Embed Size (px)
Citation preview

HDAC Inhibition Attenuates Inflammatory, Hypertrophic andHypertensive Responses in Spontaneously Hypertensive Rats
Jeffrey P Cardinale1, Srinivas Sriramula1, Romain Pariaut2, Anuradha Guggilam1, NithyaMariappan1, Carrie Elks1, and Joseph Francis11Comparative Biomedical Sciences, School of Veterinary Medicine, Louisiana State University,Baton Rouge, LA 70803, USA2Veterinary Clinical Sciences, School of Veterinary Medicine, Louisiana State University, BatonRouge, LA 70803, USA
AbstractReactive oxygen species and pro-inflammatory cytokines contribute to cardiovascular diseases.Inhibition of downstream transcription factors and gene modifiers of these components are keymediators of hypertensive response. Histone acetylases/deacetylases can modulate the geneexpression of these hypertrophic and hypertensive components. Therefore, we hypothesized thatlong-term inhibition of histone deacetylase with valproic acid might attenuate hypertrophic andhypertensive responses by modulating reactive oxygen species and pro-inflammatory cytokines inSHR rats. Seven week-old SHR and WKY rats were used in this study. Following baseline bloodpressure measurement, rats were administered valproic acid in drinking water (0.71% wt/vol), orvehicle, with pressure measured weekly thereafter. Another set of rats were treated with hydralazine(25mg/kg/day orally) to determine the pressure-independent effects of HDAC inhibition onhypertension. Following 20 weeks of treatment, heart function was measured usingechocardiography, rats were sacrificed and heart tissue collected for measurement of total reactiveoxygen species as well as pro-inflammatory cytokine, cardiac hypertrophic and oxidative stress geneand protein expressions. Blood pressure, pro-inflammatory cytokines, hypertrophic markers andreactive oxygen species were increased in SHR versus WKY rats. These changes were decreased invalproic acid treated SHR rats, while hydralazine treatment only reduced blood pressure. These dataindicate that long-term histone deacetylase inhibition, independent of the blood pressure response,reduces hypertrophic, pro-inflammatory and hypertensive responses by decreasing reactive oxygenspecies and angiotensin-type1 receptor expression in the heart, demonstrating the importance ofuncontrolled histone deacetylase activity in hypertension.
KeywordsAng II; cardiac hypertrophy; cytokines; hypertension; oxidative stress; HDAC
Corresponding author: Joseph Francis, B.V.Sc, M.V.Sc, Ph.D., Comparative Biomedical Sciences, School of Veterinary Medicine,Louisiana State University, Baton Rouge, LA 70803, USA, Phone: +1 225 578 9752, Fax: +1 225 578 9895, [email protected]'s Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customerswe are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resultingproof before it is published in its final citable form. Please note that during the production process errors may be discovered which couldaffect the content, and all legal disclaimers that apply to the journal pertain.DisclosuresNone
NIH Public AccessAuthor ManuscriptHypertension. Author manuscript; available in PMC 2011 September 1.
Published in final edited form as:Hypertension. 2010 September ; 56(3): 437–444. doi:10.1161/HYPERTENSIONAHA.110.154567.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

IntroductionEssential hypertension is a condition associated with increased expression of pro-inflammatorycytokines (PICs)1, 2. Studies from our lab and others have shown that PICs lead to an increasein reactive oxygen species (ROS), which up-regulates Nuclear Factor-kappaB (NFκB) activity,thus further increasing PIC and ROS transcription and amplifying their subsequent actions3–5. Along with renin-angiotensin system (RAS) components, PICs also activate hypertrophicmediators, which can result in cardiac hypertrophy and altered cardiac remodeling andfunction6, 7.
There are many triggers of hypertensive-induced inflammation resulting in both hypertrophicand hypertensive responses, many of which are through transcription factor NFκB activation,ultimately resulting in alterations of gene transcription and perpetuation of the hypertensivestate5, 8. In order for transcription factors such as NFκB to activate their target genes, DNAand chromatin remodeling must occur. Post-translational modifications of histone coresthrough a tightly regulated addition/removal of an acetyl tag on their N-terminal tails plays amajor role in gene expression modulation9. These additions/removals are accomplished byseveral members of the histone acetyltransferase (HAT) and histone deacetylase (HDAC)families, which either open or close DNA strands to the actions of transcription factors10, 11.
Normally, this balance is tightly controlled, but during conditions of stress and inflammation,activation of PICs can result in increased HDAC activation and histone acetylation, correlatingwith an increase in NFκB activity and further increases in PIC expression, including tumornecrosis factor-α (TNF) and the interleukins (ILs) 12, 13. Though HDACs would appear torepress inflammatory responses through reduced gene expression, this view is too simplisticin regards to their non-histone protein acetylation/deacetylation abilities, often times havingquite opposite effects in regards to the inflammatory response14–17.
Recent evidence indicates that the various HDAC classes respond differently towards inducingcardiac hypertrophy in non-hypertensive animal models16, 18, 19 and that global HDACinhibition (HDACi) can prevent these hypertrophic changes20. However, it is not knownwhether HDACi protects against cardiac hypertrophy and hypertensive response bymodulating PICs and oxidative stress in spontaneously hypertensive (SHR) rats. A previousstudy used valproate, a derivative of valproic acid (VPA), to study hypertension in SHR rats.Though they showed a reduction in systolic blood pressure21, the treatment was only carriedout to 9 weeks of age, which is still 3 weeks too young to display the full systemic changesassociated with hypertension in this animal model, including cardiac hypertrophy andinflammation. This study was established to assess the role of HDAC blockade on theinflammatory response and its effect on the pathogenesis of hypertension. Therefore, wehypothesize that chronic HDACi will attenuate the inflammatory and hypertensive responsesassociated with the hypertensive state. To test this, we administered VPA, a fairly novel HDACinhibitor22, especially Class I HDACs, as a long-term treatment in SHR rats, for assessmentof inflammatory, hypertrophic and hypertensive changes associated with essentialhypertension.
Materials and MethodsAll the procedures in this study were approved by the Louisiana State University InstitutionalAnimal Care and Use Committee and were performed in accordance with the NationalInstitutes of Health Guide for the Care and Use of Laboratory Animals.
Cardinale et al. Page 2
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Animals and Experimental DesignMale Wistar-Kyoto (WKY; n=20) and SHR (n=20) rats were randomly assigned to vehicle(water) or VPA (0.71% wt/vol23, dissolved in water, prepared and provided daily) treatmentgroups. Another set of rats were treated with hydralazine (HYD; 25mg/kg/day in drinkingwater8). Rats received drug or vehicle for 20 weeks starting from 7 weeks of age. Rats wereeuthanized at 27 weeks of age with left ventricular (LV) tissue collected for molecular analyses.We performed the following experimental procedures: blood pressure measurements,echocardiographic analysis, real time RT-PCR, western blot analysis, electron paramagneticspin resonance (EPR) studies, electrophoretic mobility shift assays (EMSA), colorimetricassays, immunofluorescence, immunohistochemical and statistical analysis. For an expandedMaterials and Methods section, please see the online Data Supplement athttp://hyper.ahajournals.org.
ResultsVPA treatment attenuates the blood pressure changes in SHR rats
Blood pressure recordings show that SHR+VPA maintained a lower blood pressure level fromthe pre-hypertensive to the more advanced hypertensive phases as compared to SHR controls(Figure 1). Following 10 weeks of treatment, SHR control MAP continued to rise while SHR+VPA rats plateaued at a lower pressure (163.1±2.32 vs. 127.5±2.35mmHg, respectively,p<0.05). The MAP of SHR+VPA rose slightly throughout the course of the study, becomingsignificantly elevated above WKY controls towards the end of the treatment period (141.2±4.5vs. 114.8±3.41, respectively, p<0.05), but still was significantly lower than the MAP of SHRcontrols (141.2±4.5 vs. 173.2±3.47, respectively, p<0.05). Mean diastolic and systolicpressures followed the same pattern as the MAP (data not shown), indicating that VPAattenuated all phases of blood pressure in SHR rats. Furthermore, VPA did not have any adverseeffects on the health of the animals used in this study.
Another set of animals (n=5 for each SHR and WKY) were treated with HYD, a direct smoothmuscle relaxant and vasodilator, to compare their effects against treatment with VPA (OnlineData Supplement, Figure S1). SHR+HYD saw a decrease similar to SHR+VPA in MAP ascompared to SHR (142.57±4.906 vs. 173.2±3.47). WKY+HYD had no change in MAP. Meandiastolic and systolic followed the same pattern as the MAP in HYD treated rats (data notshown), indicating that HYD similarly attenuated all phases of increased blood pressure seenin SHR rats.
VPA attenuates cardiac hypertrophy in SHR ratsEchocardiographic assessment showed that SHR controls had significantly more concentrichypertrophy of the left ventricular posterior wall (LVPWT) during diastole and systole (Figure2A) (2.48±0.11 and 3.43±0.2mm, respectively) when compared to WKY controls (1.73±0.02and 2.32±0.06, respectively). However, SHR+VPA (1.85±0.03, 2.52±0.04mm) rats displayedno change in ventricular thickness as compared to WKY and WKY+VPA (1.75±0.06, 2.28±0.07mm) rats, indicating that VPA had a beneficial effect on preventing the increasedLVPWTd/s that is typically observed in SHR rats.
Heart weight (HW)/body weight (BW) and lung weight (LW)/BW ratios are often used to showphenotypic changes due to hypertension such as increased heart mass due to hypertrophy (HW/BW) and increased edema of the lungs (LW/BW) due to cardiac dysfunction and increasedsystemic circulatory resistance. Treatment with VPA or HYD alone did not have any effectson BW (Figure S2). SHR+VPA normalized both HW/BW and LW/BW indices versus SHRcontrol rats (0.0032 and 0.0048 vs. 0.0039 and 0.0062, respectively, p<0.05) as compared toWKY (0.0029 and 0.0038), reinforcing that VPA reduces cardiac hypertrophy and dysfunction
Cardinale et al. Page 3
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

(Figure 2B). However, the HW/BW ratio assessed for SHR+HYD did not improve cardiachypertrophy (Figure S3) as compared to SHR controls (0.0036 vs. 0.0039), indicating thatimprovements in cardiac hypertrophy due to VPA treatment were not pressure dependent.
VPA reduces hypertrophic response elements in the LV tissue of SHR ratsTo further demonstrate the effects of VPA on hypertrophy, RT-PCR analysis was undertakenon hypertrophic response genes in the LV. Compared to WKY, untreated SHR rats had elevatedlevels of collagen IV and atrial natriuretic peptide (ANP) - two markers of LV remodelingassociated with cardiac hypertrophy (Figure 2C), as well as angiotensin type 1 receptor (AT1-R). SHR+VPA reduced these levels to that of normotensive WKY controls (p<0.05), indicatingHDACi has a positive effect on reducing molecular markers of cardiomyocyte and interstitialgrowth normally attributed to systemic hypertension. Furthermore, SHR+VPA had reduced %fibrosis staining (Figure 2D) as compared to SHR rats (3.395±0.07 vs 4.713±0.32), signifyinga reduction in total fibrosis within the heart following HDACi. The mRNA expression of AT1-R was significantly increased within the LV of SHR rats versus that of normotensive WKYcontrols (2.7±0.5 fold vs WKY) (Figure 3B), which was fully attenuated in SHR+VPA rats(2.7±0.5 vs. 0.9±0.1 fold vs WKY, respectively) and reconfirmed by western blot of the LVtissues (Figure3A). HYD had no effect on the mRNA expression of either ANP or AT1-R(Figure S4A, B), demonstrating the effect that HDACi has on controlling these locally activatedhypertrophic mediators in the LV of SHR rats.
VPA reduces HDAC activity in the LV of treated groupsTo assess the effectiveness of VPA on HDAC activity in LV tissue, a colorimetric assay kitwas used to analyze differences between the VPA treated and untreated groups (Figure 4A).Global HDAC activity was reduced in untreated WKY versus untreated SHR rats (17.87±1.06vs. 22.19±0.57 O.D./mg protein sample, respectively, p<0.05). Furthermore, WKY+VPA andSHR+VPA groups both exhibited a lower HDAC activity level (not significant and p<0.05,respectively) as compared to their own strain controls. Conversely, SHR and WKY rats treatedwith HYD did not have a decrease in HDAC activity when compared with their respectivecontrols (Figure S5).
VPA normalizes inflammatory response in the LV tissue of SHR ratsImmunohistochemistry revealed that SHR controls had increased protein expression of TNFand IL-1β in the LV as compared to WKY controls. This protein expression was reduced inSHR+VPA rats (Figure 5A). RT-PCR similarly indicated that SHR controls had increasedexpression of TNF, IL-1β, IL-6 (Figure 5B) and NFκB (Figure 4C) mRNA in LV tissue whencompared to WKY controls. This increase was attenuated in SHR+VPA rats, while WKY+VPA exhibited no change. Furthermore, HYD had no effect on the PIC mRNA expressionof TNF or IL-1β (Figure S6A, B). These combined results indicate that VPA treatment reducesand normalizes the inflammatory response observed in SHR rats.
VPA reduces ROS and gp91phox in the LV of SHR ratsAs inflammation during hypertensive response is associated with an increase in oxidativestress, both total ROS as assessed by EPR (Figure 6A) and the expression of gp91phox (Figure6B, 6C) were examined in the LV. Untreated SHR rats experienced a significant increase inROS when compared to WKY rats (0.54±0.05 vs. 0.22±0.03mM/mg protein/min). SHR+VPAnormalized this ROS increase (0.54±0.05 vs. 0.21±0.03mM/mg protein/min). Furthermore,gp91phox, the major catalytic subunit of NADPH oxidase and a ROS contributor, wasincreased in untreated SHR rats versus WKY controls. This was subsequently reduced in SHR+VPA (3.39 vs. 1.56 fold change/WKY), but not SHR+HYD (Figure S6C). The reduction inprotein expression was confirmed by immunofluorescence of the LV tissue. These results
Cardinale et al. Page 4
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

indicate that treatment of SHR rats with VPA had beneficial effects in reducing oxidative stressin the LV tissue.
DiscussionThe major findings in this study are as follows: 1) HDACs played an important role inhypertensive drive by modulating inflammatory and oxidative stress actions and contributedto hypertrophic and hypertensive responses in SHR rats, 2) HDACi attenuated MAP in SHRrats, 3) HDACi also attenuated LVPWT and HW/BW ratio in SHR rats, demonstrating thatHDACi reduces cardiac hypertrophy, possibly in part through modulation of ANP, CollagenIV and AT1-R expression, 4) untreated SHR rats had an increase in inflammatory markers,including TNF and NFκB, as well as an increase in ROS and gp91phox expression, which weredecreased through long-term HDACi using VPA, and 5) HDACi reduced AT1-R expression,thereby limiting the role that angiotensin II (Ang II) plays in mediating hypertension. Thesefindings suggest that long-term treatment with VPA reduces hypertension-induced PICs,thereby attenuating hypertrophic and hypertensive responses in SHR rats.
HDACs play an important role in inducing the structural remodeling of chromatin that exposesDNA to transcription factors, ultimately yielding changes in gene expression. Along withHATs, HDACs maintain a relative balance in cellular systems during normal physiology thatallows typical function9. However, during diseased conditions, this balance is tilted in favorof HDACs, which can lead to an elevation in inflammatory and immune responses11, 13, 24–27. In this study, we also observed an elevated HDAC activity which was accompanied byincreased PIC and oxidative stress gene expression in the hearts of SHR rats.
Cardiac hypertrophy is a well described consequence of systemic hypertension. Studies haveshown the role of HDACi on controlling cardiac growth and remodeling28. However, little isknown about the holistic role of HDACi on hypertrophy during hypertensive response. Asevidence indicates16, 18, 20, 28, 29, pathological cardiac hypertrophy and function rely uponthe balanced abundance of α- and β-myosin heavy chain (MHC) protein throughout the heart.During the progression of cardiac hypertrophy, the adult isoform of MHC (α-MHC) undergoesa stressed-trigger switch to the fetal isoform (β-MHC), contributing to cardiac hypertrophy.HDACi blunts MHC isoform switching, therefore preserving ventricular function14, 16, 19.These results are contradictory to earlier experiments showing that class II HDACs block pro-growth genes through interaction with transcription factor myocyte enhancer factor-2 (MEF2)30. More recently though, research has indicated that the pro-hypertrophic class I HDACs,when activated, are more potent and take priority over the anti-hypertrophic class IIHDACs28, thus explaining how global HDACi can attenuate cardiac hypertrophy in variousanimal models16, 29. From these changes, it has been suggested that within the hypertrophiedheart, hypertrophic stress signals cause the phosphorylation of HDACs bound to MEF2,causing their disassociation into the cytoplasm. Since these HDACs are not bound to thechromatin structure, their inhibition would have no overt effect on MEF2 transcription,indicating that HDACi has no direct effect on the depression of MEF2 controlled pro-hypertrophic genes and signifying that another mechanism must be in place16.
HDACi using Trichostatin A, an inhibitor of Class I and II HDACs, either induced31 orblunted28 agonist-induced expression of ANP, a hypertrophic growth factor, in culturedneonatal myocytes. The present study also demonstrated that HDACi in SHR rats showed asignificant reduction in ANP, suggesting that in SHR rats, ANP possibly plays an importantrole in attenuating cardiac hypertrophy, and that HDACi silences/blunts this signalingmechanism. Moreover, collagen IV, an indicator of cardiac remodeling, especially within thefailing heart32, was increased in SHR rats, but not SHR+VPA rats. Finally, AT1-R, animportant component of the RAS which, when acted upon by Ang II in the LV, causes cardiac
Cardinale et al. Page 5
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

hypertrophy33, has been shown to be attenuated with VPA treatment29, 34, but the mechanisminvolved has not been fully investigated. The present study showed significantly reduced AT1-R expression in SHR+VPA rats versus SHR controls, indicating that a possible hypertrophicmechanism more intimately involves AT1-R activation.
There are several well known modulators of pressure-independent cardiac hypertrophyincluding Ang II and sympathetic neurohormones35. Recently, it was also shown that Ang II-induced cardiac hypertrophy can be prevented using HDACi29. To determine if the currentstudy’s effects on cardiac hypertrophy were pressure-independent or -dependent of HDACi,we looked at the effect of HYD on blood pressure and cardiac hypertrophy, as well as on ANPand AT1-R expression. HYD reduced MAP in SHR rats similar to VPA; however, cardiachypertrophy was unaffected, including hypertrophic mediators ANP and AT1-R. This indicatesa possible pressure-independent mechanism regarding VPA’s effect on cardiac hypertrophy.These results are in agreement with a recent study where NFκB inhibition reduces cardiachypertrophy in a pressure-independent manner8. This may offer another mechanism wherebyHDACi reduces cardiac hypertrophy, for results herein show a decrease in NFκB activity andexpression following VPA treatment. Therefore, from these results, not only was cardiachypertrophy alleviated through long-term VPA treatment, but several possible mechanismsthat induce cardiac remodeling were also attenuated.
We and others have recently demonstrated that hypertensive drive is partially controlledthrough the over-expression of PICs, especially TNF, along with downstream alterations inNFκB, ROS and RAS components, as regulated through AT1-R activation3–5, 36. This studydemonstrates that chronic HDACi attenuates PIC response in SHR+VPA rats. Moreover, VPAreduces the presence of ROS and AT1-R, two components implicated in the inflammatoryresponse observed in hypertension. HDACi has been increasingly identified as a possibletherapeutic approach towards many inflammatory conditions11, 14, 22, 37, includingcardiovascular diseases12. Though this mechanism has not yet been entirely delineated, it issuggested that the use of a HDACi, such as VPA, blocks HDAC actions on protein functionoutside of its normal action on altering transcription17, 38, as subsets of these families possessthe ability to act on non-histone proteins, further complicating their roles in genemodulation39. A recent study showed that HDACi can deactivate Akt, a potential mediator ofcardiac hypertrophy and oxidative stress, via dephosphorylation by HDAC-protainphosphatase 1 complexes17. However, other studies have shown that specific sets of Toll-likereceptor-inducible genes are targeted by HDACi in macrophages and dendritic cells, and thatone of these is through the NFκB pathway24, 25, which concurs with the present study. Itunderscores the role of HDACi on PIC activation of NFκB, preventing a further, cyclicallydriven up-regulation of PICs, including TNF, IL-1β and IL-6.
The effect of inflammation on ROS in hypertension has been previously demonstrated3, 40. Anumber of pro-inflammatory mediators of this increased ROS have been identified, includingTNF and IL-6, which, as demonstrated here, are attenuated with HDACi, confirming the rolesof HDACs on inflammatory and oxidative stress responses on hypertension in SHR rats.Though the signaling pathway is not entirely clear on how HDACi attenuates ROS in SHRrats, either directly or indirectly through its blockade of PICs and the NFκB pathway, wepostulate that by blocking PIC activation, with its subsequent down-regulation of NFκBactivity and gp91phox expression, ROS is inhibited.
The interaction between the RAS, PICs and ROS has also been demonstrated by work in ourlab5 and others41–43. Presently we show that untreated SHR rats have increased AT1-Rexpression, an important component of the pro-hypertensive portion of the RAS. HDACithrough VPA treatment attenuated this increase concomitant with that of PICs. Thepathophysiological mechanism of hypertension intimately involves the action of Ang II,
Cardinale et al. Page 6
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

including vasoconstriction, increased aldosterone secretion, increased sympathetic nerveactivity, tissue remodeling and increased sodium and water intake, all of which are mediatedthrough AT1-Rs that are distributed throughout most organ systems, including the liver, brain,kidney, heart and blood vessels44. Reports indicate that Ang II is controlled by, and controls,HDAC-induced changes in gene and protein response29, 34. Here we show a possible newmechanism involving the regulation of Ang II responses as directed through the AT1-R.HDACi reduces AT1-R gene expression and receptor density, thereby ameliorating the actionsof Ang II. This alteration in AT1-R expression could be either through direct HDACi effectson the receptor’s production and function, or through the effects of HDACi on inflammatorygene response during hypertension. This mechanism must be further investigated in order todetermine the full effect of HDACi in attenuating hypertensive response.
In conclusion, the present study’s results show that long-term HDACi through VPA attenuatedMAP and cardiac hypertrophy, possibly through modulation of ANP, Collagen IV and AT1-R. HDAC inhibition also attenuated the increased inflammatory response, including TNF andNFκB, as well as the increase in ROS and gp91phox. These findings suggest that HDACi withVPA reduced inflammation, ROS, and AT1-R, thereby attenuating hypertension and itssecondary consequences in SHR rats.
Perspectives
We chose to use VPA due to its current use in clinical settings as an anti-seizure and bipolardrug, demonstrating its availability to patients. In our study, VPA was administered long-term without any adverse effects towards the treated animal groups. This outlines theimportance of the continuous drug administration necessary for the successful treatment ofhypertension and its consequences, including cardiac hypertrophy, systemic inflammationand end organ damage due to ROS. While we cannot rule out the effects of VPA on bloodpressure from its GABAergic actions or non-histone protein interactions, we feel confidentthat this study provides sufficient evidence that the use of HDACi can reduce not only bloodpressure, but cardiac hypertrophy and the inflammatory state associated with hypertension.The specific mechanisms involved in HDACi must be studied more closely. However, asthe quest to find new therapeutic strategies in hypertensive control is ever pressing, thiscould present a possible new approach in future treatment options.
Supplementary MaterialRefer to Web version on PubMed Central for supplementary material.
AcknowledgmentsSources of Funding
These studies were supported by National Heart, Lung and Blood Institute Grant HL-80544 (to J. Francis).
References1. Phillips MI, Kagiyama S. Angiotensin II as a pro-inflammatory mediator. Curr Opin Investig Drugs
2002;3:569–577.2. Ferrario CM, Strawn WB. Role of the renin-angiotensin-aldosterone system and proinflammatory
mediators in cardiovascular disease. Am J Cardiol 2006;98:121–128. [PubMed: 16784934]3. Elks CM, Mariappan N, Haque M, Guggilam A, Majid DS, Francis J. Chronic NF-{kappa}B blockade
reduces cytosolic and mitochondrial oxidative stress and attenuates renal injury and hypertension inSHR. Am J Physiol Renal Physiol 2009;296:F298–F305. [PubMed: 19073636]
Cardinale et al. Page 7
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

4. Mariappan N, Elks CM, Fink B, Francis J. TNF-induced mitochondrial damage: a link betweenmitochondrial complex I activity and left ventricular dysfunction. Free Radic Biol Med 2009;46:462–470. [PubMed: 19041937]
5. Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensinII-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension2008;51:1345–1351. [PubMed: 18391105]
6. Kudo H, Kai H, Kajimoto H, Koga M, Takayama N, Mori T, Ikeda A, Yasuoka S, Anegawa T, MifuneH, Kato S, Hirooka Y, Imaizumi T. Exaggerated blood pressure variability superimposed onhypertension aggravates cardiac remodeling in rats via angiotensin II system-mediated chronicinflammation. Hypertension 2009;54:832–838. [PubMed: 19704105]
7. Tokuda K, Kai H, Kuwahara F, Yasukawa H, Tahara N, Kudo H, Takemiya K, Koga M, YamamotoT, Imaizumi T. Pressure-independent effects of angiotensin II on hypertensive myocardial fibrosis.Hypertension 2004;43:499–503. [PubMed: 14699000]
8. Gupta S, Young D, Sen S. Inhibition of NF-kappaB induces regression of cardiac hypertrophy,independent of blood pressure control, in spontaneously hypertensive rats. Am J Physiol Heart CircPhysiol 2005;289:H20–H29. [PubMed: 15749748]
9. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases(HDACs): characterization of the classical HDAC family. Biochem J 2003;370:737–749. [PubMed:12429021]
10. Forsberg EC, Bresnick EH. Histone acetylation beyond promoters: long-range acetylation patternsin the chromatin world. Bioessays 2001;23:820–830. [PubMed: 11536294]
11. Halili MA, Andrews MR, Sweet MJ, Fairlie DP. Histone deacetylase inhibitors in inflammatorydisease. Curr Top Med Chem 2009;9:309–319. [PubMed: 19355993]
12. Keslacy S, Tliba O, Baidouri H, Amrani Y. Inhibition of tumor necrosis factor-alpha-inducibleinflammatory genes by interferon-gamma is associated with altered nuclear factor-kappaBtransactivation and enhanced histone deacetylase activity. Mol Pharmacol 2007;71:609–618.[PubMed: 17108260]
13. Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibitanti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiplemechanisms of action. J Pharmacol Exp Ther 2007;321:892–901. [PubMed: 17371805]
14. Bush EW, McKinsey TA. Targeting histone deacetylases for heart failure. Expert Opin Ther Targets2009;13:767–784. [PubMed: 19466913]
15. Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated byreversible acetylation. Science 2001;293:1653–1657. [PubMed: 11533489]
16. Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, Olson EN, Hill JA. Suppression of class I andII histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 2006;113:2579–2588. [PubMed: 16735673]
17. Chen CS, Weng SC, Tseng PH, Lin HP. Histone acetylation-independent effect of histone deacetylaseinhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J Biol Chem2005;280:38879–38887. [PubMed: 16186112]
18. Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases actas signal-responsive repressors of cardiac hypertrophy. Cell 2002;110:479–488. [PubMed:12202037]
19. Lee TM, Lin MS, Chang NC. Inhibition of histone deacetylase on ventricular remodeling in infarctedrats. Am J Physiol Heart Circ Physiol 2007;293:H968–H977. [PubMed: 17400721]
20. Davis FJ, Pillai JB, Gupta M, Gupta MP. Concurrent opposite effects of trichostatin A, an inhibitorof histone deacetylases, on expression of alpha-MHC and cardiac tubulins: implication for gain incardiac muscle contractility. Am J Physiol Heart Circ Physiol 2005;288:H1477–H1490. [PubMed:15388503]
21. Sasaki S, Nakata T, Kawasaki S, Hayashi J, Oguro M, Takeda K, Nakagawa M. Chronic centralGABAergic stimulation attenuates hypothalamic hyperactivity and development of spontaneoushypertension in rats. J Cardiovasc Pharmacol 1990;15:706–713. [PubMed: 1692929]
Cardinale et al. Page 8
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

22. Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, NerviC, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducingdifferentiation of transformed cells. EMBO J 2001;20:6969–6978. [PubMed: 11742974]
23. Kook H, Lepore JJ, Gitler AD, Lu MM, Wing-Man W, Mackay Yung J, Zhou R, Ferrari V, GruberP, Epstein JA. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repressionmediated by the atypical homeodomain protein Hop. J Clin Invest 2003;112:863–871. [PubMed:12975471]
24. Aung HT, Schroder K, Himes SR, Brion K, van Zuylen W, Trieu A, Suzuki H, Hayashizaki Y, HumeDA, Sweet MJ, Ravasi T. LPS regulates proinflammatory gene expression in macrophages by alteringhistone deacetylase expression. FASEB J 2006;20:1315–1327. [PubMed: 16816106]
25. Brogdon JL, Xu Y, Szabo SJ, An S, Buxton F, Cohen D, Huang Q. Histone deacetylase activities arerequired for innate immune cell control of Th1 but not Th2 effector cell function. Blood2007;109:1123–1130. [PubMed: 17008546]
26. Ito K, Charron CE, Adcock IM. Impact of protein acetylation in inflammatory lung diseases.Pharmacol Ther 2007;116:249–265. [PubMed: 17720252]
27. Wade PA. Transcriptional control at regulatory checkpoints by histone deacetylases: molecularconnections between cancer and chromatin. Hum Mol Genet 2001;10:693–698. [PubMed: 11257101]
28. Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, Schreiber K, Rindt H, GorczynskiRJ, Olson EN. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylaseinhibitors. J Biol Chem 2003;278:28930–28937. [PubMed: 12761226]
29. Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, Yin Z, Ahn Y, Jeong MH, Bang YJ, Kim N, Kim JK,Kim KK, Epstein JA, Kook H. Inhibition of histone deacetylation blocks cardiac hypertrophy inducedby angiotensin II infusion and aortic banding. Circulation 2006;113:51–59. [PubMed: 16380549]
30. McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division,differentiation and death. Trends Biochem Sci 2002;27:40–47. [PubMed: 11796223]
31. Kuwahara K, Saito Y, Ogawa E, Takahashi N, Nakagawa Y, Naruse Y, Harada M, Hamanaka I, IzumiT, Miyamoto Y, Kishimoto I, Kawakami R, Nakanishi M, Mori N, Nakao K. The neuron-restrictivesilencer element-neuron-restrictive silencer factor system regulates basal and endothelin 1-inducibleatrial natriuretic peptide gene expression in ventricular myocytes. Mol Cell Biol 2001;21:2085–2097.[PubMed: 11238943]
32. Grimm D, Huber M, Jabusch HC, Shakibaei M, Fredersdorf S, Paul M, Riegger GA, Kromer EP.Extracellular matrix proteins in cardiac fibroblasts derived from rat hearts with chronic pressureoverload: effects of beta-receptor blockade. J Mol Cell Cardiol 2001;33:487–501. [PubMed:11181017]
33. Makino N, Sugano M, Otsuka S, Hata T. Molecular mechanism of angiotensin II type I and type IIreceptors in cardiac hypertrophy of spontaneously hypertensive rats. Hypertension 1997;30:796–802.[PubMed: 9336375]
34. Lu Y, Yang S. Angiotensin II induces cardiomyocyte hypertrophy probably through histonedeacetylases. Tohoku J Exp Med 2009;219:17–23. [PubMed: 19713680]
35. Kang YM, Ma Y, Zheng JP, Elks C, Sriramula S, Yang ZM, Francis J. Brain nuclear factor-kappa Bactivation contributes to neurohumoral excitation in angiotensin II-induced hypertension. CardiovascRes 2009;82:503–512. [PubMed: 19246475]
36. Wilcox CS, Welch WJ. Oxidative stress: cause or consequence of hypertension. Exp Biol Med(Maywood) 2001;226:619–620. [PubMed: 11444093]
37. Adcock IM. HDAC inhibitors as anti-inflammatory agents. Br J Pharmacol 2007;150:829–831.[PubMed: 17325655]
38. Bode KA, Schroder K, Hume DA, Ravasi T, Heeg K, Sweet MJ, Dalpke AH. Histone deacetylaseinhibitors decrease Toll-like receptor-mediated activation of proinflammatory gene expression byimpairing transcription factor recruitment. Immunology 2007;122:596–606. [PubMed: 17635610]
39. Walkinshaw DR, Tahmasebi S, Bertos NR, Yang XJ. Histone deacetylases as transducers and targetsof nuclear signaling. J Cell Biochem 2008;104:1541–1552. [PubMed: 18425769]
40. Paravicini TM, Touyz RM. Redox signaling in hypertension. Cardiovasc Res 2006;71:247–258.[PubMed: 16765337]
Cardinale et al. Page 9
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

41. Arenas IA, Xu Y, Lopez-Jaramillo P, Davidge ST. Angiotensin II-induced MMP-2 release fromendothelial cells is mediated by TNF-alpha. Am J Physiol Cell Physiol 2004;286:C779–C784.[PubMed: 14644777]
42. Brasier AR, Li J, Wimbish KA. Tumor necrosis factor activates angiotensinogen gene expression bythe Rel A transactivator. Hypertension 1996;27:1009–1017. [PubMed: 8613256]
43. Sasamura H, Nakazato Y, Hayashida T, Kitamura Y, Hayashi M, Saruta T. Regulation of vasculartype 1 angiotensin receptors by cytokines. Hypertension 1997;30:35–41. [PubMed: 9231818]
44. Allen AM, Zhuo J, Mendelsohn FA. Localization and function of angiotensin AT1 receptors. Am JHypertens 2000;13 31S–38S.
Cardinale et al. Page 10
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 1.Effects of VPA treatment on MAP. VPA attenuated the increase in MAP seen in untreatedSHR rats. VPA: valproic acid, MAP: mean arterial pressure. n=7–8 in each group, *P<0.05 vsWKY, #P<0.05 vs SHR.
Cardinale et al. Page 11
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 2.Effects of VPA treatment on cardiac hypertrophy. VPA attenuated ventricular wall thicknessas assessed by echocardiography (2A) during both diastole and systole. VPA also attenuatedthe HW/BW (indicating reduction in cardiac hypertrophy) and LW/BW (indicating reductionin lung edema due to reduced cardiac function) ratio in SHR rats (2B). SHR rats had increasedlevels of ANP and Collagen IV mRNA expression which were reduced with VPA treatment(2C). Graphs expressed as fold change versus WKY rats. VPA reduced cardiac fibrosis versusSHR rats as indicated by picrosirius red staining collagen versus total area (2D). Stained slidesare representative of typical results for each group (n=3/group). VPA: valproic acid, LVPWT:left ventricular parietal wall thickness, d: diastole, s: systole, HW: heart weight, LW: lung
Cardinale et al. Page 12
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

weight, BW: body weight, ANP: atrial natriuretic peptide. n=7–8 in each group, *P<0.05 vsSHR, #P<0.05 vs WKY.
Cardinale et al. Page 13
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 3.Effect of VPA on AT1-R mRNA and protein expression. Untreated SHR rats showed highermRNA (3B) and protein expression (3A) levels of the AT1-R in the LV when compared toWKY controls. SHR+VPA attenuated this increase. Western blot protein expression bandswere normalized to GAPDH. VPA: valproic acid, AT1-R: angiotensin II type 1 receptor. n=7–8 in each group, *P<0.05 vs SHR.
Cardinale et al. Page 14
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
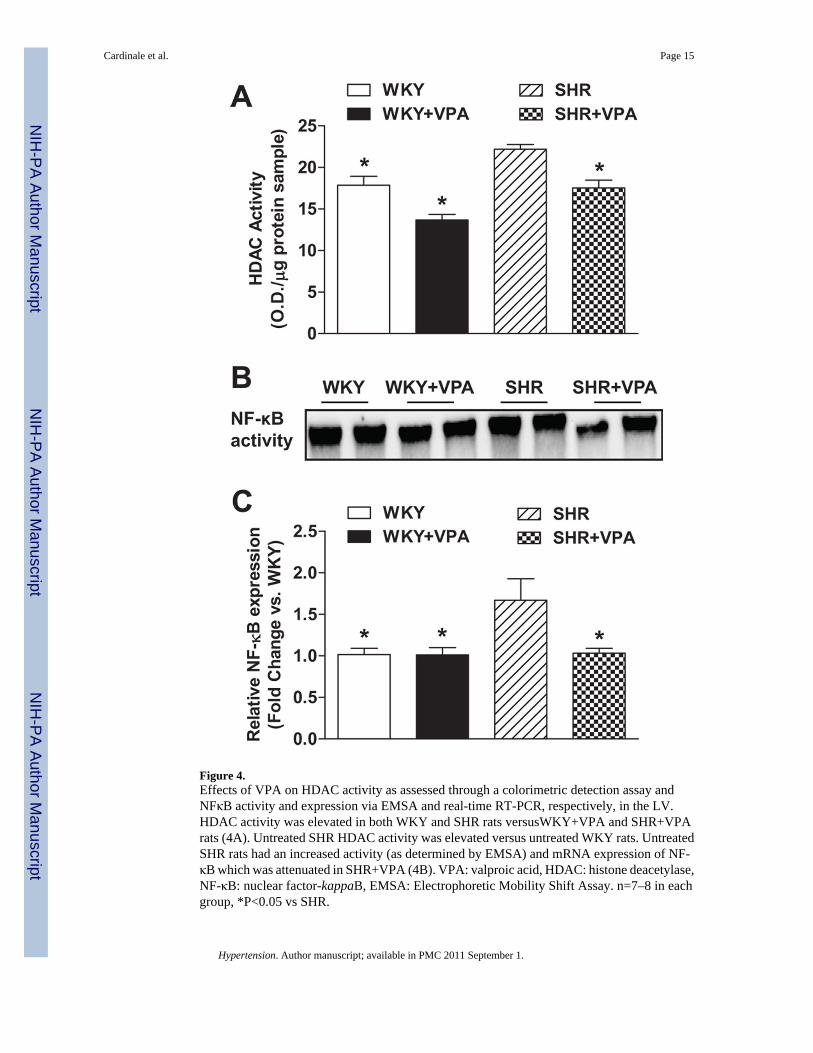
Figure 4.Effects of VPA on HDAC activity as assessed through a colorimetric detection assay andNFκB activity and expression via EMSA and real-time RT-PCR, respectively, in the LV.HDAC activity was elevated in both WKY and SHR rats versusWKY+VPA and SHR+VPArats (4A). Untreated SHR HDAC activity was elevated versus untreated WKY rats. UntreatedSHR rats had an increased activity (as determined by EMSA) and mRNA expression of NF-κB which was attenuated in SHR+VPA (4B). VPA: valproic acid, HDAC: histone deacetylase,NF-κB: nuclear factor-kappaB, EMSA: Electrophoretic Mobility Shift Assay. n=7–8 in eachgroup, *P<0.05 vs SHR.
Cardinale et al. Page 15
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 5.Effects of VPA treatment on TNF and IL-1β protein and mRNA expression. SHR rats hadnotably increased protein staining of TNF and IL-1β in the LV when compared to WKY ratsas shown through immunohistochemical staining (5A). This staining increase was markedlyreduced in SHR+VPA. Images shown represent results observed in preparations from 4 to 6rats. Untreated SHR rats had increased levels of PIC expression compared to WKY rats (5B).SHR+VPA reduced this increased expression in the LV tissue. VPA: valproic acid, TNF: tumornecrosis factor, IL-1β: interleukin-1beta, IL-6: interleukin-6, PIC: pro-inflammatorycytokines. n=7–8 in each group, *P<0.05 vs SHR.
Cardinale et al. Page 16
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 6.Effects of VPA treatment on tROS and gp91phox in the LV. SHR rats had increased tROSlevels as assessed by EPR. These levels were normalized in SHR+VPA (6A). SHR rats alsoshowed increased mRNA (6B) and protein (6C) expression of gp91phox, the catalytic subunitof NADPH oxidase, in the LV versus WKY rats. This was attenuated in SHR+VPA.Immunofluorescence of LV cardiomyocytes shows increased presence of gp91phox in SHRrats versus SHR+VPA. VPA: valproic acid, ROS: reactive oxygen species, EPR: electronparamagnetic spin resonance. n=7–8 in each group, *P<0.05 vs SHR.
Cardinale et al. Page 17
Hypertension. Author manuscript; available in PMC 2011 September 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript





























