. North America Nephrotic Syndrome Study Group.Kidney Int. 1999 Dec;56(6):2220-6. PubMed PMID: 10594798.
40. Braun N, Schmutzler F, Lange C, Perna A, Remuzzi G, Willis NS. Immunosuppressive treatment for focal segmental glomerulosclerosis in adults (review). Cochrane Database of systematic reviews nr 4, 2008
41. Heering P, Braun N, Müllejans R, et al. Cyclosporine A and chlorambucil in the treatment of idiopathic focal segmental glomerulosclerosis. Am J Kidney Dis. 2004 Jan;43(1):10-8. PubMed PMID: 14712422.

KDIGO® GN Guideline March 2011 Public Review Draft
61
42. Ittel TH, Clasen W, Fuhs M, Ket al. Long-term ciclosporine A treatment in adults with minimal change nephrotic syndrome or focal segmental glomerulosclerosis. Clin Nephrol. 1995 Sep;44(3):156-62. PubMed PMID: 8556831.
43. Ghiggeri GM, Catarsi P, Scolari F, et al. Cyclosporine in patients with steroid-resistant nephrotic syndrome: an open-label, nonrandomized, retrospective study. Clin Ther. 2004 Sep;26(9):1411-8. PubMed PMID: 15531003.
44. Meyrier A, Condamin MC, Broneer D. Treatment of adult idiopathic nephritic syndrome with cyclosporin A: minimal-change disease and focal-segmental glomerulosclerosis. Collaborative Group of the French Society of Nephrology. Clin Nephrol. 1991;35 Suppl 1:S37-42. PubMed PMID: 1860266.
45. Melocoton TL, Kamil ES, Cohen AH, Fine RN. Long-term cyclosporine A treatment of steroid-resistant and steroid-dependent nephrotic syndrome. Am J Kidney Dis. 1991 Nov;18(5):583-8. PubMed PMID: 1951339.
46. Segarra A, Vila J, Pou L, et al. Combined therapy of tacrolimus and corticosteroids in cyclosporin-resistant or -dependent idiopathic focal glomerulosclerosis: a preliminary uncontrolled study with prospective follow-up. Nephrol Dial Transplant. 2002 Apr;17(4):655-62. PubMed PMID: 11917061.
47. Senthil Nayagam L, Ganguli A, et al. Mycophenolate mofetil or standard therapy for membranous nephropathy and focal segmental glomerulosclerosis: a pilot study. Nephrol Dial Transplant. 2008 Jun;23(6):1926-30. Epub 2007 Nov 7. PubMed PMID: 17989103.

KDIGO® GN Guideline March 2011 Public Review Draft
62
CHAPTER 7: IDIOPATHIC MEMBRANOUS NEPHROPATHY
INTRODUCTION
This chapter gives treatment recommendations for patients with biopsy-proven idiopathic membranous nephropathy (IMN). The cost implications for global application of this guideline are addressed in Chapter 2.
7.1: Evaluation of IMN 7.1.1: Perform appropriate investigations to exclude secondary causes in all
cases of idiopathic membranous nephropathy. (Not Graded)
BACKGROUN D
The diagnosis of MN is made on kidney biopsy. Diagnostic features include capillary wall thickening, normal cellularity, IgG and C3 along capillary walls on immunofluorescence, and subepithelial deposits on electron microscopy. MN is often seen in association with an underlying disorder (secondary MN) [1-3]. Secondary MN is more common in children (75%) than adults (25%) (Table 7.1). The diagnosis of IMN is made by exclusion of secondary causes, using history, physical exam, and apppropriate laboratory tests (e.g., serology, imaging) and by careful examination of the kidney biopsy by light, immunofluorescence, and electron microscopy. Distinguishing secondary MN from IMN is very important, since the therapy in the former must be directed at the underlying cause and some of the recommended treatments for IMN are potentially toxic both to the patient and the kidney.A correct diagnosis is essential to avoid unnecessary drug exposure.
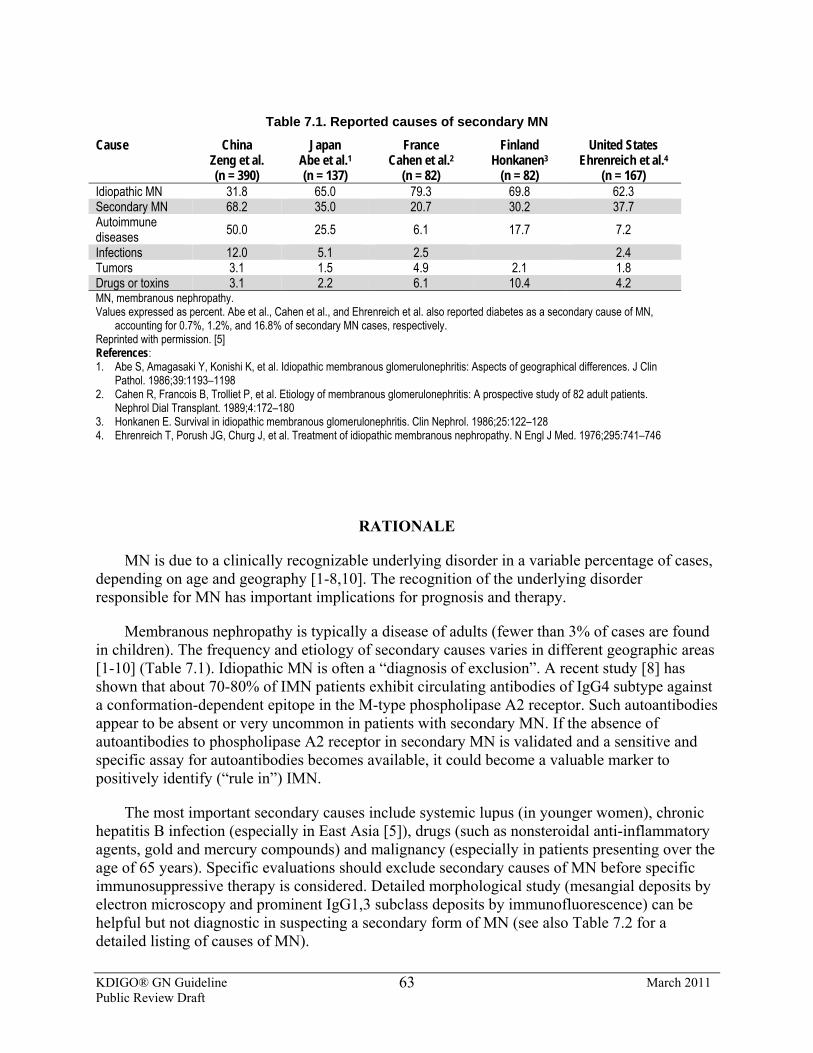
KDIGO® GN Guideline March 2011 Public Review Draft
63
Table 7.1. Reported causes of secondary MN
Cause China Japan France Finland United States Zeng et al. (n = 390)
Abe et al.1 (n = 137)
Cahen et al.2 (n = 82)
Honkanen3 (n = 82)
Ehrenreich et al.4 (n = 167)
Idiopathic MN 31.8 65.0 79.3 69.8 62.3 Secondary MN 68.2 35.0 20.7 30.2 37.7 Autoimmune diseases 50.0 25.5 6.1 17.7 7.2 Infections 12.0 5.1 2.5 2.4 Tumors 3.1 1.5 4.9 2.1 1.8 Drugs or toxins 3.1 2.2 6.1 10.4 4.2 MN, membranous nephropathy. Values expressed as percent. Abe et al., Cahen et al., and Ehrenreich et al. also reported diabetes as a secondary cause of MN,
accounting for 0.7%, 1.2%, and 16.8% of secondary MN cases, respectively. Reprinted with permission. [5] References: 1. Abe S, Amagasaki Y, Konishi K, et al. Idiopathic membranous glomerulonephritis: Aspects of geographical differences. J Clin
Pathol. 1986;39:1193–1198 2. Cahen R, Francois B, Trolliet P, et al. Etiology of membranous glomerulonephritis: A prospective study of 82 adult patients.
Nephrol Dial Transplant. 1989;4:172–180 3. Honkanen E. Survival in idiopathic membranous glomerulonephritis. Clin Nephrol. 1986;25:122–128 4. Ehrenreich T, Porush JG, Churg J, et al. Treatment of idiopathic membranous nephropathy. N Engl J Med. 1976;295:741–746
RATIONALE
MN is due to a clinically recognizable underlying disorder in a variable percentage of cases, depending on age and geography [1-8,10]. The recognition of the underlying disorder responsible for MN has important implications for prognosis and therapy.
Membranous nephropathy is typically a disease of adults (fewer than 3% of cases are found in children). The frequency and etiology of secondary causes varies in different geographic areas [1-10] (Table 7.1). Idiopathic MN is often a “diagnosis of exclusion”. A recent study [8] has shown that about 70-80% of IMN patients exhibit circulating antibodies of IgG4 subtype against a conformation-dependent epitope in the M-type phospholipase A2 receptor. Such autoantibodies appear to be absent or very uncommon in patients with secondary MN. If the absence of autoantibodies to phospholipase A2 receptor in secondary MN is validated and a sensitive and specific assay for autoantibodies becomes available, it could become a valuable marker to positively identify (“rule in”) IMN.
The most important secondary causes include systemic lupus (in younger women), chronic hepatitis B infection (especially in East Asia [5]), drugs (such as nonsteroidal anti-inflammatory agents, gold and mercury compounds) and malignancy (especially in patients presenting over the age of 65 years). Specific evaluations should exclude secondary causes of MN before specific immunosuppressive therapy is considered. Detailed morphological study (mesangial deposits by electron microscopy and prominent IgG1,3 subclass deposits by immunofluorescence) can be helpful but not diagnostic in suspecting a secondary form of MN (see also Table 7.2 for a detailed listing of causes of MN).

KDIGO® GN Guideline March 2011 Public Review Draft
64
Table 7.2. Reported causes of MN (detailed listing)
Autoimmune Autoimmune diseases Systemic lupus erythematosus Rheumatoid arthritis Mixed connective tissue disease Dermatomyositis Ankylosing spondylitis Systemic sclerosis Myasthenia gravis Bullous pemphigoid Autoimmune thyroid disease Sjögren's syndrome Temporal arteritis Crohn's disease Graft-versus-host disease
Infections Hepatitis B Hepatitis C Human immunodeficiency virus Malaria Schistosomiasis Filariasis Syphilis Enterococcal endocarditis Hydatid disease Leprosy
Malignancies Carcinomas Lung Esophageal Colon Breast Stomach Renal Ovary Prostate Oropharynx
Noncarcinomas Hodgkin's lymphoma Non-Hodgkin's lymphoma Leukemia (chronic lymphocytic leukemia) Mesothelioma Melanoma Wilm's tumor Hepatic adenoma Angiolymphatic hyperplasia Schwannoma Neuroblastoma Adrenal ganglioneuroma
Drugs/Toxins Gold Penicillamine Bucillamine Mercury compounds Captopril Probenicid Trimethadione Nonsteroidal anti-inflammatory drugs Cyclooxygenase-2 inhibitors Clopidogrel Lithium Formaldehyde Hydrocarbons
Miscellaneous Diabetes mellitus (association or cause?) Sarcoidosis Sickle cell disease Polycystic kidney disease α1-antitrypsin deficiency Weber-Christian disease Primary biliary cirrhosis Systemic mastocytosis Guillain-Barre syndrome Urticarial vasculitis Hemolytic-uremic syndrome Dermatitis herpetiformis Myelodysplasia

KDIGO® GN Guideline March 2011 Public Review Draft
65
RESEARCH RECOMMENDATIONS
• Studies are needed to validate the utility of antibody against M-type phospholipase A2 receptor in terms of its accuracy in separating primary from secondary MN.
• Studies are needed to determine the most cost-effective panel of investigations for screening for an underlying (covert) malignancy in the older patient with MN.
References
1, Shwartz MM. Membranous glomerulonephritis. In: Jennette JC, Olson JL, Schwartz MM, Silva FG (eds.). Heptinstall's Pathology of the Kidney. Vol. 1. Lippincott Williams & Wilkins 2007.
2. Abe S, Amagasaki Y, Konishi K, et al: Idiopathic membranous glomerulonephritis: Aspects of geographical differences. J Clin Pathol 1986; 39:1193-1198.
3. Cahen R, Francois B, Trolliet P, et al: Etiology of membranous glomerulonephritis: A prospective study of 82 adult patients. Nephrol Dial Transplant 1989; 4:172-180.
4. Honkanen E: Survival in idiopathic membranous glomerulonephritis. Clin Nephrol 1986;25: 122-128. 5. Zeng CH, Chen HM, Wang RS, Chen Y, Zhang SH, Liu L, Li LS, Liu ZH. Etiology and clinical
characteristics of membranous nephropathy in Chinese patients. Am J Kidney Dis. 2008 ;52: 691-8. 6. Gluck MC, Gallo G, Lowenstein J, et al: Membranous glomerulonephritis. Evolution of clinical and
pathological features. Ann Intern Med 1973;78: 1-12. 7. Bjørneklett R, Vikse BE, Svarstad E, et al: Membranous nephropathy and cancer: Epidemiologic evidence
and determinants of high-risk cancer association. Kidney Int 2006 ;70: 1510-1517. 8. Beck LH Jr, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, Klein JB, Salant DJ. M-type
phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361:11-21.
9. Burstein DM, Korbet SM, Schwartz MM. Membranous glomerulonephritis and malignancy. Am J Kidney Dis. 1993; 22: 5-10.
10. Glassock RJ. Secondary Membranous Nephropathy.Nephrol Dial Transplant. 1992;7: (Supplement 1):64-71
7.2: Selection of patients with IMN to be considered for treatment with corticosteroids and immunosuppressive agents 7.2.1: We recommend that initial therapy be started ONLY in patients with
nephrotic syndrome AND when at least ONE of the following conditions are met: • urinary protein excretion persistently exceeds 4 g/d, remains >50% of
baseline value, AND does not show progressive decline, during antihypertensive and antiproteinuric therapy (see Chapter 1) during an observation period of at least 6 months; (1B)
• the presence of severe, disabling, or life-threatening symptoms related to the nephrotic syndrome; (1C)
• SCr has risen by 30% or more within 6 to 12 months from the time of diagnosis but the eGFR is not less than 25-30 ml/min/1.73 m2 AND this change is not explained by superimposed complications. (2C)
7.2.2: Patients with a SCr persistently >3.5 mg/dl (>320 µmol/l) (or eGFR <30 ml/min per 1.73 m2) and those with marked reduction of kidney size on ultrasound should not be exposed to immunosuppressive therapy. (Not Graded)

KDIGO® GN Guideline March 2011 Public Review Draft
66
BACKGROUND
The commonest presentation of IMN is nephrotic syndrome with preserved kidney function. About 50% of patients with persistent high-grade proteinuria eventually progress to ESRD, often after many years of observation. Complete remission of nephrotic syndrome predicts excellent long-term kidney and patient survival. A partial remission also significantly reduces the risk of progression to ESRD (see Table 7.3 for definitions of complete and partial remission used in this chapter). The primary aims of treatment, therefore, are to induce a lasting reduction in proteinuria. All currently used treatment modalities have significant toxicity; therefore, selecting patients at high risk of progression is important so that exposure to treatment-related adverse events is minimized. The degree and persistence of proteinuria during a period of observation helps in selecting patients for this therapy. There is no agreed definition of the “point of no return” in the evolution of IMN after which the risks of immunosuppressive drugs become unacceptable and futile. However, the presence of severe tubular interstitial fibrosis, tubular atrophy, and glomerular obsolescence on biopsy, accompanied by persistent irreversible elevation of SCr >3.5 mg/dl (>320 µmol/l) (or eGFR <30 ml/min per 1.73 m2), may be such indicators.
Table 7.3. Definitions of complete and partial remission in IMN
Complete Remission: Urinary protein excretion <0.3 g/d (uPCR <0.3 g/g or <30 mg/mmol), confirmed by two values at least 1 week apart, accompanied by a normal serum albumin concentration. Partial Remission: Urinary protein excretion <3.5 g/d (uPCR <3.5 g/g or <350 mg/mmol) and a 50% or greater reduction from peak values; confirmed by two values at least 1 week apart, accompanied by an improvement or normalization of the serum albumin concentration. IMN, idiopathic membranous nephropathy; uPCR, urine protein:creatinine ratio. See also Chapter 1. Adapted with permission.[1A, 37]
RATIONALE
• There is low- to moderate-quality evidence to support a recommendation that patients with time-averaged proteinuria <4.0 g/d or those who achieve a complete or partial remission have an excellent long-term prognosis.
• Observational studies of the natural history of IMN have shown that male gender, persistent heavy proteinuria, and elevated SCr at diagnosis predict the risk of later progressive decline in kidney function, although these factors may not all be independent risks.
• About 30-35% of patients with IMN eventually undergo spontaneous remission of nephrotic syndrome; therefore, it is reasonable to delay specific therapy for at least 6 months utilizing supportive therapy, including RAS blockade, unless the patient has unexplained rapid deterioration in kidney function or there are complications related to uncontrolled nephrotic syndrome. The frequency of spontaneous remssion, however, is lower the higher the grade of presenting proteinuria.

KDIGO® GN Guideline March 2011 Public Review Draft
67
• It may be difficult to define precisely the onset time of a partial remission, since some patients experience a slow reduction in proteinuria, even in the absence of specific treatment, to non-nephrotic levels over several years.
• There is support for the use of predictive models for determining risk of progression in IMN (i.e., persistent proteinuria >4 g/d and/or decline in kidney function over a 6-month period of observation).
• There is low-quality evidence to support a recommendation that the period of observation may be extended in patients who exhibit a consistent progressive decline in proteinuria during observation, have stable kidney function, and no complications related to the nephrotic state.
About 80% of adults with IMN have nephrotic syndrome at presentation [1] and the
remainder have subnephrotic proteinuria (see definitions in Chapter 1). The disease course may be punctuated with spontaneous remissions and relapses [2-10]. In about 20% of patients, there is spontaneous complete remission of the nephrotic syndrome, and another 15-20% undergo partial remission. Remission may be delayed for as long as 18-24 months. In a recent study, the mean time to remission was 14.7 ± 11.4 months following presentation [11]. About 15-30% suffer one or more relapses, leaving about 50% of the patients with persistent nephrotic syndrome. Data from natural history studies and placebo arms of intervention studies show that about 30-40% of the patients with persistent nephrotic syndrome progress to ESRD over 10 years [2,13]. Those with a persistent nephrotic syndrome are also exposed to the related complications, including infections, thromboembolic events, and accelerated atherosclerotic cardiovascular disease.
The likelihood of spontaneous remission and progression is dependent upon the age, gender, degree of proteinuria, and kidney function at presentation [12,13]. The risk of progression is highest in those with proteinuria >8 g/d, persistent for 6 months. A validated algorithm allowed creation of a model based on time-averaged proteinuria over 6 months, CrCl at diagnosis, and the slope of CrCl over 6 months that correctly identified patients at risk of progression with 85-90% accuracy [14]. Based on this model, patients at low risk for progression present with a normal CrCl, proteinuria consistently <4 g/d, and have stable kidney function over a 6-month observation period. Patients at medium risk for progression (~50-55% probability of developing progressive CKD over 10 years) have normal kidney function that remains unchanged during 6 months of observation, but continue to have proteinuria between 4 and 8 g/d. Those classified as high risk for progression (65-80% probability of progression to advanced CKD within 10 years from diagnosis) have persistent proteinuria >8 g/d, independent of the degree of kidney dysfunction [15,16]. Treatment-induced remissions are associated with an improved prognosis [17,18]. The 10-year survival free of kidney failure is about 100% in complete remission, 90% in partial remission, and 50% with no remission. Patients with complete or partial remission have a similar rate of decline in CrCl: –1.5 ml/min/y for complete remission, and –2 ml/min/y for partial remission. Although spontaneous remissions are less common in those with higher baseline proteinuria, they are not unknown; a recent report [11] showed spontaneous remission in 26% among those with baseline proteinuria 8-12 g/d and 22% among those with proteinuria >12 g/d. Treatment with RAS blockade, and a 50% decline of proteinuria from baseline during the first year of follow-up, were significant independent predictors for remission. Most reported natural history studies were performed in an era before drugs that act on the RAS became available. The long-term value of RAS blockade in management of IMN has been assessed largely by

KDIGO® GN Guideline March 2011 Public Review Draft
68
observational studies and has been observed only in those patients with proteinuria (<10 g/d) at baseline. A recent small RCT (n = 27) compared an ACEi (lisinopril, up to 10 mg/d) to an ARB (losartan, up to 100 mg/d) in patients with IMN and variable-range proteinuria (2.5-7 g/d). Both agents were of comparable efficacy, reducing proteinuria on average by 2.5 g/d by 12 months. The absence of a placebo control and the failure to include patents with higher-grade proteinuria (>8-10 g/d) weaken the impact of the study [18A]. There is only low-quality evidence to support the value of other predictors, such as hypertension, histologic evidence of interstitial fibrosis and tubular atrophy, persistently elevated urinary C5b-9, and excretion of increased quantities of low- or high-molecular-weight proteins (β2-microglobulin and IgG) in urine [19,20]. Staging of MN by histologic criteria has limited utility for prediction of outcomes or response to therapy in IMN.
7.3: Initial therapy of IMN 7.3.1: We recommend that initial therapy consist of a 6-month course of
alternating monthly cycles of oral and i.v. corticosteroids, and oral alkylating agents. (1B)
7.3.2: We suggest using cyclophosphamide rather than chlorambucil for initial therapy. (2B)
7.3.3: We recommend patients be managed conservatively for at least 6 months following the completion of the initial regimen before being evaluated for remission, unless kidney function is deteriorating. (1C)
7.3.4: Perform a repeat kidney biopsy only if the patient has rapidly deteriorating kidney function (doubling of SCr over 1-2 month of observation), in the absence of massive proteinuria (>15 g/d). (Not Graded)
7.3.5: Adjust the dose of cyclophosphamide or chlorambucil according to age of the patient and eGFR. (Not Graded)
7.3.6: We suggest that continuous (noncyclical) use of alkylating agents may also be effective, but is associated with greater risk of toxicity, particularly when administered for >6 months. (2C)
BACKGROUND
Three RCTs have shown that monotherapy with oral corticosteroids are not superior to symptomatic therapy alone in IMN. Alkylating agents (cyclophosphamide or chlorambucil), most commonly in conjunction with steroids, are effective in inducing remission and preventing ESRD (Suppl Tables 20-25). The toxicity profile suggests that cyclophosphamide might be preferred over chlorambucil.
RATIONALE
• There is moderate-quality evidence to recommend a 6-month cyclical regimen of alternating alkylating agents (cyclophosphamide or chlorambucil) plus i.v. pulse and oral corticosteroids (see Table 7.4 for description of regimen) for initial therapy of IMN meeting the criteria in Statement 7.2.1 above. This evidence indicates this

KDIGO® GN Guideline March 2011 Public Review Draft
69
treatment is superior to supportive therapy alone in inducing remissions and preventing long-term decline of kidney function, including the need for dialysis, in patients with IMN and persisting nephrotic syndrome. The risks and adverse events associated with the use of cyclophosphamide in IMN are summarized in Table 7.5.
• Other combined regimens of cyclophosphamide and corticosteroids have also been used. Some omit i.v. methylprednisolone, others use alkylating agent and corticosteroids concurrently, rather than cyclically, for a longer duration [24-26]. However, the long-term efficacy and safety of these regimens are less well-established than the cyclical regimen [27].
• A complete or partial remission of nephrotic syndrome is associated with an excellent long-term prognosis; therefore, persisting remission of the nephrotic state is an acceptable surrogate end-point to assess efficacy of treatment.
• Treated patients continue to enter complete or partial remission for as long as 12-18 months following completion of the regimen, so it is reasonable to wait this period of time before deciding whether the initial treatment has been unsuccessful (see Statements 7.6.1 and 7.6.2), providing that serum albumin levels or kidney function are not deteriorating, and that morbid events have not supervened. During the period of observation, patients should continue to receive ACEi or ARBs, other antihypertensives, and other supportive therapies as clinically indicated.
• In comparative studies, cyclophosphamide has a superior safety profile compared to chlorambucil. Low-quality evidence exists that cyclophosphamide can lead to more frequent and longer remissions than chlorambucil. Cumulative toxicities of alkylating agents can be significant and require careful monitoring by the treating physician.
• Since the decline in GFR in IMN is often very gradual, especially in the absence of massive proteinuria, any acceleration of the rate of decline indicates the possibility of a superimposed disease process (such as crescentic glomerulonephritis or acute interstitial nephritis, which is often drug-related) that might dictate a change in treatment approach. A repeat kidney biopsy is necessary to identify these conditions.
• Relapses of nephrotic syndrome ocuur in about 25% of patients treated with the “Ponticelli” regimen. A similar fraction of patients with spontaneous remssions also will relapse (see treatment of relapses in IMN in Statement 7.7).
Table 7.4. Cyclical corticosteroid/alkylating-agent therapy for IMN (the “Ponticelli Regimen”)
Month 1: i.v. methylprednisolone (1 g) daily for three doses, then oral methyprednisolone (0.5 mg/kg/d) for 27 days. Month 2: Oral chlorambucil (0.15-0.2 mg/kg/d) or oral cyclophosphamide (2.0 mg/kg/d) for 30 days.* Month 3: Repeat Month 1 Month 4: Repeat Month 2 Month 5: Repeat Month 1 Month 6: Repeat Month 2 IMN, idiopathic membranous nephropathy. *Monitor every 2 weeks for 2 months, then every month for 6 months, with serum creatinine, urinary protein excretion, serum albumin, and
white blood cell count. If total leukocyte count falls to <3500/mm3, then chlorambucil or cyclophosphamide withheld until recovery to >4000/mm3.
An open-label RCT utilizing a 6-month course of chlorambucil and steroids in alternating
monthly cycles was initiated in the 1980s (see Table 7.4 for the description of the regimen) [27-29]. After 10 years of follow-up, 92% of the treated (n = 42) and 60% of the control (n = 39) patients were alive with normal kidney function (P = 0.0038). There was remission in 61% (40%

KDIGO® GN Guideline March 2011 Public Review Draft
70
complete remission) and 33% (5% complete remission) in the two groups. In another RCT [30], this same regimen was compared to one where steroids alone were used for the entire 6-month period (chlorambucil was substituted with oral prednisolone 0.5 mg/kg/d). A significantly higher proportion of patients in the chlorambucil arm were in remission in the first 3 years. The difference was lost at 4 years, probably because of a small number of at-risk cases. The duration of remission was also longer in those treated with chlorambucil. Another RCT [31] compared the same combination of chlorambucil and steroids to one in which chlorambucil had been replaced with oral cyclophosphamide (2.5 mg/kg/d). Remission of nephrotic syndrome was noted with equal frequency in the two arms (82% vs 93%; P = 0.116) (Suppl Tables 20-25). However, severe adverse effects leading to discontinuation of therapy occurred more frequently in the chlorambucil group compared to the cyclophosphamide group (12% vs. 4%). Other small trials and several meta-analyses and systematic reviews have indicated that the alkylating agents are associated with a higher remission rate, although the long-term benefits on kidney function could not be demonstrated [32-37].
A more recent open-label study gave similar results to the initial trials of Ponticelli (see details in Suppl Table 20). Quality of life, as measured by a visual analog scale, was significantly better in the treatment group throughout the follow-up period. The complication rate was not different in the two groups.
One small open-label RCT (N = 29) examined the efficacy of cyclophosphamide for 12 months plus moderate-dose steroids in IMN patients considered to be at high risk of progression (based on urinary IgG and urine β2 microglobulin levels) that previously indicated these patients would have an increase in SCr levels by >25%, and reach a SCr >135 µmol/l (>1.5 mg/dl) or have an increase of >50% from baseline. The study compared an early-start group (urinary abnormalities at baseline) vs. the group started only after SCr had risen by >25-50 %. They found a more rapid remission in proteinuria in early-start patients, but no differences between the two groups in overall remission rates,SCr levels, average proteinuria, relapse rates, or adverse events after 6 years [40].
This study agrees with earlier observational studies from the same authors, and supports an initial conservative treatment approach in IMN patients. However, toxicity with this specific approach has been reported to be substantially increased by both prolonging its duration and by selecting patients with impaired kidney function (SCr >1.5 mg/dl [>135 µmol/l]). The overall evidence for this approach is at the 2B level [38-40]. The adverse effects of alkylating-cytotoxic agents are substantial, and include gonadal toxicity, bladder carcinoma, bone marrow hypoplasia, leukemogenesis, and serious opportunistic infections (Table 7.5). The balance of risk and benefit may be altered by patient-dependent factors, such as age and comorbidities. Table 7.6 lists some of the contraindications to the use of the cyclical alkylating-agent/steroid regimen. Cyclophosphamide has a more favorable side-effect profile compared to chlorambucil. The available evidence does not suggest a beneficial effect of i.v. cyclophosphamide on the course of IMN, and its use is not recommended. Based on limited pharmacokinetic data, the dose of alkylating agents should be reduced when GFR declines, in order to avoid bone-marrow toxicity. Azathioprine does not favorably influence the course of I MN, either alone or with corticosteroids [21-23]. Evidence from studies of immunosuppressed patients with diseases other than IMN indicates that patients on corticosteroids should receive prophylaxis for Pneumocystis jevuei with trimethoprim-sulfamethoxazole. Those at risk for osteoporosis (e.g., elderly or

KDIGO® GN Guideline March 2011 Public Review Draft
71
postmenopausal females) should also receive bisphosphonates, unless these are contraindicated, such as an eGFR <30 ml/min per 1.73 m2 (see also Chapter 1).
Table 7.5. Risks and benefits of the cyclical corticosteroid/alkylating-agent regimen in IMN
Risks Benefits Enhanced risk of opportunistic infection Reactivation of viral hepatitis Alopecia Gonadal damage (aspermatogenesis, ovulation failure) Hemorrhagic cystitis (cyclophosphamide only) Neoplasia (myelodysplastic syndrome, acute myelogenous leukemia Transitional cell carcinoma of the bladder, ureter or pelvis Toxic hepatitis
Prevention of CKD and ESRD Avoidance of complications of nephrotic syndrome (thrombosis, accelerated atherogenesis) Prolongation of life; improved quality of life
CKD, chronic kidney disease; ESRD, end-stage renal disease; IMN, idiopathic membranous nephropathy.
Table 7.6. Contraindications to the use of the cyclical corticosteroid/alkylating-agent regimen in IMN
Untreated infection (HIV, hepatitis B and C, tuberculosis, fungal infection, etc.) Neoplasia (lung, skin [except squamous cell]), breast, colon, etc. Urinary retention Inabilty to comply with monitoring Pre-existing leukopenia (<4000 leukocytes/mm3) SCr >3.5mg/dl (300 μmol/L) HIV, human immunodeficiency virus; IMN, idiopathic membranous nephropathy; SCr, serum creatinine.
Deterioration of kidney function in IMN is usually slow, and development of advanced CKD most often takes several years of persistent high-level proteinuria. A rapid deterioration of kidney function in the absence of massive proteinuria (e.g., >15 g/d) usually indicates the superimposition of another pathologic process, such as acute bilateral renal-vein thrombosis, a superimposed crescentic GN, or acute interstitial nephritis. A repeat kidney biopsy is the most appropriate tool to identify any pathology changes that may require a change in treatment. In patients with severe proteinuria (>10-15 g/d), however, an acute decline in kidney function (<50% reduction in GFR) can be seen, possibly as a result of hemodynamic changes. This usually reverses with remission of the nephrotic state, and hence does not require a change in the therapeutic approach.
RESEARCH RECOMMENDATIONS
• Clinical, pathological, and biological markers are needed to identify patients who will benefit most from therapy, and also to avoid unnecessary drug exposure risk to the rest. There is a lack of evidence to guide ideal dosing to minimize drug toxicity, especially the gonadal and bladder toxicity of cyclophosphamide.
• RCTs are needed to compare alkylating agents with CNIs and/or MMF as initial therapy of IMN with nephrotic syndrome.

KDIGO® GN Guideline March 2011 Public Review Draft
72
• Studies are needed to determine the value of renal pathology and urinary biomarkers in predicting prognosis and/or treatment responsiveness.
• Serial anti-PLA2R antibodies and urinary biomarkers (such as urinary IgG, β2-microglobulin) should be measured in natural history studies, and in all future treatment trials for IMN, in order to assess their value in determining spontaneous remission, response to treatment, and prognosis.
References 1A. Passerini P and Ponticelli C. In: Ponticelli C and Glassock R (Eds). Treatment of Primary Glomerular
Disease. Oxford Medical Publishers, Oxford. 2009; pp 261-312 1. Glassock RJ. Diagnosis and natural course of membranous nephropathy. Semin Nephrol. 2003; 23: 324-32. 2. Cattran DC. Idiopathic membranous glomerulonephritis. Kidney Int. 2001; 59: 1983-94. 3. Row PG, Cameron JS, Turner DR, Evans DJ, White RH, Ogg CS, et al. Membranous nephropathy. Long-
term follow-up and association with neoplasia. Q J Med. 1975; 44: 207-39. 4. Murphy BF, Fairley KF, Kincaid-Smith PS. Idiopathic membranous glomerulonephritis: long-term follow-up
in 139 cases. Clin Nephrol. 1988; 30: 175-81. 5. Schieppati A, Mosconi L, Perna A, Mecca G, Bertani T, Garattini S, et al. Prognosis of untreated patients
with idiopathic membranous nephropathy. N Engl J Med. 1993; 329: 85-9. 6. Donadio JV, Jr., Torres VE, Velosa JA, Wagoner RD, Holley KE, Okamura M, et al. Idiopathic membranous
nephropathy: the natural history of untreated patients. Kidney Int. 1988; 33: 708-15. 7. Honkanen E. Survival in idiopathic membranous glomerulonephritis. Clin Nephrol. 1986; 25: 122-8. 8. Hopper J, Jr., Trew PA, Biava CG. Membranous nephropathy: its relative benignity in women. Nephron.
1981; 29: 18-24. 9. Erwin DT, Donadio JV, Jr., Holley KE. The clinical course of idiopathic membranous nephropathy. Mayo
Clin Proc. 1973; 48:697-712 10. Cameron JS. Membranous nephropathy and its treatment. Nephrol Dial Transplant. 1992; 7 Suppl 1: 72-9. 11. Polanco N, Gutiérrez E, Covarsí A, Ariza F, Carreño A, Vigil A, Baltar J, Fernández-Fresnedo G, Martín C,
Pons S, Lorenzo D, Bernis C, Arrizabalaga P, Fernández-Juárez G, Barrio V, Sierra M, Castellanos I, Espinosa M, Rivera F, Oliet A, Fernández-Vega F, Praga M; for the Grupo de Estudio de las Enfermedades Glomerulares de la Sociedad Española de Nefrología.Spontaneous Remission of Nephrotic Syndrome in Idiopathic Membranous Nephropathy. J Am Soc Nephrol. 2010 on-lineJan 28, 2010.
12. Cattran DC, Reich HN, Beanlands HJ, Miller JA, Scholey JW, Troyanov S. The impact of sex in primary glomerulonephritis. Nephrol Dial Transplant. 2008; 23: 2247-53.
13. Cattran D. Management of Membranous Nephropathy: When and What for Treatment. J Am Soc Nephrol 16: 1188–1194, 2005
14. Pei Y, Cattran D, Greenwood C. Predicting chronic renal insufficiency in idiopathic membranous glomerulonephritis. Kidney Int. 1992; 42: 960-6.
15. Cattran DC, Pei Y, Greenwood CM, Ponticelli C, Passerini P, Honkanen E. Validation of a predictive model of idiopathic membranous nephropathy: its clinical and research implications. Kidney Int. 1997; 51: 901-7.
16. Fervenza FC, Sethi S, Specks U. Idiopathic membranous nephropathy: diagnosis and treatment. Clin J Am Soc Nephrol. 2008; 3: 905-19.
17. Laluck BJ, Jr., Cattran DC. Prognosis after a complete remission in adult patients with idiopathic membranous nephropathy. Am J Kidney Dis. 1999; 33: 1026-32.
18. Troyanov S, Wall CA, Miller JA, Scholey JW, Cattran DC. Idiopathic membranous nephropathy: definition and relevance of a partial remission. Kidney Int. 2004; 66: 1199-205.
18A. Kosmadakis G, et al. Comparison of the influence of angiotensin-converting enzyme inhibitor lisinopril and angiotensin II receptor antagonist losartan in patients with idiopathic membranous nephropathy and nephrotic syndrome. Scand J Urol Nephrol 44:251-256, 2010).

KDIGO® GN Guideline March 2011 Public Review Draft
73
19. Reichert LJ, Koene RA, Wetzels JF. Urinary excretion of beta 2-microglobulin predicts renal outcome in patients with idiopathic membranous nephropathy. J Am Soc Nephrol. 1995; 6: 1666-9.
20. Reichert LJ, Koene RA, Wetzels JF. Urinary IgG excretion as a prognostic factor in idiopathic membranous nephropathy. Clin Nephrol. 1997; 48: 79-84.
21. Controlled trial of azathioprine in the nephrotic syndrome secondary to idiopathic membranous glomerulonephritis. Can Med Assoc J. 1976; 115: 1209-10.
22. Report by Medical Research Council Working Party.Controlled trial of azathioprine and prednisone in chronic renal disease. Br Med J. 1971; 2: 239-41.
23. Lagrue G, Bernard D, Bariety J, Druet P, Guenel J. [Treatment with chlorambucil and azathioprine in primary glomerulonephritis. Results of a "controlled" study]. J Urol Nephrol (Paris). 1975; 81: 655-72.
24. Donadio JV, Jr., Holley KE, Anderson CF, Taylor WF. Controlled trial of cyclophosphamide in idiopathic membranous nephropathy. Kidney Int. 1974; 6: 431-9.
25. Murphy BF, McDonald I, Fairley KF, Kincaid-Smith PS. Randomized controlled trial of cyclophosphamide, warfarin and dipyridamole in idiopathic membranous glomerulonephritis. Clin Nephrol. 1992; 37: 229-34.
26. Shearman JD, Yin ZG, Aarons I, Smith PS, Woodroffe AJ, Clarkson AR. The effect of treatment with prednisolone or cyclophosphamide-warfarin-dipyridamole combination on the outcome of patients with membranous nephropathy. Clin Nephrol. 1988; 30: 320-9
27. Ponticelli C, Zucchelli P, Imbasciati E, Cagnoli L, Pozzi C, Passerini P, et al. Controlled trial of methylprednisolone and chlorambucil in idiopathic membranous nephropathy. N Engl J Med. 1984; 310: 946-50.
28. Ponticelli C, Zucchelli P, Passerini P, Cagnoli L, Cesana B, Pozzi C, et al. A randomized trial of methylprednisolone and chlorambucil in idiopathic membranous nephropathy. N Engl J Med. 1989; 320: 8-13.
29. Ponticelli C, Zucchelli P, Passerini P, Cesana B, Locatelli F, Pasquali S, et al. A 10-year follow-up of a randomized study with methylprednisolone and chlorambucil in membranous nephropathy. Kidney Int. 1995; 48: 1600-4.
30. Ponticelli C, Zucchelli P, Passerini P, Cesana B. Methylprednisolone plus chlorambucil as compared to methylprednisolone alone for the treatment of idiopathic membranous nephropathy. The Italian Idiopathic Membranous Nephropathy Treatment Study Group. N Engl J Med. 1992; 327: 599-603.
31. Ponticelli C, Altieri P, Scolari F, Passerini P, Roccatello D, Cesana B, et al. A randomized study comparing methylprednisolone plus chlorambucil versus methylprednisolone plus cyclophosphamide in idiopathic membranous nephropathy. J Am Soc Nephrol. 1998; 9: 444-50.
32. West ML, Jindal KK, Bear RA, Goldstein MB. A controlled trial of cyclophosphamide in patients with membranous glomerulonephritis. Kidney Int. 1987; 32: 579-84
33. Jindal K, West M, Bear R, Goldstein M. Long-term benefits of therapy with cyclophosphamide and prednisone in patients with membranous glomerulonephritis and impaired kidney function. Am J Kidney Dis. 1992; 19: 61-7.
34. Imperiale TF, Goldfarb S, Berns JS. Are cytotoxic agents beneficial in idiopathic membranous nephropathy? A meta-analysis of the controlled trials. J Am Soc Nephrol. 1995; 5: 1553-8.
35. Hogan SL, Muller KE, Jennette JC, Falk RJ. A review of therapeutic studies of idiopathic membranous glomerulopathy. Am J Kidney Dis. 1995; 25: 862-75.
36. Perna A, Schieppati A, Zamora J, Giuliano GA, Braun N, Remuzzi G. Immunosuppressive treatment for idiopathic membranous nephropathy: a systematic review. Am J Kidney Dis. 2004; 44: 385-401.
37. Jha V, Ganguli A, Saha TK, Kohli HS, Sud K, Gupta KL, et al. A randomized, controlled trial of steroids and cyclophosphamide in adults with nephrotic syndrome caused by idiopathic membranous nephropathy. J Am Soc Nephrol. 2007; 18: 1899-904.
38. Branten AJ, Reichert LJ, Koene RA, Wetzels JF. Oral cyclophosphamide versus chlorambucil in the treatment of patients with membranous nephropathy and renal insufficiency. QJM. 1998; 91: 359-66.
39. du Buf-Vereijken PW, Branten AJ, Wetzels JF. Cytotoxic therapy for membranous nephropathy and renal insufficiency: improved renal survival but high relapse rate. Nephrol Dial Transplant. 2004; 19: 1142-8

KDIGO® GN Guideline March 2011 Public Review Draft
74
40. Hofstra JM, Branten AJ, Wirtz JJ, Noordzij TC, du Buf-Vereijken PW, Wetzels JF. Early versus late start of immunosuppressive therapy in idiopathic membranous nephropathy: a randomized controlled trial. Nephrol Dial Transplant. 2010 Jan;25(1):129-36
7.4: Alternative regimens for the initial therapy of IMN: CNIs 7.4.1: We recommend that cyclosporine or tacrolimus be used for a period of at
least 6 months in patients who meet the criteria for initial therapy (as described in Recommendation 7.1.2), but who choose not to receive the cyclical corticosteroid/alkylating-agent regimen or who have contraindications to this regimen. (See Table 7.7 for specific recommendations for dosage during therapy.) (1C)
7.4.2: We suggest that CNIs be discontinued in patients who do not achieve complete or partial remission after 6 months of treatment. (2C)
7.4.3: We suggest that the dosage of CNI be reduced at intervals of 4-8 weeks to a level of about 50% of the starting dosage and continued for at least 12 months if a complete or partial remission of proteinuria occurs, and no treatment-limiting CNI-related nephrotoxicity occurs. (2C)
7.4.4: Monitor CNI blood levels regularly during the initial treatment period, and when there is an unexplained rise in SCr (>20%) during therapy. (Not Graded). (See Table 7.7 for specific CNI-based regimen dosage recommendations.)
RATIONALE
There is low- to moderate-quality evidence to support a recommendation for CNI therapy (cyclosporine or tacrolimus) as an alternative to cyclical corticosteroid/alkylating-agent therapy in IMN (see Suppl Tables 26-29). There is low-quality evidence to suggest that a minimum of 6 months therapy with CNI should be employed, which should be continued for at least 6-12 months if there is abeneficial effect on proteinuria, based on the high relapse rates if therapy is discontinued early. (The suggested dosage regimens for CNIs in IMN are given in Table 7.7.)
Table 7.7. CNI-based regimens for IMN
Cyclosporin: 3.5-5.0 mg/kg/d given orally in two equally divided doses 12 hours apart, with prednisone 0.15 mg/kg/d, for 6 months. We suggest starting at the low range of the recommended dosage and gradually increasing to avoid acute nephrotoxicity (Sandimmune®, Neoral®, and generic cyclosporin considered equivalent). Tacrolimus: 0.05-0.075 mg/kg/d given orally in two divided doses 12 hours apart, without prednisone, for 6-12 months. We suggest starting at the low range of the recommended dosage and gradually increasing to avoid acute nephrotoxicity. IMN, idiopathic membranous nephropathy. Note: Monitoring of blood levels during therapy is discussed in the text.
Cyclosporine (See Suppl Tables 26-29.)

KDIGO® GN Guideline March 2011 Public Review Draft
75
Early uncontrolled studies suggested an initial benefit, but a high relapse rate, with cyclosporine in IMN [1, 2]. In a single-blind, randomized controlled study, 51 patients with steroid-resistant MN were treated with low-dose prednisone plus cyclosporine and compared to placebo plus prednisone [3]. Complete and partial remissions in proteinuria were seen in 69% of the patients, but the relapse rate when cyclosporine was discontinued was high, approximately 45% of the end of 1 year. Observational data from the German Cyclosporine in NS Study Group suggests that prolonging cyclosporine treatment for 1 year results in higher (34%) complete remission at 1 year, and more sustained rate of remissions [4]. Current recommendations, for patients who respond to cyclosporine, are to continue treatment for at least 1 year [5]. Prolonged low-dose cyclosporine (~1.5 mg/kg/d) could be considered for long-term maintenance of patients who achieve a compelte or partial remission, especially in patients at high risk for relapse [6]. Regular monitoring of cyclosporine blood concentration as well as kidney function is often recommended, according to data accumulated from experiences in kidney transplantation. There is no evidence in patients with IMN to indicate optimal cyclosporine blood levels. Cyclosporine levels usually regarded as nontoxic are 125-175 ng/ml (C0, trough level) or 400-600 ng/ml (C2, 2-hour post-dose level) [5]. Suppl Tables 26-29 summarize studies using cyclosporine. [1, 7-10].
There has been only one small RCT using cyclosporine in patients with high-grade proteinuria and progressive kidney failure [10]. At the time of initiation of treatment, mean CrCl was 55 ml/min, and mean proteinuria 11 g/d. After 12 months of treatment with cyclosporine, there was a significant reduction in proteinuria, and the rate of loss of kidney function decreased from –2.4 to –0.7 ml/min/mo, whereas, in those receiving placebo, there was no change: –2.2 to –2.1 ml/min/mo (P <0.02). This improvement was sustained in ~50% of the patients for up to 2 years after cyclosporine was stopped [1, 7-10].
Tacrolimus In an RCT using tacrolimus monotherapy in IMN, patients with normal kidney function (n =
25) and mean proteinuria (~8 g per 24 hours) received tacrolimus (0.05 mg/kg/d) over 12 months with a 6-month taper, and were compared to conservatively treated controls (n = 23) [11]. After 18 months, the probability of remission was 94% in the tacrolimus group but only 35%, in the control group. Six patients in the control group and only one in the tacrolimus group reached the secondary end-point of a 50% increase in SCr [11]. Almost half of the patients relapsed after tacrolimus was withdrawn, similar to patients treated with cyclosporine. There is only low-quality evidence to support prolonged use of low-dose tacrolimus to maintain remission; the safety of this approach is uncertain [12-26].
Comparison Studies of CNIs vs. Alkylating Agents
Only one RCT in MGN patients of Asian ancestry has compared tacrolimus (n = 39) for 6-9 months to oral cyclophosphamide (n = 34) for 4 months (both groups received prednisone tapered off over 8 months). The results indicated no difference between treatments in terms of partial or complete remission of proteinuria (79% vs. 69%), or adverse events at 12 months of follow-up. Relapses occurred in approximately 15% of both groups. These data support the use of tacrolimus, short-term (with or without concomitant steroids) as an alternative to an oral alkylating-agent regimen [ref]. However, the long-term efficacy of a tacrolimus-based regimen for IMN remains uncertain [27].

KDIGO® GN Guideline March 2011 Public Review Draft
76
RESEARCH RECOMMENDATIONS
• RCTs are needed in IMN to assess the efficacy, safety, and risks of long-term CNI therapy.
• Studies are needed to determine the value of monitoring blood levels of CNIs during therapy of IMN.
References
1. Ambalavanan S, Fauvel JP, Sibley RK, Myers BD: Mechanism of the antiproteinuric effect of cyclosporine in membranous nephropathy. J Am Soc Nephrol 1996;7: 290-298.
2. Guasch A, Suranyi M, Newton L, Hall BM, Myers BD: Short-term responsiveness of membranous glomerulopathy to cyclosporine.[comment]. Am J Kidney Dis 1992;20: 472-481
3. Cattran DC, Appel GB, Hebert LA, Hunsicker LG, Pohl MA, Hoy WE, Maxwell DR, Kunis CL, North America Nephrotic Syndrome Study G: Cyclosporine in patients with steroid-resistant membranous nephropathy: a randomized trial. Kidney Int 2001;59: 1484-1490
4. Meyrier A, Noel LH, Auriche P, Callard P: Long-term renal tolerance of cyclosporin A treatment in adult idiopathic nephrotic syndrome. Collaborative Group of the Societe de Nephrologie. Kidney Int 1994;45: 1446-1456
5. Cattran DC, Alexopoulos E, Heering P, Hoyer PF, Johnston A, Meyrier A, Ponticelli C, Saito T, Choukroun G, Nachman P, Praga M, Yoshikawa N: Cyclosporin in idiopathic glomerular disease associated with the nephrotic syndrome : workshop recommendations. Kidney Int 2007;72: 1429-1447
6. Alexopoulos E, Papagianni, A., Tsamelashvili, M., Leontsini, M. Memmos, D.: Induction and long-term treatment with cyclospirin A in membranous glomerulonephritis with the nephrotic syndrome. J Am Soc Nephrol 2005;16: 780A
7. DeSanto NG, Capodicasa G, Giordano C: Treatment of idiopathic membranous nephropathy unresponsive to methylprednisolone and chlorambucil with cyclosporin. Am J Nephrol 1987;7: 74-76
8. Cattran DC: Idiopathic membranous glomerulonephritis. Kidney Int 2001;59: 1983-1994 9. Rostoker G, Belghiti D, Ben Maadi A, Remy P, Lang P, Weil B, Lagrue G: Long-term cyclosporin A therapy
for severe idiopathic membranous nephropathy. Nephron 1993;63: 335-341 10. Cattran DC, Greenwood C, Ritchie S, Bernstein K, Churchill DN, Clark WF, Morrin PA, Lavoie S: A
controlled trial of cyclosporine in patients with progressive membranous nephropathy. Canadian Glomerulonephritis Study Group. Kidney Int 1995;47: 1130-1135
11. Praga M, Barrio V, Juarez GF, Luno J: Tacrolimus monotherapy in membranous nephropathy: a randomized controlled trial. Kidney Int 2007;71: 924-930
12. Donadio JV, Jr., Holley KE, Anderson CF, Taylor WF: Controlled trial of cyclophosphamide in idiopathic membranous nephropathy. Kidney Int 1974;6: 431-439
13. Ponticelli C, Zucchelli P, Imbasciati E, Cagnoli L, Pozzi C, Passerini P, Grassi C, Limido D, Pasquali S, Volpini T: Controlled trial of methylprednisolone and chlorambucil in idiopathic membranous nephropathy. N Engl J Med 1984;310: 946-950
14. Ponticelli C, Zucchelli P, Passerini P, Cesana B: Methylprednisolone plus chlorambucil as compared to methylprednisolone alone for the treatment of idiopathic membranous nephropathy. The Italian Idiopathic Membranous Nephropathy Treatment Study Group.[comment]. N Engl J Med 1992;327: 599-603
15. Murphy BF, McDonald I, Fairley KF, Kincaid-Smith PS: Randomized controlled trial of cyclophosphamide, warfarin and dipyridamole in idiopathic membranous glomerulonephritis. Clin Nephrol 1992;37: 229-234
16. Ponticelli C, Zucchelli P, Passerini P, Cesana B, Locatelli F, Pasquali S, Sasdelli M, Redaelli B, Grassi C, Pozzi C: A 10-year follow-up of a randomized study with methylprednisolone and chlorambucil in membranous nephropathy. Kidney Int 1995; 48: 1600-1604
17. Ponticelli C, Altieri P, Scolari F, Passerini P, Roccatello D, Cesana B, Melis P, Valzorio B, Sasdelli M, Pasquali S, Pozzi C, Piccoli G, Lupo A, Segagni S, Antonucci F, Dugo M, Minari M, Scalia A, Pedrini L, Pisano G, Grassi C, Farina M, Bellazzi R: A randomized study comparing methylprednisolone plus

KDIGO® GN Guideline March 2011 Public Review Draft
77
chlorambucil versus methylprednisolone plus cyclophosphamide in idiopathic membranous nephropathy. J Am Soc Nephrol 1998; 9: 444-450
18. West ML, Jindal KK, Bear RA, Goldstein MB: A controlled trial of cyclophosphamide in patients with membranous glomerulonephritis. Kidney Int 1987;32: 579-584
19. Mathieson PW, Turner AN, Maidment CG, Evans DJ, Rees AJ: Prednisolone and chlorambucil treatment in idiopathic membranous nephropathy with deteriorating kidney function. Lancet 1988;2: 869-872
20. Bruns FJ, Adler S, Fraley DS, Segel DP: Sustained remission of membranous glomerulonephritis after cyclophosphamide and prednisone. Ann Intern Med 1991;114: 725-730
21. Falk RJ, Hogan SL, Muller KE, Jennette JC: Treatment of progressive membranous glomerulopathy. A randomized trial comparing cyclophosphamide and corticosteroids with corticosteroids alone. The Glomerular Disease Collaborative Network. Ann Intern Med 1992;116: 438-445
22. Jindal K, West M, Bear R, Goldstein M: Long-term benefits of therapy with cyclophosphamide and prednisone in patients with membranous glomerulonephritis and impaired kidney function.[comment]. Am J Kidney Dis 1992;19: 61-67
23. Warwick GL, Geddes CG, Boulton-Jones JM: Prednisolone and chlorambucil therapy for idiopathic membranous nephropathy with progressive renal failure. Q J M 1994:87: 223-229
24. Branten AJ, Reichert LJ, Koene RA, Wetzels JF: Oral cyclophosphamide versus chlorambucil in the treatment of patients with membranous nephropathy and renal insufficiency. QJM 1998;91: 359-366
25. Torres A, Dominguez-Gil B, Carreno A, Hernandez E, Morales E, Segura J, Gonzalez E, Praga M: Conservative versus immunosuppressive treatment of patients with idiopathic membranous nephropathy. Kidney Int 2002;61: 219-227
26. du Buf-Vereijken PW, Branten AJ, Wetzels JF: Cytotoxic therapy for membranous nephropathy and renal insufficiency: improved renal survival but high relapse rate. Nephrol Dial Transplant 2004;19: 1142-1148
27. Chen M,et al. Tacrolimus combined with corticosteroids in treatment of nephrotic idiopathic membranous nephropathy: a multicenter randomized controlled trial. Am J Med Sci 339:233-238, 2010
7.5: Regimens not recommended or suggested for initial therapy of IMN 7.5.1: We recommend that corticosteroid monotherapy not be used for initial
therapy of IMN. (1B) 7.5.2: We suggest that monotherapy with MMF not be used for initial therapy
of IMN. (2C) 7.5.3: We suggest that rituximab not be used for initial therapy of IMN. (2D) 7.5.4: We suggest that ACTH not be used for initial therapy of IMN. (2C)
BACKGROUND
A number of treatments, other than combined therapy of corticosteroid/alkylating agents or CNIs, have been tried as initial therapy in IMN (meeting the criteria outline in Statement 7.1.2). However, none of these have been shown in appropriately sized RCTs to be consistently effective and safe, and therefore are not recommendation as “first-line” initial therapy in IMN.
RATIONALE
Corticosteroid Monotherapy There is moderate-quality evidence to recommend not using corticosteroid monotherapy for
inducing remissions or delaying the onset of progressive CKD in IMN. An early study reported

KDIGO® GN Guideline March 2011 Public Review Draft
78
that a 2- to 3-month course of high-dose, alternate-day prednisone resulted in a significant reduction compared to placebo in progression to kidney failure, although there was no sustained effect on proteinuria [1]. A subsequent RCT in patients with IMN, using an identical corticosteroid regimen vs. placebo, showed no improvement during drug exposure, or over a 3-year follow-up in either proteinuria or kidney function (SCr). An additional RCT comparing a 6-month course of prednisone given on alternate days (n = 81) to no specific treatment (n =7 7) showed no significant benefit of corticosteroid treatment alone, in either induction of remission or preservation of kidney function, even after the data were adjusted to include only patients with proteinuria at entry > 3.5 g per 24 hours [2].
MMF (see Suppl Tables 30-32) MMF as initial therapy in IMN has not been shown in RCTs to be consistently effective for
inducing remissions or delaying the onset of progressive CKD. Thirty-two patients with IMN and impairment of kidney function (SCr >1.5 mg/dl [132 μmol/l]) were treated with oral MMF 1 g twice daily for 12 months, in combination with corticosteroids, and compared to 32 patients—historical controls treated for the same duration with oral cyclophosphamide in combination with corticosteroids (cyclophosphamide; 1.5 mg/kg/d) [3]. Cumulative incidences of remission of proteinuria at 12 months were 66% with MMF vs. 72% with cyclophosphamide (P = 0.3). Adverse effects occurred at a similar rate in the two groups, but relapses were much more common with MMF [3].
In contrast, in a pilot RCT in low risk-of-progression adults that were all drug-naïve with nephrotic syndrome due to IMN, the efficacy of a MMF-based monotherapy regimen was compared to conservative therapy alone [4]. There was no significant difference in the proportion of patients achieving remission: 64% with MMF, 80% with conservative therapy (no corticosteroids or immunosuppression). The frequency of relapses and incidence of infections were similar in both groups.
An additional RCT randomized 36 patients with IMN and nephrotic syndrome to conservative therapy (RAS blockade, statins, low-salt and low-protein diet, and diuretics) plus MMF (2 g/d, without concomitant steroids) (n = 19) or conservative therapy alone (n = 17) for 12 months [5]. The probability of a complete or partial remission did not differ between the two groups after 12 months. While MMF plus steroids might have comparable efficacy to the standard regimen of cyclical alkylating agents and steroids, the present evidence is conflicting, of low quality, and only short-term.
Rituximab There are no RCTs using rituximab for therapy of IMN, although small observational pilot
studies have provided encouraging data. A pilot study used four weekly doses of rituximab (375 mg/m2) in eight nephrotic patients with IMN and followed them for 1 year [6,7]. Proteinuria significantly decreased at 12 months, and kidney function remained stable in all patients. Adverse effects were reported as mild. An observational study from the same investigators suggested that rituximab is likely to be most effective in patients with minimal degrees of tubulointerstitial injury [8].

KDIGO® GN Guideline March 2011 Public Review Draft
79
A prospective observational study in 15 patients with IMN and proteinuria >4 g per 24 hours—despite ACEi/ARB use for >3 months and systolic blood pressure <130 mmHg—has been reported [9]. At 6 months, patients who remained with proteinuria >3 g per 24 hours, and in whom total CD19+ B-cell count was >15 cells/µl, received a second identical course of rituximab. Baseline proteinuria of 13.0 ± 5.7 g per 24 hours (range 8.4-23.5) decreased to 9.1 ± 7.4 g, 9.3 ± 7.9 g, 7.2 ± 6.2 g, and 6.0 ± 7.0 g per 24 hours (range 0.2-20) at 3, 6, 9, and 12 months, respectively (mean ± SD). The mean decline in proteinuria from baseline to 12 months was 6.2 ± 5.1 g/d and was statistically significant (P = 0.002). Rituximab was well-tolerated, and was effective in reducing proteinuria in some patients with IMN. The complete and partial remission rate was almost 60%, higher than would have been expected based on known spontaneous remission rates.
Another observational study used circulating B-cell counts to guide dosing, significantly reducing total dose of rituximab [10]. At 1 year, the proportion of patients who achieved disease remission was identical to that of 24 historical patients who were given a standard rituximab protocol of four weekly doses of 375 mg/m2.
More recently, another prospective observational study in 20 patients with IMN and baseline persistent proteinuria >5.0 g/d received rituximab (375 mg/m2 weekly for four doses), with retreatment at 6 months regardless of proteinuria response [11]. Baseline proteinuria of 11.9 g/d decreased to 4.2 g/d and 2.0 g/d at 12 and 24 months, respectively, while CrCl increased from 72.4 to 88.4 ml/min per 1.73 m2 at 24 months. Among 18 patients who completed 24 months of follow-up, four achieved complete remission, 12 achieved partial remission (complete plus partial remission of 80%). One patient relapsed during follow-up. More than 50% of the patients in this pilot trial had not responded to prior therapy. No short-term toxicity of rituximab was observed. This study also reinforced the observation, made with alklylating agent/corticosteroid therapy that proteinuria declines gradually, and many months may be required for proteinuria to reach its nadir. An RCT is needed to confirm these results, but the findings are very encouraging in regards to the benefits of rituximab for “treatment-resistant” IMN. The long-term relapse rate is unknown.
Adrenocorticotrophic Hormone (ACTH)
One observational study and one small RCT provide preliminary, low-quality evidence for the use of long-acting ACTH as initial therapy in IMN.
Depot synthetic ACTH (Synacthen®) administered for 1 year in an observational study decreased proteinuria in patients with IMN [12,13]. More recently, an open-label pilot RCT compared i.v. methylprednisolone and oral corticosteroids plus a cytotoxic agent (n = 16) vs. synthetic ACTH (n = 16) as initial therapy in IMN, and found them to be of similar efficacy [14]. Side-effects associated with the use of synthetic ACTH included dizziness, glucose intolerance, diarrhea, and the development of bronze-colored skin, which resolved after the end of therapy. Larger, more-powerful RCTs are required before synthetic ACTH can be recommended for initial therapy of IMN. Preliminary and anecdotal reports of a similar effect of native, intact (porcine) ACTH in a gel formulation have very recently appeared. No RCTs have yet been conducted with this agent.

KDIGO® GN Guideline March 2011 Public Review Draft
80
RESEARCH RECOMMENDATIONS
• Larger RCTs with longer follow-up are needed to test MMF and corticosteroids vs. established regimens as initial therapy.
• An RCT is needed to compare rituximab to cyclical corticosteroid/alkylating-agent therapy or CNIs for initial treatment of IMN with nephrotic syndrome.
• An RCT is needed to compare synthetic or native (intact, porcine) ACTH in gel form with cyclical corticosteroid/alkylating-agent therapy or CNIs for initial treatment of IMN with nephrotic syndrome.
References 1. Anonymous: A controlled study of short-term prednisone treatment in adults with membranous nephropathy.
Collaborative Study of the Adult Idiopathic Nephrotic Syndrome. N Engl J Med 1979301:1301-1306 2. Cattran DC, Delmore T, Roscoe J, Cole E, Cardella C, Charron R, Ritchie S: A randomized controlled trial of
prednisone in patients with idiopathic membranous nephropathy. N Engl J Med 1989320:210-215 3. Branten AJ, du Buf-Vereijken PW, Vervloet M, Wetzels JF: Mycophenolate mofetil in idiopathic
membranous nephropathy: a clinical trial with comparison to a historic control group treated with cyclophosphamide. Am J Kidney Dis 2007;50:248-256
4. Senthil Nayagam L, Ganguli A, Rathi M, Kohli HS, Gupta KL, Joshi K, Sakhuja V, Jha V: Mycophenolate mofetil or standard therapy for membranous nephropathy and focal segmental glomerulosclerosis: a pilot study. Nephrol Dial Transplant 200823:1926-1930 5. Dussol B, Morange S, Burtey S, Indreies M, Cassuto E, Mourad G, Villar E, Pouteil-Noble C, Karaaslan H, Sichez H, Lasseur C, Delmas Y, Nogier MB, Fathallah M, Loundou A, Mayor V, Berland Y: Mycophenolate mofetil monotherapy in membranous nephropathy: a 1-year randomized controlled trial. Am J Kidney Dis 200852:699-705
6. Remuzzi G, Chiurchiu C, Abbate M, Brusegan V, Bontempelli M, Ruggenenti P: Rituximab for idiopathic membranous nephropathy. Lancet 2002360:923-924
7. Ruggenenti P, Chiurchiu C, Brusegan V, Abbate M, Perna A, Filippi C, Remuzzi G: Rituximab in idiopathic membranous nephropathy: a one-year prospective study. J Am Soc Nephrol 200314:1851-1857
8. Ruggenenti P, Chiurchiu C, Abbate M, Perna A, Cradevi P, Bontempelli M, Remuzzi G: Rituximab for idiopathic membranous nephropathy: who can benefit? Clin J Am Soc Nephrol 2006 1:738-748
9. Fervenza FC, Cosio FG, Erickson SB, Specks U, Herzenberg AM, Dillon JJ, Leung N, Cohen IM, Wochos DN, Bergstralh E, Hladunewich M, Cattran DC: Rituximab treatment of idiopathic membranous nephropathy. Kidney Int 2008 73:117-125
10. Cravedi P, Ruggenenti P, Sghirlanzoni MC, Remuzzi G: Titrating rituximab to circulating B cells to optimize lymphocytolytic therapy in idiopathic membranous nephropathy. Clin J Am Soc Nephrol 2007 2:932-937
11. Fervenza FG,Abraham RS, Erickson SB, et al. Rituximab therapy in Idiopathic Membranous Nephropathy: a 2 year study. Clin J Am Soc Nephrol 2010;5:2188-2198
12. Berg AL, Arnadottir M: ACTH-induced improvement in the nephrotic syndrome in patients with a variety of diagnoses. Nephrol Dial Transplant 2004 19:1305-1307
13 Berg AL, Nilsson-Ehle P, Arnadottir M: Beneficial effects of ACTH on the serum lipoprotein profile and glomerular function in patients with membranous nephropathy. Kidney Int 1999 56:1534-1543
14. Ponticelli C, Passerini P, Salvadori M, Manno C, Viola BF, Pasquali S, Mandolfo S, Messa P: A randomized pilot trial comparing methylprednisolone plus a cytotoxic agent versus synthetic adrenocorticotropic hormone in idiopathic membranous nephropathy. Am J Kidney Dis 2006 47:233-240

KDIGO® GN Guideline March 2011 Public Review Draft
81
7.6: Treatment of IMN resistant to recommended initial therapy 7.6.1: We suggest that patients with IMN resistant to alkylating agent–based
initial therapy be treated with a CNI. (2C) 7.6.2: We suggest that patients with IMN resistant to CNI-based initial therapy
be treated with an alkylating agent. (2C)
BACKGROUND
The results of trials using an initial cyclical treatment alternating steroids and an alkylating agent or an initial CNI have shown excellent kidney survival and a high rate of remission, even in the long–term [1-7]. However, 9-28% of patients are treatment-resistant (fail to achieve a remission) to steroids and alkylating-agent therapy, and approximately 25% of patients are treatment-resistant to CNI therapy. Patients who fail to achieve a complete or partial remission of nephrotic syndrome should be considered for additional therapy if no contraindication to such treatment exists. The response to alternative therapeutic strategies in treatment-resistant disease cannot presently be predicted with any degree of accuracy. Failure to respond to one regimen does not reliably predict a failure to respond to another regimen.
RATIONALE
Unresponsiveness to initial therapy is observed in 10-30% of patients following a complete course of treatment. There is low-quality evidence to suggest that failure to respond to one regimen does not reliably predict failure to respond to another regimen.
If there is no remission with cyclical treatment with a corticosteroid/alkylating-agent regimen, an alternative is to use CNIs. Cyclosporine is the best studied, although tacrolimus has also been shown to induce a high initial rate of remission, comparable to the overall response rate observed with combined steroids and alkylating agents, particularly after a prolonged administration and associated with moderate doses of steroids [6].
Many treatment-resistant patients also have deteriorating kidney function. There has been only one small RCT using cyclosporine in patients with high-grade proteinuria (>10 g/d) and progressive kidney failure (initial CrCl approximately 55 ml/min). It showed a significant reduction in the rate of loss of kidney function with cyclosporine [8]. For those patients that receive a CNI for initial therapy and show no response after a period of at least 6 months, we suggest treatment with an alkylating agent–based regimen, using the same regimen as for initial therapy. However, adverse effects of treatment may be more frequent in patients with established or progressing kidney impairment.
In patients with kidney impairment [8,9], bone marrow is more susceptible to the toxic effect of alkylating agents, and there may also be heightened susceptibility to infections. Therefore, it is recommended not to exceed daily doses for chlorambucil of 0.1 mg/kg and cyclophosphamide of 1.5 mg/kg in patients with SCr >2.0 mg/dl [176 μmol/l] [10] and to limit the total duration of therapy to <6 months. A higher incidence of side-effects with this regimen is to be expected.

KDIGO® GN Guideline March 2011 Public Review Draft
82
The roles of MMF, rituximab, or ACTH in patients resistant to both alkylating agent–based and CNI-based regimens remain undefined; there have been no RCTs [11-17].
Additional causative factors should be considered. Rapidly progressive kidney insufficiency also may occur from an acute hypersensitivity interstitial nephritis in IMN patients receiving diuretics, antibiotics, or nonsteroidal anti-inflammatory drugs. Kidney biopsy is often necessary to confirm the diagnosis, and a course of high-dose oral prednisone may result in complete recovery of kidney function.
Finally, pulses of i.v. methylprednisolone as monotherapy should not be used for treatment of resistant disease, unless the steady evolution of IMN is interrupted by a rapidly progressive course, and an extracapillary (crescentic) GN superimposed on IMN is shown by kidney biopsy.
RESEARCH RECOMMENDATIONS
• RCTs are needed to assess risks and benefits of rituximab in the treatment of IMN patients resistant to first-line therapy.
• RCTs are neededto assess risks and benefits of the cyclical alkylating agent/corticosteroid regimen or with a CNI regimen in IMN patients with impaired or deteriorating kidney function.
References
1. Ponticelli C, Zucchelli P, Passerini P, Cesana B. Methylprednisolone plus chlorambucil as compared to
methylprednisolone alone for the treatment of idiopathic membranous nephropathy. The Italian Idiopathic Membranous Nephropathy Treatment Study Group. N Engl J Med. 1992; 327: 599-603
2. Ponticelli C, Zucchelli P, Passerini P, et al . A 10 year follow-up of a randomized study with methylprednisolone and chlorambucil in membranous nephropathy. Kidney Int 1995;48:1600-1604
3. Ponticelli C, Altieri P, Scolari F et al . A randomized study comparing methylprednisolone plus chlorambucil versus methylprednisolone plus cyclophosphamide in idiopathic membranous nephropathy. J Am Soc Nephrol 1998;9:444-450
4. Ahmed S, Rahman M, Alam MR et al. Methylprednisolone plus chlorambucil as compared to prednisolone alone for the treatment of idiopathic membranous nephropathy. A preliminary study. Bangladesh Renal J 1994;13:51-54
5. Jha V, Ganguli A, Saha TK et al. A randomized, controlled trial of steroids and cyclophosphamide in adults with nephrotic syndrome caused by idiopathic membranous nephropathy. J Am Soc Nephrol 2007;18:1899-1904
6. Cattran DC, Appel GB, Hebert LA et al . North America Nephrotic Syndrome Study Group Cyclosporine in patients with steroid-resistant membranous nephropathy : a randomized trial. Kidney Int 2001;59: 1484-1490
7. Praga M, Barrío V, Juárez GF, Luño J. Grupo Español Estudio Nefropatía Membranosa. Tacrolimus monotherapy in membranous nephropathy: a randomized controlled trial. Kidney Int 2007;71:924-930
8. Cattran DC, Greenwood C, Ritchie S et al : A controlled trial of cyclosporine in patients with progressive membranous nephropathy. Canadian Glomerulonephritis Study Group. Kidney Int 1995; 47:1130-1135
9. du Buf-Vereijken PW, Branten AJ, Wetzels JF. Cytotoxic therapy for membranous nephropathy and renal insufficiency: improved renal survival but high relapse rate. Nephrol Dial Transplant 2004; 19:1142-1148
10. Reichter CJM, Koene KAP, Wetzels JFM, Huysmans FTM, Assman K : Preserving kidney function in patients with membranous nephropathy : daily oral chlorambucil compared to intermittent monthly pulses of cyclophosphamide. Ann Intern Med 1994; 121:328-323

KDIGO® GN Guideline March 2011 Public Review Draft
83
11. Passerini P, Ponticelli C: Membranous nephropathy. Membranous nephropathy. In Ponticelli,C Glassock,R Teatment of Primary Glomerulonephritis, Oxford University press 2009
12. Dussol B, Morange S, Burtey S et al . Mycophenolate mofetil monotherapy in membranous nephropathy : a 1 year randomized controlled trial . Am J Kidney Dis 2008; 52:699-705
13. Nayagam LS, Ganguli A, Rathi M et al : Mycophenolate mofetil or standard therapy for membranous nephropathy and focal segmental glomerulosclerosis : a pilot study. Nephrol Dial Transplant 2008;23:1926-1930
14. Branten AJ, du Buf-Vereijken PW, Vervloet M, Wetzels FJ. Mycophenolate mofetil in idiopathic membranous nephropathy : a clinical trial with comparison to a historic control group treated with cyclophosphamide. Am J Kidney Dis 2007;50:248-256
15. Bomback AS, Derebail VM, McGregor JG, Kshirsagar AV, Falk RJ, Nachman PH: Rituximab therapy for membranous nephropathy: A systematic review. Clin J Am Soc Nephrol 2009;4:734-744
16. Berg AL, Arnadottir M . ACTH induced improvement in the nephrotic syndrome in patients with a variety of diagnoses. Nephrol Dial Transplant 2004; 19:1305-1307
17. Ponticelli C, Passerini P, Salvadori M et al: A randomized pilot trial comparing methylprednisolone plus a cytotoxic agent versus synthetic adrenocorticotropic hormone in idiopathic membranous nephropathy. Am J Kidney Dis 2006; 47:233-240
7.7: Treatment for relapses of nephrotic syndrome in IMN 7.7.1: We suggest that relapses of nephrotic syndrome in IMN be treated by
reinstitution of the same therapy that resulted in the initial remission. (2D)
7.7.2: We suggest that, if a 6-month cyclical corticosteroid/alkylating-agent regimen was used for initial therapy (see Recommendation 7.2.1), it be repeated only once for treatment of a relapse. (2B)
BACKGROUND
Clinical trials using cyclical treatment of alternating steroids and alkylating agents or CNIs in IMN have shown excellent kidney survival in those subjects with complete or partial remission, even in the long term. However, relapses of nephrotic syndrome occur in 25-30% of patients within 5 years of discontinuation of therapy with alkylating agents, and 40-50% of patients within 1 year of discontinuation of CNIs. For those patients who show a complete or partial remission and then a relapse of nephrotic syndrome, a second course of treatment can be given [1].
RATIONALE
There is very low–quality evidence to suggest that responses to re-treatment of a relapse are similar to those observed after the first treatment.
There is moderate-quality evidence to suggest that there are significant risks of neoplasia induction, opportunistic infections, and gonadal damage when alkylating agents are used for an extended period.

KDIGO® GN Guideline March 2011 Public Review Draft
84
If there is a relapse of nephrotic syndrome in IMN following remission, reintroduction of a corticosteroid/alkylating-agent regimen or CNIs will often, but not uniformly, induce another remission.
Most data on repeated courses of immunosuppressive therapy relate to patients in whom relapses occurred after a partial remission, and with normal kidney function [2,3]. There are no RCTs to guide therapy for patients with IMN who relapse after a first course of therapy and have kidney impairment [4].
Cancer induction is a major concern when alkylating agents are used for an extended period. Cumulative doses of more than 36 g of cyclophosphamide (equivalent to 100 mg daily for 1 year) were associated with a 9.5-fold increased risk of bladder cancer, in patients with Wegener granulomatosis. Extended courses have also been associated with an increased risk of lymphoproliferative, myelodysplastic, and leukemic disorders [5]. Because of this, repeated courses (more than two) of cyclical alkylating-agent therapy are not advised.
Mild relapses (redevelopment of subnephrotic proteinuria after a complete remission) do not require any specific treatment, and should be managed conservatively. Blood pressure should be kept <125/75 mm Hg and an ACEi or ARB should be used as the first line of treatment (see Chapter 1).
RESEARCH RECOMMENDATIONS
• RCTs are needed to examine the efficacy and safety of MMF, rituximab, or ACTH in relapsing patients with IMN.
References
1. Waldman M, Austin H : Controversies in the treatment of idiopathic membranous nephropathy. Nature Rev Nephrol 2009; 5;469-479
2. Ponticelli C, Passerini P, Altieri P, et al : Remissions and relapses in idiopathic membranous nephropathy. Nephrol Dial Transplant 1992; 7 (suppl 1): 85-90
3. Suki WN, Trimarchi H, Frommer JP. Relapsing membranous nephropathy. Response to therapy of relapses compared to that of the original disease. Am J Nephrol 1999;19:474-479
4. Du Buf-Vereijken PW, Wetzels JF : Efficacy of a second course of immunosuppressive therapy in patients with membranous nephropathy and persistent or relapsing disease activity. Nephrol Dial Transplant 2004;19:2036-2043
5. Faurschou M, Penkowa A, Andersen CB et al: Malignancies in Wegener`s granulomatosis : incidence and relation to cyclophosphamide therapy in a cohort of 293 patients. J Rheumatol 2008;35:100-105

KDIGO® GN Guideline March 2011 Public Review Draft
85
7.8: Treatment of IMN in children 7.8.1: We suggest that treatment of IMN in children follows the
recommendations for treatment of IMN in adults. (2C) (See Recommendations 7.2.1, 7.3.1.)
7.8.2 We suggest that no more than one course of the cyclical corticosteroid/alkylating-agent regimen be given in children. (2D)
BACKGROUND
IMN in children is uncommon, and usually presents as nephrotic syndrome or asymptomatic proteinuria. IMN contributes less than 5% of cases of nephrotic syndrome in children [1,2]. Most cases (>75%) of MN in children are secondary to chronic viral infections (e.g., hepatitis B), autoimmune diseases (SLE, thyroiditis), or drugs.
RATIONALE
There is low-quality evidence to suggest children with IMN should be treated with the same regimens as adults, with appropriate dosage modification.
Most knowledge of the natural history of IMN in children, treatment options, and long-term outcome is derived from small, uncontrolled observational studies [3] that show a high spontaneous remission rate, and low incidence of ESRD. Children with IMN will not usually require more than conservative therapy, unless they are severely symptomatic, as they seem to have a higher spontaneous remission rate than adults. For children with severe symptomatic disease, the same drug combinations used in adults are suggested, with appropriate dosage adjustments [4]. Most of these protocols use chlorambucil 0.15-0.2 mg/kg/d or cyclophosphamide 2 mg/kg/d for 8-12 weeks, with alternate-day prednisone. The risk for gonadal toxicity with chlorambucil and cyclophosphamide is greater in boys than in girls, and is related to both the duration and total dose of treatment [5].
There are no data on the use of CNIs in children with IMN; the use of CNIs is based only on the evidence from adults RCTs. MMF, rituximab, or ACTH has not been studied in children (see also Table 7.8).
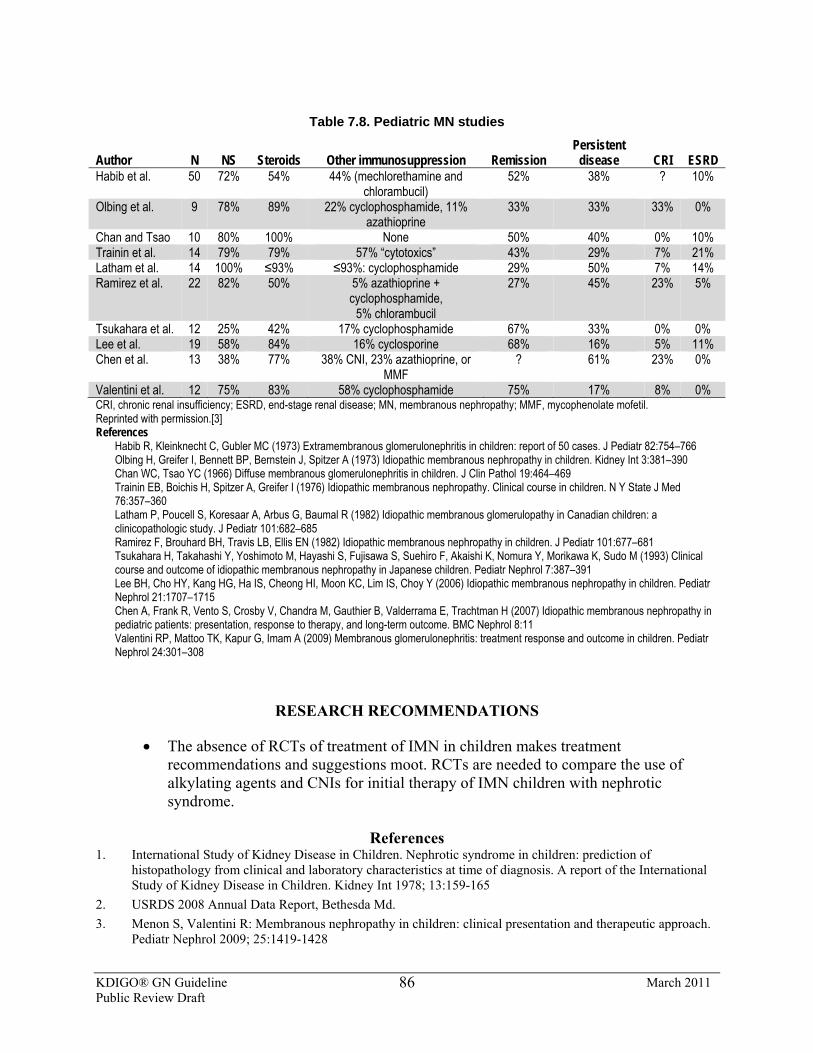
KDIGO® GN Guideline March 2011 Public Review Draft
86
Table 7.8. Pediatric MN studies
Author N NS Steroids Other immunosuppression Remission Persistent
disease CRI ESRD Habib et al. 50 72% 54% 44% (mechlorethamine and
chlorambucil) 52% 38% ? 10%
Olbing et al. 9 78% 89% 22% cyclophosphamide, 11% azathioprine
33% 33% 33% 0%
Chan and Tsao 10 80% 100% None 50% 40% 0% 10% Trainin et al. 14 79% 79% 57% “cytotoxics” 43% 29% 7% 21% Latham et al. 14 100% ≤93% ≤93%: cyclophosphamide 29% 50% 7% 14% Ramirez et al. 22 82% 50% 5% azathioprine +
cyclophosphamide, 5% chlorambucil
27% 45% 23% 5%
Tsukahara et al. 12 25% 42% 17% cyclophosphamide 67% 33% 0% 0% Lee et al. 19 58% 84% 16% cyclosporine 68% 16% 5% 11% Chen et al. 13 38% 77% 38% CNI, 23% azathioprine, or
MMF ? 61% 23% 0%
Valentini et al. 12 75% 83% 58% cyclophosphamide 75% 17% 8% 0% CRI, chronic renal insufficiency; ESRD, end-stage renal disease; MN, membranous nephropathy; MMF, mycophenolate mofetil. Reprinted with permission.[3] References Habib R, Kleinknecht C, Gubler MC (1973) Extramembranous glomerulonephritis in children: report of 50 cases. J Pediatr 82:754–766 Olbing H, Greifer I, Bennett BP, Bernstein J, Spitzer A (1973) Idiopathic membranous nephropathy in children. Kidney Int 3:381–390 Chan WC, Tsao YC (1966) Diffuse membranous glomerulonephritis in children. J Clin Pathol 19:464–469 Trainin EB, Boichis H, Spitzer A, Greifer I (1976) Idiopathic membranous nephropathy. Clinical course in children. N Y State J Med
76:357–360 Latham P, Poucell S, Koresaar A, Arbus G, Baumal R (1982) Idiopathic membranous glomerulopathy in Canadian children: a
clinicopathologic study. J Pediatr 101:682–685 Ramirez F, Brouhard BH, Travis LB, Ellis EN (1982) Idiopathic membranous nephropathy in children. J Pediatr 101:677–681 Tsukahara H, Takahashi Y, Yoshimoto M, Hayashi S, Fujisawa S, Suehiro F, Akaishi K, Nomura Y, Morikawa K, Sudo M (1993) Clinical
course and outcome of idiopathic membranous nephropathy in Japanese children. Pediatr Nephrol 7:387–391 Lee BH, Cho HY, Kang HG, Ha IS, Cheong HI, Moon KC, Lim IS, Choy Y (2006) Idiopathic membranous nephropathy in children. Pediatr
Nephrol 21:1707–1715 Chen A, Frank R, Vento S, Crosby V, Chandra M, Gauthier B, Valderrama E, Trachtman H (2007) Idiopathic membranous nephropathy in
pediatric patients: presentation, response to therapy, and long-term outcome. BMC Nephrol 8:11 Valentini RP, Mattoo TK, Kapur G, Imam A (2009) Membranous glomerulonephritis: treatment response and outcome in children. Pediatr
Nephrol 24:301–308
RESEARCH RECOMMENDATIONS
• The absence of RCTs of treatment of IMN in children makes treatment recommendations and suggestions moot. RCTs are needed to compare the use of alkylating agents and CNIs for initial therapy of IMN children with nephrotic syndrome.
References
1. International Study of Kidney Disease in Children. Nephrotic syndrome in children: prediction of histopathology from clinical and laboratory characteristics at time of diagnosis. A report of the International Study of Kidney Disease in Children. Kidney Int 1978; 13:159-165
2. USRDS 2008 Annual Data Report, Bethesda Md. 3. Menon S, Valentini R: Membranous nephropathy in children: clinical presentation and therapeutic approach.
Pediatr Nephrol 2009; 25:1419-1428

KDIGO® GN Guideline March 2011 Public Review Draft
87
4. Makker S: Treatment of membranous nephropathy in children. SeminNephrol 2003; 23:379-385. 5. Watson AR, Ranee CP, Bain J: Long-term effects of cyclophosphamide on testicular function. Br Med J
1985; 91:1457
7.9: Prophylactic anticoagulants in IMN 7.9.1: We suggest that patients with IMN and nephrotic syndrome, with
marked reduction in serum albumin (<2.5 g/dl [<25 g/l]) and additional risks for thrombosis, be considered for prophylactic anticoagulant therapy, using oral warfarin. (2C)
BACKGROUND
IMN seems to constitute a special hazard for venous thromboembolism and spontaneous arterial thrombosis (such as deep venous thrombosis or pulmonary artery embolism/thrombosis), even more so than other causes of nephrotic syndrome [1-3] (see also Chapter 1). This may also apply to other types of primary GN associated with severe nephrotic syndrome; the evidence base, however, is lacking. There have been no RCTs of prophylactic anticoagulation in IMN with nephrotic syndrome [1-3].
RATIONALE
There is very low–quality evidence to suggest the use of prophylactic anticoagulation with warfarin in patients with IMN and severe nephrotic syndrome. However, based on Markov modeling of anticipated benefits and risks derived from observational studies, prophylactic anticoagulation might be considered when the serum albumin concentration is <2.0-2.5 g/dl (<20-25 g/l) with one or more of the following: proteinuria >10 g/d; BMI >35 kg/m2; prior history of thromboembolism; family history of thromboembolism with documented genetic predisposition; NYHA class III or IV congestive heart failure; recent abdominal or orthopedic surgery; prolonged immobilization [1-3]. Treatment with warfarin should always be preceded by a short period of treatment with heparin (fractionated or unfractionated) in sufficient dosage to obtain prolongation of the clotting time. Dosage adjustments for fractionated heparin may be required if kidney function is impaired. Due to insufficent experience with the use of newer oral or parenteral anticoagulants in nephrotic syndrome, no recommendations can be made regarding their use for prophylaxis of thrombosis. The duration of prophylactic anticoagulation needed for optimal benefit compared to risk is not known, but it seems reasonable to continue therapy for as long as the patient remains nephrotic with a serum albumin <3.0 g/dl (<30 g/l).
RESEARCH RECOMMENDATIUON
• An RCT is needed of prophylactic warfarin in patients with nephrotic syndrome with/without additional risk for thromboembolism in IMN patients.
Please refer to Supplemental Tables 20-32 for additional data related to Chapter 7.

KDIGO® GN Guideline March 2011 Public Review Draft
88
References 1. Glassock RJ. Prophylactic anticoagulation in nephrotic syndrome: a clinical conundrum.. J. Am Soc Nephrol
2007; 18:2221-2225 2. Bellomo R, Atkins RC. Membranous nephropathy and thrombo-embolism: is prophylactic anticoagulation
warranted? Nephron 63:249-254, 1993 3. Sarasin FP, Schifferli JA. Prophylactic anticoagulation in nephrotic patients with idiopathic membranoius
nephropathy. Kidney Int 45:578-585, 1994

KDIGO® GN Guideline March 2011 Public Review Draft
89
CHAPTER 8: MEMBRANOPROLIFERATIVE GLOMERULONEPHRITIS
INTRODUCTION
This chapter makes treatment recommendations for MPGN in adults and children. The cost implications for global application of this guideline are addressed in Chapter 2.
8.1: Evaluation of MPGN 8.1.1: Evaluate patients with the histological (light microscopic) pattern of
MPGN for underlying diseases before considering a specific treatment regimen (see Table 8.1). (Not Graded)
BACKGROUND
MPGN is a histologic “pattern of injury” caused by many disorders (see Table 8.1) [1,2]. Patients commonly present with nephrotic syndrome, hypertension, glomerular hematuria, and progressive kidney dysfunction [1,2]. Reduction in the serum concentration of complement components (C3 and/or C4) is commonly, but not uniformly, observed [1,3].
MPGN can be further classified based on the extent and location of deposits of immunoglobulin and/or complement. Type I MPGN is associated with subendothelial and mesangial electron-dense deposits containing immunoglobulin and/or C3 [1,4,5], and is often due to an underlying chronic hepatitis C infection (see Chapter 9); type II MPGN with electron dense intramembranous deposits containing numerous complement components, but not immunoglobulin [1,5] and is now known as “dense-deposit disease”. It has a distinctive etiology based on inherited or acquired abnormalities of complement regulatory proteins [1,6]. Other rarer variants (type III and type IV MPGN) are also recognized based on abnormalities of the glomerular basement membrane and the location of electron-dense deposits. Additional variants are recognized based on deposition of C3 component of complement in glomeruli (in the absence of IgG deposits)—so-called C3 GN [1,6]. Treatment of MPGN is highly dependent on proper identification of underlying causes (see Table 8.1). In some patients C3 nephritic factor, an autoantibody to C3bBb, can be involved in the pathogenesis of type I, II, III, or C3 GN [7,8].
Idiopathic MPGN is defined by exclusion of any other identifiable cause in a patient with the pathological features of type I MPGN. Type I MPGN is uncommon in developed countries, but remains a relatively common cause of nephrotic syndrome in developing countries, especially those with a high burden of endemic infectious diseases [9].
RATIONALE
• Based on the heterogeneity of cause and pattern of histologic injury of MPGN, all patients with MPGN must be thoroughly evaluated for underlying diseases before

KDIGO® GN Guideline March 2011 Public Review Draft
90
classifying as idiopathic MPGN, and before any specific treatment decisions can be made.
Table 8.1. Underlying conditions associated with a membranoproliferative pattern of GN
Chronic infections (especially hepatitis C) Autoimmune diseases (especially LN) Monoclonal gammopathies (especially light-chain deposition disease and monoclonal IgG disease) Complement dysregulation (especially complement factor H deficiency) Chronic and healed thrombotic microangiopathies
GN, glomerulonephritis; LN, lupus nephritis.
8.2: Treatment of MPGN 8.2.1: We suggest that adults or children with idiopathic type I MPGN
accompanied by nephrotic syndrome and/or progressive decline of kidney function receive oral cyclophosphamide or MMF plus low-dose alternate-day or daily corticosteroids. (2D)
RATIONALE
• There is very low–quality evidence to suggest the benefit of an immunosuppressive agent plus corticosteroids in the treatment of idiopathic type I MPGN with nephrotic syndrome and/or deteriorating kidney function
MPGN is identified by exclusion of all other known causes of the MPGN pattern on kidney
biopsy. When there is a secondary MPGN, i.e., a defined cause for the MPGN pattern, treatment should be directed against that cause. Review of the evidence for the management of each of those conditions is outside the scope of this guideline.
Many of the early reports of treatment of idiopathic MPGN likely inadvertently included cases of secondary MPGN. Therefore, the results of these studies must be interpreted with great caution given today’s knowledge [1,2]. Truly idiopathic MPGN is now an uncommon condition, except in developing countries with a high endemic burden of infections. The few RCTs of treatment of idiopathic MPGN in children and adults have given inconsistent and largely inconclusive results [1,2]. Many of the reported trials have weak experimental design or are underpowered, and the evidence base underlying the recommendations for treatment of “idiopathic” MPGN is very weak. Early claims of benefit for a combination of aspirin and dipyridamole for adults with idiopathic MPGN were later rejected [10,11] and benefits remain in doubt [12,20]. The benefit of long-term alternate-day corticosteroid therapy for idiopathic MPGN in children was suggested by observational studies and a single RCT, but the results were equivocal; there have been no confirmatory RCTs[13-16]. The benefits of immunosuppressive therapy (cyclophosphamide or MMF) often combined with high-dose i.v. or oral steroids have never been demonstrated in RCTs. However, small, observational studies with short-term follow-up have suggested a benefit, mostly in subjects with a rapidly progressive course, often associated with extensive crescents, or in those with progressive kidney disease with persistence of severe nephrotic syndrome [17-25]. Publication bias might be operative in these reports.

KDIGO® GN Guideline March 2011 Public Review Draft
91
RESEARCH RECOMMENDATIONS
An RCT is needed to test corticosteroids in combination with an immunosuppressive agent such as cyclophosphamide, MMF, or rituximab in idiopathic MPGN in adults and children.
Please refer to Supplemental Tables 33-39 for additional data related to Chapter 8.
References 1. Glassock RJ. Membranoproliferative glomerulonephritis. In Treatment of Primary Glomerulonephritis, 2nd
Edition C. Ponticelli and R. Glassock, Editors. Oxford Medical Publications, Oxford. 2009. pgs. 375-398 2. Glassock RJ. Membranoproliferative Glomerulonephritis. In Evidence-Based Nephrology. DA Molony and
JC Craig, Editors. Wiley-Blackwell, Oxford.2009. pages 183-196 3. West CD, McAdams AJ, McConville JM, Davis NC, Holland NH. Hypocomplementemic and
normocomplementemic persistent (chronic) glomerulonephritis: Clinical and pathologic characteristics. J.Pediatr 1965 ;67:1089-1112
4. Holley KE and Donadio JV. Membranoproliferative glomerulonephritis. In Renal Pathology (with clinical and functional correlations), 2nd Edition, edited by CC Tisher and BM Brenner. JB Lippincott and Co, Philadelphia, 1994. pgs.294-329
5. Appel GB, Cook HT, Hageman G, Jennette JC, Kashgarian M, Kirschfink M, Lambris JD, Lanning L, Smoyer W, Tully HF, Tully SP, Walker P, Welsh M, Wurzner R, Zipfel PF. Membranoproliferative glomerulonephritis type II (dense deposit disease): an update. J Am Soc Nephrol 2005;16:1392-1403
6. Servais A, Fremeaux-Bacchi V, Salomon R, Lequintrec M, noel L-H, Knebelman B, Grunfeld J-P, Lesavre P, Fakhouri F. Mutations in complement regulatory genes, Factor H, I and CD46 and C3 nephritic factor predispose to membranoproliferative glomerulonephritis with isolated mesangial C3 deposits. J Am Soc Nephrol 2005;16:51A
7 Schwertz R; Rother U; Anders D; Gretz N; Scharer K; Kirschfink Complement analysis in children with idiopathic membranoproliferative glomerulonephritis: a long-term follow-up. Pediatr Allergy Immunol. 2001 (3):166-72.
8. Smith RJ, Alexander J, Barlow PN, et al New approaches to the treatment of dense deposit disease.. J Am Soc Nephrol 2007;18:2447-2456
9. Asinobi AO, Gbadegesin RA, Adeyemo AA, Akang EE, Arowolo FA, Abiola OA, Osinusi K. The predominance of membranoproliferative glomerulonephritis in childhood nephrotic syndrome in Ibadan, Nigeria West Afr J Med. 1999. ;18:203-206
10. Donadio JV, Offord KP. Reassessment of treatment results in membranoproliferative glomerulonephritis, with emphasis on life-table analysis. Am J Kidney Dis. 14: 445-451, 1989
11. Donadio JV, Anderson CF, Mitchell JC, Holley CE, Ilstrup DM, Fuster V, Chesebro JH. Membranoproliferative glomerulonephritis. A prospective clinical trial of platelet-inhibitor therapy. N Engl J Med 1984;31-1421-1426
12. Zauner I, Bohler J, Braun N, Grupp C, Herring P, Schollmeyer P. Effect of aspirin and dipyridamole on proteinuria in idiopathic membranoproliferative glomerulonephritis. Nephrol Dial Transplant 1994;9:619-622
13. Tarshish P, Bernsein J, Tobin JN, Edelmann CM. Treatement of mesangiocapillary glomerulonephritis with alternate-day prednisone—a report of the International Study of Kidney Disease in Children. Pediatr Nephrol 1992 ;6:123-130
14. McEnery PT, McAdams AJ, West CD. The effect of prednisone in a high-dose, alternate day regimen on the natural history of idiopathic membranoproliferative glomerulonephritis. Medicine (Baltimore) 1985;64:401-424

KDIGO® GN Guideline March 2011 Public Review Draft
92
15. McEnery PT. Membranoproliferative glomerulonephritis: The Cincinnati experience-cumulative renal survival from 1957-1989. J. Pediatr 1990;116:s109-114
16. Warady BA, Guggenheim SJ, Sedman A, Lum GM. Prednisone therapy of membranoproliferative glomerulonephritis in children. J. Pediatr 1985 ;107:702-707
17. Tiller D, Clarkson A, Mathew T. A prospective randomized trial fo the use of cyclophosphamide, dipyridamole and warfarin in membranous and membranoproliferative glomerulonephritis. In Proceedings of the 8th International Congress of nephrology. Edited by R. Robinson, R Glassock, C Tisher, T Andreoli and J Kokko. Karger, Basel, 1981. pgs. 345-351
18. Cattran DC, Cardella CJ, Roscoe JM, Charron, RC, Rance PC, Ritchie SM, Corey PN. Results of a controlled drug trial in membranoproliferative glomerulonephritis. Kidney Int 1985; 27:436-441
19. Bergstein JM, Andreoli SP. Response of type I membranoproliferative glomerulonephritis to pulse methyl prednisolone and alternate day prednisone therapy. Pediatr Nephrol. 1995;9:268-271
20. Emre S, Sirin A, Alpay H, Tanman F, Uysai V, Nayir A, Bilge I. Pulse methylprednisolone therapy in children with membranoproliferative glomerulonephritis. Acta Paediatr Jpn 1995;37:626-629
21. Chapman SJ, Cameron JS, Chantler C, Turner D. Treatment of mesangiocapillary glomerulonepthritis in children with combined immunosuppression and anti-coagulation. Arch Dis Child 1980;55:446-451
22. Faedda R, Satta A, Tanda F, Pirisi M, Bartole E. Immunosuppressive treatment of membranoproliferative glomerulonephritis. Nephron 1994;67:59-65
23. Choi MJ, Eustace JA, Giminez LF, Atta MG, Scheel PJ, Sothinathan R, Briggs WA. Mycophenolate mofetil treatment for primary glomerular diseases. Kidney Int 2002; 61:1098-1114
24. Jones G, Juszczak M, Kingdon E, Harber M, Sweny P, Burns A. Treatment of idiopathic membranoproliferative glomerulonephritis with mycophenolate mofetil and steroids. Nephrol Dial Transplant 2004;19:3160-3164
25. Levin A. Management of membranoproliferative glomerulonephritis: evidence-based recommendations Kidney Int 1999; (Supplement 70) s41-46

KDIGO® GN Guideline March 2011 Public Review Draft
93
CHAPTER 9: INFECTION-RELATED GLOMERULONEPHRITIS
9.1: For the following infection-related GN, we suggest appropriate treatment of the infectious disease and standard approaches to management of the kidney manifestations: (2D) • post-streptococcal GN; • infective endocarditis-related GN; • shunt nephritis.
INTRODUCTION
This chapter provides recommendations for the treatment of infection-associated GN, which may occur in association with bacterial, viral, fungal, protozoal, and helminthic infection (Table 9.1). The cost implications for global application of this guideline are addressed in Chapter 2.
9.1.1: Bacterial infection–related GN
BACKGROUND AND RATIONALE
The prototype for bacterial infection-related GN (also called postinfectious GN) is poststreptococcal GN, which most often occurs in children following a pharyngeal or cutaneous infection (impetigo) caused by a particular nephritogenic strain of Streptococci, and usually has a favorable outcome.
However, in the last decades the spectrum of postinfectious GN has changed. The incidence of poststreptococcal GN, particularly in its epidemic form, has progressively declined in industrialized countries. Recent series reported that streptococcal infections accounted for only 28-47% of acute GN, Staphylococcus aureus or Staphylococcus epidermidis being isolated in 12-24% of cases and Gram-negative bacteria in up to 22% of cases [1-3]. Bacterial endocarditis and shunt infections are also frequently associated with postinfectious GN. Moreover, the atypical postinfectious GN tends to affect mainly adults who are immunocompromised, e.g., in association with alcoholism, diabetes, and drug addiction. While spontaneous recovery within a few weeks is still the rule in children affected by the typical poststreptococcal GN, the prognosis in immunocompromised adults with postinfectious GN is significantly worse, with less than 50% in complete remission after a long follow-up [4].
9.1.2: Poststreptococcal GN
BACKGROUND AND RATIONALE
The diagnosis of poststreptococcal GN requires the demonstration of antecedent streptococcal infection in a patient who presents with acute GN. Nephritis may follow 7-15 days after streptococcal tonsillitis and 4-6 weeks after impetigo [5].

KDIGO® GN Guideline March 2011 Public Review Draft
94
The nature of the nephritogenic streptococcal antigen is still controversial [5-7]. Kidney biopsy is not indicated unless there are characteristics that make the diagnosis doubtful, or to assess prognosis and/or for potential therapeutic reasons. The kidney histology shows acute endocapillary GN with mesangial and capillary granular immune deposition.
The clinical manifestations of acute nephritic syndrome usually last less than 2 weeks. Less than 4 % of children with poststreptococcal GN have massive proteinuria, and occasionally a patient develops crescentic GN with rapidly progressive kidney dysfunction. The prognosis of the acute phase is excellent in children; however, in elderly patients, mortality in some series is as high as 20%.
Although the long term prognosis of poststreptococcal GN is debated, the incidence of ESRD in studies with 15 years of follow-up is less than 1%, with the exception being that long-term prognosis is poor in elderly patients who develop persistent proteinuria [4,5].
Streptococcal infection should be treated with penicillin, or erythromycin if the patient is allergic to penicillin, to resolve streptococcal infection and prevent the spread of the nephritogenic streptococcus among relatives or contacts. However, antibiotics are of little help for reversing GN, as the glomerular lesions induced by immune complexes are already established.
The management of acute nephritic syndrome, mainly in adults, requires hospital admission if features of severe hypertension or congestive heart failure are present. Hypertension and edema usually subside after diuresis is established. Adult patients persisting with urinary abnormalities beyond 6 months, especially if proteinuria >1 g/d, should receive ACEi or ARBs, such as used in other proteinuric glomerular diseases (see Chapter 1). The long-term prognosis is worse in those, mainly adults, that persist with proteinuria after 6 months [8].
Pulses of i.v. methylprednisolone can be considered in patients with extensive glomerular crescents and rapidly progressive GN, based on extrapolation of other rapidly progressive and crescentic GNs, although there is no evidence from RCTs.
RESEARCH RECOMMENDATIONS
• An RCT is needed to evaluate the treatment of crescentic poststreptococcal GN with corticosteroids.
• Research is needed to determine the nature of the streptococcal antigen, as a basis for developing immunoprophylactic therapy.
9.1.3: GN associated with infective endocarditis
BACKGROUND AND RATIONALE
The natural history of GN associated with infective endocarditis has been significantly altered with the changing epidemiology of the disorder, and with the use of antibiotics [8-11].

KDIGO® GN Guideline March 2011 Public Review Draft
95
In USA, infective endocarditis is diagnosed in approximately 40 cases per million every year, and the disease is increasingly frequent in elderly individuals and in patients with no underlying heart disease. i.v. drug usage, prosthetic heart valves, and structural heart disease are risk factors. Staphylococcus aureus has replaced Streptococcus viridans as the leading cause of infective endocarditis. The incidence of GN associated with Staphylococcus aureus endocarditis ranges from 22% to 78%, the highest risk being among i.v. drug users. Focal and segmental proliferative GN, often with focal crescents, is the most typical finding. Some patients may exhibit a more diffuse proliferative endocapillary lesion with or without crescents [8-11].
The immediate prognosis of the GN is good, and is related to the prompt eradication of the infection, using appropriate antibiotics for 4-6 weeks.
RESEARCH RECOMMENDATIONS
• Multicenter studies are needed to determine the incidence, prevalence, and long-term prognosis of infective endocarditis–related GN.
9.1.4: Shunt nephritis
BACKGROUND AND RATIONALE
Shunt nephritis is an immune complex–mediated GN that develops as a complication of chronic infection on ventriculoatrial or ventriculojugular shunts inserted for the treatment of hydrocephalus [12].
The diagnosis is based on clinical evidence of kidney disease (most commonly, microscopic hematuria and proteinuria, frequently in the nephrotic range, occasionally elevated SCr and hypertension) with prolonged fever or signs of chronic infection, in a patient with a ventriculovascular shunt implanted for treatment of hydrocephalus. The histologic findings are typically type 1 MPGN, with granular deposits of IgG, IgM, and C3, and electron-dense mesangial and subendothelial deposits.
The renal outcome of shunt nephritis is good if there is early diagnosis and treatment of the infection. Ventriculovascular shunts may become infected in about 30% of cases. GN may develop in 0.7-2% of the infected ventriculovascular shunts in an interval of time ranging from 2 months to many years after insertion. The infected organisms are usually Staphylococcus epidermidis or Staphylococcus aureus. In contrast to ventriculovascular shunts, ventriculoperitoneal shunts are rarely complicated with GN.
A late diagnosis, resulting in delays in initiating antibiotic therapy and in removing the shunt, results in a worse renal prognosis.

KDIGO® GN Guideline March 2011 Public Review Draft
96
RESEARCH RECOMMENDATIONS
• Multicenter observational studies are needed to determine the incidence, prevalence, and long-term prognosis of shunt nephritis.
Table 9.1: Underlying conditions associated with a membranoproliferative pattern of GN
Bacterial Mycobacterium leprae, M. tuberculosis Treponema pallidum Salmonella typhi, S. paratyphi, S. typhimurium Streptococcus pneumoniae, S. virdans,S. pyogenes Staphyloccoccus aureus, S. epidermidis, S. albus Leptospira species* Yersinia enterocolitica* Neisseria meningitidis, Neisseria gonorrhoeae* Corynebacterium diphtheriae* Coxiella burnettii* Brucella abortus* Listeria monocytogenes*
Viral Hepatitis B and C Human immunodeficiency virus Epstein-Barr virus Coxsackie B ECHO virus Cytomegalovirus Varicella zoster Mumps Rubella Influenza
Fungal Histoplasma capsulatum* Candida* Coccidiodes immitis* Protozoal Plasmodium malariae, P. falciparum Leishmania donovani Toxoplasma gondii Trypanosoma cruzei, T. brucei Toxocara canis* Strongloides stercoralis*
Helminthic Schistosoma mansoni, S. japonicum, S. haematobium Wuchereria bancrofti Brugia malayi Loa loa Onchocerca volvulus Trichinella spiralis*
ECHO, enteric cytopathic human orphan; GN, glomerulonephritis. *Only case reports documented
References
1. Montseny JJ, Meyrier A, Kleinknecht D, Callard P. The current spectrum of infectious glomerulonephritis. Experience with 76 patients and review of the literature. Medicine 1995; 74:63-73
2. Moroni G, Pozzi C, Quaglini S, et al: Long term prognosis of diffuse proliferative glomerulonephritis associated with infection in adults. Nephrol Dial Transplant 2002; 17:1204-1211
3. Nasr SH, Markowitz GS, Stokes MB et al: Acute postinfectious glomerulonephritis in the modern era. Experience with 86 adults and review of the literature. Medicine 2008; 87:21-31
4. Moroni G, Ponticelli C: Acute post-infectious glomerulonephritis. In Ponticelli C, Glassock R: treatment of primary glomerulonephritis, Oxford University Press . 2009
5. Rodríguez-Iturbe B, Musser JM : The current state of poststreptococcal glomerulonephritis. J Am Soc Nephrol 2008;19:1855-1864.
6. Yoshizawa N, Yamakami K, Fujino M et al: Nephritis associate plasmin receptor and acute poststreptococcal glomerulonephritis: characterization of the antigen and associated immune response. J Am Soc Nephrol 2004; 15:1785-1793

KDIGO® GN Guideline March 2011 Public Review Draft
97
7. Batsford SR, Mezzano S, Mihatsch M, Schiltz E, Rodríguez-Iturbe B.: Is the nephritogenic antigen in poststreptococcal glomerulonephritis pyrogenic exotoxin B (SPE B) or GAPDH? Kidney Int. 2005;68:1120-9.
8. Baldwin D: Poststreptococcal glomerulonephritis. A progressive disease? American Journal Medicine 1977; 62: 1-11
9. Guidelines on the prevention, diagnosis, and treatment of infective endocarditis (new version 2009): the Task Force on the Prevention, Diagnosis, and Treatment of Infective Endocarditis of the European Society of Cardiology (ESC). Eur Heart J 2009;30(19):2369
10. Hoen B et al: Association pour l’Etude et la Prevention de l’Endocardite Infectieuse (AEPEI) Study Group . Changing profile of infective endocarditis: results of a 1 year survey in France. JAMA 2002;288-:75 PMID 12090865
11. Koya D et al: Succesful recovery of infective endocarditis -induced rapidly progressive glomerulonephritis by steroid therapy combined with antibiotics: a case report BMC Nephrol 2004;21:18
12. Iwata Y, Ohta S, Kawai K, Yamahana J, Sugimori H, Ishida Y, Saito K, Miyamori T, Futami K, Arakawa Y, Hirota Y, Wada T, Yokoyama H, Yoshida K. : Shunt nephritis with positive titers of ANCA specific for proteinase 3. Am J Kidney Dis. 2004;43(5):e11-6.
9.2: Hepatitis C virus (HCV) infection–related GN (Please also refer to the published KDIGO Clinical Practice Guidelines for the
Prevention, Diagnosis, Evaluation, and Treatment of Hepatitis C in Chronic Kidney Disease[14].) 9.2.1: We suggest, for HCV-infected patients with CKD Stages 1 and 2,
combined antiviral treatment using pegylated interferon and ribavirin as in the general population. (2C) [based on KDIGO HCV Recommendation 2.2.1] 9.2.1.1: Titrate ribavirin dose according to patient tolerance. (Not
Graded) 9.2.2: We suggest, for HCV-infected patients with CKD Stages 3, 4, and 5 not
yet on dialysis, monotherapy with pegylated interferon, with doses adjusted to the level of kidney function. (2D) [based on KDIGO HCV Recommendation 2.2.2]
9.2.3: We suggest that patients with HCV and mixed cryoglobulinemia (IgG/IgM) with nephrotic proteinuria or evidence of progressive kidney disease or an acute flare of cryoglobulinemia be treated with one of the following options (not necessarily in the order below), in conjunction with i.v. methylprednisolone and with antiviral therapy: (2D) • cyclophosphamide; • plasma exchange; • rituximab.
BACKGROUND
HCV infection is a major public health problem, with an estimated 130-170 million people infected worldwide [1-3]. HCV frequently causes extrahepatic manifestations, including mixed cryoglobulinemia, lymphoproliferative disorders, Sjögren’s syndrome, and kidney disease. A major concern is the lack of safe and effective drugs to treat HCV-infected patients with CKD

KDIGO® GN Guideline March 2011 Public Review Draft
98
[4]. Unfortunately, there are no large-scale clinical trials in patients with HCV-associated kidney disease; thus, evidence-based treatment recommendations cannot be made in this patient population. However, we have extrapolated HCV treatment from the non-CKD population, with the appropriate and necessary dose adjustments.
Kidney involvement due to HCV is most commonly associated with type II cryoglobulinemia, and is clinically manifested by proteinuria, microscopic hematuria, hypertension, and mild to moderate kidney impairment [5,6].
On kidney biopsy, a type I MPGN pattern of injury is the most common pathological finding [7]. Vasculitis of the small- and medium-sized renal arteries can also be present. Immunofluorescence usually demonstrates deposition of IgM, IgG, and C3 in the mesangium and capillary walls. On electron microscopy, subendothelial immune complexes are usually seen and may have an organized substructure suggestive of cryoglobulin deposits [6, 8]. Besides MPGN, other forms of glomerular disease have been described in patients with HCV, including IgAN, MN, postinfectious GN, thrombotic microangiopathies, FSGS, and fibrillary and immunotactoid GN [6-12].
Patients with type II cryoglobulinemia (mixed polyclonal IgG and monoclonal IgM [Rheumatoid-factor positive] cryoglobulins) should be tested for HCV. Patients with proteinuria and cryoglobulinemia should be tested for HCV RNA even in the absence of clinical and/or biochemical evidence of liver disease. Similarly, HCV-infected patients should be tested at least annually for proteinuria, hematuria, and eGFR to detect possible HCV-associated kidney disease. Practice guidelines for treatment of HCV infection in general have been recently published [13]. For detailed information regarding treatment of HCV-mediated kidney disease the reader is also referred to the recently published KDIGO Clinical Practice Guidelines for the Prevention, Diagnosis, Evaluation, and Treatment of Hepatitis C in Chronic Kidney Disease [14].
RATIONALE
• There is low-quality evidence to recommend treatment of HCV-associated GN. Treatment should be focused on reducing or eliminating HCV replication, and reducing the formation and glomerular deposition of HCV-containing immune complexes (including cryoglobulins).
• There is low-quality evidence to recommend dose adjustments for interferon and ribavirin based on level of kidney function.
• There is very low–quality evidence to suggest that patients with HCV-associated GN and severe kidney manifestations require additional treatment with immunosuppression and/or corticosteroids and/ or plasma exchange.
The best long-term prognostic indicator of HCV-associated GN is sustained virologic
response (defined as HCV RNA clearance from serum) for at least 6 months after cessation of therapy. In patients with normal kidney function, this aim can be best achieved by the use of pegylated interferon-α-2a/2b in combination with ribavirin, which results in sustained virological response rate of 45-50% in genotypes 1 and 4, and 70-80% in genotypes 2 and 3 in HVC-monoinfected patients. This represents the current standard of care for HCV infection [13,14].

KDIGO® GN Guideline March 2011 Public Review Draft
99
Treatment regimens for HCV-associated GN and the doses of individual agents will vary with the severity of the kidney disease. No dose adjustment is needed for patients with eGFR >60 ml/min [15-17].
There is a paucity of information regarding treatment of HCV-infected patients with GFR <60 ml/min but not yet on dialysis (CKD stages 3-5). The suggested doses (based on expert opinion, not evidence) are pegylated interferon-α-2b, 1 μg/kg subcutaneously once weekly or pegylated interferon-α-2a, 135 μg subcutaneously once weekly, together with ribavirin 200-800 mg/d in two divided doses, starting with the low dose and increasing gradually, as long as side-effects are minimal and manageable (see Table 9.2). Hemolysis secondary to ribavirin very commonly limits its dosage or prevents its use in patients with CKD.
Table 9.2. Treatment of HCV infection according to stages of CKD
eGFR (ml/min) Recommended treatment >60 A
30-59 B 15-29 C <15 C
Dialysis C A: Routine combination therapy according to viral genotype. B: Pegylated interferon-α 2b, 1 μg/kg subcutaneously once
weekly, or pegylated interferon-α 2a, 135 μg subcutaneously once weekly plus ribavirin, 200-800 mg/d in two divided doses, starting with low dose and increasing gradually.
C: Controversial: Standard interferon-α (2a or 2b) 3 mU three times weekly, or pegylated interferon-α 2b, 1 μg/kg/wk, or pegylated interferon-α 2a, 135 μg/kg/wk ± ribavirin in markedly reduced daily dose.
eGFR, estimated glomerular filtration rate (MDRD formula); HCV, hepatitis C virus; CKD, chronic kidney disease.
Adapted with permission. [13]
Monotherapy with interferon-α has been used in cryoglobulinemic GN with complete
clearance of HCV RNA and improved kidney function; however, recurrence of viremia and relapses of kidney disease were universally observed after interferon was discontinued [18,19]. Subsequent studies with interferon-α monotherapy [19-22] have yielded mixed results [19]. Treatment with interferon-α may exacerbate cryoglobulinemic vasculitis [23,24]. Thus, it is recommended that interferon -α should be started after the acute flare has been controlled with immunosuppressive agents [25].
Better outcomes have been achieved by combined use of interferon -α with ribavirin [26-29] and pegylated interferon with ribavirin [25, 29-33]. In a recent meta-analysis of controlled clinical trials comparing the efficacy and safety of antiviral vs. immunosuppressive therapy (corticosteroids alone or in combination with cyclophosphamide) in patients with HCV-associated GN, proteinuria decreased more (odds ratio 3.86) after interferon therapy (3 MU thrice weekly for at least 6 months) [34]. However, both treatments failed to significantly improve kidney function. KDIGO guidelines for treatment of viral hepatitis in patients with kidney disease recently published suggest that patients with moderate proteinuria and slowly

KDIGO® GN Guideline March 2011 Public Review Draft
100
progressive kidney disease can be treated with a 12-month course of standard interferon-α or pegylated interferon-α-2a with dose adjusted as described below plus ribavirin, with or without erythropoietin support, depending on the level of hemoglobin [14]. Ribavirin dose needs to be titrated according to patient tolerance; it is not recommended for patients with an eGFR <50 ml/min per 1.73 m2.
There is very low–quality evidence that patients with nephrotic-range proteinuria and/or rapidly progressive kidney failure or an acute flare of cryoglobulinemia, should receive additional therapy with either plasma exchange (3 l of plasma thrice weekly for 2-3 weeks), rituximab (375 mg/m2 once a week for 4 weeks), or cyclophosphamide (2 mg/kg/d for 2-4 months) plus i.v. methylprednisolone 0.5-1 g/d for 3 days [14]. There are no comparative data to favour any one of these three additional therapies. Corticosteroids may lead to increases in HCV viral load [35,36].
Case reports have suggested remarkable reduction in proteinuria and stabilization of kidney function in response to rituximab in patients with cryoglobulinemic vasculitis [37, 38]. Although HCV viremia increased modestly in some patients, it remained unchanged or decreased in others and the overall treatment was considered safe [39]. Observations in 16 patients with severe refractory HCV-related cryoglobulinemia vasculitis treated with rituximab in combination with pegylated interferon-α-2b and ribavirin also showed good response [40]. Symptoms usually reappear with reconstitution of peripheral B cells. The long-term safety of multiple courses of rituximab in patients with HCV is unknown.
There is a paucity of controlled studies available in HCV-associated GN; most studies are retrospective analyses with small sample sizes. Most of the available evidence comes from studies of patients with significant proteinuria, hematuria, or reduced kidney function.
RESEARCH RECOMMENDATIONS
• Epidemiologic studies are needed to determine: o the prevalence and types of glomerular lesions in HCV-infected patients; o whether there are true associations between HCV infection and GN other than
MPGN (e.g., IgAN). • An RCT is needed to evaluate corticosteroids plus cyclophosphamide in addition to
antiviral therapy in HCV-associated GN. • An RCT is needed to evaluate rituximab in addition to antiviral therapy in HCV-
associated GN.
References 1. Williams R: Global challenges in liver disease. Hepatology 44:521-526, 2006 2. Poynard T, Yuen MF, Ratziu V, Lai CL: Viral hepatitis C. Lancet 362:2095-2100, 2003 3. Alter MJ: Epidemiology of hepatitis C virus infection. World J Gastroenterol 13:2436-2441, 2007 4. Perico N, Cattaneo D, Bikbov B, Remuzzi G: Hepatitis C infection and chronic renal diseases. Clin J Am Soc
Nephrol 4:207-220, 2009 5. Johnson RJ, Gretch DR, Yamabe H, Hart J, Bacchi CE, Hartwell P, Couser WG, Corey L, Wener MH, Alpers
CE, et al.: Membranoproliferative glomerulonephritis associated with hepatitis C virus infection. N Engl J Med 328:465-470, 1993

KDIGO® GN Guideline March 2011 Public Review Draft
101
6. Meyers CM, Seeff LB, Stehman-Breen CO, Hoofnagle JH: Hepatitis C and renal disease: an update. Am J Kidney Dis 42:631-657, 2003
7. Roccatello D, Fornasieri A, Giachino O, Rossi D, Beltrame A, Banfi G, Confalonieri R, Tarantino A, Pasquali S, Amoroso A, Savoldi S, Colombo V, Manno C, Ponzetto A, Moriconi L, Pani A, Rustichelli R, Di Belgiojoso GB, Comotti C, Quarenghi MI: Multicenter study on hepatitis C virus-related cryoglobulinemic glomerulonephritis. Am J Kidney Dis 49:69-82, 2007
8. D'Amico G: Renal involvement in hepatitis C infection: cryoglobulinemic glomerulonephritis. Kidney Int 54:650-671, 1998
9. Markowitz GS, Cheng JT, Colvin RB, Trebbin WM, D'Agati VD: Hepatitis C viral infection is associated with fibrillary glomerulonephritis and immunotactoid glomerulopathy. J Am Soc Nephrol 9:2244-2252, 1998
10. Sabry A, A EA, Sheashaa H, El-Husseini A, Mohamed Taha N, Elbaz M, Sobh M: HCV associated glomerulopathy in Egyptian patients: clinicopathological analysis. Virology 334:10-16, 2005
11. Kamar N, Izopet J, Alric L, Guilbeaud-Frugier C, Rostaing L: Hepatitis C virus-related kidney disease: an overview. Clin Nephrol 69:149-160, 2008
12. Arase Y, Ikeda K, Murashima N, Chayama K, Tsubota A, Koida I, Suzuki Y, Saitoh S, Kobayashi M, Kobayashi M, Kobayashi M, Kumada H: Glomerulonephritis in autopsy cases with hepatitis C virus infection. Intern Med 37:836-840, 1998
13. Ghany MG, Strader DB, Thomas DL, Seeff LB: Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 49:1335-1374, 2009
14. KDIGO clinical practice guidelines for the prevention, diagnosis, evaluation, and treatment of hepatitis C in chronic kidney disease. Kidney Int Suppl:S69-77, 2008
15. Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, Jr., Haussinger D, Diago M, Carosi G, Dhumeaux D, Craxi A, Lin A, Hoffman J, Yu J: Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 347:975-982, 2002
16. Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht JK: Peginterferon alfa-2b plus ribavirin compared to interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958-965, 2001
17. Hadziyannis SJ, Sette H, Jr., Morgan TR, Balan V, Diago M, Marcellin P, Ramadori G, Bodenheimer H, Jr., Bernstein D, Rizzetto M, Zeuzem S, Pockros PJ, Lin A, Ackrill AM: Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med 140:346-355, 2004
18. Johnson RJ, Gretch DR, Couser WG, Alpers CE, Wilson J, Chung M, Hart J, Willson R: Hepatitis C virus-associated glomerulonephritis. Effect of alpha-interferon therapy. Kidney Int 46:1700-1704, 1994
19. Misiani R, Bellavita P, Fenili D, Vicari O, Marchesi D, Sironi PL, Zilio P, Vernocchi A, Massazza M, Vendramin G, et al.: Interferon alfa-2a therapy in cryoglobulinemia associated with hepatitis C virus. N Engl J Med 330:751-756, 1994
20. Cresta P, Musset L, Cacoub P, Frangeul L, Vitour D, Poynard T, Opolon P, Nguyen DT, Golliot F, Piette JC, Huraux JM, Lunel F: Response to interferon alpha treatment and disappearance of cryoglobulinaemia in patients infected by hepatitis C virus. Gut 45:122-128, 1999
21. Mazzaro C, Panarello G, Carniello S, Faelli A, Mazzi G, Crovatto M, Baracetti S, Nascimben F, Zorat F, Pozzato G, Faccini L, Campanacci L: Interferon versus steroids in patients with hepatitis C virus-associated cryoglobulinaemic glomerulonephritis. Dig Liver Dis 32:708-715, 2000
22. Komatsuda A, Imai H, Wakui H, Hamai K, Ohtani H, Kodama T, Oyama Y, Miura AB, Nakamoto Y: Clinicopathological analysis and therapy in hepatitis C virus-associated nephropathy. Intern Med 35:529-533, 1996
23. Cid MC, Hernandez-Rodriguez J, Robert J, del Rio A, Casademont J, Coll-Vinent B, Grau JM, Kleinman HK, Urbano-Marquez A, Cardellach F: Interferon-alpha may exacerbate cryoblobulinemia-related ischemic manifestations: an adverse effect potentially related to its anti-angiogenic activity. Arthritis Rheum 42:1051-1055, 1999

KDIGO® GN Guideline March 2011 Public Review Draft
102
24. Suzuki T, Yonemura K, Miyaji T, Suzuki H, Takahira R, Fujigaki Y, Fujimoto T, Hishida A: Progressive renal failure and blindness due to retinal hemorrhage after interferon therapy for hepatitis C virus-associated membranoproliferative glomerulonephritis. Intern Med 40:708-712, 2001
25. Garini G, Allegri L, Lannuzzella F, Vaglio A, Buzio C: HCV-related cryoglobulinemic glomerulonephritis: implications of antiviral and immunosuppressive therapies. Acta Biomed 78:51-59, 2007
26. Garini G, Allegri L, Carnevali L, Catellani W, Manganelli P, Buzio C: Interferon-alpha in combination with ribavirin as initial treatment for hepatitis C virus-associated cryoglobulinemic membranoproliferative glomerulonephritis. Am J Kidney Dis 38:E35, 2001
27. Rossi P, Bertani T, Baio P, Caldara R, Luliri P, Tengattini F, Bellavita P, Mazzucco G, Misiani R: Hepatitis C virus-related cryoglobulinemic glomerulonephritis: long-term remission after antiviral therapy. Kidney Int 63:2236-2241, 2003
28. Bruchfeld A, Lindahl K, Stahle L, Soderberg M, Schvarcz R: Interferon and ribavirin treatment in patients with hepatitis C-associated renal disease and renal insufficiency. Nephrol Dial Transplant 18:1573-1580, 2003
29. Sabry AA, Sobh MA, Sheaashaa HA, Kudesia G, Wild G, Fox S, Wagner BE, Irving WL, Grabowska A, El-Nahas AM: Effect of combination therapy (ribavirin and interferon) in HCV-related glomerulopathy. Nephrol Dial Transplant 17:1924-1930, 2002
30. Cacoub P, Saadoun D, Limal N, Sene D, Lidove O, Piette JC: PEGylated interferon alfa-2b and ribavirin treatment in patients with hepatitis C virus-related systemic vasculitis. Arthritis Rheum 52:911-915, 2005
31. Mazzaro C, Zorat F, Caizzi M, Donada C, Di Gennaro G, Maso LD, Carniello G, Virgolini L, Tirelli U, Pozzato G: Treatment with peg-interferon alfa-2b and ribavirin of hepatitis C virus-associated mixed cryoglobulinemia: a pilot study. J Hepatol 42:632-638, 2005
32. Saadoun D, Resche-Rigon M, Thibault V, Piette JC, Cacoub P: Antiviral therapy for hepatitis C virus--associated mixed cryoglobulinemia vasculitis: a long-term followup study. Arthritis Rheum 54:3696-3706, 2006
33. Alric L, Plaisier E, Thebault S, Peron JM, Rostaing L, Pourrat J, Ronco P, Piette JC, Cacoub P: Influence of antiviral therapy in hepatitis C virus-associated cryoglobulinemic MPGN. Am J Kidney Dis 43:617-623, 2004
34. Fabrizi F, Bruchfeld A, Mangano S, Dixit V, Messa P, Martin P: Interferon therapy for HCV-associated glomerulonephritis: meta-analysis of controlled trials. Int J Artif Organs 30:212-219, 2007
35. Charles ED, Dustin LB: Hepatitis C virus-induced cryoglobulinemia. Kidney Int 76:818-824, 2009 36. Koziolek MJ, Scheel A, Bramlage C, Groene HJ, Mueller GA, Strutz F: Effective treatment of hepatitis C-
associated immune-complex nephritis with cryoprecipitate apheresis and antiviral therapy. Clin Nephrol 67:245-249, 2007
37. Cacoub P, Delluc A, Saadoun D, Landau DA, Sene D: Anti-CD20 monoclonal antibody (rituximab) treatment for cryoglobulinemic vasculitis: where do we stand? Ann Rheum Dis 67:283-287, 2008
38. Ahmed MS, Wong CF: Should rituximab be the rescue therapy for refractory mixed cryoglobulinemia associated with hepatitis C? J Nephrol 20:350-356, 2007
39. Sansonno D, De Re V, Lauletta G, Tucci FA, Boiocchi M, Dammacco F: Monoclonal antibody treatment of mixed cryoglobulinemia resistant to interferon alpha with an anti-CD20. Blood 101:3818-3826, 2003
40. Saadoun D, Resche-Rigon M, Sene D, Perard L, Karras A, Cacoub P: Rituximab combined with Peg-interferon-ribavirin in refractory hepatitis C virus-associated cryoglobulinaemia vasculitis. Ann Rheum Dis 67:1431-1436, 2008

KDIGO® GN Guideline March 2011 Public Review Draft
103
9.3: Hepatitis B virus (HBV) infection–related GN 9.3.1: We recommend that patients with HBV infection and GN receive
treatment with IFN-α or with nucleoside analogues as recommended for the general population by standard clinical practice guidelines for HBV infection (see Table 9.3). (1C)
9.3.2: We recommend that the dosing of these antiviral agents be adjusted to the degree of kidney function. (1C)
BACKGROUND
Approximately one-third of the world’s population has serological evidence of past or present infection with HBV, and 350 million people are chronically infected, making it one of the most common human pathogens [1,2]. The spectrum of disease and natural history of chronic HBV infection is diverse and variable, ranging from a low viremic inactive carrier state to progressive chronic hepatitis, which may evolve to cirrhosis and hepatocellular carcinoma. It is not possible to predict which patients with HBV infection are more likely to develop kidney disease [3].
HBV-associated patterns of GN include MN, MPGN, FSGS and IgAN. MN is the most common form of HBV-mediated GN, especially in children. The diagnosis of HBV-mediated GN requires detection of the virus in the blood and the exclusion of other causes of glomerular disease. In children, HBV-mediated GN has a favorable prognosis, with high spontaneous remission rate. In adults, HBV-mediated GN is usually progressive. Patients with nephrotic syndrome and abnormal liver function tests have an even worse prognosis, with >50% progressing to ESRD in the short term [4]. There are no RCT studies on the treatment of HBV-mediated GN, so evidence-based treatment recommendations cannot be made. Clinical practice guidelines on the management of chronic hepatitis B have been recently published in Europe and in the USA, but do not include specific recommendations on HBV-mediated kidney disease [1,2].
RATIONALE
• Treatment of HBV-associated GN with interferon or nucleoside analogues is indicated.
Several drugs are now available for the treatment of chronic HBV infection (see Table 9.3).
The efficacy of these drugs has been assessed in an RCT at 1 year (2 years with telbivudine). Longer-term follow-up (up to 5 years) are available for lamivudine, adefovir, entecavir, telbivudine, and tenofovir in patient subgroups [1]. However, there are no data to indicate the effect of these treatments for HBV infection on the natural history of HBV-related GN. Treatment of patients with HBV infection and GN should be conducted according to standard clinical practice guidelines for HBV infection.

KDIGO® GN Guideline March 2011 Public Review Draft
104
Table 9.3. Dosage adjustment of drugs for HBV infection according to kidney function
(endogenous CrCl)
Drug CrCl >50 (ml/min) 30< CrCl <50 (ml/min)
10< CrCl <30 (ml/min) CrCl <10 (ml/min)
Lamivudine 100 mg p.o. q.d. 100 mg first dose then 50 mg p.o. q.d.
100 mg first dose then 25 mg p.o. q.d.
35 mg first dose then 15 mg p.o. q.d.a
Adefovir 10mg p.o. q.d. 10 mg p.o. every 48 hours
10 mg po every 72 hours
No dosing recommended
Entecavir 0.5 mg p.o. q.d. 0.25 mg p.o. q.d. 0.15 mg p.o. q.d. 0.05 mg p.o. q.d. Entecavir in lamivudine-refractory patients
1 mg p.o. q.d. 0.5 mg p.o. q.d. 0.3 mg p.o. q.d. 0.1 mg p.o. q.d.
Telbivudine 600 mg p.o. q.d. 600 mg p.o. every 48 hours
600 mg p.o. every 72 hours
600 mg p.o. every 96 hours
Tenofovir 300 mg p.o. q.d. 300 mg p.o. q.d every 48 hours
300 mg p.o. q.d every 72-96 hours 300 mg p.o. q.w.
CrCl, creatinine clearance; HBV, hepatitis B virus; p.o., orally; q.d., every day; q.w., once a week. aLamivudine should be dosed at 35 mg initially and then 10 mg daily for patients with CrCl <15 ml/min. Adapted with permission. [3,5]
The heterogeneity of patients with HBV infection (e.g., degree of liver function impairment, extent of extrahepatic involvement) creates substantial complexity in establishing treatment guidelines in patients with HBV-mediated kidney disease.
RESEARCH RECOMMENDATIONS
• RCTs are needed to establish the most effective antiviral treatment regimen in modifying the progression of HBV-associated GN. Studies will need to account for the extrarenal disease involvement, as well as evaluate varying drug combinations, including timing and duration of therapy.
• RCTs in children should be evaluated separately in view of the higher rate of spontaneous remission in HBV-associated GN.
References 1. European Association for The Study Of The L: EASL Clinical Practice Guidelines: management of chronic
hepatitis B. J Hepatol 50:227-242, 2009 2. Sorrell MF, Belongia EA, Costa J, Gareen IF, Grem JL, Inadomi JM, Kern ER, McHugh JA, Petersen GM,
Rein MF, Strader DB, Trotter HT: National Institutes of Health Consensus Development Conference Statement: management of hepatitis B. Ann Intern Med 150:104-110, 2009
3. Olsen SK, Brown RS, Jr.: Hepatitis B treatment: Lessons for the nephrologist. Kidney Int 70:1897-1904, 2006
4. Appel G: Viral infections and the kidney: HIV, hepatitis B, and hepatitis C. Cleve Clin J Med 74:353-360, 2007
5. Lok AS, McMahon BJ: Chronic hepatitis B: update 2009. Hepatology 50:661-662, 2009

KDIGO® GN Guideline March 2011 Public Review Draft
105
9.4: Human Immunodeficiency virus (HIV) infection–related glomerular disorders 9.4.1: We recommend that antiretroviral therapy be initiated in all patients
with biopsy-proven HIV-associated nephropathy, regardless of CD4 count. (1B)
BACKGROUND
Approximately 5 million people a year are infected with HIV worldwide [1]. Kidney disease is a relatively frequent complication in patients infected with HIV.
Human immunodeficiency virus–associated nephropathy (HIVAN) is the most common cause of CKD in patients with HIV-1, and is particularly common in patients of African descent [2,3]. Untreated, HIV-associated nephropathy rapidly progresses to ESRD. Typical HIVAN pathology includes FSGS, often with a collapsing pattern, accompanied by microcystic change in tubules. There are usually many tubuloreticular structures seen on electron microscopy. In addition to HIVAN, a number of other HIV-associated kidney diseases have been described [2,4,5]. In patients with HIV, proteinuria and/or decreased kidney function is associated with increased mortality and worse outcomes [6]. Data from a number of RCTs suggest that highly active antiretroviral therapy (HAART) is beneficial in both preservation and improvement of kidney function in patients with HIV [7-9]. Patients with kidney dysfunction at start of HAART have the most dramatic improvements in kidney function [10,11]. A decrease in HIV viral load during HAART is associated with kidney function improvement, while an increase in viral load is associated with worsening kidney function [12-14].
RATIONALE
• There is low-quality evidence to suggest a kidney biopsy is necessary to define the specific type of kidney diseases present in patients with HIV infection.
• HAART may be effective in HIVAN, but it is not effective in other GN associated with HIV infection.
Causes of kidney disease, other than HIVAN, that occur in patients with HIV infection
include diabetic nephropathy, thrombotic microangiopathies, cryoglobulinemia, immune complex GN, an SLE-like GN, or amyloidosis (see Table 9.4) [4,5,15,16]. More than a third of the patients with HIV who underwent a kidney biopsy had diabetic nephropathy; or MN, MPGN, IgAN, or another pattern of immune-complex GN [5,17]. In patients with HIV infection, many of these pathologies can mimic HIVAN, but each condition requires a different therapy [2,4,5,18]. Studies in HIV-infected patients with kidney disease from Africa showed a high prevalence of HIVAN, but other forms of GN and interstitial nephritis were also present (see Table 9.4) [19,20]. Cohen and Kimmel recently reviewed the rationale for a kidney biopsy in the diagnosis of HIV-associated kidney disease [2,21].

KDIGO® GN Guideline March 2011 Public Review Draft
106
Table 9.4. The spectrum of kidney disease in HIV-infected patients
• HIVAN-collapsing FSGS • Arterionephrosclerosis • Immune-complex GN –MPGN pattern of injury –Lupus-like GN • Idiopathic FSGS • HCV and cryoglobulinemia • Thrombotic microangiopathies • Membranous nephropathy –HBV-mediated –Malignancy • Minimal-change nephropathy • IgAN • Diabetic nephropathy • Postinfectious GN –Infectious endocarditis –Other infections: Candida, Cryptococcus • Amyloidosis • Chronic pyelonephritis • Acute or chronic interstitial nephritis • Crystal nephropathy –Indinavir, atazanavir, i.v. acyclovir, sulfadiazine • Acute tubular necrosis • Proximal tubulopathy (Fanconi syndrome) –Tenofovir FSGS, focal segmental glomerulosclerosis; GN, glomerulonephritis; HBV, hepatitis B virus;
HCV, hepatitis C virus; HIVAN, human immunodeficiency virus–associated nephropathy; IgAN, immunoglobulin A nephropathy; MPGN, mesangial proliferative glomerulonephritis.
Observational studies and data from uncontrolled or retrospective studies [7,9,22-25] and
from an RCT [8] suggest that HAART (defined as combination therapy with three or more drugs) is beneficial in both preservation and improvement of kidney function in patients with HIVAN. Since the introduction of HAART in the 1990s, there has also been a substantial reduction in the incidence of HIVAN [26]. In multivariate analysis, HIVAN risk was reduced by 60% (95% CI –30% to –80%) by use of HAART, and no patient developed HIVAN when HAART had been initiated prior to the development of acquired immune deficiency syndrome [26]. The use of HAART has also been associated with improved kidney survival in patients with HIVAN [27]. Antiviral therapy has been associated with GFR improvements in HIV patients with both low CD4 lymphocyte counts and impaired baseline kidney function, supporting an independent contribution of HIV-1 replication to chronic kidney dysfunction in advanced HIV disease [9].
Early observational studies suggest a benefit for ACEi [28]. A number of retrospective, observational, or uncontrolled studies conducted before or during the initial phases of HAART reported variable success with the use of corticosteroids in patients with HIV-associated kidney diseases [29-31]. There is only one study using cyclosporine in 15 children with HIV and nephrotic syndrome [32]. These early observational studies suggest a benefit for ACEi and corticosteroids in HIV-mediated kidney disease, but the studies were prior to introduction of HAART, and in the era of modern HAART therapy it is unclear what the potential benefits are,

KDIGO® GN Guideline March 2011 Public Review Draft
107
if any, of the use of corticosteroids or cyclosporine in the treatment of patients with HIVAN or other HIV-related kidney diseases. It is not known whether this benefit remains in the context of current management [28].
There is no RCT that evaluates the value of HAART therapy in patients with HIVAN [33]. There is very low–quality evidence to suggest that HAART may be of benefit in patients with HIV-associated immune-complex kidney diseases and thrombotic microangiopathies [2,4,21]. There are recent comprehensive reviews of HIV and kidney disease that describes current knowledge and gaps therein [34,35].
RESEARCH RECOMMENDATIONS
• RCTs are needed to evaluate the efficacy of HAART in HIVAN and other HIV-associated glomerular diseases. Long-term follow-up is needed to determine if kidney damage in susceptible individuals is halted or merely slowed by HAART, particularly when control of viremia is incomplete or intermittent.
• An RCT is needed to evaluate the role of corticosteroids in combination with HAART in the treatment of HIV-associated kidney diseases.
• An RCT is needed to determine if benefits of RAS blockade are independent of HAART therapy in patients with HIVAN and other HIV-mediated kidney diseases.
References
1. Sepkowitz KA: One disease, two epidemics--AIDS at 25. N Engl J Med 354:2411-2414, 2006 2. Kimmel PL, Barisoni L, Kopp JB: Pathogenesis and treatment of HIV-associated renal diseases: lessons from
clinical and animal studies, molecular pathologic correlations, and genetic investigations. Ann Intern Med 139:214-226, 2003
3. Wyatt CM, Klotman PE: HIV-1 and HIV-Associated Nephropathy 25 Years Later. Clin J Am Soc Nephrol 2 Suppl 1:S20-24, 2007
4. Cohen SD, Kimmel PL: Immune complex renal disease and human immunodeficiency virus infection. Semin Nephrol 28:535-544, 2008
5. Szczech LA, Gupta SK, Habash R, Guasch A, Kalayjian R, Appel R, Fields TA, Svetkey LP, Flanagan KH, Klotman PE, Winston JA: The clinical epidemiology and course of the spectrum of renal diseases associated with HIV infection. Kidney Int 66:1145-1152, 2004
6. Szczech LA, Hoover DR, Feldman JG, Cohen MH, Gange SJ, Gooze L, Rubin NR, Young MA, Cai X, Shi Q, Gao W, Anastos K: Association between renal disease and outcomes among HIV-infected women receiving or not receiving antiretroviral therapy. Clin Infect Dis 39:1199-1206, 2004
7. Krawczyk CS, Holmberg SD, Moorman AC, Gardner LI, McGwin G, Jr.: Factors associated with chronic renal failure in HIV-infected ambulatory patients. Aids 18:2171-2178, 2004
8. El-Sadr WM, Lundgren JD, Neaton JD, Gordin F. et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med 355:2283-2296, 2006
9. Kalayjian RC, Franceschini N, Gupta SK, Szczech LA, Mupere E, Bosch RJ, Smurzynski M, Albert JM: Suppression of HIV-1 replication by antiretroviral therapy improves kidney function in persons with low CD4 cell counts and chronic kidney disease. Aids 22:481-487, 2008
10. Peters PJ, Moore DM, Mermin J, Brooks JT, Downing R, Were W, Kigozi A, Buchacz K, Weidle PJ: Antiretroviral therapy improves kidney function among HIV-infected Ugandans. Kidney Int 74:925-929, 2008
11. Reid A, Stohr W, Walker AS, Williams IG, Kityo C, Hughes P, Kambugu A, Gilks CF, Mugyenyi P, Munderi P, Hakim J, Gibb DM: Severe renal dysfunction and risk factors associated with renal impairment in HIV-infected adults in Africa initiating antiretroviral therapy. Clin Infect Dis 46:1271-1281, 2008

KDIGO® GN Guideline March 2011 Public Review Draft
108
12. Longenecker CT, Scherzer R, Bacchetti P, Lewis CE, Grunfeld C, Shlipak MG: HIV viremia and changes in kidney function. Aids 23:1089-1096, 2009
13. Gupta SK, Parker RA, Robbins GK, Dube MP: The effects of highly active antiretroviral therapy on albuminuria in HIV-infected persons: results from a randomized trial. Nephrol Dial Transplant 20:2237-2242, 2005
14. Gupta SK, Smurzynski M, Franceschini N, Bosch RJ, Szczech LA, Kalayjian RC: The effects of HIV type-1 viral suppression and non-viral factors on quantitative proteinuria in the highly active antiretroviral therapy era. Antivir Ther 14:543-549, 2009
15. Wyatt CM, Winston JA, Malvestutto CD, Fishbein DA, Barash I, Cohen AJ, Klotman ME, Klotman PE: Chronic kidney disease in HIV infection: an urban epidemic. Aids 21:2101-2103, 2007
16. Wyatt CM, Morgello S, Katz-Malamed R, Wei C, Klotman ME, Klotman PE, D'Agati VD: The spectrum of kidney disease in patients with AIDS in the era of antiretroviral therapy. Kidney Int 75:428-434, 2009
17. Haas M, Kaul S, Eustace JA: HIV-associated immune complex glomerulonephritis with "lupus-like" features: a clinicopathologic study of 14 cases. Kidney Int 67:1381-1390, 2005
18. Fine DM, Perazella MA, Lucas GM, Atta MG: Kidney biopsy in HIV: beyond HIV-associated nephropathy. Am J Kidney Dis 51:504-514, 2008
19. Han TM, Naicker S, Ramdial PK, Assounga AG: A cross-sectional study of HIV-seropositive patients with varying degrees of proteinuria in South Africa. Kidney Int 69:2243-2250, 2006
20. Gerntholtz TE, Goetsch SJ, Katz I: HIV-related nephropathy: a South African perspective. Kidney Int 69:1885-1891, 2006
21. Cohen SD, Kimmel PL: Renal biopsy is necessary for the diagnosis of HIV-associated renal diseases. Nat Clin Pract Nephrol 5:22-23, 2009
22. Babut-Gay ML, Echard M, Kleinknecht D, Meyrier A. Zidovudine and nephropathy with human immunodeficiency virus (HIV) infection. Ann Intern Med 1989; 111: 856-857.
23. Ifudu O, Rao TK, Tan CC, Fleischman H, et al. Zidovudine is beneficial in human immunodeficiency virus associated nephropathy. Am J Nephrol 1995; 15: 217-221.
24. Kirchner JT. Resolution of renal failure after initiation of HAART: 3 cases and a discussion of the literature. AIDS Read 2002; 12: 103-105, 110-102.
25. Szczech LA, Edwards LJ, Sanders LL, van der Horst C, et al. Protease inhibitors are associated with a slowed progression of HIV-related renal diseases. Clin Nephrol 2002; 57: 336-341.
26. Lucas GM, Eustace JA, Sozio S, Mentari EK, Appiah KA, Moore RD: Highly active antiretroviral therapy and the incidence of HIV-1-associated nephropathy: a 12-year cohort study. Aids 18:541-546, 2004
27. Atta MG, Gallant JE, Rahman MH, Nagajothi N, Racusen LC, Scheel PJ, Fine DM: Antiretroviral therapy in the treatment of HIV-associated nephropathy. Nephrol Dial Transplant 21:2809-2813, 2006
28. Kalayjian RC: The treatment of HIV-associated nephropathy. Adv Chronic Kidney Dis 17:59-71, 2010 29. Laradi A, Mallet A, Beaufils H, Allouache M, et al. HIV-associated nephropathy: outcome and prognosis
factors. Groupe d' Etudes Nephrologiques d'Ile de France. J Am Soc Nephrol 1998; 9: 2327-2335. 30. Eustace JA, Nuermberger E, Choi M, Scheel PJ, Jr., et al. Cohort study of the treatment of severe HIV-
associated nephropathy with corticosteroids. Kidney Int 2000; 58: 1253-1260. 31. Smith MC, Austen JL, Carey JT, Emancipator SN, et al. Prednisone improves kidney function and proteinuria
in human immunodeficiency virus-associated nephropathy. Am J Med 1996; 101: 41-48. 32. Ingulli E, Tejani A, Fikrig S, Nicastri A, et al. Nephrotic syndrome associated with acquired
immunodeficiency syndrome in children. J Pediatr 1991; 119: 710-716. 33. Yahaya I, Uthman AO, Uthman MM: Interventions for HIV-associated nephropathy. Cochrane Database Syst
Rev:CD007183, 2009 34. Novak JE, Szczech L. (Editors) HIV and Kidney Disease. Adv Chronic Kidney Dis. 17:1-111, 2010 35. Elewa U, Sandri, A.M., Rizza, S.A., Fervenza, F.C. Treatment of HIV-associated nephropathies. Nephron
Clin Pract 2011; 118:346-354.

KDIGO® GN Guideline March 2011 Public Review Draft
109
9.5: Schistosomal, filarial, and malarial nephropathies 9.5.1: We suggest that patients with GN and concomitant malarial,
schistosomal, or filarial infection be treated with an appropriate antiparasitic agent in sufficient dosage and duration to eradicate the organism. (Not Graded)
9.5.2: We suggest that corticosteroids or immunosuppressive agents are not used for treatment of schistosomal GN. (2D)
9.5.3: We suggest that blood culture for Salmonella be performed in all patients with hepatosplenic schistosomiasis who show urinary abnormality and/or reduced GFR. (2C) 9.5.3.1: We suggest that all patients who show a positive blood culture
for Salmonella receive anti-Salmonella therapy. (2C)
A. SCHISTOSOMAL NEPHROPATHY
BACKGROUND
Schistosomiasis (syn. Bilharziasis), a chronic infection by trematodes (blood flukes), is encountered in Asia, Africa, and South America [1,2]. S. mansoni and S. japonicum cause glomerular lesions in experimental studies, but clinical glomerular disease has been described most frequently in association with hepatosplenic schistosomiasis produced by S. mansoni. [3-11]. A classification of schistosomal glomerulopathies is given in Table 9.5.
RATIONALE
The incidence of GN in schistosomiasis is not well defined. Hospital-based studies have shown overt proteinuria in 1-10% and microalbuminuria in about 22% of patients with hepatosplenic schistosomiasis due to S. mansoni [12,13]. Sobh et al. [14] documented asymptomatic proteinuria in 20% patients with “active” S. mansoni infection. A field study in an endemic area of Brazil showed only a 1% incidence of proteinuria [15]. However, histological studies have documented glomerular lesions in 12-50% of cases [3,10].
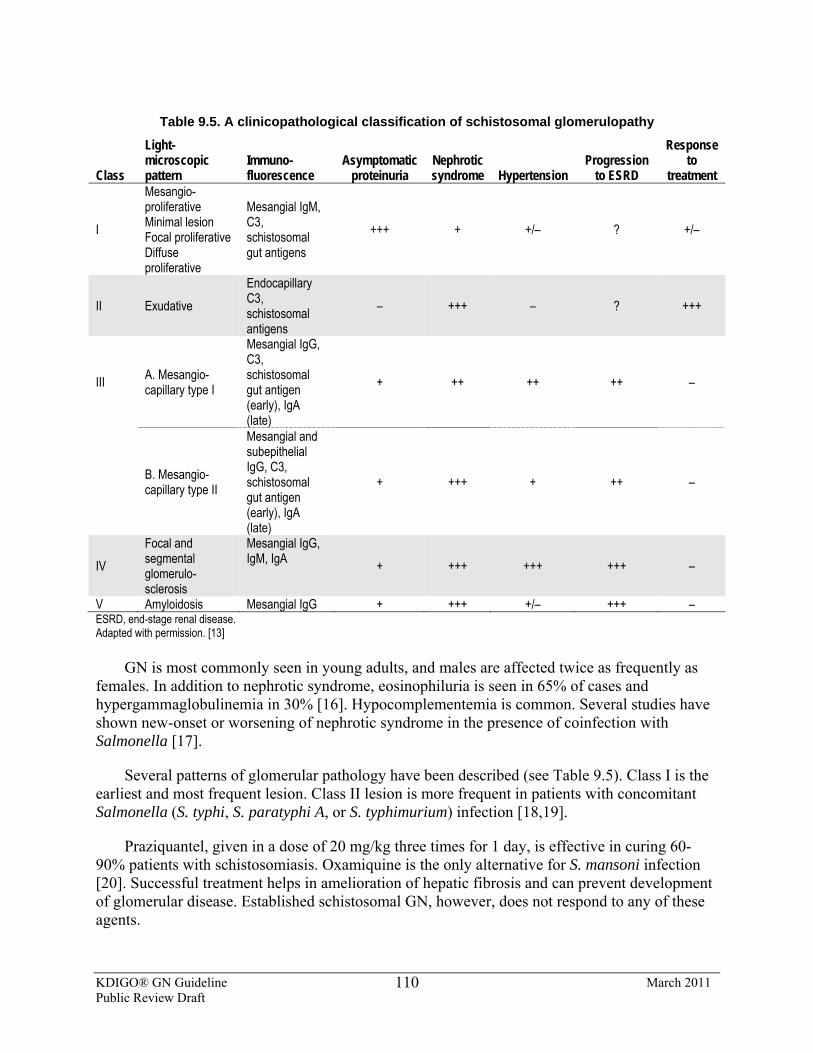
KDIGO® GN Guideline March 2011 Public Review Draft
110
Table 9.5. A clinicopathological classification of schistosomal glomerulopathy
Class
Light-microscopic pattern
Immuno-fluorescence
Asymptomatic proteinuria
Nephrotic syndrome Hypertension
Progression to ESRD
Response to
treatment
I
Mesangio-proliferative Minimal lesion Focal proliferative Diffuse proliferative
Mesangial IgM, C3, schistosomal gut antigens
+++ + +/– ? +/–
II Exudative Endocapillary C3, schistosomal antigens
– +++ – ? +++
III A. Mesangio-capillary type I
Mesangial IgG, C3, schistosomal gut antigen (early), IgA (late)
+ ++ ++ ++ –
B. Mesangio-capillary type II
Mesangial and subepithelial IgG, C3, schistosomal gut antigen (early), IgA (late)
+ +++ + ++ –
IV Focal and segmental glomerulo-sclerosis
Mesangial IgG, IgM, IgA + +++ +++ +++ –
V Amyloidosis Mesangial IgG + +++ +/– +++ – ESRD, end-stage renal disease. Adapted with permission. [13]
GN is most commonly seen in young adults, and males are affected twice as frequently as females. In addition to nephrotic syndrome, eosinophiluria is seen in 65% of cases and hypergammaglobulinemia in 30% [16]. Hypocomplementemia is common. Several studies have shown new-onset or worsening of nephrotic syndrome in the presence of coinfection with Salmonella [17].
Several patterns of glomerular pathology have been described (see Table 9.5). Class I is the earliest and most frequent lesion. Class II lesion is more frequent in patients with concomitant Salmonella (S. typhi, S. paratyphi A, or S. typhimurium) infection [18,19].
Praziquantel, given in a dose of 20 mg/kg three times for 1 day, is effective in curing 60-90% patients with schistosomiasis. Oxamiquine is the only alternative for S. mansoni infection [20]. Successful treatment helps in amelioration of hepatic fibrosis and can prevent development of glomerular disease. Established schistosomal GN, however, does not respond to any of these agents.

KDIGO® GN Guideline March 2011 Public Review Draft
111
Steroids, cytotoxic agents, and cyclosporine are ineffective in inducing remission [21]. In one RCT, neither prednisolone nor cyclosporine, given in combination with praziquantel and oxamiquine were effective in inducing remissions in patients with established schistosomal GN [22].
Treatment of coexistent Salmonella infection favorably influences the course of GN. In a study of 190 patients with schistosomiasis, 130 were coinfected with Salmonella. All of them showed improvement in serum complement levels, CrCl, and proteinuria following antibilharzial and anti-Salmonella treatment, either together or sequentially [23]. Other studies have shown disappearance of urinary abnormalities following anti-Salmonella therapy alone [17,18]. The prognosis is relatively good with class I and II schistosomal GN, provided sustained eradication of Schistosoma and Salmonella infection can be achieved, whereas class IV and V lesions usually progress to ESRD despite treatment [21,24,25].
RESEARCH RECOMMENDATIONS
• Studies are required to evaluate the precise contribution of Salmonella infection to schistosomal nephropathy, and the value of treating these two infections separately or together on the outcome.
Please refer to Suppl Tables 40-41 for additional data .
References 1. Chitsulo, L., Ingels, D., Montresor, A., Savioli, L. (2000) The global status of schistosomiasis and its control.
Acta Tropica 77:41-51 2. Chugh, K. S. et al. (1986). Urinary schistosomiasis in Maiduguri, North East Nigeria. Annals of Tropical
Medicine and Parasitology, 80, 593–9. 3. Andrade, Z. A., Andrade, S. G., and Sadigursky, M. (1971). Renal changes in patients with hepatosplenic
schistosomiasis. American Journal of Tropical Medicine and Hygiene, 20, 77–83. 4. Falcao, H. A. and Gould, D. B. (1975). Immune complex nephropathy in schistosomiasis. Annals of Internal
Medicine, 83, 148–54. 5. Abdurrahman, M. B., Attah, B., and Narayana, P. T. (1981). Clinicopathological features of hepatosplenic
schistosomiasis in children. Annals of Tropical Paediatrics, 1, 5–11. 6. Abu Romeh, S. H. et al. (1989). Renal diseases in Kuwait. Experience with 244 renal biopsies. International
Urology and Nephrology, 21, 25–9. 7. Chandra Shekhar, K., and Pathmanathan, R. (1987). Schistosomiasis in Malaysia. Review of Infectious
Diseases, 9, 1026–37. 8. Musa, A. M., Abu Asha, H., and Veress, B. (1980). Nephrotic syndrome in Sudanese patients with
schistosomiasis mansoni infection. Annals of Tropical Medicine and Parasitology, 74, 615–18. 9. Queiroz, F. P., Brito, E., Martinelli, R., and Rocha, H. (1973). Nephrotic syndrome in patients with
Schistosoma mansoni infections. American Journal of Tropical Medicine and Hygiene, 22, 622–8. 10. Rocha, H., Cruz, T., Brito, E., and Susin, M. (1976). Renal involvement in patients with hepatosplenic
schistosomiasis mansoni. American Journal of Tropical Medicine and Hygiene, 25, 108–15. 11. Sobh, M.A., Moustafa, F.E., el-Housseini, F., Basta, M.T., Deelder, A.M., Ghoniem, M.A. (1987)
Schistosomal specific nephropathy leading to end-stage renal failure. Kidney International 31:1006-11

KDIGO® GN Guideline March 2011 Public Review Draft
112
12. Barsoum, R.S., Abdel-Rahman,Y., Francis, M.R., El-Kaliour, A., Zakareya, S., El-Garem, A.A. (1992) Patterns of glomerular injury associated with hepato-splenic schistosomiasis. Proceedings of XII Egyptian Congress of Nephrology, Cairo.
13. Barsoum, R.S. (1993) Schistosomal glomerulopathies. Kidney International 44:1-12. 14. Sobh M, Moustafa F, el-Arbagy A, el-Din MS, Shamaa S, Amer G. (1990) Nephropathy in asymptomatic
patients with active Schistosoma mansoni infection. International Urology Nephrology 22:37-43 15. Rabello, A.L., Lambertucci, J.R., Freire, M.H., Garcia, M.M., Amorim, M.N., Katz, N. (1993) Evaluation of
proteinuria in an area of Brazil endemic for schistosomiasis using a single urine sample. Transactions of Royal Society of Tropical Medicine and Hygiene 87:187-9
16. Eltoum, I.A., Ghalib, H.W., Sualaiman, S., Kordofani, A., Mustafa, M.D., Homeida. M. (1989) Significance of eosinophiluria in urinary schistosomiasis. A study using Hansel's stain and electron microscopy. American Journal of Clinical Pathology 92:329-38
17. Martinelli, R., Pereira, L.J., Brito, E., Rocha, H. (1992) Renal involvement in prolonged Salmonella bacteremia: the role of schistosomal glomerulopathy. Rev Inst Med Trop Sao Paulo 34:193-8
18. Lambertucci, J.R., Godoy, P., Neves, J., Bambirra, E.A., Ferreira, M.D. (1988) Glomerulonephritis in Salmonella-Schistosoma mansoni association. American Journal of Tropical Medicine and Hygiene 38:97-102
19. Bassily, S., Farid, Z., Barsoum, R. S., Soliman, L. A., Higashi, G. I., and Miner, W. F. (1976). Renal biopsy in Schistosoma-salmonella associated nephrotic syndrome. Journal of Tropical Medicine and Hygiene, 79, 256–8.
20. Ross, A.G.P., Bartley, P.B., Sleigh, A.C., Olds, G.R., Li, Y., Williams, G.M., McManus D.P. (2002) Schistosomiasis. New England Journal of Medicine 346:1212-20.
21. Martinelli, R., Pereira, L.J., Brito, E., Rocha, H. (1995) Clinical course of focal segmental glomerulosclerosis associated with hepatosplenic schistosomiasis mansoni. Nephron 69:131-4
22. Sobh, M.A., Moustafa, F.E., Sally, S.M., Foda, M.A., Deelder, A.M., Ghoneim, M.A. (1989) A prospective, randomized therapeutic trial for schistosomal specific nephropathy. Kidney International 36:904-7
23. Abdul-Fattah MM, Yossef SM, Ebraheem ME, Nasr ME, Hassan MA and Abdul Wahab SE (1995): Schistosomal glomerulopathy: a putative role for commonly associated Salmonella infection. J Egypt Soc Parasitol 25: 165-173
24. Martinelli, R., Pereira, L.J., Rocha, H. (1987) The influence of anti-parasitic therapy on the course of the glomerulopathy associated with Schistosomiasis mansoni. Clinical Nephrology 27:229-32
25. Sobh, M.A., Moustafa, F.E., Sally, S.M., Deelder, A.M., Ghoniem, M.A. (1988) Characterisation of kidney lesions in early schistosomal-specific nephropathy. Nephrology Dialysis Transplantation 3:392-8
B. FILARIAL NEPHROPATHY
BACKGROUND AND RATIONALE
Filarial worms are nematodes that are transmitted to humans through arthropod bites, and dwell in the subcutaneous tissues and lymphatics. Clinical manifestations depend upon the location of microfilariae and adult worms in the tissues. Of the eight filarial species that infect humans, glomerular disease has been reported in association with Loa loa, Onchocerca volvulus, Wuchereria bancrofti, and Brugia malayi infections in Africa and some Asian countries [1-5].
Glomerular involvement is seen in a small number of cases. Light microscopy reveals a gamut of lesions, including diffuse GN and MPGN, membranoproliferative GN, minimal-change and chronic sclerosing GN, and the collapsing variant of FSGS [6]. Microfilariae may be found

KDIGO® GN Guideline March 2011 Public Review Draft
113
in the arterioles, glomerular and peritubular capillary lumina, tubules, and interstitium [6]. Immunofluorescence and electron microscopy show immune deposits along with worm antigens and structural components [4,7].
Urinary abnormalities have been reported in 11-25% and nephrotic syndrome is seen in 3-5% of patients with loiasis and onchocerciasis, especially those with polyarthritis and chorioretinitis [4,8]. Proteinuria and/or hematuria was detected in over 50% of cases with lymphatic filariasis; 25% showed glomerular proteinuria [9,10]. A good response (diminution of proteinuria) is observed following antifilarial therapy in patients with non-nephrotic proteinuria and/or hematuria. The proteinuria can increase and kidney functions worsen following initiation of diethylcarbamazepine or ivermectin [10,11], probably because of exacerbation of immune process secondary to antigen release into circulation after death of the parasite [12].
The response is inconsistent in those with nephrotic syndrome, and deterioration of kidney function may continue, despite clearance of microfilariae with treatment. Therapeutic apheresis has been utilized to reduce the microfilarial load before starting diethylcarbamazepine to prevent antigen release [13].
The incidence, prevalence, and natural history of glomerular involvement in various forms of filariasis are poorly documented. This condition is usually found in areas with poor vector control and inadequate health-care facilities. Similarly, the treatment strategies have not been evaluated.
RESEARCH RECOMMENDATIONS
• Epidemiological studies of kidney involvement in regions endemic for these conditions are required. The effect of population-based treatment with filaricidal agents on the course of kidney disease should be studied.
References
1. Bariéty, J. et al. (1967). Proteinurie et loase. Étude histologique, optique et electronique d’un cas. Bulletin et Mémoires de la Société Médicale des Hôpitaux de Paris, 118, 1015–25.
2. Chugh, K. S., Singhal, P. C., and Tewari, S. C. (1978). Acute glomerulonephritis associated with filariasis. American Journal of Tropical Medicine and Hygiene, 27, 630–1.
3. Date, A., Gunasekaran, V., Kirubakaran, M. G., and Shastry, J. C. M. (1979). Acute eosinophilic glomerulonephritis with Bancroftian filariasis. Postgraduate Medical Journal, 55, 905–7.
4. Pillay, V. K. G., Kirch, E., and Kurtzman, N. A. (1973). Glomerulopathy associated with filarial loiasis. (Letter.) Journal of the American Medical Association, 255, 179.
5. Ngu, J. L., Chatelanat, F., Leke, R., Ndumbe, P., and Youmbissi, J. (1985). Nephropathy in Cameroon: evidence for filarial derived immune complex pathogenesis in some cases. Clinical Nephrology, 24, 128–34.
6. Pakasa, N.M., Nseka, N.M., Nyimi, L.M. (1997) Secondary collapsing glomerulopathy associated with Loa loa filariasis. American Journal of Kidney Diseases 30:836-9
7. Ormerod, A. D., Petersen, J., Hussey, J. K., Weir, J., and Edward, N. (1983). Immune complex glomerulonephritis and chronic anerobic urinary tract infection: complication of filariasis. Postgraduate Medical Journal, 59, 730–3.
8. Hall, C.L., Stephens, L., Peat, D., Chiodini, P.L. (2001) Nephrotic syndrome due to loiasis following a tropical adventure holiday: a case report and review of the literature. Clinical Nephrology 56:247-50

KDIGO® GN Guideline March 2011 Public Review Draft
114
9. Dreyer, G., Ottesen, E.A., Galdino, E., Andrade, L., Rocha, A., Medeiros, Z., Moura, I., Casimiro, I., Beliz, F., Coutinho, A. (1992) Renal abnormalities in microfilaremic patients with Bancroftian filariasis. American Journal of Tropical Medicine and Hygiene 46:745-51
10. Langhammer, J., Birk, H.W., Zahner, H. (1997) Renal disease in lymphatic filariasis: evidence for tubular and glomerular disorders at various stages of the infection. Tropical Medicine and International Health 2:875-84
11. Cruel, T., Arborio, M., Schill, H., Neveux, Y., Nedelec, G., Chevalier, B., Teyssou, R., Buisson, Y. (1997) Nephropathy and filariasis from Loa loa. Apropos of 1 case of adverse reaction to a dose of ivermectin. Bull Soc Pathol Exot 90:179-81
12. Ngu, J. L., Adam, M., Leke, R., and Titanji, V. (1980). Proteinuria associated with diethylcarbamazine treatment of onchocerciasis. Lancet, i, 254–5.
13. Abel, L., Ioly, V., Jeni, P., Carbon, C., Bussel, A. (1986) Apheresis in the management of loiasis with high microfilariaemia and renal disease. British Medical Journal 1986 292:24
C. MALARIAL NEPHROPATHY
BACKGROUND AND RATIONALE
Infection with Plasmodium falciparum usually results in acute kidney injury or proliferative GN. Chronic infection with the protozoal malarial parasites Plasmodium malariae (and, to a lesser extent, Plasmodium vivax or ovale) has been associated with a variety of kidney lesions, including MN and membranoproliferative GN [1]. In the past, this has been known as “quartan malarial nephropathy” [1,2]. Nephrotic syndrome, sometimes with impaired kidney function, is a common clinical manifestation; it is principally encountered in young children. The glomerular lesions are believed to be caused by deposition of immune complexes containing antigens of the parasite, but autoimmunity may participate as well. The clinical and morphological manifestations vary from country to country [3]. Nowadays, the lesion is much less common, and most children in the tropics with nephrotic syndrome have either MCD or FSGS, rather than malarial nephropathy [3,4]. HBV and HIV infection and streptococcal-related diseases are also now more common causes of nephrotic syndrome than malarial nephropathy in Africa [3-5].
There are limited observational studies and no RCTs for an evidence-based treatment strategy for malarial nephropathy. Patients with GN and concomitant infection with Plasmodium species (typically Plasmodium malariae) should be treated with an appropriate antimalarial agent (such as chloroquine or hydroxychloroquine) for suffcient duration to eradicate the organism from blood and hepatosplenic sites. Observational studies have suggested improvement in clinical manifestations in some—but not all—patients, following successful eradication of the parasitic infection. There does not appear to be any role for steroids or immunosuppressant therapy in malarial nephropathy [1,2], although controlled trials are lacking. Dosage reductions of chloroquine or hydroxychloroquine may be needed in patients with impaired kidney function.
RESEARCH RECOMMENDATIONS
• Studies of the incidence and prevalence of malarial nephropathy, and its response to antimalarial therapy are needed, especially in endemic areas of West Africa.

KDIGO® GN Guideline March 2011 Public Review Draft
115
• RCTs are needed to investigate the role of corticosteroids and immunosuppressive agents when malarial nephropathy progresses, despite eradication of the malarial parasite.
References
1. Barsoum RS. Malarial nephropathies. Nephrol Dial Transplant 13:1588-1597, 1998 2. Elam-Ong S. Malarial nephropathy. Semin Nephrol 23:21-33, 2003 3. Olowu WA, Adelusola KA, Adefehinti O, Oyetunji TG. Quartan malaria-associated childhood nephrotic
syndrome: now a rare entity in malaria endemic Nigeria. Nephrol Dial Transplant 2009 4. Doe JY, Funk M, Mengel M, Doehring E, Ehrich JHH. Nephrotic syndrome in African children: lack of
evidence for “tropical nephrotic syndrome”? Nephrol Dial Transplant 21:672-676, 2006 5. Seggie J, Davies PG, Ninin D, Henry J. Patterns of glomerulonephritis in Zimbabwe: survey of disease
characterized by nephrotic proteinuria. Q J Med 53:109-118, 1984

KDIGO® GN Guideline March 2011 Public Review Draft
116
CHAPTER 10: IMMUNOGLOBULIN A NEPHROPATHY
INTRODUCTION
This chapter provides treatment recommendations for primary IgAN. Secondary IgAN will not be discussed. The cost implications for global application of this guideline are addressed in Chapter 2.
10.1: Initial evaluation including assessment of risk of progressive kidney disease 10.1.1: Assess all patients with biopsy-proven IgAN for secondary causes of
IgAN. (Not Graded) 10.1.2: Assess the risk of progression in all cases by evaluation of proteinuria,
blood pressure, and eGFR at the time of diagnosis and during follow-up. (Not Graded)
10.1.3: Pathological features may be used to assess prognosis. (Not Graded)
BACKGROUND
IgAN is diagnosed by kidney biopsy and is defined as dominant or codominant staining with IgA in glomeruli by immunohistology [1]. LN should be excluded. The intensity of IgA staining should be more than trace. The distribution of IgA staining should include presence in the mesangium, with or without capillary loop staining. IgG and IgM may be present, but not in greater intensity than IgA, except that IgM may be prominent in sclerotic areas. C3 may be present. The presence of C1q staining in more than trace intensity should prompt consideration of LN.
IgAN is the most common primary GN in the world. The prevalence rate varies across different geographical regions. Typically, it is 30-35% of all primary glomerular diseases in Asia, but can be up to 45% [2]. In Europe, this is about 30-40%. Recently in the USA, IgAN was also reported to be the most common primary glomerulopathy in young adult Caucasians [3].
Secondary IgAN is uncommon. Cirrhosis, celiac disease, and HIV infection are all associated with a high frequency of glomerular IgA deposition. IgAN has been infrequently associated with a variety of other diseases, including dermatitis herpetiformis, seronegative arthritis (particularly ankylosing spondylitis), small-cell carcinoma, lymphoma (Hodgkin lymphoma and T-cell lymphomas, including mycosis fungoides), disseminated tuberculosis, bronchiolitis obliterans, and inflammatory bowel disease (Crohn’s disease and ulcerative colitis). These are usually clinically evident at the time of biopsy. All patients should undergo viral serologies (HIV, HBV, and HCV), liver function tests, and electrophoreses of serum immunoglobulins.
IgAN has a wide spectrum of clinical presentations, varying from isolated hematuria to rapidly progressive GN. Thorough risk assessment is essential to determine management and

KDIGO® GN Guideline March 2011 Public Review Draft
117
ensure that the risks of therapy are balanced by the selection of patients at highest risk of progression. Definitive outcomes in IgAN are kidney survival and the rate of kidney function decline. Determinants of mortality in IgAN have not been addressed in previous studies, although it is reasonable to assume that CKD increases cardiovascular morbidity and mortality in these patients, as in others with CKD [4].
RATIONALE
• There is moderate-quality evidence to suggest that accelerated decline in kidney function is associated with proteinuria ≥1g/d in a dose-dependent fashion and independently of other risk factors [5-8].
• There is moderate-quality evidence to suggest a favourable outcome when time-averaged proteinuria is reduced to <1 g/d [5]. Whether long-term outcome differs in adult patients with a proteinuria between 0.5 and 1.0 g/d compared to <0.5 g/d remains uncertain. In children, expert opinion suggests a goal of a proteinuria <0.5 g/d per 1.73 m2 [9].
• There is moderate-quality evidence to recommend strict blood pressure control, as it is associated with better kidney survival in chronic proteinuric nephropathies, including IgAN.
• There is low-quality evidence to suggest GFR at presentation is associated with the risk of ESRD. However, studies that have assessed the rate of change of kidney function have questioned its association with initial GFR. Proteinuria, blood pressure, and kidney biopsy findings at presentation have been associated with both risk of ESRD and doubling of SCr.
• There is low-quality evidence to suggest kidney biopsy findings associated with a worse prognosis are the presence and severity of mesangial and endocapillary proliferation, extensive crescents, focal and segmental as well as global glomerulosclerosis, tubular atrophy, and interstitial fibrosis [10,11]. However, no single approach to the objective evaluation of biopsy findings has yet been validated or evaluated prospectively.
• There is moderate-quality evidence to suggest that IgAN that presents with hematuria and minimal proteinuria is a progressive disease, and that life-long follow-up with regular monitoring of blood pressure and proteinuria is recommended [12].
Proteinuria is the strongest prognostic factor in IgAN and has a “dose-dependent” effect that
is independent of other risk factors in multiple large observational studies, as well as prospective trials. The threshold above which the risk develops in adults is uncertain; some studies indicate 0.5 g/d [13] while others studies could only demonstrate a higher risk of ESRD and a more rapid rate of decline in kidney function when time-averaged proteinuria was above 1 g/d [5,8]. A large observational study demonstrated that a reduction of proteinuria to <1 g/d carried the same favorable impact on long-term outcome, whether the initial value was 1-2 g/d, 2-3 g/d, or >3 g/d [5]. Other surrogates of long-term outcome, such as a 50% decline in proteinuria, have been used [14]. In children, observational studies have also consistently shown a relationship between the level of proteinuria and outcome, but did not assess a threshold value. Expert opinions in children advocate a cut-off of 0.5 g/d per 1.73 m2 for partial remission, and 0.16 g/d per 1.73 m2 for complete remission; these thresholds have been used in RCTs [9,15].

KDIGO® GN Guideline March 2011 Public Review Draft
118
Uncontrolled hypertension during follow-up is associated with greater proteinuria and predicts a faster GFR decline [16,17]. As in other proteinuric chronic glomerulopathies, a blood pressure goal <130/80 mm Hg in patients with proteinuria >0.3 g/d, and <125/75 mm Hg when proteinuria is >1 g/d, is recommended [18,19].
The GFR at presentation has consistently been related to the risk of ESRD. Whether a lower GFR is also accompanied by a faster rate of kidney function decline is questionable; two observational studies have failed to show this relationship [16,20]. Proteinuria, blood pressure, and pathological features should take precedence over initial GFR in the estimation of the future rate of kidney function decline.
Numerous studies have addressed the predictive value of pathology findings. Mesangial [21,22] and endocapillary proliferation [10,23], extensive crescents [24-27], FSGS [28,29], global glomerulosclerosis, tubular atrophy, and interstitial fibrosis [10,23,24,28] are associated with a more rapid rate of deterioration and lower kidney survival using univariate and, at times, multivariate analysis adjusting for clinical assessment. The recent Oxford Classification of IgAN has demonstrated the importance of i) mesangial hypercellularity; ii) segmental glomerulosclerosis; iii) endocapillary hypercellularity; and iv) tubular atrophy/ interstitial fibrosis, as independent pathological variables predicting kidney outcome [10]. This may become the standard, but requires validation before it can be recommended in routine clinical practice. Whether classification of the disease in this manner should impact treatment choice has also not been determined.
Obesity has been identified as an independent risk factor for the appearance of ESRD [30], and weight loss induces a significant decrease in proteinuria [31]. Some observational studies [32,33] have reported an increased risk of greater proteinuria, more severe pathological lesions, and ESRD among IgAN patients with overweight (BMI >25 kg/m2).
Other risk factors have been studied. Outcomes do not differ between sexes [34]. Children are less likely to reach ESRD compared to adults, but this may be because GFR is higher at presentation in children, even though there is a similar rate of kidney function decline. Different biopsy and treatment practices in the pediatric population limit comparisons to adults. Since the risk factors presented above have been validated in both children and adults, clinicians should consider these before the age of the patient. Similarly, it is uncertain whether geographical or ethnic variations in outcomes are secondary to different biopsy and treatment practices or variations in disease severity [6]. Macroscopic hematuria is more frequent in children, and some studies have associated its presence to a favorable outcome, while others have shown this benefit to be confounded by a higher initial GFR and earlier detection with no independent value [35,36].
10.2: Antiproteinuric and antihypertensive therapy 10.2.1: We recommend long-term ACEi or ARB treatment when proteinuria is
>1 g/d. (1B) 10.2.2: We suggest ACEi or ARB treatment if proteinuria is between 0.5 to 1 g/d
(in children, between 0.5 to 1 g/d per 1.73 m2). (2D)

KDIGO® GN Guideline March 2011 Public Review Draft
119
10.2.3: We suggest the ACEi or ARB be titrated upwards as far as tolerated to achieve proteinuria <1 g/d. (2C)
10.2.4: The goal of blood pressure treatment in IgAN should be <130/80 mmHg in patients with proteinuria <1 g/d, and <125/75 mmHg when initial proteinuria is >1 g/d (see Chapter 2). (Not Graded)
RATIONALE
Many of the trials using ACEi/ARBs in IgAN recruited patients with proteinuria ≥1 g/d [9,37] while some recruited patients with proteinuria ≥ 0.5g/d [38].
In registry data [5], the rate of decline of function increased with the amount of proteinuria; those with sustained proteinuria ≥3 g/d lost kidney function 25-fold faster than those with proteinuria <1 g/d. Patients who presented with ≥3 g/d who achieved proteinuria <1 g/d had a similar course to patients who had <1 g/d throughout, and fared far better than patients who never achieved this level. There is, as yet, no evidence in IgAN that reducing proteinuria below 1 g/d in adults gives additional benefit.
Several RCTs [9, 37-39] have shown that ACEi and ARBs can reduce proteinuria and improve kidney function (assessed by reduction of the slope of GFR deterioration) (see Suppl Table 42). However, there is, as yet, no definitive study of sufficient duration to show the benefit of either ACEi or ARBs in reducing the incidence of ESRD.
There are no data to suggest preference of ACEi over ARBs, or vice versa, except in terms of a lesser side-effect profile with ARBs compared to ACEi.
One study [40] suggested the combination of ACEi and ARBs induced a 73% greater reduction of proteinuria than monotherapy (ACEi 38% and ARB 30%, respectively). A small study of seven pediatric IgAN patients also showed some benefits [41] of combination of ACEi and ARB. However, more studies are needed to determine whether the definite benefit of combination therapy is effective, leading to a better kidney outcome.
RESEARCH RECOMMENDATIONS
• RCTs are needed to compare the efficacy in proteinuric IgAN of combination therapy using ACEi and ARBs, to monotherapy using either alone.
10.3: Corticosteroids 10.3.1: We suggest that patients with persistent proteinuria ≥1 g/d, despite 3-6
months of optimized supportive care (including ACEi or ARBs and blood pressure control), and GFR >50 ml/min, receive a 6-month course of corticosteroid therapy. (2C)

KDIGO® GN Guideline March 2011 Public Review Draft
120
RATIONALE
• There is low-quality evidence that corticosteroids provide an additional benefit to optimized supportive care (see Suppl Table 45).
• A 6-month corticosteroid regimen can follow either of two regimens, which have been used in published trials (see Table 10.1).
• There is no evidence to suggest the use of corticosteroids in patients with GFR <50 ml/min.
• The available studies do not allow recommendations for preferred dosage regimens. The studies did not report serious side-effects. However, there are other studies with similar regimens in non-IgAN patients that suggest more side-effects with high-dose pulse corticosteroids, including hypothalamic-pituitary-adrenal axis suppression and acute myopathies.
Table 10.1. Corticosteroid regimens in patients with IgAN
References Pozzi C et al. [42] Manno C et al. [43]; Lu J et al. [44]
Regimen
i.v. bolus injections of 1 g methylprednisolone for 3 days each at months 1, 3, and 5, followed by oral steroid 0.5 mg/kg prednisone on alternate days for 6 months
6-month regime of oral prednisone* starting with 0.8-1 mg/kg/d for 2 months and then reduced by 0.2 mg/kg/d per month for the next 4 months
IgAN, immunoglobulin A nephropathy. *Prednisone and prednisolone are equivalent and can be used interchangeably with the same dosing regimen.
Few RCTs so far have tested the efficacy of a corticosteroid regimen vs. no
immunosuppressive therapy. In an Italian trial [42], a 6-month course of corticosteroids led to better clinical disease remission and long-term outcome [45] than no steroids. However, only about 15% of the patients had received an ACEi at randomization [42], and blood pressure control was not optimal by contemporary standards [46].
Two more recent RCTs [43,44,47] used oral prednisone added to an ACEi and compared this to an ACEi alone. In the Italian study [43], the mean annual GFR loss was reduced from about –6 ml/min to –0.6 ml/min, and in the Chinese study [44] the proportion of patients with a 50% increase in SCr decreased from 24% to 3% with corticosteroid therapy. A major limitation of both studies is that all ACEi and ARBs had to be halted for 1 month prior to study inclusion, and then an ACEi was started together with corticosteroids in the combination group. Therefore, a number of low-risk patients may have been included, who would have achieved proteinuria <1 g/d with ACEi therapy alone. A further potential confounder is that both studies included patients who had received prior immunosuppression. An American trial in adults and children, all of whom received an ACEi, also noted reduced proteinuria with corticosteroids (60 mg/m2 prednisone every other day tapered to 30 mg/m2 at 12 months) but no difference in kidney function was observed at 2 years [48].
A Japanese RCT that used low-dose corticosteroids (20 mg/d prednisolone, tapered to 5 mg/d by 2 years) observed no benefit on kidney function, despite reduced proteinuria with the corticosteroid regimen [49].

KDIGO® GN Guideline March 2011 Public Review Draft
121
Subjects with IgAN and GFR<50 ml/min were either excluded from these trials [42, 47] or were few in number [44], so that currently, there are no data to assess the value of corticosteroids in this population.
A recent meta-analysis [50] concluded that corticosteroids reduce doubling of SCr. However, in that analysis, 85% of the weight was contributed by two studies [42, 51], both of which lacked optimal antiproteinuric and antihypertensive therapy based on contemporary standards. Of note, an American RCT in children and adults with IgAN [48] noted no difference in reaching the endpoint (>40% decrease in GFR) between a group receiving ACEi only vs. ACEi plus prednisone (60 mg/m2 per 48 hours for 3 months, reduced to 30 mg/m2 per 48 hours at month 12). However, few end-points were reached in this trial; thus, it was underpowered to detect small differences
RESEARCH RECOMMENDATIONS
• Studies using immunosuppressive agents should always include rigorous blood pressure control and antiproteinuric therapy. This is currently being tested in the STOP-IgAN trial [52]. Newer immunosuppressives (alone or in combination) should be compared in RCTs to a “control” group receiving corticosteroids alone.
10.4: Immunosuppressive agents (cyclophosphamide, azathioprine, MMF, cyclosporine) 10.4.1: We suggest not treating with corticosteroids combined with
cyclophosphamide or azathioprine in IgAN patients (unless there is crescentic IgAN with rapidly deteriorating kidney function; see Recommendation 10.6.3). (2D)
10.4.2: We suggest not using immunosuppressive therapy in patients with GFR <30 ml/min unless there is crescentic IgAN with rapidly deteriorating kidney function (see Section 10.6). (2C)
10.4.3: We suggest not using MMF in IgAN. (2C)
RATIONALE
(See Suppl Table 49-57) • There is very low–quality evidence from a single RCT in high-risk adults to use a
combination of prednisolone (40 mg/d reduced to 10 mg/d by 2 years) and cyclophosphamide (1.5 mg/kg/d) for 3 months, followed by azathioprine (1.5 mg/kg/d) for a minimum of 2 years. This study showed a better kidney survival over controls in a highly selected group of patients.
• There is insufficient evidence that immunosuppressive agents other than steroids used as first-line therapy offer an advantage or equivalence compared to steroids.
• The risk-benefit assessment is strongly impacted by the potential for severe adverse effects of these drugs.
Despite retrospective studies in IgAN supporting the use of immunosuppressive therapy
other than corticosteroids, few RCTs have demonstrated a benefit. An RCT using corticosteroids

KDIGO® GN Guideline March 2011 Public Review Draft
122
combined with cyclophosphamide, followed by azathioprine, included a highly selected group of patients with SCr >130-250 µmol/L with a 15% increase within the last year, and initial proteinuria 3.9 ± 0.8 and 4.6 ± 0.4 g/d in the treatment and control groups, respectively. The active treatment group achieved lower proteinuria, a 4-fold lower rate of kidney function decline, and a much greater kidney survival (72% 5-year survival compared to 6% in controls, P = 0.006). There are limitations in the applicability of the findings: i) there was no steroid monotherapy arm; ii) the use of RAS blockade was not detailed but these agents could not be initiated after the start of the trial; iii) the follow-up blood pressure was higher than recommended by current guidelines.
Two RCTs compared cyclophosphamide, dipyridamole, and warfarin to controls and found no benefit [53,54]. Given these results and the potential side-effects, we do not suggest the use of cyclophosphamide monotherapy.
Azathioprine Two RCTs, one in children and another in children and adults, tested azathioprine and
corticosteroids in patients with preserved kidney function. They demonstrated a reduction in chronic lesions compared to controls on repeat biopsy [15,55]. Monotherapy with steroids has been shown to preserve kidney function (a plausible surrogate for reduced chronic lesions). In a recent trial in patients with IgAN [56], adding low-dose azathioprine for 6 months did not increase the benefit of corticosteroids alone, but may increase the occurrence of adverse events.
A study in 80 children with newly diagnosed IgAN [57] compared the effects of the combination of prednisolone, azathioprine, warfarin, and dipyridamole with those of prednisolone alone. There was complete remission of proteinuria (<0.1 g/m2/d) in 36 (92.3%) of the 39 patients who received the combination and 29 (74.4%) of the 39 who received prednisolone alone (P = 0.007). Some side-effects were observed including leucopenia, glaucoma, and aseptic necrosis. The percentage of sclerosed glomeruli was unchanged in the patients who received the combination, but increased in the prednisolone group. In summary of these studies, we do not suggest the addition of azathioprine to corticosteroids for the treatment of IgAN.
An RCT compared 6 months of treatment with corticosteroids plus azathioprine or corticosteroids alone in 207 IgAN patients with plasma creatinine ≤2.0 mg/dl (≤176.8 µmol/l) and proteinuria ≥1 g/d. After a median follow-up of 4.9 years, a 50% increase in plasma creatinine from baseline occurred in 13% of the combination group and 11% of the monotherapy group (P = 0.83); effects on proteinuria and 5-year cumulative kidney survival were also similar in both two groups (88% vs. 89%; P = 0.83). Treatment-related adverse events were more frequent in the combination group (17%) as compared to the monotherapy group (6%; P = 0.01). Thus, in this study, 6 months of treatment with azathioprine did not increase the benefit obtained from steroids alone, but increased the occurrence of adverse events [56].
MMF The findings from RCTs studying MMF in IgAN are variable. A Belgian study [58] assessed
MMF 2 g/d for 3 years vs. placebo in 34 patients with an average initial inulin clearance of 70 ml/min per 1.73 m2 and proteinuria of 1.8 g/d. No difference in proteinuria reduction or

KDIGO® GN Guideline March 2011 Public Review Draft
123
preservation of GFR was observed. Similarly, a North American study [14] found no benefits over 24 months using a 1-year regimen of MMF 2 g/d vs. placebo in 32 patients with an initial GFR of 40 ml/min per 1.73 m2 and a proteinuria of 2.7 g/d. In contrast, a Chinese study in 40 patients with a mean initial GFR of 72 ml/min per 1.73 m2 and mean proteinuria 1.8 g/d found a significant reduction in proteinuria at 18 months with MMF given for 6 months over controls [59]. A 6-year follow-up of the same cohort demonstrated a kidney survival benefit [60]. No steroid was given in these trials, and all patients received ACEi. The results of these studies are too heterogeneous to suggest the use of MMF at the present time. The reasons for heterogeneity of outcome require furher investigation, but different ethnicity or differences in drug levels achieved may be contributory factors.
Steroid Resistance The approach to patients, who have no benefit in response to corticosteroids added to
optimal antihypertensive and antiproteinuric therapy is unknown; no relevant RCTs have been conducted.
RESEARCH RECOMMENDATIONS
• An RCT is needed comparing MMF to corticosteroids vs. corticosteroids alone in patients receiving optimal antihypertensive and antiproteinuric therapy.
• An RCT is needed to investigate the different efficacy of MMF in Asians vs. Caucasians, including evaluation of drug and metabolite levels.
10.5: Other treatments 10.5.1: Fish oil treatment (Suppl Table 58)
10.5.1.1: We suggest using fish oil in the treatment of IgAN. (2D)
BACKGROUND
Fish oil supplements have shown a number of beneficial cardiovascular effects, including systolic blood pressure and triglyceride lowering, reduced resting heart rate, improvement in several markers of endothelial damage, and reduction in the risk of sudden cardiac death in patients with established coronary heart disease. Several RCTs have evaluated the effect of fish oil in IgAN.
RATIONALE
• There is mostly low-quality evidence that suggests using fish oil supplements in patients with IgAN, but the RCTs evaluating this therapy have reported conflicting results. However, given the very low risk profile and the potentially beneficial cardiovascular effects, fish oil can be considered a very safe treatment.
In a trial that included 106 patients, fish oil treatment (12 g/d) improved kidney survival and
retarded the rate of kidney function loss, without significant reduction of proteinuria [61]. Of

KDIGO® GN Guideline March 2011 Public Review Draft
124
note, the outcome of the control group, treated with 12 g/d of olive oil was poor (cumulative incidence of death or ESRD after 4 years was 10% in fish oil–treated patients, 40% in the control group). Longer follow-up confirmed the beneficial influence of fish oil treatment in this study [62]. Another RCT including 34 patients reported a beneficial influence of fish oil (3 g/d) on two end-points: the risk of ESRD, and ≥50% increase in SCr [63]. In this study, fish oil reduced proteinuria significantly. In a short-term (6-month) RCT, a significant proteinuria reduction was observed in patients treated with a combination of ACEi and ARB plus fish oil (3 g/d) in comparison to a control group that received only ACEi and ARB (percentage of patients with ≥50% proteinuria reduction, 80% and 20%, respectively) [64].
In contradiction to these studies, other RCTs failed to detect a significant benefit of fish oil treatment [65,66]. A meta-analysis [67] concluded that fish oils are not beneficial in IgAN. A more recent RCT compared steroids (33 patients), fish oil (4 g/d, 32 patients), and placebo (31 patients) for 2 years [48]. Neither treatment group showed benefit over the placebo group. However, patients in the placebo group had a statistically significant lower degree of proteinuria at baseline.
Trying to explain these discordant results, some authors have proposed that the effects of fish oil in IgAN patients could be dosage-dependent [68]. However, another prospective trial reported that high (6.7 g/d) and low (3.3 g/d) doses of fish oil were similar in slowing the rate of kidney function loss in high-risk IgAN patients [69]. At present, there is no evidence to support the use of high-dose fish oil in IgAN.
RESEARCH RECOMMENDATIONS
• An RCT is needed of fish oil in IgAN to examine preserved kidney function with persistent significant proteinuria, despite optimal antihypertensive and antiproteinuric therapy.
10.5.2: Antiplatelet agents (Suppl Table 62) 10.5.2.1: We suggest not using antiplatelet agents to treat IgAN. (2C)
RATIONALE
• There is low-quality evidence to recommend not using antiplatelet therapy in IgAN.
A meta-analysis [70], based on seven studies, most of them performed in Japan, concluded that antiplatelet therapy resulted in reduced proteinuria and protected kidney function in patients with moderate to severe IgAN. However, there were significant limitations of the evidence in this meta-analysis, due to suboptimal quality of individual controlled trials. Importantly, the effect of antiplatelet agents alone could not be discerned because patients received other concomitant therapies. Thus, in three studies, both treatment and control groups received other agents, including cytotoxics, steroids, antihypertensive agents, and anticoagulants. In three other studies, the intervention group received warfarin (two studies) and aspirin (one study) in addition

KDIGO® GN Guideline March 2011 Public Review Draft
125
to the antiplatelet agent (dipyridamole). Dipyridamole was the most commonly used antiplatelet agent (five studies) followed by trimetazidine and Dilazep (one study each).
RESEARCH RECOMMENDATION
• A multicenter RCT is needed to address the role of antiplatelet therapy in IgAN.
10.5.3: Tonsillectomy 10.5.3.1: We suggest that tonsillectomy not be performed for IgAN. (2C)
RATIONALE
• There is low-quality evidence to suggest not using tonsillectomy as treatment for IgAN. No RCT has been performed of tonsillectomy for IgAN.
Tonsillectomy may be indicated in those with IgAN for conventional reasons, e.g., recurrent
bacterial tonsillitis. However, only retrospective analyses [71,72] as well as one nonrandomized trial [73] have reported a better outcome for IgAN after tonsillectomy. In these studies, tonsillectomy was often combined with other— in particular, immunosuppressive—treatment [71-73]; thus, the specific value of tonsillectomy is not always apparent. Furthermore, in other retrospective series, investigators failed to note a benefit from tonsillectomy [74].
RESEARCH RECOMMENDATION
• A multicenter RCT is needed to address the role of tonsillectomy in IgAN.
10.6: Atypical forms of IgAN 10.6.1: MCD with mesangial IgA deposits
10.6.1.1: We recommend treatment as forMCD (see Chapter 5) in nephrotic patients showing pathological findings of MCD with mesangial IgA deposits on kidney biopsy. (2B)
BACKGROUND
Patients with IgAN can present with proteinuria within the nephrotic range (>3.5 g/d), and it portends a poor prognosis if this high-grade proteinuria persists during follow-up. However, the typical accompanying findings of complete nephrotic syndrome (edema, hypoalbuminemia, hyperlipidemia) are uncommon. Rarely, some patients with nephrotic syndrome have been identified in whom kidney biopsy shows minimal glomerular changes by light microscopy, diffuse podocyte foot processes effacement on electron microscopy, and predominant mesangial deposits of IgA on immunofluorescence. A coincidence of two different glomerular diseases (minimal-change nephrotic syndrome and IgAN) has been proposed as the most likely explanation for such cases.

KDIGO® GN Guideline March 2011 Public Review Draft
126
RATIONALE
• There is low-quality evidence to recommend that patients with nephrotic syndrome and coincidental histological findings of MCD and IgAN should be treated like patients with MCD.
Several series [75,76] have described prompt, complete remissions after corticosteroid
therapy in a majority of patients with nephrotic syndrome and a pathological diagnosis of coincidental MCD and IgAN. This initial treatment response and the following clinical course, with a frequent appearance of nephrotic syndrome relapses, are very reminiscent of that of patients with pure MCD. A RCT in IgAN patients with nephrotic proteinuria [77] also showed a high percentage of complete remission in patients with such characteristics.
10.6.2: AKI associated with macroscopic hematuria 10.6.2.1: Perform a repeat kidney biopsy in IgAN patients with AKI
associated with macroscopic hematuria if, after 5 days from the onset of kidney function worsening, there is no improvement. (Not Graded)
10.6.2.2: We suggest general supportive care for AKI in IgAN, with a kidney biopsy performed during an episode of macroscopic hematuria showing only ATN and intratubular erythrocyte casts. (2C)
BACKGROUND
Episodic macroscopic hematuria coinciding with mucosal (usually upper respiratory) infections are typical of IgAN. The macroscopic hematuria usually resolves spontaneously in a few days, but in some cases it can persist for several weeks [78]. The development of AKI during macroscopic hematuria episodes is uncommon [78,79] but represents the first manifestation of IgAN in some patients.
RATIONALE
• ATN and intratubular erythrocyte casts are the most common pathological findings in kidney biopsies during AKI accompanying macroscopic hematuria episodes in IgAN.
• Kidney function usually, but not always, recovers completely after the disappearance of macroscopic hematuria.
• Kidney biopsy allows differentiation of tubular damage and tubular occlusion by erythrocyte casts from crescentic IgAN or other coincidental causes of AKI.
Kidney biopsies performed during an episode of macroscopic hematuria typically show
mesangial proliferation and occasional segmental crescents [80]. In those cases with AKI coincidental with gross hematuria, the glomerular changes are usually insufficient to account for the AKI. Hematuria by itself may be responsible for the AKI, through tubular injury that is

KDIGO® GN Guideline March 2011 Public Review Draft
127
induced by intratubular erythrocytic casts and a possible nephrotoxic effect of the hemoglobin that is released from these casts. Features of ATN and tubules filled by red blood cells are the most relevant histological findings. In a majority of patients, kidney function returns to baseline after the disappearance of macroscopic hematuria [78-80], but incomplete recovery of kidney function has been described in up to 25% of affected patients [78]. Duration of macroscopic hematuria longer than 10 days is the most significant risk factor for persistent kidney impairment [78]. Continuous supportive care, as in other types of ATN, is the recommended therapeutic approach to these patients. There is no information about the usefulness of corticosteroids in patients with more severe forms of AKI or longer duration of macroscopic hematuria.
However, some patients with AKI and macroscopic hematuria exhibit a crescentic form of IgAN (crescents affecting >50% of glomeruli), whose prognosis is considerably worse [24-27]. A repeat kidney biopsy is suggested in patients with known IgAN who present a protracted AKI accompanying a new episode of gross hematuria, in order to differentiate ATN from crescentic IgAN or other types of AKI (Algorithm 1).
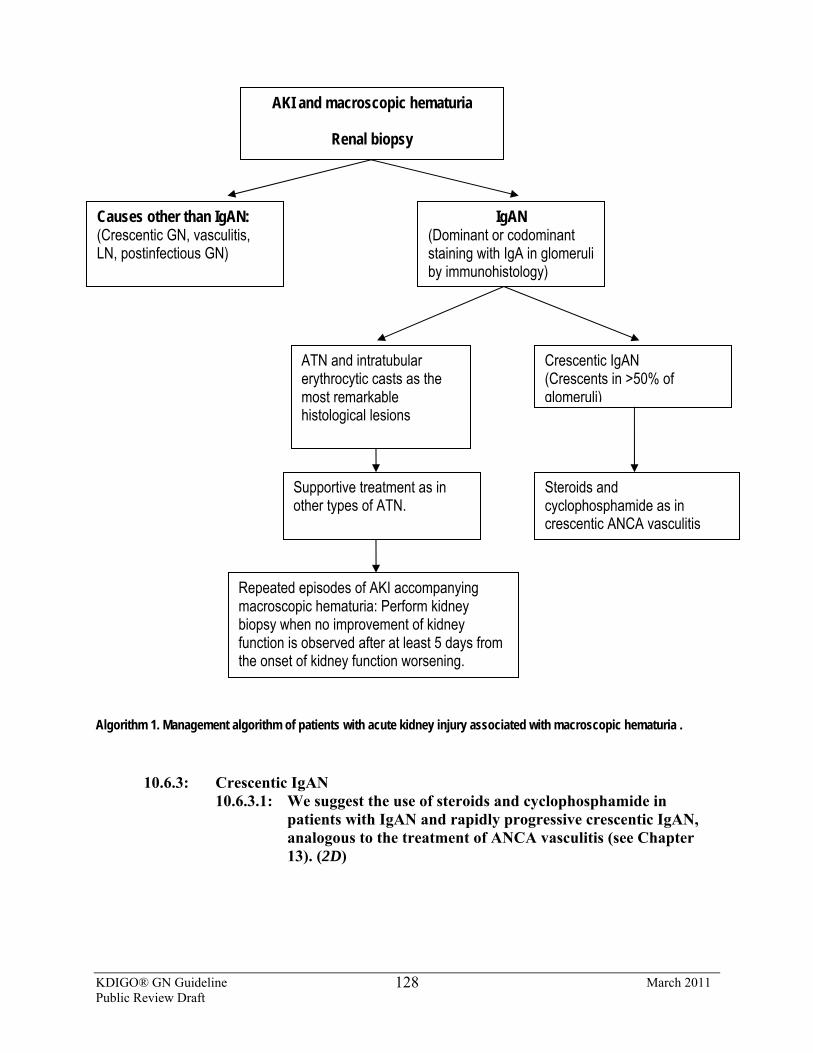
KDIGO® GN Guideline March 2011 Public Review Draft
128
Algorithm 1. Management algorithm of patients with acute kidney injury associated with macroscopic hematuria .
10.6.3: Crescentic IgAN 10.6.3.1: We suggest the use of steroids and cyclophosphamide in
patients with IgAN and rapidly progressive crescentic IgAN, analogous to the treatment of ANCA vasculitis (see Chapter 13). (2D)
AKI and macroscopic hematuria
Renal biopsy
Causes other than IgAN: (Crescentic GN, vasculitis, LN, postinfectious GN)
IgAN (Dominant or codominant staining with IgA in glomeruli by immunohistology)
ATN and intratubular erythrocytic casts as the most remarkable histological lesions
Crescentic IgAN (Crescents in >50% of glomeruli)
Supportive treatment as in other types of ATN.
Steroids and cyclophosphamide as in crescentic ANCA vasculitis
Repeated episodes of AKI accompanying macroscopic hematuria: Perform kidney biopsy when no improvement of kidney function is observed after at least 5 days from the onset of kidney function worsening.

KDIGO® GN Guideline March 2011 Public Review Draft
129
BACKGROUND
Crescentic IgAN has a poor prognosis. In a historical group of 12 untreated crescentic IgAN patients, about 42% reached ESRD in 36 months [27]. Another Japanese study showed that patients with >50% crescents developed ESRD in 75% of patients by 10 years follow-up [81].
The outcome of IgAN with diffuse crescent formation has been studied in 25 Chinese IgAN patients [82]. Most of them showed rapidly progressive GN associated with more severe pathological changes, including glomerular, tubular interstitial, and vascular lesions, than in patients with general IgAN. The infiltrates in glomeruli may contribute to the crescentic formation.
A recent study of 67 patients [83] with vasculitic IgAN (33 HSP, 34 IgAN) showed that three factors significantly affected kidney outcome: kidney function, blood pressure at presentation, and the amount of chronic damage in the biopsy.
RATIONALE
• There is no RCT of treatment in crescentic IgAN.
The three largest observational studies [27,82,83] all concluded that immunosuppression is potentially useful. In a study of 25 patients with diffuse crescentic IgAN treated with immunosuppression, 67% of patients maintained sufficient kidney function to avoid renal replacement therapy, four had SCr <124 µmol/l (<1.4 mg/dl), and only five were dialysis-dependent. In another study, although an improved outcome was seen in those receiving immunosuppression, the conclusions were cautious, as the treated and untreated groups were not comparable. The third study also suggested positive effects of immunosupression [27]. This study used i.v. methylprednisolone 15 mg/kg/d for 3 days and monthly i.v. cyclophosphamide 0.5 g/m2 for 6 months. Twelve treated patients were compared to 12 historical controls. After 36 months, the rate of ESRD in the treated group was lower (one out of 12) than in the historical controls (five out of 12).
Recommended therapeutic regimens in these reports are varied, but initial therapy has usually included high-dose oral or i.v. corticosteroids plus oral or i.v. cyclophosphamide. In one study, some patients were changed from cyclophosphamide to azathioprine at 3 months. Durations of treatment in these three series varied from 3 to 24 months.
Please refer to Supplemental Tables 42-67 for additional data related to Chapter 10.
References 1. Roberts IS, Cook HT, Troyanov S, Alpers CE, Amore A, Barratt J, Berthoux F, Bonsib S, Bruijn JA, Cattran
DC, Coppo R, D'Agati V, D'Amico G, Emancipator S, Emma F, Feehally J, Ferrario F, Fervenza FC, Florquin S, Fogo A, Geddes CC, Groene HJ, Haas M, Herzenberg AM, Hill PA, Hogg RJ, Hsu SI, Jennette JC, Joh K, Julian BA, Kawamura T, Lai FM, Li LS, Li PKT, Liu ZH, Mackinnon B, Mezzano S, Schena FP, Tomino Y, Walker PD, Wang H, Weening JJ, Yoshikawa N, Zhang H. A Working Group of the International

KDIGO® GN Guideline March 2011 Public Review Draft
130
IgAN Network and the Renal Pathology Society. The Oxford classification of IgAN: pathology definitions, correlations, and reproducibility. Kidney Int 2009; 76: 546-56
2. Li PKT, Ho KKL, Szeto CC, Yu LM, Lai FM. Prognostic indicators of IgAN in the Chinese – Clinical and pathological perspectives. Nephrol Dial Transplant 2002; 17:64-69
3. Nair R, Walker PD. Is IgAN the commonest primary glomerulopathy among young adults in the USA? Kidney Int 2006; 69: 1455-8.
4. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY: Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization, N Engl J Med 2004, 351:1296-1305
5. Reich HN, Troyanov S, Scholey JW, Cattran DC: Remission of proteinuria improves prognosis in IgAN. J Am Soc Nephrol 2007, 18:3177-3183
6. Geddes CC, Rauta V, Gronhagen-Riska C, Bartosik LP, Jardine AG, Ibels LS, Pei Y, Cattran DC. A tricontinental view of IgAN. Nephrol Dial Transplant 2003; 18:1541-8.
7. Goto M, Wakai K, Kawamura T, Ando M, Endoh M, Tomino Y. A scoring system to predict renal outcome in IgA nephropathy: a nationwide 10-year prospective cohort study. Nephrol Dial Transplant. 2009; 24: 3068-74
8. Donadio JV, Bergstralh EJ, Grande JP, Rademcher DM. Proteinuria patterns and their association with subsequent end-stage renal disease in IgA nephropathy. Nephrol Dial Transplant. 2002; 17: 1197-203
9. Coppo R, Peruzzi L, Amore A, Piccoli A, Cochat P, Stone R, Kirschstein M, Linné T. IgACE: a placebo-controlled, randomized trial of angiotensin-converting enzyme inhibitors in children and young people with IgAN and moderate proteinuria. J Am Soc Nephrol 2007; 18: 1880-8
10. Cattran DC, Coppo R, Cook HT, Feehally J, Roberts IS, Troyanov S, Alpers CE, Amore A, Barratt J, Berthoux F, Bonsib S, Bruijn JA, D'Agati V, D'Amico G, Emancipator S, Emma F, Ferrario F, Fervenza FC, Florquin S, Fogo A, Geddes CC, Groene HJ, Haas M, Herzenberg AM, Hill PA, Hogg RJ, Hsu SI, Jennette JC, Joh K, Julian BA, Kawamura T, Lai FM, Leung CB, Li LS, Li PKT, Liu ZH, Mackinnon B, Mezzano S, Schena FP, Tomino Y, Walker PD, Wang H, Weening JJ, Yoshikawa N, Zhang H. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int 2009;76: 534-45
11. Roberts, ISD., et al. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility Kidney Int 76: 546-556, 2009
12. Szeto, CC, Lai, F, To, KF, Wong TY, Chow KM, Choi PC, Lui SF, Li PKT. The natural history of immunoglobulin A nephropathy among patients with hematuria and minimal proteinuria. Am J Med 2001; 110:434-7
13. Coppo R, D'Amico G. Factors predicting progression of IgA nephropathies. J Nephrol. 2005;18:503-12. 14. Frisch G, Lin J, Rosenstock J, Markowitz G, D'Agati V, Radhakrishnan J, Preddie D, Crew J, Valeri A, Appel
G. Mycophenolate mofetil (MMF) vs placebo in patients with moderately advanced IgA nephropathy: a double-blind randomized controlled trial. Nephrol Dial Transplant. 2005; 20: 2139-45
15. Harmankaya O, Ozturk Y, Basturk T, Obek A, Kilicarslan I: Efficacy of immunosuppressive therapy in IgAN presenting with isolated hematuria, Int Urol Nephrol 2002, 33:167-171
16. Bartosik, LP, Lajoie, G, Sugar, L, Cattran, DC. Predicting progression in IgAN. Am J Kidney Dis 2001; 38:728.
17. Kanno Y, Okada H, Saruta T, Suzuki H. Blood pressure reduction associated with preservation of kidney function in hypertensive patients with IgAN: a 3-year follow-up. Clin Nephrol 54: 360-365, 2000.
18. Sarafidis PA, Khosla N, Bakris GL. Antihypertensive therapy in the presence of proteinuria. Am J Kidney Dis 2007; 49: 12-26.
19. Jafar TH, Schmid CH, Landa M et al: Angiotensin-converting enzyme inhibitors and progression of nondiabetic renal disease. A meta-Analysis of Patient-Level data. Ann Intern Med 135: 73-87, 2001.
20. Fellin G, Gentile MG, Duca G, D'Amico G. Kidney function in IgA nephropathy with established renal failure. Nephrol Dial Transplant. 1988;3: 17-23
21. Ballardie FW, Roberts IS: Controlled prospective trial of prednisolone and cytotoxics in progressive IgAN, J Am Soc Nephrol 2002, 13:142-148

KDIGO® GN Guideline March 2011 Public Review Draft
131
22. Rekola S, Bergstrand A, Bucht H. Deterioration of GFR in IgA nephropathy as measured by 51Cr-EDTA clearance. Kidney Int. 1991 Dec;40(6):1050-4
23. D'Amico G, Minetti L, Ponticelli C, Fellin G, Ferrario F, Barbiano di Belgioioso G, Imbasciati E, Ragni A, Bertoli S, Fogazzi G, et al. Prognostic indicators in idiopathic IgA mesangial nephropathy. Q J Med. 1986; 59: 363-78.
24. Freese P, Nordén G, Nyberg G. Morphologic high-risk factors in IgA nephropathy. Nephron. 1998; 79: 420-5 25. Hogg RJ, Silva FG, Wyatt RJ, Reisch JS, Argyle JC, Savino DA. Prognostic indicators in children with IgA
nephropathy--report of the Southwest Pediatric Nephrology Study Group. Pediatr Nephrol. 1994; 8: 15-20 26. Boyce NW, Holdsworth SR, Thomson NM, Atkins RC. Clinicopathological associations in mesangial IgA
nephropathy. Am J Nephrol. 1986; 6: 246-52 27. Tumlin JA, Lohavichan V, Hennigar R. Crescentic, proliferative IgA nephropathy: clinical and histological
response to methylprednisolone and intravenous cyclophosphamide. Nephrol Dial Transplant. 2003; 18: 1321-9.
28. Katafuchi R, Oh Y, Hori K, Komota T, Yanase T, Ikeda K, Omura T, Fujimi S. An important role of glomerular segmental lesions on progression of IgA nephropathy: a multivariate analysis. Clin Nephrol. 1994; 41: 191-8.
29. Packham DK, Yan HD, Hewitson TD, Nicholls KM, Fairley KF, Kincaid-Smith P, Becker GJ. The significance of focal and segmental hyalinosis and sclerosis (FSHS) and nephrotic range proteinuria in IgA nephropathy. Clin Nephrol. 1996; 46: 225-9
30. Hsu CY, McCulloch CE, Iribarren C, Darbinian J, Go AS. Body mass index and risk for end-stage renal disease. Ann Intern Med 2006; 144: 21-28
31. Navaneethan SD, Yehnert H, Moustarah F, Schreiber MJ, Schauer PR, Beddhu S. Weight Loss Interventions in Chronic Kidney Disease: A Systematic Review and Meta-analysis.. Clin J Am Soc Nephrol 2009; 4: 1565-1574.
32. Bonnet F, Deprele C, Sassolas A, Moulin P, Alamartine E, Berthezene F et al. Excessive body weight as a new independent risk factor for clinical and pathological progression in primary IgA nephritis. Am J Kidney Dis 2001; 37: 720-727.
33. Tanaka M, Tsujii T, Komiya T, Iwasaki Y, Sugishita T, Yonemoto S, Tsukamoto T, Fukui S, Takasu A, Muso E. Clinicopathological influence of obesity in IgAN: comparative study of 74 patients. Contrib Nephrol 2007; 157: 90-93.
34. Cattran DC, Reich HN, Beanlands HJ, Miller JA, Scholey JW, Troyanov S; Genes, Gender and Glomerulonephritis Group. The impact of sex in primary glomerulonephritis. Nephrol Dial Transplant. 2008; 23: 2247-53
35. Coppo R, Troyanov S, Camilla R, Hogg RJ, Cattran DC, Cook HT, Feehally J, Roberts IS, Alpers CE, Amore A, Alpers CE, Barratt J, Berthoux F, Bonsib S, Bruijn JA, D'Agati V, D'Amico G, Emancipator S, Emma F, Ferrario F, Fervenza FC, Florquin S, Fogo A, Geddes CC, Groene HJ, Haas M, Herzenberg AM, Hill PA, Hsu SI, Jennette JC, Joh K, Julian BA, Kawamura T, Lai FM, Li LS, Li PK, Liu ZH, Mezzano S, Schena FP, Tomino Y, Walker PD, Wang H, Weening JJ, Yoshikawa N, Zhang H. Does a new IgA nephropathy clinicopathological classification have the same value in children and adults? Kidney Int 2010; 77: 921-7.
36. Rauta V, Finne P, Fagerudd J, Rosenlöf K, Törnroth T, Grönhagen-Riska C. Factors associated with progression of IgA nephropathy are related to kidney function--a model for estimating risk of progression in mild disease. Clin Nephrol. 2002; 58: 85-94.
37. Li PKT, Leung CB, Chow KM, Cheng YL, Fung SK, Mak SK, Tang AW, Wong TY, Yung CY, Yung JC, Yu AW, Szeto CC. Hong Kong study using Valsartan in IgAN (HKVIN) – A double-blind randomized placebo-controlled study. Am J Kidney Dis 2006; 47: 751-60.
38. Praga M, Gutierrez E, Gonzalez E, Morales E, Hernandez E: Treatment of IgAN with ACE inhibitors: A randomized and controlled trial. J Am Soc Nephrol 2003, 14:1578-1583
39. Horita Y, Tadokoro M, Taura K, Ashida R, Hiu M, Taguchi T, Furusu A, Kohno S: Prednisolone co-administered with losartan confers renoprotection in patients with IgAN. Ren Fail 2007; 29:441-446

KDIGO® GN Guideline March 2011 Public Review Draft
132
40. Russo D, Pisani A, Balletta MM, De Nicola L, Savino FA, Andreucci M, Minutolo R., Additive antiproteinuric effect of converting enzyme inhibitor and losartan in normotensive patients with IgA nephropathy. Am J Kidney Dis 1999; 33:851-6
41. Yang Y, Ohta K, Shimizu M, Nakai A, Kasahara Y, Yachie A, Koizumi S. Treatment with low-dose angiotensin-converting enzyme inhibitor (ACEI) plus angiotensin II receptor blocker (ARB) in pediatric patients with IgA nephropathy. Clin Nephrol. 2005; 64: 35-40.
42. Pozzi C, Bolasco PG, Fogazzi GB, Andrulli S, Altieri P, Ponticelli C, Locatelli F: Corticosteroids in IgAN: a randomised controlled trial, Lancet 1999, 353:883-887
43. Manno C, Torres DD, Rossini M, Pesce F, Schena FP: Randomized controlled clinical trial of corticosteroids plus ACE-inhibitors with long-term follow-up in proteinuric IgAN, Nephrol Dial Transplant 2009, 24:3694-3701
44. Lv J, Zhang H, Chen Y, Li G, Jiang L, Singh AK, Wang H: Combination therapy of prednisone and ACE inhibitor versus ACE-inhibitor therapy alone in patients with IgAN: a randomized controlled trial, Am J Kidney Dis 2009, 53:26-32
45. Pozzi C, Andrulli S, Del Vecchio L, Melis P, Fogazzi GB, Altieri P, Ponticelli C, Locatelli F: Corticosteroid effectiveness in IgAN: long-term results of a randomized, controlled trial, J Am Soc Nephrol 2004, 15:157-163
46. Pozzi C, Locatelli F: Corticosteroids in IgAN [letter], Lancet 1999; 353:2159-2160 47. Manno C, Gesualdo L, D'Altri C, Rossini M, Grandaliano G, Schena FP: Prospective randomized controlled
multicenter trial on steroids plus ramipril in proteinuric IgAN, J Nephrol 2001, 14:248-252 48. Hogg RJ, Lee J, Nardelli N, Julian BA, Cattran D, Waldo B, Wyatt R, Jennette JC, Sibley R, Hyland K,
Fitzgibbons L, Hirschman G, Donadio JV, Jr., Holub BJ: Clinical trial to evaluate omega-3 fatty acids and alternate day prednisone in patients with IgAN: report from the Southwest Pediatric Nephrology Study Group, Clin J Am Soc Nephrol 2006, 1:467-474
49. Katafuchi R, Ikeda K, Mizumasa T, Tanaka H, Ando T, Yanase T, Masutani K, Kubo M, Fujimi S: Controlled, prospective trial of steroid treatment in IgAN: a limitation of low-dose prednisolone therapy, Am J Kidney Dis 2003, 41:972-983
50. Strippoli GF, Maione A, Schena FP, Tognoni G, Craig JC. IgAN: a disease in search of a large-scale clinical trial to reliably inform practice. Am J Kidney Dis. 2009, 53:5-8.
51. Kobayashi Y, Hiki Y, Kokubo T, Horii A, Tateno S. Steroid therapy during the early stage of progressive IgAN. A 10-year follow-up study. Nephron 1996;72(2):237-42.
52. Eitner F, Ackermann D, Hilgers RD, Floege J: Supportive Versus Immunosuppressive Therapy of Progressive IgAN (STOP) IgAN trial: rationale and study protocol, J Nephrol 2008, 21:284-289
53. Walker RG, Yu SH, Owen JE, Kincaid-Smith P. The treatment of mesangial IgA nephropathy with cyclophosphamide, dipyridamole and warfarin: a two-year prospective trial. Clin Nephrol. 1990; 34: 103-7
54. Woo KT, Lee GS. The treatment of mesangial IgA nephropathy with cyclophosphamide, dipyridamole and warfarin. Clin Nephrol. 1991; 35: 184
55. Yoshikawa N, Ito H, Sakai T, Takekoshi Y, Honda M, Awazu M, Ito K, Iitaka K, Koitabashi Y, Yamaoka K, Nakagawa K, Nakamura H, Matsuyama S, Seino Y, Takeda N, Hattori S, Ninomiya M. A controlled trial of combined therapy for newly diagnosed severe childhood IgA nephropathy. The Japanese Pediatric IgA Nephropathy Treatment Study Group. J Am Soc Nephrol. 1999; 10: 101-9
56. Pozzi C, Andrulli S, Pani A, Scaini P, Del Vecchio L, Fogazzi G, Vogt B, De Cristofaro V, Allegri L, Cirami L, Procaccini AD, Locatelli F: Addition of Azathioprine to Corticosteroids Does Not Benefit Patients with IgA Nephropathy. JASN 2010 21: 1783-1790
57. Yoshikawa N, Honda M, Iijima K, Awazu M, Hattori S, Nakanishi K, Ito H; Japanese Pediatric IgA Nephropathy Treatment Study Group. Steroid treatment for severe childhood IgA nephropathy: a randomized, controlled trial. Clin J Am Soc Nephrol. 2006;1:511-7
58. Maes BD, Oyen R, Claes K, Evenepoel P, Kuypers D, Vanwalleghem J, Van Damme B, Vanrenterghem YF. Mycophenolate mofetil in IgA nephropathy: results of a 3-year prospective placebo-controlled randomized study. Kidney Int. 2004; 65: 1842-9

KDIGO® GN Guideline March 2011 Public Review Draft
133
59. Tang S, Leung JC, Chan LY, Lui YH, Tang CS, Kan CH, Ho YW, Lai KN. Mycophenolate mofetil alleviates persistent proteinuria in IgA nephropathy. Kidney Int. 2005; 68: 802-12.
60. Tang SC, Tang AW, Wong SS, Leung JC, Ho YW, Lai KN. Long-term study of mycophenolate mofetil treatment in IgA nephropathy. Kidney Int. 2010; 77: 543-9
61. Donadio JV Jr, Bergstralh EJ, Offord KP, Spencer DC, Holley KE. A controlled trial of fish oil in IgAN. Mayo Nephrology Collaborative Group. N Engl J Med 1994; 331:1194-9.
62. Donadio JV Jr, Grande JP, Bergstralh EJ, Dart RA, Larson TS, Spencer DC. The long-term outcome of patients with IgAN treated with fish oil in a controlled trial. Mayo Nephrology Collaborative Group. J Am Soc Nephrol 1999; 10: 1772-7
63. Alexopoulos E, Stangou M, Pantzaki A, Kirmizis D, Memmos D.Treatment of severe IgAN with omega-3 fatty acids: the effect of a "very low dose" regimen. Ren Fail 2004; 26:453-9
64. Ferraro PM, Ferraccioli GF, Gambaro G, Fulignati P, Costanzi S.Combined treatment with renin-angiotensin system blockers and polyunsaturated fatty acids in proteinuric IgAN: a randomized controlled trial.Nephrol Dial Transplant. 2009 ;24:156-60
65. Bennett WM, Walker RG, Kincaid-Smith P. Treatment of IgAN with eicosapentanoic acid (EPA): a two-year prospective trial. Clin Nephrol 1989 ; 31:128-31
66. Pettersson EE, Rekola S, Berglund L, Sundqvist KG, Angelin B, Diczfalusy U, Bjorkhem I, Bergstrom J. Treatment of IgAN with omega-3-polyunsaturated fatty acids: a prospective, double-blind, randomized study. Clin Nephrol 1994; 41:183-90
67. Strippoli GF, Manno C, Schena FP. An "evidence-based" survey of therapeutic options for IgAN: assessment and criticism. Am J Kidney Dis 2003 ; 41:1129-39.
68. Hogg RJ, Fitzgibbons L, Atkins C, Nardelli N, Bay RC; North American IgAN Study Group. Efficacy of Omega-3 Fatty Acids in Children and Adults with IgAN Is Dosage- and Size-Dependent. Clin J Am Soc Nephrol 2006; 1: 1167–1172 b
69. Donadio JV, Larson TS, Bergstrahl EJ, Grande JP. A randomized trial of high-dose compared to low-dose omega-3 fatty acids in severe IgAN. J Am Soc Nephol 2001; 12:791-9.
70. Taji Y, Kuwahara T, Shikata S, Morimoto T. Meta-analysis of antiplatelet therapy for IgAN. Clin Exp Nephrol 2006; 10: 268-273.
71. Hotta O, Miyazaki M, Furuta T, Tomioka S, Chiba S, Horigome I, Abe K, Taguma Y: Tonsillectomy and steroid pulse therapy significantly impact on clinical remission in patients with IgAN, Am J Kidney Dis 2001, 38:736-743.
72. Xie Y, Nishi S, Ueno M, Imai N, Sakatsume M, Narita I, Suzuki Y, Akazawa K, Shimada H, Arakawa M, Gejyo F: The efficacy of tonsillectomy on long-term renal survival in patients with IgAN, Kidney Int 2003, 63:1861-1867
73. Komatsu H, Fujimoto S, Hara S, Sato Y, Yamada K, Kitamura K: Effect of tonsillectomy plus steroid pulse therapy on clinical remission of IgAN: a controlled study, Clin J Am Soc Nephrol 2008, 3:1301-1307
74. Rasche FM, Schwarz A, Keller F: Tonsillectomy does not prevent a progressive course in IgAN, Clin Nephrol 1999, 51:147-152
75. Lai KN, Lai FM, Chan KW, Ho CP, Leung CP, Vallance-Owen J. An overlapping syndrome of IgAN and lipoid nephrosis. Am J Clin Pathol 1986; 86: 716-723
76. Kim SM, Moon KC, Oh KH, Joo KW, Kim YS, Ahn C, Han JS, Kim S. Clinicopathologic characteristics of IgAN with steroid-responsive nephrotic syndrome. J Korean Med Sci 2009; 24 Suppl: S44-9.
77. Lai KN, Lai FM, Ho CP, Chan KW. Corticosteroid therapy in IgAN with nephrotic syndrome: a long-term controlled trial. Clin Nephrol 1986; 26: 174-180
78. Gutiérrez E, González E, Hernández E et al. Factors that determine an incomplete recovery of kidney function in macrohematuria-induced acute renal failure of IgAN. Clin J Am Soc Nephrol 2: 51-57, 2007
79. Praga M, Millet VG, Navas JJ, Ruilope LM, Mora¬les JM, Alcazar JM, Bello I, Rodicio JL. Acute worsening of kidney function during episodes of macroscopic hematuria in IgAN. Kidney Int 1985; 28: 69-74
80. Bennett WM, Kincaid-Smith P. Macroscopic hematuria in mesangial nephropathy: Correlations with glomerular crescents and renal dysfunction. Kidney Int 1983; 23: 393-400

KDIGO® GN Guideline March 2011 Public Review Draft
134
81. Abe T, Kida H, Yoshimura M, Yokoyama H, Koshino Y, Tomosugi N, Hattori N. Participation of extracapillary lesions (ECL) in progression of IgA nephropathy. Clin Nephrol 1986; 25:37-41
82. Tang Z, Wu Y, Wang QW, Yu YS, Hu WX, Yao XD, Chen HP, Liu ZH, Li LS. Idiopathic IgAN with diffuse crescent formation. Am J Nephrol. 2002 ; 22: 480-6.
83. Pankhurst T, Lepenies J, Nightingale P, Howie AJ, Adu D, Harper L. Vasculitic IgAN: prognosis and outcome. Nephron Clin Pract 2009; 112(1):c16-24.

KDIGO® GN Guideline March 2011 Public Review Draft
135
CHAPTER 11: HENOCH-SCHÖNLEIN PURPURA NEPHRITIS
INTRODUCTION
This chapter will focus on the treatment of HSP nephritis in adults and children. The cost implications for global application of this guideline are addressed in Chapter 2.
11.1: Treatment of HSP nephritis in children 11.1.1: We suggest that children with HSP nephritis and persistent proteinuria,
>0.5-1 g/d per 1.73 m2, are treated with ACEi or ARBs. (2D) 11.1.2: We suggest that children with persistent proteinuria, >1 g/d per 1.73 m2,
after a trial of ACEi or ARBs, and GFR >50 ml/min per 1.73 m2, be treated the same as for IgAN with a 6-month course of corticosteroid therapy (see Chapter 10). (2D)
11.2: Treatment of crescentic HSP nephritis in children 11.2.1: We suggest that children with crescentic HSP with nephrotic syndrome
and/or deteriorating kidney function are treated the same as for crescentic IgAN (see Recommendation 10.6.3). (2D)
BACKGROUND
HSP is an acute small-vessel vasculitis, characterized clinically by a nonthrombocytopenic purpuric rash, nondeforming arthritis, gastrointestinal involvement, and nephritis [1]. The incidence of HSP is about 10 cases per 100,000 per year. It affects all ages, but 90% of cases are found in those less than 10 years of age, with the median age at presentation being 6 years [1]. Kidney involvement occurs in 30-50% patients [1-3]. Microscopic hematuria is the most common finding. In a systematic review of 12 studies of 1133 unselected children with HSP, abnormal urinalysis occurred in 34% with the majority (79%) having isolated hematuria with or without proteinuria [3]. Only 21% of those with kidney involvement (or 7.2% of all cases) developed a nephritic and/or nephrotic syndrome. Ninety percent of children had developed kidney involvement by 8 weeks after acute presentation, while 97% developed kidney involvement by 6 months. Recurrence of rash and other symptoms occur in one-third of patients [2]. Nephritis is associated with older age at presentation, persistent rash, and recurrence of HSP, while proteinuria >20 mg/m2/h was associated with recurrence and severe abdominal pain [4]. Only 1-3% of patients progress to ESRD [1]. Long-term prognosis correlates with kidney presentation at onset. Compared to 1.6% of children with isolated hematuria with/without proteinuria, 19.5% of children with nephritic or nephrotic syndromes at initial presentation have nephrotic-range proteinuria, hypertension, and/or reduced GFR at long-term follow up [3]. Among 78 patients managed in specialized pediatric kidney units, 44% of those with a nephritic or nephrotic presentation had hypertension and/or impaired kidney function at a mean follow-up of 23.4 years, while 82% of those presenting with hematuria with or without proteinuria had normal urinalysis, kidney function, and blood pressure [5]. A recent study of 103 children found

KDIGO® GN Guideline March 2011 Public Review Draft
136
that, at final follow up, GFR correlated with GFR and proteinuria at onset and 1 year, with ISKDC pathology grade and interstitial fibrosis. Multivariate analysis identified that proteinuria at 1 year and ISKDC grade were most useful in identifying patients with a poor prognosis [6]. However, one long-term study found that severity of findings on first kidney biopsy did not correlate with the risk of a poor outcome (hypertension, persistent proteinuria, ESRD) [7].
RATIONALE
• There is no evidence for the use of RAS blockade in HSP nephritis in children, but an RCT in children and young adults with IgAN demonstrated the benefit of this therapy in reducing proteinuria and maintaining GFR.
• There is no evidence for the use of oral corticosteroids in HSP nephritis, but data from RCTs in adults with IgAN have demonstrated a benefit in reducing proteinuria and maintaining GFR.
• There is very low–quality evidence for the benefit of high-dose corticosteroids and immunosuppressive agents in HSP nephritis with deteriorating kidney function.
There is no evidence available for the use of RAS blockade in HSP nephritis. However, an
RCT in 66 children and young adults with IgAN with moderate proteinuria (>1 to <3.5 g/d per 1.73 m2) and GFR >50 ml/min per 1.73 m2 demonstrated the benefit of ACEi in reducing proteinuria and maintaining GFR [8]. There are very limited data to support the use of corticosteroids in children with established nephritis of any severity [9], though corticosteroids are widely used in children presenting with nephrotic-range proteinuria or acute nephritis. In a post-hoc analysis of one placebo-controlled RCT [10], nephritis resolved more rapidly in children treated with prednisone compared to placebo. Seven of 36 children (19%) in the prednisone group still had kidney involvement at 6 months compared to 15 of 35 (43%) in the placebo group. The trial only provided outcome data to 6 months after randomization, so it is unclear whether prednisone treatment reduced the number of patients with persistent HSP nephritis overall, or promoted more rapid resolution of kidney disease compared to placebo.
A prospective but uncontrolled study of 38 consecutive children with mean follow-up period of 5 years and 7 months showed resolution of severe nephritis (nephrotic syndrome and/or >50% crescents on biopsy) in 27 of 38 children treated with three pulses of methylprednisolone followed by oral prednisone for 4 months [11]. Seven children had residual abnormalities and four progressed to ESRD. Two recent RCTs in adults with IgAN, proteinuria ≥1 g/d, and GFR ≥30-50 ml/min per 1.73 m2 have demonstrated the benefit of 6-8 months of prednisone and ACEi, compared to ACEi alone, on reducing the rate of kidney functional deterioration and reducing proteinuria during follow-up periods of up to 48 months [12] or 96 months [13].
In the absence of sufficient long-term data in HSP nephritis, we suggest that persistent HSP nephritis can be treated as isolated IgAN (see Statement 10.3.1). However, there are no data to determine when prednisone should be commenced in children with HSP nephritis, and for how long ACEi or ARB therapy should be administered before commencing prednisone. Foster et al. [14] noted that the chronicity score (interstitial fibrosis, tubular atrophy, fibrosed crescents) on initial kidney biopsy increased with increasing delay between onset of kidney involvement and time of biopsy. Most children in their series of 20 patients were biopsied within 3 months, with a

KDIGO® GN Guideline March 2011 Public Review Draft
137
median of 30 days. Treatment with prednisone and azathioprine resulted in improvement in acuity score but not chronicity score. Therefore, in HSP nephritis, it may be appropriate to commence prednisone therapy earlier than in IgAN.
There are limited data on immunosuppressive agents, so it remains unclear whether these have any role in HSP nephritis. In a single RCT of 56 children with significant HSP nephritis (nephrotic range proteinuria, reduced kidney function, ISKDC* grades III-V on kidney biopsy [crescents <50% to >75%]) treated within 3 months of onset of HSP and followed for 5-6 years, there was no significant difference in the risk for persistent kidney involvement of any severity between cyclophosphamide and supportive treatment (RR 1.07; 95% CI 0.65-1.78) [15]. Corticosteroids were not administered to these children. A nonrandomized comparative study of 37 children with HSP nephritis and >50% crescents (ISKDC grades IV-V) on kidney biopsy found that none of 17 treated with cyclophosphamide plus corticosteroids, compared to four of 20 treated with corticosteroids alone, had persistent nephropathy (proteinuria >20 mg/m2/h with/without GFR <40 ml/min per 1.73 m2) at 6-8 years of follow-up [16]. A small RCT, in children with nephrotic-range proteinuria and/or ISKDC grades III-V on kidney biopsy, found that all of 10 children treated with cyclosporin achieved remission compared to five of nine children treated with methylprednisolone [17]. However, at the 2-year follow-up of 23 children, seven of 11 children treated with cyclosporin and seven of 12 treated with methylprednisolone had persistent proteinuria with/without decreased GFR [18].
One nonrandomized comparative study involving 20 children with nephrotic-range proteinuria and ISKDC grades II-III on kidney biopsy reported that none of 10 children treated with azathioprine and prednisone, compared to four of 10 treated with prednisone alone, had nephrotic-range proteinuria and/or GFR <60 ml/min per 1.73 m2 after 4-5 years of follow-up [19]. Observational studies have reported good outcomes with corticosteroids combined with azathioprine [14], cyclophosphamide [20], cyclosporine [21,22], and plasma exchange [23,24]. There are no data, other than small observational studies, examining the treatment of crescentic HSP nephritis with rapidly progressive kidney failure. In the absence of data, we suggest treating such patients similarly to patients with ANCA vasculitis.
A single small RCT [25] comparing 1 year of treatment with MMF to azathioprine has enrolled 17 children (ISKDC grade II and III) to date. Proteinuria resolved in all of 10 children treated with MMF, and six of eight treated with azathioprine. Seven patients treated with MMF and five treated with azathioprine showed regression of histological changes at 1 year. Children received prednisone for 6 months, but were not treated with ACEi. These data are insufficient to draw any conclusions on the value of MMF in HSP nephritis in children.
*The histological classification of HSP nephritis proposed by the International Study of Kidney Disease in Children, and still widely used.

KDIGO® GN Guideline March 2011 Public Review Draft
138
11.3: Prevention of HSP nephritis in children 11.3.1: We recommend not using corticosteroids to prevent HSP nephritis. (1B)
BACKGROUND
At first presentation with HSP, nephritis may be clinically mild or even absent. Therefore, treatment strategies at the time of presentation have been developed with the goal of preventing nephritis, or reducing the risk of severe persistent nephritis.
RATIONALE
• There is moderate-quality evidence to recommend that corticosteroids not be given at presentation of HSP, since they do not appear to influence the development of persistent kidney involvement.
A meta-analysis [26,27] of five RCTs (789 children) found no significant difference in the
number of children with evidence of persistent kidney disease (microscopic hematuria, proteinuria, hypertension, reduced kidney function) during follow-up between those treated with prednisone for 2-4 weeks and those not treated (RR 0.73; 95% CI 0.43-1.24). There were no significant differences in the risk of persistent kidney disease at 6 months (379 children; RR 0.54; 95% CI 0.25-1.18) and 12 months (498 children; RR 1.02; 95% CI 0.40-2.62). Three of the five trials (568 patients) were well designed, placebo-controlled trials; exclusion of poor-quality studies from the meta-analysis removed heterogeneity without altering the findings. Two RCTs [28,29] found no significant difference in the risk of severe nephritis (nephrotic-range proteinuria, hypertension with/without reduced kidney function) between children treated with prednisone or placebo at presentation, though the number of events was small, resulting in imprecision (261 children; RR 1.92; 95% CI 0.57-6.50). There are no data on prevention strategies for HSP nephritis in adults.
11.4: HSP nephritis in adults 11.4.1: We suggest that HSP nephritis in adults be treated the same as in
children. (2D)
RATIONALE
Outcome data from HSP nephritis in adults are from retrospective series. A Spanish retrospective study of HSP in adults suggested a higher frequency of kidney involvement than children, but the final outcome of HSP is equally good in patients of both age groups [30]. In an Italian cohort, the risk for progression of HSP nephritis was found greater in adults and was associated with increasing proteinuria during follow-up [31]. In a UK series, the risk factors for ESRD were: proteinuria ≥1 g/d during follow-up, hypertension at presentation and during follow-up, kidney impairment at presentation—very similar to the prognostic indicators in IgAN in adults [32].

KDIGO® GN Guideline March 2011 Public Review Draft
139
In a Finnish series, kidney survival 10 years after biopsy was 91% [33]. A recent cohort of HSP nephritis in Chinese adults showed a higher risk of progression to kidney impairment compared to children [34]. The biggest retrospective cohort of 250 adults with HSP was from France [35]. After a median follow-up of 14.8 y, 32% of the patients showed kidney impairment (CrCl <50 ml/min), usually associated with proteinuria and/or hematuria.
There is no RCT of HSP nephritis in adults. Therefore, we suggest treatment for HSP nephritis should use the approach proposed for HSP in children (see Statements11.1 and 11.2).
RESEARCH RECOMMENDATIONS
• An RCT comparing a 6- to 12-month course of corticosteroids to shorter-duration corticosteroids (28 days) should be performed in children with moderately severe HSP nephritis (acute nephritic syndrome or nephrotic syndrome with normal kidney function and <50% crescents or sclerosing lesions on biopsy).
• RCTs are required to determine whether immunosuppressive agents (cyclosporine, azathioprine, MMF) and corticosteroids are effective in treating children with severe HSP nephritis (acute nephritic syndrome, nephrotic syndrome with or without reduced kidney function with >50% crescents or sclerosing lesions on biopsy).
References
1. Saulsbury FT. Clinical update: Henoch-Schonlein purpura. Lancet 2007; 369(9566):976-978. 2. Saulsbury FT. Henoch-Schonlein purpura in children. Report of 100 patients and review of the literature.
Medicine 1999; 78(6):395-409. 3. Narchi H. Risk of long term renal impairment and duration of follow up recommended for Henoch-Schonlein
purpura with normal or minimal urinary findings: a systematic review. Archives of Disease in Childhood 2005; 90(9):916-920.
4. Shin JI, Park JM, Shin YH, Hwang DH, Kim JH, Lee JS. Predictive factors for nephritis, relapse, and significant proteinuria in childhood Henoch-Schonlein purpura. Scandinavian Journal of Rheumatology 2006; 35(1):56-60.
5. Goldstein AR, White RH, Akuse R, Chantler C. Long-term follow-up of childhood Henoch-Schonlein nephritis. Lancet 1992; 339(8788):280-282.
6. Edstrom Halling S., Soderberg MP, Berg UB. Predictors of outcome in Henoch-Schonlein nephritis. Pediatric Nephrology 25(6):1101-8, 2010.
7. Ronkainen J, Nuutinen M, Koskimies O. The adult kidney 24 years after childhood Henoch-Schonlein purpura: a retrospective cohort study. Lancet 2002; 360(9334):666-670.
8. Coppo R, Peruzzi L, Amore A, Piccoli A, Cochat P, Stone R et al. IgACE: a placebo-controlled, randomized trial of angiotensin-converting enzyme inhibitors in children and young people with IgA nephropathy and moderate proteinuria. Journal of the American Society of Nephrology 2007; 18(6):1880-1888.
9. Zaffanello M, Fanos V. Treatment-based literature of Henoch-Schonlein purpura nephritis in childhood. Pediatric Nephrology 2009; 24(10):1901-1911.
10. Ronkainen J, Koskimies O, la-Houhala M, Antikainen M, Merenmies J, Rajantie J et al. Early prednisone therapy in Henoch-Schonlein purpura: a randomized, double-blind, placebo-controlled trial. Journal of Pediatrics 2006; 149(2):241-247.
11. Niaudet P, Habib R. Methylprednisolone pulse therapy in the treatment of severe forms of Schonlein-Henoch purpura nephritis. Pediatric Nephrology 1998; 12(3):238-243.
12. Lv J, Zhang H, Chen Y, Li G, Jiang L, Singh AK et al. Combination therapy of prednisone and ACE inhibitor versus ACE-inhibitor therapy alone in patients with IgA nephropathy: a randomized controlled trial. American Journal of Kidney Diseases 53(1):26-32, 2009.

KDIGO® GN Guideline March 2011 Public Review Draft
140
13. Manno C, Torres DD, Rossini M, Pesce F, Schena FP. Randomized controlled clinical trial of corticosteroids plus ACE-inhibitors with long-term follow-up in proteinuric IgA nephropathy. Nephrology Dialysis Transplantation 24(12):3694-701, 2009.
14. Foster BJ, Bernard C, Drummond KN, Sharma AK. Effective therapy for severe Henoch-Schonlein purpura nephritis with prednisone and azathioprine: a clinical and histopathologic study. Journal of Pediatrics 2000; 136(3):370-375.
15. Tarshish P, Bernstein J, Edelmann CM. Henoch-Schonlein purpura nephritis: course of disease and efficacy of cyclophosphamide. Pediatric Nephrology 2004; 19(1):51-56.
16. Kawasaki Y, Suzuki J, Suzuki H. Efficacy of methylprednisolone and urokinase pulse therapy combined with or without cyclophosphamide in severe Henoch-Schoenlein nephritis: a clinical and histopathological study. Nephrology Dialysis Transplantation 2004; 19(4):858-864.
17. Ronkainen J, Ala-Houhala M, Antkainen M, Jahnukainen T, Koskimies O, Ormala T et al. Cyclosporine A (CyA) versus MP pulses (MP) in the treatment of severe Henoch-Schonlein Nephritis (HSN). Pediatric Nephrology 21, 1531. 2006.
18. Jauhola O, Ronkainen J, Ala-Houhala M, Autio-Harmainen H, Holtta T, Jahnukainen T et al. Cyclosporine A (CyA) versus MP pulses in severe Henoch-Schonlein Nephritis (HSN): Outcome after 2 year follow up. Pediatric Nephrology 23, 1584. 2008.
19. Bergstein J, Leiser J, Andreoli SP. Response of crescentic Henoch-Schoenlein purpura nephritis to corticosteroid and azathioprine therapy. Clinical Nephrology 1998; 49(1):9-14.
20. Flynn JT, Smoyer WE, Bunchman TE, Kershaw DB, Sedman AB. Treatment of Henoch-Schonlein Purpura glomerulonephritis in children with high-dose corticosteroids plus oral cyclophosphamide. American Journal of Nephrology 2001; 21(2):128-133.
21. Shin JI, Park JM, Shin YH, Kim JH, Kim PK, Lee JS et al. Cyclosporin A therapy for severe Henoch-Schonlein nephritis with nephrotic syndrome. Pediatric Nephrology 20(8)()(pp 1093-1097), 2005 Date of Publication: Aug 2005 2005;(8):1093-1097.
22. Ronkainen J, Autio-Harmainen H, Nuutinen M. Cyclosporin A for the treatment of severe Henoch-Schonlein glomerulonephritis. Pediatric Nephrology 2003; 18(11):1138-1142.
23. Shenoy M, Ognjanovic MV, Coulthard MG. Treating severe Henoch-Schonlein and IgA nephritis with plasmapheresis alone. Pediatric Nephrology 2007; 22(8):1167-1171.
24. Kawasaki Y, Suzuki J, Murai M, Takahashi A, Isome M, Nozawa R et al. Plasmapheresis therapy for rapidly progressive Henoch-Schonlein nephritis. Pediatric Nephrology 1919;(8):920-923.
25. Fuentes Y, Valverde S, Valesquez-Jones L, Romero B, Hernandez AM, Del Moral IE et al. Comparison of azathioprine vs mofetil mycophenolate for Henoch-Schönlein nephritis treatment. Pediatric Nephrology 25, 1802. 2010.
26. Chartapisak W, Opastiraku S, Willis NS, Craig JC, Hodson EM. Prevention and treatment of renal disease in Henoch-Schonlein purpura: a systematic review. Archives of Disease in Childhood 2009; 94(2):132-137.
27. Chartapisak W, Opastirakul S, Hodson EM, Willis NS, Craig JC. Interventions for preventing and treating kidney disease in Henoch-Schonlein Purpura (HSP). Cochrane Database of Systematic Reviews 2009;(3):CD005128.
28. Ronkainen J, Koskimies O, Ala-Houhala M, Antikainen M, Merenmies J, Rajantie J et al. Early prednisone therapy in Henoch-Schonlein purpura: a randomized, double-blind, placebo-controlled trial. Journal of Pediatrics 2006; 149(2):241-247.
29. Dudley J, Smith G, Llewellyn-Edwards A, Tizard EJ. Randomised placebo controlled trial to assess the role of early prednisolone on the development and progression of Henoch-Schonlein Purpura Nephritis. Pediatric Nephrology 2007; 22(9):1457.
30. Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M, Gonzalez-Gay MA. Henoch-Schonlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum 1997; 40: 859-64.
31. Coppo R, Andrulli S, Amore A, Gianoglio B, Conti G, Peruzzi L, Locatelli F, Cagnoli L. Predictors of outcome in Henoch-Schonlein nephritis in children and adults. Am J Kidney Dis. 2006; 47: 993-1003.

KDIGO® GN Guideline March 2011 Public Review Draft
141
32. Shrestha S, Sumingan N, Tan J, Althous H, McWilliam L, Ballardie F. Henoch Schonlein purpura with nephritis in adults: adverse prognostic indicators in a UK population. Q J Med. 2006; 99: 253-65
33. Rauta V, Törnroth T, Grönhagen-Riska C. Henoch-Schoenlein nephritis in adults-clinical features and outcomes in Finnish patients. Clin Nephrol. 2002 ;58: 1-8.
34. Hung SP, Yang YH, Lin YT, Wang LC, Lee JH, Chiang BL . Clinical manifestations and outcomes of Henoch-Schönlein purpura: comparison between adults and children. Pediatr Neonatol. 2009 Aug;50(4):162-8.
35. Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schonlein Purpura in adults: outcome and prognostic factors. J Am Soc Nephrol 2002; 13:1271-8.

KDIGO® GN Guideline March 2011 Public Review Draft
142
CHAPTER 12: TREATMENT OF LUPUS NEPHRITIS
INTRODUCTION
This chapter gives treatment recommendations for LN in adults and children.
BACKGROUND
Kidney involvement in systemic lupus, known asLN, is most often due to glomerular immune complex accumulation, which leads to glomerular inflammation and, if unchecked, also involves the renal interstitium. Lupus patients with LN have worse outcomes than those with no kidney involvement [1-3]. This poor prognosis is explained only in part by the risk of CKD and ESRD, suggesting that LN is a manifestation of a more severe form of systemic lupus.
The reported incidence of clinically important kidney disease in systemic lupus is about 38%. Of those who develop clinical LN, 40-60% have overt kidney disease at the time lupus is diagnosed [4-6]. The incidence of kidney involvement differs with ethnicity. Caucasians (European, European Americans; 12-33%) are less likely to have LN than black (African American, Afro-Caribbean; 40-69%), Hispanic (36-61%), or Asian (Indian, Chinese; 47-53%) patients.
Based on the United States Renal Data Service database, between 1996 and 2004 the incidence of ESRD attributed to LN in adults was 4.5 cases per million in the general population [7], but was greater in blacks (17-20/million) and Hispanics (6/million) than Caucasians (2.5/million). Similarly, a retrospective cohort from the UK found that 19% of Caucasians and 62% of blacks with LN progressed to ESRD [8]. In a Saudi Arabian population, 12% of patients with LN developed ESRD [6, 9]. The prevalence of CKD in patients with systemic lupus is difficult to estimate, but because current therapies induce complete remission in only about 50% of those with LN, CKD is likely to be common.
The presence of LN should be considered in any lupus patient with impaired kidney function, proteinuria, or an active urine sediment. LN must be confirmed by kidney biopsy. The histologic findings provide the basis for treatment recommendations for LN.
12.1: Class I LN (minimal-mesangial LN) 12.1.1: Treat patients with class I LN with corticosteroids and
immunosuppressives only as dictated by the extrarenal clinical manifestations of lupus. (Not Graded)
BACKGROUND
In class I LN, glomeruli are normal by light microscopy. Class I LN is defined by the presence of immune deposits restricted to the mesangium, and seen only by immunofluorescence or electron microscopy.

KDIGO® GN Guideline March 2011 Public Review Draft
143
RATIONALE
• Class I LN has no clinical kidney manifestations. • Class I LN is not associated with long-term impairment of kidney function.
Kidney tissue obtained for research purposes in patients with systemic lupus but without
clinical signs of kidney disease showed LN was present in about 90% of patients [10, 11], far more than the 40% or so who manifest clinical kidney disease. In some patients with clinically silent class I LN, there is transformation to more aggressive and clinically relevant forms of LN [12]. However, at present, there are no data to suggest that every patient with lupus requires a kidney biopsy, or that treatment of class I LN is clinically necessary.
12.2: Class II LN (mesangial-proliferative LN) 12.2.1: Treat patients with class II LN and proteinuria <3 g/d with
corticosteroids and immunosuppressives only as dictated by the extrarenal clinical manifestations of lupus. (Not Graded)
12.2.2: We suggest that class II LN with proteinuria >3 g/d be treated with corticosteroids or CNIs as described for MCD (see Chapter 5). (2D)
BACKGROUND
The kidney biopsy of class II LN shows mesangial hypercellularity and matrix expansion on light microscopy, and mesangial immune deposits by immunofluorescence and electron microscopy. Clinically, proteinuria and/or hematuria may be seen in class II LN, but usually not nephrotic syndrome, or kidney impairment. If nephrotic-range proteinuria is found with class II LN, this may be due to a concomitant podocytopathy.
RATIONALE
• There are no evidence-based data on the treatment of class II LN. • Podocytopathies, characterized histologically by diffuse foot process effacement in
the absence of glomerular capillary wall immune complex deposition or endocapillary proliferation, have been observed in patients with class II LN.
Podocyte injury in class II LN does not appear related to the extent of mesangial immune
complex deposition [13]. While there have been no prospective studies of the treatment of nephrotic-range proteinuria in class II LN, it is reasonable to treat such patients as for MCD/FSGS in case of nephrotic syndrome, or if proteinuria cannot be controlled using RAS blockade.

KDIGO® GN Guideline March 2011 Public Review Draft
144
12.3: Class III LN (focal LN) and class IV LN (diffuse LN)—initial therapy
12.3.1: We recommend initial therapy with corticosteroids (1A), combined with either cyclophosphamide (1B) or MMF (1B).
12.3.2: We suggest that, if patients have worsening LN (rising SCr, worsening proteinuria) during their initial treatment, a change to the alternative recommended initial therapy be instituted. (2D)
BACKGROUND
Class III and IV LN are differentiated by the percentage of affected glomeruli (class III, <50%; class IV, ≥50%). Glomerular lesions are classified as active (A) or chronic (C). The active lesions of class III and IV are endocapillary and (usually) mesangial hypercellularity, crescents, necrosis, wire loops, and hyaline thrombi. Chronic lesions include segmental and global glomerulosclerosis. Immunofluorescence and electron microscopy show significant subendothelial and mesangial immune deposits. If there are extensive subepithelial immune deposits, there is coincidental class V LN (see Rationale).
Almost all patients will have microscopic hematuria and proteinuria; nephrotic syndrome and kidney impairment are common. However, if the histologic lesions are mainly chronic (see Rationale) there may be less overt clinical activity, other than progressive kidney failure. Therapy should be adjusted according to the extent of activity or chronicity.
There is no standard definition of treatment response for class III and IV LN, which makes direct comparison of clinical trials difficult. Nonetheless, the overall goals of treatment are similar between trials, and the following definitions of response based on published trials are provided as a guide to the success of therapy:
Complete response: Return of SCr to previous baseline, plus a decline in the uPCR to <0.5 mg/mg (50 mmol/mg).
Partial response: Stabilization (±25%), or improvement of SCr, but not to normal, plus a ≥50% decrease in uPCR. If there was nephrotic-range proteinuria (uPCR ≥3 mg/mg [300 mmol/mg]), improvement requires a ≥50% reduction in uPCR, and a uPCR <3 mg/mg.
Deterioration: There is no definition of deterioration in LN to define treatment failure that has been tested prospectively as an indication to change in initial therapy. A sustained 25% increase in SCr is widely used but has not been validated.
RATIONALE
• Proliferative LN (class III or IV) is an aggressive disease. • Before 1970, kidney survival and overall patient survival in diffuse proliferative LN
were very poor, in the range of 20-25%.

KDIGO® GN Guideline March 2011 Public Review Draft
145
• Patient and kidney survival in class III and IV LN have dramatically improved through the use of intensive immunosuppression.
• The International Society of Nephrology/Renal Pathology Society classification of LN assigns activity (A) or chronicity (C) in class III and IV LN. Our treatment recommendations are for active or active plus chronic lesions. Thorough review with the nephropathologist is required to ensure accurate classification prior to starting therapy.
• Therapy for class III and IV LN has initial and maintenance phases. The objective is to rapidly decrease kidney inflammation by initial intensive treatment, and then consolidate treatment over a longer time. The initial phase is often called induction, which implies remission is achieved at its completion. This, however, is often not the case, and remissions continue to occur well into the maintenance phase. The term “initial” treatment is therefore preferred.
• The benefit of the addition of cyclophosphamide to corticosteroids for initial treatment was shown in controlled trials demonstrating that, during long term follow-up, this combination decreased the frequency of kidney relapse, CKD, and ESRD compared to corticosteroids alone.
• The evolution of initial therapy in proliferative LN has been to reduce toxicity while maintaining efficacy. This has resulted in several modifications of cyclophosphamide dosing, and the introduction of MMF as an alternative to cyclophosphamide.
• The efficacy of newer initial treatment regimens should be assessed not only by initial responses, but also by long-term effects on kidney relapse, and development of CKD.
Widely used treatment regimens are shown in Table 12.1.
*Increases in disease activity in systemic lupus in general, and in LN in particular, may be described as “flares” or “relapses”. In this guideline, we use the term “relapse”.
Corticosteroids All regimens use similar corticosteroid dosing: an initial dose of oral prednisone up to
1 mg/kg, tapering according to clinical response over 6-12 months. Additional i.v. methylprednisolone is widely used at the beginning of treatment for more severe disease. However, the dosing and duration of corticosteroids has never been subject to evaluation by RCTs.
Cyclophosphamide
i.v. cyclophosphamide (0.5-1 g/m2) given monthly for 6 months (Regimen A, sometimes called the “NIH regimen”) was the first immunosuppressive treatment shown in RCT to be superior to corticosteroids alone [14-17].
A lower-dose regimen using i.v. cyclophosphamide 500 mg every 2 weeks for 3 months (Regimen B, sometimes called the “Euro-Lupus regimen”) had equivalent efficacy to Regimen A in an RCT in Caucasians [18,19]. However, those with severe LN—defined as rapidly progressive kidney failure, typically with widespread (>50%) segmental glomerular necrosis or

KDIGO® GN Guideline March 2011 Public Review Draft
146
crescents—were not included in that study. It remains uncertain whether Regimen B has equivalent efficacy to Regimen A in severe class III/IV LN, and in patients of other ethnicities.
Oral cyclophosphamide 1.0-1.5 mg/kg/d (maximum dose 150 mg/d) for 2-4 months (Regimen C) has been used as an alternative to i.v. cyclophosphamide [20,21]. It has equivalent efficacy to i.v. cyclophosphamide in prospective observational studies [14, 22-25], and has also been shown equivalent to mycophenolate in Chinese patients [26, 27], although this has not yet been verified in other ethnicities. More adverse effects have been reported with oral compared to i.v. cyclophosphamide, but this is not a consistent finding.
Mycophenolate MMF (maximum 3g/d) for 6 months (Regimen D) has been tested in an RCT in a Chinese
population, and was equivalent in achieving remission to Regimen C; patients with severe LN were excluded from this study [27].
An RCT known as ALMS [28] recruited 370 patients with class III, IV, and V LN, and comparing MMF to Regimen A, showed that MMF had an equivalent response rate to i.v. cyclophosphamide at 6 months, and had a similar incidence of adverse events including serious infections and deaths [28].
There is more limited RCT evidence for the use of three other regimens as initial treatment: corticosteroids combined with i) azathioprine, or ii) cyclosporine, or iii) the combination of tacrolimus and MMF (sometimes called “multitarget” therapy).
Azathioprine An RCT in Europeans compared initial therapy with azathioprine combined with i.v.
methylprednisone, followed by oral prednisone, to i.v. cyclophosphamide with oral prednisone [29]. At 2 years, there was no difference in response rate, and fewer adverse effects in those receiving azathioprine. However, supplementary studies in these cohorts showed a higher late relapse rate and higher risk of doubling of SCr after azathioprine. Furthermore, there was more chronicity on later biopsies after azathioprine [30].
Cyclosporine A small (n = 40), open-label RCT compared cyclosporine to cyclophosphamide as initial
therapy combined with corticosteroids for proliferative LN [31]. Cyclosporine (4-5 mg/kg/d) was used for 9 months, and then tapered over the next 9 months. Cyclophosphamide was used in a different regimen than in most published trials: eight i.v. pulses (10 mg/kg) were given in the first 9 months, and then four to five oral pulses (10 mg/kg) over the next 9 months. There were no differences in responses or remissions at 9 or 18 months, or relapse rate after 40 months of follow-up. Infections and leukopenia did not differ between the groups.
Tacrolimus with Mycophenolate In a small RCT from China in patients with combined class IV and V LN, the combination
of tacrolimus (4 mg/d), MMF (1 g/d), and oral corticosteroids (sometimes known as “multitarget” therapy) was compared to pulse monthly i.v. cyclophosphamide (0.75 g/m2 for 6 months) plus oral corticosteroids. At 6 months, 90% of patients treated with this multitarget
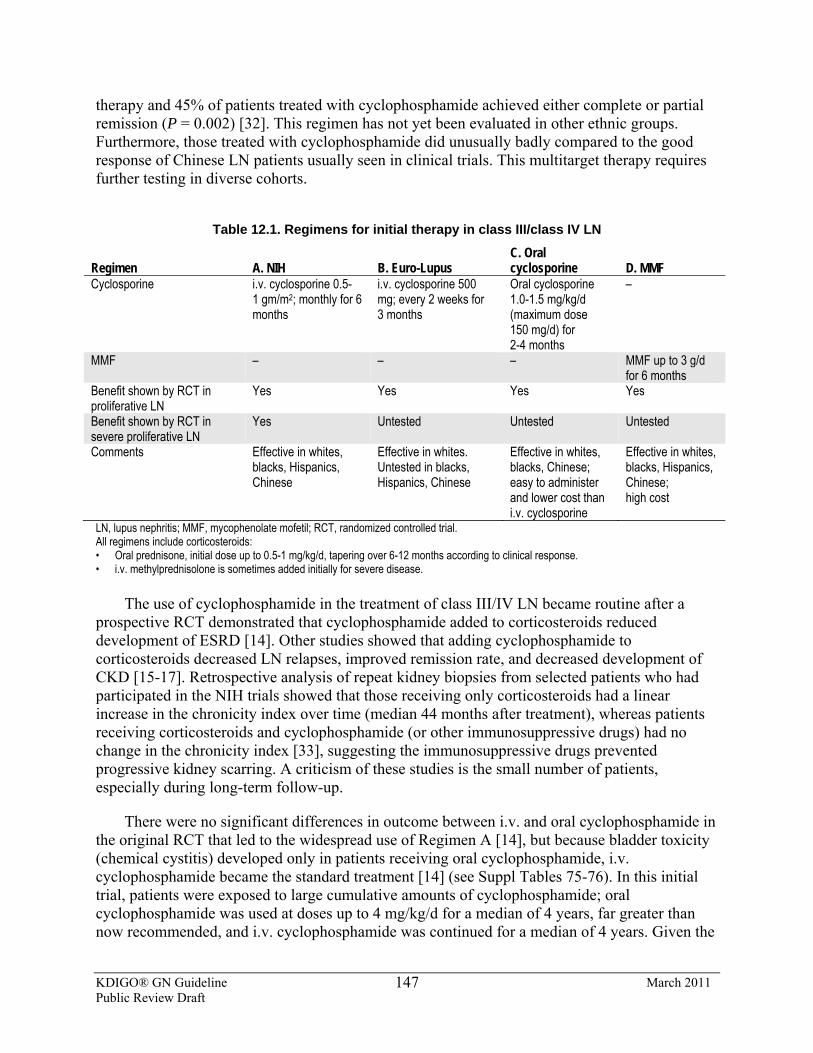
KDIGO® GN Guideline March 2011 Public Review Draft
147
therapy and 45% of patients treated with cyclophosphamide achieved either complete or partial remission (P = 0.002) [32]. This regimen has not yet been evaluated in other ethnic groups. Furthermore, those treated with cyclophosphamide did unusually badly compared to the good response of Chinese LN patients usually seen in clinical trials. This multitarget therapy requires further testing in diverse cohorts.
Table 12.1. Regimens for initial therapy in class III/class IV LN
Regimen A. NIH B. Euro-Lupus C. Oral cyclosporine D. MMF
Cyclosporine i.v. cyclosporine 0.5-1 gm/m2; monthly for 6 months
i.v. cyclosporine 500 mg; every 2 weeks for 3 months
Oral cyclosporine 1.0-1.5 mg/kg/d (maximum dose 150 mg/d) for 2-4 months
–
MMF – – – MMF up to 3 g/d for 6 months
Benefit shown by RCT in proliferative LN
Yes Yes Yes Yes
Benefit shown by RCT in severe proliferative LN
Yes Untested Untested Untested
Comments Effective in whites, blacks, Hispanics, Chinese
Effective in whites. Untested in blacks, Hispanics, Chinese
Effective in whites, blacks, Chinese; easy to administer and lower cost than i.v. cyclosporine
Effective in whites, blacks, Hispanics, Chinese; high cost
LN, lupus nephritis; MMF, mycophenolate mofetil; RCT, randomized controlled trial. All regimens include corticosteroids: • Oral prednisone, initial dose up to 0.5-1 mg/kg/d, tapering over 6-12 months according to clinical response. • i.v. methylprednisolone is sometimes added initially for severe disease.
The use of cyclophosphamide in the treatment of class III/IV LN became routine after a prospective RCT demonstrated that cyclophosphamide added to corticosteroids reduced development of ESRD [14]. Other studies showed that adding cyclophosphamide to corticosteroids decreased LN relapses, improved remission rate, and decreased development of CKD [15-17]. Retrospective analysis of repeat kidney biopsies from selected patients who had participated in the NIH trials showed that those receiving only corticosteroids had a linear increase in the chronicity index over time (median 44 months after treatment), whereas patients receiving corticosteroids and cyclophosphamide (or other immunosuppressive drugs) had no change in the chronicity index [33], suggesting the immunosuppressive drugs prevented progressive kidney scarring. A criticism of these studies is the small number of patients, especially during long-term follow-up.
There were no significant differences in outcome between i.v. and oral cyclophosphamide in the original RCT that led to the widespread use of Regimen A [14], but because bladder toxicity (chemical cystitis) developed only in patients receiving oral cyclophosphamide, i.v. cyclophosphamide became the standard treatment [14] (see Suppl Tables 75-76). In this initial trial, patients were exposed to large cumulative amounts of cyclophosphamide; oral cyclophosphamide was used at doses up to 4 mg/kg/d for a median of 4 years, far greater than now recommended, and i.v. cyclophosphamide was continued for a median of 4 years. Given the

KDIGO® GN Guideline March 2011 Public Review Draft
148
potential for developing hematologic malignancies later in life, these large cumulative doses of cyclophosphamide should be avoided. We suggest a lifetime maximum of 36 g cyclophosphamide in patients with systemic lupus [34,35]. This is reflected in Regimens A-C.
There are other important considerations, when using cyclophosphamide, to reduce its toxicity. The dose of cyclophosphamide should be decreased by 20% or 30% in patients with CrCl 25-50 and 10-25 ml/min, respectively [36]. The dose of i.v. cyclophosphamide should be adjusted to keep the day 10-14 leucocyte count nadir ≥3000/μl. When using oral cyclophosphamide, white blood cell counts should be monitored weekly and cyclophosphamide dose should be adjusted to keep leucocytes ≥3000/μl. Leukopenia requires careful evaluation, since systemic lupus, as well as cyclophosphamide, can cause suppression of bone marrow.
To minimize bladder toxicity with oral cyclophosphamide, we suggest instructing patients to take cyclophosphamide in the morning, and to drink extra fluid at each meal and at bed time. The use of sodium-2-mercaptoethane (mesna) will also minimize the risk of hemorrhagic cystitis when cyclophosphamide is given as i.v. pulses.
To protect fertility, women should be offered prophylaxis with leuprolide and men testosterone while cyclophosphamide is being given [37,38]. Administration of leuprolide must be timed carefully in relation to cyclophosphamide to maximize benefit. Ovarian tissue cryopreservation is an additional, but expensive, option. The efficacy of testosterone in preserving fertility in males is poorly established, so sperm banking should be offered.
Given the toxicity of cyclophosphamide, studies were undertaken to determine if the dosing regimen could be modified. An RCT has tested the efficacy of low-dose, short-duration cyclophosphamide (Regimen B) in Caucasians [18,19]. This regimen resulted in a higher percentage of remissions and a lower incidence of severe infections than Regimen A, although the differences were not statistically significant [18]. Importantly, this low-dose cyclophosphamide regimen had similar long-term outcomes (mean follow-up of 10 years) to Regimen A [19] (Suppl Table 74). In this trial, the majority of patients were white, and most patients did not have clinically severe disease. Therefore, it is not certain whether this protocol will be effective in patients of other ancestry, or in patients with more severe class III/IV LN.
A cyclophosphamide-free regimen has been proposed (Regimen D). MMF is used for the first 6 months of LN treatment, instead of sequential cyclophosphamide followed by MMF. The basis for this approach was three small studies of MMF in Asia, and one larger study (140 patients) from the USA [26, 39-41]. The Asian studies concluded MMF was equivalent to cyclophosphamide, but the USA trial demonstrated MMF was superior to i.v. cyclophosphamide, although many patients did not achieve the target dose of cyclophosphamide. An RCT known as the Aspreva Lupus Management Study (ALMS) [28] recruited 370 patients with class III, IV, and V LN, giving oral corticosteroids and either daily oral MMF or 6-monthly i.v. pulses of cyclophosphamide (0.5-1g/m2). The ALMS trial showed that MMF was equivalent to i.v. cyclophosphamide in inducing a response at 6 months [28]. ALMS showed a similar incidence of adverse events, serious infections, and deaths for MMF and cyclophosphamide (see Suppl Tables 68-70). Similar results were found in an all-Egyptian cohort [41A].

KDIGO® GN Guideline March 2011 Public Review Draft
149
A posthoc analysis of the ALMS trial indicated that black, Hispanic, and mixed-race patients, (generally considered to have more resistant LN [42]) had inferior outcomes with cyclophosphamide compared to MMF. Further information is required from RCTs before recommendations can be made about the efficacy of MMF in patients of specific ethnicity.
Because the kidney response rate for class III and IV LN with any of the initial therapies so far discussed is only about 60% at 6-12 months, an RCT adding rituximab or placebo to MMF plus corticosteroids for initial LN therapy was undertaken to determine if remission rates could be improved [43]. This RCT was based on several small, open-label, uncontrolled trials that suggested rituximab may be effective in proliferative LN, either for refractory disease or as initial therapy [44-50]. At 12 months, however, there were no differences between the rituximab and placebo groups in terms of complete or partial remissions. Thus, rituximab cannot be recommended as adjunctive initial therapy.
Choice of Initial Therapy The patients in the two largest studies of MMF vs. cyclophosphamide generally had less
severe LN, assessed by level of proteinuria and kidney function [28, 41], than the patients in some of the RCTs of cyclophosphamide [16,51]. Thus, in severe class III/IV LN, a cyclophosphamide-containing protocol for initial therapy may be preferred.
Additionally, the beneficial effect of cyclophosphamide in preservation of kidney function was only apparent after 3-5 years of follow-up [14,16,17]. This length of time, which was needed to show a difference between initial therapies in long-term kidney survival, must therefore be kept in mind when evaluating new, noncyclophosphamide-containing regimens as initial therapy for class III/IV LN. For example, the Dutch Working Party on systemic lupus found that azathioprine, an antimetabolite like MMF, was equivalent to cyclophosphamide as initial therapy of class III and IV LN; however, in the long term, repeat biopsies showed more chronic damage with azathioprine, as well as a higher incidence of kidney relapse and doubling of SCr [29,30] (see Suppl Tables 71-73). In some regions where cost and drug availability are an issue, it may be necessary to use azathioprine for initial treatment of class III and IV LN.
In a long-term study of continuous MMF therapy compared to initial cyclophosphamide followed by azathioprine, there were no significant differences in kidney function between the groups after a median of 64 months [27]. However, in the MMF group, more patients had relapses, prolonged proteinuria >1 g/d, and persistent SCr >2 mg/dl (176 μmol/l). These combined clinical findings have been associated, in other studies, with deterioration of kidney function over time.
After the initial 6-month treatment period, the ALMS trial was extended for 3 years to evaluate maintenance therapy with either MMF or azathioprine [52]. Although not designed to compare the long-term efficacy of initial therapy on kidney function, there was a (nonsignificant) trend toward fewer treatment failures in those who received cyclophosphamide as initial therapy as opposed to MMF. This result was independent of whether maintenance therapy was azathioprine or MMF.
Thus, it cannot yet be stated that initial therapy with MMF is equal to cyclophosphamide for proliferative LN with respect to long-term kidney function.

KDIGO® GN Guideline March 2011 Public Review Draft
150
12.4: Class III LN (focal LN) and class IV LN (diffuse LN)—maintenance therapy 12.4.1: We recommend that, after initial therapy is complete, patients with class
III and IV LN receive maintenance therapy with azathioprine (1.5-2.5 mg/kg/d) or MMF (1-3g/d in divided doses), and low-dose oral corticosteroids (≤10 mg/d prednisone equivalent). (1B)
12.4.2: We suggest that CNIs with low-dose corticosteroids can be used for maintenance therapy in patients who are intolerant of MMF and azathioprine. (2C)
12.4.3: We suggest that, after complete remission is achieved, maintenance therapy be continued for at least 1 year before tapering the immunosuppression. (2D)
12.4.4: If complete remission has not been achieved after 12 months of maintenance therapy, consider performing a repeat kidney biopsy before determining if a change in therapy is indicated. (Not Graded)
12.4.5: While maintenance therapy is being tapered, if kidney function deteriorates and/or proteinuria worsens, we suggest that treatment be increased to the previous level of immunosuppression that controlled the LN. (2D)
RATIONALE
• There is moderate-quality evidence from RCTs in patients with class III/IV LN that prolonged maintenance therapy after initial treatment is required.
• There is moderate-quality evidence that maintenance therapy with azathioprine or MMF is superior to maintenance with cyclophosphamide as judged by risk of death, and risk of development of CKD.
• There is moderate-quality evidence that azathioprine and cyclosporine A have comparable efficacy as maintenance therapies for class III/IV LN.
• There is very low–quality evidence to guide the duration of maintenance therapy after complete remission, but most randomized studies of class III/IV LN have given therapy for several years.
The need for maintenance therapy was suggested when patients treated only with short-term
(6 months) i.v. cyclophosphamide therapy were shown to have an increased frequency of kidney relapses [17].
Choice of Maintenance Therapy Presently, there are several options for maintenance therapy after the initial treatment of
proliferative LN. The data currently available do not allow a definitive recommendation as to the choice of agent for maintenance therapy. Patient-specific factors, such as desire for pregnancy or occurrence of side-effects, may be considered when making this choice.

KDIGO® GN Guideline March 2011 Public Review Draft
151
A cohort of mainly black and Hispanic patients with class III/IV LN was treated with monthly i.v. cyclophosphamide for up to seven cycles, followed by azathioprine or MMF, and compared to patients treated with 6-monthly cyclophosphamide pulses followed by quarterly cyclophosphamide pulses for 1 year beyond remission [53]. This study showed that, over 72 months, the patients treated with maintenance azathioprine or MMF were significantly less likely to reach the composite end-point of death or CKD than the cyclophosphamide maintenance group, and to experience fewer adverse effects.
The MAINTAIN Nephritis Trial compared MMF with AZA as maintenance therapy in a predominantly Caucasian population after initial treatment with low-dose (Regimen B) cyclophosphamide [54]. They had not necessarily achieved remission after initial therapy. The primary end-point was time to kdney relapse. After at least 3 years of follow-up, this trial found MMF and azathioprine to be equivalent.
The ALMS trial extension phase [52] compared MMF and AZA as maintenance therapies after the 6-month initial treatment period (Regimen D). Patients entered this extension phase only if they achieved a complete or partial remission after initial therapy. Over 3 years, the composite treatment failure end-point (death, ESRD, kidney flare, sustained doubling of SCr, or requirement for rescue therapy) was reached in 16% of MMF-treated patients compared to 32% of azathioprine-treated patients (P = 0.003). The superiority of MMF over azathioprine was not dependent on initial therapy or race of the patient.
A pilot RCT in 69 patients with class III/IV LN suggested that 2 years of CSA may be as effective as 2 years of azathioprine for maintenance, after initial treatment with prednisone and oral cyclophosphamide, in terms of relapse prevention and reduction of proteinuria [55]. Another RCT showed cyclosporine was as effective as azathioprine in terms of tapering maintenance corticosteroids in severe systemic lupus, but only 29% of the patients had LN [56].
Duration of Therapy Few patients reach complete remission by 6 months, and kidney biopsies after 6 months of
initial therapy have shown that, while active inflammation tends to improve, complete resolution of pathologic changes is unusual [39, 57-59]. Consistent with this finding, clinical improvement in class III/IV LN continues well beyond 6 months and into the maintenance phase of therapy [18, 21-23, 29, 60]. Decisions to alter therapy should not be based on urine sediment alone. A repeat kidney biopsy may be considered if kidney function is deteriorating.
There is no evidence to help determine the duration of maintenance therapy. The average duration of immunosuppression was 3.5 years in seven RCTs [14, 17-19, 24, 27, 29, 53]. We suggest that immunosuppressive therapy should usually be slowly tapered after patients have been in complete remission for a year. If a patient has a history of kidney relapses it may be prudent to extend maintenance therapy.
Immunosuppression should be continued for patients who achieve only a partial remission. However, the strategy of trying to convert a partial remission to a complete remission by increasing corticosteroids or using alternative immunosuppressive agents is not supported by evidence.

KDIGO® GN Guideline March 2011 Public Review Draft
152
There are few data on repeat biopsies after therapy. Biopsies taken two or more years after initial therapy often continue to show activity, especially when there is still significant proteinuria or an abnormal SCr [61]. Of more concern, one study found that, in patients with initial class III and IV LN, only 40% had reverted to class II LN on repeat biopsy after 2 years of immunosuppressive therapy [30]. The SCr and extent of proteinuria at the time of the second biopsy did not differentiate between the group that reverted to class II and the group that remained with class III or IV LN.
Predictors of Response to Treatment of Class III/IV LN Reported response rates are affected by variability in the definition of remission and
variability of initial treatment regimens. Although complete remission should be the goal for LN, attaining at least a partial remission significantly improves kidney prognosis and patient mortality compared to no remission [62].
The 6- to 12-month response rates (both complete and partial) from several trials involving black, white, Hispanic, Mexican, and mixed-race patients are between 20% and 85% [18, 21, 28, 29, 41, 53]. Complete remission rates at 6-12 months were between 8% and 30% in these studies. In contrast, Chinese patients in clinical trials had a consistently better response rate of about 90% and a complete remission rate of 60-80% [22, 24, 25, 26].
Multivariate analyses of retrospective studies suggest that the most important predictors for not achieving remission are SCr at the start of treatment (RR 0.21 per 1 mg/dl [88μmol/l]), the magnitude of increase in SCr during relapse, a delay in starting therapy for more than 3 months after a clinical diagnosis of LN, and severity of proteinuria (HR 0.86 per 1 g/d proteinuria [uPCR 1]) [42, 60].
In one prospective study there were no clinical variables predictive of achieving remission on multivariate analysis [24], while another prospective study showed initial SCr was a predictor of complete remission (RR = 0.96 per 1 μmol/l increase in SCr) [25].
Multivariate analysis from a prospective study showed that failure to achieve complete remission was a major risk factor for kidney relapse [22], while other studies found that no variables were independently predictive of relapse [30]. A survey of several retrospective studies shows that the one common predictor for risk of CKD, ESRD, or death is SCr at presentation [42, 63-65]. In children with LN, failure to respond to therapy and kidney relapse were risk factors for ESRD, HR 5.5 and 11.8 respectively [66].
Monitoring Therapy of Class III/IV LN The progress of LN therapy is monitored with serial measurements of proteinuria and SCr.
There are not yet any more sensitive biomarkers of kidney response in lupus of proven clinical value [67]. In LN, as in other proteinuric GN, resolution of proteinuria is the strongest predictor of kidney survival [68-70]; thus, effective treatment is expected to decrease proteinuria over time.
Effective therapy is also expected to result in reduction of an elevated SCr. A caveat is that there may be may be an acceptable increment in SCr in association with concomitant RAS blockade. Urine sediment should be monitored serially during LN therapy, specifically looking

KDIGO® GN Guideline March 2011 Public Review Draft
153
for resolution of cellular casts over time. However, hematuria may persist for months even if therapy is otherwise successful in improving proteinuria and kidney dysfunction. It is desirable to see serologic markers of lupus activity, such as complement and double-stranded DNA antibody levels, normalize with treatment. However, C3 and C4, and anti–double-stranded DNA antibodies have low sensitivity (49-79%) and specificity (51-74%) in relationship to LN activity [71-77].
RESEARCH RECOMMENDATIONS
• RCTs are needed to compare the efficacy of MMF and cyclophosphamide as initial and maintenance therapy, and in black, non-Caucasian mixed-race, and Hispanic patients.
• An RCT is needed to determine the duration of maintenance therapy in proliferative LN after complete remission.
• Studies are needed to determine if repeat biopsy of patients who achieve only partial remission can guide therapy to achieve complete remission.
• Biomarkers need to be identified that reflect response to therapy and kidney pathology. These would then need to be tested to determine whether they could be used to guide treatment withdrawal, re-treatment, and change in treatment.
12.5: Class V LN (membranous LN) 12.5.1: We recommend that patients with class V LN and non-nephrotic range
proteinuria be treated with antiproteinuria and antihypertensive medications, and only receive corticosteroids and immunosuppressives as dictated by the extrarenal manifestations of systemic lupus. (1B)
12.5.2: We suggest that patients with pure class V LN and persistent nephrotic proteinuria be treated with corticosteroids plus an additional immunosuppressive agent: cyclophosphamide (2C), or CNIs (2C), or MMF (2D), or azathioprine (2D).
BACKGROUND
In class V LN, light microscopy typically shows thickened glomerular basement membranes; immunofluorescence and electron microscopy show only subepithelial immune complexes. If class V LN is accompanied by endocapillary hypercellularity and/or subendothelial immune deposits, this adds class III or IV to the histologic diagnosis. In class V LN, the main clinical finding is proteinuria, often nephrotic-range, with or without hematuria; kidney function is usually normal. If class III or IV LN is also present, urine sediment may be more active, and kidney impairment is more likely.

KDIGO® GN Guideline March 2011 Public Review Draft
154
RATIONALE
• Pure class V LN, although regarded as indolent compared to class III and IV LN, is still associated with the development of CKD and ESRD, especially if there is heavy proteinuria.
• Nephrotic-range proteinuria in class V LN generally does not spontaneously remit. • There has only been one small RCT in class V LN, which compared corticosteroids
plus immunosuppression to corticosteroids alone. • There have been a few small, retrospective trials of MMF and azathioprine in class V
LN. • There have been no studies of the effect of treatment of class V LN on long-term
kidney outcomes. • The prognosis for patients with mixed membranous and proliferative lesions [i.e.,
class V plus class III or IV LN] is less favorable than pure class V LN, and similar to that of patients with class III or IV LN. Patients with mixed membranous and proliferative lesions should be treated similarly to those with class III and IV LN.
There are no convincing data to treat class V LN and subnephrotic proteinuria with
immunosuppression; however, given the adverse effects of proteinuria on the kidney, it is reasonable to treat these patients with antiproteinuric and antihypertensive medications (see Chapter 2). These therapies may reduce proteinuria by as much as 30-50% in class V LN [69, 78, 79]. They should also be used as an adjunct to immunosuppression for patients with nephrotic-range proteinuria.
The justifications to treat class V LN and nephrotic proteinuria with immunosuppression are as follows. Decreased GFR occurs in about 20% of cases of class V LN, and ESRD in about 8-12% after 7-12 years [80-83], with one study reporting death or ESRD in 28% of patients at 10 years [84]. Spontaneous remission of heavy proteinuria occurs in only a minority of class V LN [85, 86]. The adverse effects of sustained, heavy proteinuria include hyperlipidemia and atherosclerosis, contributing to cardiovascular morbidity and mortality [69, 87], and hypercoagulability with arterial and venous thromboses [69, 88]. Thrombotic events occur in 13-23% of class V LN, and have been associated with antiphospholipid antibodies, and/or the nephrotic syndrome [80, 82, 89].
There is only one small RCT (n = 15 in each treatment arm) examining the treatment of class V LN [90]. This study compared the addition of cyclophosphamide or cyclosporine to prednisone in a USA cohort that included blacks, Hispanics, and whites. Both cyclophosphamide and cyclosporine significantly increased response (complete remission 40-50% vs. 14% at 12 months). However, relapse after stopping therapy was much more likely in those treated with cyclosporine (40% within 1 year) compared to cyclophosphamide (no relapse in 48 months). In the same study, the only independent predictor of failure to achieve remission (by multivariate analysis) was initial proteinuria over 5 g/d. Failure to achieve sustained remission was a risk factor for decline in kidney function (see Suppl Tables 79-81).
There have been small uncontrolled retrospective, or open-label, studies of MMF and azathioprine with or without corticosteroids in class V LN [83, 89, 91, 92]. In general, these studies have shown complete remission rates of 40-60% at 6-12 months. A small open-label trial

KDIGO® GN Guideline March 2011 Public Review Draft
155
of tacrolimus in class V LN showed a complete remission rate of 39% at 6 months [93]. Before these regimens can be recommended, they will need to be tested in RCTs.
Patients with mixed class V and class III or IV LN may have a less favorable prognosis, and should be treated as for the proliferative component [80].
RESEARCH RECOMMENDATIONS
• An RCT is needed of corticosteroid monotherapy, compared to corticosteroids combined with MMF or cyclophosphamide, for induction of remission of pure class V LN.
12.6: General treatment of LN 12.6.1: We suggest that all patients with LN of any class be treated with
hydroxychloroquine unless they have a specific contraindication to this drug. (2C)
RATIONALE
• There is low-quality evidence that hydroxychloroquine may protect against the onset of LN, against relapses of LN, ESRD, vascular thrombosis, and that it has a favorable impact on lipid profiles.
In a prospective study, hydroxychloroquine was maintained or withdrawn in a cohort of
patients who had been receiving it before the diagnosis of LN [94]. Those who had been on hydroxychloroquine before developing LN had a lower frequency of ESRD, cardiovascular events, and thrombotic events than patients who had never received hydroxychloroquine; HR for ESRD 0.29 (95% CI 0.026-1.009) [95]. A large (n = 1930), retrospective study found that treatment with hydroxychloroquine protected against vascular thrombosis (OR 0.62; P <0.0005) [96]. Finally, in a prospective observational cohort, hydroxychloroquine was shown to retard kidney damage in LN; the cumulative probability of a 50% reduction in GFR or ESRD after 10 years was 38% for patients on hydroxychloroquine and 70% for those who were not (P <0.0001) [97]. Patients on hydroxychloroquine should have yearly eye examinations for retinal toxicity.
12.7: Class VI LN (advanced sclerosis LN) 12.7.1: We recommend that patients with class VI LN be treated with
corticosteroids and immunosuppressives only as dictated by the extrarenal manifestations of systemic lupus. (2D)

KDIGO® GN Guideline March 2011 Public Review Draft
156
BACKGROUND
In class VI LN, at least 90% of the glomeruli are sclerotic, usually globally, along with interstitial fibrosis and tubular atrophy, with no signs of immunologic activity; the biopsy specimen should be sufficient to be representative of the whole kidney. The dominant clinical picture in class VI LN is severe kidney impairment, usually accompanied by proteinuria and sometimes hematuria.
RATIONALE
• Class VI LN reflects chronic injury, and the consequences of the loss of functional kidney mass, without active immune-mediated injury. Therefore, immunosuppression is not indicated.
• Despite the absence of active LN, patients may still have extrarenal manifestations of systemic lupus requiring immunosuppression.
• As with AKI from any etiology, antiproteinuric and antihypertensive therapies are indicated to preserve residual kidney function and delay ESRD as long as possible.
12.8: Relapse of LN 12.8.1: We suggest that a relapse of LN after complete or partial remission be
treated with the initial therapy followed by the maintenance therapy that was effective in inducing the original remission. (2B) 12.8.1.1: If resuming the original therapy would put the patient at risk
for excessive lifetime cyclophosphamide exposure, then we suggest a non-cyclophosphamide-based initial regimen be used (Regimen D). (2B)
12.8.2: Consider a repeat kidney biopsy during relapse if there is suspicion that the histologic class of LN has changed, or there is uncertainty whether a rising SCr and/or worsening proteinuria represents disease activity or chronicity. (Not Graded)
RATIONALE
• LN is a relapsing condition. • Relapses are associated with development of CKD. • The pathologic findings in LN may change with a relapse, and such changes cannot,
with certainty, be predicted clinically.
In subjects with LN who had participated in RCTs, 40% of complete responders experienced a kidney relapse within a median of 41 months after remission, and 63% of partial responders had a kidney flare within a median of 11.5 months after response [98]. The strongest risk factor for relapse is failure to achieve complete remission (HR 6.2) [22].
Relapses are important to recognize and treat, because the kidneys sustain some chronic damage with each relapse that may culminate in CKD, or eventually ESRD. This is supported by

KDIGO® GN Guideline March 2011 Public Review Draft
157
repeat biopsy studies that showed an increase in the chronicity index at the second biopsy, even after successful treatment [30, 32, 39, 58, 59, 61, 99].
LN may spontaneously transform from one class to another. The most common transformation is from class III to IV [61]. Also, a recent retrospective study found clinically relevant class transformation to be more frequent from a nonproliferative to a proliferative class, rather than proliferative to nonproliferative transformation [100]. Clues to a change in LN class are the development of nephrotic-range proteinuria and changes in the activity of urine sediment, but definitive diagnosis requires a biopsy.
Kidney relapse is diagnosed by clinical criteria based on changes in urine sediment, rate of protein excretion, and SCr change from baseline values in an individual patient. There is no consensus on the definition of a kidney relapse; criteria used in several published studies are shown in Table 12.2 [101-105]. A fall in levels of serum complement components and a rise in anti–double-stranded DNA antibody titers also support a diagnosis of relapse but will not necessarily be present.
Table 12.2. Criteria for the diagnosis and classification of LN flares
Mild kidney flare Moderate kidney flare Severe kidney flare Increase in glomerular hematuria from <5 to >15 RBC/hpf, with ≥2 acanthocytes/hpf and/or recurrence of ≥1 RBC cast, WBC cast (no infection), or both
If baseline creatinine is: <2.0 mg/dl, an increase of 0.20-1.0 mg/dl ≥ 2.0 mg/dl, an increase of 0.40-1.5 mg/dl and/or If baseline uPCR is: <0.5, an increase to ≥1.0 0.5-1.0, an increase to ≥2.0, but absolute increase of <5.0 >1.0, an increase of ≥2-fold with absolute uPCR <5.0
If baseline creatinine is: <2 mg/dl, an increase of >1.0 mg/dl ≥2 mg/dl, an increase of >1.5 mg/dl and/or an absolute increase of uPCR >5.0
LN, lupus nephritis; RBC, red blood cell; uPCR, urine protein:creatinine ratio; WBC, white blood cell.
RESEARCH RECOMMENDATIONS
A study of repeat kidney biopsies at the time of kidney relapse is needed to determine whether it is beneficial to tailor therapy based on biopsy findings.

KDIGO® GN Guideline March 2011 Public Review Draft
158
12.9: Treatment of resistant disease 12.9.1: For patients with worsening SCr and/or proteinuria after completing one
of the initial treatment regimens, consider a repeat kidney biopsy to distinguish active LN from scarring. (Not Graded)
12.9.2: Treat patients with worsening SCr and/or proteinuria who continue to have active LN on biopsy with one of the alternative initial treatment regimens (see Section 12.4). (Not Graded)
12.9.3: We suggest that nonresponders who have failed more than one of the recommended initial regimens (see Section 12.4) may be considered for treatment with i.v. immunoglobulin, CNIs, or rituximab. (2D)
RATIONALE
• Most patients are expected to show some evidence of response to treatment after a year of therapy, although complete remission may occur beyond a year.
• There are no prospective data on patients who fail to achieve at least partial response; it is reasonable, however, to repeat biopsy and determine if there has been a change in kidney pathology that could account for treatment failure.
• There are no prospective data on patients who fail initial therapy; however, it is reasonable to try a second course of initial therapy using an alternative regimen, as dictated by repeat biopsy.
• There have been small studies of “rescue” therapies for patients who have been refractory despite multiple treatment attempts.
In both prospective and retrospective LN cohorts, despite treatment with different protocols
and follow-up under different definitions of remission, the majority of patients who remitted did so within 1 year of therapy [18, 21, 29, 32, 62]. Studies generally show that 50% of patients had a remission (complete or partial) by 12 months, with another 5-25% remitting by 24 months. Among complete remissions, about half were achieved by 12 months, and the other half by 20-24 months.
There is no consensus definition of refractory LN. A patient may be considered refractory if conventional cyclophosphamide regimens have been tried without success, and non-cyclophosphamide regimens have not worked. If repeat kidney biopsy confirms active LN is the cause of continuing clinical abnormalities, there is no definitive information to guide therapy. The following “salvage” treatments have only been evaluated in small observational studies.
The evidence for using i.v. immunoglobulin in refractory cases is of very low quality. It has been used in a handful of patients with proliferative LN, and in some has shown comparable efficacy to cyclophosphamide (reviewed by Rauova et al. [106]). Some formulations of i.v. immunoglobulin (sucrose-containing) have shown nephrotoxicity, and are therefore best avoided in patients with pre-existing kidney impairment.
There is only evidence from small prospective, open-label trials for using low-dose cyclosporine (2.5 mg/kg/d) to treat refractory LN [107, 108]. Although kidney function did not improve, most patients had a reduction in proteinuria, resolution of hematuria, and needed lower

KDIGO® GN Guideline March 2011 Public Review Draft
159
doses of corticosteroids. Similarly, a prospective trial used tacrolimus (3 mg/d) in patients with LN in whom corticosteroids could not be reduced, and demonstrated improvement in proteinuria and C3 levels [109].
The evidence that refractory LN can be treated with rituximab comes only from small, open-label studies [41, 110]. Many of these patients had failed multiple attempts at treatment with the conventional therapies described previously. Rituximab may be considered as a “rescue therapy” when usual therapeutic options have been exhausted. This use of rituximab is in contrast to its lack of utility as add-on therapy to an initial standard regimen (Regimen D) for proliferative LN [57].
RESEARCH RECOMMENDATIONS
• A globally accepted definition of nonresponse needs to be developed. • The salvage therapies discussed in the text must be subject to RCTs to determine
effect on remission and kidney outcomes.
12.10: Systemic lupus and thrombotic microangiopathy
12.10.1: We suggest that the antiphospholipid antibody syndrome (APS) involving the kidney in systemic lupus patients, with or without LN, be treated by anticoagulation (target INR 2-3). (2D)
12.10.2: We suggest that thrombotic thrombocytopenic purpura (TTP) in systemic lupus be treated as for TTP without systemic lupus. (2D)
BACKGROUND
Lupus-associated thrombotic microangiopathies (TMA) may occur alone or in combination with immune-complex LN. TMA in systemic lupus may occur in association with accelerated hypertension, systemic sclerosis, TTP, or in lupus anticoagulant/APS.
While TMA associated with APS, TTP, and accelerated hypertension is often characterized by AKI, APS can also cause slowly progressive kidney impairment with few specific clinical manifestations. In retrospective studies, kidney APS occurred in about 30% of systemic lupus patients [111, 112]. Lupus anticoagulant was present in 30-52% of those with kidney APS, while 72-95% of patients had anticardiolipin antibodies, but 15% had neither of these serologic markers [111, 113]. Routine testing does not identify all antiphospholipid antibodies; therefore, those with TMA who are antiphospholipid antibody–negative are treated in the same way as antibody-positive patients. A high index of suspicion is needed along with a kidney biopsy to confirm the diagnosis.

KDIGO® GN Guideline March 2011 Public Review Draft
160
RATIONALE
• APS occurs frequently in systemic lupus, and there is moderate-quality evidence that failure to treat it may lead to CKD or ESRD, despite adequate control of LN or other systemic lupus manifestations with immunosuppression.
• Although there are no specific studies of anticoagulation for APS with systemic lupus, there have been two RCTs of the intensity of warfarin therapy in APS [114, 115]. They provided moderate-quality evidence of no difference in thrombotic events if the INR was 2-3 or 3-4, but that bleeding complications were higher when INR was maintained greater than 3.
• TTP in lupus is associated with a high mortality [116]. There are no RCTs to guide treatment of TTP in the setting of systemic lupus, but it seems appropriate to use regimens beneficial in TTP without lupus.
RESEARCH RECOMMENDATIONS
• A clinical trial is needed to determine the effect of treating APS on long-term kidney function.
• A clinical trial is needed to determine the efficacy of plasma exchange in TTP, in the setting of systemic lupus.
12.11: Systemic lupus and pregnancy 12.11.1: We suggest that women be counseled to delay pregnancy until a complete
remission of LN has been achieved. (2D) 12.11.2: We recommend that cyclophosphamide, MMF, ACEi, and ARBs not be
used during pregnancy. (1A) 12.11.3: We suggest that hydroxychloroquine be continued during pregnancy.
(2B) 12.11.4: We recommend that LN patients who become pregnant while being
treated with MMF be switched to azathioprine. (1B) 12.11.5: We recommend that, if LN patients relapse during pregnancy, they
receive treatment with corticosteroids and, depending on the severity of the relapse, azathioprine. (1B)
12.11.6: If pregnant patients are receiving corticosteroids or azathioprine, do not taper these drugs during pregnancy or for at least 3 months after delivery. (Not Graded)
12.11.7: We suggest administration of low-dose aspirin during pregnancy to decrease the risk of fetal loss. (2C)
RATIONALE
• Data suggest that active LN or LN in partial remission is associated with an increase in fetal loss and an increased rate of kidney relapse during pregnancy.
• Cyclophosphamide, MMF, ACEi, and ARBss are teratogenic.

KDIGO® GN Guideline March 2011 Public Review Draft
161
• Hydroxychloroquine, azathioprine, and corticosteroids have been used safely during pregnancy in patients with systemic lupus; low-dose aspirin may decrease fetal loss in systemic lupus.
The risk of fetal loss in patients with LN has been examined in several retrospective series.
In a nested case-control study of 78 pregnancies, the incidence of fetal loss was not different in patients with a history of LN compared to systemic lupus patients with no history of LN [117]. In patients with LN in remission, fetal loss of 8-13% has been documented [118-120]. However, in patients with active LN, fetal loss was significantly higher at 35% [120]. In addition to the clinical activity of LN, hypocomplementemia appears to be a risk factor for fetal loss, whereas the use of low-dose aspirin may be protective. In a retrospective study of 113 pregnancies in patients with systemic lupus and LN, hypocomplementemia conferred a RR of 19 for fetal loss, and aspirin conferred a RR of 0.11 [118]. All the patients in this investigation were Caucasian, so the results may not be applicable to other ethnicities.
Hydroxychloroquine should be continued in pregnancy because its withdrawal may lead to flares of lupus, including LN [121].
There may be additional risk to the kidneys of patients with LN who become pregnant. One study noted that kidney relapses and progressive kidney dysfunction were not different between pregnant and nonpregnant patients with LN [117]. In other studies, kidney relapses were more common in pregnancies occurring when only partial remission of LN had been achieved, or in patients who had more than 1 g/d proteinuria or kidney impairment [118, 120]. Kidney relapse rates of 10-69% have been reported during or following pregnancy [117-120].
12.12: LN in children 12.12.1: We suggest that children with LN receive the same therapies as adults
with LN, with dosing based on patient size and GFR. (2D)
RATIONALE
• LN in children shows the same range of clinical and pathological phenotypes as is seen in adults.
• There are no RCTs of LN therapy in children.
Therefore, we suggest that children with LN be treated with the regimens recommended earlier in this chapter. The research recommendations made under 12.1-12.10 also apply to children.
Please refer to Supplemental Tables 68-91 for additional data related to Chapter 12.

KDIGO® GN Guideline March 2011 Public Review Draft
162
References 1. Campbell R, Jr., Cooper GS, Gilkeson GS. Two aspects of the clinical and humanistic burden of systemic
lupus erythematosus: mortality risk and quality of life early in the course of disease. Arthritis Rheum 2008; 59: 458-464.
2. Danila MI, Pons-Estel GJ, Zhang J, Vila LM, et al. Renal damage is the most important predictor of mortality within the damage index: data from LUMINA LXIV, a multiethnic US cohort. Rheumatology (Oxford, England) 2009; 48: 542-545.
3. Font J, Ramos-Casals M, Cervera R, Garcia-Carrasco M, et al. Cardiovascular risk factors and the long-term outcome of lupus nephritis. Qjm 2001; 94: 19-26.
4. Bastian HM, Roseman JM, McGwin G, Jr., Alarcon GS, et al. Systemic lupus erythematosus in three ethnic groups. XII. Risk factors for lupus nephritis after diagnosis. Lupus 2002; 11: 152-160.
5. Seligman VA, Lum RF, Olson JL, Li H, et al. Demographic differences in the development of lupus nephritis: A retrospective analysis. Am J Med 2002; 112: 726-729.
6. Arfaj ASa, Khalil N. Clinical and immunological manifestations in 624 SLE patient in Saudi Arabia. Lupus 2009; 18: 465-473.
7. Ward MM. Changes in the incidence of endstage renal disease due to lupus nephritis in the United States, 1996-2004. J Rheumatol 2009; 36: 63-67.
8. Adler M, Chambers S, Edwards C, Neild G, et al. An assessment of renal failure in an SLE cohort with special reference to ethnicity, over a 25-year period. Rheumatol 2006; 45: 1144-1147.
9. Al Arfaj AS, Khalil N, Al Saleh S. Lupus nephritis among 624 cases of systemic lupus erythematosus in Riyadh, Saudia Arabia. Rheumatology international 2009; 29: 1057-1067.
10. Gonzalez-Crespo MR, Lopez-Fernandez JI, Usera G, Poveda MJ, et al. Outcome of silent lupus nephritis. Seminars in arthritis and rheumatism 1996; 26: 468-476.
11. Valente de Almeida R, Rocha de Carvalho JG, de Azevedo VF, Mulinari RA, et al. Microalbuminuria and renal morphology in the evaluation of subclinical lupus nephritis. Clin Neprol 1999; 52: 218-229.
12. Zabaleta-Lanz ME, Munoz LE, Tapanes FJ, Vargas-Arenas RE, et al. Further description of early clinically silent lupus nephritis. Lupus 2006; 15: 845-851.
13. Kraft SW, Schwartz MM, Korbet SM, Lewis EJ. Glomerular podocytopathy in patients with systemic lupus erythematosus. J Am Soc Nephrol 2005; 16: 175-179.
14. Austin HAr, Klippel JH, Balow JE, le Riche WG, et al. Therapy of lupus nephritis. Controlled trial of prednisone and cytotoxic drugs. N Eng J Med 1986; 314: 614-619.
15. Gourley MF, Austin HA, 3rd, Scott D, Yarboro CH, et al. Methylprednisolone and cyclophosphamide, alone or in combination, in patients with lupus nephritis. A randomized, controlled trial. Annals of internal medicine 1996; 125: 549-557.
16. Donadio JV, Holley KE, Ferguson RH, Ilstrup DM. Treatment of diffuse proliferative lupus nephritis with prednisone and combined prednisone and cyclophosphamide. N Engl J Med 1978; 23: 1151-1155.
17. Boumpas DT, Austin HAr, Vaughn EM, Klippel JH, et al. Controlled trial of pulse methylprenisolone versus two regimens of pulse cyclophosphamide in severe lupus nephritis. Lancet 1992; 340: 741-745.
18. Houssiau FA, Vasconcelos C, D'Cruz D, Sebastiani GD, et al. Immunosuppressive therapy in lupus nephritis: the Euro-Lupus Nephritis Trial, a randomized trial of low-dose versus high-dose intravenous cyclophosphamide. Arthritis Rheum 2002; 46: 2121-2131.
19. Houssiau FA, Vasconcelos C, D'Cruz D, Sebastiani GD, et al. The 10-year follow-up data of the Euro-Lupus Nephritis Trial comparing low-dose versus high-dose intravenous cyclophosphamide. Annals of the rheumatic diseases 2010; 69: 61-64.
20. Moroni G, Doria A, Mosca M, Alberighi ODC, et al. A randomized pilot trial comparing cyclosporine and azathioprine for maintenance in diffuse lupus nephritis over four years. Clin J Am Soc Nephrol 2006; 1: 925-932.
21. McKinley A, Park E, Spetie DN, Hackshaw K, et al. Oral cyclophosphamide for lupus glomerulonephritis: An under-utilized therapeutic option. Clin J Am Soc Nephrol 2009; 4(11):1754-60

KDIGO® GN Guideline March 2011 Public Review Draft
163
22. Chan TM, Tse KC, Tang CSO, Lai KN, et al. Long-term outcome of patients with diffuse proliferative lupus nephritis treated with prednisolone and oral cyclophosphamide followed by azathioprine. Lupus 2005; 14: 265-272.
23. Moroni G, Quaglini S, Gallelli B, Banfi G, et al. The long-term outcome of 93 patients with proliferative lupus nephritis. Nephrol Dial Transplant 2007; 22: 2531-2539.
24. Mok CC, Ho CTK, Siu YP, Chan KW, et al. Treatment of diffuse proliferative lupus glomerulonephritis: A comparison of two cyclophosphamide-containing regimens. Am J Kidney Dis 2001; 38: 256-264.
25. Mok CC, Ho CTK, Chan KW, Lau CS, et al. Outcome and prognostic indicators of diffuse proliferative lupus glomerulonephritis treated with sequential oral cyclophosphamide and azathioprine. Arthritis Rheum 2002; 46: 1003-1013.
26. Chan TM, Li FK, Tang CSO, Wong RWS, et al. Efficacy of mycophenolate mofetil in patients with diffuse proliferative lupus nephritis. N Eng J Med 2000; 343: 1156-1162.
27. Chan TM, Tse KC, Tang CSO, Mok M-Y, et al. Long-term study of mycophenolate mofetil as continuous induction and maintenance treatment for diffuse proliferative lupus nephritis. J Am Soc Nephrol 2005; 16: 1076-1084.
28. Appel GB, Contreras G, Dooley MA, Ginzler EM, et al. Mycophenolate Mofetil versus Cyclophosphamide for Induction Treatment of Lupus Nephritis. J Am Soc Nephrol 2009; 20: 1103-1112.
29. Grootscholten C, Ligtenberg G, Hagen EC, van den Wall Bake AWL, et al. Azathioprine/methylprednisolone versus cyclophosphamide in proliferative lupus nephritis. A randomized, controlled trial. Kidney Int 2006; 70: 732-742.
30. Grootscholten C, Bajema IM, Florquin S, Steenbergen EJ, et al. Treatment with cyclophosphamide delays the progression of chronic lesions more effectively than does treatment with azathioprine plus methylprednisolone in patients with proliferative lupus nephritis. Arthritis Rheum 2007; 56: 924-937.
31. Zavada J, Pesickova SS, Rysava R, Olejarova M, Horak P, Hrncir Z, Rychlik I, Havrda M, Vitova J, Lukac J, Rovensky J, Tegzova D, Bohmova J, Zadrazil J, Hana J, Dostal C, Tesar V, Cyclosproine A or intravenous cyclophosphamide for lupus nephritis: The Cyclopfa-Lune Study. Lupus. 2010 Oct; 19(11):1281-9.
32. Bao H, Liu Z-H, Xie H-L, Hu W-X, et al. Successful treatment of class V+IV lupus nephritis with multitarget therapy. J Am Soc Nephrol 2008; 19: 2001-2010.
33. Balow JE, Austin HA, Muenz LR, Joyce KM, et al. Effect of treatment on the evolution of renal abnormalities in lupus nephritis. N Engl J Med 1084; 311: 491-495.
34. Philibert D, Cattran D. Remission of proteinuria in primary glomerulonephritis: we know the goal but do we know the price? Nature clinical practice 2008; 4: 550-559.
35. Faurschou M, Sorensen IJ, Meliemkjaer L, Loft AGR, et al. Malignancies in Wegener's granulomatosis: Incidence and realtion to cyclophosphamide therapy in a cohort of 293 patients. The Journal of rheumatology 2008; 35: 100-105.
36. Haubitz M, Bohnenstengel F, Brunkhorst R, Schwab M, et al. Cyclophosphamide pharmacokinetics and dose requirements in patients with renal insufficiency. Kidney international 2002; 61: 1495-1501.
37. Pendse S, Ginsburg E, Singh AK. Strategies for preservation of ovarian and testicular function after immunosuppression. Am J Kidney Dis 2004; 43: 772-781.
38. Sommers EC, Marder W, Christman GM, Ognenovski V, et al. Use of a gonadotropin-releasing hormone analog for protection against premature ovarian failure during cyclophosphamide therapy in women with severe lupus. Arthritis Rheum 2005; 52: 2761-2767.
39. Ong LM, Hooi LS, Lim TO, Goh BL, et al. Randomized controlled trial of pulse intravenous cyclophosphamide versus mycophenolate mofetil in the induction therapy of proliferative lupus nephritis. Nephrology (Carlton, Vic 2005; 10: 504-510.
40. Hu W, Liu Z, Chen H, Tang Z, et al. Mycophenolate mofetil vs cyclophosphamide therapy for patients with diffuse proliferative lupus nephritis. Chinese medical journal 2002; 115: 705-709.
41. Ginzler EM, Dooley MA, Aranow C, Kim MY, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Eng J Med 2005; 353: 2219-2228.

KDIGO® GN Guideline March 2011 Public Review Draft
164
41A. El-Shafey EM, Abdou SH, Shareef MM. Is mycophenolate mofetil superior to pulse intravenous cyclophosphamide for induction therapy of proliferative lupus nephritis in Egyptian patients? Clinical & Experimental Nephrology 14(3):214-21, 2010
42. Korbet SM, Schwartz MM, Evans J, Lewis EJ. Severe lupus nephritis: Racial differences in presentation and outcome. J Am Soc Nephrol 2007; 18: 244-254.
43. Rovin BH, Appel GB, Furie RA, Looney RJ, et al. Efficacy and safety of rituximab in subjects with proliferative lupus nephritis: Results fromt he randomized, double-blind, phase III LUNAR study. J Am Soc Nephrol 2009; in press.
44. Tieng AT, Peeva E. B-Cell-directed therapies in systemic lupus erythematosus. Seminars in arthritis and rheumatism 2008; 38: 218-227.
45. Karim MY, Pisoni CN, Khamashta MA. Update on immunotherapy for systemic lupus erythematosus-what's hot and what's not! Rheumatology (Oxford, England) 2009; 48: 332-341.
46. Sousa E, Isenberg D. Treating lupus: from serendipity to sense, the rise of the new biologicals and other emerging therapies. Best Practice and Clinical Rheumatology 2009; 23: 563-574.
47. Ramos-Casals M, Soto MJ, Cuadrado MJ, Khamashta MA. Rituximab in systemic lupus erythematosus: A systematic review of off-label use in 188 cases. Lupus 2009; 18: 767-776.
48. Lu TY, Ng KP, Cambridge G, Leandro MJ, et al. A retrospective seven-year analysis of the use of B cell depletion therapy in systemic lupus erythematosus at University College London Hospital: the first fifty patients. Arthritis Rheum 2009; 61: 482-487.
49. Li EK, Tam LS, Zhu TY, Li M, et al. Is combination rituximab with cyclophosphamide better than rituximab alone in the treatment of lupus nephritis? Rheumatology (Oxford, England) 2009; 48: 892-898.
50. Gunnarsson I, Sundelin B, Jonsdottir T, Jacobson SH, et al. Histopathologic and clinical outcome of rituximab treatment in patients with cyclophosphamide-resistant proliferative lupus nephritis. Arthritis Rheum 2007; 56: 1263-1272.
51. Cameron JS, Turner DR, Ogg CS, Williams DG, et al. Systemic lupus with nephritis: A long-term study. QJ Med 1979; 189: 1-24.
52. Jayne DRW, Appel GB, Dooley MA, Ginzler E, et al., Results of the Aspreva Lupus Management Study (ALMS) Maintenance Phase. J Am Soc Nephrol 2010 21: 25A.
53. Contreras G, Pardo V, Leclercq B, Lenz O, et al. Sequential therapies for proliferative lupus nephritis. N Eng J Med 2004; 350: 971-980.
54. Houssiau FA, D’Cruz D, Sangle S, Remy P, et al. Azathioprine versus mycopheolate mofetil for long-term immunosuppression in lupus nephritis : results from the MAINTAIN Nephritis Trial. Ann Rheum Dis 2010; 69:2083-2089.
55. Moroni G, Doria A, Mosca M, Alberighi ODC, et al. A randomized pilot trial comparing cyclosporine and azathioprine for maintenance in diffuse lupus nephritis over four years. Clin J Am Soc Nephrol 2006; 1: 925-932.
56. Griffiths B, Emery P, Ryan V, Isenberg D, et al. The BILAG multicentre open randomized controlled trial comparing ciclosporin vs azathioprine in patients with severe SLE. Rheumatology (Oxford, England) 2010; Apr;49(4):723-32.
57. Hill GS, Delahousse M, Nochy D, Remy P, et al. Predictive poawer of the second renal biopsy in lupus nephritis: Significance of macrophages. Kidney international 2001; 59: 304-316.
58. Gunnarsson I, Sundelin B, Heimburger M, Forslid J, et al. Repeated renal biopsy in proliferative lupus nephritis--predictive role of serum C1q and albuminuria. The Journal of rheumatology 2002; 29: 693-699.
59. Traitanon O, Avihingsanon Y, Kittikovit V, Townamchai N, et al. Efficacy of enteric-coated mycophenolate sodium in patients with resistant-type lupus nephritis: a prospective study. Lupus 2008; 17: 744-751.
60. Ioannidis JPA, Boki KA, Katsorida ME, Drosos AA, et al. Remission, relapse, and re-remission of proliferative lupus nephritis treated with cyclophosphamide. Kidney Int 2000; 57: 258-264.
61. Moroni G, Pasquali S, Quaglini S, Banfi G, et al. Clinical and prognostic value of serial renal biopsies in lupus nephritis. Am J Kidney Dis 1999; 34: 530-539.

KDIGO® GN Guideline March 2011 Public Review Draft
165
62. Chen YE, Korbet SM, Katz RS, Schwartz MM, et al. Value of a complete or partial remission in severe lupus nephritis. Clin J Am Soc Nephrol 2008; 3: 46-53.
63. Contreras G, Pardo V, Cely C, Borja E, et al. Factors associated with poor outcomes in patients with lupus nephritis. Lupus 2005; 14: 890-895.
64. Barr RG, Seliger S, Appel GB, Zuniga R, et al. Prognosis in proliferative lupus nephritis: the role of socio-economic status and race/ethnicity. Nephrol Dial Transplant 2003; 18: 2039-2046.
65. GISNEL. Lupus nephritis: Prognostic factors and probability of maintaining life-supporting kidney function 10 years after the diagnosis. Am J Kidney Dis 1992; 19: 473-479.
66. Gipson KL, Gipson DS, Massengill SA, Dooley MA, et al. Predictors of relapse and end stage kidney disease in proliferative lupus nephritis: Focus on children, adolescents, and young adults. Clin J Am Soc Nephrol 2009; 4: 1962-1967.
67. Rovin BH, Zhang X. Biomarkers for lupus nephritis: The quest continues. Clin J Am Soc Nephrol 2009; 4: 1858-1865.
68. Hebert LA, Wilmer WA, Falkenhain ME, Ladson-Wofford SE, et al. Renoprotection: One or many therapies. Kidney Int 2001; 59: 1211-1226.
69. Wilmer WA, Rovin BH, Hebert CJ, Rao SV, et al. Management of Glomerular Proteinuria: A Commentary. J Am Soc Nephrol 2003; 14: 3217-3232.
70. Reich HN, Troyanov S, Scholey JW, Cattran DC. Remission of proteinuria improves prognosis in IgA nephropathy. J Am Soc Nephrol 2007; 18: 3177-3183.
71. Rovin BH, Birmingham DJ, Nagaraja HN, Yu CY, et al. Biomarker discovery in human SLE nephritis. Bulletin of the NYU hospital for joint diseases 2007; 65: 187-193.
72. Moroni G, Radice A, Giammarresi G, Quaglini S, et al. Are laboratory tests useful for monitoring the activity of lupus nephritis? A 6-year prospective study in a cohort of 228 patients with lupus nephritis. Annals of the rheumatic diseases 2009; 68: 234-237.
73. Esdaile JM, Joseph L, Abrahamowicz M, Li Y, et al. Routine immunologic tests in systemic lupus erythematosus: Is there a need for more studies? J Rheumatol 1996; 23: 1891-1896.
74. Esdaile JM, Abrahamowicz M, Joseph L, Mackenzie T, et al. Laboratory tests as predictors of disease exacerbations in systemic lupus erythematosus. Arthritis Rheum 1996; 39: 370-378.
75. Coremans IEM, Spronk PE, Bootsma H, Daha MR, et al. Changes in antibodies to C1q predict renal relapse in system lupus erythematosus. Am J Kidney Dis 1995; 26: 595-601.
76. Ho A, Barr SG, Magder LS, Petri M. A decrease in complement is associated with increased renal and hematologic activity in patients with systemic lupus erythematosus. Arthritis Rheum 2001; 44: 2350-2367.
77. Ho A, Magder LS, Barr SG, Petri M. Decreases in anti-double-stranded DNA levels are associated with concurrent flares in patients with systemic lupus erythematosus. Arthritis Rheum 2001; 44: 2342-2349.
78. Jafar TH, Schmid CH, Landa M, Giatras I, et al. Angiotensin-converting enzyme inhibitors and progression of nondiabetic renal disease. A meta-analysis of patient-level data. Annals of internal medicine 2001; 135: 73-87.
79. Giatras I, Lau J, Levey AS. Effect of antgiotensin-converting enzyme inhibitors on the progression of non-diabetic renal disease: a meta-analysis of randomized trials. Angiotensin-converting-enzyme inhibition and progressive renal disease study group. Annals of internal medicine 1997; 127: 337-345.
80. Pasquali S, Banfi G, Zucchelli A, Moroni G, et al. Lupus membranous nephropathy: long-term outcome. Clin Nephrol 1993; 39: 175-182.
81. Mok CC. Membranous nephropathy in systemic lupus erythematosus: a therapeutic enigma. Nat Rev Nephrol 2009; 5: 212-220.
82. Mercadal L, Montcel ST, Nochy D, Queffeulou G, et al. Factors affecting outcome and prognosis in membranous lupus nephropathy. Nephrol Dial Transplant 2002; 17: 1771-1778.
83. Mok C, Ying K, Yim C, Ng W, et al. Very long-term outcome of pure membranous nephropathy treated with glucocorticoid and azathioprine. Lupus 2009; 18: 1091-1095.
84. Sloan RP, Schwartz MM, Korbet SM, Borok RZ. Long-term outcome in systemic lupus erythematosus membranous glomerulonephritis. J Am Soc Nephrol 1996; 7: 299-305.

KDIGO® GN Guideline March 2011 Public Review Draft
166
85. Donadio JV, Burgess JH, Holley KE. Membranous lupus nephropathy: A clinicopathologic study. Medicine 1977; 56: 527-536.
86. Gonzalez-Dettoni H, Tron F. Membranous glomerulonephropathy in systemic lupus erythematosus. Adv Nephrol 1985; 14: 347-364.
87. Ordonez JD, Hiatt RA, Killebrew EJ, Fireman BH. The increased risk of coronary heard disease associated with the nephrotic syndrome. Kidney international 1993; 44: 638-642.
88. Font J, Ramos-Casals M, Cervera R, Garcia-Carrasco M, et al. Cardiovascular risk factors and the long-term outcome of lupus nephritis. QJ Med 2001; 94: 19-26.
89. Mok CC, Ying KY, Lau CS, Yim CW, et al. Treatment of pure membranous lupus nephropathy with prednisone and azathioprine: an open-label trial. Am J Kidney Dis 2004; 43: 269-276.
90. Austin HAr, Illei GG, Braun MJ, Balow JE. Randomized, controlled trial of prednisone, cyclophosphamide, and cyclosporine in lupus membranous nephropathy. J Am Soc Nephrol 2009; 20: 901-911.
91. Kasitanon N, Petri M, Haas M, Magder LS, et al. Mycophenolate mofetil as the primary treatment of membranous lupus nephritis with and without concurrent proliferative disease: a retrospective study of 29 cases. Lupus 2008; 17: 40-45.
92. Spetie DN, Tang Y, Rovin BH, Nadasdy G, et al. Mycophenolate therapy of SLE membranous nephropathy. Kidney international 2004; 66: 2411-2415.
93. Szeto CC, Kwan BC, Lai FM, Tam LS, et al. Tacrolimus for the treatment of systemic lupus erythematosus with pure class V nephritis. Rheumatology (Oxford, England) 2008; 47: 1678-1681.
94. Tsakonas E, Joseph L, Esdaile JM, Choquette D, et al. A long-term study of hydroxychloroquine withdrawal on exacerbations in systemic lupus erythematosus. The Canadian hydroxychloroquine study group. Lupus 1998; 7: 80-85.
95. Siso A, Ramos-Casals M, Bove A, Brito-Zeron P, et al. Previous antimalarial therapy in patients diagnosed with lupus nephritis: influence on outcomes and survival. Lupus 2008; 17: 281-288.
96. Kaiser R, Cleveland CM, Criswell LA. Risk and protective factors for thrombosis in systemic lupus erythematosus: results from a large, multi-ethnic cohort. Annals of the rheumatic diseases 2009; 68: 238-241.
97. Pons-Estel GJ, Alarcon GS, McGwin G, Jr., Danila MI, et al. Protective effect of hydroxychloroquine on renal damage in patients with lupus nephritis: LXV, data from a multiethnic US cohort. Arthritis Rheum 2009; 61: 830-839.
98. Illei GG, Takada K, Parkin D, Austin HA, et al. Renal flares are common in patients with severe proliferative lupus nephritis treated with pulse immunosuppressive therapy: long-term followup of a cohort of 145 patients participating in randomized controlled studies. Arthritis Rheum 2002; 46: 995-1002.
99. Esdaile JM, Joseph L, Mackenzie T, Kashgarian M, et al. The pathogenesis and prognosis of lupus nephritis: Information from repeat renal biopsy. Seminars in arthritis and rheumatism 1993; 23: 135-148.
100. Daleboudt GMN, Bajema IM, Goemaere NNT, van Laar JM, et al. The clinical relevance of a repeat biopsy in lupus nephritis flares. Nephrol Dial Transplant 2009: gfp359.
101. Rovin BH, Song H, Birmingham DJ, Hebert LA, et al. Urine chemokines as biomarkers of human systemic lupus erythematosus activity. J Am Soc Nephrol 2005; 16: 467-473.
102. Rovin BH, Nadasdy G, Nuovo GJ, Song H, et al. Expression of adiponectin and its receptors in the kidney during SLE nephritis. J Am Soc Nephrol 2006; 17: 256A.
103. Birmingham DJ, Nagaraja HN, Rovin BH, Spetie L, et al. Fluctuation in self-perceived stress increases risk of flare in patients with lupus nephritis patients carrying the serotonin receptor1A-1019G allele. Arthritis Rheum 2006; 54: 3291-3299.
104. Clough JD, Lewis EJ, Lachin JM. Treatment protocols of the lupus nephritis collaborative study of plasmapheresis in severe lupus nephritis. Apheresis 1990: 301-307.
105. Linnik MD, Hu J-Z, Heilbrunn KR, Strand V, et al. Relationship between anti-double-stranded DNA antibodies and exacerbation of renal disease in patients with systemic lupus erythematosus. Arthritis Rheum 2005; 52: 1129-1137.
106. Rauova L, Lukac J, Levy Y, Rovensky J, et al. High-dose intravenous immunoglobulins for lupus nephritis-a salvage immunomodulation. Lupus 2001; 10: 209-213.

KDIGO® GN Guideline March 2011 Public Review Draft
167
107. Ogawa H, Kameda H, Nagasawa H, Sekiguchi N, et al. Prospective study of low-dose cyclosporine A in patients with refractory lupus nephritis. Mod Rheumatol 2007; 17: 92-97.
108. Ogawa H, Kameda H, Amano K, Takeuchi T. Efficacy and safety of cyclosporine A in patients with refractory systemic lupus erythematosus in a daily clinical practice. Lupus 2010; 19: 162-169.
109. Miyasaka N, Kawai S, Hashimoto H. Efficacy and safety of tacrolimus for lupus nephritis: a placebo-controlled double-blind multicenter study. Mod Rheumatol 2009; 19: 606-615.
110. Garcia-Carrasco M, Mendoza-Pinto C, Sandoval-Cruz M, Soto-Vega E, et al. Anti-CD20 therapy in patients with refractory systemic lupus erythematosus: a longitudinal analysis of 52 Hispanic patients. Lupus 2009.
111. Daugas E, Nochy D, Huong DL, Duhaut P, et al. Antiphospholipid syndrome nephropathy in systemic lupus erythematosus. J Am Soc Nephrol 2002; 13: 42-52.
112. Tektonidou MG. Renal involvement in the antiphospholipid syndrome (APS)-APS nephropathy. Clinical reviews in allergy & immunology 2009; 36: 131-140.
113. Tektonidou MG, Sotsiou F, Moutsopoulos HM. Antiphospholipid syndrome nephropathy in catastrophic, primary, and systemic lupus erythematosus-related APS. The Journal of rheumatology 2008; 35: 1983-1988.
114. Finazzi G, Marchioli R, Brancaccio V, Schinco P, et al. A randomized clinical trial of high-intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS). J Thromb Haemost 2005; 3: 848-853.
115. Crowther MA, Ginsberg JS, Julian J, Denburg J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med 2003; 349: 1133-1138.
116. Kwok SK, Ju JH, Cho CS, Kim HY, et al. Thrombotic thrombocytopenic purpura in systemic lupus erythematosus: risk factors and clinical outcome: a single centre study. Lupus 2009; 18: 16-21.
117. Tandon A, Ibanez D, Gladman D, Urowitz M. The effect of pregnancy on lupus nephritis. Arthritis Rheum 2004; 50: 3941-3946.
118. Imbasciati E, Tincani A, Gregorini G, Doria A, et al. Pregnancy in women with pre-existing lupus nephritis: predictors of fetal and maternal outcome. Nephrol Dial Transplant 2009; 24: 519-525.
119. Carvalheiras G, Vita P, Marta S, Trovao R, et al. Pregnancy and Systemic Lupus Erythematosus: Review of Clinical Features and Outcome of 51 Pregnancies at a Single Institution. Clinical reviews in allergy & immunology 2009.
120. Wagner SJ, Craici I, Reed D, Norby S, et al. Maternal and foetal outcomes in pregnant patients with active lupus nephritis. Lupus 2009; 18: 342-347.
121. Levy RA, Vilela VS, Cataldo MJ et al. Hydroxychloroquine (HCQ) in lupus pregnancy: double-blind and placebo-controlled study. Lupus. 2001;10(6):401-4.

KDIGO® GN Guideline March 2011 Public Review Draft
168
CHAPTER 13: TREATMENT OF PAUCI-IMMUNE FOCAL AND SEGMENTAL NECROTIZING GLOMERULONEPHRITIS
INTRODUCTION
This chapter applies to adults with pauci-immune focal and segmental necrotizing GN with or without systemic vasculitis, and with or without circulating ANCA.
13.1: Initial treatment of pauci-immune focal and segmental necrotizing GN 13.1.1: We recommend that cyclophosphamide and corticosteroids be used as
initial treatment for ANCA vasculitis. (1A) 13.1.2: We suggest that rituximab and corticosteroids be used as an alternative
initial treatment in patients. (1A)
13.2: Special patient populations 13.2.1: We recommend the addition of plasmapheresis for patients requiring
dialysis or with rapidly increasing SCr. (1C) 13.2.2: We suggest the addition of plasmapheresis for patients with diffuse
pulmonary hemorrhage. (2C) 13.2.3: We suggest the addition of plasmapheresis for patients with overlap
syndrome of ANCA vasculitis and anti-GBM, according to proposed criteria and regimen for anti-GBM GN (see Chapter 14). (2D)
13.2.4: We suggest discontinuing cyclophosphamide therapy after 3 months in patients who remain dialysis-dependent and who do not have any extrarenal manifestations of disease. (2C)
BACKGROUND
Small-vessel vasculitis encompasses a group of diseases characterized by necrotizing inflammation of the small vessels: arterioles, capillaries, and venules. They are characterized by little or no deposition of immune complexes in the vessel wall (pauci-immune). Medium or large vessels may occasionally be involved. Pauci-immune small vessel vasculitides include Wegener’s granulomatosis, microscopic polyangiitis, and Churg-Strauss syndrome. The characteristic kidney lesion in these conditions is pauci-immune necrotizing and crescentic glomerulonephritis (NCGN). Active pauci-immune small vessel-vasculitis is typically associated with circulating ANCA. NCGN may also occur without extrarenal manifestations of disease.
The clinical manifestations associated with NCGN include microscopic hematuria with dysmorphic red blood cells and red cell casts, and proteinuria that is usually moderate (1-3 g/d). Pauci-immune NCGN is frequently associated with a rapidly declining GFR over days or weeks. A minority of patients may present with a more indolent course with asymptomatic microscopic hematuria and minimal proteinuria, which may progress over months.

KDIGO® GN Guideline March 2011 Public Review Draft
169
Patients with systemic vasculitis may present with a variety of extrarenal clinical manifestations affecting one or several organ systems, with or without kidney involvement. Commonly involved systems are upper and lower respiratory tract, skin, eyes, and the nervous system. Severe pulmonary hemorrhage affects about 10% of patients with ANCA GN, and is associated with an increased risk of death [1]. The need to treat extrarenal vasculitis may impinge on treatment choices for renal vasculitis.
About 90% of patients with small-vessel vasculitis or pauci-immune NCGN have ANCA, directed primarily to the neutrophil granule proteins myeloperoxidase (MPO) or proteinase 3 (PR3).
The treatment recommendations in this guideline derive from studies of patients with ANCA-positive vasculitis and/or GN. About 10% of patients presenting with signs and symptoms of microscopic polyangiitis, Wegener’s granulomatosis, or pauci-immune NCGN are persistently ANCA-negative. These patients are treated similarly to ANCA-positive patients, although no study has focused specifically on the treatment of ANCA-negative patients.
RATIONALE
• Without therapy, ANCA vasculitis with GN is associated with very poor outcomes. • There is high-quality evidence for treatment with corticosteroids and
cyclophosphamide that has dramatically improved the short- and long-term outcomes of ANCA vasculitis associated with systemic disease.
• Immunosuppressive therapy may not be appropriate in patients with severe NCGN already requiring dialysis.
• All patients with extrarenal manifestations of disease should receive immunosuppressive therapy regardless of the degree of kidney dysfunction.
• There is high-quality evidence that plasmapheresis provides additional benefit in those with severe NCGN.
• There is low-quality evidence that plasmapheresis provides additional benefit for diffuse pulmonary hemorrhage.
• There is high-quality evidence that rituximab is not inferior to cyclophosphamide in induction therapy.
Recommended treatment regimens are shown in Table 13.1.
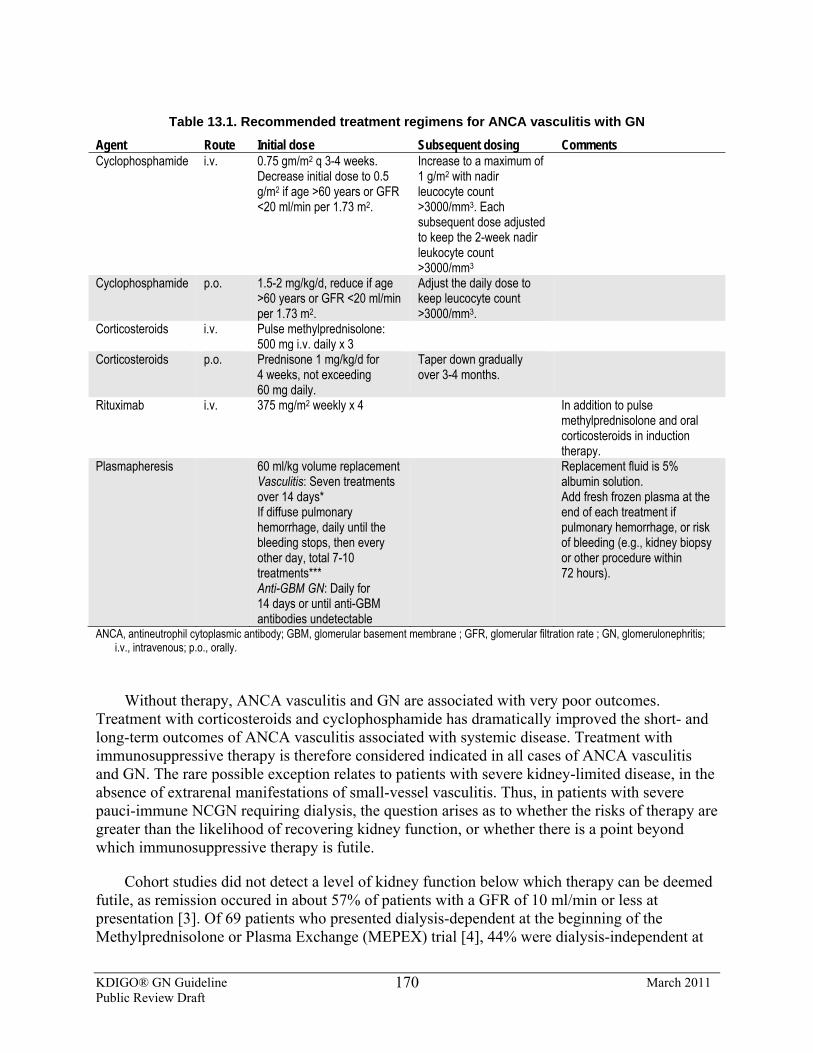
KDIGO® GN Guideline March 2011 Public Review Draft
170
Table 13.1. Recommended treatment regimens for ANCA vasculitis with GN
Agent Route Initial dose Subsequent dosing Comments Cyclophosphamide i.v. 0.75 gm/m2 q 3-4 weeks.
Decrease initial dose to 0.5 g/m2 if age >60 years or GFR <20 ml/min per 1.73 m2.
Increase to a maximum of 1 g/m2 with nadir leucocyte count >3000/mm3. Each subsequent dose adjusted to keep the 2-week nadir leukocyte count >3000/mm3
Cyclophosphamide p.o. 1.5-2 mg/kg/d, reduce if age >60 years or GFR <20 ml/min per 1.73 m2.
Adjust the daily dose to keep leucocyte count >3000/mm3.
Corticosteroids i.v. Pulse methylprednisolone: 500 mg i.v. daily x 3
Corticosteroids p.o. Prednisone 1 mg/kg/d for 4 weeks, not exceeding 60 mg daily.
Taper down gradually over 3-4 months.
Rituximab i.v. 375 mg/m2 weekly x 4
In addition to pulse methylprednisolone and oral corticosteroids in induction therapy.
Plasmapheresis 60 ml/kg volume replacement Vasculitis: Seven treatments over 14 days* If diffuse pulmonary hemorrhage, daily until the bleeding stops, then every other day, total 7-10 treatments*** Anti-GBM GN: Daily for 14 days or until anti-GBM antibodies undetectable
Replacement fluid is 5% albumin solution. Add fresh frozen plasma at the end of each treatment if pulmonary hemorrhage, or risk of bleeding (e.g., kidney biopsy or other procedure within 72 hours).
ANCA, antineutrophil cytoplasmic antibody; GBM, glomerular basement membrane ; GFR, glomerular filtration rate ; GN, glomerulonephritis; i.v., intravenous; p.o., orally.
Without therapy, ANCA vasculitis and GN are associated with very poor outcomes. Treatment with corticosteroids and cyclophosphamide has dramatically improved the short- and long-term outcomes of ANCA vasculitis associated with systemic disease. Treatment with immunosuppressive therapy is therefore considered indicated in all cases of ANCA vasculitis and GN. The rare possible exception relates to patients with severe kidney-limited disease, in the absence of extrarenal manifestations of small-vessel vasculitis. Thus, in patients with severe pauci-immune NCGN requiring dialysis, the question arises as to whether the risks of therapy are greater than the likelihood of recovering kidney function, or whether there is a point beyond which immunosuppressive therapy is futile.
Cohort studies did not detect a level of kidney function below which therapy can be deemed futile, as remission occured in about 57% of patients with a GFR of 10 ml/min or less at presentation [3]. Of 69 patients who presented dialysis-dependent at the beginning of the Methylprednisolone or Plasma Exchange (MEPEX) trial [4], 44% were dialysis-independent at

KDIGO® GN Guideline March 2011 Public Review Draft
171
12 months, and the point at which the chance of dying from therapy with plasmapheresis exceeded that of the chance of recovery was reached only in patients with severe tubular atrophy and injury of nearly all glomeruli [5]. This study suggests that, in the absence of extrarenal manifestations of disease, treatment is warranted in all but patients with extreme glomerular obsolescence and severe tubulointerstitial scarring. All patients with extrarenal manifestations of disease should receive immunosuppressive therapy, regardless of the degree of kidney dysfunction.
Disease Activity Kidney manifestations of active GN are a progressive decline in kidney function, ongoing
proteinuria with the continued presence of dysmorphic red cells in the urine, and red cell casts.
Remission is defined by the absence of manifestations of vasculitis and GN disease activity. For GN, it is defined as the absence of microscopic hematuria and a stable or improved proteinuria and GFR. Disease activity of ANCA-vasculitis represents signs or symptoms attributable to active disease is any organ system.
Cyclophosphamide The addition of cyclophosphamide to corticosteroids in induction therapy improved the
remission rate from about 55% to about 85%, and decreased the relapse rate three-fold [2,3].
Pulse i.v. and daily oral regimens for cyclophosphamide are associated with similar remission and relapse rates [7]. Considerations in choosing one approach over the other are: compliance, cost, cumulative dose of cyclophosphamide, frequency of leucopenia, and infection. For the same duration of therapy, the use of the i.v. pulse regimen resulted in a two to three times smaller cumulative dose of cyclophosphamide [7]. Pulse i.v. cyclophosphamide is also associated with a lower frequency of severe leucopenia, but not necessarily with a lower incidence of severe infections. In a meta-analysis of three RCTs, pulse i.v. cyclophosphamide compared to daily oral cyclophosphamide was associated with a significantly reduced rate of infection (OR 0.4;, 95% CI 0.23-0.89) and leucopenia (OR 0.36; 95%CI 0.17-0.78) [8]. Pulse i.v. cyclophosphamide may be associated with a trend towards a higher rate of late relapses (after 18 months) [7,8].
The recommended duration of treatment with cyclophosphamide is 2 months after remission is attained, based on the RCT of maintenance therapy comparing cyclophosphamide to azathioprine [9]. Whether this duration of treatment applies to pulse i.v. cyclophosphamide is inferred, but not tested. The only study of a short (6-month) vs. long (12-month) course of cyclophosphamide was not powered to detect a difference in outcome [10]. A retrospective cohort analysis did not indicate that longer treatment with cyclophosphamide reduces the rate of relapse [3].
Pulse Methylprednisolone The value of pulse methylprednisolone induction therapy has not been tested directly. The
rationale for pulse methylprednisolone is related to its rapid anti-inflammatory effect. High-dose methylprednisolone may also contribute to a rapid reduction in ANCA-producing plasma cells. The only randomized evaluation of pulse methylprednisolone (3 x 1000 mg) was in the setting of

KDIGO® GN Guideline March 2011 Public Review Draft
172
the MEPEX trial, where it was compared to plasmapheresis as adjunctive therapy to oral corticosteroids and oral cyclophosphamide [4]. In that trial, pulse methylprednisolone was less efficacious than plasmapheresis in preserving kidney function. There are no data that 1000 mg daily for 3 days is better than 500 mg; this lower dose is widely used in clinical practice, and the higher dose may be associated with increased short- and long-term risks of infection and other complications of steroids.
Rituximab Two RCTs examined rituximab as first-line induction therapy for ANCA vasculitis. In the
RITUXVAS trial, 44 patients with newly diagnosed ANCA vasculitis were randomized 3:1 to either rituximab (375 mg/m2 weekly x 4) in addition to cyclophosphamide (15 mg/kg i.v., 2 weeks apart for a total of two doses); or to cyclophosphamide (15 mg/kg i.v. every 2 weeks x 3, then every 3 weeks for a maximum total of 10 doses) [11]. Both groups received the same regimen of methylprednisolone 1000 mg IV followed by oral corticosteroids. Rates of remission were similar (76% with rituximab group vs. 82% with cyclophosphamide); however, the combination of rituximab and two doses of cyclophosphamide was associated with more frequent serious adverse events (infections) than i.v. cyclophosphamide for 6 months [11].
In Rituximab for the Treatment of Wegener’s Granulomatosis and Microscopic Polyangiitis (RAVE), 197 patients were randomized to treatment with either rituximab (375 mg/m2 infusions once weekly for 4 weeks) or cyclophosphamide (2 mg/kg/d orally) for months 1-3, followed by azathioprine (2 mg/kg/d orally) for months 4-6. All patients received one to three i.v. pulses of methylprednisolone (1000 mg each) followed by the same oral corticosteroid regimen. There was no difference between the two treatment groups in rates of complete remission at 6 months [14], nor in the rates of disease relapse. The RAVE trial excluded patients with severe alveolar hemorrhage or severe kidney dysfunction (SCr >4 mg/dl [390 μmol/l]), so the role of rituximab for such patients remains unknown.
Although rituximab shows equivalent efficacy to cyclophosphamide in initial therapy, there is no evidence it is associated with fewer adverse effects. Analysis of the long-term outcomes, including safety, are still awaited, and the very high cost of rituximab compared to cyclophosphamide means it cannot be considered as first-choice therapy.
Plasmapheresis The addition of plasmapheresis to initial therapy with corticosteroids and cyclophosphamide
is indicated for patients presenting with either advanced kidney failure (SCr >500 µmol/l [5.8 mg/dl]) or with diffuse alveolar hemorrhage.
In a large, multicenter controlled trial [4], 137 patients with a new diagnosis of ANCA vasculitis confirmed by kidney biopsy were randomly assigned to either seven treatments of plasmapheresis, or three doses of 1000 mg of i.v. methylprednisolone. Both groups received standard therapy with oral cyclophosphamide and oral prednisone followed by azathioprine for maintenance therapy. Plasmapheresis was associated with a significantly higher rate of kidney recovery at 3 months (69% of patients with plasmapheresis vs. 49% with i.v. methylprednisolone), and with dialysis-free survival at 12 months. Whether duration of plasmapheresis should be tailored to ANCA titers has not been studied.

KDIGO® GN Guideline March 2011 Public Review Draft
173
Studies of plasmapheresis as adjunctive therapy in patients with SCr <500 µmol/L (5.8 mg/dl) have not shown benefit, but were underpowered to provide definitive evidence [14,15]. A large RCT of adjunctive therapy with plasmapheresis is currently underway (clinicaltrials.gov identifier NCT00987389).
Duration of Therapy for Patients with ANCA Vasculitis Requiring Dialysis Among patients who require dialysis, those who recover sufficient kidney function nearly
always do so within the first 3 months of treatment [2,5]. Therefore, in patients who are still dialysis-dependent after 3 months and who have no evidence of ongoing extrarenal manifestations of active vasculitis, we suggest discontinuing cyclophosphamide therapy.
Plasmapheresis for Patients with Diffuse Alveolar Hemorrhage The impact of plasmapheresis in patients with diffuse alveolar hemorrhage is the reduction
of mortality, based on retrospective case series [6,15]. Although the strength of supportive data is low (retrospective case series without controls), the impact of such treatment is high (mortality) [2,16]. Whether patients with “mild” alveolar hemorrhage (small focal infiltrate without or with mild hypoxemia) require plasmapheresis is unknown.
Patients with ANCA Vasculitis: Anti-GBM GN Overlap Syndrome The recommendation for plasmapheresis, in addition to corticosteroids and
cyclophosphamide for patients with both circulating ANCA and anti-GBM antibodies, is based on the rationale for the treatment of anti-GBM GN. About one-third of patients with anti-GBM disease also have ANCA antibodies, usually directed against MPO. Patients with ANCA/anti-GBM overlap have a worse outcome than patients with ANCA vasculitis alone, or anti-GBM alone [17].
MMF There are insufficient data to support the use of MMF for induction therapy in ANCA
vasculitis. Although small uncontrolled studies report remission rates similar to those reported with corticosteroids and cyclophosphamide [18], relapses have been reported, despite continued use of MMF [19].
The only controlled study to date of MMF (1.5-2 g/d) vs. cyclophosphamide (monthly i.v. pulse of 0.75-1 gm/m2) includes 35 patients from China [20], four of whom were lost to follow-up (all in the cyclophosphamide group). When patients lost to follow-up were excluded from the analysis, the rates of remission were similar in the two groups. No data on follow-up beyond 6 months is provided in this study. A larger RCT of MMF vs. i.v. cyclophosphamide for induction treatment is currently underway (clinicaltrials.gov identifier NCT00414128).

KDIGO® GN Guideline March 2011 Public Review Draft
174
13.3: Maintenance therapy 13.3.1: We recommend maintenance therapy in patients who have achieved
remission. (1B) 13.3.2: We suggest continuing maintenance therapy for 12-18 months in patients
who remain in complete remission. (2D) 13.3.3: We recommend no maintenance therapy in patients who are dialysis-
dependent and have no extrarenal manifestations of disease. (1C)
13.4: Choice of agent for maintenance therapy 13.4.1: We recommend azathioprine 1-2 mg/kg/d orally as maintenance therapy.
(1B) 13.4.2: We suggest that MMF 1 g twice daily be used for maintenance therapy in
patients who are allergic to, or intolerant of, azathioprine. (2C) 13.4.3: We suggest trimethoprim-sulfamethoxazole as an adjunct to maintenance
therapy in patients with upper respiratory tract disease. (2B) 13.4.4: We suggest methotrexate (initially 0.3 mg/kg/wk, maximum 25 mg/wk)
for maintenance therapy in patients intolerant of azathioprine and MMF, but not if GFR is <60 ml/min. (1C)
13.4.5: We recommend not using etanercept as adjunctive therapy. (1A)
BACKGROUND
The indications for maintenance therapy are not well defined. The goal of maintenance therapy is to decrease the incidence and severity of relapsing vasculitis. With the exception of a small trial with trimethoprim-sulfamethoxazole (see Rationale), no placebo-controlled RCT has studied the benefit of maintenance therapy, although RCTs have compared the efficacy of different maintenance regimens. Therefore, the likely benefit of maintenance therapy depends on the assessment of the risk of relapse, which differs among various subgroups of patients. For example, the risk of low-dose maintenance immunosuppression in a frail, elderly patient has to be weighed against the very high risk for such a patient of severe relapse. Maintenance immunosuppressive therapy is justified in patients at high risk of relapse, but the potential benefit of maintenance therapy may be low in patients who have a low likelihood of relapse.
RATIONALE
• There is moderate-quality evidence that maintenance therapy is required in those at high risk of relapse or who have received less than 6 months induction treatment with cyclophosphamide.
• There is low-quality evidence that the duration of maintenance therapy should be 12-18 months.
• There is moderate-quality evidence that azathioprine is the preferred maintenance immunosuppressive agent, being equivalent in efficacy to cyclophosphamide in an RCT with a more favorable adverse-effect profile.

KDIGO® GN Guideline March 2011 Public Review Draft
175
• There is moderate-quality evidence that trimethoprim-sulfamethoxazole as an adjunct to maintenance therapy reduces the risk of relapse, but only in those with upper respiratory disease due to vasculitis.
Risk of Relapse Based on cohort studies, risk factors for relapse include persistence of PR3-ANCA
(compared to MPO-ANCA), history of upper respiratory tract disease (e.g., sinusitis, subglottic stenosis), or lower respiratory tract disease (e.g. alveolar hemorrhage, cavities, or nodules). Patients with any one of these three risk factors have an approximately 1.7-fold increased risk of relapse, and those with all three risk factors have an approximately 4.7-fold increased risk of relapse [3].
Patients with persistent PR3-ANCA–positivity at the end of cyclophosphamide therapy have a 2- to 3-fold increased risk of relapse, compared to patients who are ANCA-negative at the end of initial therapy [21]. In addition, patients who are persistently PR3-ANCA–positive are significantly more likely to relapse within 5 years after diagnosis [21]. Among patients who achieved remission and were switched from cyclophosphamide to azathioprine, those who remained PR3-ANCA–positive at the time of the switch had a 2.2-fold increased risk of suffering a relapse when compared to patients who were PR3-ANCA–negative. No similar data are available for patients with MPO-ANCA.
It is unknown whether patients with none of the risk factors for relapse need maintenance immunosuppression. The risk-benefit ratio of maintenance therapy has not been evaluated in such patients. The tailoring of maintenance therapy, based on the risk factors of relapse, has not been tested in clinical trials.
Choice of Immunosuppressive Agent for Maintenance Therapy The optimal total duration of corticosteroid therapy is unknown. Some studies have
maintained patients on a low dose of prednisone (7.5 mg daily) for >12 months [9]. In other cohort studies, corticosteroids are tapered completely off by the end of 5 months if the patient is in remission [3].
The best available data support the use of azathioprine 1-2 mg/kg/d for 6-18 months. This is inferred from an RCT of azathioprine vs. cyclophosphamide for the maintenance of remission [9]. Although not specifically designed to demonstrate the ability of azathioprine to prevent relapses (compared to placebo), the study established that introducing azathioprine after 3-6 months of cyclophosphamide, compared to continuing cyclophosphamide for 12 months, resulted in similar rates of relapse up to 18 months.
Maintenance therapy with azathioprine appears superior to MMF. In a large RCT of 155 patients with ANCA vasculitis, who attained remission with cyclophosphamide and corticosteroids, those randomized to MMF (2 g/d) vs. azathioprine (2 mg/kg/d) had a higher cumulative incidence of relapse (HR 1.7; P = 0.02) [22]. We therefore recommend azathioprine as the first choice for maintenance therapy in ANCA vasculitis. However, we suggest using MMF in patients who are allergic to or intolerant of azathioprine.

KDIGO® GN Guideline March 2011 Public Review Draft
176
In a placebo-controlled trial, the use of trimethoprim-sulfamethoxazole was associated with a decreased rate of upper airway-relapse [23]. The use of trimethoprim-sulfamethoxazole had no impact on the rate of relapse in other organs.
In a large prospective RCT, 12 months maintenance therapy with methotrexate (0.3 mg/kg/wk initially and progressively increased to 25 mg/wk) was compared to azathioprine (2 mg/kg/d) after induction of remission with cyclophosphamide and corticosteroids [24]. The study was not designed to demonstrate the superiority of methotrexate over azathioprine in preventing relapses, but to test the hypothesis that methotrexate would be safer than azathioprine. The rates of relapse were not significantly different between the azathioprine- and methotrexate-treated groups (36% and 33%, respectively; P = 0.71) with a mean randomization-to-relapse interval of 20.6 ± 13.9 months. Methotrexate was not associated with a higher rate of adverse events when compared to azathioprine (HR 1.65; 95% CI 0.65-4.18; P = 0.29). However, the severity of the adverse effects with the use of methotrexate was greater; therefore, it is not recommended in patients with a reduced GFR <30 ml/min per 1.73 m2, and the dose should be adjusted in patients with a GFR <60 ml/min per 1.73 m2.
The efficacy and safety of the tumor necrosis factor receptor–Fc fusion protein, etanercept, in the maintenance of remission among patients with Wegener’s granulomatosis was evaluated in an RCT in which etanercept or placebo was added to a regimen of daily oral cyclophosphamide or methotrexate and corticosteroids. Etanercept did not reduce the rate or the severity of relapses, and was associated with a higher rate of solid tumors and is therefore not recommended [27,28]. Although not tested, we also do not recommend the use of other anti–tumor necrosis factor agents.
Duration of Maintenance Therapy There are no direct data to support a recommendation for the duration of maintenance
therapy. The suggestion of continuing maintenance therapy for 12-18 months in patients who remain in complete remission is inferred from the duration of maintenance therapy used in the CYCAZAREM trial [9]. Some cohort studies, but not others, have suggested a higher incidence of relapse in the first 18 months after induction therapy.
In retrospective analyses of patients with ANCA vasculitis, the relapse rates of vasculitis were about 60% lower in patients with ESRD, and infections almost twice as frequent among patients maintained on immunosuppressive agents with ESRD [27,28]. In addition, infections were an important cause of death in this population. Given the lower risk of relapse and higher risk of infection and death, the risk-benefit ratio does not support the routine use of maintenance immunosuppression therapy in ANCA vasculitis patients on chronic dialysis, in the absence of active extrarenal disease.
Continued maintenance therapy is associated with the risks of immunosuppression, bone marrow suppression (leucopenia, anemia, thrombocytopenia), and possibly increased risk of cancer, notably skin cancer [29].

KDIGO® GN Guideline March 2011 Public Review Draft
177
13.5: Treatment of relapse 13.5.1: We recommend treating patients with severe relapse of ANCA vasculitis
(life- or organ-threatening) according to the same guidelines as for the initial therapy (see Section 13.1). (1C)
13.5.2: We suggest treating other relapses of ANCA vasculitis by reinstituting immunosuppressive therapy or increasing its intensity with agents other than cyclophosphamide, including instituting or increasing dose of corticosteroids, with or without azathioprine or MMF. (2C)
RATIONALE
• Relapse is associated with increased risk of ESRD. • Relapse is associated with severe or life-threatening extrarenal damage. • There is low-quality evidence that relapses are responsive to reintroduction or
increased dosing of immunosuppression, but the preferred treatment regimen has not been defined.
Impact of Relapse Relapse is defined as the occurrence of increased disease activity after a period of partial or
complete remission. Thus, a relapse can manifest as a worsening of pre-existing disease activity or the recurrence or development of active GN, or new signs or symptoms of vasculitis in any organ system.
Severe relapse is defined as life- or organ-threatening relapse. Examples of life-threatening relapse include diffuse alveolar hemorrhage and severe subglottic stenosis. Examples of organ-threatening disease are active GN, or a retro-orbital mass threatening vision.
In a cohort study, patients who had a relapse of GN were 4.7 times more likely to progress to ESRD compared to those who did not relapse. This increased risk of ESRD associated with relapse was independent of age, gender, race, ANCA specificity, and kidney function at the time of initial biopsy [3].
Relapses respond to immunosuppression with corticosteroids and cyclophosphamide with a similar response rate as the initial disease [2]. The repeated use of cyclophosphamide should be based on the severity of the relapse, taking into account the cumulative dose previously received by the patient. Severe relapses should be treated with cyclophosphamide, corticosteroids and plasmapheresis (when indicated) as described in Statement 13.1 and Table 13.1
Although a “safe” dose of cyclophosphamide has not been precisely determined, a recent retrospective study suggests that the risk of malignancy (other than nonmelanoma skin cancer) increases with cumulative doses of cyclophosphamide above 36 g [29]. Therefore, for patients who have previously received a 36 g cumulative dose of cyclophosphamide, we suggest treating subsequent relapses with a rituximab-based regimen.
For patients with relapse that is not severe (as defined earlier), immunosuppressive therapy should be increased while avoiding, if possible, more cyclophosphamide. If such relapse occurs

KDIGO® GN Guideline March 2011 Public Review Draft
178
when the patient is not receiving maintenance therapy, treatment may include the reinstitution of corticosteroids, azathioprine, or MMF, alone or in combination; however, there is no RCT evidence to support any of these regimens. In patients who suffer a relapse while on maintenance therapy with azathioprine or MMF, one treatment option is i.v. immunoglobulin. In an uncontrolled study, the addition of 6-monthly pulses of i.v. immunoglobulin (0.5 g/kg/d x 4 days) over background maintenance immunosuppression was associated with rates of complete or partial remission of 83% and 63% at 6 and 9 months, respectively [30]. In patients with kidney dysfunction, it is preferable to use a sucrose-free formulation of i.v. immunoglobulin in order to minimize the risk of osmotic-induced AKI [31].
Rituximab has not been tested in relapse, but appears to be of similar efficacy to cyclophosphamide in induction therapy [12]. Although the long-term safety of rituximab remains uncertain, its use in relapse may provide an opportunity to minimize cumulative dosage and potential toxicity of cyclophosphamide.
13.6: Treatment of resistant disease 13.6.1: In ANCA GN resistant to induction therapy with cyclophosphamide and
corticosteroids, we suggest the addition of i.v. immunoglobulin (2C) or rituximab (2D), or plasmapheresis (2D).
BACKGROUND
Resistance is defined as the persistence of or appearance of kidney and/or systemic manifestation of vasculitis, while receiving treatment equal in intensity to initial immunosuppressive therapy. Kidney manifestations of resistance include the continued presence of dysmorphic erythrocyturia and red blood cell casts, associated with a progressive decline in kidney function. Disease resistance to corticosteroids and cyclophosphamide occurs in approximately 20% of patients.
RATIONALE
Adjunctive therapy with i.v. immunoglobulin (single course of a total of 2 g/kg) was evaluated in an RCT in patients with resistant ANCA vasculitis. Patients treated with i.v. immunoglobulin had a more rapid decline in disease activity (as measured by a 50% reduction in Birmingham vasculitis activity score) and C-reactive protein at 1 and 3 months, but there was no significant difference between the two groups after 3 months, with respect to disease activity or frequency of relapse [35].
Several small, uncontrolled case series suggest a role for rituximab in resistant ANCA vasculitis [32-34]. In these reports, rituximab (375 mg/m2 i.v. weekly x 4, or 500 mg i.v. weekly x 4 fixed doses), in conjunction with corticosteroids, resulted in remission in the majority of patients, and was generally well-tolerated.

KDIGO® GN Guideline March 2011 Public Review Draft
179
There has been no trial of plasmapheresis in resistant ANCA vasculitis, but its value in this setting has been inferred from the MEPEX study, which demonstrated improved kidney outcome with plasmapheresis in patients with severe kidney dysfunction, and studies suggesting decreased mortality with plasmapheresis in patients with diffuse alveolar hemorrhage (see Statement 13.1.3.1).
13.7: Monitoring 13.7.1: We recommend not changing immunosuppression based on changes in
ANCA titer alone. (2D)
RATIONALE
Available data mostly report the assessment of PR3-ANCA, with limited data for MPO-ANCA. The data do not support the contention that PR3-ANCA is clinically useful in predicting relapse and should not be used (alone) to alter immunosuppression [36,37]. A persistently positive PR3-ANCA, at the time of switch to maintenance therapy with azathioprine, is associated with a 2- to 3-fold increased risk of relapse, and warrants close follow up [21]. For patients who are in clinical remission but remain PR3-ANCA–positive after 3-4 months of cyclophosphamide and corticosteroids, continuing cyclophosphamide for up to 6 months may be considered; however, there are no data on the risks or benefits of such an approach.
13.8: Transplantation 13.8.1: We recommend delaying transplantation until patients are in complete
extrarenal remission for 12 months. (1C) 13.8.2: We recommend not delaying transplantation for patients who are in
complete remission but are still ANCA-positive. (1C)
RATIONALE
No prospective data are available to assess the likelihood of recurrent ANCA vasculitis after kidney transplantation, or the impact of disease activity or that of a positive ANCA test at the time of transplantation, on patient outcome. The frequency of recurrent ANCA vasculitis after kidney transplantation has been assessed in several retrospective case series. These have revealed a frequency of relapse around 15-20%, although the frequency of recurrent pauci-immune necrotizing GN is only around 5% [38, 39]. In the largest retrospective study of 107 kidney transplant recipients in the UK, relapses occurred in only 5% of patients [40]. By multivariate analysis, kidney transplantation within 12 months of achieving remission was associated with increased mortality; the causes of death were not related to recurrent vasculitis. ANCA positivity at the time of transplantation does not appear to affect graft or patient survival, or the frequency of relapse after transplantation.
Please refer to Supplemental Tables 92-95 for additional data related to Chapter 13.

KDIGO® GN Guideline March 2011 Public Review Draft
180
References 1. Hogan SL, Nachman PH, Wilkman AS, Jennette JC et al. Prognostic markers in patients with antineutrophil
cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol 1996; 7: 23-32.
2. Nachman PH, Hogan SL, Jennette JC, Falk RJ. Treatment response and relapse in antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol 1996; 7: 33-39.
3. Hogan SL, Falk RJ, Chin H, Cai J et al. Predictors of relapse and treatment resistance in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis. Ann Intern Med 2005; 143: 621-631.
4. Jayne DR, Gaskin G, Rasmussen N, Abramowicz D et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol 2007; 18: 2180-2188.
5. de Lind van Wijngaarden RA, Hauer HA, Wolterbeek R, Jayne DR et al. Chances of renal recovery for dialysis-dependent ANCA-associated glomerulonephritis. J Am Soc Nephrol 2007; 18: 2189-2197.
6. Klemmer PJ, Chalermskulrat W, Reif MS, Hogan SL et al. Plasmapheresis therapy for diffuse alveolar hemorrhage in patients with small-vessel vasculitis. Am J Kidney Dis 2003; 42: 1149-1153.
7. de Groot K, Harper L, Jayne DR, Flores Suarez LF et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med 2009; 150: 670-680.
8. de Groot K, Adu D, Savage CO. The value of pulse cyclophosphamide in ANCA-associated vasculitis: meta-analysis and critical review. Nephrol Dial Transplant 2001; 16: 2018-2027.
9. Jayne D, Rasmussen N, Andrassy K, Bacon P et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003; 349: 36-44.
10. Guillevin L, Cohen P, Mahr A, Arene JP et al. Treatment of polyarteritis nodosa and microscopic polyangiitis with poor prognosis factors: a prospective trial comparing glucocorticoids and six or twelve cyclophosphamide pulses in sixty-five patients. Arthritis Rheum 2003; 49: 93-100.
11. Jones R, Walsh M, Jayne DR, European Vasculitis Study Group. Randomised trial of rituximab versus cyclophosphamide for ANCA-associated renal vasculitis: RITUXVAS [Abstract]. J Am Soc Nephrol 2008; 19:61A.
12. Specks U, Stone JH, ITN RAVE (ITN021A1) Study Group. Rituximab for ANCA-associated vasculitis (RAVE) Trial. In: 14th International Vasculitis and ANCA Workshop. Rasmussen N, Segelmark M, Westman K (editors). Copenhagen: Wiley-Blackwell; 2009.
13. Pusey CD, Rees AJ, Evans DJ, Peters DK et al. Plasma exchange in focal necrotizing glomerulonephritis without anti-GBM antibodies. Kidney Int 1991; 40: 757-763.
14. Cole E, Cattran D, Magil A, Greenwood C et al. A prospective randomized trial of plasma exchange as additive therapy in idiopathic crescentic glomerulonephritis. The Canadian Apheresis Study Group. Am J Kidney Dis 1992; 20: 261-269.
15. Lauque D, Cadranel J, Lazor R, Pourrat J et al. Microscopic polyangiitis with alveolar hemorrhage. A study of 29 cases and review of the literature. Groupe d'Etudes et de Recherche sur les Maladies "Orphelines" Pulmonaires (GERM"O"P). Medicine (Baltimore) 2000; 79: 222-233.
16. Holguin F, Ramadan B, Gal AA, Roman J. Prognostic factors for hospital mortality and ICU admission in patients with ANCA-related pulmonary vasculitis. Am J Med Sci 2008; 336: 321-326.
17. Rutgers A, Slot M, van PP, van B, V et al. Coexistence of anti-glomerular basement membrane antibodies and myeloperoxidase-ANCAs in crescentic glomerulonephritis. Am J Kidney Dis 2005; 46: 253-262.
18. Silva F, Specks U, Kalra S, Hogan MC et al. Mycophenolate mofetil for induction and maintenance of remission in microscopic polyangiitis with mild to moderate renal involvement--a prospective, open-label pilot trial. Clin J Am Soc Nephrol 2010; 5: 445-453.
19. Stassen PM, Cohen Tervaert JW, Stegeman CA. Induction of remission in active anti-neutrophil cytoplasmic antibody-associated vasculitis with mycophenolate mofetil in patients who cannot be treated with cyclophosphamide. Ann Rheum Dis 2007; 66: 798-802.
20. Hu W, Liu C, Xie H, Chen H et al. Mycophenolate mofetil versus cyclophosphamide for inducing remission of ANCA vasculitis with moderate renal involvement. Nephrol Dial Transplant 2008; 23: 1307-1312.

KDIGO® GN Guideline March 2011 Public Review Draft
181
21. Sanders JS, Huitma MG, Kallenberg CG, Stegeman CA. Prediction of relapses in PR3-ANCA-associated vasculitis by assessing responses of ANCA titres to treatment. Rheumatology (Oxford) 2006; 45: 724-729.
22. Hiemstra T, Walsh M, de Groot K, Hauser T, Mahr A, Pagnoux C, et al. Randomized trial of mycophenolate mofetil vs. azathioprine for maintenance therapy in ANCA-associated vasculitis (IMPROVE) [Abstract]. APMIS 2009; 117(Suppl 127):77-78.
23. Stegeman CA, Tervaert JW, De Jong PE, Kallenberg CG. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener's granulomatosis. Dutch Co-Trimoxazole Wegener Study Group. N Engl J Med 1996; 335: 16-20.
24. Pagnoux C, Mahr A, Hamidou MA, Boffa JJ et al. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med 2008; 359: 2790-2803.
25. WGET Research Group. Etanercept plus standard therapy for Wegener's granulomatosis. N Engl J Med 2005; 352: 351-361.
26. Stone JH, Holbrook JT, Marriott MA, Tibbs AK et al. Solid malignancies among patients in the Wegener's Granulomatosis Etanercept Trial. Arthritis Rheum 2006; 54: 1608-1618.
27. Weidanz F, Day CJ, Hewins P, Savage CO et al. Recurrences and infections during continuous immunosuppressive therapy after beginning dialysis in ANCA-associated vasculitis. Am J Kidney Dis 2007; 50: 36-46.
28. Lionaki S, Hogan SL, Jennette CE, Hu Y et al. The clinical course of ANCA small-vessel vasculitis on chronic dialysis. Kidney Int 2009; 76: 644-651.
29. Faurschou M, Sorensen IJ, Mellemkjaer L, Loft AG et al. Malignancies in Wegener's granulomatosis: incidence and relation to cyclophosphamide therapy in a cohort of 293 patients. J Rheumatol 2008; 35: 100-105.
30. Martinez V, Cohen P, Pagnoux C, Vinzio S et al. Intravenous immunoglobulins for relapses of systemic vasculitides associated with antineutrophil cytoplasmic autoantibodies: results of a multicenter, prospective, open-label study of twenty-two patients. Arthritis Rheum 2008; 58: 308-317.
31. Dickenmann M, Oettl T, Mihatsch MJ. Osmotic nephrosis: acute kidney injury with accumulation of proximal tubular lysosomes due to administration of exogenous solutes. Am J Kidney Dis 2008; 51: 491-503.
32. Eriksson P. Nine patients with anti-neutrophil cytoplasmic antibody-positive vasculitis successfully treated with rituximab. J Intern Med 2005; 257: 540-548.
33. Keogh KA, Wylam ME, Stone JH, Specks U. Induction of remission by B lymphocyte depletion in eleven patients with refractory antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005; 52: 262-268.
34. Stasi R, Stipa E, Poeta GD, Amadori S et al. Long-term observation of patients with anti-neutrophil cytoplasmic antibody-associated vasculitis treated with rituximab. Rheumatology (Oxford) 2006; 45: 1432-1436.
35. Jayne DR, Chapel H, Adu D, Misbah S et al. Intravenous immunoglobulin for ANCA-associated systemic vasculitis with persistent disease activity. QJM 2000; 93: 433-439.
36. Nowack R,Grab I, Flores-Suarez LF, Schnulle P, Yard B, van der Woude FJ. ANCA titers, even of IgG subclasses, and soluble CD14 fail to predict relapses in patients with ANCA-associated vasculitis. Nephrol Dial Transplant 2001;16:1631-1637.
37. Finkielman JD, Merkel PA, Schroeder D, Hoffman GS, Spiera R, St. Clair EW, Davis Jr, JC, McCune WJ, Lears AK, Ytterberg RS, Hummel AM, Viss MA, Peikert T, Stone JH, Specks U, for the WGET Research Group. Antiproteinase 3 Antineutrophil Cytoplasmic Antibodies and Disease Activity in Wegener Granulomatosis. Ann Intern Med. 2007;147:611-619.
38. Nachman PH, Segelmark M, Westman K, Hogan SL, Satterly KK, Jennette JC and Falk RJ. Recurrent ANCA-small vessel vasculitis after transplantation: a pooled analysis. Kidney Int. 1999;56:1544-50
39. Gera M, Griffin MD, Specks U, Leung N, Stegall MD, Fervenza FC: Recurrence of ANCA-associated vasculitis following renal transplantation in the modern era of immunosuppression. Kidney Int 71:1296-1301, 2007
40. Little MA, Hassan B, Jacques S, Game D, Salisbury E, Courtney AE , Brown C, Salama AD, Harper L. Renal transplantation in systemic vasculitis: when is it safe? Nephrol Dial Transplant. 2009 Oct;24(10):3219-25

KDIGO® GN Guideline March 2011 Public Review Draft
182
CHAPTER 14: TREATMENT OF ANTI-GLOMERULAR BASEMENT MEMBRANE ANTIBODY GLOMERULONEPHRITIS
INTRODUCTION
This chapter gives treatment recommendations for GN mediated by antibodies against the GBM (i.e., anti-GBM GN) whether or not it is associated with pulmonary hemorrhage (Goodpasture’s disease).
14.1: Treatment of anti-GBM GN 14.1.1: We recommend initiating immunosuppression using plasmapheresis,
cyclophosphamide, and corticosteroids (see Table 14.1) in all patients with anti-GBM GN except those who are dialysis-dependent at presentation and have 100% crescents in an adequate biopsy sample, and do not have pulmonary hemorrhage. (1B)
14.1.2: We recommend no maintenance immunosuppressive therapy for anti-GBM GN. (1C)
14.1.3: Start treatment for anti-GBM GN without delay once the diagnosis is confirmed. If the diagnosis is highly suspected, it would be appropriate to begin high-dose corticosteroids (Table 14.1) while waiting for confirmation. (Not Graded)
14.1.4: Defer kidney transplantation after anti-GBM GN until anti-GBM antibodies have been undetectable for a minimum of 6 months. (Not Graded)
BACKGROUND
Anti-GBM GN is generally a fulminant and rapidly progressive disease that is caused by autoantibodies to the noncollagenous domain of the α3 chain of type IV collagen. Anti-GBM GN is relatively rare, with an estimated annual incidence of 0.5-1 per million population. It can present as an isolated GN, or as a pulmonary-renal syndrome with severe lung hemorrhage. Prior to the introduction of intense immunosuppression for anti-GBM GN, patient survival was very poor. Although mortality has improved, kidney survival remains poor, possibly because of delays in making the diagnosis and initiating treatment. The strategy for treating anti-GBM GN is to remove the pathogenic autoantibodies from the circulation, and simultaneously prevent further autoantibody production and attenuate existing glomerular inflammation and injury.

KDIGO® GN Guideline March 2011 Public Review Draft
183
Table 14.1: Therapy of anti-GBM GN
Corticosteroids Week Prednisone dose
0-2 Methylprednisolone 500-1000 mg/d i.v., followed by prednisone, 1 mg/kg/d IBW (maximum 80 mg/d, two divided doses)
2-4 0.6 mg/kg/d 4-8 0.4 mg/kg/d 8-10 30 mg/d 10-11 25 mg/d 11-12 20 mg/d 12-13 17.5 mg/d 13-14 15 mg/d 14-15 12.5 mg/d 15-16 10 mg/d 16- IBW <70 kg: 7.5 mg/d
IBW ≥70 kg: 10 mg/d Discontinue after 6 months
Cyclophosphamide: 2 mg/kg/d orally for 3 months Plasmapheresis: One 4-liter exchange per day with 5% albumin. Add 150-300 ml fresh frozen plasma at the end of each pheresis session if patients have pulmonary hemorrhage, or have had recent surgery, including kidney biopsy. Plasmapheresis should be continued for 14 days or until anti-GBM antibodies are no longer detectable. GBM, glomerular basement membrane; GN, glomerulonephritis; IBW, ideal
body weight. There is no evidence to support these dosing schedules, which are based on
regimens associated with good outcome in observational studies.1
RATIONALE
• Patient and kidney survival in untreated anti-GBM GN is poor. • There is moderate-quality evidence that intense immunosuppression plus
plasmapheresis improves patient and kidney survival; this evidence comes from one small RCT, one large, and several smaller retrospective series. All of these studies demonstrate good patient survival and moderate kidney survival, providing a compelling rationale to use immunosuppression and plasmapheresis.
• Many patients at presentation have severe kidney failure, and require dialysis. This is usually correlated with the number of glomeruli that show crescents on kidney biopsy. Despite intense immunosuppression, patients who are dialysis-dependent at the start of treatment and have 85-100% glomerular crescents do not recover kidney function, and generally will require long-term RRT.
• Because the progression of anti-GBM GN can be very rapid, and outcome is related to the severity at presentation, it is appropriate to start treatment immediately with high-dose corticosteroids. After the diagnosis is confirmed, cyclophosphamide and plasmapheresis must be started. Patients should be free of infection or receiving appropriate antimicrobial therapy.

KDIGO® GN Guideline March 2011 Public Review Draft
184
• Patients with pulmonary hemorrhage as well as anti-GBM GN (Goodpasture’s disease) should receive treatment with corticosteroids, cyclophosphamide, and plasmapheresis, even in the setting of severe kidney failure and extensive glomerular crescent formation. Without such therapy, Goodpasture’s disease has a very high mortality. There is, however, no definite evidence that plasmapheresis is beneficial when there are only minor clinical signs of pulmonary hemorrhage.
• Because anti-GBM antibodies are pathogenic, it is prudent to wait until they are undetectable before considering a kidney transplant for those with ESRD.
As the pathogenesis of anti-GBM GN became clear, treatment regimens were designed to
remove the circulating pathogenic antibody that caused the disease, suppress further synthesis of this pathogenic antibody, and attenuate the glomerular inflammatory response initiated by the anti-GBM antibody. The best summary of this approach is a large retrospective study of anti-GBM GN from the Hammersmith Hospital [1], including 85 patients seen over 25 years. Seventy-one patients were treated with high-dose prednisone* (1 mg/kg/d) tapered over 6-9 months, oral cyclophosphamide for 2-3 months, and daily plasmapheresis for 14 days, or until the anti-GBM antibody was no longer detectable. The kidney outcome for this cohort was influenced by kidney function at presentation. Patients who had an initial SCr <5.7 mg/dl (500 μmol/l) had a 1-year overall survival of 100% and a kidney survival of 95%, and at 5 years patient and kidney survival were both 94%. If the initial SCr was >5.7 mg/dl (500 μmol/l) but dialysis was not required immediately, the patient and kidney survivals were 83% and 82% at 1 year, and 80% and 50% at 5 years, respectively. However, among patients who needed dialysis at presentation, patient and kidney survival were reduced to 65% and 8% at 1 year, and 44% and 13% at 5 years, respectively. Compared to nearly 100% mortality from pulmonary hemorrhage and kidney failure in historical series, this treatment strategy represented a significant improvement.
The role of plasmapheresis in addition to immunosuppression has been questioned, and was tested in a small RCT (n = 17) [2]. Although this study used prednisone and cyclophosphamide for immunosuppression, there were slight differences in dose and duration compared to the Hammersmith study. Most importantly, plasmapheresis was done every 3 days instead of daily and a mean of nine treatments was completed. All patients received prednisone and cyclophosphamide, and half were randomized to additional plasmapheresis. In those receiving plasmapheresis, anti-GBM antibodies disappeared about twice as fast as in the control group (all within 50 days, P <0.05). At the end of therapy, SCr in those receiving plasmapheresis was 4.1 ± 0.5 mg/dl (360 ± 44 μmol/l) compared to 9.2 ± 0.7 mg/dl (810 ± 62 μmol/l) in the controls (P <0.05); only two patients receiving plasmapheresis needed chronic dialysis vs. six in the controls. Although the two treatment groups were well-matched clinically at the beginning of the study, kidney biopsies showed a higher percentage of glomerular crescents in controls. Because of this difference in histology and the small study size, the evidence for better kidney outcome with plasmapheresis cannot be regarded as definitive.
Anti-GBM antibody titers should be regularly monitored [3]. Plasmapheresis may be stopped when the circulating antibody is no longer detectable, usually after 10-14 treatments. Corticosteroids have generally been continued for at least 6 months, and cyclophosphamide for 2-3 months. This immunosuppression must be sufficient both to prevent further antibody production, and to treat kidney inflammation.

KDIGO® GN Guideline March 2011 Public Review Draft
185
About 20-30% of patients with anti-GBM disease will also have ANCA, usually with anti-MPO specificity, but the double-antibody–positive patients do not appear to have a different prognosis or disease course, according to most studies [4-8].
The outcomes of the Hammersmith cohort [1] are representative of what can be expected with a uniform, aggressive approach to therapy as outlined. In other series of anti-GBM GN, not all necessarily using the same treatment regimens, and encompassing patients from the USA, Europe, China, and Japan, patient survival at 6-12 months was approximately 67-94%, and kidney survival was about 15-58% [2, 8-11].
The predictors of kidney survival in anti-GBM GN are SCr at presentation, the need for dialysis at presentation, and the percentage of glomerular crescents [1, 2, 7]. In two studies, patients with an initial SCr > 5.7 mg/dl (500 μmol/l) or 9.7 mg/dl (850 μmol/l) all became chronically dialysis-dependent despite aggressive treatment [4, 6]. Two studies found that patients who required dialysis at presentation were never able to come off dialysis, despite aggressive treatment [6, 8]. The most optimistic study observed that all patients with a combination of dialysis at presentation plus 100% crescents on kidney biopsy never recovered kidney function sufficiently to come off dialysis [1]. A survey of several studies shows dialysis dependence at diagnosis in a median of 55% (range 12-83%) of patients, 100% crescents on kidney biopsy in 20.5% (range 7-50%) of patients, and a median initial SCr of 6.9 mg/dl (600 μmol/l) (range 4.9-7.2 mg/dl [430-630 μmol/l]), underscoring the importance of early diagnosis and intervention [1, 2, 4, 6, 8, 9, 11, 12]. These findings, along with the patient’s general condition, will help in deciding how aggressive to be in treating the kidney manifestations of anti-GBM GN. However, in the presence of pulmonary hemorrhage, aggressive treatment should be undertaken, regardless of the kidney prognosis [13].
In contrast to most other autoimmune kidney diseases, anti-GBM GN is not characterized by a frequently relapsing course; the autoantibodies seem to disappear spontaneously after 12-18 months [14]. Nonetheless, relapses of anti-GBM GN have been reported in the literature, can manifest as recurrent clinical kidney disease or pulmonary hemorrhage, and are often associated with a reappearance of circulating anti-GBM antibodies [14-17]. It has been estimated that the mean time to recurrence is 4.3 years, with a range of 1-10 years, and that late recurrences may occur with a frequency of 2-14% [11, 14, 17]. Retreatment with intense immunosuppression and plasmapheresis is generally successful in reinducing remission [14].
There is very little information on the treatment of refractory anti-GBM GN. Some case reports have used MMF or rituximab, but no firm recommendation can be made.
There is very little evidence as to the timing of transplant after anti-GBM disease has caused ESRD. Most transplant centers require at least 6 months of undetectable anti-GBM antibody levels before kidney transplantation [18, 19]. Recurrent anti-GBM disease in a kidney allograft is very unusual [18, 19].
*Prednisone and prednisolone are interchangeable according to local practice, with equivalent dosing

KDIGO® GN Guideline March 2011 Public Review Draft
186
RESEARCH RECOMMENDATIONS
• A study is needed to compare rituximab to cyclophosphamide, both combined with prednisone plus plasmapheresis for induction of remission.
• A study is needed to compare MMF plus prednisone plus plasmapheresis to standard treatment—cyclophosphamide plus prednisone plus plasmapheresis—for induction of remission.
References 1. Levy JB, Turner N, Rees AJ, Pusey CD. Long-term outcome of anti-glomerular basement membrane
antibody disease treated with plasma exchange and immunosuppression. Annals of internal medicine 2001; 134: 1033-1042.
2. Johnson JP, Moore J, Jr., Austin HA, Balow JE, et al. Therapy of anti-glomerular basement membrant antibody disease: Analysis of prognostic significance of clinical, pathologic, and treatment factors. Medicine 1985; 64: 219-227.
3. Jindal KK. Management of idiopathic crescentic and diffuse proliferative glomerulonephritis: Evidence-based recommendations. Kidney international 1999; 55: S33-S40.
4. Segelmark M, Hellmark T, Wieslander J. The prognostic significance in Goodpasture's disease of specificity, titre, and affinity of anti-glomerular-basement-membrane antibodies. Nephron Clin Pract 2003; 94: c59-c88.
5. Lindic J, Vizjak A, Ferluga D, Kovac D, et al. Clinical outcome of patients with coexistent antineutrophil cytoplasmic antibodies and antibodies against glomerular basement membrane. Therap Apher Dialy 2009; 13: 278-281.
6. Levy JB, Hammad T, Coulhart A, Dougan T, et al. Clinical features and outcome of patients with both ANCA and anti-GBM antibodies. Kidney international 2004; 66: 1535-1540.
7. Cui Z, Zhao MH, Xin G, Wang HY. Characteristics and prognosis of Chinese patients with anti-glomerular basement membrane diseasse. Nephron Clin Pract 2005; 99: c49-c55.
8. Li FK, Tse KC, Lam MF, Yip TP, et al. Incidence and outcome of antiglomerular basement membrane disease in Chinese. Nephrology (Carlton, Vic 2004; 9: 100-104.
9. Shah MK, Hugghins SY. Characteristics and outcomes of patients with Goodpasture's syndrome. Southern Med J 2002; 95: 1411-1418.
10. Balow JE, Austin HA, Muenz LR, Joyce KM, et al. Effect of treatment on the evolution of renal abnormalities in lupus nephritis. N Engl J Med 1084; 311: 491-495.
11. Hirayama K, Yamagata K, Kobayashi M, Koyama A. Anti-glomerular basement membrane antibody disease in Japan: part of the nationwide rapidly progressive glomerulonephritis survey in Japan. Clinical and experimental nephrology 2008; 12: 329-347.
12. Stegmayr BG, Almroth G, Berlin G, Ferhrman I, et al. Plasma exchange or immunoadsorption in patients with rapidly progressive crescentic glomerulonephritis: A Swedish multi-center study. Int J Artificial Org 1999; 22: 81-87.
13. Proskey AJ, Weatherbee L, Easterling RE, Greene JA, et al. Goodpasture's syndrome: A report of five cases and review of the literature. The American journal of Medicine 1970; 48: 162-173.
14. Levy JB, Lachmann RH, Pusey CD. Recurrent Goodpasture's disease. Am J Kidney Dis 1996; 27: 573-578. 15. Adler S, Bruns FJ, Fraley DS, Segel DP. Rapid progressive glomerulonephritis: Relpase after prolonged
remission. Arch Intern Med 1981; 141: 852-854. 16. Klasa RJ, Abboud RT, Ballon HS, Grossman L. Goodpasture's syndrome: Recurrence after a five-year
remission. The American journal of medicine 1988; 84: 751-755. 17. Hind CRK, Bowman C, Winearls CG, Lockwood CM. Recurrence of circulating anti-glomerular basement
membrane antibody three years after immunosuppressive treatment and plasma exchange. Clin Nephrol 1984; 21: 244-246.

KDIGO® GN Guideline March 2011 Public Review Draft
187
18. Joshi K, Nada R, Minz M, Sakhuja V. Recurrent glomerulopathy in the renal allograft. Transplant Proc 2007; 39: 734-736.
19. Choy BY, Chan TM, Lai KN. Recurrent glomerulonephritis after kidney transplantation. Am J Transplant 2006; 6: 2535-2542.

KDIGO® GN Guideline March 2011 Public Review Draft
188
APPENDIX: METHODS FOR GUIDELINE DEVELOPMENT
AIM
The overall aim of the project was to create a clinical practice guideline with recommendations for GN, using an evidence-based approach. After topics and relevant clinical questions were identified, the pertinent scientific literature on those topics was systematically searched and summarized.
OVERVIEW OF PROCESS
The development of the guideline included sequential and concurrent steps: • Appoint the Work Group and Evidence Review Team (ERT), which were
responsible for different aspects of the process. • Confer to discuss process, methods, and results. • Develop and refine topics. • Triage topics to systematic review or narrative review. • Define specific populations, interventions or predictors, and outcomes of
interest for systematic review topics. • Create and standardize quality assessment methods. • Create data-extraction forms. • Develop literature search strategies and run searches. • Screen abstracts and retrieve full articles based on predetermined eligibility
criteria. • Extract data and perform critical appraisal of the literature. • Incorporate existing systematic reviews and underlying studies. • Grade quality of the outcomes of each study. • Tabulate data from articles into summary tables. • Update the systematic review search. • Grade the quality of evidence for each outcome, and assess the overall quality
and findings of bodies of evidence with the aid of evidence profiles. • Write recommendations and supporting rationale statements. • Grade the strength of the recommendations based on the quality of the evidence
and other considerations.
The Work Group, KDIGO Co-Chairs, ERT, and NKF support staff met for three 3-day meetings for training in the guideline development process, topic discussion, and consensus development.
Creation of Groups The KDIGO Co-Chairs appointed the Co-Chairs of the Work Group, who then assembled
the Work Group to be responsible for the development of the guidelines. The Work Group included individuals with expertise in adult and pediatric nephrology, epidemiology, and kidney pathology. For support in evidence review, expertise in methods, and guideline development, the

KDIGO® GN Guideline March 2011 Public Review Draft
189
NKF contracted with the ERT based primarily at the Tufts Center for Kidney Disease Guideline Development and Implementation at Tufts Medical Center in Boston, Massachusetts, USA. The ERT consisted of physician-methodologists with expertise in nephrology and internal medicine, and research associates and assistants. The ERT instructed and advised Work Group members in all steps of literature review, critical literature appraisal, and guideline development. The Work Group and the ERT collaborated closely throughout the project.
Systematic Review: General Process The first task of the Work Group was to define the overall topics and goals for the guideline.
The Work Group Co-Chairs drafted a preliminary list of topics. The Work Group identified the key clinical questions and triaged topics for systematic review and narrative review. The Work Group and ERT further developed and refined each systematic review topic, specified screening criteria, literature search strategies, and data extraction forms.
The ERT performed literature searches, and organized abstract and article screening. The ERT also coordinated the methodological and analytic processes of the report. In addition, it defined and standardized the methodology in relation to these searches and data extraction, and produced summaries of the evidence. Throughout the project, the ERT offered suggestions for guideline development, led discussions on systematic review, literature searches, data extraction, assessment of quality and applicability of articles, evidence synthesis, grading of evidence and recommendations, and consensus development. With input from the Work Group, the ERT finalized eligible studies, performed all data extraction, and summarized data into summary tables. They also created preliminary evidence profiles (described below), which were completed by the Work Group members. The Work Group members reviewed all included articles, data extraction forms, and summary tables for accuracy and completeness. The Work Group took the primary role of writing the recommendations and rationale statements, and retained final responsibility for the content of the recommendation statements and the accompanying narrative.
For questions of treatments in GN, systematic reviews of the eligible RCTs were undertaken (Table A). For these topics, the ERT created detailed data-extraction forms, and extracted information on baseline data for the populations, interventions, study design, results, and provided an assessment of quality of study and outcomes. The ERT then tabulated studies in summary tables, and assigned grades for the quality of the evidence in consultation with the Work Group.
Refinement of Topics At the first 3-day meeting, Work Group members added comments to the scope-of-work
document as prepared by the Work Group Chairs and ERT, until the initial working document included all topics of interest to the Work Group. The inclusive, combined set of questions formed the basis for the deliberation and discussion that followed. The Work Group aimed to ensure that all topics deemed clinically relevant and worthy of review were identified and addressed. The major topic areas of interest for the care of GN included IgAN, lupus and vasculitis, MCD and FSGS, and MN, MPGN, and infection.
At the initiation of the guideline development process, it was agreed that these guidelines would focus on patients who have GN. Thus, all topics, systematic reviews, and study eligibility

KDIGO® GN Guideline March 2011 Public Review Draft
190
criteria were restricted to patients with a biopsy-proven diagnosis of GN, with exceptions for diseases that do not require biopsy confirmation.
Based on the list of topics, the Work Group and ERT developed a list of specific research questions for which systematic review would be performed. For each systematic review topic, the Work Group Co-Chairs and the ERT formulated well-defined systematic review research questions using a well-established system [1]. For each question, explicit criteria were agreed on for the population, intervention or predictor, comparator, outcomes of interest, and study design features. A list of outcomes of interest was generated.
The Work Group and the ERT agreed upon specific outcomes of interest: all-cause mortality, ESRD, disease remission, relapse, proteinuria, kidney function, and adverse events. ESRD and mortality were ranked as being of critical importance. The Work Group ranked patient-centered clinical outcomes (such as death, ESRD, remission and categorical proteinuria and kidney function changes) as more important than intermediate outcomes (such as continuous outcomes of proteinuria and kidney function). Categorical outcomes are those that describe when a patient moves from one health state (e.g., macroalbuminuria) to another (e.g., no albuminuria). Continuous outcomes would be evaluations of the laboratory values alone (e.g, change in proteinuria in mg/dl). The outcomes were further categorized as being of critical, high, or moderate clinical importance to patients with GN. The specific criteria used for each topic are described below in the description of the review topics. In general, eligibility criteria were determined based on clinical value, relevance to the guidelines and clinical practice, determination whether a set of studies would affect recommendations or the strength of evidence, and practical issues, such as available time and resources.
Literature Searches and Article Selection A list of pertinent published systematic reviews relevant to GN guidelines was generated,
organized by topic, and reviewed with the Work Group. If an existing systematic review adequately addressed a question of interest as determined by the Work Group, this was used instead of the ERT conducting a de novo systematic review. This systematic review was then used as the starting point for building the evidence base and supplemented with articles from the ERT’s own searches. The searches were updated on January 20, 2011. All searches were also supplemented by articles identified by Work Group members through December 2010.
Editorials, letters, stand-alone abstracts, unpublished reports, and articles published in non–peer-reviewed journals were excluded. The Work Group also decided to exclude publications from journal supplements.
Potentially relevant existing systematic reviews were reviewed. If these reviews were deemed to adequately address topics of interest (even if only selected outcomes were reviewed), de novo searches on these topics were limited to the time period since the end of literature search within the systematic reviews. MEDLINE and Cochrane search results were screened by the ERT for relevance using predefined eligibility criteria, described below. For questions related to treatment, the systematic search aimed to identify RCTs with sample sizes as described in Table A. For some topics, nonrandomized comparative trials were also reviewed, in addition to RCTs, to strengthen the evidence base. For most topics, the minimum sample size was >10. For MCD and FSGS, because of sparse data, smaller studies were included.

KDIGO® GN Guideline March 2011 Public Review Draft
191
For most topics, the minimum duration of follow-up of 6 months was chosen based on clinical reasoning. For the treatments of interest, the proposed effects on patient-important clinical outcomes require long-term exposure and, typically, would not be expected to become evident before several months of follow-up.
In addition, a search was conducted for data on predictors of kidney failure, kidney function, and remission. Only associations from multivariable regression analyses were considered. These “predictor studies” were not graded for quality. For these topics, the ERT completed its search in October 5, 2009 and did not update the search.
Included were studies of all patients with glomerular diseases, excluding those with diabetic nephropathy, thrombotic microangiopathy, amyloidosis, Alport’s and other hereditary glomerular diseases, paraproteinemia, and recurrence of GN following kidney transplantation.
Interventions of interest included all treatments for GN, including drugs, herbs, dietary supplements, tonsillectomy, infection prophylaxis, and postdiagnosis tests to determine treatment.
Literature yield for systematic review topics Table B summarizes the numbers of abstracts screened, articles retrieved, data extracted, and
included in summary tables.
Data extraction The ERT designed data-extraction forms to tabulate information on various aspects of the
primary studies. Data fields for all topics included study setting, patient demographics, eligibility criteria, type of GN, numbers of subjects randomized, study design, study funding source, descriptions of interventions (or predictors), description of outcomes, statistical methods used, results, quality of outcomes (as described below), limitations to generalizability, and free-text fields for comments and assessment of biases.
Summary tables Summary tables were developed to tabulate the data from studies pertinent to each question
of intervention. Each summary table contains a brief description of the outcome, baseline characteristics of the population, intervention, comparator results, and methodological quality of each outcome. Baseline characteristics include a description of the study size, country of residence, and baseline kidney function and proteinuria. Intervention and concomitant therapies, and the results, were all captured. The studies were listed by outcome within the table, based on the hierarchy of important outcomes (Table C). Categorical and continuous outcomes were summarized in separate sets of tables. Work Group members were asked to proof all data in summary tables on RCTs and non-RCTs. Separate sets of summary tables were created for predictor studies. Summary tables are available at www.kdigo.org.
Evaluation of individual studies Study size and duration: The study (sample) size is used as a measure of the weight of the
evidence. In general, large studies provide more precise estimates. Similarly, longer-duration studies may be of better quality and more applicable, depending on other factors.

KDIGO® GN Guideline March 2011 Public Review Draft
192
Methodological quality: Methodological quality (internal validity) refers to the design, conduct, and reporting of the outcomes of a clinical study. A three-level classification of study quality was used (Table D). Given the potential differences in quality of a study for its primary and other outcomes, the methodological quality was assessed for each outcome. Variations of this system have been used in most KDOQI and all KDIGO guidelines, and have been recommended for the US Agency for Healthcare Research and Quality Evidence-based Practice Center program (http://effectivehealthcare.ahrq.gov/repFiles/2007_10DraftMethodsGuide.pdf). Each study was given an overall quality grade. Each reported outcome was then evaluated and given an individual quality grade depending on reporting and methodological issues specific to that outcome. However, the quality grade of an individual outcome could not exceed the quality grade for the overall study.
Results: The results data for each outcome of interest were extracted including baseline values (when relevant), final values (or number of events), and net differences (between interventions). These included net change in values, RR, OR, HR, and risk difference, as reported by the studies. The CI values of the net differences and their statistical significance were also extracted. When necessary, for categorical outcomes,RR and their 95% CI were calculated based on available data. The calculated data were distinguished from the reported data in the summary tables.
Evidence profiles Evidence profiles were constructed by the ERT and reviewed and confirmed with the Work
Group members. These profiles serve to make transparent to the reader the thinking process of the Work Group in systematically combining evidence and judgments. Each evidence profile was reviewed by Work Group members. Decisions were based on facts and findings from the primary studies listed in corresponding summary tables, as well as selected existing systematic reviews, and judgments of the Work Group. Judgments about the quality, consistency, and directness of evidence were often complex, as were judgments about the importance of an outcome or the summary of effects sizes. The evidence profiles provided a structured transparent approach to grading, rather than a rigorous method of quantitatively summing up grades.
Evidence profiles were constructed for research questions addressed by at least two studies. When the body of evidence for a particular comparison of interest consisted of only one study, either a RCT or a systematic review, the summary table provides the final level of synthesis.
Grading the quality of evidence and the strength of a recommendation A structured approach, based on GRADE [2-4], and facilitated by the use of evidence
profiles, was used in order to grade the quality of the overall evidence and the strength of recommendations. For each topic, the discussion on grading of the quality of the evidence was led by the ERT, and the discussion regarding the strength of the recommendations was led by the Work Group Co-Chairs. The “strength of a recommendation” indicates the extent to which one can be confident that adherence to the recommendation will do more good than harm. The “quality of a body of evidence” refers to the extent to which our confidence in an estimate of effect is sufficient to support a particular recommendation [4].
Grading the quality of evidence for each outcome: Following the GRADE method, the quality of a body of evidence pertaining to a particular outcome of interest was initially

KDIGO® GN Guideline March 2011 Public Review Draft
193
categorized based on study design. For questions of interventions, the initial quality grade was “High” when the body of evidence consisted of RCTs. In theory, the initial grade would have been “Low” if the evidence consisted of observational studies or “Very Low” if it consisted of studies of other study designs; however, the quality of bodies of evidence was formally determined only for topics where we performed systematic reviews of RCTs. The grade for the quality of evidence for each intervention/outcome pair was decreased if there were serious limitations to the methodological quality of the aggregate of studies, if there were important inconsistencies in the results across studies, if there was uncertainty about the directness of evidence including limited applicability of the findings to the population of interest, if the data were imprecise (a low event rate [0 or 1 event] in either arm or CI spanning a range <0.5 to >2.0) or sparse (only one study or total N <100), or if there was thought to be a high likelihood of bias. The final grade for the quality of the evidence for an intervention/outcome pair could be one of the following four grades: “High”, “Moderate”, “Low”, or “Very Low” (Table E). The quality of grading for topics relying on systematic reviews are based on quality items recorded in the systematic review.
Grading the overall quality of evidence: The quality of the overall body of evidence was then determined based on the quality grades for all outcomes of interest, taking into account explicit judgments about the relative importance of each outcome, weighting critical outcomes more than high or moderate. The resulting four final categories for the quality of overall evidence were: “A”, “B”, “C” or “D” (Table F). This evidence grade is indicated within each recommendation.
Assessment of the net health benefit across all important clinical outcomes: The net health benefit was determined based on the anticipated balance of benefits and harm across all clinically important outcomes. The assessment of net medical benefit was affected by the judgment of the Work Group. The assessment of net health benefit is summarized in Table G.
Grading the strength of the recommendations: The strength of a recommendation is graded as Level 1 or Level 2. Table H shows the KDIGO nomenclature for grading the strength of a recommendation, and the implications of each level for patients, clinicians, and policy-makers. Recommendations can be for or against doing something. Table I shows that the strength of a recommendation is determined not just by the quality of the evidence, but also by other—often complex—judgments regarding the size of the net medical benefit, values, and preferences, and costs. Formal decision analyses including cost analysis were not conducted.
Ungraded statements: This category was designed to allow the Work Group to issue general advice. Typically an ungraded statement meets the following criteria: it provides guidance based on common sense, it provides reminders of the obvious, it is not sufficiently specific to allow application of evidence to the issue and, therefore, it is not based on systematic evidence review. Common examples include recommendations about frequency of testing, referral to specialists, and routine medical care. We strove to minimize the use of ungraded recommendations.
This grading scheme with two levels for the strength of a recommendation together with four levels of grading the quality of the evidence, and the option of an ungraded statement for general guidance, was adopted by the KDIGO Board in December 2008. The Work Group took the primary role of writing the recommendations and rationale statements, and retained final

KDIGO® GN Guideline March 2011 Public Review Draft
194
responsibility for the content of the guideline statements and the accompanying narrative. The ERT reviewed draft recommendations and grades for consistency with the conclusions of the evidence review.
Format for recommendations Each section contains one or more specific recommendations. Within each recommendation,
the strength of recommendation is indicated as level 1 or level 2, and the quality of the supporting evidence is shown as A, B, C, or D. These are followed by a brief background with relevant definitions of terms, then the rationale starting with a “chain of logic”, which consists of declarative sentences summarizing the key points of the evidence base, and the judgments supporting the recommendation. This is followed by a narrative in support of the rationale. In relevant sections, research recommendations suggest future research to resolve current uncertainties.
Limitations of Approach While the literature searches were intended to be comprehensive, they were not exhaustive.
MEDLINE and various Cochrane databases were the only databases searched. Hand searches of journals were not performed, and review articles and textbook chapters were not systematically searched. However, important studies known to the domain experts that were missed by the electronic literature searches were added to retrieved articles and reviewed by the Work Group. Not all topics and subtopics covered by these guidelines could be systematically reviewed. Decisions to restrict the topics were made to focus the systematic reviews on those topics where existing evidence was thought to be likely to provide support for the guidelines. Although nonrandomized studies were reviewed, the majority of the ERT and Work Group resources were devoted to review of the randomized trials, since these were deemed to be most likely to provide data to support level 1 recommendations with very high– or high- (A or B) quality evidence. Where randomized trials were lacking, it was deemed to be sufficiently unlikely that studies previously unknown to the Work Group would result in higher-quality level 1 recommendations.
Review of the Guideline Development Process Several tools and checklists have been developed to assess the quality of the methodological
process for guideline development. These include the Appraisal of Guidelines for Research and Evaluation (AGREE) criteria [5] and the Conference on Guideline Standardization (COGS) checklist [6]. Table J shows the COGS criteria that correspond to the AGREE checklist, and how each one of them is addressed in these guidelines.

KDIGO® GN Guideline March 2011 Public Review Draft
195
Table A. Screening criteria for systematic review topics of nontreatment and treatment
PICOD Criteria Chapter 3: SSNS in Children Population Steroid sensitive (Any definition), Children Bx not required, Define Nephrotic Syndrome:
Urine Prot:Cr Ratio & Serum albumin Intervention Long course or alternate day prednisone Comparator Short course or daily prednisone Outcomes Proteinuria, Complete Remission, Relapse Study design RCTs; No minimum follow-up Minimum N of Subjects No minimum N FRNS in Children Population Steroid resistance, Children, Bx not required, Define Nephrotic Syndrome: Urine Prot:Cr
Ratio & Serum albumin Intervention Cyclosporine, Cytoxan, Chlorambucil, Tacrolimus (Prograf), Rituximab, MMF, Levamisole,
Plasmapharesis, Mizoribine, AZA Comparator Prednisone and Other comparators depending on the study Outcomes Proteinuria, Complete Remission, Relapse Study design RCTs
No minimum follow-up Nonrandomized comparative studies Retrospective comparative or prospective or retrospective single arm cohort Minimum duration: 6 months
Minimum N of Subjects No minimum N SDNS in Children Population Steroid resistance, Children, Bx not required, Define Nephrotic Syndrome: Urine Prot:Cr
Ratio & Serum albumin Intervention Cyclosporine, Cytoxan, Chlorambucil, Tacrolimus (Prograf), Rituximab, MMF, Levamisole,
Plasmapharesis, Mizoribine, AZA Comparator Prednisone and Other comparators depending on the study Outcomes Proteinuria, Complete Remission, Relapse Study design RCTs
No minimum follow-up Nonrandomized comparative studies Retrospective comparative or prospective or retrospective single arm cohort Minimum duration: 6 months
Minimum N of Subjects No minimum N Chapter 4: SRNS in Children Population Steroid resistance (define), Children, Bx not required, Define Nephrotic Syndrome: Urine
Prot:Cr Ratio & Serum albumin Intervention Cyclosporine, Cytoxan, Chlorambucil, Tacrolimus (Prograf), Rituximab, MMF, Levamisole,
Plasmapharesis, Mizoribine, AZA Comparator Prednisone, Other comparators depending on the study Outcomes Proteinuria, Complete Remission, Relapse Study design RCTs
No minimum follow-up Nonrandomized comparative studies Retrospective comparative or prospective or retrospective single arm cohort Minimum duration: 6 months
Minimum N of Subjects No minimum N

KDIGO® GN Guideline March 2011 Public Review Draft
196
Chapter 5: MCD in Adults (biopsy proven) Population Minimal Change Disease, Bx proven, Define Nephrotic Syndrome: Urine Prot:Cr Ratio &
Serum albumin Intervention Short course prednisone and Long course prednisone and Cyclosporine, Cytoxan,
Chlorambucil, Tacrolimus (Prograf), Rituximab, MMF, Levamisole, Plasmapharesis, Mizoribine, AZA
Comparator No treatment, Short course prednisone, Prednisone and other comparators depending on study
Outcomes Change in Proteinuria, Complete Remission, Partial Remission, Relapse, GFR, SCr doubling, ESRD, Death
Study design RCTs No minimum follow-up Nonrandomized comparative studies Retrospective comparative or prospective or retrospective single arm cohort Minimum duration: 6 months
Minimum N of Subjects N ≥10/arm Chapter 6: FSGS in Adults Population Population FSGS, by biopsy and list FSGS subtypes, Adults, Define Nephrotic Syndrome:
Urine Prot:Cr Ratio & Serum albumin Intervention Long course prednisone, Cyclosporine +/-ACEi, MMF +/- ACEi, Prograf +/- ACEi,
Rituximab +/- ACEi, Lamivudine +/- ACEi, Plasmapharesis +/- ACEi-levamisole, Mizoribine, AZA
Comparator Any treatment Outcomes Change in Proteinuria, Complete Remission , Partial Remission, Relapse, GFR, SCr
doubling, ESRD, Death Study design RCTs
No minimum follow-up Nonrandomized comparative studies Retrospective comparative or prospective or retrospective single arm cohort Minimum duration: 6 mo
Minimum N of Subjects N ≥10/arm Chapter 7: MN Population Bx proven MN Intervention Steroids alone (any regimen), Alkylating agent (Cyclophosphamide or Chlorambucil), CNI
(Cyclosporin or Tacrolimus +/- steroids), IVIG, ACEi or ARBs (+/-steroids), AZA or Mizoribine (+/- steroids), Alkylating agent, MMF (+/- steroids), ACTH, Rituximab, Eculizumab, Sirolimus, Pentoxyphlline, any combination
Comparator Steroids, No treatment, ACEi or ARBs, Calcineurin inhibitor (Tac, CsA), Alkylating agents CNI, steroids only, no treatment, any combination, any other treatment
Outcomes All cause mortality, ESRD, CKD 5, RRT, etc., Progression of CKD, SCr increase/GFR decrease, Change in CKD stage, Disease remission, Partial disease remission, Protocol-driven additional treatment of GN, Disease relapse, Quality of life, Proteinuria, Adverse Events: Including cancer, thromboembolic complications, pulmonary embolism, CVD especially acute MI
Study design RCTs; Minimum duration ≥6 months for remissions/AE, 5 years for survival Minimum N of Subjects N ≥10/arm
KDIGO® GN Guideline March 2011 Public Review Draft
197
Chapter 8: MPGN Population Bx-proven MPGN Intervention Rituximab, Eculizumab, CNI (CsA, Tac), MMF, Sirolimus, ACEi & ARBs,
Pentoxyphylline, IVIG, Treatment of relapse (any), Steroid therapy (any regimen) Comparator Any Outcomes Complete & partial remission, Relapse, Categorical changes in proteinuria,
Categorical changes in kidney function (Cr, GFR), ESRD, Death/survival, Adverse events
Study design RCTs Minimum follow-up 6 mo Nonrandomized comparative studies Prospective or retrospective Minimum duration: 12 mo
Minimum N of Subjects N ≥20 Chapter 9: Infection-Related MN Population Patients with infection associated GN, Bx proven, Postinfectious GN Intervention Antiviral (lamuvidine, ribavirin or interferon) for HBV, HCV, Anti-parasitic agents
for malaria or other helminthic/protozoal infections. For post infectious GN: any intervention
Comparator Any treatment Outcomes All cause mortality, ESRD, CKD 5, RRT, etc., Progression of CKD, SCr
increase/GFR decrease, Change in CKD stage, Disease remission, Partial disease remission, Protocol-driven additional treatment of GN, Disease relapse, Quality of life, Proteinuria, AE: Including cancer, thromboembolic complications, pulmonary embolism, CVD especially acute MI
Study design RCTs; No minimum duration of follow-up Minimum N of Subjects For post-infectious: N ≥10 for RCTs, N ≥20 for observational Chapter 10: IgAN Population Bx proven IgAN, Primary disease only (exclude secondary disease) Intervention Any Comparator Any, regardless of ACEi use, BP control, etc. Outcomes All cause mortality, ESRD, CKD 5, RRT, etc., Progression of CKD, SCr
increase/GFR decrease, Change in CKD stage, Disease remission, Protocol-driven additional treatment of GN, Disease relapse, Proteinuria
Study design RCTs; Minimum follow-up: 6 months Minimum N of Subjects N ≥10 Chapter 11: HSP Nephropathy Population Bx proven HSP Intervention Any (for RCTs and nonrandomized comparative studies) Comparator Any (for RCTs and nonrandomized comparative studies) Outcomes All cause mortality, ESRD, CKD 5, RRT, etc., Progression of CKD, SCr
increase/GFR decrease, Change in CKD stage, Disease remission, Protocol-driven additional treatment of GN, Disease relapse, Proteinuria
Study design RCTs and nonrandomized comparative studies Minimum N of Subjects N ≥10

KDIGO® GN Guideline March 2011 Public Review Draft
198
Chapter 12: LN Induction Therapy Population Bx proven Lupus nephritis, class III, IV , V, (also any combination of class V + III or V
+ IV ), Adults and pediatric Intervention MMF, Cyclophosphamide, Rituximab, Long duration Cyclophosphamide, i.v.
cyclophosphamide, Cyclosporine/Tacrolimus + MMF + Prednisone Hydroxychloroquine (class V) as a concomitant therapy with other drug therapies
Comparator Cyclophosphamide (p.o. or i.v.), Azathioprine, Cyclophosphamide, EURO protocol cyclophosphamide, p.o. cyclophosphamide, Cyclophosphamide. No addition of hydroxychloroquine
Outcomes Mortality, Need for RRT/ renal survival, Proteinuria, Kidney function preservation in terms of SCr/eGFR such as doubling of SCr – as categorical outcome, Remission and Relapse, Preservation of menses (fertility), Thrombotic and thromboembolic events, Alopecia and other adverse events
Study design RCTs Minimum follow-up: 6 months Nonrandomized comparative studies Prospective study design Minimum follow-up: 6 months
Minimum N of Subjects N ≥10/arm for RCTs and N ≥30 for nonrandomized comparative studies Chapter 12: LN Maintenance Therapy Population Bx proven Lupus nephritis, class III, IV, V, (also any combination of class V + III or V
+ IV ), Both adults and pediatric Intervention RCTs:
Maintenance therapy 1. MMF, 2. MMF, 3. Steroids, Hydroxychloroquine Nonrandomized comparative studies: Etanercept, TNF alpha antagonists ( eg infliximab, etc), CTLA4-Ig and derivatives, Campath, Abetimus LJP394
Comparator Cyclophosphamide, Azathioprine, Placebo/ No Rx, Placebo/ No Rx Outcomes Mortality, Need for RRT/ renal survival, Proteinuria, Kidney function preservation in
terms of SCr/eGFR such as doubling of SCr – as categorical outcome, Remission and Relapse, Preservation of menses (fertility) Thrombotic and thromboembolic events, Alopecia and other adverse events
Study design RCTs Minimum follow-up: 12 months Nonrandomized comparative studies Prospective study design Minimum follow-up: 12 months
Minimum N of Subjects N ≥10/arm for RCTs and N ≥30 for nonrandomized comparative studies

KDIGO® GN Guideline March 2011 Public Review Draft
199
Chapter 13: Treatment of Pauci-immune Focal and Segmental Necrotizing GN Population Adults or pediatric population , ANCA Vasculitis, Bx proven, Positive ANCA, Wegener’s
granulomatosis, microscopic polyangiitis, pauci-immune GN). Churg Strauss syndrome Intervention RCTs:
Cyclophosphamide + steroids, Cyclophosphamide + steroids + Plasmapheresis/ IVIG, MMF, i.v. cyclophosphamide regimens, Pulsed cyclophosphamide, Rituximab Maintenance: Azathioprine, MMF, Cyclophosphamide, Methotrexate, Cyclosporine, Leflunomide For nonrandomized comparative studies: MMF, Rituximab, Infliximab, Campath, Abetacept, Cyclosporine, IVIG, Leflunomide Plasmapheresis or immunoadsorption
Comparator Cyclophosphamide, Cyclophosphamide + steroids, Cyclophosphamide, p.o. cyclophosphamide regimens, Continuous p.o. cyclophosphamide, Maintenance Any comparator
Outcomes Mortality, Kidney survival, Relapse, Disease free survival, Thromboembolism, Proteinuria Coming off dialysis
Study design RCTs: Minimum follow up: 6 months; For maintenance therapy trials, duration at least 1 year Nonrandomized comparative studies: Prospective or Retrospective study design Minimum follow-up: 6 months
Minimum N of Subjects Any N for RCTs and N ≥30 for nonrandomized comparative studies Chapter 14: Treatment of Anti-GBM GN Population Anti GBM disease, Bx proven, Anti GBM antibody, Adults or pediatric population
Either renal or combined pulmonary renal involvement Intervention Prednisone + Cyclophosphamide + Plasmapheresis, Prednisone + cyclophosphamide +
Immunoadsorption Comparator Prednisone + Cyclophosphamide, Prednisone + Cyclophosphamide Outcomes Mortality, Recovery of kidney function, Proteinuria Study design Any; No minimum follow-up Minimum No. of Subjects No minimum N
Abbreviations: ACTH, Adrenocorticotropic hormone; ACEi, Angiotensin-converting enzyme inhibitors; AE, Adverse events; ANCE, Anti-neutrophil cytoplasmic antibody; ARB, Angiotensin receptor blockade; AZA, Azathioprine; BP, Blood pressure; Bx; Biopsy; CKD, Chronic Kidney Disease; CNI, Calcineurin inhibitors; Cr, Creatinine; CsA, Cyclosporine; CVD, Cardiovascular disease; Cyc, Cyclophosphamide; eGFR, Estimated glomerular filtration rate; ESRD, End-stage renal disease; FSGS, Focal segmental glomerulonephritis, GFR, Glomerular filtration rate; GN, Glomerulonephritis; HBV, Hepatitis B Virus; HCV, Hepatitis C Virus; HSP, Henoch-Schonlein Purpura; IgAN, IgA Nephropathy; i.v., Intravenous; IVIG, Intravenous immunoglobulin; MI, Myocardial infarction; MMF, Mycophenolate mofetil; mo, Month; MPGN, Membranoproliferative glomerulonephritis; N, Number; p.o., Oral; Prot, Proteinuria; RCT, Randomized controlled trials; RRT, Renal replacement therapy; Rx, Treatment; SCr, Serum creatinine; Tac, Tacrolimus
KDIGO® GN Guideline March 2011 Public Review Draft
200
Table B: Literature search yield of RCTs
Topic Abstracts identifieda
Studies retrieved
Studies data-extracted
# of systematic reviews
# of summary tablesb
# of evidence profiles
IgA
13,516 406 82 11 65 22
Lupus MCD & FSGS MN & Infection Total
FSGS, focal segmental glomerulosclerosis; MCD, minimal-change disease; MN, membranous nephropathy; RCTs, randomized controlled trials.
a. All topics and all study designs combined. b. Available at: www.kdigo.org
Table C: Hierarchy of outcomes
Hierarchya Outcomesb Critical importance Mortality, ESRD, CKD 5, RRT High importance Progression of CKD, Disease remission, Protocol-driven additional treatment of GN, Disease
relapse, Quality of life Moderate importance Partial disease remission, Proteinuria
CKD, chronic kidney disease; ESRD, end-stage renal disease; GN, glomerulonephritis; RRT, renal replacement therapy. a. Outcomes of lesser importance are excluded from review. b. This categorization was the consensus of the Work Group for the purposes of this GN guideline only. The lists are not meant to reflect
outcome ranking for other areas of kidney disease management. The Work Group acknowledges that not all clinicians, patients or families, or societies would rank all outcomes the same.
Table D: Classification of study quality
Good quality: Low risk of bias and no obvious reporting errors, complete reporting of data. Must be prospective. If study of intervention, must be RCT.
Fair quality: Moderate risk of bias, but problems with study/paper are unlikely to cause major bias. If study of intervention, must be prospective.
Poor quality: High risk of bias or cannot exclude possible significant biases. Poor methods, incomplete data, reporting errors. Prospective or retrospective.
RCT, randomized controlled trial.

KDIGO® GN Guideline March 2011 Public Review Draft
201
Table E. GRADE system for grading quality of evidence
Step 1: Starting grade for quality of evidence based on study design Step 2: Reduce grade Step 3: Raise grade
Final grade for quality of evidence and definition
Randomized trials = High Study quality –1 level if serious limitations –2 levels if very serious limitations Consistency –1 level if important inconsistency Directness –1 level if some uncertainty –2 levels if major uncertainty Other: –1 level if sparse or imprecise datac –1 level if high probability of reporting bias
Strength of association +1 level if stronga, no plausible confounders +2 levels if very strongb, no major threats to validity Other +1 level if evidence of a dose-response gradient +1 level if all residual plausible confounders would have reduced the observed effect
High = Further research is unlikely to change confidence in the estimate of the effect
Observational study = Low
Moderate = Further research is likely to have an important impact on confidence in the estimate of effect, and may change the estimate Low = Further research is very likely to have an important impact on confidence in the estimate, and may change the estimate
Any other evidence = Very Low
Very Low = Any estimate of effect is very uncertain
GRADE, Grading of Recommendations Assessment, Development, and Evaluation. a. Strong evidence of association is defined as “significant relative risk of >2 (<0.5)” based on consistent evidence from two or more
observational studies, with no plausible confounders. b. Very strong evidence of association is defined as “significant relative risk of >5 (<0.2)” based on direct evidence with no major threats to
validity. c. Sparse if there is only one study or if total N <100. Imprecise if there is a low event rate (0 or 1 event) in either arm or confidence interval
spanning a range <0.5 to >2.0. Reproduced with permission [3]; adapted with permission. [2,7]
Table F. Final grade for overall quality of evidence
Grade Quality of Evidence Meaning
A High We are confident that the true effect lies close to that of the estimate of the effect. B Moderate The true effect is likely to be close to the estimate of the effect, but there is a possibility that
it is substantially different. C Low The true effect may be substantially different from the estimate of the effect. D Very Low The estimate of effect is very uncertain, and often will be far from the truth.
Table G. Balance of benefits and harm
When there was evidence to determine the balance of medical benefits and harm of an intervention to a patient, conclusions were categorized as follows:
• Net benefits = the intervention clearly does more good than harm. • Trade-offs = there are important trade-offs between the benefits and harm. • Uncertain trade-offs = it is not clear whether the intervention does more good than harm. • No net benefits = the intervention clearly does not do more good than harm.
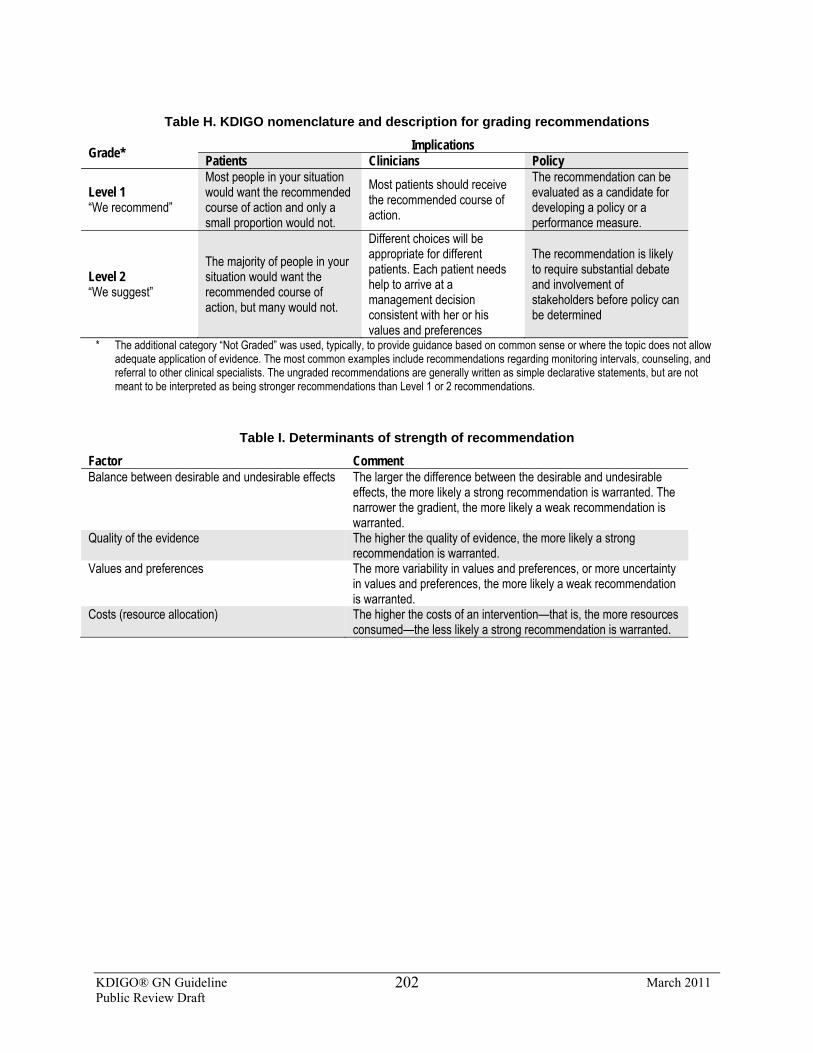
KDIGO® GN Guideline March 2011 Public Review Draft
202
Table H. KDIGO nomenclature and description for grading recommendations
Grade* Implications Patients Clinicians Policy
Level 1 “We recommend”
Most people in your situation would want the recommended course of action and only a small proportion would not.
Most patients should receive the recommended course of action.
The recommendation can be evaluated as a candidate for developing a policy or a performance measure.
Level 2 “We suggest”
The majority of people in your situation would want the recommended course of action, but many would not.
Different choices will be appropriate for different patients. Each patient needs help to arrive at a management decision consistent with her or his values and preferences
The recommendation is likely to require substantial debate and involvement of stakeholders before policy can be determined
* The additional category “Not Graded” was used, typically, to provide guidance based on common sense or where the topic does not allow adequate application of evidence. The most common examples include recommendations regarding monitoring intervals, counseling, and referral to other clinical specialists. The ungraded recommendations are generally written as simple declarative statements, but are not meant to be interpreted as being stronger recommendations than Level 1 or 2 recommendations.
Table I. Determinants of strength of recommendation
Factor Comment Balance between desirable and undesirable effects The larger the difference between the desirable and undesirable
effects, the more likely a strong recommendation is warranted. The narrower the gradient, the more likely a weak recommendation is warranted.
Quality of the evidence The higher the quality of evidence, the more likely a strong recommendation is warranted.
Values and preferences The more variability in values and preferences, or more uncertainty in values and preferences, the more likely a weak recommendation is warranted.
Costs (resource allocation) The higher the costs of an intervention—that is, the more resources consumed—the less likely a strong recommendation is warranted.
KDIGO® GN Guideline March 2011 Public Review Draft
203
Table J. The Conference on Guideline Standardization (COGS) checklist for reporting clinical practice guidelines
Topic Description Discussed in KDIGO GN Guideline 1. Overview material Provide a structured abstract that includes the
guideline’s release date, status (original, revised, updated), and print and electronic sources.
Executive Summary.
2. Focus Describe the primary disease/condition and intervention/service/technology that the guideline addresses. Indicate any alternative preventative, diagnostic or therapeutic interventions that were considered during development.
The “Guideline Scope” is addressed in a separate section in the introduction.
3. Goal Describe the goal that following the guideline is expected to achieve, including the rationale for development of a guideline on this topic.
This clinical practice guideline is intended to assist the practitioner caring for patients with biopsy-proven GN, and to improve kidney survival and the patients’ quality of life.
4. User/setting Describe the intended users of the guideline (e.g., provider types, patients) and the settings in which the guideline is intended to be used.
This guideline is primarily for the practicing nephrologist but does have application for other medical profession that directly care for kidney patients with GN. Policy Makers: Those in related health fields.
5. Target population Describe the patient population eligible for guideline recommendations and list any exclusion criteria.
All patients with the most common variants of primary and secondary GN in both the adult and pediatric populations. Exclusions are listed in the introductory chapter.
6. Developer Identify the organization(s) responsible for guideline development and the names/credentials/potential conflicts of interest of individuals involved in the guideline’s development.
Organization: KDIGO Names/credentials/potential conflicts of interest of individuals involved in the guideline’s development are disclosed in the Biographic and Disclosure Statements.
7. Funding source/sponsor
Identify the funding source/sponsor and describe its role in developing and/or reporting the guideline. Disclose potential conflict of interest.
KDIGO is supported by the following consortium of sponsors: Abbott, Amgen, Belo Foundation, Coca-Cola Company, Dole Food Company, Genzyme, Hoffmann-LaRoche, JC Penney, NATCO—The Organization for Transplant Professionals, National Kidney Foundation—Board of Directors, Novartis, Robert and Jane Cizik Foundation, Shire, Transwestern Commercial Services, and Wyeth. No funding is accepted for the development or reporting of specific guidelines. All stakeholders could participate in open review. Refer to Biographic and Disclosure Statements.
8. Evidence collection Describe the methods used to search the scientific literature, including the range of dates and databases searched, and criteria applied to filter the retrieved evidence.
The MEDLINE, Cochrane Central Registry for trials, and Cochrane database of systematic reviews were searched by the ERT to capture all citations relevant to the topic of GN, including original articles, systematic reviews, and previous guidelines. The search was updated through January 2011 and supplemented by articles identified by Work Group members through December 2010.
9. Recommendation grading criteria
Describe the criteria used to rate the quality of evidence that supports the recommendations and the system for describing the strength of the recommendations. Recommendation strength communicates the importance of adherence to a recommendation and is based on both the quality of the evidence and the magnitude of anticipated benefits and harm.
Quality of individual studies was graded in a three-tiered grading system (see Table D). Quality of evidence and strength of recommendations were graded following the GRADE approach (Tables F and H). The Work Group could provide general guidance in ungraded statements.
KDIGO® GN Guideline March 2011 Public Review Draft
204
10. Method for synthesizing evidence
Describe how evidence was used to create recommendations, e.g., evidence tables, meta-analysis, decision analysis.
1) Topics were triaged either to a) systematic review; b) systematic search followed by narrative summary; or c) narrative summary. For systematic review topics, summary tables and evidence profiles were generated. 2) For recommendations on treatment interventions, the steps outlined by GRADE were followed.
11. Prerelease review Describe how the guideline developer reviewed and/or tested the guidelines prior to release.
The guideline will undergo internal and external review.
12. Update plan State whether or not there is a plan to update the guideline and, if applicable, expiration date for this version of the guideline.
There is no date set yet for updating. The updating of the guideline will depend on the publication of new evidence that would change the quality of the evidence or the estimates for effect sizes. Results from registered ongoing studies and other publications will be reviewed periodically to evaluate their potential to impact on the recommendations in this guideline.
13. Definitions Define unfamiliar terms and those critical to correct application of the guideline that might be subject to misinterpretation.
See List of Abbreviations and Acronyms.
14. Recommendations and rationale
State the recommended action precisely and the specific circumstances under which to perform it. Justify each recommendation by describing the linkage between the recommendation and its supporting evidence. Indicate the quality of evidence and the recommendation strength, based on the criteria described in 9.
Recommendations for management of each of the GN variants described in the guideline are provided within each focused chapter. Each recommendation builds on a supporting rationale with evidence tables if available. The strength of the recommendation and the quality of evidence is provided in parenthesis within each recommendation.
15. Potential benefits and harm
Describe anticipated benefits and potential risks associated with implementation of guideline recommendations.
The benefits and harm for each intervention are provided in summary tables and summarized in evidence profiles. The estimated balance between potential benefits and harm was considered when formulating the recommendations.
16. Patient preferences Describe the role of patient preferences when a recommendation involves a substantial element of personal choice or values.
Level 2 recommendations inherently indicate a greater need to help each patient arrive at a management decision consistent with her or his values and preferences.
17. Algorithm Provide (when appropriate) a graphical description of the stages and decisions in clinical care described by the guideline.
These were not provided in the guideline in each chapter but, where available, they have been included.
18. Implementation considerations
Describe anticipated barriers to application of the recommendations. Provide reference to any auxiliary documents for providers or patients that are intended to facilitate implementation. Suggest review criteria for measuring changes in care when the guideline is implemented.
These recommendations are global. Review Criteria were not suggested because implementation with prioritization and development of review criteria has to proceed locally. Suggestions were provided for future research.
ERT, Evidence Review Team; GN, glomerulonephritis; GRADE, Grading of Recommendations Assessment, Development, and Evaluation; KDIGO, Kidney Disease: Improving Global Outcomes.
Adapted with permission. [6]

KDIGO® GN Guideline March 2011 Public Review Draft
205
References
1. Counsell, C. Formulating questions and locating primary studies for inclusion in systematic reviews. Ann Int Med 1997; 127: 380-387.
2. Atkins D, et al.; GRADE Working Group., Grading quality of evidence and strength of recommendations. BMJ. 2004 Jun 19;328(7454):1490-4.
3 Uhlig K, et al. Grading evidence and recommendations for clinical practice guidelines in nephrology. A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006 Dec; 70(12):2058-65.
4. Guyatt GH, Oxman AD, Kunz R et al. Going from evidence to recommendations. BMJ 2008; 336: 1049-1051.
5. The AGREE Collaboration. Development and validation of an international appraisal instrument for assessing the quality of clinical practice guidelines: the AGREE project. Qual Saf Health Care 2003;12: 18-23.
6. Shiffman RN, et al. Standardized Reporting of Clinical Practice Guideline: A Proposal from the Conference on Guideline Standardization. Annals of Internal Medicine. 2003; 139 (6): 493-498.
7. Farquhar C, Kunz K. Grading and the GRADE instrument. Second Guidelines International Network Conference: Evidence in Action, Wellington, NZ. 2004

KDIGO® GN Guideline March 2011 Public Review Draft
206
BIOGRAPHIC AND DISCLOSURE INFORMATION
Daniel C. Cattran, MD, FRCP (Work Group Co-Chair), is a graduate of the University
of Toronto Medical School and completed his postgraduate training both in Toronto, Canada and Sydney, Australia. He is currently a Professor of Medicine at the University of Toronto and a senior scientist at the Toronto General Research Institute. His administrative roles have included Chairman of the Royal College of Canada specialty program in nephrology, the renal transplant program of the Toronto General Hospital, and Director of the postgraduate education program in nephrology at the University of Toronto. Dr. Cattran is a member of postgraduate education committee of the American Society of Nephrology and he also currently serves on the KDIGO Board.
Dr. Cattran’s major research work has been in field of GN. He was the principal organizer and present Chair of the Toronto Glomerulonephritis Registry, which currently includes over 11,000 cases of biopsy-proven GN. His current search activities also include chairing the steering committee of NEPTUNE, an NIH-sponsored North American Consortium of 15 Centers focused on investigating the molecular mechanisms of the nephrotic syndrome and in particular the histologic variants, MN and FSGS. His own research is currently examining the role of ACTH as new therapy for IMN, and Dr. Cattran is lead investigator of a North American validation study of the new Oxford classification of IgAN. Grant support for his work has come from the Kidney Foundation of Canada, PSI, the National Institutes of Health, and the Canadian Institutes for Health Research. He has published over 190 articles in peer-reviewed journals and has been the principal author of more than 20 book chapters, as well as multiple reviews related to the natural history, outcome, and treatment studies in GN.
In recognition of his career in clinical investigation, the Kidney Foundation of Canada recently awarded him their prestigious Medal for Research Excellence. He has also been active in volunteer work, in particular, the Kidney Foundation of Canada and has held a number of positions including the chair of their Medical Advisory Board. The Foundation's award, “Volunteer of the Year”, acknowledged his contributions to this organization. He has also held a number of positions in the Canadian Society of Nephrology, including a term as President.
Advisor/Consultant: Genentech; Osprey; Questcor; Roche; Vifor Speaker: Novartis Grant/Research Support: Amgen

KDIGO® GN Guideline March 2011 Public Review Draft
207
John Feehally, DM, FRCP (Work Group Co-Chair), is Professor of Renal Medicine at The John Walls Renal Unit, Leicester General Hospital, UK. He received training in nephrology in Manchester and Leicester, UK, and in Boston, MA, USA, and was appointed Consultant Nephrologist at Leicester General Hospital in 1988. In 1999 he became Honorary Professor of Renal Medicine at the University of Leicester, UK. His particular clinical interests include topics such as GN and kidney transplantation. He has an active laboratory research program with a focus on immune renal disease, particularly IgAN, and maintains an epidemiological and clinical research interest in ethnic variations in renal disease.
Professor Feehally is Co-Editor of Comprehensive Clinical Nephrology, 4th Edition which has been the best-selling single-volume nephrology text worldwide. He is President-Elect of the International Society of Nephrology (ISN), served as President of the Renal Association (2004-2007), a professional body of nephrologists and renal scientists in the UK, and was a member of the Renal Advisory Group at the UK Department of Health in 2003. In 2005, he became Secretary General of the ISN and Chairman of the ISN Fellowship Committee.
Dr. Feehally reported no relevant financial relationships
H. Terence Cook, MBBS, MRCP, MRCPath, FRCPath, FMedSci, is Professor of Renal Pathology at Imperial College of Medicine and Deputy Director of the Centre for Complement and Inflammation Research at Imperial College, London, UK. Dr. Cook completed his undergraduate studies at New College, University of Oxford and received his medical degree from St. Mary’s Hospital Medical School in London, UK. His major research interests include the studying of pathological mechanisms of injury in glomerulonephritis and the role of macrophages, antibodies, Fc receptors, complement, and other mediators in the disease process. He also maintains an active interest in the use of histological features from human renal biopsies to predict response to treatments. In particular, he is presently conducting transcriptome analysis from renal biopsies for the diagnosis of transplant rejection. Dr. Cook is currently the President of Renal Pathology Society and a member of committees including, ISN COMGAN Subcommittee on Renal Pathology; Royal College of Pathologists Ethics Committee; and Renal Association International Committee. He has authored more than 200 publications and served as a consensus working group member of the International IgA Nephropathy Network, which recently developed the Oxford Classification of IgA Nephropathy.
Grant/Research Support: GSK
Fernando C. Fervenza, MD, PhD, is Professor of Medicine and Section Head, Parenchymal Renal Division at Mayo Clinic College of Medicine in Rochester, MN, USA. He received his medical training at the Medical School of the Pontificia Universidade, Brazil and completed his doctoral degree from University of Oxford, UK and fellowship studies from Stanford University. His primary research interest lies in the area of parenchymal renal disease with a focus on the glomerulopathies. Specifically, Dr. Fervenza is a current principal investigator in a number of clinical trials examining the efficacy of ocrelizumab in class III or IV nephritis; the use of rituximab as induction therapy in patients with lupus membranous patients; and the effectiveness of ACTH in lowering proteinuria in patients with IMN. In addition to

KDIGO® GN Guideline March 2011 Public Review Draft
208
authoring more than 100 peer-reviewed publications, Dr. Fervenza has been actively involved in the mentoring of residents, nephrology fellows, and visiting physicians. Since 2001, he has organized an annual nephrology up-to-date course in Brazil with the aim of fostering scientific improvement in the Brazilian nephrology community. In recognition for his multitude of contributions, Dr. Fervenza was awarded the Career Development Award in 2007 by the Mayo Foundation and most recently received the Laureate Award from the Mayo Clinic.
Advisor/Consultant: Genentech; Questcor Grant/Research Support: Genentech; Novartis; Questcor; Roche; Teva
Jürgen Floege, MD, graduated from the Hannover Medical School in Germany in 1984 and subsequently received his clinical training at the Hannover Medical School. His particular interest in renal diseases and renal replacement therapies developed during various research periods in physiology, pharmacology, nephrology, and pathology at the Hannover Medical School, Germany; the Albert Einstein College of Medicine, New York; and the University of Washington, Seattle, WA, USA. He was appointed as head of the Division of Nephrology and Immunology at the University of Aachen, Germany in 1999. In 2001, he became the Vice Dean of the Medical School, and since 2004, a Director of a government-funded center grant on chronic inflammation.
Professor Floege is an active member of several nephrology societies, including the American Society of Nephrology, ISN, and the European Renal Association (ERA-EDTA). He is a current member of the ERA-EDTA scientific advisory board, Co-Chair of the World Congress of Nephrology (Vancouver 2011) and former ERA-EDTA and ISN council member. In addition, he is a founding member and vice president of the German Society of Nephrology, since 2008. Together with Professors Richard Johnson and John Feehally he is editor of the best-selling textbook Comprehensive Clinical Nephrology. Finally, Professor Floege is one of two deputy editors of the ERA-EDTA journal Nephrology Dialysis Transplantation and a member of the editorial board of several other journals, including Journal of the American Society of Nephrology, Kidney International, Nature Clinical Practice Nephrology, the Journal of Nephrology, and Clinical Nephrology.
His research interests encompass both basic research (i.e., studies on growth factors, cytokines, angiogenesis, stem cells and fibrosis in the course of renal disease) as well as clinical problems such as immune-mediated renal disease, bone and mineral disorders and cardiovascular risk factors in dialysis patients. Dr. Floege’s scientific work encompasses 300 original papers, reviews and editorials, and 40 book chapters.
Advisor/Consultant: Amgen; Curagen; Fresenius; Gambro; Genzyme Speaker: Amgen; Fresenius; Genzyme Grant/Research Support: Amgen

KDIGO® GN Guideline March 2011 Public Review Draft
209
Debbie Gipson, MD, MS, is Associate Professor, Division of Nephrology in the Department of Pediatrics at University of Michigan, MI, USA, and Director for Research Design, Epidemiology, Biostatistics and Clinical Research Ethics Core at Michigan Institute for Clinical and Health Research. After obtaining a medical degree from Indiana University School of Medicine, Dr. Gipson completed her fellowship in pediatric nephrology at University of Washington, Seattle, WA, USA, where she also received her master’s degree in epidemiology. She is currently a member of NIH FSGS Clinical Steering Committee and the Chair of its Clinical Management Committee. In addition, she is a research investigator in several clinical trials which strive to better assess risk factors for progression of chronic kidney disease in children and identify therapeutic options for patients with multi-drug resistant FSGS. Dr. Gipson is also Co-Director for Pediatric Clinical Research, Nephrotic Syndrome Study Network (NEPTUNE) whose goal is to bring clinical and translational scientists together with the aim to educate patients with FSGS, MN, and MCD.
Grant/Research Support: FibroGen; Genzyme
Richard J. Glassock, MD, MACP, is currently Professor Emeritus at the David Geffen School of Medicine at UCLA and an internationally recognized expert in the field of glomerular diseases and clinical nephrology. He has published over 400 original papers, books, book chapters, reviews, and served as Co-Editor in a recent text, Treatment of Primary Glomerulonephritis (2nd Edition). Professor Glassock has lectured in more than 90 countries and has been a visiting professor at over 100 academic institutions. Throughout his career, he has trained over 50 nephrologists.
Professor Glassock has received many awards, including the David Hume Memorial Award of the National Kidney Foundation, and the Robert Narins Award of the American Society of Nephrology. He is the founding Editor- in-Chief of the NephSAP and sits on the editorial board of Journal of the American Society of Nephrology and Journal of Nephrology.
Advisor/Consultant: Aspreva (Vifor Pharma); BioMarin; Bristol-Myers-Squibb; FibroGen; Genentech; La Jolla Pharmaceutical; Novartis; Wyeth
Elisabeth M. Hodson, MBBS, FRACP, is a consultant pediatric nephrologist at the Children’s Hospital at Westmead in Sydney, Australia and was Head of the Department of Nephrology from 1995 to 2008. Her research interests have included CKD-metabolic bone disease, antenatal diagnosis of urinary tract abnormalities, growth in children with chronic kidney disease, and since 2002, a cohort study of antecedents of kidney disease in Aboriginal and non-Aboriginal children and young people. Since 2000, she has been an editor for the Cochrane Collaboration’s Renal Review Group, which is based in the Centre for Kidney Research at the Children’s Hospital at Westmead. Dr. Hodson has written and/or updated fourteen systematic reviews of RCTs of therapies for kidney disease for the Cochrane Renal Group. These include systematic reviews on the management in children of SSNS with corticosteroids and corticosteroid-sparing agents, of SRNS, and of HSP nephritis.
Dr. Hodson reported no relevant financial relationships

KDIGO® GN Guideline March 2011 Public Review Draft
210
Vivekanand Jha, MD, DM, FRCP, FAMS, is Additional Professor of Nephrology and Coordinator of Stem Cell Research Facility at the Postgraduate Institute of Medical Education & Research, Chandigarh, India. Dr Jha has held numerous committee positions in professional bodies such as The Transplantation Society, International Society of Nephrology and, most recently, a Steering Committee member of the World Health Organization initiative on data harmonization in transplantation. His ongoing research projects include the development of optimal strategies of immunosuppressive drug use after kidney transplantation by pharmacogenomic approaches, and the studying of BMD and histomorphometry in chronic kidney failure and its evolution after transplantation. He is Editor of The Cochrane Renal Group and a frequent peer reviewer for 14 journals. Dr. Jha has authored over 160 publications and 25 book chapters, and serves as an editor of an upcoming textbook, Management of Kidney Transplant Recipient. He was awarded Fellowships from The Royal Society of Physicians (London) and The National Academy of Medical Sciences (India) in 2009 and 2010, respectively.
Dr. Jha reported no relevant financial relationships
Philip Kam-Tao Li, MD, FRCP, FACP, is the Chief of Nephrology & Consultant Physician of the Department of Medicine and Therapeutics at the Prince of Wales Hospital, Hong Kong. He is also the Honorary Professor of Medicine at the Chinese University of Hong Kong.
Prof. Li dedicates his efforts to promoting nephrology both locally and internationally. He is the Past Chairman of the Hong Kong Society of Nephrology and presently the Executive Committee Member of the ISN and current President of the Hong Kong Transplantation Society. Prof. Li serves on the Board of Directors for KDIGO and is a Steering Committee Member for World Kidney Day 2010 as well as the Executive Committee Member of the Asian Forum for CKD Initiatives. He also sits on the Executive Council of the Asian Pacific Society of Nephrology, the Council of International Society for Peritoneal Dialysis, and serves as the Honorary Secretary at the Hong Kong College of Physicians.
Prof. Li had been President of the Organizing Committees for the ISN 2004 Conference on Prevention of Progression of Kidney Diseases and the 11th International Congress of International Society for Peritoneal Dialysis in 2006. He was also the Scientific Vice-President and Program Chair for the 2nd Congress of the International Society for Hemodialysis in 2009. Prof. Li is now the Chairman of the Organizing Committee for the World Congress of Nephrology 2013, which will be held in Hong Kong.
Prof. Li is the founding Editor-in-Chief of the Hong Kong Journal of Nephrology, Deputy Editor of Nephrology, and Editor of Nephrology Dialysis & Transplantation and the International Journal of Artificial Organs. He is on the Editorial Boards of Clinical Nephrology, Peritoneal Dialysis International, Nephron Clinical Practice, Chinese Journal of Nephrology, Dialysis & Transplantation, Medical Progress, Indian Journal of Peritoneal Dialysis, and is a regular reviewer for all the major nephrology journals. He has published over 380 original and review articles in peer-reviewed journals, two books and 17 book chapters, and has given lectures to over 100 international congresses and meetings.

KDIGO® GN Guideline March 2011 Public Review Draft
211
His research interests include many aspects of peritoneal dialysis, such as residual renal function, cardiovascular disease, connectology, peritonitis, biocompatible solutions adequacy; cardiovascular mortality in dialysis patients; IgAN; prevention of progression of CKD; diabetes in kidney failure; immunogenetics of nephropathies; and drug pharmacokinetics and complications after transplantation.
Speaker: Baxter Healthcare
Zhi-Hong Liu, MD, earned her medical degree from Xinjiang University School of Medical in 1982, and postgraduate degree at University of Secondary Military School of Medicine, Shanghai, China, in 1989. She worked as a research associate at the NIH, NIDDK, USA (1993-95) and a visiting scholar in the Division of Nephrology, University of Washington, Seattle, WA, USA (1996).
Dr. Liu was appointed Professor of Medicine at Nanjing University in 1996 and became Adjunct Professor of Medicine at Brown University in 2008. In 2003, she was also elected as Academician in Chinese Academy of Engineering. Her primary interest is in the field of renal disease, with special interest in GN, including IgAN, LN, MN, FSGS, and diabetic and metabolic renal disease. She has published 390 articles, authored two books, and contributed chapters to nephrology textbooks. Dr. Liu also served on the editorial boards of several peer-reviewed journals, including as editor-in-chief of the Chinese Journal of Nephrology, Dialysis & Transplantation. She was honored with the National Science and Technology Progress Award of China; the National Young Investigator Award and National Outstanding Individual Award in Science and Technology from the China Association for Science and Technology; and the Guanghua Engineering & Technological Science Award from Chinese Academy of Engineering. She is the President-Elect of the Chinese Society of Nephrology.
Dr. Liu directs one of the most productive renal patient care and research programs in China, the Research Institute of Nephrology, Jinling Hospital at the Nanjing University School of Medicine. She is the Board member of Academic Degrees Committee, Nanjing University. Dr. Liu has served on several international committees related to scientific programs and global scientific interactions. She worked as Scientific Program Committee member of the World Congress of Nephrology in 2007. She has been a KDIGO Board Member since 2009 and presently chairs the annual meeting of “Forefronts in Glomerular Disease—Nanjing Forum”, which has been endorsed as ISN GO Continuing Medical Education Course since 2006.
Dr. Liu reported no relevant financial relationships

KDIGO® GN Guideline March 2011 Public Review Draft
212
Sergio A. Mezzano, MD, FASN, FACP, is Cathedratic Professor of Medicine and Chairman, Division of Nephrology at Universidad Austral in Valdivia, Chile. He completed his medical studies at Universidad de Chile and nephrology fellowship at University of Cincinnati, Ohio, USA. Dr. Mezzano has authored over 110 publications and book chapters on topics spanning immunopathogenesis of glomerular diseases, role of proteinuria and RAS activation in progression of CKD and mechanisms of renal fibrosis, epithelial mesenchymal transdifferentiation, and interstitial inflammatory infiltration. He has served as Past President of both the Chilean Society of Nephrology (1996-1998) and Latin American Society of Nephrology (2002-2004). In 2002, he was honored with an International Medal Award by the NKF and received the Laureate Award from the American College of Physicians in 2007.
Advisor/Consultant: Fresenius
Patrick H. Nachman, MD, is Professor of Medicine at the University of North Carolina at Chapel Hill, NC, USA. He graduated from Boston University School of Medicine and received fellowship training at University of North Carolina at Chapel Hill. His research interests encompass many areas, including the participation of a number of clinical trials for treatment of FSGS, polycystic kidney disease, IMN, diabetic nephropathy, LN, and ANCA vasculitis. As an author of over 50 publications, he maintains a firm commitment to postgraduate education both at the national and international level. Dr. Nachman was awarded the Internal Medicine Housestaff Faculty Award in 2007 and acknowledged in Best Doctors in America from 2008-2010.
Advisor/Consultant: Questcor
Manuel Praga MD, PhD, is Professor of Medicine at the Complutense University, and Chief of the Nephrology Division at the Hospital 12 de Octubre in Madrid, Spain. He is member of the Scientific Committee at the “Instituto de Investigación Hospital 12de Octubre”. He obtained his medical degree from the University of Valladolid and completed a nephrology fellowship at Hospital Puerta de Hierro, Madrid, Spain. Dr Praga is a founding member and coordinator of the Group for the Study of Glomerular Diseases of the Spanish Society of Nephrology (GLOSEN). He has authored more than 200 peer-reviewed publications and numerous book chapters, and has received many awards, including the “Iñigo Alvarez de Toledo” award to Clinical Investigation in 2000 and 2008. Dr. Praga has served on the Directory Board of the Spanish Society of Nephrology, and also sits as an editorial board member and reviewer for international journals. His current research activities focus on primary and secondary glomerular diseases, renal complications of obesity, diabetic nephropathy, role of proteinuria in the progression of renal damage, interstitial renal diseases, and renal transplantation.
Advisor/Consultant: Aspreva (Vifor Pharma); Novartis; Roche Speaker: Astellas; Novartis; Roche Grant/Research Support: Astellas; Novartis; Roche; Wyeth

KDIGO® GN Guideline March 2011 Public Review Draft
213
Jai Radhakrishnan, MD, MS, MRCP, FACC, FASN, is Associate Professor of Clinical Medicine and the Director of the Nephrology Fellowship Training Program at Columbia University, NY, USA. He acquired his medical school and internal medicine training in India, Great Britain, and the USA, and later completed a nephrology fellowship at the Massachusetts General Hospital in Boston and Columbia-Presbyterian Medical Center in New York. His clinical and research interests are therapy of glomerular diseases and intensive-care nephrology. Dr. Radhakrishnan is an associate editor of Kidney International and the editor-in-chief of the Nephrology Gateway (the ISN website). His commitment to medical education and patient care is exemplified by his numerous teaching awards and his inclusion in the Who’s Who Among America’s Teachers & Educators and America’s Best Doctors.
Advisor/Consultant: Genentech
Brad H. Rovin, MD, FACP, FASN, is Professor of Medicine and Pathology, Vice-Chairman of Research for Internal Medicine, and Director, Division of Nephrology at The Ohio State University College of Medicine, OH, USA. He received his medical degree from University of Illinois and completed nephrology fellowship training at Washington University School of Medicine in St. Louis, Missouri, USA. As a leading authority on treatment for lupus, Dr. Rovin has conducted a randomized controlled study (LUNAR) examining the effectiveness of rituximab in lupus nephritis patients already receiving MMF and corticosteroids. In addition, he is also a primary investigator in a multicenter RCT evaluating the efficacy and safety of rituximab in patients with moderate to severe SLE. With over 120 authored publications, Dr. Rovin has served on the editorial boards of American Journal of Kidney Disease, Clinical Nephrology, JASN, Kidney International and as a manuscript reviewer for 16 other journals. He is also a member of numerous professional societies, including the Scientific Advisory Board of the Lupus Foundation of America and various NIDDK Study Sections. Dr. Rovin is a past recipient of the NKF Young Investigator Award and Clinical Scientist Award, and has appeared in multiple editions of America's Best Doctors since 2005.
Advisor/Consultant: Centocor; Genetech; Osprey; Questcor; Teijan Pharmaceuticals
Stéphan Troyanov, MD, is Assistant Professor of Clinical Medicine at Université de Montréal, Quebec, Canada and nephrologist at Hôpital du Sacré-Coeur de Montréal. Dr. Troyanov completed his medical studies at Université de Montréal and received fellowship training at University of Toronto, Canada. His earlier research centered on ascertaining the determinants of remission in proteinuria in primary glomerular disease. Currently, he is investigating the use of urinary inflammatory markers in predicting the outcome of progressive glomerular disease, and is conducting an European validation study on the Oxford Classification of IgAN. Dr. Troyanov received the Clinician Research Award twice from Fonds de la Recherche en Santé du Québec, an agency of the Government of Québec, and the Young Nephrologist Prize from Société Québécoise de Néphrologie.
Dr. Troyanov reported no relevant financial relationships

KDIGO® GN Guideline March 2011 Public Review Draft
214
Jack F. M. Wetzels, MD, PhD, was born in Heerlen, The Netherlands. He studied Medicine at the University of Nijmegen, and received his MD in 1980. He undertook his training in Internal Medicine and Nephrology at the Radboud University Nijmegen Medical Center and later received his Ph.D. in 1989. From 1990 to 1992, he studied ischemic renal tubular injury as a postdoctoral fellow under the supervision of Robert W Schrier at the University of Colorado Health Sciences Center in Denver, CO, USA. Since 1992, he has been working as a nephrologist and was appointed Professor of Nephrology in the Department of Nephrology at Radboud University Nijmegen in 2002. Professor Wetzels is committed to teaching and research, with an emphasis on the diagnosis and treatment of patients with glomerular diseases.
Advisor/Consultant: Genzyme Speaker: Amgen; Genzyme Grant/Research Support: Amgen; Genzyme; Roche KDIGO Chairs
Kai-Uwe Eckardt, MD, is Professor of Medicine and Chief of Nephrology and Hypertension at the University of Erlangen–Nuremberg, Germany. He received his MD from the Westfälische Wilhelms-Universität Münster, Germany. In 1993, following postgraduate training in internal medicine, pathology and physiology, he was appointed Assistant Professor of Physiology at the University of Regensburg, Germany. Subsequently, he continued his training in internal medicine and nephrology at the Charité, Humboldt University in Berlin, where he was appointed Associate Professor of Nephrology in 2000. His major scientific interests are in the molecular mechanisms and physiological/pathophysiological relevance of oxygen sensing and the management of anemia. As such, he has contributed to the development of the European Best Practice Guidelines for Anemia Management and participated in the CREATE and TREAT trial studies. Professor Eckardt is Subject Editor of Nephrology, Dialysis & Transplantation, and serves on the editorial board of several other journals. He has also authored book chapters and most recently served as a Co-Editor of the text, Studies on Renal Disorders. Dr. Eckardt is a member of the executive committee of KDIGO.
Advisor/Consultant: Affymax; Amgen; Hexal Sandoz; Johnson & Johnson; Roche Speaker: Amgen; Jansen Cilag; Johnson & Johnson; Roche Grant/Research Support: Roche
Bertram L. Kasiske, MD, is Professor of Medicine and Medical Director of Kidney and Pancreas Transplantation at University of Minnesota, MN, USA. He received his medical degree from the University of Iowa and completed his Internal Medicine residency and fellowship training in Nephrology at Hennepin County Medical Center, where he is also currently Director of Nephrology and Medical Director of Kidney Transplantation. His primary research interests encompass immunosuppression, dyslipidemia, and cardiovascular disease in transplant recipients. He is a Co-Investigator in a RCT of homocysteine (FAVORIT), Study of Heart and Renal Protection, and the USRDS. Dr. Kasiske has served as Medical/Scientific Representative to the Board of Directors of United Network for Organ Sharing, and was formerly Editor-in-Chief of American Journal of Kidney Diseases. He has published over 200 journal articles and

KDIGO® GN Guideline March 2011 Public Review Draft
215
has recently contributed book chapters in Brenner and Rector’s The Kidney, Comprehensive Clinical Nephrology, The Kidney: Physiology and Pathophysiology, Kidney Transplantation: Principles and Practice and Primer on Kidney Diseases. Dr. Kasiske is also a recipient of the Outstanding Research Accomplishment Award from University of Minnesota School of Medicine in 2002 and the NKF’s Garabed Eknoyan Award in 2003.
Advisor/Consultant: LithoLink (Labcorp); Wyeth Grant/Research Support: Bristol-Myers Squibb; Genzyme; Merck/Schering Plough Evidence Review Team
Ethan M. Balk, MD, MPH, is the Director, Evidence-based Medicine at the Tufts Center for Kidney Disease Guideline Development and Implementation, in Boston, MA, USA, and Assistant Professor of Medicine at Tufts University School of Medicine. Dr Balk completed a fellowship in Clinical Care Research. His primary research interests are evidence-based medicine, systematic review, clinical practice guideline development, and critical literature appraisal.
Dr. Balk reported no relevant financial relationships
Gowri Raman, MD, MS, is Assistant Professor of Medicine at Tufts University School of Medicine, Boston, MA, USA and Assistant Director, Tufts Evidence Practice Center at the Center for Clinical Evidence Synthesis. She completed her Clinical and Translational Science Research fellowship in the Institute for Clinical Research and Health Policy Studies at Tufts Medical Center. Her primary research interests are health technology assessment, systematic review and clinical practice guideline development.
Dr. Raman reported no relevant financial relationships
Dana C. Miskulin, MD, MS, is Assistant Professor of Medicine at Tufts University School of Medicine, Boston, MA, USA. She completed a fellowship in Clinical Care Research and participated in the conduct of systematic reviews and critical literature appraisals for this guideline. Her primary research interests are in comparative effectiveness research in dialysis patients, blood pressure treatment in dialysis patients, and autosomal dominant polycystic kidney disease.
Dr. Miskulin receives salary support from Dialysis Clinic Inc., a not-for-profit dialysis organization

KDIGO® GN Guideline March 2011 Public Review Draft
216
Aneet Deo, MD, MS, served as a research fellow at the Tufts Center for Kidney Disease Guideline Development and Implementation in Boston, MA, USA, and participated in the conduct of systematic reviews and critical literature appraisals for this guideline.
Dr. Deo reported no relevant financial relationships
Amy Earley, BS, is a project coordinator at the Tufts Center for Kidney Disease Guideline Development and Implementation in Boston, MA, USA. She assists in the development of clinical practice guidelines and conducts systematic reviews and critical literature appraisals.
Ms. Earley reported no relevant financial relationships
Shana Haynes, MS, DHSc, is a research assistant at the Tufts Center for Kidney Disease Guideline Development and Implementation in Boston, MA, USA. She assists in the development of clinical practice guidelines and conducts systematic reviews and critical literature appraisals.
Dr. Haynes reported no relevant financial relationships








![[Practice guideline 'Premature ejaculation']](https://img.dokumen.tips/doc/110x75/634814bbde40dd034d09061e/practice-guideline-premature-ejaculation.jpg)




















