Embed Size (px)
Citation preview

Elasticity of Model Poly( oxypropylene) Networks
ANTHONY L. ANDRADY, Center for Physical Sciences, Research Triangle Institute, Research Triangle Park, North Carolina and
MIGUEL A. LLORENTE, Departurnento ck General Y Macromoleculus, Universidad a Distuncia (UNED),
28040 Madrid, Spain
Synopsis
The equilibrium stress-strain properties and the swelling behavior of moderately cross-linked model networks of poly(oxypropy1ene) were studied. Results were in general agreement with the theory of rubber elasticity due to Flory. However, data on the highly cross-linked networks (ac 3 725) could not be satisfactorily described by the recent theories of elasticity or swelling. This is believed to be primarily due to the marked non-Gaussian character of the very short network chains and the substantial chemical modification of the polymer by the cross-linking moiety which inevitably occurs at high cross-link densities.
INTRODUCTION
Model networks prepared by stoichiometric end-linking of suitable tele- chelic polymer chains with a multifunctional cross-linking agent have been widely used in experimental studies on rubber elasti~ity.'-~ Provided that the end-linking reaction is complete, the networks so prepared have minimal network defects, are easily characterized, and allow the variation of network topology to a degree quite unattainable in networks prepared by conventional cross-linking techniques. This approach has been used to control important network parameters such as junction-point functionality, average network- chain length,3 network chain-length di~tribution,~ and the extent of network defects such as dangling chain^.^
Although most of the research on elasticity of model network systems has been carried out with poly(dimethylsi1oxane) networks, 2 - 5 model poly- urethane networks prepared with polyether diols have also been These are simple homogeneous networks prepared by end-linking a polyether macrodiol with a suitable triisocyanate. Alternatively, a macrodiol-diiso- cyanate condensate with isocyanate end groups might be stoichiometrically cross-linked by a low-molecular-weight trio1 or a tetrol. Systems where poly(oxypropy1ene)diol is the polyether component is of particular interest because they yield networks not prone to strain-induced crystallization. This is due to the stereochemical irregularity of the polyether macromolecule. Commercially available poly(oxypropy1ene)diols have relatively narrow molec- ular-weight distributions and contain about 1 to 3% of monofunctional material." The range of average molecular weights of the commercially available diols (pn ca. 4000 to 425) allows the achievement of moderate to
Journal of Polymer Science: Part B: Polymer Physics, Vol. 25, 195-204 (1987) 0 1987 John Wiley & Sons, Inc. CCC 0098-1273/87/010195-10$04.00

196 ANDRADY AND LLORENTE
high cross-link densities in the model networks. However, with most cross- linking agents, the networks prepared with the low-molecular-weight diols yield glassy networks due to the contribution of the cross-linking agent to the glass-transition temperature of the network.
Mark and Sungg studied the elastic behavior of a series of poly(oxypropy- 1ene)diol-based networks prepared using an aromatic triisocyanate as the cross-linking agent. The moduli of their networks were in good agreement with those reported for the corresponding model networks of poly(dimethy1- siloxane) of comparable cross-link density. The values obtained for the struc- ture factor A, ranged from 0.266 to 0.505, in good agreement with the theoretical prediction of 1/3. At the affine limit where the modulus is given by 2C1 + 2C2, the structure factor A; has a theoretically expected value of unity, a value much larger than 0.50 (*0.12) obtained by Mark and Sung.g The lower value they obtained is, a t least in part, due to the shortness of the network chains which results in insufficient “embedding” or interpenetration to cause affine deformation.
Ddek and Ilavsky,” using poly(oxypropy1ene trio1)-based networks pre- pared with 4,4’-diphenylmethanediisocyanate cross-linking agent, obtained experimental equilibrium moduli which suggested A, values between the theoretical limits of 1/3 and 1, for networks prepared with macrodiol with a,, about 700. However, for networks having an average a,, of about 2600, the experimental moduli yielded values of A, greater than unity; the discrepancy was explained in terms of a contribution to the elastic modulus by trapped chain entanglements.
The data obtained with the present networks will be interpreted by the Mooney-Rivlin approacH2i l3 and the recent elasticity theory of Flory.14* l5
EXPERIMENTAL
This work is on model networks of poly(oxypropy1ene)diol end-linked with tris( p-phenylisocyanate)thiophosphate (Desmodur RF, Mobay). The use of this particular cross-linking agent allows the networks, for the most part, to be cured a t room temperature without the use of catalysts. This effectively minimizes the possibilities of thermooxidative degradation of the network and the occurrence of side reactions such as allophonate formation and biuret formation during elevated temperature curing. The network systems used in previous studies used different cross-linking agents and were cured at temper- atures of 80” to 95°C for a period of 1.5 to 7 d a y ~ . ~ . ’ ~
The poly(propy1ene glyco1)s used were the NIAX PPG series of prepolymers from Union Carbide Corporation. These polymers, prepared by a nonstereo- specific polymerization, have narrow molecular-weight distributions.16 The prepolymer was dried at room temperature in vacuo for 24 h before use. The cross-linking agent was Desmodur RF. Dissolved in methylene chloride i t was mixed in stoichiometric proportions with the polymer diol, and the mixture was warmed at about 50°C to drive off the solvent. The viscous reaction mixture was then placed in a Teflon-lined steel mold under about 300 psi a t ambient temperature for 24 h. A t the end of 24 h, the sheets of volcanizates were placed in an oven at 50°C under dry nitrogen for 3 to 4 h. The sol fractions for networks were less than about 3%.

POLY(OXYPR0PYLENE) NETWORKS
TABLE I Composition of the Model Networks
197
Composition* -
Network 425 725 1025 2000 3000 Me
R-1 1 .o 725 R-2 1 .o 1,025 R-3 1.0 2,000 R-4 1 .o 3,000 M- 1 0.52 0.26 0.14 0.06 0.02 730 M-2 0.32 0.51 0.12 0.04 0.01 740 M-3 0.81 0.19 725
*Molar fraction of the component having the indicated average molecular weight. Note: is assumed to be equal to a,, before cross-linking.
Mixed-chain networks were prepared by mixing the appropriate weight fractions of the polymer diols of different chain lengths and reacting them with stoichiometric quantities of the cross-linking agent as described. Table I gives the comparison of the various networks.
The experimental procedure for stress-strain measurements a t 25°C and equilibrium swelling measurements has been described elsewhere.'
RESULTS AND DISCUSSION
The equilibrium stress-strain behavior of cross-linked elastomers is conve- niently represented by the Mooney-Rivlin equation: 12,
where f is the tensile force on a network having an area of cross section A* (in the undeformed state), and a is the uniaxial extension ratio. The rela- tionship between the reduced stress [ f *] and the extension ratio is de- termined by the values of two strain-independent constants 2C1 and 2C2. The constant 2C, may be further interpreted', in terms of the number average molecular weight ii?, between the junction points of the network:
A3pV,2,/,RT
@C
2c1 =
In eq. (2), V,, is the volume fraction of polymer incorporated into the network, p is the density, and A , is the structure factor. The RT term has its usual meaning. The numerical value of the structure factor A , may be calculated from known values of 2C, (obtained as the modulus at the limit of high extension approximating phantom network conditions). Theoretically, a value of 1/3 is expected for A , of a trifunctional network.'
The stress-strain data on the networks plotted in the usual manner sug- gested by eq. (1) are shown in Figure 1. The measurements are reversible, as indicated by the data points represented by filled circles. This observation is usually taken as evidence of a lack of reinforcement from strain-induced
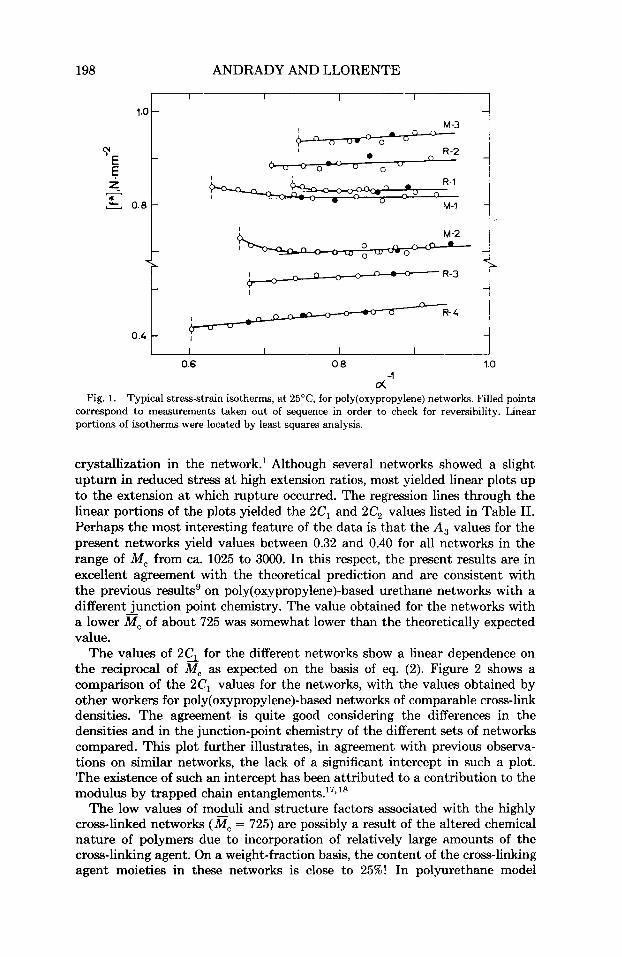
198 ANDRADY AND LLORENTE
1.0 c 7 E
c E
L 0.8 . =- 1
M -3
0.6 0.8 1.0
o( -I Fig. 1. Typical stress-strain isotherms, at 25"C, for poly(oxypropy1ene) networks. Filled points
correspond to measurements taken out of sequence in order to check for reversibility. Linear portions of isotherms were located by least squares analysis.
crystallization in the network.' Although several networks showed a slight upturn in reduced stress a t high extension ratios, most yielded linear plots up to the extension at which rupture occurred. The regression lines through the linear portions of the plots yielded the 2C1 and 2C2 values listed in Table 11. Perhaps the most interesting feature of the data is that the A , values for the present networks yield values between 0.32 and 0.40 for all networks in the range of M, from ca. 1025 to 3000. In this respect, the present results are in excellent agreement with the theoretical prediction and are consistent with the previous resultsg on poly(oxypropy1ene)-based urethane networks with a different junction point chemistry. The value obtained for the networks with a lower il?, of about 725 was somewhat lower than the theoretically expected value.
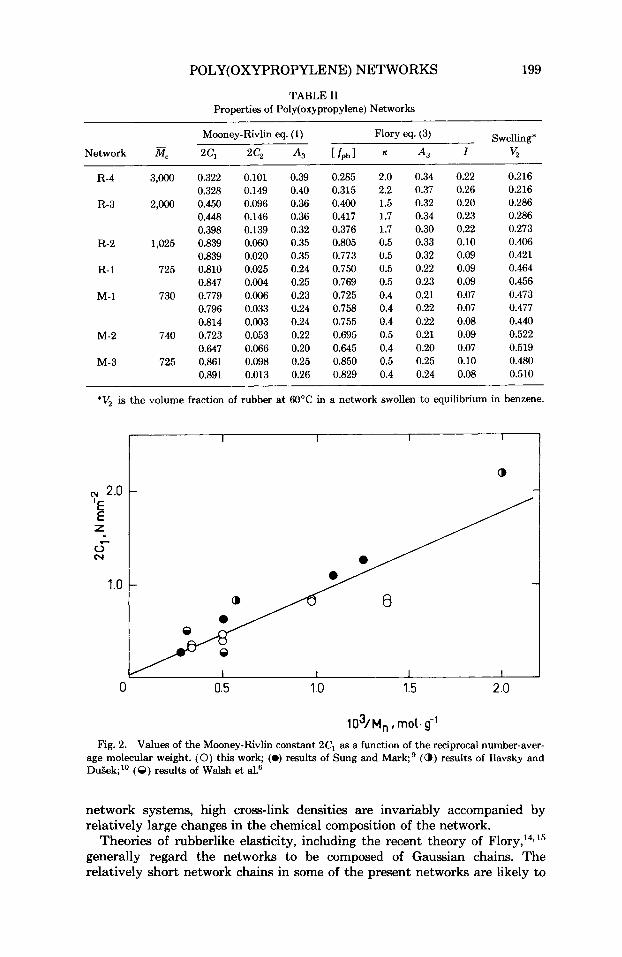
The values of 2% for the different networks show a linear dependence on the reciprocal of M, as expected on the basis of eq. (2). Figure 2 shows a comparison of the 2C1 values for the networks, with the values obtained by other workers for poly(oxypropy1ene)-based networks of comparable cross-link densities. The agreement is quite good considering the differences in the densities and in the junction-point chemistry of the different sets of networks compared. This plot further illustrates, in agreement with previous observa- tions on similar networks, the lack of a significant intercept in such a plot. The existence of such an intercept has been attributed to a contribution to the modulus by trapped chain
The low values of moduli and structure factors associated with the highly cross-linked networks (He = 725) are possibly a result of the altered chemical nature of polymers due to incorporation of relatively large amounts of the cross-linking agent. On a weight-fraction basis, the content of the cross-linking agent moieties in these networks is close to 25%! In polyurethane model
POLY(OXYPR0PYLENE) NETWORKS 199
TABLE I1 Properties of Poly(oxypropy1ene) Networks
I /
Flory eq. (3) Swelling* Mooney-Rivlin eq. (1)
Network Bc 2C1 2C2 A, [/,,,I K A, I v, R-4 3,000 0.322
0.328 R-3 2,000 0.450
0.448 0.398
R-2 1,025 0.839 0.839
R-1 725 0.810 0.847
M-1 730 0.779 0.796 0.814
M-2 740 0.723 0.647
M-3 725 0.861 0.891
0.101 0.39 0.149 0.40 0.096 0.36 0.146 0.36 0.139 0.32 0.060 0.35 0.020 0.35 0.025 0.24 0.004 0.25 0.006 0.23 0.033 0.24 0.003 0.24 0.053 0.22 0.066 0.20 0.098 0.25 0.013 0.26
0.285 2.0 0.315 2.2 0.400 1.5 0.417 1.7 0.376 1.7 0.805 0.5 0.773 0.5 0.750 0.5 0.769 0.5 0.725 0.4 0.758 0.4 0.755 0.4 0.695 0.5 0.645 0.4 0.850 0.5 0.829 0.4
0.34 0.37 0.32 0.34 0.30 0.33 0.32 0.22 0.23 0.21 0.22 0.22 0.21 0.20 0.25 0.24
0.22 0.216 0.26 0.216 0.20 0.286 0.23 0.286 0.22 0.273 0.10 0.406 0.09 0.421 0.09 0.464 0.09 0.456 0.07 0.473 0.07 0.477 0.08 0.440 0.09 0.522 0.07 0.519 0.10 0.480 0.08 0.510
~~
*V2 is the volume fraction of rubber at 60°C in a network swollen to equilibrium in benzene.
% 2.0 E
0-
z
h(
1.0
I I
Y I I 1 1
0 0.5 1.0 1.5 2 .o
103/M,, mol. g-' Fig. 2. Values of the Mooney-Rivlin constant 2C1 as a function of the reciprocal number-aver-
age molecular weight. (0) this work; (0) results of Sung and Mark9 (a,) results of Ilavsky and DuBek;" (8) results of Walsh et al.6
network systems, high cross-link densities are invariably accompanied by relatively large changes in the chemical composition of the network.
Theories of rubberlike elasticity, including the recent theory of Flory,l4, l5 generally regard the networks to be composed of Gaussian chains. The relatively short network chains in some of the present networks are likely to

200
% E
t. z- 0.8 c
u
ANDRADY AND LLORENTE
'-O- L
R- 2 - R-1 -
R- 4
-
R- 4
0.2 I 1 I I 1
h
0.0 0.2 0.4 0.6 0.0 1.0
Gt-'
Fig. 3. Typical stress-strain results fitted with the molecular theory of F10ry.l~ The theoret- ical lines were calculated with the parameters K given in Table I1 and 5 = 0 (see text).
be non-Gaussian. Thus, the networks dealt with here do not afford an ideal system for testing the recent theories of elasticity, in spite of the advantage of being well characterized and having a relatively wide range of cross-link densities. It is, nevertheless, of interest to determine the extent to which the theory is applicable to highly cross-linked systems used in the present work.
According to the recent theory of rubber elasticity, the uniaxial stress-strain data in Figure 1 might be fitted with a nonlinear function, as shown in Figure 3. The procedure for analysis of the data, described fully else~here, '~ involves the fitting of the nominal reduced stress [ f *] and the reciprocal of extension with a curve uniquely defined by the parameters K and [ fJ. The parameter c, generally set at zero for model network systems, is relatively unimportant. The theory itself rests on the assumption that the deviations in the elastic properties of real networks from those of ideal phantom networks are a result of the strain-dependent constraints placed on the junction-point fluctuations by the copious interpenetration of the network chains. The parameter K is a measure of the severity of such constraints. The values of K and [ f *] which best fit the data are listed in Table 11. The relationship between ths pair of parameters may be derived following F10ry.l~ Taking into account the func- tionality 3 (as opposed to 4 in Flory's work) in the networks used in the present study, the following corresponding equation can be derived:
9h
Here N is Avogadro's number, I is a constant, ( r 2 ) o is the mean square

POLY(OXYPR0PYLENE) NETWORKS
z - Y *,a
0.5
201
.
1.0 1 I 1 1
0 0.5 1 .o EkT/V,,N.m-2
Fig. 4. Limiting values of the nominal reduced stress against [kT/V, from the molecular weight of the chains.
length of an unperturbed chain, M is the molecular weight, and kT have their usual meanings.
The good fit of the experimental data to the theoretical function when 5 is zero suggests the networks are relatively defect free.l49l5 Therefore the values of [ fpt] obtained from the plots in Figure 3 should correlate well with the cycle rank (or with the cross-link density) of the networks, calculated from known chemical cross-link densities of the networks, provided there is no contribution of trapped entanglements to the elastic stress. For a trifunctional network, it can be shown that
[kT pRT - - - - v, 3ac (4)
where is the cycle rank of the network, V, is the volume of the network in the reference state, and KT, RT have the usual meanings (note: 5 = 2p, where p is the cross-link density of the network). Figure 4 shows a plot of the data in Table I1 in the manner suggested by the foregoing discussion. It is clear - that the data (with the exception of highly cross-linked networks with M, 5: 725) agree with the linear relationship and show no significant intercept. Equation (4) identifies the cycle rank (and therefore the cross-link density) of the network with an expression which depends only on the chemical constitu- tion of the network. This can only be valid when the effective cycle rank of the network is not affected by physical processes such as specific network chain entanglements. The lack of a significant intercept in both Figures 2 and 4 suggests that this is indeed the case for the networks dealt with here. Flory has demonstrated the same for two other network system^.'^*^^
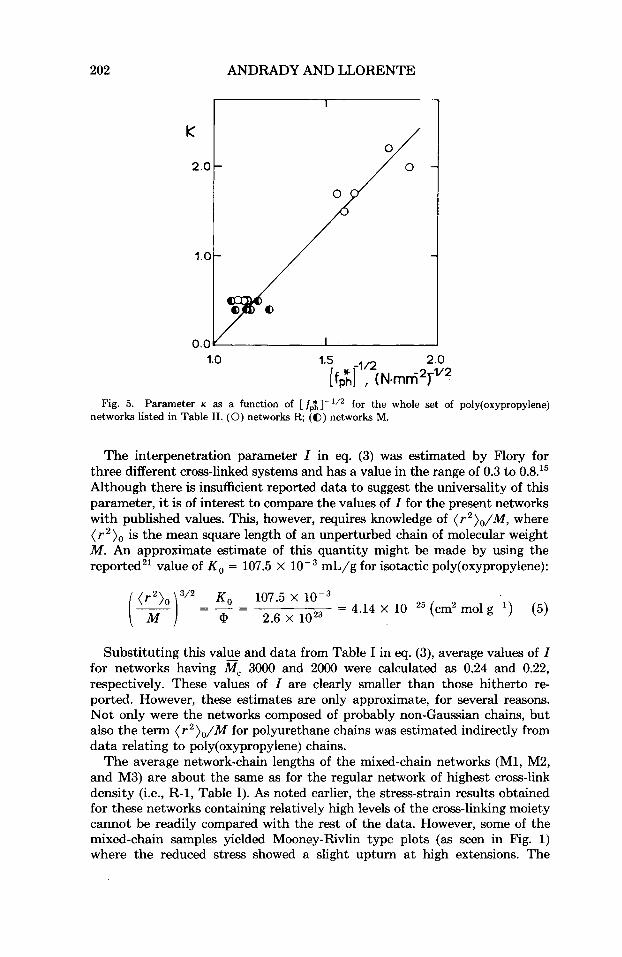
As might be expected on the basis of eq. (4), the parameter K (see Table 11) correlates reasonably well with [ fpt] with a correlation coefficient of 0.98 for the R series of networks. The data for all networks are shown in Figure 5. Some degree of scatter is inevitable a t low ac values where the network chains are markedly non-Gaussian, yielding smaller-than-expected values of K .
202 ANDRADY AND LLORENTE
1.0
Fig. 5. Parameter K as a function of [ fp1]- ' /2 for the whole set of poly(oxypropy1ene) networks listed in Table 11. (0) networks R; (@) networks M.
The interpenetration parameter I in eq. (3) was estimated by Flory for three different cross-linked systems and has a value in the range of 0.3 to 0.8.15 Although there is insufficient reported data to suggest the universality of this parameter, i t is of interest to compare the values of I for the present networks with published values. This, however, requires knowledge of ( T ~ ) ~ / M , where ( T ~ ) ~ is the mean square length of an unperturbed chain of molecular weight M . An approximate estimate of this quantity might be made by using the reported21 value of KO = 107.5 X mL/g for isotactic poly(oxypropy1ene):
( (z)0)3'2 - 107.5 X - = 4.14 X (cm2 mol gg') (5)
2.6 x 1023
Substituting this value and data from Table I in eq. (3), average values of I for networks having 3000 and 2000 were calculated as 0.24 and 0.22, respectively. These values of I are clearly smaller than those hitherto re- ported. However, these estimates are only approximate, for several reasons. Not only were the networks composed of probably non-Gaussian chains, but also the term (r2), /M for polyurethane chains was estimated indirectly from data relating to poly(oxypropy1ene) chains.
The average network-chain lengths of the mixed-chain networks (Ml, M2, and M3) are about the same as for the regular network of highest cross-link density (i.e., R-1, Table I). As noted earlier, the stress-strain results obtained for these networks containing relatively high levels of the cross-linking moiety cannot be readily compared with the rest of the data. However, some of the mixed-chain samples yielded Mooney-Rivlin type plots (as seen in Fig. 1) where the reduced stress showed a slight upturn a t high extensions. The

POLY(OXYPR0PYLENE) NETWORKS 203
ultimate extensions of such networks were relatively high in comparison to those for the regular networks of about the same aC. Similar behavior in poly(dimethylsi1oxane) networks". 23 has been explained in terms of limited chain extensibility of the short-chain component in a mixed-chain network.
The results from the equilibrium swelling experiments on the networks were also difficult to interpret by using the recent theories of swelling behavior because of the same limitations discussed in relation to the stress-strain data. An additional difficulty is that the interpretation of the data requires a knowledge of the polymer-solvent interaction parameter x, a t the relevant temperature, over the range of v2 (volume fraction of rubber in the swollen network) values observed. The value of this parameter used in the interpreta- tion of the present data is that reported by Booth and DevoyZ4 for the poly(oxypropylene)/benzene system a t 60°C.
The values of vz for the networks listed in Table I agree well with the values reported by Mark and Sungg for similar networks over a comparable range of crosslink densities. They interpreted their results using the version of Flory's theory of swelling available a t that time, which required calculating K values at each extent of swelling. Since K ( X ) was a function of parameters p and K as well as v2, reasonable estimates of these parameters were made ( p = 2 and K = 20) in the calculation. The following equation was then used to obtain the theoretical curve, which showed very good agreement with the data. In view of the agreement between the previously reported data and the present data on swelling measurements, the latter would also be consistent with the theory, subject to the selection of appropriate values of p , K, and F3( P, K, 02):
2/3 1/3 -F,PVV,, 0 2
In(1 - v2) + u2 + xv,Z ac =
Here, V is the molar volume of benzene at 60"C, and x is the polymer-solvent interaction parameter.
The most recent development of Flory's the0ryl47l5 does not require the estimation of parameters for interpretation of equilibrium swelling data. The K(X) values may be directly obtained from values of v2, and the values of K
and [ f J ] can be determined in the equilibrium stress-strain study. Then by equating the contributions to the chemical potential from mixing of polymer and diluent with that from the elastic deformation of the network and substituting [ f$ ] = SkTV,, it may be shown that
RT (ln(1 - v2) + v2 + xv;}h [ f * ] = --
ph V 1 + K(X') (7)
Thus the swelling data may be directly translated to values of [ fP;] by using bvailable stress-strain data and the relevant functional dependence of x on the degree of swelling v2. The present results when treated in this manner yield overestimates of [ fP$] compared to the values obtained by the stress- strain study and shown in Figure 3. The only exception was the most lightly cross-linked set of networks with ac = 3000, which yielded values of [ /$I,

204 ANDRADY AND LLORENTE
agreeing closely with the values suggested by the stress-strain study. The foregoing calculation, however, is h o d 5 to be sensitive to errors in the estimate of x used, leading to overestimates of [ f J ] , particularly at high cross-link densities where the system has a relatively large fraction of the polyurethane linkages. Furthermore, several previous studies on model net- works of poly(dimethylsi1oxane); systems strongly suggest that the shorter network chains contribute even less to the swelling response than they do to the modulus in uniaxial e ~ t e n s i o n . ~ ~ - ~ ~ This might, at least in part, explain the discrepancy between the equilibrium stress-strain data and the swelling data for the present networks.
Thanks are due t o Mr. H. Perucha for his technical assistance.
References 1. J. E. Mark, in Elastomers and Rubber Elasticity, J. E. Mark and J. Lal, Eds., American
2. M. A. Llorente and J. E. Mark, Macromolecules, 13, 681 (1980). 3. J. E. Mark, Rubber Chem. Technol., 55, 762 (1982). 4. A. L. Andrady, M. A. Llorente, and J. E. Mark, J . Chem. Phys., 72, 2282 (1980). 5. A. L. Andrady, M. A. Llorente, M. A. Sharaf, R. R. Rahalkar, J. E. Mark, J. L. Sullivan,
6. D. J. Walsh, J. S. Higgins, and R. H. Hall, Polymer, 20, 951 (1979). 7. A. L. Andrady and M. D. Sefcik, J. Polym. Sci. Polym. Phys. Ed., 21, 2453 (1983). 8. A. L. Andrady and M. D. Sefcik, J. Appl. Polym. Sci., 29(11), 3561 (1984). 9. P. H. Sung and J. E. Mark, J. Polym. Sci. Polym. Phys. Ed., 19, 507 (1981).
Chemical Society, Washington, DC, 1982.
C. U. Yu, and J. R. Falender, J. Appl. Polym. Sci., 26, 1829 (1981).
10. M. Ilavsky and K. Dusek, Polymer, 24, 981 (1983). 11. 0. G. Tarakanov and N. A. Vakhtina, Sint. Fiz-Khim. Polim., 21, 28 (1977). 12. L. R. G. Treloar, The Physics of Rubber Elasticity, 3rd ed., Clarendon Press, Oxford, 1975. 13. M. J. Mooney, J. Appl. Phys., 19, 434 (1948). 14. P. J. Flory and B. Erman, Macromolecules, 15, 800 (1982). 15. P. J. Flory, Polymer J., 17, 1 (1985). 16. R. J. Morris and H. E. Persinger, J. Polym. Sci. A , 1, 1041 (1963). 17. W. Langley, Macromolecules, 1, 348 (1968). 18. D. S. Pearson and W. W. Greassley, Macromolecules, 13, 1001 (1980). 19. B. Erman, W. Wagner, and P. J. Flory, Macromolecules, 13, 1554 (1980). 20. J. E. Mark and J. L. Sullivan, J . Chem. Phys., 66, 1006 (1977). 21. Polymer Handbook, Section IV-24, 2nd ed. J. Brandup and E. H. Immergut, Eds.,
22. M. A. Llorente, A. L. Andrady, and J. E. Mark, J. Polym. Sci. Polym. Phys. Ed., 19, 621
23. M. A. Llorente, A. L. Andrady, and J. E. Mark, Colloid and Polymer Sci., 259, 1056 (1981). 24. C. Booth and C. J. Devoy, Polymer, 12, 320 (1971). 25. J. R. Falender, G. S. Y. Yeh, and J. E. Mark, J . Am. Chem. SOC., 101, 7353 (1979). 26. J. R. Falender, G. S. Y. Yeh, and J. E. Mark, Macromolecules, 12, 1207 (1979). 27. M. A. Llorente and J. E. Mark, Rubber Chem. Technol., 53(5), 988 (1980).
Wiley-Interscience, 1974.
(1981).
Received March 7, 1986 Accepted May 22, 1986





























