Embed Size (px)
Citation preview

ORIGINAL INVESTIGATION
Gabor G. Kovacs Æ Maria Puopolo Æ Anna Ladogana
Maurizio Pocchiari Æ Herbert Budka
Cornelia van Duijn Æ Steven J. Collins Æ Alison Boyd
Antonio Giulivi Æ Mike Coulthart
Nicole Delasnerie-Laupretre Æ Jean Philippe Brandel
Inga Zerr Æ Hans A. Kretzschmar
Jesus de Pedro-Cuesta Æ Miguel Calero-Lara
Markus Glatzel Æ Adriano Aguzzi Æ Matthew Bishop
Richard Knight Æ Girma Belay Æ Robert Will
Eva Mitrova
Genetic prion disease: the EUROCJD experience
Received: 18 March 2005 / Accepted: 15 June 2005 / Published online: 27 September 2005� Springer-Verlag 2005
Abstract A total of 10–15% of human transmissiblespongiform encephalopathies (TSEs) or prion diseasesare characterised by disease-specific mutations in theprion protein gene (PRNP). We examined the pheno-
type, distribution, and frequency of genetic TSEs(gTSEs) in different countries/geographical regions. Wecollected standardised data on gTSEs between 1993 and2002 in the framework of the EUROCJD collaborative
I. ZerrDepartment of Neurology, Georg-August-Universitat Gottingen,Robert-Koch Strasse 40, 37075 Gottingen, Germany
H. A. KretzschmarInstitute of Neuropathology, University of Munich,Marchioninistr. 17, 81377 Munich, Germany
J. de Pedro-CuestaDepartamento de Epidemiologia Aplicada, Instituto de SaludCarlos III, Centro Nacional de Epidemiologia, Calle SinesioDelgado 6, 28029 Madrid, Spain
M. Calero-LaraCentro National de Microbiologia Unidad de EncefalopatiasEspongiformes, Ctra Majadakonda—Pozuelo km2,28220 Majadakonda, Madrid, Spain
M. Glatzel Æ A. AguzziSwiss National Reference Centre for Prion Diseases, UniversityHospital of Zurich, Schmelzbergstrasse 12,CH-8091 Zurich, Switzerland
M. Bishop Æ R. KnightNational CJD Surveillance Unit, Western General Hospital,Edinburgh, EH4 2XU UK
G. Belay Æ R. Will Æ E. Mitrova (&)Institute of Preventive and Clinical Medicine, Research Base ofSlovak Medical University, National Reference Centre of prionDiseases, Limbova 14, 833 01 Bratislava, SlovakiaE-mail: [email protected].: +421-2-59-36-9564Fax: +421-2-59-36-9585
Gabor G. Kovacs and Maria Propolo Contributed equally
G. G. Kovacs Æ H. BudkaAustrian Reference Centre for Human Prion Diseases (OERPE)and Institute of Neurology, Medical University AKH 4J,Waehringer Guertel 18-20, 1097 Vienna, Austria
M. Puopolo Æ A. Ladogana Æ M. PocchiariDepartment of Cell Biology and Neurosciences,Istituto Superiore di Sanita, Viale Regina Elena 299, 00161Rome, Italy
C. van DuijnDepartment of Epidemiology and Biostatistics, Erasmus MC,PO Box 1738, 3000 DR, Rotterdam, The Netherlands
S. J. Collins Æ A. BoydDepartment of Pathology, The University of Melbourne, Parkville,Victoria, 3052 Australia
A. GiuliviBlood Safety Surveillance and Health Care Acquired InfectionsDivision, The Centre for Infectious Disease Preventionand Control, LCDC Building, PL 0601E2,Tunney’s Pasture, Ottawa, ON,K1A 0L2 Canada
M. CoulthartNational Laboratory for Host Genetic and Prion Diseases,NML, PHAC, Health, Winnipeg, MB, Canada
N. Delasnerie-Laupretre Æ J. P. BrandelU.360 INSERM, Hopital de la Salpetriere, 75651 Paris,Cedex 13, France
Hum Genet (2005) 118: 166–174DOI 10.1007/s00439-005-0020-1

surveillance project. Our results show that clinicopath-ological phenotypes include genetic Creutzfeldt–Jakobdisease (gCJD), fatal familial insomnia (FFI), andGerstmann–Straussler–Scheinker disease (GSS). GeneticTSE patients with insert mutation in the PRNP repre-sent a separate group. Point and insertional mutations inthe PRNP gene varies significantly in frequency betweencountries. The commonest mutation is E200K. Absenceof a positive family history is noted in a significantproportion of cases in all mutation types (12–88%). FFIand GSS patients develop disease earlier than gCJD.Base pair insertions associated with the Creutzfeldt–Ja-kob disease (CJD) phenotype, GSS, and FFI cases havea longer duration of illness compared to cases with pointmutations and gCJD. Cerebrospinal fluid 14-3-3immunoassay, EEG, and MRI brain scan are useful inthe diagnosis of CJD with point mutations, but are lesssensitive in the other forms. Given the low prevalence offamily history, the term ‘‘gTSE’’ is preferable to‘‘familial TSE’’. Application of genetic screening inclinical practice has the advantage of early diagnosis andmay lead to the identification of a risk of a TSE.
Keywords Prion protein gene Æ Creutzfeldt-Jakobdisease Æ Fatal familial insomnia Æ Gerstmann-Straussler-Scheinker disease Æ Pointmutation Æ Insertional mutation
Introduction
Human transmissible spongiform encephalopathies(TSEs) or prion diseases are characterised by neurolog-ical and psychiatric symptoms and a progressive fatalcourse. Accumulation in the central nervous system(CNS) of the pathological prion protein (PrPSc) is acommon disease marker (Prusiner 2001). The most fre-quent human TSE is Creutzfeldt–Jakob disease (CJD).The majority of cases (85%) present as a sporadic dis-order (sCJD) without defined aetiology (Masters et al.1979; WHO 2003). Acquired forms, including iatrogenicCJD due to transmission of infection in the course ofmedical or surgical treatment, and variant CJD (vCJD),which has been linked to infection with bovine spongi-form encephalopathy, are less frequent (Masters et al.1979; Prusiner 2001; WHO 2003). The familial occur-rence of cases has been reported with a broad range offrequency: 6% of TSE cases in France (Brown et al.1979), 25.5% in Israel (Kahana et al. 1974), 26% in Chile(Galvez et al. 1980), 53.6% in Slovakia (Mitrova andBelay 2002), and world-wide 15% (Masters et al. 1979).
Current clinical and neuropathological diagnosticcriteria distinguish familial or genetic TSEs (gTSEs),including familial/genetic CJD (gCJD), fatal familialinsomnia (FFI), and Gerstmann–Straussler–Scheinkerdisease (GSS) (Budka et al. 1995; Prusiner 2001; WHO2003). Experimental transmissibility of all the major
subtypes has been established, albeit not with all muta-tions (Masters et al. 1981; Tateishi et al. 1979, 1995). IngTSEs, disease-specific point or insertional mutations inthe prion protein gene (PRNP) have been demonstrated(Goldfarb et al. 1990a, b, 1992; Goldgaber et al. 1989;Haltia et al. 1991; Owen et al. 1989), with individualPRNP mutations showing variable geographical distri-bution and frequency. While certain mutations are rare(Kovacs et al. 2002), the E200K mutation has been re-ported not only in Europe but also in Chile, Israel, Japan,and USA (Goldfarb et al. 1990a, b; Goldgaber et al.1989; Miyakawa et al. 1998). Geographic or ethnic clus-ters of cases of gTSEs have been found in Israel, Slovakia,Chile, and Italy (Chapman et al. 1994; D’Alessandroet al. 1998; Kahana et al. 1974; Mayer et al. 1977;Mitrova and Belay 2002; Mitrova and Bronis 1991).
In parallel with the increasing numbers of PRNPmutations, it has been recognised that, unexpectedly, notall patients withPRNPmutations appear to have affectedfamily members (Chapman et al. 1994; D’Alessandroet al. 1998; EuroCJD group 2001; Goldman et al. 2004;Mitrova and Belay 2002). Therefore the terms ‘‘familial’’,‘‘hereditary’’, or ‘‘inherited’’ TSEmay not be appropriatein cases without a family history and the inclusive term‘‘genetic TSE’’ may be preferable for cases associatedwith a mutation, whether or not there is a family history.
The negative family history in some gTSE cases andthe identification of ‘‘healthy’’ carriers has drawnattention to the issue of penetrance, i.e., the proportionof carriers who will eventually develop the disease(Chapman et al. 1994; D’Alessandro et al. 1998; Gold-farb et al. 1990a, b; Mitrova and Belay 2002). Pene-trance of the E200K mutation shows considerablevariability. While in Israeli carriers, penetrance appearsto be almost complete (89%), in Slovakian and ItalianE200K carriers, penetrance is partial (54–59%) (Chap-man et al. 1994; D’Alessandro et al. 1998; Goldfarbet al. 1990a, b; Mitrova and Belay 2002).
The EUROCJD project, funded by the EuropeanCommission, started in 1993 and compares data fromnational registries in Australia, Austria, Canada,France, Germany, Italy, The Netherlands, Slovakia,Spain, Switzerland, and the UK. The present studyanalyses the EUROCJD data on gTSE cases. Recentlyclinicopathological data of more than 500 publishedgenetic cases of the literature has been reviewed (Kovacset al. 2002) and some of the data on genetic epidemiol-ogy of CJD in Europe has been published (EuroCJDgroup 2001). The present paper includes standardiseddata from a large number of cases not previously re-ported in the context of a study carried out over a periodof years in a defined population. Our aims are: (1) todescribe the distribution and frequency of gTSEs, (2) todefine and describe specific features of subgroups ofgTSEs, (3) to contribute to a better understanding onhow mutations and polymorphisms of PRNP influenceclinical phenotypes, and (4) to assess the terms geneticand familial according to epidemiological and molecularbiological data.
167

Patients and methods
The EUROCJD database contains data on sporadic,variant, iatrogenic, and gTSE cases collected between1993 and 2002 (for detailed methodology see Ladoganaet al. 2005a). In this paper we have analysed data of 455gTSE cases from the following groups: gCJD cases(including patients with PRNP analysis and those withno available PRNP analysis but positive family historyfor TSE), FFI, GSS, and gTSE patients carrying basepair insertions (insert gTSE): referred to in previouspublications as BPI (Kovacs et al. 2002) because thesemutations are associated with repeat base pair insertionsin the octarepeat region. No deletions in this region wereidentified in this study. We evaluated E200K gCJD andV210I gCJD separately as there were sufficient numbersof cases to allow statistical analyses. Cases of gCJDwithout either an E200K or V210I mutation are classi-fied as ‘‘other CJD’’. Cases were classified either asdefinite or probable according to recent surveillance cri-teria (WHO 2003), which for the diagnosis of ‘‘familial’’CJD require definite or probable CJD in the index caseand a first-degree relative or a neuropsychiatric disorderin association with a PRNP mutation.
Statistical analysis
Differences among distinct forms of gTSE cases withrespect to age at onset or clinical duration were assessedby Mann–Whitney test; variation in clinical signs by thechi-square test or the Fisher’s exact probability test. TheBonferroni correction for multiple testing was adoptedwithin the six subgroups of gTSE (E200K, V210I, andother forms of gCJD, GSS, FFI, and insert mutations)at an experimental probability type 1 error of 0.05.
Age at onset was given as mean, standard deviation(SD), and range; clinical duration as median and halfinterquartile range (IQR) because of highly skeweddistribution of data. Box-plots were used for the graphicrepresentations of continuous variables. Statisticalanalyses were performed using BMDP and STATA.
Crude and sex-specific mortality rates were calculatedusing as denominator populations data for 1998 pro-vided from National or Federal Statistics Bureau ofparticipating countries. For Canada, population data(2001) were taken from the Canadian Statistics website.The annual mortality rates were calculated using datacollected from 1999–2002 when the surveillance systemwas well established in all countries.
Results
Distribution of cases and mutations
The distribution and frequency of PRNP mutationsamong countries participating in the study are sum-marised in Table. 1 and 2. The proportion of all gTSEcases with respect to the total number of TSE cases(including sporadic, iatrogenic, and variant CJD) was10.2%, but, this varied significantly among participatingcountries ranging from 69.5% in Slovakia to 1.2% inSwitzerland. The overall annual mortality rate of gTSEcases was 0.17 patients per million people for the period1999–2002. It is of note that Slovakia had an overallmortality rate for gTSE diseases 3.5 times that of thesecond highest country (Italy). However, while theE200K mutation was the only mutation present in theSlovak population, several mutations were present in theItalian population, including the E200K and V210IgCJD, FFI, and GSS. Switzerland reported a single
Table 1 EUROCJD 1993–2002: number of reported cases in each EUROCJD country
All genetic TSE diseases gCJD GSS FFI Insert
n gTSE (%) gTSE (%)with respectto all TSEin eachcountry
Mortality rates(million people)a
Australia 22/215 4.8 10.2 0.14 14 3 4 1Austria 13/90 2.9 14.4 0.28 9 0 3 1Canada 16/189 3.5 8.5 0.12 7 9 0 0France 84/938 18.5 9.0 0.18 68 5 6 5Germany 68/900 14.9 7.6 0.13 31 8 17 12Italy 115/662 25.3 17.4 0.30 94 8 10 3Netherlands 3/142 0.7 2.1 0.02 1 0 0 2Slovakia 41/59 9.0 69.5 1.07 41 0 0 0Spain 44/429 9.7 10.3 0.23 18 0 25 1Switzerland 1/85 0.2 1.2 0.04 1 0 0 0UK 48/732 10.5 6.6 0.07 11 19 1 17Total 455/4,441 – – 0.17 295 52 66 42% Total – 100 10.2 – 64.9 11.4 14.5 9.2
Abbreviations: gTSE, genetic transmissible spongiform encephalopathy; gCJD, genetic Creutzfeldt–Jakob disease; FFI, fatal familialinsomnia; GSS, Gerstmann–Straussler–Scheinker disease; Insert, gTSE patients with insert mutations.a For the period 1999–2002.
168

gTSE patient for the period 1996–2002 while the Neth-erlands had the lowest mortality rate for gTSE diseases.More than 90% of FFI and 75% of GSS patientsunderwent post-mortem examination and were classifiedas definite gTSE cases, while 52% of V210I gCJD pa-tients did not undergo autopsy (see Table 3) and wereclassified as probable gTSE based on clinical and labo-ratory features. There was an excess of females in gCJD(more pronounced for E200K than V210I or ‘‘other’’gCJD patients) and, to a lesser extent, in GSS or insertgTSE cases, and an excess of males in FFI cases (Ta-ble 3). Data on the polymorphic codon 129 of the PRNPgene were available in 87% of all cases (95% in E200K,97% in V210I, 75% in ‘‘other’’ gCJD, 95% in FFI, 56%in GSS, and 79% in insert gTSE). The distribution of thepolymorphic codon 129 in all forms of gTSE is shown inTable 3. Differences between codon 129 distribution incontrols (39% methionine homozygous (MM), 50%heterozygous (MV), 11% valine homozygous (VV), Al-perovitch et al. 1999) and in individual gTSE groupshave been evaluated by chi-sqared test with the follow-ing results: E200K gCJD, V210I gCJD and FFI,P<0.0001, ‘‘other’’ gTSE, P=0.0010, Insert gTSE,P=0.0577 and GSS, P=0.3825. The majority of PRNPmutations co-segregate with methionine at the poly-morphic codon 129 (see Table 2). Thus, the majority ofgTSE patients overall were either MM (67.9%) or MV(25.8%) with only a few valine homozygotes (6.3%).
Family history
About 47% of all gTSE cases were reported to have noTSE or other neurological disorder in family members.Almost 90% of V210I gCJD patients had a negativefamily history implying that a correct classification ofthese cases would not have been possible without PRNPgenetic analysis. Interestingly, a positive family historyfor TSE was reported in only about two-thirds of GSSpatients (Table 3).
Age at onset
There were significant differences in the age at onset ingTSE forms (Fig. 1a). Patients with FFI (mean51.2 years, SD 12.3, range 19–83) and GSS (51.6, 12.8,26–87) developed disease significantly earlier thanE200K gCJD (60.4, 10.2, 33–84, p<0.0001), V210IgCJD (59.3, 9.8, 39–82, p=0.0001 and 0.0009, respec-tively), and ‘‘other’’ gCJD cases (60.4, 14.7, 31–87,p=0.0004, 0.0036). There was no significant differencein the age at onset between FFI and GSS patients andamong distinct forms of gCJD. Age at onset in patientswith insert mutations (57.2, 14.8, 32–85) did not signif-icantly differ from that of gCJD, GSS, or from FFI(p=0.0193) after Bonferroni’s correction. The youngestage at onset was 19 years (FFI, MM at codon 129),while the two oldest were 87 years (gCJD with missinginformation on PRNP mutation, and A117V GSS, VVat codon 129). The age at onset was earlier in valinehomozygotes in comparison to methionine homozygotes(P=0.0001) and, to a lesser extent, in MV vs MM(P=0.0059, not significant after Bonferroni’s correction)in ‘‘other’’ gCJD patients. The codon 129 polymorphismdid not significantly influence the age at onset in all theother forms of gTSEs (Table 4). Gender did not haveany significant effect on the age at onset (data notshown).
Duration of illness
The duration of disease varied between different formsof gTSE (Fig. 1b). Clinical durations of E200K (median5.0 months, IQR 2.5) and V210I (4.0, 1.5) gCJDpatients were shorter than ‘‘other’’ gCJD (7.0, 7.0,p=0.0031 and 0.0012, respectively), FFI (12.4, 4.2,P<0.0001 and 0.0001), GSS (40.0, 25.0, P<0.0001 and0.0001) and insert gTSE (13.0, 35.5, P=0.0001 and0.0001). GSS patients had a longer survival than FFI(P<0.0001) and ‘‘other’’ gCJD (P<0.0001) patients.
Table 2 EUROCJD 1993–2002: number of reported cases and distribution of PRNP mutations in each EUROCJD country
Country gCJD
P105T-129? N171S-129V D178N-129V V180I-129M T188A-129M E196K-129M/V E200K-129M/V V203I-129M R208H-129M V210I-129M E211Q-129M NS
Australia 1 – – – 1 – 8 – – 2 – 2Austria – – 1 – – 1 5 – – 1 – 1
Canada – – 2 – – – 4 1 – – – –France – 1 8 1 – 1 46 3 – 6 2 –
Germany – – – – 2 3 15 – 1 9 1 –Italy – – – – – – 35 1 1 50 1 6
Netherlands – – 1 – – – – – – – – –Slovakia – – – – – – 40 – – – – 1
Spain – – 4 – – – 14 – – – – –Switzerland – – – – – – 1 – – – – –
UK – – – – – – 7 – – 1 – 3Total 1 1 16 1 3 5 175 5 2 69 4 13
% Total 0.2 0.2 3.5 0.2 0.7 1.1 38.5 1.1 0.4 15.2 0.9 2.9
Abbreviations: NS, not specified; others as in Table 1.
169

The longest duration of illness was 216 months in aninsert gTSE case, the group with the highest variabilityin clinical duration. However, in all other groups therewere a few cases with an exceptional long clinicalduration for their group (outliers in Fig. 1b). This in-cluded 11 cases in E200K gCJD (five MM at codon 129:16, 19, 20 and two 24 months; 6 MV: 17, two 18, 19,and two 36 months), four cases in V210I gCJD (MM:11, 15, 31, and 34 months), four in ‘‘other’’ gCJD(43 months, D178N-129VV, 46 months, V180I-129MV,52 months, V203I-129MV, 59 months, missing muta-tion and codon 129), and one in FFI (MV: 97 months),and two in insert gTSE (216 months 120 bp insert-129MV, and 192 months 168 bp insert-129 MV).
Investigations
Data on CSF 14-3-3 immunoassay were available in57% of cases (60% in E200K, 72% in V210I, 59% in‘‘other’’ gCJD, 58% in FFI, 31% in GSS, and 50% ininsert gTSE), EEG in 82% of cases (90% in E200K,96% in V210I, 86% in ‘‘other’’ gCJD, 77% in FFI, 50%in GSS, and 67% in insert gTSE), and MRI brain scanin 43% of cases (38% in E200K, 67% in V210I, 31% in‘‘other’’ gCJD, 52% in FFI, 33% in GSS, and 36% ininsert gTSE). In the various forms of gTSEs, there weresignificant differences in the frequency in positivity ofthe 14-3-3 test in the CSF (P<0.0001, Fisher’s exacttest), the EEG (P<0.0001, chi-squared test) and theMRI brain scan (P=0.0038, Fisher’s exact test) (WHO2003). A positive 14-3-3 test was present in the majorityof gCJD patients and insert gTSE patients (Fig. 2). Atypical EEG (WHO 2003) was more frequent in gCJDthan in other forms of gTSE diseases. About 50% ofGSS cases had a positive 14-3-3 test in the CSF, whilethe EEG showed typical pseudoperiodic activity in only2 of 26 patients with available information (Fig. 2). InGSS and FFI, neither the EEG nor the 14-3-3 tests wereof help in the clinical diagnosis of disease. MRI brainscan was performed in a small proportion of cases andwas positive (WHO 2003) in about 50% of E200K and
‘‘other’’ gCJD and in about 30% of GSS and insertgTSE cases. In FFI and V210I gCJD patients, the MRIbrain scan was positive in only 18 and 15%, respectively.
Classification of protease-resistant PrP
Results were available in only 43 cases. Twenty-twoE200K, 6 V210I gCJD, 9 ‘‘other’’ gCJD, 1 FFI, 2 GSS,and 3 insert gTSE cases (Table 5). Thirty-four patientshad type 1 protease-resistant prion protein (according tothe system described by Parchi et al.) (Parchi et al. 1999)in brain samples, six had type 2A, and three patients hadboth type 1 and type 2A.
Discussion
EUROCJD has data on 23 specified PRNP mutations.Currently more than 30 mutations have been reported inthe world literature, but many of these mutations arevery rare or are restricted to specific populations (Kov-acs et al. 2002). Comparison of genetic and sporadicTSEs shows striking differences in their frequency andgeographic distribution. While sCJD has a similar inci-dence in all participating countries (Ladogana et al.2005a), PRNP mutations show significant variability.Some mutations (P105L, N171S, V180I, T188A, E196K,R208H, V203I, 168 BPI) are extremely rare, whileE200K is recognised in 9 out of 10 reporting countries.The high absolute and proportionate incidence of theV210I mutation in Italy is striking; most affected fami-lies lived in three adjacent areas (Ladogana et al. 2005b).It is a pertinent point that a single report on the N171Smutation suggested a psychiatric phenotype, whereas inour series this mutation was associated with CJD(Samaia et al. 1997). A high proportion of all cases ineach category underwent genetic analysis. The threelowest percentages of PRNP tests were 42.3% (Canada),32% (Australia) and 31.7% (Netherlands). The vari-ability in the frequency of mutations countrywise isunexpected and is not related to variation countrywise in
GSS FFI Insert
P102L-129M A117V-129V G131V-129M NS D178N-129M NS 24-129M 48-129M 72-129? 96-129M/V 120-129M/V 144-129M 168-129M 192-129M/V NS
1 – 1 1 3 1 – – – – – – 1 – –– – – – 3 – – – – – – 1 – – –
1 – – 8 – – – – – – – – – – –3 2 – – 6 – 2 – – 1 – 1 – 1 –
3 5 – – 17 – 1 – 1 2 8 – – – –8 – – – 10 – 2 – – 1 – – – – –
– – – – – – 1 – – – – – – 1 –– – – – – – – – – – – – – – –
– – – – 24 1 – 1 – – – – – – –– – – – – – – – – – – – – – –
8 5 – 6 1 – – – – 1 4 9 1 – 224 12 1 15 64 2 6 1 1 5 12 11 2 2 2
5.3 2.6 0.2 3.3 14.1 0.4 1.3 0.2 0.2 1.1 2.6 2.4 0.4 0.4 0.4
170

the availability of genetic data. Focal accumulations ofgenetic patients (Mayer et al. 1977) may be due to ge-
netic isolation but the explanation for the overall inter-country variability in this study is uncertain.
The ratio of female to male mortality rates in Table 3indicates that there is an excess of females in all mutationsexcept FFI. Taking account of varying distribution byage, the weighted female to male ratio (1.59 for E200K,1.62 for V210I, 1.90 for other gCJD, 1.43 for GSS, 1.62for insert gTSE, and 0.62 for FFI) strongly confirms theexcess of female cases in all forms of gCJD except FFI.Although consistent with similar findings in sporadicCJD (Ladogana et al. 2005a), the explanation for thisgender bias in mortality is uncertain. Possible explana-tion include sex-linked genetic factors influencing diseaseexpression, bias in case ascertainment linked to gender ordifferential susceptibility/exposure to a co-factor.
Clinical and laboratory parameters show consider-able similarity between the most frequent gCJD (e.g.,E200K mutation) and sCJD cases. Although gCJD caseswith point mutations have an earlier mean age at death,there is no difference between gCJD cases with pointmutations and sCJD in the mean duration of the disease(Alperovitch et al. 1999; Pocchiari et al. 2004). As insCJD (Zerr et al. 2000), analysis of CSF 14-3-3 protein,EEG, and MRI is helpful in the diagnosis of gTSEs and
Table 3 Characteristics of genetic transmissible spongiform encephalopathies
Forms of gTSE n Percentage ofdefinite cases(n)
Gender Codon 129 polymorphism Percentage ofpositive familyhistory for TSE(available data)
F M F/M ratiosof mortalityratesa
MM % (n) MV % (n) VV % (n)
E200K gCJD 175 68.0 (119) 107 68 1.69 78.3 (130) 19.9 (33) 1.8 (3) 49.1 (114)V210I gCJD 69 47.8 (33) 39 30 1.42 73.1 (49) 26.9 (18) – 12.3 (57)Other gCJD 51 58.8 (30) 28 23 1.38 36.8 (14) 34.2 (13) 29.0 (11) 75.9 (29)FFI 66 92.4 (61) 29 37 0.62 71.4 (45) 28.6 (18) – 88.0 (50)GSS 52 76.9 (40) 29 23 1.17 51.7 (15) 34.5 (10) 13.8 (4) 69.7 (33)Insert gTSE 42 69.0 (29) 21 21 1.23 48.5 (16) 30.3 (10) 21.2 (7) 48.0 (25)
Abbreviations: gTSE, genetic transmissible spongiform encephalopathy; gCJD, genetic Creutzfeldt–Jakob disease; FFI, fatal familialinsomnia; GSS, Gerstmann–Straussler–Scheinker disease; Insert, gTSE patients with insert mutations.a For the period 1999–2002.
Fig. 1 Box- and whisker-plots of age at onset (a) and clinicalduration (b) in different forms of genetic TSE diseases. The line inthe middle of the box represents median. The box extends from the25th percentile (x[25]) to the 75th percentile (x[75]), the so-calledinterquartile range (2IQR). The lines emerging from the box arecalled the whiskers and they extend to the upper and lower adjacentvalues. The upper adjacent value is defined as the largest data pointless than or equal to x[75]+1.52IQR. The lower adjacent value isdefined as the smallest data point greater than or equal to x[25]�1.52IQR. Filled circles represent values more extreme than theadjacent values (referred to as outliers)
Table 4 Influence of codon 129 polymorphism of the PRNP genein determining the age at onset
Forms of gTSE Codon 129
Met/ Metmean (SD)
Met/Valmean (SD)
Val/Valmean (SD)
E200K gCJD 60.4 (10.8) 60.7 (8.6) 55.7 (9.0)V210I gCJD 59.3 (9.7) 57.9 (9.8) –Other gCJD 70.9 (7.5) 56.2 (14.2) 48.1 (12.6)FFI 50.8 (13.4) 52.5 (10.0) –GSS 54.0 (11.1)a 49.0 (12.1) 66.2 (17.9)Insert gTSE 60.9 (14.0)a 56.2 (15.2) 62.7 (11.3)
Abbreviations: gTSE, genetic transmissible spongiform encepha-lopathy; gCJD, genetic Creutzfeldt–Jakob disease; FFI, fatalfamilial insomnia; GSS, Gerstmann–Straussler–Scheinker disease;Insert, gTSE patients with insert mutations.a Two patients had missing data on age at onset.
171
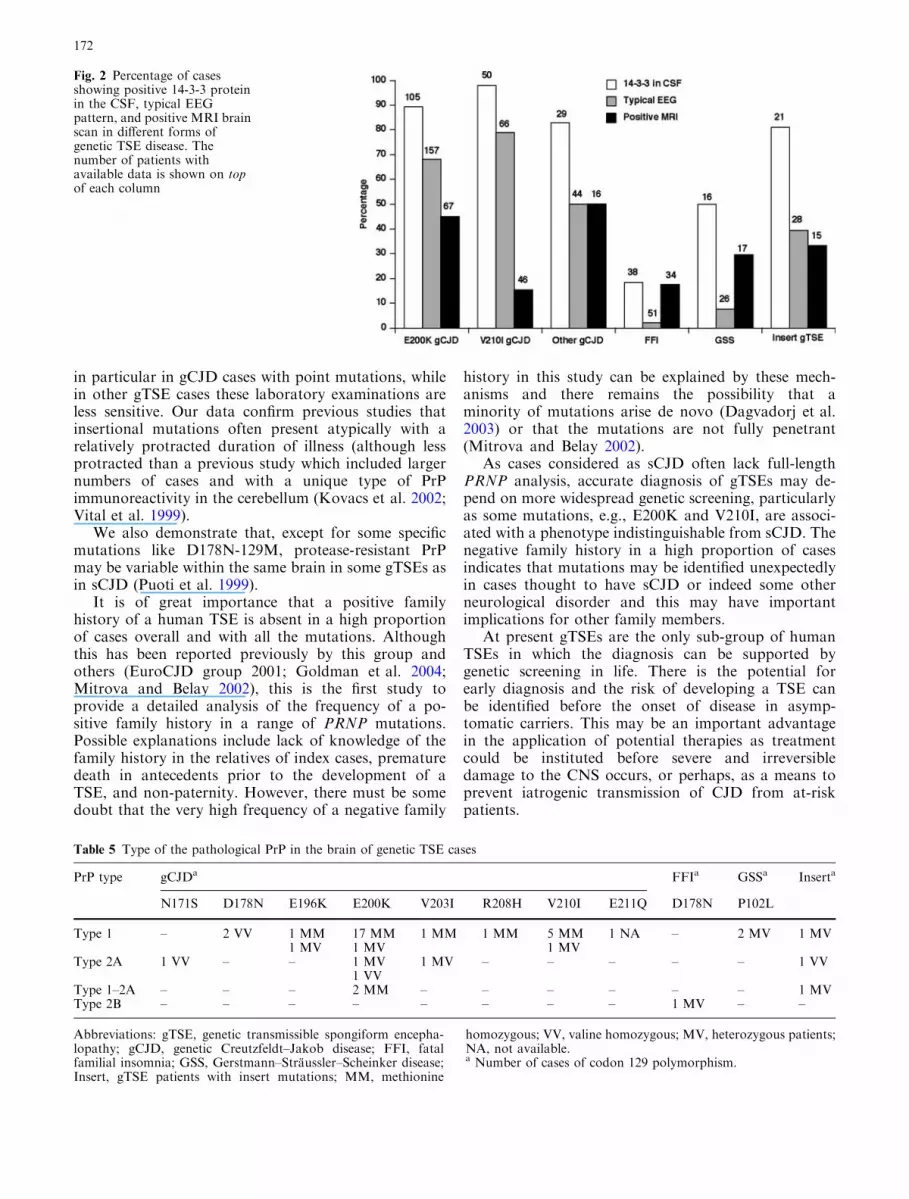
in particular in gCJD cases with point mutations, whilein other gTSE cases these laboratory examinations areless sensitive. Our data confirm previous studies thatinsertional mutations often present atypically with arelatively protracted duration of illness (although lessprotracted than a previous study which included largernumbers of cases and with a unique type of PrPimmunoreactivity in the cerebellum (Kovacs et al. 2002;Vital et al. 1999).
We also demonstrate that, except for some specificmutations like D178N-129M, protease-resistant PrPmay be variable within the same brain in some gTSEs asin sCJD (Puoti et al. 1999).
It is of great importance that a positive familyhistory of a human TSE is absent in a high proportionof cases overall and with all the mutations. Althoughthis has been reported previously by this group andothers (EuroCJD group 2001; Goldman et al. 2004;Mitrova and Belay 2002), this is the first study toprovide a detailed analysis of the frequency of a po-sitive family history in a range of PRNP mutations.Possible explanations include lack of knowledge of thefamily history in the relatives of index cases, prematuredeath in antecedents prior to the development of aTSE, and non-paternity. However, there must be somedoubt that the very high frequency of a negative family
history in this study can be explained by these mech-anisms and there remains the possibility that aminority of mutations arise de novo (Dagvadorj et al.2003) or that the mutations are not fully penetrant(Mitrova and Belay 2002).
As cases considered as sCJD often lack full-lengthPRNP analysis, accurate diagnosis of gTSEs may de-pend on more widespread genetic screening, particularlyas some mutations, e.g., E200K and V210I, are associ-ated with a phenotype indistinguishable from sCJD. Thenegative family history in a high proportion of casesindicates that mutations may be identified unexpectedlyin cases thought to have sCJD or indeed some otherneurological disorder and this may have importantimplications for other family members.
At present gTSEs are the only sub-group of humanTSEs in which the diagnosis can be supported bygenetic screening in life. There is the potential forearly diagnosis and the risk of developing a TSE canbe identified before the onset of disease in asymp-tomatic carriers. This may be an important advantagein the application of potential therapies as treatmentcould be instituted before severe and irreversibledamage to the CNS occurs, or perhaps, as a means toprevent iatrogenic transmission of CJD from at-riskpatients.
Fig. 2 Percentage of casesshowing positive 14-3-3 proteinin the CSF, typical EEGpattern, and positive MRI brainscan in different forms ofgenetic TSE disease. Thenumber of patients withavailable data is shown on topof each column
Table 5 Type of the pathological PrP in the brain of genetic TSE cases
PrP type gCJDa FFIa GSSa Inserta
N171S D178N E196K E200K V203I R208H V210I E211Q D178N P102L
Type 1 – 2 VV 1 MM 17 MM 1 MM 1 MM 5 MM 1 NA – 2 MV 1 MV1 MV 1 MV 1 MV
Type 2A 1 VV – – 1 MV 1 MV – – – – – 1 VV1 VV
Type 1–2A – – – 2 MM – – – – – – 1 MVType 2B – – – – – – – – 1 MV – –
Abbreviations: gTSE, genetic transmissible spongiform encepha-lopathy; gCJD, genetic Creutzfeldt–Jakob disease; FFI, fatalfamilial insomnia; GSS, Gerstmann–Straussler–Scheinker disease;Insert, gTSE patients with insert mutations; MM, methionine
homozygous; VV, valine homozygous; MV, heterozygous patients;NA, not available.a Number of cases of codon 129 polymorphism.
172

The occurrence of asymptomatic carriers of PRNPmutations (Mitrova and Belay 2002) underlines theimportance of genetic testing in all suspect cases of hu-man TSEs and, in positive cases, relatives may be offeredgenetic testing, provided appropriate ethical protocolsare followed. Although there may be incomplete pene-trance and an absence of presymptomatic neuropatho-logical changes or accumulation of disease-associatedPrP (Sasaki et al. 2003), healthy carriers of PRNPmutations are at greater risk of developing a TSE andmay be subject to restrictions, such as donating bloodand tissues (e.g., cornea, dura mater), because of the riskof iatrogenic transmission.
It is not known why there is partial penetrance insome mutations, or why there is marked variability inclinical phenotype, including age at onset, both betweenand within pedigrees. There is a need to analyse eventspreceding the onset of clinical manifestation in carriersof PRNP mutations as possible triggering factors. InE200K, psychological stress (divorce, death of a closerelative, retirement, loss of a job), complicated surgerywith prolonged anaesthesia, serious accidents, or currentinfectious disease have been frequently noted (Brandeland Delasnerie-Laupretre 1997; Mitrova and Belay2002).
In conclusion, the term gTSE, which includes gCJD,FFI, and GSS, is more appropriate than familial TSEfor all patients with TSE-specific genetic marker,regardless of the number of affected family members,while familial TSE designates only genetic cases withother (one or more) TSE-affected relative. The distri-bution of gCJD is geographically heterogeneous. Thecodon 129 polymorphism influences phenotype. IngTSEs, PRNP analysis may allow early or pre-symp-tomatic diagnosis and this may be important if specifictherapies become available. Further studies are requiredto identify whether PRNP mutations are sufficient inthemselves to cause disease.
Acknowledgements This study was funded through an EU Con-certed Action (BIOMED2 Contract No. BMH4-CT97-2216).Australia: The Australian National CJD Registry is funded by theCommonwealth Department of Health and Ageing. We aregrateful to the following people involved in the Australian NationalCJD Registry: C.L. Masters, A. Boyd, G. Klug, and J. Lee. Aus-tria: The Austrian Reference Centre for Human Prion Diseases(OERPE, Head: Prof. Herbert Budka) acknowledges the help ofDrs Christa Jarius, Ellen Gelpi, Christine Haberler, ThomasStrobel, and Till Voigtlander; DI Dita Drobna; and Ms. HelgaFlicker, Brigitte Millan-Ruiz, and Monika Richter. Canada: TheCanadian Surveillance System is funded by Health Canada. Othercollaborators on the project are Dr C Bergeron, neuropathologist(University of Toronto), Dr N Cashman, neurologist and one ofthe principal investigators for CJD-SS, Dr D Westaway, consultingscientist (University of Toronto). France: We would like toacknowledge all reporting physicians and the members of the Re-seau National de surveillance de maladies de Creutzfeldt-Jakob etmaladies apparentees. Germany: The German surveillance system isfunded by the Federal Ministry of Health 9BMG, 325-4471-02/15.We are grateful to all reporting physicians throughout Germanywho contributed to the German surveillance system and especiallyto Maja Schneider-Dominico for her excellent support in the co-ordination of surveillance. We also acknowledge the help of Drs
Otto Windl and Walter Sculz-Schaeffer. Italy: We would like toacknowledge the Ministry of Health and the Istituto Superiore diSanita for supporting the surveillance of CJD in Italy and S. Al-monti, V. Mellina, and L. Ingrosso for help in collecting data andadvice. The Netherlands: CJD surveillance in the Netherlands isfunded by the Dutch Ministry of Health, Welfare, and Sports. Weacknowledge the help of colleagues at the Department of Neurol-ogy at the Academic Medical Centre, Amsterdam and theDepartment of Pathology at the University Medical Centre, Utr-echt. Slovakia: The Slovak Surveillance System was supported bythe Slovak Ministry of Health and by grants from the EuropeanUnion. We would like to acknowledge the help of Dr. Dana Sliv-arichova, Dr. Vladimıra Verchovodkova, all reporting physiciansand collaborating pathologists. Spain: We are grateful to allreporting physicians and to members of the Spanish TSE studygroup at Consejo Interterritorial and co-workers at CNE andISCIII and laboratories, particularly to N. Cuadrado and J.Yague.Switzerland: This work was supported by the Kanton of Zurich andby grants from the European Union. The Swiss Reference Centerfor Prion Diseases is being funded by the Swiss Federal Office ofPublic Health. UK: The UK CJD Surveillance System is funded bythe Department of Health and the Scottish Executive HealthDepartment. We are grateful to all the members of staff at theNational CJD Surveillance Unit and in particular to James Iron-side for neuropathological expertise and to clinicians throughoutthe UK for their co-operation with the study.
References
Alperovitch A, Zerr I, Pocchiari M, Mitrova E, de Pedro Cuesta J,Hegyi I, Collins S, Kretzschmar H, van Duijn C, Will RG(1999) Codon 129 prion protein genotype and sporadic Cre-utzfeldt–Jakob disease. Lancet 353:1673–1674
Brandel JF, Delasnerie-Laupretre N (1997) Creutzfeldt–Jakobdisease and stress. J Neurol 62:541
Brown P, Cathala F, Sadowsky D, Gajdusek DC (1979) Creutz-feldt–Jakob disease in France: III. Epidemiological study of 170patients dying during the decade 1968–1977. Ann Neurol 6:438–446
Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, Gullotta F,Haltia M, Hauw JJ, Ironside JW, Jellinger K et al (1995)Neuropathological diagnostic criteria for Creutzfeldt–Jakobdisease (CJD) and other human spongiform encephalopathies(prion diseases). Brain Pathol 5:459–466
Chapman J, Ben-Israel J, Goldhammer Y, Korczyn AD (1994) Therisk of developing Creutzfeldt–Jakob disease in subjects withthe PRNP gene codon 200 point mutation. Neurology 44:1683–1686
Dagvadorj A, Petersen RB, Lee HS et al (2003) Spontaneousmutations in the prion protein gene causing transmissiblespongiform encephalopathy. Ann Neurol 52:355–359
D’Alessandro M, Petraroli R, Ladogana A, Pocchiari M (1998)High incidence of Creutzfeldt–Jakob disease in rural Calabria,Italy. Lancet 352:1989–1990
EuroCJD group (2001) Genetic epidemiology of Creutzfeldt–Jakobdisease in Europe. Rev Neurol (Paris) 157:633–637
Galvez S, Masters C, Gajdusek C (1980) Descriptive epidemiologyof Creutzfeldt–Jakob disease in Chile. Arch Neurol 37:11–14
Goldfarb LG, Korczyn AD, Brown P, Chapman J, Gajdusek DC(1990a) Mutation in codon 200 of scrapie amyloid precursorgene linked to Creutzfeldt–Jakob disease in Sephardic Jews ofLibyan and non-Libyan origin. Lancet 336:637–638
Goldfarb LG, Mitrova E, Brown P, Toh BK, Gajdusek DC(1990b) Mutation in codon 200 of scrapie amyloid protein genein two clusters of Creutzfeldt–Jakob disease in Slovakia. Lancet336:514–515
Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC,Montagna P, Cortelli P, Julien J, Vital C, Pendelbury WW et al(1992) Fatal familial insomnia and familial Creutzfeldt–Jakobdisease: disease phenotype determined by a DNA polymor-phism. Science 258:806–808
173

Goldgaber D, Goldfarb LG, Brown P, Asher DM, BrownWT, Lin S,Teener JW, Feinstone SM, Rubenstein R, Kascsak RJ et al (1989)Mutations in familial Creutzfeldt–Jakob disease and Gerstmann–Straussler–Scheinker’s syndrome. Exp Neurol 106:204–206
Goldman JS, Miller BL, Safar J, de Tourreil S, Martindale JL,Prusiner SB, Geschwind MD (2004) When sporadic disease isnot sporadic: the potential for genetic etiology. Arch Neurol61:213–216
Haltia M, Kovanen J, Goldfarb LG, Brown P, Gajdusek DC(1991) Familial Creutzfeldt–Jakob disease in Finland: epide-miological, clinical, pathological and molecular genetic studies.Eur J Epidemiol 7:494–500
Kahana E, Alter M, Braham J, Sofer D (1974) Creutzfeldt–Jakob disease: focus among Libyan Jews in Israel. Science183:90–91
Kovacs GG, Trabattoni G, Hainfellner JA, Ironside JW, KnightRSG, Budka H (2002) Mutations of the human prion proteingene: phenotypic spectrum. J Neurol 249:1567–1582
Ladogana A, Puopulo M, Croes EA, Budka H, Jarius C, Collins S,Masters C, Sutcliffe T, Giulivi A, Alperovitch A, Delasnerie-Laupretre N, Brandel JP, Poser S, Kretzschmar H, Rietvald I,Mitrova E, De Pedro Cuesta J, Martinez-Martin P, Glatzel M,Aguzzi A, Knight RS, Ward H, Pocchiari M, van Duijn C, WillRG, Zerr I (2005a) Mortality from Creutzfeldt–Jakob diseaseand related disorders in Europe, Australia and Canada. Neu-rology 64:1586–1591
Ladogana A, Puopulo M, Poleggi D, Almonti S, Mellina V,Equestre M, Pocchiari M (2005b) High incidence of genetichuman transmissible spongiform encephalopathies in Italy.Neurology 64:1592–1597
Masters CL, Harris JO, Gajdusek DC, Gibbs CJ Jr, Bernoulli C,Asher DM (1979) Creutzfeldt–Jakob disease: patterns ofworldwide occurrence and the significance of familial andsporadic clustering. Ann Neurol 5:177–188
Masters CL, Gajdusek DC, Gibbs CJ Jr (1981) Creutzfeldt–Jakobdisease virus isolations from the Gerstmann–Straussler syn-drome with an analysis of the various forms of amyloid plaquedeposition in the virus-induced spongiform encephalopathies.Brain 104:559–588
Mayer V, Orolin D, Mitrova E (1977) Cluster of Creutzfeldt–Jakobdisease and presenile dementia. Lancet 2:256
Mitrova E, Belay G (2002) Creutzfeldt–Jakob disease with E200Kmutation in Slovakia: characterization and development. ActaVirol 46:31–39
Mitrova E, Bronis M (1991) ‘‘Clusters’’ of CJD in Slovakia: thefirst statistically significant temporo-spatial accumulations ofrural cases. Eur J Epidemiol 7:450–456
Miyakawa T, Inoue K, Iseki E, Kawanishi C, Sugiyama N, OnishiH, Yamada Y, Suzuki K, Iwabuchi K, Kosaka K (1998) Jap-anese Creutzfeldt–Jakob disease patients exhibiting high inci-dence of the E200K PRNP mutation and located in the basin ofa river. Neurol Res 20:684–688
Owen F, Poulter M, Lofthouse R, Collinge J, Crow TJ, Risby D,Baker HF, Ridley RM, Hsiao K, Prusiner SB (1989) Insertionin prion protein gene in familial Creutzfeldt–Jakob disease.Lancet 1:51–52
Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W,Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S,Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B,Gambetti P, Kretzschmar H (1999) Classification of sporadicCreutzfeldt–Jakob disease based on molecular and phenotypicanalysis of 300 subjects. Ann Neurol 46:224–233
Pocchiari M, Puopolo M, Croes EA, Budka H, Gelpi E, Collins S,Lewis V, Sutcliffe T, Guilivi A, Delasnerie-Laupretre N,Brandel JP, Alperovitch A, Zerr I, Poser S, Kretzschmar HA,Ladogana A, Rietvald I, Mitrova E, Martinez-Martin P, dePedro-Cuesta J, Glatzel M, Aguzzi A, Cooper S, Mackenzie J,van Duijn CM, Will RG (2004) Predictors of survival in spo-radic Creutzfeldt–Jakob disease and other human transmissiblespongiform encephalopathies. Brain 127:2348–2359
Prusiner SB (2001) Shattuck lecture—neurodegenerative diseasesand prions. N Engl J Med 344:1516–1526
Puoti G, Giaccone G, Rossi G, Canciani B, Bugiani O, TagliaviniF (1999) Sporadic Creutzfeldt–Jakob disease: co-occurrence ofdifferent types of PrP(Sc) in the same brain. Neurology53:2173–2176
Samaia HB, Mari JJ, Vallada HP, Moura RP, Simpson AJ,Brentani RR (1997) A prion-linked psychiatric disorder. Nature390:241
Sasaki K, Doh-Ura K, Furuta A, Nakashima S, Morisada Y,Tateishi J, Iwaki T (2003) Neuropathological features of a casewith schizophrenia and prion protein gene P102L mutationbefore onset of Gerstmann–Straussler–Scheinker disease. ActaNeuropathol 106:92–96
Tateishi J, Ohta M, Koga M, Sato Y, Kuroiwa Y (1979)Transmission of chronic spongiform encephalopathy withkuru plaques from humans to small rodents. Ann Neurol5:581–584
Tateishi J, Brown P, Kitamoto T, Hoque ZM, Roos R, Woll-man R, Cervenakova L, Gajdusek DC (1995) First experi-mental transmission of fatal familial insomnia. Nature376:434–435
Vital C, Gray F, Vital A, Ferrer X, Julien J (1999) Prion diseasewith octapeptide repeat insertion. Clin Exp Pathol 47:153–159
WHO (2003) WHOmanual for surveillance of human transmissiblespongiform encephalopathies including variant Creutzfeldt–Jakob disease
Zerr I, Pocchiari M, Collins S, Brandel JP, de Pedro Cuesta J,Knight RS, Bernheimer H, Cardone F, Delasnerie-LaupretreN, Cuadrado Corrales N, Ladogana A, Bodemer M, FletcherA, Awan T, Ruiz Bremon A, Budka H, Laplanche JL, Will RG,Poser S (2000) Analysis of EEG and CSF 14-3-3 proteins as aidsto the diagnosis of Creutzfeldt–Jakob disease. Neurology55:811–815
174










![A Non-Q/N-Rich Prion Domain of a Foreign Prion, [Het-s], Can Propagate as a Prion in Yeast](https://img.dokumen.tips/doc/110x75/63498a64d6804e97060ada39/a-non-qn-rich-prion-domain-of-a-foreign-prion-het-s-can-propagate-as-a-prion.jpg)

















![Structure-based view on [PSI(+)] prion properties](https://img.dokumen.tips/doc/110x75/6358c9d1debc1859f60474ed/structure-based-view-on-psi-prion-properties.jpg)
