Embed Size (px)
Citation preview

HMG‐CoA Reductase Inhibitor Simvastatin Mitigates VEGF‐Induced “Inside‐Out” Signaling to Extracellular Matrix by Preventing RhoA Activation
Hanshi Xu1, Lixia Zeng1, Hui Peng1, Sheldon Chen1, Jonathan Jones2, Teng‐Leong Chew2, Mehran M. Sadeghi3, Yashpal S. Kanwar4, and Farhad R. Danesh1
1Division of Nephrology/Hypertension, 2Dept. of Cell and Molecular Biology,
Feinberg School of Medicine, Chicago, IL, 3Division of Cardiovascular Medicine, Yale School of Medicine, 4Dept. of Pathology, Northwestern University, Chicago, IL.
Running title: VEGF‐induced inside‐out signaling
Address correspondence: Farhad R. Danesh, M.D. Feinberg School of Medicine Northwestern University 303 E. Chicago Avenue Searle Building 10‐440 Chicago, IL 60611 Tel: 312‐5034753 e‐mail: f‐[email protected]
1
Page 1 of 40Articles in PresS. Am J Physiol Renal Physiol (June 13, 2006). doi:10.1152/ajprenal.00092.2006
Copyright © 2006 by the American Physiological Society.

ABSTRACT
The 3‐hydroxy‐3‐methylglutaryl coenzyme A (HMG‐CoA) reductase inhibitors exert
modulatory effects on a number of cell signaling cascades by preventing the
synthesis of various isoprenoids derived from the mevalonate pathway. In the
present study, we describe a novel pleiotropic effect of HMG‐CoA reductase
inhibitors, also commonly known as statins, on vascular endothelial growth factor
(VEGF)‐induced type IV collagen accumulation. VEGF is an angiogenic polypeptide
that is also known to play a central role in endothelial cell permeability and
differentiation. Recently, VEGF has also been implicated in promoting extracellular
matrix (ECM) accumulation, although the precise signaling mechanism that mediates
VEGF‐induced ECM expansion remains poorly characterized. Elucidation of the
mechanisms through which VEGF exerts its effect on ECM is clearly a prerequisite
for both understanding the complex biology of this molecule as well as targeting
VEGF in several pathological processes. To this end, this study explored the
underlying molecular mechanisms mediating VEGF‐induced ECM expansion in
mesangial cells. Our findings show that VEGF stimulation elicits a robust increase in
ECM accumulation that involves RhoA activation, an intact actin cytoskeleton, and
β1 integrin activation. Our data also indicate that simvastatin, via mevalonate
depletion, reverses VEGF‐induced ECM accumulation by preventing RhoA
activation.
Key words: HMG‐CoA reductase inhibitor, extracellular matrix, VEGF, focal
adhesion, mevalonate
2
Page 2 of 40

INTRODUCTION
Many of the cells in tissues are embedded in an extracellular matrix (ECM) that
provides not only scaffolding support for the cell, but it also presents environmental
cues to the cell which affect ultimately many aspects of a cell’s fate, including its
proliferation, differentiation, motility, and death. In the kidneys, the mesangial cells
are embedded in mesangial matrix, a basement membrane‐like PAS‐positive ECM.
The mesangial matrix, although composed of the same protein macromolecules as
the glomerular basement membrane, is more coarsely fibrillar and less electron dense
than the latter. Despite the importance of mesangial matrix in health and disease,
little is known regarding the cross‐talk between mesangial cells and mesangial
matrix and the signaling events that contribute to mesangial ECM expansion at the
present time. For instance, expansion of the mesangial matrix is considered to be a
major hallmark of diabetic nephropathy (23, 26). However, the signaling events that
mediate mesangial matrix expansion in the diabetic milieu remain controversial.
Interestingly, a number of recent observations have suggested that the vascular
endothelial growth factor (VEGF), a multifunctional cytokine, may play a central role
in the pathogenesis of microvascular complications of diabetes including diabetic
nephropathy through its modulatory effects on ECM (5, 8, 9, 28, 40, 45). Therefore, in
the current study, we explored the underlying molecular mechanisms of VEGF‐
induced ECM expansion in mesangial cells. In particular, we tested the role of RhoA
activation in actin cytoskeletal remodeling leading to aberrant ECM synthesis and
accumulation.
3
Page 3 of 40

The guanine nucleotide‐binding protein Rho family, consisting of Rho, Rac, and
Cdc42, are 20‐ to 40‐kD monomeric G proteins that can cycle between two inter‐
convertible forms: GDP‐bound (inactive) and GTP‐bound (active) states (34, 46).
Several growth factors can promote the exchange of GDP to GTP on Rho proteins
resulting in membrane translocation and activation of GTP‐bound Rho proteins. The
Rho family of small GTPases are involved in a number of essential biological
activities of the cell including actin stress fiber formation, cell motility, and cell
aggregation (1, 37). More recently, Rho GTPases have also emerged as key regulators
in the regulation of integrin‐mediated signaling (4, 11, 12).
Integrins are heterodimeric transmembrane molecules that consist of α and β
subunits. The main ligands for integrins are extracellular matrix adhesive proteins
and cellular counter‐receptors. High‐affinity ligand binding requires that integrins to
become activated by undergoing conformational changes regulated by “inside‐out”
signaling. In turn, integrin ligation triggers “outside‐in” signaling that regulate,
among many other functions, gene regulation and cell motility (13, 14, 18, 42, 44).
Integrin β subunit cytoplasmic domains are required for integrin activation, whereas
α cytoplasmic domain usually plays a regulatory role. Once integrins are activated,
one of the earliest changes initiated by their activation is tyrosin phosphorylation of
proteins such as talin, paxilin, and the cytosolic focal adhesion kinase (FAK) (31, 38).
FAK phosphorylation is considered to be a critical step in the integrin‐mediated
signaling events. In this study, we report a critical role for β1 integrin activation and
FAK phosphorylation in VEGF signaling to ECM.
4
Page 4 of 40

A growing body of evidence is suggesting that HMG‐CoA reductase inhibitors
(statins), by inhibiting mevalonate (MEV) biosynthesis, may also exert modulatory
effects on several signaling pathways beyond their cholesterol‐lowering properties
(27, 36, 47). For instance, we and others have previously provided evidence that some
of the beneficial effects of statins in the diabetic milieu may be mediated through
their modulatory effects on several signaling pathways (6, 7, 55). In the present study,
we extend our previous observations and describe the underlying signaling
mechanism through which VEGF leads to increased type IV collagen accumulation
in mesangial cells. We also explored the effect of mevalonate depletion by using
simvastatin (SMV), a hydrophobic statin, on VEGF‐induced signaling pathway.
5
Page 5 of 40

MATERIALS & METHODS
Reagents and antibodies
Recombinant rat VEGF165 was obtained from R&D Systems (Minneapolis, MN).
DMEM/F12, FBS, and PBS were purchased from Invitrogen (Carlsbad, CA).
Mevalonate (MEV) was purchased from Sigma Chemicals (St. Louis, MO). Rho A
Activation Assay Kit was obtained from Upstate Biotechnology (Lake Placid, NY).
Rho A antibody was purchased from Santa Cruz (Santa Cruz, CA). The monoclonal
anti‐β1 integrin subunit (active conformation) antibody, clone HUTS‐4, and function‐
blocking monoclonal mouse anti‐β1 integrin antibody were purchased from
Chemicon Inc. (Temecula, CA). The monoclonal anti‐β1 integrin subunit (active
conformation) antibody, clone HUTS‐21 was purchased from (BD PharMingen, San
Diego, CA) and Collagen IV antibody was from Novus Biologicals (Littleton, CO),
and cytochalasin D was obtained from Sigma Chemicals (ST. Louis, MO). Rhodamine
phalloidin was purchased from Molecular Probes (Eugene, OR). SMV was kindly
provided by Merck & Co, Inc. (West Point, PA). Both SMV and MEV were chemically
activated as described previously (6, 7, 55).
Cell Culture and transfection
Early passaged (3‐10th passage) rat glomerular mesangial cells (6, 7, 55) were grown
in DMEM/F12 medium containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml
streptomycin in a humidified incubator at 37°C under 5% CO2. Transfections of RhoA
mutants were performed as described previously (6, 7, 55). Briefly, in transfection
studies, cells were grown to 50%‐‐60% confluence and then transfected with 1 μg of
6
Page 6 of 40

fusion plasmid DNA using LipofectAMINE Reagent according to the manufacturer’s
protocol (Invitrogen). Subsequently, the transfected colonies were grown in growth
medium containing 800 μg/ml G418 until the cells achieve ~70% confluence.
Plasmids containing wild type RhoA (pcDNA3 RhoA) and dominant negative RhoA
construct (pcDNA3 RhoA N17) were generous gifts of Dr. Jacob Sznajder
(Northwestern University, Chicago, IL). In transfection studies, control experiments
were performed using lipofectAMINE alone as an additional control group.
Rho A activity pull‐down assay
RhoA activity was measured by a pull‐down assay according to the instructions by
the manufacturer (Rho Activity Assay Kit, Upstate Biotechnology). Briefly, 107 cells
were grown in 100‐mm dishes, washed in ice‐cold PBS twice, and lysed in ice‐cold
MLB buffer (25 mM HEPES [pH 7.5], 150 mM NaCl, 1% NP‐40, 10 mM MgCl2, 1.0
mM EDTA, and 2 % glycerol).The samples were centrifuged and incubated for 45
min at 4°C with 10 μl of rhotekin agarose to precipitate GTP‐bound Rho. Precipitated
complexes were washed three times in MLB buffer and resuspended in 30 μl of 2 x
Laemmli buffer. Total and precipitates were analyzed by performing SDS‐PAGE and
Western blot analysis using monoclonal anti‐RhoA antibody at a dilution of 1:500.
Confocal laser scanning fluorescence microscopy
MCs were grown on glass coverslips. The cells were fixed with 3%
paraformaldehyde and permeabilized with 0.2% Triton X‐100 in PBS for 10 min at
room temperature. The cells were incubated with anti‐phospho‐FAK antibody for 1 h
at room temperature, and then incubated with FITC‐conjugated secondary antibody
7
Page 7 of 40

(Zymed Laboratories, San Francisco, CA). To detect F‐actin, cells were incubated
with rhodamine phalloidin (1:100) for 30 m at 37°C after fixation and
permeabilization. The coverslips were mounted on glass slides with antifade
mounting media (Molecular Probes) and examined using a confocal fluorescence
microscope (Zeiss LSM510, Thornwood, NY).
Flow‐cytometric analysis
Near‐confluent growth‐arrested mesangial cells were pre‐treated with simvastatin or
vehicle control for 12‐18 hrs, after which VEGF (50 ng/ml) was added to the medium.
At the end of the indicated times, cells were harvested by HyQTase® (Hyclone,
Logan, UT) in their culture media and washed twice with 1X DPBS without Ca+2 and
Mg+2 (400 X g, 5 min, room temperature). Mesangial ells were then resuspended, and
fixed with 3.7% formaldehyde. Cells were then incubated with total anti‐integrin
antibody (Chemicon), monoclonal anti‐β1 integrin subunit (active conformation)
antibodies, clones HUTS‐21 (BD Pharmingen) and HUTS‐4 (Chemicon), in blocking
buffer (2% goat serum in DPBS without Ca+2 and Mg+2) for 45 min on ice. Cells were
then incubated with secondary antibodies on ice for 45 min. Flow‐cytometric analysis
was performed using a Beckman Coulter EPICS®XL‐MCL (Becton Dickinson, CA).
[3H]‐proline incorporation
The collagen synthesis was examined by [3H]‐proline incorporation. For these
experiments, 5x104 cells/well were seeded in 24‐well plates in DMEM/F12 medium
containing 10% FBS. At confluence, cells were starved with serum‐free medium for
48 hrs, and then the cells were exposed to various concentrations of VEGF in the
8
Page 8 of 40

presence or absence of SMV for 24 hrs. 2 μCi of [3H]‐proline (Amersham, Piscataway,
NJ) and ascorbic acid (50 μg/ml) were added to each well for the last 12 h of
incubation. The cells were washed twice with ice‐cold PBS, precipitated twice with
ice‐cold 10% TCA (Sigma), solubilized in 2ml 0.2N NaOH containing 0.1% Triton X‐
100. The samples were taken to liquid scintillation counting. Additional cells seeded
in parallel were scraped off the plate to detect the protein concentration. Proline
incorporation was expressed as counts per minute (cpm)/μg protein.
Western Blotting
For each experiment, a total of 5x105 cells were seeded, and at subconfluence (~70%),
cells were made quiescent for 48 h. Cells were rinsed twice with ice‐cold PBS and
added 0.5 ml of the ice‐cold lysis buffer [50 mM Tris Cl, pH 7.5, 150 mM NaCl, 100
μg/ml PMSF, 0.1% SDS, 1% Nonidet P‐40 (NP‐40), 0.5% sodium deoxycholate, 10
μg/ml aprotinin, 2 μg/ml leupeptin, 10 mM EDTA], incubated for 20 min on ice, and
then were scraped and centrifuged. Protein concentrations were determined by the
BCA protein assay (Pierce, Rockford, IL), and then separated by SDS‐polyacrylamide
gel electrophoresis and transferred to nitrocellulose membranes. The membranes
were probed with primary antibodies diluted in TBS‐T containing 5% nonfat milk at
4°C overnight. The membranes were incubated with the appropriate secondary
antibodies for 1 h at room temperature. Immunoreactive bands were visualized by
enhanced chemiluminescence (ECL) reaction. Each blot is representative of at least
three similar independent experiments.
For detection of occupied β1 integrins, cells were plated on collagen‐coated slides
9
Page 9 of 40

prior to VEGF stimulation. Cells were incubated with HUTS‐21 or HUTS‐4
antibodies for 30 min at 370. Cells were rinsed with PBS, lysed in SDS sample buffer
and bound antibody was detected by Western blot analysis.
Statistical analyses
Data are expressed as mean±SEM. ANOVA with a Student‐Newman‐Keuls test was
used to evaluate differences between two or more different experimental groups. A
value of p≤0.05 was considered significant.
10
Page 10 of 40

RESULTS
Effects of VEGF and SMV on cell‐associated collagen synthesis
VEGF‐induced cell‐associated collagen synthesis in mesangial cells (MC) was
measured by [3H]‐proline incorporation. Serum‐starved cultured rat MCs were
exposed to incremental concentrations of VEGF for 24 hrs. As shown in Figure 1,
VEGF stimulation caused a significant increase in [3H]‐proline incorporation in a
dose‐dependent manner. Co‐treatment of cells with SMV (1 μM) prevented VEGF‐
induced (50 ng/ml) increase in [3H]‐proline uptake. Addition of MEV (300 μM)
reversed the effect of SMV on [3H]‐proline incorporation indicating that the
inhibitory effect of SMV on VEGF‐induced [3H]‐proline incorporation was MEV‐
dependent. We next tested whether the inhibitory effect of SMV on VEGF signaling
was mediated by isoprenoids derived from the mevalonate pathway. To this end,
VEGF‐stimulated MCs were incubated with SMV (1 μM) and co‐treated with either
geranyl geranyl pyrophosphate (GGPP) or squalene (SQ), two important isoprenoids
derived from the mevalonate pathway. GGPP was used since post‐translational
modification of GGPP is negatively affected by statin‐mediated inhibition of HMG‐
CoA reductase. We also investigated the effect of SQ, an immediate precursor of
cholesterol, to test whether the inhibitory effect of SMV on VEGF signaling is
independent of cholesterol biosynthesis. The data on figure 1 show that co‐treatment
of cells with GGPP but not with SQ reversed the inhibitory effect of SMV, suggesting
that the modulatory effect of statins on VEGF signaling is geranyl geranyl‐dependent
but independent of cholesterol synthesis since co‐treatment of cells with SQ failed to
11
Page 11 of 40

reverse the effect of SMV. Taken together, these findings indicate that the inhibitory
effect of SMV on VEGF signaling is MEV and geranyl geranyl‐dependent.
We next examined the effect of VEGF and SMV on type IV collagen protein levels by
Western blot analysis. As shown in Figure 2, cells exposed to incremental
concentrations of VEGF exhibited significant increase in type IV collagen protein
levels. Co‐treatment of VEGF‐stimulated cells with SMV (1 μM) abrogated the effect
of VEGF (50 ng/ml) on cell‐layer collagen type IV protein levels. The inhibitory effect
of SMV was reversed when cells were co‐treated with MEV (300 μM).
Effects of VEGF and SMV on RhoA activity
We have recently reported that SMV modulates the activation of the Rho family of
small GTPases in the diabetic milieu (6, 7, 55). To decipher whether RhoA, a member
of the Rho family of small GTPases, mediates VEGF‐induced collagen accumulation,
MCs were exposed to VEGF (50 ng/ml), and RhoA activity was measured by an
affinity pull‐down assay using GST fusion protein rhotekin, which recognizes only
the active form of RhoA (GTP‐RhoA). VEGF stimulation significantly increased RhoA
activity in MCs after 20 min (Figure 3). To examine the effect of SMV on VEGF‐
induced RhoA activity, cells stimulated with VEGF were co‐treated with SMV (1 μM).
As shown in figure 3, co‐treatment of VEGF‐stimulated cells with SMV inhibited
VEGF‐induced increase in RhoA activity without significantly changing total RhoA
protein levels. The inhibitory effect of SMV on RhoA activity was reversed when cells
were co‐treated with MEV (300 μM), indicating that the effect of SMV on VEGF‐
induced RhoA activation was MEV‐dependent.
12
Page 12 of 40

To establish a link between RhoA activation and VEGF‐induced type IV collagen
protein levels, MCs were transfected with dominant‐negative mutant (N17RhoA)
and wild‐type (wt) RhoA. The data in Figure 4 show that VEGF (50 ng/ml)
stimulation failed to increase cell‐layer collagen IV protein levels in dominant
negative RhoA transfected cells, indicating a critical role for RhoA activation in
VEGF‐induced collagen synthesis.
Effects of VEGF and SMV on the actin cytoskeleton remodeling
To examine whether VEGF signaling in MCs involves actin cytoskeleton remodeling
and to elucidate the modulatory effect of SMV on VEGF‐induced cytoskeletal
remodeling, MCs were incubated with VEGF (50 ng/ml) for 30 min in the presence or
absence of SMV (1 μM) and the actin cytoskeleton was visualized by rhodamine
phalloidin staining using scanning confocal electron microscopy. MCs treated with
VEGF exhibited a significant increase in actin stress fiber formation (Figure 5‐b).
Upon treatment with SMV (1 μM), the density of stress fibers was significantly
decreased (Figure 5‐c). Because RhoA is one of the major regulator of actin stress
fiber formation, we further examined whether RhoA mediates VEGF‐induced stress
fiber formation. As shown in Figure 5‐d, MCs transfected with dominant negative
RhoA also showed a significant reduction in VEGF‐induced density of stress fibers,
indicating that VEGF‐induced actin stress fiber formation was mediated by a RhoA‐
dependent pathway.
13
Page 13 of 40

Effects of VEGF and SMV on tyrosine phosphorylation of focal adhesion
kinase
Focal adhesion kinase (FAK), a cytoplasmic protein tyrosine kinase, binds directly to
the cytoplasmic domain of β1 integrin subunits and plays an important role in the
integrin‐mediated signaling pathway (31, 38). To examine the potential involvement
of FAK phosphorylation in VEGF‐induced signaling, we performed laser scanning
confocal immunofluorescent microcopy using anti‐phosphorylated FAK (Tyr397)
antibody. MCs stimulated with VEGF (50 ng/ml) exhibited a significant increase in
FAK Tyr397 phosphorylation (Figure 6). However, in cells co‐treated with SMV (1
μM), VEGF‐induced FAK phosphorylation was significantly reduced (Figure 6‐c).
MCs transfected with dominant negative mutant of RhoA also showed a significant
decrease in VEGF‐induced FAK phosphorylation (Figure 6‐d) indicating the central
role of RhoA in VEGF‐induced FAK phosphorylation.
We also assessed VEGF‐induced total and phosphorylated FAK protein expression
by Western blot analysis. Whereas VEGF stimulation (50 ng/ml) significantly
increased phospho‐FAK protein levels, SMV (1 μM) and MCs transfected with
N17RhoA exhibited significantly lower protein levels of phospho‐FAK. Total FAK
protein levels were unchanged as shown in Figure 7. Thus, our data indicate that
VEGF signaling pathway involves FAK phosphorylation, and RhoA mediates VEGF‐
induced FAK phosphorylation.
For determining whether actin cytoskeleton and FAK phosphorylation are involved
in VEGF‐induced cell‐associated collagen synthesis, MCs were treated with
14
Page 14 of 40

cytochalasin D (Cyto D, 1.0 μg/ml). Cyto D, a specific inhibitor of filament actin
cytoskeleton, has also been shown to abolish FAK phosphorylation (25). As shown in
Figure 8, VEGF (50 ng/ml) stimulation did not increase the levels of cell‐layer
collagen type IV in MCs treated with Cyto D, indicating that VEGF‐induced actin
cytoskeleton reorganization is necessary for VEGF‐induced collagen synthesis.
Effects of VEGF and SMV on VEGF‐induced β1 integrin activation
Growing evidence has indicated that β1 integrins mediate growth factor‐dependent
expansion of ECM in MCs (23, 26). However, whether VEGF‐induced mesangial
matrix expansion is also mediated by β1 integrin activation and whether RhoA
activation is necessary for VEGF‐induced β1 integrin activation have not yet been
explored. Accordingly, we examined the effect of VEGF and SMV on β1 integrin
activation. Integrins can switch between active and inactive conformations. In the
inactive state, integrins have a low affinity for ligands. Intracellular signaling events
such as protein kinase C stimulation can prime the integrins, which result in a
conformational change that makes the extracellular domain competent for ligand
binding by exposing the ligand‐binding site. To address the effect of VEGF and SMV
on β1 integrin activation, serum‐starved MCs were stimulated with VEGF (50 ng/ml)
and β1 integrin activation was detected by flow cytometry using an antibody (HUTS‐
21) specific for the active conformation of β1 integrin (16, 25, 41, 48). The data
obtained from flow cytometry in figure 9‐a show that VEGF clearly enhances the
expression of activated β1 integrins in MCs. The results in figure 9‐a further show
that SMV ameliorates activation of β1 integrins by VEGF. To test if SMV also
15
Page 15 of 40

modulates total β1 integrin levels, MCs were stimulated with VEGF (50 ng/ml) and
total β1 integrin levels were analyzed. The results of flow cytometry indicate that
VEGF stimulation increased both the total and activated β1 integrin. However, SMV
only prevented conformational activation of β1 integrin without significantly altering
total β1 integrin levels (Figure 9‐b).
To further explore the role of RhoA activation and to examine the effects of VEGF
and SMV on β1 integrin‐ligand binding, MCs were plated on collagen‐coated slides
prior to VEGF (50 ng/ml) stimulation. Cells were incubated with HUTS‐21 or HUTS‐4
antibodies, which specifically recognize the expression of the active conformation of
β1 integrins, for 30 min at 370. Cells were then lysed in SDS sample buffer and bound
antibody was detected by Western blot analysis as previously described (48). The
results in figure 9‐c indicate that stimulation of MCs with VEGF caused a significant
increase in binding of HUTS‐4 as well as HUTS‐21 (data not shown). Co‐treatment of
cells with SMV (1 μM) inhibited VEGF‐induced increase in β1 integrin activation. The
inhibitory effect of SMV was reversed when the cells were co‐treated with MEV (300
μM). The data in figure 9‐c also indicate that RhoA activation is necessary to
stimulate β1 integrin activation since VEGF stimulation failed to activate β1 integrin
in MCs transfected with a dominant negative mutant of RhoA, N17RhoA, indicating
that VEGF‐induced β1 integrin activation is mediated by a RhoA‐dependent
pathway. This experiment also provides strong evidence for the proposed VEGF‐
induced “inside‐out” signaling pathway since our data indicate that RhoA activation
is necessary prior to β1 integrin activation.
16
Page 16 of 40

To further ascertain that β1 integrin activation is necessary for VEGF‐induced
collagen synthesis, MCs were treated with a monoclonal function‐blocking anti‐β1
integrin antibody (20 μg/ml) and stimulated with VEGF (50 ng/ml). As shown in
Figure 10, VEGF stimulation did not increase the protein levels of cell‐layer collagen
type IV in MCs when β1 integrin activation was blocked, indicating that β1 integrin
activation is necessary for VEGF‐induced cell‐layer collagen synthesis.
17
Page 17 of 40

DISCUSSION
This study describes the signaling events that are responsible for VEGF‐induced
collagen IV accumulation in MCs. Our data suggest that VEGF triggers an inside‐out
signaling pathway in MCs which is characterized by RhoA activation, actin
cytoskeleton remodeling, intracellular FAK phosphorylation, and β1 integrin
activation leading to increased type IV collagen synthesis. Thus, the results of the
present study provide a missing piece of the mechanistic puzzle concerning VEGF‐
induced mesangial matrix expansion. Furthermore, our data also provide strong
evidence that SMV, by preventing RhoA activation, inhibits VEGF signaling pathway
and enhanced collagen synthesis in MCs.
Statins are commonly used drugs in the treatment of patients with
hypercholesterolemia that have been suggested to exert significant pleiotropic effects
on cell signaling pathways largely by preventing mevalonic acid biosynthesis (27, 36,
47, 50, 51) . Mevalonate is necessary for the posttranslational lipid modification
(isoprenylation) of small GTPase proteins, a process essential for the proper
translocation of Rho GTPases from the cytosol to the membrane where activation of
these proteins takes place (3, 15, 43). Previous studies from our laboratory and others
have indicated that statins modulate several cellular processes by preventing
prenylation of small Rho GTPases such as RhoA and Rac1 (6, 7, 20, 27, 29, 36, 49, 55).
In support of a modulatory effect of statins on ECM accumulation, Kim et al.
reported that lovastatin inhibited high glucose‐induced overexpression of fibronectin
(20). Similarly, Nishimura et al. showed that pravastatin prevented serum‐induced
18
Page 18 of 40

type IV collagen secretion (29). The data presented in this study are consistent with
these previous results by establishing the inhibitory effects of statins on ECM
expansion. In addition, we have clearly characterized the sequential activation of
various components of VEGF‐induced signaling pathway in the current study and
identified the modulatory effect of HMG‐CoA reductase inhibitors and mevalonate
depletion on VEGF signaling.
To decipher VEGF signaling, we initially explored the role of RhoA activation on
VEGF‐induced signaling pathway in MCs. We showed that RhoA activity is required
for VEGF‐induced collagen synthesis since MCs transfected with dominant negative
RhoA mutant failed to increase cell‐layer type IV collagen protein levels in response
to VEGF. Co‐treatment of MCs with SMV also inhibited VEGF‐induced collagen
accumulation by preventing RhoA activation. Addition of MEV reversed the
inhibitory effect of SMV on VEGF‐induced RhoA activity and collagen accumulation,
indicating that the effect of SMV on VEGF‐induced collagen accumulation was MEV‐
dependent. Taken together, our data suggest that RhoA mediates VEGF‐induced
collagen synthesis; and SMV inhibits VEGF signaling to ECM by preventing RhoA
inactivation.
The list of cellular events instigated by actin cytoskeletal reorganization is rapidly
growing. For instance, Hubchak et al. showed that TGF β1‐induced collagen
accumulation is associated with cytoskeleton reorganization (17). Moreover, Cyto D,
an inhibitor of actin cytoskeletal assembly, has been previously reported to disrupt
the formation of fibronectin networks (30, 54). In this study, we report that VEGF
19
Page 19 of 40

induces actin cytoskeletal rearrangement in MCs, and disruption of actin
cytoskeleton with Cyto D reduces VEGF‐stimulated collagen type IV synthesis.
Furthermore, our data indicate that VEGF‐induced actin cytoskeletal remodeling is
mediated by RhoA activation which leads to ß1 integrin activation. Thus, the results
of this study show that actin cytoskeletal remodeling, regulated by RhoA activation,
is required for VEGF‐induced collagen synthesis.
Integrins, a group of heterodimeric transmembrane receptors, play a pivotal role in
ECM assembly and cell‐ECM interactions (32, 35). A critical role of integrins is to
provide a link between ECM and the cytoskeleton, whereby ECM ligand‐integrins
interactions can activate intracellular signaling transduction cascades resulting in
enhanced gene expression, and cellular differentiation (“outside‐in” signaling) (13,
42). Interestingly, several recent studies have indicated that signals from within the
cells can also propagate through integrins and regulate ECM remodeling through an,
as yet, incompletely understood mechanism termed “inside‐out” signaling (14, 18,
19, 39, 44). Activation of integrins through inside‐out signaling seems to be mediated
by increased affinity and/or avidity state of integrins (44). The affinity state of
integrin is regulated by its conformational changes induced by a number of signaling
pathways or proteins that interact with the cytoplasmic domains of the integrins.
Structural studies suggest that modulation of integrin affinity involves changes in the
spatial relationship of the cytoplasmic and/or transmembrane domains of the α and ß
subunits (52). The ß cytoplasmic domain is critical for recruitment of integrins to
focal contacts since its truncation/mutation impairs this process (33). Several
20
Page 20 of 40

previous studies have suggested a central role for β1 integrin activation in mesangial
matrix accumulation (21, 22, 24). In this study, we assessed the contribution of β1
integrin activation on VEGF signaling to ECM. Our data indicate that VEGF
stimulation significantly increases β1 integrins activity. Moreover, the results of this
study suggest that SMV inhibits increased affinity of β1 integrins via a MEV‐
dependent pathway. Thus, the data presented in this study provides strong evidence
indicating that β1 integrins activation is required for VEGF‐induced collagen
synthesis since a specific β1 integrin blocker prevented increased protein levels of
cell‐layer collagen type IV. Our study also provides the first evidence that VEGF‐
induced activation of β1 integrins in MCs is mediated by RhoA activation because
VEGF stimulation failed to increase β1 integrin activity in cells transfected with
dominant negative mutant of RhoA. Furthermore, our data clearly demonstrate that
RhoA activation precedes β1 integrin activation consistent with an inside‐out
signaling. This adds to the growing body of evidence of the importance of RhoA
activation in inside‐out signaling.
Several cytoplasmic proteins including talin, α‐actin, and FAK bind to the β1
cytoplasmic domain of integrins and contribute to integrin‐cytoskeletal interactions
(2, 10, 31, 38, 53). However, earlier studies on integrin‐dependent cell adhesion and
signaling demonstrated that integrin clustering could also trigger increased tyrosine
phosphorylation of a120 kD tyrosine kinase known as FAK (2, 10, 53). FAK activation
as demonstrated by an increase in phosphorylation on Tyr397 is an integral
component of the integrin signaling pathway (10, 53). In order to explore whether
21
Page 21 of 40

FAK phosphorylation was involved in VEGF‐induced signaling pathway, we
examined the effect of VEGF and SMV on FAK phosphorylation. The data presented
in this study suggest that FAK phosphorylation is involved in the inside‐out VEGF‐
dependent signaling by transmitting RhoA activation and actin cytoskeletal cues to
ECM leading ultimately to collagen accumulation. Our data indicate that VEGF
stimulation increased FAK Tyr397 phosphorylation, and FAK phosphorylation was
inhibited by SMV. We also showed that VEGF failed to increase FAK
phosphorylation in MCs transfected with dominant negative mutant of RhoA. Thus,
our results suggest that VEGF, via a RhoA‐dependent pathway, mediates tyrosine
phosphorylation of FAK.
In conclusion, we have demonstrated that VEGF‐induced collagen accumulation
involves several mediators that include activation of small GTPase protein, RhoA,
reorganization of the actin cytoskeleton, FAK phosphorylation and activation of β1
integrins. Based on these data, we propose an inside‐out signaling model for VEGF‐
induced mesangial matrix expansion. This model provides several novel targets in
the therapeutic approach to the abnormal mesangial matrix expansion observed in
the diabetic milieu. Our study also provides a new rationale to the use of statins,
independent of their cholesterol‐lowering properties, in the early stages of DN.
ACKNOWLEDGEMENT:
This study was supported by grants from the National Institutes of Health (DK67604 and DK064106), and American Diabetes Association (1‐03‐RA‐15).
22
Page 22 of 40

REFERENCES
1. Burridge K, Wennerberg, K. Rho and Rac take center stage. Cell 116: 167-179, 2004. 2. Calderwood DA, Zent R, Grant R, Rees DJG, Hynes RO, and Ginsberg MH. The Talin Head Domain Binds to Integrin beta Subunit Cytoplasmic Tails and Regulates Integrin Activation. J Biol Chem 274: 28071-28074, 1999. 3. Casey PJ. Protein lipidation in cell signaling. Science 268:: 221-225, 1995. 4. Clark EA, King, W.G., Brugge, J.S., Symons, M., Hynes, R.O. Integrin-mediated Signals Regulated by members of the Rho Family of GTPases. J Cell Biol 142: 573-586, 1998. 5. Cooper ME, Vranes, D., Youssef, S., Stacke, S.A. Cox, A.J., Rizkalla, B., Casley, D.J., Bach, L.A., Kelly, D.J., Gilbert, R.E. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptors in experimental diabetes. Diabetes 48: 2229-2239, 1993. 6. Danesh FR, Sadeghi MM, Amro N, Philips C, Zeng L, Lin S, Sahai A, and Kanwar YS. 3-Hydroxy-3-methylglutaryl CoA reductase inhibitors prevent high glucose-induced proliferation of mesangial cells via modulation of Rho GTPase/ p21 signaling pathway: Implications for diabetic nephropathy. PNAS 99: 8301-8305, 2002. 7. Danesh FR and Y.S. K. Modulatory effects of HMG-CoA reductase inhibitors in diabetic microangiopathy. FASEB J 18: 805-815, 2004. 8. de Vriese AS, Tilton, R.G., Elger, M., Stephan, C.C., Kriz, W., Lameire, N.H. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J Am Soc Nephrol 12: 993-1000, 2001. 9. Flyvbjerg A, Dagnes-Hansen, F., Flyvbjerg, A., Dagnaes-Hansen, F., De Vriese, A.S., Schrijvers, B.F., Tilton, R.G., Rasch, R. Amelioration of long-term renal changes in obese type 2 diabetic mice by a neutralizing vascular endothelial growth factor antibody. Diabetes 51, 2002. 10. Frisch S, Vuori K, Ruoslahti E, and Chan-Hui P. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol 134: 793-799, 1996. 11. Fukata M, Nakagawa M, Kuroda S, and Kaibuchi K. Cell adhesion and Rho small GTPases. J Cell Sci 112: 4491-4500, 1999. 12. Geiger B, Bershadsky, A., Pankov, R., Yamada, K.M. Transmembrane crosstalk between the extracellular matrix-cytoskeleton crosstalk. Nat Rev Mol Cell Biol 2:: 793-805, 2001. 13. Giancotti FG and Ruoslahti E. Integrin Signaling. Science 285: 1028-1033, 1999. 14. Ginsberg M, Partridge A, and Shattil S. Integrin regulation. Curr Opin Cell Biol 17: 509-516, 2005. 15. Goldstein J and Brown M. Regulation of the mevalonate pathway. Nature 343: 425-430, 1990. 16. Gomez M, Luque, A., del Pozo, M.A., Hogg, N., Sanchez-Madrid, F., Cabanas, C. Functional relevance during lymphocyte migration and cellular localization of activated beta1 integrins. Eur J Immunol 27: 8-16, 1997. 17. Hubchak SC, Runyan CE, Kreisberg JI, and Schnaper HW. Cytoskeletal Rearrangement and Signal Transduction in TGF-{beta}1-Stimulated Mesangial Cell Collagen Accumulation. J Am Soc Nephrol 14: 1969-1980, 2003. 18. Hughes P and Pfaff M. Integrin affinity modulation. Trends Cell Biol 8: 359-364, 1998. 19. Hynes R. Integrins: bidirectional, allosteric signaling machines. Cell 110: 673-687, 2002.
23
Page 23 of 40

20. IL KIM S, HAN DC, and LEE HB. Lovastatin Inhibits Transforming Growth Factor-{beta}1 Expression in Diabetic Rat Glomeruli and Cultured Rat Mesangial Cells. J Am Soc Nephrol 11: 80-87, 2000. 21. Jin D, Fish A, Wayner E, Mauer M, Setty S, Tsilibary E, and Kim Y. Distribution of integrin subunits in human diabetic kidneys. J Am Soc Nephrol 7: 2636-2645, 1996. 22. Kagami S, Border W, Ruoslahti E, and Noble N. Coordinated expression of beta 1 integrins and transforming growth factor-beta-induced matrix proteins in glomerulonephritis. Lab Invest 69: 68-76, 1993. 23. Kanwar YS, Wada, J., Lin, S., Danesh, F.R., Chugh, S.S., Yang, Q., Banerjee, T., Lomasney, J.W. Update of extracellular matrix, its receptors, and cell adhesion molecules in mammalian nephrogenesis. Am J Physiol Renal Physiol 286: F202-215, 2004. 24. Kuhara T, Kagami S, and Kuroda Y. Expression of beta 1-integrins on activated mesangial cells in human glomerulonephritis. J Am Soc Nephrol 8: 1679-1687, 1997. 25. Luque A, Gómez M, Puzon W, Takada Y, Sánchez-Madrid F, and Cabañas C. Activated Conformations of Very Late Activation Integrins Detected by a Group of Antibodies (HUTS) Specific for a Novel Regulatory Region(355-425) of the Common beta1 Chain. J Biol Chem 271: 11067-11075, 1996. 26. Mason RM, Wahab, N.A. Extracellular matrix metabolism in diabetic nephropathy. J Am Soc Nephrol 14: 1358-1373, 2003. 27. McFarlane SI, Muniyappa R, Francisco R, and Sowers JR. Pleiotropic Effects of Statins: Lipid Reduction and Beyond. J Clin Endocrinol Metab 87: 1451-1458, 2002. 28. Neufeld G, Cohen, T., Gengrinovitch, S., Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J 13, 1999. 29. Nishimura M, Tanaka T, Yasuda T, Kurakata S, Kitagawa M, Yamada K, Saito Y, and Hirai A. Collagen secretion and growth of mesangial cells require geranylgeranylpyrophosphate. Kidney Int 55: 520-528, 1999. 30. Ohashi T, Kiehart, D.P., Erickson, H.P. Dual labeling of the fibronectin matrix and actin cytoskeleton with green fluorescent protein variants. J Cell Sci 115: 1221-1229, 1997. 31. Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci 116: 1409-1416, 2003. 32. Pedchenko VK, Chetyrkin SV, Chuang P, Ham A-JL, Saleem MA, Mathieson PW, Hudson BG, and Voziyan PA. Mechanism of Perturbation of Integrin-Mediated Cell-Matrix Interactions by Reactive Carbonyl Compounds and Its Implication for Pathogenesis of Diabetic Nephropathy. Diabetes 54: 2952-2960, 2005. 33. Reszka A, Hayashi Y, and Horwitz A. Identification of amino acid sequences in the integrin beta 1 cytoplasmic domain implicated in cytoskeletal association. J Cell Biol 117: 1321-1330, 1992. 34. Ridley AJ, Hall, A. The small GTP-binding protein Rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70: 389-400, 1992. 35. Ruoslahti E and Engvall E. Integrins and Vascular Extracellular Matrix Assembly. J Clin Invest 99: 1149-1152, 1997. 36. Sadeghi MM, Collinge M, Pardi R, and Bender JR. Simvastatin Modulates Cytokine-Mediated Endothelial Cell Adhesion Molecule Induction: Involvement of an Inhibitory G Protein. J Immunol 165: 2712-2718, 2000. 37. Sahai E, Marshall, C.J. RHO-GTPases and cancer. Nat Rev Cancer 2: 133-142, 2002.
24
Page 24 of 40

38. Schaller M. Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim Biophys Acta 1540: 1-21, 2001. 39. Schoenwaelder S and Burridge K. Bidirectional signaling between the cytoskeleton and integrins. Curr Opin Cell Biol 11: 274-286, 1999. 40. Schrijvers BF, Flyvbjerg A., De Vriese, A.S. The role of vascular endothelial growth factor (VEGF) in renal pathophysiology. Kidney Int 65: 2003-2017, 2004. 41. Schwartz M. Integrin signaling revisited. Trends Cell Biol 11: 466-470, 2001. 42. Schwartz M, Schaller M, and Ginsberg M. Integrins: emerging paradigms of signal transduction. Annu Rev Cell Dev Biol 11: 549-599, 1995. 43. Scit G, Tenca, P., Frittoli, E. Signaling from Ras to Rac and beyond: Not just a matter of GEFs. EMBO J 19: 2393-2398, 2000. 44. Shimaoka M, Takagi J, and Springer T. Conformational regulation of integrin structure and function. Annu Rev Biophys Biomol Struct 31: 485-516, 2002. 45. Sugimoto H, Hamano, Y., Charytan, D., Cosgrove, D., Kieran, M., Sudhakar, A., Kalluri, R. Neutralization of circulating vascular endothelial growth factor (VEGF) by anti-VEGF antibodies and soluble VEGF receptor 1 (sFlt-1) induces proteinuria. J Biol Chem 278: 12605-12608, 2003. 46. Takai Y, Sasaki, T., Matozaki, T. (2001) . Small GTP-binding proteins. Physiol Rev 81: 153-208, 2001. 47. Takemoto M and Liao JK. Pleiotropic Effects of 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Inhibitors. Arterioscler Thromb Vasc Biol 21: 1712-1719, 2001. 48. Tzima E, del Pozo M, Shattil S, Chien S, and Schwartz M. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J 20: 4639-4647, 2001. 49. Usui H, Shikata K, Matsuda M, Okada S, Ogawa D, Yamashita T, Hida K, Satoh M, Wada J, and Makino H. HMG-CoA reductase inhibitor ameliorates diabetic nephropathy by its pleiotropic effects in rats. Nephrol Dial Transplant 18: 265-272, 2003. 50. Vincent L, Albanese P, Bompais H, Uzan G, Vannier J, Steg P, Soria J, and Soria C. Insights in the molecular mechanisms of the anti-angiogenic effect of an inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Thromb Haemost 89: 530-537, 2003. 51. Vincent L, Soria C, Mirshahi F, Opolon P, Mishal Z, Vannier J-P, Soria J, and Hong L. Cerivastatin, an Inhibitor of 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase, Inhibits Endothelial Cell Proliferation Induced by Angiogenic Factors In Vitro and Angiogenesis in In Vivo Model Arterioscler Thromb Vasc Biol 22: 623-629, 2002. 52. Vinogradova O, Haas T, Plow EF, and Qin J. A structural basis for integrin activation by the cytoplasmic tail of the alpha IIb-subunit. PNAS 97: 1450-1455, 2000. 53. Wozniak MA, Modzelewska, K., Kwong, L., Keely, P.J. Focal adhesion regulation of cell behavior. Biochim Biophys Acta 1692: 103-119, 2004. 54. Wu C, Keivens V, O'Toole T, McDonald J, and Ginsberg M. Integrin activation and cytoskeletal interaction are essential for the assembly of a fibronectin matrix. Cell 83: 715-724, 1995. 55. Zeng L, Xu H, Chew T-L, Chisholm R, Sadeghi MM, Kanwar YS, and Danesh FR. Simvastatin Modulates Angiotensin II Signaling Pathway by Preventing Rac1-Mediated Upregulation of p27. J Am Soc Nephrol 15: 1711-1720, 2004.
25
Page 25 of 40

FIGURE LEGENDS
Figure 1: Graph illustrating the effects of VEGF and SMV on [3H]‐proline
incorporation. MCs were exposed to various concentrations of VEGF for 24 hrs. [3H]‐
proline incorporation was measured as previously described in Materials & Methods.
The amount of [3H]‐proline incorporation was measured in a scintillation counter
and corrected for the cellular protein content. The results are expressed as
percentages of the control and the mean±SE of three independent experiments.
*p<0.05, **p<0.01 vs. control. SMV: simvastatin, MEV: mevalonate, GGPP: geranyl
geranyl pyrophosphate, SQ: squalene.
Figure 2: (a) The protein levels of cell‐layer type IV collagen were assessed by
Western blot analysis. Cells exposed to incremental concentrations of VEGF exhibited
significant increase in cell‐layer collagen type IV protein levels. Co‐treatment of
VEGF‐stimulated cells with SMV abrogated the effect of VEGF on cell‐layer collagen
type IV protein levels, and addition of MEV reversed the inhibitory effect of SMV. (b)
Densitometric analysis of cell‐associated type IV collagen synthesis (n=3, *p<0.05 vs.
control)
Figure 3: (a) RhoA‐GTP was pulled down using the fusion protein GST‐RBD, and
samples were then subjected to Western blot analysis using a monoclonal anti‐RhoA
antibody. RhoA activity significantly increased in cells exposed to VEGF (50 ng/ml)
for 20 minutes. Co‐treatment of VEGF‐stimulated cells with SMV significantly
attenuated the effect of VEGF on RhoA activity, and the addition of MEV reversed
26
Page 26 of 40

the inhibitory effect of SMV. (b) Densitometric analysis of GTP‐RhoA (n=3, *p<0.05 vs.
control)
Figure 4: (a) MCs were transfected with wild type (wt), and dominant negative
mutants of RhoA (N19RhoA). Exposure to VEGF (50 ng/ml) in N19RhoA transfected
cells caused a significant reduction in VEGF‐induced collagen IV protein levels.
However, VEGF stimulation in transfected MCs with wtRhoA did not significantly
alter type IV collagen protein levels. (b) Densitometric analysis (n=4, *p<0.05 vs.
control)
Figure 5: Confocal microscopy of the actin cytoskeleton network in MCs. Panel a:
Control cells. Panel b: MCs exposed to VEGF exhibited significant stress fiber
formation. However, in cells co‐treated with SMV (Panel c) as well as in dominant
negative RhoA‐transfected cells (Panel d), VEGF stimulation failed to induce actin
cytoskeletal remodeling and significant stress fiber formation. The figure is
representative of 3 separate experiments.
Figure 6: FAK‐phosphorylation (Tyr397) was evaluated by laser scanning confocal
immunofluorescent microcopy. Panel a: Control cells. Panel b: MCs stimulated with
VEGF exhibited significant increase in FAK tyrosine phosphorylation. However, co‐
treatment of cells with SMV prevented VEGF‐induced FAK phosphorylation (Panel
c). MCs transfected with N19RhoA also showed a significant decrease in VEGF‐
induced FAK phosphorylation (Panel d).
Figure 7: (a) VEGF‐induced total and phosphorylated FAK protein levels were
27
Page 27 of 40

assessed by Western blot analysis. VEGF stimulation significantly increased
phospho‐FAK protein levels. Cells co‐treated with SMV and MCs transfected with
N17RhoA exhibited significant decrease in phospho‐FAK in response to VEGF.
However, total FAK protein levels were unchanged. (b) Densitometric analysis (n=3,
*p<0.05 vs. control)
Figure 8: Effect of cytochalasin D (cyto D) on cell‐layer collagen IV protein levels.
Cells were treated with either DMSO as control or 1.0 μg/ml of cyto D. VEGF (50
ng/ml) stimulation failed to increase collagen type IV protein levels in MCs treated
with Cyto D. The figure is representative of 3 separate experiments.
Figure 9: (a) Flow‐cytometric analysis of the active conformation of β1 integrin
activity in MCs. Flow cytometry was performed by using HUTS‐21, an antibody
specific for the active conformation of β1 integrin. Mean fluorescence intensity (±SE)
are calculated from three independent experiments (*p<0.05 vs. control unstimulated
cells). (b) Flow‐cytometric analysis of the total β1 integrin levels. Taken together,
these data show that VEGF (50 ng/ml) caused a significant increase in both β1
integrin activity and the total β1 integrin levels, and SMV reversed the β1 integrin
activation without significantly changing the total β1 integrin levels (**p<0.001 vs.
control unstimulated cells). (c) Quantification of integrin‐ligand occupancy when
cells were incubated with HUTS‐4 antibody. Bound antibody was assessed by
Western blotting. VEGF stimulation failed to increase β1 integrin activity in cells co‐
treated with SMV and in cells transfected with N17RhoA. (d) Densitometric analysis
28
Page 28 of 40

(n=3, *p<0.05 vs. control)
Figure 10: (a) β1 integrin activation was blocked by a functional blocking antibody.
MCs were treated with either IgG as control or with anti‐β1 functional blocking
antibody. MCs stimulated with VEGF failed to increase cell‐layer collagen type IV
protein levels when β1 integrin activation was blocked as well as in cells treated with
SMV. (b) Densitometric analysis (n=3, *p<0.05 vs. control)
29
Page 29 of 40
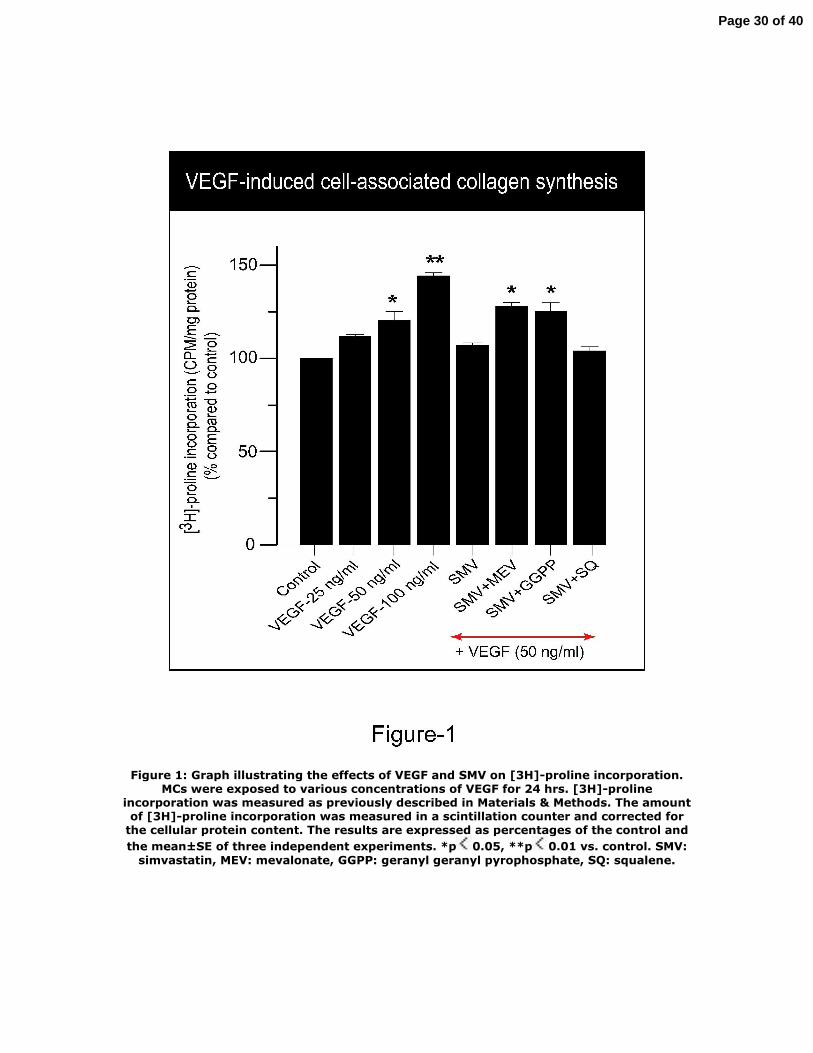
Figure 1: Graph illustrating the effects of VEGF and SMV on [3H]-proline incorporation. MCs were exposed to various concentrations of VEGF for 24 hrs. [3H]-proline
incorporation was measured as previously described in Materials & Methods. The amount of [3H]-proline incorporation was measured in a scintillation counter and corrected for
the cellular protein content. The results are expressed as percentages of the control and the mean±SE of three independent experiments. *p 0.05, **p 0.01 vs. control. SMV:
simvastatin, MEV: mevalonate, GGPP: geranyl geranyl pyrophosphate, SQ: squalene.
Page 30 of 40
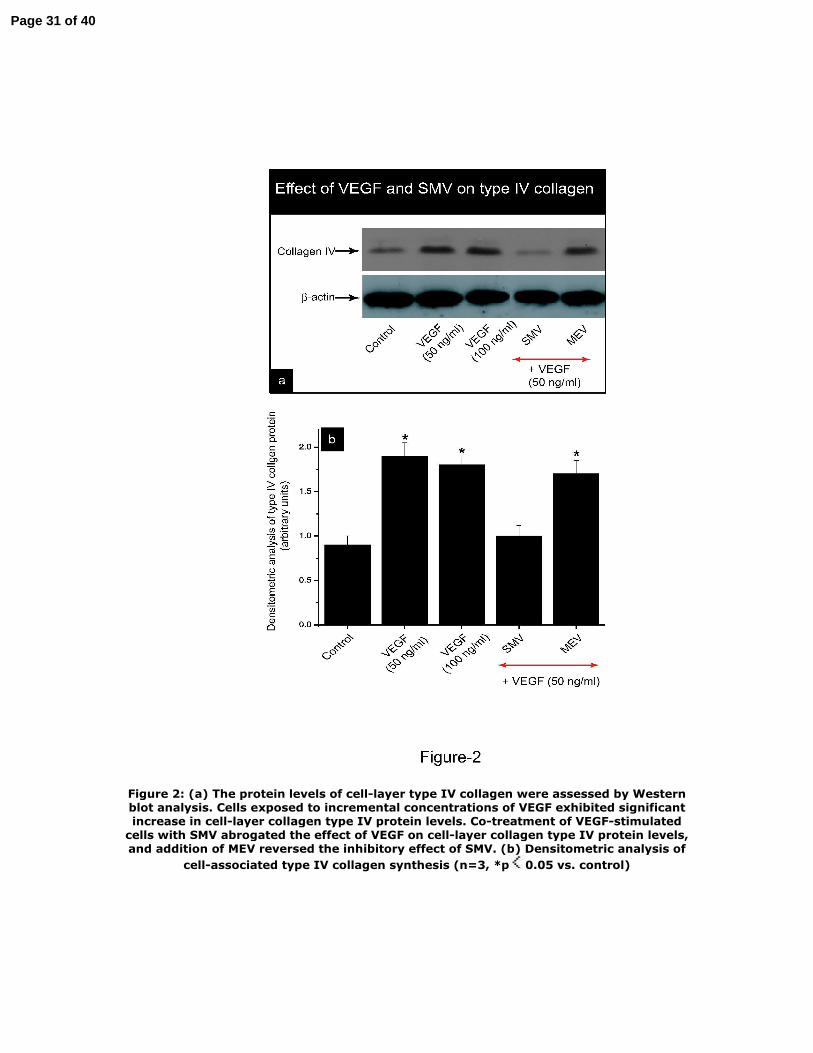
Figure 2: (a) The protein levels of cell-layer type IV collagen were assessed by Western blot analysis. Cells exposed to incremental concentrations of VEGF exhibited significant increase in cell-layer collagen type IV protein levels. Co-treatment of VEGF-stimulated
cells with SMV abrogated the effect of VEGF on cell-layer collagen type IV protein levels, and addition of MEV reversed the inhibitory effect of SMV. (b) Densitometric analysis of
cell-associated type IV collagen synthesis (n=3, *p 0.05 vs. control)
Page 31 of 40
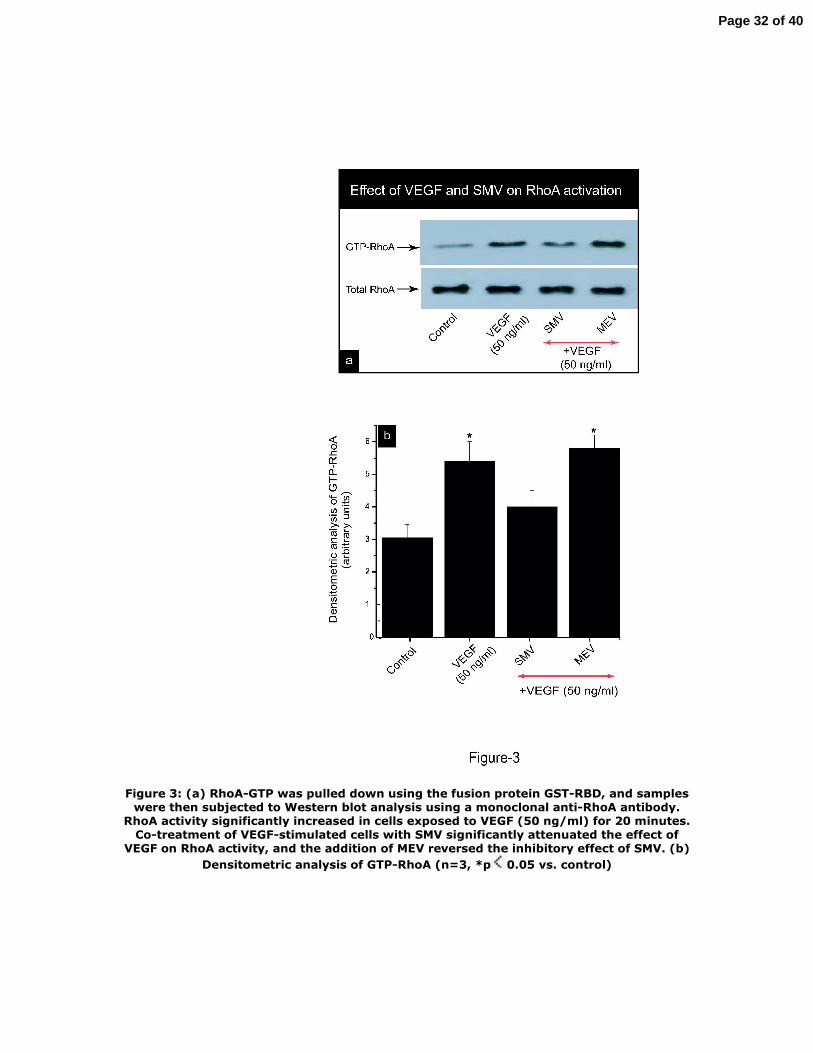
Figure 3: (a) RhoA-GTP was pulled down using the fusion protein GST-RBD, and samples were then subjected to Western blot analysis using a monoclonal anti-RhoA antibody.
RhoA activity significantly increased in cells exposed to VEGF (50 ng/ml) for 20 minutes. Co-treatment of VEGF-stimulated cells with SMV significantly attenuated the effect of
VEGF on RhoA activity, and the addition of MEV reversed the inhibitory effect of SMV. (b) Densitometric analysis of GTP-RhoA (n=3, *p 0.05 vs. control)
Page 32 of 40
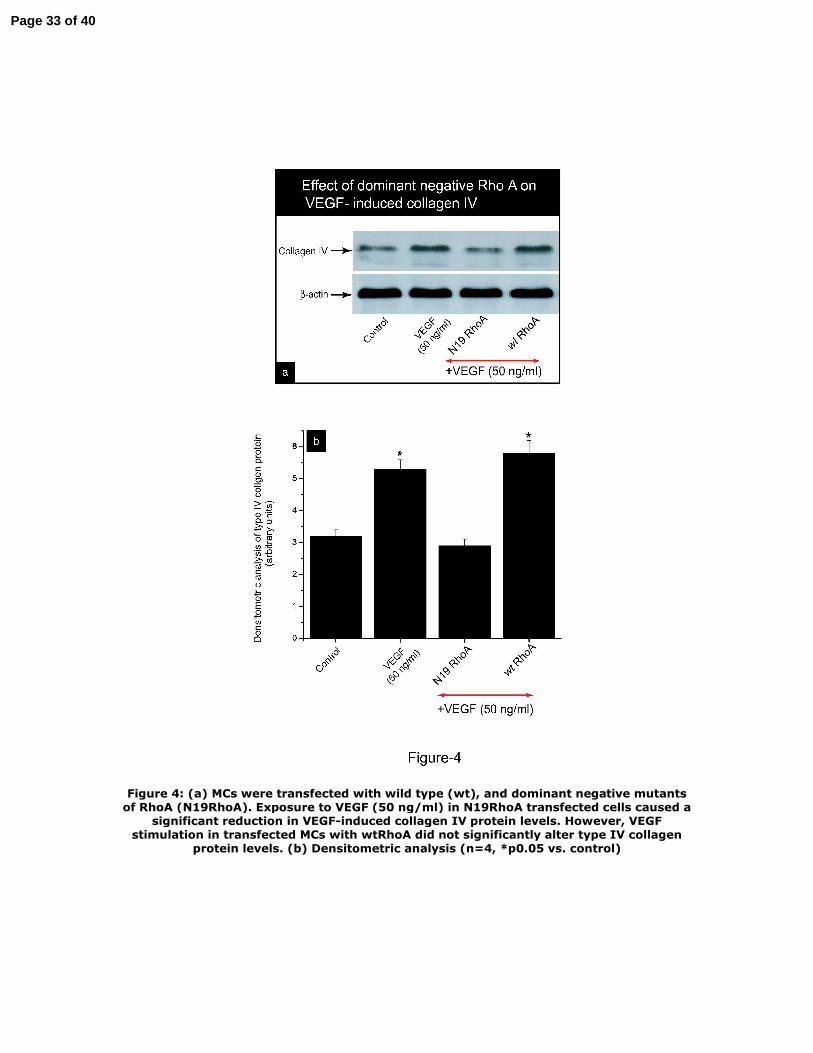
Figure 4: (a) MCs were transfected with wild type (wt), and dominant negative mutants of RhoA (N19RhoA). Exposure to VEGF (50 ng/ml) in N19RhoA transfected cells caused a
significant reduction in VEGF-induced collagen IV protein levels. However, VEGF stimulation in transfected MCs with wtRhoA did not significantly alter type IV collagen
protein levels. (b) Densitometric analysis (n=4, *p0.05 vs. control)
Page 33 of 40
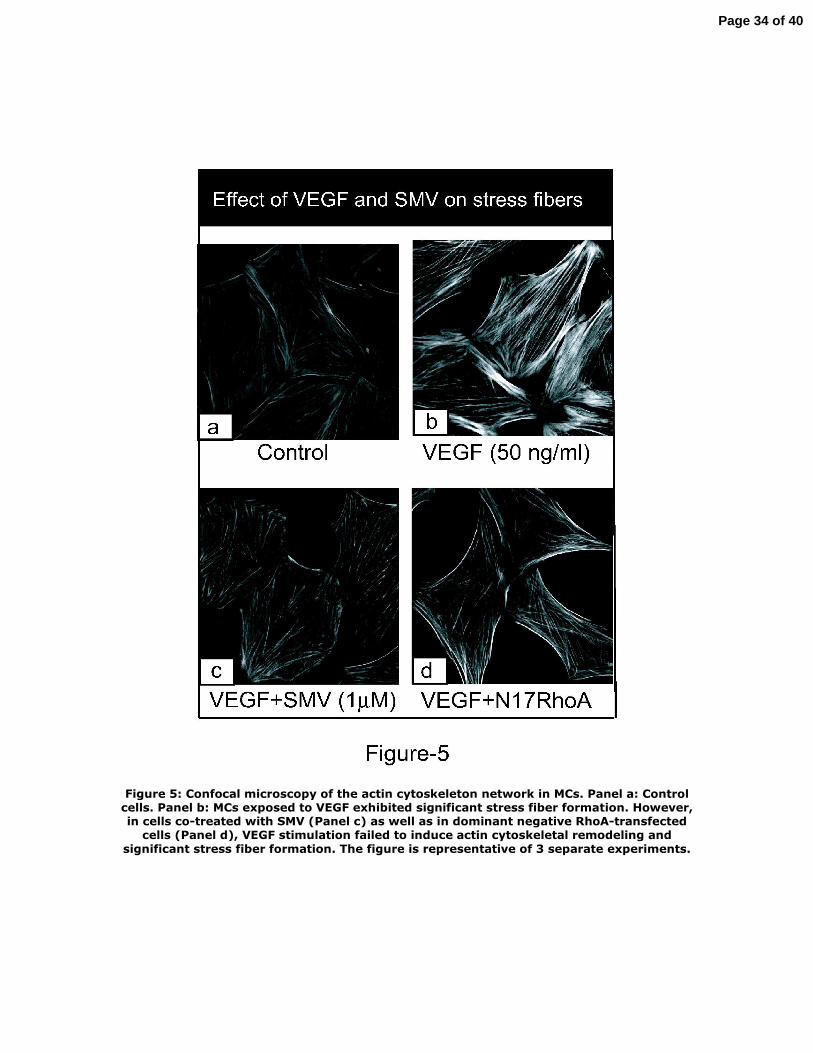
Figure 5: Confocal microscopy of the actin cytoskeleton network in MCs. Panel a: Control cells. Panel b: MCs exposed to VEGF exhibited significant stress fiber formation. However, in cells co-treated with SMV (Panel c) as well as in dominant negative RhoA-transfected
cells (Panel d), VEGF stimulation failed to induce actin cytoskeletal remodeling and significant stress fiber formation. The figure is representative of 3 separate experiments.
Page 34 of 40
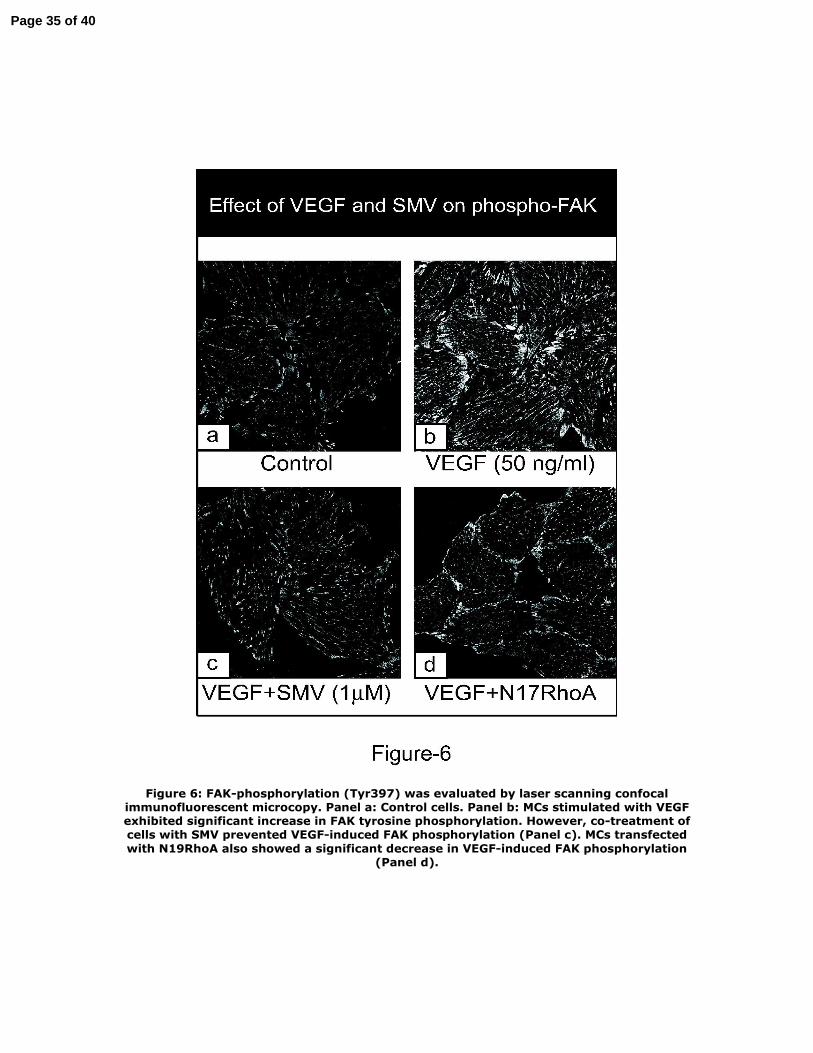
Figure 6: FAK-phosphorylation (Tyr397) was evaluated by laser scanning confocal immunofluorescent microcopy. Panel a: Control cells. Panel b: MCs stimulated with VEGF exhibited significant increase in FAK tyrosine phosphorylation. However, co-treatment of cells with SMV prevented VEGF-induced FAK phosphorylation (Panel c). MCs transfected with N19RhoA also showed a significant decrease in VEGF-induced FAK phosphorylation
(Panel d).
Page 35 of 40
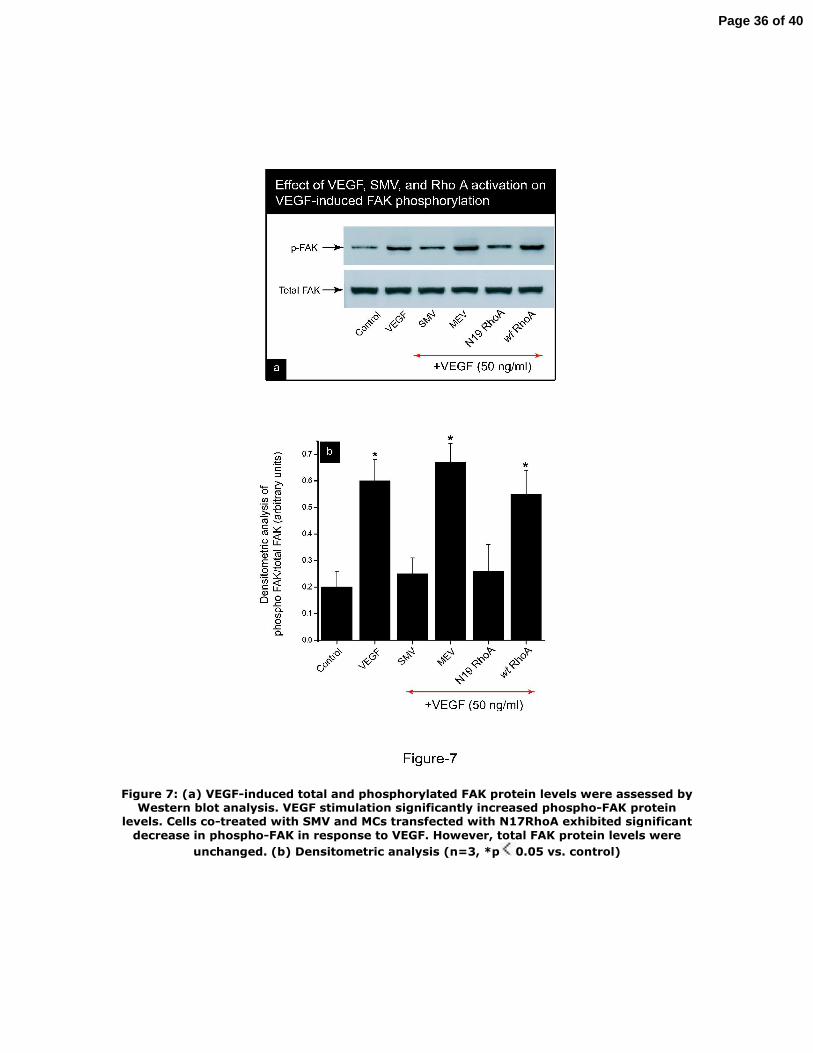
Figure 7: (a) VEGF-induced total and phosphorylated FAK protein levels were assessed by Western blot analysis. VEGF stimulation significantly increased phospho-FAK protein
levels. Cells co-treated with SMV and MCs transfected with N17RhoA exhibited significant decrease in phospho-FAK in response to VEGF. However, total FAK protein levels were
unchanged. (b) Densitometric analysis (n=3, *p 0.05 vs. control)
Page 36 of 40
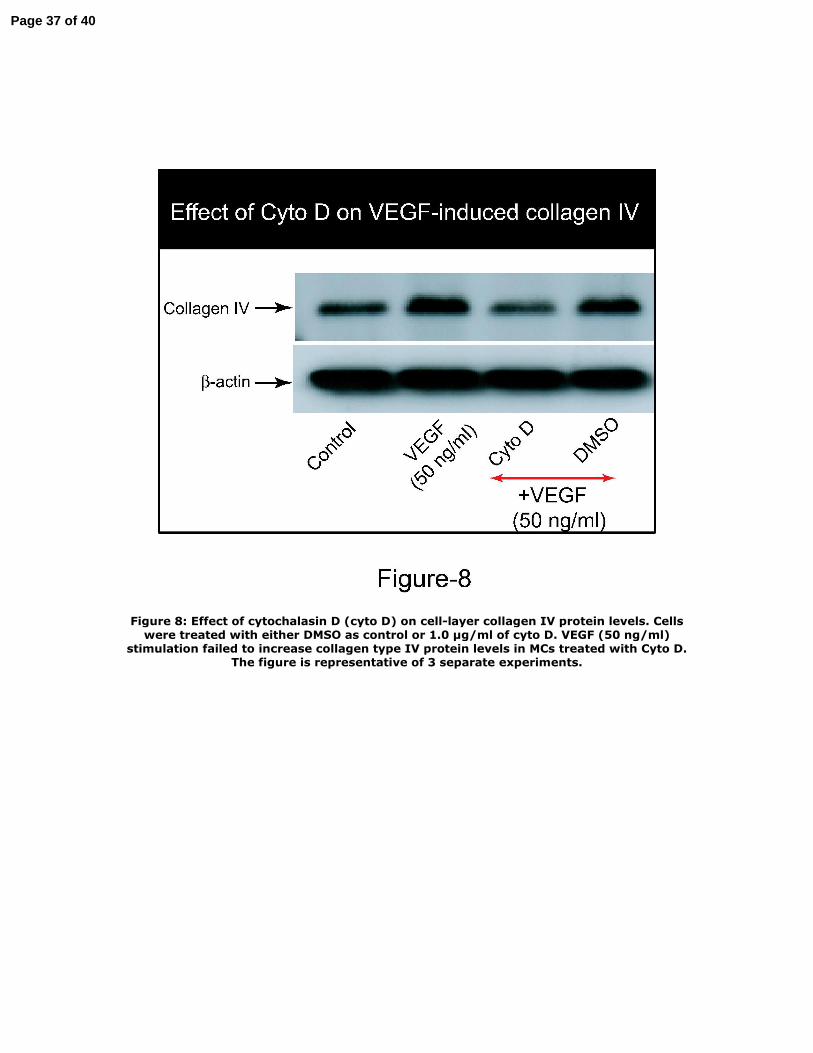
Figure 8: Effect of cytochalasin D (cyto D) on cell-layer collagen IV protein levels. Cells were treated with either DMSO as control or 1.0 µg/ml of cyto D. VEGF (50 ng/ml)
stimulation failed to increase collagen type IV protein levels in MCs treated with Cyto D. The figure is representative of 3 separate experiments.
Page 37 of 40
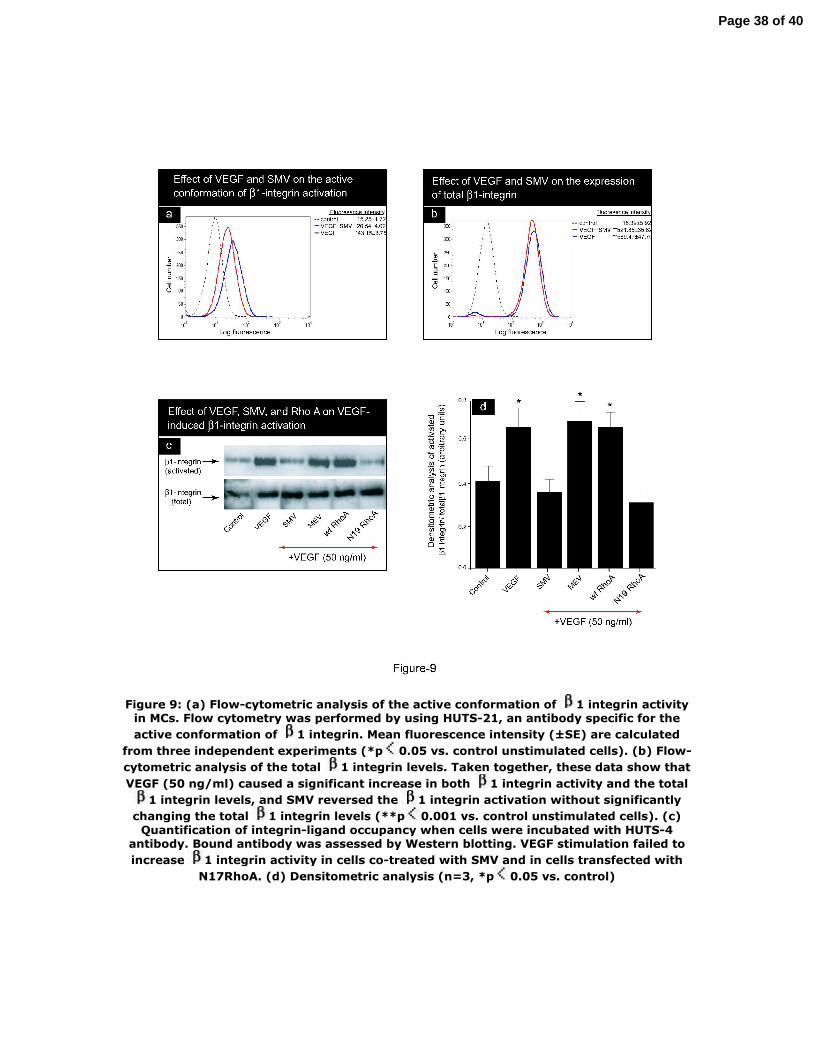
Figure 9: (a) Flow-cytometric analysis of the active conformation of 1 integrin activity in MCs. Flow cytometry was performed by using HUTS-21, an antibody specific for the active conformation of 1 integrin. Mean fluorescence intensity (±SE) are calculated
from three independent experiments (*p 0.05 vs. control unstimulated cells). (b) Flow-cytometric analysis of the total 1 integrin levels. Taken together, these data show that VEGF (50 ng/ml) caused a significant increase in both 1 integrin activity and the total
1 integrin levels, and SMV reversed the 1 integrin activation without significantly changing the total 1 integrin levels (**p 0.001 vs. control unstimulated cells). (c)
Quantification of integrin-ligand occupancy when cells were incubated with HUTS-4 antibody. Bound antibody was assessed by Western blotting. VEGF stimulation failed to increase 1 integrin activity in cells co-treated with SMV and in cells transfected with
N17RhoA. (d) Densitometric analysis (n=3, *p 0.05 vs. control)
Page 38 of 40
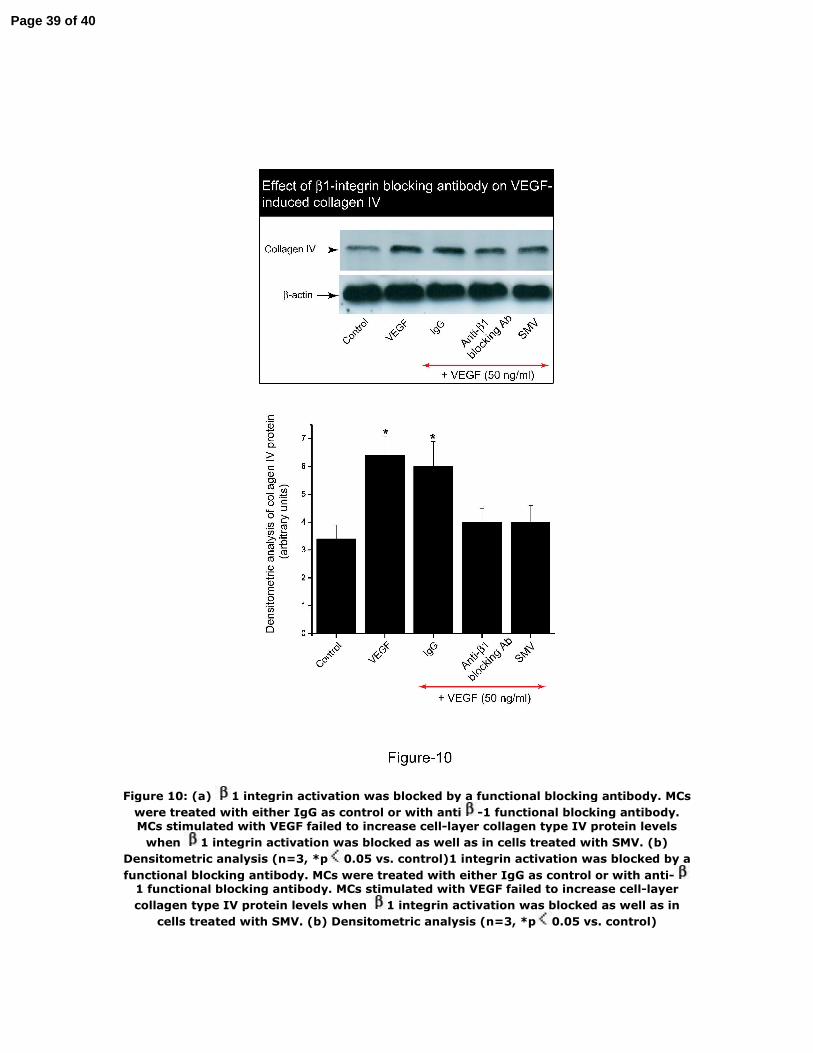
Figure 10: (a) 1 integrin activation was blocked by a functional blocking antibody. MCs were treated with either IgG as control or with anti -1 functional blocking antibody. MCs stimulated with VEGF failed to increase cell-layer collagen type IV protein levels
when 1 integrin activation was blocked as well as in cells treated with SMV. (b) Densitometric analysis (n=3, *p 0.05 vs. control)1 integrin activation was blocked by a functional blocking antibody. MCs were treated with either IgG as control or with anti-
1 functional blocking antibody. MCs stimulated with VEGF failed to increase cell-layer collagen type IV protein levels when 1 integrin activation was blocked as well as in
cells treated with SMV. (b) Densitometric analysis (n=3, *p 0.05 vs. control)
Page 39 of 40

Page 40 of 40





























