Embed Size (px)
Citation preview

www.elsevier.com/locate/ijpara
International Journal for Parasitology 37 (2007) 605–615
Identification of basic transcriptional elements required for rifgene transcription
Wai-Hong Tham *, Paul D. Payne, Graham V. Brown, Stephen J. Rogerson
Department of Medicine, University of Melbourne, Royal Melbourne Hospital, Parkville, Victoria 3050, Australia
Received 9 October 2006; received in revised form 16 November 2006; accepted 19 November 2006
Abstract
The rif gene family is the largest multi-gene family in the malaria parasite Plasmodium falciparum. The gene products of rif genes,rifins, are clonally variant and transported to the surface of the infected erythrocyte where they are targets of the human immuneresponse. Maximal rif transcription occurs during the late ring to early trophozoite stages of the intra-erythrocytic cycle. The factorsinvolved in the transcriptional activation and repression of rif genes are not known. In this paper, we characterize several DNA elementsinvolved in the regulation of rif transcription. We identify the upstream region that contains a functional promoter and the transcrip-tional start site of a rif gene. In addition, we identify two distinct regions within the rif upstream region involved in the transcriptionalrepression of these genes. These repressor sites are bound by nuclear protein factors expressed in different stages of the Plasmodium lifecycle. We propose that the differential timing of binding provides a mechanism for the temporal repression of rif genes. In addition, wefind that transcription profiles of upsA var genes and their neighbouring rif genes are unlinked.� 2006 Australian Society for Parasitology Inc. Published by Elsevier Ltd. All rights reserved.
Keywords: rif; var; Multi-gene families; Transcription regulation; Repressor; Plasmodium falciparum
1. Introduction
Mortality and disease symptoms associated with Plas-
modium falciparum malaria infections occur exclusivelyduring the blood stage of infection. As the parasites matureto early pigmented trophozoites within the infected eryth-rocyte, parasite-derived molecules are expressed on theerythrocyte surface. These surface antigens are recognizedby the host immune system and are implicated in host-pro-tective immunity (Bull et al., 1998). However, by switchingthe proteins expressed, the parasite has the ability to gener-ate novel antigenic properties on the surface of infectederythrocytes in response to pressure from the humanimmune system, thus providing a mechanism for immuneevasion.
0020-7519/$30.00 � 2006 Australian Society for Parasitology Inc. Published b
doi:10.1016/j.ijpara.2006.11.006
* Corresponding author. Present address: Walter and Eliza HallInstitute of Medical Research, 1G Royal Parade, Parkville, Victoria3050, Australia. Tel.: +61 3 9345 2456; fax: +61 3 9347 0852.
E-mail address: [email protected] (W.-H. Tham).
With 149 members, the rif gene family represents thelargest multi-copy gene family in P. falciparum whose pro-tein products, rifins, are located at the Maurer’s clefts andindirect evidence also supports their expression at theinfected erythrocyte surface (Fernandez et al., 1999; Kyeset al., 1999; Marti et al., 2004; Khattab and Klinkert,2006). Several studies have shown that rifins are immuno-genic in natural infections and are recognized by humanimmune sera (Fernandez et al., 1999; Abdel-Latif et al.,2002, 2003, 2004). Although the functional role of rifinson the surface of the infected erythrocyte remains specula-tive, it is possible these proteins play an accessory role inrosette formation or cytoadherence (Kyes et al., 1999).
There is compelling evidence that rifins are clonally var-iant, as different subsets of rifins are expressed on the eryth-rocyte surface by sibling clones derived from an isogenicbackground (Fernandez et al., 1999; Kyes et al., 1999).The heterogeneity of rifins on the erythrocyte surfacemay arise from post-translational modification of rifinsdestined for the surface or from a selective repression of
y Elsevier Ltd. All rights reserved.

606 W.-H. Tham et al. / International Journal for Parasitology 37 (2007) 605–615
transcription of individual rif genes. By analogy to the mul-ti-copy var gene family whose protein products are trans-ported to the erythrocyte surface, clonal variation couldbe controlled on a gene transcriptional level. Regulationof var gene transcription results in the expression of onefull-length transcript from a single var gene and the tran-scriptional repression of all other var genes. The parasiteswitches expression to a different member through in situactivation of a transcriptionally silent var gene (Scherfet al., 1998).
Very little is known about the mechanisms that regulaterif transcription. Northern blot analysis using a complex rif
probe shows that maximal rif gene transcription occursduring the intra-erythrocytic cycle corresponding with thelate ring to early pigmented trophozoite stage in the para-site clone Palo Alto (Kyes et al., 1999). Mechanisms musttherefore be in place for the regulation of rif expression.
To understand rif transcriptional regulation, we wereinterested in identifying the basic DNA sequence elementsinvolved in rif gene expression using the genome referenceclone 3D7. We focused our attention on a subset of eightrif genes that share a common upstream region with avar gene as they are orientated in a head-to-head fashion.Var genes are categorized into five different groups (upsA
to upsE) from the alignment of their upstream regionsand only upsA var genes are in a head-to-head orientationwith a neighbouring rif gene (Lavstsen et al., 2003). UpsA
var genes and their associated rif genes are located primar-ily in the subtelomeric regions of P. falciparum. Identifica-tion of regulatory elements within the shared upstreamregion would shed light on the elements involved in the reg-ulation of rif and var gene transcription.
In this paper, we identify the transcription start site of arif gene and show that the region also possesses functionalpromoter activity. We believe we provide the first charac-terization of two distinct DNA sequence elements involvedin the transcriptional repression of a rif gene. As expected,mutation of one of the repressor elements and the completeremoval of the other lead to a decrease in repression. Wealso show that these repressor elements bind nuclear pro-teins expressed in different stages of the Plasmodium lifecycle. The differential timing of binding of the repressor ele-ments leads us to propose a model where one repressor ele-ment is involved in the selective repression of rif genes earlyin the life cycle and another separate repressor element isresponsible for the repression of rif genes in the later stagesof the life cycle. We also provide evidence that the tran-scription profile of upsA var and their neighbouring rif
genes are not linked in unselected lines or in 3D7sir2D.
2. Materials and methods
2.1. Transcription analysis of rif genes and rapid
amplification of cDNA ends (5 0RACE)
The 3D7 strain was cultured and synchronized using stan-dard methods (Lambros and Vanderberg, 1979; Trager and
Jenson, 1978). Parasites were harvested at 0, 6, 12, 18, 24, 30,32, 34, 36 and 40 h post invasion and total RNA and genomicDNA were extracted using the Trizol method (Kyes et al.,2000). RNA samples were treated with DNaseI accordingto the manufacturer’s instructions (GIBCO/BRL). cDNAwas primed using random hexamers and the first strandcDNA synthesis kit from Invitrogen. To amplify genomicand cDNA transcript, two rounds of PCR were performed.The first PCR cycle used primers RINT1.5 (ATCCATTATACTAATATATTATTGTTTCCT) and 0009.3 (ACCTTACCTGCACTACCTAATGT) and the second nestedPCR cycle was performed using RINT2.5 (TATATTATTGTTTCCTCTAAAATTAAATAT) and 0009.3.
5 0RACE was carried out using the 5 0RACE system forrapid amplification of cDNA Ends, Version 2.0 kit fromInvitrogen. Parasites were harvested at 18–24 h post inva-sion to coincide with the period of rif gene transcriptionand total RNA was extracted using the Trizol method(Kyes et al., 2000). All amplified products were cloned intoa pGEM-T vector (Promega) and sequenced.
2.2. Vector construction for firefly luciferase reporters
Upstream regions of rif genes were amplified from the3D7 genome using a set of primers specific to the rif genePF11_0009. The primers contain an introduced HindIII sitein the 5 0 end and BamHI site in the 3 0 end. Primers 393F(CCCAAGCTTTAGAAGTTCTTACTAAATCGTAACGTA), 562F (CCCAAGCTTATAACAAATATTATGATATGCAATGA), 700F (CCCAAGCTTAGATACATTAGAGAGAAAGGTATAACAT), and 870F (CCCAAGCTTCGCACAACACTACATAAAACTATACAATAGT) were used as the 5 0 end primers together with 3R(CGCGGATCCTTATTGTGATACGTATATTATTTTATGATA; restriction sites are italized) as the 3 0 end primer toclone pRIF393, pRIF562, pRIF700 and pRIF870, respec-tively. Amplified products were cloned into a pGEM-T vec-tor (Promega) and sequenced. In all cases, the sequenceswere 99% identical to PF11_0009. These sequences were sub-sequently cloned in the BamHI–HindIII site of pHHI-pac/luc plasmid (a kind gift from Brendan Crabb, The Walterand Eliza Hall Institute of Medical Research), thereby fusingthe rif upstream region to the Firefly luciferase gene.
To clone pRIF870mut, we amplified the region spanning�3 to �870 from 3D7 genomic DNA using the primers870mutF(GCGCTCGAGAAAACAACAAAACATAAAACTATACAATAGT) and 54.3 (GCGCCATGGTTATTGTGATACGTATATTATTTTA TGATA; XhoI and NcoIsites are in italics). This fragment was inserted into a PGEM-T vector to create 870mutPGEM-T. The mutated rif
upstream region was excised from 870mutPGEM-T andinserted into the XhoI and NcoI sites of Pf86 (Militello andWirth, 2003) to create pRIF870mut, thereby fusing themutated upstream region to the Firefly luciferase gene. Toclone pRIF870wt, we amplified the region spanning �3 to�870 from 3D7 genomic DNA using the primers 870wt.5(GCGCTCGAGCGCACAACACTACATAAAACTATAC

W.-H. Tham et al. / International Journal for Parasitology 37 (2007) 605–615 607
AATAGT) and 54.3. We followed the same cloning proce-dure as above to create pRIF870wt. Both pRIF870wt andpRIF870mut were sequenced to ensure that no DNA modi-fications were introduced during the cloning procedure.
To clone pRIF562mut and pRIF486, primers 562mutF(CCCAAGCTTATAACAAATATTATGATAAAAAATGA) and 486F (CCCAAGCTTACTAATAGGTTTTTCCAAATTGT) were used, together with 3R to amplify the relevantupstream regions. Amplified products were cloned into apGEM-T vector (Promega) and sequenced. These sequenceswere subsequently cloned in the BamHI–HindIII site ofpHHI-pac/luc plasmid, thereby fusing the rif upstreamregion to the Firefly luciferase gene.
2.3. Transient transfection
To control for transfection efficiency, we used plasmidpPfrluc which has the 5 0 untranslated region (UTR) ofthe calmodulin gene fused to the Renilla luciferasereporter (a kind gift from Kevin Militello; Militello andWirth, 2003). Ring stage parasites were transfected with100 lg of rif transfection constructs and 75 lg of pPfr-Luc at 0.310 kV and 950 lF using a Biorad Gene PulserII.
2.4. Luciferase assays
Parasites were harvested at the early trophozoitestage in the following cell cycle post-transfection andlysates were extracted as described in the manufacturer’sinstructions for the DLR Assay Kit (Promega). Twentymicroliters of lysate was mixed with 100 ll of LARIIreagent to activate Firefly luminescence and 100 ll ofStop and Glo reagent to activate Renilla luminescence.Luminescence measurements were done on an EG&GBerthold Lumat LB9507. For each transfection con-struct, transfections were repeated three times and lumi-nescence measurements were an average value obtainedfrom three independent readings. To control for back-ground luminescence, a no-DNA control was includedin every experiment and its luminescence measurementswere subtracted from the measurements obtained forevery transfection. The ratio of Firefly to Renilla lumi-nescence was calculated for each transfection and thehighest ratio in each experiment was normalized to100%.
2.5. Electrophoretic mobility shift assays (EMSA)
Electrophoretic mobility shift assays (EMSAs) were per-formed as described in Voss et al. (2003). For all competi-tion experiments, unlabeled double-stranded oligos wereincubated with the nuclear proteins for 10 min at roomtemperature prior to the addition of a radiolabeled probe.Binding reactions were loaded under current onto 4% or6% native polyacrylamide gels, dried and exposed over-night onto Kodak BioMax Film.
2.6. Quantitative real-time PCR
RNA was harvested from each parasite line at earlyrings and at late rings/early trophozoites using the Trizolmethod (Kyes et al., 2000) and treated with DNaseI (GIB-CO/BRL) as described in the manufacturer’s instructions.3D7sir2D was a gift from Dr. Alan Cowman (The Walterand Eliza Hall Institute of Medical Research). Reversetranscription was carried out using Superscript III and ran-dom hexamers (Invitrogen) and the cDNA was diluted andstored as single use aliquots in �80 �C.
Primer sets 7, 8, 20, 25, 35, 96 and 97 as published inSalanti et al. (2003) were used to amplify the upsA var genesin head-to-head orientation with a rif gene. Primer sets 60and 61 from Salanti et al. (2003) were also used to amplifytwo housekeeping genes; seryl-tRNA synthetase and fruc-tose-biphosphate aldolase. In addition, we designed aunique primer pair to PFF0020c. Primer pairs to the neigh-bouring rif genes were designed using Primer Express soft-ware (Applied Biosystems) and all primers were made byGeneworks (Australia).
Each PCR reaction used 5 ll of SYBR Green PCR mas-ter Mix (PE Biosystems) in a 10 ll reaction that was ampli-fied using the ABI Prism 7900HT Sequence DetectionSystem. The PCR cycling conditions were 95 �C for10 min, then 40 cycles of 94 �C for 30 s, 54 �C for 40 sand 68 �C for 50 s with a 10 min extension at 68 �C fol-lowed by a dissociation step. The specificity of each primerpair was determined by dissociation curve analysis and gelelectrophoresis. Absolute quantitation was performedusing in vitro transcribed RNA standards that were diluted10-fold seven times to generate an eight point standardcurve to enable quantitation of 1011–104 molecules ofcDNA. Absolute quantification was performed by plottingof threshold cycle values of the unknown samples ontostandard curves of quantity plotted against Ct. The quan-tity of seryl-tRNA synthetase was used to normalize eachvar and rif transcript.
3. Results
3.1. Stages of rif gene transcription and identification of the
transcriptional start site of a rif gene
To determine when the upsA var-associated rif genes aretranscribed in 3D7, we harvested RNA and genomic DNAfrom synchronized parasites at 0, 6, 12, 18, 24, 30, 32, 34,36 and 40 h post invasion. To amplify the cDNA tran-scripts, we designed gene-specific primers to all eight rif
genes neighbouring an upsA var gene. These primer pairsspan the rif intron, allowing us to differentiate betweengenomic DNA versus cDNA amplification. All gene-specif-ic primers could amplify the predicted products using geno-mic DNA as a template (data not shown). However, weonly managed to amplify cDNA transcript (predicted sizeof 594 bp) for rif gene PF11_0009 (Fig. 1A). Maximal genetranscription for this gene occurred between 18 and 24 h

Fig. 1. The stages of rif gene transcription and transcription start site of a rif gene identified. (A) Using RNA harvested at 0, 6, 12, 18, 24, 30, 32, 34, 36and 40 h post invasion, amplification of cDNA transcript show that maximal rif transcription occurs at 18 and 24 h post invasion. Genomic DNAamplification is shown in lane g. A 100 bp ladder (New England Biolabs) was loaded in the first and last lane of the gel. (B) Gel analysis of 5 0RACEproducts obtained from the first and second PCR amplification. The + and � symbols refer to reverse transcription reactions which did and did notcontain Superscript II, respectively. A 100 bp ladder (New England Biolabs) was loaded and the 400 bp fragment is marked with an asterisk. (C) A clonedupstream region of a 5 0RACE product is shown. The three gene-specific primers are shown as GSP1, GSP2 and GSP3. First initiation codon and splicejunction are shown in bold as ATG and GG, respectively. Arrows mark the three different transcriptional start sites identified from the 5 0RACE products.
608 W.-H. Tham et al. / International Journal for Parasitology 37 (2007) 605–615
post invasion, which corresponds with the late ring to earlytrophozoite stage (lanes 18 and 24, Fig. 1A). From 30 honwards we observed a band that corresponds to genomicDNA amplification (predicted size of 748 bp, lane g,Fig. 1A) which is a likely consequence of incomplete DNa-seI digestion.
To identify the transcriptional start site of a rif gene, weused a 5 0RACE technique. Reverse transcription of the rif
gene was performed using a gene-specific primer (GSP1)located downstream of the rif intron (Fig. 1C). The gene-specific primers were designed based on sequence align-ments of the upstream region of eight rif genes that arein a head-to-head orientation with an upsA var gene butthe final sequence for the primers was based on the rif genePF11_0009. In order to visualize the cDNA transcripts, thereverse transcription products were subjected to two ampli-fication cycles of PCR using nested gene-specific primers(GSP2 and 3) located downstream of a rif intron in combi-nation with anchored primers (Fig. 1C). We used gene-spe-cific primers located downstream of the rif intron to allowidentification of properly spliced cDNA transcripts uponsequencing of the 5 0RACE products. The first PCR cycleyielded no amplified product whereas the second PCR cycleyielded amplified product only in reactions that containedSuperscript II (Invitrogen) during the reverse transcriptionreaction (Fig. 1B).
Eleven genuine spliced cDNA transcripts were identifiedupon sequencing and found to be co-linear with the 3D7
genomic sequence but heterogeneous in their 5 0 ends. ThesecDNA transcripts were >98% identical to the following rifgenes; PFD0050w, PFD0025w, PFI0065w, PFI0020w,PF11_0009, PF11_0520, PF13_0004 and PFA0020w. Threedifferent transcriptional start sites were found clusteredwithin a 47 nucleotide region (Fig. 1C, arrows). The pres-ence of three different transcriptional start sites may be aresult of reverse transcriptase pausing due to the AT rich-ness of the upstream region, rather than the existence ofmultiple transcriptional start sites. Of the 11 RACE prod-ucts analyzed, five indicated initiation at position �245 andwe propose this site to be the major transcription start site.
3.2. The rif upstream region possesses promoter andtranscriptional repression activities
To assess whether the upstream regions of rif genespossess functional promoter and repressor activity, weused transient transfection luciferase assays. Episomallymaintained plasmids require passage through S-phasefor the proper assembly of silent chromatin so parasiteswere harvested in the trophozoite stage in the followingcell cycle post-transfection in all of our experiments (Dei-tsch et al., 2001). Transient luciferase assays were used toexamine var intron silencing and therefore, we believe thistechnique would also facilitate the identification of repres-sor activity of rif genes (Calderwood et al., 2003; Deitschet al., 2001).
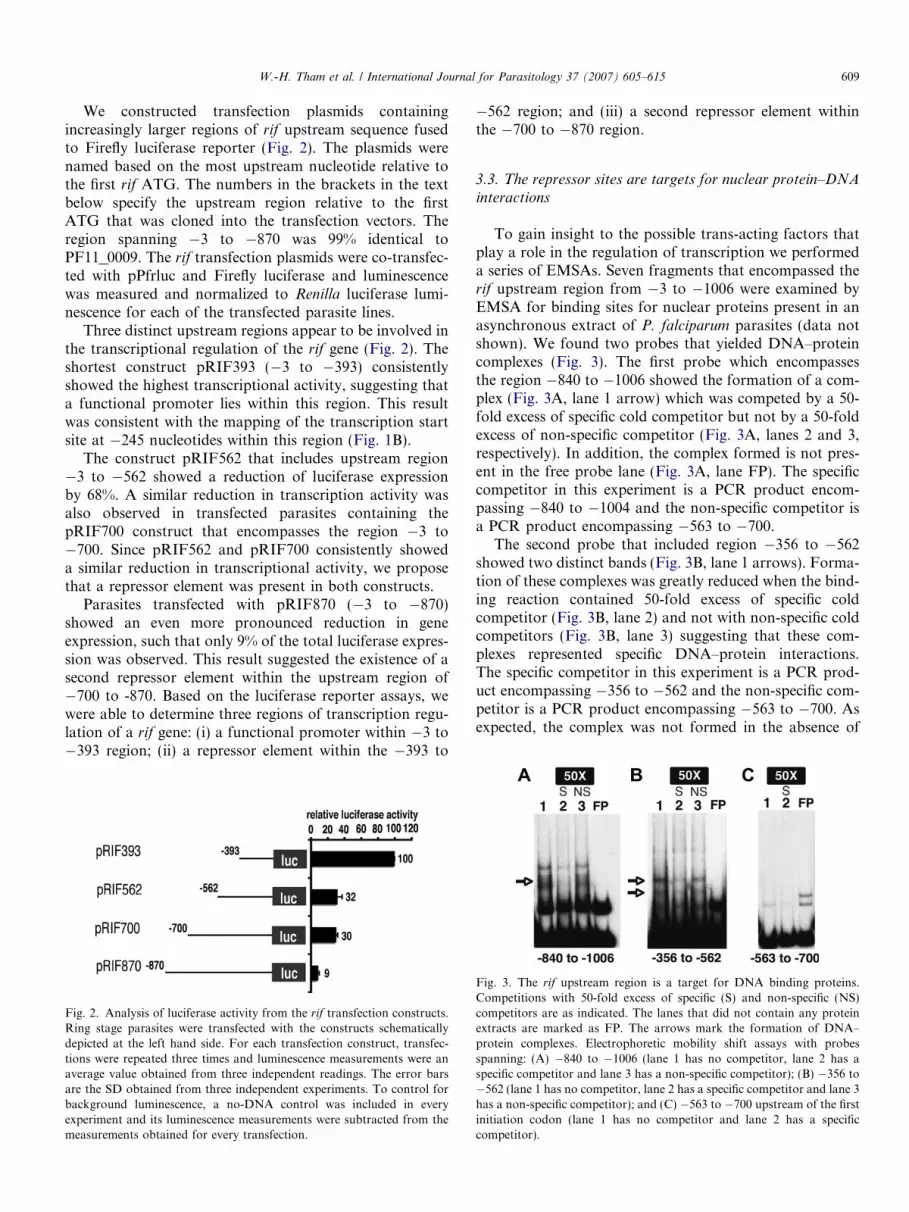
W.-H. Tham et al. / International Journal for Parasitology 37 (2007) 605–615 609
We constructed transfection plasmids containingincreasingly larger regions of rif upstream sequence fusedto Firefly luciferase reporter (Fig. 2). The plasmids werenamed based on the most upstream nucleotide relative tothe first rif ATG. The numbers in the brackets in the textbelow specify the upstream region relative to the firstATG that was cloned into the transfection vectors. Theregion spanning �3 to �870 was 99% identical toPF11_0009. The rif transfection plasmids were co-transfec-ted with pPfrluc and Firefly luciferase and luminescencewas measured and normalized to Renilla luciferase lumi-nescence for each of the transfected parasite lines.
Three distinct upstream regions appear to be involved inthe transcriptional regulation of the rif gene (Fig. 2). Theshortest construct pRIF393 (�3 to �393) consistentlyshowed the highest transcriptional activity, suggesting thata functional promoter lies within this region. This resultwas consistent with the mapping of the transcription startsite at �245 nucleotides within this region (Fig. 1B).
The construct pRIF562 that includes upstream region�3 to �562 showed a reduction of luciferase expressionby 68%. A similar reduction in transcription activity wasalso observed in transfected parasites containing thepRIF700 construct that encompasses the region �3 to�700. Since pRIF562 and pRIF700 consistently showeda similar reduction in transcriptional activity, we proposethat a repressor element was present in both constructs.
Parasites transfected with pRIF870 (�3 to �870)showed an even more pronounced reduction in geneexpression, such that only 9% of the total luciferase expres-sion was observed. This result suggested the existence of asecond repressor element within the upstream region of�700 to -870. Based on the luciferase reporter assays, wewere able to determine three regions of transcription regu-lation of a rif gene: (i) a functional promoter within �3 to�393 region; (ii) a repressor element within the �393 to
Fig. 2. Analysis of luciferase activity from the rif transfection constructs.Ring stage parasites were transfected with the constructs schematicallydepicted at the left hand side. For each transfection construct, transfec-tions were repeated three times and luminescence measurements were anaverage value obtained from three independent readings. The error barsare the SD obtained from three independent experiments. To control forbackground luminescence, a no-DNA control was included in everyexperiment and its luminescence measurements were subtracted from themeasurements obtained for every transfection.
�562 region; and (iii) a second repressor element withinthe �700 to �870 region.
3.3. The repressor sites are targets for nuclear protein–DNAinteractions
To gain insight to the possible trans-acting factors thatplay a role in the regulation of transcription we performeda series of EMSAs. Seven fragments that encompassed therif upstream region from �3 to �1006 were examined byEMSA for binding sites for nuclear proteins present in anasynchronous extract of P. falciparum parasites (data notshown). We found two probes that yielded DNA–proteincomplexes (Fig. 3). The first probe which encompassesthe region �840 to �1006 showed the formation of a com-plex (Fig. 3A, lane 1 arrow) which was competed by a 50-fold excess of specific cold competitor but not by a 50-foldexcess of non-specific competitor (Fig. 3A, lanes 2 and 3,respectively). In addition, the complex formed is not pres-ent in the free probe lane (Fig. 3A, lane FP). The specificcompetitor in this experiment is a PCR product encom-passing �840 to �1004 and the non-specific competitor isa PCR product encompassing �563 to �700.
The second probe that included region �356 to �562showed two distinct bands (Fig. 3B, lane 1 arrows). Forma-tion of these complexes was greatly reduced when the bind-ing reaction contained 50-fold excess of specific coldcompetitor (Fig. 3B, lane 2) and not with non-specific coldcompetitors (Fig. 3B, lane 3) suggesting that these com-plexes represented specific DNA–protein interactions.The specific competitor in this experiment is a PCR prod-uct encompassing �356 to �562 and the non-specific com-petitor is a PCR product encompassing �563 to �700. Asexpected, the complex was not formed in the absence of
Fig. 3. The rif upstream region is a target for DNA binding proteins.Competitions with 50-fold excess of specific (S) and non-specific (NS)competitors are as indicated. The lanes that did not contain any proteinextracts are marked as FP. The arrows mark the formation of DNA–protein complexes. Electrophoretic mobility shift assays with probesspanning: (A) �840 to �1006 (lane 1 has no competitor, lane 2 has aspecific competitor and lane 3 has a non-specific competitor); (B) �356 to�562 (lane 1 has no competitor, lane 2 has a specific competitor and lane 3has a non-specific competitor); and (C) �563 to �700 upstream of the firstinitiation codon (lane 1 has no competitor and lane 2 has a specificcompetitor).

610 W.-H. Tham et al. / International Journal for Parasitology 37 (2007) 605–615
protein extract (Fig. 3B, lane FP). Fig. 3C shows an exam-ple of a region that showed no protein binding.
Interestingly, the sites of protein binding (�840 to�1006 and �356 to �562) were located within upstreamregions that were identified to contain repressor elementsfrom the luciferase reporter assays (Fig. 2, pRIF870 andpRIF562) and therefore we propose that these protein-binding sites function as transcriptional repressionelements.
3.4. The repressor site at �840 to �1004 is mapped to a 10
nucleotide GC rich sequence
To characterize the exact region of binding, we intro-duced shorter oligonucleotides that span the entire regionof the bound fragment. For the �840 to �1004 fragment,four double-stranded competitor probes labeled 8A–8Dwere incubated with the binding reaction to determinewhich of these probes could compete specifically(Fig. 4A). Lanes 1 and 10 in Fig. 4A (arrow) show the pro-tein complexes formed on the �840 to �1004 fragmentwithout the addition of any competitor. As expected, com-plex formation did not occur without the addition of pro-tein extract (lane FP). For the specific competitionexperiments, only competitor 8C and 8D could reducethe level of protein binding to the �840 to �1004 fragmentat 20-fold excess (Fig. 4A, lanes 7 and 9, respectively). The8C probe has sequence overlap with the 8D probe, suggest-
Fig. 4. The 10 bp GC rich sequence CGCACAACAC is responsible for protelabeled as FP. The arrows mark the formation of DNA–protein complexes. Unlcompetitor. (A) The binding to �840 to �1006 is isolated to a 31 bp site. Lanesstranded competitor. Lanes 2, 4, 6 and 8 are competitions performed withcompetitions performed with the indicated competitors at 20-fold excess. ComOverlap between competitor 8C and 8D is underlined. (B) A 10 bp GC richradiolabeled probe is shown above the gel with the competitor fragments markereactions in lanes 2–6 contain competitor 8.1–8.5, respectively. (C) Strict sequeare shown below the figure. Lane 1 is a binding reaction that contains no coBinding reactions in lanes 3–6 contain competitor 8.3mut1 to 8.3mut4, respec
ing that the region of binding might be present within thisoverlap (Fig. 4A, underlined).
To test this hypothesis, we radiolabeled the 49 nucle-otide region that encompassed probes 8C and 8D, incu-bated it with protein extract and competed the bindingwith smaller double-stranded competitors within thatregion, labeled 8.1–8.5 (Fig. 4B). Lane 1 in Fig. 4B(arrow) shows the formation of a DNA–protein complexin the absence of a competitor and lane FP did not con-tain any protein extract and therefore shows the absenceof this complex (Fig. 4B, lane FP). At 50-fold excess, theonly competitor that managed to compete for bindingwas competitor 8.3 (Fig. 4B, lane 4). This competitorcontained a 10 bp GC rich sequence CGCACAACAC,which was the only sequence that overlapped the 8Cand 8D sequence.
To determine the sequence requirements for the binding,we repeated the competition experiments with mutated ver-sions of oligonucleotide 8.3 that introduced three adeninesconsecutively (Fig. 4C). These mutated oligos were named8.3mut1 to 8.3mut4. Lane 1 in Fig. 4C shows the formationof the complex when no competitor is added. However,when the EMSAs were repeated with these mutated oligos,none of them could successfully compete for binding, evenat 50-fold excess (Fig. 4C, lanes 3–6) although 50-foldexcess of wild-type 8.3 oligonucleotide could compete spe-cifically (Fig. 4C, lane 2). This result suggested that bindingsite CGCACAACAC has relatively stringent nucleotide
in binding at �840 to �1006. The lanes without any protein extracts areess otherwise noted, all competitions were performed with 50-fold excess of1 and 10 are binding reactions that contain no competitor except for single-the indicated competitors at 10-fold excess. Lanes 3, 5, 7 and 9 werepetitors 8A–8D encompass the whole region between �840 and �1006.
sequence is the site of protein binding. The entire sequence used for thed. Lanes 1 and 7 are binding reactions that contain no competitor. Bindingnce requirements for the 10 bp binding site. The mutated competitors usedmpetitor. Lane 2 contains specific competitor 8.3 in the binding reaction.tively.
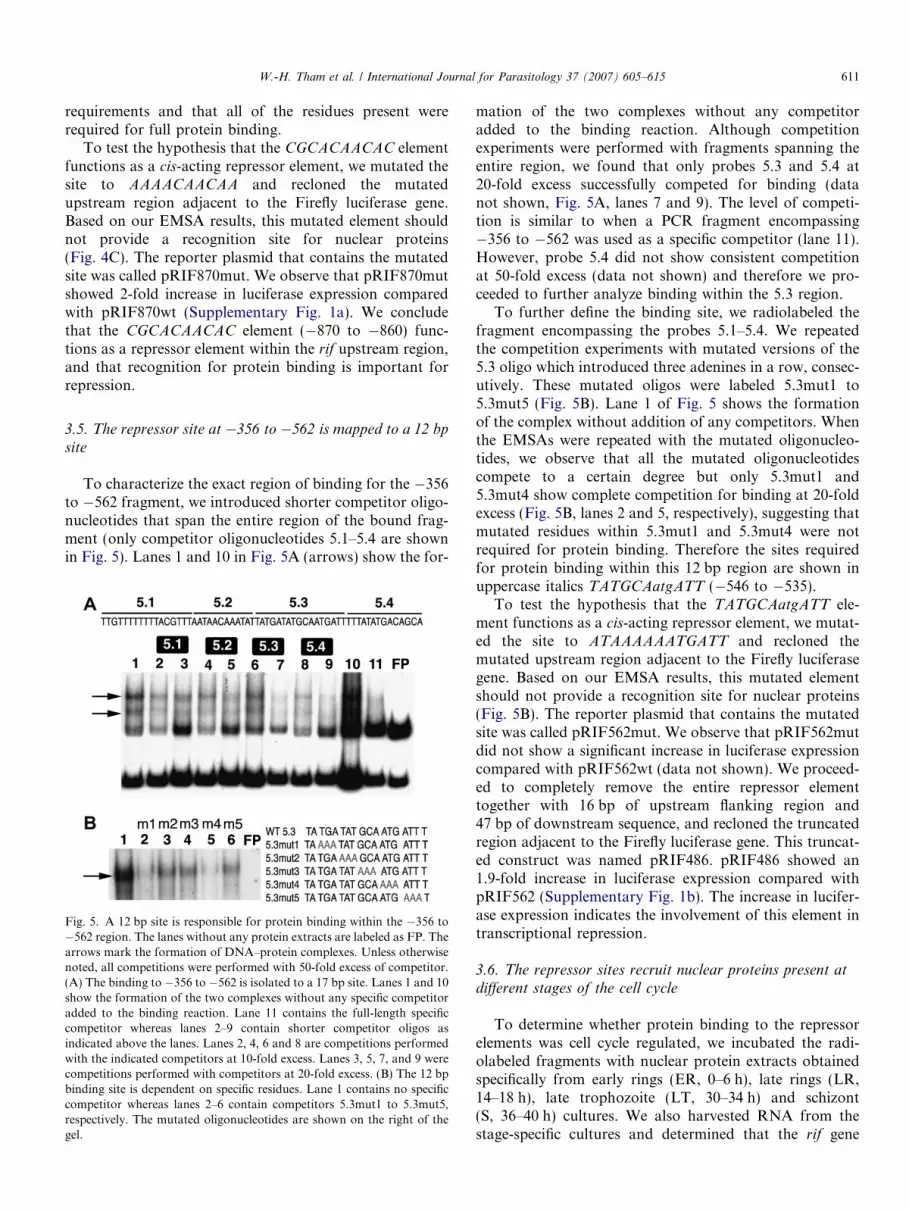
W.-H. Tham et al. / International Journal for Parasitology 37 (2007) 605–615 611
requirements and that all of the residues present wererequired for full protein binding.
To test the hypothesis that the CGCACAACAC elementfunctions as a cis-acting repressor element, we mutated thesite to AAAACAACAA and recloned the mutatedupstream region adjacent to the Firefly luciferase gene.Based on our EMSA results, this mutated element shouldnot provide a recognition site for nuclear proteins(Fig. 4C). The reporter plasmid that contains the mutatedsite was called pRIF870mut. We observe that pRIF870mutshowed 2-fold increase in luciferase expression comparedwith pRIF870wt (Supplementary Fig. 1a). We concludethat the CGCACAACAC element (�870 to �860) func-tions as a repressor element within the rif upstream region,and that recognition for protein binding is important forrepression.
3.5. The repressor site at �356 to �562 is mapped to a 12 bp
site
To characterize the exact region of binding for the �356to �562 fragment, we introduced shorter competitor oligo-nucleotides that span the entire region of the bound frag-ment (only competitor oligonucleotides 5.1–5.4 are shownin Fig. 5). Lanes 1 and 10 in Fig. 5A (arrows) show the for-
Fig. 5. A 12 bp site is responsible for protein binding within the �356 to�562 region. The lanes without any protein extracts are labeled as FP. Thearrows mark the formation of DNA–protein complexes. Unless otherwisenoted, all competitions were performed with 50-fold excess of competitor.(A) The binding to �356 to �562 is isolated to a 17 bp site. Lanes 1 and 10show the formation of the two complexes without any specific competitoradded to the binding reaction. Lane 11 contains the full-length specificcompetitor whereas lanes 2–9 contain shorter competitor oligos asindicated above the lanes. Lanes 2, 4, 6 and 8 are competitions performedwith the indicated competitors at 10-fold excess. Lanes 3, 5, 7, and 9 werecompetitions performed with competitors at 20-fold excess. (B) The 12 bpbinding site is dependent on specific residues. Lane 1 contains no specificcompetitor whereas lanes 2–6 contain competitors 5.3mut1 to 5.3mut5,respectively. The mutated oligonucleotides are shown on the right of thegel.
mation of the two complexes without any competitoradded to the binding reaction. Although competitionexperiments were performed with fragments spanning theentire region, we found that only probes 5.3 and 5.4 at20-fold excess successfully competed for binding (datanot shown, Fig. 5A, lanes 7 and 9). The level of competi-tion is similar to when a PCR fragment encompassing�356 to �562 was used as a specific competitor (lane 11).However, probe 5.4 did not show consistent competitionat 50-fold excess (data not shown) and therefore we pro-ceeded to further analyze binding within the 5.3 region.
To further define the binding site, we radiolabeled thefragment encompassing the probes 5.1–5.4. We repeatedthe competition experiments with mutated versions of the5.3 oligo which introduced three adenines in a row, consec-utively. These mutated oligos were labeled 5.3mut1 to5.3mut5 (Fig. 5B). Lane 1 of Fig. 5 shows the formationof the complex without addition of any competitors. Whenthe EMSAs were repeated with the mutated oligonucleo-tides, we observe that all the mutated oligonucleotidescompete to a certain degree but only 5.3mut1 and5.3mut4 show complete competition for binding at 20-foldexcess (Fig. 5B, lanes 2 and 5, respectively), suggesting thatmutated residues within 5.3mut1 and 5.3mut4 were notrequired for protein binding. Therefore the sites requiredfor protein binding within this 12 bp region are shown inuppercase italics TATGCAatgATT (�546 to �535).
To test the hypothesis that the TATGCAatgATT ele-ment functions as a cis-acting repressor element, we mutat-ed the site to ATAAAAAATGATT and recloned themutated upstream region adjacent to the Firefly luciferasegene. Based on our EMSA results, this mutated elementshould not provide a recognition site for nuclear proteins(Fig. 5B). The reporter plasmid that contains the mutatedsite was called pRIF562mut. We observe that pRIF562mutdid not show a significant increase in luciferase expressioncompared with pRIF562wt (data not shown). We proceed-ed to completely remove the entire repressor elementtogether with 16 bp of upstream flanking region and47 bp of downstream sequence, and recloned the truncatedregion adjacent to the Firefly luciferase gene. This truncat-ed construct was named pRIF486. pRIF486 showed an1.9-fold increase in luciferase expression compared withpRIF562 (Supplementary Fig. 1b). The increase in lucifer-ase expression indicates the involvement of this element intranscriptional repression.
3.6. The repressor sites recruit nuclear proteins present at
different stages of the cell cycle
To determine whether protein binding to the repressorelements was cell cycle regulated, we incubated the radi-olabeled fragments with nuclear protein extracts obtainedspecifically from early rings (ER, 0–6 h), late rings (LR,14–18 h), late trophozoite (LT, 30–34 h) and schizont(S, 36–40 h) cultures. We also harvested RNA from thestage-specific cultures and determined that the rif gene

612 W.-H. Tham et al. / International Journal for Parasitology 37 (2007) 605–615
PF11_0009 transcription was only present in late ringculture (data not shown). Although in Fig. 1A we showthat PF11_0009 transcription was present in early tro-phozoite stage (lane 24, Fig. 1A), the trophozoite stagefor this experiment consisted mostly of late pigmentedtrophozoites (� past 30 h post invasion) and thereforeshowed no rif transcription.
For the �840 to �1004 fragment, proteins that boundthis fragment were present in the late trophozoite andschizont extracts (Fig. 6A, lanes 4 and 5, respectively).The complex formed with these staged cultures was similarto the complex formed using an asynchronous nuclearextract (Fig. 6A, lane 1). Although we did observe a singlecomplex in the ring extracts (Fig. 6A, lanes 2 and 3), thiscomplex appeared to be non-specific as it could not becompeted by 50-fold excess of a specific competitor(Fig. 6B, lane 2 asterisk). However, the DNA–protein com-plexes found in the late trophozoite extracts were competedby 50-fold excess of a specific competitor (Fig. 6B, lane 4)suggesting the late trophozoite and schizont stage DNA–protein complexes formed were genuine.
Binding of the �356 to �562 fragment occurred only inthe ring stage extracts (Fig. 6C, lanes 1 and 2). Interesting-ly, early ring extracts showed the formation of one complexrather than two complexes previously seen in the asynchro-nous nuclear extracts and with the late ring extracts(Fig. 3B, lane 1; Fig. 6C, lane 2). However, both set ofcomplexes found in early and late rings were genuine asthey were competed using specific competitors (Fig. 6D,lanes 2 and 4, specifically). From these results, we proposethat proteins bind the TATGCAatgATT element early inthe parasite life cycle whereas proteins bind the moreupstream repressor element CGCACAACAC in later stagesof the parasite life cycle.
Fig. 6. Protein binding to the repressor elements is cell cycle regulated. Radextracts obtained from early rings (ER), late rings (LR), late trophozoite (LT)extracts are marked as FP. The arrows mark the formation of DNA–proteicompetitor. (A) Binding to the �840 to �1006 region only occurs in late troextracted from an asynchronous culture. Lanes 2–5 are incubated with proteincultures, respectively. (B) Protein complex formed by late trophozoite extractnuclear proteins obtained from late rings and lanes 3 and 4 are incubated wispecific competitors. The non-specific complex formed by late ring stage extractoccurs in the ring stages. Lanes 1–4 are binding reactions incubated with earrespectively. (D) The ring stage complexes are competed using a specific compeprotein extracts and lanes 3 and 4 are incubated with late ring nuclear protein
3.7. The rif transcription profile is unlinked to the upsA var
transcription profile in unselected lines
To determine if transcription profiles between upsA var
and their neighbouring rif genes were linked, we harvestedRNA from the early ring (for var transcripts) and late ring/early trophozoite stages within the cell cycle (for rif tran-scripts) from three different batches of 3D7 (labeled A–C,Fig. 7). All three lines showed a different transcription pro-file for both upsA var and their neighbouring rif genes. Inline A, the var gene PFA0015c showed the highest levelof transcript but both its neighbouring rif PFA0020w andnon-neighbouring rif PFF0025w showed almost similarlevels of transcript. For the B line, the neigbouring varand rif genes PFA0015c and PFA0020w showed highestlevels of transcripts but so did the non-neighbouring var
PF11_0521. In the C line, the var gene PFA0015c onceagain has the highest level of transcript but its neighbour-ing rif gene PFA0020w showed no increase in transcriptlevel. Therefore, from these analyses we conclude that wecannot observe any evidence for transcriptional co-regula-tion of upsA var and their neighbouring rif genes.
3.8. A subset of upsA related rif genes is regulated by PfSir2
PfSIR2 is a homolog of budding yeast SIR2p that playsa central role in epigenetic gene silencing at telomeres, mat-ing type loci and rDNA locus (Gasser and Cockell, 2001).Disruption of PfSir2 leads to a dramatic up-regulation ofupsA var gene transcription as shown by microarray anal-yses (Duraisingh et al., 2005). To determine whether ornot PfSir2 also played a role in the regulation of the neigh-bouring rif genes, we harvested RNA from synchronizedparasites from wild-type and 3D7sir2D strains at both early
iolabeled probes as indicated below the gels were incubated with protein, schizont (S) and asynchronous (AS) cultures. Lanes without any proteinn complexes and all competitions were performed with 50-fold excess ofphozoite and schizont stages. Lane 1 is incubated with nuclear proteins
extracts obtained from early rings, late rings, late trophozoite and schizontis competed using specific competitors. Lanes 1 and 2 are incubated with
th nuclear proteins from late trophozoite cultures. Lanes 2 and 4 contains is highlighted by an asterisk. (C) Binding to the �356 to �562 region onlyly ring, late ring, late trophozoite and schizont nuclear protein extracts,
titor. Lanes 1 and 2 are binding reactions incubated with early ring nuclearextracts. Lanes 2 and 4 contain specific competitor.
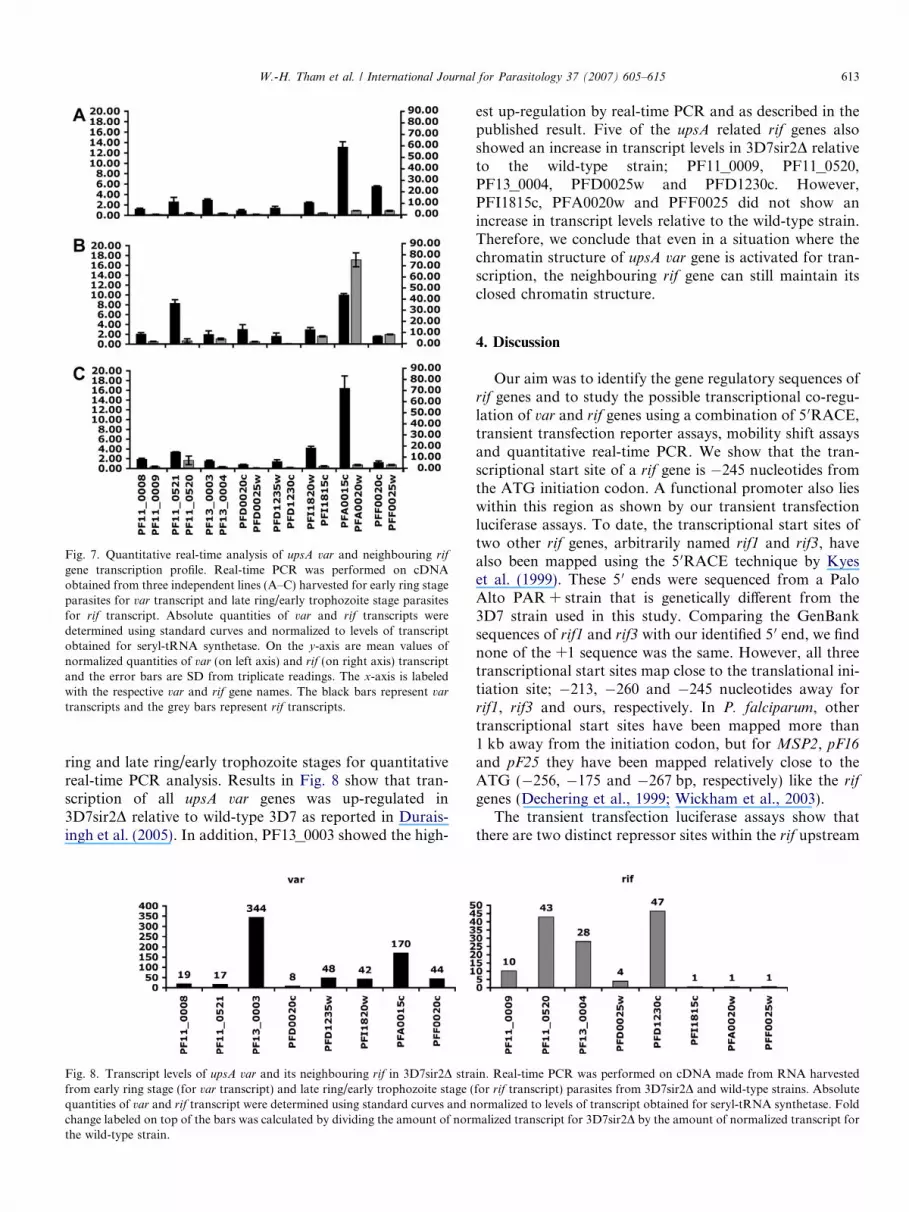
Fig. 7. Quantitative real-time analysis of upsA var and neighbouring rif
gene transcription profile. Real-time PCR was performed on cDNAobtained from three independent lines (A–C) harvested for early ring stageparasites for var transcript and late ring/early trophozoite stage parasitesfor rif transcript. Absolute quantities of var and rif transcripts weredetermined using standard curves and normalized to levels of transcriptobtained for seryl-tRNA synthetase. On the y-axis are mean values ofnormalized quantities of var (on left axis) and rif (on right axis) transcriptand the error bars are SD from triplicate readings. The x-axis is labeledwith the respective var and rif gene names. The black bars represent var
transcripts and the grey bars represent rif transcripts.
W.-H. Tham et al. / International Journal for Parasitology 37 (2007) 605–615 613
ring and late ring/early trophozoite stages for quantitativereal-time PCR analysis. Results in Fig. 8 show that tran-scription of all upsA var genes was up-regulated in3D7sir2D relative to wild-type 3D7 as reported in Durais-ingh et al. (2005). In addition, PF13_0003 showed the high-
Fig. 8. Transcript levels of upsA var and its neighbouring rif in 3D7sir2D strafrom early ring stage (for var transcript) and late ring/early trophozoite stage (quantities of var and rif transcript were determined using standard curves and nchange labeled on top of the bars was calculated by dividing the amount of normthe wild-type strain.
est up-regulation by real-time PCR and as described in thepublished result. Five of the upsA related rif genes alsoshowed an increase in transcript levels in 3D7sir2D relativeto the wild-type strain; PF11_0009, PF11_0520,PF13_0004, PFD0025w and PFD1230c. However,PFI1815c, PFA0020w and PFF0025 did not show anincrease in transcript levels relative to the wild-type strain.Therefore, we conclude that even in a situation where thechromatin structure of upsA var gene is activated for tran-scription, the neighbouring rif gene can still maintain itsclosed chromatin structure.
4. Discussion
Our aim was to identify the gene regulatory sequences ofrif genes and to study the possible transcriptional co-regu-lation of var and rif genes using a combination of 5 0RACE,transient transfection reporter assays, mobility shift assaysand quantitative real-time PCR. We show that the tran-scriptional start site of a rif gene is �245 nucleotides fromthe ATG initiation codon. A functional promoter also lieswithin this region as shown by our transient transfectionluciferase assays. To date, the transcriptional start sites oftwo other rif genes, arbitrarily named rif1 and rif3, havealso been mapped using the 5 0RACE technique by Kyeset al. (1999). These 5 0 ends were sequenced from a PaloAlto PAR + strain that is genetically different from the3D7 strain used in this study. Comparing the GenBanksequences of rif1 and rif3 with our identified 5 0 end, we findnone of the +1 sequence was the same. However, all threetranscriptional start sites map close to the translational ini-tiation site; �213, �260 and �245 nucleotides away forrif1, rif3 and ours, respectively. In P. falciparum, othertranscriptional start sites have been mapped more than1 kb away from the initiation codon, but for MSP2, pF16
and pF25 they have been mapped relatively close to theATG (�256, �175 and �267 bp, respectively) like the rif
genes (Dechering et al., 1999; Wickham et al., 2003).The transient transfection luciferase assays show that
there are two distinct repressor sites within the rif upstream
in. Real-time PCR was performed on cDNA made from RNA harvestedfor rif transcript) parasites from 3D7sir2D and wild-type strains. Absoluteormalized to levels of transcript obtained for seryl-tRNA synthetase. Foldalized transcript for 3D7sir2D by the amount of normalized transcript for

614 W.-H. Tham et al. / International Journal for Parasitology 37 (2007) 605–615
region. Through a series of EMSAs, we were able to deter-mine that a CGCACAACAC element (within pRIF870)and a TATGCAatgATT element (within pRIF562) wereresponsible for protein binding to the repressor sites. Inaddition, mutation of CGCACAACAC and the completeremoval of the TATGCAatgATT element resulted in anincrease in luciferase expression as expected if these sitesfunction as repressor elements.
Genome-wide BLAST analyses show that the CGC
ACAACAC element and the TATGCAatgATT elementare exclusively associated with rif genes. Three distinctrepressor elements, CPE1, SPE1 and SPE2, are presentin the upstream regions of upsB and upsC var genes (Vosset al., 2003). Sequence comparison between CPE1, SPE1and the rif repressors show no significant similarity. WithinSPE2 there is a CG rich motif TATAAATTCGCAC
CACTATGCACAATAAAG that is similar to the rif
CGCACAACAC element (shown in italics). However, webelieve that SPE2 functions in a different context to theCGCACAACAC element for the following reasons. First,SPE2 has a conserved sequence motif consisting of a direct(T/G)GTGC(A/G) repeat spaced by four nucleotides thatis required for protein binding. We do not see such a con-served motif within the CGCACAACAC element. Second,SPE2 protein binding occurs only in schizont stageswhereas the CGCACAACAC element bound proteinsexpressed in both trophozoite and schizont stages. Howev-er, if SPE2 and the CGCACAACAC element were found tobind the same proteins, it would provide an intriguingmechanism for the co-regulation of rif and var repression.
From our studies in 3D7, the expression profile of the rif
gene PF11_0009 shows maximal transcription at the latering to early trophozoite stage. Therefore this gene requiresat least two elements of gene regulation to account for theactivation of transcription in the early ring stage and therepression of transcription past the early trophozoite stage.Our identification of two distinct repressor elements thatshow differential timing of protein binding provides amechanism for the separation of these two transcriptionalfunctions. Binding of the TATGCAatgATT elementoccurred during the early ring stage and also during thelate ring stage when gene transcription is activated forthe rif gene. However, deletion of this element results inan increase in luciferase expression, suggesting that this ele-ment functions as a repressor site rather than an activator.We propose that protein binding to the TATGCAatgATTelement which is present in all rif genes orientated head-to-head with an upsA var gene, can selectively repress tran-scription of this subset of rif genes early in the cycle. Themobility shift assays show that the CGCACAACAC ele-ment was bound by proteins expressed in the late trophozo-ite and schizont stages, when transcription is repressed forthe rif genes. Therefore, proteins bound to the CGCACAA-
CAC element may serve to repress transcription of rif genesin the later stages of the cell cycle.
The upsA var genes are the only var genes in a head-to-head orientation with a rif gene. It is possible that this subset
of var and rif genes may be transcriptionally co-regulated asthey share a common upstream region. Var gene transcrip-tion occurs earlier in the life cycle compared with rif genetranscription, and a possible mechanism that allows tran-scription co-regulation is that var gene expression wouldestablish an open and transcriptionally active chromatinstructure that would spread to the entire upstream region,thus allowing transcription of the neighbouring rif gene.However, using real-time PCR analyses in three separateunselected lines, we did not find any evidence that the upsA
var and their neighbouring rif genes were transcriptionallyco-regulated. Although the transcription profile of var andrif genes were different among the three independent lines,high level expression of a var gene did not correlate withhigh level expression of its neighbouring rif nor vice versa.Furthermore, transcription of upsA var genes is regulatedby PfSir2 but only five of the eight neighbouring rif genesshow that same dependence on PfSir2. Therefore, theother three rif genes may not be epigenetically regulatedand their transcription regulation is dependent on some-thing other than PfSir2. In terms of co-regulation of othermulti-gene families, Sharp et al. (2006) also did not findany evidence of transcriptional co-regulation between ste-
vor and var genes in both sexual and asexual parasites.However, it is still possible that stevor and rif transcriptionare co-regulated with var transcription for specific adhe-sion phenotypes in which they play a role, but as yet noshared adhesion phenotype has been described for thesemulti-gene families, making such transcription analysesimpossible.
We believe the data presented in this paper constitutethe first detailed description of the cis-acting elementsinvolved in rif gene transcription. Future analyses that leadto the identification of the proteins that bind to these ele-ments will yield more insight into the mechanism of rif generegulation. Studies of var gene regulation find that thesilencing of var genes is associated with the presence of avar intron (Deitsch et al., 2001). A single var gene escapesthe default transcriptional down-regulation of all othervar genes and is exclusively transcribed by localization ofits promoter at a transcriptionally permissive perinuclearsite (Duraisingh et al., 2005; Ralph et al., 2005 Vosset al., 2006; Dzikowski et al., 2006; Marty et al., 2006).Further studies are needed to determine if rif genes alsoemploy some of these mechanisms. By understanding thetranscriptional mechanism behind clonal variation, greaterprogress can be made in the understanding of immune eva-sion and malaria pathogenesis.
Acknowledgements
We thank Alan Cowman, Brendan Crabb, Michael Duf-fy and Till Voss for critical reading of this manuscript. Wethank Joanne McCoubrie for invaluable advice on tran-sient transfection, and Jean Baptiste Boule, David Katzand Till Voss for advice with EMSA. This work was sup-ported by an Australian Research Council APD fellowship

W.-H. Tham et al. / International Journal for Parasitology 37 (2007) 605–615 615
to WHT and a Wellcome Trust Fellowship to SJR. GVB isfunded from a Program Grant of the National Health andMedical Research Council.
Appendix A. Supplementary data
Supplementary data associated with this article can befound, in the online version, at doi:10.1016/j.ijpara.2006.11.006.
References
Abdel-Latif, M.S., Cabrera, G., Kohler, C., Kremsner, P.G., Luty, A.J.,2004. Antibodies to rifin: a component of naturally acquired responsesto Plasmodium falciparum variant surface antigens on infected eryth-rocytes. Am. J. Trop. Med. Hyg. 71, 179–186.
Abdel-Latif, M.S., Dietz, K., Issifou, S., Kremsner, P.G., Klinkert, M.Q.,2003. Antibodies to Plasmodium falciparum rifin proteins are associ-ated with rapid parasite clearance and asymptomatic infections. Infect.Immun. 71, 6229–6233.
Abdel-Latif, M.S., Khattab, A., Lindenthal, C., Kremsner, P.G., Klinkert,M.Q., 2002. Recognition of variant rifin antigens by human antibodiesinduced during natural Plasmodium falciparum infections. Infect.Immun. 70, 7013–7021.
Bull, P.C., Lowe, B.S., Kortok, M., Molyneux, C.S., Newbold, C.I.,Marsh, K., 1998. Parasite antigens on the infected red cell surface aretargets for naturally acquired immunity to malaria. Nat. Med. 4, 358–360.
Calderwood, M.S., Gannoun-Zaki, L., Wellems, T.E., Deitsch, K.W.,2003. Plasmodium falciparum var genes are regulated by two regionswith separate promoters, one upstream of the coding region and asecond within the intron. J. Biol. Chem. 278, 34125–34132.
Dechering, K.J., Kaan, A.M., Mbacham, W., Wirth, D.F., Eling, W.,Konings, R.N., Stunnenberg, H.G., 1999. Isolation and functionalcharacterization of two distinct sexual-stage-specific promoters of thehuman malaria parasite Plasmodium falciparum. Mol. Cell Biol. 19,967–978.
Deitsch, K.W., Calderwood, M.S., Wellems, T.E., 2001. Malaria. Coop-erative silencing elements in var genes. Nature 412, 875–876.
Duraisingh, M.T., Voss, T.S., Marty, A.J., Duffy, M.F., Good, R.T.,Thompson, J.K., Freitas-Junior, L.H., Scherf, A., Crabb, B.S.,Cowman, A.F., 2005. Heterochromatin silencing and locus reposi-tioning linked to regulation of virulence genes in Plasmodium
falciparum. Cell 121, 13–24.Dzikowski, R., Frank, M., Deitsch, K., 2006. Mutually exclusive
expression of virulence genes by malaria parasites is regulatedindependently of antigen production. PLoS Pathog. 2, e22.
Fernandez, V., Hommel, M., Chen, Q., Hagblom, P., Wahlgren, M., 1999.Small, clonally variant antigens expressed on the surface of thePlasmodium falciparum-infected erythrocyte are encoded by the rif
gene family and are the target of human immune responses. J. Exp.Med. 10, 1393–1404.
Gasser, S.M., Cockell, M.M., 2001. The molecular biology of the SIRproteins. Gene 279, 1–16.
Khattab, A., Klinkert, M.Q., 2006. Maurer’s clefts-restricted localization,orientation and export of a Plasmodium falciparum RIFIN. Traffic.(Epub ahead of print).
Kyes, S., Pinches, R., Newbold, C., 2000. A simple RNA analysis methodshows var and rif multigene family expression patterns in Plasmodium
falciparum. Mol. Biochem. Parasitol. 105, 311–315.Kyes, S.A., Rowe, J.A., Kriek, N., Newbold, C.I., 1999. Rifins: a second
family of clonally variant proteins expressed on the surface of red cellsinfected with Plasmodium falciparum. Proc. Natl. Acad. Sci. 96, 9333–9338.
Lambros, C., Vanderberg, J.P., 1979. Synchronization of Plasmodium
falciparum erythrocytic stages in culture. J. Parasitol. 65, 418–420.Lavstsen, T., Salanti, A., Jensen, A.T., Arnot, D.E., Theander, T.G., 2003.
Sub-grouping of Plasmodium falciparum 3D7 var genes based onsequence analysis of coding and non-coding regions. Malar. J. 10, 27–31.
Marti, M., Good, R.T., Rug, M., Knuepfer, E., Cowman, A.F., 2004.Targeting malaria virulence and remodeling proteins to the hosterythrocyte. Science 306, 1897–1898.
Marty, A.J., Thompson, J.K., Duffy, M.F., Voss, T.S., Cowman, A.F.,Crabb, B.S., 2006. Evidence that Plasmodium falciparum chromosomeend clusters are cross-linked by protein and are the sites of bothvirulence gene silencing and activation. Mol. Microbiol. 62, 72–83.
Militello, K.T., Wirth, D.F., 2003. A new reporter gene for transienttransfection of Plasmodium falciparum. Parasitol. Res. 89, 154–157.
Ralph, S.A., Scheidig-Benatar, C., Scherf, A., 2005. Antigenic variation inPlasmodium falciparum is associated with movement of var locibetween subnuclear locations. Proc. Natl. Acad. Sci. USA 102,5414–5419.
Salanti, A., Staalsoe, T., Lavstsen, T., Jensen, A.T., Sowa, M.P., Arnot,D.E., Hviid, L., Theander, T.G., 2003. Selective upregulation of asingle distinctly structured var gene in chondroitin sulphate A-adheringPlasmodium falciparum involved in pregnancy – associated malaria.Mol. Microbiol. 49, 179.
Scherf, A., Hernandez-Rivas, R., Buffet, P., Bottius, E., Benatar, C.,Pouvelle, B., Gysin, J., Lanzer, M., 1998. Antigenic variation inmalaria: in situ switching, relaxed and mutually exclusive transcriptionof var genes during intra-erythrocytic development in Plasmodium
falciparum. EMBO J. 17, 5418–5426.Sharp, S., Lavstsen, T., Fivelman, Q.L., Saeed, M., McRobert, L.,
Templeton, T.J., Jensen, A.T.R., Baker, D.A., Theander, T.G.,Sutherland, C.J., 2006. Programmed transcription of the var genefamily, but not of stevor, in Plasmodium falciparum gametocytes.Eukaryot. Cell 5, 1206–1214.
Trager, W., Jenson, J.B., 1978. Cultivation of malarial parasites. Nature273, 621–622.
Voss, T.S., Healer, J., Marty, A.J., Duffy, M.F., Thompson, J.K., Beeson,J.G., Reeder, J.C., Crabb, B.S., Cowman, A.F., 2006. A var genepromoter controls allelic exclusion of virulence genes in Plasmodiumfalciparum malaria. Nature 439, 1004–1008.
Voss, T.S., Kaestli, M., Vogel, D., Bopp, S., Beck, H.P., 2003. Identifi-cation of nuclear proteins that interact differentially with Plasmodium
falciparum var gene promoters. Mol. Microbiol. 48, 1593–1607.Wickham, M.E., Thompson, J., Cowman, A.F., 2003. Characterisation of
the merozoite surface protein-2 promoter using stable and transienttransfection in Plasmodium falciparum. Mol. Biochem. Parasitol. 129,147–156.





























