Embed Size (px)
Citation preview

lable at ScienceDirect
Neuropharmacology 105 (2016) 533e542
Contents lists avai
Neuropharmacology
journal homepage: www.elsevier .com/locate/neuropharm
Failure and rescue of preconditioning-induced neuroprotection insevere stroke-like insults
Joseph S. Tauskela a, Amy Aylsworth a, Melissa Hewitt a, Eric Brunette a,Nicolas Blondeau b, c, *
a Department of Translational Bioscience, Human Health Therapeutics, National Research Council Canada, 1200 Montreal Road,Ottawa, Ontario, Canada K1A 0R6b Universit�e de Nice Sophia Antipolis, IPMC, Sophia Antipolis, F-06560, Francec CNRS, IPMC, Sophia Antipolis, F-06560, France
a r t i c l e i n f o
Article history:Received 18 November 2015Received in revised form4 February 2016Accepted 5 February 2016Available online 8 February 2016
Keywords:NeuroprotectionExcitotoxicityToleranceNMDA receptorIschemiaPreconditioning
Chemical compounds:4-Aminopyridine (Pubmed CID: 1727)Bicuculline (Pubmed CID: 104871)2-Deoxyglucose (Pubmed CID: 24894205)Desferoxamine ((Pubmed CID: 24894304)Dimethyloxallyl glycine (Pubmed CID:560326)Diazoxide (Pubmed CID: 3019)Lovastatin (Pubmed CID: 16760544)N-Methyl-D-aspartate (Pubmed CID: 22880)Tunicamycin (Pubmed CID: 16760689)
The abbreviations used are: 4-AP, 4-aminopyridine;extracellular glutamate; MEM, minimal essential medmethyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,1oxygen-glucose deprivation; PI, propidium iodide;yaspartic acid.* Corresponding author. Institut de Pharmacologie M
7275, CNRS/Universit�e de Nice Sophia Antipolis, 66Antipolis, 06560, Valbonne, France.
E-mail address: [email protected] (N. Blonde
http://dx.doi.org/10.1016/j.neuropharm.2016.02.0070028-3908/© 2016 Elsevier Ltd. All rights reserved.
a b s t r a c t
Preconditioning is a well established neuroprotective modality. However, the mechanism and relativeefficacy of neuroprotection between diverse preconditioners is poorly defined. Cultured neurons werepreconditioned by 4-aminopyridine and bicuculline (4-AP/bic), rendering neurons tolerant to normallylethal (sufficient to kill most neurons) oxygen-glucose deprivation (OGD) or a chemical OGD-mimic,ouabain/TBOA, by suppression of extracellular glutamate (glutamateex) elevations. However, subjectingpreconditioned neurons to longer-duration supra-lethal insults caused neurotoxic glutamateex eleva-tions, thereby identifying a ‘ceiling’ to neuroprotection. Neuroprotective ‘rescue’ of neurons could beobtained by administration of an NMDA receptor antagonist, MK-801, just before glutamateex rose duringthese supra-lethal insults. Next, we evaluated if these concepts of glutamateex suppression during lethalOGD, and a neuroprotective ceiling requiring MK-801 rescue under supra-lethal OGD, extended to thepreconditioning field. In screening a panel of 42 diverse putative preconditioners, neuroprotectionagainst normally lethal OGD was observed in 12 cases, which correlated with glutamateex suppression,both of which could be reversed, either by the inclusion of a glutamate uptake inhibitor (TBOA, to in-crease glutamateex levels) during OGD or by exposure to supra-lethal OGD. Administrating MK-801during the latter stages of supra-lethal OGD again rescued neurons, although to varying degreesdependent on the preconditioning agent. Thus, ‘stress-testing’ against the harshest ischemic-like insultsyet tested identifies the most efficacious preconditioners, which dictates how early MK-801 needs to beadministered during the insult in order to maintain neuroprotection. Preconditioning delays a neurotoxicrise in glutamateex levels, thereby ‘buying time’ for acute anti-excitotoxic pharmacologic rescue.
© 2016 Elsevier Ltd. All rights reserved.
1. Introduction
Preconditioning of the brain is an established experimental
bic, bicuculline; glutamateex,ium; MK-801, (5S,10R)-(þ)-5-0-imine maleate; OGD,TBOA, DL-threo-b-benzylox-
ol�eculaire et Cellulaire, UMR0 route des Lucioles, Sophia
au).
neuroprotective modality against cerebral ischemia (Gidday, 2006).Preconditioning activates an endogenous neuroprotective ‘reserve’,the nature of which may considerably vary, since numerous anddiverse stimuli can function as preconditioners, each of which re-sults in myriad changes to the brain at the genomic, proteomic andpost-translational level. With interest in preconditioning nowemerging at the clinical level (Hougaard et al., 2014; Keep et al.,2014), it becomes important to consider questions important totranslation. Conventional neuroprotective drug therapy usuallyinhibits one (or more) well defined components in the neurotoxicsignaling cascade activated by ischemia, but the neuronal target orend effector in preconditioning is generally undefined. The failure

J.S. Tauskela et al. / Neuropharmacology 105 (2016) 533e542534
of so many neuroprotective drugs in clinical stroke trials e whichostensibly succeeded in in vitro and in vivo preclinical models ofcerebral ischemia e may reflect a lack of stringency in preclinicaltesting. It seems prudent to ‘stress test’ any emergent neuro-protective modality against more severe ischemic insults to deter-mine if a neuroprotective ‘ceiling’ exists, at the earliest preclinicallevel possible. This would lead to a ranking of diverse precondi-tioners according to how they fare in stress testing. Moreover,understanding the mechanism of how preconditioning might failunder severe conditions may yield solutions which allow phar-macologic ‘rescue’ at the point during ischemiawhen failure occurs(i.e., combination therapy). It is also important to understand if themechanism of failure is common to all preconditioners, whichwould dictate if one or more different types rescue therapies arerequired.
Preconditioning achieved via a chronic elevation in electricalactivity provides potent and pleotropic neuroprotection againstischemic-like insults, primarily via exposure to 4-aminopyridineand bicuculline (4-AP/bic) in vitro (Papadia et al., 2005; Sorianoet al., 2006; Tauskela et al., 2008, 2012) and via epileptiform in-ducers in vivo (Blondeau et al., 2000). We have previously shownthat 4-AP/bic preconditions cultured neurons against OGD andpharmacologic OGD-mimetics - insults sufficient to otherwise killmost neurons - primarily by suppressing neurotoxic glutamateexelevations during these insults, likely involving a pre-synapticbased mechanism (Tauskela et al., 2012). However, circumventingthis mechanism by exposing neurons to exogenous glutamate (orNMDA) resulted in reversal of neuroprotection. This raises apotentially key flaw which may emerge under stress testing: if thispre-synaptic based mechanism of [glutamate]ex suppression is notsustained or permanent during more severe ischemic-like condi-tions, then preconditioning may yet fail to provide tolerance by apost-synaptic based excitotoxic mechanism. If so, it would beimportant to determine if an appropriately timed administration ofa glutamate receptor antagonist to block the neurotoxic effect of theelevated glutamateex could rescue neurons. Such analyses couldyield earlier and better predictions of the ultimate success e orfailure e of preconditioning mono- or duo-therapy to more com-plex, time-consuming and expensive preclinical animal models ofischemia.
In the current study, we examine if 4-AP/bic preconditionedneurons encounter a neuroprotective ‘ceiling’ against ‘supra-lethal’OGD and OGD-mimic insults; i.e., insults sufficiently long to killneurons many times over, achieved by extending the insult dura-tionwell past the threshold required to kill all neurons. By focusingon the role of glutamateex, an anti-excitotoxic pharmacologic‘rescue’ protocol was developed. We then screened 42 putativepreconditioning stimuli, resulting in a ranking of preconditionersaccording to an ability to withstand severe OGD. We evaluated ifthe basis for a neuroprotective ceiling was common to all pre-conditioners, and if the same rescue therapy could be applied.Whatresults is an experimentally driven rationale for combinationtherapy utilizing pre-emptive preconditioning and acute inter-vention with anti-excitotoxic pharmacology during severe OGD.
2. Materials and methods
2.1. Materials
Tissue culture plates were purchased from Du Pont-Life Tech-nologies (Burlington, ON, Canada) or VWRCanlab (Mississauga, ON,Canada). Fetal bovine serum and horse serum were bought fromGemini Bio (Woodland, CA, U.S.A.) and Hyclone Laboratories(Logan, UT, U.S.A.), respectively. MEM was obtained from WisentCanadian Laboratories (St-Bruno, QC, Canada). A glutamate assay
kit was bought from Molecular Probes (Eugene, OR, U.S.A.). 4-AP,MK-801 and DL-TBOA were purchased from Tocris Bioscience(Ellisville, MO, U.S.A.). Bicuculline, NMDA, ouabain, PI, ouabain andall other reagents were purchased from Sigma (St. Louis, MO,U.S.A.). Sources of other putative preconditioning chemicals areprovided in Supplementary Table 1.
2.2. Preparation of cultures
Cultures of E18 rat cortical neurons were prepared as describedpreviously, with all experiments performed on cultures grown14e18 days in vitro (Tauskela et al., 2003). Cultures contain corticaland hippocampal neurons, and astrocytes. The Animal Care Com-mittee in Human Health Therapeutics at the National ResearchCouncil Canada approved the use of animals.
2.3. Preconditioning and neurotoxic stimuli
Cultures were preconditioned by exposure to 500 mM 4-AP with50 mM bicuculline for 48 h, followed by a 24 h recovery periodbefore being subjected to an insult (Tauskela et al., 2008). OGD wasperformed as described previously by placing cultures in a 37 �Cincubator housed in an anaerobic glovebox (Forma Scientific,Marjetta, OH, U.S.A.) under 95% N2/5% CO2, producing an O2 partialpressure equal to 10e15 Torr, as measured with an oxygen micro-electrode (Microelectrodes, Londonberry, N.H., U.S.A.) (Tauskelaet al., 2003).
Cultures were exposed to OGD in durations ranging from 60min(lethal) to 120 min (supra-lethal), in the presence or absence of aglutamate uptake inhibitor TBOA (40 mM), or to the OGD-mimeticouabain (5 mM) þ TBOA (40 mM), in durations ranging from20 min (lethal) to 60 min (supra-lethal). The NMDA receptorantagonist MK-801 (1 mM) was applied where indicated. All ex-periments were performed on at least three different cultureplatings, with each experimental condition comprising at least 3wells per plate in a minimum of 2 plates per plating period. In allexperiments examining a preconditioner, experimental data wasnormalized relative to data obtained from 3 to 4 wells within each12-well plate devoted to vehicle control, in order to eliminate anyinter-plate variability in responses to insults.
2.4. Assessment of neuronal injury
Neuronal injury was assessed 24 h following treatments asdescribed previously using PI, with fluorescence quantitated by aCytofluor 2350 fluorescence plate reader (Millipore Corp., Bedford,MA, U.S.A.) (Tauskela et al., 2003).
2.5. Measurement of glutamateex levels
Immediately prior to washout of a toxic stimulus, extracellularbuffer was collected and combined from several wells and storedat �80 �C. The glutamateex concentration was subsequentlydetermined using a commercially available kit (Tauskela et al.,2012).
2.6. Statistical analyses
Data are presented as the mean ± S.E.M. Statistical comparisonswere made by analysis of variance (ANOVA) or the Student's t-test.When significant differences were observed using an ANOVA,Bonferroni's test was employed for multiple comparisons. Statisti-cal significance was inferred at P < 0.05.
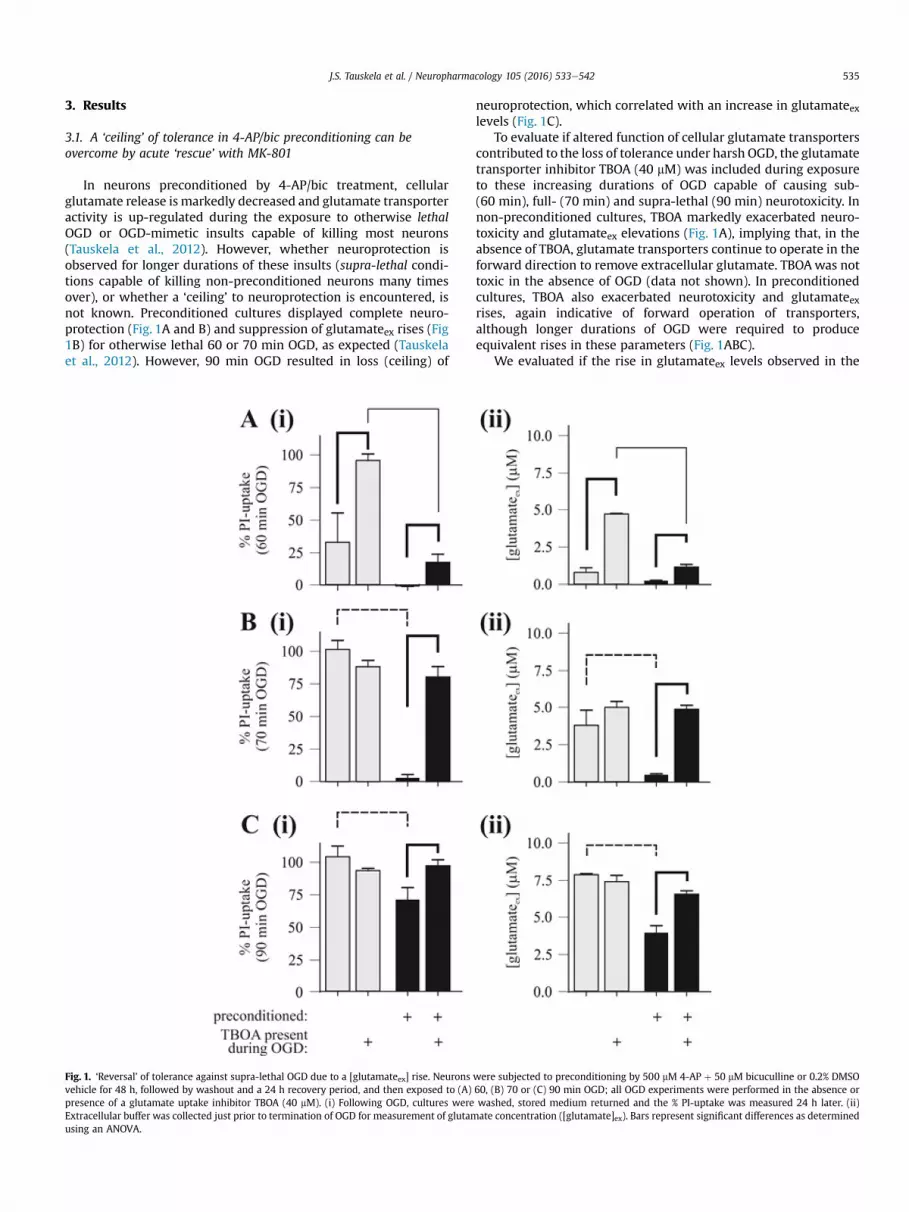
J.S. Tauskela et al. / Neuropharmacology 105 (2016) 533e542 535
3. Results
3.1. A ‘ceiling’ of tolerance in 4-AP/bic preconditioning can beovercome by acute ‘rescue’ with MK-801
In neurons preconditioned by 4-AP/bic treatment, cellularglutamate release is markedly decreased and glutamate transporteractivity is up-regulated during the exposure to otherwise lethalOGD or OGD-mimetic insults capable of killing most neurons(Tauskela et al., 2012). However, whether neuroprotection isobserved for longer durations of these insults (supra-lethal condi-tions capable of killing non-preconditioned neurons many timesover), or whether a ‘ceiling’ to neuroprotection is encountered, isnot known. Preconditioned cultures displayed complete neuro-protection (Fig. 1A and B) and suppression of glutamateex rises (Fig1B) for otherwise lethal 60 or 70 min OGD, as expected (Tauskelaet al., 2012). However, 90 min OGD resulted in loss (ceiling) of
Fig. 1. ‘Reversal’ of tolerance against supra-lethal OGD due to a [glutamateex] rise. Neuronsvehicle for 48 h, followed by washout and a 24 h recovery period, and then exposed to (A)presence of a glutamate uptake inhibitor TBOA (40 mM). (i) Following OGD, cultures wereExtracellular buffer was collected just prior to termination of OGD for measurement of glutamusing an ANOVA.
neuroprotection, which correlated with an increase in glutamateexlevels (Fig. 1C).
To evaluate if altered function of cellular glutamate transporterscontributed to the loss of tolerance under harsh OGD, the glutamatetransporter inhibitor TBOA (40 mM) was included during exposureto these increasing durations of OGD capable of causing sub-(60 min), full- (70 min) and supra-lethal (90 min) neurotoxicity. Innon-preconditioned cultures, TBOA markedly exacerbated neuro-toxicity and glutamateex elevations (Fig. 1A), implying that, in theabsence of TBOA, glutamate transporters continue to operate in theforward direction to remove extracellular glutamate. TBOAwas nottoxic in the absence of OGD (data not shown). In preconditionedcultures, TBOA also exacerbated neurotoxicity and glutamateexrises, again indicative of forward operation of transporters,although longer durations of OGD were required to produceequivalent rises in these parameters (Fig. 1ABC).
We evaluated if the rise in glutamateex levels observed in the
were subjected to preconditioning by 500 mM 4-AP þ 50 mM bicuculline or 0.2% DMSO60, (B) 70 or (C) 90 min OGD; all OGD experiments were performed in the absence orwashed, stored medium returned and the % PI-uptake was measured 24 h later. (ii)ate concentration ([glutamate]ex). Bars represent significant differences as determined

J.S. Tauskela et al. / Neuropharmacology 105 (2016) 533e542536
latter stage of supra-lethal OGD was responsible for the loss oftolerance in preconditioned cultures by examining if an NMDAreceptor antagonist was neuroprotective (Fig. 2A). Including MK-801 (1 mM) during lethal (70 min) or supra-lethal OGD (90 min or120 min) completely preserved neuronal viability (Fig. 2A) andmorphology (data not shown), in preconditioned and non-preconditioned cultures. Hence, the rise in [glutamateex] causedthe neurotoxicity.
We considered the possibility that MK-801 could still be neuro-protective even if administered quite late during the insult; i.e.,immediately prior to when glutamateex starts to rise (Fig. 2B). Cul-tureswere exposed to OGD (80, 90,100 or 115min)±MK-801 (1 mM)added 15 min before the end of OGD (at the 65, 75, 85 or 100 minmark during the OGD insults, respectively). In non-preconditionedcultures, MK-801 provided significant neuroprotection (~50%) only
Fig. 2. Acute ‘rescue’ of preconditioned neurons by the NMDA receptor antagonist MK-801 awere subjected to preconditioning by 500 mM 4-AP þ 50 mM bicuculline or 0.2% DMSO vehOGD (70, 90 or 120 min) in the presence or absence of MK-801 (1 mM) and the % PI-uptake w115 min ± MK-801 (1 mM) added to cultures within the anaerobic glovebox 15 min prior towas measured 24 h following OGD. (C) Cultures were exposed to OGD (75, 85, 95 or 110 min)uptake 24 h later. Bars represent significant differences as determined using an ANOVA.
against the two shortest OGD durations. In contrast, MK-801 treat-ment provided significant neuroprotection (~50%) in preconditionedcultures for all four OGD durations examined, although withdiminished efficacy for the longest OGD duration. For each OGDduration examined, MK-801 treatment consistently preserved >50%more preconditioned than non-preconditioned neurons, except forthe most severe OGD insult.
The feasibility of delaying MK-801 administration until imme-diately following OGD (re-perfusion therapy) was also considered(Fig. 2C). In non-preconditioned cultures, MK-801 (1 mM) treatmentfollowing lethal 75 min OGD preserved ~50% of the neuronalpopulation, confirming a post-OGD excitotoxic component (Hartleyand Choi, 1989; Fogal et al., 2005; Norris et al., 2006), but MK-801did not provide further neuroprotection against longer OGDdurations. MK-801 improved survival of preconditioned neurons
dministered before, during (the last 15 min) or after supra-lethal OGD insults. Neuronsicle for 48 h, followed by washout and recovery for 24 h. (A) Cultures were exposed toas measured 24 h following OGD. (B) Cultures were exposed to OGD for 80, 90, 100 or
terminating OGD (at the 65, 75, 85 or 100 min mark, respectively) and the % PI-uptake±MK-801 (1 mM) added immediately after OGD, followed by measurement of the % PI-
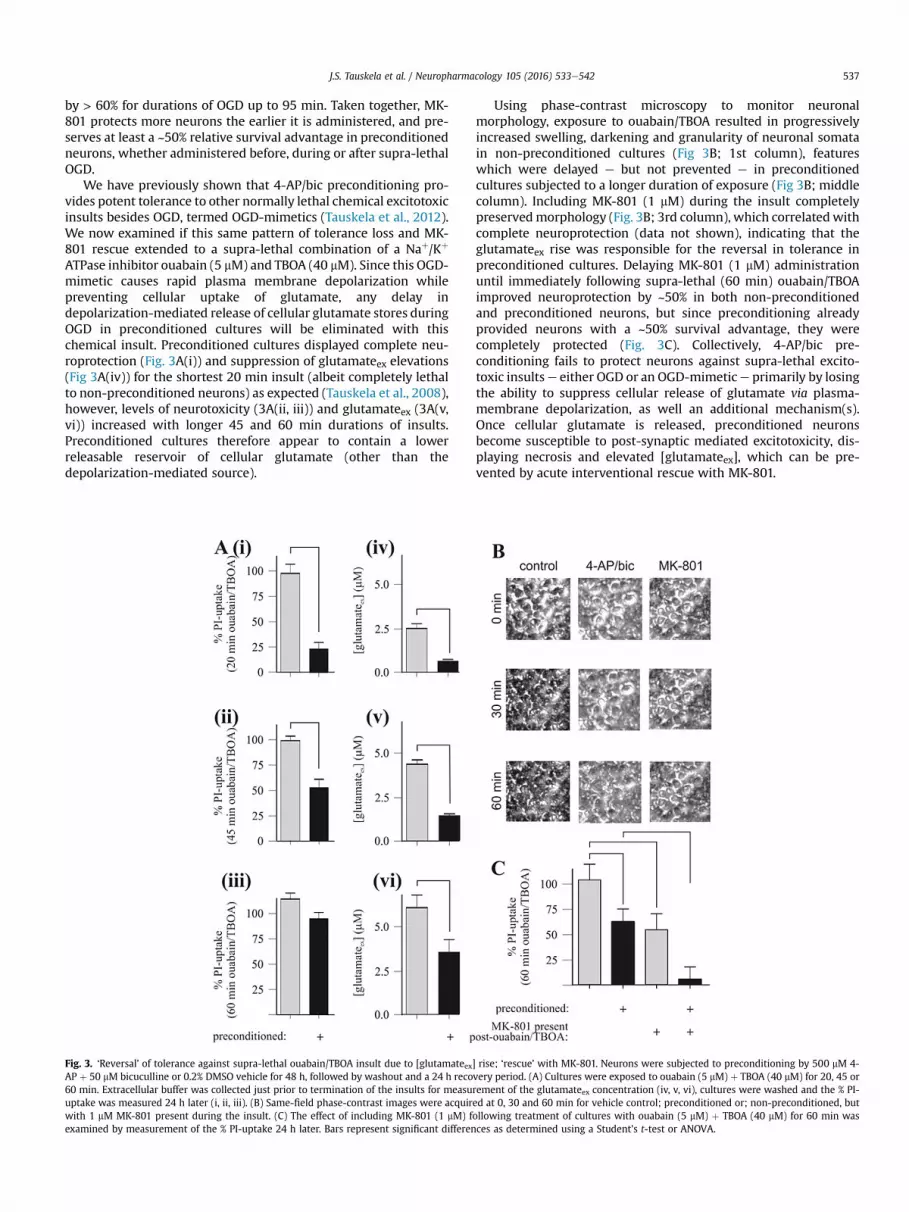
J.S. Tauskela et al. / Neuropharmacology 105 (2016) 533e542 537
by > 60% for durations of OGD up to 95 min. Taken together, MK-801 protects more neurons the earlier it is administered, and pre-serves at least a ~50% relative survival advantage in preconditionedneurons, whether administered before, during or after supra-lethalOGD.
We have previously shown that 4-AP/bic preconditioning pro-vides potent tolerance to other normally lethal chemical excitotoxicinsults besides OGD, termed OGD-mimetics (Tauskela et al., 2012).We now examined if this same pattern of tolerance loss and MK-801 rescue extended to a supra-lethal combination of a Naþ/Kþ
ATPase inhibitor ouabain (5 mM) and TBOA (40 mM). Since this OGD-mimetic causes rapid plasma membrane depolarization whilepreventing cellular uptake of glutamate, any delay indepolarization-mediated release of cellular glutamate stores duringOGD in preconditioned cultures will be eliminated with thischemical insult. Preconditioned cultures displayed complete neu-roprotection (Fig. 3A(i)) and suppression of glutamateex elevations(Fig 3A(iv)) for the shortest 20 min insult (albeit completely lethalto non-preconditioned neurons) as expected (Tauskela et al., 2008),however, levels of neurotoxicity (3A(ii, iii)) and glutamateex (3A(v,vi)) increased with longer 45 and 60 min durations of insults.Preconditioned cultures therefore appear to contain a lowerreleasable reservoir of cellular glutamate (other than thedepolarization-mediated source).
Fig. 3. ‘Reversal’ of tolerance against supra-lethal ouabain/TBOA insult due to [glutamateex]AP þ 50 mM bicuculline or 0.2% DMSO vehicle for 48 h, followed by washout and a 24 h reco60 min. Extracellular buffer was collected just prior to termination of the insults for measuuptake was measured 24 h later (i, ii, iii). (B) Same-field phase-contrast images were acquirwith 1 mM MK-801 present during the insult. (C) The effect of including MK-801 (1 mM) fexamined by measurement of the % PI-uptake 24 h later. Bars represent significant differen
Using phase-contrast microscopy to monitor neuronalmorphology, exposure to ouabain/TBOA resulted in progressivelyincreased swelling, darkening and granularity of neuronal somatain non-preconditioned cultures (Fig 3B; 1st column), featureswhich were delayed e but not prevented e in preconditionedcultures subjected to a longer duration of exposure (Fig 3B; middlecolumn). Including MK-801 (1 mM) during the insult completelypreservedmorphology (Fig. 3B; 3rd column), which correlatedwithcomplete neuroprotection (data not shown), indicating that theglutamateex rise was responsible for the reversal in tolerance inpreconditioned cultures. Delaying MK-801 (1 mM) administrationuntil immediately following supra-lethal (60 min) ouabain/TBOAimproved neuroprotection by ~50% in both non-preconditionedand preconditioned neurons, but since preconditioning alreadyprovided neurons with a ~50% survival advantage, they werecompletely protected (Fig. 3C). Collectively, 4-AP/bic pre-conditioning fails to protect neurons against supra-lethal excito-toxic insultse either OGD or an OGD-mimetice primarily by losingthe ability to suppress cellular release of glutamate via plasma-membrane depolarization, as well an additional mechanism(s).Once cellular glutamate is released, preconditioned neuronsbecome susceptible to post-synaptic mediated excitotoxicity, dis-playing necrosis and elevated [glutamateex], which can be pre-vented by acute interventional rescue with MK-801.
rise; ‘rescue’ with MK-801. Neurons were subjected to preconditioning by 500 mM 4-very period. (A) Cultures were exposed to ouabain (5 mM) þ TBOA (40 mM) for 20, 45 orrement of the glutamateex concentration (iv, v, vi), cultures were washed and the % PI-ed at 0, 30 and 60 min for vehicle control; preconditioned or; non-preconditioned, butollowing treatment of cultures with ouabain (5 mM) þ TBOA (40 mM) for 60 min wasces as determined using a Student's t-test or ANOVA.

J.S. Tauskela et al. / Neuropharmacology 105 (2016) 533e542538
3.2. Extension of ‘ceiling’ and ‘rescue’ concepts to thepreconditioning field
We next investigated if the concepts of a ceiling to neuro-protection and MK-801-based rescue is a feature common to thepreconditioning field. First, 42 putative preconditioning stimuliwere screened against normally lethal OGD to determine if neu-roprotection correlated with suppression of the glutamateex rise
Table 1Effect of Preconditioning on % Neurotoxicity and % [Glutamate]ex Rise. Experimental meCultures were subjected to preconditioning stimuli or vehicle within each 12-well plate (iresulting in >75% PI-uptake in nonpreconditioned wells. Exceptions were phenformin (#exposed to 65 min OGD (65 min OGD caused ~30% PI-uptake in nonpreconditioned plattoxicity was calculated as a percentage of the % PI-uptake of the preconditioned relativerise was calculated as a percentage of the [glutamateex] in preconditioned relative to notoxicity is plotted in Fig. 4 for all preconditioners listed in this Table, except for phenformthese treatments markedly exacerbated neurotoxicity and glutamateex in tandem.
# Preconditioner Class
1 oxygen-glucose deprivation(OGD)
substrate deprivation
2 NMDA NMDA receptor agonist3 4-aminopyridine/bicuculline Kþ channel blocker/GABAA
receptor agonist4 cyclothiazide AMPAR desens/GABAAR antagonist5 high KCl buffer plasma membrane depolarization6 NS 1619 BKCa channel opener7 bepridil stable angina pectoris treatment;
affects many channels8 diazoxide mitochondrial KATP agonist
9 clonidine А2-adrenergic receptor agonist10 sulforaphane Dietary phytochemical11 butylated hydroxyanisole Phenolic antioxidant; preservative12 tert-butylhydro-quinone active metabolite of butylated
hydroxyanisole; preservative13 dithione-3-thione Chemopreventive thiol14 DL-kawain Kavalactone15 desferrioxamine Fe chelator, prolyl 4-hydroxylase
inhibitor16 dimethyloxallyl glycine Prolyl hydroxylase inhibitor17 ethyl 3,4-dihydrobenzoate Prolyl hydroxylase inhibitor18 tilirone Immunomodulatory agent19 salubrinal PP1-GADD34 phosphatase inhibitor
20 2-deoxyglucose Nonmetabolizeable glucose analog21 phenformin Biguanide (decrease glucose utilization)22 tunicamycin protein N-glycosylation inhibitor23 geldanamycin Ansamycin antibiotic24 resveratrol Phytoalexin25 cycloheximide Protein synthesis inhibitor26 nordihydro-guaiaretic acid 5-lipooxygenase inhibitor27 ceftriaxone b-lactam antibiotic28 losartan Angiotensin type 1 receptor antagonist29 rosiglatazone PPARg inhibitor30 Li Bipolar disorder treatment31 Valproic acid Bipolar disorder treatment32 Li þ valproic acid Bipolar disorder treatment33 trichostatin A Natural product34 Na butyrate antitumor35 phenylbutyrate Fatty acid, chemical chaperone36 scriptaid antitumor37 suberoylanilide hydroxamic acid
(suberoyl bis-hydroxamic acid)Synthetic, antitumor
38 DL-amino-glutethimide Suppresses steroid synthesis39 ursodeoxycholic acid Endogenous bile acid40 epigallocatechin gallate Green tea polyphenol flavonoid41 bilobalide component of ginkgo biloba
extract EGb 76142 lovastatin statin
43 perillic acid geranylgeranyl transferase inhibitor
during OGD (75 min). Preconditioners were chosen based onliterature reports to span multiple signaling activators and trans-duction pathways. Table 1 lists the preconditioning agent, putativesignaling target and mechanism, the % neurotoxicity (measured24 h later using PI) and the % [glutamateex] rise (measured fromextracellular buffer collected just before termination of OGD);Supplementary Table 1 provides the experimental protocols, n,commercial source and relevant references. In our hands, only 12
thods employed for preconditioning stimuli are outlined in Supplementary Table 1.n order to remove any inter-plate variability), and thenwere exposed to 75 min OGD,21), valproic acid (#31), Li þ valproic acid (#32), trichostatin A (#33), which were
es), since these preconditioning stimuli markedly exacerbated neurotoxicity. The %to nonpreconditioned wells within each 12-well plate. Similarly, the % [glutamateex]npreconditioned wells within each 12-well plate. The % [glutamateex] rise versus %in (# 21), valproic acid (#31), Li þ valproic acid (#32), and trichostatin A (#33), since
Putative mechanism % Neuro-toxicity % [glutamate]exincrease
NMDA receptors 3 ± 9* 13 ± 10*
NMDA receptors 13 ± 5* 12 ± 4*electrical activity; synapticNMDA receptors
4 ± 3* 7 ± 2*
convulsant 100 ± 4 91 ± 18NMDA receptors, connexin 36 95 ± 20 124 ± 25mitochondrial KATP 81 ± 9 106 ± 20mitochondrial KATP 92 ± 3 60 ± 16
mito KATP/AMPAR desens/succinatedehydrogenase
6 ± 2* 0 ± 29*
Anti-excitotoxic 98 ± 5 96 ± 4Nrf2 83 ± 9 96 ± 8Nrf2 64 ± 7* 61 ± 21*Nrf2 104 ± 3 94 ± 26
Nrf2 66 ± 4* 48 ± 10*Nrf2 94 ± 5 74 ± 9*HIF-1a 11 ± 5* 28 ± 16*
HIF-1a-dependent/independent 5 ± 1* 1 ± 2*HIF-1a-dependent/independent 92 ± 4 20 ± 4HIF-1a 93 ± 4 98 ± 35ER stress/eIF2a dephosphory-lationinhibitor
88 ± 6 83 ± 18
ER stress/dietary restriction 6 ± 3* 4 ± 2*ER stress/Dietary restriction 138 ± 7* 181 ± 81ER stress 2 ± 1* 3 ± 4*HSP90 95 ± 9 78 ± 12SIRT1 92 ± 7 67 ± 19Bcl-2, GSH 7 ± 3* 17 ± 20*GLT1 104 ± 10 86 ± 27GLT1, Nrf2, xCT 98 ± 4 109 ± 21GLT-1 90 ± 8 107 ± 3GLT-1 99 ± 3 107 ± 15GSK-3 inhibitor/HDAC inhibitor 92 ± 2 81 ± 2*GSK-3 inhibitor/HDAC inhibitor 448 ± 33* 289 ± 72*GSK-3 inhibitor/HDAC inhibitor 244 ± 32* 237 ± 60*HDAC inhibitor 364 ± 16* 524 ± 83*HDAC inhibitor 102 ± 4 120 ± 11HDAC inhibitor 104 ± 4 118 ± 24HDAC inhibitor 108 ± 4 122 ± 7HDAC inhibitor 105 ± 7 117 ± 12
Anti-excitotoxic 62 ± 6* 53 ± 14*ER stress, Bcl-2 106 ± 4 153 ± 32anti-oxidant response element 110 ± 4 89 ± 18
130 ± 6* 121 ± 8
cholesterol/geranylgeranylpyrophosphate depletion
9 ± 2* 6 ± 2*
geranylgeranyl pyrophosphatedepletion
97 ± 17 98 ± 21

Table 2Tolerance Induced by Preconditioning Agents is Reversed by TBOA Present DuringOGD. Experimental methods employed for preconditioning stimuli are outlined inSupplementary Table 1.* significant difference between preconditioning and controlfor OGD alone (TBOA not present) (p < 0.05).** significant difference between pre-conditioning performed in the absence and presence of TBOA (p < 0.05).
Preconditioner (PC) % PI-uptake
OGD þ TBOA OGD
PC Control PC Control
OGD 76 ± 17** 95 ± 4 16 ± 2* 76 ± 6NMDA 84 ± 18** 101 ± 11 0 ± 0* 84 ± 104-aminopyridine/bicuculline 81 ± 9** 106 ± 9 5 ± 3* 81 ± 9cycloheximide 88 ± 1** 103 ± 2 11 ± 2* 69 ± 72-deoxyglucose 76 ± 3** 82 ± 2 0 ± 0* 64 ± 4tunicamycin 105 ± 10** 103 ± 2 2 ± 3* 82 ± 9desferrioxiamine 91 ± 12** 95 ± 7 0 ± 0* 100 ± 6DMOG 93 ± 7** 102 ± 5 4 ± 0* 101 ± 2diazoxide 84 ± 8** 105 ± 7 3 ± 2* 102 ± 6Butylated hydroxyanisole 83 ± 7** 115 ± 6 45 ± 8* 90 ± 5Dithione-3-thione 83 ± 7** 107 ± 4 57 ± 4* 87 ± 7DL-aminoglutethimide 97 ± 6** 94 ± 5 53 ± 4* 73 ± 3lovastatin 94 ± 6** 99 ± 3 9 ± 2* 88 ± 3
J.S. Tauskela et al. / Neuropharmacology 105 (2016) 533e542 539
preconditioning stimuli provided significant tolerance against le-thal OGD, despite extensive testing of different doses, durations andrecovery periods; in most cases testing included evaluatingnumerous conditions and, since many preconditioners representsubstantial stressors, these conditions included those whichapproached but did not surpass the death threshold(Supplementary Table 1). A plot of the % [glutamateex] rise versus%-neurotoxicity (values were normalized relative to data valuesobtained for non-preconditioned samples examined in the sameexperiment), yielded a linear regression best fit with a slope equalto 0.96 ± 0.07 and an R2 equal to 0.81 (Fig. 4). Thus, this screeningclearly demonstrates a strong correlation between glutamateexsuppression during OGD and neuroprotection for all pre-conditioning stimuli examined.
To elucidate if this suppression of glutamateex rises underliestolerance to lethal OGD (75 min) for each of the 12 neuroprotectivepreconditioners, TBOA (40 mM) was included during this insult toprevent cellular glutamate uptake. This resulted in completeelimination of OGD tolerance for all preconditioning agents(Table 2). This implies that cellular glutamate uptake during OGD isnecessary to remove extracellular glutamate; otherwise, with TBOApresent glutamateex levels build and neuroprotective tolerance islost. Importantly, despite vastly different inductionmechanisms, allpreconditioning nevertheless converges to a common convergencepoint or end effector, which is the suppression of [glutamate]exelevations during OGD.
We tested if the strongest preconditioners failed when exposedto supra-lethal 100 min OGD and, if so, whether administration ofMK-801 incrementally applied at different intervals between the70e100 min stages of this insult rescued neurons (Fig. 5). Onehundredmin OGD is substantially longer than durations required tocause 100% neurotoxicity in naive cultures (supra-lethal). MK-801-induced neuroprotection in non-preconditioned neurons was lostwhen applied at or after the 80 min time-point of 100 min OGD. Incontrast, adding MK-801 at the 80 min time-point in any of thepreconditioned cultures resulted in >80% neuroprotection. AddingMK-801 at 90 min during the 100 min OGD insult nonetheless stillprovided significant neuroprotection except for preconditioning by
Fig. 4. Correlation between OGD tolerance and suppression of [glutamate]ex rise for allpreconditioners. Cultured cortical neurons were subjected to 43 putative pre-conditioning stimuli (detailed in Supplementary Table 1). Cultures were exposed tolethal 75 min OGD. Extracellular buffer was collected just prior to termination of theinsults for measurement of the glutamateex concentration; cultures were then washedand the % PI-uptake was measured 24 h later. The % [glutamateex] rise was plottedversus % toxicity, each of which were normalized relative to values obtained for thenon-preconditioned samples performed in the same companion experiment. The solidline represents a linear regression best fit with a slope of 0.96 ± 0.07 and an R2 equal to0.81 and the dotted lines represent 95% confidence intervals.
desferrioxamine. Adding MK-801 immediately after the 100 minOGD insult still preserved 40e70% levels of neuroprotection for allother preconditioners. Tunicamycin and DMOG provided thehighest (~70%) level of neuroprotection. Thus, MK-801 adminis-tration could be delayed until the latter stages of OGD or during re-perfusion, while still providing substantial rescue ofpreconditioning-induced tolerance/protection of neurons thatotherwise would have died.
4. Discussion
In this study, several concepts are identified that are common tothe field of preconditioning. Preconditioning results in suppressionof neurotoxic glutamateex rises during ischemic-like insults suffi-cient to kill neurons (lethal), but not if the duration of the insult isextended (supra-lethal), thereby identifying a ceiling to neuro-protection. Timely administration of an NMDA receptor antagonistcan rescue neurons, the degree to which depends on the strength ofthe preconditioner. Thus, preconditioning delays an excitotoxiconslaught, thereby ‘buying time' by extending the therapeuticwindow for administration of therapies targeting post-synapticNMDA receptors. This is a particularly crucial advantage, giventhat excitotoxicity is regarded as the earliest and most deleteriousconsequence of cerebral ischemia.
4.1. Suppression of glutamateex levels during lethal insults as thecommon end effector of preconditioning
Neuroprotection provided by preconditioners against lethalOGD strongly correlated with the suppression of intracellular glu-tamateex rise that normally accompanies this insult (Fig. 4). Thissuppression was crucial to tolerance, since inclusion of TBOA toprevent cellular glutamate uptake during OGD completely reversedneuroprotection by all preconditioners (Table 2). A limited body ofdata indicates that OGD (Tauskela et al., 2001) or ischemic tolerancecorrelates with suppressed [glutamateex] and/or [Ca2þ]i (seeTauskela and Morley, 2004 for review). The current study consid-erably extends and validates this trend, having the advantage thatcomparisons of diverse preconditioning stimuli were performed onthe same neuronal system subjected to the same insult. Given thediversity of signaling activated by the preconditioning agents, thiscommon convergence point or end effector of suppressed

Fig. 5. ‘Rescue’ achieved by MK-801 added at different points during 100 min OGD in cultures preconditioned by various stimuli. Cortical neuron cultures subjected to pre-conditioning by various stimuli (procedures described in Supplementary Table 1) or vehicle, and subjected to MK-801 (1 mM) applied at different intervals between 70 and 100 minof a 100 min OGD insult. Following OGD, the % PI-uptake was measured 24 h later.
J.S. Tauskela et al. / Neuropharmacology 105 (2016) 533e542540
[glutamateex] was unexpected; it may represent a combination of adirect effect(s) on glutamatergic neurotransmission - direct sup-pression of cellular glutamate release or up-regulated cellularglutamate uptake e and possibly an indirect effect resulting fromupstream interruption of some component of the excitotoxiccascade.
4.2. Elevation of glutamateex levels during supra-lethal insultsunderlies a ‘ceiling’ of neuroprotection in preconditioning
We considered the possibility that, although conferring statis-tically significant survival (lethal insults), perhaps preconditioningdoes not provide sufficient neuroprotection against supra-lethalinsults, first as a proof of concept in 4-AP/bic preconditioning andthen for a panel of diverse preconditioners. Indeed, 4-AP/bic pre-conditioning fails to protect neurons against supra-lethal OGD, thusidentifying a ceiling of neuroprotection. This failure correlated witha rise in glutamateex levels, indicating that the initial suppression ofglutamate cellular release observed under lethal condition was notpermanent. We evaluated if reversal of glutamate uptake trans-porters was responsible for this [glutamateex] increase, particularlywith the higher Naþ influx expected with the profound plasmamembrane depolarization that accompanies severe OGD. However,TBOA never decreased glutamateex levels or neurotoxicity, soglutamate transporters never reverse. In supra-lethal conditions,the capacity for cellular uptake by glutamate transporters must beexceeded, resulting in a neurotoxic rise in glutamateex levels.
‘Ceilings’ of neuroprotection by preconditioning have beenidentified in limited instances with extended durations of the testOGD in vitro (Meloni et al., 2002) (Weller et al., 2008); we andothers also demonstrated this concept in in vivomodels of ischemia(Blondeau et al., 2002) (Abe and Nowak, 2004) (Ueda and Nowak,2005). The basis for a neuroprotective tolerance ‘ceiling’ identi-fied in the current study appears common in one important regard,since Nowak and co-workers have reported that ischemic pre-conditioning delays e but does not prevent e ischemic depolari-zation (which is associated with a rapid and massive glutamateexrise) measured by monitoring of a DC potential shift (Abe andNowak, 2004; Ueda and Nowak, 2005). This was termed ‘pseudo-preconditioning’, since it reflected attenuation of the effectiveinsult severity during ischemia rather than an improved responseto a consistent metabolic challenge (Ueda and Nowak, 2005).However, we previously demonstrated that 4-AP/bic induced
tolerance to normally lethal ouabain/TBOA - which represents aconsistent metabolic challenge, since ouabain will cause rapidplasma membrane depolarization and cellular glutamate release,and TBOAwill prevent cellular uptake. Tolerance to this lethal formof this insult implies lower levels of depolarization-induciblecellular glutamate release (Tauskela et al., 2012). Glutamaterelease from dying neurons can be ruled out, since MK-801 rescuedall neurons. Consequently, the [glutamateex] rise that accompaniessupra-lethal ouabain/TBOA suggests activation of an additional(non-depolarization mediated) cellular glutamate release pathway.
4.3. Combination therapy to overcome the ‘ceiling’ ofneuroprotection
It is well known that some neurons survive if an NMDA receptorantagonist is applied after exposure to exogenous NMDA or gluta-mate in primary (non-preconditioned) neuron culture (Rothmanet al., 1987; Choi et al., 1988; Hartley and Choi, 1989; Levy andLipton, 1990; Shalaby et al., 1992; Fogal et al., 2005; Norris et al.,2006). This post-insult component has been termed secondaryexcitotoxic injury, reflecting pre-synaptic and/or post-synapticmediated excitotoxicity (Fogal et al., 2005; Norris et al., 2006).Given the glutamateex rise that accompanies the failure of tolerancein the current study, these studies formed the basis for determiningif this delayed-MK-801 protocol(s) could be extended to pre-conditioned neurons undergoing failure.
Based on key similarities in neuroprotection profiles conferredbyMK-801, 4-AP/bic preconditioned neurons appear to ‘fail’ againstharsh OGD in amanner similar to non-preconditioned neurons; i.e.,by excitotoxicity. That is, once a rise in glutamateex occurs (albeit forlonger durations of OGD in preconditioned cultures), precondi-tioned and non-preconditioned cultures respond similarly: (i) In a‘head-to-head’ competition, when present during the harshestinsult investigated (120 min OGD), MK-801 protected all neurons;(ii) MK-801 added 15 min prior to the end of OGD maximally res-cues similar ~50% proportions of neurons; (iii) such an interventionis not neuroprotective if the duration of OGD is too long; (iv) post-OGD exposure to MK-801 maximally rescues ~50% similar pro-portions of neurons; (v) post-OGDMK-801 is not neuroprotective ifthe duration of OGD is too long, and; (vi) TBOA exacerbates gluta-mateex elevations and neurotoxicity. Similarly, several of thesesame points hold for the OGD mimic ouabain/TBOA insult. Conse-quently, a major mechanistic advantage of preconditioning is to

J.S. Tauskela et al. / Neuropharmacology 105 (2016) 533e542 541
prevent a glutamateex elevation, but if an insult is sufficiently se-vere, glutamateex levels rise, so this advantage is lost and neuronsdie in the same manner as if they were not preconditioned.
This bimodal approach to neuroprotection is synergistic inseveral respects. Preconditioning activates endogenous neuro-protective cellular signaling, while MK-801 represents an exoge-nous agent. Preconditioning causes myriad changes at the genomic,proteomic and post-translational level (moreover, this profile canbe quite distinct among the quite variable stimuli able to act aspreconditioners), while MK-801 represents a mono-modal phar-macological approach. Each modality targets distinct mechanisticcomponents in the neurotoxic signaling cascade activated byischemic-like insults, with preconditioning and MK-801 primarilytargeting pre-synaptic (cellular glutamate release) andpost-synaptic (glutamate receptor activation) excitotoxicity,respectively. Finally, this therapy is temporally distinct, sincepreconditioning is pre-emptive and MK-801 therapy is acutelydelivered during or following the insult.
4.4. Identification of best-in-class preconditioners to be used incombination therapy
We identified 12 additional preconditioning stimuli able toprovide tolerance to normally lethal OGD (Table 1). Only primaryneurons were considered and most methods were previouslypublished, although differences in tissue (in vivo, brain slices, anddissociated culture), neuron type, species, age and other factorsexist. It was beyond the scope of this study to exhaustively inves-tigate reasons for why tolerancewas not observed in themajority ofcases, although we did investigate numerous iterations in dose,duration and recovery period, particularly including conditionswhich represented a substantial stress to neurons, since manypreconditioners bring neurons to the brink of death(Supplementary Table 1). We also employed a relatively harsh le-thal OGD insult (�75% neurotoxicity), perhaps resulting inscreening out of relatively ineffectual preconditioners. Nonetheless,the successful preconditioners were sufficient in number and var-iable enough in signaling mechanisms to provide a basis fordetermining if major concepts identified for 4-AP/bic pre-conditioning extended to preconditioning in general.
The progression in the current study was to screen 42 additionalpreconditioners against lethal OGD, of which 12 provided strongneuroprotection; seven of the most efficacious were subsequentlysubjected to rescue experiments performed under supra-lethalOGD, from which two candidates (tunicamycin and DMOG)emerged slightly strongest. In addition to being a prolyl-4-hydroxylase domain (PHD) enzyme inhibitor (and thereforehypoxia-inducible factor), DMOG inhibits 2-OG oxygenases andother enzymes, so its exact target(s) is not fully defined. Novel PHDinhibitors have been identified and may be worthy of furtherinvestigation (Smirnova et al., 2010; Speer et al., 2013). This survivaladvantage declined with successive delays in MK-801 administra-tion but, nonetheless, even re-perfusion treatment with MK-801conferred neuroprotection for most preconditioners (Fig. 5).
4.5. A neuroprotective framework
In summary, a novel neuroprotective framework for develop-ment of combination therapy is provided, one which can beimplemented in a variety of guises in stroke research laboratories.At the in vitro dissociated neuron culture or brain slice level,different types of preconditioners can be examined, and the con-ditions of each can be optimized to provide the greatest efficacy.The nature of the rescue agent, and the timing of its administrationcan be investigated. For instance, it would be of interest to
substitute MK-801 with the clinically approved NMDA receptorantagonist memantine in further investigations. The nature of theinsult and its severity can be changed. No matter the choices, thefundamental concept that a neuroprotective ‘ceiling’ exists allowsranking of preconditioners according to their relative efficacythough stress-testing. Supra-lethal OGD or OGD-mimic insults maybe more representative of a cerebral infarct in vivo e existinginitially in the core, but expanding into at-risk penumbral regions.These in vitro approaches should assist in identifying the mostpromising candidate(s) for preconditioning and the rescue agent toin vivo models of ischemia, potentially as a gateway to humanclinical trials. Clinical stroke trials increasingly recognize theimportance of decreasing ‘door to needle’ time in the administra-tion of neuroprotectants, as reflected by paramedics in the fielddelivering neuroprotectants in phase 3 clinical trials of stroke (theFAST-MAG trial which testedmagnesium sulfate (Saver et al., 2015),as well as the ongoing FRONTIER trial testing NA-1 (Tat-NR2B9c), apeptide which interrupts neurotoxic NMDA receptor-mediatedneurotoxicity). Activation of preconditioning on its own may beof benefit in mild strokes and, in more severe strokes, by delayingwhen treatment is required (‘buying time’). Therefore finding newpreconditioners may offer a mechanistic supplement to theselogistical efforts to extend the window of opportunity for acutetreatment of stroke.
Conflicts of interest
The authors declare no conflicts of interest with the contents ofthis article.
Author contributions
JST and NB conceived the experiments, coordinated the studyand wrote the paper. JST, AA, MH and EB performed the experi-ments. All authors analyzed the results and approved the finalversion of the manuscript.
Acknowledgments
This work was supported in part by a grant from FRM, France(DPM 20121125559) and the Centre National de la Recherche Sci-entifique (CNRS). The authors are grateful to Dr Catherine Heur-teaux for helpful discussions.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.neuropharm.2016.02.007.
References
Abe, H., Nowak Jr., T.S., 2004. Induced hippocampal neuron protection in an opti-mized gerbil ischemia model: insult thresholds for tolerance induction andaltered gene expression defined by ischemic depolarization. J. Cereb. BloodFlow. Metab. 24, 84e97.
Blondeau, N., Plamondon, H., Richelme, C., Heurteaux, C., Lazdunski, M., 2000.K(ATP) channel openers, adenosine agonists and epileptic preconditioning arestress signals inducing hippocampal neuroprotection. Neuroscience 100,465e474.
Blondeau, N., Lauritzen, I., Widmann, C., Lazdunski, M., Heurteaux, C., 2002.A potent protective role of lysophospholipids against global cerebral ischemiaand glutamate excitotoxicity in neuronal cultures. J. Cereb. Blood Flow. Metab.22, 821e834.
Choi, D.W., Koh, J.Y., Peters, S., 1988. Pharmacology of glutamate neurotoxicity incortical cell culture: attenuation by NMDA antagonists. J. Neurosci. 8, 185e196.
Fogal, B., Trettel, J., Uliasz, T.F., Levine, E.S., Hewett, S.J., 2005. Changes in secondaryglutamate release underlie the developmental regulation of excitotoxicneuronal cell death. Neuroscience 132, 929e942.
Gidday, J.M., 2006. Cerebral preconditioning and ischaemic tolerance. Nat. Rev.

J.S. Tauskela et al. / Neuropharmacology 105 (2016) 533e542542
Neurosci. 7, 437e448.Hartley, D.M., Choi, D.W., 1989. Delayed rescue of N-methyl-D-aspartate receptor-
mediated neuronal injury in cortical culture. J. Pharmacol. Exp. Ther. 250,752e758.
Hougaard, K.D., Hjort, N., Zeidler, D., Sorensen, L., Norgaard, A., Hansen, T.M., vonWeitzel-Mudersbach, P., Simonsen, C.Z., Damgaard, D., Gottrup, H., Svendsen, K.,Rasmussen, P.V., Ribe, L.R., Mikkelsen, I.K., Nagenthiraja, K., Cho, T.H.,Redington, A.N., Botker, H.E., Ostergaard, L., Mouridsen, K., Andersen, G., 2014.Remote ischemic perconditioning as an adjunct therapy to thrombolysis inpatients with acute ischemic stroke: a randomized trial. Stroke 45, 159e167.
Keep, R.F., Wang, M.M., Xiang, J., Hua, Y., Xi, G., 2014. Full steam ahead with remoteischemic conditioning for stroke. Transl. Stroke Res. 5, 535e537.
Levy, D.I., Lipton, S.A., 1990. Comparison of delayed administration of competitiveand uncompetitive antagonists in preventing NMDA receptor-mediatedneuronal death. Neurology 40, 852e855.
Meloni, B.P., Majda, B.T., Knuckey, N.W., 2002. Evaluation of preconditioningtreatments to protect near-pure cortical neuronal cultures from in vitroischemia induced acute and delayed neuronal death. Brain Res. 928, 69e75.
Norris, C.M., Blalock, E.M., Thibault, O., Brewer, L.D., Clodfelter, G.V., Porter, N.M.,Landfield, P.W., 2006. Electrophysiological mechanisms of delayed excitotox-icity: positive feedback loop between NMDA receptor current anddepolarization-mediated glutamate release. J. Neurophysiol. 96, 2488e2500.
Papadia, S., Stevenson, P., Hardingham, N.R., Bading, H., Hardingham, G.E., 2005.Nuclear Ca2þ and the cAMP response element-binding protein family mediate alate phase of activity-dependent neuroprotection. J. Neurosci. 25, 4279e4287.
Rothman, S.M., Thurston, J.H., Hauhart, R.E., 1987. Delayed neurotoxicity of excit-atory amino acids in vitro. Neuroscience 22, 471e480.
Saver, J.L., FAST-MAG Investigators, Coordinators, 2015. Prehospital use of magne-sium sulfate as neuroprotection in acute stroke. N. Engl. J. Med. 372, 528e536.
Shalaby, I.A., Chenard, B.L., Prochniak, M.A., Butler, T.W., 1992. Neuroprotective ef-fects of the N-methyl-D-aspartate receptor antagonists ifenprodil and SL-82,0715 on hippocampal cells in culture. J. Pharmacol. Exp. Ther. 260, 925e932.
Smirnova, N.A., Rakhman, I., Moroz, N., Basso, M., Payappilly, J., Kazakov, S.,
Hernandez-Guzman, F., Gaisina, I.N., Kozikowski, A.P., Ratan, R.R., Gazaryan, I.G.,2010. Utilization of an in vivo reporter for high throughput identification ofbranched small molecule regulators of hypoxic adaptation. Chem. Biol. 17,380e391.
Soriano, F.X., Papadia, S., Hofmann, F., Hardingham, N.R., Bading, H.,Hardingham, G.E., 2006. Preconditioning doses of NMDA promote neuro-protection by enhancing neuronal excitability. J. Neurosci. 26, 4509e4518.
Speer, R.E., Karuppagounder, S.S., Basso, M., Sleiman, S.F., Kumar, A., Brand, D.,Smirnova, N., Gazaryan, I., Khim, S.J., Ratan, R.R., 2013. Hypoxia-inducible factorprolyl hydroxylases as targets for neuroprotection by “antioxidant” metalchelators: from ferroptosis to stroke. Free Radic. Biol. Med. 62, 26e36.
Tauskela, J.S., Morley, P., 2004. On the role of Ca2þ in cerebral ischemic pre-conditioning. Cell Calcium 36, 313e322.
Tauskela, J.S., Comas, T., Hewitt, K., Monette, R., Paris, J., Hogan, M., Morley, P., 2001.Cross-tolerance to otherwise lethal N-methyl-D-aspartate and oxygen-glucosedeprivation in preconditioned cortical cultures. Neuroscience 107, 571e584.
Tauskela, J.S., Brunette, E., Monette, R., Comas, T., Morley, P., 2003. Preconditioningof cortical neurons by oxygen-glucose deprivation: tolerance induction throughabbreviated neurotoxic signaling. Am. J. Physiol. Cell Physiol. 285, C899eC911.
Tauskela, J.S., Fang, H., Hewitt, M., Brunette, E., Ahuja, T., Thivierge, J.P., Comas, T.,Mealing, G.A., 2008. Elevated synaptic activity preconditions neurons against anin vitro model of ischemia. J. Biol. Chem. 283, 34667e34676.
Tauskela, J.S., Aylsworth, A., Hewitt, M., Brunette, E., Mealing, G.A., 2012. Pre-conditioning induces tolerance by suppressing glutamate release in neuronculture ischemia models. J. Neurochem. 122, 470e481.
Ueda, M., Nowak Jr., T.S., 2005. Protective preconditioning by transient globalischemia in the rat: components of delayed injury progression and lastingprotection distinguished by comparisons of depolarization thresholds for cellloss at long survival times. J. Cereb. Blood Flow. Metab. 25, 949e958.
Weller, M.L., Stone, I.M., Goss, A., Rau, T., Rova, C., Poulsen, D.J., 2008. Selectiveoverexpression of excitatory amino acid transporter 2 (EAAT2) in astrocytesenhances neuroprotection from moderate but not severe hypoxia-ischemia.Neuroscience 155, 1204e1211.





























