Embed Size (px)
Citation preview

University of Groningen
Host cell responses to dengue virus infectionDiosa Toro, Mayra
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2017
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Diosa Toro, M. (2017). Host cell responses to dengue virus infection. University of Groningen.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 27-08-2022

8SUMMARIZING DISCUSSION AND
FUTURE PERSPECTIVES

176 | Chapter 8 Discussion and future perspectives | 177
8
Dengue virus (DENV) causes the most common arthropod-borne viral disease in humans, with an estimated number of cases of 390 million per year. Approximately one-quarter of the infected individuals develop symptoms ranging from mild-febrile illness to hemorrhages and potentially fatal hypotensive shock1. Four DENV serotypes have been described and all of them can cause disease. Importantly, individuals experiencing a secondary infection with a heterologous DENV serotype are at higher risk for the development of severe disease2. The increased hyper-endemicity observed in many regions in the last years will therefore probably result in an increment of the number of severe cases3,4.
The complex pathogenesis of severe disease hampered the design of vaccines for several decades until, at the end of the year 2015, the first dengue vaccine became commercially available5. This vaccine is currently licensed in 11 endemic countries though some concerns regarding the efficacy and safety remain6–8. Furthermore, specific antiviral therapies are not available against DENV. Treatment of severe cases depend on supportive care in medical facilities, which can represent an economic hurdle in many DENV-endemic countries9.
Because the disease and socio-economic burden of dengue is very high and likely to increase, it is imperative to continue the search for better vaccines and specific antivirals. This can only be accomplished inasmuch as we fully comprehend the cellular and molecular mechanisms that lead to disease. Therefore, unraveling the intricate network of interactions between DENV and its human host cells is of outmost importance. Understanding the mechanisms by which DENV hijacks host cell factors and how different cell targets respond to infection, will provide cues for the development of effective therapeutics.
The work presented in this thesis provides insight into the virus-host interactions that occur during infection. Emphasis was on the role of different target cells in viral infection, the molecular mechanisms involved in antibody dependent enhancement of infection, the contribution of microRNAs to promote/restrict infection and the molecular mechanism of virus-induced cellular stress. In addition, we explored the antiviral potential of tomatidine towards dengue virus. The results of my thesis are summarized in Fig. 1 and they will be discussed under the umbrella of four main topics which continue to be of great interest in the field of DENV research:
I) Contribution of macrophages to primary and secondary dengue virus infectionsII) Mammalian miRNAs during viral infection: fulfilling the expectations?III) Viral strategies aimed at the control of cellular stress responsesIV) Antivirals towards DENV: can we find the perfect drug?
Discussion and future perspectives | 177
8
DENV-infected MΦ DENV-negative MΦ
miR-3960miR-4508miR-4301miR-181a
miR-3614-5p
Infectious DENVNon-infectious DENV
mDC iDC MΦ
miR-3614-5p
ADAR1
ADAR1
Tomatidine
ATF4
Binding
Entry
FusionInterferon
viral RNA
i)
ii)
iii)
iv)
A.
B.
C.
Figure 1.
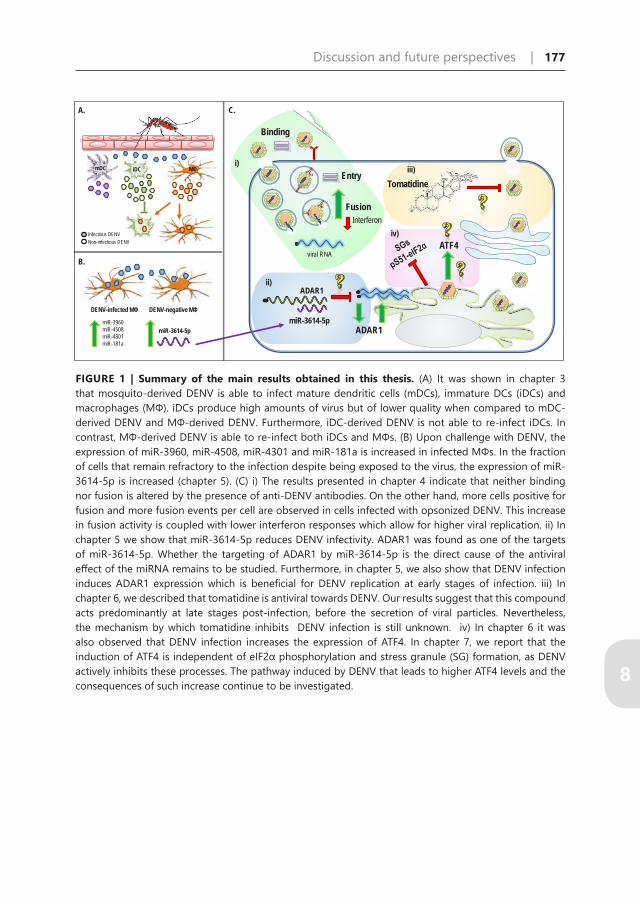
FIGURE 1 | Summary of the main results obtained in this thesis. (A) It was shown in chapter 3 that mosquito-derived DENV is able to infect mature dendritic cells (mDCs), immature DCs (iDCs) and macrophages (MΦ). iDCs produce high amounts of virus but of lower quality when compared to mDC-derived DENV and MΦ-derived DENV. Furthermore, iDC-derived DENV is not able to re-infect iDCs. In contrast, MΦ-derived DENV is able to re-infect both iDCs and MΦs. (B) Upon challenge with DENV, the expression of miR-3960, miR-4508, miR-4301 and miR-181a is increased in infected MΦs. In the fraction of cells that remain refractory to the infection despite being exposed to the virus, the expression of miR-3614-5p is increased (chapter 5). (C) i) The results presented in chapter 4 indicate that neither binding nor fusion is altered by the presence of anti-DENV antibodies. On the other hand, more cells positive for fusion and more fusion events per cell are observed in cells infected with opsonized DENV. This increase in fusion activity is coupled with lower interferon responses which allow for higher viral replication. ii) In chapter 5 we show that miR-3614-5p reduces DENV infectivity. ADAR1 was found as one of the targets of miR-3614-5p. Whether the targeting of ADAR1 by miR-3614-5p is the direct cause of the antiviral effect of the miRNA remains to be studied. Furthermore, in chapter 5, we also show that DENV infection induces ADAR1 expression which is beneficial for DENV replication at early stages of infection. iii) In chapter 6, we described that tomatidine is antiviral towards DENV. Our results suggest that this compound acts predominantly at late stages post-infection, before the secretion of viral particles. Nevertheless, the mechanism by which tomatidine inhibits DENV infection is still unknown. iv) In chapter 6 it was also observed that DENV infection increases the expression of ATF4. In chapter 7, we report that the induction of ATF4 is independent of eIF2α phosphorylation and stress granule (SG) formation, as DENV actively inhibits these processes. The pathway induced by DENV that leads to higher ATF4 levels and the consequences of such increase continue to be investigated.

178 | Chapter 8 Discussion and future perspectives | 179
8
Contribution of macrophages to primary and secondary dengue virus infections
The complexity of DENV infection in humans and the absence of a suitable animal model to study it, has hindered the development of specific antiviral therapies and safe/effective vaccines. Most of our understanding of dengue pathogenesis comes from studies conducted in cells lines. Moreover, a number of studies have used primary human target cells for their studies, such as monocytes, macrophages and dendritic cells (DCs)10–13. These cells are important during DENV infection because, upon the mosquito bite, they are the first targets of DENV14–17. Furthermore, DCs, monocytes and macrophages are thought to produce high numbers of progeny virions thereby contributing significantly to the high viral loads characteristic of the acute phase of the infection. A high viral load early in infection is, in turn, associated with an increased chance to develop of severe disease17–19. However, a direct comparison between the infectious properties of DENV in primary cells derived from the same donor is lacking. Due to the inherent differences between blood donors it remains unclear which cells contribute the most to DENV viremia, dissemination and antibody-dependent enhancement during natural infection.
In chapter 3, we studied the contribution of macrophages and DCs to DENV infection by performing side-by-side comparisons between these cells derived from the same blood donor. Initially, monocytes were also included in our study given their importance in DENV infection, yet, the rapid and high mortality rates that we observed in DENV-infected monocytes impeded us to include these cells in our studies.
The results show that mosquito cell-derived DENV is more infectious in immature DCs (iDCs) than in mature DCs (mDCs) and macrophages as demonstrated by the higher percentages of infection at MOI 1. In addition, and in line with previous studies20, iDCs produce more progeny DENV particles. However, iDC-virus progeny was found to be 10-100 times less infectious when compared to virus derived from mDCs and macrophages. Thus, although iDCs produce more virus, the overall quality of iDC-derived DENV progeny is lower. Furthermore, our results also indicate that iDCs are not susceptible to iDC-derived DENV, whereas macrophage-derived DENV is able to infect both, iDCs and macrophages. Collectively, the above observations suggest that during primary infection, iDCs are likely the first target cell of DENV whereas macrophages and mDCs contribute to sustain subsequent rounds of infection and spread of the infection.
An explanation for the low infectivity of iDC-derived DENV remains elusive. The presence of defective interfering particles has been observed in the sera from dengue patients21,22. Defective interfering particles have deletions within the viral genome; thus, these particles are not infectious because they lack a coding region that is

Discussion and future perspectives | 179
8
important for infection. However, defective interfering particles are still phagocytized by antigen-presenting cells, thereby amplifying the number of activated cells that can prime adaptive immunity without the risk of spreading infection. Therefore, it is tempting to speculate that iDCs secrete large numbers of non-infectious particles to facilitate priming of the adaptive immunity and thus actively help to clear the infection. Similar observations have been made for vesicular stomatitis virus23,24 and future studies should address whether this hypothesis also holds true for DENV.
During secondary infection with a heterologous serotype, the presence of cross-reactive non-neutralizing antibodies has been shown to enhance DENV infectivity on Fc-receptor expressing cells, a phenomenon referred to as antibody-dependent enhancement (ADE). ADE has been associated with increased infection of host cells and a higher production of virus particles per infected cell. In vitro studies revealed that the extent of ADE is dependent on the antibody characteristics (concentration, avidity and the viral epitope targeted) and the susceptibility of the Fc-receptor-expressing cell in absence of antibodies20,25,26. For example, iDCs do not support ADE and the authors describe that the high expression levels of the DENV-receptor DC-SIGN in iDCs negatively correlates with the enhancement of infection through Fc receptors26. Indeed, previous studies from our group confirm that iDCs do not support ADE27. In chapter 3 it was found that macrophages and mDCs do elicit ADE, albeit the power of enhancement is higher in macrophages. Importantly, ADE in macrophages was observed at high serum concentrations whereas peak enhancement for mDC was observed at extremely low serum concentrations. Therefore, we hypothesize that macrophages are particularly important during the acute phase of secondary infections as a high antibody concentration is rapidly reached in these cases28. At these conditions, ADE in mDCs is most likely suppressed due to high antibody concentrations. Furthermore, macrophage-derived DENV is more prone to be enhanced than iDC-derived DENV. Thus, our results suggest that macrophages are the main contributors to the increased viral burden observed during re-infection with heterologous DENV serotypes. The susceptibility and contribution of monocytes during primary and secondary DENV infection remains to be elucidated.
So far our results indicate that macrophages play an important role during primary and secondary DENV infections. During primary infection, macrophages are less susceptible to DENV compared to iDCs, yet the virus produced is more infectious and is able to re-infect macrophages and iDCs. During heterologous secondary infection, i.e. in the presence of non-neutralizing antibodies, the infectivity of mosquito-derived DENV is enhanced in macrophages and macrophage-derived DENV is prone to cause enhancement in cell types that support ADE. Thus, in chapter 4, we aimed to better understand the response of macrophages to DENV infection and the molecular mechanisms behind ADE.

180 | Chapter 8 Discussion and future perspectives | 181
8
The dogma is that the increased number of infected cells during ADE is due to increased DENV cell binding and entry (extrinsic ADE)29–31. Furthermore, activation of the Fc receptor has been associated with an induction of a immunosuppressive state which allows higher virus particle production per infected cell (intrinsic ADE)32,33. Our initial results suggested that both extrinsic and intrinsic ADE might occur in primary macrophages because, at peak-enhancement, the number of infected cells increased 2-fold, whereas virus particle production increased 7-fold. However, during ADE conditions, we did not observe increased virus binding nor enhanced virus cell entry. Also, no enhancement of virus-cell binding was observed in monocyte/macrophage-like THP-1cells32. Intriguingly, however, our group did observe enhancement of virus-cell binding in the murine macrophage-like cell line P388D134. Thus, the mechanism by which antibodies lead to higher percentage of infection is cell type specific. We hypothesize that this may be related to the extent of virus binding in the absence of antibodies, in a similar way to that has been described for iDCs26. In such case, it is likely that in human macrophages, the interaction of DENV with its native receptors is as effective as the interaction of immune complexes (DENV-antibody) with Fc-receptors thereby less robust ADE is seen.
Then, why do we observe a higher percentage of infection in primary macrophages? This can be explained by the observed enhancement of membrane fusion. Our results show 65% more fusion events per cell and 40% more cells positive for fusion. Furthermore, although the increase in fusion activity was variable between donors, on a per donor basis, the enhancement of membrane fusion correlated with the increased production of DENV particles. Therefore, the increased fusion during ADE conditions most likely leads to the increased number of infected cells during ADE. Why enhanced fusion is seen remains to be investigated. Our group recently found that DENV cell entry into murine P388D1 macrophages in the presence of antibodies occurs via a phagocytosis-like pathway, whereas in the absence of antibodies entry is mediated by a clathrin-mediated endocytosis-like pathway34. Furthermore, DENV-immune complexes were found to fuse faster than non-opsonized DENV34. This shows that the receptor dictates the route of cell entry used by the virus. In other words, entry via a different receptor may deliver the particles to a different endocytic pathway whose environment is more favorable for fusion35. The observed increase in the fusion events per cell favors this hypothesis. Nevertheless, due to the differences earlier described between P388D1 cells and human macrophages regarding the enhancement of binding, this premise requires caution and further investigation.
I have stated above that our results suggested the existence of extrinsic and intrinsic ADE because we did not observe a linear correlation between the increased number of infected cells and the increased number of produced virus particles. Nevertheless, our data contradict the basic argument of the intrinsic ADE theory.

Discussion and future perspectives | 181
8
Earlier studies suggest that higher virus particle production per infected cell is due to an immunosuppressive state induced by the activation of Fc-receptors33,36. Our transcriptome analysis, however, shows that ADE does not lead to the establishment of an immunosuppressive state, neither at early nor at late stages of infection (2 vs 24 hours post infection (hpi)). Furthermore, treatment with IFN-α at early stages of infection resulted in the inhibition of DENV infection, regardless of the presence of antibodies. Consequently, the antibody-Fc-receptor interaction is not able to antagonize the immune responses elicited by IFN-α at the beginning of the infection. Likewise, other reports have also shown that treatment with IFN inhibits antibody-mediated infection of primary monocytes and monocyte-like cell lines37,38. Altogether, these data disprove the immunosuppressive action of antibodies and the current explanation of intrinsic ADE.
A possible explanation for the higher production of infectious particles in comparison to the extent of fusion during ADE conditions may come from the differential expression of IFN-stimulated genes. DENV is sensitive to IFN-mediated antiviral effects early in infection, but as the infection progresses the effect of IFN becomes limited. Indeed, several non-structural proteins have been described to hijack the production and cascade signaling of IFN39–41. Our results show that an antiviral state is triggered in all infection conditions (MOI 1, MOI 1-ADE and high MOI), yet, more IFN-related genes were induced at high MOI. At high MOI we observed more fusion, yet lower viral replication compared to MOI 1-ADE. We hypothesize that, the enhanced IFN activity triggered at high MOI leads to a lower replication efficiency of fused particles.
Final remarks: the snowball effect of ADE of infection
Collectively, our results indicate that antibodies enhance DENV infection of human macrophages by increasing the total extent of membrane fusion and the number of fusion events per cell. The increased number of particles fused per cell results in a higher number of genome molecules delivered into the cytoplasm and consequently, higher replication. This, next to the lower activation of the IFN-responses early in infection by DENV-inmune complexes, have great consequences on viral output. Thus, the process of ADE of infection as a whole can be seen as the result of the snowball effect. An initial moderate effect in fusion translates into more efficient DENV replication thereby increasing the virus burden.
Our results suggest that, contrary to what has been observed in murine macrophages, in human primary macrophages antibodies do not enhance DENV- binding and entry. We postulate these results might be linked to the expression of the mannoser receptor (CD206) on macrophages (Chapter 3). Because CD206 is a well-recognized DENV

182 | Chapter 8 Discussion and future perspectives | 183
8
receptor42, it is likely that the expression of CD206 inversely correlates with ADE, in a similar manner as the expression of DC-SIGN in iDCs has been negatively correlated with enhancement of infection26,27. Furthermore, a detailed characterization of the routes employed by opsonized DENV virus will problably help to clarify whether specific differences, e.g., lipid composition and pH of the trafficking vesicles, correlate with the extent of fusion. Lastly, a deeper understanding of the mechanisms of IFN evasion/inhibition by DENV and how these differ in the presence of antibodies, will help to reveal approaches to bust the antiviral effect of the innate immunity without exacerbating cytokine responses. Importantly, the mechanism of ADE in human primary cells is different from that observed in cell lines, highlighting the importance of selecting relevant cell types when studying the pathogenesis of complex diseases such as dengue.
Mammalian miRNAs during viral infection: fulfilling the expectations?
The concept of RNA interference (RNAi), originally introduced by Fire et al. in 199843, refers to sequence-specific gene silencing induced by the presence of foreign double stranded RNAs (dsRNAs). RNAi-dependent silencing is facilitated by small RNA molecules (~22 nt) among them microRNAs (miRNAs) and small interfering RNAs (siRNAs). miRNAs and siRNAs differ in origin (Chapter 2, Fig. 2). MiRNAs are encoded in the genome of all metazoan species whereas siRNAs are derived from dsRNA molecules generated by replicating viruses and integrated transposons.
MiRNA biogenesis begins with the transcription of long RNA molecules that fold in stem-loop structures known as primary miRNAs (pri-miRNAs)44. Pri-miRNAs are cleaved by the nuclear RNase Drosha, resulting in a precursor miRNA (pre-miRNA) hairpin of about 60-70 n45. Thereafter, pre-miRNAs are exported to the cytoplasm in an Ran-GTPase-dependent manner by the nuclear transport receptor exportin-546. The dsRNA structure of the pre-miRNA is recognized by the RNase Dicer and its activity leads to the generation of short miRNA duplexes47. These duplexes are loaded into the RNA-induced silencing complex (RISC), whose core components are the Argonaute (Ago) proteins. Ago mediates the recognition of complementary mRNA transcripts, which are not 100% complementary to the miRNA molecule. Thus, their binding results in translational repression and/or mRNA decay48. On the other hand, siRNAs are generated by the presence of foreign dsRNA molecules in the cytoplasm, where they are also recognized by Dicer. Dicer processing leads to the formation of siRNA duplexes that mediate target recognition through RISC. siRNAs target the same sequence from which they were generated. Due to the 100% complementarity, siRNA-mediated silencing is associated with the degradation of the target sequence49.

Discussion and future perspectives | 183
8
RNAi is a conserved mechanism among eukaryotes and is believed to have evolved as a defense mechanism against invasion by mobile genetic elements, such as viruses, transposons, and possibly other types of highly repetitive genomic sequences50. Thus, RNAi is central to the innate immune response. In fact, RNAi is considered as the main antiviral response in nematodes, insects and plants as indicated by three pieces of evidence. First, during infection, viral genomic dsRNA or dsRNA intermediates (present during infection with all RNA viruses) are processed by the host RNAi pathway to generate hundreds of virus-derived siRNAs, known as viRNAs. viRNAs target the viral genome, to which they are completely complementary, resulting in its degradation. Second, as evidence of an evolutionary race to defend from RNAi, many plant and insect viruses encode RNA silencing suppressors (RSSs). RSSs are diverse in structure and mode of action thereby inhibiting the RNAi signaling cascade at various stages. Lastly, loss-of-function mutations in key components of the RNAi, e.g. Dicer, results in increased infection due to impairment of the antiviral function51–54.
Due to the presence of the RNAi components in vertebrates, it was anticipated that an RNA-mediated defense mechanism against mammalian viruses would also exist. Thus, extensive effort has been made to try to demonstrate antiviral RNAi responses in mammalian cells. However, the naturally occurring antiviral activity of RNAi in mammalian cells remains controversial. In mammals, foreign dsRNA molecules induce a potent protein-based antiviral immunity orchestrated by type I interferons (IFN)55–58. The IFN response results in the activation of several effector mechanisms that induce global inhibition of mRNA translation and mRNA decay, among others. Thus, the detection of a specific antiviral response mediated by RNAi is masked by the nonspecific IFN response induced by the same long dsRNA molecules59. In recent years, researchers have postulated that RNAi is not part of the antiviral arsenal of mammalian cells based on three main points; 1) deep sequencing of small RNA populations in somatic mammalian cells infected with a variety of RNA viruses, including dengue virus (DENV), West Nile virus (WNV), influenza and vesicular stomatitis virus failed to detect viRNAs or detected them at exceptionally low levels60–64. 2) viruses replicate to a similar extent in Dicer wild type and Dicer knockout cells60,65 and 3) in some mammalian cells the activation of the IFN pathway leads to a reduction in RNAi activity due to the inactivation of RISC66.
For DENV, I also question the existence of viral siRNAs in mammalian cells as we did not observe these molecules in our deep-sequencing approach (unpublished results). However, increasing evidence suggests that miRNAs can contribute to the outcome of virus-infected mammalian cells thereby having both antiviral and pro-viral effects. miRNA-mediated regulation occurs either by sequence-specific binding of miRNAs to viral genomes or by modulation of the host proteome67. Binding of cellular miRNAs to the viral genome generally reduces viral translation/replication68–72.

184 | Chapter 8 Discussion and future perspectives | 185
8
The first example of miRNA-mediated virus regulation was given by Lecellier and co-workers in 2005, who showed that human miR-32 restricts primate foamy virus type 1 (PFV-1) accumulation by targeting two sequences on the viral genome68. Importantly, as a counter defense, PFV-1 encodes Tas, a broadly effective RSS. Other examples have followed and showed that cellular miRNAs are able to restrict the replication of important human pathogens such as human immunodeficiency virus, influenza A and hepatitis C virus69–72. Yet, the existence of miRNAs whose sole purpose is to bind to invading viruses has been challenged by the fact that high viral mutation rates will contribute to the escape of miRNA-mediated regulation. In addition, most miRNAs are conserved across a wide range of vertebrate species, thus it is unlikely that a given miRNA could have specifically evolved to counter a virus, since viral host ranges are typically more restricted73. Therefore, due to the short nucleotide complementarity needed for miRNA-regulation (positions 2 – 8 from the 5’UTR of the miRNA), it is likely that fortuitous recognition of viral genomes occurs in a widespread manner. In the latter case, the chance of sufficient complementarity will increase correspondingly with the size of the viral genome68. Intriguingly, binding of human miR-122 to hepatitis C virus (HCV) genome has been shown to be required for viral replication74. Of note, this is the only miRNA described so far to promote the expression of its target instead of reducing it. Because HCV mainly infects hepatic cells and miR-122 expression is liver-specific, the development of miravirsen, a specific inhibitor of miR-122, represents the first proof of concept of the utility of miRNA-targeted drugs. Currently, miravirsen clinical trials are being conducted75.
MiRNAs can also influence viral infection through modulation of the host proteome. This possibility is somehow easier to envision as miRNAs are well-known regulators of gene expression and viruses are susceptible to changes in the intracellular environment of their hosts. Therefore, miRNA activity, which is also altered by infection, can result in either a more favorable or less permissible environment for replication. Furthermore, miRNAs may influence the cellular tropism of viruses and contribute to the induction/inhibition of antiviral immune responses.
In light of these interesting possibilities, at the end of the year 2011, we became interested in the potential miRNA-mediated regulation of DENV infection. At that time, very little was known about the interactions between DENV and cellular miRNAs and as the field of miRNA research was developing by leaps and bounds we decided to explore the contribution of these regulatory molecules to DENV infection. We anticipated that due to the large cellular changes induced upon infection, DENV would significantly alter the biogenesis and effector mechanisms of cellular miRNAs.
In chapter 5, we investigated the changes induced in the microRNAome of macrophages infected with DENV. As described earlier, macrophages are important

Discussion and future perspectives | 185
8
natural targets of DENV. Interestingly, infection of macrophages with high doses of DENV does not result in the infection of the whole cell population (Chapters 3 and 4). This phenomenon gave us the opportunity to study the microRNAome of infected and non-infected macrophages in the same culture conditions. Importantly, given the high dose of particles (92 genome equivalents per cell) we anticipate that all cells in culture were exposed to DENV.
The results showed that only a handful of miRNAs are differentially expressed upon DENV challenge. This was at first surprising to us because DENV-infected cells show an altered transcriptome when compared to mock-infected cells76–78 and we expected that part of these changes could be attributable to the action of cellular miRNAs. However, data from other groups, published at the time we were conducting our experiments, also suggested that the expression of miRNAs upon DENV infection is only mildly altered (Chapter 1, Table 1 and79,80). By now, the data suggest that RNA viruses in general do not profoundly change the small RNA landscape of the mammalian host cell81–84. Therefore, the overall impact of miRNAs on the cellular environment is rather limited and these observations dim the original enthusiasm that miRNAs serve as potent antiviral molecules. Furthermore, there was no overlap between the differentially expressed miRNAs in our study compared to the microRNA profiles already published79,80. Although this is easily explained by the use of different experimental systems (endothelial cells, hepatic cells and macrophages) and the strong cellular-context dependency of miRNA activity, it does highlight the lack of a universal miRNA targeted in all DENV natural host cells.
Nevertheless, in our system using primary macrophages, miR-3614-5p was found upregulated in non-infected cells that were exposed to DENV. In order to determine the effect of miR-3614-5p expression on DENV infectivity, we overexpressed miR-3614-5p by transfecting miRNA mimics. Because transfecting miRNA mimics into macrophages influenced DENV infection (Chapter 5), we conducted the overexpression experiments in Huh7 cells. We found that miR-3614-5p slightly but significantly reduced DENV infectivity and also West Nile virus infectivity in Huh7 cells. While conducting our study, several other human miRNAs were reported to restrict DENV infection (listed in Table 2, Chapter 1) and in most of the cases these miRNAs reduced the production of infectious virus particles by half80,85,86. The relatively small reduction in infectivity can be explained as miRNAs are fine-tuners of gene expression and their individual impact on protein levels is not as large as initially expected87,88.
The limited capacity of regulation represents an additional constraint in the fight against viruses, particularly if we envision miRNAs as potential therapeutics. Nevertheless, the lack of potent effects does not disregard the possibility that during

186 | Chapter 8 Discussion and future perspectives | 187
8
naturally occurring infections, viral replication or viral load is kept in check due to the action of antiviral miRNAs. Unfortunately, assessing the real contribution of miRNAs to virus control in mammalians remains challenging because transfection of RNA mimics (which is the most widely used methodology) results in increased non-physiological levels of the miRNA. Hence, the development, improvement and general implementation of methodologies by which miRNA expression can be controlled in a dosage-dependent manner, for example by the use of drug-inducible vectors89, will significantly expand our understanding of miRNA function. Another limitation to study the impact of miRNAs on the antiviral response, is the potent effect of the IFN system that can mask the more subtle effects of other molecules. In this case, studying models in which the IFN response is naturally decreased could shed light on the role of other players of the antiviral response. A proof of concept of this possibility was given recently by the demonstration that inactivation of the IFN pathway in mammalian somatic cells unveils the presence of long dsRNA-mediated RNAi90.
Interestingly, adenosine deaminase acting on RNA 1 (ADAR1), a previously reported pro-DENV host factor, was identified as one of the targets of miR-3614-5p. It is tempting to speculate that human miR-3614-5p negatively regulates DENV and WNV infectivity through the downregulation of ADAR1. However, our results do not support a black and white role for ADAR1 during DENV infection. Rather, we found that ADAR1 facilitates DENV replication at early stages of infection, but at later time points it controls virus production. Therefore, it is more likely that a synergistic effect of all the proteins modulated by miR-36145-p exists, since we found that not just ADAR1, but 28 additional proteins were experimentally regulated by the expression of miR-3614-5p. Of these, 8 were down- and 20 were upregulated. In general, one should be careful to link the antiviral capacity of a miRNA to the regulation of one single host factor (as is done in most publications), because an individual miRNA is known to directly target multiple transcripts. Moreover, the reduced levels of those targeted transcripts can in turn alter the expression of many other proteins, as we observed in our analysis (i.e, the upregulated proteins). If we were to dissect the molecular mechanism by which miR-3614-5p and other published miRNAs reduce DENV infectivity, then the action of all the proteins regulated by those miRNAs (directly and indirectly) must be taken into consideration. Certainly, such types of holistic approaches add another layer of complexity to the already difficult to study miRNA field.
It was not possible to determine the effect of miR-3614-5p on the infection of macrophages, but we would like to think that it reduces DENV infectivity as observed in the Huh7 cells. However, due to the cell type specificity of miRNA activity, further experiments are needed to confirm the role of miRNA-3614-5p in restricting the

Discussion and future perspectives | 187
8
infection of macrophages. In addition, it remains to be elucidated whether the increased expression of miR-3614-5p renders cells refractory to infection, or whether miRNA-3614-5p expression is associated with a particular subpopulation of macrophages that is not susceptible to DENV infection.
The results of our study favor the notion of the existence of mammalian miRNAs whose activity aids in the control of infecting viruses. A deeper understanding of the interactions occurring between viruses and the host miRNA machinery will help us elucidate the real extent of miRNA contribution to the antiviral response.
Final remarks: miRNAs towards the slope of enlightenment?
The discovery of miRNAs represented a breakthrough in the modern era of biology. How the field has developed since then fits very well –to my eyes- Gartner’s hype cycle91. In the hype cycle, used to describe the adoption and social application of new technologies, five phases are clearly visible: 1) technology breakthrough; 2) peak of inflated expectations; 3) trough of disillusionment; 4) slope of enlightenment and; 5) plateau of productivity. Here I want to make use of this trademark cycle to depict my point of view with regard to the development of miRNA research; it is not intended as a historical compilation. A summary of this view is depicted in Fig. 2.
The first phase of miRNA research is undisputable and began in 1993 with the discovery of the first miRNA, Lin-4, by the joint efforts of Ambro’s and Ruvkun’s laboratories92,93. What you find in the following phases will depend on your particular field of research. In light of the potential antiviral activity of miRNAs, the way to the peak of inflated expectations was paved by the discovery of the conserved nature of miRNAs and by the description of RNAi as a mechanism to defend from foreign dsRNA. Moving up to the peak, the ratification of miRNAs as key regulators of gene expression and deciphering the role they play in controlling cellular processes (such as differentiation, stress and oncogenesis) were of great importance. For me, as a virologist, the peak’s zenith was the discovery that miR-122 is an indispensable host factor for hepatitis C virus replication74. The entry into clinical trials of miravirsen75, an anti-microRNA molecule, to treat a viral infection opened a new window of opportunity to study the interactions between viruses and cellular miRNAs. However, in the years that followed the high expectations surrounding miRNAs were not met, and the fall through the trough of disillusionment began. Certain methodological hurdles anticipated this descent through the trough. For example, the difficulty of predicting reliable miRNA targets, both among the pool of endogenous transcripts and in viral sequences, became apparent94,95. Furthermore, coming to a consensus on how to analyze small RNA-seq data proved challenging96–98. Other setbacks came from unraveling the details of the miRNA-silencing mechanism. It was discovered

188 | Chapter 8 Discussion and future perspectives | 189
8
that the suppression of an individual transcript by a given miRNA results in less than a twofold decrease in protein output, which makes experimentally difficult to prove the biological effect of a given miRNA87,88. Finally, the disillusionment was complete with the heated debates regarding the antiviral potential of miRNAs.
Fortunately, in the last years more evidence in favor of the antiviral role of mammalian miRNAs has been accumulating. Although the effect of these molecules in controlling viral infection seems limited, the development of newer technologies to study miRNA function would probably help to unravel the exact mechanisms by which viruses interact with the cellular miRNA machinery. I believe in the years to come we will witness the slope of enlightenment of miRNA research, as we will get a better notion of the true role they play during viral infection. As to whether miRNAs would ever reach a plateau of productivity in the field of virology, miravirsen would probably give us an answer very soon.
Peak of inflated expectations
Plateau of productivity
Trough of disillusionment
Trigger
Discovery of miR-122
miRNA discovery
miRNAs are well conserved
miRNA target predictionmiRNAs regulate
development, cancer, etc Small RNA-seq analysis
Antiviral microRNAs?
RNAi is anantiviral mechanism
More evidence of thepotential of miRNAs
Newer technologies tostudy miRNA function
Limited silencing
Miravirsen?
Figure 2.
Time
Expe
ctat
ions
FIGURE 2 | miRNA research field follows the Gartner’s hype cycle. Upon their discovery, miRNAs changed the conception of gene-expression regulation. Important findings such as their conservation among eukaryotic cells and their participation in many cellular processes opened a window of curiousity on their contribution to viral infection. A few years later, it was clear that miRNA research is more compex than anticipated as technical and biological limitations to study miRNA activity were revealed. Further understanding of the role of miRNAs and the development of better technologies is warranted to study the real potential of miRNAs.

Discussion and future perspectives | 189
8
Viral strategies aiming at the control of cellular stress responses
Intricate networks of interactions occur between viruses and their host cells. Viruses hijack many different cellular components within infected cells in order to maintain host metabolism and allow virus particle production. In parallel, cells activate antiviral responses aiming at the elimination of the virus in infected cells as well as protection of surrounding non-infected cells. Thus, a productive infection is the result of a tight balance between the virus-induced cellular changes aimed at promoting replication and the cellular responses to it.
Although it is generally accepted that DENV infection induces endoplasmic reticulum (ER) stress and oxidative stress, little is known about the mechanisms by which DENV copes with these responses to allow efficient production of progeny virions. A master regulator of cell fate that integrates signaling from various stress stimuli is activating transcription factor 4 (ATF4). By controlling the expression of numerous stress-responsive genes, ATF4 determines whether cell homeostasis can be restored or whether cell death programs must be set in place99,100. While the precise molecular mechanism behind the cellular stress response is not fully understood, it is known that increased translation of ATF4 can be triggered by the presence of phosphorylated eukaryotic translation initiation factor 2 alpha (p-eIF2α). eIF2α in turn is phosphorylated by the action of four distinct kinases as response to the recognition of stress signals: proteinase kinase R (PKR) recognizes dsRNA, PKR-like ER kinase (PERK) senses ER stress, heme-regulated eIF2α kinase (HRI) is activated by oxidative stress and general control non-derepressible (GCN2) recognizes nutrient deprivation101. Because the expression of ATF4 is a convergent point that integrates the signaling triggered by the eIF2α kinases, the eIF2α/ATF4 pathway is commonly referred to as the integrated stress response (ISR)99.
In chapter 6, we showed that DENV causes an increase in the expression level of ATF4 over time. Therefore, in chapter 7, we decided to study this phenomenon in more detail. We aimed for deciphering the mechanism by which ATF4 expression is induced during DENV infection and hypothesized that ATF4 alleviates cellular stress levels induced by DENV.
ATF4 expression is predominantly regulated by the presence of p-eIF2α99. Currently there is no a consensus as to whether DENV alters p-eIF2α levels (Chapter 1). Initial reports suggest that the levels of p-eIF2α in DENV-infected cells are comparable to those of mock-infected cells102. However, it has also been shown that DENV increases eIF2α phosphorylation in a time- and serotype-dependent manner103,104. These contrasting results prompted us to re-investigate the status of eIF2α phosphorylation in Huh7 cells infected with DENV-2. No differences were found

190 | Chapter 8 Discussion and future perspectives | 191
8
in the p-eIF2α levels between DENV-infected and mock-infected Huh7 cells at any evaluated time point (3,6,12,18,24 and 30 hpi). Importantly, during the course of our study Roth and coworkers published similar results for DENV-infected Huh7 cells105. It is not clear how DENV avoids activation of the ISR. It has been suggested that DENV avoids activation of PKR by concealing dsRNA intermediates in compartmentalized membranes106,107. Yet, the recently published study from Roth and coworkers showed that DENV induces PKR activation105. Furthermore, and in line with Roth’s study, we found reduced levels of p-eIF2α in DENV-infected cells treated with the HRI inducer NaAs when compared to NaAs-treated mock-infected cells (Chapter 7). Collectively, these data strongly suggest that DENV actively inhibits eIF2α phosphorylation. Yet, the above observations also suggest that the increased expression of ATF4 in DENV-infected cells is not due to eIF2α phosphorylation.
Subsequent experiments revealed higher levels of ATF4 mRNA upon DENV infection and therefore we now propose that there is either an increase in ATF4 mRNA transcription or a decrease of ATF4 mRNA turnover in DENV-infected cells. In the first case, several transcription factors have been shown to regulate ATF4 mRNA synthesis. Interestingly, one of them, nuclear factor erythroid 2-related factor (NRF2), was shown to be activated in response to the oxidative stress induced in DENV-infected DCs108,109. Secondly, mRNA turnover is mainly regulated by the activity of processing bodies (P-bodies)110,111. Intriguingly, DENV was found to decrease the formation of P-bodies in baby hamster kidney cells102. Whether reduced P-bodies assembly results in the specific reduction of ATF4 turnover or whether it alters mRNA degradation in a more general manner remains to be investigated. Alternatively, the expression of ATF4 during DENV infection could be driven by both, NRF2 activation and reduced mRNA turnover. Ongoing experiments in our group are aimed at testing this hypothesis.
Antagonizing eIF2α phosphorylation is a strategy deployed by viruses such as influenza, vesicular stomatitis virus and herpes simplex virus to promote the translation of viral proteins112–115. The mechanisms by which these viruses hinder eIF2α phosphorylation include the expression of chaperons that inhibit PKR and PERK and the increased activity of the phosphatase GADD34/PP1 which de-phosphorylates eIF2α112–115. However, in case of DENV, PKR is activated105. The expression of GADD34 is regulated by ATF4 and therefore we hypothesized that the high levels of ATF4 observed during DENV infection might correlate with higher GADD34 levels and consequently eIF2α de-phosphorylation. However, this is not the case. We did not observe increased protein levels of GADD34 and siRNA-mediated silencing of ATF4 did not increase eIF2α phosphorylation in DENV-infected cells. These results were unexpected, because GADD34 is a well-known downstream target of ATF4 and ATF4 is a key regulator of cellular stress. A possible explanation for what we observed

Discussion and future perspectives | 191
8
could rely on the timing of our analysis. The levels of GADD34 were assessed at the same time point at which we found the highest ATF4 expression level (at 30 hpi, a 3.6-fold increase in ATF4 compared to mock-infected cells). However, we did not analyze ATF4 nor GADD34 levels at later time points. It is a possibility that ATF4-mediated expression of GADD34 occurs at later time points during infection. Despite of this reasoning, GADD34 does not appear to control the levels of p-eIF2α in DENV-infected cells as salubrinal-mediated inhibition of GADD34 activity had no effect in restoring p-eIF2α levels in chemically stressed cells.
Although there is clear evidence that DENV induces ER and oxidative stress, ATF4, key regulator during cellular stress, does not appear to play an active role in controlling the stress response in DENV-infected cells. Instead, it appears that DENV actively inhibits the ISR upstream of ATF4 through inhibition of eIF2α phosphorylation. It is likely that one of the DENV viral proteins directly impairs eIF2α phosphorylation as is the case for human papillomavirus and herpes simplex virus 112,114. Future studies should address whether a similar mechanism is used by DENV. Understanding the molecular basis for the inhibition eIF2α phosphorylation will increase our knowledge on the replication cycle of DENV and might open new avenues for intervention.
Final remarks: what are the consequences of increased ATF4 expression?
Why is there such a robust increase in ATF4 expression in DENV-infected cells? We discard that it is the result of an unspecific response because the protein levels as well as the mRNA levels were increased in a time-dependent manner. Yet, given the above discussed results together with the moderate effect of siRNA-mediated ATF4 silencing on DENV production (Chapter 6) we conclude that ATF4 is not a crucial factor in DENV multiplication. Is it possible that the induction of ATF4 plays a role in the pathogenesis of dengue? In this regard, I would like to describe two pathogenic events that are potentially linked to ATF4. First, ATF4 activation has been associated with age-related muscle weakness and atrophy116. Myalgia (muscle pain) is a common symptom in dengue patients, especially in the elderly117,118. Is ATF4 perhaps involved in this event? To this end it would be interesting to evaluate whether ATF4 expression is sustained for longer time periods thereby activating cell death pathways. Second, ATF4 is a regulator of monocyte chemoattractant protein-1 (MCP-1) production in microvascular endothelial cells119. MCP-1 is a major chemokine which recruits monocytes/macropages to the sites of injury thereby exacerbating inflammation120. Patients with severe forms of dengue have increased levels of MCP-1121,122. Is ATF4 expression correlated with MCP-1 production in cells and in patients? Activation of cellular stress pathways have indeed been linked to increased inflammatory responses during flavivirus infection123. Thus, future studies should address how the stress status of DENV-infected cells relates to increased/pathologic immune responses.

192 | Chapter 8 Discussion and future perspectives | 193
8
Antivirals towards DENV: can we find the perfect drug?
Despite the high socio-economic burden of dengue disease and years of trying to develop effective antiviral drugs there are still no specific therapies available. As a result, the treatment of symptomatic severe dengue disease relies on supportive care in medical units.
The rationale behind the development of anti-dengue compounds relies on the observation that high levels of viremia in the early stages of infection correlate with the development of severe disease18,124,125. Consequently, a drug that is able to decrease viremia early in infection could prevent the progression to dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS). However, several difficulties have to be considered when trying to meet this premise. First, reducing viral titers early in infection is difficult to achieve as the viremia drops rapidly upon the onset of symptoms and the severe signs manifest when the viral load has significantly decreased126. Therefore, therapy is likely only successful when combined with early diagnosis of infection. Second, anti-DENV drugs should act against all four serotypes as all serotypes lead to (severe) disease. Third, given the importance of ADE in severe disease, anti-DENV drugs should also inhibit the infectivity of DENV-immune complexes.
In chapter 6 we explored the antiviral potential of tomatidine. Tomatidine is an aglycon metabolite of tomatine, a steroidal alkaloid found in the skin and leaves of tomatoes. In nature, tomatine is an important defense mechanism for phytopathogens such as bacteria fungi and viruses127. Our results showed that tomatidine exerts potent antiviral activity towards DENV in Huh7 cells, with EC50 values of 0.82 and 0.97 µM for MOI 1 and MOI 10, respectively. These EC values places tomatidine among the most potent DENV antivirals described to date128. Time-of-addition experiments suggest that tomatidine predominantly acts at late stages post-infection. In addition, the comparable reduction in the number of produced infectious and physical virus particles when the drug was added at 12 hpi indicates that tomatidine interferes with a step prior to virus secretion. Compounds acting at late time points in the replication cycle are attractive from the therapeutic point of view as these are likely to more drastically reduce the viral load when administered upon the onset of the symptoms.
It remains to be investigated whether tomatidine acts as a direct antiviral agent (DAA) or whether it targets a crucial cellular factor that is required for productive infection. The latest results conducted in our group point towards an action at the level of the host. This as we recently observed antiviral effects towards Chikungunya, a non-related arthropod-borne virus (unpublished data). Because tomatidine was previously shown to reduce ATF4 levels, we investigated whether the antiviral effect of

Discussion and future perspectives | 193
8
tomatidine was caused by the inhibition of ATF4. Indeed, treatment with tomatidine reduced the levels of ATF4 expression in DENV-infected cells. Nevertheless, silencing of ATF4 expression only modestly (2-fold) altered the production of infections DENV particles. Therefore, ATF4 might contribute to the observed effect of tomadine yet it does not fully explain the potent antiviral effect observed.
There is not a consensus as to what is the preferred action (virus vs host) for the ideal anti-dengue therapy. Screening for DAA towards DENV is an intense research field and during the past decade great progress has been made in developing DENV inhibitors. However, DAA possess two main hurdles, first, the inherent differences between serotypes (69-78% identity at the amino acid level129) which hinders the development of pan-dengue-acting drugs. Secondly, there is an increased chance for the emergence of resistant variants. On the other hand, it is likely that dengue serotypes use the same host factors for replication and assembly; thus, a drug targeting cellular components may easier achieve pan-serotype antiviral activity. Furthermore, resistance to host-factor-targeted drugs is harder to accomplish. The disadvantage of targeting host factors is the higher potential to induce cytotoxicity and thereby, adverse secondary effects. Therefore, compounds which achieve potent antiviral effects at low doses are preferred, particularly if they can be used in children, as they account for the vast majority of severe dengue cases130. The high selectivity index we found for tomatidine in Huh7 cells (97.7) and the fact that it has been used safely in several mouse models, including pregnant mice131–133, is suggestive of a safe profile of the compound. Nevertheless, a proper assessment of safety in humans is currently lacking.
Final remarks: is tomatidine a potential antiviral drug?
So far, our results show a potent antiviral effect of tomatidine against DENV serotype 2 in cell lines. As mentioned earlier, an effective anti-DENV drug should act against all serotypes. Thus, the next step is to assess the antiviral effect against the other three DENV-serotypes. Furthermore, ADE experiments should be performed to test the effectivity of tomatidine against DENV-immune complexes. Along the course of our studies, we also observed that the response elicited in cell lines might be distinct from that in primary cells. Therefore, the antiviral effects of tomatidine should also be investigated in primary macrophages and DCs. Also, the duration of the antiviral effect of tomatidine should be investigated by prolonging the incubation times. Likewise, to receive more insight in the mode of action, the potential antiviral activity of tomatine derivatives should be tested. In parallel, detailed genetic and proteome analysis of tomatidine-treated cells may shed light on the mode of action of this compound. Alternatively, atemps to develop viral resistance might aid to dissect the mechanism of action.

194 | Chapter 8 Discussion and future perspectives | 195
8
Another, important feature to be investigated is the assessment of the safety and effectiveness of tomatidine in vivo. In vivo studies (animal models or clinical trials), should assess the ability of tomatidine to reduce viral loads early in infection and investigate whether it helps to alleviate the symptoms of severe disease late in infection. Tomatidine is known to reduce inflammation by inhibiting the NF-κB and JNK signaling (involved in the production of cytokines) and it was shown to reduce the age-related muscle weakness and atrophy in a mouse model116,134. Therefore, it is possible that next to reducing viral loads, tomatidine also acts late in infection thereby relieving the symptoms associated with inflammation and muscle pain.
Finally, gaining a better understanding of the interactions between DENV and its human host cells will pave the way for the further development of tomatidine and will lead to the discovery of new compounds with great antiviral potential. Finding the perfect anti-DENV drug will be feasible inasmuch as we fully decipher the molecular details of DENV replication and the cellular responses elicited upon infection.
References
1. Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, et al. The global distribution and burden of dengue. Nature. 2013 Apr 25;496(7446):504–7.
2. Halstead SB. Antibody-enhanced dengue virus infection in primate leukocytes. Nature. 1977;265:739–41.
3. Kraemer MUG, Sinka ME, Duda KA, Mylne AQN, Shearer FM, Barker CM, et al. The global distribution of the arbovirus vectors Aedes aegypti and Ae. Albopictus. Elife. 2015;4(JUNE2015):1–18.
4. Kyle JL, Harris E. Global Spread and Persistence of Dengue. Annu Rev Microbiol. 2008 Oct;62(1):71–92.
5. Sanofi Pasteur. Dengvaxia®, World’s First Dengue Vaccine, Approved In Mexico [Internet]. 2015 [cited 2017 Apr 6]. Available from: http://www.sanofipasteur.com/en/articles/dengvaxia-world-s-first-dengue-vaccine-approved-in-mexico.aspx
6. Sanofi Pasteur. First Dengue Vaccine Approved in More Than 10 Countries [Internet]. 2016 [cited 2017 Apr 6]. p. 4–7. Available from: http://www.sanofipasteur.com/en/articles/first_dengue_vaccine_approved_in_more_than_10_countries.aspx
7. Halstead SB. Critique of World Health Organization Recommendation of a Dengue Vaccine. J Infect Dis. 2016;214:jiw340.
8. Halstead SB. Licensed dengue vaccine: Public health conundrum and scientific challenge. Am J Trop Med Hyg. 2016;95(4):741–5.
9. Castro MC, Wilson ME, Bloom DE. Disease and economic burdens of dengue. Lancet Infect Dis. 2017;3099(16):1–9.
10. Chen Y, Wang S. Activation of Terminally Differentiated Human Monocytes / Macrophages by Dengue Virus : Productive Infection , Hierarchical Production of Innate Cytokines and Chemokines , and the Synergistic Effect of Lipopolysaccharide Activation of Terminally Different. J Virol. 2002;76(19):9877–87.
11. Wati S, Li P, Burrell CJ, Carr JM. Dengue Virus (DV) Replication in Monocyte-Derived Macrophages Is Not Affected by Tumor Necrosis Factor Alpha (TNF- ), and DV Infection Induces Altered Responsiveness to TNF- Stimulation. J Virol. 2007;81(18):10161–71.
12. Libraty D, Pichyangkul S, Ajariyakhajorn C, Endy T, Ennis F. Human Dendritic Cells Are Activated by Dengue Virus Infection :

Discussion and future perspectives | 195
8
Enhancement by Gamma Interferon and Implications for Disease Pathogenesis. J Virol. 2001;75(8):3501–3508.
13. Alayli F, Scholle F. Dengue virus NS1 enhances viral replication and pro-inflammatory cytokine production in human dendritic cells. Virology. 2016;496:227–36.
14. Schaeffer E, Flacher V, Papageorgiou V, Decossas M, Fauny J-D, Krämer M, et al. Dermal CD14(+) Dendritic Cell and Macrophage Infection by Dengue Virus Is Stimulated by Interleukin-4. J Invest Dermatol. 2015;135(7):1743–51.
15. Wu SJ, Grouard-Vogel G, Sun W, Mascola JR, Brachtel E, Putvatana R, et al. Human skin Langerhans cells are targets of dengue virus infection. Nat Med. 2000;6(7):816–20.
16. Cerny D, Haniffa M, Shin A, Bigliardi P, Tan BK, Lee B, et al. Selective Susceptibility of Human Skin Antigen Presenting Cells to Productive Dengue Virus Infection. PLoS Pathog. 2014;10(12).
17. Schmid M a., Harris E. Monocyte Recruitment to the Dermis and Differentiation to Dendritic Cells Increases the Targets for Dengue Virus Replication. PLoS Pathog. 2014;10(12):e1004541.
18. Libraty DH, Young PR, Pickering D, Endy TP, Kalayanarooj S, Green S, et al. High Circulating Levels of the Dengue Virus Nonstructural Protein NS1 Early in Dengue Illness Correlate with the Development of Dengue Hemorrhagic Fever. J Infect Dis. 2002;186(8):1165–8.
19. Rothman AL. Immunity to dengue virus: a tale of original antigenic sin and tropical cytokine storms. Nat Rev Immunol. 2011 Aug;11(8):532–43.
20. Boonnak K, Dambach KM, Donofrio GC, Tassaneetrithep B, Marovich M a. Cell type specificity and host genetic polymorphisms influence antibody-dependent enhancement of dengue virus infection. J Virol. 2011;85(4):1671–83.
21. Li D, Lott WB, Lowry K, Jones A, Thu HM, Aaskov J. Defective interfering viral particles in acute dengue infections. PLoS One. 2011;6(4).
22. Li D, Aaskov J. Sub-genomic RNA of defective interfering (D.I.) dengue viral particles is
replicated in the same manner as full length genomes. Virology. 2014;468:248–55.
23. Honke N, Shaabani N, Cadeddu G, Sorg UR, Zhang D-E, Trilling M, et al. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat Immunol. 2011;13(1):51–7.
24. Freigang S, Probst HC, Van Den Broek M. DC infection promotes antiviral CTL priming: The “Winkelried” strategy. Trends Immunol. 2005;26(1):13–8.
25. Pierson TC, Xu Q, Nelson S, Oliphant T, Nybakken GE, Fremont DH, et al. The Stoichiometry of Antibody-Mediated Neutralization and Enhancement of West Nile Virus Infection. Cell Host Microbe. 2007;1(2):135–45.
26. Boonnak K, Slike BM, Burgess TH, Mason RM, Wu S-J, Sun P, et al. Role of Dendritic Cells in Antibody-Dependent Enhancement of Dengue Virus Infection. J Virol. 2008;82(8):3939–51.
27. Richter MKS, Da Silva Voorham JM, Torres Pedraza S, Hoornweg TE, Van De Pol DPI, Rodenhuis-Zybert IA, et al. Immature dengue virus is infectious in human immature dendritic cells via interaction with the receptor molecule DC-SIGN. PLoS One. 2014;9(6).
28. Centers for Disease Control and Prevention (CDC). Dengue Laboratory Guidance and Diagnostic Testing [Internet]. 2016 [cited 2017 May 19]. Available from: https://www.cdc.gov/dengue/clinicallab/laboratory.html
29. Gollins SW, Porterfield JS. Flavivirus infection enhancement in macrophages: Radioactive and biological studies on the effect of antibody on viral fate. J Gen Virol. 1984;65(8):1261–72.
30. Gollins SW, Porterfield JS. Flavivirus Infection Enhancement in Macrophages : an Electron Microscopic Study of Viral Cellular Entry. J Gen Virol. 1985;66(May):1969–82.
31. Guzman MG, Vazquez S. The complexity of antibody-dependent enhancement of dengue virus infection. Viruses. 2010;2(12):2649–62.
32. Chareonsirisuthigul T, Kalayanarooj S, Ubol S. Dengue virus (DENV) antibody-dependent enhancement of infection upregulates the production of anti-inflammatory cytokines, but suppresses anti-DENV free radical and

196 | Chapter 8 Discussion and future perspectives | 197
8
pro-inflammatory cytokine production, in THP-1 cells. J Gen Virol. 2007;88(2):365–75.
33. Halstead SB, Mahalingam S, Marovich MA, Ubol S, Mosser DM. Intrinsic antibody-dependent enhancement of microbial infection in macrophages: Disease regulation by immune complexes. Lancet Infect Dis. 2010;10(10):712–22.
34. Ayala-Nunez N V., Hoornweg TE, van de Pol DPI, Sjollema KA, Flipse J, van der Schaar HM, et al. How antibodies alter the cell entry pathway of dengue virus particles in macrophages. Sci Rep. 2016;6(June):28768.
35. Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol. 2007 Aug;8(8):603–12.
36. Ubol S, Halstead SB. How innate immune mechanisms contribute to antibody-enhanced viral infections. Clin Vaccine Immunol. 2010 Dec;17(12):1829–35.
37. Yang KD, Yeh W-T, Yang M-Y, Chen R-F, Shaio M-F. Antibody-dependent enhancement of heterotypic dengue infections involved in suppression of IFNgamma production. J Med Virol. 2001;63(2):150–7.
38. Diamond MS, Roberts TG, Edgil D, Lu B, Ernst J, Harris E. Modulation of Dengue virus infection in human cells by alpha, beta, and gamma interferons. J Virol. 2000;74(11):4957–66.
39. Muñoz-Jordan JL, Sánchez-Burgos GG, Laurent-Rolle M, García-Sastre A. Inhibition of interferon signaling by dengue virus. Proc Natl Acad Sci U S A. 2003 Nov;100(24):14333–8.
40. Yu CY, Chang TH, Liang JJ, Chiang RL, Lee YL, Liao CL, et al. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog. 2012;8(6).
41. Angleró-Rodríguez YI, Pantoja P, Sariol CA. Dengue virus subverts the interferon induction pathway via NS2B/3 protease-IκB kinase ε interaction. Clin Vaccine Immunol. 2014;21(1):29–38.
42. Miller JL, de Wet BJM, deWet BJM, Martinez-Pomares L, Radcliffe CM, Dwek R a, et al. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog. 2008 Feb 8;4(2):e17.
43. Fire, Andrew; Xu, SiQun; Montgomery,
Mary K; Kostas, Steven A; Driver, Samuel E; Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Lett to Nat. 1998;391(February):806–11.
44. Lee Y, Kim M, Han J, Yeom K-H, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. Embo J. 2004;23(20):4051–60.
45. Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425(6956):415–9.
46. Lund E, Güttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004 Jan 2;303(5654):95–8.
47. Bernstein E, Caudy a a, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001 Jan 18;409(6818):363–6.
48. Liu J, Carmell MA, Rivas F V, Marsden CG, Thomson JM, Song J-J, et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305(5689):1437–41.
49. Rand TA, Petersen S, Du F, Wang X. Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell. 2005;123(4):621–9.
50. Sidahmed A, Abdalla S, Mahmud S, Wilkie B. Antiviral innate immune response of RNA interference. J Infect Dev Ctries. 2014;8(7):804–10.
51. Song L, Gao S, Jiang W, Chen S, Liu Y, Zhou L, et al. Silencing suppressors: viral weapons for countering host cell defenses. Protein Cell. 2011 Apr;2(4):273–81.
52. Hamilton AJ, Baulcombe DC. A Species of Small Antisense RNA in Posttranscriptional Gene Silencing in Plants. Science (80- ). 1999 Oct 29;286(5441):950–2.
53. Ding SW, Voinnet O. Antiviral Immunity Directed by Small RNAs. Cell. 2007;130(3):413–26.
54. Wu Q, Wang X, Ding SW. Viral suppressors of RNA-Based viral immunity: Host targets. Cell Host Microbe. 2010;8(1):12–5.
55. Nanduri S, Carpick BW, Yang Y, Williams BRG, Qin J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J.

Discussion and future perspectives | 197
8
1998;17(18):5458–65. 56. Alexopoulou L, Czopik Holt A, Medzhitov R,
Flavell RA. Recognition of double-stranded RNA and activation of NF-kappa B by Toll-like receptor 3. Nature. 2001;413(6857):732–8.
57. Hornung V, Kato H, Poeck H, Akira S, Conzelmann K, Schlee M. 5’- Triphosphate RNA is the ligand for RIG-I. Science (80- ). 2006;314:994–7.
58. Zust R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, et al. Ribose 2’-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol. 2011;12(2):137–43.
59. Cullen BR. Viruses and RNA interference: issues and controversies. J Virol. 2014;88(22):12934–6.
60. Otsuka M, Jing Q, Georgel P, New L, Chen J, Mols J, et al. Hypersusceptibility to vesicular stomatitis virus infection in Dicer1-deficient mice is due to impaired miR24 and miR93 expression. Immunity. 2007 Jul;27(1):123–34.
61. Parameswaran P, Sklan E, Wilkins C, Burgon T, Samuel M a, Lu R, et al. Six RNA viruses and forty-one hosts: viral small RNAs and modulation of small RNA repertoires in vertebrate and invertebrate systems. PLoS Pathog. 2010 Feb;6(2):e1000764.
62. Backes S, Langlois R a., Schmid S, Varble A, Shim J V., Sachs D, et al. The Mammalian Response to Virus Infection Is Independent of Small RNA Silencing. Cell Rep. 2014;8(1):114–25.
63. Hussain M, Asgari S. MicroRNA-like viral small RNA from Dengue virus 2 autoregulates its replication in mosquito cells. Proc Natl Acad Sci. 2014 Feb 3;111(7).
64. Skalsky RL, Olson KE, Blair CD, Garcia-Blanco M a, Cullen BR. A “microRNA-like” small RNA expressed by Dengue virus? Proc Natl Acad Sci U S A. 2014 Jun 10;111(23):E2359.
65. Bogerd HP, Skalsky RL, Kennedy EM, Furuse Y, Whisnant AW, Flores O, et al. Replication of Many Human Viruses Is Refractory to Inhibition by Endogenous Cellular MicroRNAs. J Virol. 2014 Jul 15;88(14):8065–76.
66. Seo GJ, Kincaid RP, Phanaksri T, Burke JM, Pare JM, Cox JE, et al. Reciprocal inhibition
between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe. 2013;14(4):435–45.
67. Haasnoot J, Berkhout B. RNAi and cellular miRNAs in infectiions by mammalian viruses. In: Rij RP, editor. Antiviral RNAi: Concepts, Methods, and Applications, Methods in Molecular Biology,. Totowa, NJ: Humana Press; 2011. (Methods in Molecular Biology; vol. 721).
68. Lecellier C, Dunoyer P, Arar K, Lehmann-che J, Eyquem S, Voinnet O. A Cellular MicroRNA Mediates Antiviral Defense in Human Cells. Science (80- ). 2005;308:557.
69. Ahluwalia JK, Khan SZ, Soni K, Rawat P, Gupta A, Hariharan M, et al. Human cellular microRNA hsa-miR-29a interferes with viral nef protein expression and HIV-1 replication. Retrovirology. 2008 Jan;5:117.
70. Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, et al. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med. 2007 Oct;13(10):1241–7.
71. Song L, Liu H, Gao S, Jiang W, Huang W. Cellular microRNAs inhibit replication of the H1N1 influenza A virus in infected cells. J Virol. 2010 Sep;84(17):8849–60.
72. Cheng J-C, Yeh Y-J, Tseng C-P, Hsu S-D, Chang Y-L, Sakamoto N, et al. Let-7b is a novel regulator of hepatitis C virus replication. Cell Mol Life Sci. 2012;69(15):2621–33.
73. Umbach JL, Cullen BR. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. 2009;1151–64.
74. Jopling C, Yi M, Lancaster A. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science (80- ). 2005;309(September):1577–81.
75. Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368(18):1685–94.
76. Flipse J, Diosa-Toro MA, Hoornweg TE, van de Pol DPI, Urcuqui-Inchima S, Smit JM. Antibody-Dependent Enhancement of Dengue Virus Infection in Primary Human Macrophages; Balancing Higher Fusion against Antiviral Responses. Sci Rep. 2016;6(January):29201.

198 | Chapter 8 Discussion and future perspectives | 199
8
77. Fischl W, Bartenschlager R. Exploitation of cellular pathways by Dengue virus. Curr Opin Microbiol. 2011 Aug;14(4):470–5.
78. Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature. 2016;535(7610):159–63.
79. Wu N, Gao N, Fan D, Wei J, Zhang J, An J. miR-223 inhibits dengue virus replication by negatively regulating the microtubule-destabilizing protein STMN1 in EAhy926 cells. Microbes Infect. 2014 Nov;16(11):911–22.
80. Escalera-Cueto M, Medina-Martínez I, Del Angel RM, Berumen-Campos J, Gutiérrez-Escolano AL, Yocupicio-Monroy M. Let-7c overexpression inhibits dengue virus replication in human hepatoma Huh-7 cells. Virus Res. 2014 Nov 20;196C:105–12.
81. Qi Y, Li Y, Zhang L, Huang J. microRNA expression profiling and bioinformatic analysis of dengue virus-infected peripheral blood mononuclear cells. Mol Med Rep. 2013 Mar;7(3):791–8.
82. Egana-Gorrono L, Escriba T, Boulanger N, Guardo AC, Leon A, Bargallo ME, et al. Differential MicroRNA expression profile between stimulated PBMCs from HIV-1 infected elite controllers and viremic progressors. PLoS One. 2014;9(9).
83. Zhao L, Zhu J, Zhou H, Zhao Z, Zou Z, Liu X, et al. Identification of cellular microRNA-136 as a dual regulator of RIG-I-mediated innate immunity that antagonizes H5N1 IAV replication in A549 cells. Sci Rep. 2015;5(October):14991.
84. tenOever BR. RNA viruses and the host microRNA machinery. Nat Rev Microbiol. 2013 Mar;11(3):169–80.
85. Wu S, He L, Li Y, Wang T, Feng L, Jiang L, et al. miR-146a facilitates replication of dengue virus by dampening interferon induction by targeting TRAF6. J Infect. 2013 Oct;67(4):329–41.
86. Wen W, He Z, Jing Q, Hu Y, Lin C, Zhou R, et al. Cellular microRNA-miR-548g-3p modulates the replication of dengue virus. J Infect. 2014 Dec 11;1–10.
87. Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on
protein output. Nature. 2008;455(7209):64–71.
88. Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455(7209):58–63.
89. Badalà F, Nouri-mahdavi K, Raoof DA. microRNA Therapeutics. Gene Ther. 2011;18(12):1104–10.
90. Maillard P V, Van der Veen AG, Deddouche-Grass S, Rogers NC, Merits A, Reis e Sousa C. Inactivation of the type I interferon pathway reveals long double-stranded RNA-mediated RNA interference in mammalian cells. EMBO J. 2016;35(23):2505–18.
91. Gartner Inc. Gartner Hype Cycle [Internet]. 2017 [cited 2017 May 9]. Available from: http ://www.gartner .com/technology/research/methodologies/hype-cycle.jsp
92. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993 Dec 3;75(5):843–54.
93. Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993 Dec 3;75(5):855–62.
94. Yue D, Liu H, Huang Y. Survey of Computational Algorithms for MicroRNA Target Prediction. Curr Genomics. 2009;10(7):478–92.
95. Witkos TM, Koscianska E, Krzyzosiak WJ. Practical Aspects of microRNA Target Prediction. Curr Mol Med. 2011;11(2):93–109.
96. Pritchard CC, Cheng HH, Tewari M. MicroRNA profiling: approaches and considerations. Nat Rev Genet. 2012 May;13(5):358–69.
97. Meyer SU, Pfaffl MW, Ulbrich SE. Normalization strategies for microRNA profiling experiments: a “normal” way to a hidden layer of complexity? Biotechnol Lett. 2010 Dec;32(12):1777–88.
98. Williamson V, Kim A, Xie B, McMichael GO, Gao Y, Vladimirov V. Detecting miRNAs in deep-sequencing data: a software performance comparison and evaluation. Brief Bioinform. 2013 Jan;14(1):36–45.
99. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism

Discussion and future perspectives | 199
8
and resistance to oxidative stress. Mol Cell. 2003;11(3):619–33.
100. Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17(10):1374–95.
101. Donnelly N, Gorman AM, Gupta S, Samali A. The eIF2 alpha kinases: Their structures and functions. Cell Mol Life Sci. 2013;70(19):3493–511.
102. Emara MM, Brinton M a. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc Natl Acad Sci U S A. 2007;104(21):9041–6.
103. Peña J, Harris E. Dengue virus modulates the unfolded protein response in a time-dependent manner. J Biol Chem. 2011;286(16):14226–36.
104. Umareddy I, Pluquet O, Wang QY, Vasudevan SG, Chevet E, Gu F. Dengue virus serotype infection specifies the activation of the unfolded protein response. Virol J. 2007;4:91.
105. Roth H, Magg V, Uch F, Mutz P, Klein P, Haneke K, et al. Flavivirus Infection Uncouples Translation Suppression from Cellular Stress Responses. MBio. 2017;8(1):e02150-16.
106. Uchida L, Espada-Murao LA, Takamatsu Y, Okamoto K, Hayasaka D, Yu F, et al. The dengue virus conceals double-stranded RNA in the intracellular membrane to escape from an interferon response. Sci Rep. 2014;4:7395.
107. Courtney SC, Scherbik S V., Stockman BM, Brinton M a. West Nile Virus Infections Suppress Early Viral RNA Synthesis and Avoid Inducing the Cell Stress Granule Response. J Virol. 2012;86(7):3647–57.
108. Olagnier D, Peri S, Steel C, van Montfoort N, Chiang C, Beljanski V, et al. Cellular Oxidative Stress Response Controls the Antiviral and Apoptotic Programs in Dengue Virus-Infected Dendritic Cells. PLoS Pathog. 2014;10(12):1–18.
109. Cheng Y-L, Lin Y-S, Chen C-L, Tsai T-T, Tsai C-C, Wu Y-W, et al. Activation of Nrf2 by the dengue virus causes an increase in CLEC5A, which enhances TNF-α production by mononuclear phagocytes. Sci Rep. 2016;6(August):32000.
110. Sheth U. Decapping and Decay of Messenger
RNA Occur in Cytoplasmic Processing Bodies. Science (80- ). 2003;300(5620):805–8.
111. Cougot N, Babajko S, Séraphin B. Cytoplasmic foci are sites of mRNA decay in human cells. J Cell Biol. 2004;165(1):31–40.
112. Kazemi S, Papadopoulou S, Li S, Su Q, Wang S, Yoshimura A, et al. Control of α Subunit of Eukaryotic Translation Initiation Factor 2 ( eIF2 α ) Phosphorylation by the Human Papillomavirus Type 18 E6 Oncoprotein : Implications for eIF2 α -Dependent Gene Expression and Cell Death Control of ␣ Subunit of Eukaryotic Transla. 2004;2(8):3415–29.
113. Goodman AG, Smith JA, Balachandran S, Perwitasari O, Proll SC, Thomas MJ, et al. The cellular protein P58IPK regulates influenza virus mRNA translation and replication through a PKR-mediated mechanism. J Virol. 2007;81(5):2221–30.
114. He B, Gross M, Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activa. Proc Natl Acad Sci U S A. 1997;94(3):843–8.
115. Li Y, Zhang C, Chen X, Yu J, Wang Y, Yang Y, et al. ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2α (eIF2α) and protein phosphatase 1. J Biol Chem. 2011;286(28):24785–92.
116. Ebert SM, Dyle MC, Bullard SA, Dierdorff JM, Murry DJ, Fox DK, et al. Identification and small molecule inhibition of an activating transcription factor 4 (ATF4)-dependent pathway to age-related skeletal muscle weakness and atrophy. J Biol Chem. 2015;290(42):25497–511.
117. Unnikrishnan R, Faizal BP, Vijayakumar P, George P, Sharma R. Clinical and laboratory profile of dengue fever. J Fam Med Prim Care. 2015;4(3):369–72.
118. Rajesh V, Holla V V, Vijay K, Amita J, Nuzhat H, Kiran PM, et al. A study of acute muscle dysfunction with particular reference to dengue myopathy. Ann Indian Acad Neurol. 2017;20(1):13–22.

200 | Chapter 8 Discussion and future perspectives | 201
8
119. Huang H, Jing G, Wang JJ, Sheibani N, Zhang SX. ATF4 is a novel regulator of MCP-1 in microvascular endothelial cells. J Inflamm (Lond). 2015;12(1):31.
120. Barker CE, Thompson S, O’Boyle G, Lortat-Jacob H, Sheerin NS, Ali S, et al. CCL2 nitration is a negative regulator of chemokine-mediated inflammation. Sci Rep. 2017;7(October 2016):44384.
121. Sierra B, Perez AB, Vogt K, Garcia G, Schmolke K, Aguirre E, et al. MCP-1 and MIP-1?? expression in a model resembling early immune response to dengue. Cytokine. 2010;52(3):175–83.
122. Lee YR, Liu MT, Lei HY, Liu CC, Wu JM, Tung YC, et al. MCP1, a highly expressed chemokine in dengue haemorrhagic fever/dengue shock syndrome patients, may cause permeability change, possibly through reduced tight junctions of vascular endothelium cells. J Gen Virol. 2006;87(12):3623–30.
123. Valadao ALC, Aguiar RS, de Arruda LB. Interplay between inflammation and cellular stress triggered by Flaviviridae viruses. Front Microbiol. 2016;7(AUG):1–19.
124. Libraty DH, Endy TP, Houng HH, Green S, Kalayanarooj S, Suntayakorn S, et al. Differing Influences of Virus Burden and Immune Activation on Disease Severity in Secondary Dengue-3 Virus Infections. J Infect Dis. 2002;185(9):1213–21.
125. Vaughn DW, Green S, Kalayanarooj S, Innis BL, Nimmannitya S, Suntayakorn S, et al. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J Infect Dis. 2000;181(1):2–9.
126. Perng GC, Lei H-Y, Lin Y-S, Chokephaibulkit K. Dengue Vaccines: Challenge and Confrontation. World J Vaccines. 2011;1(4):109–30.
127. Friedman M. Tomato glycoalkaloids: Role in the plant and in the diet. J Agric Food Chem. 2002;50(21):5751–80.
128. Lim SP, Wang QY, Noble CG, Chen YL, Dong H, Zou B, et al. Ten years of dengue drug discovery: Progress and prospects. Antiviral Res. 2013;100(2):500–19.
129. Urcuqui-inchima S, Patin C, Dı FJ. Recent developments in understanding dengue virus replication. Adv Virus Res. 2010;77(10).
130. Hammond SN, Balmaseda A, Pérez L, Tellez Y, Saborío SI, Mercado JC, et al. Differences in dengue severity in infants, children, and adults in a 3-year hospital-based study in Nicaragua. Am J Trop Med Hyg. 2005;73(6):1063–70.
131. Friedman M, Henika PR, Mackey BE. Effect of feeding solanidine, solasodine and tomatidine to non-pregnant and pregnant mice. Food Chem Toxicol. 2003;41(1):61–71.
132. Fujiwara Y, Kiyota N, Tsurushima K, Yoshitomi M, Horlad H, Ikeda T, et al. Tomatidine, a tomato sapogenol, ameliorates hyperlipidemia and atherosclerosis in ApoE-deficient mice by inhibiting acyl-CoA:cholesterol acyl-transferase (ACAT). J Agric Food Chem. 2012;60(10):2472–9.
133. Dyle MC, Ebert SM, Cook DP, Kunkel SD, Fox DK, Bongers KS, et al. Systems-based discovery of tomatidine as a natural small molecule inhibitor of skeletal muscle atrophy. J Biol Chem. 2014;289(21):14913–24.
134. Chiu FL, Lin JK. Tomatidine inhibits iNOS and COX-2 through suppression of NF-κB and JNK pathways in LPS-stimulated mouse macrophages. FEBS Lett. 2008;582(16):2407–12.
Discussion and future perspectives | 201
8






























