Embed Size (px)
Citation preview

ORIGINAL INVESTIGATION
Influence of !-Endorphin on anxious behaviorin mice: interaction with EtOH
Judith E. Grisel & Jessica L. Bartels & Stephani A. Allen &
Victoria L. Turgeon
Received: 31 July 2007 /Accepted: 3 April 2008 / Published online: 5 July 2008# Springer-Verlag 2008
AbstractRationale The opioid peptide !-endorphin (!-E) is synthe-sized by the pro-opiomelanocortin gene in response toenvironmental stressors and alcohol administration and isimplicated in the behavioral sequelae associated with thesestimuli.Objectives We sought to determine the influence of !-E onthe stress response by evaluating basal measures of anxietyas well as on EtOH-induced anxiolytic behavior usingtransgenic mice that differ with respect to !-E.Methods Anxious behavior was evaluated for male andfemale heterozygous, wild-type, and !-E knockout miceusing the Light–Dark Box and Plus Maze assays. Subse-quent tests evaluated behavior 20 min after administrationof intraperitoneal saline or EtOH (0.5, 1.0, and 1.5 g/kg).Results We observed a direct relationship between !-Elevels and the percentage of entries into open arms of thePlus Maze as well as the time spent in either the open armsor the light compartment of the Light–Dark box duringbasal conditions, suggesting that this peptide normallyinhibits anxious behavior. However, mice lacking !-E
demonstrated an exaggerated anxiolytic response to EtOHin these assays.Conclusions These data suggest that !-E moderates theresponse to stressful stimuli and supports the hypothesisthat this peptide influences the behavioral effects of EtOH.
Keywords Addiction . Ethanol . Anxiety . Opioids .Mice .
Transgenic . Self-medication
Introduction
!-endorphin (!-E) is a 31 amino-acid peptide that iscleaved from the carboxyl terminus of the Proopiomelano-cortin (POMC) gene. As a member of the large family ofopioid peptides that are widely and differentially distributedthroughout the nervous system, it has been implicated in avariety of behaviors including the regulation of pain andreward, as well as in modulating neurocircuitry involved inlearning and memory, motivation and processes associatedwith stress, fear, or anxiety (e.g., Bloom 1980). Theresponse to stressors involves a complex cascade ofendocrine, autonomic, and behavioral changes that seemto be generally aimed at maintaining or restoring homeo-stasis. At least in part because of its complexity, neither aprecise definition nor thorough knowledge of the neurobi-ological underpinnings of the stress response has beenelucidated (Pacak and Palkovitis 2001). Nonetheless, nearlyany operational understanding includes activation of thehypothalamic-pituitary-adrenal (HPA) axis. This reactioninvolves secretion of corticotrophin-releasing hormone(CRH) from the hypothalamus leading to adrenocorticotro-pic hormone (ACTH) release into the bloodstream andsubsequent glucocorticoid secretion from the adrenalglands, contributing to the behavioral fight or flight
Psychopharmacology (2008) 200:105–115DOI 10.1007/s00213-008-1161-4
This publication was made possible by NIH Grant Numbers P20RR-016461 from the National Center for Research Resources,AA13259 (through the INIA Stress Consortium) and AA13641 fromthe National Institute on Alcohol Abuse and Alcoholism.
J. E. Grisel (*) : S. A. Allen :V. L. TurgeonFurman University,3300 Poinsett Hwy,Greenville, SC 29613, USAe-mail: [email protected]
J. L. BartelsNeural Signals Incorporated,3400 McClure Bridge Road, Bldg. D Suite B,Duluth, GA 30096, USA

response. Corticotropin-releasing hormone in both thehypothalamus and pituitary stimulates POMC gene expres-sion which is initially cleaved into two subunits, one ofwhich eventually gives rise to ACTH and the other whichmay yield !-E (Pfaff et al. 2004).
The most well-studied effect of !-E is its ability tomodulate pain, but an early report by Fratta et al. (1981)suggested that central administration of !-E producedopposite effects to that of ACTH administration (“recipro-cal antagonism”) on several behaviors in rats includingcataplexy, rigidity, and analgesia and supported the notionthat !-E is directly implicated in the homeostatic regulationof ACTH response. Indeed, !-E synthesis and release isprecipitated by stressful stimuli (Guillemin et al. 1977;Oltras et al. 1987), and low doses of !-E have been shownto decrease stress responding (Panksepp 2003). Modulationof the behavioral response to stressful stimuli by !-E isdiscussed in a recent review by Ribiero et al. (2005) andpreviously by Yamada and Nabeshima (1995).
Corticotropin-releasing hormone is the primary regulatorof POMC peptides, and ethanol (EtOH) exposure leads toCRH release in vivo (Redei et al. 1988) and in vitro(deWaele and Gianoulakis 1993). Thus, acute EtOH, likeexposure to classic stressors, increases the synthesis andrelease of ACTH and !-E (Froehlich et al. 1990, 2000;Gianoulakis 1990; Marinelli et al. 2004; Millan 1981; Oliveet al. 2001; Rivier 1996; Sarkar et al. 2007; Scanlon et al.1992; Schulz et al. 1980; Thiagarajan et al. 1988, 1989).Paradoxically though, one of the primary factors influenc-ing alcohol ingestion is thought to be stress reduction,particularly in genetically or environmentally prone indi-viduals (see Cappell and Herman 1972; Pohorecky 1991 forreviews).
Although the relationship between EtOH consumptionand the sequelae associated with stress is naturally complex(as each, independently, are highly multidimensional), thereis a large body of empirical research evincing the anxiolyticeffects of EtOH (Eckardt et al. 1998; Gianoulakis et al.2003; Grobin et al. 1998; LaBuda and Fuchs 2001).Furthermore, some stressors have been shown to increaseself-administration of EtOH (e.g., Eckardt et al. 1998;Mollenauer et al. 1993; Wolffgramm 1990) and inhibiting thestress axis via CRH antagonism reduces EtOH consumption(Lê et al. 2000; Funk et al. 2006). In the clinic, alcoholismand anxiety disorders are frequently comorbid (Kushner etal. 1990; Bradizza et al. 2006; Conway et al. 2006; Fein etal. 2007; Vendruscolo et al. 2006), and a significantproportion of alcohol-dependent individuals report thatalcohol is reinforcing because it reduces anxiety (Barrenhaand Chester 2007; Hill and Angel 2005; Koven et al. 2005;Lawyer et al. 2002). Thus, the stress-attenuating properties ofEtOH likely contribute to its reinforcing effects, and thesemay be mediated by !-E.
We chose to directly assess the impact of !-E onbehavior associated with anxiety, as well as the anxiolyticeffects of EtOH, by evaluating transgenic mice that havebeen engineered to lack !-E. These mice were developedover a decade ago in the laboratory of Malcolm Low(Rubinstein et al. 1996) and have been evaluated fordifferences in pain responses and energy balance (Appleyardet al. 2003; Hayward et al. 2006; Mogil et al. 2000) aswell as sensitivity to cocaine and morphine (Marquezet al. 2008). In addition, we have looked at oral self-administration of EtOH (Grisel et al. 1999 and Williamset al. 2007) where heterozygote mice, possessing half of thenormal levels of !-E, generally drink slightly more thaneither wild-type controls or mice totally lacking thispeptide. Beside the direct correlation between gene andpeptide concentration, no differences have been found inother POMC peptides or in opioid receptor levels acrossgenotypes (Mogil et al. 2000; Rubinstein et al. 1996). Inorder to further evaluate the effects of !-E on behaviorand pharmacology, we studied three lines of mice withdiffering levels of !-E in the Plus Maze and Light–Darktests, both under basal conditions and following EtOHadministration.
Method
Subjects were adult male and female wild-type controls(C57BL/6J; B6), !-E deficient (KO), and heterozygous(HT) mice bred in-house from progenitors obtained fromthe Jackson Laboratories (Bar Harbor, ME, USA). The genemutation has been fully backcrossed to the C57BL/6J strain(>20 generations). HT mice were bred from KO males andB6 females; others were bred under identical conditionsfrom genotype-matched pairs. These subjects were housedtwo to five per Plexiglas cage following weaning at 20 to21 days with sex-matched siblings. The ventilated colonyroom maintained a constant light schedule (12-h reverselight–dark cycle with lights on 1,900 h) and a temperatureof 22±2°C. Water and food (Lab Diet 5015) were availablead libitum. All subjects were between 50 and 75 days ofage at the time of testing, and each subject was tested onlyone time. Throughout experimentation, subject order wascounterbalanced with regard to genotype, sex, and drug.Behavioral assessment of genotypic differences may befacilitated by testing during the animal’s active phase(Hossain et al. 2004) and so occurred between 9 a.m. and6 p.m. In all experiments, data were recorded by anexperimenter blind to drug condition and genotype. Inaddition, all procedures were carried out in accordance withthe National Institutes of Health guidelines and wereapproved by the Animal Care and Use Committee ofFurman University.
106 Psychopharmacology (2008) 200:105–115

Experimental apparatus
The elevated Plus Maze, made from Plexiglas, was raisedapproximately 45 cm from ground level. The perimeter ofthe base of the maze was enclosed with a clear 15 cm tallwall; the area of the base was filled with wood shavings.The floor of the elevated portion of the maze was black.Two opposite arms (30!5 cm each) of the maze wereenclosed by a clear 10 cm high Plexiglas wall, and theremaining two arms were “open”, with a very short (2 mm)wall. A center space (5 cm2) between these four arms wasalso not enclosed. The elevated portion of the apparatuswas cleaned with a dilute detergent between each session.
The Light–Dark Box was composed of two Plexiglaschambers of equal size (25 cm3) that shared a commonwall. The bottom of the entire apparatus was constructed ofblack, opaque Plexiglas, as were the four walls of the darkchamber. A 27-cm2 panel of the same black Plexiglasserved as a top for the dark side of the apparatus. The threewalls of the light side were constructed of clear Plexiglas,as was the top panel; the fourth wall of the light chamberwas black, as it was shared with the dark chamber. A squarehole (2.5 cm2) in the bottom center of the wall shared byboth chambers allowed subjects to pass freely between lightand dark chambers. Between each subject’s test session, theapparatus was cleaned using a sponge filled with a dilute,low-residue detergent.
Experiment 1
Animals (KO n=10, HT n=11, B6 n=11) were removedfrom the colony room, marked and weighed, and allowed45–60 min to acclimate to conditions in an experimentalroom before testing. This room was lit with fluorescentceiling lights as well as natural light from a single windowand, thus, contrasted with the colony room (dark phase).Though this abrupt transition may have altered chemical orbehavioral function, we wanted to test subjects during theiractive phase; moreover, maze behavior is routinely assessedin well-lit conditions as results may depend upon lighting(Costall et al. 1989). Experiment 1 subjects received noinjections but were placed in individual housing for 20 minafter habituation and before testing (to parallel methodsused in experiments involving drug administration; becausesaline and EtOH-injected animals are generally taken fromthe same cage, we wanted to avoid possible confoundsfrom differential interaction between cage mates followinginjection). After this 20-min period, they were lifted by thetail and placed gently into a closed arm facing the center ofthe maze. For the following 5 min, the number of open armentries, closed arm entries, time spent in open arms, closedarms, and center area were recorded by a blind observerseated about 1 M from the maze. Entry into an arm was
defined as all four feet crossing into the region. From thesedata, the percentage of arm entries in to open arms wascalculated as a measure of anxious behavior corrected forgeneral locomotor activity.
Experiment 2
The procedure for this experiment mimicked that of“Experiment 1” with the exception that animals receivedintraperitoneal (IP) injections of either saline, 0.5, 1.0, or1.5 g/kg 20% EtOH v/v (KO n=11, 10, 11, and 9,respectively, B6 n=9, 9, 7, and 7) after about 45 min ofacclimation. Immediately following injection, subjects wereindividually housed for 20 min and then placed onto thePlus Maze where behavior was evaluated, as in “Experiment1,” for 5 min. Because the primary purpose of this study wasto evaluate a range of EtOH doses to inform subsequenttesting (Exp 3 and 5), heterozygote mice were not assessed.
Experiment 3
The procedure for this experiment paralleled those used in“Experiment 2” with the exception that each of the threegenotypes was tested following IP injections of 1.5 g/kg20% EtOH v/v (KO n=13, HT n=10, B6 n=14) orequivolume saline (KO n=13, HT n=12, B6 n=14) after45 min of acclimation and before 20 min of individualhousing. This dose was chosen based on the results of“Experiment 2.”
Experiment 4
Experiment 4 was similar to “Experiment 1” except thatbehavioral testing took place in the Light–Dark box.Subjects (KO n=10, HT n=8, B6 n=7) were removedfrom the colony room, marked, and weighed and allowed45 min to acclimate to experimental conditions beforetesting. The testing room was lit with fluorescent ceilinglights as well as natural light from a single window and,thus, contrasted with the colony room (dark phase). A 60-Whalogen lamp was also affixed above the light side of theapparatus to ensure optimal contrast between light and darkchambers (to facilitate discrimination between the twochambers). Again, we wanted to test subjects during theiractive phase, and this test is conventionally done in brightconditions. Experiment 4 subjects received no injections butwere individually housed for 20 min before testing. After thisperiod, they were placed into the dark side of the Light–DarkBox facing the wall opposite the opening into the lightchamber. The dark chamber was immediately covered andbehavior was recorded over a 5-min period by an experimen-tally blind, silent observer seated approximately 1 M from theexperimental apparatus. Latency to emerge from the dark
Psychopharmacology (2008) 200:105–115 107
chamber, number of crossings between chambers (defined asall four feet across the boundary), and total time spent in thelight chamber were recorded.
Experiment 5
Procedure for this experiment was as described for“Experiment 4” with the exception that all subjects receivedIP injections of either 1.5 g/kg 20% EtOH v/v (KO n=9,HT n=11, B6 n=11) or equivolume saline (KO n=12, HTn=14, B6 n=11) before being placed into individualhousing for 20 min. A dose of 75 g/kg EtOH was alsoevaluated but produced no effects substantially differingfrom saline; those data are not included.
Experiment 6
As an additional measure of stress reactivity, we assessedadrenal gland weight in mice of each genetic line. Briefly,mice were sacrificed by cervical dislocation, and bothadrenal glands were located, removed, and weighed(together). The experimenter was blind to genotype in allcases. There was a total N of 143: 56 KOs, 59B6s, and 28HTs. Many of the adrenal glands were taken afterexperimental manipulation (at least 1–2 weeks post manip-ulation), but about 30% were experimentally naïve (ap-proximately equally divided across genotypes).
Statistical analysis
Data were analyzed separately for each experiment byfactorial analysis of variance (ANOVA) in SYSTAT: firstby genotype, sex, and drug (where appropriate) and, then,in the absence of interactions with sex, collapsing acrossthis factor. Significant main effects and interactions wereinvestigated further using Tukey’s HSD test for post hoccomparisons. In all cases, the criterion for significance(! level) was set at p<0.05.
Results
There were no significant interactions with sex in any of thedata analyses, and so this factor was excluded fromsubsequent analysis and results.
Experiment 1
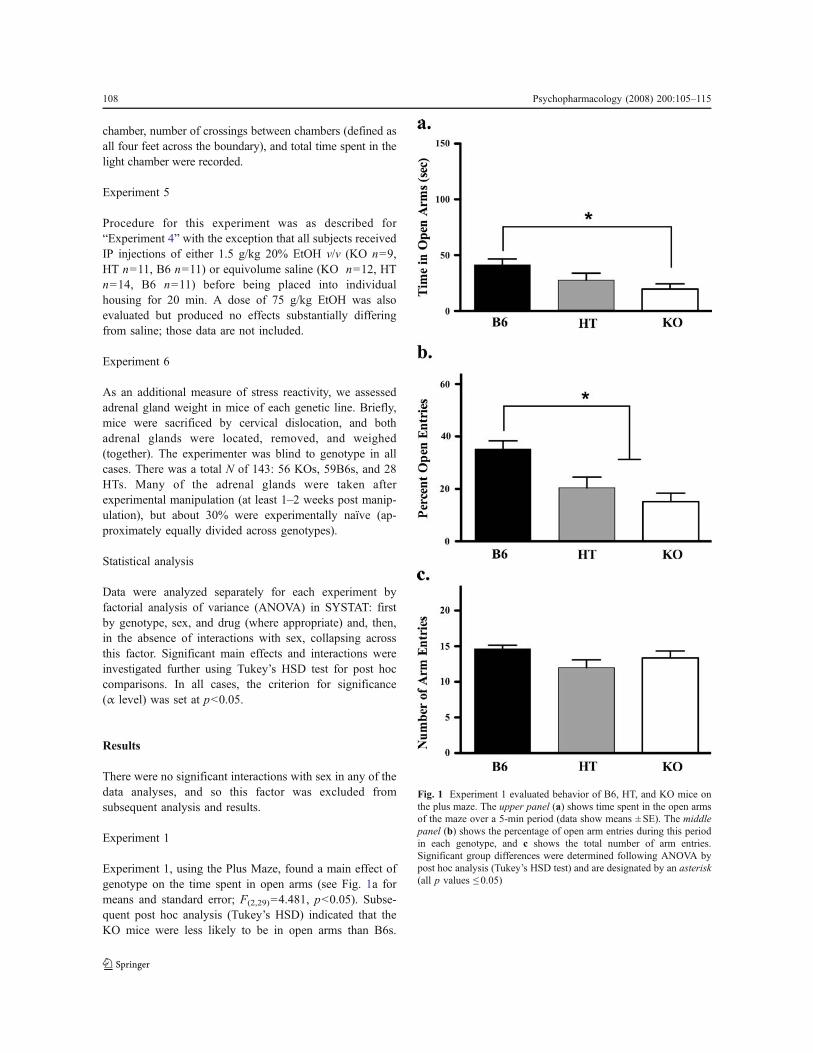
Experiment 1, using the Plus Maze, found a main effect ofgenotype on the time spent in open arms (see Fig. 1a formeans and standard error; F(2,29)=4.481, p<0.05). Subse-quent post hoc analysis (Tukey’s HSD) indicated that theKO mice were less likely to be in open arms than B6s.
Fig. 1 Experiment 1 evaluated behavior of B6, HT, and KO mice onthe plus maze. The upper panel (a) shows time spent in the open armsof the maze over a 5-min period (data show means ± SE). The middlepanel (b) shows the percentage of open arm entries during this periodin each genotype, and c shows the total number of arm entries.Significant group differences were determined following ANOVA bypost hoc analysis (Tukey’s HSD test) and are designated by an asterisk(all p values ! 0.05)
108 Psychopharmacology (2008) 200:105–115
Another way of looking at the data is to evaluate thepercentage of time spent in open arms as a function of totalarm time (in other words, controlling for time spent in thecenter of the maze). In our analysis, the F statistic wasvirtually identical in both cases (for percentage of opentime, F(2,29)=4.917, p<0.05), so the more conservative rawdata are depicted in Fig. 1a. KOs also evidenced a smallerpercentage of entries into open arms than B6s (as did HTs;Fig. 1b, F(2,29)=10.416, p<0.05), but there were nosignificant genotypic differences in total arm entries(Fig. 1c, p=0.136) a measure of general locomotor activity.
Experiment 2
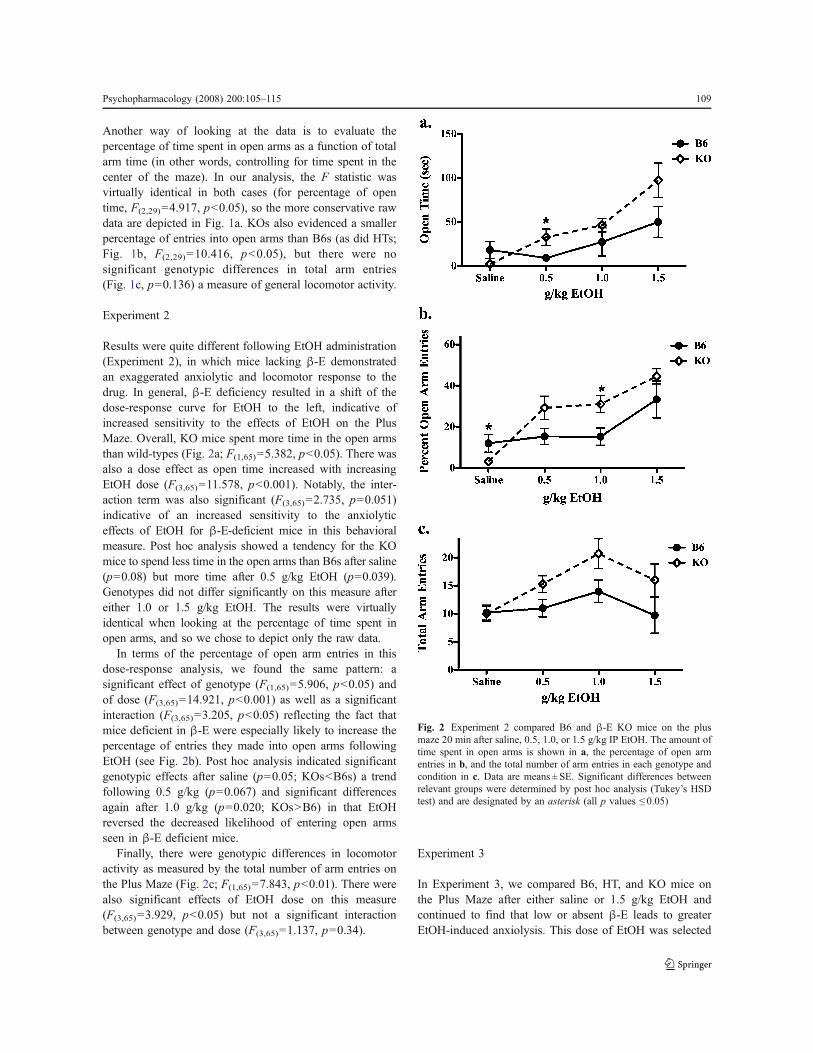
Results were quite different following EtOH administration(Experiment 2), in which mice lacking !-E demonstratedan exaggerated anxiolytic and locomotor response to thedrug. In general, !-E deficiency resulted in a shift of thedose-response curve for EtOH to the left, indicative ofincreased sensitivity to the effects of EtOH on the PlusMaze. Overall, KO mice spent more time in the open armsthan wild-types (Fig. 2a; F(1,65)=5.382, p<0.05). There wasalso a dose effect as open time increased with increasingEtOH dose (F(3,65)=11.578, p<0.001). Notably, the inter-action term was also significant (F(3,65)=2.735, p=0.051)indicative of an increased sensitivity to the anxiolyticeffects of EtOH for !-E-deficient mice in this behavioralmeasure. Post hoc analysis showed a tendency for the KOmice to spend less time in the open arms than B6s after saline(p=0.08) but more time after 0.5 g/kg EtOH (p=0.039).Genotypes did not differ significantly on this measure aftereither 1.0 or 1.5 g/kg EtOH. The results were virtuallyidentical when looking at the percentage of time spent inopen arms, and so we chose to depict only the raw data.
In terms of the percentage of open arm entries in thisdose-response analysis, we found the same pattern: asignificant effect of genotype (F(1,65)=5.906, p<0.05) andof dose (F(3,65)=14.921, p<0.001) as well as a significantinteraction (F(3,65)=3.205, p<0.05) reflecting the fact thatmice deficient in !-E were especially likely to increase thepercentage of entries they made into open arms followingEtOH (see Fig. 2b). Post hoc analysis indicated significantgenotypic effects after saline (p=0.05; KOs<B6s) a trendfollowing 0.5 g/kg (p=0.067) and significant differencesagain after 1.0 g/kg (p=0.020; KOs>B6) in that EtOHreversed the decreased likelihood of entering open armsseen in !-E deficient mice.
Finally, there were genotypic differences in locomotoractivity as measured by the total number of arm entries onthe Plus Maze (Fig. 2c; F(1,65)=7.843, p<0.01). There werealso significant effects of EtOH dose on this measure(F(3,65)=3.929, p<0.05) but not a significant interactionbetween genotype and dose (F(3,65)=1.137, p=0.34).
Experiment 3
In Experiment 3, we compared B6, HT, and KO mice onthe Plus Maze after either saline or 1.5 g/kg EtOH andcontinued to find that low or absent !-E leads to greaterEtOH-induced anxiolysis. This dose of EtOH was selected
Fig. 2 Experiment 2 compared B6 and !-E KO mice on the plusmaze 20 min after saline, 0.5, 1.0, or 1.5 g/kg IP EtOH. The amount oftime spent in open arms is shown in a, the percentage of open armentries in b, and the total number of arm entries in each genotype andcondition in c. Data are means ± SE. Significant differences betweenrelevant groups were determined by post hoc analysis (Tukey’s HSDtest) and are designated by an asterisk (all p values ! 0.05)
Psychopharmacology (2008) 200:105–115 109
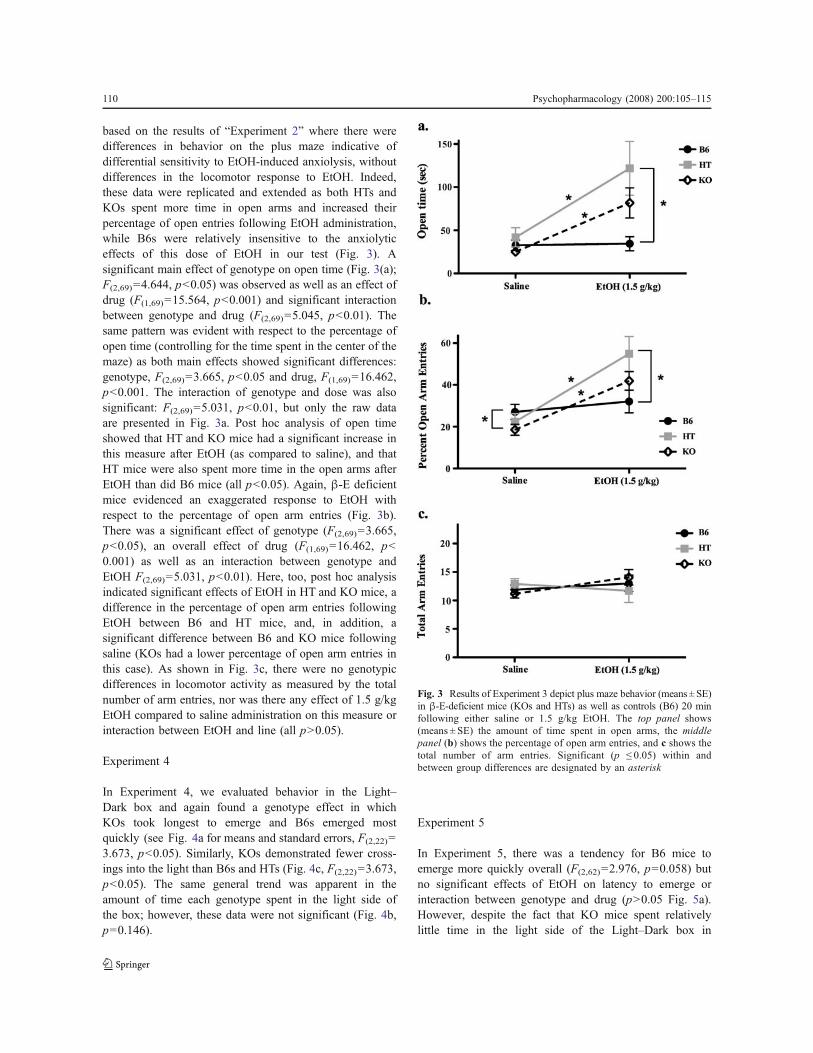
based on the results of “Experiment 2” where there weredifferences in behavior on the plus maze indicative ofdifferential sensitivity to EtOH-induced anxiolysis, withoutdifferences in the locomotor response to EtOH. Indeed,these data were replicated and extended as both HTs andKOs spent more time in open arms and increased theirpercentage of open entries following EtOH administration,while B6s were relatively insensitive to the anxiolyticeffects of this dose of EtOH in our test (Fig. 3). Asignificant main effect of genotype on open time (Fig. 3(a);F(2,69)=4.644, p<0.05) was observed as well as an effect ofdrug (F(1,69)=15.564, p<0.001) and significant interactionbetween genotype and drug (F(2,69)=5.045, p<0.01). Thesame pattern was evident with respect to the percentage ofopen time (controlling for the time spent in the center of themaze) as both main effects showed significant differences:genotype, F(2,69)=3.665, p<0.05 and drug, F(1,69)=16.462,p<0.001. The interaction of genotype and dose was alsosignificant: F(2,69)=5.031, p<0.01, but only the raw dataare presented in Fig. 3a. Post hoc analysis of open timeshowed that HT and KO mice had a significant increase inthis measure after EtOH (as compared to saline), and thatHT mice were also spent more time in the open arms afterEtOH than did B6 mice (all p<0.05). Again, !-E deficientmice evidenced an exaggerated response to EtOH withrespect to the percentage of open arm entries (Fig. 3b).There was a significant effect of genotype (F(2,69)=3.665,p<0.05), an overall effect of drug (F(1,69)=16.462, p<0.001) as well as an interaction between genotype andEtOH F(2,69)=5.031, p<0.01). Here, too, post hoc analysisindicated significant effects of EtOH in HT and KO mice, adifference in the percentage of open arm entries followingEtOH between B6 and HT mice, and, in addition, asignificant difference between B6 and KO mice followingsaline (KOs had a lower percentage of open arm entries inthis case). As shown in Fig. 3c, there were no genotypicdifferences in locomotor activity as measured by the totalnumber of arm entries, nor was there any effect of 1.5 g/kgEtOH compared to saline administration on this measure orinteraction between EtOH and line (all p>0.05).
Experiment 4
In Experiment 4, we evaluated behavior in the Light–Dark box and again found a genotype effect in whichKOs took longest to emerge and B6s emerged mostquickly (see Fig. 4a for means and standard errors, F(2,22)=3.673, p<0.05). Similarly, KOs demonstrated fewer cross-ings into the light than B6s and HTs (Fig. 4c, F(2,22)=3.673,p<0.05). The same general trend was apparent in theamount of time each genotype spent in the light side ofthe box; however, these data were not significant (Fig. 4b,p=0.146).
Experiment 5
In Experiment 5, there was a tendency for B6 mice toemerge more quickly overall (F(2,62)=2.976, p=0.058) butno significant effects of EtOH on latency to emerge orinteraction between genotype and drug (p>0.05 Fig. 5a).However, despite the fact that KO mice spent relativelylittle time in the light side of the Light–Dark box in
Fig. 3 Results of Experiment 3 depict plus maze behavior (means ± SE)in !-E-deficient mice (KOs and HTs) as well as controls (B6) 20 minfollowing either saline or 1.5 g/kg EtOH. The top panel shows(means ± SE) the amount of time spent in open arms, the middlepanel (b) shows the percentage of open arm entries, and c shows thetotal number of arm entries. Significant (p ! 0.05) within andbetween group differences are designated by an asterisk
110 Psychopharmacology (2008) 200:105–115
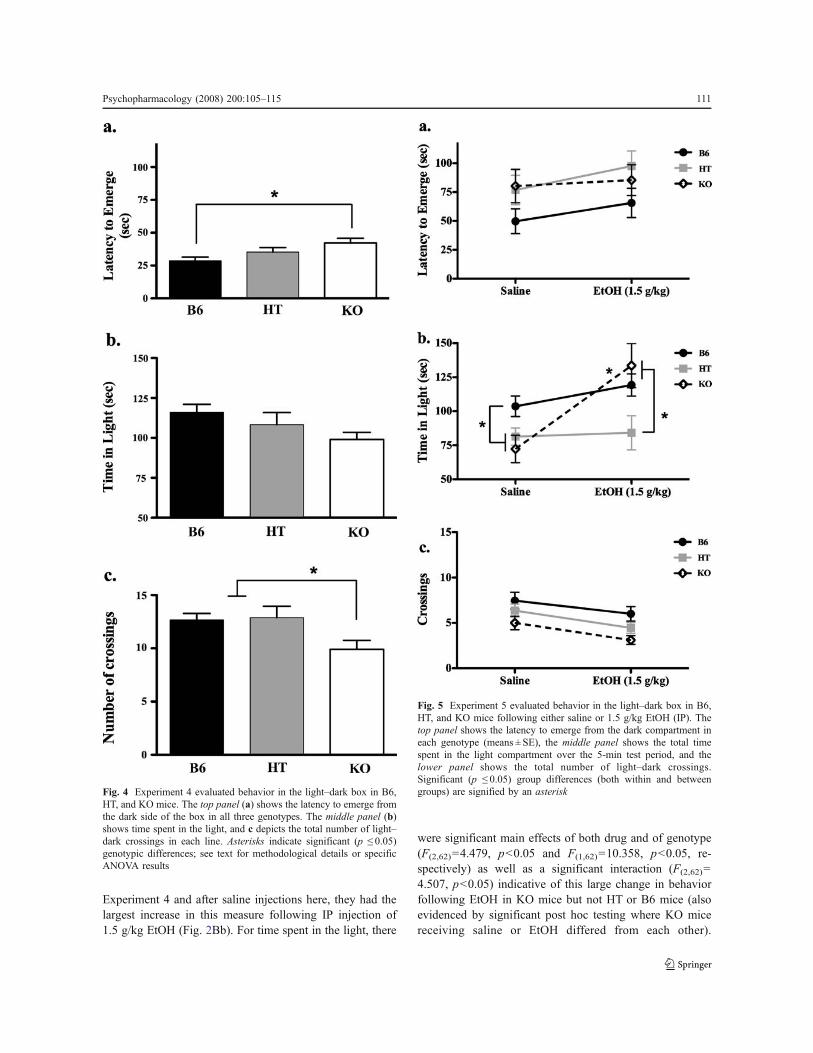
Experiment 4 and after saline injections here, they had thelargest increase in this measure following IP injection of1.5 g/kg EtOH (Fig. 2Bb). For time spent in the light, there
were significant main effects of both drug and of genotype(F(2,62)=4.479, p<0.05 and F(1,62)=10.358, p<0.05, re-spectively) as well as a significant interaction (F(2,62)=4.507, p<0.05) indicative of this large change in behaviorfollowing EtOH in KO mice but not HT or B6 mice (alsoevidenced by significant post hoc testing where KO micereceiving saline or EtOH differed from each other).
Fig. 4 Experiment 4 evaluated behavior in the light–dark box in B6,HT, and KO mice. The top panel (a) shows the latency to emerge fromthe dark side of the box in all three genotypes. The middle panel (b)shows time spent in the light, and c depicts the total number of light–dark crossings in each line. Asterisks indicate significant (p ! 0.05)genotypic differences; see text for methodological details or specificANOVA results
Fig. 5 Experiment 5 evaluated behavior in the light–dark box in B6,HT, and KO mice following either saline or 1.5 g/kg EtOH (IP). Thetop panel shows the latency to emerge from the dark compartment ineach genotype (means ± SE), the middle panel shows the total timespent in the light compartment over the 5-min test period, and thelower panel shows the total number of light–dark crossings.Significant (p ! 0.05) group differences (both within and betweengroups) are signified by an asterisk
Psychopharmacology (2008) 200:105–115 111
Significant main effects of drug and genotype wereobserved on the variable crossings into light (F(2,62)=6.1664, p<0.05 and F(1,62)=8.3028, p<0.05, respectively)though the interaction was not significant, indicative of thefact that KOs were generally less active than B6 mice inthis test, and EtOH had a nonspecific tendency to decreaseactivity in this test.
Experiment 6
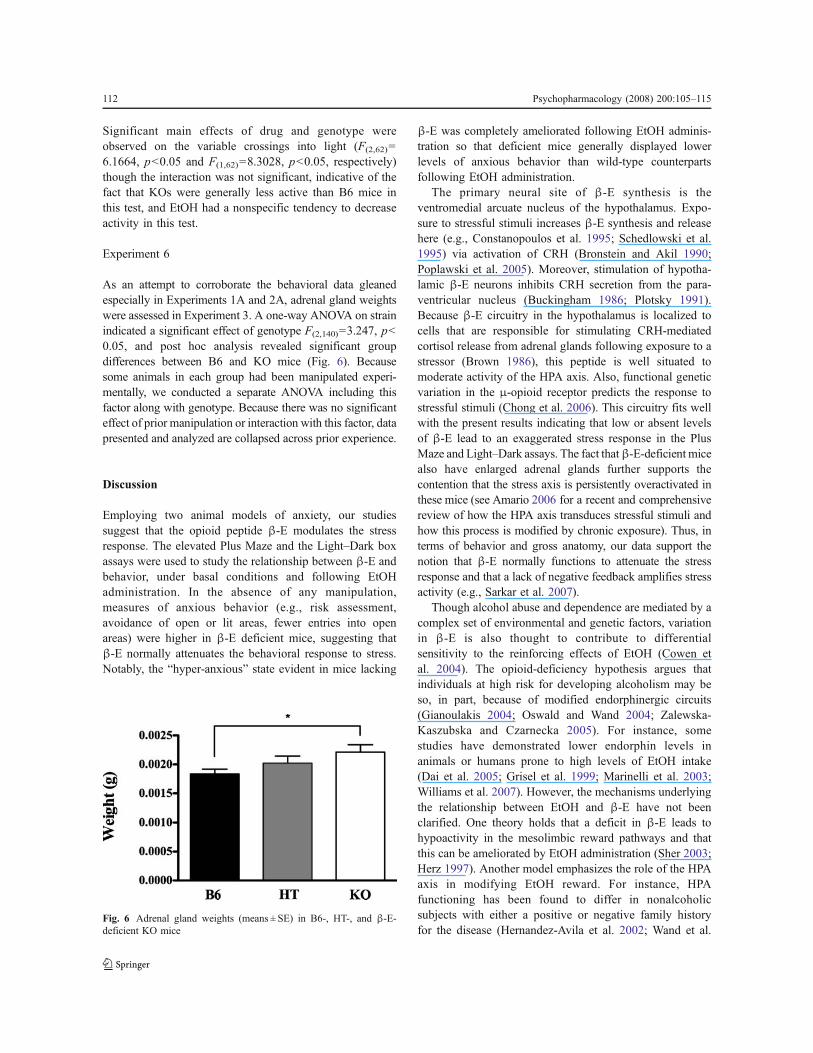
As an attempt to corroborate the behavioral data gleanedespecially in Experiments 1A and 2A, adrenal gland weightswere assessed in Experiment 3. A one-way ANOVA on strainindicated a significant effect of genotype F(2,140)=3.247, p<0.05, and post hoc analysis revealed significant groupdifferences between B6 and KO mice (Fig. 6). Becausesome animals in each group had been manipulated experi-mentally, we conducted a separate ANOVA including thisfactor along with genotype. Because there was no significanteffect of prior manipulation or interaction with this factor, datapresented and analyzed are collapsed across prior experience.
Discussion
Employing two animal models of anxiety, our studiessuggest that the opioid peptide !-E modulates the stressresponse. The elevated Plus Maze and the Light–Dark boxassays were used to study the relationship between !-E andbehavior, under basal conditions and following EtOHadministration. In the absence of any manipulation,measures of anxious behavior (e.g., risk assessment,avoidance of open or lit areas, fewer entries into openareas) were higher in !-E deficient mice, suggesting that!-E normally attenuates the behavioral response to stress.Notably, the “hyper-anxious” state evident in mice lacking
!-E was completely ameliorated following EtOH adminis-tration so that deficient mice generally displayed lowerlevels of anxious behavior than wild-type counterpartsfollowing EtOH administration.
The primary neural site of !-E synthesis is theventromedial arcuate nucleus of the hypothalamus. Expo-sure to stressful stimuli increases !-E synthesis and releasehere (e.g., Constanopoulos et al. 1995; Schedlowski et al.1995) via activation of CRH (Bronstein and Akil 1990;Poplawski et al. 2005). Moreover, stimulation of hypotha-lamic !-E neurons inhibits CRH secretion from the para-ventricular nucleus (Buckingham 1986; Plotsky 1991).Because !-E circuitry in the hypothalamus is localized tocells that are responsible for stimulating CRH-mediatedcortisol release from adrenal glands following exposure to astressor (Brown 1986), this peptide is well situated tomoderate activity of the HPA axis. Also, functional geneticvariation in the "-opioid receptor predicts the response tostressful stimuli (Chong et al. 2006). This circuitry fits wellwith the present results indicating that low or absent levelsof !-E lead to an exaggerated stress response in the PlusMaze and Light–Dark assays. The fact that!-E-deficient micealso have enlarged adrenal glands further supports thecontention that the stress axis is persistently overactivated inthese mice (see Amario 2006 for a recent and comprehensivereview of how the HPA axis transduces stressful stimuli andhow this process is modified by chronic exposure). Thus, interms of behavior and gross anatomy, our data support thenotion that !-E normally functions to attenuate the stressresponse and that a lack of negative feedback amplifies stressactivity (e.g., Sarkar et al. 2007).
Though alcohol abuse and dependence are mediated by acomplex set of environmental and genetic factors, variationin !-E is also thought to contribute to differentialsensitivity to the reinforcing effects of EtOH (Cowen etal. 2004). The opioid-deficiency hypothesis argues thatindividuals at high risk for developing alcoholism may beso, in part, because of modified endorphinergic circuits(Gianoulakis 2004; Oswald and Wand 2004; Zalewska-Kaszubska and Czarnecka 2005). For instance, somestudies have demonstrated lower endorphin levels inanimals or humans prone to high levels of EtOH intake(Dai et al. 2005; Grisel et al. 1999; Marinelli et al. 2003;Williams et al. 2007). However, the mechanisms underlyingthe relationship between EtOH and !-E have not beenclarified. One theory holds that a deficit in !-E leads tohypoactivity in the mesolimbic reward pathways and thatthis can be ameliorated by EtOH administration (Sher 2003;Herz 1997). Another model emphasizes the role of the HPAaxis in modifying EtOH reward. For instance, HPAfunctioning has been found to differ in nonalcoholicsubjects with either a positive or negative family historyfor the disease (Hernandez-Avila et al. 2002; Wand et al.
Fig. 6 Adrenal gland weights (means ± SE) in B6-, HT-, and !-E-deficient KO mice
112 Psychopharmacology (2008) 200:105–115

1998). In these studies, genetically prone individuals showedan exaggerated HPA response to challenge by opiateantagonists. This relationship was also found in a recentlaboratory study in which two lines of mice selectively bredfor high alcohol preference demonstrated exaggerated fearpotentiated startle relative to their low-alcohol-preferringcounterparts (Barrenha and Chester 2007). Furthermore, thewithdrawal state experienced following chronic EtOHexposure, which is associated with high anxiety, is thoughtto result in part from downregulation of !-E (Aguirre et al.1995; Diana et al. 1993; Scanlon et al. 1992; Valdez et al.2004). Although these theories are not mutually exclusive,the present data especially support the latter idea bysuggesting that !-E modifies sensitivity to both stress andEtOH. Specifically, low !-E leads to increased activity of theHPA axis, and by some unknown mechanism, this deficitfacilitates EtOH-mediated anxiolysis.
Although transgenic animals like the ones used in thisstudy can provide useful insight into neural mechanismsunderlying behavior, the differences evident in our mouselines may not be directly related to an effect of missing !-E. For example, in a study of "-receptor knockout mice(a primary site of !-E action), no differences were found inEtOH-mediated anxiolysis on the plus maze (LaBuda andFuchs 2001). Furthermore, a review of the literature failedto find precedent for the notion that absent !-E shouldexaggerate EtOH effects, and the most straightforwardinterpretation seems unlikely (that !-E acts as a functionalEtOH antagonist). It is plausible, however, that theenhanced sensitivity to EtOH’s anxiolytic effect seen in!-E deficient mice reflects compensatory changes in relatedneural systems that occur as a result of absent !-Ethroughout development. Especially in the case of inbredstrains, gene knockout studies can help elucidate the func-tional interplay between contributing neural factors (e.g.,Mogil and Grisel 1998) and epistatic interactions betweengenes and their products now seems to be the norm ratherthan the exception in neurobiology. Thus, consequentadaptation of the nervous system to experimental transgenicmanipulation may provide a window into the complexsystems underlying behavioral states such as anxiety andalcoholism (e.g., Bowers et al. 1999).
Alcoholism and anxiety disorders are frequently comor-bid, and a significant percentage of alcoholics report thatthey ingest alcohol to reduce anxiety (Compton et al. 2007;Zimmerman et al. 2003). The fact that low !-E levels havebeen linked to a higher risk for excessive alcohol intake andalcoholism (Dai et al. 2005; Gianoulakis 1996; Wand et al.1998) and, in our study, to predict higher levels of anxiousbehavior, suggest that !-E may be a contributing factor tothis relationship. Future efforts employing these mice maybe useful in elucidating the neural substrates mediatingthese relationships.
Acknowledgements The authors would like to acknowledge theforesight of Dr. John C. Crabbe at Oregon Health Sciences Universitythat lead to this series of studies, the Psychology Department at ReedCollege, in Portland, OR, USA where the studies began andChristopher Smith, Gregory Cloonan, Amanda Lee, and CarmenSanchez who assisted with experiments at Furman University.
References
Aguirre JC, del Arbol JL, Rico J, Raya J, Mirand MT (1995)Classification of alcoholics on the basis of plasma b-endorphinconcentration. Alcohol 12:531–534
Appleyard SM, Hayward M, Young JI, Butler AA, Cone RD,Rubinstein M, Low MJ (2003) A role for the endogenous opioidbeta-endorphin in energy homeostasis. Endocrinology 144:1753–1760
Amario A (2006) The hypothalamic-pituitary-adrenal axis: what can ittell us about stressors. CNS Neurol Disord Drug Targets 5:485–501
Barrenha GD, Chester JA (2007) Genetic correlation between innatealcohol preference and fear-potentiated startle in selected mouselines. Alcohol Clin Exp Res 31:1081–1088
Bloom FE (1980) The endorphins: natural peptides for pain, pleasure,and other purposes. Psychopharmacol Bull 16:51–52
Boyadjieva NI, Sarkar DK (1997) The role of cAMP in ethanol-regulated beta-endorphin release from hypothalamic neurons.Alcohol Clin Exp Res 21:728–31
Bowers BJ, Owen EH, Collins AC, Abeliovich A, Tonegawa S,Wehner JM (1999) Decreased ethanol sensitivity and tolerancedevelopment in gamma-protein kinase C null mutant mice isdependent on genetic background. Alcohol Clin Exp Res23:387–397
Bradizza CM, Stasiewicz PR, Paas ND (2006) Relapse to alcohol anddrug use among individuals diagnosed with co-occurring mentalhealth and substance use disorders: a review. Clin Psychol Rev26:162–178
Bronstein DM, Akil H (1990) In vitro release of hypothalamic beta-endorphin (beta E) by arginine vasopressin, corticotropin-releasinghormone and 5-hydroxytryptamine: evidence for release of opioidactive and inactive beta E forms. Neuropeptides 16:33–40
Brown M (1986) Corticotropin releasing factor: central nervoussystem sites of action. Brain Res 99:10–14
Buckingham JC (1986) Stimulation and inhibition of corticotrophinreleasing factor secretion by endorphin. Neuroendocrinology42:148–152
Cappell H, Herman CP (1972) Alcohol and tension reduction. Areview. Q J Stud Alcohol 33:33–64
Chong RY, Oswald L, Yang X, Uhart M, Lin PI, Wand GS (2006) TheMicro-opioid receptor polymorphism A118G predicts cortisolresponses to naloxone and stress. Neuropsychopharmacology31:204–211
Compton WM, Thomas YF, Stinson FS, Grant BF (2007) Prevalence,correlates, disability, and comorbidity of DSM-IV drug abuse anddependence in the United States: results from the nationalepidemiologic survey on alcohol and related conditions. ArchGen Psychiatry 64:566–576
Constantopoulos A, Papadaki-Papandreou U, Papaconstantinou E(1995) Increased beta-endorphin but not Leu-enkephalin inplasma due to preoperative stress. Experientia 51:16–18
Conway KP, Compton W, Stinson FS, Grant BF (2006) Lifetimecomorbidity of DSM-IV mood and anxiety disorders and specificdrug use disorders: results from the National EpidemiologicSurvey on Alcohol and Related Conditions. J Clin Psychiatry67:247–57
Psychopharmacology (2008) 200:105–115 113

Costall B, Jones BJ, Kelly ME, Naylor RJ, Tomkins DM (1989)Exploration of mice in a black and white test box: validation as amodel of anxiety. Pharmacol Biochem Behav 32:777–785
Cowen MS, Chen F, Lawrence AJ (2004) Neuropeptides: implicationsfor alcoholism. J Neurochem 89:273–285
Dai X, Thavundayil J, Gianoulakis C (2005) Differences in theperipheral levels of !-E in response to alcohol and stress as afunction of alcohol dependence and family history of alcoholism.Alcohol Clin Exp Res 29:1965–1975
De Waele JP, Gianoulakis C (1993) Effects of single and repeatedexposures to ethanol on hypothalamic beta-endorphin and CRHrelease by the C57BL/6 and DBA/2 strains of mice. Neuroendo-crinology 57:700–709
Diana M, Pistis M, Carboni S, Gessa GL, Rossetti ZL (1993)Profound decrement of mesolimbic dopaminergic neuronalactivity during ethanol withdrawal syndrome in rats: Electro-physiological and biochemical evidence. Proc Natl Acad Sci U SA 90:7966–7969
Eckardt MJ, File SE, Gessa GL, Grant KA, Guerri C, Hoffman PL,Kalant H, Koob Gf, Li TK, Tabakoff B (1998) Effects ofmoderate alcohol consumption on the central nervous system.Alcohol Clin Exp Res 22:998–1040
Fein G, Di Sclafani V, Finn P, Scheiner DL (2007) Sub-diagnosticpsychiatric comorbidity in alcoholics. Drug Alcohol Depend 87(2–3):139–145
Fratta W, Rossetti ZI, Poggioli R, Gessa GL (1981) Reciprocalantagonism between ACTH1-24 and b-endorphin in rats. Neuro-sci Lett 24:71–74
Froehlich JC, Harts J, Lument L, Li TK (1990) Naloxone attenuatesvoluntary ethanol intake in rats selectively bred for high ethanolpreference. Pharmacol Biochem Behav 35:385–390
Froehlich JC, Zink RW, Li TK, Christian JC (2000) Analysis ofheritability of hormonal responses to alcohol in twins: beta-endorphin as a potential biomarker of genetic risk for alcoholism.Alcohol Clin Exp Res 24:265–277
Funk CK, Zorrilla EP, Lee MJ, Rice KC, Koob GF (2007)Corticotropin-releasing factor 1 antagonists selectively reduceethanol self-administration in ethanol-dependent rats. Biol Psychiatry61:78–86
Gianoulakis C (1990) Characterization of the effects of acute ethanoladministration on the release of beta-endorphin peptides by therat hypothalamus. Eur J Pharmacol 180:21–29
Gianoulakis C (1996) Implications of endogenous opioids anddopamine in alcoholism: human and basic science studies.Alcohol 1:33–42
Gianoulakis C (2004) Endogenous opioids and addiction to alcoholand other drugs of abuse. Curr Top Med Chem 4:39–50
Gianoulakis C, Dai X, Brown T (2003) Effect of chronic alcoholconsumption on the activity of the hypothalamic-pituitary-adrenal axis and pituitary !-E as a function of alcohol intake,age, and gender. Alcohol Clin Exp Res 27:410–423
Grisel JE, Mogil JS, Grahame NJ, RubinsteinM, Belknap JK, Crabbe JC,Low MJ (1999) Ethanol oral self-administration is increased inmutant mice with decreased !-E expression. Brain Res 835:62–67
Grobin AC, Matthews DB, Devaud LL, Morrow AL (1998) The roleof GABA(A) receptors in the acute and chronic effects ofethanol. Psychopharmacology 139:2–19
Guillemin R, Vargo T, Rossier J, Minick S, Ling N, Rivier C, Vale W,Bloom F (1977) beta-Endorphin and adrenocorticotropin are selectedconcomitantly by the pituitary gland. Science. 197:1367–1369
Hayward MD, Schaich-Borg A, Pintar JE, Low MJ (2006) Differentialinvolvement of endogenous opioids in sucrose consumption andfood reinforcement. Pharmacol Biochem Behav 85:601–611
Hernandez-Avila CA, Oncken C, Van Kirk J, Wand G, Kranzler HR(2002) Adrenocorticotropin and cortisol responses to a naloxonechallenge and risk of alcoholism. Biol Psychiatry 51:652–658
Herz A (1997) Endogenous opioid systems and alcohol addiction.Psychopharmacology 129:99–111
Hill TD, Angel RJ (2005) Neighborhood disorder, psychologicaldistress, and heavy drinking. Soc Sci Med 61:965–975
Hossain SM, Wong BK, Simpson EM (2004) The dark phaseimproves genetic discrimination for some high throughput mousebehavioral phenotyping. Genes Brain Behav 3:167–177
Koven NS, Heller W, Miller GA (2005) The unique relationshipbetween fear of cognitive dyscontrol and self-reports of prob-lematic drinking. Addict Behav 30:489–499
Kushner MG, Sher KJ, Beitman BD (1990) The relation betweenalcohol problems and anxiety disorders. Am J Psychiatry147:685–695
LaBuda CJ, Fuchs PN (2001) The anxiolytic effect of acute ethanol orDiazepam exposure is unaltered in u-opioid receptor knockoutmice. Brain Res Bull 55:755–760
Lawyer SR, Karg RS, Murphy JG, McGlynn FD (2002) Heavydrinking among college students is influenced by anxietysensitivity, gender, and contexts for alcohol use. J AnxietyDisord 16:165–173
Lê AD, Harding S, Juzytsch W, Watchus J, Shalev U, Shaham Y (2000)The role of corticotrophin-releasing factor in stress-inducedrelapse to alcohol-seeking behavior in rats. Psychopharmacology150:317–324
Marinelli PW, Quirion R, Gianoulakis C (2003) A microdialysisprofile of beta-endorphin and catecholamines in the rat nucleusaccumbens following alcohol administration. Psychopharmacol-ogy 169:60–67
Marinelli PW, Quirion R, Gianoulakis C (2004) An in vivo profile ofbeta-endorphin release in the arcuate nucleus and nucleusaccumbens following exposure to stress or alcohol. Neuroscience127:777–784
Marquez P, Baliram R, Dabaja I, Gajawada N, Lufty K (2008) The roleof beta-endorphin in the acute motor stimulatory and rewardingactions of cocaine in mice. Psychopharmacology 197:443–448
Millan MJ (1981) Stress and endogenous opioid peptides: a review.Mod Probl Pharmacopsychiatry 17:49–67
Mogil JS, Grisel JE (1998) Transgenic studies of pain. Pain 77:107–28Mogil JS, Grisel JE, Hayward MD, Bales JR, Rubinstein M, Belknap
JK, Low MJ (2000) Disparate spinal and supraspinal opioidantinociceptive responses in beta-endorphin-deficient mutantmice. Neuroscience 101:709–717
Mollenauer S, Bryson R, Robison M, Sardo J, Coleman C (1993)EtOH self-administration in anticipation of noise stress inC57BL/6J mice. Pharmacol Biochem Behav 46:35–38
Oltras CM, Mora F, Vives F (1987) Beta-endorphin and ACTH inplasma: effects of physical and psychological stress. Life Sci40:1683–1686
Olive MF, Koenig HN, Nannini MA, Hedge CW (2001) Stimulationof endorphin neutrotransmission in the nucleus accumbens byethanol, cocaine, and amphetamine. J Neurosci L21:RC184
Oswald LM, Wand GS (2004) Opioids and alcoholism. Physiol Behav81:339–358
Pacak K, Palkovits M (2001) Stressor specificity of central neuroen-docrine responses: Implications for stress-related disorders.Endocrine Reviews 22:502–548
Panksepp J (2003) Feeling the pain of social loss. Science 302:237–239Pfaff DW, Phillips MI, Rubin RT (2004) Principles of Hormone/
Behavior Relations. Elsevier, Amsterdam, pp 55–56Plotsky PM (1991) Pathways to the secretion of adrenocorticotropin: a
review from the portal. J Endocrinol 3:1–18Pohorecky LA (1991) Stress and alcohol interaction: an update of
human research. Alcohol Clin Exp Res 15:438–459Poplawski MM, Boyadjieva N, Sarkar DK (2005) Vasoactive
intestinal peptide and corticotropin-releasing hormone increasebeta-endorphin release and proopiomelanocortin messenger RNA
114 Psychopharmacology (2008) 200:105–115

levels in primary cultures of hypothalamic cells: effects of acuteand chronic ethanol treatment. Alcohol Clin Exp Res 29:648–655
Redei E, Branch BJ, Gholami S, Lin EY, Taylor AN (1988) Effectsof ethanol on CRF release in vitro. Endocrinology 123:2736–2743
Ribeiro SC, Kennedy SE, Smith YR, Stohler CS, Zubieta JK (2005)Interface of physical and emotional stress regulation through theendogenous opioid system and mu-opioid receptors. Prog Neuro-psychopharmacol Biol Psychiatry 29:1264–1280
Rivier C (1996) Alcohol stimulates ACTH secretion in the rat:mechanisms of action and interactions with other stimuli.Alcohol Clin Exp Res 20:240–254
Rubinstein M, Mogil JS, Japón M, Chan EC, Allen RG, Low MJ(1996) Absence of opioid stress-induced analgesia in micelacking beta-endorphin by site-directed mutagenesis. Proc NatlAcad Sci U S A 93:3995–4000
Sarkar DK, Kuhn P, Marano J, Chen C, Boyadjieva N (2007) Alcoholexposure during the developmental period induces beta-endor-phin neuronal death and causes alteration in the opioid control ofstress axis function. Endocrinology 148:2828–2834
Scanlon MN, Lazar WE, Grant KA, Kunos G (1992) Proopiomela-nocortin messenger RNA is decreased in medio-basal hypothal-amus of rats made dependent upon ethanol. Alc Clin Exp Res16:1147–1151
Schedlowski M, Flüge T, Richter S, Tewes U, Schmidt RE, WagnerTO (1995) Beta-endorphin, but not substance-P, is increased byacute stress in humans. Psychoneuroendocrinology 20:103–110
Schulz R, Wuster M, Duka T, Herz A (1980) Acute and chronicethanol treatment changes endorphin level in brain and pituitary.Psychopharmacology 68:221–227
Sher L (2003) Alcoholism, anxiety, and opioid-dopaminergic inter-actions. Psychopharmacology 165:202–203
Thiagarajan AB, Mefford IN, Eskay RL (1988) Acute effect ofintragastric ethanol administration on plasma levels of stresshormones. Adv Alcohol Subst Abuse 7:227–130
Thiagarajan AB, Mefford IN, Eskay RL (1989) Single-dose ethanoladministration activates the hypothalamic-pituitary-adrenal axis:exploration of the mechanism of action. Neuroendocrinology50:427–432
Valdez GR, Sabino V, Koob GF (2004) Increased anxiety-likebehavior and ethanol self-adminstration in dependent rats:reversal via corticotrophin-releasing factor-2 receptor activation.Alcohol Clin Exp Res 28:865–872
Vendruscolo LF, Terenina-Rigaldie E, Raba F, Ramos A, TakahashiRN, Mormede P (2006) Evidence for a female-specific effect of achromosome 4 locus on anxiety-related behaviors and ethanoldrinking in rats. Genes Brain Behav 5:441–450
Wand GS, Mangold D, El Deiry S, McCaul ME, Hoover D (1998)Family history of alcoholism and hypothalamic opioidergicactivity. Arch Gen Psychiatry 55:1114–1119
Williams SB, Holloway A, Karwan K, Allen SA, Grisel JE (2007)Oral self-administration of Ethanol in transgenic mice lacking b-endorphin. Published online, Impulse. Accessed at http://impulse.schc.sc.edu/articles/2007-9-14-Williams.pdf
Wolffgramm J (1990) Free choice ethanol intake of laboratory rats underdifferent social conditions. Psychopharmacology 101:233–239
Yamada K, Nabeshima T (1995) Stress-induced behavioral responses andmultiple opioid systems in the brain. Behav Brain Res 67:133–145
Zalenska-Kaszubska J, Czarnecka E (2005) Deficit in beta-endorphinpeptide and tendency to alcohol abuse. Peptides 26:701–705
Zimmerman P, Wittchen H–U, Hofler M, Pfister H, Kessler RC, LiebR (2003) Primary anxiety disorders and the development ofsubsequent alcohol use disorders: a 4-year community study ofadolescents and young adults. Psychol Med 33:1211–1222
Psychopharmacology (2008) 200:105–115 115





























